
Transduction, Expression, and Secretion of Human Glucocerebrosidase by Murine Myoblasts
14
Transduction, Expression, and Secretion of Human Glucocerebrosidase by Murine Myoblasts VIJAYA BANSAL,%b PATRICIA MOWERY-RUSHTON, LORRIE LUCHT," JUAN LI," ALFRED BAHNSON," SIMON C. WATKINS,' AND JOHN A. BARRANGER&" aDeparhnent of Human Genetics Graduate School of Public Health bDeparhnent of Reproductive Genetics w e e Ww's Hospital and CDeparhnent of Neumbiolom Unbwrity of Pittsburgh Pimbutyh, Penqlvania 15261 INTRODUCTION Gaucher disease is caused by deficiency of a lysosomal enzyme, glucocere- brosidase, which hydrolyzes glucosylceramide to glucose and ceramide. Defi- ciency leads to abnormal lysosomal storage of glucosylceramide primarily in macrophages. Consequently, the disease is manifested by hepatosplenomegaly and painful bone lesions with (type I1 and 111) or without (type I) neurologc involvement. Currently, there are two accepted treatments for Gaucher disease, enzyme replacement and allogeneic bone marrow transplantation. Early studies on en- zyme replacement using the mature native form of enzyme were disappointing until the enzyme was modified to target the macrophages. To achieve this targeting, the oligosaccharide side chains were enzymatically cleaved by exo- glycosidases to expose nacent mannose residues for specific binding to mannose receptors on macrophages. This alteration led to a tenfold increase in enzyme delivery to Kupffer cells in animal experiments.' Administration of large amounts of this preparation has produced clinical improvement in patients, resulting in regression of organomegaly and improvement in blood counts. "Address for correspondence: John A. Barranger, M.D., Ph.D., Department ofHuman Genetics, University of Pittsburgh, El650 Biomedical Science Tower, Pittsburgh, Penn- sylvania 15261. 307
Transcript of Transduction, Expression, and Secretion of Human Glucocerebrosidase by Murine Myoblasts

Transduction, Expression, and Secretion of
Human Glucocerebrosidase by Murine Myoblasts
VIJAYA BANSAL,%b PATRICIA MOWERY-RUSHTON, LORRIE LUCHT," JUAN LI,"
ALFRED BAHNSON," SIMON C. WATKINS,' AND JOHN A. BARRANGER&"
aDeparhnent of Human Genetics Graduate School of Public Health
bDeparhnent of Reproductive Genetics w e e W w ' s Hospital
and CDeparhnent of Neumbiolom
Unbwrity of Pittsburgh Pimbutyh, Penqlvania 15261
INTRODUCTION
Gaucher disease is caused by deficiency of a lysosomal enzyme, glucocere- brosidase, which hydrolyzes glucosylceramide to glucose and ceramide. Defi- ciency leads to abnormal lysosomal storage of glucosylceramide primarily in macrophages. Consequently, the disease is manifested by hepatosplenomegaly and painful bone lesions with (type I1 and 111) or without (type I) neurologc involvement.
Currently, there are two accepted treatments for Gaucher disease, enzyme replacement and allogeneic bone marrow transplantation. Early studies on en- zyme replacement using the mature native form of enzyme were disappointing until the enzyme was modified to target the macrophages. To achieve this targeting, the oligosaccharide side chains were enzymatically cleaved by exo- glycosidases to expose nacent mannose residues for specific binding to mannose receptors on macrophages. This alteration led to a tenfold increase in enzyme delivery to Kupffer cells in animal experiments.' Administration of large amounts of this preparation has produced clinical improvement in patients, resulting in regression of organomegaly and improvement in blood counts.
"Address for correspondence: John A. Barranger, M.D., Ph.D., Department ofHuman Genetics, University of Pittsburgh, El650 Biomedical Science Tower, Pittsburgh, Penn- sylvania 15261.
307

308 ANNALS NEW YORK ACADEMY OF SCIENCES
However, the treatment is expensive. Enzyme infusion at a dose of 60 units per kilogram of body weight costs $14,700 per infusion for a 70-kg adult. At the recommended frequency of one infusion every 2 weeks, the annual cost per patient for the enzyme alone is $382,200. The alternative treatment of allogeneic bone marrow transplantation is unavailable to most patients, as only 25 to 30% of patients have an =-matched sibling. It is also a high-risk procedure, with associated mortality varying from 5 to 40%. The availability of a cDNA and efficient vectors makes somatic gene therapy an exciting prospect for Gaucher disease. The target cells for gene transfer are hematopoietic stem cells with the potential to provide a long-term source of enzyme-competent macrophages in the patient.
An alternative approach is a cellular transport system that can stably produce and deliver the recombinant enzyme into the systemic circulation, where it would be available for uptake by macrophages, the pathobiologically important cell in Gaucher disease. Normally, glucocerebrosidase is synthesized as a single polypeptide (58 kDa) with a signal sequence (2 m a ) at the amino terminal.2 Cotranslationally the signal sequence is cleaved off and the enzyme is core glycosylated. This “high mannose” precursor is posttranslationally processed in the Golgi apparatus to a 66-kDa intermediate and further tailored in the lysosome to the mature enzyme (59 m a ) . The mature enzyme is membrane bound, and very little is secreted extracellularly. However, when a cell is produ- cing enzyme at a rate several times normal, as could be expected of a successfidly transduced cell with a high copy number, the posttranslational processes of oligosaccharide side chain modification and membrane incorporation would be overloaded, leading to extracellular secretion of the surplus enzyme, a signif- icant fraction of which may be expected to be in the “high mannose” precursor form.
The ideal recombinant protein delivery system would use a cell that can be easily isolated from the recipient, grown in culture, transduced in vim, and reimplanted in the host. These genetically modified cells should survive for long periods of time and synthesize large quantities of the recombinant protein avail- able to the pathobiologically important sites. Myoblasts are an excellent candi- date for such a system. Myoblasts can be readily isolated from muscle biopsies, expanded in culture, and transduced in pitro.3 Myoblasts injected intramuscularly survive and fuse into adjacent muscle fibers at the site of injection.* As skeletal muscle is highly vascular, proteins secreted from myoblasts readily enter the circulation. In vim and in vivo expression of the transferred gene have been demonstrated in mouse myoblast cell line C2C12.536
To explore the feasibility of developing a “myogenic organoid” for use in Gaucher disease, we transduced murine C2C12 myoblasts with human GC cDNA using a replication-incompetent retrovirus vector, MFG-GC. The trans- duced myoblasts were transplanted syngeneically into C3H mice for studies of in vivo expression. Here we report the efKciency of the retrovirus vector MFG- GC in transducing murine myoblasts with the human glucocerebrosidase gene, and expression of the transferred gene in vim and in vivo, and in extracellular secretion of the recombinant enzyme.

BANSAL et al.: MURINE MYOBLASTS 309
MATERIALS AND METHODS
Animals and Cell Culture Conditions
C2C12 cells (obtained from ATCC) are clonally derived myoblasts from leg skeletal muscles of an adult C3H mouse and have all the characteristics of myoblasts. C2C12 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 20% (vol/vol) fetal bovine serum, 1 % glutamine, penicillin (50 units/ml), and streptomycin (50 ~g/ml). The retroviral packaging cell lines psi-cre and psi-crip and the cell line NIH 3T3 were maintained in DMEM supplemented with 10% fetal bovine serum, 1% glutamine, and 1% pen/strept. Adult C3H mice (Jackson Laboratories, ME), 6 to 8 weeks old were used for syngeneic transplantation of MFG-GC-infected C2C12 myoblasts.
Vector Construction
The vector MFG-GC is a retrovirus derived from the moloney murine leu- kemia virus in which the polymerase (pol) and envelope (env) sequences are deleted to render it incapable of replication. Gag gene sequences up to base 1035 have been retained to increase the packaging efficiency of the unspliced tran- script. Viral donor and acceptor sites are utilized for mRNA splicing. (The MFG retrovirus vector was constructed by P. Robbins and B. Guild in R. C. Mulligan’s laboratory, Whitehead Institute, Cambridge, MA.) The human GC cDNA was cloned into the MFG vector such that the translational start site for the GC cDNA is precisely at the start site of the deleted env gene at the Nco I site. The inserted GC cDNA is transcribed from promotor/enhancer sequences in the retroviral long terminal repeat (LTR). The MFG-GC vector was cloned by T. Ohashi (University of Pittsburgh, Pittsburgh, PA), and the authenticity of the construct has been verified by DNA sequencing, which revealed no PCR or cloning artifacts. The proviral structure of the vector is shown in FIGURE 1.
Production of Virus-Containing Supernatant
The MFG-GC plasmid was cotransfected with pSV2neo to the ecotropic packaging cell line psi-cre at a molecular ratio of 20:l by calcium phosphate coprecipitation. After G418 selection (400 pg/ml), 30 clones were isolated and grown to near confluence in 100-mm dishes. The supernatant from these clones was titrated by targeting NIH 3T3 cells. Culture media from psi-cre clone #4 with a viral titer of 1 x lo6 colony-forming units (&)/ml was used to infect the amphotropic producer cell line psi-crip. Following titration by NIH 3T3 cell targeting, the clone cc2 with a viral titer of 1 x lo7 &/ml was used in these experiments. Both the ecotropic producer psi-cre #4 and amphotropic producer cc2 were tested for the presence of helper virus using the BAG mobilization assay as Briefly, the indicator line (3T3-BAG) was prepared by trans-

310 ANNALS NEW YORK ACADEMY OF SCIENCES
X S K N B X S K **
N w GCcDNA
~~
1.7 W _ _ ... -. -. -. -. ~ ~
i x : m I S D : W m :S:Sstl scl.#p/b&xep& : rc.@nr ~ Nd:Nde/ i N : W I ! B:BamHI ._ _ _ _ _ . . _.
FIGURE 1. Provirus structure of MFG-GC. Human GC cDNA was cloned into the MFG plasmid by creating a NcoI(N) site in a 79-bp PCR product, usin the full-length cDNA as the template. A NcoI-~indl l~ fragment was ligatexi to a Hin&-Bcn fragment from the human cDNA and then cloned into a NcoI(N)-BarnHI(B) site in the MFG vector.
ducing NM-3T3 cells with the BAG virus, a Moloney murine leukemia virus containing both the bacterial @-galaaosidase gene at the viral gag gene (@-gal at gag or BAG) and the neomycin resistance gene, neo. The 3T3-BAG cells (2 x lo5) were cocultured with supernatants from the viral producer line (psi-crip #cc2 or psi-cre #4) in 10-cm tissue culture dishes. Supernatants from the BAG cocultures were analyzed for the presence of helper virus by infecting fresh 3T3 cells, which were assayed for lacZ expression by 5-bromo-4-chloro-3-indolyl B-D-galactoside (X-Gal) staining and for neo resistance by replating 5 x lo5 cells in the presence of G418 at 400 &mI. Both were found to be free of helper virus.
Transduction of C2C12 Myoblasts with MPG-GC
Myoblasts (1 x lo5) were plated on a tissue culture dish (60 mm x 15-mm) in DMEM with 4.9 g/l glucose, 10% fetal bovine serum, 1% glutamine, and 1% penicillin/streptomycin and incubated for 24 h at 3T" C in 5% C02. The media was then replaced with 1 ml of virus-containing supernatant (VCS) and poly- brene added to a final concentration of 8 &d. The plates were incubated at 37" C in 5% C 0 2 for 2 h and gently shaken every 15 min to ensure uniform dispersal of the retrovirus vector. After 2 h VCS was removed and replaced with fresh culture media. Infection was conducted in triplicate with two infections in 2

BANSAL et al.: MURINE MYOBLASTS 311
days, three infections in 2 days, and four infections in 2 days each. Infection with supernatant from the amphotropic producer of MFG-lacZ and non-virus-con- taining supernatant from NIH 3T3 cells were used as controls.
Southern Blot
Genomic DNA was extracted by proteinase K treatment, phenol extraction, and ethanol precipitation. DNA was digested with Sst I, which cuts in both LTR regions of the construct and produces a 4.4-kb band. Following electrophoresis in 1% agarose DNA was blotted onto a nylon membrane and probed with radiolabeled human GC cDNA. Hybridization and washing were performed using standard techniq~es.~ A known amount of plasmid DNA from MFG-GC mixed with the control animal genome was used for copy number estimation by comparison of band intensities per blot. Ten picograms of the plasmid DNA equals one copy of the human gene.
Pofymerase Chain Reaction
DNA samples from fresh or frozen pellets of lo5 cells or less were prepared as described by Kim and Smithies. lo Ten microliters of the extract was added to the PCR reagent mixture, with a final volume of 50 pl. The final PCR mixture contained 200 $t4 each dNTP, 2.5 units Taq polymerase, 0.2 $t4 of each primer in a commercial buffer (Perkin-Elmer), 22.5 mM MgCI,, and 7% DMSO. The primers hybridize one in the GC cDNA region and the other to the viral se- quence, to yield a unique 403-bp amplification product (AB1: 5' ACG GCA TCG CIT GGA TA 3'; AB2: 5' AGT AGC AAA T'IT TGG GCA GG 3'). Thermal cycling was performed on Gene Amp PCR system 9600 as follows: 94" C x 10 min to denature followed by 94" C x 45 min, 58" C x 2 min/30 cycles. The PCR products were separated in a 2% agarose gel in the presence of ethidium bromide and the band visualized under W light. To detect smaller quantities, a Southern blot was prepared and hybridized with a radiolabeled human GC cDNA probe.
Immunochemical Staining
Immunocytochemical staining of the C2C12 myoblasts and immunohisto- chemical staining of tissues from transplanted animals was performed with the Vecta stain ABC kit (Vector Laboratories) according to the manufacturer's instructions. Briefly, slides (cytospins and frozen sections) were washed twice in 1 x phosphate-buffered saline (PBS) and thrice in bovine serum albumin (BSA) solution (0.5 g bovine serum albumin, 0.15 g glycine in 100 ml PBS) in a humidity chamber. Nonspecific binding sites were blocked with 20% goat serum for 30 min followed by goat antimouse IgG (150 dilution) for another 30 min and then washed thrice in BSA solution. Slides were then incubated with the primary antibody, 8E4, ( 1: 1000 dilution in BSA solution) in a humidity cham-

312 ANNALS NEW YORK ACADEMY OF SCIENCES
ber at room temperature for 60 min or overnight at 4"C, washed with BSA solution, then incubated with the secondary antibody, biotinylated antimouse IgG, for 60 min, washed again with BSA, incubated with 0.1% H202 in meth- anol for 30 min, and washed again with 1 x PBS. Slides were then exposed to Vectastain ABC reagent at RT for 60 min, washed with 1 x PBS, and then incubated with the peroxidase substrate DAB (diamino benzidine tetrahydro- chloride) for 5 min. Finally, counterstaining was done with 1% toluidine blue.
hunofluorescent Staining
Frozen sections were fixed and held in PBS to prevent drymg. Nonspecific binding sites were blocked with 5% goat serum at room temperature for 30 min. Monoclonal antibody 8E4-L, flouorolabeled with C43 fluorochrome, was then added dropwise, and slides were incubated at room temperature for 60 min in a humidity chamber, followed by waslung three times with 1 x PBS.
Enzyme Activity Assay
Cells and tissues were rinsed flee of serum with PBS, frozen rapidly, and stored at -80°C until analysis. For the assay, cells were lysed with cold potassium phosphate buffer (pH 6.5) containing 2.5 m g / d Triton X-100; ultrason- ification followed, then centdigation at 4°C for 15 min. Supernatant was anal- yzed for enzyme activity by incubating one part lysate with three p m synthetic substrate, 4-methylumbelliferyl glucopyranoside (Sigma Chemical Co.) at 10 mM in citric acid-sodium phosphate (0.12 M) buffer (pH 5.4) containing 2.5 m g / d sodium taurocholate, 2 mg/ml Triton X-100, and 10 mg/ml bovine serum albumin, at 37°C for 30 min. Glycine-carbonate buffer (0.17M, pH 10.4) was added to terminate the reaction, and 4methylumbelliferone, the product of the enzymatic reaction, was measured with a fluorimeter. Protein concentration in the sample was measured using bicinchoninic acid (Pierce). Glucocerebrosi- dase activity is expressed in nmoles per hour mg of protein (U/mg).
Immunoblot
Western blots were done by standard techniques.8 Human GC protein was detected by immunostaining with monoclonal antibody (mAb) 8E4 as the pri- mary antibody and alkaline phosphatas-onjugated antimouse IgG goat anti- body (Bio-Rad) as the second antibody. The 8E4 mAb is specific for human GC and does not cross-react with endogenous mouse GC.11,12
Mpbfast Implantation
Six- to eight-week-old adult C3H mice were anesthetized by intraperitoneal injection of 0.015-0.017 ml/g body weight of 2.4% Avertin (2,2,2,mbromo-

BANSAL et af.: MURINE MYOBLASTS 313
ethyl alcohol) in PBS. The dorsal surfaces of both hind h b s were shaved and prepped with 75% ethanol. The transduced C2C12 myoblasts were harvested well before confluence and differentiation into myotubes, were suspended at a concentration of 2 x lo7 cells per ml of serum-free media, and were loaded into a 1-ml tuberculin syringe. Posterior thigh muscles were exposed via a small skin incision, and 250 4 of cell suspension was injected by multiple injections into each hind leg. Skin was closed with surgical steel clips. Transplanted mice were monitored for adverse reactions and euthanized 10 days or 8 weeks after trans- plantation. Muscle from injection sites was examined by Southern blot and immunohistochemistry for human GC cDNA carriage and in Pivo expression.
RESULTS
Transduction of C2C12 Myoblasts with MFG-GC
To evaluate the efficiency of transduction with human GC cDNA, three separate C2C12 myoblast cultures were infected with MFG-GC. MFG-lacZ infection and mock infection with non-virus-containing supernatant from NIH 3T3 cells were used as controls. Southern blot analysis of C2C12 cells following infection is shown in FIGURE 2. Human GC cDNA was detected in all three of
1 2 3 4 5 6 7
FIGURE 2. Southern blot analysis ofC2C12 murine myoblasts following transduction with MFG-GC. Human GC cDNA was used as probe. A known amount of MFG-GC plasmid mixed with control mouse genome was run simultaneously, and copy number was estimated by comparison of band intensities (10 picograms of plasmid DNA e uals one co y of the human GC gene per cell). Lane 1. MFG-GGtransduced myobqasts infecteg once per day for two days. Lane 2. MFG-GC-transduced myoblasts infected thrice over two days. Lane 3. MFG-GC-transduced m oblasts infected twice per day for two days. Lane 4. Uninfected C2C12 murine myobi/asts. Lane 5. MFG-GC plasmid DNA representing one copy of the human GC gene per cell. Lane 6. Plasmid DNA representing two co ies per cell. Lane 7. Plasmid DNA representing three copies of the human gene per celf:

3 14 ANNALS NEW YORK ACADEMY OF SCIENCES
the MFG-GC clones and in none of the controls. Copy number was estimated at greater than 10 copies of the transferred human GC gene per cell.
In Vim Expression of the T k a n s M Human GC cDNA
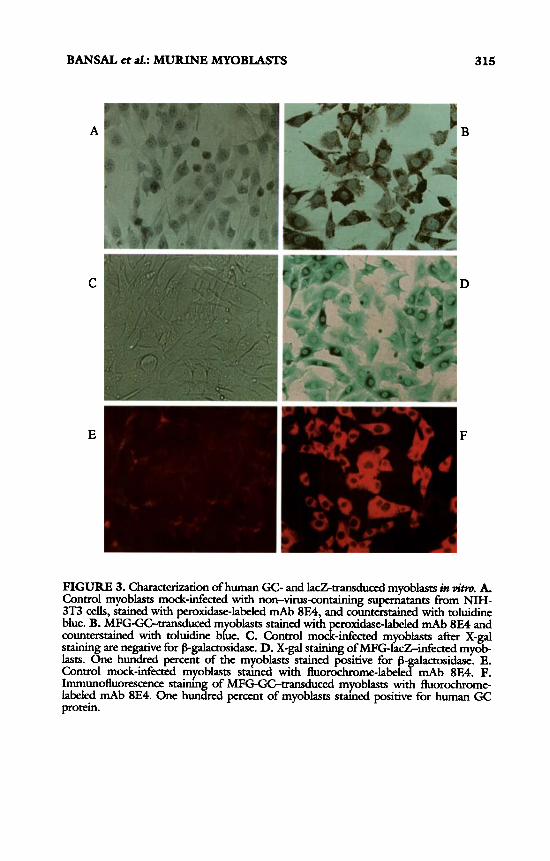
Expression of human glucocerebrosidase gene was examined by immun- Ocytochemical staining and intracelldar enzyme activity assay. On immunostain- ing with 8E4, and mAb specific for human glucocerebrosidase protein without sigdicant cross-reaction with endogenous mouse protein, 100% of the MFG- GC-infected myoblasts stained positive for the recombinant human protein (FIG. 3) . Similarly, MFG-IacZinfected myoblasts all stain positive with X-gal staining, whereas the control cells mock-infected with non-virus-containing su- pernatants were consistently negative. The MFG-GC-transduced myoblasts were next analyzed for glucocerebrosidase activity. As sh own in FIGURE^, intracelldar enzyme activity was 10-fold greater in MFG-GGtransduced myob- lasts as compared to MFG-lacZ-transduced or md-infected control cells as well as uninfected normal human or C2C12 mucine myoblasts. Thus viral LTR promotor-controlled expression of glucocerebrosidase and B-galactosidase genes is robust in myoblasts.
In Vivo Expression of Human Glucocerebmidase
Mice transplanted with MFG-GC-transduced myoblasts were euthanized 10 days or 8 weeks after transplantation. Hind limb muscle from the injection site was stained with fluorochrome-labeled mAb 8E4. Human glucocerebrosidase expression was observed in muscle from all the mice transplanted with h4FG- GC-transduced myoblasts and in none of the control animals injected with mock-infected myoblasts. As seen in FIGURE 5, myoblasts expressing recombi- nant human glucocerebrosidase protein were seen at both 10 days and 8 weeks. Examination of muscle distant from the injection site fded to reveal GC-ex- pressing cells, suggesting that the transduced myoblasts remain localized at injection sites. No tumorigenic overgrowth of transplanted myoblasts was ob- served.
Extracellular Secretion of Recombinant Glucocere6rasida.w
Cell culture media was collected from MFG-GC-transduced myoblasts as well as from control cell cultures and analyzed for enzyme activity and, by Western blot, for the presence of glucocerebrosidase protein (59-63 m a ) . As expected, very little GC activity was measured in supernatants from normal human or murine C2C12 myoblasts, normal human or Gaucher fibroblasts, or from MFG-IacZinfected or mock-infected (infected with non-virus-containing
BANSAL et af.: MURINE MYOBLASTS
A
C
E
B
D
F
315
FIGURE 3. Chcterization of human GC- and IacZtransdd myoblasts in vim. A. Control myoblasts mock-infected with non-virus-containing supernatants from N M - 3T3 cells, stained with peroxidase-labeled mAb 8E4, and counterstained with toluidine blue. B. MFG-GC-transduced m oblasts stained with roxidase-labeled mAb 8E4 and counterstained with toluidine b&e. C. Control mocf&kted myoblasts after X-gal stainin are negative for f5-galactasidase. D. X-gal staining of MFG-IacZinfected myob- lasts. %ne hundred percent of the myoblasts stained positive for f5 actosidase. E. Control mock-infected myoblasts s m e d with fluorochmrne-label P mAb 8E4. P. Immunofluorescence s m h n of MFG-GC-transduced myoblasts with fluorochrome- labeled mAb 8E4. One hunkd percent of myoblasts s m e d positive for human GC protein.

316 ANNALS NEW YORK ACADEMY OF SCIENCES
Hu c2cl2 c2Cu c2cl2 C2CU MY0 m Mz cc2
FIGURE 4. Intracellular glucocerebrosidase enzyme activity of myoblasts. Hu Myo: Activity in uninfected human myoblasts. C2C12: GC activity in uninfected C2C12 murine myoblasts. C2C12 3T3: Activity in C2C12 myoblasts modc-infected with super- natants from NIH-3T3 cells. C2C12 MZ: Activity in C2C12 m oblasts transduced with MFG-lacZ. C2C12 CC2: Activity in C2C12 myoblasts infectecywith supernatants from MFG-GC producer cells, psi-crip clone #cc2.
supernatant from NM-3T3 cells) C2C12 myoblasts. However, the MFG-GC- infected C2C12 myoblasts producing large quantities of glucocerebrosidase in- tracellularly (10-fold higher) did, in fact, exhibit a sigdicant amount of gluco- cerebrosidase activity in the cell culture media (FIG. 6). This was further confirmed when cell culture supernatants were examined by immunoblot. As shown in FIGURE 7, a protein with 59-63-kDa molecular mass, consistent with
A B
FIGURE 5. Immunofluorescent staining with fluorochrome labeled mAb 8E4 of mouse skeletal muscle injected with MFG-Gtransduced myoblasts. A. Muscle from injection sites 8 weeks after transplantation with 5 x 106 myoblasts mock-mfected wlth super- natants from NIH-3T3 cells. B. Muscle at injection sites from mice transplanted with MFG-GGtransduced myoblasts, 8 weeks after injection.

BANSAL et al.: MURINE MYOBLASTS 317
Hu HuPib Gau C2Cl2 C2Cl2 C2Cl2 C2Cl2 MY0 Fb 3T3 M-Z CCZ
FIGURE 6. Extracellular secretion of glucocerebrosidase by MFG-GGtransduced myoblasts. Levels of GC activity in supernatants from uninfected human myoblasts (Hu Myo), uninfected human fibroblasts (Hu Fib), uninfected Gaucher fibroblasts (Gau Fib), uninfected C2C12 myoblasts (C2C12), C2C12 myoblasts mock-infected with NIH-3T3 supernatants (C2Cl2 3T3), C2C12 myoblasts transduced with MFG-lacZ (C2C12 M- Z), and C2C12 myoblasts uansduced with MFG-GC (C2C12 CC2).
the molecular mass of different forms of glucocerebrosidase, was seen. As serum in the media contains albumin with a molecular mass of 66 ma, the supernatants were fractionated with Aftigel blue to remove albumin and were reexamined by Western blot. As can be seen in FIGURE 7, a residual band of fair intensity was still visible, signifying extracellular secretion of glucocerebrosidase by the GC- transduced myoblasts.
1 2 3 4 5 6 7 8
FIGURE 7. Immunoblot analysis for Gc protein in the cell culture supernatants. Lane 1. Glucocerebrosidase e m e. Lane 2. GC-transduced myoblasts. Lane 3. Mannose- terminated glucocerebrosiEe. Lane 4. Cell culture supernatants from uninfected myob- lasts. Lane 5. ( X U culture supernatants from GC-uansduced myoblasts. Lane 6. Man- nose-terminated glucocerebrosidase after Atfigel blue fractionation. Lane 7. GC-transduced media after AfKgel blue fractionauon. Lane 8. Nontransduced C2C12 media after Atfigel blue fractionation.

318 ANNALS NEW YORK ACADEMY OF SCIENCES
DISCUSSION
Development of a “myogenic organoid” of therapeutic benefit in Gaucher disease requires isolation and expansion of myoblasts in culture, stable trans- duction with glucocerebrosidase gene at a high copy number, vigorous long- term expression of the transferred gene, and systemic secretion of the recombi- nant protein in a form capable of efiicient receptor- (mannose receptors) mediated uptake by macrophages. In the initial experiments reported here, we demonstrated that myoblasts can be efficiently transduced with human GC cDNA using the retrovirus vector MFG-GC. The presence of the recombinant human protein in all of the cells, as shown by immunocytochemical staining, suggests a 100% transduction efficiency, with several copies of the gene per cell (as estimated from Southern blot analysis). As transduction efficiency of retro- virus vectors depends not only on the vector and its titer but also on the growth characteristics of the cells, use of a rapidly growing cell line, C2C12, may partly account for the very high transduction efficiency seen. We are currently testing the transduction efiiciency of this vector in primary myoblasts derived from murine and human sources.
MFG-GC has been previously demonstrated to cause sustained, high expres- sion of GC in hematopoietic cells.13 We now demonstrate that expression is simi- larly robust in myoblasts. In MFG-GC, the retroviral LTR promotor/enhancer sequences are used for the expression of human glucocerebrosidase. It has been reported that the retroviral LTR prornotor is gradually inactivated in PiPo in trans- planted skin f ibr~blasts .~~J~ However, in myoblasts, for unknown reasons, th is appears not to be the cause, at least for 3 4 m ~ n t h ~ . ~ ? ~ Our initial experiments confirm that expression is sustained for at least 8 weeks in Pipg. Further studies are planned to determine the duration of in PiPo expression of the human glucocere- brosidase gene. Extracellular secretion of recombinant glucocerebrosidase by the transduced myoblasts is critical to an organoid approach. The finding of an al- most 350-fold increment in glucocerebrosidase activity in the cell culture super- natant from MFG-GC-transduced myoblasts suggests that overproduction of glucocerebrosidase does, indeed, lead to its extracellular secretion.
The initial results from the experiments reported here demonstrate that retro- virus-mediated transduction of myoblasts can lead to high expression of gluco- cerebrosidase for at least 8 weeks, with sigdcant extracellular secretion of the recombinant protein, which is thus potentially available for uptake by macro- phages. Whether or not the secreted enzyme is taken up by macrophages in sficient quantities to ameliorate disease is not yet clear. Expression with ex- tracellular secretion in human primary myoblasts and longevity of the system need to be determined. Thus, several important questions remain to be answered before an organoid approach can be considered a valid therapeutic option for Gaucher disease.
REFERENCES
1. MURRAY, G. J., T. W. DOEBBER, M. S. Wu, M. M. POMPIPPOM, R. L. BUGIANESE, T. Y. SHEN & J. A. BARRANGER. 1985. Targeting of the synthdcally gl cosylated human placental glucocerebrosidase. Biochem. Med. Metab. Biol. 34: 141-245.

BANSAL et al.: MURINE MYOBLASTS 3 19
2. ERICKSON, A. H., E. I. GINNS& J. A. BARRANGER. 1985. Biosynthesis of the lysosomal enzyme glucocerebrosidase. J. Biol. Chem. 260: 14319-14324.
3. BLAU, H. & C. WEBSTER. 1981. Proc. Natl. Acad. Sci. USA 78: 5623-5625. 4. PARTRIGE, T. A., J. E. MORGAN, G. R. COULTON, E. P. HOFFMAN & L. M. KUNKEL.
1989. Conversion of mdx myofibers from dystro hin negative to -positive by injection of normal myoblasts. Nature 337: 176-1!9. -
5. DHAWAN, J., L. C. PAN, G. K. PALATH, M. A. TRAVIS, A. M. LANCLOT& H. M. BLAU. 1991. Systemic delivery of human growth hormone by injection of genet-
injection of genetically modified myobt)asts. Proc. Natl. Acad. Sci. USA 89: 3357- 3361.
7. DANOS, 0. & R. C. MULLIGAN. 1988. Safe and efficient generation of recombinant retroviruses with am hotropic and ecotropic host ranges. Proc. Natl. Acad. Sci. USA 85: 6460-6468
8. STANLEY, E. R. & P. M. HEARD. 1977. Factors regulating macrophage production and growth. Purification and some properties of the colony stimulating factor from medium conditioned by mouse L cells. 1977. J. Biol. Chem. 252: 4305- 4312.
9. AUSBEL, F. M., R. BRENT, R. E. KINGSTON, D. D. MOORE, J. G. SEDMEN, J. A. SMITH & K. STRUHL. 1989. In Current Protocols in Molecular Biology: 2.9.1- 2.9.10. Greene and Wdey. New York, NY.
10. KIM, H. S. & 0. SMITHIES. 1988. Recombinant fragment assay for gene targeting based on the polymerase chain reaction. Nucleic Acids Res. 16: 88874903.
11. OHASHI, T., C. M. HONG, S. WEILER, J. TOMICH, J. M. F. G. AERTS, J. T. TAGER & J. A. BARRANGER. 1991. Characterization of human glucocerebrosidase from dif- ferent mutant alleles. J. Biol. Chem. 266: 3661-3667.
12. BARNEVALD, R. A., F. P. W. TAGELAERS, E. I. GINNS, P. BISSER, A. LEANER, J. A. BARRANGER, R. 0. BRADY, A. J. J. REUSER, H. GALJAARD & J. M. TAGER. Mono- clonal antibodies against human beta-glucocerebrosidase. Eur. J. Biochem. 134:
13. OHASHI, T., S. Boccs, P. ROBBINS, A. BHANSON, K. PATRENE, F. S. WEI, J. F. WEI, L. Lucm, Y. FEI, S. CLARK, M. KIMAK, H. HUILING, P. MOWERY-RUSHTON & J. A. BARRANGER. Efficient transfer and sustained high expression of the human glucocerebrosidase gene in mice and their functional macrophages following trans- plantation of bone marrow transduced by a retroviral vector. 1992. Proc. Natl. Acad. Sci. USA 89: 11332-11336.
14. PALMER, T. D., A. R. THOMPSON& A. D. MILLER. Production of factor IX in animals by eneticdy modified skin fibroblasts: Potential therapy for hemophilia B. Blood 78: 4 3 8 4 5 .
15. PALMER, T. D., G. J. ROSMAN, W. R. A. OSBORNE&A. D. MILLER. 1991. Genet- ically modified s k i n fibroblasts persist longer after transplantation b u t r d d y inactivate introduced genes. Proc. Natl. Acad. Sci. USA 88: 1330-133 .
eered myoblasts. Science 254 1509-1512. 6. YAO, ically S. er & K. KURACHI. 1992. Ex ression of human factor IX in mice after
585-589.
DISCUSSION OF THE PAPER
J. S. LAZO (Univm3y ofRmbu&, Pimbugh, Pa.): I guess the assumption is that the CD3Cpositive cells in the normal individual are fairly similar to those in a patient with Gaucher's disease with respect to their ability to take up a retrovirus?. . .
BARRANGER: That's right. As a matter of fact, the CD34-positive cells from

320 ANNALS NEW YORK ACADEMY OF SCIENCES
the bloodstream seem to be better targets for gene transfer, even in the same CD34-positive population from bone marrow. And their ability to generate BF-UGM BFUE out of the clonogenic assays seems to be uneffected by the enzyme deficiency. Those cells only become storage cells in the peripheral tissues; they become sick once they accumulate this lipid.




















