Toward consistent assignment of structural domains in proteins
32
Toward Consistent Assignment of Structural Domains in Proteins Stella Veretnik 1 *, Philip E. Bourne 1,2 , Nickolai N. Alexandrov 3 and Ilya N. Shindyalov 1 1 San Diego Supercomputer Center, University of California San Diego, 9500 Gilman Dr. La Jolla, CA 92093-0537, USA 2 Department of Pharmacology University of California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093, USA 3 Ceres Inc, 3007 Malibu Canyon Road, Malibu, CA 90265, USA The assignment of protein domains from three-dimensional structure is critically important in understanding protein evolution and function, yet little quality assurance has been performed. Here, the differences in the assignment of structural domains are evaluated using six common assign- ment methods. Three human expert methods (AUTHORS (authors’ anno- tation), CATH and SCOP) and three fully automated methods (DALI, DomainParser and PDP) are investigated by analysis of individual methods against the author’s assignment as well as analysis based on the consensus among groups of methods (only expert, only automatic, com- bined). The results demonstrate that caution is recommended in using current domain assignments, and indicates where additional work is needed. Specifically, the major factors responsible for conflicting domain assignments between methods, both experts and automatic, are: (1) the definition of very small domains; (2) splitting secondary structures between domains; (3) the size and number of discontinuous domains; (4) closely packed or convoluted domain–domain interfaces; (5) structures with large and complex architectures; and (6) the level of significance placed upon structural, functional and evolutionary concepts in consider- ing structural domain definitions. A web-based resource that focuses on the results of benchmarking and the analysis of domain assignments is available at http://pdomains.sdsc.edu q 2004 Elsevier Ltd. All rights reserved. Keywords: assignment of structural domains; consensus approach; benchmarking; discontinuous domains; domain boundaries *Corresponding author Introduction Domains can be viewed as the basic components of proteins and the division of a protein structure into domains often precedes its functional characterization. 1 The number of such domains is surprisingly limited, being in the range of 1000 – 6000, 2,3 which makes systematic characterization feasible. Moreover, domain characterization is a key component of proteomics efforts. 4 The defi- nition of protein domains varies widely across the discipline of biology. Domains are defined simul- taneously as: (1) regions that display a significant level of sequence homology; (2) a minimal part of the gene that is capable of performing a function; (3) a region of the protein with an experimentally assigned function; (4) parts of structures that have significant structural similarity; and (5) compact spatially distinct units of protein structure. This work addresses structural domains, thus it spans definitions (3) – (5). The concept of domains as structural com- ponents of proteins has been around for the last 30 years, starting with the papers by Wetlaufer 5 and Rossman & Liljas. 6 It began with the notion of structurally separate regions of the molecule, 7 which was extended to describe folding units, whose origins are kinetically favored nucleation sites along the polypeptide chain. 5 A limited reper- toire of structures assumed by folding units was observed, 8 and a relationship between recurrent folds and domains was proposed. 9 A possible correlation between the organization of the DNA (intron/exon) and the organization of the protein (structural domains) has been debated but remains an open issue. 1 Compactness and relative spatial 0022-2836/$ - see front matter q 2004 Elsevier Ltd. All rights reserved. E-mail address of the corresponding author: [email protected] Abbreviations used: PDB, Protein Data Bank. doi:10.1016/j.jmb.2004.03.053 J. Mol. Biol. (2004) 339, 647–678
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Toward consistent assignment of structural domains in proteins

Toward Consistent Assignment of Structural Domainsin Proteins
Stella Veretnik1*, Philip E. Bourne1,2, Nickolai N. Alexandrov3 andIlya N. Shindyalov1
1San Diego SupercomputerCenter, University of CaliforniaSan Diego, 9500 Gilman Dr.La Jolla, CA 92093-0537, USA
2Department of PharmacologyUniversity of California, SanDiego, 9500 Gilman Dr., LaJolla, CA 92093, USA
3Ceres Inc, 3007 MalibuCanyon Road, Malibu, CA90265, USA
The assignment of protein domains from three-dimensional structure iscritically important in understanding protein evolution and function, yetlittle quality assurance has been performed. Here, the differences in theassignment of structural domains are evaluated using six common assign-ment methods. Three human expert methods (AUTHORS (authors’ anno-tation), CATH and SCOP) and three fully automated methods (DALI,DomainParser and PDP) are investigated by analysis of individualmethods against the author’s assignment as well as analysis based on theconsensus among groups of methods (only expert, only automatic, com-bined). The results demonstrate that caution is recommended in usingcurrent domain assignments, and indicates where additional work isneeded. Specifically, the major factors responsible for conflicting domainassignments between methods, both experts and automatic, are: (1) thedefinition of very small domains; (2) splitting secondary structuresbetween domains; (3) the size and number of discontinuous domains; (4)closely packed or convoluted domain–domain interfaces; (5) structureswith large and complex architectures; and (6) the level of significanceplaced upon structural, functional and evolutionary concepts in consider-ing structural domain definitions. A web-based resource that focuses onthe results of benchmarking and the analysis of domain assignments isavailable at http://pdomains.sdsc.edu
q 2004 Elsevier Ltd. All rights reserved.
Keywords: assignment of structural domains; consensus approach;benchmarking; discontinuous domains; domain boundaries*Corresponding author
Introduction
Domains can be viewed as the basic componentsof proteins and the division of a protein structureinto domains often precedes its functionalcharacterization.1 The number of such domains issurprisingly limited, being in the range of 1000–6000,2,3 which makes systematic characterizationfeasible. Moreover, domain characterization is akey component of proteomics efforts.4 The defi-nition of protein domains varies widely across thediscipline of biology. Domains are defined simul-taneously as: (1) regions that display a significantlevel of sequence homology; (2) a minimal part ofthe gene that is capable of performing a function;(3) a region of the protein with an experimentally
assigned function; (4) parts of structures that havesignificant structural similarity; and (5) compactspatially distinct units of protein structure. Thiswork addresses structural domains, thus it spansdefinitions (3)–(5).
The concept of domains as structural com-ponents of proteins has been around for the last30 years, starting with the papers by Wetlaufer5
and Rossman & Liljas.6 It began with the notion ofstructurally separate regions of the molecule,7
which was extended to describe folding units,whose origins are kinetically favored nucleationsites along the polypeptide chain.5 A limited reper-toire of structures assumed by folding units wasobserved,8 and a relationship between recurrentfolds and domains was proposed.9 A possiblecorrelation between the organization of the DNA(intron/exon) and the organization of the protein(structural domains) has been debated but remainsan open issue.1 Compactness and relative spatial
0022-2836/$ - see front matter q 2004 Elsevier Ltd. All rights reserved.
E-mail address of the corresponding author:[email protected]
Abbreviations used: PDB, Protein Data Bank.
doi:10.1016/j.jmb.2004.03.053 J. Mol. Biol. (2004) 339, 647–678

separation of the domains were the key factors inthe first generation of algorithms, which performedautomatic domain decomposition.10 – 12 A structuraldomain was described broadly as a segment orseveral segments of the polypeptide that forms acompact and stable structure, with an associatedhydrophobic core, and that can fold and functionindependently from other parts of the polypeptide.It was defined also as a region of the structure thatrecurs in different contexts in different proteins.8,9
Structural domains are often associated withidentifiable cellular/biochemical functions.13,14
As more structures were solved, contradictionsin these definitions of domains became apparent.Some domains were clearly not compact or globu-lar, others were compact and could be separatedeasily from each other. Some recurrent structuralunits contained more than one compact module,yet others had a defined function, but were toosmall to form a stable domain and were lacking ahydrophobic core. The result was a divergence inthe definitions of a structural domain, each reflect-ing a particular approach to the concept of whatconstitutes a domain. In short, the fundamentalpremises underlying domain definition, thermo-dynamic stability, structural independence,evolutionary history, functional/biochemicalsignificance, took different precedence in differentapproaches. At the present time, the concept of astructural domain is well accepted, yet itsdefinition is more ambiguous than ever.15 – 18 Theawareness of this ambiguity is confined mostly tothe groups involved directly in domain assign-ments, while the wider biological communityremains uninformed. Here, we address the needsof the wider community by systematically analyz-ing the issues with current domain analysismethods. This work points the way for developingimproved methods of assignment through asystematic analysis of the work done thus far.
The methods for decomposition of structuresinto domains can be divided into two categories;those that require human expertise for evaluationof each structure and those that are completelyautomated. The first type we refer to as expertmethods, the second group we call algorithmicmethods. In our analysis, we consider three expertmethods:
(1) Annotation of domains provided by theauthors or crystal structures, as assembled byIslam et al.,19 and referred to here as theAUTHORS method.
(2) Assignments of domains by CATH.20
(3) Assignment of domains by SCOP.21
Unlike the SCOP and AUTHORS methods,which rely mainly on experts, CATH uses threealgorithms for domain decomposition as a firststep in the assignment process. However, all finaldomain assignments are made by an expert, whoeither accepts the assignments for which there is aconsensus among the three algorithms or assignsdomains manually when there is no consensus
among three algorithms or when the expert dis-agrees with the consensus.15 The AUTHORSmethod is probably the most diverse and leastsystematic, as different investigators are guidedby somewhat different ideas about the nature ofstructural domains. SCOP imposes the evolution-ary definition of structural domains as foldingunits that appear in different contexts.21
Algorithmic methods to define structuraldomains appeared soon after the introduction ofthe concept itself,10 – 12 and a steady stream of newalgorithmic methods continues to appear.16,17,19 – 26
While approaches taken by different methods varygreatly, they have the same underlying principle:finding the best spatial partitioning of the proteinstructure. Since the idea of a compact structuredoes not always correspond to other notions of astructural domain, such as presence of a specificfunction or of an evolutionary recurrence, thesuccess of these algorithms is only partial. Further-more, the ambiguity of defining structural domainsis found also in the automatic methods, as auto-matic methods use datasets of expert-annotatedstructures as training data to tune the performanceof the algorithm. The choice of training datasetdetermines, to a great extent, the tendencies of thealgorithm, particularly in ambiguous cases, inwhich the expert-defined domain is not a single,autonomous, compact structure. Different methodsuse different datasets to tune their performance:some methods compare the results to AUTHORS,19
others to CATH,22 and yet others to manuallyannotated datasets.16 Each algorithm then reflectsthe “philosophical” approach and associated biastowards the training dataset used. The variety ofautomatic approaches is not necessarily a draw-back; however, it is important that the user beinformed about the tendencies of the individualalgorithms. Different automatic methods usedifferent datasets to benchmark their performance;this makes comparisons of performance of differ-ent methods difficult and sometimes impossible.
Here, we address the problem of structuraldomain ambiguity by providing a detailed com-parison between different domain assignmentmethods, in order to understand: (1) ambiguitiesand significant issues in the expert assignments ofstructural domains; (2) limitations of current algor-ithms and their possible remediation; (3) trendsfound in individual assignment methods (bothexpert and automatic); and (4) realistic benchmark-ing of automatic assignment methods.
Results and Discussion
How do we approach the analysis of domainassignment methods?
Three expert assignment methods, SCOP,21
CATH20 and AUTHORS,19 and three algorithmicassignments DALI,24 DomainParser17 and PDP,23
are investigated. The dataset of chains used
648 Consistent Assignment of Protein Structural Domains

throughout this work is referred to as the 467 chaindataset; it is a comprehensive dataset for whichresults for all six assignment methods were avail-able. The details of its construction can be foundin Materials and Methods. We should point outthat the 467 chain dataset used in our analysis isnot current with respect to the RCSB Protein DataBank (PDB). That is, many domain assignmentsthat are available for CATH and SCOP do notexist for the AUTHORS method. Depositors ofPDB data are not required to provide domainassignments. Thus, our source of AUTHORSassignments is limited to the collection by Islamet al.19 and its updates. This collection constitutes749 chains, when chains with sequence identityover 90% and chains with ambiguous assignmentsare removed (almost threefold reduction from theoriginal size of the dataset). Of these 749 chains,467 were annotated by CATH and SCOP. Currently,this 467 chain dataset provides the most compre-hensive dataset available for the three expertassignment methods.
Several types of analysis are pursued and com-bined in order to get a comprehensive view of thesuccess and failure of domain assignmentmethods. First, we explore the basic properties ofthe six individual assignment methods. We thenlook at classes of methods to tease out typicalbehaviors that are common to each class of assign-ment method, only to expert methods, or only toalgorithmic methods. We investigate cases that aredifficult for all or most of the assignment methods.We then explore in detail each assignment methodto better understand its underlying assumptions.Finally, we discuss the central issues required fora comprehensive definition of a structural domain.A roadmap (Table 1) is designed to help younavigate through the analysis below.
Major features of the assignment methods
This section deals with basic properties of eachof the assignment methods, specifically:
(1) The distribution of single and multipledomains
(2) The ratio of continuous to non-continuousdomains
(3) The distribution of domain sizes(4) The distribution of fragment sizes in non-
continuous domains.
Comparing performance of assignment methods
We evaluated the relative accuracy of eachdomain assignment method using the AUTHORSmethod as a reference (Figure 1). It should benoted that benchmarking analysis was performedfor two versions of SCOP data: one considersdomain boundaries as provided by the Astral, theother considers manual annotations, which areincluded in the individual SCOP entries. Theseresults are reflected in Figure 1(A). Throughout
the rest of the analysis, we consider SCOP domainsas defined by the Astral (see discussion of SCOPbelow). The fraction of correct assignments (whenonly the number of domains is considered)ranges from 90% for SCOP (manual annotationsfor domains) to 80% for DALI (Figure 1(A)).Among misassigned proteins, the fraction ofundercut predictions (a method predicts fewerdomains than the reference method) is moreprevalent than overcut predictions (predictingmore domains than the reference method). Theexception is DALI, where there is roughly anequal fraction of undercut and overcut misassign-ments (10% of each).
When accuracy is further evaluated using thecriterion of correct assignment of individual resi-dues to a domain, the performance of all methodsdiminishes. We use a cutoff of 75% for residueassignments; that is, the chain is assigned correctlyif 75% (or more) of its residues are assigned to thecorrect domains. In the case of residue assignment,the performance ranges from 80.5% using CATH to67.5% using DALI (Figure 1(B)). The decrease inperformance is proportional to the stringency ofthe cutoff imposed on the level of residue assign-ment, as discussed in subsection “Percentage ofdomain overlap” (below).
Single domain versus multi-domain chains
The fraction of single and multiple-domainchains in the 467 chain dataset varies dependingon the assignment method (Figure 2(A)). Single-domain proteins dominate the dataset; the contentranges from 69.5% for AUTHORS assignment to81% for SCOP. The majority of multi-domain pro-teins contain two or three domains; the fraction oftwo-domain chains ranges from 22% forAUTHORS to 12% for SCOP. The fraction of three-domain protein chains ranges from 7.3% forAUTHORS to 3.9% for SCOP. DALI tends to createthe largest number of domains 23% for four-domain proteins versus less than 1% for all othermethods; and partitions five protein chains intofive domains.
Continuous versus discontinuous domains
Both, continuous domains (consisting of onecontinuous segment of the polypeptide chain) andnon-continuous domains (consisting of two ormore fragments from the same polypeptide chain)are present in the dataset. The fraction of con-tinuous domains varies significantly by assignmentmethod (Figure 2(B)). It is the highest in SCOP,where 97% of all tested chains consist of single-fragment domains and is lowest for DALI, where77% of chains have single domains. For eachassignment method, approximately 10–15% ofchains have one or more domain composed ofnon-continuous fragments. The exception is SCOP,where only 2.6% of the chains contain domainscomprising more than one fragment. The number
Consistent Assignment of Protein Structural Domains 649

of fragments comprising a domain also varies byassignment method. Domains comprising threenon-continuous fragments are present in 3.6% ofchains assigned by DomainParser and in 5.6% ofchains assigned by DALI. DALI also partitions
nine (1.4% of the entire dataset) domains into fournon-continuous fragments and five (0.76% of theentire dataset) into five non-continuous fragments.The number of domains per chain combined withthe number of fragments per domain affects the
Table 1. Roadmap for navigation through the Results and Discussion
Question addressed Relevant sectionsRelevant Figures/Tables
What is the overall performance of individualassignment methods?
Major features of the assignment methods (page 649) Figures 1–3
Comparing performance of assignment methodsSingle versus multi-domain chainsContinuous versus discontinuous domainsDomain and fragment sizes
What is the domain boundary overlap? Considering domain boundaries (page 654) Figures 4,5 Table 2, 3Percentage of domain overlapWhat causes disagreement on boundary assignmentAssignments can disagree on the number of domains
and still meet domain overlap requirement
How are assignment methods grouped andcompared?
What we can learn from comparing assignmentmethods? The philosophy behind the consensusapproach (page 656)
Figure 6
Distribution of single and multi-domain chains insubsets
What do all methods capture well? What do all methods capture well? (page 657) Figure 7Cases of chains with complete consensus among
methodsCases of chains in which expert consensus is different
from algorithmic consensus
What do only experts’ methods capture well? What do only experts’ methods capture well? (page 660) Figure 8Cases of expert only consensus versus algorithmic
methods
What do only algorithmic methods capture well? What do only algorithmic methods capture well?(page 660)
Figure 9
Cases of algorithmic only consensus versus expertmethods
What is difficult for all methods? What is difficult for all methods? (page 663) Figure 10 Table 4Cases of chains with no expert or algorithmic
consensus
Why do experts disagree? What is difficult for expert methods: why methodsdisagree (page 665)
Figures 9–12
Cases of algorithmic only consensus versus expertmethods
Cases of chains with no expert or algorithmicconsensus
AUTHORS assignment methodSCOP assignment methodCATH assignment method
What is difficult for algorithmic methods? What is difficult for algorithmic methods (page 670) Figures 8 and 10,Table 5
Cases of expert only consensus versus algorithmicmethods
Cases of chains with no expert or algorithmicconsensus
What are algorithmic methods capable of? Analysis of algorithmic methods: benchmarking using aconsensus approach (page 670)
Figures 12–14
PDP methodDomainParser methodDALI method
How can algorithmic methods be improved? How can we improve algorithmic methods? (page 675)
What is a structural domain? What are the central issues in the definition ofstructural domains? (page 675)
650 Consistent Assignment of Protein Structural Domains
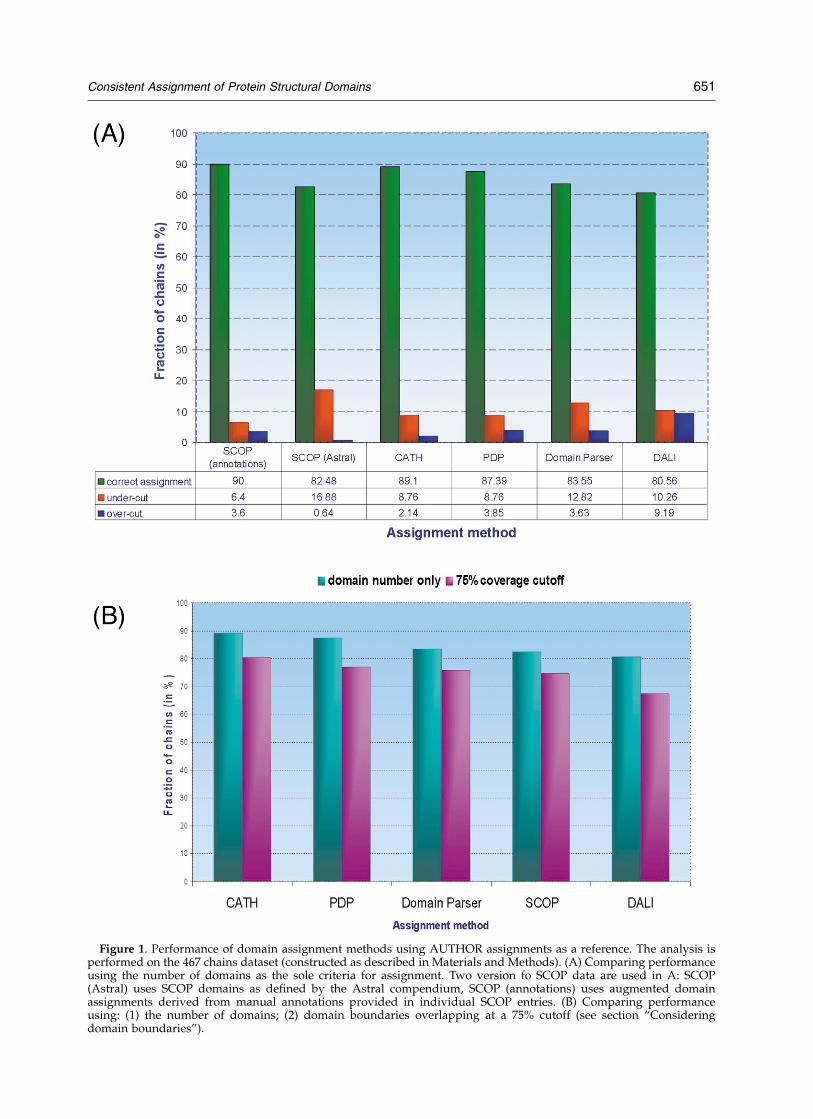
Figure 1. Performance of domain assignment methods using AUTHOR assignments as a reference. The analysis isperformed on the 467 chains dataset (constructed as described in Materials and Methods). (A) Comparing performanceusing the number of domains as the sole criteria for assignment. Two version fo SCOP data are used in A: SCOP(Astral) uses SCOP domains as defined by the Astral compendium, SCOP (annotations) uses augmented domainassignments derived from manual annotations provided in individual SCOP entries. (B) Comparing performanceusing: (1) the number of domains; (2) domain boundaries overlapping at a 75% cutoff (see section “Consideringdomain boundaries”).
Consistent Assignment of Protein Structural Domains 651
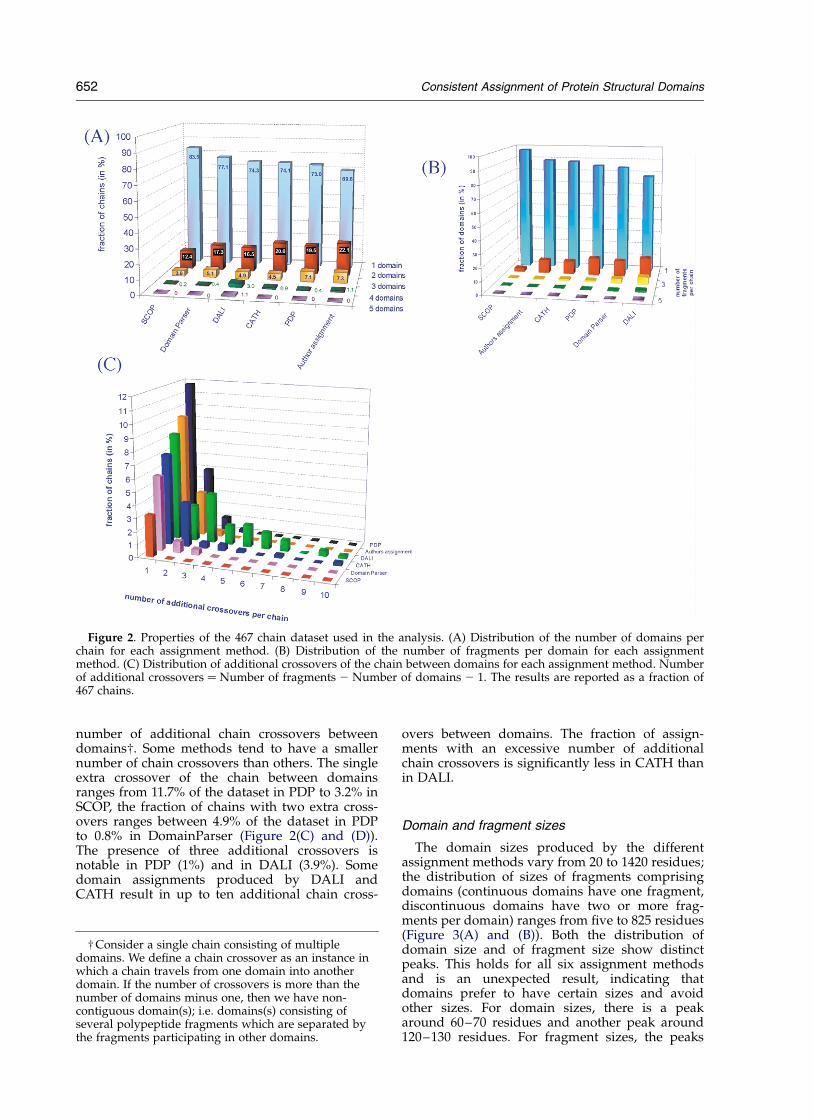
number of additional chain crossovers betweendomains†. Some methods tend to have a smallernumber of chain crossovers than others. The singleextra crossover of the chain between domainsranges from 11.7% of the dataset in PDP to 3.2% inSCOP, the fraction of chains with two extra cross-overs ranges between 4.9% of the dataset in PDPto 0.8% in DomainParser (Figure 2(C) and (D)).The presence of three additional crossovers isnotable in PDP (1%) and in DALI (3.9%). Somedomain assignments produced by DALI andCATH result in up to ten additional chain cross-
overs between domains. The fraction of assign-ments with an excessive number of additionalchain crossovers is significantly less in CATH thanin DALI.
Domain and fragment sizes
The domain sizes produced by the differentassignment methods vary from 20 to 1420 residues;the distribution of sizes of fragments comprisingdomains (continuous domains have one fragment,discontinuous domains have two or more frag-ments per domain) ranges from five to 825 residues(Figure 3(A) and (B)). Both the distribution ofdomain size and of fragment size show distinctpeaks. This holds for all six assignment methodsand is an unexpected result, indicating thatdomains prefer to have certain sizes and avoidother sizes. For domain sizes, there is a peakaround 60–70 residues and another peak around120–130 residues. For fragment sizes, the peaks
Figure 2. Properties of the 467 chain dataset used in the analysis. (A) Distribution of the number of domains perchain for each assignment method. (B) Distribution of the number of fragments per domain for each assignmentmethod. (C) Distribution of additional crossovers of the chain between domains for each assignment method. Numberof additional crossovers ¼ Number of fragments 2 Number of domains 2 1. The results are reported as a fraction of467 chains.
† Consider a single chain consisting of multipledomains. We define a chain crossover as an instance inwhich a chain travels from one domain into anotherdomain. If the number of crossovers is more than thenumber of domains minus one, then we have non-contiguous domain(s); i.e. domains(s) consisting ofseveral polypeptide fragments which are separated bythe fragments participating in other domains.
652 Consistent Assignment of Protein Structural Domains

are less distinct, the first peak corresponds to frag-ments 50–70 residue long and the second peak cor-responds to fragments 110–130 residue long. Thereis an additional peak corresponding to fragmentsof 15–25 residues, contributed mostly by DALI(Figure 3(B)). The distribution of domain and frag-ment sizes was tested also on the dataset withlower domain/fragment redundancy (Figure 3(C)and (D)). The original dataset of 467 chains waspurged so that each of the SCOP superfamilies inthe original dataset was represented by only onechain; this reduction analysis was performed alsoat the level of SCOP folds. The resulting data stillreveal peaks around 60–70 residues and 110–130residues. The intensity of the peak correspondingto 60–70 residue domains diminishes (due toover-representation of small proteins, SCOP classG) and the 120–130 residue peak increases in mag-nitude (Figure 3(C)). When we superimpose thedistribution of chain sizes with that of domainsizes, we observe that the majority of the 60–70residue peak and a substantial fraction of 110–130residue peak consists of single-domain chains (the
size of the domain and the size of the chains arethe same). We conclude that the remaining fractionof 110–130 residue domains are contributed bymulti-domain chains, since the fraction of chainsthat are over 300 residues long is several-foldlarger than the fraction of domains of the samesize (i.e. the majority of the chains over 300 resi-dues are multi-domain). Multiple peaks in thedomain size distribution are an interestingphenomenon. Inspection of the literature indicatesthat multiple peaks are present when the incre-mental steps through the distribution are small(10–20 residue).27,28 The domain size of 110–130residues, the largest peak in our data, is in agree-ment with the proposed “single standard unit sizeof fossil proteins”, a concept based on the optimalsize of the circular DNA fragments, which werepresent at early stages of genome evolution.29
Even in our purged datasets, some of the domainswere present more than once. This is consistentwith the observation that some domains occurmore frequently in a dataset due to their pairingwith unique domains.30 The consequence is that
Figure 3. Distribution of domain and fragment sizes in the 467 chain dataset. (A) Distribution of domain sizes in theentire 467 chain dataset shown for each method. (B) Distribution of fragment sizes in the entire 467 chain datasetshown for each method. (C) Distribution of domain sizes in the purged dataset (purged dataset, as described in thetext, at the level of unique superfamilies or at the level of unique folds): shown for all chains (blue), unique SCOPsuperfamilies (red) and unique SCOP folds (green). Superimposed is the distribution of chain sizes (magenta). (D) Dis-tribution of fragment sizes in the purged dataset shown for all chains (blue), unique SCOP superfamilies (red) andunique SCOP folds (green). Superimposed is the distribution of chain sizes (magenta). Data are shown for AUTHORSassignment in (C) and (D).
Consistent Assignment of Protein Structural Domains 653
the purged datasets still have some over-represen-tation of “frequently used domains”, since theevaluation is performed on the entire chain. Thisover-representation alone cannot account for theobserved phenomenon.
In summary, the agreement between individualassignment methods in assigning the number ofdomains is within the range of 80–89% for the testset. Individual assignment methods varysignificantly in their tendencies to: (1) partition thestructure into domains, as indicated by the singleversus multi-domain distribution; and (2) partitionthe domain into fragments, as indicated by thecontinuous versus discontinuous domain distri-bution, as well as the domain and fragmentdistribution.
Considering domain boundaries
The analysis of the correspondence betweendomain boundaries assigned by different methodsis given using the AUTHORS assignment as areference.
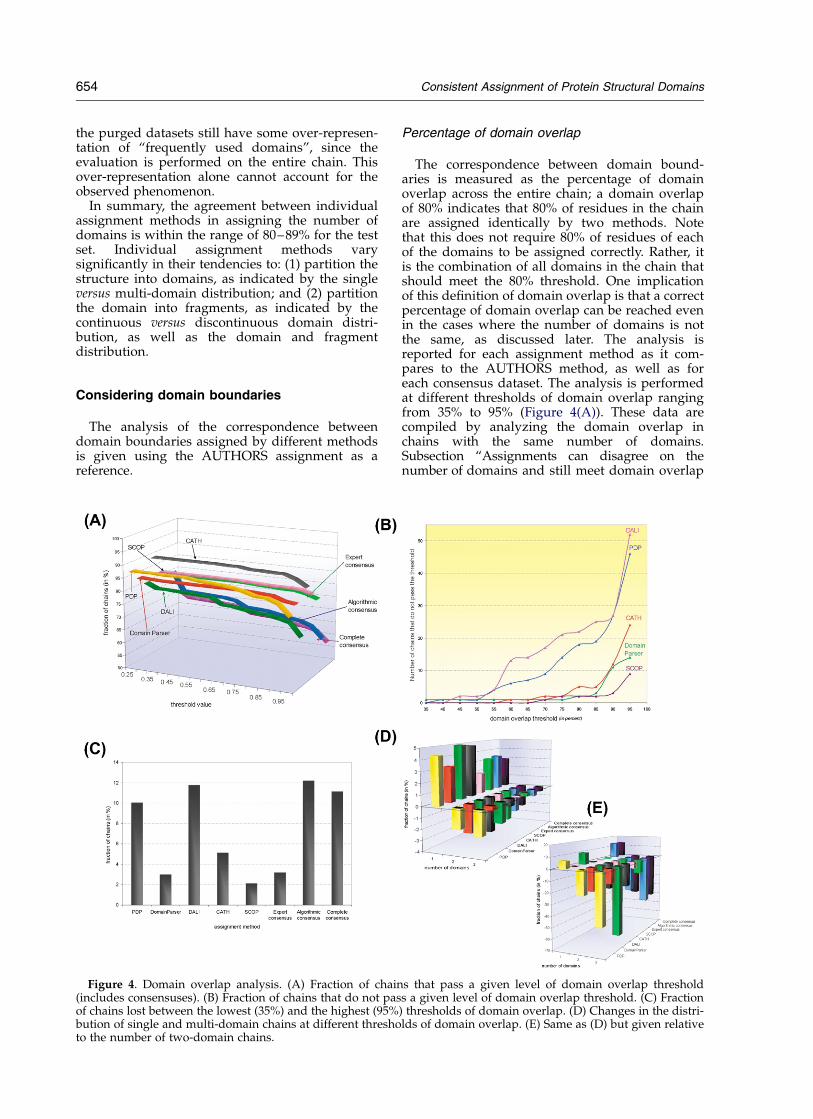
Percentage of domain overlap
The correspondence between domain bound-aries is measured as the percentage of domainoverlap across the entire chain; a domain overlapof 80% indicates that 80% of residues in the chainare assigned identically by two methods. Notethat this does not require 80% of residues of eachof the domains to be assigned correctly. Rather, itis the combination of all domains in the chain thatshould meet the 80% threshold. One implicationof this definition of domain overlap is that a correctpercentage of domain overlap can be reached evenin the cases where the number of domains is notthe same, as discussed later. The analysis isreported for each assignment method as it com-pares to the AUTHORS method, as well as foreach consensus dataset. The analysis is performedat different thresholds of domain overlap rangingfrom 35% to 95% (Figure 4(A)). These data arecompiled by analyzing the domain overlap inchains with the same number of domains.Subsection “Assignments can disagree on thenumber of domains and still meet domain overlap
Figure 4. Domain overlap analysis. (A) Fraction of chains that pass a given level of domain overlap threshold(includes consensuses). (B) Fraction of chains that do not pass a given level of domain overlap threshold. (C) Fractionof chains lost between the lowest (35%) and the highest (95%) thresholds of domain overlap. (D) Changes in the distri-bution of single and multi-domain chains at different thresholds of domain overlap. (E) Same as (D) but given relativeto the number of two-domain chains.
654 Consistent Assignment of Protein Structural Domains

requirement” (below) considers the results whenthe requirement for the same number ofdomains is removed. The domain overlapanalysis is performed at increments of 5% which,therefore, defines the level of accuracy. In ouranalysis, we refer to the lower end of thethreshold range. For example, if the domainassignment passes the 80% threshold, but does notpass the 85% threshold, we assign domain overlapas 80%.
As the percentage of domain overlap increases,the number of chains that can meet this thresholddecreases. The decrease is smooth throughoutmost of the range of thresholds; however, whenthe threshold reaches 90% or 95%, the drop in thenumber of chains that can meet it becomes moredramatic (Figure 4(A) and (B)). This may indicatethat a reasonable threshold for domain overlapshould be set close to, but below 90%. The fractionof chains that are lost during the process of increas-ing the domain overlap threshold depends on theassignment method (Figure 4(C)). This loss is rela-tively low for the expert methods SCOP (2%) andCATH (5%) as well as for the algorithmic methodDomainParser (3%). For the other two algorithmicmethods, the fraction of chains lost is several-foldhigher; DALI loses 12% of chains and PDP loses10% of chains between the lowest and highestlevels of domain overlap threshold. The fraction ofchains that makes up algorithmic consensus(consensus is described in section “What can welearn from comparing assignment methods?”,(below) decreases more rapidly than the fractionof chains in the experts’ consensus. This indicatesthat expert methods are more consistent in place-ment of domain boundaries, than the algorithmicmethods.
An increase in the threshold of domain overlapaffects the distribution of single and multi-domainchains in the set of chains that passes a giventhreshold. Similarly, for consensus subsets, thefraction of multi-domain chains decreases, whilethe fraction of single-domain chains increases(Figure 4(D)) as the stringency of domain overlapgoes up. This decrease in the multi-domain chainsis even more apparent relative to its own class (i.e.
two-domain or three-domain fraction) (Figure4(E)).
What causes disagreement onboundary assignment?
To better understand how different methodsassign domain boundaries, we take a detailed lookat the overall statistics as well as some specificexamples. The plot of the number of chains thatdo not meet the progressively increasing domainoverlap threshold indicates that the region between60% and 85% shows a stable and gradual increasein the number of chains failing to meet thethreshold (Figure 4(B)). We focused on two levelsof thresholds for domain overlap: 95% (as themost stringent level of domain assignment) and80% (as a realistic and stable level of domainassignment). We observe three major causes ofdomain boundary disagreement (Table 2): (1)assigning less than an entire chain to a domain inthe case of single-domain chains; (2) different num-ber of fragments per domain assigned by twomethods; and (3) everything else, that is, disagree-ments in multi-domain chains with continuousdomains or non-continuous domains with identicalnumbers of fragments. This last type of disagree-ment is caused by a mismatch in the domain/frag-ment boundaries. The situation is similar forsingle-domain chains where the N-terminal and/or C-terminal boundary of the domain does notmatch the beginning and the end of the polypep-tide chain. When the number of fragments perdomain differs between a given method and thereference method, we record whether it is causedby assigning too many or too few fragments rela-tive to the reference method. Different assignmentmethods vary significantly in both (1) the numberof chains that do not pass a given level of domainoverlap threshold and (2) the type of misassign-ment (Table 2 and Figure 4). We notice that as thestringency of domain overlap decreases from 95%to 80%, the number of chains that passes thethreshold increases dramatically, indicating thatthe majority of disagreements are minor.
Table 2. Disagreement in assignment of domain boundaries by each method using AUTHORS assignment as a reference
Multi-domain chains (identicalnumber of fragments/domain)
Multi-domain chains (differentnumber of fragments/domain)
Assignmentmethod
Number of chains(total)
Single-domainchains
Continuousdomains
Non-continuousdomains
More thanreference
Less thanreference
PDP 47 (20) 20 (11) 2 (1) 7 (2) 15 (4) 3 (2)DomainParser 14 (2) 0 (0) 4 (1) 2 (0) 3 (0) 5 (1)DALI 52 (22) 30 (16) 5 (1) 6 (0) 10 (4) 1 (1)CATH 24 (5) 1 (1) 8 (1) 4 (0) 11 (3) 0 (0)SCOP 9 (2) 1 (1) 2 (0) 0 (0) 1 (0) 5 (1)
Values represent the number of chains that do not agree on domain boundaries. Values are given for two thresholds: 95% domainoverlap threshold (first value) and 80% domain overlap threshold (value in parentheses). Analysis is limited to the chains for whicha given method and the reference method agree on the number of domains.
Consistent Assignment of Protein Structural Domains 655

Exceptions are groups of single-domain chainsassigned by PDP and DALI.
Assignments can disagree on the number ofdomains and still meet domain overlap requirement
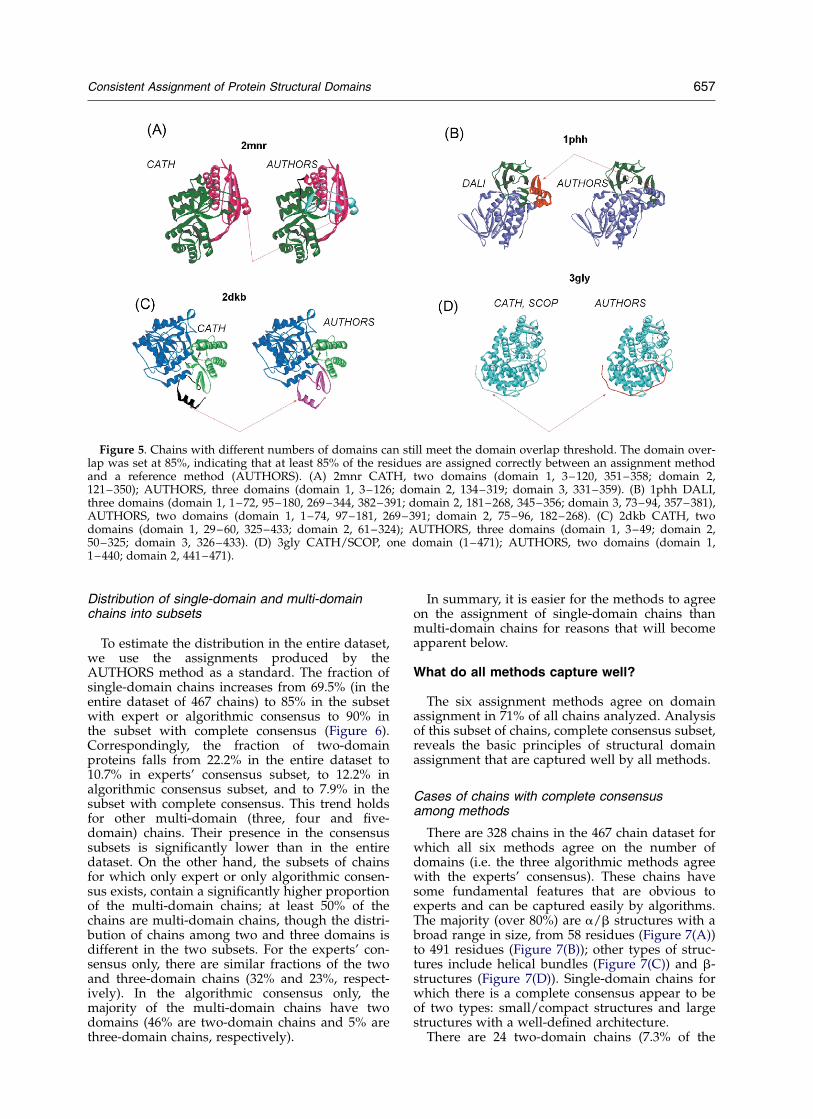
The above analysis was performed for chains inwhich there was agreement between the numberof assigned domains. However, if one or more ofthe assigned domains represents a small fractionof the entire chain, then domain overlap require-ment can be attained even when the number ofdomains assigned by methods differs. We find 12cases in which the number of domains differs, butthe domain overlap still meets the 85% threshold(Table 3). In seven cases, the reference (AUTHORS)has one more domain than the other method(s); infive cases, one less (four for DALI, one for PDP).The additional domains are small and usuallyrecruited in two ways: (1) from a fragment of anexisting discontinuous domain (Figure 5(A) and(B)); and (2) from a part of a continuous domain(and/or unassigned region; Figure 5(C) and (D))that is promoted to a full domain. In the lattercase, the part of a domain is always taken fromthe end of the domain, not by generatingadditional discontinuities within the domain. Thenew domain can appear at the ends of a poly-peptide chain or in the middle.
In summary, given a reasonable range of domainoverlap, such as 81–85%, the agreement on domainboundaries is very good.
What can we learn from comparing assignmentmethods? The philosophy behind theconsensus approach
One of our goals is to understand what the sixassignment methods under investigation have incommon and how they differ. Are there basicfeatures that are restricted to expert methods oralgorithmic methods? What are the features thatall methods capture well and what are the features
that are difficult for all the methods? To addressthese questions, the entire 467 chain dataset is firstpartitioned into four mutually exclusive subsets: acomplete consensus, no consensus, algorithmiconly consensus, and expert only consensus(Figure 6). A “complete consensus” (328 chains,71% of the dataset) is the subset of chains forwhich all six methods agreed on the number ofdomain assignments. “No consensus” (49 chains,10.4% of the dataset) is the subset where neitheralgorithmic methods nor experts could come to anagreement. An “algorithmic only consensus”(43 chains, 9.2% of the dataset) represents the sub-set of chains for which algorithms agree, butexperts disagree. An “expert only consensus”(45 chains, 9.4% of the dataset) represents the sub-set of chains for which expert, but no algorithmicconsensus exists. Finally, there are three chains(0.6% of the dataset) for which experts’ consensusis different from that of algorithmic consensus.These three chains represent an interestingphenomenon (see subsection “Cases of chains inwhich expert consensus is different from algorith-mic consensus” (below), but are excluded fromfurther consideration in the consensus analysis.
Subsequently, additional categories are defined:an “algorithmic consensus” (371 chains, 80% ofthe dataset), the subset of chains for which thethree algorithmic methods agree, it is a combi-nation of a “complete consensus” and the “algo-rithmic only consensus” subsets; the “experts’consensus” (373 chains, 80% of the dataset), forwhich three expert methods agree (a combinationof “complete consensus” and “expert only consen-sus” subsets). Finally, a subset called no “experts’consensus” is defined; it contains the chains forwhich the three expert methods cannot reach aconsensus (a combination of no consensus andalgorithmic only consensus subsets). Analyzingthe chains that belong to each of the above subsetswill help us to understand what type of chains allalgorithms assign similarly and what type ofchains present problem to some or all methods.
Table 3. Enumeration of chains in the 467 chain dataset for which 85% of residues are assigned to correct domains evenin the absence of agreement on the number of domains
Type of domain number mismatchChains that pass 85% domain overlap threshold, but havedifferent number of domains
Reference method (AUTHORS) have more domains thancurrent method
3gly: AUTHORS, 2; DomainParser, CATH and SCOP, 1
4icd: AUTHORS, 3; DomainParser and DALI, 22mnr: AUTHORS, 3; DomainParser, DALI, CATH andSCOP, 21hex: AUTHORS, 3; DomainParser and PDP, 22dkb: AUTHORS, 3; CATH, 21tpla: AUTHORS, 3; CATH, 21chra: AUTHORS, 3; CATH and SCOP, 2
Current method has more domains than referencemethod (AUTHORS)
1xima: AUTHORS, 1; DALI and PDP, 2
1ddt: AUTHORS, 3; DALI, 41phh: AUTHORS, 2; DALI, 31aoza: AUTHORS, 3; DALI, 42pcdm: AUTHORS 1; DALI, 2
656 Consistent Assignment of Protein Structural Domains
Distribution of single-domain and multi-domainchains into subsets
To estimate the distribution in the entire dataset,we use the assignments produced by theAUTHORS method as a standard. The fraction ofsingle-domain chains increases from 69.5% (in theentire dataset of 467 chains) to 85% in the subsetwith expert or algorithmic consensus to 90% inthe subset with complete consensus (Figure 6).Correspondingly, the fraction of two-domainproteins falls from 22.2% in the entire dataset to10.7% in experts’ consensus subset, to 12.2% inalgorithmic consensus subset, and to 7.9% in thesubset with complete consensus. This trend holdsfor other multi-domain (three, four and five-domain) chains. Their presence in the consensussubsets is significantly lower than in the entiredataset. On the other hand, the subsets of chainsfor which only expert or only algorithmic consen-sus exists, contain a significantly higher proportionof the multi-domain chains; at least 50% of thechains are multi-domain chains, though the distri-bution of chains among two and three domains isdifferent in the two subsets. For the experts’ con-sensus only, there are similar fractions of the twoand three-domain chains (32% and 23%, respect-ively). In the algorithmic consensus only, themajority of the multi-domain chains have twodomains (46% are two-domain chains and 5% arethree-domain chains, respectively).
In summary, it is easier for the methods to agreeon the assignment of single-domain chains thanmulti-domain chains for reasons that will becomeapparent below.
What do all methods capture well?
The six assignment methods agree on domainassignment in 71% of all chains analyzed. Analysisof this subset of chains, complete consensus subset,reveals the basic principles of structural domainassignment that are captured well by all methods.
Cases of chains with complete consensusamong methods
There are 328 chains in the 467 chain dataset forwhich all six methods agree on the number ofdomains (i.e. the three algorithmic methods agreewith the experts’ consensus). These chains havesome fundamental features that are obvious toexperts and can be captured easily by algorithms.The majority (over 80%) are a/b structures with abroad range in size, from 58 residues (Figure 7(A))to 491 residues (Figure 7(B)); other types of struc-tures include helical bundles (Figure 7(C)) and b-structures (Figure 7(D)). Single-domain chains forwhich there is a complete consensus appear to beof two types: small/compact structures and largestructures with a well-defined architecture.
There are 24 two-domain chains (7.3% of the
Figure 5. Chains with different numbers of domains can still meet the domain overlap threshold. The domain over-lap was set at 85%, indicating that at least 85% of the residues are assigned correctly between an assignment methodand a reference method (AUTHORS). (A) 2mnr CATH, two domains (domain 1, 3–120, 351–358; domain 2,121–350); AUTHORS, three domains (domain 1, 3–126; domain 2, 134–319; domain 3, 331–359). (B) 1phh DALI,three domains (domain 1, 1–72, 95–180, 269–344, 382–391; domain 2, 181–268, 345–356; domain 3, 73–94, 357–381),AUTHORS, two domains (domain 1, 1–74, 97–181, 269–391; domain 2, 75–96, 182–268). (C) 2dkb CATH, twodomains (domain 1, 29–60, 325–433; domain 2, 61–324); AUTHORS, three domains (domain 1, 3–49; domain 2,50–325; domain 3, 326–433). (D) 3gly CATH/SCOP, one domain (1–471); AUTHORS, two domains (domain 1,1–440; domain 2, 441–471).
Consistent Assignment of Protein Structural Domains 657

Figure 6. Partitioning the dataset using a consensus approach; distribution of the single and multi-domain chainswithin each consensus subset. The consensus among methods is defined as an agreement on the number of domainsassigned to a protein chain. Expert and algorithmic consensus is reached for 80% of the entire 467 chain dataset.Among the chains with consensus, 80.5% (331 chains) have an identical consensus, 9.2% of the chains have algorithmicconsensus only and 9.4% of the chains have experts’ consensus only, and 0.9% of the chains have a disagreement inautomatic and manual consensuses. The distribution of single and multi-domain fractions varies significantly amongdifferent subsets and is presented for each type of consensus as well as for the entire dataset and the subset withoutconsensus. For the cases without consensus, the calculation of the domain distribution was performed as follows. Forthe entire dataset, the domain number distribution is predicted on the basis of AUTHORS, for the subset without con-sensus, the domain number distribution was calculated on the basis of the average number of chains predicted by allmethods (the value was rounded down if the fraction was less than 0.51, thus the calculation is somewhat biasedtowards a smaller number of domains).
658 Consistent Assignment of Protein Structural Domains

complete consensus subset), ranging in size from145 to 265 residues. Most of the two-domain chainsin this group have two very distinct lobescontaining a b-structure component or are all-b(Figure 7(E) and (F)). Less trivial cases consist oftwo a/b domains that are not separated clearly(Figure 7(G) and (H)) and in order to definedomains, individual secondary structures have tobe cut. In all cases of complete consensus there isgood agreement among assignments with respectto the domain boundaries as well as domainnumber.
There are four, three-domain chains (1.2% of thecomplete consensus subset), ranging in size from381 to 585 residues. All cases are a/b structurewith 3-fold symmetry. The boundaries betweendomains are more difficult to draw: in at least onecase individual helices close to the domain bound-aries are assigned differently by different experts(Figure 7(I)). The only four-domain chain forwhich all methods agree is 171 residues long and
consists of four very small b-structures connectedby loops (Figure 7(J)).
Cases of chains in which expert consensus isdifferent from algorithmic consensus
There are three chains in the 467 chain datasetfor which both experts and algorithms reach con-sensus; however, the consensuses are different. Allthree cases involve short DNA-binding proteinsconsisting of 2 (2drpa Figure 7(K), 1bboFigure 7(L)) or three consecutively repeated (1zaaFigure 7(M)) Zn-finger motif. Because individualdomains are less than 35 residues long (which isprobably a minimum size of domain as defined byalgorithms), algorithmic methods leave the chainuncut, in spite of the well-recognized architectureof the Zn-finger motif. Expert methods however,identify each Zn-finger as an individual domain.This indicates that current automatic methods are
Figure 7. Examples of complete consensus among methods and conflicting consensuses. (A–J) All methods agreedon the number of domains. Domain boundaries are shown for the AUTHORS assignments. In the case of one domainassignment, the entire chain is considered a domain. (K–M) Algorithmic consensus is different from expert consensus.(A) 8pti bovine pancreatic trypsin inhibitor, one domain. (B) 1btc soybean b-amylase, one domain. (C) 1mrr E. coli ribo-nucleotide reductase,one domain. (D) 5nn9 influenza virus neuroamidase, one domain. (E) 1hil specific antibody Fab17/9, two domains (domain1, 1–104; domain 2, 110–211). (F) 1gdh NAD-dependent D-glycerate dehydrogenase, twodomains (domain1, 2–99, 291–321; domain 2, 100–290). (G) 8atc aspartate carbonyltransferase, two domains(domain1, 1–137, 288–310; domain 2, 144–283). (H) tbpa yeast TATA-binding protein, two domains (domain 1, 61-71,159–240; domain 2, 72–158). (I) 1npx NADH peroxidase, three domains (domain 1, 1–115, 242–325; domain 2,116–241; domain 3, 326–447). (J) 9wga wheat–germ agglutinin, four domains (domain 1, 2–43; domain 2, 44–83;domain 3, 84–120; domain 4, 121–171). (K) 2drpa D. melanogaster tramtrack protein; algorithmic consensus, onedomain; expert consensus, two domains (domain 1, 103–138; domain 2, 139–165). (L) 1bbo human enhancer bindingprotein Mbp-1; algorithmic consensus, one domain; expert consensus, two domains (domain 1, 1–25; domain 2,29–57). (M) 1zaa mouse Zif268 immediatealy early gene; algorithmic consensus, one domain; expert consensus, threedomains (domain 1, 3–31; domain 2, 33–60; domain 3, 61–87).
Consistent Assignment of Protein Structural Domains 659

not sophisticated enough to catch very small, butwell-characterized domains.
What do only expert methods capture well?
The subset “expert only consensus” is a group ofchains for which all expert methods agree on theassignment, while algorithmic methods do not.
Cases of expert only consensus versusalgorithmic methods
Of the 375 chains of experts’ consensus, there are44 chains (9.5% of the 467 chain dataset) for whichone or more of the algorithmic methods disagreewith the experts’ consensus. This category isreferred to as expert only consensus. This subsetcontains a considerable fraction of multi-domainchains, 32% (14 chains) are two-domain chains and23% (ten chains) are three-domain chains, while45% (20 chains) are single-domain chains. Thesingle-domain cases that trick the algorithms arelarge structures with low contact density(Figure 8(A) and (B)). Conversely, there are compactstructures that experts consistently divide intodomains (Figure 8(C) and (D)), but some algorithmsleave the chain uncut. We observe that occasionallyall experts partition chains into domains with verylittle secondary structure (Figure 8(E) and (F)), test-ing the definition of what constitutes a structuraldomain.
In summary, it would appear that the cases inwhich only expert methods can reach consensusrequire additional functional or evolutionaryknowledge.
What do only algorithmic methodscapture well?
There is also a set of structures, an “algorithmiconly consensus”, for which algorithmic methodsreach consensus, while expert methods do not.
Cases of “algorithmic only consensus” versusexpert methods
The subset “algorithmic only consensus” (Figure 6)contains 43 chains and may provide some insightinto the reasons for disagreements among expertmethods. This subset contains 49% (21) single-domain chains, 46.5% (20) two-domain chains and4.5% (2) three-domain chains. In the majority ofcases, only one of the expert methods (SCOP orAUTHORS) disagrees with the algorithmic consen-sus, while the other two expert methods agree withit (61% of 43 chains). CATH in most of the casessides either with SCOP or with AUTHORS andrarely disagrees with both of them.
The largest fraction, 40% (17 chains) comprisescases in which algorithmic consensus agrees withAUTHORS and CATH, but disagrees with SCOP.In the majority of cases (15 out of 17), algorithmicconsensus partitions chains into two domains,
Figure 8. Experts’ consensus versus algorithmic assign-ments. Cases where experts reach consensus on the num-ber of domains, however, there is no agreement amongalgorithmic methods. Domain boundaries for experts’consensus are shown using AUTHORS data; domainboundaries are given for each algorithmic method. Inthe case of single domain assignments, the entire chainis considered a domain. (A) 2bpa bacteriophage Phix174coat protein Gpf, experts’ consensus, one domain; PDP,three domains (domain 1, 1–173, 210–297,358–426;domain 2, 298–357; domain 3, 174–209); DomainParser,two domains (domain 1, 1–166, 214–296, 357–426;domain 2, 167–213, 297–356); DALI, two domains(domain 1, 1–166, 234–297, 357–426; domain 2, 167–213, 297–356). (B) 1pho E. coli phosphoporin,experts’consensus, one domain; PDP and DomainParser, twodomains (domain 1, 1–100; domain 2, 101–340); DALI,four domains (domain 1, 17–40, 304–340; domain 2,103–121, 180–191, 220–233, 260–280, 296–303; domain3, 1–16, 41–102, 133–158; domain 4, 122–132, 159–179,192–219, 234–259,281–295). (C) 1emd malate dehydro-genase, experts’ consensus, two domains (domain 1,1–148; domain 2, 149–309); PDP and DALI, one domain;DomainParser, two domains (domain 1, 1–144; domain2, 145–312). (D) 1gof galactose oxidase, experts’ consen-sus, three domains (domain 1, 1–152; domain 2, 153–532, domain 3, 542–639); PDP and DomainParser, twodomains (domain 1, 1–150; domain 2, 151–639); DALI,three domains (domain 1, 1–151; domain 2, 152–528,575–588; domain 3, 529–574, 589–639). (E) 4rcr photo-synthetic reaction center, experts’ consensus, twodomains (domain 1, 12–39; domain 2, 40–248), PDP,two domains (domain 1, 25–60; domain 2, 83–248);DomainParser, one domain, DALI, two domains(domain 1, 31–80; domain 2, 101–248). (F) 1psp pan-creatic spasmolitic polypeptide, experts’ consensus, twodomains (domain 1, 1–56, 99–106; domain 2, 57–98);DomainParser and DALI, one domain; PDP, twodomains (domain 1, 1–11, 49–106; domain 2, 12–48).
660 Consistent Assignment of Protein Structural Domains
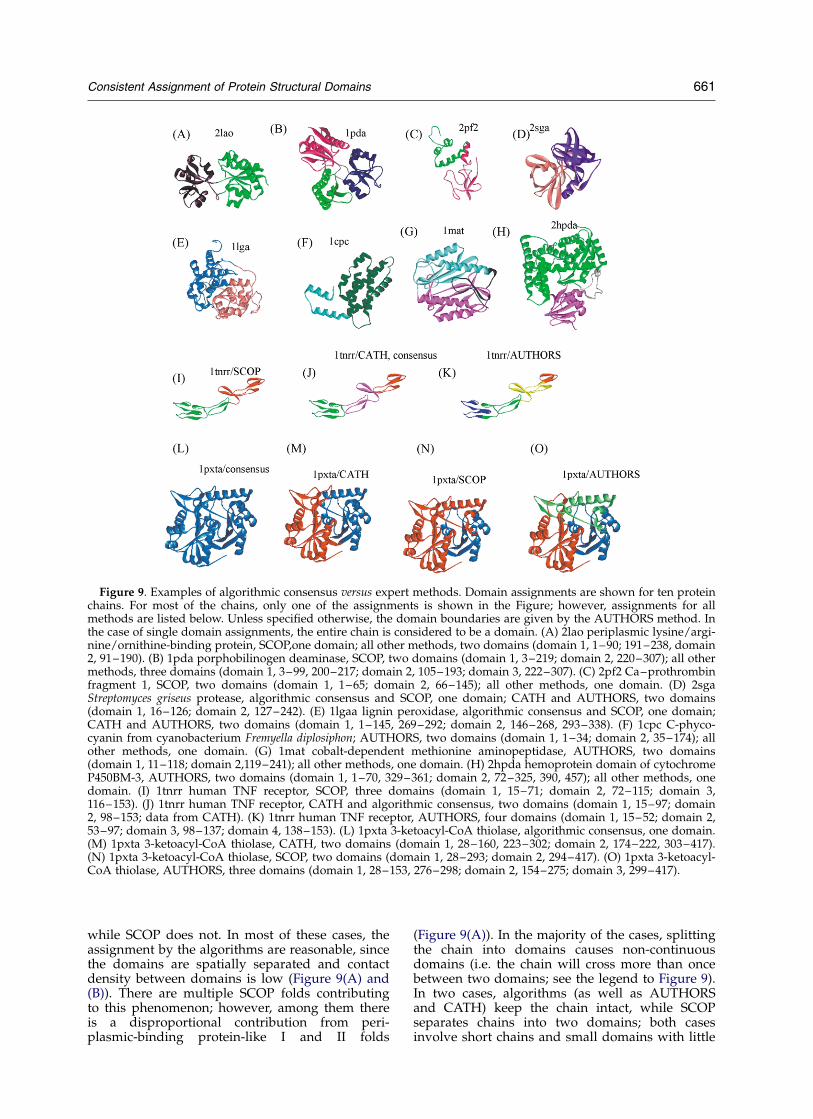
while SCOP does not. In most of these cases, theassignment by the algorithms are reasonable, sincethe domains are spatially separated and contactdensity between domains is low (Figure 9(A) and(B)). There are multiple SCOP folds contributingto this phenomenon; however, among them thereis a disproportional contribution from peri-plasmic-binding protein-like I and II folds
(Figure 9(A)). In the majority of the cases, splittingthe chain into domains causes non-continuousdomains (i.e. the chain will cross more than oncebetween two domains; see the legend to Figure 9).In two cases, algorithms (as well as AUTHORSand CATH) keep the chain intact, while SCOPseparates chains into two domains; both casesinvolve short chains and small domains with little
Figure 9. Examples of algorithmic consensus versus expert methods. Domain assignments are shown for ten proteinchains. For most of the chains, only one of the assignments is shown in the Figure; however, assignments for allmethods are listed below. Unless specified otherwise, the domain boundaries are given by the AUTHORS method. Inthe case of single domain assignments, the entire chain is considered to be a domain. (A) 2lao periplasmic lysine/argi-nine/ornithine-binding protein, SCOP,one domain; all other methods, two domains (domain 1, 1–90; 191–238, domain2, 91–190). (B) 1pda porphobilinogen deaminase, SCOP, two domains (domain 1, 3–219; domain 2, 220–307); all othermethods, three domains (domain 1, 3–99, 200–217; domain 2, 105–193; domain 3, 222–307). (C) 2pf2 Ca–prothrombinfragment 1, SCOP, two domains (domain 1, 1–65; domain 2, 66–145); all other methods, one domain. (D) 2sgaStreptomyces griseus protease, algorithmic consensus and SCOP, one domain; CATH and AUTHORS, two domains(domain 1, 16–126; domain 2, 127–242). (E) 1lgaa lignin peroxidase, algorithmic consensus and SCOP, one domain;CATH and AUTHORS, two domains (domain 1, 1–145, 269–292; domain 2, 146–268, 293–338). (F) 1cpc C-phyco-cyanin from cyanobacterium Fremyella diplosiphon; AUTHORS, two domains (domain 1, 1–34; domain 2, 35–174); allother methods, one domain. (G) 1mat cobalt-dependent methionine aminopeptidase, AUTHORS, two domains(domain 1, 11–118; domain 2,119–241); all other methods, one domain. (H) 2hpda hemoprotein domain of cytochromeP450BM-3, AUTHORS, two domains (domain 1, 1–70, 329–361; domain 2, 72–325, 390, 457); all other methods, onedomain. (I) 1tnrr human TNF receptor, SCOP, three domains (domain 1, 15–71; domain 2, 72–115; domain 3,116–153). (J) 1tnrr human TNF receptor, CATH and algorithmic consensus, two domains (domain 1, 15–97; domain2, 98–153; data from CATH). (K) 1tnrr human TNF receptor, AUTHORS, four domains (domain 1, 15–52; domain 2,53–97; domain 3, 98–137; domain 4, 138–153). (L) 1pxta 3-ketoacyl-CoA thiolase, algorithmic consensus, one domain.(M) 1pxta 3-ketoacyl-CoA thiolase, CATH, two domains (domain 1, 28–160, 223–302; domain 2, 174–222, 303–417).(N) 1pxta 3-ketoacyl-CoA thiolase, SCOP, two domains (domain 1, 28–293; domain 2, 294–417). (O) 1pxta 3-ketoacyl-CoA thiolase, AUTHORS, three domains (domain 1, 28–153, 276–298; domain 2, 154–275; domain 3, 299–417).
Consistent Assignment of Protein Structural Domains 661
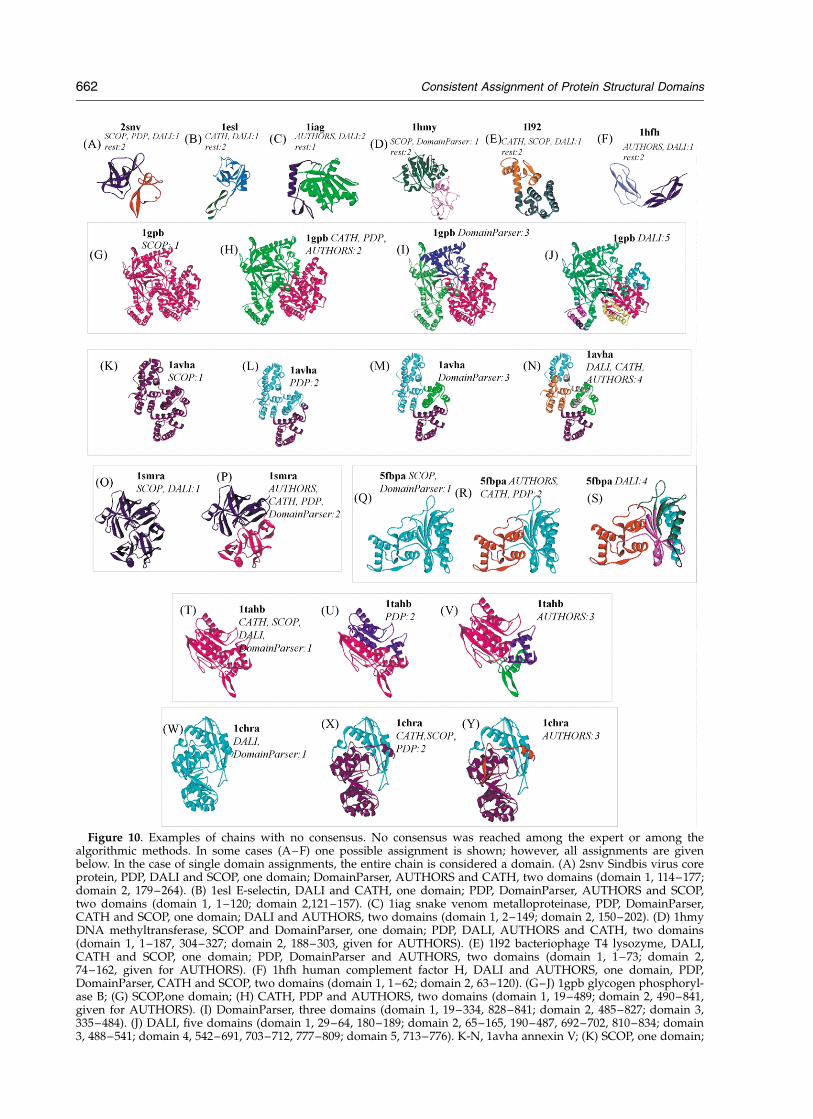
Figure 10. Examples of chains with no consensus. No consensus was reached among the expert or among thealgorithmic methods. In some cases (A–F) one possible assignment is shown; however, all assignments are givenbelow. In the case of single domain assignments, the entire chain is considered a domain. (A) 2snv Sindbis virus coreprotein, PDP, DALI and SCOP, one domain; DomainParser, AUTHORS and CATH, two domains (domain 1, 114–177;domain 2, 179–264). (B) 1esl E-selectin, DALI and CATH, one domain; PDP, DomainParser, AUTHORS and SCOP,two domains (domain 1, 1–120; domain 2,121–157). (C) 1iag snake venom metalloproteinase, PDP, DomainParser,CATH and SCOP, one domain; DALI and AUTHORS, two domains (domain 1, 2–149; domain 2, 150–202). (D) 1hmyDNA methyltransferase, SCOP and DomainParser, one domain; PDP, DALI, AUTHORS and CATH, two domains(domain 1, 1–187, 304–327; domain 2, 188–303, given for AUTHORS). (E) 1l92 bacteriophage T4 lysozyme, DALI,CATH and SCOP, one domain; PDP, DomainParser and AUTHORS, two domains (domain 1, 1–73; domain 2,74–162, given for AUTHORS). (F) 1hfh human complement factor H, DALI and AUTHORS, one domain, PDP,DomainParser, CATH and SCOP, two domains (domain 1, 1–62; domain 2, 63–120). (G–J) 1gpb glycogen phosphoryl-ase B; (G) SCOP,one domain; (H) CATH, PDP and AUTHORS, two domains (domain 1, 19–489; domain 2, 490–841,given for AUTHORS). (I) DomainParser, three domains (domain 1, 19–334, 828–841; domain 2, 485–827; domain 3,335–484). (J) DALI, five domains (domain 1, 29–64, 180–189; domain 2, 65–165, 190–487, 692–702, 810–834; domain3, 488–541; domain 4, 542–691, 703–712, 777–809; domain 5, 713–776). K-N, 1avha annexin V; (K) SCOP, one domain;
662 Consistent Assignment of Protein Structural Domains

secondary structure (Figure 9(C)). In seven cases(15% of chains), the algorithmic consensus agreeswith SCOP assignment and disagrees withAUTHORS and CATH assignments. Most of thechains belong to the trypsin-like serine proteasefold (Figure 9(D)). We conclude that in these casesthe domains are packed too closely and cannot besplit successfully by any one of the algorithms.
Another fraction, 24% of chains (ten chains), arethose in which algorithmic consensus disagreewith AUTHORS, but agrees with SCOP andCATH. This fraction is interesting, since fivemethods (CATH, SCOP and three algorithmicmethods) form consensus yet disagree with theAUTHORS assignment. Further discussion can befound in “What is difficult for expert methods:why experts disagree?” (below).
For the majority of these cases, it appears thatAUTHORS overcuts the chain either by cutting offsmall domains (Figure 9(F)) or by cutting througha densely packed structure (Figure 9(G)). This indi-cates that algorithms capture well some aspects ofwhat does not constitute a structural domain.Some assignment cases are less clear-cut: in thecase of chain 2hpda, a hemoprotein domain ofcytochrome, the additional domain can be justified(Figure 9(H)), since the structure consists of twospatially distinct parts. Thus, the fact that all algor-ithms and two expert methods (SCOP and CATH)assign this chain as one domain is surprising. It isnot clear whether this case indicates an error byall the algorithms or captures a fundamentalprinciple.
In one case, each of the experts produces a differ-ent assignment: for 1tnrr (tumor necrosis factor,chain R) SCOP assigns two domains (Figure 7(I)),CATH assigns three domains (Figure 9(J)) andAUTHORS assign four domains (Figure 9(K));algorithmic consensus agrees with the CATHassignment. This is a clear example of algorithms,guided by somewhat distinct principles, conver-ging on a domain assignment. Expert assignmentsmay incorporate additional knowledge (besidesdomain compactness) producing discord.
Finally, there is an interesting case, 1pxta,3-ketoacyl-CoA thiolase, for which algorithmicconsensus does not agree with any of the expertmethods: algorithmic methods do not cut the
chain (Figure 9(L)), CATH and SCOP predict twodomains (Figure 9(M) and (N)) and AUTHORSpredicts three domains (Figure 9(O)). This struc-ture is quite unusual and no two experts agreecompletely: SCOP and CATH both assign thesame number of domains, but partitioning of thedomains is different (Figure 9(M) and (N)). This isan example of a complex architecture that doesnot lend itself to unambiguous partitioning.
The consensus among algorithmic assignmentscan be subdivided into three general classes.
(1) Algorithmic consensus has more domainsthan one of the expert methods (usually SCOP),the assignment make sense, algorithms arecorrect.
(2) Algorithmic consensus has fewer domainsthan one of the expert methods (frequentlyAUTHORS, but others too) and introduction ofadditional domains is difficult to justify,algorithms are correct.
(3) Algorithmic consensus does not cut thestructure that could or should have been cut(some or all experts cut it), algorithms havedifficulties.
Our criteria for correctness of assignments(above) are subjective; we assume that the assign-ment is correct if the assigned domains conform tothe basic principles of independent structuralunits (compact structures with hydrophobic core,which can be clearly separated from each other).
In summary, consensus among algorithmsfrequently gives reasonable partitioning of chainsinto domains. This consistency indicates that thedriving force behind partitioning is retaining thestructural integrity of a domain. The lack ofconsensus among experts implies additionalknowledge is coming into play. A fraction ofalgorithmic consensus results from the algorithms’universal inability to partition complex architec-tures (algorithms undercut).
What is difficult for all methods?
For a significant fraction (10.4%) of chains,neither algorithms nor experts are able to come toagreement on partitioning of the chain. This subset,
(L) PDP, two domains (domain 1, 3–140, 247–320; domain 2, 141–246); (M) DomainParser, three domains (domain 1,3–89, 247–320; domain 2, 90–145; domain 3, 146–246); (N) DALI, CATH and AUTHORS, four domains (domain 1,3–87; domain 2, 88–167; domain 3, 168–246; domain 4, 247–320, given for AUTHORS); (O) and (P) 1smra mouserenin; (O) SCOP and DALI, one domain; (P) AUTHORS, CATH, PDP and DomainParser, two domains (domain 1,2–174, domain 2, 175–326, given for AUTHORS). (Q–S) 5fbpa fructose-1,6-biphosphatase; (Q) SCOP and Domain-Parser, one domain; (R) AUTHORS, CATH and PDP, two domains (domain 1, 6–201; domain 2, 202–335, given forAUTHORS); (S) DALI, four domains (domain 1, 9–51; domain 2, 72–117, 137–161; domain 3, 118–136, 162–201;domain 4, 202–335). (T–V) 1tahb triacylglycerol lipase; (T) CATH, SCOP, DALI and DomainParser, one domain;(U) PDP, two domains (domain 1, 2–104, 142–163; domain 2, 105–141, 164–319); (V) AUTHORS, three domains(domain 1, 2–117, 166–213,272–319; domain 2, 118–165; domain 3, 214–271). (W–Y) 1chra chloromuconate cyclo-isomerase; (W) DALI and DomainParser, one domain; (X) CATH, SCOP and PDP, two domains (domain 1, 1–127;domain 2, 128–329); (Y) AUTHORS, three domains (domain 1, 1–122; domain 2, 130–327; domain 3, 339–367).
Consistent Assignment of Protein Structural Domains 663

“no consensus” subset, represents chains that arechallenging for most or all methods. Inspection ofthis subset reveals unresolved problems in domainassignments.
Cases of chains with no expert oralgorithmic consensus
“No consensus” (49 chains, 10.4% of the dataset)is the subset where neither algorithmic nor expertmethods could come to an agreement. This set ofchains represents the most diverse set ofjudgments among methods; thus, the tendenciesof individual methods may be discernible. Weobserved that methods that generally tend to haveopposite propensities toward domain partitioning,such as SCOP (which tends to undercut) andDALI (which tends to overcut), agree for this sub-set while they disagree with all other methods(1ppke, 2cts, 1mpp, 3psg, 1smv, 1tahb; 1smv(Figure 10(O) and (P)) and 1tahb (Figure 10(T)–(V))). There are other chains in this subset inwhich SCOP assigns more domains than DALI, anuncharacteristic result (1dsba, 1chra;Figure 10(W)–(Y)). We find that it is possible tofind almost any combination of two methods (oneexpert and one algorithmic) that will agree amongthemselves in this subset. We observe also thatCATH, which appears to be “conciliatory” up tonow, agreeing with either with SCOP orAUTHORS (or both), often disagrees with bothSCOP and AUTHORS in this subset (1dsba, 1gky,1prcc, 1hvc, 1mioa, 1miob, 2dkb, 1tpla, 2hhmb,3mdda, 1lla, 1esl; Figure 10(B)).
The subset of chains with no consensus can bepartitioned into two principal groups. The firstgroup comprises chains in which some methodscut a small fragment off a structure and/or dividesmall structures into domains. The second groupconsists of chain with complex architecture, ofteninvolving two or more a/b structures that are diffi-cult to separate unambiguously into domains.
The tendency to cut off small domains is acharacteristic of several methods and the under-lying logic for each method appears to be different;
Figure 10(A)–(D) and Table 4 highlight some ofthese cases. DALI cuts off small structures thatconsist of a helix (Figure 10(C)) or an unstructuredregion (Figure 10(D)); however, it does not cut offsmall b-sheets (Figure 10(A) and (B)). DomainPar-ser is the opposite: it cuts off small b-sheets(Figure 10(A) and (B)), but leaves helices andunstructured regions uncut. AUTHORS cuts offsmall domains in all four cases (Figure 10(A)–(D)), whereas PDP cuts off small structures whenthere appears to be very little contact between thestructure and the rest of the chain, regardless ofthe domain size (Figure 10(B) and (D)). SCOP cutsthe structure in one case only (1esl, Figure 10(B)).Paradoxically, this is the smallest domain amongthe four cases (38 residues, EGF domain). CATHcuts off domains that are spatially well separatedand are not too small (Figure 10(A) and (D)).
We analyzed two cases where a small structure iscut into two domains of similar size. Chain 1hfh, acomplementary control module, comprises twowell-separated lobes; each is a small b-structure(Figure 10(F)). DALI and AUTHORS leave thisstructure uncut, while other methods divide itinto two similar domains. Chain 1l92 is a lyso-zyme; it has an all-a structure with a high densityof residue contacts (Figure 10(E)), yet AUTHORS,PDP and DomainParser split the structure. In bothof the above cases, AUTHORS predict assignmentsthat are counter-intuitive: it does not cut a chainwith two distinct lobes (1hfh, Figure 10(F)); how-ever, it does cut through the helix to subdivide ahelical structure into two domains. Thus,AUTHORS, which we use as a reference, occasion-ally tends to produce an irrational partitioning ofthe chains (see “Analysis of algorithmic methods:benchmarking using the consensus approach”,(below).
Complex architectures dominate the subset “noconsensus” (38 chains, 79% of the subset). In somecases, the six methods produce three or even fourdifferent domain assignments, which are difficultto discern. Consider six examples.
The first example is an annexin (1avha) with anall-a structure. This chain has four distinct domainassignments: SCOP assigns a single domain(Figure 10(K)), although it also indicates in thecomments that it consist of the four domains ofthe same fold; PDP assigns two domains(Figure 10(L)); DomainParser assigns three domains(Figure 10(M)); AUTHORS, CATH and DALI assignfour domains, although the boundaries are differentfor the three methods. In this case none of the algor-ithms produces a completely correct assignment(assuming four domains to be correct), DALI pre-dicts the correct number of domains, but leaves 45residues (53% of the domain) between domain 1and 2 unassigned. If we include the SCOP annotationincluded in the fold (which is not available in com-puter-readable form), all three expert assignmentmethods come to a consensus.
The second example is a glycogen phosphoryl-ase (1gpb). This is a large chain (841 residues)
Table 4. Analysis of six chains with small domains
Number of domains in
Assignment method1esl(8B)
2snv(8A)
1iag(8C)
1hmy(8D)
1l92(8E)
1hfh(8F)
PDP 2 1 1 2 2 2DomainParser 2 2 1 1 2 2DALI 1 1 2 2 1 1AUTHORS 2p 2p 2p 2p 2p 1CATH 1 2 1 2 1 2p
SCOP 2 1 1 1 1 2
For each chain the number of domains assigned appears inthe corresponding method row. An asterisk ( p ) indicates themethod used for displaying domain boundaries of the corre-sponding chain in Figure 10.
664 Consistent Assignment of Protein Structural Domains

with a complex a/b architecture (Figure 10(G)–(J)). SCOP keeps it intact, although in the com-ments it indicates two non-similar domains withthree layers each (Figure 10(G)); CATH, PDP andAUTHORS divide it into two domains: a three-layer transferase domain and a complex glycogenphosphorylase domain (CATH nomenclature,Figure 10(H)), DomainParser assigns threedomains (Figure 10(I)); DALI assigns five domains(Figure 10(J)). Similar to the previous example,expert methods would agree on the domain assign-ments if we consider SCOP annotation (SCOP’smethodology is discussed below).
The third example is an acid protease (1smr), anall-b structure. It has two similar domains, identifiedby all methods (Figure 10(P)), with the exception ofSCOP and DALI (Figure 10(O)), which keep thechain uncut. This is one of the cases where twomethods with generally opposing tendencies agree.
The fourth example is a sugar phosphatase(5fbp). This is an example of a relatively smallchain with a confusing architecture, which is diffi-cult for algorithmic methods (three methodsproduce three different assignments). SCOP andDomainParser keep the chain uncut (Figure 10(Q));AUTHORS, CATH and PDP predict two domains(a/b and a þ b, Figure 10(R)); DALI predicts fourdomains through splitting the a/b domain intothree parts (Figure 10(S)).
The fifth example is a carboxylic esterase (1tahb;Figure 10(T)–(V)); its 319 residues form a compactstructure. Most methods leave it uncut; however,PDP cuts it into two domains of similar size alongthe b-sheet axis. AUTHORS partitions the chaininto three domains: (1) a core domain, which con-sists of six b-sheets flanked by a-helices on bothsides; (2) a small a-helical structure that representa movable lid to the active site; and (3) a smalla/b structure that extends from the body of theprotein and contains two of the three catalyticresidues.
The final example is a chloromuconate cyclo-isomerase (1chr) with a complex architecture. Twoof the algorithmic methods, DALI and Domain-Parser, keep it intact (Figure 10(W)), SCOP andPDP predict two domains (Figure 10(X)),AUTHORS predicts three domains by promoting avery short C-terminal end to a status of domain(Figure 10(Y)). This is one of the examples wherenone of the assignment methods agree withAUTHORS.
In summary, as the structures get more complex,domain partitioning diverges further. Hence, thecomplex structures that populate the “no consen-sus” subset are possibly the best grounds toexplore the full range of an individual method’scapabilities and tendencies. Analysis of the alterna-tive partitioning produced by expert methodsshould stimulate a discussion about the set ofworkable principles for partitioning a chain intodomains. An initial discussion is presented in thesection “What are the central issues in thedefinition of structural domains?”
What is difficult for expert methods: whyexperts disagree?
Difficult cases for experts are those structures forwhich experts disagree on the number of assigneddomains and/or on domains boundaries. Whenwe consider the number of assigned domains,experts’ consensus cannot be reached for 93 chains(20% of 467 chain consensus): 43 chains belong tothe subset for which algorithmic methods agreeon the domain assignment (algorithmic only con-sensus); the remaining 49 chains represent caseswhere there is no consensus (neither amongalgorithmic nor among expert methods). We detailindividual cases for which there is no experts’consensus.
AUTHORS19
AUTHORS assignment represents the least con-sistent dataset, since assignments are done bymany individuals with varying opinions onstructural domain definitions. When AUTHORSdisagrees with other expert methods, the tendencyof AUTHORS assignments is to partition a rela-tively compact substructure into two domains.Such partitioning contradicts the basic concepts ofstructural domains, as the partitioning disturbsmany contacts between secondary structures(1mat, Figure 9(G); 1lga, Figure 9(E); 1tahb,Figure 10(V)). Often the extra domains are alsoquite small (in addition to being in close proximityto the main body of the domain), thus, assignmentof such domains contradicts the basic principles ofstructural domains as compact and semi-indepen-dent units (1iag, Figure 10(C); 1l92, Figure 10(E);1tahb, Figure 10(V); 1chra, Figure 10(Y); 1cpca/cpcl, Figure 9(F); 1pxta, Figure 9(O); 3gly,Figure 5(D)). There is only one case for whichAUTHORS assigns fewer domains than otherexpert methods and in this case the assignment ismost likely wrong (1hfh, Figure 10(F)), since thestructure has two clear lobes. Some of the caseswhere AUTHORS disagree with other expertmethods appear quite plausible (1caub, 2hpda,1ppn; Figure 11(A)–(C)).
Domain boundaries. Not applicable, sinceAUTHORS is a reference method for calculation ofdomain boundaries.
SCOP31
SCOP tends to leave large structures uncut,while other experts cut them into two or moredomains. The underlying reason for this is the factthat SCOP uses evolutionary and structuralrelationships to define domains. According toSCOP’s authors, “A domain is defined as anevolutionary unit, in the sense that it is eitherobserved in isolation in nature, or in more thanone context in different multi-domain proteins”.31
Thus, SCOP tends to define a domain as the largestrecurrent unit, rather than smallest independent
Consistent Assignment of Protein Structural Domains 665
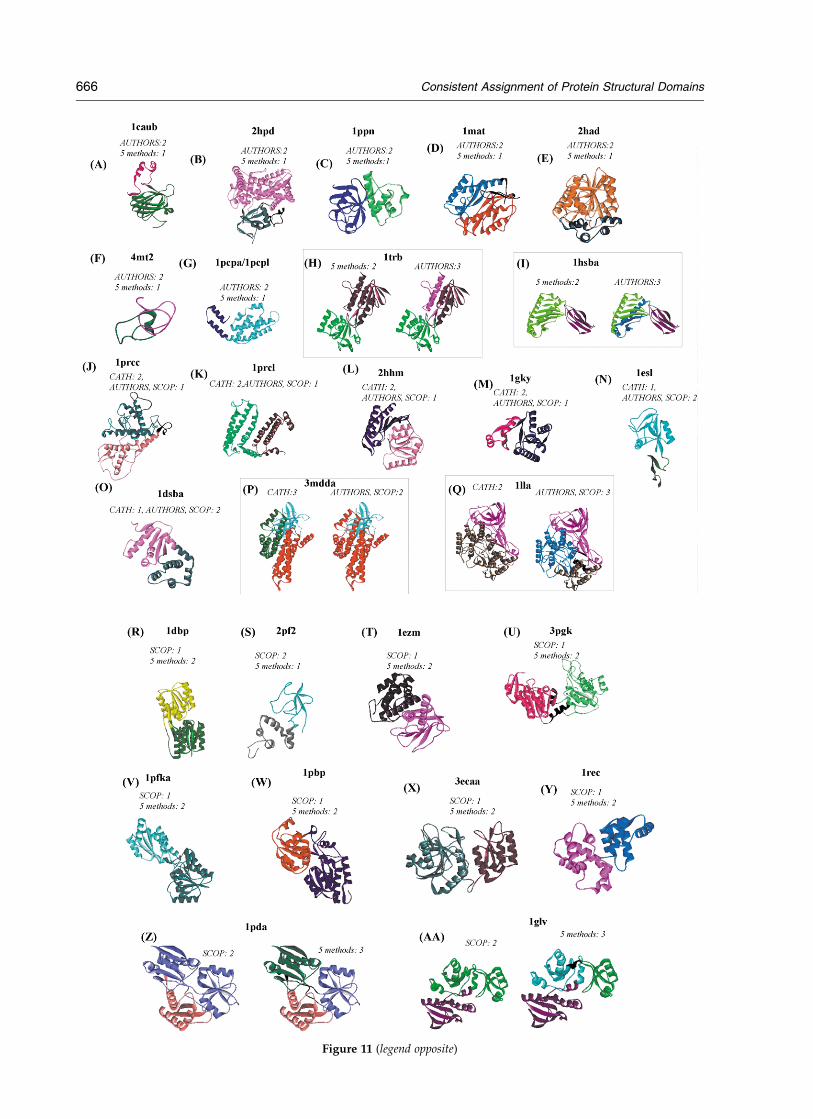
Figure 11 (legend opposite)
666 Consistent Assignment of Protein Structural Domains
structural unit. This approach constitutes the basisof SCOP classification and it is at this level ofrecurrent fold (the Astral compendium) thatvirtually everybody in structural biology usesSCOP domains.18,32 – 34 A recurrent fold may consistof two or more structurally independent units; inmany of such cases SCOP entries are annotatedwith topological description (but no boundariesare provided) for each structural unit, referred as“domains”. This annotation is consistent with thedescription of a domain as an independentstructural unit. If we were to consider SCOP dataat the level of annotation, there would be fewerdisagreement cases among the experts and SCOPbenchmarking results would improve (seeFigure 1(A)). This is not, however, how the vastmajority of structural biologists use SCOP data,because it is inconsistent with SCOP notion of adomain as a recurrent evolutionary unit andbecause partitioning on the level of structuralunits in not provided by SCOP in any systematicform. Therefore, our analysis, with the exception
of benchmarking results, uses SCOP domains asdefined by Astral.
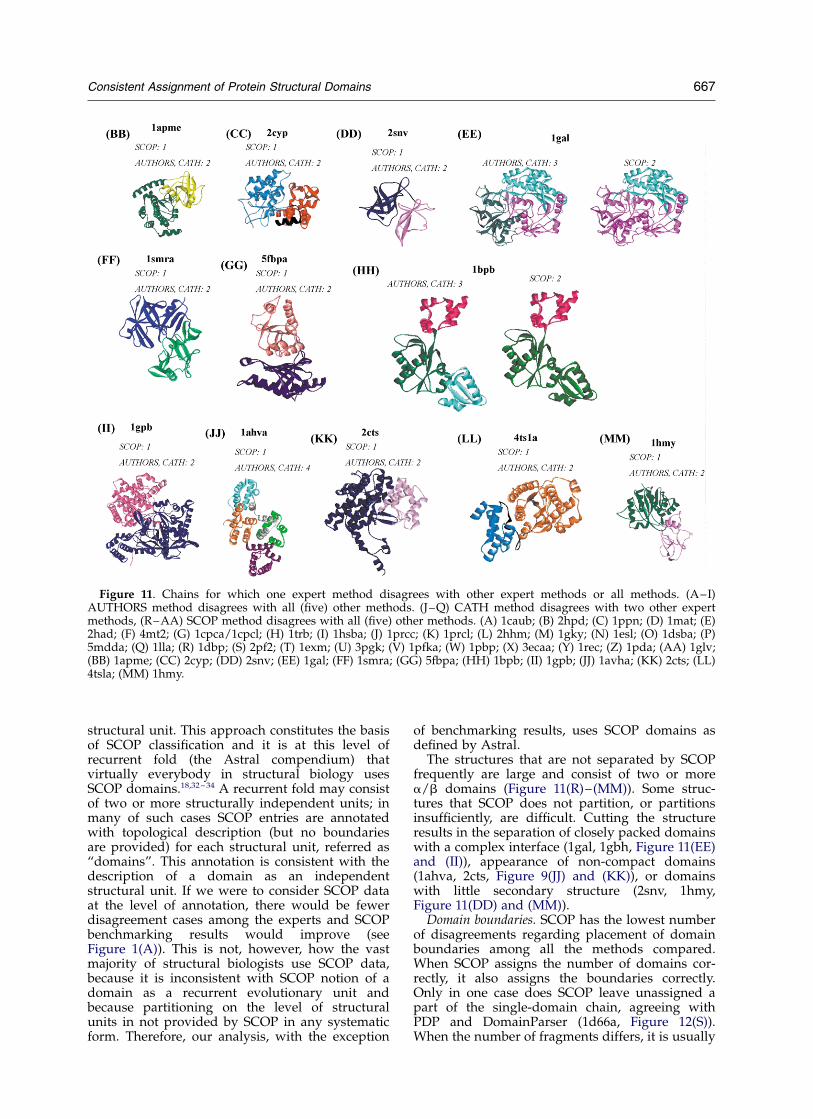
The structures that are not separated by SCOPfrequently are large and consist of two or morea/b domains (Figure 11(R)–(MM)). Some struc-tures that SCOP does not partition, or partitionsinsufficiently, are difficult. Cutting the structureresults in the separation of closely packed domainswith a complex interface (1gal, 1gbh, Figure 11(EE)and (II)), appearance of non-compact domains(1ahva, 2cts, Figure 9(JJ) and (KK)), or domainswith little secondary structure (2snv, 1hmy,Figure 11(DD) and (MM)).
Domain boundaries. SCOP has the lowest numberof disagreements regarding placement of domainboundaries among all the methods compared.When SCOP assigns the number of domains cor-rectly, it also assigns the boundaries correctly.Only in one case does SCOP leave unassigned apart of the single-domain chain, agreeing withPDP and DomainParser (1d66a, Figure 12(S)).When the number of fragments differs, it is usually
Figure 11. Chains for which one expert method disagrees with other expert methods or all methods. (A–I)AUTHORS method disagrees with all (five) other methods. (J–Q) CATH method disagrees with two other expertmethods, (R–AA) SCOP method disagrees with all (five) other methods. (A) 1caub; (B) 2hpd; (C) 1ppn; (D) 1mat; (E)2had; (F) 4mt2; (G) 1cpca/1cpcl; (H) 1trb; (I) 1hsba; (J) 1prcc; (K) 1prcl; (L) 2hhm; (M) 1gky; (N) 1esl; (O) 1dsba; (P)5mdda; (Q) 1lla; (R) 1dbp; (S) 2pf2; (T) 1exm; (U) 3pgk; (V) 1pfka; (W) 1pbp; (X) 3ecaa; (Y) 1rec; (Z) 1pda; (AA) 1glv;(BB) 1apme; (CC) 2cyp; (DD) 2snv; (EE) 1gal; (FF) 1smra; (GG) 5fbpa; (HH) 1bpb; (II) 1gpb; (JJ) 1avha; (KK) 2cts; (LL)4tsla; (MM) 1hmy.
Consistent Assignment of Protein Structural Domains 667
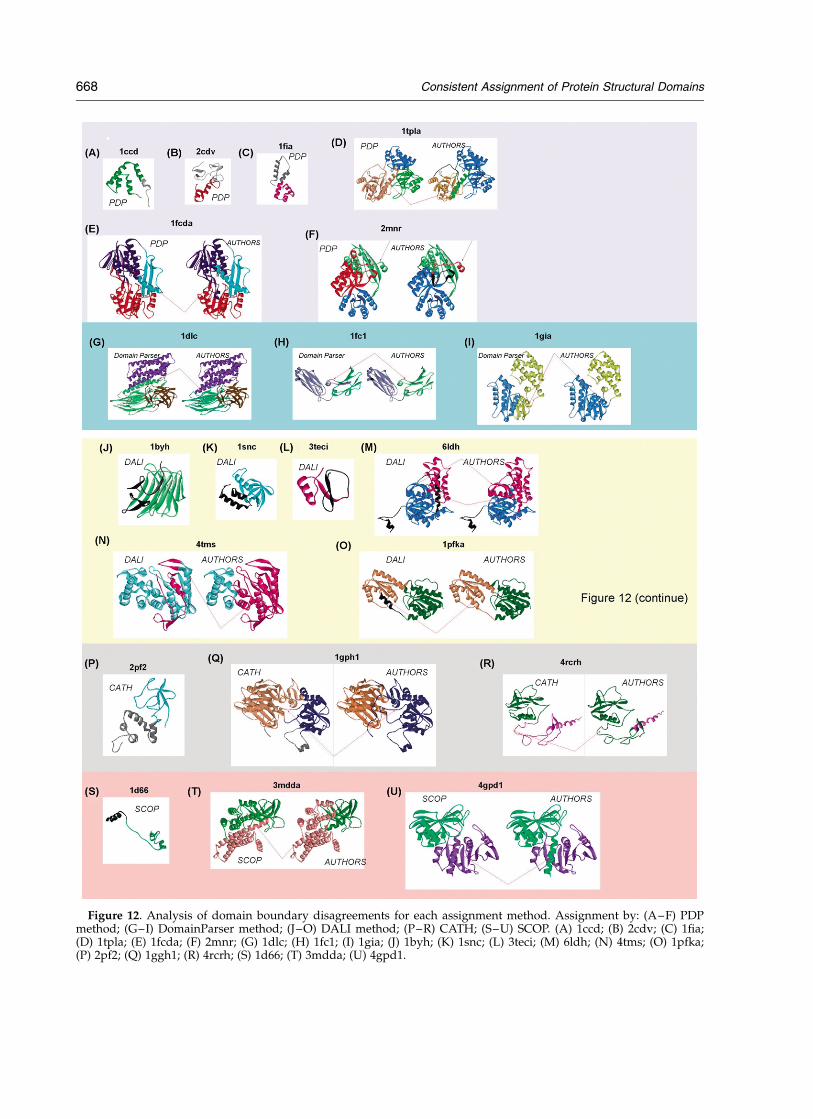
Figure 12. Analysis of domain boundary disagreements for each assignment method. Assignment by: (A–F) PDPmethod; (G–I) DomainParser method; (J–O) DALI method; (P–R) CATH; (S–U) SCOP. (A) 1ccd; (B) 2cdv; (C) 1fia;(D) 1tpla; (E) 1fcda; (F) 2mnr; (G) 1dlc; (H) 1fc1; (I) 1gia; (J) 1byh; (K) 1snc; (L) 3teci; (M) 6ldh; (N) 4tms; (O) 1pfka;(P) 2pf2; (Q) 1ggh1; (R) 4rcrh; (S) 1d66; (T) 3mdda; (U) 4gpd1.
668 Consistent Assignment of Protein Structural Domains

because SCOP assigns fewer fragments and as aresult the partitioning of the domains is less thanoptimal (3mdda, Figure 12(T)). Finally, there is acase for which SCOP (along with PDP and DALImethods) splits a continuous domain (in thereference method) into fragments in order toachieve compactness of the resulting domains(4gpd1, Figure 12(U)).
CATH20
CATH appears in this analysis as the most“reasonable” method, agreeing with one of thetwo remaining expert assignments (and usuallysiding with a more “intuitive” partitioning of thechain). CATH rarely disagrees with both expertmethods and in those cases one or two algorithmicassignments side with CATH (the exception is 1lla,where CATH disagrees with all the algorithmicassignments). There are eight chains for whichCATH dissents from both AUTHORS and SCOP(Figure 11(J)–(Q)). These cases are intriguing,because they are the chains for which methodswith opposing tendencies (AUTHORS and SCOP)agree while CATH disagrees. Chains in thiscategory are either all-a or a/b, consisting mostlyof a-helices. We do not detect an obviouspreference on the part of CATH to overcut or toundercut: in three cases, CATH splits the a-struc-ture into two domains (1prcl, 2hhm, 3mdda,Figure 11(K), (L) and (P)), while the other twoexpert methods keep it intact. In another case itdoes not split the a-structure when other expertsdo (1lla, Figure 11(Q)). In two cases, CATH splitthe a/b structure into two domains (other expertskeep it as one domain; 2hhm, 1gky, Figure 11(L)and (M)). Yet in two other cases CATH keeps thechain intact, while other experts divide it into twodomains (1esl, 1dsba, Figure 11(N) and (O)). Weobserve that in both cases of multi-domain chains,when alternative assignments produce two-domain or three-domain chains, the additionaldomain “evolves” from one fragment of a dis-continuous domain (3mdda, 1lla Figure 11(P) and(Q)). Most of the above assignments are not clear-cut and may point to another genuine problem indomain definition, i.e. difficulty deciding what todo with large all-a or mostly-a structures.
Domain boundaries. CATH disagrees withAUTHORS on the placement of boundaries in 24cases at the stringency level of 95% and in fivecases at the stringency level of 80%. CATH tendsto produce more fragments per domain thanAUTHORS. There is only one case of the single-domain chain for which CATH agrees with PDPand DALI and leaves 30% of the chain, comprisingseveral small helices, unassigned (2pf2,Figure 12(P)). In this particular case, SCOP givesthe unassigned region the status of a separatedomain. Some of the misassignments by CATHinvolve different positioning of the boundarywithin the loop region (4rcrh, Figure 12(R));however, several methods differ in boundary
placement in this particular case. In the case of4gph (Figure 12(Q)), CATH introduces severaladditional fragments within each of two domainsto accommodate the correct assignment of loopsthat cross multiple times between the two maindomains.
Of the three expert methods, AUTHORS, CATHand SCOP, there is a strong tendency of AUTHORSto overcut, a strong tendency of SCOP to undercut,while CATH keeps the middle ground, agreeingeither with SCOP or with AUTHORS. AUTHORSdisagrees with the other two experts (which agreeamong themselves) in 23 cases; SCOP disagrees(under similar circumstances) in 33 cases; whileCATH disagrees with both methods (under similarcircumstances) in eight cases. There is one case inwhich all expert methods disagree among them-selves (1tnrr, Figure 91-K). An alternative (morestringent) way to look at the statistics of agreementamong expert methods is to check the number ofcases for which all other methods (two expert andthree algorithmic methods) agree among them-selves and disagree with a given expert method.There are ten chains for which five methods dis-agree with AUTHORS; 17 chains for which fivemethods disagree with SCOP; and no instance inwhich five methods disagree with CATH (Table 5).Correspondence between domain boundaries isvery good for all expert methods.
In summary, differences among experts’methods result from conceptually differentapproaches to domain definition. AUTHORS,while the most diverse, exhibits a strong tendencytoward identifying small functional regions asdomains. This approach results in some domainsthat are structurally unstable or inseparable fromthe rest of the structure. SCOP partitions structureonly to the largest recurring unit, which often canbe, but is not, separated further into smaller
Table 5. Chains for which all or most methods disagreewith expert method
Criteria for selection Chains
Five methods disagreewith AUTHORS
1ppn(1:2), 1caub(1:2), 2hpda(1:2),1hsba (2;3), 2had (1:2), 1trb(2:3),1cpca (1:2), 1cpcl (1:2), 4mt2 (1:2),1mat (1:2)
Five methods disagreewith SCOP
1dbp(2:1), 2pf2 (1:2), 2liv (2:1), 1ezm(2:1), 3gbp (2:1), 3pgk (2:1), 1rec(2:1), 8abp (2:1), 1glv (3:2), 1pbp(2:1), 1osa (2:1), 1pfka (2:1), 1pda(3:2), 1lao (2:1), 3tmne (2:1), 3ecaa(2:1), 1sbp (2:1)
Five methods disagreewith CATH
None
Following the name of the chain, the number in parenthesesindicates the number of domains: the first number indicates thenumber of domains agreed to by five methods, the secondnumber is the number of domains predicted by the dissentingexpert method. The underlined chains for SCOP are those forwhich SCOP provides manual annotation that matches domainassignment by other methods.
Consistent Assignment of Protein Structural Domains 669
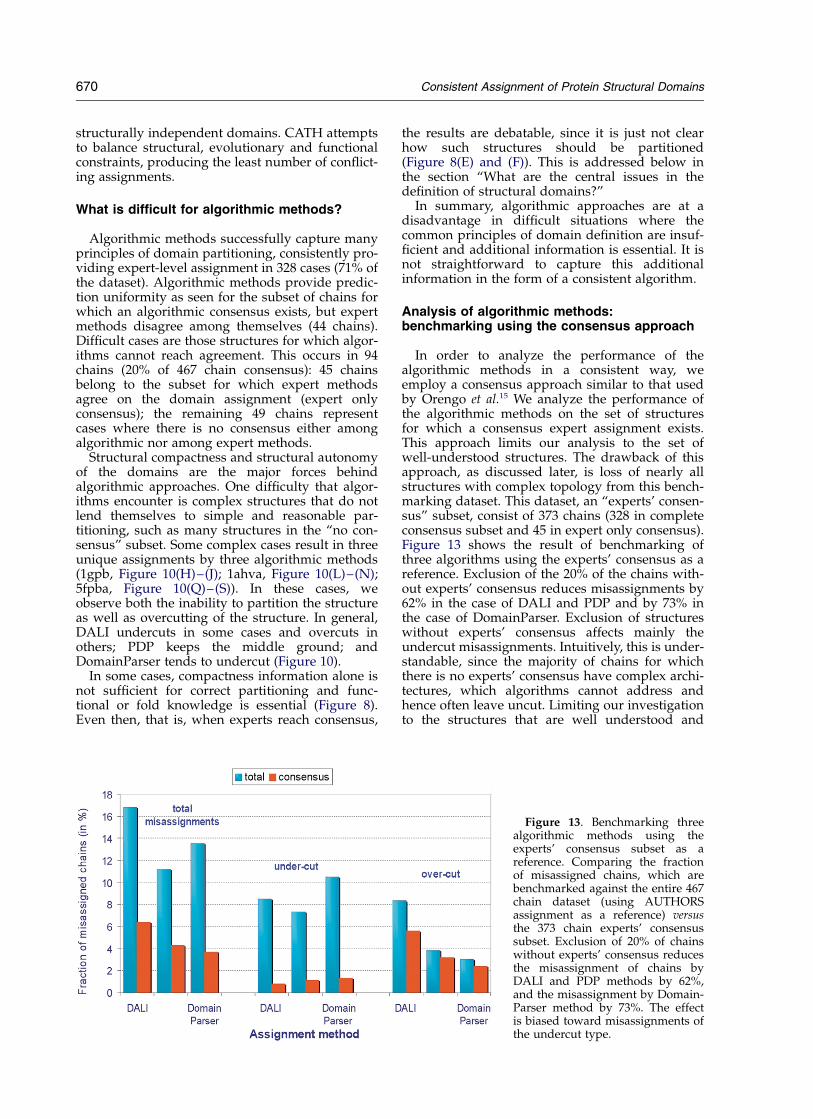
structurally independent domains. CATH attemptsto balance structural, evolutionary and functionalconstraints, producing the least number of conflict-ing assignments.
What is difficult for algorithmic methods?
Algorithmic methods successfully capture manyprinciples of domain partitioning, consistently pro-viding expert-level assignment in 328 cases (71% ofthe dataset). Algorithmic methods provide predic-tion uniformity as seen for the subset of chains forwhich an algorithmic consensus exists, but expertmethods disagree among themselves (44 chains).Difficult cases are those structures for which algor-ithms cannot reach agreement. This occurs in 94chains (20% of 467 chain consensus): 45 chainsbelong to the subset for which expert methodsagree on the domain assignment (expert onlyconsensus); the remaining 49 chains representcases where there is no consensus either amongalgorithmic nor among expert methods.
Structural compactness and structural autonomyof the domains are the major forces behindalgorithmic approaches. One difficulty that algor-ithms encounter is complex structures that do notlend themselves to simple and reasonable par-titioning, such as many structures in the “no con-sensus” subset. Some complex cases result in threeunique assignments by three algorithmic methods(1gpb, Figure 10(H)–(J); 1ahva, Figure 10(L)–(N);5fpba, Figure 10(Q)–(S)). In these cases, weobserve both the inability to partition the structureas well as overcutting of the structure. In general,DALI undercuts in some cases and overcuts inothers; PDP keeps the middle ground; andDomainParser tends to undercut (Figure 10).
In some cases, compactness information alone isnot sufficient for correct partitioning and func-tional or fold knowledge is essential (Figure 8).Even then, that is, when experts reach consensus,
the results are debatable, since it is just not clearhow such structures should be partitioned(Figure 8(E) and (F)). This is addressed below inthe section “What are the central issues in thedefinition of structural domains?”
In summary, algorithmic approaches are at adisadvantage in difficult situations where thecommon principles of domain definition are insuf-ficient and additional information is essential. It isnot straightforward to capture this additionalinformation in the form of a consistent algorithm.
Analysis of algorithmic methods:benchmarking using the consensus approach
In order to analyze the performance of thealgorithmic methods in a consistent way, weemploy a consensus approach similar to that usedby Orengo et al.15 We analyze the performance ofthe algorithmic methods on the set of structuresfor which a consensus expert assignment exists.This approach limits our analysis to the set ofwell-understood structures. The drawback of thisapproach, as discussed later, is loss of nearly allstructures with complex topology from this bench-marking dataset. This dataset, an “experts’ consen-sus” subset, consist of 373 chains (328 in completeconsensus subset and 45 in expert only consensus).Figure 13 shows the result of benchmarking ofthree algorithms using the experts’ consensus as areference. Exclusion of the 20% of the chains with-out experts’ consensus reduces misassignments by62% in the case of DALI and PDP and by 73% inthe case of DomainParser. Exclusion of structureswithout experts’ consensus affects mainly theundercut misassignments. Intuitively, this is under-standable, since the majority of chains for whichthere is no experts’ consensus have complex archi-tectures, which algorithms cannot address andhence often leave uncut. Limiting our investigationto the structures that are well understood and
Figure 13. Benchmarking threealgorithmic methods using theexperts’ consensus subset as areference. Comparing the fractionof misassigned chains, which arebenchmarked against the entire 467chain dataset (using AUTHORSassignment as a reference) versusthe 373 chain experts’ consensussubset. Exclusion of 20% of chainswithout experts’ consensus reducesthe misassignment of chains byDALI and PDP methods by 62%,and the misassignment by Domain-Parser method by 73%. The effectis biased toward misassignments ofthe undercut type.
670 Consistent Assignment of Protein Structural Domains
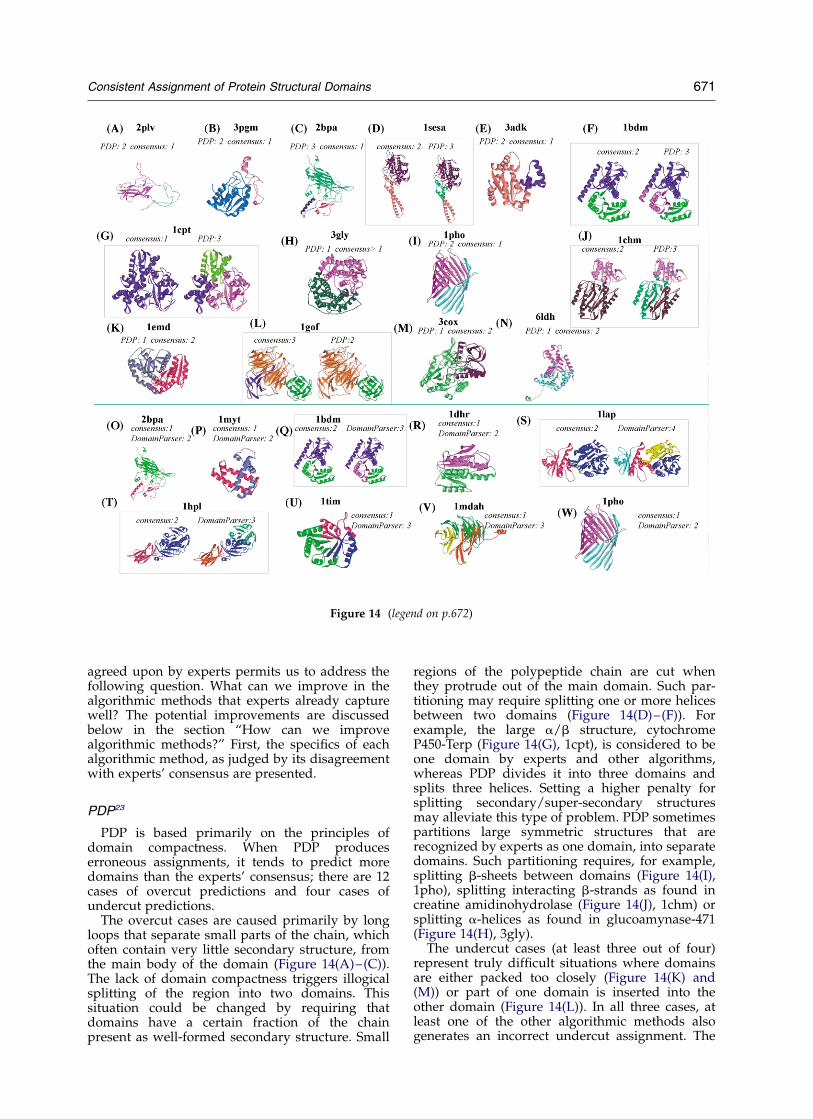
agreed upon by experts permits us to address thefollowing question. What can we improve in thealgorithmic methods that experts already capturewell? The potential improvements are discussedbelow in the section “How can we improvealgorithmic methods?” First, the specifics of eachalgorithmic method, as judged by its disagreementwith experts’ consensus are presented.
PDP23
PDP is based primarily on the principles ofdomain compactness. When PDP produceserroneous assignments, it tends to predict moredomains than the experts’ consensus; there are 12cases of overcut predictions and four cases ofundercut predictions.
The overcut cases are caused primarily by longloops that separate small parts of the chain, whichoften contain very little secondary structure, fromthe main body of the domain (Figure 14(A)–(C)).The lack of domain compactness triggers illogicalsplitting of the region into two domains. Thissituation could be changed by requiring thatdomains have a certain fraction of the chainpresent as well-formed secondary structure. Small
regions of the polypeptide chain are cut whenthey protrude out of the main domain. Such par-titioning may require splitting one or more helicesbetween two domains (Figure 14(D)–(F)). Forexample, the large a/b structure, cytochromeP450-Terp (Figure 14(G), 1cpt), is considered to beone domain by experts and other algorithms,whereas PDP divides it into three domains andsplits three helices. Setting a higher penalty forsplitting secondary/super-secondary structuresmay alleviate this type of problem. PDP sometimespartitions large symmetric structures that arerecognized by experts as one domain, into separatedomains. Such partitioning requires, for example,splitting b-sheets between domains (Figure 14(I),1pho), splitting interacting b-strands as found increatine amidinohydrolase (Figure 14(J), 1chm) orsplitting a-helices as found in glucoamynase-471(Figure 14(H), 3gly).
The undercut cases (at least three out of four)represent truly difficult situations where domainsare either packed too closely (Figure 14(K) and(M)) or part of one domain is inserted into theother domain (Figure 14(L)). In all three cases, atleast one of the other algorithmic methods alsogenerates an incorrect undercut assignment. The
Figure 14 (legend on p.672)
Consistent Assignment of Protein Structural Domains 671
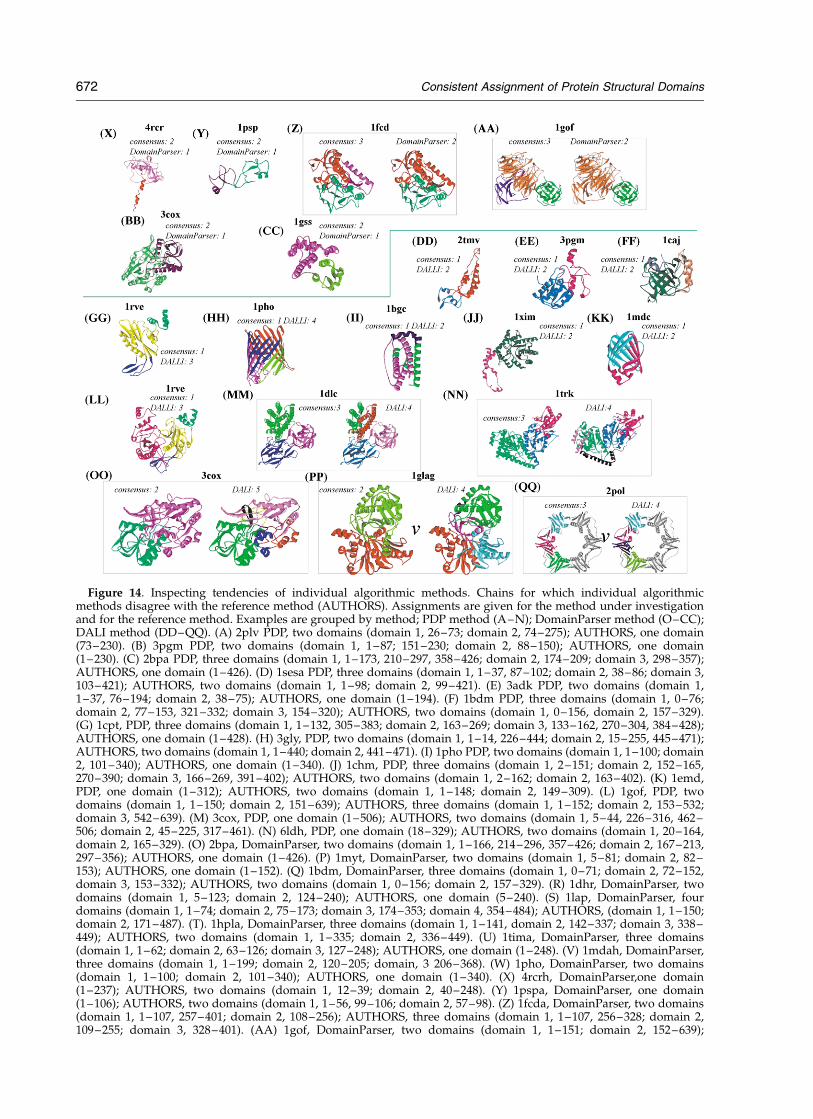
Figure 14. Inspecting tendencies of individual algorithmic methods. Chains for which individual algorithmicmethods disagree with the reference method (AUTHORS). Assignments are given for the method under investigationand for the reference method. Examples are grouped by method; PDP method (A–N); DomainParser method (O–CC);DALI method (DD–QQ). (A) 2plv PDP, two domains (domain 1, 26–73; domain 2, 74–275); AUTHORS, one domain(73–230). (B) 3pgm PDP, two domains (domain 1, 1–87; 151–230; domain 2, 88–150); AUTHORS, one domain(1–230). (C) 2bpa PDP, three domains (domain 1, 1–173, 210–297, 358–426; domain 2, 174–209; domain 3, 298–357);AUTHORS, one domain (1–426). (D) 1sesa PDP, three domains (domain 1, 1–37, 87–102; domain 2, 38–86; domain 3,103–421); AUTHORS, two domains (domain 1, 1–98; domain 2, 99–421). (E) 3adk PDP, two domains (domain 1,1–37, 76–194; domain 2, 38–75); AUTHORS, one domain (1–194). (F) 1bdm PDP, three domains (domain 1, 0–76;domain 2, 77–153, 321–332; domain 3, 154–320); AUTHORS, two domains (domain 1, 0–156, domain 2, 157–329).(G) 1cpt, PDP, three domains (domain 1, 1–132, 305–383; domain 2, 163–269; domain 3, 133–162, 270–304, 384–428);AUTHORS, one domain (1–428). (H) 3gly, PDP, two domains (domain 1, 1–14, 226–444; domain 2, 15–255, 445–471);AUTHORS, two domains (domain 1, 1–440; domain 2, 441–471). (I) 1pho PDP, two domains (domain 1, 1–100; domain2, 101–340); AUTHORS, one domain (1–340). (J) 1chm, PDP, three domains (domain 1, 2–151; domain 2, 152–165,270–390; domain 3, 166–269, 391–402); AUTHORS, two domains (domain 1, 2–162; domain 2, 163–402). (K) 1emd,PDP, one domain (1–312); AUTHORS, two domains (domain 1, 1–148; domain 2, 149–309). (L) 1gof, PDP, twodomains (domain 1, 1–150; domain 2, 151–639); AUTHORS, three domains (domain 1, 1–152; domain 2, 153–532;domain 3, 542–639). (M) 3cox, PDP, one domain (1–506); AUTHORS, two domains (domain 1, 5–44, 226–316, 462–506; domain 2, 45–225, 317–461). (N) 6ldh, PDP, one domain (18–329); AUTHORS, two domains (domain 1, 20–164,domain 2, 165–329). (O) 2bpa, DomainParser, two domains (domain 1, 1–166, 214–296, 357–426; domain 2, 167–213,297–356); AUTHORS, one domain (1–426). (P) 1myt, DomainParser, two domains (domain 1, 5–81; domain 2, 82–153); AUTHORS, one domain (1–152). (Q) 1bdm, DomainParser, three domains (domain 1, 0–71; domain 2, 72–152,domain 3, 153–332); AUTHORS, two domains (domain 1, 0–156; domain 2, 157–329). (R) 1dhr, DomainParser, twodomains (domain 1, 5–123; domain 2, 124–240); AUTHORS, one domain (5–240). (S) 1lap, DomainParser, fourdomains (domain 1, 1–74; domain 2, 75–173; domain 3, 174–353; domain 4, 354–484); AUTHORS, (domain 1, 1–150;domain 2, 171–487). (T). 1hpla, DomainParser, three domains (domain 1, 1–141, domain 2, 142–337; domain 3, 338–449); AUTHORS, two domains (domain 1, 1–335; domain 2, 336–449). (U) 1tima, DomainParser, three domains(domain 1, 1–62; domain 2, 63–126; domain 3, 127–248); AUTHORS, one domain (1–248). (V) 1mdah, DomainParser,three domains (domain 1, 1–199; domain 2, 120–205; domain, 3 206–368). (W) 1pho, DomainParser, two domains(domain 1, 1–100; domain 2, 101–340); AUTHORS, one domain (1–340). (X) 4rcrh, DomainParser,one domain(1–237); AUTHORS, two domains (domain 1, 12–39; domain 2, 40–248). (Y) 1pspa, DomainParser, one domain(1–106); AUTHORS, two domains (domain 1, 1–56, 99–106; domain 2, 57–98). (Z) 1fcda, DomainParser, two domains(domain 1, 1–107, 257–401; domain 2, 108–256); AUTHORS, three domains (domain 1, 1–107, 256–328; domain 2,109–255; domain 3, 328–401). (AA) 1gof, DomainParser, two domains (domain 1, 1–151; domain 2, 152–639);
672 Consistent Assignment of Protein Structural Domains

last undercut case, M4 apo-lactate dehydrogenase(6ldh, Figure 14(N)) is unique to PDP, all otheralgorithms are able to partition the chain into twodomains. PDP fails as a result of the large numberof contacts between domains as well as a lack ofcompactness.
Domain boundary. PDP exhibits 21 cases ofboundary differences in continuous domains and25 cases in non-continuous domains compared toAUTHORS assignments. The largest group (20chains) comprises single-domain chains in whichN-terminal (13 cases) or C-terminal (seven cases)boundaries do not match the end of the chain.This set contains two single-domain chains thatconsist of two fragments each, with the middlefragment of approximately 20 residues unassigned(2cy3 and 2tmvp). This problem highlights one ofthe basic inconsistencies between methods: in thesingle-domain cases some methods (SCOP, CATHand DomainParser) assign the entire chain as adomain, while other methods (PDP, DALI andAUTHORS) may leave part of the chainunassigned. PDP assignments sometimes leaveshort stretches unassigned, but in 11 cases at least20% of residues are left unassigned. Of these 11,two cases leave 45% and 60% of residues at one ofthe end of the chain unassigned (Figure 12(A)–(C)). Another type of disagreement is partitioningof non-continuous domains; such partitioningoften results in an excessive number of fragmentsper domain. The creation of extra fragments doesnot necessarily cause severe misassignment. Thedifference in the number of fragments betweentwo assignments can result in rather minor differ-ences of domain boundaries, as in the case of1fcda (Figure 12(E)). On the other hand, the mis-placement of the boundaries among the domains
with the same number of fragments can havedramatic impact, as in the case of 1tpla(Figure 12(D)). It is possible to have identicaldomain assignment in terms of domain number,but very different assignment when domainboundaries are considered. In the case ofbacterioops in fragment (2 nmr; Figure 12(F)), bothPDP and AUTHORS partition the chain into threedomains; however, the domains are quite different.The C-terminal domain in the AUTHORS assign-ment consists of a short loop and a helix, compris-ing 29 residues. In the PDP assignment, this smalldomain appears as a part of the larger domain. Atthe same time, PDP splits a large a/b domain(define by AUTHORS as one) into two domains.
In summary, PDP requires compact domains.Small to medium (35-50 residue) protrudingsegments are assigned to separate domains andsecondary structures are often split. Domainsoften have multiple fragments and unassignedregions.
DomainParser35
DomainParser is based on finding the minimalcut through domain interfaces and gives ninecases of overcut predictions and six cases of under-cut predictions when compared to the experts’ con-sensus. Generally, DomainParser does not splitsecondary structures nor does it generate domainswith very little or no secondary structure. Overcutcases are observed in: (1) sprawled structure withlong loops, such as 2bpa (Figure 14(O)), whichgenerates overcut predictions in all three algorith-mic methods (DomainParser and DALI predicttwo domains, while PDP predicts three domains);(2) helical structures with little interaction between
AUTHORS, three domains (domain 1, 1–152; domain 2, 153–532; domain 3, 542–639). (BB) 3cox, DomainParser, onedomain (1–500); AUTHORS, two domains (domain 1, 5–44, 226–316, 462–506; domain 2, 45–225, 317–461). (CC)1gssa, DomainParser, one domain (1–209); AUTHORS, two domains (domain 1, 1–78; domain 2, 81–207). (DD) 2tmv,DALI, two domains (domain 1, 5–11, 55–68, 139–153; domain 2, 14–54, 69–138); AUTHORS, one domain (1–154).(EE) 3pgm, DALI, two domains (domain 1, 1–10, 27–81, 159–230; domain 2, 11–26, 82–158); AUTHORS, one domain(1–230). (FF) 1caj, DALI, two domains (domain 1, 26–148, 173–216, 242–261; domain 2, 149–172, 217–241);AUTHORS, one domain (3–261). (GG) 1rvea, DALI, three domains (domain 1, 2–18, 36–139, 165–205; domain 2,19–35, 140–164; domain 3, 206–245); AUTHORS, one domain (2–245). (HH) 1pho, DALI, four domains (domain 1,17–40, 304–340; domain 2, 103–121, 180–191, 220–233,260–280, 296–303; domain 3, 1–16, 41–102, 133–158; domain4, 122–132, 159–179, 192–219, 234–259, 281–295); AUTHORS, one domain (1–340). (II) 1bgc, DALI, two domains(domain 1, 9–35, 88–111; domain 2, 57–87, 122–173); AUTHORS, one domain (9–173). (JJ) 1xima, DALI, two domains(domain 1, 3–320; domain 2, 321–394); AUTHORS, one domain (3–328). (KK) 1mdc, DALI, two domains (domain 1,1–13, 35–67, 104–131; domain 2, 14–33, 68–103); AUTHORS, one domain (1–132). (LL) 1rvea, DALI, three domains(domain 1, 2–18, 36–139, 165–205; domain 2, 19–35, 140–164; domain 3, 206–245); AUTHORS, one domain (2–245).(MM) 1dlc, DALI, four domains (domain 1, 80–184, domain 2, 185–289; domain 3, 290–301, 502–644; domain 4,302–501); AUTHORS, three domains (domain 1, 61–292; domain 2, 293–300, 497–644, domain 3, 301–496). (NN)1trka, DALI, four domains (domain 1, 30–264; domain 2, 3–29, 265–288; domain 3, 3345–534; domain 4, 535–680);AUTHORS, three domains (domain 1, 3–322; domain 2, 323–538; domain 3, 539–680). (OO) 3cox, DALI, five domains(domain 1, 10–48, 109–119, 227–317, 450–480, 493–506; domain 2, 176–190, 318–332, 346–449; domain 3,157–175,191–226, 337–345; domain 4, 55–96; domain 5, 120–156, 481–492); AUTHORS, two domains (domain 1,5–44, 226–316, 462–506; domain 2, 45–225, 317–461). (PP) 1glag, DALI, four domains (domain 1, 4–36, 48–84,216–249, 436–462; domain 2, 262–271, 304–435, 463–488; domain 3, 250–261, 272–303; domain 4, 37–47, 84–215);AUTHORS, two domains (domain 1, 5–253; domain 2, 254–501). (QQ) 2pol, DALI, four domains (domain 1, 1–15,26–45, 56–116; domain 2, 16–25, 46–55, 117–132, 198–236; domain 3, 133–197, 237–249; domain 4, 250–366);AUTHORS, three domains (domain 1, 1–121; domain 2, 121–249; domain 3, 250–366).
Consistent Assignment of Protein Structural Domains 673

helices: (1myt, Figure 14(P) and 1bmdFigure 14(Q)); and (3) the majority, cases of largedomains with extensive b-structures(Figure 14(R)–(W)). This last group comprisesdifferent architectures, but in all cases overcutpredictions disregard interactions betweensb-strands and other secondary structures (neigh-boring b-strand, helix or loop). Five out of sixcases in this category are unique to DomainParser(1dhr, 1lap, 1hpl, 1tim and 1mda; Figure 14(S)–(V)). This may indicate that some assumptionsabout interactions involving b-structures shouldbe re-evaluated or fine-tuned.
The undercut cases are truly difficult situationsin which expert decisions seem to go against thebasic principles of domain definition. In two cases,one or more of the domains is too small (4rcr,Figure 14(X); and 1psp, Figure 14(Y)); in anothercase, the proposed domain contains little second-ary structure and is in close contact with the restof the structure (1fcd, Figure 14(Z)). Two morecases involve closely interacting domains (1gof,Figure 14(AA); and 3cox, Figure 14(BB)). One caseof undercuting by DomainParser (1gss,Figure 14(CC)) is clearly wrong; it involves achain with two small but spatially separateddomains and it is not clear why DomainParsermisses it.
Domain boundaries. DomainParser has a surpris-ingly small number of chains with domain overlapdisagreements (11 chains at 95% stringency; twochains at 80% stringency). All the chains that dis-agree on domain boundaries are multi-domainchains and for the majority of the cases, at leastone domain is non-continuous, that is, split intotwo or more non-linear fragments. Different place-ment of the fragment boundaries can result insomewhat different partitioning of the chain intodomains, such as separating a single helix out of ahelical bundle in 1dlc (Figure 12(G)). The tendencyof DomainParser is to avoid splitting domains intofragments, and this results in a suboptimal domainseparation (1gia, Figure 12(I)). A curious case isthat of the human Fc fragment 1fc1 (Figure 12(H)),in which DomainParser assigns more fragmentsthan the AUTHORS reference method, but thespatial separation of domains is worse than oneachieved in AUTHORS assignment.
In summary, DomainParser respects secondarystructures and rarely splits them between domains.DomainParser sometimes misses domains of smallsize or with low secondary structure/size ratio.Overcuts are confined primarily to the chains withextensive b-structures.
DALI36
DALI is based on local compactness of the struc-ture as well as recurrence of the structures. Thereare 21 cases of overcut predictions and three casesof undercut predictions when compared to theexperts’ consensus. The overcut predictions arechiefly the result of cutting small segments of the
structure that are protruding from an otherwisecompact domain. The deviation from a compactstructure is either caused by loops that movesmall secondary structures away or by long helicesthat extend away from the body of the domain.The tendency to overcut is evident in the chainsthat are predicted by experts’ consensus to besingle-domain chains (2tmv, Figure 14(DD); 3pgm,Figure 14(EE); 1caj, Figure 14(FF); 1rve,Figure 14(GG); 2bpa, Figure 14(O); and 1xim,Figure 14(JJ)) as well as those that are predictedby experts’ consensus to be multi-domain chains(1dlc, Figure 14(MM); 1trk, Figure 14(NN); 3cox,Figure 14(OO); and 1gla, Figure 14(PP)). Usuallythe small regions that are split off, larger domainsare assigned a status of separate domains; how-ever, there are cases when such regions are leftunassigned (1xima, Figure 14(JJ); 1bgc, Figure14(II); 1trk, Figure 14(NN); and 3grs).
Some overcut cases involve b or mainly b struc-tures (1mdc, Figure 14(KK); 2pol, Figure 14(QQ);and 1pho, Figure 14(HH)). If we consider cases forwhich there is no consensus, we observe thatDALI tends to undercut in the cases whereb-structures are present (2snv, Figure 8(A); 12sl,Figure 8(B); 1hfh, Figure 8(F); and smra,Figure 8(O)). Frequently, DALI partitioninginvolves excessive fragmentation of the individualdomains into multiple, often very short, fragments(3cox, Figure 14(OO); 1gla Figure 14(PP); 2tpr,1pho, Figure 14(HH); 2tmv, Figure 14(DD); 2pol,Figure 14(DD); 1caj, Figure 14(FF); 2por and3pgm, Figure 14(EE); 1mdc, Figure 14(KK); and3grs and 3lad). There is a tendency to cut throughthe secondary structures (2pol, 1bcg, 1mdc, 1gla,3adk, 1pho, 3pgm and 2trp). The overcut casescould be reduced drastically if a more stringentrule regarding the partitioning of a domain intofragments was implemented. A higher penalty forsplitting secondary structure between two domainscould reduce the fraction of overcut-typepredictions.
None of the three undercut predictions is uniqueto DALI. One case, 1psp (Figure 14(Y)) involves asmall domain with little secondary structure thatAUTHORS divides into two domains, while DALIand DomainParser keep it intact. Another caseinvolves assigning a helix and a very short b-sheetto a separate domain by AUTHORS (1fcd,Figure 14(Z)), which DALI and DomainParser donot do. This case is surprising, since DALIfrequently separates a few small structures into aseparate domain. The last case (1emd,Figure 14(K)) involves closely packed and inter-acting domains, which cannot be separated byDALI or PDP. All these cases of undercut predic-tions can be justified as they contradict one ormore basic designs of structural domainarchitecture.
Domain boundaries. DALI produces the largestnumber of chains with disagreement on the place-ment of the domain boundaries (52 cases at 95%stringency, 22 cases at 80% stringency). The largest
674 Consistent Assignment of Protein Structural Domains

fraction of misplaced boundaries is for single-domain assignments, where DALI often leaves asignificant section of the domain, large (1byh,Figure 12(J)) or small (1snc, Figure 12(J); 3teci,Figure 12(L)), unassigned. In over 50% of thecases, the unassigned portions exceeds 15% of thechain length. In the cases of non-continuousdomains, DALI tends to split domains into morefragments as well as leave some segmentsunassigned, often attaining cleaner separation ofdomains (6ldh, Figure 12(M)). In some cases,additional fragments produced by DALI in combi-nation with very different placement of the domainboundaries results in drastically different domainassignments (4tms, Figure 12(N)). The only caseswhere DALI partitions domains into fewer frag-ments than the reference method is when noassignment is made (1pfka, Figure 12(O)).
In summary, DALI favors compact domains,resulting in frequent overcutting of the chain andnon-continuous domains that consist of many(often small) fragments that also split secondarystructures. Excessive fragmentation may leave thenumber of domains intact, but results in disagree-ment on their precise position. The effect is morepronounced in single-domain chains, where asignificant fraction of the chain may be leftunassigned.
How can we improve algorithmic methods?
From the analysis of the structures for whichexperts’ consensus exist, we observe that overcutpredictions are more frequent than undercut pre-dictions for all algorithmic methods. Both PDPand DALI emphasize compact domains, thus theyhave a tendency to cut off portions of the chainthat break this compactness and assign the cut-offsegment to a separate domain. Unlike DALI, PDPdoes not partition the chains into domains whenthe partitioning results in domains with many frag-ments (three or more). Consequently, PDP has 60%less overcut predictions than DALI. The tendencyof the algorithms to overcut structures is likelyfrom two effects: (1) the initial assumptions of thealgorithms regarding structural compactness; and(2) the large fraction of small domains present inthe training data. DomainParser has a significantlylower fraction of overcuts. Overcuts in Domain-Parser occur when chains (usually large) getdivided into multiple domains probably due tothe underestimation of the interaction of b-strandswith other secondary structures. Cases of undercutpredictions are less frequent for all methods; mostof the chains that cause undercuts are exceptionsfrom standard structural domain definitions andthus require additional knowledge for successfulprediction. Undercuts are common in the cases ofvery complex architectures, where it is not necess-arily the domain compactness, but the shear com-plexity of the domain interfaces that presents aproblem. To tackle this issue, it is necessary tohave more examples of structures of complex
architectures with experts’ consensus on domainassignment. Given the current dearth of complexstructures, algorithms cannot be trained or vali-dated. It would be possible, however, for the algo-rithmic method to either opt out of partitioning ofcomplex structures (when recognized as such) orto provide a level of confidence associated withassignments.
In summary, algorithmic methods place exagger-ated importance on domain compactness and this,combined with misinterpretation of b-sheet inter-actions, causes overcutting of structures by allmethods. The incorporation of training from largefolds might prevent overcutting. Conversely,undercutting due to the closely packed domainsmight be improved if functional information canbe incorporated. Further improvements, particu-larly in the cases of complex architectures, willrequire a significantly larger dataset of multi-domain structures for which experts’ consensusexists.
What are the central issues in the definition ofstructural domains?
The first issue is what indeed defines a structuraldomain? Certainly, the three current experts’methods discussed here define a domain differ-ently. AUTHORS often gives over-riding import-ance to the function information; SCOPemphasizes evolutionary recurrence of the fold;while CATH attempts to balance function,structure and evolutionary factors. Algorithmicmethods approach the problem mainly from thestructural perspective.
Agreeing on a “unified” definition of a structuraldomain is critical for the improvement of auto-matic methods of domain assignment. In thisspirit, we begin on the consistent definition of astructural domain by providing a list of issuesencountered in this work and the questions thatneed to be addressed.
Issue 1. Very small domains. Small, functionallyimportant regions can be assigned the status ofdomain. Most frequently this happens inAUTHORS, but all expert methods do thisoccasionally. What are the requirements for a shortsegment of the chain to be called a structuraldomain? We propose the following criteria: distinctfunction, a fraction of secondary structures in thesegment, spatial separation from the rest of thechain, evolutionary precedence and minimal size.If the short segment does not warrant status ofseparate domain, when should it be leftunassigned and when should it be part of theother domain? This particularly pertains to indi-vidual or small group of helices, which appear asa separate domain in almost every method.
Issue 2. Very large domains. Some single domainsare very large, although infrequent. What shouldbe the determining factors when considering par-titioning the chain into domains? To what extentshould the fold, size and compactness play a role?
Consistent Assignment of Protein Structural Domains 675

Issue 3. Discontinuous domains. When is it reason-able to have domains consisting of more than onecontinuous fragment? What should determine thenumber of fragments within a domain (and there-fore crossovers in and out of the domain)? Weobserved two types of fragmentation: cases whenactual secondary structures are cut to achieveclear separation; and cases in which there ismultiple loop traversals between two domains.
Issue 4. Unassigned regions in the chain. Allmethods, with the exception of SCOP and Domain-Parser, occasionally leave some residuesunassigned. This is particularly poignant whenmany residues are left unassigned in a single-domain chain. Can some regions of the chain beleft unassigned and if so, what fraction? Underwhich conditions should leaving residuesunassigned be considered (e.g., to avoid excessivepartitioning of the domain, to avoid generation ofadditional domains, etc.)? Can a single domainchain have unassigned residues?
Issue 5. Splitting a secondary structure. In somecases the interface between domains is complicatedand in order to separate them one or more of thesecondary structures have to be split. This is par-ticularly relevant with respect to b-sheets, sincedifferent algorithmic methods treat themdifferently. When is it justifiable to split secondarystructure between two domains?
Issue 6. Mostly a-structures. We observeinconsistent treatment of large all-a or mostly-astructures by all experts’ methods. What shouldaffect the decision of splitting an a -structure versuskeeping it together? How is it different from otherstructures, such as a/b?
Issue 7. Domain interfaces. Sometimes functionaldomains are very closely packed; however, ifseparated each one can be a structurally stableautonomous unit. Should the domains be separ-ated in such cases, and if so, what can helpalgorithmic methods be able to do so? Somedomains are relatively well separated, but thereare many loops that criss-cross the interface.Cutting loops introduces many fragmented dis-continuous domains. How should short loopswithin a domain interface be treated? In complexarchitectures, interfaces are frequently long andconvoluted. They can be discerned by eye, but aredifficult to formalize. What set of rules can beused to achieve reasonable separation of complexinterfaces by algorithms?
Issue 8. Complex architectures. For some complexarchitectures there is more than one reasonableassignment of domains or domain boundaries. Isit possible to discern between two partitions ofcomplex architectures when both make sensestructurally? What should be considered in suchcases?
In summary, further improvement of automaticdomain assignments will require new workingdefinitions of structural domains based on existingdata, which encompasses evolutionary, structuraland functional approaches. In the absence of such
comprehensive and robust domain definitions, thecommunity of structural biologists should at leastbe aware of the discord and understand the con-ceptual underpinnings of structural domains asdetermined by each assignment method. Exercisesin quality assurance such as that undertaken hereare a first step.
Materials and Methods
Construction of the 467 chain dataset
The dataset of polypeptide chains for which assign-ment of domains is available for the three expertmethods was assembled as follows. AUTHORS assign-ments originally compiled by Islam et al.19 and updatedperiodically, were downloaded from the web†. As indi-cated on the site, these domain assignments are takenprimarily from the literature. If domain assignment wasnot provided in the publication (about 50% of cases) itwas made by visual inspection. Domain assignments forproteins homologous to this set were assigned by pro-jecting the author assignment onto a multiple sequencealignment. These assignments were made for a non-redundant representative set of the PDB of April 1996.We extracted domain boundary assignments using asuite of Perl scripts. There were 2363 chains in this collec-tion, comprising 1530 single-domain, 720 two-domain, 85three-domain, 26 four-domain, and two five-domainchains. The collection was then refined in two consecu-tive steps. First, chains with more than 90% sequenceidentity are removed, leaving 749 chains (in-housemethod for purging sequences based on identity).Second, chains with inconsistent annotation of domainboundaries were removed, leaving 563 chains (some ofthe chains had domains with no left, right or bothboundaries).
Each of the 563 chains was tested for its presence inCATH (CATH version 2.4‡) and SCOP (Astral SCOP1.59§). The results for DALI, an automatic domainassignment method, were from the website(DaliDomainDefinitionsk).
The resulting dataset for which assignments byAUTHORS, CATH, DALI and SCOP exist comprised467 chains. For the remaining two automatic domainassignment methods, DomainParser and PDP, the pro-grams were obtained from the authors and run locallyon the 467-chain dataset.
Analyzing number of domains anddomain boundaries
Domain assignments from each method as well as allfurther automatic analysis were performed usingdomain_assign, a Cþþ analysis package written forthis purpose. The detailed inspection of individual caseswas done manually using WebLabViewer (Accelrys){.
† http://www.bmm.icnet.uk/~domains/test/dom-rr.html
‡ ftp://ftp.biochem.ucl.ac.uk/pub/cathdata/v2.4/§ http://astral.stanford.edu/khttp://www.ebi.ac.uk/dali/domain/3.1beta/{www.accelrys.com
676 Consistent Assignment of Protein Structural Domains

Calculation of domain number distribution
The distribution of each single and multi-domainchain predicted by each assignment method was calcu-lated automatically. In two cases, entire dataset and “noconsensus” dataset, calculation of the domain numberdistribution is complicated, as each method assigns adifferent number of domains. In the case of the entiredataset, a reference method (AUTHORS) was used as arepresentative of the distribution. In the case of no con-sensus, the distribution of number of domains wastaken as an average value predicted by all six methods.The average was rounded down if the fractional part ifthe value was less than 0.51 (rather than 0.50), thus thedistribution is biased towards a lower number ofdomains.
Calculating domain overlap
Domain overlap between two assignment methodswas evaluated by calculating the fraction of residues inthe entire chain for which assignments by a givenmethod and the reference method were identical (i.e.assigned to the same domain, Figure 15(A)). No require-ment was set for percentage overlap among individualdomains, which would be a more restrictive approach.The intuitive approach for finding the best domain over-lap in the case of multi-domain chains is to determinethe domain in the second assignment that best overlapsthe largest domain in the first assignment. This initialmapping between domains in the two assignments isthen propagated by considering the remaining domains.Starting with the overlap for the largest domain doesnot always lead to finding the best overlap of domainsin the entire chain. As shown in a hypothetical examplein Figure 15(B), starting with the largest domain A, thebest mapping is found, which is domain A to domainA0. This forces the mapping of domain B to domain B0.The total overlap across the entire chain in this case is140 or 48%. However, if domain A is mapped to domainB0 and domain B to domain A0, then the total overlapover the entire chain is 150 or 52%. In order to find thebest possible overlap between domains across the entirechain, we calculated domain overlaps by consideringevery possible mapping between domains in the twoassignments. The mapping that produced the maximalscore for the entire chain was used. The analysis ofdomain overlap was performed at incrementally increas-ing stringency: beginning at 35% threshold (more than35% and less than 40% of all residues in the chain agreedon domain assignment) and ending at 95% threshold(more than 95% and less than 100% of residues in thechain agreed on domain assignment). The incrementwas set to 5%.
Acknowledgements
We are grateful to the authors of DomainParser,Ying Xu, Dong Xu & Juntao Guo, for providing uswith a version of the latest algorithm as well astheir willingness to customize the user interface.This work was supported by NIH grant1P01GM63208-01A1.
References
1. Ponting, C. & Russell, R. (2002). The natural historyof protein domains. Annu. Rev. Biophys. Biomol.Struct. 31, 45–71.
2. Brenner, S. E., Chothia, C. & Hubbard, T. (1997).Population statistics of protein structures: lessonsfrom structural classification. Curr. Opin. Struct. Biol.7, 369–376.
3. Orengo, C., Jones, D. T. & Thornton, J. M. (1994).Protein superfamilies and domain superfolds.Nature, 372, 631–634.
4. Fields, S. (2001). Proteomics. Proteomics in genome-land. Science, 291, 1221–1224.
5. Wetlaufer, D. (1973). Nucleation, rapid folding, andglobular intrachain regions in proteins. Proc. NatlAcad. Sci. USA, 70, 697–701.
6. Rossman, M. G. & Liljas, A. (1974). Letter: Recog-nition of structural domains in globular proteins.J. Mol. Biol. 85, 177–181.
7. Cunningham, B., Gottlieb, P., Pflumm, M. &
Figure 15. Hypothetical examples of domain overlapbetween two assignment methods. (A) A hypotheticalexample of 80% domain overlap across the chain: 80 resi-dues out of 100 are mapped similarly by two methods(50 residues are mapped to domain A by both methods,30 residues are mapped to domain B by both methods,20 residues are mapped differently by two methods). (B)A hypothetical example in which starting with the lar-gest domain does not result in a maximum possible over-lap of domains across the entire chain. If we start withdomain A (which is the larger of the two in both assign-ments), the best mapping (or correspondence) is domainA to domain A0 and domain B to domain B0. In thiscase, the overlap across the entire chain is 140 residues(48% of the chain). Mapping of domain A to domain B0
produces a better overlap of 150 residues (53% of thechain). This second mapping can be found during anexhaustive search of all possible domain mappings (inthis case there are only two possibilities).
Consistent Assignment of Protein Structural Domains 677

Edelman, G. (1991). ????? Progress in Immunology(Amos, B., ed.), pp. 3–24, Academic Press, NewYork.
8. Levitt, M. & Chothia, C. (1976). Structural patterns inglobular proteins. Nature, 261, 552–558.
9. Rossmann, M. G. & Argos, P. (1976). Exploring struc-tural homology of proteins. J. Mol. Biol. 105, 75–95.
10. Richards, F. M. (1977). Areas, volumes, packing andprotein structure. Annu. Rev. Biophys. Bioeng. 6,151–176.
11. Zehfus, M. H. & Rose, G. D. (1986). Compact units inproteins. Biochemistry, 25, 5759–5765.
12. Wodak, S. J. & Janin, J. (1981). Location of structuraldomains in protein. Biochemistry, 20, 6544–6552.
13. Baron, M., Norman, D. G. & Campbell, I. D. (1991).Protein modules. Trends Biochem. Sci. 16, 13–17.
14. Campbell, I. D. & Baron, M. (1991). The structure andfunction of protein modules. Philos. Trans. Roy. Soc.ser. B, 332, 165–170.
15. Jones, S., Stewart, M., Michie, A., Swindells, M. B.,Orengo, C. & Thornton, J. M. (1998). Domain assign-ment for protein structures using a consensusapproach: characterization and analysis. Protein Sci.7, 233–242.
16. Taylor, W. (1999). Protein structure domain identifi-cation. Protein Eng. 12, 203–216.
17. Xu, Y., Xu, D. & Gabow, H. N. (2000). Protein domaindecomposition using a graph-theoretic approach.Bioinformatics, 16, 1091–1104.
18. Day, R., Beck, D. A. C., Armen, R. S. & Daggett, V.(2003). A consensus view of fold space: combiningSCOP, CATH and the Dali domain dictionary. ProteinSci. 12, 2150–2160.
19. Islam, S. A., Luo, J. & Sternberg, M. J. (1995). Identifi-cation and analysis of domains in proteins. ProteinEng. 8, 513–525.
20. Orengo, C. A., Michie, A. D., Jones, S., Jones, D. T.,Swindells, M. B. & Thornton, J. M. (1997). CATH—ahierarchic classification of protein domain structures.Structure, 5, 1093–1108.
21. Murzin, A. G., Brenner, S. E., Hubbard, T. & Chothia,C. (1995). SCOP: a structural classification of proteinsdatabase for the investigation of sequences andstructures. J. Mol. Biol. 247, 536–540.
22. Wernisch, L., Hunting, M. & Wodak, S. J. (1999).Identification of structural domains in proteins by agraph heuristic. Proteins: Struct. Funct. Genet. 35,338–352.
23. Alexandrov, N. & Shindyalov, I. (2003). PDP: proteindomain parser. Bioinformatics, 19, 429–430.
24. Holm, L. & Sander, C. (1996). Mapping the proteinuniverse. Science, 273, 595–602.
25. Siddiqui, A. S. & Barton, G. J. (1995). Continuous anddiscontinuous domains: an algorithm for the auto-matic generation of reliable protein domaindefinitions. Protein Sci. 4, 872–884.
26. Swindells, M. B. (1995). A procedure for the auto-matic determination of hydrophobic cores in proteinstructures. Protein Sci. 4, 93–102.
27. Wheelan, S., Marchler-Bauer, A. & Bryant, S. (2000).Domain size distribution can predict domainboundaries. Bioinformatics, 16, 613–618.
28. Heger, A. & Holm, L. (2003). Exhaustiveenumeration of protein domain families. J. Mol. Biol.328, 749–767.
29. Trifonov, E. N. (1995). Segmented structure of proteinsequences and early evolution of genome bycombinatorial fusion of DNA elements. J. Mol. Evol.40, 337–342.
30. Park, J., Lappe, M. & Teichmann, S. A. (2001). Map-ping protein family interactions: intramolecular andintermolecular protein family interaction repertoiresin the PDB and yeast. J. Mol. Biol. 307, 929–938.
31. Lo Conte, L., Brenner, S. E., Hubbard, T. J., Chothia,C. & Murzin, A. G. (2002). SCOP database in 2002:refinements accommodate structural genomics.Nucl. Acids Res. 30, 264–267.
32. Hadley, C. & Jones, D. T. (1999). A systematic com-parison of protein structure classifications: SCOP,CATH and FSSP. Struct. Fold. Des. 7, 1099–1112.
33. Dietmann, S. & Holm, L. (2001). Identification ofhomology in protein structure classification. NatureStruct. Biol. 8, 953–957.
34. Gough, J., Karplus, K., Hughey, R. & Chothia, C.(2001). Assignment of homology to genomesequences using a library of hidden Markov modelsthat represent all proteins of known structure. J. Mol.Biol. 313, 903–919.
35. Guo, J. T., Xu, D., Kim, D. & Xu, Y. (2003). Improvingthe performance of DomainParser for structuraldomain partition using neural network. Nucl. AcidsRes. 31, 944–952.
36. Holm, L. & Sander, C. (1998). Dictionary of recurrentdomains in protein structures. Proteins: Struct. Funct.Genet. 1998, 88–96.
Edited by B. Honig
(Received 17 November 2003; received in revised form 23 March 2004; accepted 23 March 2004)
678 Consistent Assignment of Protein Structural Domains