
Thermodynamics of Uranium Minerals and Related Materials
18
NAVROTSKY ET AL. 1 CHAPTER 4: THERMODYNAMICS OF URANIUM MINERALS AND RELATED MATERIALS Navrotsky, Alexandra, Tatiana Shvareva and Xiaofeng Guo Peter A. Rock Thermochemistry Laboratory and NEAT ORU University of California Davis Davis CA 95616 USA e-mail: [email protected] Mineralogical Association of Canada Short Course 43, Winnipeg MB, May 2013, p. xx-xx INTRODUCTION Uranium in natural systems exists in two oxidation states: tetravalent and hexavalent. The aqueous chemistry and geologic and environmental transport of U is dominated by U 6+ because of its much greater aqueous solubility as the uranyl (UO 2 2+ ) ion. However, its solid state chemistry is rich in compounds of both U 4+ and U 6+ , as well as some materials containing U 5+ . Thorium, geologic- ally more abundant than U, is associated with it in U 4+ minerals and also forms some specific structures on its own. Thorium cannot be oxidized to the 5+ or 6+ state and thus lacks the aqueous mobility of U and does not substitute significantly into uranyl solid phases. Because of radioactive decay as well as the general multi-component nature of geochemical systems, U minerals accommodate many impurities, including fission products dominated by the rare earth elements. Interest in the thermodynamics of U minerals and associated compounds is motivated by four main concerns: the mining and processing of U ore (the front end of the nuclear fuel cycle), the operation of nuclear reactors, the disposal and environmental transport of nuclear materials (the back end of the nuclear fuel cycle), and the use of U and Th as geochemical indicators, particularly in age dating. The purpose of this review is to summarize available thermo- chemical data for U minerals and related materials, to identify systematic crystal chemical trends in the data, and to point out areas where further research is needed. URANINITE AND RELATED MATERIALS Uranium dioxide, UO 2 , by far is the most environmentally abundant U oxide, however it is not found in nature in its pure form; it is partially oxidized and contains additional elements (impurities and fission products). With composition varying in individual geological deposits, especially as a function of age, UO 2 forms the mineral uraninite, approximated by the formula M 2+ a RE 3+ b UO 2+x . Both oxidation and doping influence its stability and affect its formation. Therefore we first consider thermodynamic effects of oxidation and doping prior to discussion of mineral formation and its behaviour in the environment. Formation of uranium oxides The phase diagram for the UO 2 –UO 3 system has been developed by Hoekstra et al. (1961) (Fig. 4-1) and shows UO 2 , U 4 O 9 , U 3 O 8 and UO 3 as major stable phases at room and elevated temperatures, with several phases occurring in more than one polymorphic form. The thermodynamic properties of these oxides have been studied extensively and are summarized in Table 4-1. For UO 2 , U 3 O 8 and –UO 3 the preferred data are recommended by CODATA (1989) and the data for several U oxide phases are most recently reviewed by Guillaumont et al. (2008). UO 2 with cubic fluorite structure, space group Fm3m, a = 5.470 Å, where U is in the 4+ oxidation state, is stable at room temperature in air. Several datasets obtained by different methods have been reported with the CODATA accepted value determined by combustion calorimetry of UO 2 to U 3 O 8 under O atmosphere performed by Huber et al. (1969). Later Johnson et al. (1981) confirmed the recommended value by combustion calorimetry in F. Upon increasing temperature above 300 ºC under higher O partial pressure, the oxidation of U occurs. At lower O/U ratios it is accompanied by the incorporation of the additional O atoms into the fluorite crystal lattice. Extra O occupies interstitial sites and displaces the lattice anions from their crystallographic positions, forming the anionic cuboctahedral sublattice (Desgranges et al. 2009). Both UO 2.25 and UO 2.33 phases are characterized by the presence of such stabilizing cuboctahedra, and are more exothermic in enthalpy of formation compared to UO 2 . When the concentration of the excess O atoms exceeds a critical level, cubocta-
Transcript of Thermodynamics of Uranium Minerals and Related Materials

NAVROTSKY ET AL.
1
CHAPTER 4: THERMODYNAMICS OF URANIUM MINERALS AND RELATED MATERIALS Navrotsky, Alexandra, Tatiana Shvareva and Xiaofeng Guo Peter A. Rock Thermochemistry Laboratory and NEAT ORU University of California Davis Davis CA 95616 USA e-mail: [email protected]
Mineralogical Association of Canada Short Course 43, Winnipeg MB, May 2013, p. xx-xx
INTRODUCTION Uranium in natural systems exists in two
oxidation states: tetravalent and hexavalent. The aqueous chemistry and geologic and environmental transport of U is dominated by U6+ because of its much greater aqueous solubility as the uranyl (UO2
2+) ion. However, its solid state chemistry is rich in compounds of both U4+ and U6+, as well as some materials containing U5+. Thorium, geologic-ally more abundant than U, is associated with it in U4+ minerals and also forms some specific structures on its own. Thorium cannot be oxidized to the 5+ or 6+ state and thus lacks the aqueous mobility of U and does not substitute significantly into uranyl solid phases. Because of radioactive decay as well as the general multi-component nature of geochemical systems, U minerals accommodate many impurities, including fission products dominated by the rare earth elements. Interest in the thermodynamics of U minerals and associated compounds is motivated by four main concerns: the mining and processing of U ore (the front end of the nuclear fuel cycle), the operation of nuclear reactors, the disposal and environmental transport of nuclear materials (the back end of the nuclear fuel cycle), and the use of U and Th as geochemical indicators, particularly in age dating. The purpose of this review is to summarize available thermo-chemical data for U minerals and related materials, to identify systematic crystal chemical trends in the data, and to point out areas where further research is needed. URANINITE AND RELATED MATERIALS
Uranium dioxide, UO2, by far is the most environmentally abundant U oxide, however it is not found in nature in its pure form; it is partially oxidized and contains additional elements (impurities and fission products). With composition varying in individual geological deposits, especially as a function of age, UO2 forms the mineral uraninite, approximated by the formula M2+
aRE3+bUO2+x. Both oxidation and doping
influence its stability and affect its formation. Therefore we first consider thermodynamic effects of oxidation and doping prior to discussion of mineral formation and its behaviour in the environment. Formation of uranium oxides
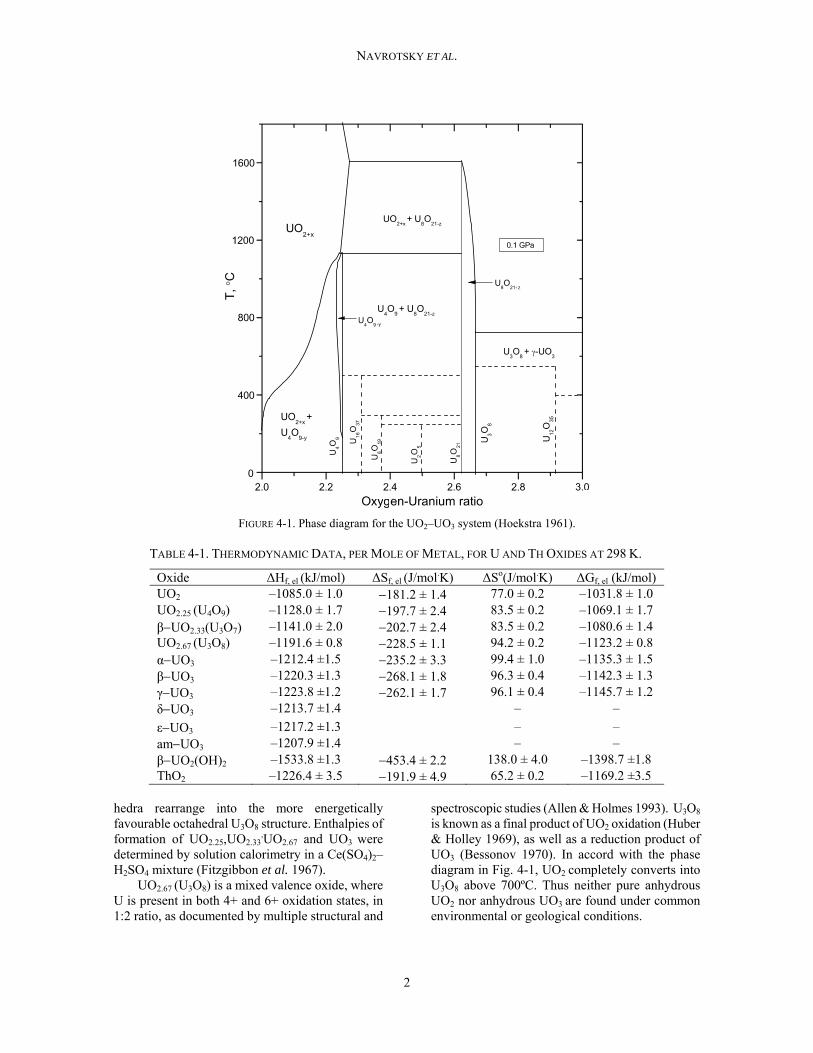
The phase diagram for the UO2–UO3 system has been developed by Hoekstra et al. (1961) (Fig. 4-1) and shows UO2, U4O9, U3O8 and UO3 as major stable phases at room and elevated temperatures, with several phases occurring in more than one polymorphic form.
The thermodynamic properties of these oxides have been studied extensively and are summarized in Table 4-1. For UO2, U3O8 and –UO3 the preferred data are recommended by CODATA (1989) and the data for several U oxide phases are most recently reviewed by Guillaumont et al. (2008).
UO2 with cubic fluorite structure, space group Fm3m, a = 5.470 Å, where U is in the 4+ oxidation state, is stable at room temperature in air. Several datasets obtained by different methods have been reported with the CODATA accepted value determined by combustion calorimetry of UO2 to U3O8 under O atmosphere performed by Huber et al. (1969). Later Johnson et al. (1981) confirmed the recommended value by combustion calorimetry in F.
Upon increasing temperature above 300 ºC under higher O partial pressure, the oxidation of U occurs. At lower O/U ratios it is accompanied by the incorporation of the additional O atoms into the fluorite crystal lattice. Extra O occupies interstitial sites and displaces the lattice anions from their crystallographic positions, forming the anionic cuboctahedral sublattice (Desgranges et al. 2009). Both UO2.25 and UO2.33 phases are characterized by the presence of such stabilizing cuboctahedra, and are more exothermic in enthalpy of formation compared to UO2. When the concentration of the excess O atoms exceeds a critical level, cubocta-
NAVROTSKY ET AL.
2
+
FIGURE 4-1. Phase diagram for the UO2–UO3 system (Hoekstra 1961).
TABLE 4-1. THERMODYNAMIC DATA, PER MOLE OF METAL, FOR U AND TH OXIDES AT 298 K.
Oxide ΔHf, el (kJ/mol) ΔSf, el (J/mol.K) ΔSo(J/mol.K) ΔGf, el (kJ/mol) UO2 –1085.0 ± 1.0 181.2 ± 1.4 77.0 ± 0.2 –1031.8 ± 1.0 UO2.25 (U4O9) –1128.0 ± 1.7 197.7 ± 2.4 83.5 ± 0.2 –1069.1 ± 1.7 βUO2.33(U3O7) –1141.0 ± 2.0 202.7 ± 2.4 83.5 ± 0.2 –1080.6 ± 1.4 UO2.67 (U3O8) –1191.6 ± 0.8 228.5 ± 1.1 94.2 ± 0.2 –1123.2 ± 0.8 αUO3 –1212.4 ±1.5 235.2 ± 3.3 99.4 ± 1.0 –1135.3 ± 1.5 βUO3 –1220.3 ±1.3 268.1 ± 1.8 96.3 ± 0.4 –1142.3 ± 1.3 γUO3 –1223.8 ±1.2 262.1 ± 1.7 96.1 ± 0.4 –1145.7 ± 1.2 δUO3 –1213.7 ±1.4 – –
εUO3 –1217.2 ±1.3 – – amUO3 –1207.9 ±1.4 – – βUO2(OH)2 –1533.8 ±1.3 453.4 ± 2.2 138.0 ± 4.0 –1398.7 ±1.8 ThO2 –1226.4 ± 3.5 191.9 ± 4.9 65.2 ± 0.2 –1169.2 ±3.5
hedra rearrange into the more energetically favourable octahedral U3O8 structure. Enthalpies of formation of UO2.25,UO2.33
,UO2.67 and UO3 were determined by solution calorimetry in a Ce(SO4)2–H2SO4 mixture (Fitzgibbon et al. 1967). UO2.67 (U3O8) is a mixed valence oxide, where U is present in both 4+ and 6+ oxidation states, in 1:2 ratio, as documented by multiple structural and
spectroscopic studies (Allen & Holmes 1993). U3O8 is known as a final product of UO2 oxidation (Huber & Holley 1969), as well as a reduction product of UO3 (Bessonov 1970). In accord with the phase diagram in Fig. 4-1, UO2 completely converts into U3O8 above 700ºC. Thus neither pure anhydrous UO2 nor anhydrous UO3 are found under common environmental or geological conditions.

NAVROTSKY ET AL.
3
UO3 is the polymorph most exothermic in formation enthalpy. It has U in two different octahedral environments, one with two short uranyl distances and the other without (Engmann 1963). The structure is nominally considered a “uranyl uranate” and consists of uranyl chains inter-connected by regular U octahedra. In an aqueous alteration sequence, the first formed and the most abundant product is the uranyl oxide hydrate schoepite, UO3·H2O, with a uranyl cation-based layered structure (Finch & Ewing 1992). The complete oxidation of U4+ to U6+ and the hydration give this mineral a much more negative enthalpy of formation as shown in Table 4-1. A summary plot of enthalpies of formation as a function of O content is shown in Figure 4-2.
FIGURE 4-2. Enthalpies of formation for various U oxides as a function of O/U ratio.
2
2
T,
Co
FIGURE 4-3. Phase diagram for Th–ThO2 system (Benz 1969).
In contrast to U, Th forms only one stable oxide, ThO2 (Fig. 4-3). Adopting the fluorite structure, it is isostructural with UO2, PuO2, CeO2, and the high temperature forms of ZrO2 and HfO2. ThO2 is very refractory and melts above 3390ºC (Benz 1969). Enthalpy and Gibbs energies of formation of UO2 and ThO2 from the elements are similar (Table 4-1).
Effects of doping The choice of the dopant cations in uraninite structure strongly depends on environmental conditions and age of the deposit (rare earth fission products build up with age). The most common impurities are Th4+ and Ce4+, trivalent rare-earth cations RE3+, Pb2+ and Ca2+. Phase diagrams for these systems have shown that there are extensive compositional ranges where solid solutions with cubic fluorite structure can be formed. However experimental thermodynamic data are scarce, mostly due to the complex synthesis pathways and refractory nature of the materials and difficulty in controlling and determining the exact O stoichiometry. The thermodynamic properties of doped urania were studied mainly through their O potentials, as reviewed by Fujino et al. (1988) and have strong dependence on the O/M ratio. Recently high temperature oxide melt solution calorimetry has been applied to the investigation of mixing energetics for stoichiometric ceria-doped urania and thoria with results supported by DFT calculations (Hanken et al. 2012, Shvareva et al. 2011).UO2–CeO2 forms an essentially ideal solid solution in the entire compositional range with nearly zero mixing enthalpies (Hanken et al. 2012), due to very small difference in U4+ and Ce4+ ionic radii. The data also argue against significant charge transfer to form U5+ (or U6+) and Ce3+ in this system, at least at room temperature. For the ThO2–CeO2 system, mixing enthalpies are slightly positive with an interaction parameter of 15.1 ± 2.2 kJ/mol. and a calculated critical temperature of 428 K for demixing (Shvareva et al. 2011). Consistently, for the solid solution of Ce4+ and Zr4+ oxides with even larger ionic radius difference, the mixing enthalpies are more positive resulting in interaction parameter of 51.0 ± 8.0 kJ/mol in the cubic fluorite structure (Lee et al. 2008). Fluorite-structured oxides doped with trivalent ions contain O vacancies to balance the lower cation charge. These vacancies lend mobility to the O sublattice, and materials such as yttria-doped

NAVROTSKY ET AL.
4
zirconia and gadolinia-doped ceria, to name a few, have received extensive attention as ionic conductors in applications ranging from O sensors in automobile exhausts (Yao & Yu 1984) to solid electrolytes for solid oxide fuel cells (SOFC) (Steele et al. 1994) to gas separation membranes (Dyer et al. 2000). RE-doped urania and thoria are relevant to the nuclear fuel cycle and Ce4+ is considered a useful analog or surrogate for Pu4+ because of similar ionic radii. Motivated by these materials science applications, enthalpies of formation of an extensive series of doped ZrO2, HfO2, CeO2, and ThO2 materials have been measured. The main results are discussed in detail in recent reviews (Navrotsky et al., Navrotsky 2010) and are summarized together with newly obtained data in Table 4-2. The doping of ZrO2, HfO2, CeO2 and ThO2 with aliovalent rare-earth cations creates O vacancies. In the zirconia and hafnia solid solutions these vacancies tend to associate with smaller Zr4+ and Hf4+ cation and stabilize 7-fold coordination, characteristic for monoclinic zirconia and hafnia. The binary C-type rare earth oxides REO1.5 (RE2O3) have the bixbyite structure and contain an ordered array of O vacancies that are located nearest neighbour to the trivalent cations. The change in location of vacancies on formation of the doped fluorite phase is correlated with strongly exothermic formation enthalpies for all zirconia and hafnia solid solutions with the largest heat effect associated with
the largest doping cations. An example of thermodynamic data for ZrO2, HfO2 and CeO2 doped with YO1.5 is shown in Figure 4-4 and discussed in detail by Navrotsky et al. (2007) and Simoncic & Navrotsky (2007). In the case of 8-coordinated cations in ceria and thoria, vacancies form, due to Coulomb interactions, near the dopant. Overall, formation enthalpies for CeO2 and ThO2 systems are positive and reflect heat effects of two independent phenomena: the regular solid solution behaviour at lower doping content and formation of energetically favorable vacancy– dopant associates at higher dopant concentration (Fig. 4-5). From enthalpies of formation and mixing, the enthalpies of vacancy clustering have been experimentally evaluated (Buyukkilic et al. 2012, Aizenshtein et al. 2010). There are strong correl-ations between the composition of maximum ionic conductivity and least stable enthalpy for RE-doped thoria and ceria, linking the energetics, defect equilibria, and transport properties. Mazeina et al. (2008) have shown for the UO2–CaO and UO2–YO1.5 systems that partial oxidation of U and forming UO2–UO3–CaO and UO2–UO3–YO1.5 solid solutions leads to negative enthalpies of formation which significantly stabilize the structure. This is consistent with the observation of Janeczek & Ewing (1992) that instead of slow post-formation partial oxidation of U in uraninite, it is likely that uraninite initially crystallizes as a complex U+4U+6 oxide, even under reducing conditions. In such a
TABLE 4-2. INTERACTION PARAMETERS FOR SOLID SOLUTIONS WITH FLUORITE STRUCTURE.
System Interaction parameter, kJ/mol
System Interaction parameter, kJ/mol
ZrO2–SmO1.5 –107.5 ± 8.4 a,h CeO2–LaO1.5 106.3 ± 24.3 i ZrO2–GdO1.5 –104.2 ± 5.7 a,h CeO2–GdO1.5 27.0 ± 10.9 i ZrO2–DyO1.5 –94.0 ± 4.9 a,h CeO2–SmO1.5 179.2 ± 2.8 c,i ZrO2–YO1.5 –90.3 ± 6.9 a,h CeO2–NdO1.5 166.9 ± 4.6 c,i ZrO2–YbO1.5 –89.3 ± 9.7 a,h CeO2–SmO1.5–NdO1.5 61.8 ± 1.7 c,i HfO2–SmO1.5 –138.1 ± 16.0 a,h ThO2–LaO1.5 27.8 ± 4.1 d,i HfO2–GdO1.5 –115.0 ± 11.7 a,h ThO2–YO1.5 119.3 ± 11.4 d ZrO2–DyO1.5 –119.8 ± 11.4 a,h ThO2–CeO2 15.1± 2.2 e ZrO2–YO1.5 –111.8 ± 13.6 a,h UO2–CeO2 0 f ZrO2–YbO1.5 –93.7 ± 14.7 a,h BiO1.5–DyO1.5 –73.0 ± 1.4 g ZrO2–CeO2 51.0 ± 8.0 b,h
a Simoncic & Navrotsky (2007); b Lee et al. (2008); c Buyukkilic et al. (2012); d Aizenshtein et al. (2010); e Shvareva et al. (2011); f Hanken et al. (2012); g Tran & Navrotsky (2012); h. These negative values reflect strong ordering of cations and vacancies into clusters; i. Positive values of the interaction parameter for low dopant concentrations reflect size mismatch, and the association of defects, with an exothermic effect, at higher dopant concentrations (see text for details).

NAVROTSKY ET AL.
5
FIGURE 4-4. Thermodynamic data for ZrO2, HfO2 and CeO2 doped with YO1.5. Enthalpies of monoclinic– cubic transitions Htrs for ZrO2 and HfO2, as well as enthalpies of transition from C or A-type to cubic structure for CeO1.5,Htrs(CeO1.5), are reflected in the plot Navrotsky et al. (2007)
scenario, the doping of U oxide with lower valence cations enables the formation of O vacancies in the fluorite structure, which in turn accommodate excess O atoms associated with U oxidation, resulting in the most stable arrangement of RE-doped partially oxidized U oxide, in the form of uraninite, abundantly occurring in nature. Detailed oxide melt solution calorimetric studies of synthetic RE-doped U oxides are in
progress in our laboratory. Though the calorimetry is straight forward, the determination of exact O stoichiometry in the samples is critical and methodology is being fine-tuned to accomplish this routinely and accurately. It is anticipated that thermochemical data for materials relevant to natural complex uraninite will become available over the next several years.
Zircon-related silicates: USiO4, ThSiO4, CeSiO4, ZrSiO4, HfSiO4
Uranium(IV) silicate (coffinite) is a secondary important alteration reduced uranium mineral from an economic point of view (Finch & Murakami 1999). Thorite (ThSiO4) and coffinite are iso-structural with zircon (ZrSiO4) and Th and U have extensive solubility in zircon (Ioudintsev et al. 1998). Properties of the higher tetravalent actinide silicates has been predicted from the thermodynamic systematics as discussed by Mazeina et al. (2005).
With positive enthalpies of formation from the oxides, thorite and coffinite are metastable at room temperature but may persist from high temperature conditions. Coffinite is commonly found in reducing low temperature environments rich in organic matter, suggesting its formation by reduction from U(VI) phases. Such coffinite is commonly very fine grained (nanophase) and may be hydrated (Frondel 1958, Speer 1980, Smits 1989, Janeczek & Ewing 1992). Thorite needs much higher temperature to form (Hazen et al. 2009).
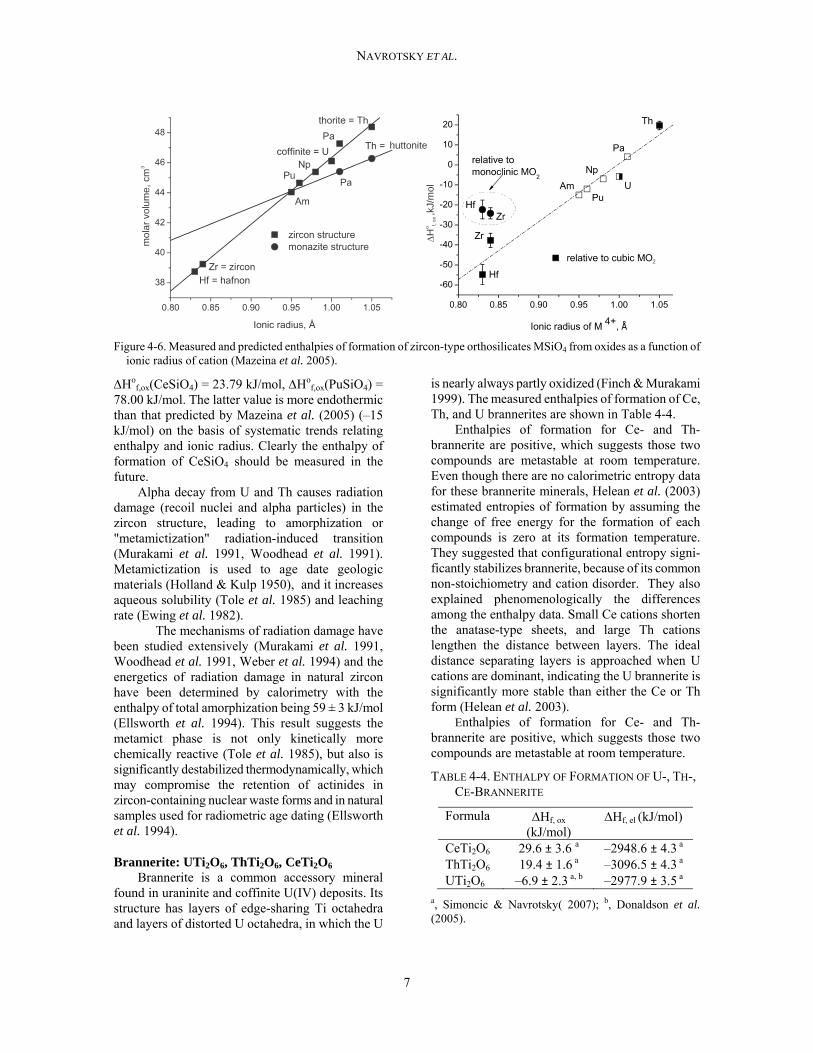
Mazeina et al. (2005) suggested that all actinide orthosilicates show a linear dependence of molar volume on ionic radius of tetravalent cations in eightfold coordination, as shown in Figure 4-6 and Table 4-3. Based on the enthalpies of formation and ionic radius for USiO4, ThSiO4, ZrSiO4, HfSiO4, they were able to predict the enthalpies of formation of AmSiO4, PuSiO4, NpSiO4, PaSiO4. Some computational work has been done on the thermodynamic properties of zircon-type actinide silicates. The total energy of ASiO4 (A = Hf, Th, U, Pu, Ce) and their solid solutions have been calculated via density functional theory (DFT) with local density approximation (LDA) (Ferriss et al. 2010). Total energy of CeSiO4 and PuSiO4 at 0 K are –5259.13 kJ/mol and –5914.60 kJ/mol, respect-ively (Ferriss et al. 2010). Although experimental thermodynamic data for CeSiO4 and PuSiO4 are not available, Ho
f,ox can be estimated from the total energy by looking into the relationship between the total energies of other ASiO4 with their known enthalpies of formation, which gives

NAVROTSKY ET AL.
6
FIGURE 4-5. Regular solution parameters (Ω) and experimentally determined enthalpies of vacancies clustering (ΔHassociation) for selected RE doped ThO2 and CeO2 systems (Aizenshtein et al. 2010).
TABLE 4-3. ENTHALPY OF FORMATION OF ZIRCON-TYPE ORTHOSILICATES
Phase Hf, ox
(kJ/mol) Sf, ox (kJ/mol
K) Hf, el
(kJ/mol) Gf, el
(kJ/mol) Sf, el
(kJ/mol K)
Thorite ThSiO4
19.6 ± 2.0 a 148.9 ± 10.4 e –2117.5 ± 4.2 a –2050.3 ± 3.9 a –225.4 ± 10.5 e
Coffinite USiO4
4.3 ± 5.6 a –0.4 ± 12.8 e –1991.3 ± 5.4 a –1883.6 ± 4.0 a –361.3 ± 12.3 e
Zircon ZrSiO4
–24.2 ± 2.8 b,c –12.4 ± 8.5 e –2034.2 ± 3.1 a –1919.7 ± 3.1 a –384.0 ± 8.0 e
Hafnon HfSiO4
–22.3 ± 4.7 a,d – – –2050.6 ± 5.1 a,b – –
a Simoncic & Navrotsky (2007); b Robie & Hemingway (1995); c Molodetsky et al. (1998); d Lee & Navrotsky (2004); e calculated values from formation of enthalpy and free energy.
NAVROTSKY ET AL.
7
Ho f,
ox ,k
J/m
ol
mo
lar
volu
me
, cm
3huttonite
2
Figure 4-6. Measured and predicted enthalpies of formation of zircon-type orthosilicates MSiO4 from oxides as a function of ionic radius of cation (Mazeina et al. 2005).
Hof,ox(CeSiO4) = 23.79 kJ/mol, Ho
f,ox(PuSiO4) = 78.00 kJ/mol. The latter value is more endothermic than that predicted by Mazeina et al. (2005) (–15 kJ/mol) on the basis of systematic trends relating enthalpy and ionic radius. Clearly the enthalpy of formation of CeSiO4 should be measured in the future.
Alpha decay from U and Th causes radiation damage (recoil nuclei and alpha particles) in the zircon structure, leading to amorphization or "metamictization" radiation-induced transition (Murakami et al. 1991, Woodhead et al. 1991). Metamictization is used to age date geologic materials (Holland & Kulp 1950), and it increases aqueous solubility (Tole et al. 1985) and leaching rate (Ewing et al. 1982). The mechanisms of radiation damage have been studied extensively (Murakami et al. 1991, Woodhead et al. 1991, Weber et al. 1994) and the energetics of radiation damage in natural zircon have been determined by calorimetry with the enthalpy of total amorphization being 59 ± 3 kJ/mol (Ellsworth et al. 1994). This result suggests the metamict phase is not only kinetically more chemically reactive (Tole et al. 1985), but also is significantly destabilized thermodynamically, which may compromise the retention of actinides in zircon-containing nuclear waste forms and in natural samples used for radiometric age dating (Ellsworth et al. 1994). Brannerite: UTi2O6, ThTi2O6, CeTi2O6
Brannerite is a common accessory mineral found in uraninite and coffinite U(IV) deposits. Its structure has layers of edge-sharing Ti octahedra and layers of distorted U octahedra, in which the U
is nearly always partly oxidized (Finch & Murakami 1999). The measured enthalpies of formation of Ce, Th, and U brannerites are shown in Table 4-4.
Enthalpies of formation for Ce- and Th-brannerite are positive, which suggests those two compounds are metastable at room temperature. Even though there are no calorimetric entropy data for these brannerite minerals, Helean et al. (2003) estimated entropies of formation by assuming the change of free energy for the formation of each compounds is zero at its formation temperature. They suggested that configurational entropy signi-ficantly stabilizes brannerite, because of its common non-stoichiometry and cation disorder. They also explained phenomenologically the differences among the enthalpy data. Small Ce cations shorten the anatase-type sheets, and large Th cations lengthen the distance between layers. The ideal distance separating layers is approached when U cations are dominant, indicating the U brannerite is significantly more stable than either the Ce or Th form (Helean et al. 2003).
Enthalpies of formation for Ce- and Th-brannerite are positive, which suggests those two compounds are metastable at room temperature.
TABLE 4-4. ENTHALPY OF FORMATION OF U-, TH-, CE-BRANNERITE
Formula Hf, ox
(kJ/mol) Hf, el (kJ/mol)
CeTi2O6 29.6 ± 3.6 a –2948.6 ± 4.3 a ThTi2O6 19.4 ± 1.6 a –3096.5 ± 4.3 a UTi2O6 –6.9 ± 2.3 a, b –2977.9 ± 3.5 a
a, Simoncic & Navrotsky( 2007); b, Donaldson et al. (2005).

NAVROTSKY ET AL.
8
Even though there are no calorimetric entropy data for these brannerite compounds, Helean et al. (2003) estimated entropies of formation by assuming the change of free energy for the form-ation of each compound is zero at its formation temperature. They suggested that configurational entropy significantly stabilizes brannerite, because of its common non-stoichiometry and cation disorder. They also explained phenomenologically the differences among the enthalpy data. Small Ce cations shorten the anatase-type sheets, and large Th cations lengthen the distance between layers. The ideal distance separating layers is approached when U cations are dominant, indicating the U brannerite is significantly more stable than either the Ce or Th form (Helean et al. 2003).
Furthermore, these authors connected the energetics to irradiation studies by comparing their critical amorphization doses Dc: Dc(CeTi2O6) < Dc(ThTi2O6) < Dc(UTi2O6) (Lumpkin et al. 2001, Lian et al. 2001), which suggests the resistance of a phase to radiation damage is related to its thermo-dynamic stability (Helean et al. 2003). Such relations between radiation resistance and thermo-dynamic stability have been studied extensively in pyrochlore structures not containing U (Lang et al. 2010).
Ce-brannerite can be used to estimate the properties of Pu-brannerite due to similarity in ionic radii of Ce4+ and Pu4+. Thus Helean et al. (2003) predicted that PuTi2O6 will be energetically meta-stable with respect to its binary oxides, and can be stabilized only at high temperature (> 1500oC), which is consistent with the difficulties in its synthesis (Vance et al. 2001)
Substituted monazite: synthetic cheralite
The mineral monazite, LnPO4 (Ln3+ = Ce3+, La3+, Nd3+, Sm3+, or Pr3+) has remarkable capacity of retaining tetravalent actinides, especially U4+ and Th4+ (McCarthy et al. 1978, Beall et al. 1981, Sales et al. 1983, Meldrum et al. 1996). Among the many Ln3+–Ac4+substitutional ceramics, cheralite is one of the synthetic monazite-based compounds that has been extensively studied. Popa et al. (2008) reported
the full thermodynamic data set, based on cryogenic heat capacity measurements and oxide melt solution calorimetric determination of enthalpy of formation, for cheralite CaTh(PO4)2 (Popa et al. 2008) see Table 4-5. These data reflect the enthalpy and free energy of formation from binary oxides from cheralite are ~55 ± 10 kJ/mol less exothermic than those for monazite LaPO4 (Ushakov et al. 2001, Ushakov et al. 2004). Popa et al. (2008) explained this more positive enthalpy value as originating from structural disorder of Ca2+ and Th4+. After considering several solid state reactions related to cheralite formation and the strong acid–base interaction between CaO and P2O5, Popa et al. (2008) concluded that appropriate mineralogical conditions (with other phases present) have great impact on incorporation of actinides into monazite for nuclear waste disposal. Uranium in garnet
In a recent discovery in Northern Caucasus, Russia, Galuskina et al. (2010) have shown that natural garnet can accommodate up to 27 wt.% U. Also as high as 18 wt.% of U has been reported in synthetic Ca–Zr–Fe-based garnet (Yudintsev et al. 2002). The latter study suggested that U substitutes more easily in garnet with larger lattice parameters. These findings suggest that a specifically tailored garnet composition may be a good host for U in nuclear waste. Study of U substitution in a simple garnet system may shed light on the substitution mechanisms and their energetics. Charge balance for Y3+–M4+ substitution can be maintained by two mechanisms. One is that charges are balanced by introducing other divalent cations: Y3+ + Fe3+ = M4+ + N2+. In the second mechanism proposed by Rak et al. (2011) from computational studies, which is in a sense a special case of the first, the divalent cation is Fe2+, produced by reduction of the Fe3+ already in the garnet structure. Such reduction is of course coupled with oxidation, presumably of species elsewhere in the environment. A series of Ce and of
TABLE 4-5. THERMODYNAMIC FUNCTIONS FOR CHERALITE (BASED ON ONE PHOSPHATE UNIT)
Formula Hf, ox (kJ/mol) Hf, el (kJ/mol) Gf, el (kJ/mol) Sf, ox (kJ/mol K) Ca0.5Th0.5PO4 –253.2 ± 4.8 –1936.4 ± 4.9 –1817.7 ± 5.1* Sf = –13.7 ± 0.8 Sf,total = –8.0 ± 0.8**
* Calculated values from enthalpy and entropy of formation; **Sf,total includes the configurational entropy while Sf does not.
NAVROTSKY ET AL.
9
Th-substituted YIG garnets has been synthesized, and thermodynamic properties obtained for the Ce-substituted garnet suggest this substitution is energetically favourable. Uranyl Minerals
Most minerals containing U6+ incorporate the hexavalent U as the uranyl ion UO2
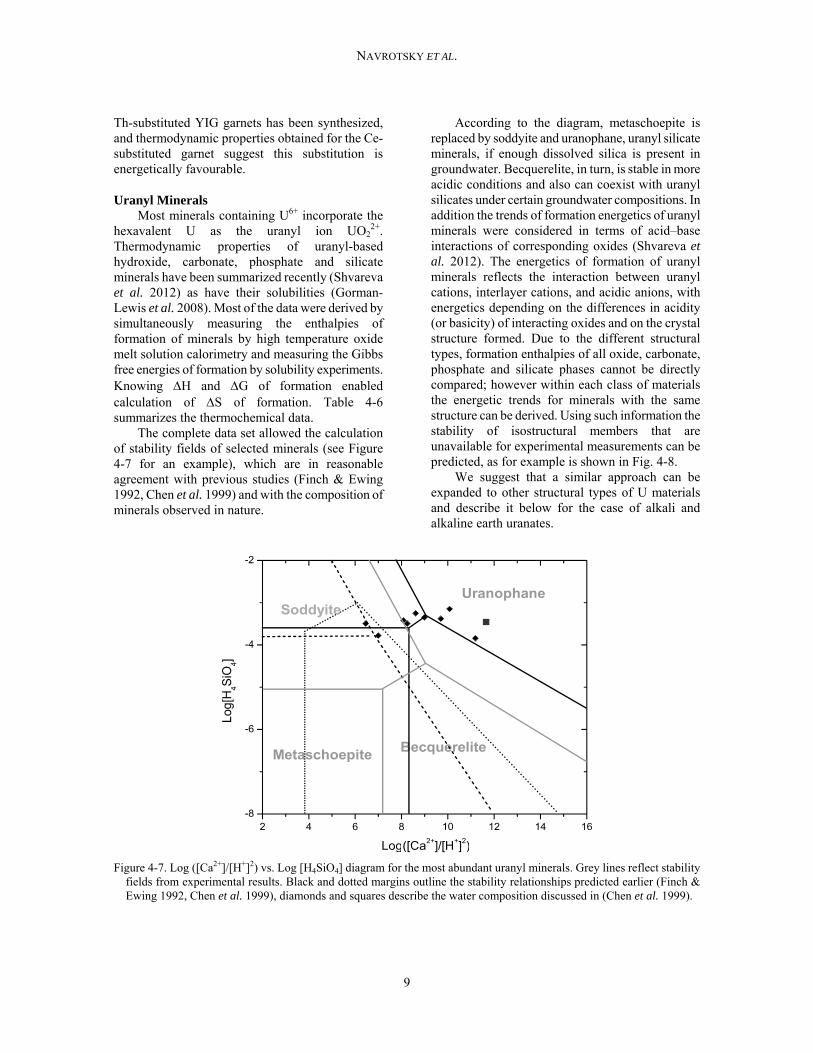
2+. Thermodynamic properties of uranyl-based hydroxide, carbonate, phosphate and silicate minerals have been summarized recently (Shvareva et al. 2012) as have their solubilities (Gorman-Lewis et al. 2008). Most of the data were derived by simultaneously measuring the enthalpies of formation of minerals by high temperature oxide melt solution calorimetry and measuring the Gibbs free energies of formation by solubility experiments. Knowing H and G of formation enabled calculation of S of formation. Table 4-6 summarizes the thermochemical data. The complete data set allowed the calculation of stability fields of selected minerals (see Figure 4-7 for an example), which are in reasonable agreement with previous studies (Finch & Ewing 1992, Chen et al. 1999) and with the composition of minerals observed in nature.
According to the diagram, metaschoepite is replaced by soddyite and uranophane, uranyl silicate minerals, if enough dissolved silica is present in groundwater. Becquerelite, in turn, is stable in more acidic conditions and also can coexist with uranyl silicates under certain groundwater compositions. In addition the trends of formation energetics of uranyl minerals were considered in terms of acid–base interactions of corresponding oxides (Shvareva et al. 2012). The energetics of formation of uranyl minerals reflects the interaction between uranyl cations, interlayer cations, and acidic anions, with energetics depending on the differences in acidity (or basicity) of interacting oxides and on the crystal structure formed. Due to the different structural types, formation enthalpies of all oxide, carbonate, phosphate and silicate phases cannot be directly compared; however within each class of materials the energetic trends for minerals with the same structure can be derived. Using such information the stability of isostructural members that are unavailable for experimental measurements can be predicted, as for example is shown in Fig. 4-8.
We suggest that a similar approach can be expanded to other structural types of U materials and describe it below for the case of alkali and alkaline earth uranates.
Figure 4-7. Log ([Ca2+]/[H+]2) vs. Log [H4SiO4] diagram for the most abundant uranyl minerals. Grey lines reflect stability fields from experimental results. Black and dotted margins outline the stability relationships predicted earlier (Finch & Ewing 1992, Chen et al. 1999), diamonds and squares describe the water composition discussed in (Chen et al. 1999).

NAVROTSKY ET AL.
10
TA
BL
E 4
-6. T
HE
RM
OD
YN
AM
IC F
UN
CT
ION
S F
OR
FO
RM
AT
ION
FR
OM
OX
IDE
S A
ND
EL
EM
EN
TS
OF
UR
AN
YL
MIN
ER
AL
S, U
ND
ER
ST
AN
DA
RD
TE
MP
ER
AT
UR
E A
ND
PR
ES
SU
RE.
S°,
J/m
ol·K
*
Ura
nyl o
xide
s hy
drat
es a
nd p
erox
ides
291.
8± 1
4.4
–––
203.
9± 9
.1
276.
6± 4
3.9
283.
0± 8
.8
––––
–
Ura
nyl c
arbo
nate
s
––––
–
Ura
nyl p
hosp
hate
s
169.
6± 2
1.2
196.
3± 6
.8
Ura
nyl s
ilic
ates
53.2
± 5.
9
1020
.2±
7.2
681.
9± 7
.2
336.
8± 1
2.6
*dat
a on
S0 w
ere
reca
lcul
ated
fro
m S
hare
va e
t al.
(201
2), a
nd c
alcu
late
d us
ing
data
fro
m a
, Kub
atko
et a
l, (2
006)
; b, G
orm
an-L
ewis
et a
l. (2
008)
; c,
Kub
atko
et a
l, (2
003)
; d, K
ubat
ko e
t al,
(200
5); e
, Gor
man
-Lew
is e
t al.
(200
9); f
, Sha
reva
et a
l. (2
012)
; g, G
orm
an-L
ewis
et a
l. (2
007)
; h, S
hare
va e
t al
. (20
11).
S f
, el,
J/m
ol·K
–532
.5±8
.1
–605
.8±5
.0
–300
.5±
23.9
–497
.9±5
.0
–130
2.3
±21.
2f
–964
.3 ±
6.2f
–635
.4 ±
10.9
g
–27.
5 ±7
.3 h
–352
.5 ±
7.2 h
–100
7.6±
12.0
h
G
f, el, k
J/m
ol
–163
2.2
±7.4
–171
7.6
±4.4
–163
5.1±
23.4
–167
4.3
±4.1
–307
2.3
±4.8
e
–204
6.3
±12.
2e
–182
6.1
±2.1
g
–275
8.6
± 3.
5 h
–272
5.2
± 2.
6 h
–309
9.3
± 5.
6 h
H
f, el, k
J/m
ol
–179
1.0
± 3.
2 a
–153
6.2
± 2.
8 a
–189
8.2
± 2.
3 a
–172
4.7
± 5.
1 a
–182
2.7
± 2.
4 a
–164
5.4
±4.3
a
–234
4.7
± 4.
0 c
–171
6.4
± 4.
2d
–559
3.6
± 9.
1d
–443
1.6
±15.
3d
–322
3.2
± 4.
0 e
–233
3.7
± 4.
6 e
–202
2.7±
2.5
g
–276
8.1
± 6.
5 h
–294
7.2±
4.0
h
–339
9.7±
4.0
h
H
f, o
x, k
J/m
ol
4.4±
3.1a
–26.
6±2.
8 a
–44.
6±2.
2 a
–150
.6±4
.9 a
–53.
5±2.
4 a
–161
.5±4
.3 a
22.3
±3.9
c
–75.
7±4.
1c
–99.
1±4.
2d
–710
.4±9
.1d
–989
.3±1
4.0d
–241
.0 ±
3.9
e
–227
.2 ±
2.3
e
–117
.8 ±
4.3
g
–238
.5 ±
6.0
h
–215
.9 ±
6.5
–150
.0
4.3
For
mul
a pe
r on
e ur
anyl
cat
ion
UO
3(H
2O) 2
-U
O2(
OH
) 2
Ca 0
.17(
UO
2) O
0.67
(OH
) (H
2O) 1
.3
Na(
UO
2)O
(OH
)
Na 0
..34(
UO
2)O
0.67
(OH
)(H
2O) 1
.2
Pb0.
38(U
O2)
O (
OH
) 0.7
6(H
2O) 0
.3
(UO
2)O
2(H
2O) 4
UO
2CO
3
Na 2
Ca[
(UO
2)(C
O3)
3](H
2O) 5
K3N
aUO
2(C
O3)
3(H
2O)
UO
2HPO
4(H
2O) 3
UO
2(P
O4)
2/3(
H2O
) 4/3
(UO
2)(S
iO4)
1/2(
H2O
)
K(U
O2)
(SiO
3OH
)(H
2O)
Na(
UO
2)(S
iO3O
H)(
H2O
)
Ca 0
.5(U
O2)
(S
iO3O
H0.
5)(H
2O) 2
.5
Phas
e, f
orm
ula
Met
asch
oepi
teU
O3·
2H2O
-U
O2(
OH
) 2
Beq
uere
lite
C
a[(U
O2)
3O2(
OH
) 3] 2·8
H2O
Cla
rkei
te N
a(U
O2)
O(O
H)
Na-
Com
prei
gnac
ite
Na 2
[(U
O2)
3O2(
OH
) 3] 2·7
H2O
Cur
ite
Pb 3
(UO
2)8O
8(O
H) 6·2
H2O
Stu
dtit
e (U
O2)
O2·
4H2O
, ox
ide
+ H
2O
Stu
dtit
e (U
O2)
O2·
4H2O
, ox
ide
+ H
2O2
Rut
herf
ordi
ne U
O2C
O3
And
erso
nite
N
a 2C
a[(U
O2)
(CO
3)3]·5
H2O
Gri
mse
lite
K3N
aUO
2(C
O3)
3·H
2O
UO
2HP
O4
·3H
2O
(UO
2)3(
PO
4)2
·4H
2O
Sod
dyit
e (U
O2)
2(S
iO4)
·2H
2O
K-b
oltw
oodi
te
K(U
O2)
(SiO
3OH
)·1.
3H2O
N
a-bo
ltw
oodi
te
Na(
UO
2)(S
iO3O
H)·
H2O
U
rano
phan
e C
a(U
O2)
2(S
iO3O
H)·
5H2O

NAVROTSKY ET AL.
11
Alkali and Alkaline Earth Uranates Though these are not minerals, uranates are
potentially of interest to the nuclear fuel cycle, reactor accidents, and nuclear waste disposal. For example, alkaline earth uranates are important phases for incorporation of Ba and Sr as major fission products into the U matrix (Sali et al. 2000). Furthermore, uranates show clear thermodynamic trends of interest from a fundamental chemical point of view and that may guide predictions for more complex mineral phases.
The alkali and alkaline earth uranates have various stable compounds, reflecting different stoichiometries and, in some cases, U oxidation states. Griffiths & Volkovich (1999) have reviewed the structures of known alkali metal uranates as containing a modified uranyl group, [(UO2)O2]
2– in the case of monouranates, and [(UO2)O1.5]
2– or [(UO1.5)O2]
2–in case of diuranates. In each of these modifications, U is surrounded by two strongly bonded axial "uranyl" O atoms and four equatorial O atoms, less strongly bonded to U. Such fundamental building blocks can be connected in multiple ways to form planar layers or chains in
uranate structures. The three basic types of alkali metal uranates,
categorized by metal to uranium ratio of 4, 2, and 1, are M4UO5, M2UO4, and M2U2O7, respectively. Table 4-7 gives Ho
f,ox, the enthalpies of formation from oxides, for different types of alkali uranates at room temperature. These data have been normalized to per one uranyl unit per formula, analogous to the approach by Shvareva et al. (2012) in order to be able to compare the energetic trends among different stoichiometries.
Contributions of acid–base interactions to energetics of formation must be considered to compare formation enthalpies within similar structural types and among various classes of uranates. These acid–base interactions are between alkali and alkaline earth cations and "uranyl" groups. Thus, the reported data are plotted as functions of acidity of the alkali oxide, which can be indexed in the Smith scale (Smith 1987). These linear relationships shown in Figure 4-9 suggesting that formation enthalpies from oxides directly correlate with acidity of oxides, similar to data
h k
J/m
olf,
ox.
Figure 4-8. Observed linear experimental trends (uranyl silicates and hydroxide oxides) and predicted from linearity data for uranyl hydroxide oxide as a function of cation oxide acidity in Smith scale (Shvareva et al. 2012).

NAVROTSKY ET AL.
12
TABLE 4-7. ENTHALPY OF FORMATION OF ALKALI URANATES
Formula ΔHf, ox (kJ/mol) ΔHf, el (kJ/mol) ΔGf, el (kJ/mol) ΔSf, ox (kJ/mol K)
Li2U2O7 –84.05±2.96 a –1606.80 ± 2.65 a Na2U2O7 –170.70±2.16 a –1601.90 ± 2.00 a –1505.73 ± 2.01 a 4.3 ± 5.8 d K2U2O7 –219.85±2.61 a –1625.25 ± 2.25 a Li2UO4 –146.50±2.60 a –1968.20 ± 1.30 a –1853.19 ± 2.22 a –0.8 ± 7.7 d Na2UO4 –259.10±3.60 a –1897.70 ± 3.50 a –1779.30 ± 3.51 a –5.2 ± 9.5 d K2UO4 –333.70±3.14 a –1920.70 ± 2.20 a –1798.50 ± 3.25 a –10.2 ± 10.0 d Rb2UO4 –347.58±7.98 a –1922.70 ± 2.20 a –1800.14 ± 3.25 a –26.4 ± 7.5 c Cs2UO4 –358.55±2.62 a, b –1920.78 ± 2.24 a, b –1790.07 ± 2.71 a, c –25.9 ± 7.9 d Li4UO5 –219.80±4.60 a –2639.40 ± 1.70 a Na4UO5 –403.20±1.97 a –2456.60 ± 1.70 a
a Simoncic & Navrotsky (2007); b Cordfunke et al. (1986); c O'Hare & Hoekstra (1974); d calculated values from formation of enthalpy and free energy.
reported in Shvareva et al. (2012) and Le & Navrotsky (2008). A different alkali to U ratio in each type of uranate results in different slopes of the energetic trends in Figure 4-9. The data listed in Table 4-7 suggest that the higher alkali/U ratio leads to more exothermic formation enthalpies from oxides, Ho
f,ox, apparently due to the stronger contribution of acid–base interactions.
Though MUO3 uranates(V) have been categorized as perovskite-type structure with specified crystallographic position for U in +5 oxidation state (Chippindale et al. 1989, Dickens & Powell 1991), there were controversial results from XANES (Allen et al. 1974, Van et al. 2002), in which a doublet of U4f peaks is present indicating the existence of mixed valence states. Lately several attempts have been made to determine an oxidation state by X-ray absorption analysis (Soldatov et al. 2007, Liu et al. 2009). Results suggest that only one valence state (5+) presents in MUO3 species, while
hkJ
/mol
f, ox
5
4
7
3
Figure 4-9. Enthalpies of formation of selected alkali uranates as a function of oxides acidity.
the formation of doublet structure may be due to the susceptible oxidation of U5+and can be removed by carefully controlled etching process (Soldatov et al. 2007). Formation of uranates (V) can be achieved by reduction of M2UO4 or M2U2O7 at 450 to about 600oC (Griffiths & Volkovich 1999, Galkin et al. 1961, Hinatsu et al. 1992). Therefore, the enthalpies of formation from binary oxides for uranates(V)MUO3 are expected to be less exothermic compared to those for mono- and diuranates, as shown in Table 4-8 (Cordfunke & Ouweltjes 1981). Thermodynamic data for the alkaline earth uranates have been collected and plotted in a similar manner, and the energetics follow the trends found for alkali uranates, shown in Table 4-9 and Figure 4-10.
Uranyl Peroxide Materials
Simple uranyl peroxide complexes contain a single uranyl ion coordinated by as many as three peroxide groups that, each being bidentate, define the equatorial edges of hexagonal bipyramidal coordination polyhedra (Alcock 1968). When peroxide bridges uranyl ions, the configuration is bent (Miro et al. 2010, Sigmon et al. 2009, Vlaisavljevich et al. 2010) and nanoscale cage clusters containing as many as 60 uranyl ions self-assemble in aqueous systems (Burns 2011). These soluble clusters carry negative charges, are associated with counterions in solution, and can be crystallized. Under acidic conditions in deionized water, the combination of uranyl and peroxide causes the precipitation of studtite, [(UO2)(O2) (H2O)2](H2O)2 (Burns & Hughes 2003). Thus there

NAVROTSKY ET AL.
13
TABLE 4-8. ENTHALPY FORMATION OF MUO3 URANATES(V)
Formula Hf, ox (kJ/mol) Hf, el (kJ/mol) Gf, el (kJ/mol) Sf, ox (kJ/mol K)
LiUO3 –69.00 ± 2.18 –1522.3 ± 1.8 NaUO3 –133.15 ± 1.73 –1494.9 ± 1.6 –1412.50 ± 1.61 –8.8 ± 4.6* KUO3 –186.95 ± 2.10 –1522.9 ± 1.7 RbUO3 –190.89 ± 4.26 –1520.9 ± 1.8 * Calculated values from formation of enthalpy and free energy
TABLE 4-9. ENTHALPY OF FORMATION OF ALKALINE EARTH URINATES AT 298 K
Formula Hf, ox (kJ/mol) Hf, el (kJ/mol) Gf, el (kJ/mol) Sf, ox (kJ/mol K)
MgUO4 –31.90 ± 1.73 a –1857.3 ± 1.5 a –1749.6 ± 1.5 a 8.9 ± 4.8 d CaUO4 –143.10 ± 2.67 b –2002.00 ± 2.53 b –1888.71 ± 2.42 a –11.8 ± 7.1 d SrUO4 –175.70 ± 3.08 a –1990.8 ± 2.8 a –1881.36 ± 2.80 a 1.4 ± 8.1 d BaUO4 –221.90 ± 3.99 a –1993.8 ± 3.3 a –1883.3 ± 3.4 a –14.2 ± 10.6 d
Sr3UO6 –254.70 ± 3.75 c –3252.4 ± 2.1 c Ba3UO6 –342.30 ± 10.21 a –3210.4 ± 8.0 a –3044.95 ± 9.20 a –14.4 ± 27.8 d a Simoncic & Navrotsky (2007); b Kubatko et al. (2006); c Cordfunke et al. (1999); d calculated values from formation of enthalpy and free energy.
Figure 4-10. Enthalpies of formation of alkaline earth uranates .
is a richness of uranyl peroxide cluster chemistry in both aqueous solution and the solid state, the thermodynamics of which has just begun to be explored. Recently measured enthalpies of form-ation of a few of these uranyl peroxide solids (normalized to one mole of U) are summarized in Table 4-10 and shown as a function of charge-balancing cation ionic radius in Figure 4-11.
Insight can be gained by directly comparing formation reactions under environmentally relevant conditions, i.e., equilibria involving UO2(c), aqueous systems, and pertinent secondary mineral phases. In contrast to studtite, which has limited
thermodynamic stability and requires the continued presence of peroxide in solution to stabilize it (Kubatko 2003) alkali uranyl peroxide formation from the oxides features highly exothermic ΔHo
f,ox
values (Armstrong et al. 2012) and these phases could remain thermodynamically stable even in the absence of significant peroxide, perhaps even forming spontaneously by reaction of uranyl solutions with the O in air. Thus it is possible that such alkali U peroxide clusters, either as nanophases or as individual clusters in solution, can persist for
FIGURE 4-11. Comparison of enthalpies of formation from the oxides for studtite (Kubatko 2003), LiUT, NaUT, KUT and the U60 nanocluster solids (Armstrong et al. 2012), normalized per mole of U.

NAVROTSKY ET AL.
14
TABLE 4-10. SUMMARY OF THERMODYNAMIC DATA FOR URANYL PEROXIDE COMPOUNDS.
Formula Hf, ox (kJ/mol) Hf, el (kJ/mol)
Li4[UO2(O2)3](H2O)10 –260.9 ± 13.2 –5540.2 ± 14.0 Na4[UO2(O2)3](H2O)9 –515.5 ± 8.9 –5147.4 ± 9.9 K4[UO2(O2)3](H2O)9 –453.4 ± 9.3 –4975.8 ± 9.3
Li40K20[UO2(O2)(OH)]60(H2O)214 –138.1 ± 5.0 –2784.3 ± 5.1 Data refer to one mole of U (Burns & Hughes 2003, Armstrong et al. 2012)
long times and be transported away from the uraninite or nuclear fuel from which they form by oxidation under conditions of high salinity such as seawater. This may form an additional pathway for U transport in both the natural environment and in a reactor accident. The cluster compounds are metastable with respect to common uranyl mineral phases discussed above but may represent long-lived intermediates in their formation. More research in the thermochemical stability of this large class of uranyl peroxide cluster materials is under way.
Concluding Remarks This review has attempted to summarize the current state of thermochemical data for uranium (and thorium) minerals and related materials, with an emphasis on the structural and bonding factors which determine the systematic trends seen in thermodynamic properties. Although knowledge of these properties has improved substantially in the past decade, much work remains to be done, especially in the study of complex phases and solid solutions. To apply these thermochemical data for solids to calculate equilibria with aqueous fluids and silicate melts, the speciation and thermodynamics of actinides in the latter phases must be better constrained, especially at high temperature. ACKNOWLEDGEMENTS
Much of the work presented here is based upon work supported as part of Materials Science of Actinides, an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001089. REFERENCES
AIZENSHTEIN, M. SHVAREVA, T.Y. & NAVROTSKY, A. (2010): Thermochemistry of lanthana- and yttria-doped thoria, J Amer. Ceramic Soc. 93, 4142-4147.
ALCOCK, N.W. (1968): Crystal and molecular structures of sodium uranyl triperoxide, J. Chem. Soc., A, 1588-1594.
ALLEN, G.C. & HOLMES, N.R. (1993): Mixed valency behavior in some uranium oxides studies by x-ray photoelectron spectroscopy. Can. J. Appl. Spectrosc. 38, 124-130.
ALLEN, G.C., CROFTS, J.A., CURTIS, M.T., TUCKER, P.M., CHADWICK, D. & HAMPSON, P.J. (1974): X-ray photoelectron spectroscopy of uranium oxide phases. J. Chem. Soc., Dalton Trans.1296-1301.
ARMSTRONG, C.R., NYMAN, M., SHVAREVA, T., SIGMON, G.E., BURNS, P.C. & NAVROTSKY, A. (2012): Uranyl peroxide enhanced nuclear fuel corrosion in seawater. Proc. Natl. Acad. Sci. U.S.A. 109, 1874-1877, S1874/1871-S1874/1875.
BEALL, G.W., BOATNER, L.A., MULLICA, D.F. &
MILLIGAN, W.O. (1981): The structure of cerium orthophosphate, a synthetic analog of monazite. J Inorg. Nuclear Chem. 43, 101-105.
BENZ, R. (1969): Thorium-thorium dioxide phase equilibriums. J. Nucl. Mater. 29, 43-49.
BESSONOV, A.F. (1970): High-temperature x-ray diffraction study of uranium trioxide, Kristallografiya 15, 62-67.
BURNS, P.C. & HUGHES, K.-A. (2003): Studtite, [(UO2)(O2)(H2O)2](H2O)2: The first structure of a peroxide mineral, Amer. Mineral. 88, 1165-1168.
BURNS, P.C. (2011): Nanoscale uranium-based cage clusters inspired by uranium mineralogy. Mineral. Mag. 75, 1-25.
BUYUKKILIC, S. SHVAREVA, T. & NAVROTSKY, A. (2012): Enthalpies of formation and insights into defect association in ceria singly and doubly doped with neodymia and samaria. Solid State Ionics 227,17-22.
CHEN, F., EWING, R.C. & CLARK, S.B. (1999): The Gibbs free energies and enthalpies of formation of U6+ phases: An empirical method of prediction. [Erratum to document cited in CA130:354818], Amer. Mineral. 84, 1208.

NAVROTSKY ET AL.
15
CHIPPINDALE, A.M., DICKENS, P.G. & HARRISON, W.T.A. (1989): A structural study of the sodium uranate(V) (NaUO3) by time-of-flight powder neutron diffraction. J Solid State Chem. 78, 256-261.
CORDFUNKE, E.H.P. & OUWELTJES, W. (1981): Standard enthalpies of formation of uranium compounds. VI. Lithium, sodium, potassium, and rubidium trioxouranates. J.Chem. Thermodyn. 1, 3187-192.
CORDFUNKE, E.H.P., BOOIJ, A.S. & HUNTELAAR, M.E. (1999): The standard enthalpies of formation of uranium compounds. XV. Strontium urinates. J Chem. Thermodyn. 31, 1337-1345.
CORDFUNKE, E.H.P., OUWELTJES, W. & PRINS, G. (1986): Standard enthalpies of formation of uranium compounds. XIII. Cesium uranate (Cs2UO4). J. Chem. Thermodyn. 18, 503-509.
COX, J.D., WAGMAN, D.D., & MEDVEDEV, V.A. (1989): CODATA Key Values for Thermodynamics, Hemisphere Publ. Corp.
DESGRANGES, L., BALDINOZZI, G., ROUSSEAU, G., NIEPCE, J.-C. & CALVARIN, G. (2009): Neutron diffraction study of the in situ oxidation of UO2. Inorg. Chem., 48, 7585-7592.
DICKENS, P.G. & POWELL, A.V. (1991): Powder neutron diffraction study of potassium uranate(V) (KUO3). J. Mater. Chem. 1, 137-138.
DONALDSON, M., STEVENS, R., LANG, B.E., BOERIO-GOATES, J., WOODFIELDA, B.F., PUTNAM, R.L. & NAVROTSKY, A. (2005): Heat capacities and absolute entropies of UTi2O6 and CeTi2O6. J. Therm. Anal. Calorim. 81, 617-625.
DYER, P.N., RICHARDS, R.E., RUSSEK, S.L. &
TAYLOR, D.M. (2000): Ion transport membrane technology for oxygen separation and syngas production. Solid State Ionics, 134, 21-33.
ELLSWORTH, S., NAVROTSKY, A. & EWING, R.C. (1994): Energetics of radiation damage in natural zircon (ZrSiO4). Phys. Chem. Minerals 21, 140-149.
ENGMANN, R.W.P.M. de (1963): The crystal structure of γ-UO3. Acta Crystallogr. 16, 993-996.
EWING, R.C., HAAKER, R.F. & LUTZE, W. (1982): Leachability of zircon as a function of alpha dose. Mater. Res. Soc. Symp. Proc. 11, 389-397.
FERRISS, E.D.A., EWING, R.C. & BECKER, U. (2010): Simulation of thermodynamic mixing properties of actinide-containing zircon solid solutions. Amer. Mineral. 95, 229-241.
FINCH, R.J. & EWING, R.C. (1992): The corrosion of uraninite under oxidizing conditions. J. Nucl. Mater. 190, 133-156.
FINCH, R. & MURAKAMI, T. (1999): Systematics and paragenesis of uranium minerals, Rev. Mineral. 38, 91-179.
FITZGIBBON, G.C., PAVONE, D., HOLLEY, C.E., JR. (1967): Enthalpies of solution and formation of some uranium oxides. J. Chem. Eng. Data, 12, 122-125.
FRONDEL, C. (1958): Systematic Mineralogy of Uranium and Thorium. U.S. Gov't. Printing Office.
FUJINO, T. (1988): Thermodynamics of fluorite-type solid solutions containing plutonium, lanthanide elements or alkaline earth metals in uranium dioxide host lattices. J. Nucl. Mater. 154, 14-24.
GALKIN, N.P., SHUBIN, V.A. & KRYLOV, A.S. (1961): Reduction of uranium chemical concentrates. Zh. Neorg. Khim. 6, 2325-2328.
GALUSKINA, I.O., GALUSKIN, E.V., ARMBRUSTER, T., LAZIC, B., KUSZ, J., DZIERZANOWSKI, P., GAZEEV, V.M., PERTSEV, N.N., PRUSIK, K., ZADOV, A.E., WINIARSKI, A., WRZALIK, R. &
GURBANOV, A.G. (2010: Elbrusite-(Zr)-A new uranian garnet from the Upper Chegem caldera, Kabardino-Balkaria, Northern Caucasus, Russia. Amer. Mineral. 95, 1172-1181.
GORMAN-LEWIS, D., BURNS, P.C. & FEIN, J.B. (2008): Review of uranyl mineral solubility measurements, J Chem. Thermodyn. 40, 335-352.
GORMAN-LEWIS, D., FEIN, J.B., BURNS, P.C. &
SZYMANOWSKI, J.E.S. (2008): Converse, solubility measurements of the uranyl oxide hydrate phases metaschoepite, compreignacite, Na-compreignacite, becquerelite, and clarkeite. J Chem. Thermodyn. 40, 980-990.
GORMAN-LEWIS, D., MAZEINA, L., FEIN, J.B., SZYMANOWSKI, J.E.S., Burns, P.C. & Navrotsky, A. (2007): Thermodynamic properties of soddyite from solubility and calorimetry measurements. J Chem. Thermodyn. 39, 568-575.
GORMAN-LEWIS, D., SHVAREVA, T., KUBATKO, K.-A., BURNS, P.C., WELLMAN, D.M., MCNAMARA, B., SZYMANOWSKI, J.E.S., NAVROTSKY, A. &
FEIN, J.B. (2009): Thermodynamic properties of autunite, uranyl hydrogen phosphate, and uranyl orthophosphate from solubility and calorimetric measurements. Envir. Sci. & Technol. 43, 7416-7422.

NAVROTSKY ET AL.
16
GRIFFITHS, T.R. & VOLKOVICH, V.A. (1999): A review of the high temperature oxidation of uranium oxides in molten salts and in the solid state to form alkali metal uranates, and their composition and properties. J. Nucl. Mater. 274, 229-251.
GUILLAUMONT, R., ED. (2003): Update on the Chemical Thermodynamics of Uranium, Neptunium, Plutonium, Americium and Technetium, Elsevier, 2003.
HANKEN, B.E., SHVAREVA, T.Y., GRONBECH-JENSEN, N., STANEK, C.R., ASTA, M. &
NAVROTSKY, A. (2012): Energetics of cation mixing in urania-ceria solid solutions with stoichiometric oxygen concentrations. Phys. Chem. Chem. Phys. 14, 5680-5685.
HAZEN, R.M., EWING, R.C. & SVERJENSKY, D.A. (2009): Evolution of uranium and thorium minerals. Amer. Mineral. 94, 1293-1311.
HELEAN, K.B., NAVROTSKY, A., LUMPKIN, G.R., COLELLA, M., LIAN, J., EWING, R.C., EBBINGHAUS, B. & CATALANO, J.G. (2003): Enthalpies of formation of U-, Th-, Ce-brannerite: implications for plutonium immobilization. J. Nucl. Mater., 320, 231-244.
HINATSU, Y., FUJINO, T. & EDELSTEIN, N. (1992): Studies on magnetic susceptibility, electron paramagnetic resonance, and absorption spectrum of lithium uranate (Li3UO4), an octahedral uranium(5+) compound with a small tetragonal distortion, J Solid State Chem. 99, 95-102.
HOEKSTRA, H.R. (1961): Phase relationships in the uranium-oxygen and binary oxide series, In J. Belle, Ed. Uranium Dioxide: Properties and Nuclear Applications, U.S. Atomic Energy Commission, Washington, D. C., 229-304.
HOLLAND, H.D. & KULP, J.L. (1950): Geologic age from metamict minerals. Science 111, 312.
HUBER, E.J. JR. & HOLLEY, C.E. Jr. (1969): Enthalpies of formation of triuranium octaoxide and uranium dioxide. J Chem. Thermodyn., 1, 267-272.
IOUDINTSEV, S.V., OMELIANENKO, B.I. & LAPINA, M.I. (1998): Study of uranium incorporation into zircon: solubility limits and durability of fixation. Mater. Res. Soc. Symp. Proc. 506, 185-189.
JANECZEK, J. & EWING, R.C. (1992a): Coffinitiz-ation - a mechanism for the alteration of uranium dioxide under reducing conditions. Mater. Res. Soc. Symp. Proc. 257, 497-504.
JANECZEK, J. & EWING, R.C. (1992b): Dissolution and alteration of uraninite under reducing conditions. J. Nucl. Mater. 190, 157-173.
JOHNSON, G.K. & STEELE, W.V. (1981): The standard enthalpy of formation of uranium dioxide by fluorine bomb calorimetry. J Chem. Thermodyn., 13, 717-723.
KUBATKO, K.-A., HELEAN, K., NAVROTSKY, A. &
BURNS, P.C. (2006): Thermodynamics of uranyl minerals: enthalpies of formation of uranyl oxide hydrates. Amer. Mineral. 91, 658-666.
KUBATKO, K.-A.H., HELEAN, K.B., NAVROTSKY, A. & BURNS, P.C. (2003): Stability of peroxide-containing uranyl minerals. Science 302, 1191-1193.
KUBATKO, K.-A., HELEAN, K.B., NAVROTSKY, A. &
BURNS, P.C. (2005): Thermodynamics of uranyl minerals: Enthalpies of formation of rutherfordine, UO2CO3, andersonite, Na2CaUO2(CO3)3(H2O)5, and grimselite, K3NaUO2(CO3)3H2O, Amer. Mineral. 90, 1284-1290.
LANG, M.-I., ZHANG, F.-X,. ZHANG, J.-M., WANG, J.-W. LIAN, J., WEBER, W.J., SCHUSTER, B., TRAUTMANN, C., NEUMANN, R. & EWING, R.C. (2010): Review of A2B2O7 pyrochlore response to irradiation and pressure. Nucl. Instrum. Methods Phys. Res., Sect. B 268, 2951-2959.
LE, S.-N. & NAVROTSKY, A. (2008): Energetics of formation of alkali and ammonium cobalt and zinc phosphate frameworks. J Solid State Chem. 181, 20-29.
LEE, T.A. & NAVROTSKY, A. (2004): Enthalpy of formation of cubic yttria-stabilized hafnia. J. Mater. Res. 19, 1855-1861.
LEE, T.A., STANEK, C.R., MCCLELLAN, K.J., MITCHELL, J.N. & NAVROTSKY, A. (2008): Enthalpy of formation of the cubic fluorite phase in the ceria-zirconia system, J Mater. Res. 23, 1105-1112.
LIAN, J., WANG, L., LUMPKIN, G.R. & EWING, R.C. (2001): Heavy ion irradiation of brannerite-type ceramics. Mater Res. Soc. Symp. Proc. 650 R3.17/11-R13.17/16.
LIU, J.H., VAN, D.B.S. & KONSTANTINOVIC, M.J. (2009): XPS spectra of the U5+ compounds KUO3, NaUO3 and Ba2U2O7. J Solid State Chem. 182, 1105-1108.
LUMPKIN, G.R., SMITH, K.L. & BLACKFORD, M.G. (2001): Heavy ion irradiation studies of

NAVROTSKY ET AL.
17
columbite, brannerite, and pyrochlore structure types. J. Nucl. Mater. 289, 177-187.
MAZEINA, L., NAVROTSKY, A. & GREENBLATT, M. (2008): Calorimetric determination of energetics of solid solutions of UO2
+x with CaO and Y2O3. J. Nucl. Mater. 373, 39-43.
MAZEINA, L., USHAKOV, S.V., NAVROTSKY, A. &
BOATNER, L.A. (2005): Formation enthalpy of ThSiO4 and enthalpy of the thorite → huttonite phase transition. Geochim. et Cosmochim. Acta 69, 4675-4683.
MCCARTHY, G.J., WHITE, W.B. & PFOERTSCH, D.E. (1978): Synthesis of nuclear waste monazites, ideal actinide hosts for geologic disposal. Mater. Res. Bull. 13, 1239-1245.
MELDRUM, A., WANG, L.M. & EWING, R.C. (1996): Ion beam induced amorphization of monazite. Nucl. Instrum. Methods Phys. Res., Sect. B 116, 220-224.
MIRO, P., PIERREFIXE, S., GICQUEL, M., GIL, A. &
BO, C. (2010): On the origin of the cation templated self-assembly of uranyl-peroxide nanoclusters, J Amer. Chem. Soc., 132, 17787-17794.
MOLODETSKY, I., NAVROTSKY, A., LAJAVARDI, M. & BRUNE, A. (1998): The energetics of cubic zirconia from solution calorimetry of yttria- and calcia-stabilized zirconia. Z. Phys. Chem. (Munich) 207, 59-65.
MURAKAMI, T., CHAKOUMAKOS, B.C., EWING, R.C., LUMPKIN, G.R. & WEBER, W.J. (1991): Alpha-decay event damage in zircon. Amer. Mineral. 76, 1510-1532.
NAVROTSKY, A. (2010): Thermodynamics of solid electrolytes and related oxide ceramics based on the fluorite structure. J. Mater. Chem. 20, 10577-10587.
NAVROTSKY, A. SIMONCIC, P. YOKOKAWA, H. CHEN, W. & LEE, T. (2007): Calorimetric measurements of energetics of defect interactions in fluorite oxides. Faraday Discuss. 134, 171-180.
O'HARE, P.A.G. & HOEKSTRA, H.R. (1974): Thermochemistry of uranium compounds. V. Standard enthalpies of formation of the monouranates of lithium, potassium, and rubidium. J Chem. Thermodyn. 6, 1161-1169.
POPA, K., SHVAREVA, T., MAZEINA, L., COLINEAU, E., WASTIN, F., KONINGS, R.J.M. & NAVROTSKY, A. (2008): Thermodynamic properties of
CaTh(PO4)2 synthetic cheralite. Amer. Mineral. 93, 1356-1362.
RAK, Z., EWING, R.C. & BECKER, U. (2011): Role of iron in the incorporation of uranium in ferric garnet matrices, Phys. Rev. B: Condens. Matter Mater. Phys. 84, 155128/155121-155128/ 155110.
ROBIE, R.A. & HEMINGWAY, B.S. (1995): Thermodynamic properties of minerals and related substances at 298.15 K and 1 bar pressure and at higher temperatures. U.S. Geol. Surv. Bull. 2131, 461 p.
SALES, B.C., WHITE, C.W. & BOATNER, L.A. (1983): A comparison of the corrosion characteristics of synthetic monazite and borosilicate glass containing simulated nuclear defense waste. Nucl. Chem. Waste Manage. 4, 281-289.
SALI, S.K., SAMPATH, S. & VENUGOPAL, V. (2000): Thermal studies on alkaline earth urinates. J. Nucl. Mater. 277, 106-112.
SHVAREVA, T.Y., ALEXANDROV, V., ASTA, M. &
NAVROTSKY, A. (2011): Energetics of mixing in ThO2-CeO2 fluorite solid solutions. J. Nucl. Mater. 419, 72-75.
SHVAREVA, T.Y., FEIN, J.B. & NAVROTSKY, A. (2012): Thermodynamic properties of uranyl minerals: constraints from calorimetry and solubility measurements. Ind. Eng. Chem. Res. 51, 605-611.
SHVAREVA, T., MAZEINA, L., GORMAN-LEWIS, D., BURNS, P.C., SZYMANOWSKI, J.E.S., FEIN, J.B. &
NAVROTSKY, A. (2011 submitted) Thermo-dynamic characterization of boltwoodite and uranophane: Enthalpy of formation and aqueous solubility. Geochim. Cosmochim. Acta.
SIGMON, G.E., LING, J., UNRUH, D.K., MOORE-SHAY, L., WARD, M., WEAVER, B. & BURNS, P.C. (2009): Uranyl-peroxide interactions favor nanocluster self-assembly. J.Amer. Chem. Soc. 131, 16648-16649.
SIMONCIC, P. & NAVROTSKY, A. (2007): System-atics of phase transition and mixing energetics in rare earth, yttrium, and scandium stabilized zirconia and hafnia. J Amer. Ceramic Soc. 90, 2143-2150.
SMITH, D.W. (1987): An acidity scale for binary oxides. J. Chem. Educ. 64, 480-481.
SMITS, G. (1989): Uranium, thorium-bearing silicates in reefs of the Witwatersrand, South

NAVROTSKY ET AL.
18
Africa. Can. Mineral., 27, 643-656.
SOLDATOV, A.V., LAMOEN, D., KONSTANTINOVIC, M.J., VAN, D.B.S., SCHEINOST, A.C. &
VERWERFT, M. (2007): Local structure and oxidation state of uranium in some ternary oxides: X-ray absorption analysis. J Solid State Chem. 180, 54-61.
SPEER, J.A. (1980): Orthosilicates. The actinide orthosilicates. Rev. Mineral. 5, 113-135.
STEELE, B.C.H., LANE, J.A., ZHENG, K. & BAE, J. (1994): Ceramic materials for intermediate temperature solid oxide fuel cells. Proc. 2nd Internat. Conf. Ceramics in Energy Applications, The Institute of Energy, London, 109.
TOLE, M.P. (1985): The kinetics of dissolution of zircon (ZrSiO4). Geochim. et Cosmochim. Acta 49, 453-458.
TRAN, T.B. & NAVROTSKY, A. (2012): Energetics of dysprosia-stabilized bismuth oxide electrolytes. Chem. Mater. 24, 4185-4191.
USHAKOV, S.V., HELEAN, K.B., NAVROTSKY, A. &
BOATNER, L.A. (2001): Thermochemistry of rare-earth orthophosphates. J Mater. Res. 16, 2623-2633.
USHAKOV, S.V., NAVROTSKY, A., FARMER, J.M. &
BOATNER, L.A. (2004): Thermochemistry of the alkali rare-earth double phosphates, A3RE(PO4)2. J Mater. Res. 19, 2165-2175.
VAN, D.B.S., VERWERFT, M., LAVAL, J.P., GAUDREAU, B., ALLEN, P.G. & VAN, W.A. (2002): The local uranium environment in cesium
uranates: A combined XPS, XAS, XRD, and neutron diffraction analysis. J Solid State Chem. 166, 320-329.
VANCE, E.R., WATSON, J.N., CARTER, M.L., DAY, R.A. & BEGG, B.D. (2001): Crystal chemistry and stabilization in air of brannerite, UTi2O6. J Amer. Ceram. Soc. 84, 141-144.
VLAISAVLJEVICH, B., GAGLIARDI, L. & BURNS, P.C. (2010): Understanding the structure and formation of uranyl peroxide nanoclusters by quantum chemical calculations. J Amer. Chem. Soc. 132, 14503-14508.
WEBER, W.J., EWING, R.C. & WANG, L.M. (1994): The radiation-induced crystalline-to-amorphous transition in zircon. J Mater. Res. 9, 688-698.
WOODHEAD, J.A., ROSSMAN, G.R. & SILVER, L.T. (1991): The metamictization of zircon: Radiation dose-dependent structural characteristics. Amer. Mineral. 76, 74-82.
YAO, H.C. & YU, Y.Y.F. (1984): Ceria in automotive exhaust catalysts. I. Oxygen storage. J. Catal. 86, 254-265.
YUDINTSEV, S.V., LAPINA, M.I., PTASHKIN, A.G., IOUDINTSEVA, T.S., UTSUNOMIYA, S., WANG, L.M. & EWING, R.C. (2002): Accommodation of uranium into the garnet structure. Mater. Res. Soc. Symp. Proc. 713, 477-480.




















