
The risks associated with short-term placebo-controlled antihypertensive clinical trials: a...
10
ORIGINAL ARTICLE The risks associated with short-term placebo-controlled antihypertensive clinical trials: a descriptive meta-analysis A DeFelice 1 , J Willard 1 , J Lawrence 2 , J Hung 2 , MA Gordon 1 , A Karkowsky 1 , S Targum 1 , DC Throckmorton 1 , J Girton 3 , B Stertz 3 , SP Glasser 4 and RJ Lipicky 5 1 Division of Cardio-Renal Drug Products, FDA/CDER, White Oak, MD, USA; 2 Division of Biometrics, FDA/CDER, White Oak, MD, USA; 3 Ischemia Research and Education Foundation, San Francisco, CA, USA; 4 Division of Preventive Medicine, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, USA and 5 Lipicky LLC, North Potomac, MD, USA Short-term (4–8 weeks) placebo-controlled trials are used to evaluate new antihypertensive drug treatment. To evaluate the consequences of such practice, a descriptive meta-analysis was conducted, consisting of blinded review of original case report forms for all patients who died or left a study before its completion for all short-term, placebo-controlled hypertension trials submitted to the Food and Drug Administration from 1973 through 2001. There were 93 marketing applications or supplements involving 590 individual trials that involved 86 137 randomized patients (64 438 randomized to experimental drug and 21699 randomized to placebo) with 12 658 patient years of observation. There were 9636 dropouts (mean time to dropout was 28 days) and relative risk (RR (placebo/drug)) ¼ 1.33 (95% confidence limits, 1.28, 1.39; Po10 16 ). As expected, lack of blood pressure (BP) control was far more common in patients randomized to placebo; therapeutic failure, RR ¼ 2.53 (2.35, 2.73; Po10 15 ) and hypertensive emergency, RR ¼ 2.75 (2.19, 3.57; Po10 15 ). When administrative dropouts and dropouts resulting from inadequate BP control were excluded, the remaining 38% of dropouts were disproportionately more from drug (2810 drug, 816 placebo), RR ¼ 0.80 (0.74, 0.86; Po10 8 ). There were 43 deaths, RR ¼ 0.72 (0.33, 1.45; P ¼ 0.37); 40 strokes, RR ¼ 1.43 (0.68, 2.81; P ¼ 0.33) and 77 myocardial infarc- tions, RR ¼ 1.06 (0.62, 1.75; P ¼ 0.82). Irreversible harm (a combination of death, stroke and myocardial infarction, 160 total events) was equally distributed between the drug and placebo groups, RR ¼ 1.03 (0.71, 1.47; P ¼ 0.86). Journal of Human Hypertension (2008) 22, 659–668; doi:10.1038/jhh.2008.51; published online 5 June 2008 Keywords: placebo; hypertension trials; risk of placebo Introduction Long-term treatment of hypertension, even mild hypertension, protects patients from irreversible harm. 1–9 Approval of a new antihypertensive drug is primarily based upon short-term trials (usually 4–8 weeks), in which patients are randomly as- signed to the new antihypertensive treatment or to a placebo, with blood pressure (BP) the principal end point. It has been presumed that such short-term exposures should not have adverse morbid/mortal consequences, but to date, there has been no formal analysis of all available data addressing that presumption. Some data from long-term trials suggest that early lack of BP control might be harmful. 10,11 Yet, Al-Khatib et al. 12 —from a meta-analysis of pub- lished data from 25 placebo-controlled short-term trials (about 6409 patients and about 1602 patient- years of study)—concluded that harm associated with such assignments must be small or non- existent. A much larger body of pertinent trial data is available to the US Food and Drug Administra- tion, Division of Cardio-Renal Drug Products (FDA, DCRDP), in applications for marketing ap- proval of new antihypertensive agents, combina- tions of drugs and new formulations of approved drugs. From all trial reports submitted to FDA between 1973 and 2001, the Division of Cardio- Renal Drug Products and collaborators reviewed the complete case report forms (CRFs) for each patient to describe the consequences of randomization to placebo. The patients were all those who withdrew (dropped out) for any reason from short-term antihypertensive trials (mean time to dropout was 28 days). The meta-analysis of these data is the Placebo in Hypertension Adverse Reaction Meta- analysis (PHARM) project. Received 19 September 2007; revised 12 December 2007; accepted 14 February 2008; published online 5 June 2008 Correspondence: Dr RJ Lipicky, 15201 Apricot Lane, North Potomac, MD 20878, USA. E-mail: [email protected] Journal of Human Hypertension (2008) 22, 659–668 & 2008 Macmillan Publishers Limited All rights reserved 0950-9240/08 $32.00 www.nature.com/jhh
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of The risks associated with short-term placebo-controlled antihypertensive clinical trials: a...

ORIGINAL ARTICLE
The risks associated with short-termplacebo-controlled antihypertensiveclinical trials: a descriptive meta-analysis
A DeFelice1, J Willard1, J Lawrence2, J Hung2, MA Gordon1, A Karkowsky1, S Targum1,DC Throckmorton1, J Girton3, B Stertz3, SP Glasser4 and RJ Lipicky5
1Division of Cardio-Renal Drug Products, FDA/CDER, White Oak, MD, USA; 2Division of Biometrics,FDA/CDER, White Oak, MD, USA; 3Ischemia Research and Education Foundation, San Francisco, CA, USA;4Division of Preventive Medicine, Department of Medicine, University of Alabama at Birmingham,Birmingham, AL, USA and 5Lipicky LLC, North Potomac, MD, USA
Short-term (4–8 weeks) placebo-controlled trials areused to evaluate new antihypertensive drug treatment.To evaluate the consequences of such practice, adescriptive meta-analysis was conducted, consisting ofblinded review of original case report forms for allpatients who died or left a study before its completion forall short-term, placebo-controlled hypertension trialssubmitted to the Food and Drug Administration from1973 through 2001. There were 93 marketing applicationsor supplements involving 590 individual trials thatinvolved 86 137 randomized patients (64 438 randomizedto experimental drug and 21 699 randomized to placebo)with 12 658 patient years of observation. There were 9636dropouts (mean time to dropout was 28 days) andrelative risk (RR (placebo/drug))¼ 1.33 (95% confidencelimits, 1.28, 1.39; Po10�16). As expected, lack of blood
pressure (BP) control was far more common in patientsrandomized to placebo; therapeutic failure, RR¼ 2.53(2.35, 2.73; Po10�15) and hypertensive emergency,RR¼ 2.75 (2.19, 3.57; Po10�15). When administrativedropouts and dropouts resulting from inadequate BPcontrol were excluded, the remaining 38% of dropoutswere disproportionately more from drug (2810 drug, 816placebo), RR¼ 0.80 (0.74, 0.86; Po10�8). There were 43deaths, RR¼ 0.72 (0.33, 1.45; P¼ 0.37); 40 strokes,RR¼ 1.43 (0.68, 2.81; P¼ 0.33) and 77 myocardial infarc-tions, RR¼ 1.06 (0.62, 1.75; P¼ 0.82). Irreversible harm (acombination of death, stroke and myocardial infarction,160 total events) was equally distributed between thedrug and placebo groups, RR¼ 1.03 (0.71, 1.47; P¼ 0.86).Journal of Human Hypertension (2008) 22, 659–668;doi:10.1038/jhh.2008.51; published online 5 June 2008
Keywords: placebo; hypertension trials; risk of placebo
Introduction
Long-term treatment of hypertension, even mildhypertension, protects patients from irreversibleharm.1–9 Approval of a new antihypertensive drugis primarily based upon short-term trials (usually4–8 weeks), in which patients are randomly as-signed to the new antihypertensive treatment or to aplacebo, with blood pressure (BP) the principal endpoint. It has been presumed that such short-termexposures should not have adverse morbid/mortalconsequences, but to date, there has been no formalanalysis of all available data addressing thatpresumption.
Some data from long-term trials suggest thatearly lack of BP control might be harmful.10,11 Yet,
Al-Khatib et al.12—from a meta-analysis of pub-lished data from 25 placebo-controlled short-termtrials (about 6409 patients and about 1602 patient-years of study)—concluded that harm associatedwith such assignments must be small or non-existent. A much larger body of pertinent trial datais available to the US Food and Drug Administra-tion, Division of Cardio-Renal Drug Products(FDA, DCRDP), in applications for marketing ap-proval of new antihypertensive agents, combina-tions of drugs and new formulations of approveddrugs. From all trial reports submitted to FDAbetween 1973 and 2001, the Division of Cardio-Renal Drug Products and collaborators reviewed thecomplete case report forms (CRFs) for each patientto describe the consequences of randomization toplacebo. The patients were all those who withdrew(dropped out) for any reason from short-termantihypertensive trials (mean time to dropout was28 days). The meta-analysis of these data is thePlacebo in Hypertension Adverse Reaction Meta-analysis (PHARM) project.
Received 19 September 2007; revised 12 December 2007; accepted14 February 2008; published online 5 June 2008
Correspondence: Dr RJ Lipicky, 15201 Apricot Lane, NorthPotomac, MD 20878, USA.E-mail: [email protected]
Journal of Human Hypertension (2008) 22, 659–668& 2008 Macmillan Publishers Limited All rights reserved 0950-9240/08 $32.00
www.nature.com/jhh

Methods
Screening for relevant trialsFrom databases maintained by the FDA centraladministration and by the FDA’s Division of Car-dio-Renal Drug Products, we identified all new drugapplications (NDAs) or NDA supplements (sNDAs)involving the treatment of hypertension. A supple-ment is an application that allows changes to aproduct that already has an approved NDA, such asa new indication. Only randomized, double blindand placebo-controlled trials were eligible forinclusion. Because these data had been submittedto FDA only for the purpose of regulatory approval,written permission to include all trials was obtainedfrom each individual sponsor of each application.
Definition of dropout-classification criteriaand combinations of primary eventsAs listed in Table 1, we predefined 13 categoriesto which patients withdrawing from trials wereassigned.
Anticipating that individual events would beinfrequent, in the original protocol, we identifiedthree combinations of events of special interest; totalserious events (death, angina pectoris, arrhythmia,congestive heart failure (CHF), cerebrovascularaccident, cardiovascular accident (CVA), hyperten-sive emergency, myocardial infarction (MI) andtransient ischaemic attacks (TIAs)), serious cardio-vascular events, (angina, arrhythmia, CHF, MI),serious neurological events, (CVA, hypertensiveemergency, TIA). Cardiovascular events other thanthe above were not included in any of thosecombinations.
Three retrospective analyses of combinations ofevents were performed. These were all dropoutsexcluding administrative and treatment failure butincluding hypertensive emergency, all dropoutsexcluding administrative and therapeutic failure(TF) and also excluding hypertensive emergency,and a combination of events that includedonly documented events of irreversible harm(death, MI, CVA). There were no end-stage renaldisease events.
Review of case report formsFor each study, tabulation of the number of patientswho die or leave a study prior to study terminationbecause of an adverse event must be submitted inthe NDA or sNDA. For each of these patients acomplete set (each study visit as well as theterminating visit and usually a dropout form) ofCRFs must also be provided in each applicationsubmitted to the FDA (exceptions can be allowed byprearrangement). If the patient drops out of the trial,the investigator is usually expected to indicate why,in his or her opinion, the patient was withdrawn.
We retrieved all (every study visit) CRFs for allreported dropouts (for any reason) from all of the
trials included in our study (for crossover trials, weretrieved only the CRFs for the first period of thecrossover, as the second period is hampered bycarryover effects and other controversial aspects ofthis design). When CRFs were not present in thearchived NDA or sNDA submission, duplicates wereobtained from the drug sponsor. A team of healthprofessionals then reviewed all of the CRFs of everypatient who had dropped out of all trials. Thereviewers were blinded to the patient assignment(placebo/drug).
From the CRFs, each of two reviewers (by patientand by study), separately generated a 62-field Excelspreadsheet record that included the patient’s
Table 1 Dropout definitions
Category Definition
OT Inclusion criteria violation, other protocolviolations, loss to follow-up, withdrawalof consent, non-compliance with dosingor scheduled study visits, relocation,transportation impediments, inter-currentillness, non-cardiovascular surgery oremergent physical limitations from accidentsor the like
AP Typical chest pain or convincing anginaequivalent, without evidence of myocardialinfarction
AR Non-fatal arrhythmia other than ventriculartachycardia
CHF Conventional signs or symptoms of left- orright-sided heart failure
Non-fatal CVA Permanent neurological findings of abruptonset
Death Death between enrolment and 24 h afterdropout or completion
HE 1. Plausibly related new end-organ signsor symptomsa
and/or2. DBP4110 mm Hg or DBPincreased410 mm Hg from baseline
MI As assessed by the original investigatorOAE Signs or symptoms regarded as medical
events by the investigator, but not meetingany of our other criteria
OC Events judged to be cardiovascular, but notmeeting our criteria for any of AP, AR, CVA,MI, TIA or VT
TF Dropout because of excess BP, but not meetingour criteria for hypertensive emergency
TIA Neurological change with resolution within48 h
VTb Three or more successive ventricular ectopicbeats
Abbreviations: AP, angina pectoris, AR, arrhythmia; CHF, congestiveheart failure; CVA, cerebrovascular accident; HE, hypertensiveemergency; MI, myocardial infarction; OAE, other adverse events;OC, other cardiac; OT, administrative; TIA, transient ischaemic attack;VT, ventricular tachycardia.The 13 prospective categories for the primary cause for drop out fromany of the trials reviewed. Each patient had only one primary causefor dropout. There were a total of only eight VT events (seven drug,one placebo), so this category was merged into AR for the finalanalysis.aBut not stroke, MI or CHF.bEvents of VT were collapsed into AR in the final analysis.
Placebo in antihypertensive trialsA DeFelice et al
660
Journal of Human Hypertension

demographics, patient’s BPs at baseline and at lastroutine visit, any adverse events observed andwhether the dropout had been associated withadmission to an emergency department or a hospi-tal. The reviewers independently assigned thepatient’s primary reason (only one per patient) forwithdrawal to one of the 13 categories listed inTable 1. Reviewers were instructed that if two ormore categories seemed equally applicable, the moreserious category should be chosen as the primaryreason. Some deference was given to the assessmentthat had been made by the investigator, butreviewers were free to assign a different categorywhen it seemed to be more consistent with the otherdata in the CRFs, and/or in the definitions providedin the PHARM protocol. The reviewers’ assessmentswere independently entered directly onto indepen-dent spreadsheets. The two reviewers’ independentspreadsheets were then compared by ad hoccomputer methods. Discrepancies of demography,vital signs and so on were resolved informally, butdisagreements as to the primary reason for droppingout were referred to a 9-physician Dispute Resolu-tion Executive Committee (DREC). In each of thesecases, the patient’s CRFs (with the patient’s drug orplacebo assignment still masked) were reviewedfirst by a 3-member subcommittee of FDA medicalreviewers, and then, if consensus had not beenreached, by the full Committee (nine FDA medicalreviewers). After any discrepancies had been re-solved as just described, ad hoc software was usedto transcribe the spreadsheets into a form accessibleto standard statistical software.
Statistical analysisThis is a descriptive, by patient (not by study) meta-analysis. Our primary target for estimation was therelative risk (RR, placebo/drug) of dropout for anycause, comparing the patients assigned to placeboto those assigned to active treatment (drug). For RR,P-values were determined and are reported withoutcorrection for multiple comparisons. If a Bonferronicorrection is applicable (Table 2 has 22 compari-sons), for the sake of framework, a P-value equiva-lent to a standard P of 0.05 would require anuncorrected P of about 2� 10�3, and that of astandard P of 0.01 would require an uncorrectedP of about 4� 10�4.
We anticipated that most events of importancewould be of low incidence, and would not occur atall in many of the included trials. Assumption of anasymptotic normal model might then have led tomeaningless confidence intervals and other anoma-lies,13 so our primary methodology was the max-imum-likelihood method stratified by protocol.13,14
For completeness, we also used the conventionalmethods of Mantel and Haenszel15,16 (and foundalmost identical results compared to the maximum-likelihood method, Supplementary Information isavailable at Journal of Human Hypertension’s
website). After analysis of individual events, weconducted several analyses that aggregated events insome combination, including those prospectivelydefined in the protocol, but also three others asdescribed above.
Absolute event rates were calculated as (no. ofevents/(patient years))� 1000 for each group andthe 95% confidence intervals for the difference inrates (placebo/drug) were calculated assuming aconstant risk across protocols and time of observa-tion; maximum likelihood was not used for thesecalculations.
Other aggregate statistics were evaluated usingpaired comparisons and one-way analysis of var-iance; standard descriptive statistics were usedas appropriate. Except for the ad hoc spreadsheetsoftware noted above, we used standard softwarepackages, including SAS and JMP (SAS Institute,Cary, NC, USA), Access (Microsoft Corporation,Redmond, WA, USA), R (http://cran.r-project.org)and S-Plus (Insightful Software, Seattle, WA, USA).
Establishment of contextWe compared our findings from the short-term trialsreported in FDA applications to the findings of long-term trials reported in the medical literature andplan to report this complicated comparison sepa-rately. Briefly, despite the fact that PHARM evalu-ated only short-term trials (weeks), the absolute riskof events (events per 1000 patient years) observed inPHARM fell well within the range of the absoluteevent rates in long-term trials,17–29 for patientswithin the chronological age and magnitude ofsystolic BP defined by the PHARM populationbaseline values. For example, the total all causemortality rate in PHARM was 3.4 per 1000 patientyears, only slightly less than that reported by theCDC Division of Vital Statistics30 for the year 2003for this age group in the general population of theUnited States and total stroke events in PHARM was3.1 per 1000 patient years, slightly greater than thatseen in the MRC trial20 and the Australian trial19 inhypertensive populations.
Results
Eligible trialsReview of the FDA databases found 590 trials from atotal of 93 different applications that met ourinclusion criteria. There were 86 137 patients ran-domized in the trials, (64 438 assigned to activetreatment, 21 699 to placebo, with 12 658 patient-years of observation time). Forty-two differentchemical entities were represented, includinga-1 antagonists, a-2 agonists, angiotensin-receptorblockers, angiotensin-converting-enzyme inhibitors(one with neutral endopeptidase inhibition),b-adrenergic blockers, calcium-channel blockersand diuretics.
Placebo in antihypertensive trialsA DeFelice et al
661
Journal of Human Hypertension
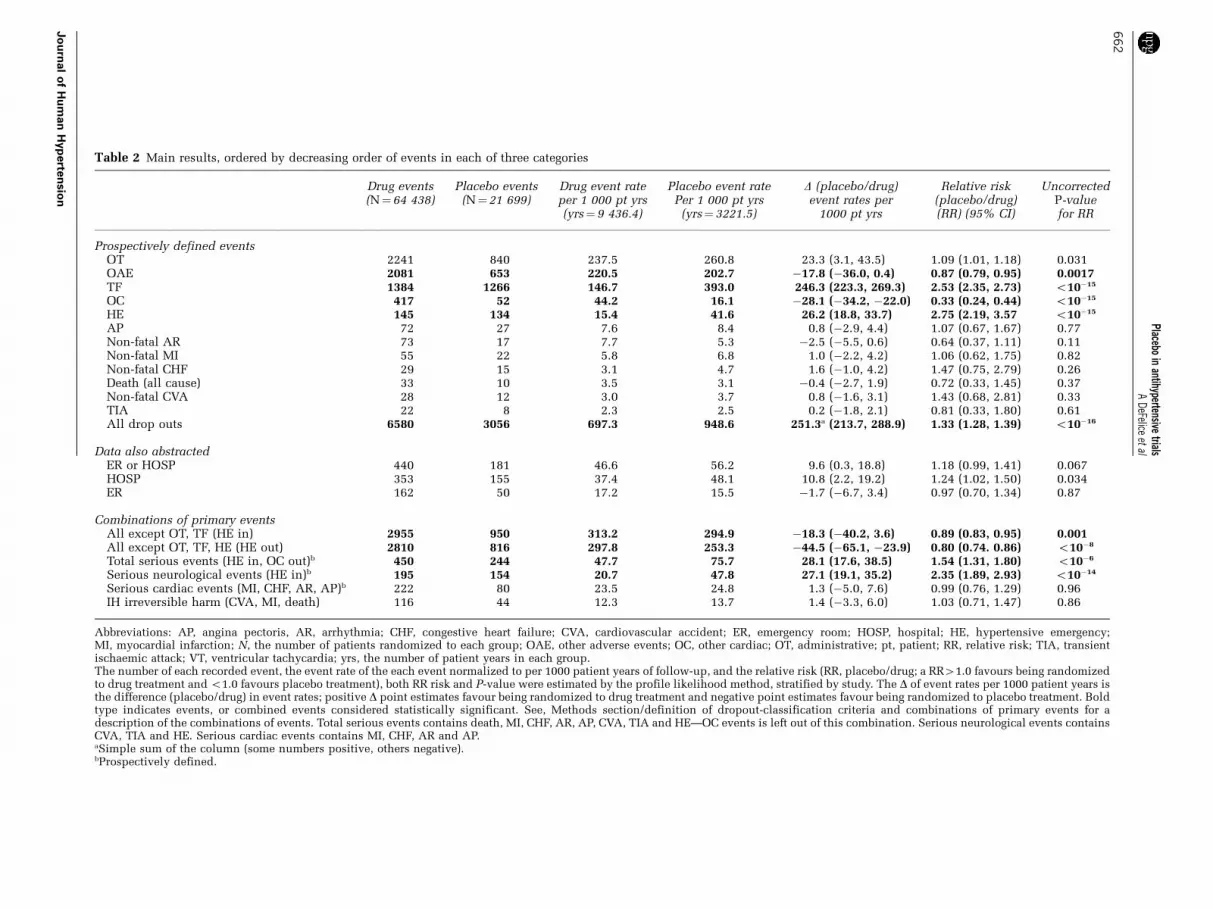
Table 2 Main results, ordered by decreasing order of events in each of three categories
Drug events(N¼64 438)
Placebo events(N¼21 699)
Drug event rateper 1 000 pt yrs(yrs¼ 9 436.4)
Placebo event ratePer 1 000 pt yrs
(yrs¼ 3221.5)
D (placebo/drug)event rates per
1000 pt yrs
Relative risk(placebo/drug)(RR) (95% CI)
UncorrectedP-valuefor RR
Prospectively defined eventsOT 2241 840 237.5 260.8 23.3 (3.1, 43.5) 1.09 (1.01, 1.18) 0.031OAE 2081 653 220.5 202.7 �17.8 (�36.0, 0.4) 0.87 (0.79, 0.95) 0.0017TF 1384 1266 146.7 393.0 246.3 (223.3, 269.3) 2.53 (2.35, 2.73) o10�15
OC 417 52 44.2 16.1 �28.1 (�34.2, �22.0) 0.33 (0.24, 0.44) o10�15
HE 145 134 15.4 41.6 26.2 (18.8, 33.7) 2.75 (2.19, 3.57 o10�15
AP 72 27 7.6 8.4 0.8 (�2.9, 4.4) 1.07 (0.67, 1.67) 0.77Non-fatal AR 73 17 7.7 5.3 �2.5 (�5.5, 0.6) 0.64 (0.37, 1.11) 0.11Non-fatal MI 55 22 5.8 6.8 1.0 (�2.2, 4.2) 1.06 (0.62, 1.75) 0.82Non-fatal CHF 29 15 3.1 4.7 1.6 (�1.0, 4.2) 1.47 (0.75, 2.79) 0.26Death (all cause) 33 10 3.5 3.1 �0.4 (�2.7, 1.9) 0.72 (0.33, 1.45) 0.37Non-fatal CVA 28 12 3.0 3.7 0.8 (�1.6, 3.1) 1.43 (0.68, 2.81) 0.33TIA 22 8 2.3 2.5 0.2 (�1.8, 2.1) 0.81 (0.33, 1.80) 0.61All drop outs 6580 3056 697.3 948.6 251.3a (213.7, 288.9) 1.33 (1.28, 1.39) o10�16
Data also abstractedER or HOSP 440 181 46.6 56.2 9.6 (0.3, 18.8) 1.18 (0.99, 1.41) 0.067HOSP 353 155 37.4 48.1 10.8 (2.2, 19.2) 1.24 (1.02, 1.50) 0.034ER 162 50 17.2 15.5 �1.7 (�6.7, 3.4) 0.97 (0.70, 1.34) 0.87
Combinations of primary eventsAll except OT, TF (HE in) 2955 950 313.2 294.9 �18.3 (�40.2, 3.6) 0.89 (0.83, 0.95) 0.001All except OT, TF, HE (HE out) 2810 816 297.8 253.3 �44.5 (�65.1, �23.9) 0.80 (0.74. 0.86) o10�8
Total serious events (HE in, OC out)b 450 244 47.7 75.7 28.1 (17.6, 38.5) 1.54 (1.31, 1.80) o10�6
Serious neurological events (HE in)b 195 154 20.7 47.8 27.1 (19.1, 35.2) 2.35 (1.89, 2.93) o10�14
Serious cardiac events (MI, CHF, AR, AP)b 222 80 23.5 24.8 1.3 (�5.0, 7.6) 0.99 (0.76, 1.29) 0.96IH irreversible harm (CVA, MI, death) 116 44 12.3 13.7 1.4 (�3.3, 6.0) 1.03 (0.71, 1.47) 0.86
Abbreviations: AP, angina pectoris, AR, arrhythmia; CHF, congestive heart failure; CVA, cardiovascular accident; ER, emergency room; HOSP, hospital; HE, hypertensive emergency;MI, myocardial infarction; N, the number of patients randomized to each group; OAE, other adverse events; OC, other cardiac; OT, administrative; pt, patient; RR, relative risk; TIA, transientischaemic attack; VT, ventricular tachycardia; yrs, the number of patient years in each group.The number of each recorded event, the event rate of the each event normalized to per 1000 patient years of follow-up, and the relative risk (RR, placebo/drug; a RR41.0 favours being randomizedto drug treatment and o1.0 favours placebo treatment), both RR risk and P-value were estimated by the profile likelihood method, stratified by study. The D of event rates per 1000 patient years isthe difference (placebo/drug) in event rates; positive D point estimates favour being randomized to drug treatment and negative point estimates favour being randomized to placebo treatment. Boldtype indicates events, or combined events considered statistically significant. See, Methods section/definition of dropout-classification criteria and combinations of primary events for adescription of the combinations of events. Total serious events contains death, MI, CHF, AR, AP, CVA, TIA and HE—OC events is left out of this combination. Serious neurological events containsCVA, TIA and HE. Serious cardiac events contains MI, CHF, AR and AP.aSimple sum of the column (some numbers positive, others negative).bProspectively defined.
Placeboin
antihypertensivetrials
ADeFelice
etal
66
2
Jo
urn
al
of
Hu
man
Hyp
erte
nsio
n

Assignment and collapse of categoriesApproximately 15% of the CRFs were referred to theDREC because of discrepant primary event categor-ization by the primary reviewers. After unblinding,we noted that only eight ventricular tachycardiaevents had been recorded, so this category wasmerged into the arrhythmia category.
Dropout ratesOverall. Of the 86 137 patients randomized in thePHARM trials, 9636 (11.2%) dropped out for anyreason. The majority (62%) of reasons for droppingout were either administrative or because of lackof BP control. Approximately 6% of the dropoutshad more than one reason recorded. At the timeof randomization the mean age was 54.1 years(median, 54.4; 95% confidence limits: 53.9, 54.3)and the mean supine BP was 159.2/102.4 mm Hg. In
total 60% were male, 40% female, 70% white, 20%black, 4% Hispanic and 4% were other. The mediantime-to-dropout was 28 days (mean 33.8 days), withno significant differences across drug classes ordropout categories (data not shown). Additionally,the RR and the time to dropout did not noticeablyvary with age, sex, ethnicity or study year (data notshown).
As shown in Table 2 and Figure 1 (a representa-tion of the RR data, which was the primary analysisand the only one with P-values), and Figure 2(a representation of absolute rate data) the overalldropout rate was 949 dropouts per 1000 patient-years among patients randomized to placebo and697 dropouts per 1000 patient-years among patientsrandomized to active treatment (RR¼ 1.33,Po10�16). For all dropouts, the difference of 251per 1000 patient years excess in the placebo groupwas mainly driven by the 246 per 1000 patient years
Figure 1 The figure is a plot of the relative risk (RR; x axis is on a log scale) of dropping out for all events and combination of eventsshown in Table 2. The centre of each circle is the RR point estimate; the area of the circle is proportional to the number of events in thecategory (the largest circle being all dropouts, total events¼9636 events). As in Table 2, individual events and combinations of events areshown in decreasing total event order. Tips of arrows are at the 95% confidence limits for the estimation. Those items that are statisticallysignificant are shown bolded, in comparison to those items that are not statistically significant. See, Methods section/definition ofdropout-classification criteria and combinations of primary events for description of these combinations of events. For combinationsof events, *¼prospectively defined of interest in the protocol.
Placebo in antihypertensive trialsA DeFelice et al
663
Journal of Human Hypertension

excess in TF. In the placebo group, there was also anexcess of hypertensive emergency, discussed furtherbelow. With respect to major events (for example,stroke, MI, CHF and death), there were neithersignificant differences nor strong trends.
Some additional description of selected indivi-dual event categories and combined event analysesis as follows.
Administrative. The most frequent reason fordropping out was administrative, and this wasslightly more common in the placebo group (RR,1.09; P¼ 0.031, uncorrected, see Statistical analysis
section). In total 4% of this population were seen inthe emergency rooms (ERs) or were hospitalized(7.8% placebo and 2.2% drug); interestingly, anincidence 4 times greater than that for TF. Thereasons for dropout in this category were protocoldeviations, loss to follow up, withdrawal of consent,non-compliance with dosing or scheduled studyvisits, relocation, transportation impediments, inter-current illness, non-cardiovascular surgery or emer-gent physical limitations from accidents and so on.
Other adverse events. This was the second mostfrequent event type (2734 events, 28% of all
Figure 2 This figure is a plot of the difference in absolute rate (placebo/drug) of dropping out for each category shown in Table 2. Plot a(inset) shows only all dropouts (All), therapeutic failure (TF) and hypertensive emergency (HE), as the x-axis scale needs to include closeto 300. Plot b shows the remainder (except that administrative dropouts (OT) are not plotted) on a different x axis scale. The centre ofeach circle is the D point estimate shown in Table 2; the area of the circle is proportional to the number of events in the category (thelargest circle being all dropouts, total events¼9636 events in plot a; the largest circle in plot b is other adverse events (OAE), totalevents¼ 2734). Tips of arrows are at the 95% confidence limits for the estimation. Those items that are statistically significant (forrelative risk, RR) are shown bolded, in comparison to those items that are not statistically significant. See, Methods section/definition ofdropout-classification criteria and combinations of primary events for description of these combinations of events. For combinations ofevents, *¼prospectively defined of interest in the protocol.
Placebo in antihypertensive trialsA DeFelice et al
664
Journal of Human Hypertension

dropouts) and was more common on drug. Althoughan expected outcome, the RR of 0.87 was only ofborderline significance (P¼ 0.0017). Most patientsin this category had three or more different descrip-tive events recorded. For example, in a sample of thefirst 300 patients in this category, headache wasmost common, and in decreasing order wereasthenia and lethargy, dizziness, laboratory abnorm-alities, skin rash, drowsiness, sexual dysfunctionand others. There were 222 emergency-department/hospital visits, but with equal frequency betweendrug and placebo groups (8% placebo, 8% drug),less than that associated with other cardiac causes(13% placebo, 11% drug) and greater than that ofadministrative.
Therapeutic failure. As expected, TF was muchmore common in the patients assigned to placebo(RR¼ 2.53, Po10�15). Most patients had some kindof symptom (for example, headache, nausea, vomit-ing and so on) in addition to lack of BP control. Atthe time of their last visit, this group had mean BPsof 166/108 and 172/109 mm Hg for systolic/diastolic,sitting and supine, respectively, representing a meanincrease, compared to baseline, of 6 and 4 mm Hg,systolic and diastolic, respectively. Very few ofthese patients were seen in an emergency depart-ment (12 patients, 0.4%) or admitted to a hospital(10 patients, 0.4%) in association with their studywithdrawal.
Cardiac adverse events. In total, 90% of the 469patients with cardiac adverse events had a singledescriptive event, and 10% had more than one event(for example, hypotension and dizziness) and wasmuch more common on drug (RR, 0.33; Po10�15).These events were, in decreasing order, posturalhypotension (8 placebo, 156 drug), bradycardiaand/or palpitations (8 placebo, 88 drug), hypoten-sion (4 placebo, 67 drug), angio-oedema (1 placebo,57 drug), non-descript electrocardiogram changesand/or ischaemia and non-descript chest pain(14 placebo, 43 drug), pitting oedema of extremities(6 placebo, 38 drug), dizziness (1 placebo, 17 drug),syncope (1 placebo, 17 drug) and fatigueand shortness of breath (5 placebo, 9 drug). Therewere 54 (13%) unscheduled visits in this population(25 placebo, 29 drug). Of the 54 unscheduledvisits, 27 (50%) were due to hypotension/posturalhypotension and 10 (19%) were due to angio-oedema.
Hypertensive emergency. As a priori defined,hypertensive emergency (HE) was much more commonin the placebo group (RR¼ 2.75; Po10�15), a RRalmost identical to that of TF, and hypertensiveemergency contributed 26 (per 1000 patient years)excess dropouts in the placebo group. The HEcategory was originally intended to only includepatients who had signs or symptoms of end-organ damage, but we also predefined an absolute
diastolic BP (DBP) or change of DBP. The protocol-specified absolute BP criterion for HE(DBP4110 mm Hg), a strictly enforced rule duringadjudication, was already satisfied at baseline byabout 10% of all PHARM patients. Similar topatients assigned to the TF category, patientsassigned to hypertensive emergency had one oranother symptom, but even if the symptoms wereheadache, non-anginal chest pain, erectile dysfunc-tion, rashes, flu-like syndromes and so on, yet if theDBP was greater than 110 mm Hg, the patientwas placed in the HE category rather than TF. Only17 patients (out of the 279 in the HE category)were declared as hypertensive emergencies bythe principal investigator of the trial; the other262 patients were placed in this category onlybecause of the authors’ deliberations. Our prospec-tive intent to qualitatively distinguish between TFand hypertensive emergency failed, so that thecategory of hypertensive emergency in this study ismisleading.
On the other had, at the time of their last regularstudy visit, the hypertensive emergency group hadmean BPs of 182/118 and 190/119 mm Hg forsystolic/diastolic, sitting and supine, respectively,representing a mean increase, compared to baselineof 17 and 12 mm Hg, systolic and diastolic, respec-tively; both being appreciably greater than that forthe patients considered TF. In addition, 37 of the279 patients in this category did have clinicalfindings, such as retinopathy, neurological findingsnot quite qualifying as TIA or CVA (or they wouldhave been placed in serious neurological events),internal carotid dissection and other findings ofsimilar severity.
Although the point estimate for the RR of hyper-tensive emergency is a reliable estimate (almostidentical to that of TF), the point estimate of 26 per1000 patient years excess dropouts in the placebogroup is in our opinion an inflated number and ofunreliable magnitude (the PHARM incidence beingat least an order of magnitude greater than most long-term intervention trials report). Around the time oftheir withdrawal, 26 of the hypertensive emergencygroup (7.5% placebo, 11.0% drug) were seen in anemergency department and another 26 were hospita-lized (9.0% placebo, 9.7% drug). Unscheduled visitswere more frequent in the hypertensive emergencygroup than the rate of 0.8% in the TF category, butthe signs and/or symptoms appeared reversible, andthere was no irreversible harm observed in thehypertensive emergency group.
Arrhythmia. There were only 90 (17 placebo, 73drug) such events, and the RR of 0.64 was notstatistically significant, but they are noted herebecause 43% of the arrhythmia group was hospita-lized or had an ER visit related to the event. Themost frequent (58) of these events was atrialdysrhythmia (13 placebo, 39 drug) and 35 (39% ifall arrhythmias) of these were atrial fibrillation/
Placebo in antihypertensive trialsA DeFelice et al
665
Journal of Human Hypertension

flutter (10 placebo, 59% of the arrhythmia popula-tion, 25 drug, 34% of the arrhythmia population).
Combinations of events of interest. As shown inTable 2, serious cardiovascular events (angina,arrhythmia, CHF or MI) were about equally commonin the drug and placebo groups, and the same is trueof the irreversible-harm group (death, stroke ormyocardial infarction), but there were few eventsin either group, and the confidence limits for theseestimates were fairly wide (for example, for seriouscardiovascular events the 95% confidence limits forRR were 0.76–1.29; for irreversible harm, 0.71–1.47).It is worth noting that eliminating dropouts foradministrative reasons, TF or hypertensive emer-gency gave a RR of 0.80 (Po10�8), favouring beingrandomized to the placebo group.
Discussion
As expected, there were few individual majorclinical events, resulting in point estimates withrather wide confidence limits, and confidence limitsremained wide even for combined events. Forexample, the RR for irreversible harm was 1.03 (a3% excess risk), if randomized to placebo, butconfidence intervals ranged from a 29% decreasedrisk (drug benefit) to 47% increased risk (placeboharm). The irreversible harm point estimate of anabsolute incidence of 1.4 excess events per 1000patient years places this in the rare category andeach of its components (death, MI and CVA) evenrarer in absolute incidence.
Of the six combined event analyses that wereconducted, there were only two that suggest beingrandomized to placebo is harmful. Both of thesetwo, contained events identified as hypertensiveemergency, a category that, as noted above, had afaulty prospective definition and confoundPHARM’s interpretation. For hypertensive emer-gency, the absolute rate of 26.2 per 1000 patientyears point estimate (Table 2 and Figure 2), excess ifrandomized to placebo, is almost identical to theexcess for total serious events and serious neurolo-gical events (28.1 and 27.1, respectively). All fourother point estimates of absolute rates are eitherneutral or favour being randomized to placebo.
We think the most appropriate descriptor of thePHARM combined events is all events absentadministrative, TF and hypertensive emergency.The RR¼ 0.80 (0.74, 0.86, Po10�8) for this afore-mentioned category rather strongly favours beingrandomized to placebo. There were 3626 suchevents (2810 placebo, 816 drug) with an excess of45 per thousand patient years in the group rando-mized to drug. Even if the inflated absolute risk ofhypertensive emergency is included, the result stillfavours being randomized to placebo, although lesscompellingly. These observations, along with thelack of obvious evidence for causing irreversible
harm, suggest that the continued use of placebo inshort-term antihypertensive is reasonably safe.
For the group of patients that had irreversibleharm (CVA, MI and death), lack of BP control wasnot obviously related to the events, as reflected byinterim visit BPs and the last routine visit BPs. Forthe entire irreversible harm group, compared tobaseline, there was a decrease in BP during thetrials; the group median difference was �12 mm Hgsystolic and �9 Hg diastolic for supine BP. In eachindividual event group there was a somewhatgreater decrease in BP for patients randomized todrug, but there was a decrease in median BPs ineach individual event category (with both placeboand drug), except for CVA where the median BPsincreased a couple of mm Hg (in both placebo anddrug groups). Further, antihypertensive therapy hadno appreciable treatment benefit, and when irrever-sible harm events did occur there was no obviousrelationship to lack of BP control. These observa-tions do not argue against the desirability of long-term (months and years) BP control, as that is wellestablished from both observational and placebo-controlled randomized clinical trial data. However,whether there is a long-term consequence of short-term (weeks) lack of BP could not be analysed in ourstudy, and may deserve systematic evaluation. Weare aware of only two small, placebo-controlledtrials,31,32 designed to evaluate very short-term(days) BP control in patients with very high baselineBPs, neither of which measured clinical end points.From those studies as well as others,33,34 it appearsthat rapid control of BP could be more harmfulthan beneficial, but a definitive conclusion is notpossible.
PHARM is the largest meta-analysis of short-termplacebo-controlled antihypertensive trials yet re-ported, and randomization to placebo was notassociated with detectable irreversible harm, despitea total of 160 events. It is unlikely that in theforeseeable future that a larger database will eitherbe available or be analysed. From the PHARManalysis, although there are clear risks associatedwith participation in a short-term antihypertensivetrial (some for drug, others for placebo), there isno objective reason to conclude that the use ofplacebo in short-term antihypertensive trails needbe discouraged.
Limitations
Publication bias, a problem for most meta-analyses,is essentially non-existent in the PHARM study; alloriginal CRFs for each patient were reviewed fromevery study submitted in support of an NDA orsNDA (whether approved or not). Studies performedunder an Investigational New Drug (IND), however,that never progressed to submission of an NDA orsupplement is not part of the PHARM database(there might be a few, but we are aware of only one,
Placebo in antihypertensive trialsA DeFelice et al
666
Journal of Human Hypertension

moxonidine). Also, PHARM has no informationwith respect to the placebo run-in phase of anystudy and has no information with respect to eventsafter the studies were concluded.
Additionally, ascertainment bias did occur. Weare aware of four errors in classification out of the9636 events recorded (but we did not correct for theknown four errors of classification in this report).Finally, and in retrospect, our definition of hyper-tensive emergency (the inclusion of absolute BP inthe definition) was a significant flaw in ourprospectively defined classification.
Acknowledgements
This study was enabled by, and could not havebeen completed without, support (by two primaryreviewers and data entry personnel) from TheIschemia Research Foundation (IREF), DennisMangano, MD, Director who was also a co-principalinvestigator. We appreciate the permission (bysigned letter) from each pharmaceutical company(too numerous to list) for us to use the data they hadsubmitted to NDAs. The original idea for the studycame from Stephen P Glasser MD, who hadgenerated a previous publication related to anti-angina trials.35 The entire staff and documentcontrol support of the Division of Cardio-Renal DrugProducts (DCRDP), FDA worked long hours to makethe study possible. Albert DeFelice, PhD and JimWillard, PhD (DCRDP) undertook major supervisorytasks in addition to their regular regulatory work-load. James Hung, PhD and John Lawrence, PhD(Division of Biostatistics, FDA) provided the initialstatistical analysis plan and performed the max-imum likelihood and Mantel Haenszel analyses.Douglas C Throckmorton, MD, prepared the originalprotocol and negotiated the IREF support. The paperwas originally drafted by Raymond J Lipicky andRobert Fenichel, MD, PhD and Stephen Glasser, MD
made major revisions and final suggestions weremade by Robert Temple, MD (Director of the Officeof Medical Policy—CDER FDA). The results wereinitially reported in abstract form36 shortly after theoriginal analysis.
Conflict of interest
There are no conflict of interest to declare. RobertFenichel, MD, PhD and Raymond J Lipicky, MD,were employees of FDA at the time of the study;each has now left the FDA and is functioning as apaid consultant to many pharmaceutical companies.Stephen Glasser, MD, has been involved withhypertension drug intervention trials, includingsome of those analysed in PHARM. IREF as apersonnel source did not conceive the project, norwas it involved in any supervisory capacity or anyoriginal analyses.
References
1 Collins R, Peto R, MacMahon S, Hebert P, Fiebach NH,Eberlein KA et al. Blood pressure, stroke, and coronaryheart disease. Part 2, short-term reductions in bloodpressure: overview of randomised drug trials in theirepidemiological context. Lancet 1990; 335: 827–838.
2 Collins R, MacMahon S. Blood pressure, antihyperten-sive drug treatment and the risks of stroke andof coronary heart disease. Br Med Bull 1994; 50: 272–298.
3 Thijs L, Fagard RH, Lijnen P, Staessen JA, Van Hoof R,Amery A. A meta-analysis of outcome trials in elderlyhypertensives. J Hypertens 1992; 10: 1103–1109.
4 Staessen JA, Gasowski J, Wang JG, Lutgarde T, Hond ED,Boissel J-P et al. Risks of untreated and treated isolatedsystolic hypertension in the elderly: meta-analysis ofoutcome trials. Lancet 2000; 355: 865–872.
5 Wang JG, Staessen JA. Conventional therapy and newerdrug classes for cardiovascular protection in hyperten-sion. J Am Soc Nephrol 2002; 13(Suppl 3): S208–S215.
6 Staessen JA, Wang JG, Thijs L. Cardiovascular preven-tion and blood pressure reduction: a quantitativeoverview updated until 1 March 2003. J Hypertens2003; 21: 1055–1076.
7 Turnbull F. Effects of different blood-pressure-loweringregimens on major cardiovascular events: results ofprospectively-designed overviews of randomisedtrials. Lancet 2003; 362: 1527–1535.
8 Chaudhry SI, Krumholz HM, Foody JM. Systolichypertension in older persons. JAMA 2004; 292:1074–1080.
9 White WB. Update on the drug treatment of hyperten-sion in patients with cardiovascular disease. Am J Med2005; 118: 695–705.
10 Julius S, Kjeldsen SE, Weber M, Brunner HR, Ekman L,Ekman S et al. Outcomes in hypertensive patients athigh cardiovascular risk treated with regimens basedon valsartan or amlodipine: the VALUE randomisedtrial. Lancet 2004; 363: 2022–2031.
11 Staessen JA, Thijs L, Fagard RH, Celis H, Birkenhager WH,Bulpit CJ et al. Effects of immediate versus delayedantihypertensive therapy on outcome in the SystolicHypertension in Europe Trial. J Hypertens 2004; 22:847–857.
What is known about the topicK Only one meta-analysis related to outcome data from
short-term, placebo-controlled, antihypertensive drug trialshas been published.12
K This reviewed only published data from 25 trials, 6049randomized patients and concluded that harm associatedwith these assignments must be small or non-existent.
What this study addsK A meta-analysis based upon case report forms submitted
to new drug applications or supplements to new drugapplications from 590 short-term, placebo-controlled,antihypertensive drug trials involving 86 137 patients.
K Enumeration of the risks associated with such trials (somefavouring being randomized to placebo, others favouringbeing randomized to drug).
K A somewhat more precise estimation of the RR ofirreversible harm (defined as all cause death, non-fatalstroke and non-fatal myocardial infarction), RR (placebo/drug)¼1.03.
Placebo in antihypertensive trialsA DeFelice et al
667
Journal of Human Hypertension

12 Al-Khatib SM, Califf RM, Hasselblad V, Alexander JH,McCrory DC, Sugarman J. Medicine. Placebo-controlsin short-term clinical trials of hypertension. Science2001; 292: 2013–2015.
13 Newcombe RG. Interval estimation for the differencebetween independent proportions: comparison ofeleven methods. Stat Med 1998; 17: 873–890.
14 Hasselblad V, Mosteller F, Littenberg B, Chalmers TC,Hunink MG, Turner JA et al. A survey of currentproblems in meta-analysis. Discussion from theAgency for Health Care Policy and Research inter-PORT Work Group on Literature Review/Meta-Analy-sis. Med Care 1995; 33: 202–220.
15 Mantel N, Haenszel W. Statistical aspects of theanalysis of data from retrospective studies of disease.J Natl Cancer Inst 1959; 22: 719–748.
16 Yusuf S, Peto R, Lewis J, Collins R, Sleight P. Betablockade during and after myocardial infarction: anoverview of the randomized trials. Prog Cardiovasc Dis1985; 27: 335–371.
17 The Veterans Administration Cooperative Study Groupon Antihypertensive Agents. Effects of treatment onmorbidity in hypertension. II. Results in patients withdiastolic blood pressure averaging 90 through114 mm Hg. JAMA 1970; 213: 1143–1152.
18 Helgeland A. Treatment of mild hypertension: a fiveyear controlled drug trial. The Oslo Study. Am J Med1980; 69: 725–732.
19 Report by the Management Committee. The Australiantherapeutic trial in mild hypertension. Lancet 1980;1: 1261–1267.
20 Medical Research Council Working Party. MRC trial oftreatment of mild hypertension: principal results. BMJ(Clin Res Ed) 1985; 291: 97–104.
21 Wang JG, Staessen JA, Gong L, Liu L. Chinese trial onisolated systolic hypertension in the elderly. SystolicHypertension in China (Syst-China) CollaborativeGroup. Arch Intern Med 2000; 160: 211–220.
22 Coope J, Warrender TS. Randomised trial of treatmentof hypertension in elderly patients in primary care.BMJ (Clin Res Ed) 1986; 293: 1145–1151.
23 Staessen JA, Fagard RH, Thijs L, Celis H, Arabidze GG,Birkenhager WH et al. Randomised double-blindcomparison of placebo and active treatment for olderpatients with isolated systolic hypertension. TheSystolic Hypertension in Europe (Syst-Eur) TrialInvestigators. Lancet 1997; 350: 757–764.
24 SHEP Cooperative Research Group. Prevention ofstroke by antihypertensive drug treatment in older
persons with isolated systolic hypertension. Finalresults of the Systolic Hypertension in the ElderlyProgram (SHEP). JAMA 1991; 265: 3255–3264.
25 Amery A, Birkenhager WH, Brixko P, Bulpitt C,Clement D, Derutterre M. Mortality and morbidityresults from the European Working Party on HighBlood Pressure in the Elderly trial. Lancet 1985; 1:1349–1354.
26 MRC Working Party. Medical Research Council trial oftreatment of hypertension in older adults: principalresults. BMJ 1992; 304: 405–412.
27 Dahlof B, Lindholm LH, Hansson L, Schersten B,Ekbom T, Wester PO. Morbidity and mortality in theSwedish Trial in Old Patients with Hypertension(STOP-Hypertension). Lancet 1991; 338: 1281–1285.
28 Bulpitt CJ, Beckett NS, Cooke J, Dumitrascu DL, Gil-Extremera B, Nachev C et al. Results of the pilot studyfor the Hypertension in the Very Elderly Trial.J Hypertens 2003; 21: 2409–2417.
29 MacMahon S, Peto R, Cutler J, Collins R, Sorlie P,Neaton J et al. Blood pressure, stroke, and coronary heartdisease. Part 1, Prolonged differences in blood pressure:prospective observational studies corrected for theregression dilution bias. Lancet 1990; 335: 765–774.
30 Hoyert DL, Kung HC, Smith BL. Deaths: preliminarydata for 2003. Natl Vital Stat Rep 2005; 53: 1–48.
31 Rutledge J, Ayers C, Davidson R, DiPette D, Gordon G,Fisher M et al. Effect of intravenous enalaprilat inmoderate and severe systemic hypertension. AmJ Cardiol 1988; 62: 1062–1067.
32 Habib GB, Dunbar LM, Rodrugues R, Neale AC, FridayKJ. Evaluation of the efficacy and safety of oralnicaradipine in the treatment of urgent hypertension:A multicenter, randomized, double-blind, parallel,placebo-controlled clinical trial. Am Heart J 1995;129: 917–923.
33 Handler J. Hypertensive urgency. J Clin Hypertens(Greenwich) 2006; 8(1): 61–64.
34 Cherney D, Straus S. Management of patients withhypertenisve urgencies and emergencies. J Gen InternMed 2002; 17: 927–945.
35 Glasser SP, Clark PI, Lipicky RJ, Olive ES, Hubbard JM,Yusuf S. Exposing patients with chronic, stable,exertional angina to placebo periods in drug trials.JAMA 1991; 265: 1550–1554.
36 DeFelice A, Gordon MA, Hung J, Karkowsky A,Lawrence J, Pelayo JC et al. Placebo in HypertensionAdverse Reaction Meta-analysis (PHARM). Circulation2003; 108(17) (Suppl): IV-452.
Supplementary Information accompanies the paper on the Journal of Human Hypertension website (http://www.nature.com/jhh)
Placebo in antihypertensive trialsA DeFelice et al
668
Journal of Human Hypertension




















