
The magnetic exchange interaction via N–H⋯O-bonding in copper(II) complex with...
7
The magnetic exchange interaction via N–HO-bonding in copper(II) complex with 1-phenyl-3-methyl-4-formylpyrazol-5-one 2-quinolylhydrazone Sergey I. Levchenkov a , Igor N. Shcherbakov b,⇑ , Leonid D. Popov b , Vladimir V. Lukov b , Vadim V. Minin c , Zoya A. Starikova d , Elena V. Ivannikova b , Arshak A. Tsaturyan b , Victor A. Kogan b a Southern Scientific Centre of Russian Academy of Sciences, Rostov-on-Don, Russia b Department of Chemistry, Southern Federal University, Rostov-on-Don, Russia c Kurnakov Institute of General and Inorganic Chemistry of Russian Academy of Sciences, Moscow, Russia d Nesmeyanov Institute of Organoelement Compounds of Russian Academy of Sciences, Moscow, Russia article info Article history: Received 5 October 2012 Received in revised form 17 May 2013 Accepted 24 May 2013 Available online 1 June 2013 Keywords: Exchange interaction DFT calculations X-ray crystal structure Copper complexes abstract The mononuclear copper(II) complex [CuL(CH 3 COO)], where L is monoanion of 1-phenyl-3-methyl-4- formylpyrazol-5-one 2-quinolylhydrazone has been synthesized. The weak intermolecular antiferromag- netic exchange interaction between copper(II) ions has been determined with the data of ESR spectros- copy and cryomagnetic measurements (2J = 3.25 cm 1 ). XRD data in combination with DFT calculations of exchange parameter within broken symmetry approach indicates that intermolecular exchange cou- pling is going through NHO hydrogen bonds in centrosymmetric H-bonded dimers. Ó 2013 Elsevier B.V. All rights reserved. 1. Introduction Study of hydrazones and azomethines of polyfunctional car- bonyl compounds as the potential ligand systems is one of the leading research areas in modern coordination as well as supramo- lecular chemistry since these ligands easily form the coordination compounds with the majority of transition metals [1–4]. Binuclear complexes based on these ligands are convenient models for devel- oping of the magneto-structural correlations, which attract interest from the viewpoint of novel molecular magnetic materials design [5–8]. During the last few decades much effort have been focused on the study of fundamental and applied aspects of coordination chemistry of acylpyrazolones and their derivatives mainly due to biological activity of these compounds and their strong complex formation ability towards metal ions [9,10]. Though the coordina- tion compounds based on 4-acylpyrazolone acylhydrazones and thiosemicarbazones are studied extensively [11–18], the complex formation ability of hetarylhydrazones of 4-formylpyrazolone and its derivatives gain less attention. In the present paper we report the results of structural character- ization, magnetic measurements and quantum-chemical modeling of copper(II) complex with 1-phenyl-3-methyl-4-formylpyrazol-5- one 2-quinolylhydrazone with unexpected intermolecular magnetic exchange interaction between two copper ions via NHO hydrogen bond. 2. Experimental All chemicals used for the preparative work were of reagent grade. Solvents were dried and distilled before use according to standard procedures. The infrared spectra were recorded on a Varian Scimitar 1000 FT-IR spectrophotometer in the range 4000–400 cm 1 . 1 H NMR spectra were recorded on a Varian Unity-300 spectrometer using TMS as internal standard. Microanalysis on C, H and N was performed on a Perkin-Elmer 240C Analyzer. ESR spectra were registered on a Bruker ELEXYS E-680X spectrometer in the X-range at room temperature and the temperature of liquid nitrogen. 2.1. Ligand synthesis (HL) To a hot solution of 1-phenyl-3-methyl-4-formylpyrazol-5- one (5 mmol) in ethanol (10 ml) was added a hot solution of 0020-1693/$ - see front matter Ó 2013 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.ica.2013.05.032 ⇑ Corresponding author. Tel./fax: +7 863 2975148. E-mail address: [email protected] (I.N. Shcherbakov). Inorganica Chimica Acta 405 (2013) 169–175 Contents lists available at SciVerse ScienceDirect Inorganica Chimica Acta journal homepage: www.elsevier.com/locate/ica
Transcript of The magnetic exchange interaction via N–H⋯O-bonding in copper(II) complex with...

Inorganica Chimica Acta 405 (2013) 169–175
Contents lists available at SciVerse ScienceDirect
Inorganica Chimica Acta
journal homepage: www.elsevier .com/locate / ica
The magnetic exchange interaction via N–H� � �O-bonding in copper(II)complex with 1-phenyl-3-methyl-4-formylpyrazol-5-one2-quinolylhydrazone
0020-1693/$ - see front matter � 2013 Elsevier B.V. All rights reserved.http://dx.doi.org/10.1016/j.ica.2013.05.032
⇑ Corresponding author. Tel./fax: +7 863 2975148.E-mail address: [email protected] (I.N. Shcherbakov).
Sergey I. Levchenkov a, Igor N. Shcherbakov b,⇑, Leonid D. Popov b, Vladimir V. Lukov b, Vadim V. Minin c,Zoya A. Starikova d, Elena V. Ivannikova b, Arshak A. Tsaturyan b, Victor A. Kogan b
a Southern Scientific Centre of Russian Academy of Sciences, Rostov-on-Don, Russiab Department of Chemistry, Southern Federal University, Rostov-on-Don, Russiac Kurnakov Institute of General and Inorganic Chemistry of Russian Academy of Sciences, Moscow, Russiad Nesmeyanov Institute of Organoelement Compounds of Russian Academy of Sciences, Moscow, Russia
a r t i c l e i n f o a b s t r a c t
Article history:Received 5 October 2012Received in revised form 17 May 2013Accepted 24 May 2013Available online 1 June 2013
Keywords:Exchange interactionDFT calculationsX-ray crystal structureCopper complexes
The mononuclear copper(II) complex [CuL(CH3COO)], where L is monoanion of 1-phenyl-3-methyl-4-formylpyrazol-5-one 2-quinolylhydrazone has been synthesized. The weak intermolecular antiferromag-netic exchange interaction between copper(II) ions has been determined with the data of ESR spectros-copy and cryomagnetic measurements (2J = �3.25 cm�1). XRD data in combination with DFT calculationsof exchange parameter within broken symmetry approach indicates that intermolecular exchange cou-pling is going through NH� � �O hydrogen bonds in centrosymmetric H-bonded dimers.
� 2013 Elsevier B.V. All rights reserved.
1. Introduction
Study of hydrazones and azomethines of polyfunctional car-bonyl compounds as the potential ligand systems is one of theleading research areas in modern coordination as well as supramo-lecular chemistry since these ligands easily form the coordinationcompounds with the majority of transition metals [1–4]. Binuclearcomplexes based on these ligands are convenient models for devel-oping of the magneto-structural correlations, which attract interestfrom the viewpoint of novel molecular magnetic materials design[5–8].
During the last few decades much effort have been focused onthe study of fundamental and applied aspects of coordinationchemistry of acylpyrazolones and their derivatives mainly due tobiological activity of these compounds and their strong complexformation ability towards metal ions [9,10]. Though the coordina-tion compounds based on 4-acylpyrazolone acylhydrazones andthiosemicarbazones are studied extensively [11–18], the complexformation ability of hetarylhydrazones of 4-formylpyrazoloneand its derivatives gain less attention.
In the present paper we report the results of structural character-ization, magnetic measurements and quantum-chemical modelingof copper(II) complex with 1-phenyl-3-methyl-4-formylpyrazol-5-one 2-quinolylhydrazone with unexpected intermolecular magneticexchange interaction between two copper ions via NH� � �O hydrogenbond.
2. Experimental
All chemicals used for the preparative work were of reagent grade.Solvents were dried and distilled before use according to standardprocedures. The infrared spectra were recorded on a Varian Scimitar1000 FT-IR spectrophotometer in the range 4000–400 cm�1. 1H NMRspectra were recorded on a Varian Unity-300 spectrometer using TMSas internal standard. Microanalysis on C, H and N was performed on aPerkin-Elmer 240C Analyzer. ESR spectra were registered on a BrukerELEXYS E-680X spectrometer in the X-range at room temperature andthe temperature of liquid nitrogen.
2.1. Ligand synthesis (HL)
To a hot solution of 1-phenyl-3-methyl-4-formylpyrazol-5-one (5 mmol) in ethanol (10 ml) was added a hot solution of

170 S.I. Levchenkov et al. / Inorganica Chimica Acta 405 (2013) 169–175
2-hydrazinoquinoline (5 mmol) in ethanol (10 ml). The mixturewas refluxed for 4 h and stayed overnight. The yellow-orange pre-cipitate was filtered out and recrystallized from the mixture buth-anol – DMF (4:1). Yield 1.17 g (68%). M.p. 176 �C.
1H NMR (DMSO-d6): 2.18 (s, 3H, CH3), 6.99 (d, 1H, J 9.41 Hz, 3-Hquinoline), 7.10 (t, 1H, J 7.3 Hz, p-Ph), 7.203 (s, 1H, CH@N), 7.34–7.40 (m, 3H, m-Ph, 8-H quinoline), 7.66 (t, 1H, J 7.7 Hz, 7-H quino-line), 7.81–7.86 (m, 2H, 5-H, 6-H quinoline), 7.99 (d, 2H, J 7.8 Hz, o-Ph), 8.15 (d, 1H, J 9.4 Hz, 4-H quinoline), 12.83 (s, 1H, NH), 17.67 (s,1H, NH).
IR (KBr, cm�1): m(NH) 3188, 3290 m(C = N) 1640, 1589.Anal. Calc. for C20H17N5O: C, 69.96; H, 4.99; N, 20.39. Found: C,
70.11; H, 4.84; N, 20.52%.
2.2. Synthesis of I
To a hot suspension of ligand HL (1 mmol) in 20 ml of methanola hot solution of copper(II) acetate (1 mmol) in 10 ml of methanolwas added. The mixture was refluxed for 5 h, the black precipitateformed was filtered off, washed with hot methanol and dried invacuum at 100 �C. Yield 0.26 g (56%). M.p. >250 �C. The single crys-tal samples of the complex suitable for X-ray analysis were ob-tained by the recrystallization from DMSO solution.
IR (KBr, cm�1): m(NH) 3194, m(C@N) 1632, 1596.Anal. Calc. for C22H19CuN5O3: C, 56.83; H, 4.12; N, 15.06. Found:
C, 56.62; H, 4.03; N, 15.38%.
2.3. X-ray diffraction study
X-ray diffraction study of complex I was carried out on a BrukerSmart Apex2 CCD diffractometer (Mo Ka, k = 0.71073 Å, graphitemonochromator). The initial array of measured intensities wasprocessed by the SAINT [19] and SADABS [20] programs. The structureswere solved by direct method and refined by the full-matrix least-squares method in the anisotropic approximation for non-hydro-gen atoms on F2. All calculations were performed by the SHELXTL
Table 1Selected crystallographic data for [CuL(CH3COO)].
Empirical formula C22H19N5O3CuFormula weight 464.96Crystal size (mm) 0.35 � 0.23 � 0.20T (K) 100(2)Wavelength (Å) 0.71073Crystal system monoclinicSpace group P21/na (Å) 8.5478(6)b (Å) 96.8170(10)c (Å) 19.4256(14)a (�) 90b (�) 104.674(3)c (�) 90V (Å3) 1969.3(2)Z 4Dcalc (Mg/m3) 1.568Absorption coefficient (mm�1) 1.146F (000) 956h Range (�) 3–30Reflections collected 24464Independent reflections 5737Reflections with I > 2r(I) 4271Index ranges �12 < h < 12
�16 < k < 16�27 < l < 27
Number of refined parameters 284Final R indices [I > 2r(I)] R1 = 0.0400, wR2 = 0.0914R indices (all data) R1 = 0.0634, wR2 = 0.1016Goodness-of-fit (GOF) on F2 1.002Largest difference in peak and hole (e �3) 0.512/�0.461
program package [21]. Details of the experiment and crystallo-graphic parameters are shown in Table 1. The hydrogen atom ofNH group was found in difference Fourier synthesis. The H(C) atompositions were calculated. All hydrogen atoms were refined in iso-tropic approximation in riding model with the Uiso(H) parametersequal to 1.2 Ueq(Ci), for methyl groups equal to 1.5 Ueq(Cii), whereU(Ci) and U(Cii) are respectively the equivalent thermal parame-ters of the atoms to which corresponding H atoms are bonded. Se-lected crystallographic data are gathered in Table 1.
2.4. Magnetic measurements
The molar magnetic susceptibility of the powder crystallinesample of complex was determined with Quantum DesignMPMSXL SQUID magnetometer in constant magnetic field of 1kOe between 2 and 300 K. The data were corrected on the diamag-netic contribution of the sample according to Pascal constants [22]and temperature independent paramagnetism. The magnetic prop-erties were interpreted using the isotropic Heisenberg–Dirac–vanVleck spin Hamiltonian (1):
H ¼ �2J � S1 � S2 ð1Þ
2.5. Computational details
All calculations within this study were performed employingthe DFT approach. For quantum-chemical modeling of the HL tau-tomers B3LYP hybrid exchange–correlation functional [23] withthe exchange component proposed in [24] and the Lee–Yang–Parrcorrelation functional [25] was used. The geometric parameters ofall tautomers were preliminarily optimized without any symmetryconstrains. The Pople’s 6–311+G(d,p) split-valence basis set wasemployed. Solvation energies were calculated within polarizablecontinuum model (PCM) of Tomasi and coworkers [26]. The GAUSS-
IAN’03 program package was utilized for these calculations [27].Quantitative description of magnetic exchange interactions re-
quires very accurate calculation of the energies of the high spin(HS) and low spin (LS) states arising from the parallel or antiparal-lel alignment of the interacting electron’s spin moments,correspondingly.
For two one-electron interacting paramagnetic centers withS1 = S2 = ½:
2J ¼ ELS � EHS
While HS state can be readily described by single determinantwave function, for LS state very accurate treatment of electron–electron correlation is required. The best results for magnetic spincoupling modeling are obtained within multireference wavefunction approaches such as complete active space second-orderperturbation theory CASPT2 [28,29] and multireference configura-tion interaction methods (difference dedicated CI, DDCI [30,31]and spectroscopy-oriented configuration interaction, SORCI [32]).These methods are very demanding towards computationalrecourses (especially multireference CI methods) and, therefore,applicable only for small to medium size systems.
Much less computational time is required for the alternativeDFT ‘‘broken symmetry‘‘ approach (DFT–BS) of Ginsberg, Noodle-man and Yamaguchi [33–35], within which LS state is approxi-mated by broken symmetry state (BS), which is described byunrestricted Kohn Sham single-determinant wave function. Bro-ken-symmetry solution for this state is obtained by allowing theelectron distribution symmetry to be lower than the actual spatialsymmetry of the molecule. DFT–BS approach is currently de factomainstream method for large scale exchange interaction modelingand the subject of critical discussion and development [36–40].

A
NN O
Ph
CH3 N
N
H
NH
NN O
Ph
CH3 NN
H
NHB
C
NN O
Ph
CH3 N
NN
HH
NN O
Ph
CH3 NN
NH
H
D
Scheme 1. Tautomeric forms A–D of HL.
Table 2Relative energies of most stable tautomeric forms of HL (kcal/mol).
Tautomer Relative energy (kcal/mol)
Gas phase CHCl3 DMSO
A 0 0 0B 0.95 0.62 0.05C 2.59 5.89 6.64D 4.57 7.21 7.99
Fig. 1. ESR spectrum of polycrystalline sample of I at T = 100 K; the inset shows thehalf-field signal.
S.I. Levchenkov et al. / Inorganica Chimica Acta 405 (2013) 169–175 171
For extraction of 2J values from the energies of BS and HS statesthree expressions (2)–(4) were proposed. Well grounded for non-overlapping single occupied magnetic orbitals (SOMOs) formula,originally obtained using spin projection operator [33,34,41,42]:
2J ¼ EBS � EHS
2S1S2ð2Þ
was criticized by Ruiz and others [43,44] to overestimate magneticcoupling due to presence of self-interaction error in the commonlyused DFT functionals. They proposed to employ non-spin projectedformula (3), derived with assumption of strong overlapping SOMOs:
2J ¼ EBS � EHS
2S1S2 þ S2; S1 P S2 ð3Þ
Some compromise between (2) and (3) is the formula (4) byYamaguchi (4) [45] for calculation of 2J, which is approximatelyvalid for different values of overlap integral:
2J ¼ 2ðEBS � EHSÞhS2
HSi � hS2BSi
ð4Þ
where hS2HSi, hS
2BSi – expectation values of total spin squared for the
HS and BS states, correspondingly.All three expressions are used in the present DFT–BS applica-
tions and there is still no general consensus found on their use[46]. In the present study we arbitrary choose expression (4) for2J calculations, though for weak interactions (2) and (4) expres-sions give the same results (in the case of two one-electron para-magnetic cites case hS2
HSi � 2; hS2BSi � 1 and (4) is equivalent to (2)).
Important problem in application of DFT–BS approach is properchoice of the DFT functional. Many benchmark studies has shownthat hybrid functionals including contribution of exact Hartree–Fock exchange give the best results in description of the magneticspin coupling [47–49], oppose to pure DFT functionals which tendto overestimate antiferromagnetic character of exchange interac-tion. Very informative review and discussion of the methods usedfor exchange interaction modeling can be found in recent paper ofBandeira and Le Guenic [48].
In the current study exchange parameter computation was per-formed with several hybrid exchange–correlation functionals,namely B3LYP [23], O3LYP [50], PBE0 [51], TPSSh [52] and doublehybrid functional B2PLYP [53] in which besides contribution of ex-act HF exchange Møller�Plesset-type perturbation part is included,at second order, on the correlation functional expression.
Basis set used for all atoms in complex molecules was 6–311G(d). Atomic coordinates were fixed at the values obtainedfrom XRD data for calculation of both high- and low-spin states.Tight convergence criteria were used in SCF procedure. Programcode ORCA v. 2.8 [54] on a cluster at Computer Centre of SFedUwas employed.
3. Results and discussion
Azomethine and hydrazone derivatives of acylpyrazolonesshow manifold possibilities of keto-enol and prototropic tautomer-ism [9,55–57]. For hydrazone HL a variety of tautomeric forms canbe suggested due to mobility of two protons which can be distrib-uted over several donor centers. The quantum-chemical calcula-tions of possible tautomers total energy in gas phase and in thesolvents of different polarity (DMSO and CHCl3) have been carriedout in order to compare their relative stability. The most stablestructures of hydrazone HL can be represented by the A–D tautom-ers (Scheme 1). Calculated relative energies are gathered in Table 2.Other isomers are much more destabilized (9–15 kcal/mol in gasphase, see Table SI.1 in Supplementary information).
The quinoline–pyrazolone tautomer B is destabilized by0.95 kcal/mol compared to A tautomer in gas phase, but the ener-gies of these tautomers become almost the same in DMSO solution.The energy difference is 0.05 kcal/mol which allows suggesting theexistence of both forms in the solution.
Nevertheless the 1H NMR spectrum of DMSO solutions of HLshow the presence of the quinolone–pyrazolone isomer A only.This conclusion is supported by the value of spin–spin couplingconstant between 3-H and 4-H protons of quinolone fragment. Thisvalue is 9.5 Hz, whereas for quinoline form it should be �8 Hz. Pyr-azole tautomers C and D are destabilized over A by 2.57 and4.57 kcal/mol, correspondingly, in gas phase. In solvents of bothpolarities their relative energies increase significantly due to lesssolvation energies compared with pyrazolone-type isomers A andB, especially in DMSO solution.
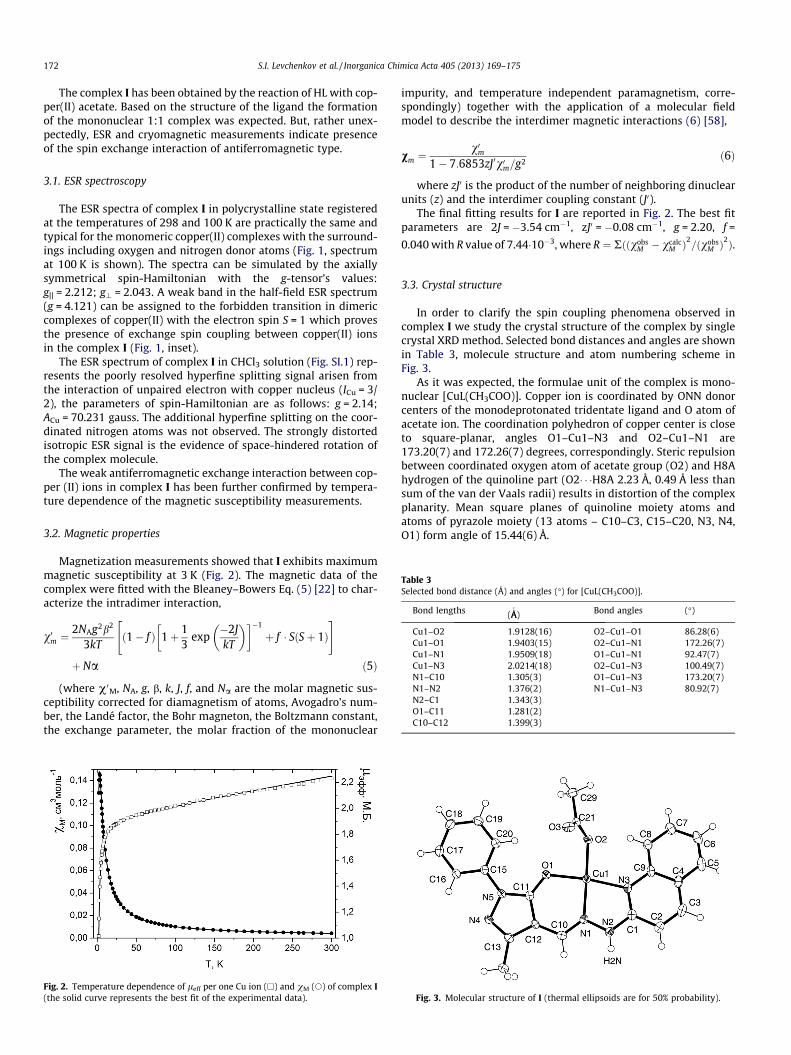
Table 3Selected bond distance (Å) and angles (�) for [CuL(CH3COO)].
Bond lengths (ÅA0
) Bond angles (�)
Cu1–O2 1.9128(16) O2–Cu1–O1 86.28(6)Cu1–O1 1.9403(15) O2–Cu1–N1 172.26(7)Cu1–N1 1.9509(18) O1–Cu1–N1 92.47(7)Cu1–N3 2.0214(18) O2–Cu1–N3 100.49(7)N1–C10 1.305(3) O1–Cu1–N3 173.20(7)N1–N2 1.376(2) N1–Cu1–N3 80.92(7)N2–C1 1.343(3)O1–C11 1.281(2)C10–C12 1.399(3)
172 S.I. Levchenkov et al. / Inorganica Chimica Acta 405 (2013) 169–175
The complex I has been obtained by the reaction of HL with cop-per(II) acetate. Based on the structure of the ligand the formationof the mononuclear 1:1 complex was expected. But, rather unex-pectedly, ESR and cryomagnetic measurements indicate presenceof the spin exchange interaction of antiferromagnetic type.
3.1. ESR spectroscopy
The ESR spectra of complex I in polycrystalline state registeredat the temperatures of 298 and 100 K are practically the same andtypical for the monomeric copper(II) complexes with the surround-ings including oxygen and nitrogen donor atoms (Fig. 1, spectrumat 100 K is shown). The spectra can be simulated by the axiallysymmetrical spin-Hamiltonian with the g-tensor’s values:g|| = 2.212; g\ = 2.043. A weak band in the half-field ESR spectrum(g = 4.121) can be assigned to the forbidden transition in dimericcomplexes of copper(II) with the electron spin S = 1 which provesthe presence of exchange spin coupling between copper(II) ionsin the complex I (Fig. 1, inset).
The ESR spectrum of complex I in CHCl3 solution (Fig. SI.1) rep-resents the poorly resolved hyperfine splitting signal arisen fromthe interaction of unpaired electron with copper nucleus (ICu = 3/2), the parameters of spin-Hamiltonian are as follows: g = 2.14;ACu = 70.231 gauss. The additional hyperfine splitting on the coor-dinated nitrogen atoms was not observed. The strongly distortedisotropic ESR signal is the evidence of space-hindered rotation ofthe complex molecule.
The weak antiferromagnetic exchange interaction between cop-per (II) ions in complex I has been further confirmed by tempera-ture dependence of the magnetic susceptibility measurements.
3.2. Magnetic properties
Magnetization measurements showed that I exhibits maximummagnetic susceptibility at 3 K (Fig. 2). The magnetic data of thecomplex were fitted with the Bleaney–Bowers Eq. (5) [22] to char-acterize the intradimer interaction,
v0m ¼2NAg2b2
3kTð1� f Þ 1þ 1
3exp
�2JkT
� �� ��1
þ f � SðSþ 1Þ" #
þ Na ð5Þ
(where v0M, NA, g, b, k, J, f, and Na are the molar magnetic sus-ceptibility corrected for diamagnetism of atoms, Avogadro’s num-ber, the Landé factor, the Bohr magneton, the Boltzmann constant,the exchange parameter, the molar fraction of the mononuclear
Fig. 2. Temperature dependence of leff per one Cu ion (h) and vM (s) of complex I(the solid curve represents the best fit of the experimental data).
impurity, and temperature independent paramagnetism, corre-spondingly) together with the application of a molecular fieldmodel to describe the interdimer magnetic interactions (6) [58],
vm ¼v0m
1� 7:6853zJ0v0m=g2ð6Þ
where zJ0 is the product of the number of neighboring dinuclearunits (z) and the interdimer coupling constant (J0).
The final fitting results for I are reported in Fig. 2. The best fitparameters are 2J = �3.54 cm�1, zJ0 = �0.08 cm�1, g = 2.20, f =
0.040 with R value of 7.44�10�3, where R ¼ RððvobsM � vcalc
M Þ2=ðvobs
M Þ2Þ.
3.3. Crystal structure
In order to clarify the spin coupling phenomena observed incomplex I we study the crystal structure of the complex by singlecrystal XRD method. Selected bond distances and angles are shownin Table 3, molecule structure and atom numbering scheme inFig. 3.
As it was expected, the formulae unit of the complex is mono-nuclear [CuL(CH3COO)]. Copper ion is coordinated by ONN donorcenters of the monodeprotonated tridentate ligand and O atom ofacetate ion. The coordination polyhedron of copper center is closeto square-planar, angles O1–Cu1–N3 and O2–Cu1–N1 are173.20(7) and 172.26(7) degrees, correspondingly. Steric repulsionbetween coordinated oxygen atom of acetate group (O2) and H8Ahydrogen of the quinoline part (O2� � �H8A 2.23 Å, 0.49 Å less thansum of the van der Vaals radii) results in distortion of the complexplanarity. Mean square planes of quinoline moiety atoms andatoms of pyrazole moiety (13 atoms – C10–C3, C15–C20, N3, N4,O1) form angle of 15.44(6) Å.
Fig. 3. Molecular structure of I (thermal ellipsoids are for 50% probability).
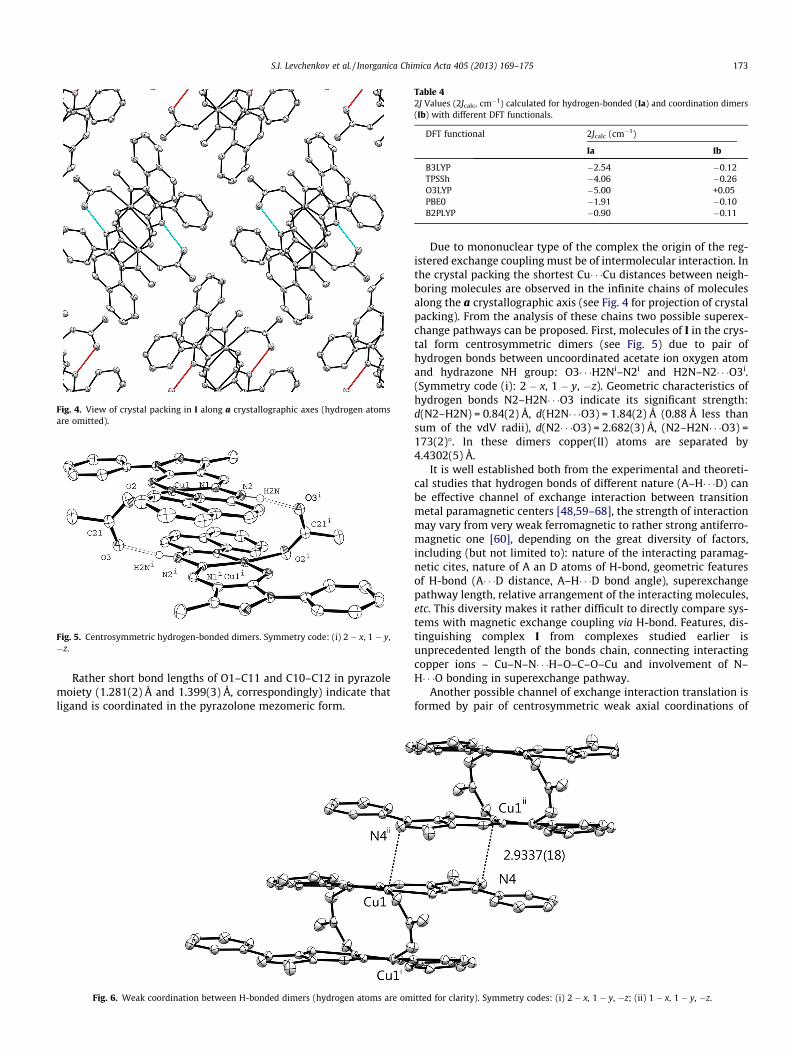
Fig. 4. View of crystal packing in I along a crystallographic axes (hydrogen atomsare omitted).
Fig. 5. Centrosymmetric hydrogen-bonded dimers. Symmetry code: (i) 2 � x, 1 � y,�z.
Table 42J Values (2Jcalc, cm�1) calculated for hydrogen-bonded (Ia) and coordination dimers(Ib) with different DFT functionals.
DFT functional 2Jcalc (cm�1)
Ia Ib
B3LYP �2.54 �0.12TPSSh �4.06 �0.26O3LYP �5.00 +0.05PBE0 �1.91 �0.10B2PLYP �0.90 �0.11
S.I. Levchenkov et al. / Inorganica Chimica Acta 405 (2013) 169–175 173
Rather short bond lengths of O1–C11 and C10–C12 in pyrazolemoiety (1.281(2) Å and 1.399(3) Å, correspondingly) indicate thatligand is coordinated in the pyrazolone mezomeric form.
Fig. 6. Weak coordination between H-bonded dimers (hydrogen atoms are om
Due to mononuclear type of the complex the origin of the reg-istered exchange coupling must be of intermolecular interaction. Inthe crystal packing the shortest Cu� � �Cu distances between neigh-boring molecules are observed in the infinite chains of moleculesalong the a crystallographic axis (see Fig. 4 for projection of crystalpacking). From the analysis of these chains two possible superex-change pathways can be proposed. First, molecules of I in the crys-tal form centrosymmetric dimers (see Fig. 5) due to pair ofhydrogen bonds between uncoordinated acetate ion oxygen atomand hydrazone NH group: O3� � �H2Ni–N2i and H2N–N2� � �O3i.(Symmetry code (i): 2 � x, 1 � y, �z). Geometric characteristics ofhydrogen bonds N2–H2N� � �O3 indicate its significant strength:d(N2–H2N) = 0.84(2) Å, d(H2N� � �O3) = 1.84(2) Å (0.88 Å less thansum of the vdV radii), d(N2� � �O3) = 2.682(3) Å, (N2–H2N� � �O3) =173(2)�. In these dimers copper(II) atoms are separated by4.4302(5) Å.
It is well established both from the experimental and theoreti-cal studies that hydrogen bonds of different nature (A–H� � �D) canbe effective channel of exchange interaction between transitionmetal paramagnetic centers [48,59–68], the strength of interactionmay vary from very weak ferromagnetic to rather strong antiferro-magnetic one [60], depending on the great diversity of factors,including (but not limited to): nature of the interacting paramag-netic cites, nature of A an D atoms of H-bond, geometric featuresof H-bond (A� � �D distance, A–H� � �D bond angle), superexchangepathway length, relative arrangement of the interacting molecules,etc. This diversity makes it rather difficult to directly compare sys-tems with magnetic exchange coupling via H-bond. Features, dis-tinguishing complex I from complexes studied earlier isunprecedented length of the bonds chain, connecting interactingcopper ions – Cu–N–N� � �H–O–C–O–Cu and involvement of N–H� � �O bonding in superexchange pathway.
Another possible channel of exchange interaction translation isformed by pair of centrosymmetric weak axial coordinations of
itted for clarity). Symmetry codes: (i) 2 � x, 1 � y, �z; (ii) 1 � x, 1 � y, �z.

Fig. 7. Localized SOMOs of Ia in HS state (contour value 0.025).
Fig. 8. Distribution of the Mulliken spin density over the atoms of exchangepathway in the BS state of Ia (B3LYP/6–311G(d)). Symmetry code: (i) 2 � x, 1 � y,�z.
174 S.I. Levchenkov et al. / Inorganica Chimica Acta 405 (2013) 169–175
copper ion with nitrogen atom of pyrazolone moiety of the neigh-boring molecule (see Fig. 6), d(Cu1-N4ii) = d(N4-Cu1ii) =2.9337(18) Å (symmetry code (ii): 1 � x, 1 � y, �z). This coordina-tion link hydrogen-bonded dimers into infinite chains along a crys-tallographic axis. Copper(II) atom is slightly shifted from the meansquare plane, defined by the nearest donor atoms (O1, O2, N1, N3)by 0.0544(3) Å towards N4ii atom of the neighboring molecule.
The distance between copper(II) ions for this pathway is muchlonger (d(Cu1–Cu1ii) = 6.0598(6) Å) than for the previous one.
3.4. Computational study
To further clarify the origin of the exchange coupling and probethe possible superexchange pathways we perform quantum-chem-ical modeling of the exchange interaction within ‘‘broken symme-try’’ approach for both hydrogen-bonded (Ia) and coordination (Ib)dimers. The binuclear molecular models for calculations were cho-sen exactly matching the structures in the crystal according to XRDdata.
2J Values calculated within DFT–BS approach strongly dependon the nature of the DFT functional employed. In order to testthe stability of the quantum chemical modeling in the presentstudy we use several hybrid functionals, namely B3LYP, PBE0,O3LYP and TPSSh and double-hybrid functional B2PLYP for 2J cal-culation in Ia and Ib binuclear units. Calculated 2J values are givenin Table 4 (energies and <S2> values calculated for HS and BS statesare listed in Table SI.2). All of the functionals considered in thepresent study predict that spin coupling in hydrogen-bonded di-mers Ia is much higher than in coordination dimers Ib and thus,exchange interaction experimentally observed in complex I, canbe attributed to the Ia binuclear unit, while interdimer exchange– to exchange through axial coordination. Values calculated withB3LYP and TPSSh functionals for Ia dimer are in remarkable agree-ment with the experimentally observed exchange coupling param-eter. The largest negative deviation for Ia is observed in case of
B2PLYP functional that can be explained by the known tendencyof it to overestimate stability of the HS states [48,69]. Calculatedwith B3LYP, PBE0, B2PLYP functionals 2J values for Ib are close toexperimentally observed interdimer exchange coupling, whileTPSSh somewhat overestimates it and O3LYP predict very small,but ferromagnetic coupling.
Localized magnetically active SOMOs of Ia dimer in HS state areshown on Fig. 7. Each of them is of r-type and is localized in one ofthe monomeric units with contributions of corresponding copperdx2�y2 atomic orbital (AO) and AOs of the nearest ligands atoms.
Delocalization of the magnetically active SOMOs over the com-plex molecule lead to non-zero spin-densities (SD) on the oxygenatom of acetate group (O3) and hydrogen atom of hydrazone frag-ment (H2N), resulting in rather high value of the exchange param-eter in the Ia dimer (if we consider the length of the pathwayconnecting the paramagnetic centers). It is clearly illustrated bythe distribution of the Mulliken SD over the atoms of exchangepathway in the BS state of Ia (Fig. 8).
Approximately 1/3 of the copper(II) spin density is delocalizedto the ligands. When moving along the exchange pathway fromthe copper atom, the SD gradually decreases in absolute value. Be-tween the molecules spin density change its sign between O3 atom(SD +0.0008) and H2N hydrogen (SD �0.0013). Strength of the H-bond (short length and favorable geometry) determines enoughoverlap for observed weak antiferromagnetic exchange coupling.
Shape of the localized SOMOs of monomeric units in Ib is verysimilar to that in Ia. Spin coupling in that case is very small due tothe fact that monomeric unit SOMOs are formally orthogonal toeach other. Yet small antiferromagnetic interaction can be attrib-uted to minor rehybridization of the mentioned orbitals due togeometrical distortions of the complex molecules.
4. Conclusion
Due to complementary combination of experimental (XRD, ESRand magnetic susceptibility measurements) and DFT calculationsof exchange parameter within broken symmetry approach it isshown that noticeable intermolecular antiferromagnetic exchangecoupling in the crystals of mononuclear copper(II) complex I is dueto NH� � �O hydrogen bonds in centrosymmetric H-bonded dimerswith unusually long superexchange pathway Cu–N–N� � �H–O–C–O–Cu.
Appendix A. Supplementary data
CCDC 904128 contains the supplementary crystallographic datafor I. These data can be obtained free of charge from The Cambridge

S.I. Levchenkov et al. / Inorganica Chimica Acta 405 (2013) 169–175 175
Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. Supplementary data associated with this article can befound, in the online version, at http://dx.doi.org/10.1016/j.ica.2013.05.032. These data include MOL files and InChiKeys of themost important compounds described in this article.
References
[1] T.S. Lobana, R. Sharma, G. Bawa, S. Khanna, Coord. Chem. Rev. 253 (2009) 977.[2] A.-M. Stadler, J. Harrowfield, Inorg. Chim. Acta 362 (2009) 4298.[3] L.D. Popov, A.N. Morozov, I.N. Shcherbakov, Y.P. Tupolova, V.V. Lukov, V.A.
Kogan, Russ. Chem. Rev. 78 (2009) 643.[4] Y.E. Alexeev, B.I. Kharisov, T.C.H. García, A.D. Garnovskii, Coord. Chem. Rev. 254
(2010) 794.[5] O. Kahn, Acc. Chem. Res. 33 (2000) 647.[6] M.N. Leuenberger, D. Loss, Nature 410 (2001) 789.[7] M.-C. Dul, E. Pardo, R. Lescouëzec, Y. Journaux, J. Ferrando-Soria, R. Ruiz-García,
J. Cano, M. Julve, F. Lloret, D. Cangussu, C.L.M. Pereira, H.O. Stumpf, J. Pasán, C.Ruiz-Pérez, Coord. Chem. Rev. 254 (2010) 2281.
[8] V.A. Kogan, V.V. Lukov, I.N. Shcherbakov, Russ. J. Coord. Chem. 36 (2010) 401.[9] A.D. Garnovskii, A.L. Nivorozhkin, V.I. Minkin, Coord. Chem. Rev. 126 (1993) 1.
[10] A.D. Garnovskii, I.S. Vasil’chenko, Russ. Chem. Rev. 74 (2005) 193.[11] F. Marchetti, C. Pettinari, R. Pettinari, Coord. Chem. Rev. 249 (2005) 2909.[12] L. Zhang, L. Liu, D. Jia, G. Xu, K. Yu, Inorg. Chem. Commun. 7 (2004) 1306.[13] B.-H. Peng, G.-F. Liu, L. Liu, D.-Z. Jia, K.-B. Yu, J. Mol. Struct. 692 (2004) 217.[14] Y. Yang, L. Zhang, L. Liu, G. Liu, J. Guo, D. Jia, Struct. Chem. 18 (2007) 909.[15] J. Lu, L. Zhang, L. Liu, G. Liu, D. Jia, D. Wu, G. Xu, Spectrochim. Acta, Part A 71
(2008) 1036.[16] V. Shul’gin, A. Obukh, E. Rusanov, V. Zub, V. Minin, Russ. J. Inorg. Chem. 54
(2009) 1223.[17] R.J. Yadav, K.M. Vyas, R.N. Jadeja, J. Coord. Chem. 63 (2010) 1820.[18] C.K. Modi, D.H. Jani, H.S. Patel, H.M. Pandya, Spectrochim. Acta, Part A 75
(2010) 1321.[19] SMART and SAINT. Release 5.0, Area Detector control and Integration Software,
Bruker AXS, Analytical X-Ray Instruments, Madison, Wisconsin, USA, 1994.[20] G.M. Sheldrick, SADABS: A Program for Exploiting the Redundancy of Area-
Detector X-Ray Data, University of Göttingen, Göttingen, Germany, 1999.[21] G.M. Sheldrick, Acta Crystallogr., Sect. A 64 (2008) 112.[22] B. Bleaney, K.D. Bowers, Proc. R. Soc. London, A 214 (1952) 451.[23] A.D. Becke, J. Chem. Phys. 98 (1993) 5648.[24] A.D. Becke, Phys. Rev. A 38 (1988) 3098.[25] C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 785.[26] J. Tomasi, B. Mennucci, R. Cammi, Chem. Rev. 105 (2005) 2999.[27] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman,
J.J.A. Montgomery, T. Vreven, K.N. Kudin, J.C. Burant, J.M. Millam, S.S. Iyengar, J.Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G.A. Petersson,H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T.Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J.E. Knox, H.P. Hratchian,J.B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J.Austin, R. Cammi, C. Pomelli, J.W. Ochterski, P.Y. Ayala, K. Morokuma, G.A.Voth, P. Salvador, J.J. Dannenberg, V.G. Zakrzewski, S. Dapprich, A.D. Daniels,M.C. Strain, O. Farkas, D.K. Malick, A.D. Rabuck, K. Raghavachari, J. B. Foresman,J.V. Ortiz, Q. Cui, A.G. Baboul, S. Clifford, J. Cioslowski, B.B. Stefanov, G. Liu, A.Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D.J. Fox, T. Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, M. Challacombe, P.M.W. Gill, B. Johnson, W.Chen, M.W. Wong, C. Gonzalez, J.A. Pople, GAUSSIAN 03 Revision E.1, 2003.
[28] K. Andersson, P.A. Malmqvist, B.O. Roos, A.J. Sadlej, K. Wolinski, J. Phys. Chem.94 (1990) 5483.
[29] K. Andersson, P.-A. Malmqvist, B.O. Roos, J. Chem. Phys. 96 (1992) 1218.[30] J. Miralles, J.-P. Daudey, R. Caballol, Chem. Phys. Lett. 198 (1992) 555.[31] J.M. Maestre, X. Lopez, C. Bo, J.M. Poblet, N. Casañ-Pastor, J. Am. Chem. Soc. 123
(2001) 3749.[32] F. Neese, T. Petrenko, D. Ganyushin, G. Olbrich, Coord. Chem. Rev. 251 (2007)
288.[33] A.P. Ginsberg, J. Am. Chem. Soc. 102 (1980) 111.[34] L. Noodleman, J. Chem. Phys. 74 (1981) 5737.[35] K. Yamaguchi, Y. Takahara, T. Fueno, K.N. Houk, Theor. Chim. Acta 73 (1988)
337.[36] C. Blanchet-Boiteux, J.-M. Mouesca, J. Phys. Chem. A 104 (2000) 2091.[37] A. Bencini, F. Totti, J. Chem. Theory Comput. 5 (2008) 144.[38] J.-M. Mouesca, J. Chem. Phys. 113 (2000) 10505.[39] N. Onofrio, J.-M. Mouesca, J. Phys. Chem. A 114 (2010) 6149.[40] N. Onofrio, J.-M. Mouesca, Inorg. Chem. 50 (2011) 5577.[41] R. Caballol, O. Castell, F. Illas, J. Phys. Chem. A 101 (1997) 7860.[42] F. Illas, Phys. Rev. B 70 (2004) 132414.[43] E. Ruiz, J. Cano, S. Alvarez, P. Alemany, J. Comput. Chem. 20 (1999) 1391.[44] E. Ruiz, S. Alvarez, J. Cano, V. Polo, J. Chem. Phys. 123 (2005) 164110.[45] K. Yamaguchi, Y. Takahara, T. Fueno, in: J.V.H. Smith, H.F. Schaefer III, K.
Morokuma (Eds.), Applied Quantum Chemistry, D. Reidel, Dordrecht, 1986, p.155.
[46] A. Bencini, Inorg. Chim. Acta 361 (2008) 3820.[47] I. Ciofini, C.A. Daul, Coord. Chem. Rev. 238–239 (2003) 187.[48] N.A.G. Bandeira, B. Le Guennic, J. Phys. Chem. A 116 (2012) 3465.[49] P. Comba, S. Hausberg, B. Martin, J. Phys. Chem. A 113 (2009) 6751.[50] A.J. Cohen, N.C. Handy, Mol. Phys. 99 (2001) 607.[51] C. Adamo, V. Barone, J. Chem. Phys. 110 (1999) 6158.[52] J. Tao, J.P. Perdew, V.N. Staroverov, G.E. Scuseria, Phys. Rev. Lett. 91 (2003)
146401.[53] S. Grimme, J. Chem. Phys. 124 (2006) 034108.[54] F. Neese, ORCA, an ab initio, DFT and Semiempirical Electronic Structure
Package, Version 2.8, Revision 15, Max–Planck-Institute für BioanorganischeChemie, Mulheim, Germany, 2009.
[55] J.S. Casas, M.S. García-Tasende, A. Sánchez, J. Sordo, Á. Touceda, Coord. Chem.Rev. 251 (2007) 1561.
[56] A. Amarasekara, O. Owereh, K. Lyssenko, T. Timofeeva, J. Struct. Chem. 50(2009) 1159.
[57] L. Liu, D. Jia, Synth. React. Inorg. Met.-Org. Chem. 35 (2005) 269.[58] O. Kahn, Molecular Magnetism, Wiley-VCH, New York, 1993.[59] J.S. Costa, N.A.G. Bandeira, B. Le Guennic, V. Robert, P. Gamez, G. Chastanet, L.
Ortiz-Frade, L. Gasque, Inorg. Chem. 50 (2011) 5696.[60] C. Desplanches, E. Ruiz, A. Rodríguez-Fortea, S. Alvarez, J. Am. Chem. Soc. 124
(2002) 5197.[61] W. Plass, A. Pohlmann, J. Rautengarten, Angew. Chem., Int. Ed. 40 (2001) 4207.[62] J. Tang, J.S. Costa, A. Golobic, B. Kozlevcar, A. Robertazzi, A.V. Vargiu, P. Gamez,
J. Reedijk, Inorg. Chem. 48 (2009) 5473.[63] D. Valigura, J. Moncol, M. Korabik, Z. Púceková, T. Lis, J. Mrozinski, M. Melník,
Eur. J. Inorg. Chem. 2006 (2006) 3813.[64] Z. Vasková, J. Moncol, M. Korabik, D. Valigura, J. Švorec, T. Lis, M. Valko, M.
Melník, Polyhedron 29 (2010) 154.[65] B. Kozlevcar, N. Kitanovski, Z. Jaglicic, N.A.G. Bandeira, V. Robert, B. Le Guennic,
P. Gamez, Inorg. Chem. 51 (2012) 3094.[66] G.A. Craig, O. Roubeau, J. Ribas-Ariño, S.J. Teat, G. Aromí, Polyhedron 52 (2013)
1369.[67] M. Peric, M. Zlatar, S. Grubišic, M. Gruden-Pavlovic, Polyhedron 42 (2012) 89.[68] R.D. Willett, C.J. Gómez-García, B. Twamley, S. Gómez-Coca, E. Ruiz, Inorg.
Chem. 51 (2012) 5487.[69] S. Ye, F. Neese, Inorg. Chem. 49 (2010) 772.



![N -[4-( N -Cyclohexylsulfamoyl)phenyl]acetamide](https://static.fdokumen.com/doc/165x107/632f4f4de68feab59a0210b7/n-4-n-cyclohexylsulfamoylphenylacetamide.jpg)

![N-[2-Methyl-5-(triazol-1-yl)phenyl]pyrimidin-2-amine as a Scaffold for the Synthesis of Inhibitors of Bcr-Abl](https://static.fdokumen.com/doc/165x107/63359563b5f91cb18a0b780c/n-2-methyl-5-triazol-1-ylphenylpyrimidin-2-amine-as-a-scaffold-for-the-synthesis.jpg)

![Antitubercular effect of 8-[(4-Chloro phenyl) sulfonyl]-7-Hydroxy-4-Methyl-2H-chromen-2-One in guinea pigs](https://static.fdokumen.com/doc/165x107/63336ec5b6829c19b80c6a0b/antitubercular-effect-of-8-4-chloro-phenyl-sulfonyl-7-hydroxy-4-methyl-2h-chromen-2-one.jpg)
![Synthesis and crystal structure of 2, 4-dihydro-4-[(5-hydroxy-3-methyl-1-phenyl-1H-pyrazol-4-yl) imino]-5-methyl-2-phenyl-3H-pyrazol-3-one and its copper (II) …](https://static.fdokumen.com/doc/165x107/6332751d83bb92fe98046bdb/synthesis-and-crystal-structure-of-2-4-dihydro-4-5-hydroxy-3-methyl-1-phenyl-1h-pyrazol-4-yl.jpg)

![N-[(4Z )-1-(3-Methyl-5-oxo-1-phenyl-4,5-dihydro-1Hpyrazol- 4-ylidene)hexyl]benzenesulfonohydrazide](https://static.fdokumen.com/doc/165x107/631d41f1f26ecf94330a76af/n-4z-1-3-methyl-5-oxo-1-phenyl-45-dihydro-1hpyrazol-4-ylidenehexylbenzenesulfonohydrazide.jpg)





![Vibrational spectroscopic (FT-IR and FT-Raman) studies, HOMO-LUMO, NBO analysis and MEP of 6-methyl-1-({[(2E)-2-methyl-3-phenyl-prop-2-en-1-yl]oxy}methyl)-1,2,3,4-tetra-hydroquinazoline-2,4-dione,](https://static.fdokumen.com/doc/165x107/633494f441100cab3c07ce05/vibrational-spectroscopic-ft-ir-and-ft-raman-studies-homo-lumo-nbo-analysis.jpg)



