
The genome of flax ( Linum usitatissimum ) assembled de novo from short shotgun sequence reads
13
The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads Zhiwen Wang 1 , Neil Hobson 2 , Leonardo Galindo 2 , Shilin Zhu 1 , Daihu Shi 1 , Joshua McDill 2 , Linfeng Yang 1 , Simon Hawkins 3 , Godfrey Neutelings 3 , Raju Datla 4 , Georgina Lambert 5 , David W. Galbraith 5 , Christopher J. Grassa 6 , Armando Geraldes 6 , Quentin C. Cronk 6 , Christopher Cullis 7 , Prasanta K. Dash 8 , Polumetla A. Kumar 8 , Sylvie Cloutier 9,10 , Andrew G. Sharpe 4 , Gane K.-S. Wong 1,2,11,* , Jun Wang 1,12,13,* and Michael K. Deyholos 2,* 1 BGI-Shenzen, Bei Shan Industrial Zone, Yantian District, Shenzhen 518083, China, 2 Department of Biological Sciences, University of Alberta, Edmonton, Alberta, T6G 2E9, Canada, 3 Universite ´ Lille-Nord de France, Lille 1 Unite ´ Mixte de Recherche Institut National de la Recherche Agronomique 1281, Stress Abiotiques et Diffe ´ renciation des Ve ´ ge ´ taux, F-59650 Villeneuve d’Ascq Cedex, France, 4 National Research Council of Canada, Plant Biotechnology Institute, Saskatoon, Saskatchewan, S7N 0W9, Canada, 5 University of Arizona, School of Plant Sciences and BIO5 Institute, Tucson, AZ 85721, USA, 6 Department of Botany, University of British Columbia, Vancouver, British Columbia, V6T 1Z4, Canada, 7 Case Western Reserve University, Cleveland, OH 44106, USA, 8 National Research Centre on Plant Biotechnology, Indian Agricultural Research Institute, Pusa Campus, New Delhi 110012, India, 9 Agriculture and Agri-Food Canada, 195 Dafoe Road, Winnipeg, Manitoba, R3T 2M1, Canada, 10 Department of Plant Science, University of Manitoba, Winnipeg, Manitoba, R3T 2N2, Canada, 11 Department of Medicine, University of Alberta, Edmonton, Alberta, T6G 2E1, Canada, 12 The Novo Nordisk Foundation Center for Basic Metabolic Research, University of Copenhagen, Denmark, and 13 Department of Biology, University of Copenhagen, Denmark Received 28 June 2011; revised 25 May 2012; accepted 26 June 2012; published online 14 August 2012. *For correspondence (e-mails [email protected], [email protected] and [email protected]). SUMMARY Flax (Linum usitatissimum) is an ancient crop that is widely cultivated as a source of fiber, oil and medicinally relevant compounds. To accelerate crop improvement, we performed whole-genome shotgun sequencing of the nuclear genome of flax. Seven paired-end libraries ranging in size from 300 bp to 10 kb were sequenced using an Illumina genome analyzer. A de novo assembly, comprised exclusively of deep-coverage (approx- imately 94· raw, approximately 69· filtered) short-sequence reads (44–100 bp), produced a set of scaffolds with N 50 = 694 kb, including contigs with N 50 = 20.1 kb. The contig assembly contained 302 Mb of non- redundant sequence representing an estimated 81% genome coverage. Up to 96% of published flax ESTs aligned to the whole-genome shotgun scaffolds. However, comparisons with independently sequenced BACs and fosmids showed some mis-assembly of regions at the genome scale. A total of 43 384 protein-coding genes were predicted in the whole-genome shotgun assembly, and up to 93% of published flax ESTs, and 86% of A. thaliana genes aligned to these predicted genes, indicating excellent coverage and accuracy at the gene level. Analysis of the synonymous substitution rates (K s ) observed within duplicate gene pairs was consistent with a recent (5–9 MYA) whole-genome duplication in flax. Within the predicted proteome, we observed enrichment of many conserved domains (Pfam-A) that may contribute to the unique properties of this crop, including agglutinin proteins. Together these results show that de novo assembly, based solely on whole- genome shotgun short-sequence reads, is an efficient means of obtaining nearly complete genome sequence information for some plant species. Keywords: whole-genome shotgun, DNA sequencing, Illumina, flax, Malpighiales, industrial crops. INTRODUCTION Flax (Linum usitatissimum) has been used by humans for at least nine millennia and possibly for as long as 30 millennia (van Zeist and Bakker-Heeres, 1975; Zohary and Hopf, 2000; Kvavadze et al., 2009). Varieties of this species are now cultivated for either fibers or seed, on approximately 3 Mha in over 20 countries (http://faostat.fao.org/). The bast fibers ª 2012 The Authors 461 The Plant Journal ª 2012 Blackwell Publishing Ltd The Plant Journal (2012) 72, 461–473 doi: 10.1111/j.1365-313X.2012.05093.x
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of The genome of flax ( Linum usitatissimum ) assembled de novo from short shotgun sequence reads

The genome of flax (Linum usitatissimum) assembledde novo from short shotgun sequence reads
Zhiwen Wang1, Neil Hobson2, Leonardo Galindo2, Shilin Zhu1, Daihu Shi1, Joshua McDill2, Linfeng Yang1, Simon Hawkins3,
Godfrey Neutelings3, Raju Datla4, Georgina Lambert5, David W. Galbraith5, Christopher J. Grassa6, Armando Geraldes6,
Quentin C. Cronk6, Christopher Cullis7, Prasanta K. Dash8, Polumetla A. Kumar8, Sylvie Cloutier9,10, Andrew G. Sharpe4,
Gane K.-S. Wong1,2,11,*, Jun Wang1,12,13,* and Michael K. Deyholos2,*
1BGI-Shenzen, Bei Shan Industrial Zone, Yantian District, Shenzhen 518083, China,2Department of Biological Sciences, University of Alberta, Edmonton, Alberta, T6G 2E9, Canada,3Universite Lille-Nord de France, Lille 1 Unite Mixte de Recherche Institut National de la Recherche Agronomique 1281, Stress
Abiotiques et Differenciation des Vegetaux, F-59650 Villeneuve d’Ascq Cedex, France,4National Research Council of Canada, Plant Biotechnology Institute, Saskatoon, Saskatchewan, S7N 0W9, Canada,5University of Arizona, School of Plant Sciences and BIO5 Institute, Tucson, AZ 85721, USA,6Department of Botany, University of British Columbia, Vancouver, British Columbia, V6T 1Z4, Canada,7Case Western Reserve University, Cleveland, OH 44106, USA,8National Research Centre on Plant Biotechnology, Indian Agricultural Research Institute, Pusa Campus, New Delhi 110012, India,9Agriculture and Agri-Food Canada, 195 Dafoe Road, Winnipeg, Manitoba, R3T 2M1, Canada,10Department of Plant Science, University of Manitoba, Winnipeg, Manitoba, R3T 2N2, Canada,11Department of Medicine, University of Alberta, Edmonton, Alberta, T6G 2E1, Canada,12The Novo Nordisk Foundation Center for Basic Metabolic Research, University of Copenhagen, Denmark, and13Department of Biology, University of Copenhagen, Denmark
Received 28 June 2011; revised 25 May 2012; accepted 26 June 2012; published online 14 August 2012.
*For correspondence (e-mails [email protected], [email protected] and [email protected]).
SUMMARY
Flax (Linum usitatissimum) is an ancient crop that is widely cultivated as a source of fiber, oil and medicinally
relevant compounds. To accelerate crop improvement, we performed whole-genome shotgun sequencing of
the nuclear genome of flax. Seven paired-end libraries ranging in size from 300 bp to 10 kb were sequenced
using an Illumina genome analyzer. A de novo assembly, comprised exclusively of deep-coverage (approx-
imately 94· raw, approximately 69· filtered) short-sequence reads (44–100 bp), produced a set of scaffolds
with N50 = 694 kb, including contigs with N50 = 20.1 kb. The contig assembly contained 302 Mb of non-
redundant sequence representing an estimated 81% genome coverage. Up to 96% of published flax ESTs
aligned to the whole-genome shotgun scaffolds. However, comparisons with independently sequenced BACs
and fosmids showed some mis-assembly of regions at the genome scale. A total of 43 384 protein-coding
genes were predicted in the whole-genome shotgun assembly, and up to 93% of published flax ESTs, and 86%
of A. thaliana genes aligned to these predicted genes, indicating excellent coverage and accuracy at the gene
level. Analysis of the synonymous substitution rates (Ks) observed within duplicate gene pairs was consistent
with a recent (5–9 MYA) whole-genome duplication in flax. Within the predicted proteome, we observed
enrichment of many conserved domains (Pfam-A) that may contribute to the unique properties of this crop,
including agglutinin proteins. Together these results show that de novo assembly, based solely on whole-
genome shotgun short-sequence reads, is an efficient means of obtaining nearly complete genome sequence
information for some plant species.
Keywords: whole-genome shotgun, DNA sequencing, Illumina, flax, Malpighiales, industrial crops.
INTRODUCTION
Flax (Linum usitatissimum) has been used by humans for at
least nine millennia and possibly for as long as 30 millennia
(van Zeist and Bakker-Heeres, 1975; Zohary and Hopf, 2000;
Kvavadze et al., 2009). Varieties of this species are now
cultivated for either fibers or seed, on approximately 3 Mha
in over 20 countries (http://faostat.fao.org/). The bast fibers
ª 2012 The Authors 461The Plant Journal ª 2012 Blackwell Publishing Ltd
The Plant Journal (2012) 72, 461–473 doi: 10.1111/j.1365-313X.2012.05093.x

that grow in the outer layers of the flax stem are made up of
remarkably long cells that are rich in crystalline cellulose and
have a very high tensile strength (Mohanty et al., 2000).
These fibers are the source of linen textiles, and their use in
composite materials is an area of active research (Bodros
et al., 2007). The seed of flax (i.e. linseed) produces oil that is
rich in unsaturated fatty acids, especially a-linolenic acid
(C18:3), polymers of which are used in linoleum, paints and
other finishes. Consumption of the oil or seed has been
reported to have beneficial effects on cardiovascular health
and in the treatment of certain cancers and inflammatory
diseases (Singh et al., 2011). Health benefits are derived
from both the a-linolenic acid and other components of the
seed, including lignans such as secoisolariciresinol dig-
lucoside (SDG), which is an antioxidant and the precursor of
several phytoestrogens (Toure and Xu, 2010). Flax seed is
also used in animal feed to increase levels of a-linolenic acid
in meat or eggs (Simmons et al., 2011). In total, flax is a
source of at least three classes of commercial products: fiber,
seed oil, and nutraceuticals. Increased understanding of the
genes affecting the quality and yield of these bioproducts will
contribute to crop improvement for flax and other oil- or fiber-
producing species.
In addition to its economic uses, flax has several charac-
teristics that make it an interesting subject for scientific
inquiry. Among these are the diversity of the flax family and
its relatives. The genus Linum comprises an estimated 180
species distributed over six continents (McDill et al., 2009).
The genus displays interesting variation in evolutionarily
significant characteristics such as breeding system (disty-
lous and homostylous species are known), flower color
(ranging from white to yellow, red and blue), pollen
morphology (tricolpate or multiporate) and edaphic ende-
mism (i.e. restriction to specific soil types). The family
Linaceae has impressive ecological and morphological
diversity, with more than 270 species in 14 genera ranging
from tiny, serpentine-endemic annuals in the California
chaparral (the genus Hesperolinon) to canopy trees in South
American black-water forests (Hebepetalum) and tendrillate
vines in Indonesian jungles (Indorouchera). Linum and the
Linaceae have recently been the subject of molecular
phylogenetic investigations using chloroplast markers, the
nuclear ribosomal internal transcribed spacer (ITS), and
retrotransposon sequences (McDill et al., 2009; Fu and
Allaby, 2010; McDill and Simpson, 2011; Smykal et al.,
2011). The Linaceae family belongs to the order Malpighi-
ales, of which three species have been fully sequenced:
Populus trichocarpa (black cottonwood, Salicaceae), Ricinus
communis (castor bean, Euphorbiaceae) and Manihot escul-
enta (cassava, Euphorbiaceae) (Tuskan et al., 2006; Chan
et al., 2010; http://phytozome.net. The phylogenetic relation-
ships among families in the Malpighiales are not entirely
resolved (Wurdack and Davis, 2009), but recent estimates
suggest that the Linaceae diverged as a lineage distinct from
other families of Malpighiales approximately 100 MYA
(Davis et al., 2005). The lineage leading to Arabidopsis
thaliana, whose genome is commonly used in comparative
genome analyses, is thought to have diverged from the
lineage leading to Malpighiales between 100 and 120 MYA
(Tuskan et al., 2006).
Flax is also of general scientific interest because some
varieties of flax (e.g. Stormont Cirrus) exhibit rapid changes
in nuclear DNA content (Cullis, 1981b, 2005). Differences in
nuclear C-value of up to 15% have been reported among
first-generation progeny of self-pollinated individuals that
were subjected to specific temperature or fertilizer regimes
(Evans et al., 1966). Altered copy numbers of many types of
genetic elements contribute to the observed changes in
genome size, including rRNA genes and an unusual inser-
tional element named LIS-1 (Goldsbrough et al., 1981; Chen,
1999). Increased genomic sequence information for flax may
help to further elucidate the basis of genome plasticity.
Patterns of genetic inheritance in L. usitatissimum are
typical of a diploid. L. usitatissimum has n = 15 chromo-
somes, which is common in certain lineages of the genus.
Other species of Linum have been reported to have n = 8, 9,
10, 13, 14, 18 or 27 chromosomes (Rogers, 1982), consistent
with a history of repeated genome duplications (polyploidy)
and possible instances of aneuploid reductions or increases.
Published estimates of the nuclear DNA content of flax range
from C = 538 Mb to C = 685 Mb, although reassociation
kinetics analyses have produced estimates as low as
C = 350 Mb (Evans et al., 1972; Cullis, 1981a; Marie and
Brown, 1993). The reassociation kinetics studies also show
that approximately 50% of the genome is composed of
low-copy-number sequences, with 35% highly repetitive
sequences, and the remaining approximately 15% in the
middle-repetitive fraction, which typically represents trans-
posable elements (Cullis, 1980). Genomic sequence
resources for L. usitatissimum in public databases include
286 294 ESTs and 80 339 BAC end sequences in GenBank
(http://www.ncbi.nlm.nih.gov/nucleotide/). The majority of
these sequences were obtained from the cultivar CDC
Bethune (Venglat et al., 2011). A linkage map based on
SSRs (Cloutier et al., 2009) and a 368 Mb BAC physical map
of CDC Bethune (Ragupathy et al., 2011) have been pub-
lished recently. Microarray gene expression and proteome
studies of flax have also been reported (Lynch and Conery,
2000; Roach and Deyholos, 2007; Fenart et al., 2010).
To further increase the available sequence resources for
flax, we have performed whole-genome sequencing of the
CDC Bethune cultivar of Linum usitatissimum, which is a
highly inbred, elite linseed cultivar grown on the majority of
flax acreage in Canada (Rowland et al., 2002). CDC Bethune
does not exhibit any of the phenotypic plasticity that
accompanies the rapid genome change observed in Stor-
mont Cirrus. We used a whole-genome shotgun approach to
sequence CDC Bethune, using de novo assembly of exclusively
462 Zhiwen Wang et al.
ª 2012 The AuthorsThe Plant Journal ª 2012 Blackwell Publishing Ltd, The Plant Journal, (2012), 72, 461–473
short reads, a strategy that has previously proven effective in
vertebrates (Li et al., 2010) and date palm (Phoenix dactylif-
era) (Al-Dous et al., 2011). The primary objective was to
characterize the gene space of flax, in support of gene
discovery, marker development and reverse genetics activ-
ities that are already underway. The analysis of the WGS
assembly is presented here.
RESULTS
To obtain a whole-genome shotgun (WGS) assembly of flax,
we extracted DNA from axenically grown, etiolated seedlings
of a linseed cultivar of L. usitatissimum, CDC Bethune, to
produce size-selected sequencing libraries based on five
insert sizes ranging from 300 bp to 10 kb in length (Table 1).
Each library was used as a template in paired-end or mate-
pair sequencing reactions in an Illumina GAII genome
analyzer, producing a total of 34.98 Gb of reads that ranged
between 44 and 100 bp in length. After filtering low-quality
sequences, the remaining 25.88 Gb were used in assembly.
Using flow cytometry, we estimated the nuclear DNA content
of CDC Bethune to be 2N = 2C = 0.764 pg (r�x ¼ 0:07 pg),
corresponding to a haploid nuclear genome of 373 Mb. The
sum of the WGS sequence reads obtained therefore repre-
sented approximately 94· coverage (raw reads) and 69·coverage (filtered reads) of the nuclear genome of flax.
SOAPdenovo is a de Bruijn graph-based assembly pro-
gram designed to use short WGS reads as its input (Li et al.,
2009). Applying this software to filtered Illumina reads, we
generated a de novo assembly in which 50% of the assembly
(N50) was contained in 4427 contigs of 20 125 bp or larger
(Table 2). All contigs were further assembled into 88 384
scaffolds (‡100 bp), of which 132 scaffolds containing 50%
of the assembly were 693.5 kb or larger (N50 = 693.5 kb).
The contigs and scaffolds each contained a total of 302 and
318 Mb of unique sequence, respectively. Therefore, as a
proportion of the nuclear DNA content estimated by flow
cytometry, the WGS assembly contained an equivalent of
81% genome coverage in contigs and 85% genome coverage
in scaffolds. The modal sequencing depth in the assembly
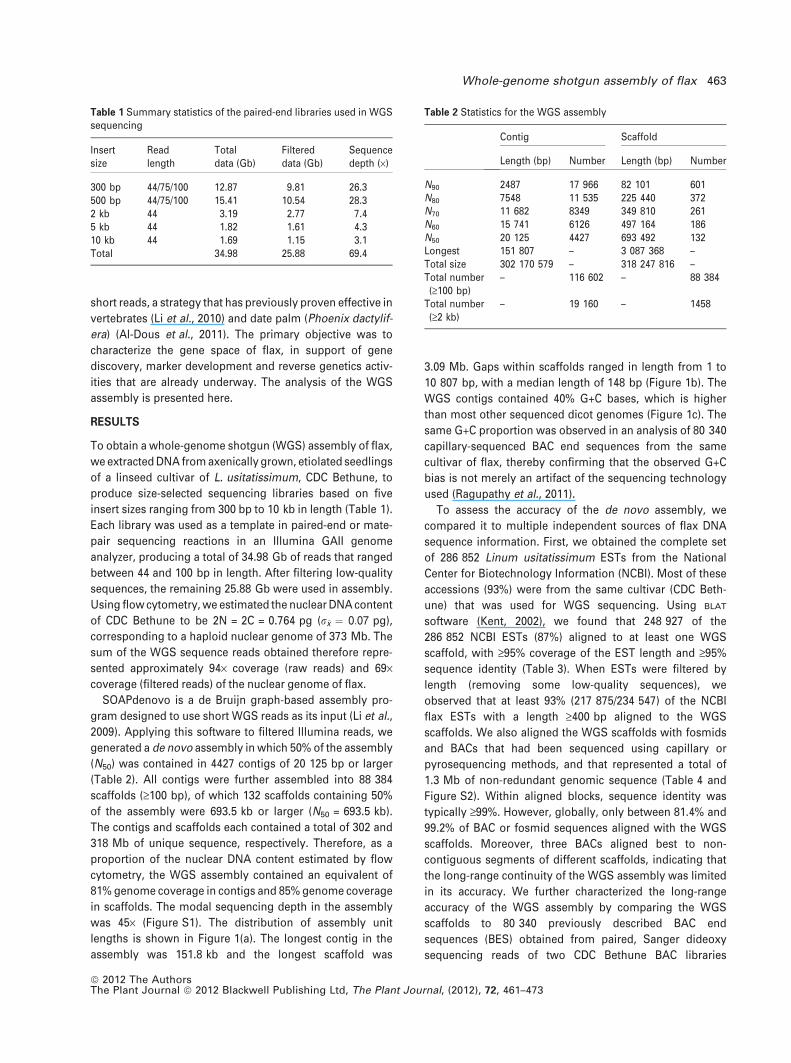
was 45· (Figure S1). The distribution of assembly unit
lengths is shown in Figure 1(a). The longest contig in the
assembly was 151.8 kb and the longest scaffold was
3.09 Mb. Gaps within scaffolds ranged in length from 1 to
10 807 bp, with a median length of 148 bp (Figure 1b). The
WGS contigs contained 40% G+C bases, which is higher
than most other sequenced dicot genomes (Figure 1c). The
same G+C proportion was observed in an analysis of 80 340
capillary-sequenced BAC end sequences from the same
cultivar of flax, thereby confirming that the observed G+C
bias is not merely an artifact of the sequencing technology
used (Ragupathy et al., 2011).
To assess the accuracy of the de novo assembly, we
compared it to multiple independent sources of flax DNA
sequence information. First, we obtained the complete set
of 286 852 Linum usitatissimum ESTs from the National
Center for Biotechnology Information (NCBI). Most of these
accessions (93%) were from the same cultivar (CDC Beth-
une) that was used for WGS sequencing. Using BLAT
software (Kent, 2002), we found that 248 927 of the
286 852 NCBI ESTs (87%) aligned to at least one WGS
scaffold, with ‡95% coverage of the EST length and ‡95%
sequence identity (Table 3). When ESTs were filtered by
length (removing some low-quality sequences), we
observed that at least 93% (217 875/234 547) of the NCBI
flax ESTs with a length ‡400 bp aligned to the WGS
scaffolds. We also aligned the WGS scaffolds with fosmids
and BACs that had been sequenced using capillary or
pyrosequencing methods, and that represented a total of
1.3 Mb of non-redundant genomic sequence (Table 4 and
Figure S2). Within aligned blocks, sequence identity was
typically ‡99%. However, globally, only between 81.4% and
99.2% of BAC or fosmid sequences aligned with the WGS
scaffolds. Moreover, three BACs aligned best to non-
contiguous segments of different scaffolds, indicating that
the long-range continuity of the WGS assembly was limited
in its accuracy. We further characterized the long-range
accuracy of the WGS assembly by comparing the WGS
scaffolds to 80 340 previously described BAC end
sequences (BES) obtained from paired, Sanger dideoxy
sequencing reads of two CDC Bethune BAC libraries
Table 1 Summary statistics of the paired-end libraries used in WGSsequencing
Insertsize
Readlength
Totaldata (Gb)
Filtereddata (Gb)
Sequencedepth (·)
300 bp 44/75/100 12.87 9.81 26.3500 bp 44/75/100 15.41 10.54 28.32 kb 44 3.19 2.77 7.45 kb 44 1.82 1.61 4.310 kb 44 1.69 1.15 3.1Total 34.98 25.88 69.4
Table 2 Statistics for the WGS assembly
Contig Scaffold
Length (bp) Number Length (bp) Number
N90 2487 17 966 82 101 601N80 7548 11 535 225 440 372N70 11 682 8349 349 810 261N60 15 741 6126 497 164 186N50 20 125 4427 693 492 132Longest 151 807 – 3 087 368 –Total size 302 170 579 – 318 247 816 –Total number(‡100 bp)
– 116 602 – 88 384
Total number(‡2 kb)
– 19 160 – 1458
Whole-genome shotgun assembly of flax 463
ª 2012 The AuthorsThe Plant Journal ª 2012 Blackwell Publishing Ltd, The Plant Journal, (2012), 72, 461–473
(Figure 1d) (Ragupathy et al., 2011). Of the 40 099 BES
accessions that aligned to masked WGS scaffolds with high
stringency (‡99% sequence identity, ‡400 bp alignment
length and ‡95% BES coverage), 9668 were pairs (i.e. from
opposite ends of the same BAC) in which both members
aligned to the same WGS scaffold. In each case, the length
(a) (b)
(c) (d)
Figure 1. Genome assembly results.
(a) Assembly units (i.e. contigs or scaffolds) were
sorted by descending length, and the cumulative
DNA content was plotted as a function of the
total number of assembly units. The total
genome size of 373 Mb (measured by flow
cytometry) is represented by a dashed line, and
the N50 and N90 assembly units are indicated by
filled and open circles, respectively.
(b) Distribution of gap length within the scaffold
assembly.
(c) G+C content as a proportion of total bases
measured in 500 bp bins throughout the flax
(Lus) WGS assembly, compared to genomes of
nine other fully sequenced dicots: Arabidopsis
thaliana, Cucumis sativa, Glycine max, Ricinus
communis, Populus trichocarpa, Malus domes-
tica, Manihot esculentum, Medicago truncatula
and Vitis vinifera.
(d) Distribution of the length of intervening
sequence in scaffold/BES alignments. The length
of scaffold DNA between paired BES that aligned
to the same scaffold was calculated, and is
plotted as a frequency distribution for each of
the pairs of aligned BES.
Table 3 Alignment of capillary-sequenced ESTs to WGS scaffolds
EST coveragein alignment (%)
Alignmentidentity (%)
EST length(bp)
Aligned ESTs(number)
Total ESTs queried(number)
ESTsaligned (%)
‡90 ‡95 >0 262 748 286 852 91.6‡95 ‡95 >0 248 927 286 852 86.8‡90 ‡95 ‡400 225 579 234 547 96.2‡95 ‡95 ‡400 217 875 234 547 92.9
Table 4 Alignment of capillary-sequenced BACs and fosmids to WGS scaffolds
BAC/fosmidBAC/fosmidsize (kb)
Matchingscaffold
Scaffoldsize (kb)
Alignment(kb)
Identity(%)
Gaps(%)
JX174446 214.6 156 2281.8 214.6 95.6 3.1RL10-contig00001 190.5 107 560 190.5 97.7 1.5JX174448 184.9 28 1933.6 119.2 98 1.4
23 781.8 64.2 99.2 0.6JX174447 180.1 1036 248.3 23.5 94 2.5
33 2127.3 156.5 94.9 3.3JX174445 179.1 155 809.2 126 95.4 2.2
177 934 53.1 95 1.7JX174444 172.5 96 1083.7 172.5 91.8 5.3JX174449 130.3 931 463.5 130.3 93.9 5.2HQ902252 39.8 1486 346.5 39.8 91.2 7.7JN133299 34.6 465 878.6 34.6 81.4 2.1JN133300 31.5 898 1045 31.5 98.6 1.1JN133301 26.2 1856 140.6 26.2 86.7 6.2
464 Zhiwen Wang et al.
ª 2012 The AuthorsThe Plant Journal ª 2012 Blackwell Publishing Ltd, The Plant Journal, (2012), 72, 461–473
of scaffold region between the aligned BES pairs was
calculated, and the distribution of these lengths is plotted in
Figure 1(d). The median length of scaffold sequence was
133.4 kb, and the mean length was 140 kb. These results
are consistent with the 135 and 150 kb mean insert lengths
reported for the two original CDC Bethune BAC libraries.
However, a minority of alignments (1448/9668; 15%) had
intervening scaffold sequence lengths that deviated greatly
from the mean (<100 kb or >200 kb), indicating that addi-
tional studies are required to improve the long-range
accuracy of the assembly.
Repetitive elements
To identify various types of middle-repetitive DNA
sequences within the CDC Bethune WGS assembly, the
complete set of WGS scaffolds was submitted to REPEAT-
MASKER version 3.3.0 (http://www.repeatmasker.org/), which
identified 15.8 Mb of sequence (5.2% of a total of 302 Mb
WGS contigs) as having significant similarity to the subjects
in RepBase (http://www.girinst.org/repbase/). The set of
repeats identified by this homology-based analysis was
expanded substantially by application of de novo repeat
identification methods, resulting in a total of 73.8 Mb (24.4%
of WGS contig assembly) being annotated as sequence with
similarity to mobile elements (Table 5). As expected,
retroelements were the most common mobile elements
found in the genome (20.6% of WGS contigs), and these
were represented primarily by LTR type retroelements
(18.4%) followed by long interspersed elements (LINEs)
(2.2%). All DNA transposons represented a much smaller
proportion of the WGS contig sequences (3.8%), and most
were Mutator-type elements (2.1% of WGS contig
sequence). The proportions of the various types of mobile
elements are similar to what has been reported in other full-
sequenced dicot genomes of comparable size. The unusual
5.8kb LIS-1 insertion sequence (GenBank accession AF104351)
that has been described in other varieties of flax was not
found in its entirety in the CDC Bethune assembly; the larg-
est LIS-1 fragment detected was 543 bp (Chen et al., 2005).
Non-coding RNAs
Non-coding RNAs comprise the majority of cellular RNAs
and play an important role in translation (tRNA and rRNA),
synthesis of the translational apparatus (snRNA), and gene
regulation (miRNA). We searched the WGS assembly for
sequences characteristic of these four types of non-coding
Table 5 Transposable elements identified in the flax WGS assembly
Class Order SuperfamilyNumber ofelements
Elementspercentage (%)
Sequenceoccupied (bp)
Sequence percentageof transposableelements (%)
Sequencepercentage ofgenome (%)
Retrotransposons LTR Copia 89 951 38.31 29 594 882 40.08 9.79Gypsy 72 626 30.93 25 123 127 34.02 8.31Unclassified 2797 1.19 902 298 1.22 0.30
DIRS DIRS 2 0.00 102 0.00 0.00PLE Penelope 548 0.23 30 214 0.04 0.01LINE RTE 11 0.00 618 0.00 0.00
L1 27 632 11.77 6 684 243 9.05 2.21SINE Unclassified 1 0.00 49 0.00 0.00
DNA transposons TIR Tc1-Mariner 191 0.08 38 231 0.05 0.01hAT 7935 3.38 1 986 522 2.69 0.66Mutator 21 124 9.00 6 320 424 8.56 2.09P 2 0.00 96 0.00 0.00Harbinger 1384 0.59 344 876 0.47 0.11En-Spm/CACTA 8330 3.55 2 372 592 3.21 0.79
Helitron Helitron 2154 0.92 434 859 0.59 0.14Unclassified Unclassified 95 0.04 8981 0.01 0.00
Total 234 783 100.00 7 384 2114 100.00 24.29
LTR, Long Terminal Repeat; DIRS, Dictyostelium Intermediate Repeat Sequence; PLE, Penelope; LINE, Long Interspersed Nuclear Element; SINE,Short Interspersed Nuclear Element; TIR, Terminal Inverted Repeat.
Table 6 Non-coding RNA species identified in the flax WGSassembly
Type Sub-typeCopynumber
Meanlength(bp)
Totallength(bp)
Percentageof genome
miRNA 297 120.49 35 785 0.01tRNA 1100 74.75 82 224 0.03rRNA 1100 90.99 100 088 0.03
18S 65 186.97 18 653 0.0028S 14 121.43 1700 0.005.8S 11 123.18 1355 0.005S 1010 77.60 78 380 0.02
snRNA 462 120.05 55 462 0.02CD box 264 102.57 27 079 0.01HACA box 41 119.85 4914 0.00Splicing 157 149.48 23 469 0.01
Whole-genome shotgun assembly of flax 465
ª 2012 The AuthorsThe Plant Journal ª 2012 Blackwell Publishing Ltd, The Plant Journal, (2012), 72, 461–473
RNAs (Table 6) (Griffiths-Jones et al., 2003; Nawrocki et al.,
2009). At least 297 putative miRNA precursor loci with
similarity to known miRNAs were identified (Table S1), and
more than 1000 copies were found of both tRNAs and 5S
RNAs. These frequencies are similar to what has been
reported in other species of comparable genome size.
However, an unexpectedly low number of 45S RNAs were
identified, with as few as 11 copies of the 5.8S RNA com-
ponent of the 45S locus found in the WGS assembly.
Plastid sequences
We searched the WGS assembly for fragments with similarity
to plastid DNA. We identified 31 scaffolds that consisted pri-
marily of chloroplast DNA, but we did not detect a complete
assembly of the plastid genome among the flax WGS scaf-
folds. However, many occurrences of plastid-derived
sequences were observed within the scaffolds of nuclear
DNA. Using BLASTN (e-value £10)4 or lower) (Altschul et al.,
1997), we compared the WGS assembly to a draft of the
L. usitatissimum chloroplast genome that was indepen-
dently sequenced (J. McDill, unpublished results). This
search revealed 1356 regions with significant similarity to flax
chloroplast DNA, ranging from 23 to 5899 bp in length in 416
scaffolds. In total, these regions represented an approxi-
mately 89% coverage of the draft flax chloroplast genome
(including only a single copy of the chloroplast inverted
repeat region). The size distribution of these regions con-
forms to the distributions of NUPT (nuclear plastid DNA)
observed in otherspecies (Figure S3) (Richly andLeister,2004).
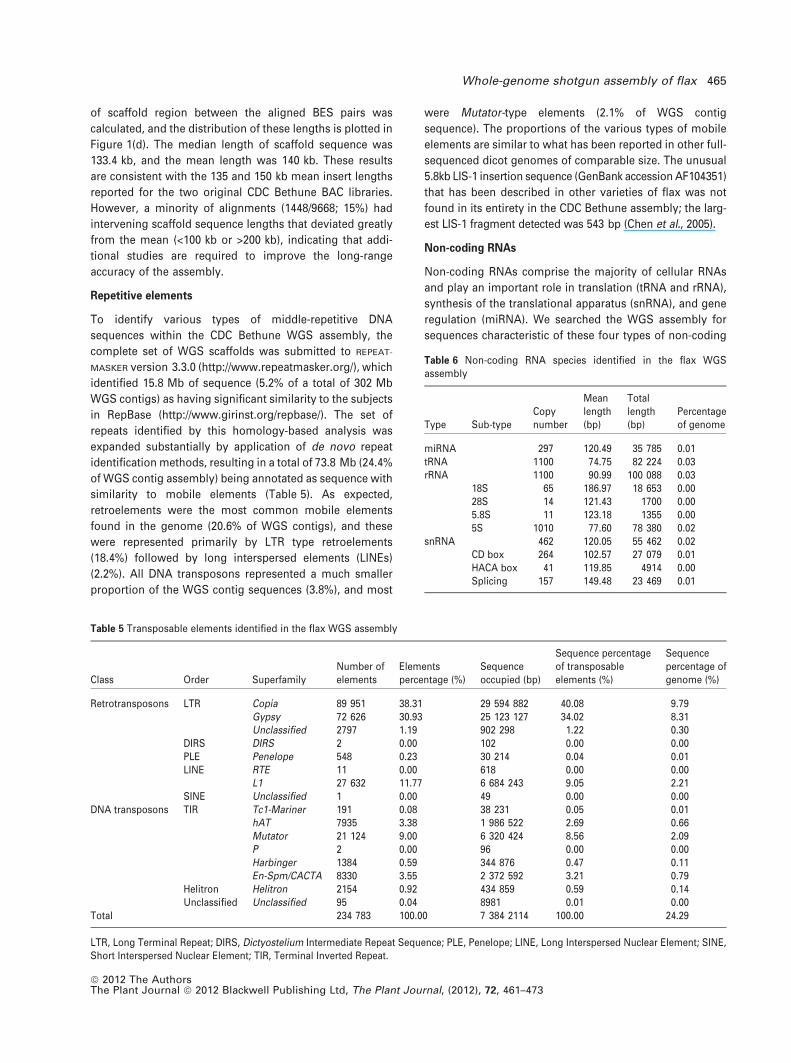
Gene prediction
To predict the locations of protein-coding genes within the
repeat-masked WGS assembly, we used two de novo hidden
Markov model-based gene-finding programs: Glimmer-
HMM and Augustus (Majoros et al., 2004; Stanke et al.,
2008). The output of these programs was integrated with
WGS alignments of flax ESTs and other empirically estab-
lished plant transcript sequences to produce a consensus
gene set using GLEAN (Elsik et al., 2007). The consensus gene
set contained a total of 43 484 genes, with one transcript
model per gene. The mean and median gene lengths [coding
sequences (CDS), no introns] of the consensus gene set
were 1200 and 987 bp, respectively. A total of 40 971 genes
had a predicted length ‡300 bp. The distributions of CDS,
exon and intron lengths are shown in Figure 2 for the pre-
dicted flax genes and genes from five other representative
genomes.
To assess the coverage and accuracy of the gene predic-
tion, we compared the filtered set of 43 484 predicted flax
genes (which excluded most transposable elements) to the
complete set of flax ESTs from the NCBI databases (Table 7).
We aligned the CDS regions of the predicted genes to the
286 852 NCBI EST sequences using BLASTN. We observed
that 248 210/286 852 NCBI flax ESTs (86.5%) aligned to one
or more WGS CDS (BLASTN e-value £10)20). Because many
ESTs potentially included UTRs, which are not part of CDS
datasets, we repeated the comparison, using only the
239 220 NCBI EST accessions that were ‡400 bp long. Of
the length-filtered NCBI flax ESTs, 222 846/239 220 (93.2%)
aligned to one or more CDS from the predicted flax WGS
proteome (BLASTN e-value £10)20) (Table 7). Therefore, the
coverage of ESTs within the genes predicted from the WGS
assembly was very high. Conversely, 28 783/43 484 CDS
sequences (66%) aligned to one or more NCBI ESTs
(BLASTN e-value £10)20) (Table 6). The relatively low
proportion of predicted WGS CDS sequences that aligned
Distribution of mRNA length
Gene length (bp) (window = 50 bp)
Perc
ent o
f gen
es(%
)
0 3000 6000 9000 12 0000
1
2
3
4
5L.usitatissimumA.thalianaC.sativusG.maxP.trichocarpaR.communis
Distribution of CDS length
CDS length (bp) (window = 50 bp)
Perc
ent o
f gen
es(%
)
0 1000 2000 3000 4000 50000
3
6
9L.usitatissimumA.thalianaC.sativusG.maxP.trichocarpaR.communis
Distribution of exon length
Exon length (bp)(window = 10 bp)
Perc
ent o
f exo
ns(%
)
0 100 200 300 400 5000
3
6
9L.usitatissimumA.thalianaC.sativusG.maxP.trichocarpaR.communis
Distribution of intron length
Intron length (bp)(window = 10 bp)
Perc
ent o
f int
rons
(%)
0 100 200 300 400 5000
7
14
21
28L.usitatissimumA.thalianaC.sativusG.maxP.trichocarpaR.communis
(a) (b)
(c) (d)
Figure 2. Gene prediction.
Distribution of the length of (a) mRNA, (b) CDS, (c)
exon and (d) intron features within the complete
set of predicted genes for flax and five other
representative dicots.
466 Zhiwen Wang et al.
ª 2012 The AuthorsThe Plant Journal ª 2012 Blackwell Publishing Ltd, The Plant Journal, (2012), 72, 461–473
to flax ESTs may indicate that the coverage of flax genes in
EST databases is incomplete and/or that a considerable
proportion of the predicted CDS sequences do not represent
components of real transcripts. Both possibilities were
examined in the subsequent analyses described below.
We compared the predicted WGS proteins with peptide
sequences in the NCBI databases, and separately with
peptide sequences from the well-characterized Arabidopsis
and poplar genomes (Tuskan et al., 2006; Swarbreck et al.,
2008). We observed that 89.5% (38 920/43 484) of flax WGS
proteins aligned to one or more proteins from the NCBI nr
protein database (http://www.ncbi.nlm.nih.gov/protein), and
nearly the same proportion aligned with Arabidopsis
(38 876/43 484; 89.4%) or poplar proteins (39 746/43 484;
91.4%; BLASTP e-value £10)5). Because sequences from
different species were not expected to be identical, a lower
level of stringency was used in these cross-species compar-
isons than was used in the comparisons of flax ESTs to flax
CDS sequences. Overall, the high proportion of predicted
proteins from the flax WGS that aligned with proteins from
Arabidopsis, poplar and the NCBI nr database indicates that
the majority of genes predicted in the flax WGS are
legitimate protein-coding sequences. We also made con-
verse comparisons to determine what proportion of Arabi-
dopsis and poplar proteins were represented by at least one
flax protein. We found that 86.0% (23 575/27 416) of Arabi-
dopsis proteins and 86.4% (35 135/40 688) of poplar proteins
aligned to one or more predicted flax proteins (BLASTP
e-value £10)5). This level of coverage is consistent with the
alignment of predicted poplar and Arabidopsis proteins
reported after sequencing of the poplar genome (Tuskan
et al., 2006).
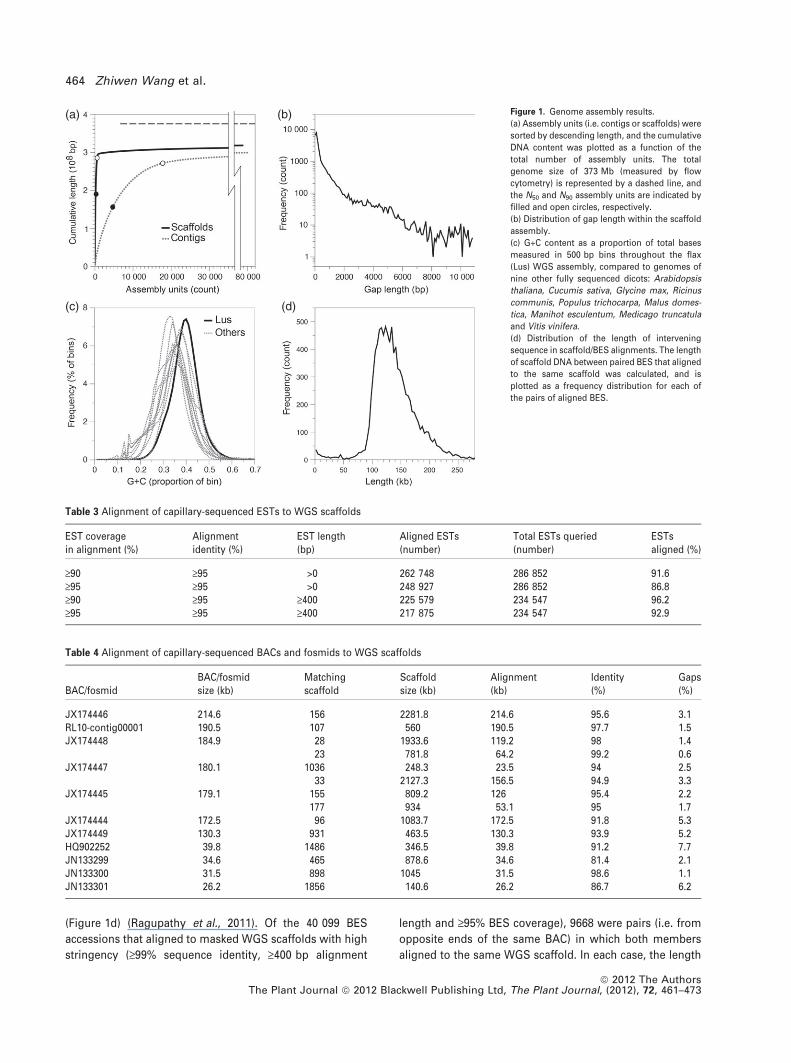
Functional annotation of predicted proteins
The Pfam-A database provides profile hidden Markov mod-
els of over 13 672 conserved protein families (Finn et al.,
2010). Approximately 79% of known proteins contain one or
more Pfam domains. We used PfamScan/HMMer3 to iden-
tify Pfam domains within the predicted genes of the flax
WGS assembly (ftp://ftp.sanger.ac.uk/pub/databases/Pfam/
Tools/; Eddy, 2011). As shown in Table 7, 77% (33 459/
43 484) of the proteins in the flax WGS assembly contained
one or more Pfam domains (Pfam-A 26.0, e-value £10)5).
Almost all of the 5312 Pfam-A domains were found to occur
at the same frequency in the predicted flax genes as in genes
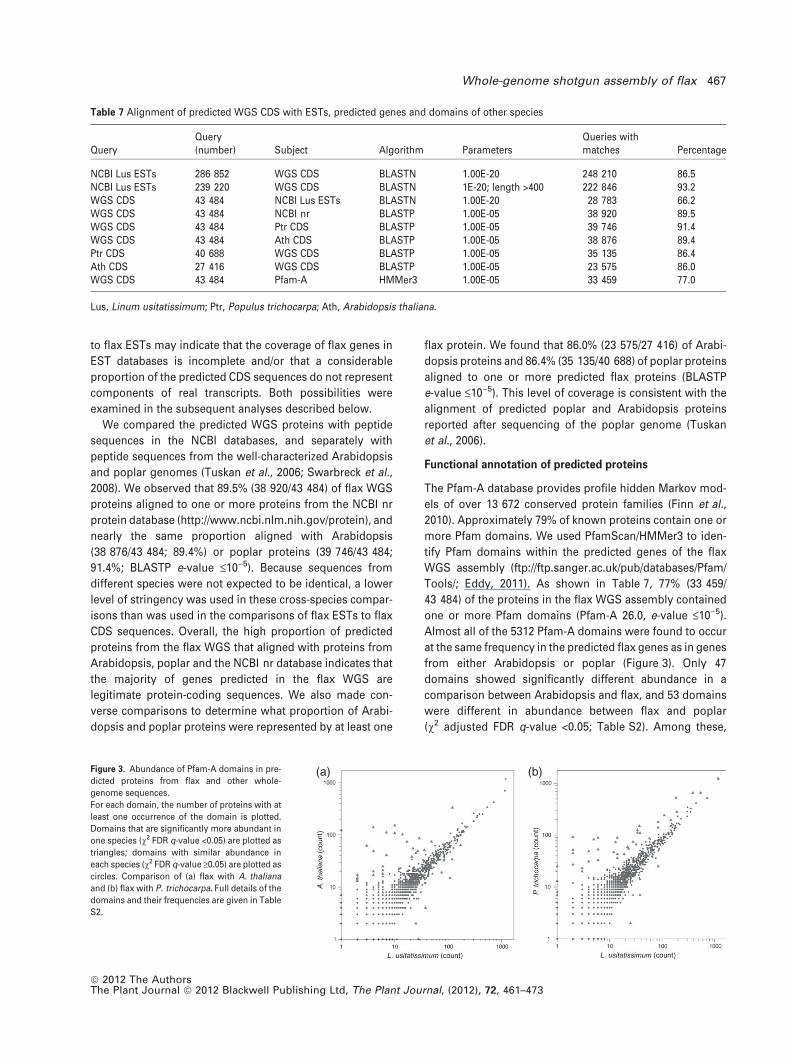
from either Arabidopsis or poplar (Figure 3). Only 47
domains showed significantly different abundance in a
comparison between Arabidopsis and flax, and 53 domains
were different in abundance between flax and poplar
(v2 adjusted FDR q-value <0.05; Table S2). Among these,
Table 7 Alignment of predicted WGS CDS with ESTs, predicted genes and domains of other species
QueryQuery(number) Subject Algorithm Parameters
Queries withmatches Percentage
NCBI Lus ESTs 286 852 WGS CDS BLASTN 1.00E-20 248 210 86.5NCBI Lus ESTs 239 220 WGS CDS BLASTN 1E-20; length >400 222 846 93.2WGS CDS 43 484 NCBI Lus ESTs BLASTN 1.00E-20 28 783 66.2WGS CDS 43 484 NCBI nr BLASTP 1.00E-05 38 920 89.5WGS CDS 43 484 Ptr CDS BLASTP 1.00E-05 39 746 91.4WGS CDS 43 484 Ath CDS BLASTP 1.00E-05 38 876 89.4Ptr CDS 40 688 WGS CDS BLASTP 1.00E-05 35 135 86.4Ath CDS 27 416 WGS CDS BLASTP 1.00E-05 23 575 86.0WGS CDS 43 484 Pfam-A HMMer3 1.00E-05 33 459 77.0
Lus, Linum usitatissimum; Ptr, Populus trichocarpa; Ath, Arabidopsis thaliana.
(a) (b)Figure 3. Abundance of Pfam-A domains in pre-
dicted proteins from flax and other whole-
genome sequences.
For each domain, the number of proteins with at
least one occurrence of the domain is plotted.
Domains that are significantly more abundant in
one species (v2 FDR q-value <0.05) are plotted as
triangles; domains with similar abundance in
each species (v2 FDR q-value ‡0.05) are plotted as
circles. Comparison of (a) flax with A. thaliana
and (b) flax with P. trichocarpa. Full details of the
domains and their frequencies are given in Table
S2.
Whole-genome shotgun assembly of flax 467
ª 2012 The AuthorsThe Plant Journal ª 2012 Blackwell Publishing Ltd, The Plant Journal, (2012), 72, 461–473
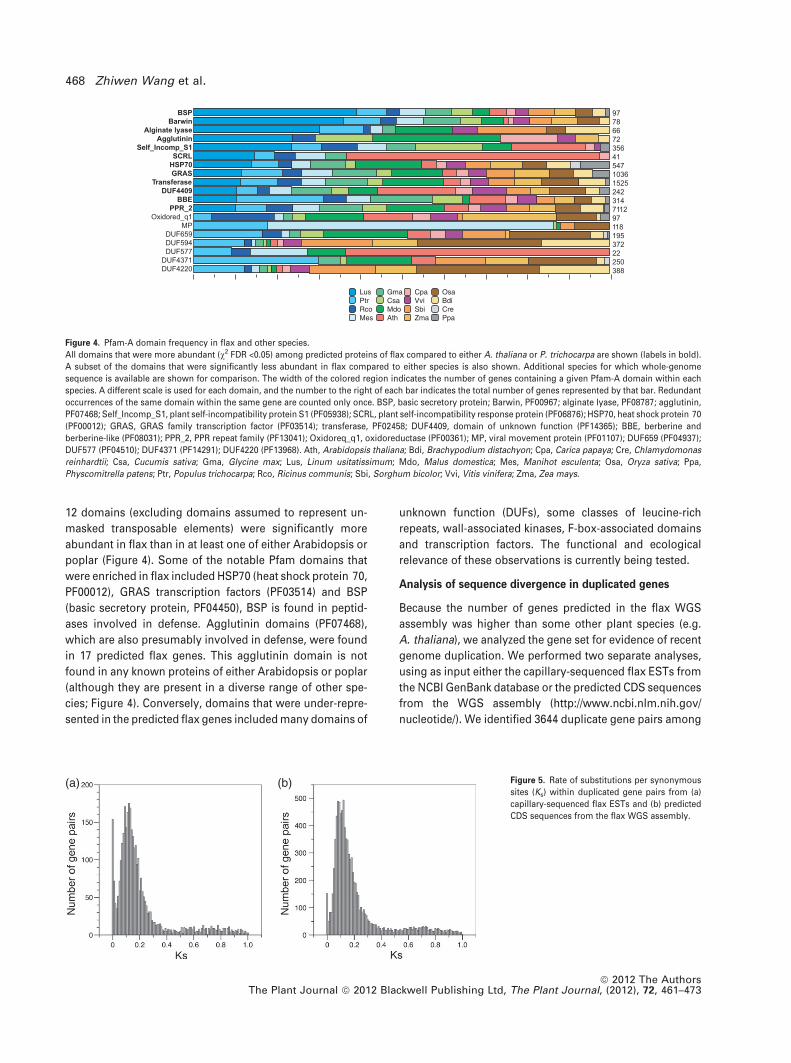
12 domains (excluding domains assumed to represent un-
masked transposable elements) were significantly more
abundant in flax than in at least one of either Arabidopsis or
poplar (Figure 4). Some of the notable Pfam domains that
were enriched in flax included HSP70 (heat shock protein 70,
PF00012), GRAS transcription factors (PF03514) and BSP
(basic secretory protein, PF04450), BSP is found in peptid-
ases involved in defense. Agglutinin domains (PF07468),
which are also presumably involved in defense, were found
in 17 predicted flax genes. This agglutinin domain is not
found in any known proteins of either Arabidopsis or poplar
(although they are present in a diverse range of other spe-
cies; Figure 4). Conversely, domains that were under-repre-
sented in the predicted flax genes included many domains of
unknown function (DUFs), some classes of leucine-rich
repeats, wall-associated kinases, F-box-associated domains
and transcription factors. The functional and ecological
relevance of these observations is currently being tested.
Analysis of sequence divergence in duplicated genes
Because the number of genes predicted in the flax WGS
assembly was higher than some other plant species (e.g.
A. thaliana), we analyzed the gene set for evidence of recent
genome duplication. We performed two separate analyses,
using as input either the capillary-sequenced flax ESTs from
the NCBI GenBank database or the predicted CDS sequences
from the WGS assembly (http://www.ncbi.nlm.nih.gov/
nucleotide/). We identified 3644 duplicate gene pairs among
LusPtrRcoMes
GmaCsaMdoAth
CpaVviSbiZma
OsaBdiCrePpa
BSPBarwin
Alginate lyaseAgglutinin
Self_Incomp_S1SCRL
HSP70GRAS
TransferaseDUF4409
BBEPPR_2
Oxidored_q1MP
DUF659DUF594DUF577DUF4371DUF4220
97786672356415471036152524231471129711819537222250388
Figure 4. Pfam-A domain frequency in flax and other species.
All domains that were more abundant (v2 FDR <0.05) among predicted proteins of flax compared to either A. thaliana or P. trichocarpa are shown (labels in bold).
A subset of the domains that were significantly less abundant in flax compared to either species is also shown. Additional species for which whole-genome
sequence is available are shown for comparison. The width of the colored region indicates the number of genes containing a given Pfam-A domain within each
species. A different scale is used for each domain, and the number to the right of each bar indicates the total number of genes represented by that bar. Redundant
occurrences of the same domain within the same gene are counted only once. BSP, basic secretory protein; Barwin, PF00967; alginate lyase, PF08787; agglutinin,
PF07468; Self_Incomp_S1, plant self-incompatibility protein S1 (PF05938); SCRL, plant self-incompatibility response protein (PF06876); HSP70, heat shock protein 70
(PF00012); GRAS, GRAS family transcription factor (PF03514); transferase, PF02458; DUF4409, domain of unknown function (PF14365); BBE, berberine and
berberine-like (PF08031); PPR_2, PPR repeat family (PF13041); Oxidoreq_q1, oxidoreductase (PF00361); MP, viral movement protein (PF01107); DUF659 (PF04937);
DUF577 (PF04510); DUF4371 (PF14291); DUF4220 (PF13968). Ath, Arabidopsis thaliana; Bdi, Brachypodium distachyon; Cpa, Carica papaya; Cre, Chlamydomonas
reinhardtii; Csa, Cucumis sativa; Gma, Glycine max; Lus, Linum usitatissimum; Mdo, Malus domestica; Mes, Manihot esculenta; Osa, Oryza sativa; Ppa,
Physcomitrella patens; Ptr, Populus trichocarpa; Rco, Ricinus communis; Sbi, Sorghum bicolor; Vvi, Vitis vinifera; Zma, Zea mays.
(a) (b) Figure 5. Rate of substitutions per synonymous
sites (Ks) within duplicated gene pairs from (a)
capillary-sequenced flax ESTs and (b) predicted
CDS sequences from the flax WGS assembly.
468 Zhiwen Wang et al.
ª 2012 The AuthorsThe Plant Journal ª 2012 Blackwell Publishing Ltd, The Plant Journal, (2012), 72, 461–473

the flax ESTs and 9920 pairs within the WGS CDS sequences,
and analyzed their divergence as nucleotide substitutions
per synonymous site per year (Ks). In both datasets, a dis-
tinct peak in the Ks distribution was observed at Ks = 0.15
(Figure 5). Based on this modal Ks value, a time of diver-
gence of 5–9 MYA was inferred depending on whether a
synonymous mutation rate per base of 1.5 · 10)8 or
8.1 · 10)9 is assumed (Koch et al., 2000; Lynch and Conery,
2000).
DISCUSSION
Assembly quality
The pace of plant genome sequencing is accelerating, aided
by advances in instrumentation and software. The clone-
by-clone sequencing strategy used to sequence the Arabid-
opsis genome has been supplanted by whole-genome
shotgun (WGS) approaches. WGS de novo assemblies have
typically relied on at least some component of long
sequencing reads (e.g. from dideoxy or pyrosequencing),
rather than the shorter (but less costly) reads produced by
Illumina or SOLiD sequencers (Applied Biosystems). How-
ever, a WGS de novo assembly of date palm was recently
reported that was based entirely on 36–84 bp Illumina reads,
with an N50 scaffold size of 30 kb (Al-Dous et al., 2011). Here
we describe de novo WGS assembly of flax based exclu-
sively on short reads (Table 1). The descriptive statistics for
the flax WGS assembly (Table 2), with scaffold N50 = 693 kb,
85% genome coverage in scaffolds, and coverage of up to
97% of the gene space as defined by ESTs (Table 3), provide
further evidence of the utility of the short-read de novo WGS
approach in plants. The occurrence of transposable ele-
ments (Table 5), NUPTs (Figure S3) and most types of non-
coding RNA (ncRNA) (Table 6) was typical of other plant
genomes sequenced to date (Richly and Leister, 2004; Tus-
kan et al., 2006). As a whole, our analyses showed that the
coverage and accuracy of the assembly was high, especially
at the scale of individual genes (Tables 3 and 7), but with
some limitations regarding long-range accuracy of the
assembly (Table 4 and Figure S1). Further improvements in
assembly accuracy will require the use of additional sources
of data such as the physical maps recently published for CDC
Bethune (Ragupathy et al., 2011). Nevertheless, we conclude
that the short-read de novo WGS approach is a highly effi-
cient strategy for characterization of the nearly complete
gene space of flax.
Predicted genes and protein domains
The masked WGS assembly supported prediction of a
consensus set of 43 484 genes (Table 7). This gene num-
ber is comparable to what has been reported in soybean,
poplar, and rice, but is substantially larger than Arabid-
opsis, Brachypodium or cucumber (Velasco et al., 2010).
The length distribution of exons of predicted flax proteins
was consistent with other species, although flax intron and
mRNA length tended to be shorter than other species in
the comparison (Figure 2). The proportion of predicted flax
genes that aligned with Arabidopsis genes (89.4%), that
contain Pfam domains (77%), and that align with one or
more proteins in the NCBI nr database (89.5%) is high
(Table 7). Moreover, the distribution of conserved protein
domains in flax is nearly equivalent when compared to
either Arabidopsis or poplar (Figure 3), and 93.2% of
known ESTs of at least 400 bp align with high stringency
to the predicted CDS sequences. Together these data
support our conclusion that the predicted WGS genes are
highly accurate and that the WGS assembly (and sub-
sequent gene prediction) produced very good coverage of
the gene space of flax. Therefore, the relatively low cov-
erage of the predicted WGS flax genes by ESTs (66.2%) is
probably due primarily to incomplete gene representation
in the ESTs, rather than systematic errors in gene predic-
tion.
Only a few functional groups appeared to be significantly
over-represented in flax compared to other species (Fig-
ure 4 and Table S2). The increased in abundance of
domains that are often related to plant defense responses
(e.g. lectins and basic secretory proteins), and the
decreased abundance of leucine-rich repeat domain-con-
taining proteins, raise the possibility of additional biotic
stress response strategies in flax. The increased abundance
of two domains nominally associated with self-incompati-
bility (PF05938 and PF06876; Figure 4) is probably not
functionally relevant to pollination control in this self-
compatible species, as we note that one of these domains
(SCRL) is also highly represented in Arabidopsis.
Evolution of the flax genome
Analysis of the rates of divergence of duplicated genes
(Figure 5) indicated that a whole-genome duplication event
probably occurred 5–9 MYA in the lineage of L. usitatissi-
mum. The same Ks distribution was observed in both CDS
sequences predicted from the Illumina WGS shotgun
assembly (Figure 5b) and capillary-sequenced ESTs (Fig-
ure 5a), demonstrating that the distribution is not an artifact
of the sequencing or assembly technique used. A recent
whole-genome duplication is also consistent with the large
gene number predicted for flax, compared for example to
Arabidopsis (Table 7), the relatively high number of dupli-
cated genes (9920) identified, and with the numbers of
chromosomes reported in L. usitatissimum and related
species (Figure 6). L. usitatissimum and the closely related
L. bienne both have n = 15 chromosomes, while sister
clades containing species such as L. grandiflorum and
L. lewisii have n = 8 and n = 9 chromosomes, respectively.
Therefore, the evolution of L. usitatissimum probably
involved chromosome doubling followed by loss of one or
more chromosomes. Comparison of patterns of gene family
Whole-genome shotgun assembly of flax 469
ª 2012 The AuthorsThe Plant Journal ª 2012 Blackwell Publishing Ltd, The Plant Journal, (2012), 72, 461–473

expansion in L. usitatissimum and L. bienne and other
congeners is ongoing.
In conclusion, de novo assembly of exclusively short,
paired reads obtained by approximately 94· raw coverage of
the flax genome was an efficient method of obtaining near-
complete and highly accurate information about the gene
space of this crop. A limitation to this approach is the
accuracy of the long-range assembly, as demonstrated by
imperfect alignments to BAC sequences. Nevertheless, the
gene-scale data are sufficient for further investigations into
the function and evolution of this species.
EXPERIMENTAL PROCEDURES
Plant growth and DNA isolation
Seeds of the L. usitatissimum linseed cultivar CDC Bethune wereobtained from its breeder, Gordon Rowland (Department of PlantSciences, University of Saskatchewan, Saskatoon, Canada). Theseseeds were the F19 generation of the original cross of variety Nor-Man · genotype FP857 (Rowland et al., 2002). All generations wereself-pollinated, and the final eight generations were obtained bysingle-seed descent. Seeds were surface-sterilized and grown axe-nically in polycarbonate vessels. DNA was extracted from etiolatedseedlings using a variation of a published protocol (Dellaporta et al.,1983), in which the concentration of NaCl in the extraction bufferwas 1 M. Upon resuspension of DNA in TE buffer (50 mM Tris and10 mM EDTA, pH 8), 12 ll of 10 mg/ml RNase A was added, and themixture was allowed to incubate at 37�C for 1 h. Subsequently aphenol/chloroform/isoamyl alchohol (25:24:1) extraction was per-formed to remove impurities. DNA was precipitated with 0.1volumes of 3 M NaOAc and 0.6 volumes of isopropanol, washed in70% EtOH, resuspended in 400 ll TE (10:0.1), and subsequentlyre-precipitated with 0.1 volumes of 3 M NaOAc and two volumes95% EtOH. DNA was washed and resuspended in TE. Aliquots of thesame DNA preparation were used for genome sequencing andfosmid library construction.
Flow cytometry
DNA content measurements (Galbraith et al., 1983) were performedon ten CDC Bethune seedlings germinated from the same source ofseeds used for WGS and fosmid sequencing. The measurementswere performed using an Accuri C6 flow cytometer (Becton, Dicksonhttp://www.bdbiosciences.com), with propidium iodide/RNase asthe nuclear DNA stain (Bharathan et al., 1994). The nuclear DNA
contents were calibrated to an external standard, Raphanus sativacv. Saxa (Dolezel et al., 2007), with a DNA content of 1.11 pg/2Cnucleus. A single preparation of the standard was run five times toprovide a mean DNA fluorescence peak position that was then usedas the reference for the DNA content calculations.
Whole-genome shotgun sequencing and assembly
Seven paired-end sequencing libraries were constructed accordingto the manufacturer’s protocols (Illumina, http://www.illumina.com/)at BGI-Shenzen. The nominal insert sizes of the libraries were300 bp, 500 bp, 2 kb, 5 kb and 10 kb. For libraries with an insert sizebetween 200 and 500 bp, library preparation proceeded as follows:genomic DNA fragmentation, end repair, adapter ligation, sizeselection and PCR. For the longer (‡2 kb) mate-paired libraries, DNAcircularization, digestion of linear DNA, fragmentation of circular-ized DNA, and purification of biotinylated DNA were performedbefore adapter ligation. After the libraries were constructed, thetemplate DNA fragments of the constructed libraries were hybrid-ized to the surface of flow cells, amplified to form clusters, and thensequenced using an Illumina GAII genome analyzer.
Raw reads were filtered to remove fragments that contained>2% unknown bases or poly(A) structure, or for which >60% ofbases for the large insert-size library data and 40% of bases for theshort-insert data showed a quality score £7. Reads were alsoremoved if more than 10 bp aligned to the adapter sequence(allowing a mismatch of £3 bp). The filtered reads were assembledusing SOAPdenovo as previously described (Li et al., 2009). Theassembly comprised three stages. In the first stage, short-insertlibrary data were split into k-mers, a de Bruijn graph wasconstructed and simplified, and then the k-mer path was con-nected to generate the contig file. In the second stage, all theusable reads were re-aligned onto the contig sequences, then thenumber of shared paired-end relationships between each pair ofcontigs was calculated and used to identify consistent andconflicting paired-ends used to construct the scaffolds. Finally,the paired-end information was used to retrieve read pairs that hadone end mapped to a unique contig and the other located in a gapregion, and then a local assembly was performed for thesecollected reads to fill the gaps.
Fosmid library construction and sequencing
Fosmid and BAC libraries were constructed from flax variety CDCBethune, and selected clones were sequences as described previ-ously (Ragupathy et al., 2011; Roach et al., 2011). Fosmids weredeposited in NCBI GenBank under accessions HQ902252 andJN133299–JN133301.
Annotation of transposable elements and ncRNA
Putative transposable element regions of the flax genome wereidentified using REPEATMASKER version 3.3.0 (http://www.repeat-masker.org/), RMBlast was used as the search algorithm with aSmith–Waterman cut-off of 225 (this cut-off was used for allRepeatMasker analyses), and a database with annotated de novorepeats was combined with the Viridiplantae database of trans-posable elements (update 20110920) for use as a library for com-parison to the shotgun assembly. To annotate the masked bases intheir respective transposable element superfamilies, a custom Perlscript was used (kindly provided by Robert Hubley, Institute forSystems Biology, Seattle, WA). De novo identification of transpos-able elements was performed using RepeatScout (Price et al., 2005),PILER (Edgar and Myers, 2005), LTR_finder (Xu and Wang, 2007) andLTR_STRUC (McCarthy and McDonald, 2003). RepeatScout was
Figure 6. Phylogram of flax (L. usitatissimum) and related species showing
haploid chromosome number (in black or shaded circle).
The star indicates the approximate placement of the whole-genome duplica-
tion that we infer occurred in the common ancestor of L. bienne and
L. usitatissimum. The tree and dated nodes are based on data from McDill
et al. (2009).
470 Zhiwen Wang et al.
ª 2012 The AuthorsThe Plant Journal ª 2012 Blackwell Publishing Ltd, The Plant Journal, (2012), 72, 461–473

used under the default parameters. Repeats identified by Repeat-Scout were filtered for low complexity using tandem repeats finder(Benson, 1999) and nseg (Wootton and Federhen, 1993), and thefiltered library was used to mask the flax genome. Repeats withfewer than ten hits in the genome were eliminated from the library.For PILER-DF analysis, the full genome was compared to itself usingPALS (part of the PILER implementation) with default parameters.Families of dispersed repeats were created using a minimum familysize of three members and a maximum length difference of 5%between all family members. The consensus sequence for eachfamily was created after aligning the sequences using MUSCLE
(Edgar, 2004). LTR transposable elements were found usingLTR_finder with option –w2 to obtain a table output that was parsedto obtain the sequences corresponding to the elements.LTR_STRUC (McCarthy and McDonald, 2003) was used underdefault parameters.
Four types of non-coding RNAs were detected by searching theunmasked scaffold assemblies using tRNASCAN-SE (version 1.23)(Lowe and Eddy, 1997), and BLAST alignment with RFAM database(release 9.1) (Griffiths-Jones et al., 2003) and Arabidopsis thalianaand Oryza sativa full-length rRNAs, followed by INFERNAL (version0.81) (Nawrocki et al., 2009).
Gene prediction and annotation
Gene prediction was performed using a combination of de novo andhomology-based methods. De novo predictions were performed onthe repeat-masked WGS assembly using AUGUSTUS (version 2.5.5)(Stanke et al., 2008) and GLIMMERHMM (version 3.0.1) (Majoroset al., 2004). Flax ESTs were also aligned to the WGS assembly usingBLAT (blat-34, identity ‡0.98, coverage ‡0.98) to generate splicedalignments, which were linked according to their overlap using PASA
(Haas et al., 2003). Plant proteins of other species were also mappedto the WGS assembly using TBLASTN (Blastall 2.2.23, Altschul et al.,1997) with an e-value cut-off of 10)5. The aligned sequences as wellas their query proteins were then filtered and passed to GENEWISE
(version 2.2) (Birney et al., 2004). Gene models from de novo pre-dictions and EST and protein alignments were integrated usingGLEAN (Elsik et al., 2007) to produce a consensus gene set.
Duplicate gene pair analysis
All analyses were performed independently on two datasets. First,we downloaded all 286 852 ESTs available for L. usitatissimum inNovember 2011 from the NCBI database (http://www.ncbi.nlm.nih.gov/nucleotide/) and assembled them into unigenes using CAP3
with default settings (Huang and Madan, 1999). Second, we used all43 484 predicted CDSs from the flax genome project.
Analyses were performed with the DupPipe pipeline (Barkeret al., 2008) using default parameters. In short, sequences wereclustered into gene family members by parsing the results of adiscontiguous MegaBlast (Zhang et al., 2000; Ma et al., 2002) toretain those that showed at least 40% sequence similarity over aminimum of 300 bp. Reading frames for each sequence pair wereidentified by comparison with all available plant protein sequencesof GenBank (Wheeler et al., 2007) using BLASTX (Altschul et al.,1997). Best-hit proteins were then paired with each nucleotidesequence at a minimum cut-off of 30% sequence similarity over atleast 150 sites. Sequences that did not meet these criteria wereremoved from further analyses. Each gene was aligned against itsbest-hit protein using GENEWISE 2.2.2 (Birney et al., 2004) to deter-mine the reading frame. Amino acid sequences for each gene wereestimated by using the highest scoring Genewise DNA–proteinalignments. Amino acid sequences for each duplicate pair werethen aligned using MUSCLE 3.6 (Edgar, 2004), and used to align their
corresponding DNA sequences using REVTRANS 1.4 (Wernerssonand Pedersen, 2003). Ks values (synonymous substitution rates) foreach duplicate pair were calculated using the maximum-likelihoodmethod implemented in codeml of the PAML package (Yang, 1997)under the F3-4 model (Goldman and Yang, 1994). Further cleaningof the dataset was then performed to remove duplication eventsthat may bias the results. To reduce the possibility that identicalgenes were represented in the dataset, but were missed by TGICLclustering (Pertea et al., 2003) due to alternative splicing, all Ks
values from one member of a duplicate pair with Ks = 0 wereremoved. Further, to reduce the multiplicative effects of multi-copygene families on Ks values, simple hierarchical clustering was usedto construct phylogenies for each gene family, identified as single-linked clusters, and the nodal Ks values were calculated.
Data access
WGS assembly data were deposited at NCBI GenBank underGenomeProject ID #68161 and Sequence Read Archive accessionSRA038451. Fosmid and BAC sequences were submitted to Gen-Bank as accessions HQ902252, JN133299–JN133301 and JX174444–JX174449. Additional resources, bulk data downloads and aGbrowse (Stein et al., 2002) implementation are available at http://www.linum.ca and http://phytozome.org.
ACKNOWLEDGEMENTS
Funding was provided by Genome Alberta/Genome Canada, theGovernment of Alberta, Alberta Innovates Technology Futures-iCORE, Institut National de la Recherche Agronomique France andthe Indian Council of Agricultural Research.
SUPPORTING INFORMATION
Additional Supporting Information may be found in the onlineversion of this article:Figure S1. Sequencing depth distribution.Figure S2. Diagrams showing alignment of BACs and fosmids withflax WGS scaffolds.Figure S3. Size distribution of putative NUPT regions in the flaxWGS assembly.Table S1. List of putative miRNAs identified in the flax WGSassembly.Table S2. Representation of Pfam-A domains in predicted genes ofthe flax WGS assembly and in genomes of other selected species.Please note: As a service to our authors and readers, this journalprovides supporting information supplied by the authors. Suchmaterials are peer-reviewed and may be re-organized for onlinedelivery, but is not copy-edited or typeset. Technical support issuesarising from supporting information (other than missing files)should be addressed to the authors.
REFERENCES
Al-Dous, E.K., George, B., Al-Mahmoud, M.E. et al. (2011) De novo genome
sequencing and comparative genomics of date palm (Phoenix dactylifera).
Nat. Biotechnol. 29, 521–527.
Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J.H., Zhang, Z., Miller, W.
and Lipman, D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation
of protein database search programs. Nucleic Acids Res. 25, 3389–3402.
Barker, M.S., Kane, N.C., Matvienko, M., Kozik, A., Michelmore, W., Knapp,
S.J. and Rieseberg, L.H. (2008) Multiple paleopolyploidizations during the
evolution of the Compositae reveal parallel patterns of duplicate gene
retention after millions of years. Mol. Biol. Evol. 25, 2445–2455.
Benson, G. (1999) Tandem repeats finder: a program to analyze DNA
sequences. Nucleic Acids Res. 27, 573–580.
Bharathan, G., Lambert, G. and Galbraith, D.W. (1994) Nuclear DNA content of
monocotyledons and related taxa. Am. J. Bot. 81, 381–386.
Whole-genome shotgun assembly of flax 471
ª 2012 The AuthorsThe Plant Journal ª 2012 Blackwell Publishing Ltd, The Plant Journal, (2012), 72, 461–473

Birney, E., Clamp, M. and Durbin, R. (2004) GeneWise and genomewise.
Genome Res. 14, 988–995.
Bodros, E., Pillin, I., Montrelay, N. and Baley, C. (2007) Could biopolymers
reinforced by randomly scattered flax fibre be used in structural applica-
tions? Compos. Sci. Technol. 67, 462–470.
Chan, A.P., Crabtree, J., Zhao, Q. et al. (2010) Draft genome sequence of the
oilseed species Ricinus communis. Nat. Biotechnol. 28, 951–953.
Chen, Y. (1999) An Insertion Sequence in Flax Induced by the Environment.
Cleveland, OH: Case Western Reserve University.
Chen, Y.M., Schneeberger, R.G. and Cullis, C.A. (2005) A site-specific insertion
sequence in flax genotrophs induced by environment. New Phytol. 167,
171–180.
Cloutier, S., Niu, Z.X., Datla, R. and Duguid, S. (2009) Development and
analysis of EST-SSRs for flax (Linum usitatissimum L.). Theor. Appl. Genet.
119, 53–63.
Cullis, C.A. (1980) DNA sequence organization in flax. Heredity, 44, 292.
Cullis, C.A. (1981a) DNA sequence organization in the flax genome. Biochim.
Biophys. Acta, 652, 1–15.
Cullis, C.A. (1981b) Environmental induction of heritable changes in flax –
defined environments inducing changes in rDNA and peroxidase isoen-
zyme band pattern. Heredity, 47, 87–94.
Cullis, C.A. (2005) Mechanisms and control of rapid genomic changes in flax.
Ann. Bot. 95, 201–206.
Davis, C.C., Webb, C.O., Wurdack, K.J., Jaramillo, C.A. and Donoghue, M.J.
(2005) Explosive radiation of malpighiales supports a mid-Cretaceous ori-
gin of modern tropical rain forests. Am. Nat. 165, E36–E65.
Dellaporta, S., Wood, J. and Hicks, J. (1983) A plant DNA minipreparation:
version II. Plant Mol. Biol. Rep. 1, 19–21.
Dolezel, J., Greilhuber, J. and Suda, J. (2007) Estimation of nuclear DNA
content in plants using flow cytometry. Nat. Protoc. 2, 2233–2244.
Eddy, S.R. (2011) Accelerated Profile HMM Searches. PLoS Comput. Biol. 7,
e1002195.
Edgar, R.C. (2004) MUSCLE: a multiple sequence alignment method with re-
duced time and space complexity. BMC Bioinformatics, 5, 1–19.
Edgar, R.C. and Myers, E.W. (2005) PILER: identification and classification of
genomic repeats. Bioinformatics, 21, I152–I158.
Elsik, C.G., Mackey, A.J., Reese, J.T., Milshina, N.V., Roos, D.S. and Wein-
stock, G.M. (2007) Creating a honey bee consensus gene set. Genome Biol.
8, R13.
Evans, G.M., Durrant, A. and Rees, H. (1966) Associated nuclear changes in
induction of flax genotrophs. Nature, 212, 697–699.
Evans, G., Rees, H., Snell, C. and Sun, S. (1972) The relationship between
nuclear DNA amount and the duration of the mitotic cycle. Chromosomes
Today, 3, 24–31.
Fenart, S., Ndong, Y.P.A., Duarte, J. et al. (2010) Development and validation
of a flax (Linum usitatissimum L.) gene expression oligo microarray. BMC
Genomics, 11, 592.
Finn, R.D., Mistry, J., Tate, J. et al. (2010) The Pfam protein families database.
Nucleic Acids Res. 38, D211–D222.
Fu, Y.B. and Allaby, R.G. (2010) Phylogenetic network of Linum species as
revealed by non-coding chloroplast DNA sequences. Genet. Resour. Crop
Evol. 57, 667–677.
Galbraith, D.W., Harkins, K.R., Maddox, J.M., Ayres, N.M., Sharma, D.P. and
Firoozabady, E. (1983) Rapid flow cytometric analysis of the cell cycle in
intact plant tissues. Science, 220, 1049–1051.
Goldman, N. and Yang, Z.H. (1994) Codon-based model of nucleotide
substitution for protein-coding DNA sequences. Mol. Biol. Evol. 11, 725–
736.
Goldsbrough, P.B., Ellis, T.H.N. and Cullis, C.A. (1981) Organization of the 5S
RNA genes in flax. Nucleic Acids Res. 9, 5895–5904.
Griffiths-Jones, S., Bateman, A., Marshall, M., Khanna, A. and Eddy, S.R.
(2003) Rfam: an RNA family database. Nucleic Acids Res. 31, 439–441.
Haas, B.J., Delcher, A.L., Mount, S.M. et al. (2003) Improving the Arabidopsis
genome annotation using maximal transcript alignment assemblies.
Nucleic Acids Res. 31, 5654–5666.
Huang, X.Q. and Madan, A. (1999) Cap3: a DNA sequence assembly program.
Genome Res. 9, 868–877.
Kent, W.J. (2002) BLAT – the BLAST-like alignment tool. Genome Res. 12,
656–664.
Koch, M.A., Haubold, B. and Mitchell-Olds, T. (2000) Comparative evolution-
ary analysis of chalcone synthase and alcohol dehydrogenase loci in Ara-
bidopsis, Arabis, and related genera (Brassicaceae). Mol. Biol. Evol. 17,
1483–1498.
Kvavadze, E., Bar-Yosef, O., Belfer-Cohen, A., Boaretto, E., Jakeli, N., Mats-
kevich, Z. and Meshveliani, T. (2009) 30,000-year-old wild flax fibers. Sci-
ence, 325, 1359.
Li, R.Q., Yu, C., Li, Y.R., Lam, T.W., Yiu, S.M., Kristiansen, K. and Wang, J.
(2009) SOAP2: an improved ultrafast tool for short read alignment. Bioin-
formatics, 25, 1966–1967.
Li, R.Q., Fan, W., Tian, G. et al. (2010) The sequence and de novo assembly of
the giant panda genome. Nature, 463, 311–317.
Lowe, T.M. and Eddy, S.R. (1997) tRNAscan-SE: a program for improved
detection of transfer RNA genes in genomic sequence. Nucleic Acids Res.
25, 955–964.
Lynch, M. and Conery, J.S. (2000) The evolutionary fate and consequences of
duplicate genes. Science, 290, 1151–1155.
Ma, B., Tromp, J. and Li, M. (2002) PatternHunter: faster and more sensitive
homology search. Bioinformatics, 18, 440–445.
Majoros, W.H., Pertea, M. and Salzberg, S.L. (2004) TigrScan and Glimmer-
HMM: two open source ab initio eukaryotic gene-finders. Bioinformatics,
20, 2878–2879.
Marie, D. and Brown, S.C. (1993) A cytometric exercise in plant DNA histo-
grams, with 2C values for 70 species. Biol. Cell, 78, 41–51.
McCarthy, E.M. and McDonald, J.F. (2003) LTR_STRUC: a novel search and
identification program for LTR retrotransposons. Bioinformatics, 19, 362–367.
McDill, J.R. and Simpson, B.B. (2011) Molecular phylogenetics of Linaceae
with complete generic sampling and data from two plastid genes. Bot. J.
Linn. Soc. 165, 64–83.
McDill, J., Repplinger, M., Simpson, B.B. and Kadereit, J.W. (2009) The phy-
logeny of Linum and Linaceae subfamily Linoideae, with implications for
their systematics, biogeography, and evolution of heterostyly. Syst. Bot.
34, 386–405.
Mohanty, A.K., Misra, M. and Hinrichsen, G. (2000) Biofibres, biodegradable
polymers and biocomposites: an overview. Macromol. Mater. Eng. 276, 1–24.
Nawrocki, E.P., Kolbe, D.L. and Eddy, S.R. (2009) Infernal 1.0: inference of
RNA alignments. Bioinformatics, 25, 1335–1337.
Pertea, G., Huang, X., Liang, F., et al. (2003) TIGR Gene Indices clustering tools
(TGICL): a software system for fast clustering of large EST datasets.
Bioinformatics, 19, 651–652.
Price, A.L., Jones, N.C. and Pevzner, P.A. (2005) De novo identification of
repeat families in large genomes. Bioinformatics, 21, I351–I358.
Ragupathy, R., Rathinavelu, R. and Cloutier, S. (2011) Physical mapping and
BAC-end sequence analysis provide initial insights into the flax (Linum
usitatissimum L.) genome. BMC Genomics, 12, 217.
Richly, E. and Leister, D. (2004) NUPTs in sequenced eukaryotes and their
genomic organization in relation to NUMTs. Mol. Biol. Evol. 21, 1972–
1980.
Roach, M.J. and Deyholos, M.K. (2007) Microarray analysis of flax (Linum
usitatissimum L.) stems identifies transcripts enriched in fibre-bearing
phloem tissues. Mol. Genet. Genomics, 278, 149–165.
Roach, M.J., Mokshina, N.Y., Badhan, A., Snegireva, A.V., Hobson, N.,
Deyholos, M.K. and Gorshkova, T.A. (2011) Development of cellulosic
secondary walls in flax fibers requires b-galactosidase. Plant Physiol. 156,
1351–1363.
Rogers, C.M. (1982) The systematics of Linum-sect Linopsis (Linaceae). Plant
Syst. Evol. 140, 225–234.
Rowland, G.G., Hormis, Y.A. and Rashid, K.Y. (2002) CDC Bethune flax. Can.
J. Plant Sci. 82, 101–102.
Simmons, C.A., Turk, P., Beamer, S., Jaczynski, J., Semmens, K. and Matak,
K.E. (2011) The effect of a flaxseed oil-enhanced diet on the product quality
of farmed brook trout (Salvelinus fontinalis) fillets. J. Food Sci. 76, S192–
S197.
Singh, K.K., Mridula, D., Rehal, J. and Barnwal, P. (2011) Flaxseed: a
potential source of food, feed and fiber. CRC Crit. Rev. Food Sci. Nutr.
51, 210–222.
Smykal, P., Bacova-Kerteszova, N., Kalendar, R., Corander, J., Schulman, A.H.
and Pavelek, M. (2011) Genetic diversity of cultivated flax (Linum usita-
tissimum L.) germplasm assessed by retrotransposon-based markers.
Theor. Appl. Genet. 122, 1385–1397.
Stanke, M., Diekhans, M., Baertsch, R. and Haussler, D. (2008) Using native
and syntenically mapped cDNA alignments to improve de novo gene
finding. Bioinformatics, 24, 637–644.
472 Zhiwen Wang et al.
ª 2012 The AuthorsThe Plant Journal ª 2012 Blackwell Publishing Ltd, The Plant Journal, (2012), 72, 461–473

Stein, L.D., Mungall, C., Shu, S. et al. (2002) The generic genome browser: a
building block for a model organism system database. Genome Res. 12,
1599–1610.
Swarbreck, D., Wilks, C., Lamesch, P. et al. (2008) The Arabidopsis Informa-
tion Resource (TAIR): gene structure and function annotation. Nucleic
Acids Res. 36, D1009–D1014.
Toure, A. and Xu, X.M. (2010) Flaxseed lignans: source, biosynthesis,
metabolism, antioxidant activity, bio-active components, and health ben-
efits. Compr. Rev. Food Sci. Food Saf. 9, 261–269.
Tuskan, G.A., DiFazio, S., Jansson, S. et al. (2006) The genome of
black cottonwood, Populus trichocarpa (Torr& Gray). Science, 313,
1596–1604.
Velasco, R., Zharkikh, A., Affourtit, J. et al. (2010) The genome of the
domesticated apple (Malus domestica Borkh.). Nat. Genet. 42, 833–839.
Venglat, P., Xiang, D., Qiu, S. et al. (2011) Gene expression analysis of flax
seed development. BMC Plant Biol. 11, 74.
Wernersson, R. and Pedersen, A.G. (2003) RevTrans: multiple alignment of
coding DNA from aligned amino acid sequences. Nucleic Acids Res. 31,
3537–3539.
Wheeler, D.L., Barrett, T., Benson, D.A. et al. (2007) Database resources of the
National Center for Biotechnology Information. Nucleic Acids Res. 35,
D5–D12.
Wootton, J.C. and Federhen, S. (1993) Statistics of local complexity in amino-
acid-sequences and sequence databases. Comput. Chem. 17, 149–163.
Wurdack, K.J. and Davis, C.C. (2009) Malpighiales phylogenetics: gaining
ground on one of the most recalcitrant clades in the angiosperm tree of life.
Am. J. Bot. 96, 1551–1570.
Xu, Z. and Wang, H. (2007) LTR_FINDER: an efficient tool for the prediction
of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268.
Yang, Z.H. (1997) PAML: a program package for phylogenetic analysis by
maximum likelihood. Comput. Appl. Biosci. 13, 555–556.
van Zeist, W. and Bakker-Heeres, J.A.H. (1975) Evidence for linseed cultivation
before 6000 BC. J. Archaeol. Sci. 2, 215–219.
Zhang, Z., Schwartz, S., Wagner, L. and Miller, W. (2000) A greedy algorithm
for aligning DNA sequences. J. Comput. Biol. 7, 203–214.
Zohary, D. and Hopf, M. (2000) Domestication of Plants in the Old World: The
Origin and Spread of Cultivated Plants in West Asia, Europe and the Nile
Valley. Oxford, UK: Oxford University Press.
Whole-genome shotgun assembly of flax 473
ª 2012 The AuthorsThe Plant Journal ª 2012 Blackwell Publishing Ltd, The Plant Journal, (2012), 72, 461–473




















