
The Complete Genomic Sequence of a BLV Strain from a Holstein Cow from Argentina
8
The Complete Genomic Sequence of a BLV Strain from a Holstein Cow from Argentina Syamalima Dube,* Guillermina Dolcini,† Lynn Abbott,* Sonali Mehta,* Dipak Dube,* Silvina Gutierrez,† Carolina Ceriani,† Eduardo Esteban,† Jorge Ferrer,‡ and Bernard Poiesz* ,1 *Department of Medicine, Upstate Medical University, Syracuse, New York 13210; †Universidad Nacional del Centro de la Provincia de Buenos Aires, Facultad de Ciencias Veterinarias, Tandil, Argentina; and ‡Comparative Leukemia and Retroviruses Unit, New Bolton Center, University of Pennsylvania, Kennett Square, Pennsylvania 19348 Received June 12, 2000; returned to author for revision August 9, 2000; accepted September 1, 2000 DNA was extracted from the peripheral blood of a seropositive, PCR-positive, BLV-infected Holstein cow (No. 38) from Argentina. The DNA was amplified via PCR with a series of overlapping primers encompassing the entire BLV proviral DNA. The amplified BLV ARG 38 DNA was cloned, sequenced, and compared phylogenetically to three other full-length BLV sequences. Characterization of its deduced proteins and its relationship to other members of the PTLV/BLV genus of retroviruses are discussed. © 2000 Academic Press Bovine leukemia virus (BLV) is an infectious agent of cattle that can cause lymphomas and benign disorders that, directly or indirectly, have a financial impact on the cattle industry (Ferrer, 1980; Brenner et al., 1989; Burny et al., 1990). It is estimated that more than 10 and 30% of the dairy and beef cattle in the United States and Argentina, respectively, are infected with BLV (Ferrer, 1980; Brenner et al., 1989; Trono et al., 1999). BLV, together with the primate T-cell leukemia/lymphoma viruses (PTLV), form a separate genus of retroviruses that exhibit in vivo lym- photropism and are characterized by the transforming property of a unique virus regulatory protein, Tax, that can transactivate both viral and cellular genes (for review see Poiesz, 1995). We are engaged in several in vitro and in vivo studies on the biology and epidemiology of BLV in a commercial herd of Holstein dairy cattle maintained near the Facultad de Ciencias Veterinarias de Tandil, Argentina (FCV-UNCP BA). We decided to fully sequence a strain of BLV infecting this herd in order to compare it phylogenetically to other members of its genus and to have a structural blueprint for correlation with observed in vitro and in vivo functional phenomena. RESULTS Sequence of BLV ARG 38 Quantitative analyses indicated that there were ap- proximately 5000 copies of BLV pol DNA per mg of cow No. 38 peripheral blood leukocyte DNA. PCR amplifica- tion and Southern blot hybridization were successful for each of the primer pair/probe groups shown in Fig. 1. Some primer pair/probe systems not listed were not successful, usually because of BLV ARG 38-specific mu- tations (data not shown). The complete sequence of BLV ARG 38 was obtained from these amplified products (GenBank Accession No. AF257515). No variability was observed among the many overlapping clones se- quenced from each of the regions shown, indicating that cow No. 38 was infected with one unique strain of BLV. Comparative analyses indicate that BLV ARG 38 is approximately 95.1% homologous to BLV A from Austra- lia, 95.6% to BLV GAGA from Belgium, and 95.9% to BLV CG from Japan. Phylogenetic analyses (Fig. 2) confirm that it is most homologous to BLV CG from Japan and markedly divergent from the primate T-cell lymphoma/ leukemia viruses. The availability of now a fourth fully sequenced BLV strain allows for a more precise estimate of the node of separation (node A in Fig. 2) among the PTLV. In the gag/pol sequences compared, the PTLV-I [human and simian T-cell lymphoma/leukemia viruses type I (HTLV-I and STLV-I)] diverge from a separate group comprising the PTLV-II [human and simian T-cell lymphoma/leuke- mia viruses type II (HTLV-II and STLV-II)] and primate T-cell lymphoma/leukemia viruses, long (PTLV-L). Inter- estingly, if one measures the distance of each PTLV strain from node A, it is apparent that the PTLV-II and PTLV-L have diverged at approximately 1.5 times the rate of the PTLV-I [mean of 1367 bases (6135) vs mean of 805 bases (630); P , 0.001]. Comparison of the LTR of BLV ARG 38 with that of the other full-length BLV sequences is shown in Fig. 3. It contains the RNA transcription promoter and enhancer elements, polyadenylation signal, and REX response el- ement typical of BLV. The only change of possible con- sequence in BLV ARG 38 is a G ➜ A at base 58 in the 1 To whom correspondence and reprint requests should be ad- dressed at the Department of Medicine, SUNY Upstate Medical Uni- versity, 750 E. Adams Street, Syracuse, NY 13210. Virology 277, 379–386 (2000) doi:10.1006/viro.2000.0622, available online at http://www.idealibrary.com on 0042-6822/00 $35.00 Copyright © 2000 by Academic Press All rights of reproduction in any form reserved. 379
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of The Complete Genomic Sequence of a BLV Strain from a Holstein Cow from Argentina

adre
Nte
dv
Virology 277, 379–386 (2000)doi:10.1006/viro.2000.0622, available online at http://www.idealibrary.com on
The Complete Genomic Sequence of a BLV Strain from a Holstein Cow from Argentina
Syamalima Dube,* Guillermina Dolcini,† Lynn Abbott,* Sonali Mehta,* Dipak Dube,* Silvina Gutierrez,†Carolina Ceriani,† Eduardo Esteban,† Jorge Ferrer,‡ and Bernard Poiesz*,1
*Department of Medicine, Upstate Medical University, Syracuse, New York 13210; †Universidad Nacional del Centro de la Provinciade Buenos Aires, Facultad de Ciencias Veterinarias, Tandil, Argentina; and ‡Comparative Leukemia and Retroviruses Unit,
New Bolton Center, University of Pennsylvania, Kennett Square, Pennsylvania 19348
Received June 12, 2000; returned to author for revision August 9, 2000; accepted September 1, 2000
DNA was extracted from the peripheral blood of a seropositive, PCR-positive, BLV-infected Holstein cow (No. 38) fromArgentina. The DNA was amplified via PCR with a series of overlapping primers encompassing the entire BLV proviral DNA.The amplified BLV ARG 38 DNA was cloned, sequenced, and compared phylogenetically to three other full-length BLVsequences. Characterization of its deduced proteins and its relationship to other members of the PTLV/BLV genus of
retroviruses are discussed. © 2000 Academic PressBovine leukemia virus (BLV) is an infectious agent ofcattle that can cause lymphomas and benign disordersthat, directly or indirectly, have a financial impact on thecattle industry (Ferrer, 1980; Brenner et al., 1989; Burny et
l., 1990). It is estimated that more than 10 and 30% of theairy and beef cattle in the United States and Argentina,
espectively, are infected with BLV (Ferrer, 1980; Brennert al., 1989; Trono et al., 1999). BLV, together with the
primate T-cell leukemia/lymphoma viruses (PTLV), form aseparate genus of retroviruses that exhibit in vivo lym-photropism and are characterized by the transformingproperty of a unique virus regulatory protein, Tax, thatcan transactivate both viral and cellular genes (for reviewsee Poiesz, 1995). We are engaged in several in vitro andin vivo studies on the biology and epidemiology of BLV ina commercial herd of Holstein dairy cattle maintainednear the Facultad de Ciencias Veterinarias de Tandil,Argentina (FCV-UNCP BA). We decided to fully sequencea strain of BLV infecting this herd in order to compare itphylogenetically to other members of its genus and tohave a structural blueprint for correlation with observedin vitro and in vivo functional phenomena.
RESULTS
Sequence of BLV ARG 38
Quantitative analyses indicated that there were ap-proximately 5000 copies of BLV pol DNA per mg of cow
o. 38 peripheral blood leukocyte DNA. PCR amplifica-ion and Southern blot hybridization were successful forach of the primer pair/probe groups shown in Fig. 1.
1 To whom correspondence and reprint requests should be ad-ressed at the Department of Medicine, SUNY Upstate Medical Uni-
ersity, 750 E. Adams Street, Syracuse, NY 13210.379
Some primer pair/probe systems not listed were notsuccessful, usually because of BLV ARG 38-specific mu-tations (data not shown). The complete sequence of BLVARG 38 was obtained from these amplified products(GenBank Accession No. AF257515). No variability wasobserved among the many overlapping clones se-quenced from each of the regions shown, indicating thatcow No. 38 was infected with one unique strain of BLV.
Comparative analyses indicate that BLV ARG 38 isapproximately 95.1% homologous to BLV A from Austra-lia, 95.6% to BLV GAGA from Belgium, and 95.9% to BLVCG from Japan. Phylogenetic analyses (Fig. 2) confirmthat it is most homologous to BLV CG from Japan andmarkedly divergent from the primate T-cell lymphoma/leukemia viruses.
The availability of now a fourth fully sequenced BLVstrain allows for a more precise estimate of the node ofseparation (node A in Fig. 2) among the PTLV. In thegag/pol sequences compared, the PTLV-I [human andsimian T-cell lymphoma/leukemia viruses type I (HTLV-Iand STLV-I)] diverge from a separate group comprisingthe PTLV-II [human and simian T-cell lymphoma/leuke-mia viruses type II (HTLV-II and STLV-II)] and primateT-cell lymphoma/leukemia viruses, long (PTLV-L). Inter-estingly, if one measures the distance of each PTLVstrain from node A, it is apparent that the PTLV-II andPTLV-L have diverged at approximately 1.5 times the rateof the PTLV-I [mean of 1367 bases (6135) vs mean of 805bases (630); P , 0.001].
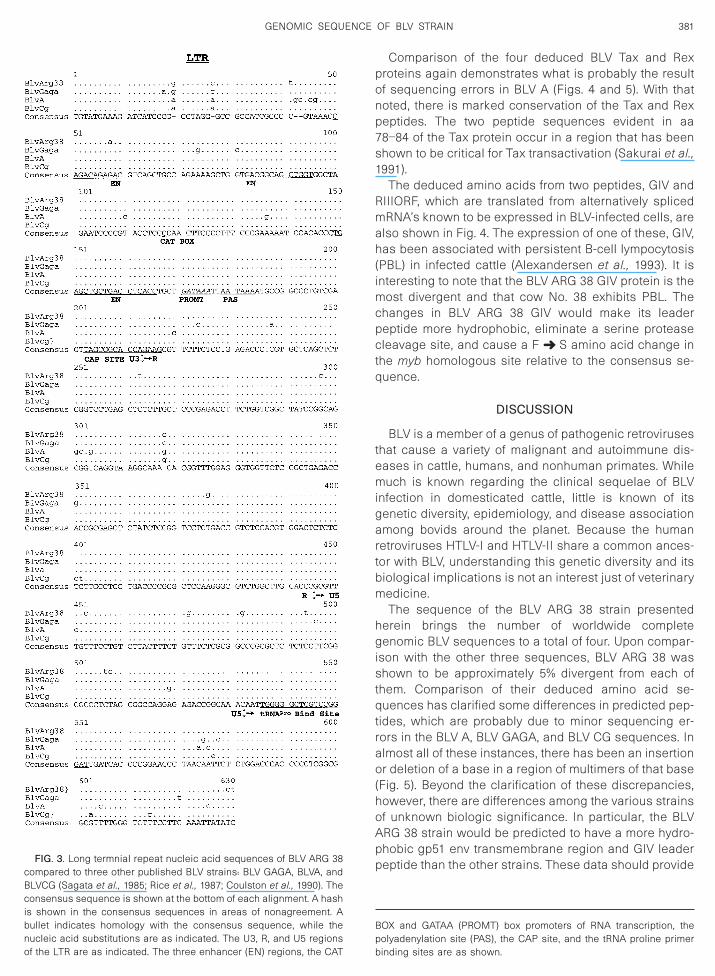
Comparison of the LTR of BLV ARG 38 with that of theother full-length BLV sequences is shown in Fig. 3. Itcontains the RNA transcription promoter and enhancerelements, polyadenylation signal, and REX response el-ement typical of BLV. The only change of possible con-
sequence in BLV ARG 38 is a G ➜ A at base 58 in the0042-6822/00 $35.00Copyright © 2000 by Academic PressAll rights of reproduction in any form reserved.

asiprc
f nd min
380 DUBE ET AL.
middle of the first enhancer element. The tRNA prolineprimer binding site is also absolutely conserved.
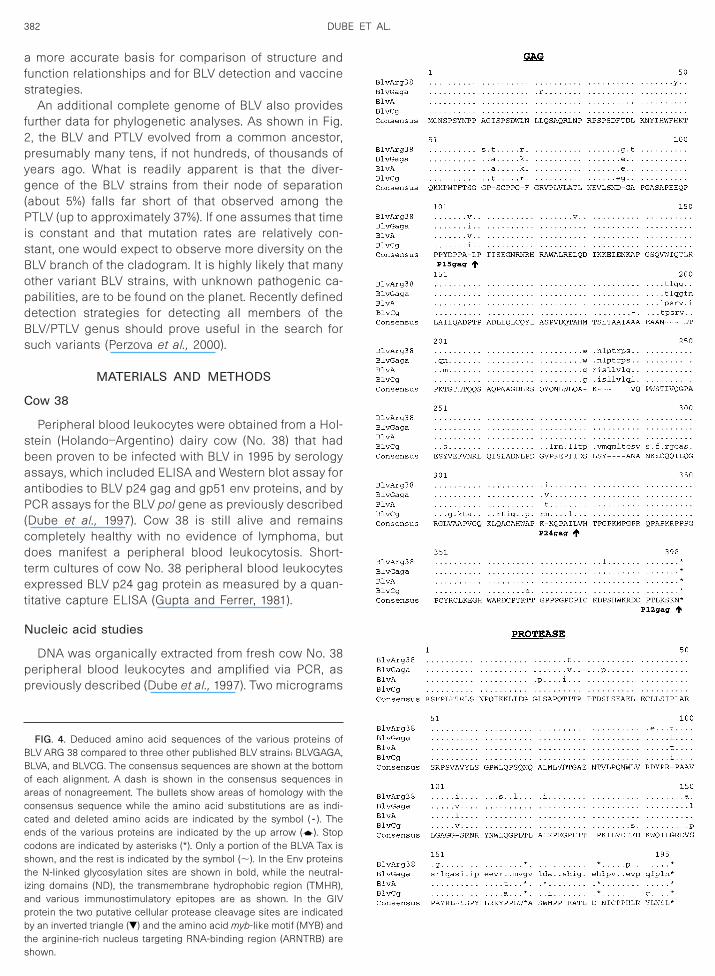
While the BLV gag region is highly conserved, it ispparent (Fig. 4) that there are two different peptideequences preferred for p15 and p24. The BLV CG strain
s markedly divergent over the last 40 amino acids of its24 protein. Whether this is a real observation or the
esult of sequencing error (Fig. 5) and whether thesehanges have functional consequences are unknown.
FIG. 1. A diagram of BLV proviral DNA showing its various functionathe BLV A isolate from Australia (Coulston et al., 1990). They representurther below the diagram indicate the bases of the overlapping plus- a
FIG. 2. A maximum parsimony distance tree comparing the phylo-genetic relationships of approximately 4.2 kb of gag and pol sequencesof the various BLV and PTLV strains shown. The relative distances areshown on the x axis and the bootstrap values for those branches areas indicated. The bar in the lower-left-hand corner shows a lengthequal to 500 unique base changes. It is equivalent to an actual se-
quence divergence of approximately 12%.However, this is a highly conserved, immunodominantregion in the BLV/PTLV genus, suggesting that such achange would have fundamental biological conse-quences (Rice et al., 1987; Perzova et al., 2000). Thededuced protease proteins were also highly conservedamong the four BLV strains (Fig. 4), except for the factthat the published sequence of BLV GAGA contains aninsertion that causes a frame shift at amino acid (aa) 150and eliminates a stop codon at amino acid 169 (Fig. 5).Again, it is highly likely that this difference may be due toa sequencing error in BLV GAGA. The deduced RNase,reverse transcriptase, and integrase amino acids en-coded by the pol gene of all four BLV strains are highlyconserved (Fig. 4), with there being again two slightlydifferent peptide sequences among the four strains.
The env leader peptides and the transmembrane gp30env proteins of all four strains are also highly conserved,as are the gp51 surface env proteins of three of thestrains (Fig. 4). BLV ARG 38, however, demonstratessignificant divergence at the carboxyl terminus of itsgp51 env protein. This is the transmembrane hydropho-bic region of gp51, believed to be responsible for anchor-ing the surface env protein in the viral membrane (Kett-man et al., 1994). Hydrophobicity plots (data not shown)indicate that this region of BLV ARG 38 would be morehydrophobic than the other three strains and theoreti-cally more stably embedded in the viral envelope.
All N-linked glycosylation sites and cysteine residuesin both env proteins are conserved, as are importantfunctional domains, including the putative cell surfacereceptor binding sites and neutralizing domains on thesurface gp51 env protein, the peptide region in gp51 thatinduces a CD81 cytotoxic T-cell response in the hostcow, the highly immunogenic epitope GD21 that is con-served in all members of the PTLV/BLV genus, the tet-rapeptide WAPE (aa 222–225 in Fig. 4) that has beenshown to be critical for infection, and the residues P andD (aa 210 and 211 in Fig. 4) that have been shown toinduce T-helper proliferative responses in cattle (Kett-
tructural genes. The bases shown directly above and below are fromd or beginning of important regions of the BLV genome. The numbersus-strand oligonucleotide primers used to amplify the BLV Arg38 DNA.
l and sthe en
mann et al., 1994; Perzova et al., 2000; Rice et al., 1987).
imcpctq
temigartbm
hgistqtrao(hoApp
of the LTR are as indicated. The three enhancer (EN) regions, the CAT
381GENOMIC SEQUENCE OF BLV STRAIN
Comparison of the four deduced BLV Tax and Rexproteins again demonstrates what is probably the resultof sequencing errors in BLV A (Figs. 4 and 5). With thatnoted, there is marked conservation of the Tax and Rexpeptides. The two peptide sequences evident in aa78–84 of the Tax protein occur in a region that has beenshown to be critical for Tax transactivation (Sakurai et al.,1991).
The deduced amino acids from two peptides, GIV andRIIIORF, which are translated from alternatively splicedmRNA’s known to be expressed in BLV-infected cells, arealso shown in Fig. 4. The expression of one of these, GIV,has been associated with persistent B-cell lympocytosis(PBL) in infected cattle (Alexandersen et al., 1993). It isnteresting to note that the BLV ARG 38 GIV protein is the
ost divergent and that cow No. 38 exhibits PBL. Thehanges in BLV ARG 38 GIV would make its leadereptide more hydrophobic, eliminate a serine proteaseleavage site, and cause a F ➜ S amino acid change in
he myb homologous site relative to the consensus se-uence.
DISCUSSION
BLV is a member of a genus of pathogenic retroviruseshat cause a variety of malignant and autoimmune dis-ases in cattle, humans, and nonhuman primates. Whileuch is known regarding the clinical sequelae of BLV
nfection in domesticated cattle, little is known of itsenetic diversity, epidemiology, and disease associationmong bovids around the planet. Because the human
etroviruses HTLV-I and HTLV-II share a common ances-or with BLV, understanding this genetic diversity and itsiological implications is not an interest just of veterinaryedicine.The sequence of the BLV ARG 38 strain presented
erein brings the number of worldwide completeenomic BLV sequences to a total of four. Upon compar-
son with the other three sequences, BLV ARG 38 washown to be approximately 5% divergent from each of
hem. Comparison of their deduced amino acid se-uences has clarified some differences in predicted pep-
ides, which are probably due to minor sequencing er-ors in the BLV A, BLV GAGA, and BLV CG sequences. Inlmost all of these instances, there has been an insertionr deletion of a base in a region of multimers of that base
Fig. 5). Beyond the clarification of these discrepancies,owever, there are differences among the various strainsf unknown biologic significance. In particular, the BLVRG 38 strain would be predicted to have a more hydro-hobic gp51 env transmembrane region and GIV leadereptide than the other strains. These data should provide
BOX and GATAA (PROMT) box promoters of RNA transcription, thepolyadenylation site (PAS), the CAP site, and the tRNA proline primer
FIG. 3. Long termnial repeat nucleic acid sequences of BLV ARG 38compared to three other published BLV strains: BLV GAGA, BLVA, andBLVCG (Sagata et al., 1985; Rice et al., 1987; Coulston et al., 1990). Theconsensus sequence is shown at the bottom of each alignment. A hashis shown in the consensus sequences in areas of nonagreement. Abullet indicates homology with the consensus sequence, while thenucleic acid substitutions are as indicated. The U3, R, and U5 regions
binding sites are as shown.
(cdtet
N
pp
382 DUBE ET AL.
a more accurate basis for comparison of structure andfunction relationships and for BLV detection and vaccinestrategies.
An additional complete genome of BLV also providesfurther data for phylogenetic analyses. As shown in Fig.2, the BLV and PTLV evolved from a common ancestor,presumably many tens, if not hundreds, of thousands ofyears ago. What is readily apparent is that the diver-gence of the BLV strains from their node of separation(about 5%) falls far short of that observed among thePTLV (up to approximately 37%). If one assumes that timeis constant and that mutation rates are relatively con-stant, one would expect to observe more diversity on theBLV branch of the cladogram. It is highly likely that manyother variant BLV strains, with unknown pathogenic ca-pabilities, are to be found on the planet. Recently defineddetection strategies for detecting all members of theBLV/PTLV genus should prove useful in the search forsuch variants (Perzova et al., 2000).
MATERIALS AND METHODS
Cow 38
Peripheral blood leukocytes were obtained from a Hol-stein (Holando–Argentino) dairy cow (No. 38) that hadbeen proven to be infected with BLV in 1995 by serologyassays, which included ELISA and Western blot assay forantibodies to BLV p24 gag and gp51 env proteins, and byPCR assays for the BLV pol gene as previously describedDube et al., 1997). Cow 38 is still alive and remainsompletely healthy with no evidence of lymphoma, butoes manifest a peripheral blood leukocytosis. Short-
erm cultures of cow No. 38 peripheral blood leukocytesxpressed BLV p24 gag protein as measured by a quan-
itative capture ELISA (Gupta and Ferrer, 1981).
ucleic acid studies
DNA was organically extracted from fresh cow No. 38eripheral blood leukocytes and amplified via PCR, asreviously described (Dube et al., 1997). Two micrograms
FIG. 4. Deduced amino acid sequences of the various proteins ofBLV ARG 38 compared to three other published BLV strains: BLVGAGA,BLVA, and BLVCG. The consensus sequences are shown at the bottomof each alignment. A dash is shown in the consensus sequences inareas of nonagreement. The bullets show areas of homology with theconsensus sequence while the amino acid substitutions are as indi-cated and deleted amino acids are indicated by the symbol ( - ). Theends of the various proteins are indicated by the up arrow (d). Stopcodons are indicated by asterisks (*). Only a portion of the BLVA Tax isshown, and the rest is indicated by the symbol (;). In the Env proteinsthe N-linked glycosylation sites are shown in bold, while the neutral-izing domains (ND), the transmembrane hydrophobic region (TMHR),and various immunostimulatory epitopes are as shown. In the GIVprotein the two putative cellular protease cleavage sites are indicatedby an inverted triangle (�) and the amino acid myb-like motif (MYB) andthe arginine-rich nucleus targeting RNA-binding region (ARNTRB) are
shown.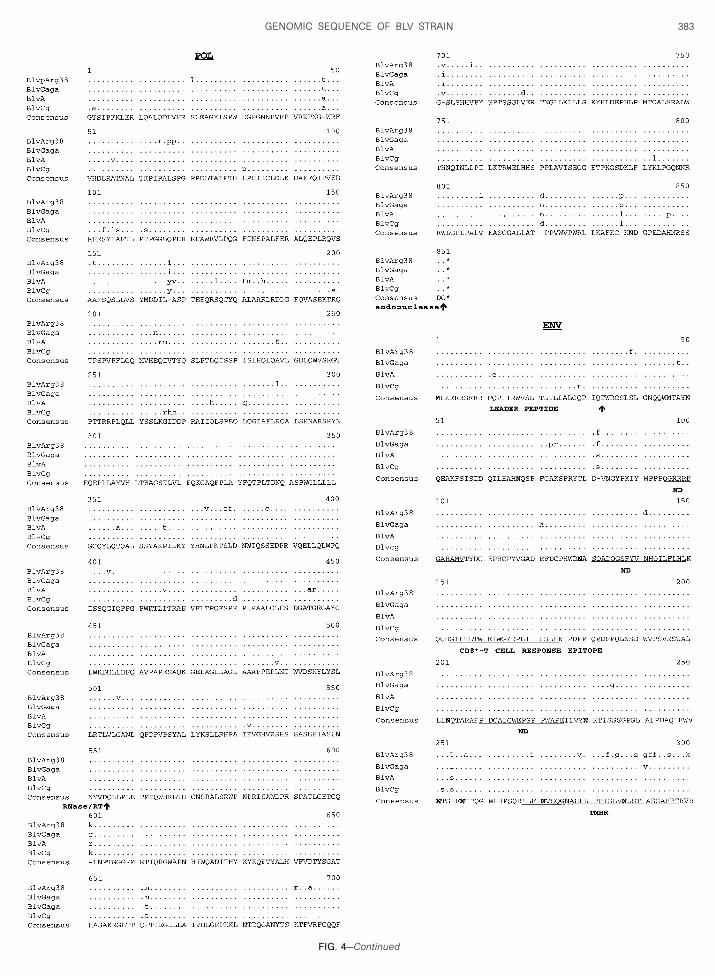
FIG. 4—Conti
383GENOMIC SEQUENCE OF BLV STRAIN
nued

384 DUBE ET AL.
FIG. 4—Conti
nued
B
B
wrA
385GENOMIC SEQUENCE OF BLV STRAIN
of each DNA sample was amplified with one of theoverlapping BLV primer sets shown in Fig. 1. The ampli-fied products were detected by Southern blot hybridiza-tion using 32P-labeled oligonucleotide probes locatedbetween the flanking primers. Amplified specific prod-ucts were cloned into a TA cloning vector (Invitrogen,San Diego, CA) and sequenced using an automatedsequencer (Applied Biosystems, Foster City, CA). Severalclones were sequenced for each primer pair and se-quences were obtained for both strands of DNA. Theoverlapping sequences were used to obtain the BLV ARG38-specific regions homologous to the primers and todouble check the accuracy of the sequences. Sequenceswere aligned (Needleman and Wunsch, 1970), and amaximum parsimony program (Swofford, 1991) was usedto compare new and previously published retroviral se-quences. Both distance and bootstrap (100 replications)trees were generated. The other strains included BLV A;BLV GAGA; BLV CG; HTLV-I ATK; HTLV-I ATL; HTLV-I EL;STLV-I TAN 90; STLV-I MACA; HTLV-II MOT; HTLV-II
FIG. 5. Alignments of nucleic acids within the p24 gag, protease, andTax/Rex regions of the BLV genome that contain deletions and/orinsertions that distort the reading frames in some of the BLV strains(see Fig. 4). The numbers at the top of the sequences indicate theterminal bases according to the BLV A sequence. Conserved se-quences are indicated by a bullet (●) and deletions are indicated by ahyphen ( - ). The reading frames are shown for the BLV Arg38 strain,
hich we believe to be the correct reading frame (Fig. 4). The Taxeading frame, which is as shown, overlaps with the Rex reading frame.s can be seen, a deletion of a C at base 1441 in the BLV Cg strain
causes a frame shift in the translation of gag p24. Through a series ofsubsequent deletions and insertions, the sequence returns to theproper reading frame by base 1566. Similarly, BLV Gaga contains anextra base in its protease gene, which distorts its reading frame relativeto the other three strains. Finally, BLV A contains an inserted A afterbase 7474, which alters both the Tax and Rex reading frames.
KAYAPO; HTLV-II NRA; HTLV-II G2; STLV-II; STLV-II pp;
and PTLV-L; the sequences of these were obtained fromGenBank.
The number of each unique base of each PTLV-I,PTLV-II, and PTLV-L strain from node A (Fig. 2) wascalculated, and the means and standard deviations ofthe PTLV-I vs the PTLV-II and the PTLV-L distances werecalculated and analyzed statistically using the Student ttest. Hydrophobicity plots of the BLV ARG 38 and theconsensus gp51 env proteins were generated using theNetwork Protein Sequence @ nalysis program (Pole Bio-Informatique Lyonnais).
ACKNOWLEDGMENTS
This work was supported by the Barbara Kopp Cancer ResearchFund, Comision de Investigaciones Cientificas de la Provincia deBuenos Aires (CIC) Resolucion N 1611/98, and Secretaria de Ciencia yTecnica de la Universidad Nacional del Centro de la Provincia deBuenos Aires (SECYT) Ord. N 2298/98.
REFERENCES
Alexandersen, S., Carpenter, S., Christensen, J., Storgaard, T., Viuff, B.,Wannemuehler, Y., Belousov, J., and Roth, J. A. (1993). Identification ofalternatively spliced mRNAs encoding potential new regulatory pro-teins in cattle infected with bovine leukemia virus. J. Virol. 67(1),39–52.
renner, J., Van Hamm, M., Savir, D., and Trainin, Z. (1989). The implicationof BLV infection in the productivity, reproductive capacity and survivalrate of a dairy cow. Vet. Immunol. Immunopathol. 22, 299–305.
urny, A., Cleuter, Y., Kettman, R., Mammericx, M., Marbaix, G., Porte-telle, D., Van den Broeke, A., Willems, L., and Thomas, R. (1990).Bovine leukemia: Facts and hypotheses derived from the study of aninfectious cancer. In “Retrovirus Biology and Human Disease” (R.Gallo and F. Wong-Staal, Eds.), pp. 9–32. Dekker, New York.
Coulston, J., Naif, H., Brandon, R., Kumar, S., Khan, S., Daniel, R. C., andLavin, M. F. (1990). Molecular cloning and sequencing of an Austra-lian isolate of proviral bovine leukaemia virus DNA: Comparison withother isolates. J. Gen. Virol. 71, 1737–1746.
Dube, S., Bachman, S., Spicer, T., Love, J., Choi, D., Esteban, E., Ferrer,J. F., and Poiesz, B. J. (1997). Degenerate and specific PCR assays forthe detection of bovine leukaemia virus and primate T cell leukae-mia/lymphoma virus pol DNA and RNA: Phylogenetic comparisons ofamplified sequences from cattle and primates from around the world.J. Gen. Virol. 78, 1389–1398.
Ferrer, J. F. (1980). Bovine lymphosarcoma. Adv. Vet. Sci. Comp. Med. 24,1–68.
Gupta, P., and Ferrer, J. F. (1981). Comparison of various serological anddirect methods for the diagnosis of BLV infection in cattle. Cancer 20,179–184.
Kettman, R., Burny, A., Callebaut, I., Droogmans, L., Mammericx,Wilems, L., and Portetelle, D. (1994). Bovine leukemia virus. In “TheRetroviridae, Volume 3” (J. Levy, Ed.), pp 39–81. Plenum, New York.
Needleman, S. B., and Wunsch, C. D. (1970). A general method appli-cable to the search for similarities in the amino acid sequence of twoproteins. J. Mol. Biol. 48, 443–453.
Network Protein Sequence @ nalysis. Poe Bio-Informatique Lyonnais.World Wide Web (URL: http://pbil.ibcp.fr/cgi-in/primanal_pcpnof.pl).
Perzova, R. N., Loughran, T. P., Dube, S., Ferrer, J., Esteban, E., andPoiesz, B. J. (2000). Lack of BLV and PTLV DNA sequences in themajority of patients with large granular lymphocyte leukemia. Br. J.Haematol. 109, 64–70.
Poiesz, B. J. (1995). Etiology of acute leukemia: Molecular genetics and
viral oncology. In “Neoplastic Diseases of the Blood” (P. H. Wiernik,
386 DUBE ET AL.
G. P. Canellos, J. P. Dutcher, and R. A. Kyle, Eds.), 3rd ed., pp. 159–175.Churchill–Livingstone, New York.
Rice, N. R., Stephens, R. M., and Gilden, R. V. (1987). Sequence analysisof the bovine leukemia virus genome. In “Enzootic Bovine Leukosisand Bovine Leukemia Virus” (A. B. a. M. Mammerichx, Ed.), pp.115–144. Nijhoff, Boston.
Sagata, N., Yasunaga, T., Tsuzuku-Kawamura, J., Ohishi, K., Ogawa, Y.,and Ikawa, Y. (1985). Complete nucleotide sequence of the genomeof bovine leukemia virus: Its evolutionary relationship to other retro-
viruses. Proc. Natl. Acad. Sci. USA 82, 677–681.Sakurai, M., Taneda, A., Nagoya, H., and Sekikawa, K. (1991). Con-struction and functional characterization of mutants of the bovineleukaemia virus trans-activator protein p34tax. J. Gen. Virol. 72,2527–2531.
Swofford, D. L. (1991). “PAUP: Phylogenetic Analysis Using Parsimony,”Version 3.05. Illinois Natural History Survey, Champaign.
Trono, K., Perez-Filgueira, D., Castelli, M., Lager, I., Duffy, S., Borca,M. V., and Camillo, C. (1999). “Virus de la Leucosis Bovina: Epide-
miologia en Argentina.” VI Congresso Argentino de Virologia.



















