
Simplifying Fatty Acid Analyses in Multicomponent Foods with ...
Upload
independentCategory
view
17download
0
Tailored fatty acid synthesis via dynamic controlof fatty acid elongationJoseph P. Torellaa,1, Tyler J. Forda,1, Scott N. Kima, Amanda M. Chena, Jeffrey C. Wayb, and Pamela A. Silvera,b,2
aDepartment of Systems Biology, Harvard Medical School, Boston, MA 02115; and bWyss Institute for Biologically Inspired Engineering, Harvard University,Boston, MA 02115
Edited by Benjamin F. Cravatt, The Scripps Research Institute, La Jolla, CA, and accepted by the Editorial Board May 30, 2013 (received for reviewApril 15, 2013)
Medium-chain fatty acids (MCFAs, 4–12 carbons) are valuable as
precursors to industrial chemicals and biofuels, but are not canon-
ical products of microbial fatty acid synthesis. We engineered mi-
crobial production of the full range of even- and odd-chain–length
MCFAs and found that MCFA production is limited by rapid, irre-
versible elongation of their acyl-ACP precursors. To address this
limitation, we programmed an essential ketoacyl synthase to de-
grade in response to a chemical inducer, thereby slowing acyl-ACP
elongation and redirecting flux from phospholipid synthesis to
MCFA production. Our results show that induced protein degrada-
tion can be used to dynamically alter metabolic flux, and thereby
increase the yield of a desired compound. The strategy reported
herein should be widely useful in a range of metabolic engineering
applications in which essential enzymes divert flux away from
a desired product, as well as in the production of polyketides,
bioplastics, and other recursively synthesized hydrocarbons for
which chain-length control is desired.
biochemicals | synthetic biology
Long-chain (16–18 carbon) fatty acids are required to synthe-size the essential lipids that maintain bacterial membrane
integrity (1). In-depth genetic and biochemical understanding offatty acid synthesis in bacteria (2, 3) has made it possible toengineer microbes for increased fatty acid production, and cat-alyzed efforts to industrialize the process (4). As fatty acids canbe transformed into a variety of biofuels and industrially usefuloleo-chemicals, engineered microbes are a promising renewablealternative to petroleum-based chemical feedstocks (4–8).Recent efforts have focused on engineering Escherichia coli
fatty acid synthesis for production of free fatty acids (FFAs)(reviewed in ref. 4), alcohols (5), esters (6), and alkanes (7).Although most such studies have focused on long-chain (C14–C18) compounds because of their natural abundance, medium-chain fatty acids (MCFAs, 4–12 carbons), which are derived fromlow-abundance acyl-ACPs, are nevertheless valuable industrialchemicals (9–12), and their shorter chain lengths are associatedwith improved fuel quality (8, 13). Tailoring chain-length speci-ficity in fatty-acid–producing microbes is therefore an importantchallenge in the production of renewable chemicals and biofuels.Chain-length control is exerted at many steps throughout fatty
acid synthesis. Fatty acid synthesis begins with the condensationof acetyl-CoA and malonyl-ACP by the ketoacyl synthase (KAS)FabH to yield acetoacetyl-ACP, a four-carbon β-ketoacyl-ACPthat is reduced to yield a four-carbon acyl-ACP (Fig. 1A). Insubsequent rounds of fatty acid synthesis, this acyl-ACP is con-densed with additional malonyl-ACP molecules by long-chainKAS enzymes FabB and FabF, then reduced to an acyl-ACP withtwo additional carbons. In this way, E. coli fatty acid synthesisbuilds acyl-ACPs two carbons at a time, in a recursive processyielding a range of even-chain–length acyl-ACPs (2). Odd-chainacyl-ACPs can also be produced when cellular propionyl-CoAlevels are high, because of their incorporation in place of acetyl-CoA in the initial step of fatty acid synthesis (14–16) (Fig. 1A).In part because of KAS chain-length specificity, acyl-ACP
elongation ends with the production of long-chain (16–18 car-bon) acyl-ACPs, which are used by the cell to synthesize mem-brane lipids (reviewed in ref. 2).Engineered fatty acid production in E. coli is typically achieved
by expressing a cytosolic derivative of the E. coli thioesteraseTesA (5), which hydrolyzes long-chain acyl-ACPs to yield long-chain free fatty acids (Fig. 1A) (17). High yields are a result ofboth the natural abundance of long-chain acyl-ACPs in the celland thioesterase-mediated depletion of the long chain acyl-ACPpool, which feedback-inhibits upstream enzymes in fatty acidsynthesis, such as FabH (16, 17). Although MCFAs can be pro-duced by expressing thioesterases with substrate specificity formedium-chain acyl-ACPs (5, 18–21), yields are generally lowerthan for long-chain fatty acids, likely because medium-chain acyl-ACPs are not abundant (22, 23) and their depletion does not re-solve an existing buildup of inhibitory long-chain acyl-ACPs.In this work, we engineered E. coli to produce free fatty acids
with all even and odd chain lengths from 4 to 13 carbons, andrationally modified fatty acid synthesis to favor the production ofmedium-chain acyl-ACPs. Treatment of MCFA-producing cellswith the KAS inhibitor cerulenin (22) increased yields, demon-strating that MCFA production is limited by overly rapid acyl-ACP elongation. To test whether this strategy could be im-plemented genetically to increase the production of a specificMCFA, octanoic acid, we replaced the KAS FabF with a mutantincapable of elongating beyond eight carbons (24). We then en-gineered FabB to degrade in response to a chemical inducer(25), such that elongation beyond C8 acyl-ACP could be slowedon-demand, and flux redirected from lipid synthesis to octanoateproduction. These interventions increased octanoate yield anddemonstrated the utility of inducible degradation to degrade es-sential genes and redirect metabolic flux (26). Our results dem-onstrate that altering the chain-length specificity of microbial fattyacid synthesis requires concerted changes in both the thioesteraseand fatty acid synthesis machinery itself, and suggest strategies forthe production of intermediate-length products in other recursivebiosynthetic systems (27, 28).
Results
Our strategy for engineering MCFA synthesis was to first dem-onstrate the production of MCFAs with all even and odd chainlengths from 4 to 13 carbons, then identify factors that limitMCFA yield. After finding that elongation rates were too rapidfor optimal MCFA production, we genetically engineered fatty
Author contributions: J.P.T., T.J.F., J.C.W., and P.A.S. designed research; J.P.T., T.J.F., S.N.K.,
and A.M.C. performed research; J.P.T., T.J.F., S.N.K., A.M.C., J.C.W., and P.A.S. analyzed
data; and J.P.T., T.J.F., J.C.W., and P.A.S. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. B.F.C. is a guest editor invited by the
Editorial Board.
1J.P.T. and T.J.F. contributed equally to this work.
2To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.
1073/pnas.1307129110/-/DCSupplemental.
11290–11295 | PNAS | July 9, 2013 | vol. 110 | no. 28 www.pnas.org/cgi/doi/10.1073/pnas.1307129110

acid synthesis to slow elongation in response to a chemical in-ducer, and thereby direct fatty acid synthesis toward the pro-duction of a specific MCFA, octanoic acid. Finally, we tested a setof knockout mutants for their ability to increase carbon flux intofatty acid synthesis and maximize yields in our engineered strain.
Comprehensive Production of all Even- and Odd-Chain MCFAs by
Engineered E. coli. We engineered production of MCFAs with4–13 carbons in E. coli by varying the expressed thioesterase andadding propionate to the culture medium. Three thioesterasescapable of hydrolyzing a range of medium-chain acyl-ACPs werechosen: BfTES [EET61113 from Bryantella formatexigens (21),CpFatB1 (20), and UcFatB2 (18)]. Strain S001 [BL21*(DE3)∆fadD] (Table S1) was transformed with plasmids encodingcodon-optimized versions of each thioesterase (pEET, pTJF010,and pJT208, respectively), grown in M9 + 0.5% glycerol, andinduced with isopropyl-β-D-thiogalactopyranoside (IPTG) for24 h (Materials and Methods). GC-MS analysis of free fatty acidsshowed that thioesterase expression increased the total moles offatty acid produced (Fig. S1), and that each even-chain fatty acidfrom 4 to 12 carbons was produced by at least one thioesterase,with BfTES, CpFatB1, and UcFatB2 producing primarily buta-noic, octanoic, and dodecanoic acids, respectively (Fig. 1B andFig. S1). Propionate supplementation has been shown to causeodd-chain fatty acid production via incorporation of propionyl-CoA into the initial step of fatty acid synthesis (14, 19) (Fig. 1A).Addition of 100 mM propionate to the culture medium causedall three thioesterases to produce odd-chain fatty acids, witheach odd-chain fatty acid in the 5–13 carbon range produced byat least one thioesterase (Fig. 1B and Fig. S1). These experi-ments demonstrated that the full range of 4–13 carbon MCFAscould be produced by engineered E. coli.Odd-chain MCFAs could be produced without the need for
propionate supplementation by genetically encoding a propionyl-
CoA production pathway. Building on recent work in whichendogenous propionyl-CoA production was engineered to syn-thesize a series of valuable small molecules (15, 29), we trans-formed S001-pJT208 with plasmid pCOLA-thrAfrBCilvAfr (15),which produces propionyl-CoA primarily by increasing flux throughthe isoleucine biosynthetic pathway (Fig. 1A). Strain S001-pJT208-pCOLA-thrAfrBCilvAfr produced the odd-chain fatty acids unde-canoate and tridecanoate from glycerol as a sole carbon source(Fig. 1C), with similar selectivity to S001-pJT208 with addedpropionate (Fig. 1 B, iii). Thus, E. coli can be engineered to pro-duce both even- and odd-chain fatty acids from a single carbonsource, without the need for propionate supplementation.
Wild-Type Fatty Acid Elongation Rates Are Nonoptimal for MCFA
Production. One challenge in MCFA production is the low con-centration of medium-chain acyl-ACPs in growing cells (22, 23).Treatment of E. coli with cerulenin, an antibiotic that targets theKAS enzymes FabB and FabF, inhibits acyl-ACP elongationwithout inhibiting the initial condensing enzyme FabH (Fig. 2A)and causes accumulation of medium-chain acyl-ACPs in vivo(22). This accumulation is likely a result of two factors: (i) de-creased elongation of medium-chain acyl-ACPs to long-chainacyl-ACPs; and (ii) the resulting decrease in long-chain acyl-ACPs, which would relieve feedback inhibition of FabH (16, 17)and increase flux into fatty acid synthesis (Fig. 2A). We hy-pothesized that there should be an optimal level of cerulenin atwhich elongation is slow enough to increase the concentration ofmedium-chain acyl-ACPs and rate of MCFA production, but notso slow that elongation itself becomes rate-limiting.Adding cerulenin to MCFA-producing cultures demonstrated
that FFA yields produced by the shorter-chain thioesterasesBfTES and CpFatB1 could be increased by inhibiting elongation(Fig. 2B). Yield increases came primarily from MCFAs ratherthan other fatty acids, because long-chain fatty acids were
Fig. 1. Engineering production of all even- and odd-length MCFAs in E. coli. (A) The KAS FabH elongates acetyl-CoA to form a four-carbon β-ketoacyl ACP,
which is reduced to C4 acyl-ACP. In subsequent rounds of fatty acid synthesis, the KAS’s FabB and FabF elongate acyl-ACPs two carbons at a time to yield
a range of even-chain–length fatty acyl-ACPs. Incorporation of propionyl-CoA in place of acetyl-CoA causes production of odd-chain acyl-ACPs; propionyl-CoA
can be produced from propionate (prpE) or from expression of a genetic cassette that increases flux through the isoleucine pathway (thrAfrBC ilvAfr) (15).
Acyl-ACPs can be hydrolyzed to FFAs by an appropriate thioesterase (TE). Fatty acid degradation can be blocked by knocking out the β-oxidation enzymes
fadD or fadE. (B) GC-MS analysis of FFA production by strain S001 [BL21*(DE3) ∆fadD] containing plasmid pEET (BfTES) (i), pTJF010 (CpFatB1) (ii), or pJT208
(UcFatB2) (iii) in M9 +0.5% glycerol alone (solid bars) or supplemented with 100 mM propionate (striped bars) 24 h after IPTG induction (n = 3, error bars =
SEM). FFAs shown for the no-propionate experiments accounted for 67% (i), 85% (ii) and 72% (iii) of total FFAs (full chain-length profiles in Fig. S1). FFAs
shown for the propionate experiments accounted for (i) 79%, (ii) 65%, and (iii) 84% of the total (Fig. S1). (C) Production of odd-chain FFAs by S001-pJT208-
pCOLAthrAfrBCilvAfr (n = 3, error bars = SEM). FFAs shown accounted for 81% of total. Asterisks in B and C indicate significantly increased odd-chain pro-
duction (P < 0.05, one-tailed Student t test). (D) Final OD595 of strains in this figure, with or without propionate supplementation (n = 3, error bars = SEM).
Asterisks indicate decreased OD595 compared with strain S001 alone (P < 0.05, one-tailed Student t test).
Torella et al. PNAS | July 9, 2013 | vol. 110 | no. 28 | 11291
APPLIEDBIOLO
GICAL
SCIENCES

detected at < 12 mg/L in all samples, and biomass generally didnot change with cerulenin concentration (Fig. S2). Ceruleninalso increased the MCFA production rate (Fig. S3A). Given thatcerulenin has been previously shown to increase the concentra-tion of medium-chain acyl-ACPs (22), and assuming the MCFAproduction rate increases with the concentration of its substrateacyl-ACPs, one possible explanation of this result is that theincreased MCFA production rate is because of an increasedcellular concentration of medium-chain acyl-ACPs. These datasuggested that wild-type fatty acid elongation rates are notoptimal for the production of MCFAs in our strains and dem-onstrated, counterintuitively, that inhibiting fatty acid synthesiscan increase the production of fatty acids.The maximal increase in FFA yield was greater for shorter
chain-length–specific thioesterases, as was the amount of cer-ulenin needed to achieve it (Fig. 2C); this is consistent with amodel in which the distribution of acyl-ACPs is a function ofelongation rate, with slower elongation rates yielding shorteracyl-ACP pools and enhancing production of shorter fatty acids.GC-MS analysis offered further evidence for this model. First,cerulenin treatment caused all three thioesterases to shift theirproduction toward shorter products (Fig. 2D), which is mosteasily explained by an increase in the fraction of shorter acyl-ACPs in the cell. Second, although cerulenin decreased total andC12-specific FFA yields in the UcFatB2 strain, it increased theyield of C10 FFAs (Fig. S4), suggesting differential effects ofcerulenin on shorter and longer acyl-ACP pools.These results suggest that BfTES and CpFatB1’s FFA yields
increase in response to cerulenin treatment because of a shiftof the acyl-ACP distribution toward shorter (4–10 carbon) acyl-ACPs, which are more effectively hydrolyzed by these thio-esterases; FFA production by UcFatB2, however (which prefersC12 acyl-ACPs), may fail to increase for the same reason. Theseapparent shifts in the acyl-ACP distribution are consistent withthe expectation that cerulenin would simultaneously increase therate at which acyl-ACPs are initiated by FabH and decrease therate at which they elongate, and with previous results demon-strating that medium-chain acyl-ACPs accumulate in response tocerulenin treatment (22). In all, these results suggest that in-hibiting fatty acid elongation increases the concentration of
medium-chain acyl-ACPs with ≤ 10 carbons, a useful strategy forincreasing MCFA yield.
Targeted Genetic Production of MCFAs via Inducible Degradation.
Having shown that inhibiting elongation can increase MCFAyield, we attempted to engineer production of a specific MCFA,octanoic acid, by implementing two genetic interventions: (i)replacing FabF with a mutant, FabF*, incapable of elongatingbeyond C8 because of the insertion of a bulky phenylalanine intoits fatty acid binding pocket (24); and (ii) engineering FabB witha C-terminal SsrA DAS+4 tag, enabling inducible degradationby the E. coli ClpXP system via overexpression of the adaptorprotein SspB (25, 30) (Fig. 3A). The inducible degradation sys-tem was necessary because fabB is required for unsaturated fattyacid synthesis, and is essential for growth on minimal media (31).We hypothesized that FabF* would allow high rates of octanoyl-ACP production, and that inducing degradation of FabB wouldslow elongation beyond C8 acyl-ACP, similar to the effect ofcerulenin. This process should cause octanoyl-ACP to accumu-late and increase fatty acid yields in strains expressing CpFatB1,which primarily produces octanoic acid (Fig. 1B and Fig. S1).Expressing CpFatB1 (pTJF038) and SspB (SspBpET21b)
(25) in strain S003 [BL21*(DE3)ΔfadE fabF*] did not result insignificantly increased FFA yields over those of S002 [BL21*(DE3)ΔfadE] (Fig. 3C), suggesting that the octanoyl-ACP gen-erated by FabF* was not accumulating sufficiently to increaseoctanoate production, likely because of elongation by FabB.We therefore appended an SsrA DAS+4 degradation tag (25)to the endogenous fabB in both S002 and S003, generating strainsS004 [BL21*(DE3) ΔfadE fabBDeg] and S005 [BL21*(DE3)ΔfadE fabF* fabBDeg], respectively. SspB-induced degrada-tion of FabBDeg was demonstrated via Western blot of strainS007, a derivative of S004 in which FabBDeg is appended with anN-terminal strep tag to facilitate detection (Fig. 3B and Fig. S5).Inducing SspB and CpFatB1 expression in strain S005, which
contains both fabF* and fabBDeg, resulted in a significant, 32%increase in FFA yield to 118 mg/L and a 12% decrease in finalOD595 compared with parent strain S002 (Fig. 3C). This findingsuggests that FabB degradation shunts fatty acid synthesis toFabF* and increases the pool of octanoyl-ACP for hydrolysis by
Fig. 2. Chemical inhibition of fatty acid elongation can increase MCFA production. (A) Schematic diagram of fatty acid elongation. FabH performs the slow
initial elongation step in fatty acid biosynthesis to generate C4-ACP; FabB and FabF more rapidly elongate the acyl-ACP 2 carbons at a time to produce 16–18
carbon acyl-ACPs, which are precursors to lipid synthesis and inhibit FabH more strongly than other acyl-ACPs (16, 17). Cerulenin inhibits FabB and FabF, but
not FabH. (B) Total FFA produced by strain S001 expressing the indicated thioesterases over a range of cerulenin concentrations from 0 to 12.8 μg/mL, 24 h
after IPTG induction, as measured by GC-MS (n = 3, error bars = SEM). Long-chain FFAs (C14–C18) accounted for ≤ 12 mg/L in any given measurement.
Asterisks indicate the point at which mean fatty acid production is highest; the increase in production is significant for BfTES and CpFatB1 (P < 0.05, one-tailed
Student t test), but not for UcFatB2. OD595 data are provided in Fig. S2. (C) Filled circles: cerulenin concentration at which fatty acid production is maximal as
a function of each thioesterase’s preferred chain length. C4 for BfTES (blue); C8 for CpFatB1 (green); C12 for UcFatB2 (orange). Filled triangles indicate the
maximum fold-increase in FFA production over the no-cerulenin control (calculated as the ratio of maximal FFA production to that of the no-cerulenin
control). (D) Ratio of the two most abundant FFAs produced by each thioesterase (shorter/longer), as a function of cerulenin concentration (n = 3, error bars =
SEM). Single asterisks indicate that the given bar is significantly different from the no-cerulenin control with P < 0.05 (two-tailed Student t test). Double
asterisks indicate that the given bar is significantly different from both other bars in the panel.
11292 | www.pnas.org/cgi/doi/10.1073/pnas.1307129110 Torella et al.

CpFatB1. Consistent with this, strain S005 (fabF* fabBDeg) hada greater rate of FFA production than strain S003 (fabF* alone)(Fig. S3B). Importantly, because fabBDeg alone (S004) does notsignificantly alter yield or OD595, it is unlikely that yield increasesin the fabF* fabBDeg strain (S005) are because of general effectson growth or fatty acid synthesis caused by SspB synthesis orFabB degradation.
Optimization of the Engineered Strain. We further optimized octa-noic acid production in our engineered strain by: (i) screening aseries of knockout mutations for their ability to increase yield, and(ii) titrating the level of SspB induction to optimize FabB degra-dation and FFA production by CpFatB1.We expressed CpFatB1 in BL21*(DE3) derivatives with knock-
outs in fatty acid degradation (fadD, fadE), fermentation ofundesirable products (pta, poxB, adhE, ldhA, pflB) (3), and thestringent response (relA), which inhibits lipid synthesis duringstarvation (32), and measured the total FFA production. Be-cause strains with fadD and pta showed the greatest improve-ment in FFA among the single-knockout strains (Fig. 4A, green),we constructed all possible double mutants in a ΔfadD back-ground (Fig. 4A, blue), as well as all possible triple mutants ina ΔfadD Δpta background (Fig. 4A, orange). The strain with the
highest yield (ΔfadD Δpta ΔldhA) showed little increase in FFAproduction beyond that of the ΔfadD Δpta double knockout.ΔfadD and Δpta were therefore used to further improve FFAyields in our strains engineered with fabF* and fabBDeg.Our improved strain S006 [BL21*(DE3) ΔfadD Δpta ΔlacY
fabf* fabBDeg], which included a ΔlacY mutation to allow titrat-able IPTG induction (33), increased FFA production ∼twofoldover S005 to 263mg/L (Fig. 4B, solid bars) at the optimal level ofIPTG induction of SspBpET21b and pTJF038 (CpFatB1). GC-MS analysis showed that FFA production by S006 was highly se-lective for octanoate (92.1 ± 0.2%) (Fig. S7), with a > 130 mg/Lincrease in octanoate production over the nonoptimized parentstrain (S002). We estimated from OD595 data that S006 producedonly 15 mg/L less lipid-bound long-chain fatty acid than S002 (SIMaterials and Methods); our genetic interventions therefore in-creased the total moles of fatty acid produced primarily by in-creasing MCFA production.Because IPTG induction controlled both CpFatB1 and SspB
expression, it was unclear whether the increase in yield was aresult of altering CpFatB1 expression, altering degradation, or acombination of the two; we therefore generated a strain in whichwe could induce CpFatB1 and SspB separately. Strain S006 was
Fig. 3. Engineering KAS for increased octanoic acid production. (A) Schematic indicating the elongation reactions carried out by ketoacyl synthases FabB and
FabF. Wild-type FabB and FabF elongate acyl-ACPs with ≥ 4 carbons up to a length of 16–18 carbons. In our engineered system, FabBDeg can elongate up to
16–18 carbons but is degraded by the E. coli ClpXP system upon SspB expression. FabF* can only elongate up to 8 carbons. (B) StrepFabBDeg degradation in
strain S007-SspBpET21b. Strain S007-SspBpET21b was induced (+IPTG, 1 mM) or not (−IPTG) at time 0 and StrepFabBDeg detected via Western blotting and
densitometry at 0 and 8 h (n = 4, error bars = SEM). Loading was normalized to OD600, and reported values are normalized to StrepFabBDeg levels at t = 0 h.
StrepFabBDeg bands are shown below the bar graph; original blots are shown in Fig. S5. An asterisk indicates a significant decrease in StrepFabBDeg between
the ± IPTG samples at 8 h (P < 0.05, one-tailed Student t test). (C) FFA production [as determined by the Free Fatty Acids, Half Micro Test (Roche)] and final
culture OD595 44 h after induction, for strains expressing CpFatB1 and SspB in S002 [BL21*(DE3) ΔfadE] with the indicated modifications (n = 24; error bars =
SEM). FFA production is represented as the percent increase over FFA production in the parent strain (S002). An asterisk indicates a significant increase in FFA
production compared with S002 (P < 0.05 by one-tailed Student t test).
Fig. 4. Optimizing octanoic acid yields via metabolic knockouts and SspB titration. (A) Total FFA production in BL21*(DE3) knockout strains. Each strain was
grown in M9 + 0.5% glucose, CpFatB1 was induced from pTJF010 with 1 mM IPTG for 44 h, and total FFA measured with the Free Fatty Acids, Half Micro Test
(Roche) (n = 3, error bars = SEM). Green, blue, and orange bars indicate single knockouts, double knockouts, and triple knockouts, respectively. Dashed lines
indicate fatty acid production in the parent strains BL21*(DE3), BL21*(DE3) ∆fadD, and BL21*(DE3) ∆fadD ∆pta. An asterisk indicates FFA production sig-
nificantly greater than the parent strain (P < 0.05 by one-tailed Student t test). OD595 for these strains is shown in Fig. S6. (B) FFA production and OD595 as
a function of IPTG concentration (0, 0.125, 0.25, 0.5 or 1.0 mM). CpFatB1 and SspB expressed from pTJF038 and SspBpET21b respectively were cotitrated in
S006 [BL21*(DE3)ΔfadD Δpta ΔlacY fabF* fabBDeg] (solid gray bars) with IPTG for 24 h in M9 + 0.5% glucose (n = 18, error bars = SEM). SspB alone was
titrated with IPTG in S006 expressing SspB from SspBpET21b and CpFatB1 from aTc-inducible pJT255 induced with 400 ng/mL aTc (striped bars, n = 18, error
bars = SEM). An asterisk indicates significantly different FFA production, or altered OD595, versus the 0 mM IPTG control (P < 0.05 by two-tailed Student t test).
Torella et al. PNAS | July 9, 2013 | vol. 110 | no. 28 | 11293
APPLIEDBIOLO
GICAL
SCIENCES

transformed with an anhydrotetracycline (aTc)-inducible, CpFatB1-expressing plasmid (pJT255) and an IPTG-inducible, SspB-expressing plasmid (SspBpET21b). This strain was induced with400 ng/mL aTc, and IPTG titrated from 0 to 1 mM (Fig. 4B,striped bars). FFA yields increased significantly at lower doses ofIPTG (110–166 mg/L) and OD595 decreased, but plateaued be-yond this point. Our highest yield in this strain was 166 mg/L.Although this is lower than the yields achieved with pTJF038 andSspBpET21b, these results nevertheless demonstrate that opti-mizing inducible degradation of FabB can increase yields overthose achieved through more traditional knockout strategies,and that it is important to tune degradation of FabB to achieveoptimal production while limiting effects on cell growth.
Discussion
MCFAs and their derivatives are important precursors to high-quality fuel and industrial molecules (3, 8–12). In this work, weengineered the production of a range of MCFAs, and showedthat their yields increase in response to chemical inhibition offatty acid elongation. By engineering ketoacyl synthases to se-lectively block long-chain acyl-ACP elongation, we demonstratedthat we can substantially increase the yield of a particular MCFA,octanoic acid. In principle, this strategy can be adapted for pro-duction of any MCFA with 4–13 carbons.In addition to even-chain MCFA production, we were able to
produce odd-chain MCFAs, which are industrially useful as plas-ticizers, as herbicides, and in the fragrance industry (11, 12, 34).Odd-chain fatty acid production has previously been achieved, butrequired propionate supplementation (14, 19). Endogenous pro-pionyl-CoA production has also been achieved (15, 29), but not inconjunction with fatty acid production. We synthesized thesestrategies to engineer production of odd-chain MCFAs froma single carbon source in the absence of propionate. Importantly,this result was achieved using thioesterases known only to produceeven-chain fatty acids, and some thioesterases produced oddchains better than others. Among the wide range of known even-chain thioesterases (21), some may therefore be capable of ef-fectively or even selectively producing odd-chain fatty acids.An important finding of our work was that application of the
KAS inhibitor cerulenin increased the yield of some MCFAs, sug-gesting that the optimal rate of acyl-ACP elongation for MCFAproduction is slower than the wild-type elongation rate. Ourresults suggest that this yield increase is because of accumulationof medium-chain acyl-ACPs, which may be a consequence ofKAS inhibition both (i) slowing loss of medium-chain acyl-ACPsto elongation, and (ii) indirectly increasing the rate of new acyl-ACP synthesis by FabH. Although the goal of metabolic engi-neering is often to increase flux through a naturally occurringpathway, our results suggest that in recursive systems like fattyacid synthesis, intermediate-length products may be producedmore efficiently by slowing the native pathway.We demonstrated that we could genetically engineer KASs to
inhibit unwanted elongation of medium-chain acyl-ACPs, andthereby increase the yield of a specific MCFA, octanoic acid.Although a large body of work has been amassed on attempts toincrease flux to fatty acid precursors (8, 35, 36), to prevent fattyacid degradation (5, 35), and to optimize the thioesterase (5, 8)(reviewed in refs. 3 and 4), there have been few attempts toengineer the fatty acid synthesis machinery itself. Reasons forthis may include the toxicity of FabF overexpression (37) and theessentiality of FabB on minimal medium (31). Nevertheless, wewere able to mimic the effect of cerulenin supplementation bycoupling expression of the octanoyl-ACP–specific FabF* (24) withinducible degradation of FabB. These interventions presumablyslowed the elongation of octanoyl-ACP produced by FabF*,thereby increasing octanoyl-ACP levels and octanoate productionby CpFatB1. Through these means we achieved 12% theoreticalyield of octanoate (Fig. 4B). This strategy should be generally
applicable to other MCFAs, although the optimal degradationrate and specificity of FabF is likely to be chain-length–dependent.Degradation tags have previously been used to decrease the
basal level of metabolic enzymes (38) and to dynamically degraderegulatory proteins (39). We engineered our strains to induciblydegrade an essential enzyme and thereby redirect metabolicflux on demand. Inducible degradation is an attractive metabolicengineering strategy because it allows an essential gene’s activityto be decreased or knocked out entirely, without blocking thestrain’s ability to grow. Inducible degradation of FabB allowed usto achieve our highest yields. This approach should be generallyapplicable to all essential metabolic genes in E. coli, opening upadditional opportunities for flux modulation.This study presents a strategy to increase fatty acid production
that combines KAS manipulation and more traditional metabolicengineering. It demonstrates the utility of engineering the fatty acidsynthesis machinery itself, and not just the thioesterase or otherenzymes that feed into the pathway, to enhance the yield of specificfatty acids. Given the interest in producing diverse fatty acid-derived compounds for use as biofuels and industrial chemicals,it is likely that the engineering of KASs and the other enzymes offatty acid synthesis will become increasingly important in micro-bial fatty acid engineering. We expect the strategy of dynamicallyinhibiting essential enzymes to be broadly useful as a tool formetabolic engineering and synthetic biology. In particular, dynam-ically inhibiting polymer elongation may be a useful approach forincreasing production or tuning the chain length of other valuablemolecules synthesized by recursive biosynthetic pathways, such asiteratively synthesized polyketides, polysaccharides, and bioplastics.
Materials and MethodsPlasmid and Strain Construction. pEET and SspBpET21b were obtained from
refs. 21 and 25, respectively. All other plasmids and strains used in this work,
and the primers used to generate them, are listed in Tables S1 and S2. Details
on plasmid and strain construction are provided in SI Materials and Methods.
Growth and Induction. Briefly, fatty acid production experiments were per-
formed at 30 °C either in shake flasks or 96-well plates inoculated from
overnight LB cultures. Shake flasks (Fig. 1) (M9 + 0.5% glycerol) were grown
to OD 0.4–0.6 and induced with 1 mM IPTG. The 96-well plates (M9 + 0.5%
glucose) were inoculated, grown for 3.5 h, and induced with 1 mM IPTG
(unless otherwise noted) and aTc (where indicated). Full experimental details
are provided in SI Materials and Methods.
OD and Enzymatic FFA Measurements. The OD595 of 100 μL of each culture was
measured in 200-μL 96-well flat-bottom, nontreated, sterile polystyrene
plates (Corning) on a Victor3V 1420 Multilabel Counter (Perkin-Elmer) using
a 595/60-nm filter. FFAs in 5 μL of culture were measured using the enzy-
matic Free Fatty Acid Half Micro Test (Roche) according to the manu-
facturer’s instructions, scaled down to 1/10 volume in a 96-well plate. OD600
was measured using an Ultrospec 10 spectrophotometer (Amersham).
Fatty Acid Identification and Quantification Using GC-MS. Fatty acids were
extracted from 400 μL of acidified culture with ethyl acetate and esterified
with ethanol. Fatty ethyl esters were extracted with hexane and run on an
Agilent GC-MS 5975/7890 on an HP-5MS column. Compounds were identi-
fied via GC retention times and mass spectra, and quantified by normalizing
to an internal standard (ethyl pentadecanoate) and comparing against
a standard curve. See SI Materials and Methods for details.
ACKNOWLEDGMENTS. We thank R. T. Sauer, K. L. Prather, P. J. Reilly, B. J.Nikolau, D. F. Savage, and D. C. MacKellar for plasmids and reagents; G.Webster, D. C. MacKellar, and D. C. Ducat for reading the manuscript; D. C.Ducat for helpful conversations throughout this work; and C P. Johnson foradvice and assistance with mass spectrometry. This work was conductedwith support from the Advanced Research Projects Agency–Energy ‘Electro-fuels’ Collaborative Agreement DE-AR0000079 (to P.A.S.); National ScienceFoundation Graduate Research fellowships (to J.P.T. and T.J.F.); a HerchelSmith Graduate Research Fellowship (to J.P.T.); and the Ruth L. KirschteinNationalesearch Service Award program of Harvard Catalyst j The HarvardClinical and Translational Science Center Award UL1 RR 025758 and financialcontributions from Harvard University and its affiliated academic health carecenters (to T.J.F.).
11294 | www.pnas.org/cgi/doi/10.1073/pnas.1307129110 Torella et al.

1. Cronan JE (2003) Bacterial membrane lipids: Where do we stand? Annu Rev Microbiol
57:203–224.
2. Magnuson K, Jackowski S, Rock CO, Cronan JE, Jr. (1993) Regulation of fatty acid
biosynthesis in Escherichia coli. Microbiol Rev 57(3):522–542.
3. Handke P, Lynch SA, Gill RT (2011) Application and engineering of fatty acid bio-
synthesis in Escherichia coli for advanced fuels and chemicals. Metab Eng 13(1):28–37.
4. Lennen RM, Pfleger BF (2012) Engineering Escherichia coli to synthesize free fatty
acids. Trends Biotechnol 30(12):659–667.
5. Steen EJ, et al. (2010) Microbial production of fatty-acid-derived fuels and chemicals
from plant biomass. Nature 463(7280):559–562.
6. Zhang F, Carothers JM, Keasling JD (2012) Design of a dynamic sensor-regulator
system for production of chemicals and fuels derived from fatty acids. Nat Biotechnol
30(4):354–359.
7. Schirmer A, Rude MA, Li X, Popova E, del Cardayre SB (2010) Microbial biosynthesis of
alkanes. Science 329(5991):559–562.
8. Liu T, Vora H, Khosla C (2010) Quantitative analysis and engineering of fatty acid
biosynthesis in E. coli. Metab Eng 12(4):378–386.
9. Ohlrogge JB (1994) Design of new plant products: Engineering of fatty acid metab-
olism. Plant Physiol 104(3):821–826.
10. Yang ST (2006) Bioprocessing for Value-Added Products from Renewable Resources:
New Technologies and Applications (Elsevier Science, Amsterdam).
11. Braude GL (1967) Preparation of Polymeric Plasticizers from Tall Oil Fatty Acids, US
Patent 3337594.
12. Bauer K, Garbe D, Surburg H (2008) Common Fragrance and Flavor Materials (Wiley-
VCH, Weinheim, Germany), pp 8–20.
13. Knothe G (2009) Improving biodiesel fuel properties by modifying fatty ester com-
position. Energy Environ Sci 2(7):759–766.
14. Ingram LO, Chevalier LS, Gabba EJ, Ley KD, Winters K (1977) Propionate-induced
synthesis of odd-chain-length fatty acids by Escherichia coli. J Bacteriol 131(3):
1023–1025.
15. Tseng HC, Prather KL (2012) Controlled biosynthesis of odd-chain fuels and chemicals
via engineered modular metabolic pathways. Proc Natl Acad Sci USA 109(44):
17925–17930.
16. Heath RJ, Rock CO (1996) Inhibition of beta-ketoacyl-acyl carrier protein synthase III
(FabH) by acyl-acyl carrier protein in Escherichia coli. J Biol Chem 271(18):
10996–11000.
17. Jiang P, Cronan JE, Jr. (1994) Inhibition of fatty acid synthesis in Escherichia coli in the
absence of phospholipid synthesis and release of inhibition by thioesterase action.
J Bacteriol 176(10):2814–2821.
18. Voelker TA, Davies HM (1994) Alteration of the specificity and regulation of fatty acid
synthesis of Escherichia coli by expression of a plant medium-chain acyl-acyl carrier
protein thioesterase. J Bacteriol 176(23):7320–7327.
19. Dellomonaco C, Clomburg JM, Miller EN, Gonzalez R (2011) Engineered reversal of
the β-oxidation cycle for the synthesis of fuels and chemicals. Nature 476(7360):
355–359.
20. Dehesh K, Edwards P, Hayes T, Cranmer AM, Fillatti J (1996) Two novel thioesterases
are key determinants of the bimodal distribution of acyl chain length of Cuphea
palustris seed oil. Plant Physiol 110(1):203–210.
21. Jing F, et al. (2011) Phylogenetic and experimental characterization of an acyl-ACP
thioesterase family reveals significant diversity in enzymatic specificity and activity.
BMC Biochem 12:44.
22. Jackowski S, Rock CO (1987) Acetoacetyl-acyl carrier protein synthase, a potential
regulator of fatty acid biosynthesis in bacteria. J Biol Chem 262(16):7927–7931.
23. Rock CO, Jackowski S (1982) Regulation of phospholipid synthesis in Escherichia coli.
Composition of the acyl-acyl carrier protein pool in vivo. J Biol Chem 257(18):
10759–10765.
24. Val D, Banu G, Seshadri K, Lindqvist Y, Dehesh K (2000) Re-engineering ketoacyl
synthase specificity. Structure 8(6):565–566.
25. McGinness KE, Baker TA, Sauer RT (2006) Engineering controllable protein degra-
dation. Mol Cell 22(5):701–707.
26. Way JC, Davis JH (2010) Methods and Molecules for Yield Improvement Involving
Metabolic Engineering, US Patent Appl PCT/US2010/036902.
27. Staunton J, Weissman KJ (2001) Polyketide biosynthesis: A millennium review. Nat
Prod Rep 18(4):380–416.
28. Suriyamongkol P, Weselake R, Narine S, Moloney M, Shah S (2007) Biotechnological
approaches for the production of polyhydroxyalkanoates in microorganisms and
plants—A review. Biotechnol Adv 25(2):148–175.
29. Tseng HC, Harwell CL, Martin CH, Prather KL (2010) Biosynthesis of chiral 3-hydrox-
yvalerate from single propionate-unrelated carbon sources in metabolically en-
gineered E. coli. Microb Cell Fact 9:96.
30. Farrell CM, Grossman AD, Sauer RT (2005) Cytoplasmic degradation of ssrA-tagged
proteins. Mol Microbiol 57(6):1750–1761.
31. Cronan JE, Jr., Birge CH, Vagelos PR (1969) Evidence for two genes specifically in-
volved in unsaturated fatty acid biosynthesis in Escherichia coli. J Bacteriol 100(2):
601–604.
32. Heath RJ, Jackowski S, Rock CO (1994) Guanosine tetraphosphate inhibition of fatty
acid and phospholipid synthesis in Escherichia coli is relieved by overexpression of
glycerol-3-phosphate acyltransferase (plsB). J Biol Chem 269(42):26584–26590.
33. Jensen PRWH, Westerhoff HV, Michelsen O (1993) The use of lac-type promoters in
control analysis. Eur J Biochem 211(1–2):181–191.
34. Miller TW (2006) Natural herbicides and amendments for organic weed control. Crop
Protection Products for Organic Agriculture, eds Felsot AS, Racke KD (ACS Pub-
lications, Washington, DC), pp 174–175.
35. Zhang F, et al. (2012) Enhancing fatty acid production by the expression of the reg-
ulatory transcription factor FadR. Metab Eng 14(6):653–660.
36. Davis MS, Solbiati J, Cronan JE, Jr. (2000) Overproduction of acetyl-CoA carboxylase
activity increases the rate of fatty acid biosynthesis in Escherichia coli. J Biol Chem
275(37):28593–28598.
37. Subrahmanyam S, Cronan JE, Jr. (1998) Overproduction of a functional fatty acid
biosynthetic enzyme blocks fatty acid synthesis in Escherichia coli. J Bacteriol 180(17):
4596–4602.
38. Doroshenko VG, et al. (2010) Construction of an L-phenylalanine-producing tyrosine-
prototrophic Escherichia coli strain using tyrA ssrA-like tagged alleles. Biotechnol Lett
32(8):1117–1121.
39. Huang D, Holtz WJ, Maharbiz MM (2012) A genetic bistable switch utilizing nonlinear
protein degradation. J Biol Eng 6(1):9.
Torella et al. PNAS | July 9, 2013 | vol. 110 | no. 28 | 11295
APPLIEDBIOLO
GICAL
SCIENCES

Supporting Information
Torella et al. 10.1073/pnas.1307129110
SI Materials and Methods
Plasmids and Strains. Plasmids pTJF010, pTJF038, pJT208, andpJT255 were constructed by conventional restriction cloning (seebelow). Plasmids and primers used in this work are listed in TablesS1 and S2. Genomic modifications in BL21*(DE3) (Invitrogen)were performed via transduction, using P1 phage generated froman appropriate TB10 or Keio collection (1) knockout strain,followed by FLP recombinase excision of the selective marker(see below). All genomic integrations were verified via sequencing.Strains generated in this work are listed in Table S1.
Plasmid Construction and Transformation. N-terminally his-taggedCpfatB1, codon optimized for expression in Synechococcus elon-gatus, was PCR-amplified from plasmid pDFS705 (constructed byDave F. Savage, University of California, Berkeley, CA) usingprimers TF0013 and TF0014, digested with SpeI and HindIII, andligated into the XbaI and HindIII sites of plasmid pETDuet-1(Merck) to generate pTJF010. The resulting plasmid was alsoPCR-amplified with primers TF0133 and TF0014, digested withNcoI and HindIII, and inserted into the NcoI and HindIII sites ofpACYCDuet-1 (Merck) to generate pTJF038. To generate pJT255,CpFatB1 was amplified with JT014 and JT015, digested with SpeI/BglII, and ligated into pWW308 [a gift from Weston Whitaker(Stanford University, Palo Alto, CA) while in the DueberLaboratory (University of California, Berkeley, CA) carrying theaTc-inducible Pzt promoter] digested with NheI/BamHI. Theresulting plasmid, pJT180, was then digested with HpaI/XhoI andthe Pzt-CpFatB1 fragment ligated into pCDFDuet-1 digested withHpaI/XhoI. UcfatB2 codon-optimized for expression in S. elon-gatus was PCR-amplified from pDFS703 using primers JT0138and JT0134, digested with XbaI and AflII, and inserted into thesame sites of pETDuet-1 to generate pJT208. Transformationswere performed using the TSS competent cell protocol (2).
Growth and Induction. Fatty acid production experiments wereperformed either in shake flasks or 96-well plates. For shake flaskcultures (data in Fig. 1), individual colonies were grown overnightin 5 mL LB in a 15-mL falcon tube at 37 °C, then diluted 1:100into 10 mLM9 + 0.5% glycerol (unless otherwise noted) in a 50-mLflask, grown at 30 °C with 250 rpm shaking until an OD600 of 0.4–0.6 was reached, and induced with 1 mM isopropyl-β-D-thio-galactopyranoside (IPTG). Cells were harvested 24 h or 48 hlater, for even- and odd-chain production experiments respectively.For 96-well plates (data in Figs. 2–4), individual colonies wereinoculated into 1 mL LB with appropriate antibiotics in a 2-mLdeep-well plate (Thermo Scientific), and shaken at at 30 °C, 1,200rpm on a Titramax 1000 platform shaker (Heidolph) for 12–18 h.Cultures were then diluted 1:20 into M9 + 0.5% glucose and grownfor 3.5 h before induction with 1 mM IPTG (unless otherwisenoted). Growth was allowed to proceed for 24 or 44 h beforeharvesting and analysis.
Western Blotting. Strains S007 and S002 were streaked out on anLB amp plate, grown overnight, and single colonies were pickedinto 5 mL LB/amp. LB cultures were allowed to grow for 18 h at30 °C and 300 μL diluted into 6 mL M9 0.5% glucose with 50 μg/mL ampicillin. M9 cultures were grown for 3.5 h at 30 °C andeither induced (+IPTG) or not (−IPTG) with 1 mM IPTG. Next,1-mL samples were taken just before induction and 2, 4, 6, and8 h after induction; 100 μL of each 1 mL sample was used tomeasure OD595. The remaining 900 μL was centrifuged at topspeed for 10 min in a table-top centrifuge; the cell pellet re-
suspended in 100 μL 3% (wt/vol) SDS and boiled for 10 min at100 °C. Samples volumes used in Western blotting were nor-malized to OD595 and prepared in 1× Tris·Glycine SDS Samplebuffer (Novex) with 1× NuPAGE sample reducing agent (No-vex). Samples were run in a Novex 4–20% Tris·glycine gel for 1.5h at 120 V, transferred to a nitrocellulose membrane using theiblot transfer system (Novex), and blotted with the Strep·Tag IIAntibody HRP Conjugate at a 1:4,000 dilution in TBS-tweenwith 1% BSA (EMDMillipore). Band intensities were quantifiedin Image J (W. S. Rasband, ImageJ, National Institutes ofHealth, Bethesda, MD).
P1 Transduction. Each Keio collection knockout strain contains aninsertion in a given gene, replacingmost of its coding sequence witha kanamycin cassette, bracketed by FRT sites. Phage generatedfrom a given Keio strain was used to transduce the desired strain aspreviously described (3). Successfully transduced strains werestreaked out serially 3× on LB + 5 mM sodium citrate plates toeliminate phage contamination. These strains were then trans-formed with the temperature-sensitive FLP recombinase plasmidpCP20 (4) and plated at 30 °C on LB-Amp plates. Restreaking thetransformants at 37 °C on LB simultaneously cured the plasmidand induced expression of FLP recombinase, excising the kana-mycin cassette as previously described (4). Strains streaked out at37 °C were restreaked on both LB + Kan and LB + Amp plates totest for retention of either the kanamycin cassette or the pCP20plasmid; colonies with neither resistance marker were then PCR-amplified at the appropriate locus, and the PCR product se-quenced to ensure successful knockout or allelic replacement.
Generation of Ketoacyl Synthase Mutant Strains. To generate thefabF* mutant (5), fabF was cloned into the BamH1 and HindIIIsites of cloning vector pUC19 to generate plasmid pUC19-FabF*. Site-directed mutagenesis was performed with primersTF0111 and TF0113 using the QuikChange Multi Site-DirectedMutagenesis Kit (Agilent Technologies) according to the man-ufacturer’s instructions. fabf* was amplified using primers TF0121and TF0122. kanR was then amplified using primers TF0119 andTF0120 which both FRT-flanked kanR and added 20 bp of ho-mology to fabF to the 5′ end of kanR and 40 bp of homology to thegenomic region downstream of fabF to the 3′ end of kanR. ThefabF* and kanR constructs were then used in a third PCR con-taining TF0121 and TF0120. The product of this reaction was theportion of fabF* containing the I-F mutation linked to a FRT-flanked kanR.Attempts to use λ red-mediated recombineering to insert this
construct directly into the Escherichia coli strain TB10 (6), aspreviously described (7), failed to result in strains containing thefabF I-F mutation. A region of fabF starting 48 bp upstream of theI-F mutation and ending 40 bp downstream of the fabF gene wastherefore first replaced with an ampicillin resistance gene (bla)amplified from plasmid pETDuet-1 using primers TF0134 andTF0135. bla was then replaced with the fabF*_kanR cassette inTB10 and clones that could grow on kanamycin and not ampicillinwere sequenced for the fabF I-F mutation. A strain positive for themutation was then used to generate P1 phage and the phage usedto insert the fabF* mutation into the strains indicated in the text.Although sequencing results showed that the fabF*_kanR con-struct was properly incorporated into the genome, the kanR cas-sette could not be removed using the pCP20 plasmid.To generate fabBDeg, a DNA construct composed (from 5′ to
3′) of 40 bp of the 3′ end of fabB, an ssrA DAS+4 tag (8), an
Torella et al. www.pnas.org/cgi/content/short/1307129110 1 of 7

FRT-flanked kanR, and 40 bp downstream of the genomic copyof fabB was synthesized (Intergraded DNA Technologies). Thisconstruct was PCR-amplified using primers TF0101 and TF0102.Purified PCR products were then used for λ red mediated re-combination in E. coli strain TB10, as previously described (6, 7).The genomic copy of fabBDeg with the downstream kanR wasthen P1 transduced into the E. coli strains indicated in the text.The FRT-flanked kanamycin cassette was then excised usingplasmid pCP20 (4).To construct strain S007, a linear DNA fragment containing
N-terminally strep-tagged FabBDeg with a C-terminal kanamycinresistance cassette was constructed by PCR of the DAS+4 taggedFabB from genomic DNA from S006 using primers TF0102 andTF0222. Once made, this construct was inserted into the TB10genome and transferred into strain BL21*(DE3) ΔfadE using P1transduction, as described above.
Fatty Acid Identification and Quantification Using GC-MS. Fatty acidswere extracted from 400 μL of culture by adding 50 μL 10%(wt/vol) NaCl, 50 μL glacial acetic acid, 20 μL of 800 mg/Lpentadecanoate (as internal standard), and 200 μL ethyl acetate,then vortexing for 20 s. The mixture was centrifuged at 16,000 ×
g for 10 min. Ethyl esters were generated by mixing 100 μL of theorganic phase with 900 μL of a 30:1 mixture of EtOH and 37%
(vol/vol) HCl, and incubated at 55 °C for 1 h (based on theprotocol in ref. 9). After cooling to room temperature, 500 μLdH2O and 500 μL hexane were added. The mixture was vortexedfor 10 s, and 150 μL taken from the top (hexane) layer foranalysis via GC-MS. Extracts were run on an Agilent GC-MS5975/7890 (Agilent Technologies) using an HP-5MS (length: 30m; diameter: either 0.25 or 0.50 mm; film: 0.25 μm) column; themethod ramped from 75 to 325 °C at 30 °C per minute. Com-pound identities were determined via GC retention times(compared with known standards) and verified by mass spectra.Where quantification was necessary, standard curves of thequantified compound were generated by extracting them fromM9 media and esterifying them as described. The area undereach peak of a single-ion trace selective for ethyl esters (88 m/z)was integrated and normalized to pentadecanoate before usingit to quantify the desired species.
Estimation of Lipid-Incorporated Fatty Acids. The amount of fattyacid incorporated into lipids was estimated from OD600 at 24 h byassuming the following: an OD600 of 1.0 equals 0.3 gDCW/L(10); 9.1% of E. coli dry cell weight is lipid (11); fatty acids makeup 71% of lipid mass (calculated as the percent molecular weightof dipalmitoyl phosphatidylglycerol that comes from its palmiticacid moieties).
1. Baba T, et al. (2006) Construction of Escherichia coli K-12 in-frame, single-gene
knockout mutants: The Keio collection. Mol Syst Biol 2:2006 0008.
2. Chung CT, Niemela SL, Miller RH (1989) One-step preparation of competent
Escherichia coli: Transformation and storage of bacterial cells in the same solution.
Proc Natl Acad Sci USA 86(7):2172–2175.
3. Thomason LC, Costantino N, Court DL (2007) E. coli genome manipulation by P1
transduction. Curr Protoc Mol Biol, Chapter 1:Unit 1.17.
4. Cherepanov PP, Wackernagel W (1995) Gene disruption in Escherichia coli: TcR and
KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance
determinant. Gene 158(1):9–14.
5. Val D, Banu G, Seshadri K, Lindqvist Y, Dehesh K (2000) Re-engineering ketoacyl
synthase specificity. Structure 8(6):565–566.
6. Johnson JE, Lackner LL, Hale CA, de Boer PA (2004) ZipA is required for targeting of
DMinC/DicB, but not DMinC/MinD, complexes to septal ring assemblies in Escherichia
coli. J Bacteriol 186(8):2418–2429.
7. Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes
in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97(12):
6640–6645.
8. McGinness KE, Baker TA, Sauer RT (2006) Engineering controllable protein degradation.
Mol Cell 22(5):701–707.
9. Ichihara K, Fukubayashi Y (2010) Preparation of fatty acid methyl esters for gas-liquid
chromatography. J Lipid Res 51(3):635–640.
10. Panula-Perälä J, et al. (2008) Enzyme controlled glucose auto-delivery for high cell
density cultivations in microplates and shake flasks. Microb Cell Fact 7:31.
11. Neidhardt FC (1996) Escherichia coli and Salmonella: Cellular and Molecular Biology.
(ASM Press, Washington, DC), pp. 14.
Torella et al. www.pnas.org/cgi/content/short/1307129110 2 of 7

Fig. S1. Fatty acid chain-length profile of medium-chain fatty acid (MCFA)-producing strains. Strain S001 alone (i) or bearing either pEET (BfTES, ii), pTJF010
(CpFatB1, iii), or pJT208 (UcFatB2, iv) was grown in M9 + 0.5% glycerol and induced with IPTG for 24 h (n = 3, error bars = SEM). Extraction and esterification of
free fatty acids (FFA) with ethanol, as well as detection by GC-MS and quantitation, were performed as described in Materials and Methods. The FFA chain-
length profile is shown for (i) S001, (ii) S001-pEET, (iii) S001-pJTF010, and (iv) S001-pJT208; odd chains were produced at negligible quantities (<2 mg/L) and are
not shown. Analysis of the fate of all produced fatty acids, including those incorporated into lipids, is shown in v. MCFAs refers to all FFAs from 4 to 12 carbons,
but long-chain fatty acids (LCFAs) refers to all FFAs from 14 to 18 carbons. The concentration of lipid-incorporated fatty acids was estimated as described in
SI Materials and Methods. Panel v shows that thioesterase expression increases the total amount of carbon flowing into fatty acid synthesis, as the sum of
MCFAs + LCFAs + Lipid-FAs increases. This result is primarily because of an increase in MCFA yield. Panels vi to viii show the FFA chain-length profile for S001-
pEET (vi), S001-pTJF010 (vii), and S001-pJT208 (viii) during supplementation with 100 mM propionate.
Fig. S2. Final OD595 measurements from cerulenin titration experiments in Fig. 2B. Experiments were performed as described in the legend to Fig. 2B. OD595
was measured for each strain at each concentration of cerulenin tested (n = 3, error bars = SEM) for the same samples used to generate Fig. 2B. BfTES, CpFatB1,
and UcFatB2 indicate strains S001-pEET, S001-pTJF010, and S001-pJT208, respectively.
Torella et al. www.pnas.org/cgi/content/short/1307129110 3 of 7

Fig. S3. Time courses of FFA production with and without ketoacyl synthase (KAS) inhibition. (A) Strain S001-pTJF010 (CpFatB1) was grown in M9 + 0.5%
glucose and induced with 1 mM IPTG, as previously described, with or without the addition of 0.4 μg/mL cerulenin. At the indicated time points (0 h = time of
induction), cultures were tested for OD595 and for total FFAs via the Roche Half-Micro test (n = 3, error bars = SEM). At all time points other than t = 0, samples
with added cerulenin had greater FFA production than those without cerulenin (P < 0.05, one-tailed Student t test). The initial rate of FFA production (first four
datapoints) was also greater in the samples containing cerulenin: 6.7 ± 0.6 mgL-1·h−1 for 0 μg/mL cerulenin vs. 13.2 ± 2.9 mgL-1·h−1 for 0.4 μg/mL cerulenin. (B)
Time course (performed as in A) of strains S003 [BL21*(DE3) ∆fadE fabF*] and S005 [BL21*(DE3) ∆fadE fabF* fabBDeg] both expressing SspBpET21b and
pTJF038. At all time points other than t = 0, fatty acids produced by S005 were significantly greater than those produced by S003 (P < 0.05, one-tailed Student
t test). The initial rate of fatty acid production (first four datapoints) was also greater: 9.6 ± 0.7 mgL-1·h−1 for S003 vs. 14.5 ± 2.4 mgL-1·h−1 for S005.
Fig. S4. Production of C10 and C12 FFAs by UcFatB2 as a function of cerulenin concentration. Strain S001-pJT208 was grown in M9 + 0.5% glucose and
induced with 1 mM IPTG and different concentrations of cerulenin for 24 h (as described for Fig. 2B). Shown are the concentrations of C10 and C12 FFAs as
a function of cerulenin concentration, as determined by GC-MS analysis (n = 3, error bars = SEM). Whereas C10 increases significantly from 0 to 0.4 0.4 μg/mL
cerulenin (*P < 0.05 by one-tailed Student t test), C12 decreases consistently with increasing concentrations of cerulenin.
Fig. S5. (A) Western blotting of StrepFabBDeg following SspB induction. Strain S007 [BL21*(DE3) ΔfadE strepfabBDeg] + SspBpET21b was grown in M9 +0.5%
glucose and either induced (+IPTG) or not induced (−IPTG) with 1 mM IPTG. StrepFabBDeg levels were monitored over time as described in SI Materials and
Methods (numbers indicate the time in hours postinduction); loading was normalized to OD600. Blue triangles indicate StrepFabBDeg and black triangles
indicate an unidentified background band. The band corresponding to StrepFabBDeg ran at the expected size (45 kDa) and did not appear in control strain
S002 (lane C on the gel). Strain S002 encodes wild-type fabB without a strep or degradation tag, and was loaded at t = 0 without IPTG induction. (B)
Quantification of the Western blot in A, normalized to StrepFabBDeg levels at t = 0 h, shows that StrepFabBDeg levels drop about 54% of their original level
over 8 h of induction. An asterisk indicates significantly lower StrepFabBDeg levels compared with −IPTG at 8 h (P < 0.05, one-tailed Student t test).
Torella et al. www.pnas.org/cgi/content/short/1307129110 4 of 7
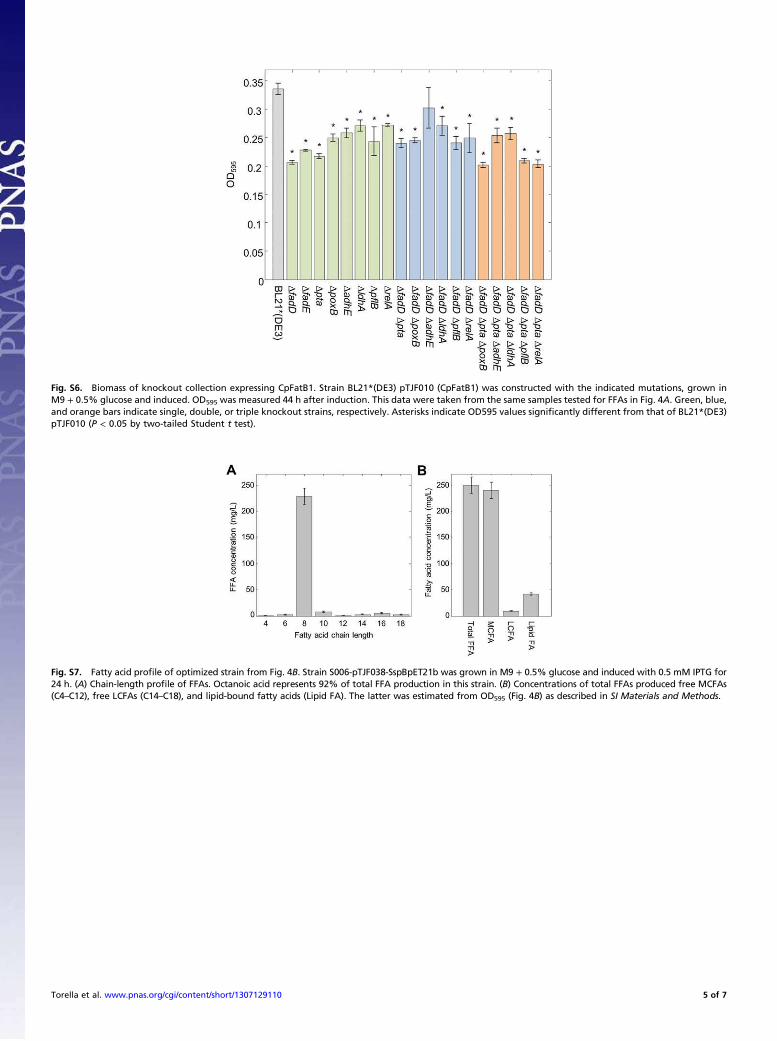
Fig. S6. Biomass of knockout collection expressing CpFatB1. Strain BL21*(DE3) pTJF010 (CpFatB1) was constructed with the indicated mutations, grown in
M9 + 0.5% glucose and induced. OD595 was measured 44 h after induction. This data were taken from the same samples tested for FFAs in Fig. 4A. Green, blue,
and orange bars indicate single, double, or triple knockout strains, respectively. Asterisks indicate OD595 values significantly different from that of BL21*(DE3)
pTJF010 (P < 0.05 by two-tailed Student t test).
Fig. S7. Fatty acid profile of optimized strain from Fig. 4B. Strain S006-pTJF038-SspBpET21b was grown in M9 + 0.5% glucose and induced with 0.5 mM IPTG for
24 h. (A) Chain-length profile of FFAs. Octanoic acid represents 92% of total FFA production in this strain. (B) Concentrations of total FFAs produced free MCFAs
(C4–C12), free LCFAs (C14–C18), and lipid-bound fatty acids (Lipid FA). The latter was estimated from OD595 (Fig. 4B) as described in SI Materials and Methods.
Torella et al. www.pnas.org/cgi/content/short/1307129110 5 of 7

Table S1. Plasmid and strain list
Plasmid/strain
designation in text Description Promoter Inducer Resistance Origin Source
BL21*(DE3) F- ompT hsdSB (rB-mB-) gal dcm rne131(DE3) Invitrogen
S001 BL21*(DE3)ΔfadD Present study
S002 BL21*(DE3)ΔfadE Present study
S003 BL21*(DE3)ΔfadE fabF* Present study
S004 BL21*(DE3)ΔfadE fabBDeg Present study
S005 BL21*(DE3)ΔfadE fabF* fabBDeg Present study
S006 BL21*(DE3)ΔfadD Δpta ΔlacY fabF* fabBDeg Present study
S007 BL21*(DE3)ΔfadE strepfabBDeg Present study
Δpta BL21*(DE3)Δpta Present study
ΔpflB BL21*(DE3)ΔplfB Present study
ΔadhE BL21*(DE3)ΔadhE Present study
ΔldhA BL21*(DE3)ΔldhA Present study
ΔpoxB BL21*(DE3)ΔpoxB Present study
ΔrelA BL21*(DE3)ΔrelA Present study
ΔfadD BL21*(DE3)ΔfadD Present study
ΔfadΔpta BL21*(DE3)ΔfadDΔpta Present study
ΔfadDΔpflB BL21*(DE3)ΔfadDΔpflB Present study
ΔfadDΔadhE BL21*(DE3)ΔfadDΔadhE Present study
ΔfadDΔldha BL21*(DE3)ΔfadDΔldhA Present study
ΔfadDΔpoxB BL21*(DE3)ΔfadDΔpoxB Present study
ΔfadDΔrelA BL21*(DE3)ΔfadDΔrelA Present study
ΔfadDΔptaΔpflB BL21*(DE3)ΔfadDΔptaΔpflB Present study
ΔfadDΔptaΔadhE BL21*(DE3)ΔfadDΔptaΔadhE Present study
ΔfadDΔptaΔldhA BL21*(DE3)ΔfadDΔptaΔldhA Present study
ΔfadDΔptaΔpoxB BL21*(DE3)ΔfadDΔptaΔpoxB Present study
ΔfadDΔptaΔrelA BL21*(DE3)ΔfadDΔptaΔrelA Present study
TB10 Recombineering strain MG1655, nadA::Tn10
λcI857 Δ(cro-bioA)
(1)
sspBpET21b E. coli sspB cloned into pET21b, obtained
from Robert T. Sauer’s Lab at MIT
T7lac IPTG Amp ColE1 (2)
pTJF010 C. palustris fatb1 codon-optimized for
S. elongatus, containing N-terminal 6x-his
tag cloned into the XbaI and HindIII sites
of pETDuet-1
T7lac IPTG Amp ColE1 Present study
pTJF038 Cuphea palustris fatb1 codon-optimized for
S. elongatus, containing N-terminal 6x-his
tag cloned into the NcoI and HindIII sites
of pACYCDuet-1
T7lac IPTG Cam P15A Present study
pEET Bryantella formatexigens thioesterase
EET61113, codon-optimized for E. coli,
cloned into pUC57.
lacZ IPTG Amp pMB1 (3)
pJT208 Umbellularia californica fatB2 codon
optimized for S. elongatus, cloned into
pETDuet-1
T7lac IPTG Amp ColE1 Present study
pJT255 CpFatB1 from pTJF010 cloned into an
anhydrotetracycline-inducible vector.
Pzt aTc Spec CDF Present study
pDFS705 Syneccochocus expression vector containing
codon-optimized, N-terminally his-tagged
C. palustris fatB1
Trc IPTG Cam pMB1 Gift from David F. Savage
(University of California,
Berkeley, CA)
pUC19_FabF pUC19 containing E. coli fabF lacZ N/A Amp pMB1 Gift from Drew C. MacKellar
(Harvard Medical School, Boston)
pCP20 contains heat shock inducible flippase and
is itself heat curable
λpR Heat Shock Amp repA101-ts (4)
pCOLA-thrAfrBCilvAfr pCOLADuet-1 expressing thrABC (thrA
is feedback-resistant) and ilvA
(feedback-resistant), obtained from
Kristala Prather’s laboratory at MIT.
T7lac IPTG Kan ColA (5)
1. Johnson JE, Lackner LL, Hale CA, de Boer PA (2004) ZipA is required for targeting of DMinC/DicB, but not DMinC/MinD, complexes to septal ring assemblies in Escherichia coli. J Bacteriol
186(8):2418–2429.
2. McGinness KE, Baker TA, Sauer RT (2006) Engineering controllable protein degradation. Mol Cell 22(5):701–707.
3. Jing F, et al. (2011) Phylogenetic and experimental characterization of an acyl-ACP thioesterase family reveals significant diversity in enzymatic specificity and activity. BMC Biochem 12:44.
4. CherepanovPP,WackernagelW(1995)Genedisruption inEscherichia coli: TcRandKmRcassetteswith theoptionofFlp-catalyzedexcisionof theantibiotic-resistancedeterminant.Gene158(1):9–14.
5. Tseng HC, Prather KL (2012) Controlled biosynthesis of odd-chain fuels and chemicals via engineered modular metabolic pathways. Proc Natl Acad Sci USA 109(44):17925–17930.
Torella et al. www.pnas.org/cgi/content/short/1307129110 6 of 7

Table S2. Primer list
Primer Description Sequence
TF0013 CpfatB1 forward primer with RBS, 6x His-tag, 5′ SpeI used to
amplify CpfatB1 from DFS705
CACACTAGTAGGAGGAAAAACATATGCATCACCATCATCATC
ACAGTTCGTTGTTGACCGCTATCACTAC
TF0014 CpfatB1 Rev primer with 3′ XbaI and HindII restriction sites used
to amplify CpfatB1 from DFS705
CTGCAAGCTTTCTAGATTACGTTTTTCCGGTCG
TF0101 Forward primer for fabBDeg_Kan ACCAACGCCACGCTGGTAATG
TF0102 Reverse primer for fabBDeg_Kan GCGACGCTGGCGCGTCTAC
TF0111 sense fabF I-F mutatgenic primer ATTGGCTCCGGGTTTGGCGGCCTCG
TF0113 antisense fabF I-F mutagenic primer CGAGGCCGCCAAACCCGGAGCCAAT
TF0119 forward kanR primer containing 5′ frt site and homology to the
3′ end of fabF
GATCTAAGTCGACCTGCAGGGAAGTTCCTATTCTCTAGAAAG
TATAGGAACTTCCTTTAAGAAGGAGATATACCATGAGCCATA
TTCAACGGGAAACGTCTTGC
TF0120 Reverse kanR primer containing 3′ frt site and 40bp of homology
downstream of fabF
AAGCTAAGAAAAAAGGCCCGCAAGCGGACCTTTTATAAGGG
AAGTTCCTATACTTTCTAGAGAATAGGAACTTCTTAGAAAAA
CTCATCGAGCATCAAATGAAACTGC
TF0121 Forward fabF primer containing 40bp homology upstream of the
fabF I-F mutation for replacing fabF with fabF*
AATATGGAATTGTCGCTGGCGTTCAGGCCATGCAGGATTC
TF0122 Reverse fabF primver for amplifying fabF from pUC19_fabF CCTGCAGGTCGACTTAGATC
TF0133 fatB1 forward primer with 6x His-tag and 5′ NcoI site used to
amplify Cpfatb1 from DF705
CACCCATGGGCCATCACCATCATCATCACAGTTCGTTGTTGA
CCGCTATCACTACTG
TF0134 Forward bla primer containing 40bp of homology to the region
upstream of the fabF I-F mutation
ATAACGGAAGAGAACGCAACCCGCATTGGTGCCGCAATTGATG
AGTATTCAACATTTCCGTGTCG
TF0135 Reverse bla primer containing 40bp of homology downstream
of fabF
AAGCTAAGAAAAAAGGCCCGCAAGCGGACCTTTTATAAGGGA
AGTTCCTATACTTTCTAGAGAATAGGAACTTCTTACCAATGCTT
AATCAGTGAGGCAC
TF0222 Forward primer containing the N-terminal strep tag for
strepfabBDeg_kan
ATTCGAAACTTACTCTATGTGCGACTTACAGAGGTATTGAATGT
GGAGCCACCCGCAGTTCGAAAAAAAACGTGCAGTGATTACTGG
JT0138 Forward primer for codon-optimized UcfatB2 TCAGTCTAGACGCAATCTCACGCAAATCACGGTAATCCCTAAA
TGACGAATCTCGAATGGAAACC
JT0134 Reverse primer for codon-optimized UcfatB2 GACTCTTAAGCTAGACCCGGGGTTCAGCTGG
JT014 Forward primer for cloning CpFatB1into pWW308 CATCAGCTCGAGTTGACAATTAATCATCGGCTCGTATAATGTG
TGGAATTGTGAGCGGATAACAATTTCAACTAGTCACAGGAGA
TATCATATGCATCACCATCATCATCACAGTTCGTTGTTGACCGCT
JT015 Reverse primer for cloning CpFatB1into pWW308 CATGACAGATCTCGGCCGCAAGCTTTCTAGATTACGTTTTTCCG
GTC
Torella et al. www.pnas.org/cgi/content/short/1307129110 7 of 7
Copyright © 2022 FDOKUMEN




















