
Synthesis of microbial elastomers based on soybean oil. Autoxidation kinetics, thermal and...
11
ORIGINAL PAPER Synthesis of microbial elastomers based on soybean oil. Autoxidation kinetics, thermal and mechanical properties Baki Hazer & Derya Burcu Hazer & Burak Çoban Received: 27 May 2009 / Accepted: 17 September 2009 / Published online: 13 October 2009 # Springer Science + Business Media B.V. 2009 Abstract Microbial bioelastomers prepared by the autox- idation of the unsaturated medium-long chain length co- poly-3-hydroxyalkanoate’ s (mlcl-PHAs) based on soybean oily acids (Sy) have been reported. Pseudomonas oleovor- ans were grown on a series of the mixture of octanoic acid (OA) and Sy with the weight ratio of 20:80, 28:72 and 50:50 in order to obtain unsaturated mlcl-copolyesters coded PHO-Sy-2080, PHO-Sy-2872, and PHO-Sy-5050, respectively. The microorganism was also grown on the mixture of Sy and 10-undecenoic acid (UA) with the weight ratio of 50:50 to obtain unsaturated copolyester coded PHU-Sy-5050. The PHAs obtained were character- ized by 1 H NMR and GC-MS techniques. Double bond contents of the unsaturated PHAs obtained were varying between 0.8 to 20 mol %. Autoxidation of the unsaturated copolyesters were carried out on exposure to air at room temperature in order to obtain new biomaterials whose mechanical strength was improved. Autoxidation kinetics, shelf life, mechanical and thermal properties of these biomaterials were evaluated. Keywords Soybean oil . Poly(3-hydroxyalkanoates) . Elastomer . Autoxidation Introduction Poly-3-hydroxy alkanoates (PHA) s with the chemical formulae shown below, are a class of reserve polyesters produced by a large number of bacteria when subjected metabolic stress [1–7]. In this equation R is the alkyl side chain depending on the substrates and the type of the bacteria. PHAs can be classified into three groups based on the number of carbon atoms in the monomer units: short-chain-length (sclPHA) containing 3–5 carbon atoms, medium-chain-length (mclPHA) containing 6–14 carbon atoms, and long-chain- length (lclPHAs), with more than 14 carbon atoms [8]. Pseudomonas oleovorans is a very versatile for PHA production because it can produce medium chain length polyesters (mclPHA) [9–17] and long chain length poly- esters (lclPHA) [18–21] from a wide variety of carbon substrates, including derivatives of alkanoic acids and fatty acids. In fact, there is a strain of P. oleovorans that can produce sclPHA from glycerol [22]. PHAs are biologically degradable materials on the basis of the green chemistry [23–25] and therefore they currently receive considerable attention. For the industrial and medical applications, sclPHAs are brittle; mclPHAs are thermoplastic indicating plastic deformation while lclPHAs are very soft and sticky. Therefore, the physical and mechanical properties of the PHAs need to be diversified and improved [26]. Among them, crosslinking reactions of the PHAs have been important in preparation of the B. Hazer (*) : B. Çoban Department of Chemistry, Zonguldak Karaelmas University, 67100 Zonguldak, Turkey e-mail: [email protected] e-mail: [email protected] D. B. Hazer Department of Neurosurgery, Faculty of Medicine, School of Medicine, Hacettepe University, Sıhhiye, 06100 Ankara, Turkey J Polym Res (2010) 17:567–577 DOI 10.1007/s10965-009-9345-0
Transcript of Synthesis of microbial elastomers based on soybean oil. Autoxidation kinetics, thermal and...

ORIGINAL PAPER
Synthesis of microbial elastomers based on soybean oil.Autoxidation kinetics, thermal and mechanical properties
Baki Hazer & Derya Burcu Hazer & Burak Çoban
Received: 27 May 2009 /Accepted: 17 September 2009 /Published online: 13 October 2009# Springer Science + Business Media B.V. 2009
Abstract Microbial bioelastomers prepared by the autox-idation of the unsaturated medium-long chain length co-poly-3-hydroxyalkanoate’s (mlcl-PHAs) based on soybeanoily acids (Sy) have been reported. Pseudomonas oleovor-ans were grown on a series of the mixture of octanoic acid(OA) and Sy with the weight ratio of 20:80, 28:72 and50:50 in order to obtain unsaturated mlcl-copolyesterscoded PHO-Sy-2080, PHO-Sy-2872, and PHO-Sy-5050,respectively. The microorganism was also grown on themixture of Sy and 10-undecenoic acid (UA) with theweight ratio of 50:50 to obtain unsaturated copolyestercoded PHU-Sy-5050. The PHAs obtained were character-ized by 1H NMR and GC-MS techniques. Double bondcontents of the unsaturated PHAs obtained were varyingbetween 0.8 to 20 mol %. Autoxidation of the unsaturatedcopolyesters were carried out on exposure to air at roomtemperature in order to obtain new biomaterials whosemechanical strength was improved. Autoxidation kinetics,shelf life, mechanical and thermal properties of thesebiomaterials were evaluated.
Keywords Soybean oil . Poly(3-hydroxyalkanoates) .
Elastomer . Autoxidation
Introduction
Poly-3-hydroxy alkanoates (PHA) s with the chemicalformulae shown below, are a class of reserve polyestersproduced by a large number of bacteria when subjectedmetabolic stress [1–7].
In this equation R is the alkyl side chain depending onthe substrates and the type of the bacteria. PHAs can beclassified into three groups based on the number of carbonatoms in the monomer units: short-chain-length (sclPHA)containing 3–5 carbon atoms, medium-chain-length(mclPHA) containing 6–14 carbon atoms, and long-chain-length (lclPHAs), with more than 14 carbon atoms [8].Pseudomonas oleovorans is a very versatile for PHAproduction because it can produce medium chain lengthpolyesters (mclPHA) [9–17] and long chain length poly-esters (lclPHA) [18–21] from a wide variety of carbonsubstrates, including derivatives of alkanoic acids and fattyacids. In fact, there is a strain of P. oleovorans that canproduce sclPHA from glycerol [22].
PHAs are biologically degradable materials on the basisof the green chemistry [23–25] and therefore they currentlyreceive considerable attention. For the industrial andmedical applications, sclPHAs are brittle; mclPHAs arethermoplastic indicating plastic deformation while lclPHAsare very soft and sticky. Therefore, the physical andmechanical properties of the PHAs need to be diversifiedand improved [26]. Among them, crosslinking reactions ofthe PHAs have been important in preparation of the
B. Hazer (*) :B. ÇobanDepartment of Chemistry, Zonguldak Karaelmas University,67100 Zonguldak, Turkeye-mail: [email protected]: [email protected]
D. B. HazerDepartment of Neurosurgery, Faculty of Medicine,School of Medicine, Hacettepe University,Sıhhiye,06100 Ankara, Turkey
J Polym Res (2010) 17:567–577DOI 10.1007/s10965-009-9345-0

elastomeric PHAs. Because cross-linking of crystallinethermoplastic polymers interfere with molecular packing,reduce the level of crystallization and consequently result ina polymer with a lower hardness and yield strength [27].There are several fine attempts to prepare cross-linkedmclPHAs (saturated) via chemical modification reactionsincluding vulcanization [28, 29], radical polymerization[30–34], acrylate grafting reactions [35], UV, γ andelectron beam irradiation processes [36, 37].
PHAs having unsaturated side chains can be obtainedfrom P.oleovorans cofed with 10-undecenoic acid and polyunsaturated oily acids. Crosslinking reactions of theunsaturated side chains of these PHAs can make themnon-sticky material with higher mechanical strength. Thereare few reports on the crosslinking of the unsaturated PHAson exposure to air [38], free radical polymerization usingbenzoyl peroxide [39, 40] and UV irradiation [39–41].Recently kinetic studies of the autooxidation of poly (3-hydroxy 10-undecenoate, PHU, and poly (3-hydroxyoctanoate-co-10-undecenoate), PHOU, at 60 °C have beenreported by Schmid et al. [42]. Higher content of the doublebonds causes higher cross-linking density which makes thePHAs brittle. These cross-linked polymers obtained by thecross-linking reactions of the double bonds of the unsatu-rated mlcl-PHAs were not considered as a sustainablematerial for industrial and medical applications because oftheir poor mechanical properties.
Briefly, to prepare elastomer films, mlcl-PHAs wereprepared containing both saturated and unsaturated units.Then, highly elastomeric materials were obtained by theautoxidation leading to cross linking of the double bonds inthe mlcl-PHAs. In this work, Pseudomonas oleovoranswere grown on a series of the mixture of octanoic acid(OA) and soybean oily acids (Sy) with the weight ratio of20:80, 28:72 and 50:50 in order to obtain unsaturated mlcl-copolyesters with the double bond contents varyingbetween 0.8 to 2.2 mol %; coded PHO-Sy-2080, PHO-Sy-2872, and PHO-Sy-5050, respectively. Additionally, themixture of Sy and 10-undecenoic acid (UA) with theweight ratio of 50:50 was also used to obtain PHU-Sy-5050with the double bond content 20%. Autoxidation kinetics,biocompatibility and shelf life of the PHA-copolyestershave been reported.
Experimental
Materials
Soybean oil is a commercial product obtained from soybeangrown in Western Turkey. Soybean oil is hydrolysed in a10% solution of KOH in ethanol, after which the solution isneutralised with a 10% solution of sulphuric acid in water
to obtain the carboxylic acid substrates consisting of oleicacid (25 wt%), linoleic acid (51 wt%) and linolenic acid (9wt%) [43]. Bacterial polyesters produced [44] by feedingPseudomonas oleovorans from the soybean oily acids, octanoicacid and/or undecenoic acid coded as PHA-Sy (polyesterobtained pure soybean oily acids), PHO-Sy’s, PHU-Sy, andPHU, PHO and PHOU were supplied from the TUBITAK-MAM Food Research Institute, Gebze-Kocaeli Turkey.
Bromination of the PHAs for determination of the doublebond inclusion using 1H NMR
The same procedure reported in the literature [44] for thebrominating reactions of the unsaturated co-polyesters wasapplied. As an example, 0.5 g of unsaturated co-polyesterwas added in 2 mL of stock solution of bromine (1 mL) infreshly distilled CCl4 (10 mL) in a schlenck tube and leftfor 2 h at room temperature in the dark. Brominated PHAwas precipitated from 50 mL of methanol and dried undervacuum at room temperature.
Auto oxidation of the unsaturated PHAs
Auto oxidation of the unsaturated PHAs was performedaccording to the same procedure for the auto oxidation ofsoybean oil which was reported in our previous work [43].For this purpose, the 20 mL of CHCl3 solution containing2.3 g of PHA was filtered from the glass-wool to a Petridish (ϕ=9 cm) and solvent evaporation was allowed slowlyfor a day at room temperature to get a smooth polymer film(thickness is at around 0.03 mm) which was left onexposure to air. Autoxidation was performed in summer(May–June, 2008) when the ratio of the sunny days was80%. During the autoxidation process, temperature of thepolymer film surfaces were changing between from 32 °C(day time) to 26 °C (night). In case of [PHU + PHA-Sy](1:1) polymer blend, the autoxidation process was carriedout using a solution containing PHU (1.0 g) and PHA-Sy(1.0 g) in 30 mL of CHCl3. After 40 days auto oxidation,the films were all in good elastic condition and ready forthe characterization. The autoxidized polymer film wastaken out from the Petri dish by washing with metanol. Apiece of vacuum dried film (0.35 g) was soaked in chlo-roform for 24 h at room temperature for sol–gel analysis.
Polymer characterization
1H NMR 1H NMR spectra were recorded in CDCl3 at 17 °Cwith a tetramethylsilane internal standard using a 400 MHzNMR AC 400 L. 1H NMR spectra of the swollen cross-linked copolyesters in CDCl3 were also taken. FT-IRspectra were recorded of the PHA films cast from CHCl3,using a Jasco model 300E FT-IR spectrometer.
568 B. Hazer et al.
The molecular weight Themolecular weight of the polymericsamples was determined by gel permeation chromatography(GPC) with a Knauer GPC in CHCl3 solution at 35 °C, at alow rate of solution, 1 mL/min, using ChromGate software, aWellChrom Interface Box, RI Detector K-2301, and Well-Chrom HPLC pumpK-501. A calibration curve was generatedwith six polystyrene standards obtained from Polyscience(MW’s: 3×106, 2.33×105, 2.2×105, 2,150 and 580).
Stress-strain measurements Stress-strain measurements ofthe PHA samples were performed on a SDL Testometricmodel Universal Tensile Testing Machine using 50 kg loadcell with a stretch speed 100 mm/min. The film sampleshad a rectangular shape with size (0.3–0.4)×10×50 mm. Atleast three samples for each experiment were used in themeasurement.
Thermogravimetric analysis Thermogravimetric analysis(TGA) was performed with a DuPont 2910 to determinethe glass transition temperatures (Tg), the melting transi-tions (Tm), and decomposition (Td). Samples were heatedfrom −50 to 200 °C in a nitrogen atmosphere at a rate of10 °C/min.
Swelling experiments Swelling experiments of the purecrosslinked PHA samples were carried out in chloro-form. The polymer films were allowed to remain insolvent for 24 h at room temperature and the swollenfilms were weighed as soon as they were taken outfrom the solvent. Swelling degrees of polymers atequilibrium [45] were determined using the equation ofswelling ratio, qv.
qv Volume of swollen polymer(Vswollen polymer)/Volume of dry polymer(Vdry polymer)
Vdry polymer m dry polymer/density of dry polymerVswollen
polymer
[(mswollen polymer- m dry polymer)/density ofchloroform] + Vdry polymer
Densities of the dry PHAs were measured in a 25 cm3
uncertified BLAUBRAND picnometer NS10/19 with ther-mometer at 29 °C using water. The densities measured werelisted below:
PHO, PHU-Sy-5050, and PHU: 1.000 g/cm3; PHA-Sy: 1.010 g/cm3; PHOU-5050: 1.021 g/cm3; PHO-Sy-2080, PHO-Sy-2872, and PHO-Sy-5050:1.030 g/cm3.
Hydrolysis of the autoxidized PHA gels 0.10 g ofautoxidized PHA gel was put in a vial containing10 mL of 1 N NaOH and left in a cupboard at roomtemperature. Three weeks later, the gel became complete-ly soluble. The solution was neutralized using 1 N HCl,extracted with chloroform and washed with water.
Solvent was evaporated and dried in a vacuum oven atroom temperature overnight.
Methanolysis and GC-MS Analysis For determination ofthe polymer composition, 2 mg of the polymer wasdegraded by methanolysis by heating for 140 min at100 °C with a solution of 1 mL of 15 wt% solution ofsulfuric acid in methanol containing 1.0 mL of chloroform.The solution was washed with 1.0 mL of water, and thechloroform layer was analyzed by gas chromatography(GC) connected with mass spectrometry (MS) using aThermo Trace GC Ultra and a Thermo Finnigan TraceDSQ Mass Spectrometry and a colon Thermo TR 5MS(60 m–0.25 mm–0.25 μm) (carrier gas He, 1.5 mL/min;temperature program: 50 °C for 4 min, then the temperaturewas increased at 5 °C/min to 250 °C and held for 10 min).
Results and discussion
Synthesis of the unsaturated copolyesters
Copolyesters from the mixture of octanoic acid and soyaoily acids in three different compositions and copolyesterfrom the mixture of 10-undecenoic acid and soya oily acids
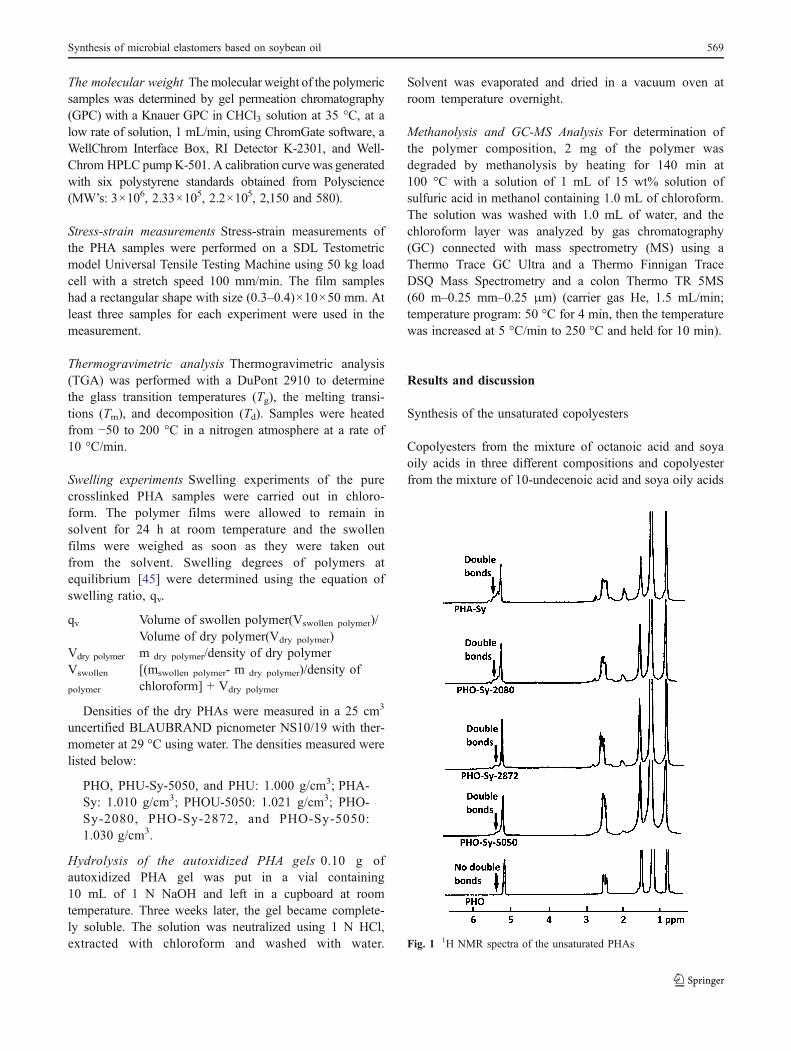
Fig. 1 1H NMR spectra of the unsaturated PHAs
Synthesis of microbial elastomers based on soybean oil 569
(1:1) were biosynthesized using Pseudomonas oleovorans.A series of the copolyesters were coded as PHO-Sy-2080, -2872, -5050 and PHU-Sy-5050 (numbers indicate thefeeding weight ratio of the related acids. PHO-Sy andPHU-Sy copolyesters were obtained in cell yield betweenfrom 16 to 25%. When we compare the physical propertiesof the unsaturated copolyesters before auto oxidation, PHO-Sy-5050 was only good enough for film preparation whilethe others were all soft and/or sticky and not suitable formaking film to use in industrial and medical application.Mechanical strength of the copolyester films increases asPHO content of the copolyester increases because PHO is athermoplastic material having a good mechanical strength.When the unsaturated soybean oily acids are introducedinto the PHO-copolyesters, it is possible to obtain crosslinked elastomer like PHAs which can be promisingbiomaterials in tissue engineering. The difference inmechanical strength between initial and cross linked PHAswill be discussed later. GPC measurements copolyesters gavethe molecular weight (Mn) of the co-polyesters at around60,000 with the molecular weight distribution (MWD) 3.0.
1H NMR analysis
Figure 1 shows 1H NMR spectra of the PHA-Sy and PHO-Sy copolyesters. Characteristic signals of the double bondswere observed at δ: 5.2–5.3 ppm (next to the CH-O- signalat 5.1 ppm). In order to calculate double bond content ofthe PHAs, since bromine addition reaction to double bondsis quantitative, unsaturated PHAs were brominated in darkat room temperature. 1H NMR spectra of the brominatedPHAs confirmed that the double bonds completely con-verted to the brominated moities. Results and conditions ofthe bromination reactions and the calculated double bondcontent of the unsaturated copolyesters have been listed inTable 1. Double bond of polyester obtained from soya oilwas 3.3 mol% while soya oil, in the beginning had double
bonds 11.5 mol%. During biosynthetic production of thepolyester, microorganism does not produce the polyesterwith double bonds which are the same as the ones in thesubstrate, the soya oily acids substrate has been transformedto saturated biopolyester together with the unsaturated ones
Entry# Molecular weight Unsatu-rationa
(mol%)Initial feeding PHA PHA-Br
Substrate Ratio, Mn MWD Mn MWD(%) (×104) (×104)
PHA-Sy Sy 100 7.2 1.80 0.80 2.29 3.3
PHO-Sy-2080 Sy / O 80/20 5.1 3.07 1.25 2.42 2.2
PHO-Sy-2872 Sy / O 72/28 6.3 2.57 1.88 2.79 1.5
PHO-Sy-5050 Sy / O 50/50 6.0 2.66 3.81 1.87 0.8
PHU-Sy-5050 U / Sy 50/50 6.3 2.80 2.16 1.72 20
PHOU-5050b O / U 50/50 6.4 3.11 11
PHUb U 100 3.6 1.92 18
PHO (control) O 100 5.1 3.69 – –
Table 1 Molecular weight anddouble bond content of thebiosynthesised unsaturatedcopolyesters
Sy soya oily acids, O octanoicacid, U 10-undecenoic acid,PHA-Br brominated PHAa Calculated from their 1 H NMRspectra using the methoddescribed in Ref [46]b Ref [44]
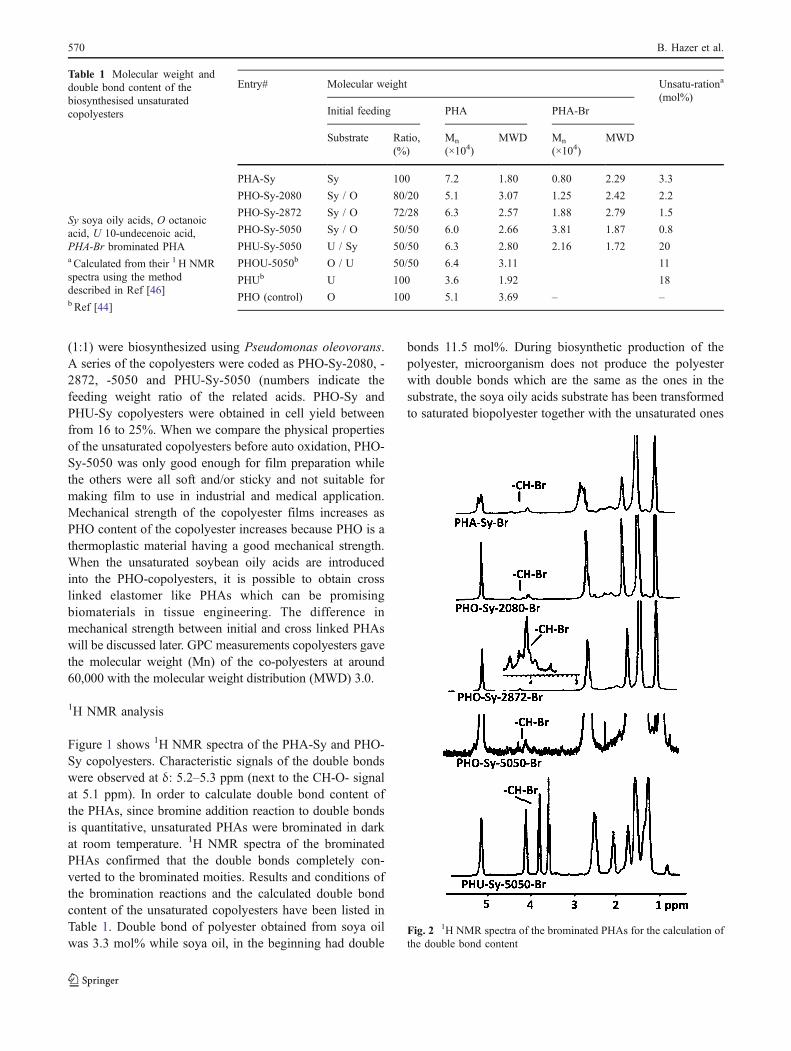
Fig. 2 1H NMR spectra of the brominated PHAs for the calculation ofthe double bond content
570 B. Hazer et al.

[38]. Because of this, double bond content of thebiopolyester obtained from soya oily acids (PHA-Sy) was3.3 mol% instead of 11.5 mol%. In this manner, thepolyesters obtained from the mixture of octanoic acid andsoya oily acid contained double bonds of 2.2, 1.5 and0.8 mol% according to the initial feeding ratios of the soyaoily and octanoic acid mixture which were less thancalculated values (Table 1).
Calculated double bond contents of the PHAs werevaried from 0.8 to 20 mol %,of which is proportional to thefeeding ratio of the olefinic substrate (Table 1). As wediscussed below, PHO-Sy copolyesters had lower doublebond content varying from 0.8 to 2.2 and gave theelastomer film when they crosslinked via autoxidation.
Figure 2 indicates 1H NMR spectra of the brominatedPHAs. The signals of the halides in the brominated PHAsappeared at δ: 3.5–4.5 ppm while the double bond signalsat δ: 5.2–5.3 ppm disappeared. The integral ratio of thehalide to that of the whole molecule gave the mol % ofdouble bond inclusion in the unsaturated PHAs [46].Molecular weight of the brominated PHAs was relativelylower than the original ones. The bromination reactionsdropped the molecular weight of the PHAs roughly from60,000 to 20,000 (Table 1).
FTIR spectra of the unsaturated PHAs have alsocharacteristic signals of the double bonds at 910 (strong),1,642 (strong) and 3,080 cm−1 (strong) for PHU copo-lyester; 910 (weak) and 1,642 cm−1 (weak) for PHA-Sy andPHO-Sy copolyesters (Spectra were not shown).
These new unsaturated copolyesters and a mixture ofPHU and PHA-Sy (1:1) were autoxidized for 40 days atroom temperature in order to obtain elastoplastic materials.
The autoxidation of the unsaturated PHAs obtained fromsoybean oil was similar to the oxidation process of precursorsoybean oil [47–49]. Autoxidation of poly unsaturated acids/oils in the air involves hydrogen abstraction from a methylenegroup between two double bonds in a polyunsaturated fattyacid chain and then cross linking via radical recombinationtogether with the peroxide derivatives. Scheme 1 shows thesimple oxidation process of the linoleic acid inclusion of thecopolyesters. Oxidation of the allylic hydrogen in the un-saturated side chain of the copolyester gives new –C-O-
bonds creating new signals in its 1H NMR spectrum at δppm):3.6–4.4. Because of the cross linking, double bond content ofthe copolyesters was also decreased during autoxidation.
Hydrolysis of the cross-linked gel
To confirm the oxide groups formed by the air oxygen inthe copolyester, the gels were hydrolyzed into a 1 N NaOHsolution at room temperature and the oxide groups werepartially transformed to hydroxyl groups. The cross-linkedpolymer was completely soluble after 3 weeks of hydroly-sis. Acidified aqueous solution was extracted with chloro-form and hydrolyzed products were isolated. 1H NMRspectrum of the hydrolyzed PHO-Sy-2872-ox was contain-ing the characteristic –CH-OH signals at 3.6–4.3 ppm asshown in Fig. 3.
Sol–gel analysis
Formation of the cross linked structure of the autoxidized un-saturated copolyesters was observed by using sol–gel analysisin chloroform. Sol–gel analysis of the autoxidized copolyestersindicated that the crosslinked polymer inclusion changed from
Scheme 1 Autoxidationprocess of the unsaturatedcopolyester, PHA-Sy
Fig. 3 1H NMR spectra of the NaOH solubilized PHO-Sy-2872-ox
Synthesis of microbial elastomers based on soybean oil 571

77% to 99%. As the octanoic acid content in the copolyesterincreases, the swelling degree increases as well. Increasingswelling degree means less crosslinking density which can beattributed to increasing elasticity and longer elongation [50].Increasing the cross linking density generally makes materialsmore stiff and decreases the swelling degree in a good solvent.In this manner, autoxidized PHU indicating the smallestswelling degree (qv=5.5) was brittle because of its highcrosslink density. On the other hand, the other cross-linkedPHAs indicated elastoplastic properties because of their higherswelling degrees (ca. qv varied from 8 to 18).
When analyzed by 1H NMR, soluble part of the autoxidizedPHAs was found to be mostly similar to that of the PHO. As a
result of this, it may be attributed that the copolyester havingless unsaturation with higher ratio of PHO units does notincorporate to the cross linking reaction. Molecular weights ofthe soluble fractions of the autoxidized PHO-Sy-copolyesterswere found to be at around Mn 18,000 with MWD 3.0, whichwas nearly three times lower than those of the precursors.Figure 4 shows GPC chromatograms of the precursorunsaturated copolyesters and the soluble parts of the autoxi-dized ones which were all unimodal except that of soluble partof the PHOU-5050-ox. In case of PHOU-5050, autoxidationcaused a significant additional shoulder in the high molecularregion (e.g. Mn=280,000 g/mol). This signal indicated thechain extension leading to cross-linking [42].
GC-MS analysis of the PHAs
Both unsaturated and autoxidized PHAs were also analyzedby the GC-MS technique. Each hydrolyzed and esterifiedsample of biopolyester was syringed to the instrument. Themain GC-spectra of each sample were drawn. Then everypeak in the main spectrum was analyzed by means of massspectrum. Then the computer program matched this massspectrum to the reference compound into the library.Figure 5 shows the GC-MS spectra of the unsaturated(a) and autooxidized (b) PHAs, respectively. All of thePHAs were containing both unsaturated and saturatedunits. Each of these consisted of mainly C8, C10 and C16
saturated carboxylic acid units (retention times: 11.4,14.6 and 19.5, respectively). Additionally they haveunsaturated units containing more than 12 carbon atomsappearing at the retention times, mostly, 20.2, 26.0, 30.6and 40–42 min which partially change when they areautoxidized.
Stress-strain measurements
Stress-strain measurements [51] tell us a material elastomeror thermoplastic indicating plastic deformation. Plasticdeformation was calculated using the following equation:
Plastic deformation;% ¼ Final length of the PHA� film after strain at break
Initial length of the PHA� film� 100
Young’s modulus (E) is an important term indicating themechanical strength of the material and E is defined as
E ¼ stres½ �strain½ � ¼
LoF
ΔLA
where Lo is the equilibrium length, ΔL is the length changeunder the applied stress, F is the force applied, and A is thearea over which the force applied.
Original films of the unsaturated PHAs before autoxida-tion process were very sticky and proper films for stress-strain measurement could not be obtained. Only the properfilm of original PHO-Sy-5050 was obtained for themechanical tests in order to be compared with theautoxidized cross linked sample.
Mechanical properties of the auto oxidized unsaturatedPHAs and the PHO-control, PHO-Sy-5050-original have
Fig. 4 GPC chromatograms of the unsaturated PHAs and PHA solsextracted from the autoxidized PHAs (PHA-ox-soluble)
572 B. Hazer et al.

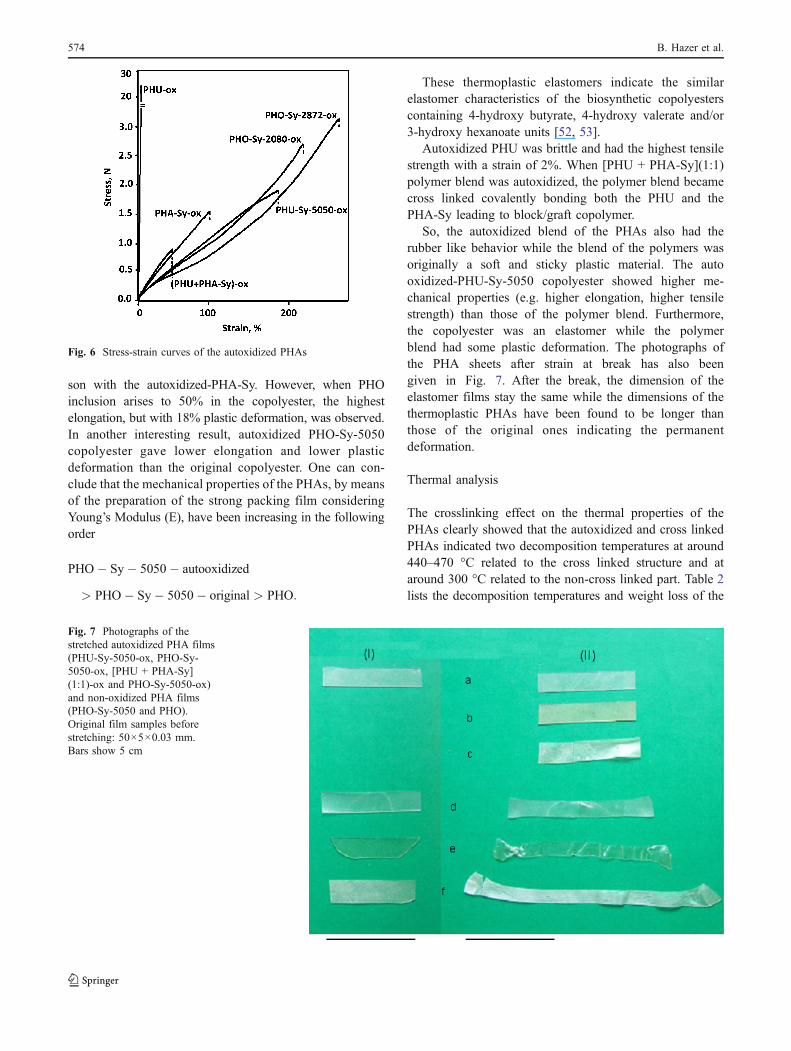
been listed in Table 2. PHO-control had the highest strain(843%) and a quite high tensile strength (0.89 MPa).Because of the effect of high tensile strength and highelongation of PHO, the insertion of PHO into the PHA-Syresulted in elastomers. In addition, the insertion of the PHOinto the unsaturated PHAs lowered the double bond contentwhich leads to the lower cross linking density and thehigher elongation. Figure 6 shows the stress-strain behav-iors of the PHA samples. Auto oxidized PHA-Sy was anelastomer with a strain of 100% and tensile strength of0.5 MPa. Some PHO insertion into this polyester may come
from the preculture using octanoic acid. While the PHOcontent in the copolyester gradually increases, the elongationand stress at the peak of the copolyesters also increase as inthe following order:
PHO� Sy� 2872� ox > PHO� Sy� 2080� ox
> PHA� Sy� ox:
Autoxidized-PHO-Sy-2080 and PHO-Sy 2872 had theelastomeric properties having higher elongation in compari-
Fig. 5 a GC-MS spectra of theunsaturated PHAs, b GC-MSspectra of the autoxidized PHAs
Table 2 Mechanical properties of the 40 days autoxidized PHAs
PHA samples Stress @ Peak, Strain @ Break, E, Young’s Modulusa, Plastic deformationb,N/mm2 % N/mm2 %
PHA-Sy-ox 0.48 100 0.52 0 (elastomer)
PHO-Sy-2080-ox 0.70 203 0.30 0 (elastomer)
PHO-Sy-2872-ox 1.03 233 0.34 0 (elastomer)
PHU-Sy-5050-ox 0.53 170 0.34 0 (elastomer)
(PHU + PHA-Sy)-ox 0.34 81 0.80 4 (elastoplastic)
PHO-Sy-5050-ox 10.5 299 2.36 18 (elastoplastic)
PHO-Sy-5050 14.1 596 1.86 132 (elastoplastic)
PHO (control) 7.0 843 0.89 220 (elastoplastic)
PHOU-5050-ox 3.3 347 0.77 20 (elastoplastic)
PHU-ox 17.0 4.1 837 2 (brittle)
a Young’s modulus E is defined as E = [stress] / [strain] which is equal to E = (Lo/ΔL)(F/A) where Lo is the equilibrium length, ΔL is the lengthchange under the applied stres, F is the force applied, and A is the area over which the force appliedb Plastic deformation % = (Final length of the PHA-film after elongated / Initial length of the PHA-film)×100
Synthesis of microbial elastomers based on soybean oil 573
son with the autoxidized-PHA-Sy. However, when PHOinclusion arises to 50% in the copolyester, the highestelongation, but with 18% plastic deformation, was observed.In another interesting result, autoxidized PHO-Sy-5050copolyester gave lower elongation and lower plasticdeformation than the original copolyester. One can con-clude that the mechanical properties of the PHAs, by meansof the preparation of the strong packing film consideringYoung’s Modulus (E), have been increasing in the followingorder
PHO� Sy� 5050� autooxidized
> PHO� Sy� 5050� original > PHO:
These thermoplastic elastomers indicate the similarelastomer characteristics of the biosynthetic copolyesterscontaining 4-hydroxy butyrate, 4-hydroxy valerate and/or3-hydroxy hexanoate units [52, 53].
Autoxidized PHU was brittle and had the highest tensilestrength with a strain of 2%. When [PHU + PHA-Sy](1:1)polymer blend was autoxidized, the polymer blend becamecross linked covalently bonding both the PHU and thePHA-Sy leading to block/graft copolymer.
So, the autoxidized blend of the PHAs also had therubber like behavior while the blend of the polymers wasoriginally a soft and sticky plastic material. The autooxidized-PHU-Sy-5050 copolyester showed higher me-chanical properties (e.g. higher elongation, higher tensilestrength) than those of the polymer blend. Furthermore,the copolyester was an elastomer while the polymerblend had some plastic deformation. The photographs ofthe PHA sheets after strain at break has also beengiven in Fig. 7. After the break, the dimension of theelastomer films stay the same while the dimensions of thethermoplastic PHAs have been found to be longer thanthose of the original ones indicating the permanentdeformation.
Thermal analysis
The crosslinking effect on the thermal properties of thePHAs clearly showed that the autoxidized and cross linkedPHAs indicated two decomposition temperatures at around440–470 °C related to the cross linked structure and ataround 300 °C related to the non-cross linked part. Table 2lists the decomposition temperatures and weight loss of the
Fig. 6 Stress-strain curves of the autoxidized PHAs
Fig. 7 Photographs of thestretched autoxidized PHA films(PHU-Sy-5050-ox, PHO-Sy-5050-ox, [PHU + PHA-Sy](1:1)-ox and PHO-Sy-5050-ox)and non-oxidized PHA films(PHO-Sy-5050 and PHO).Original film samples beforestretching: 50×5×0.03 mm.Bars show 5 cm
574 B. Hazer et al.
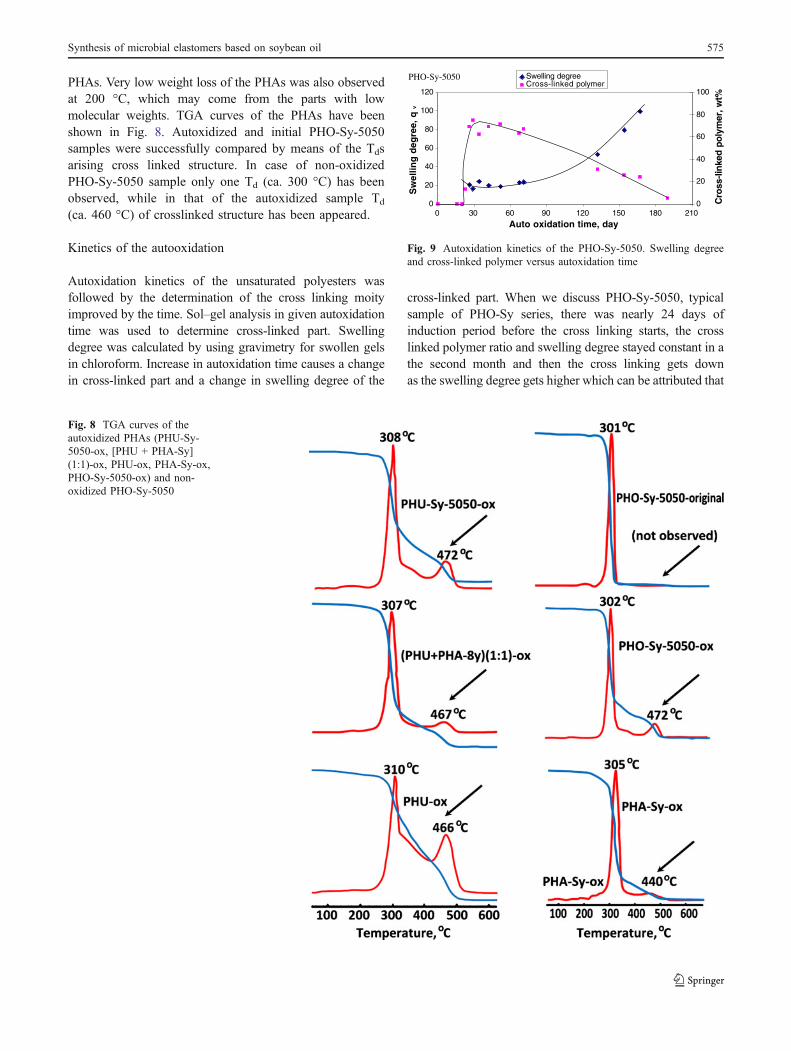
PHAs. Very low weight loss of the PHAs was also observedat 200 °C, which may come from the parts with lowmolecular weights. TGA curves of the PHAs have beenshown in Fig. 8. Autoxidized and initial PHO-Sy-5050samples were successfully compared by means of the Tdsarising cross linked structure. In case of non-oxidizedPHO-Sy-5050 sample only one Td (ca. 300 °C) has beenobserved, while in that of the autoxidized sample Td
(ca. 460 °C) of crosslinked structure has been appeared.
Kinetics of the autooxidation
Autoxidation kinetics of the unsaturated polyesters wasfollowed by the determination of the cross linking moityimproved by the time. Sol–gel analysis in given autoxidationtime was used to determine cross-linked part. Swellingdegree was calculated by using gravimetry for swollen gelsin chloroform. Increase in autoxidation time causes a changein cross-linked part and a change in swelling degree of the
cross-linked part. When we discuss PHO-Sy-5050, typicalsample of PHO-Sy series, there was nearly 24 days ofinduction period before the cross linking starts, the crosslinked polymer ratio and swelling degree stayed constant in athe second month and then the cross linking gets downas the swelling degree gets higher which can be attributed that
0
20
40
60
80
100
120
0 30 60 90 120 150 180 210
Auto oxidation time, day
Sw
ellin
g d
egre
e, q
v
0
20
40
60
80
100
Cro
ss-li
nked
pol
ymer
, wt%
Swelling degreeCross-linked polymer
PHO-Sy-5050
Fig. 9 Autoxidation kinetics of the PHO-Sy-5050. Swelling degreeand cross-linked polymer versus autoxidation time
Fig. 8 TGA curves of theautoxidized PHAs (PHU-Sy-5050-ox, [PHU + PHA-Sy](1:1)-ox, PHU-ox, PHA-Sy-ox,PHO-Sy-5050-ox) and non-oxidized PHO-Sy-5050
Synthesis of microbial elastomers based on soybean oil 575

the degradation has started. Therefore one can conclude that6 months can be accepted as shelf life of this PHO-Sy-5050.The other members of the PHO-Sy series (PHO-Sy-2080 andPHO-Sy-2872) gave the same results. Figure 9 shows thechange in cross-linked part and swelling degree by the autooxidation time for PHO-Sy-5050.
In case of PHU-copolyesters, smooth decrease in swellingdegree was observed without induction period while cross-linked part ratio stayed constant by the autoxidation time. Itseems to be that the cross linking gets denser regarding to thedecrease in swelling degree. The higher the degree ofcrosslinking is, the higher will be the stiffness. So the sameshelf life (ca. 6 month) can also be acceptable for these PHU-copolyesters PHOU-5050.
Smooth decrease in swelling degrees versus the autoxida-tion time was used to calculate the first order reaction rateconstant, k. Taking the first swelling degree qv
o and that ofthe following qv, from the slope of the Lnqv
o/qv versusautoxidation time, k calculated as 5.3×10−7 s−1 for PHU-Sy-5050 and 2.06×10−7 s−1 for PHOU-5050 which is in goodagreement with the k value for autoxidation of PHOU at60 °C found by Schmid et al. [42].
As conclusion, autoxidation is a very useful tool toprepare the bio elastomers from the unsaturated PHAs basedon the soybean oil, which can be promising bio-basedmaterials for the industrial and medical applications. By usingwide range of compositions of the substrate mixtures of soyaoily acids with octanoic acid or undecenoic acid, some morenew thermoplastic elastomers such as PHO-Sy-5050 and bioelastomers such as autoxidized-PHO-Sy-2080, autoxidized-PHO-Sy-2872; autoxidized-PHU-Sy-5050 and a mixture ofPHU and PHA-Sy copolyesters can be obtained. The mixtureof the PHU and PHO-Sy copolyester can also give a largevariety of the bio elasto plastic materials with a wide variety ofshelf life. The decomposition temperature of the cross-linkedPHAs formed by the autoxidation was observed in the higherrange. Autoxidized PHA had a second Td at around 460 °Ccoming from the cross-linked moieties, while an initial PHAhad only one Td at around 300 °C. The biosynthesis of thePHAs and the autoxidation process to obtain elastomers canalso be important for the environment-green chemistry- andreplacing petroleum with plant oils. In addition, theelastomers obtained by autoxidation have not containedany additional inorganic catalyst which may be harmful forliving systems in the medical applications. In this manner,the in vivo experiments indicated that these PHA-copolyesters were biocompatible [54].
Acknowledgement This work financially supported by grants fromZonguldak Karaelmas University Scientific Research Projects Comissiongrant# 2008-70-01-01. We also thank Taner Erdogan for his technicalassistance.
References
1. Anderson AJ, Dawes EA (1990) Microbiol Rev 54:4502. Steinbüchel A, Valentin HE (1995) FEMS Microbiol Lett 128:2193. Orts WJ, Nobes GAR, Kawada J, Nguyen S, Yu G, Ravenelle F
(2008) Can J Chem-Rev Can de Chim 6:6284. Lenz RW, Marchessault RH (2005) Biomacromolecules 6:15. Sudesh K, Abe H, Doi Y (2000) Progr Polym Sci 25:15036. Schmack G, Gorenflo V, Steinbüchel A (1998) Macromolecules
31:6447. Kim DY, Kim HW, Chung MG, Rhee YH (2007) J Microbiol
45:878. Ruth K, de Roo G, Egli T, Ren Q (2008) Biomacromolecules
9:16529. Hazer B, Lenz RW, Fuller RC (1994) Macromolecules 27:45
10. Kim YB, Lenz RW, Fuller RC (1992) Macromolecules 25:185211. Curley JM, Hazer B, Lenz RW, Fuller RC (1996) Macromolecules
29:176612. Gross RA, DeMello C, Lenz RW, Brandle H, Fuller RC (1989)
Macromolecules 22:110613. Koçer H, Borcaklı M, Demirel S, Hazer B (2003) Turk J Chem
27:36514. Lageveen RG, Huisman GW, Preusting H, Ketelaar P, Eggink G,
Witholt B (1998) Appl Environ Microbiol 54:292415. Hartmann R, Hany R, Pletscher E, Ritter A, Witholt B, Zinn M
(2006) Biotechnol Bioeng 93:73716. Sparks J, Scholz C (2008) Biomacromolecules 9:209117. Hazer B, Lenz RW, Fuller RC (1996) Polymer 37:595118. Ashby RD, Foglia TA (1998) Appl Microbiol Biotechnol 49:43119. Ballistreri A, Giuffrida M, Guglielmino SPP, Carnazza S, Ferreri
A, Impallomeni G (2001) Int J Biol Macromol 29:10720. Eggink G, van der Wal H, Huijberts GNM, de Waard P (1993) Ind
Crops Prod 1:15721. Ashby RD, Foglia TA, Solaiman DKY, Liu CK, Nunez A, Eggink
G (2000) Int J Biol Macromol 27:35522. Ashby RD, Solaiman DKY, Foglia TA (2005) Biomacromolecules
6:210623. Mecking S (2004) Angew Chem Int Ed 43:107824. Poliakoff M, Licence P (2007) Nature 450:81025. Poliakoff M, Anastas P (2001) Nature 413:25726. Hazer B, Steinbüchel A (2007) Appl Microbiol Biotechnol 74:127. Robertson GL (2006) Food packaging. principles and practice,
2nd edn. CRC Press-Taylor and Francis Group, Boca Raton28. Gagnon KD, Lenz RW, Farris RJ, Fuller RC (1994) Polymer
35:435829. Gagnon KD, Lenz RW, Farris RJ, Fuller RC (1994) Polymer
35:436830. Hazer B (1996) Macromol Chem Phys 197:43131. Ilter S, Hazer B, Borcakli M, Atici M (2001) Macromol Chem
Phys 202:228132. Hazer B (1994) Polymer Bulletin 33:43133. Hazer B, Lenz RW, Çakmaklı B, Borcaklı M, Koçer H (1999)
Macromol Chem Phys 200:190334. Cakmakli B, Hazer B, Borcakli M (2001) Macromol Biosci
1:348–35435. Kim HW, Chung MG, Kim YB, Rhee YH (2008) Int J Biol
Macromol 43:30736. Dufresne A, Reche L, Marchessault RH, Lacroix M (2001) Int J
Biol Macromol 29:7337. Konig GJM, van Bilsen HMM, Lemstra PJ, Hazenberg W,
Witholt B, Preusting H, Galien JG, Schirmer A, Jendrossek D(1994) Polymer 35:2090
38. Hazer B, Torul O, Borcakli M, Lenz RW, Fuller RC, Goodwin SD(1998) J Environ Polym Degrad 6:109
576 B. Hazer et al.

39. Hazer B, Demirel SI, Borcakli M, Eroğlu MS, Cakmak M, ErmanB (2001) Polym Bull 46:389
40. Ashby RD, Foglia TA, Solaiman DKY, Liu C-K, Nuñez A,Eggink G (2000) Int J Biol Macromol 27:355
41. Bassas M, Diaz J, Rodriguez E, Espuny MJ, Prieto MJ, ManresaA (2008) Appl Microbiol Biotechnol 78:587
42. SchmidM, Ritter A, Grubelnik A, ZinnM (2007) Biomacromolecules8:579
43. Çakmaklı B, Hazer B, Tekin İÖ, Cömert FB (2005) Biomacro-molecules 6:1750
44. ErduranlıH,Hazer B, BorcaklıM(2008)Macromol Symposia 269:16145. Hamurcu EE, Hazer B, Baysal BM (1997) Polymer 38:298146. Schneider Y, Azoulay JD, Coffin RC, Bazan GC (2008) J Am
Chem Soc 130:10464
47. Yin H, Porter NA (2003) Anal Biochem 313:319–32648. Marshall GL, Lander JA (1985) Eur Polym J 21:95949. Çakmaklı B, Hazer B, Tekin İÖ, Açıkgöz Ş, Can M (2007) J
Amer Oil Chem Soc 84:7350. Flory PJ (1971) Principles of polymer chemistry, 8th edn. Cornell
University Press, Ithaca51. Ward IM (1983) Mechanical properties of solid polymers, 2nd
edn. Wiley, New York52. Schmack G, Gorenflo V, Steinbüchel A (1998) Macromolecules
31:64453. Doi Y, Kitamura S, Abe H (1995) Macromolecules 28:482254. Hazer DB, Hazer B, Kaymaz F (2009) In vivo biocompatibility of
the elastoplastic poly-3-hydroxy alkanoates based on soya oil.Biomed Mater 4:035011
Synthesis of microbial elastomers based on soybean oil 577




















