
Syntheses of Bisoxazolidines and Morpholones
10
SvMlIETICCOMMUNICATIONS, 29(8), 1277 - 1286 (1999) SYNTHESES OF BISOXAZOLIDINES AND MORPHOLONES Victor Santes,"Aurelio Ortiz,"Rosa Santillan, " Atilano Guti6rrez b and Norberto Farfha* "Departamentode Quimica, Centro de Investigaci6n y de Estudios Avanzados del IPN, Apdo. Postal 14-740,07000 Mexico D.F., Mexico, Email [email protected]. Lab. de RMN, Universidad Aut6noma Metropolitana, Av. Michoach y la Purfsima dn, Col. Vicentina, 09340 Ixtapalapa, M6xico D. F., Mexico. Abstract: The preparation of two new bisoxazolidines, two N-(2-hydroxyethy1)- N-alkylglycine derivatives and two morpholones is described. The structure of (5S,6R)-N-isopropyl-5-methyl-6-phenyl-1,4-oxazin-2-one was established by X- ray crystallographicanalysis. In previous studies we have reported that the reaction of secondary aminoalcohols with glyoxal provides a route to aminoacids,' oxazino-oxazines and bisoxazolidines,2 while secondary anilines afford indol derivative^.^ In contrast, the reaction of primary aminoalcohols with 12-diketones yield heteropropellanes 4 and 2-hydroxy-5,6-dihydro-2H-[1,4]-oxazines? or bi(benzothiazoly1) and benzothiazino-benzothiazine, if o-aminothiophenol is reacted with gy~xal.~ Recently we have applied this sequence to the preparation of 1,4-piperazines by reaction of the N,N'-bis-(norephedrine)ethylendiamine derivatives with 1,Zdiketones and subsequent reduction with dib~rane.~ * To whom correspondence should be addressed Copyright Q 1999 by Marcel Dekker, Inc.
Transcript of Syntheses of Bisoxazolidines and Morpholones

SvMlIETIC COMMUNICATIONS, 29(8), 1277-1286 (1999)
SYNTHESES OF BISOXAZOLIDINES AND MORPHOLONES
Victor Santes," Aurelio Ortiz," Rosa Santillan," Atilano Guti6rrezb and Norberto Farfha*
"Departamento de Quimica, Centro de Investigaci6n y de Estudios Avanzados del IPN, Apdo. Postal 14-740,07000 Mexico D.F., Mexico,
Email [email protected]. Lab. de RMN, Universidad Aut6noma Metropolitana, Av. Michoach y la
Purfsima dn, Col. Vicentina, 09340 Ixtapalapa, M6xico D. F., Mexico.
Abstract: The preparation of two new bisoxazolidines, two N-(2-hydroxyethy1)- N-alkylglycine derivatives and two morpholones is described. The structure of
(5S,6R)-N-isopropyl-5-methyl-6-phenyl-1,4-oxazin-2-one was established by X- ray crystallographic analysis.
In previous studies we have reported that the reaction of secondary
aminoalcohols with glyoxal provides a route to aminoacids,' oxazino-oxazines and
bisoxazolidines,2 while secondary anilines afford indol derivative^.^ In contrast, the
reaction of primary aminoalcohols with 12-diketones yield heteropropellanes4 and
2-hydroxy-5,6-dihydro-2H-[1,4]-oxazines? or bi(benzothiazoly1) and
benzothiazino-benzothiazine, if o-aminothiophenol is reacted with gy~xa l .~ Recently
we have applied this sequence to the preparation of 1 ,4-piperazines by reaction of
the N,N'-bis-(norephedrine)ethylendiamine derivatives with 1,Zdiketones and
subsequent reduction with dib~rane.~
* To whom correspondence should be addressed
Copyright Q 1999 by Marcel Dekker, Inc.

SANTES ET AL.
In continuing with our interest in this type of reactions we report herein the
condensation of four N-alkylnorephedrines ( la- ld) with glyoxal. The
alkylnorephedrine derivatives (la-lc) were prepared by reaction of (-)-
norephedrine with the corresponding alkylbrornide: while compound I d was
obtained by reaction of pivaloyl chloride with (-)-norephedrine and subsequent
reduction with borane THF?
Unequivocal 'H and I3C spectral assignment for compounds 2a-2d was
achieved by 2D 'H and I3C correlated experiments. Reaction of l a and l b with
glyoxal afforded [5R,5'R,4S,4'S,2R,2'R]-5,5'-diphenyl-4,4'-dimethyl-N,N9-
diethyl-2,2'-bisoxazolidine (2a) and [5R,5'R,4S,4'S,2R,2'S]-5,5'-diphenyl-4,4'-
dimethyl-N,N'-dibenzyl-2,2'-bisoxazolidine (2b), respectively (Scheme I), as
established by NMR experiments. The 'H and "C NMR spectra of 2a show signals
for half of the molecule due the existence of a Cz symmetry axis in the molecule.
Determination of the bisoxazolidine type structure for 2a was based on
measurement of the "C satellite coupling constantsZ for H-2 (k4.53 ppm J,,= 154
Hz, 'JHH=3.66 Hz) and the NOESY spectrum which shows through space
interaction between H-2 and CH3-6, thus allowing to establish the configuration for
C-2 as R. In contrast, the 'H NMR spectrum of 2b shows a different coupling
pattern since the protons at the ring fusion give rise to an AF3 system at 6=4.69
ppm and 4.55 ppm (3JHH=5.6 Hz) for H-2 and H-2,' respectively, thus evidencing
that the new stereogenic centers are different. Complete assignment of the 'H NMR spectra for both compounds was based on the COSY and NOESY experiments. The
NOESY spectrum shows interaction between H-2' and H-2, H-4', H-4, the AB system due to the methylene at the position 7', H-5' and H-5; in addition, H-2
shows interaction with the AB ascribed to H,-7 and H-2', establishing the
configuration at the C2 and C2' ring fusion carbons are R and S, respectively. This
in turn allowed assignment of the I3C NMR spectrum based on the HETCOR
spectrum.
Reaction of compounds l c and I d with glyoxal afforded morpholones 2c
and 2d, respectively (Scheme 1) as evidenced by the "C NMR signal at 6=169.1
and k168.8 ppm for the carbonyl group of 2c and 26, respectively. The 'H NMR
spectra show the characteristic AF3 system for the diastereotopic protons alpha to

BISOXAZOLIDINES AND MORPHOLONES
Scheme 1
the carbonyl group at 6=3.79 and k3.34 ppm for 2c and 6=3.69 and 6=3.54 ppm
for 2d.
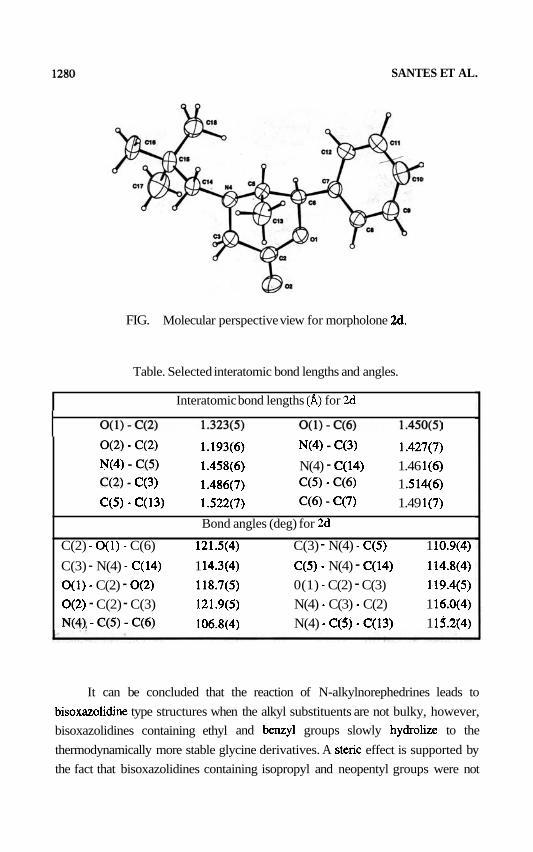
The structure of 2d was established by X-ray crystallografic analysis (Figure
1) which shows that the morpholone has a pseudochair conformation, the phenyl
and neopentyl groups occupy pseudoequatorial positions and the methyl group
occupies a pseudoaxial position. Unit cell parameters and basic information about
data collection and structure refinement are summarized in the experimental
section.1° Selected interatomic bond lengths and angles are summarized in the
Table.
Upon standing in ethanoYchoroform solution 2a and 2b hydrolyze to glycine
derivatives 3a and 3b (Scheme 1). The infrarred spectra show the characteristic
bands at 3 196 and 3268 cm" for the OH groups, and the carbonyl groups at 1628
and 1624 cm-I for 3a and 3b, respectively. The 'H NMR spectrum of 3a shows an
AB system for the H-4 methylene group at 6=3.59 ppm and 3.33 ppm ('J,,= 16.1
Hz), while that of 3b is shifted to 6=3.37 ppm and 3.31 ppm ('JH,=17.5 Hz).
Complete 13C spectral assignment was based on HETCOR experiments.
SANTES ET AL.
FIG. Molecular perspective view for morpholone 26.
Table. Selected interatomic bond lengths and angles.
I Interatomic bond lengths (A) for 2d
o(2) - c(2) 1.193(6) N(4) - C(3) 1.427(7) N(4) - C(5) 1.458(6) N(4) - C(14) 1.46 l(6) c(2) - c(3) 1.486(7) c(5) - c(6) 1 .514(6) C(5) - C(13) 1.522(7) C(6) - c(7) 1.49 l(7)
Bond angles (deg) for 26
C(2) - O(1) - C(6) 121.5(4) C(3) - N(4) - C(5) 1 10.9(4)
C(3) - N(4) - C(14) 1 14.3(4) C(5) - N(4) - C(14) 114.8(4)
O(1) - C(2) - O(2) 118.7(5) 0(1) - C(2) - C(3) 119.4(5)
O(2) - C(2) - C(3) 121.9(5) N(4) - C(3) - C(2) 1 16.0(4)
N(4), - C(5) - C(6) 106.8(4) N(4) - C(S) - C(13) 1 13.2(4)
It can be concluded that the reaction of N-alkylnorephedrines leads to
bisoxazolidine type structures when the alkyl substituents are not bulky, however,
bisoxazolidines containing ethyl and benzyl groups slowly hydrolize to the
thermodynamically more stable glycine derivatives. A steric effect is supported by
the fact that bisoxazolidines containing isopropyl and neopentyl groups were not

BISOXAZOLIDINES AND MORPHOLONES 1281
obtained, the major product being the morpholone derivatives which showed good
stability in methanol solution, under the same reaction conditions.
EXPERIMENTAL
'H and I3C NMR spectra were recorded on Jeol Eclipse +400 and Jeol-270
spectrometers. Chemical shifts (ppm) are relative to (CH,),Si. Coupling constants
are quoted in Hz. The HETCOR and COSY standard pulse sequence, which
incorporates quadrature detection in both domains, was used. NOESY experiments
were recorded on a Bruker DMX500. Infrared spectra were recorded on a Perkin
Elmer 16F spectrophotometer. Mass spectra were obtained with an HP 5989A mass
spectrometer. Melting points were obtained on a Gallenkamp MFB-595 apparatus
and are uncorrected. Elemental microanalyses were performed by Oneida Research
Services, Whitesboro, NY 13492. Data for X-ray crystal structure determination of
26 was collected on an Enraf Nonius CAD4 diffractometer, h (MoKa)=0.71069 A,
monochromator: graphite, T= 293 K, / 28 scan, range lo < 8 < 25"
(5R,5'R,4S,4'S,2R,2'R)-N,N'-diethyl-5,5'-diphenyl-4,4'-dimethyl-2,2'-
bisoxazolidine (2a).
To a solution of la (1.0 g, 5.6 rnrnol) in THF (40 rnl) a 40 wt. % solution of
glyoxal in water (0.41 g, 3.90 mrnol) was added and the reaction mixture was
stirred at room temperature for 6 h. The solution was concentrated under vacuum,
ethyl ether was added and the resulting white precipitated was filtered off and dried
to give 2a (1.2 g, 56%); mp 149-151 "C; 'H NMR (399.78 MHz, CDCI,) 6: 7.40
(2H, d, J=7.3 Hz, H-o), 7.33 (2H, t, J=7.3 Hz, H-m), 7.26 (lH, t, J=7.3Hz,
H-p), 5.48 (lH, d, J=2.6 HZ, H-S), 4.53 (lH, s, H-2). 3.47 (lH, dq, J=2.6, 6.6
Hz, H-4), 2.94 (lH, dq, J=7.2, 12.5 Hz, H-7a), 2.36 (lH, dq, J=7.2, 12.5 Hz,
H-7b), 1 .OO (3H, dd, J=7.2 Hz, H-8), 0.74 (3H, d, J=6.6 Hz, H-6) ppm; 13C
NMR (100.54 MHz, CDC13) 6: 140.05 (C-i), 128.02 (C-m), 126.8 (C-p), 126.23
(C-0), 81.93 (C-2), 72.70 (C-5), 54.54 (C-4), 42.1 1 (C-7), 12.26 (C-8), 5.46
(C-6) ppm; MS m/z (%): M+ -C,H, 303 (25), 288 (loo), 273 (27), 243 (24), 165
(41), 136 (43, 91 (13), 65 (31); IR v,(KBr): 3330, 1470, 1450, 1445, 1088, 1034,630,622,590,538,520 cm:' Elemental analysis: Calc.: C, 75.78; H, 8.42;
N, 7.36. Found: C, 75.85; H, 8.49; N, 7.29.

1282 SANTES ET AL.
(5R,5'R,4S,4'S,2R,2'S)-N,N'-dibenzyl-5,5'-diphenyl-4,4'-dimethyl-2,2'-
bisoxazolidine (2b).
To a solution of l b (2.5 g, 10.4 mmol) in ethanol (40 ml) a 40 % wt. solution of
glyoxal in water (0.30 g, 5.18 mmol) was added, the reaction mixture was refluxed
for 2 h and cooled to room temperature. The solvent was removed under vacuum,
ethyl ether was added and the resulting white precipitate was filtered off and dried to
give 2b (1.07 g, 41%); mp 134-135 "C; 'H NMR (270 MHz, CDC13) 6: 7.19-7.49
(20 H, m, H-arom.), 5.40 (lH, d, J 4 . 6 Hz, H-5), 5.04 (lH, d, J 4 . 7 Hz, H-57,
4.69 and 4.55 (2H, AB, Jc5.6 Hz, H-2 and H-2'), 4.49 and 3.84 (2H, AB, J=3.9
HZ, H-7), 4.28 and 3.88 (2H, AB, J=13.8 Hz, H-7'), 3.51 (lH, dq, J 4 . 6 , 6.6
HZ, H-4), 3.21 (lH, dq, J=4.7, 5.6 HZ, H-47, 0.60 (3H, d, J=6.6 Hz, H-6),
0.53 (3H, d, J=5.6 Hz, H-6') ppm; 13C NMR (67.8 MHz, CDC13) 6: 139.9 (C-8),
139.6 (C-12), 138.9 (C-87, 138.5 (C-12'), 129.0, 128.2, 128.1, 127.9, 127.1,
126.8 and 126.1 ( C-10, C-9, C-14, (2-13, C-lo', C-9', C-14' and C-13'), 128.6,
128.0, 127.0 and 126.5 (C-1 1, C-15, C-11' and C-15'), 97.7 (C-2'), 95.1 (C-2),
81.5 (C-5), 80.9 (C-5'), 61.7 (C-4'), 58.7 (C-7'), 57.5 (C-4), 50.9 (C-7), 17.7
(C-6'), 8.7 (C-6) ppm; MS, m/z (%): 254 (3), 253 (33), M'D 252 (100), 225 (3),
224 (15), 120 (3), 118 (6), 92 (3), 91 (34); IR v-(KBr): 3334, 1464, 1034,
1010, 636, 620, 602, 556 cm.-' Elemental analysis: Calc.: C, 80.95; H, 7.14; N,
5.55. Found: C, 80.54; H, 7.24; N, 5.45.
(5S, 6R)- 6-phenyl-5-methyl-N-isopropyl-1,4-oxazin-2-one (2c).
To a solution of l c (1.0 g, 5.2 mrnol) in ethanol (40 ml) a 40 % wt. solution of
glyoxal in water (0.30 g, 5.2 mrnol) was added, the reaction mixture was refluxed
for 2 h and cooled to room temperature. Removal of the solvent under vacuum
yielded morpholone 212 as a yellow oil (1.15 g, 95%); bp 100 "C/300rnrn (dec.); 'H
NMR (270 MHz, CDCl,) 6: 7.25-7.39 (5H, m, H-arom.), 5.60 (lH, d, J=3.1 Hz, H-6), 3.79 and 3.34 (2H, AB, J=18.1 Hz, H-3), 3.40 (lH, dq, J=3.1, 6.6 Hz,
H-5), 2.70 (1 H, heptet, J=6.3 Hz, CH-8), 1.12 (3H, d, J=6.3 Hz, CH3-9), 1.10 (3H, d, J=6.3 Hz, CH3-Y), 0.70 (3H. d, J=6.6 Hz, H-7) ppm; I3C NMR (67.8
MHz, CDC13) 6: 169.1 (C=O), 136.9 (C-i), 128.4 (C-m), 128.0 (C-p), 125.8
(CO), 83.8 (C-6), 52.6 (C-5), 50.8 (CH-8), 48.0 (C-3), 20.7 and 19.5 (CH3-9'

BISOXAZOLIDINES AND MORPHOLONES 1283
and CH3-9), 5.7 (C-7) ppm; MS m/z (%): M', 233 (32), 118 (47), 99 (100),
91(27), 86 (66), 85 (65), 84 (53, 83 (72), 77 (4% 56 (93), 44 (43), 28 (28); IR
v , (film, NaCl): 2972, 1732, 1682 (CO), 1634, 1456, 1258, 1002, 916, 700
cm.-'
(5S, 6R)-6-phenyl-5-methyl-N-neopentyl-1, 4-oxazin-Zone (26). To a solution of I d (1.0 g, 4.5 mmol) in ethanol (40 rnl) a 40% wt. solution of
glyoxal in water (0.26 g, 4.5 mmol) was added and the reaction mixture was
refluxed for 2 h and cooled to room temperature. Removal of the solvent under
vacuum afforded morpholone 2d as a white solid (0.7 g, 63%); mp 106-108 "C;
'H NMR (270 MHz, CDCI,) 6: 7.26-7.39 (SH, m, H-arom.), 5.76 (lH, d, J=3.3
Hz, H-6), 3.69 and 3.54, (2H, AB, J=18.8 Hz, H-3), 3.09 (1 H, dq, J=3.3 Hz,
6.6 HZ, H-5), 2.40 and 2.10 (2H, AB, J=13.8 HZ, CH,-8), 0.93 (9H, S,
CH3-lo), 0.75 (3H, d, J=6.6 Hz, H-7) ppm; I3C NMR (67.8 MHz, CDC1,) 6:
168.8 (C=O), 136.9 (C-i), 128.4 (C-m), 127.9 (C-p), 125.4 (C-o), 83.5 (C-6),
67.1 (CH,-8). 58.1 (C-5), 52.2 (C-3), 33.5 (C-9), 27.7 (CH3-10). 5.3 (C-7) ppm.
MS m/t (%): M', 261 (1 I), 246 (7), 205 (14). 204 (100), 146 (13), 117 (8), 112
(48), 77 (S), 71 (25), 56 (20), 43 (14). 42 (84). 41 (16), 29 (1 l), 28 (5); IR
v ,(KBr): 2976,2950,2918,2864, 1740 (CO), 1380, 1372, 1288, 1258, 1240,
1136, 1004,738,704 cm-'; Elemental analysis: calc.: C, 73.56; H, 8.81; N, 5.36.
Found: C, 73.75; H, 8.98; N, 5.34.
N-ethyl-N-(l-(S)-2-(R)-phenyl-2-hydroxyethyl)glycine (3a).
Compound 2a hydrolyzed in chloroform/methanol solution to give 3a. The same
compound was obtained when l a was heated with glyoxal in ethanol for 8 h, white
solid mp 163-165 "C; 'H NMR (399.78 MHz, DMSO-4) 6: 7.37 (2H, d, J=7.3
Hz, H-o), 7.32 (2H. t, J=7.3 Hz, H-m), 7.22 (lH, t, J=7.3 Hz, H-p), 5.05 (lH,
d, Jz1.8 HZ, H-1), 3.59 and 3.33 (2H, AB, Jz16.1 HZ, H-4), 3.26 (lH, dq,
J=1.8, 6.6 Hz, H-2), 3.00 (2H, m, CH,-7), 1 .O9 (3H, t, J=7.1 Hz, H-8). 0.94
(3H, d, J=6.6 Hz, H-6) ppm; "C NMR (100.54 MHz, DMSO-4) 6: 169.7 (C-5),

1284 SANTES ET AL.
143.7 (C-i), 128.4 (C-m), 127.4 (C-p), 126.4 (C-o), 71.8 (C-1), 63.7 (C-2), 53.2
(C-4), 48.4 (C-7), 11.6 (C-8), 8.4 (C-6) ppm; MS m/z (%): [M+ -H20, 219 (OS)],
192 (0.3), 130 (loo), 105 (8), 85 (20), 77 (30), 58 (46), 56 (82), 28 (18); IR v,,
(KBr): 3196 (OH), 3022,2852,1628 (CO), 1388, 1330, 1062, 1044, 1004, 750,
706 cm:' Elemental analysis: calc.: C, 65.82; H, 8.01; N, 5.90. Found: C, 65.85;
H, 8.03; N, 5.78.
N-benzyl-N-(l-(S)-2-(R)-phenyl-2-hydroxyethyl)glycine (3b).
Compound 2b was allowed to stand in chloroform for three weeks giving 3b as
white solid mp 155-157 "C; 'H NMR (270 MHz, DMSO-d,) 6: 7.26-7.21 ( 1 OH,
m,H-arom),4.81 (lH, d, J=3.9 Hz, H-1), 3.82 and3.73 (2H, AB, J=14.0 Hz,
CH,-7). 3.37 and 3.31 (2H, AB, J=17.5 Hz, H-4), 2.83 (lH, dq, J=3.9, 6.7 Hz,
H-2), 0.92 (3H, d, J=6.7 Hz, H-6) ppm; I3C NMR (67.8 MHz, DMSO-d,) 6:
173.7 (C-5), 145.4 (C-i), 140.1 (C-i'), 129.0, 128.6, 128.2 and 126.6 (C-m,
C-m', C-o' and C-o), 127.3 and (126.9 (C-p' and C-p), 74.2 (C-1), 61.2 (C-2),
55.8 (CH2-7), 52.1 (C-4), 9.9 (C-6) ppm; MS, m/z (%): [M' -H20, 28 1 (1 2)], 192
(47), 147 (43), 91 (87), 56 (loo), 29 (46 ); IR v ,,,(KBr): 3268 (OH), 3064,
2998, 1624 (CO), 1458, 1450, 1390, 1344, 1328, 764, 748 and 704 cm:'
Elemental analysis: calc.: C, 72.24; H, 7.02; N, 4.68. Found: C, 71.87; H, 7.00;
N, 4.62.
Acknowledgment. Financial support from CONACYT is acknowledged.
REFERENCES AND NOTES
Farfhn, N., CuCllar, L., Aceves, J. M. and Contreras, R., Synthesis, 1987,
927.
2, a) Farfh, N., Santillan, R. L., Castillo, D., Cruz, R., Joseph-Nathan, P.
and Daran. J. C., Can. J. Chem., 1992,70, 2664; c) Farfh, N., Santillan,

BISOXAZOLIDINES AND MORPHOLONES 1285
R., Guzmin, J.B., Castillo, B., Ortiz, A., Daran, J. C., Robert, F. and
Halut, S., Tetrahedron, 1994, 50, 9951.
Farfin, N., Hernindez, J. M., Joseph-Nathan, P. and Contreras, R.,
J.Heterocyclic Chem., 1990, 27, 1745.
Ortiz, A., Carrasco, J., Hopfl, H., Santillan, R. and N. Farfh Synth.
Commun., 1998, 28, 1293.
Ortiz, A., Farfh, N., Santillan, R., Rosales, M. J., Garcia-BaCz, E., Daran,
J. C. and Halut, S., Tetrahedron Asymmetry, 1995, 6, 2715.
Farfh, N., Santillan, R., Castillo, B., Carretero, P., Rosales, M. de J.,
Garcia-Bbz, E., Flores-Vela, A., Daran, J. C. and Halut, S., J. Chem.
Res.(S), 1994, 458, ( M ) , 1994, 2521.
Ortiz, A., Farfin, N., Hopfl, H., Santillan, R. and GutiCrrez A.,
Tetrahedron:Asymmetry ,1998 (in press).
Chaloner, P. A., Langadianou, E. and Renuka Perera S. A., J. Chem. Soc.
Perkin Trans. 1, 1991, 273 1.
Tlihuext, H. and Contreras, R., Tetrahedron: Asymmetry, 1992, 3,727.
Crystal data for compound 2d: Colourless crystals, Cl,H,3N02 (M=261.36
gmol-I), crystallized in the orthorrombic space group P 2,2,2,, a=8. 39O(4),
b=10.404(5), c=17.467(4) A, V=1524(1) A? 2=4, pcalcd= 0.70 ~ g m . " A total of
1572 reflections was measured of which 1551 were independent, absorption
correction: DIFABS (rnin: 0.82, max: 1.20), corrections were made for Lorentz and
polarization effects. Solution and refinement: direct methods (SHELXS-86) for
structure solution. Nonhydrogen atoms were refined anisotropically, hydrogen
atoms were found by difference Fourier maps and refined with an overall isotropic
thermal parameter, least squares refinements were carried out by minimizing the
function CW(IF,,I-IF,~)~, where F, and F, are the observed and calculated structure
factors. Unit weight was used. Models reached convergence with
R=X(IIFJ-IFJI)/CIFJ and RW=[ZW(IF,I-IF,~)~IC~(F,,)~]'~ with values R= 0.040,
Rw= 0.037 from 847 reflections with F>3a(F) for 173 variables against IF/,
w= 1 .O, s= 1.87. Largest residual electron density peak / hole in the final difference
map: pmax=0.18, Pmin= -0.36 e - / A3. (CRYSTALS, Program for Refinement of
Crystal Structures, D. J. Watkin, J. R Caruthers, P.V. Betteridge, Univ. Oxford,
version 9, 1994).

1286 SANTES ET AL.
Supplementary Material available: Tables of atomic coordinates, thermal parameters, bond lengths and angles and observed and calculated structure factors have been
deposited at the Cambridge Crystallographic Data Center.
(Received in the USA 06 October 1998)




















