
Zinc-Coated Urea for Enhanced Zinc Biofortification, Nitrogen ...
Upload
khangminh22Category
view
0download
0
Syntheses and Structures of Copper and Zinc Complexes with
N � 0 Donor Ligands
by
CHAN Sau Han
A thesis submitted in partial fulfillment of the requirements
for the degree of Master of Philosophy
in Chemistry
© The Chinese University of Hong Kong August 2001
The Chinese University of Hong Kong holds the copyright of this thesis. Any person(s) intending to use a part or whole of the materials in the thesis in a proposed publication must seek copyright release from the Dean of the Graduate School.

统 系 書 口 \ 气
I [ " 婦 • ) i
N pXlibrary

Thesis Committee:
Prof. Wai Kee L I
Prof. Thomas C. W. MAK
Prof. Hung Kay LEE (Supervisor)
Prof. Xiao-Ming CHEN (External Examiner, School of Chemistry and
Chemical Engineering, Zhongshan University)

ABSTRACT
This thesis describes the syntheses, structures and characterization of a series of
copper and zinc complexes derived from tripodal tetradentate N3O ligands,
containing phenolate pendants.
Chapter 1 gives a general introduction to the roles of copper in biological
systems. The chemistry of radical-copper proteins is also introduced.
Chapter 2 describes the syntheses and characterization of a series of
monomeric copper(II) and analogous zinc(II) complexes of the type [M(L)(X)] (M =
Cu, Zn; X = OAc, CI), where L being pyridine-based tripodal tetradentate N3O donor
ligands containing phenolate pendants. Subsequent oxidation of these complexes
with cerium(IV) ammonium nitrate has been studied.
Chapter 3 describes the syntheses and characterization of several novel mono-
and dimeric copper(I) complexes containing benzimidazole- and pyridine-based
tripodal tetradentate N3O donor ligands. The reactivities of these complexes toward
dioxygen have also been investigated.
Chapter 4 gives the experimental details of compounds described in the
preceding chapters.
i

摘要
本論文敘述了一系列由利三氮單氧四齒配體配位的具有三角支架特點的銅
鋅絡合物的合成、鑒定及結構特性。
第一章敘述了銅配合物在生物體內的作用,同時,還對自由基銅蛋白化學
進行簡要介紹。
第二章敘述了由卩a:卩定衍生的包含酌驢側基的三氮單氧四齒配體配位、具有
三角支架特點的單聚銅鋅絡合物[M(L)(X)] (M = Cu, Zn; X = OAc, CI)
的合成及鑒定o同時’還對該類絡合物在确酸鈽錢作用下的氧化反應進行了硏
究°
第三章敘述幾個新型的由苯并咪嗖和rtfc卩定衍生的三氮單氧四齒配體配位具
有三角支架特點的單聚和二聚的一價銅絡合物的合成及結構鑒定,還對該類絡
合物跟雙氧的氧化反應進行了硏究。
第四章敘述了以上各章的實驗步驟。
ii

ACKNOWLEDGMENT
I would like to express my sincere thanks to my supervisor, Prof. Hung Kay
LEE for his advice and guidance in my research work and in the preparation of this
thesis.
Thanks are given to Dr. Yee Lok WONG for his special contribution and
helpful discussion on my research project.
I would like to express my sincere gratitute to Dr. Tze Lock CHAN for
providing me a bench space in the first year of my M.Phil, study.
We thank Prof. Sunney L Chan, Vice-President of Academia Sinica, Taiwan,
for provision of a low-temperature accessory for taking all low-temperature UV-Vis
spectra. Thanks should also go to Dr. Svetlana Pavlova of the Institute of Chemistry
at Academia Sinica for her assistance in taking low-temperature UV-Vis spectra.
Thanks are also due to Prof. Thomas C. W. MAK and Miss Hung Wing L I for
carrying out all X-ray crystallographic analyses, and Mr. Kwok Wai KWONG and
Mr. Chung Hei LAM for measuring all mass spectra.
Special thanks are given to Dr. Chunmei WAI, Dr. Gulice Y. C. LEUNG, Mr. <i
Jie JIANG,Mr. Guo Xin WONG, Mr. Kai For MO, Mr. Leung HOU, Dr. Stanton H.
L. KWOK and all the group members for their helpful discussion.
Sau Han CHAN
Department of Chemistry
The Chinese University of Hong Kong
July, 2001.
iii

CONTENTS
Page
ABSTRACT i 摘要 i i
ACKNOWLEDGMENT iii CONTENTS iv ABBREVIATIONS vi
CHAPTER 1 General Introduction 1-A. Role of Copper in Biology 1
1-B. A Brief Review on Radical Copper Proteins 4
1-C. Objectives of This Work 12
1-D. References 13
CHAPTER 2 Copper(II) and Zinc(II) Complexes containing N3O Tetradentate Ligands 2-A. Introduction 14
Results and Discussion
2-B. Preparation of Tetradentate Ligands and 27
Complexes
2-C. Characterization 36
2-D. Generation of Metal Phenoxyl Radical Species 51
2-E. Summary 57
2-F. References 59
CHAPTER 3 Copper(I) Complexes with N3O Tetradentate Ligands 3-A. Introduction 62
Results and Discussion
3-B. Preparation of Copper(I) Complexes with N3O 75
Tetradentate Ligands
iv

3-C. Characterization 79
3-D. Reactivities of 86,87 and 88 toward Dioxygen 88
3-E. Summary 93
3-F. References 94
CHAPTER 4 Experimental Sections 4-A. General Preparations and Physical 97
Measurements
4-B. Compounds Described in Chapter 2 99
4-C. Compounds Described in Chapter 3 113
4-D. Oxo-Transfer to Triphenylphosphine as 117
Described in Chapter 3
4-E. References 119
APPENDIX A 1h and NMR Spectra A-1. Compounds Described in Chapter 2 120
A-2. Compounds Described in Chapter 3 127
APPENDIX B Crystallographic Data B-1. X-ray Crystal Structure Data for Complexes in 131
Chapter 2
B-2. X-ray Crystal Structure Data for Complexes in 133
Chapter 3
APPENDIX C GC-MS Spectra C-1. GC-MS Spectra for Standard Samples 134
C-2. GC-MS Spectra for the Reactions with 136
Triphenylphosphine Described in Chapter 3
V

ABBREVIATIONS
°C degree Celsius min. minute(s)
d chemical shift in parts per mol. mole(s)
million downfield from mp melting point
tetramethylsilane (spectral) MS mass spectrometry
Anal. analysis m/z mass to charge ratio
Ar aryl M+ molecular ion
br broad (spectral) NMR nuclear magnetic resource
/-Bu tert-butyl Ph phenyl
calcd. calculated ppm parts per million
cone. concentrated Py pyridine
d doublet (spectral) q quartet (spectral)
dec. decomposed r.t. room temperature
DMSO dimethylsulfoxide s singlet (spectral)
EI electron impact solv. solvent
Et ethyl t triplet (spectral), tertiary
EXAFS extended X-ray absorption THF tetrahydrofliran
fine structure TMS tetramethylsilane
FAB fast atom bombardment
g gram(s)
HRMS high resolution mass
spectrometry
Hz hertz
h hour(s)
J coupling constant
LSI liquid secondary ion
m multiplet (spectral), milli-
Me methyl
vi

CHAPTER 1
General Introduction
l-A. Role of Copper in Biology
Copper has been known to be an essential element in biological systems since
1925.1 With a rapid development in trace element analysis, protein crystallography,
spectroscopy, and molecular biology in the last two decades, the biological relevance
of copper has been d i sc losed ,� The emergence of bioinorganic chemistry has
created an interaction between inorganic chemistry and various aspects of biology,
providing insights to the understanding of the roles of copper in various biological
systems.
The occurrence of two oxidation states, viz. +1 and +2, renders copper to be an
important cofactors in a number of redox proteins, electron-transfer proteins, and
dioxygen carriers. Apart from these, copper is also an important element in a
number of non-organometallic redox catalysts, e.g. in organic syntheses and
industrial processes (Figure I ”
Although copper is an important element in biology, detailed information of its
biological functions has not been completely understood. From a structural and
spectroscopic point of view, three main types of copper centers, namely Type 1,Type
1

2,and Type 3,are classified in biological systems (Table 1).4,5 By now, corelations
with analytical features such as optical absorption, electron paramagnetic resonance
spectroscopy, and magnetism have been largely understood due to an increase in
available structural and spectroscopic information. Recent studies have established
several special combinations (e.g. Type 2+3 trimer. See Table 1) and other copper
centers such as C u a and MT-Cu.
JCXq 一 melanins
2 。 2 + i 7 JOC0H+H2O O22- + 02 RCH2NH2 + 02
2 RCHO + H2O2 + NH3
tyrosinase HO o CO + depsid i / ^ ^ ^
I 、 i . a s c o r b a t e . O ,
h c T ^ ^ c ^ Y " ^ +O2 — V r S ^ ^ ^ ^ dehydroascorbate + 2H2O
r ; N2 + H z O ^ N2O r e d u c t a s e ^ 广 . • ^"^^^blue' copper proteins ® , , , 、
) 。 U (C (electron transfer)
NCV / gaactose 、H2O
。 2 l P h 。 矿 +02 (dioxygen transport) O2 OH
RCHO RCH2OH Y y ^ +H0O + + HO-^ 2
H2O2 H2O Figure 1. The essential metabolic functions of copper-containing proteins are summarized in the 'bicopper dial'. The close association of this bioelement with dioxygen and its secondary products is obvious. Not included are transport, regulatory, and storage proteins for copper.
2

Tabl
e 1.
Pro
perti
es o
f var
ious
bio
logi
cal c
oppe
r cen
ters
.
Type
1 Ty
pe 2
Type
3 Ty
pe (2+
3) trim
er C^
MT
-Cu
Aggre
gation
state
monon
uclear
mo
nonucl
ear
dinucl
ear
trinucl
ear
dinucl
ear
monon
uclear
to
multi-
nuclea
r Sp
ecific
ity, b
io-
electr
on tra
nsfer
cataly
sis an
d redo
x O2
activ
ation f
or O2
activ
ation f
or ele
ctron
transf
er reg
ulatio
n, sto
rage,
logica
l funct
ion
reactiv
ity
transp
ort an
d oxi
dase f
uncti
on and
transp
ort fo
rm
oxygen
ation
Occur
rence
'blue'
coppe
r prot
eins a
mine
oxidas
e, hem
ocyani
n, asc
orbate
oxida
se,
N2O
reduct
ase,
CU
P2, m
etallo
-(ex
ample
s) gal
actose
oxida
se,
tyrosi
nase
laccas
e cyt
ochrom
e c
thione
in (M
T), Cu
sup
eroxid
e dis-
oxidas
e tra
nsport
ATP
ase
mutas
e, cyto
chrom
e c
oxidas
e Str
uctura
l char
acteri
stics
typica
l coor
din-
3 (trig
onal-p
lanar)
4 o
r 5 (s
quare-
planar
Cu丨:
3 (trig
onal-
Cu丨:
3 (trig
onal)
4 3 (
trigona
l-plan
ar)
ation n
umber
or
-pyram
idal)
planar
) Cu
": 4
coordi
nated
atoms
S"
(Cys")
, 2 N
(His),
3 N
(His)
# 2x3
N (H
is); Q
jH:
N (H
is), pa
rtly
2 S" (C
ys"),
3 S"
(Cys*)
S (
Met)
'-Oi
(0)0H
- 2 N
(His),
S (M
et)
or O
(pepti
de)
Spectr
oscopi
c char
acteri
stics o
f the o
xidize
d or o
xygena
ted fo
rm
EPR
small
"Cu/^
'Cu
‘norma
l,Cu"
EPR
no sig
nal (s
trong
‘norma
l,Cu"
EPR
very s
mall C
u no
oxidiz
ed for
m hyp
erfme
splitt
ing
param
eters
antipa
rallel
spin-s
pin p
arame
ters
hyper
fme s
plittin
g (A/
/)’ low
g fac
tor (g
") cou
pling)
due
to tw
o equi
valent
Cu
, low g
facto
r ligh
t absor
ption
intens
e abso
rption
in n
o inte
nse ab
sorp-
intens
e abso
rption
; int
ense a
bsorpt
ion;
absorp
tion i
n the
near
t^e vi
sible
LMCT
tion,
only
LMCT
((-一
匚!!之十)
LMCT (
(V.—
Cu?,
infrar
ed reg
ion
(Cys-
—Cu2
� ‘fo
rbidde
n, lig
and
(MMC
T) fie
ld tra
nsitio
ns fre
e coor
dinatio
n site,
speci
al lig
ands (o
rganic
radic
als), o
r adja
cent m
etal c
enters
(Zn,
Fe).
“

1-B. A Brief Review on Radical Copper Proteins
In recent years, studies of protein systems containing metal-radical arrays have
7 8
attracted considerable interest., As shown in Figure 1,three types of copper-
containing enzymes, namely, galactose oxidase (GAO), copper amine oxidase (CAO)
and dopamine monooxygenase (D/3M) have been known to catalyze the
oxidation of substrate by involving a metal-radical species in their catalytic cycles/
Among these copper enzymes, galactose oxidase (GAO) is the one that received the
most extensive studies. GAO is a secretory fungal enzyme which has been isolated
from culture filtrates of Dactylium dendroides, Gibberella fujikuroi, and Fusarium
graminearum^ This enzyme is monomeric with a relative molecular mass of 68
kDa. It catalyzes the two-electron oxidation of galactose (and a broad range of
primary alcohols) to the corresponding aldehydes together with a concomitant
formation of hydrogen peroxide. It is noteworthy that the enzyme catalyzes the
oxidation of alcohols in a regioselective, but not substrate-specific manner (Eq.l).4
CH2OH CHO 0 \ y \ 0 \ 0 H OH/^ O OH N O H i ^ + 〇2 X O H i ^ + H2O2
OH OH
R-CH2OH + O2 R-CHO + H2O2 (Eq 1)
4

In fact, the biological role of the enzyme is not fully understood. One possible
function of this enzyme is degradation of rotting wood by oxidation of
oligosaccharide intermediates, which are preferred substrates of the enzyme.
Besides, the coupled formation of hydrogen peroxide is also important as it serves as
a bacteriostatic agent and an essential cosubstrate for peroxidases
The catalytic mechanism on how GAO catalyzes the oxidation of substrates has
been a controversial topic over years. In 1978,Hamilton et al suggested a reaction
mechanism which involved a redox pair.^^ However, with detailed
spectroscopic and magnetic studies of the enzyme, together with a recent report on
its crystal structure, a catalytic mechanism for the enzymatic turnover has been
proposed (Figure 2)}^
5

Tyr495
His496 ^ ― ( .His581 His496 V " ' ^ xHis581
)= \ 9 /=< >=\ \ r=\ Active form . c u T 一 . c u T 一
r A ' H 尸 FA""H / H ^ ^ H ^
^ ‘ Cys228 Cys228
O2 + RCH2OH ) H atom abstraction 广 H2O2 + RCHO
1 r 丁 y r ^ Tyr495
Q , Q His49a. (?^ /His581 His496 .His581
> = \ i / = \ > = \ [ / = \ H N ^ N v : Z*、N^NH H N ^ N v ^ i ^ N ^ ^ N H
Reduced form ^ c ' ^ 一
Cys228 (^s228
Figure 2. Proposed catalytic mechanism for galactose oxidase.
The active site of the enzyme consists of a mononuclear copper ion in a square-
pyramidal coordination geometry (Figure 3). At pH 7.0,the copper ion is
coordinated to two histidine residues (His 496, His 581), a tyrosine residue (Tyr
495),a water molecule (which is replaced by an acetate ion in acidic media), and a
cysteinate-substituted tyrosine residue (Tyr 272) of which the ortho position is linked
to a cysteinate side chain (Cys 228). This arrangement is believed to stabilize the
corresponding tyrosyl radical by lowering its redox potential and allowing for spin
delocalization. Another feature that help to stabilize such radical species is the
6

presence of the tryptophan residue, Tip 290. The indole part of which, being located
ca. 3.4 A above the cysteinate-substituted tyrosine residue, is believed to exert a
stabilizing effect to the tyrosyl radical through a t t to t t stacking interaction.
Tyr495
5 8 1 H i s ^ \ j N I
H N ; 2 N Z 、 〇 s〉
产 acetate or Cys228 His496 water
Figure 3. A schematic view of the active site of galactose oxidase in its inactive form.
Copper amine oxidase (CAO) belongs to a family of quinoproteins. It catalyzes
a two-electron oxidation of primary amines to the corresponding aldehydes, with the
formation of a reduced form of the cofactor. Subsequently, the reduced cofactor is
reoxidized by oxygen, resulting in the formation of both ammonia and hydrogen
peroxide. Such reactions can be formally divided into a reductive and an oxidative
half-reaction (Eq. 2 and 3).
7

Reductive half-reaction:
0 OH G ^ o r r V N H 2
:少 + RCH2NH2 • I . + RCHO
^ ^ (Eq. 2)
Oxidative half-reaction:
OH O
1 J + O2 + H2O • X J + H2O2 + NH3
^ ^ (Eq. 3)
CAO can be isolated in bacteria, yeast, plants, and mammals. It plays an
important role in the regulation of biogenic amine levels through oxidative
metabolism. However, the exact number, location, and regulation of this enzyme in
cells remain unclear. This enzyme is dimeric in nature with each monomer
consisting of a 75-80 kDa polypeptide, each of which contains a single curpic ion7
The copper center in the active site is ligated by three histidine residues (His 442, His
444, and His 603) and two water molecules in a distorted square-pyramidal
coordination geometry. Besides, an organic cofactor, 2,4,5-trihydroxyphenylalanine
(TPQ) is also present. X-Ray crystallography revealed that the latter cofactor is
located close to the copper center. Figure 4 depicts a proposed catalytic mechanism
for CAO.8
8

⑷ RCH2NH2 H20
^ X c u " " I V > ^ T^
裕 V U % V 0 Asp/Glu 0 H
1 r RCHO H2O
1 / I fast J L I o h R h o
OH O一 H
(b)
7—— , ~ O2 , — — , - i • • O s ^
cull C u i Y V ^ N H , OH OH OH
n h ^ r " " ' t i ^ Cu" ^ o Cu" \ A S H 2
-O 一 0 O
Figure 4. Proposed mechanism for copper amine oxidase: (a) reductive half-reaction, (b) oxidative half-reaction.
As GAO and CAO are specific for the oxidation of primary alcohols and
amines, respectively, considerable interests have been attracted to these enzymes for
biosensor applications.
9

Dopamine 曰 monooxygenase (D/3M) catalyzes the conversion of neuro-
transmitter dopamine to norephinephrine (Eq. 4).^ Therefore, this enzyme plays an
important role in controlling the levels of these neurotransmitters in higher
organisms.
OH
_ ascorbate i + O2 • I ^ + H2O
H O " H C A ^ (Eq.4)
During enzymatic turnover, an oxygen atom is inserted into the benzylic
position of the ethylamine side chain of dopamine (or other phenethylamine
analogues), with a concomitant four-electron reduction of oxygen to water. Two of
these electrons originate from the substrate, while the other two come from the
exogenous electron donor, ascorbic acid. The latter undergoes oxidation in two
sequential steps to produce semidehydroascorbate^
Interestingly, D/5M contains two coppers in each subunit. One of the copper
center exits in an active dimeric form while the other one exits in a tetrameric form.
These two copper centers are separated by 4 A apart. However, they can be
categorized as mononuclear copper proteins because these two copper centers
10

perform different functions during the enzymatic turnover. One of them, C u a ,
catalyzes an electron transfer from ascorbate to the active site, while the other one,
Cub, catalyzes the hydroxylation of the substrate. In 1994, Klinman and co-workers
proposed a mechanism for the C u b site (Figure 5) which involves a tyrosyl radical in
the hydroxylation of dopamine. ^ However, no direct evidence has been obtained to
support this model.
广 H20 CUB(II) / f J ^
\ V ^ CUB\(") L J U i、u X O T CUB(II) H.. T 、H H/0 H H + .0 ..。• + 乂 7NH3 •
Ar〜NH3 Ar 〜 Ar〜NH3
T X — - v V A r 二 N 、
Figure 5. Proposed mechanism for dopamine 曰 monooxygenase in substrate hydroxylation.
11

1-C. Objectives of This Work
The first part of this research work focuses on model studies of the copper-
containing protein, galactose oxidase (GAO). A series of copper(II) and zinc(II)
complexes of the type [M(L")(X)] (M = Cu, Zn; X = OAc,CI), where V being N3O-
type tripodal tetradentate ligands containing phenolate pendants, have been
synthesized as mimics to the active center of GAO. Subsequent oxidation of some of
these complexes with cerium(IV) ammonium nitrate gave the corresponding copper-
phenoxyl radical species, which are thermally stable only at low temperatures. The
model complexes and the corresponding oxidized radical species have been
characterized by EPR and UV-Vis. They elicit similar spectroscopic properties as
those observed for the native enzymes.
The second part of this work deals with the chemistry of copper(I) complexes.
Copper(I) complexes of the type [M(HL”)( -I)]2 and [M(HL”)I] have been prepared
and structurally characterized. Their reactivities toward dioxygen have also been
investigated.
12

1-D. References
1. Linder,M. C.; Goode, C. A. Biochemistry of Copper, Plenum, New York, 1991.
2. Karlin, K. D.; Tyeklar,Z. Bioinorganic Chemistry of Copper, Chapman & Hall,
New York,1993.
3. Lippard, S. J.; Berg, J. M. Principles of Bioinorganic Chemistry, University
Science Books, Mi l l Valley, 1994.
4. Kaim, W.; Rail, J. Angrew. Chem. Int. Ed. Engl 1996,35, 43-60.
5. Solomon, E.; Baldwin, M. J.; Lowery, M. D. Chem. Rev. 1992, 92, 521.
6. Stubbe, J. Biochemistry 1998,27, 3893-3900.
7. Klinman, J. P. Chem. Rev. 1996, 96, 2541-2561.
8. Stubbe, J.; Donk, W. A. Chem. Rev. 1998,98, 705-762.
9. Whittaker, M. M.; Whittaker, J. W. J. Biol Chem. 1988,263, 6074-6080.
10. Whittaker, M. M.; Ballou, D. P.; Whittaker, J. W. Biochemistry 1998,57, 8426-
8436.
11. Hamilton, G. A.; Adolf, P. K.; De Jersey, J.; DuBois, G. C.; Dyrkacz, G. R.;
Libby,R. D. J. Am. Chem. Soc. 1978,100, 1899-1912.
12. Ito, N.; Philips, S. E. V.; Stevens, C.; Ogel,Z. B.; McPherson, M. J.; Keen, J. N.;
Yadav, K. D. S.; Knowles, P. F. Nature 1991, 350, 87-90.
13

CHAPTER 2
Copper(II) and Zinc(II) Complexes containing N3O Tetradentate Ligands
2-A. Introduction
2-A-I. The Active Site Structure of Galactose Oxidase
In recent years, the structures, spectroscopic properties, and catalytic mechanism
of GAO have been extensively studied. Ito and co-workers have reported the crystal
structure of GAO at 1.7 人 resolution. The protein crystals were obtained from an
acetate buffer (0.8 M) at pH 4.3 - 4.7 in the presence of the precipitant, ammonium
sulfate. The structure was consistent with the information from spectroscopic
studies. The copper active site is coordinated by five ligands in a square pyramidal
coordination geometry. The equatorial positions are occupied by Tyr 272,His 496,
His 581,and a water molecule or an acetate ion, whilst the axial position consists of
Tyr 495 (Figure 6 ) }
Electron paramagnetic resonance, optical absorption, and circular dichroism
studies of GAO have been carried out by Whittaker and co-workers.^ The EPR
spectra consist of a typical copper signal with g// = 2.28 and g丄=2.05.2 The optical
14
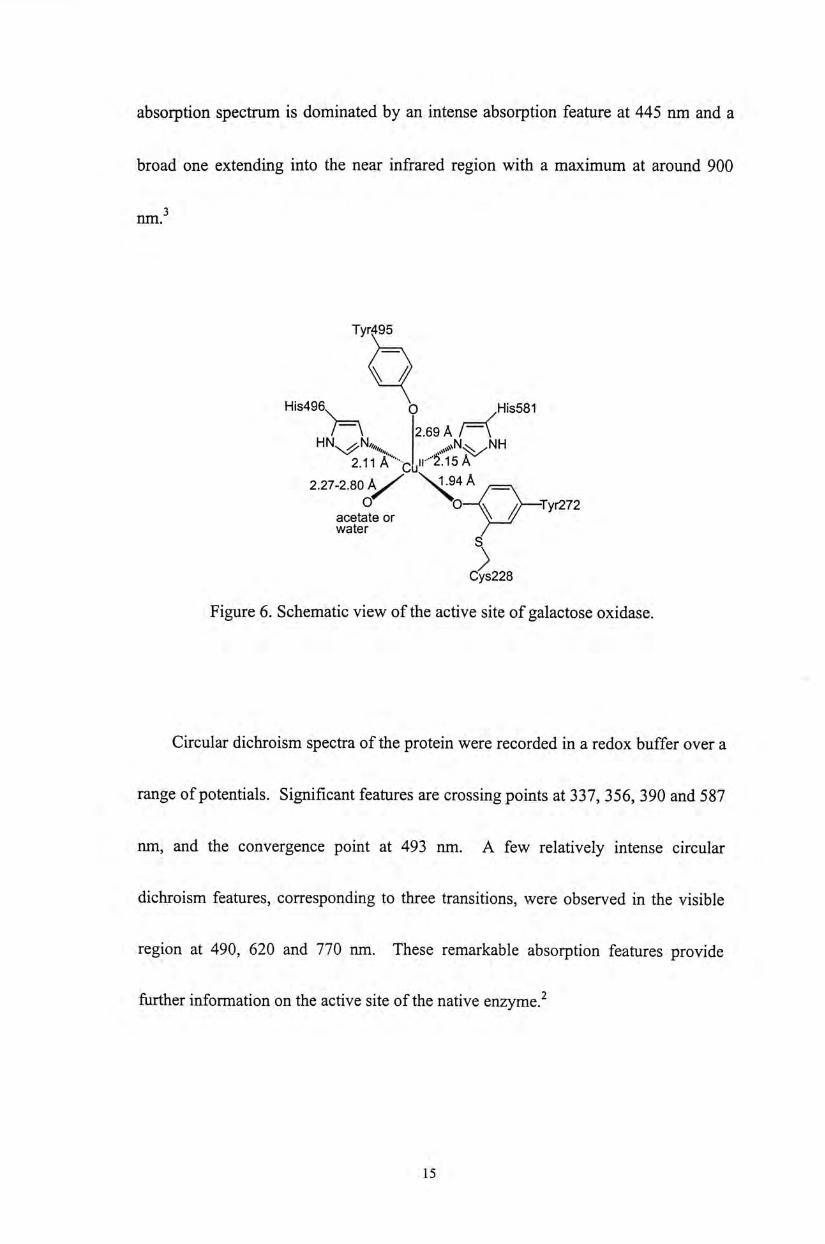
absorption spectrum is dominated by an intense absorption feature at 445 nm and a
broad one extending into the near infrared region with a maximum at around 900
nm.3
Tyr495
His496^ O His581
r ^ 2.69 A f = \ h n ^ N , . .N^NH
2.11 A...Cu".么 15 A 2.27-2.80
O ^ ― T y r 2 7 2 acetate or __(J water /
s〉
Cys228
Figure 6. Schematic view of the active site of galactose oxidase.
Circular dichroism spectra of the protein were recorded in a redox buffer over a
range of potentials. Significant features are crossing points at 337,356, 390 and 587
nm, and the convergence point at 493 nm. A few relatively intense circular
dichroism features, corresponding to three transitions, were observed in the visible
region at 490,620 and 770 nm. These remarkable absorption features provide
further information on the active site of the native enzyme.^
15

2-A-II. Biomimetic Models for Galactose Oxidase
A variety of metal complexes are synthesized as mimic to the active site of
various metalloenzymes in order to understand their structures and catalytic
mechanisms. However, most of these model complexes serve only as structural
models and very few of them show catalytic reactivities similar to those of the native
enzymes. Thus, biomimetic models that mimic both structure and function of an
enzyme remain rare.
Stack and co-workers have prepared a series of copper(II) complexes with non-
square planar N2O2 donor ligands (Scheme 1).4 It is believed that the binaphthyl
unit of the ligands can enforce a non-square planar coordination geometry, which is
OH o 1- TMEDA. EtOH, 25°C OH O
I ^ A y ^ H 2. n-BuLi (2 equiv.), TMEDA, EtzO, 25°C 3. R"-R" K ^
R 4.2M HCI, H2O R
^ 8 5 1 H2N NH2 , EtOH, reflux
2. Cu(0Ac〉2, MeOH, reflux
R" R"
1 R" = f-Bu; R 二 t-Bu 2 R" = f-Bu; R = SPh 3 R" = f-Bu; R = SPK 4 R" = t-Bu\ R = H 5 R" = H; R = SPh 6 R" = H ; R = H (Scheme 1 )
16

favourable to copper(II) ions. It is also believed that this distortion may enhance the
stability of the corresponding copper(I) species and facilitate the coordination of
alcoholate substrates to the copper center during the catalytic turnovers. The alkyl
substituents, R and R" at the ortho and para positions of the phenolate rings help to
stabilize the phenoxyl-radical species formed upon oxidation. It is noteworthy that
oxidation of complexes 1 and 2 gave thermally stable copper(II) phenoxyl-radical
species. These complexes can catalyze the oxidation of benzylic and allylic alcohols
to the corresponding aldehydes by molecular oxygen at room temperature with
turnover numbers in the range 30 to 1300 (Figure 7).
Chaudhuri, Wieghardt, and co-workers have reported two copper complexes (7
and 8) which showed reactivities similar to that of galactose oxidase, though the
coordination environment around the copper center in these complexes is far from
that of the native enzyme (Scheme In the solid state, compound 7 is a
monomer, whilst compound 8 is a dimer. The copper centers in both compounds
exhibit a distort square planar coordination geometry.
17

^ f ^ R = SPh, f-Bu t-Bu PhCHzO" latmO:
H2。V 'Bu \
PhCHjOH—
S i : p T ^ 器 於 (-Bu t-Bu
Figure 7. Proposed mechanism of alcohol oxidation by complexes 1 and 2.
|-Bu |-Bu
air f-Bu-'^^S^^^f-Bu 0、c|u,0
-Bu -Bu j 、
f f ^ CuCI, EtgN 7 NBu- Y S MeCH’ Ar ⑷
OH OH f-Bu___ dry O2. THF ‘ [bu
L ^"Vbu 一 8
(Scheme 2)
18

Compounds 7 and 8 catalyze the oxidation of primary and secondary alcohols
to the corresponding aldehydes and glycol derivatives, respectively, together with
the concomitant conversion of dioxygen to hydrogen peroxide. Compound 8
catalyzes the oxidation of primary alcohols to aldehydes with a turnover number of
630. It has been shown that only one alcoholate substrate is bound to one of the
copper centers of the complex during the reaction (Figure 8). On the other hand,
both copper centers of 8 are bound by two alcoholate substrates during the catalytic
oxidation of primary alcohols. It is believed that the two alcoholate moieties bind in
a syn-fdiCidX way that C-C bond formation takes place between two coordinated ketyl
radical species.
2+ f-B^_ •““] 2+ > = < • RCH^OH
沾 u R / ^ H ^ ^ B u
H202-K '
o A
他 ~ I 2 + f - B ^ _ “ I 2+
H � � B U / 々 絶
Figure 8. Proposed mechanism for the catalytic oxidation of primary alcohols by complex 8.
19

Cyclic voltammetry of 7 in dichloromethane shows two reversible one-electron
processes at £"1/2= -0.14 and —0.16 V (versus Fc+/Fc) while that of 8 shows two
reversible one-electron reduction waves at Ei/2== -1.26 and -0.29 V (versus Fc+/Fc).
Wieghardt and co-workers have also reported a series of copper(II) and zinc(II)
complexes derived from the macrocyclic ligands 9-14,which contain phenolate
pendants.6 Besides copper(II) phenolate compounds and their analogous zinc(II)
compounds have also been prepared in order to understand the chemistry of metal-
phenoxyl radical species. The reasons for studying the zinc(II) analogues are: (1)
zinc(II) ion is redox-innocent within a wide range of potentials, (2) the
corresponding phenoxyl radical species can be studied by EPR spectroscopy without
interference from paramagnetic metal centers, and (3) the electronic spectra of the
radical species are not affected by d-d transitions due to the metal center. As a
result, many research groups have studied both copper(II) and analogous zinc(II)
complexes as structural models for galactose oxidase.
T W Y Y h 3
11 1 2 R = t-Bu] R" = CH3
13 R = OCH3; R" = CH3
9 R = t-Bu 14 R = -Bu; R" = /-Pr 10 R = OCH3
20

Jean-Louis Pierre and co-workers have reported the dimeric copper complex
15, which was obtained by treating copper(II) perchlorate with the iV2<^2-type donor
ligand, bis(3 '-/-butyl-2 '-hydroxybenzyl)(2-pyridylmethyl)amine, and triethylamine
Q
at room temperature (Eq. 5). Complex 15 could be converted to the monomelic
complex 16 upon addition of acetonitrile (Eq. 5).
A
代 一 於 Z � - 3 f-Bd I r NBi! W 16
15 (Eq. 5)
Apart from these, the same research group also reported the mononuclear
copper complex 17,which could catalyze the oxidation of primary alcohols to
aldehydes with more than 30 turnovers in a basic medium.^ The electrochemical
feature of 17 in dimethylformamide was studied with cyclic voltammetry. Three
successive redox processes have been observed in the anodic region. The first one
was quasi-reversible with Em = 0.11 V and the other two appeared to be irreversible
with £pa = 0.45 and 0.76 V. However, the catalytic mechanism for the oxidation of
alcohols by this compound remains unclear.
21

t-Bu t-Bu
17
Tolman and co-workers have prepared a series of macrocyclic ligands 18-23,
containing isopropyl substituents, by modification of ligands 9-14 (Eq.6).^^
A CH30H ^ 人
令u令 + —q.) + - ^ Y 18 R = R' = CH3
19 R = R' = t-Bu 20 R = CH3; R = OCH3 21 R = CH3; R. = SCH3 22 R = /-Bu; R. = SCH3
H R OH _ _ OH
> + HCHO(aq.) + 2 O I J { > M
丫 U H • X ^ X 23
(Eq. 6)
Deprotonation of 18-23 with sodium hydride followed by treatment with
anhydrous metal salts [CuCl2, Cu(03SCF3)2,Cu(02CCH3)2’ and ZnCy in
tetrahydrofuran gave a series of monomelic metal-phenolate complexes. Copper(II)
22

and zinc(II) complexes derived from ligands 19 and 23 could be chemically oxidized
to give the corresponding metal-phenoxyl radicals by treatment with stoichiometric
amounts of either cerium ammonium nitrate in acetonitrile at -40°C or
thianthryljtetrafluoroborate in dichloromethane at -80 。C. Spectroscopic
characterization of these metal-phenolate complexes and the corresponding metal-
radical species have also been carried out. With ligand 19, the copper complex 24
has been prepared. ^ Treatment of 24 with NaOCHsPh gave the corresponding
copper benzyl alkoxide complex, which could be oxidized electrochemically to give
benzaldehyde, 1,4-di-isopropyl-1,4,7-triazacyclononane and 4,6-di-,孙 butyl-2-
formylphenol (Figure 9). These reaction products have been identified by ^NMR
and GC-MS analysis.
r n +
CP3SO3- N a O C H 2 P h 、 〖 - B u ^ ^ ^ ^Q!^
24
CHO , D ?H electrochemical 人 广 V V ^ " ^ * ^
• [1 I + \ / + 、IJ + unidentified Cu product(s) reaction ^ N ^ ^ 丫
~‘ 丁 t-Bu 46% < 5% < 5%
Figure 9. The catalytic reaction of complex 24.
23

Whittaker et al. have reported a dimeric copper(II) complex (25) by the
reaction of tetrakis(acetonitrile)copper(II) tetrafluoroborate with one equivalent of
the duncamine ligand and two equivalents of tetramethylammonium hydroxide.
Upon addition of pyridine, compound 25 could be converted to the monomeric
copper(II) species 26 (Eq. 1)}^
A y
丫 26 25 (Eq. 7)
A number of structural models for galactose oxidase have also been reported.
Fenton and co-workers have prepared a series of copper(II) complexes which
derived from the bispyridine ligands l l - Z l P Interestingly, the spectroscopic
features of the copper complexes were similar to those of the native enzyme,
although they did not show any catalytic behaviour.
24

/ = < N > = \ / = < N > = \
G n ( p h O G n ( / O H W
P P H O2N
27 X = y = 1 30 X = y = 1 28 X = y = 2 31 x = y = 2 29 X = 1; y = 2 32 x = 1; y = 2
By modification of ligand 28 with the incorporation of a thioether functionality
at the ortho position of the phenolate ring, Itoh and co-workers have reported a new
ligand 33.14
r y
k W Vbu t-Bu 33
Compound 33 reacted with copper(II) and zinc(II) salts to form the
corresponding metal complexes in which the metal centers have a five-coordinate
N3O environment. Although the coordination environment around the metal center
in these complexes are different from those observed in the native enzyme, both
metal complexes could oxidize benzyl alcohol to benzaldehyde upon chemical
25

oxidation with eerie ammonium nitrate. The involvement of a metal radical species
in the catalytic reaction cycles has been confirmed by resonance Raman
spectroscopy, electrospray ionization mass spectroscopy and EPR spectroscopy. It
was found that the oxidation of benzyl alcohol by the copper-phenolate complex
obeyed a first-order kinetics, whilst that of the zinc analog obeyed a second-order
kinetics. 14b
In this research work, with a modification of ligand 33 reported by Itoh and co-
workers, a series of N3O tetradentate ligands have been prepared. The preparation
of the ligands, the syntheses and structures of copper(II) and zinc(II) complexes, and
their characterization will be explained in the following sections.
26

Results and Discussion
2-B. Preparation of Tetradentate Ligands and Complexes
2-B-I. Preparation of A jO-type Tetradentate Ligands
A series of pyridine- and benzimidazole-based A(jO-type tetradentate proligands
34-39 (Scheme 3),which contain substituted phenolate pendant, were used in our
studies.
n r ^ N, /口 " Y ^
V V R ' J T X ^ R R R
34 R = R '=f.Bu ( H L ” 3 7 R = R' = ^ B u (HL^)
35 R = f-Bu,R' = H (HL2 ) 38 R = f-Bu, R'= H (HL^ )
36 R = H,R = t -Bu (HL3 ) 39 R= H. R' = / -Bu (HL® ) (Scheme 3)
(a) Preparation of the Pyridine-based Proligands 34-36
The proligands 34-36 were prepared by reductive amination of substituted benzyl
bromides (46-48) with A^,A^-bis(2-pyridylmethyl)amine (BPMA) 49,in the presence
of tributylamine, in refluxing tetrahydrofliran (Scheme 4).
27

R n n 2. Et3N. r e f l u x ~ “ H O ^ ^ R .
46 R = R' = f-Bu R 47 R = f -Bu, R' = H 34 R = R' =t -Bu (HlJ) 84% 48 R = H, R' = f-Bu 35 R = f-Bu, R' = H (HL^) 80%
36 R = H.R' = f -Bu (hl3 ) 70% (Scheme 4)
The substituted benzyl bromides 46-48 were prepared from the corresponding
substituted phenols. Reaction of a refluxing solution of the appropriate substituted
phenols, tributylamine, tin(IV) chloride and paraformaldehyde gave the
corresponding benzaldehydes 40-42. Subsequent reduction of 40-42 with sodium
borohydride gave the respective benzyl alcohols 43-45. Bromination of 43-45 with
phosphorus tribromide led to the formation of a series of substituted benzyl bromides
46-48 in excellent yields (Scheme 5)7
f? f?
n-BuaN • (TS^OH
R ^ ^ -(CH20)n- R J 40 R = R-= f-Bu (80%) 41 R = f-Bu. R' = H(72%) 42 R = H, R' = NBu (70%)
睡 4 , 久 O H PBra, 久 O H MeOH I ^ CHCI3 I J [ o,
43 R = R' = t -Bu (95%) 46 R = R' = f -Bu (95%) 44 R = f-Bu. R' = H (93%) 47 R = f-Bu. R '= H (96%) 45 R = H, R. = f -Bu (96%) 48 R = H, R_ = f -Bu (96%) (Scheme 5)
28

iV,iV-Bis(2-pyridylmethyl)amine 49 was prepared by treating a solution of 2-
pyridinecarboxaldehyde and 2-aminomethylpyridine, followed by reduction of the
resulting solution with sodium borohydride (Scheme 6).
f S + f S NaBH4, i T ^ ^ �N^"^NH2 + MeOH * S / ^ Y ^ ^ ^ N " ^ ^
H H 49 (90%) (Scheme 6)
(b) Preparation of the Benzimidazole-based Proligands 37-39
Reaction of iminodiacetic acid and o-phenylenediamine in refluxing methanol
gave 7V,A^-bis(2-benzimidazolymethyl)amine (BBA) 50 in 50% yield (Scheme 7).
^COOH H H
C ^ ^ N H U reflux ^ ^ ^ N ^ J ^ N " - ^
^COOH 刚 2 n SO ( 5 0 % ) (Scheme 7)
BBA 50 was prepared according to a modified procedure based on the one
reported by Stephan and co-workers/^ The reaction time has been shortened from
72 hours to 18 hours and the reaction yield has been improved from 28% to 50%.
29

The proligands 37-39 were prepared by an analogous procedure as for 34-36,by
treating BBA with the appropriate benzyl bromides 46-48 and triethylamine in
refluxing tetrahydrofliran (Scheme 8).
R O c V - J Y ^ ^ O H 1.5�’THF I … L Y ^ R "
R . J ^ ^ A / B r 2. EtsN, reflux H O ^ ^ R
46 R = R' = f -Bu 37R = R'=f-Bu (Hi / ) 51% 47 R = f -Bu, R' = H 3BR = t -Bu, R'= H (HL®) 40% 48 R = H, R' = f -Bu 39 R= H,R' = t -Bu (HL®) 36% (Scheme 8)
The crystal structure of galactose oxidase revealed that the equatorial tyrosinate
residue (Tyr 272) is linked to a nearby cysteine via an ortho C-S bond. However, an
S-alkyl substituent has not been introduced on the phenolate pendants of proligands
34-39 in order to avoid the formation of Cu-S b o n d s I n fact, bulky electron-
donating 〜butyl substituents have been introduced at the ortho and para postitions of
the phenolate moieties of 34-39 because (1) they help to stabilize the phenoxyl
radical species formed by electronic effect; (2) their bulkiness help to prevent
dimerization of the resulting copper complexes by a steric mean; (3) they help to
protect the phenoxyl radical species from attacks by other reagents in the
1 1 surroundings. These substituents exert different electronic and steric effects on the
30

ligands and hence their effects on the formation and properties of the resulting
complexes can be studied.
By comparing the structure of proligands 34-36 with that of 37-39, it should be
noteworthy that the latters provide a closer structural mimic to the histidine
coordination environment around the copper active site of the native enzyme. These
two series of ligands, with different steric and electronic properties, are expected to
exert different effects on the catalytic reactivities of the resulting complexes.
31

2-B-II. Preparation of Copper(II) and Zinc(II) Complexes
The copper(II) salts, Cu(C104)2.6H205b,6a’8’i8, Q i C O s S C F])]。, ,
Cu(BF4)2.H20i3b,i9 and CuCb^^''^^''^^ are common starting materials for the
preparation of model complexes of GAO. With the exception of CUCI2, these
copper(II) salts consists of weakly coordinating anions. Interestingly, some
copper(I) compounds, [Cu(NCCH3)4](C104)6c and CuCP'io," may be used to prepare
copper(II) complexes by subsequent oxidation. For the preparation of zinc(II)
analogs, Zn(BF4)2 ^HsO^ Zn(C104)2 ^HsO^^ and ZnCl2^® are commonly employed.
In our research work, both CUCI2 and Cu(0Ac)2 have been used as starting
materials for the preparation of the copper(II) complexes. For the preparation of the
zinc(II) analogs, ZnCl� and Zn(0Ac)2 were employed. We reason that acetate is
present in the coordination sphere of the copper center of GAO in acidic media.
Accordingly, both Cu(0Ac)2 and Zn(0Ac)2 have been used as starting materials to
the corresponding metal complexes in our work, although reports on their use are
rare in the literatures.
The preparation of copper(II) complexes derived from proligands 34-39 have
been attempted. However, only copper(II) complexes supported by 34 have been
successfully prepared (Scheme 9). Although triethylamine is commonly used to
deprotonate phenol ligands, it is not basic enough to generate the phenolate ligands
32
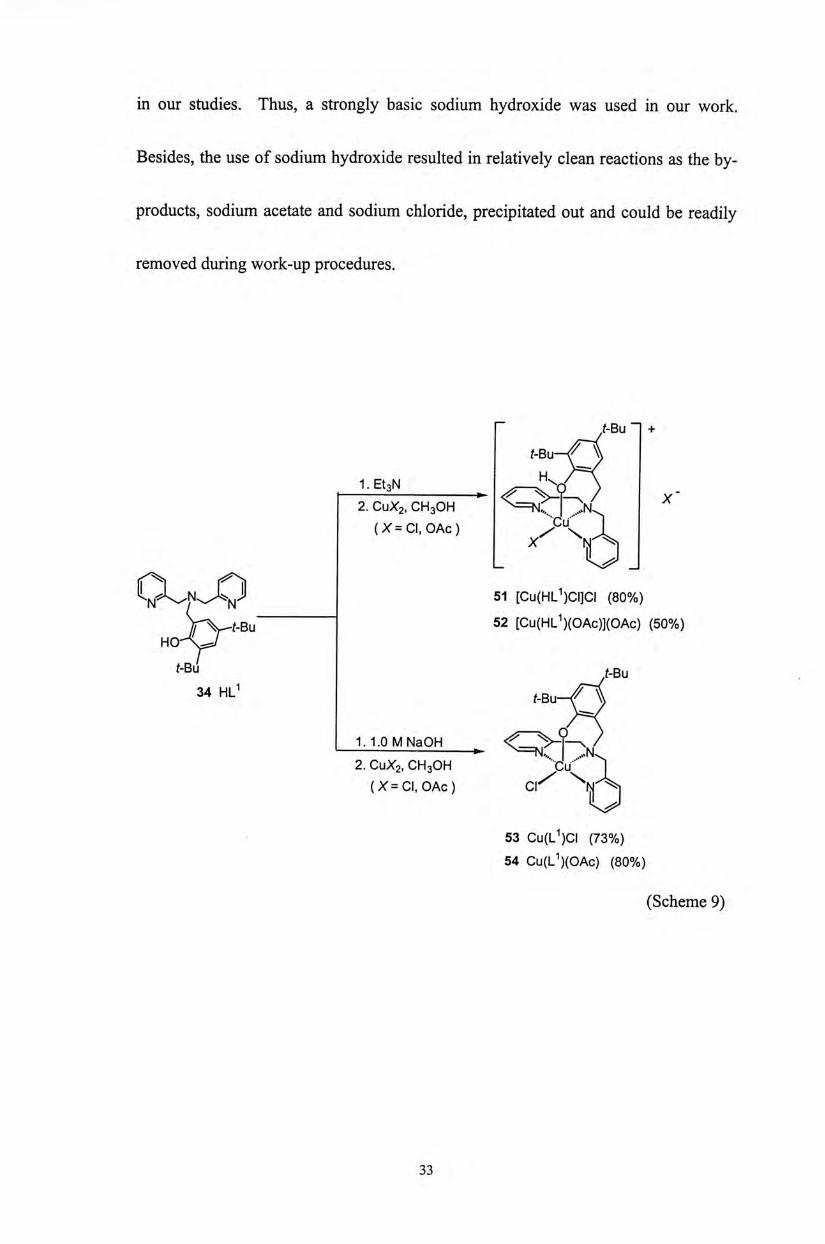
in our studies. Thus, a strongly basic sodium hydroxide was used in our work.
Besides, the use of sodium hydroxide resulted in relatively clean reactions as the by-
products, sodium acetate and sodium chloride, precipitated out and could be readily
removed during work-up procedures.
一 t-Bu +
f - B u ^ Q _ X-
2. CuX2, CH3OH ^ = = < 7 T^N. ^
( X = C I . OAc) 力 \ X
S M ^ ^ ^ / N ^ ^ N 』 51 [CU(HL1)CI]CI (80%)
V V f - B u 52 [Cu(HlJ)(OAc)](OAc) (50%) HO-^W (-Bu f-Bu
34 HLI f - B u ^ Q 1.1.0MNaOH < C ^ � 飞 /
• z N � 2. CUX2, CH3OH Cu..
{X=CI, OAc ) C | Z
53 Cu(L1)CI (73%)
54 Cu(lJ)(OAc) (80%)
(Scheme 9)
33

Attempts to prepare copper(II) complexes supported by proligands 37-39 were
unsuccessful. Only an intractable oil was obtained after the reactions. Further
attempts to repeat the reactions in tetrahydrofliran with sodium hydroxide as a base
were also unsuccessful.
A series of zinc(II) complexes 55-59 with proligands 34-36 have been
successfully prepared using either triethylamine or sodium hydroxide as the base
(Scheme 10). The results are different from those observed for the copper(II)
analogs.
r i n ^ N ^ I ^ ^ N ^ I .E tgN or NaOH ^
V V R ' 2. ZnX2. CH3OH
( X = C I . OAc)
34 R = R'=t -Bu (HlJ) ^ ^
35 R = f -Bu, R' = H (HL2) 55 [Zn(L^)CI] (71%)
36 R = H’ R, = f -Bu (HL3) 56 [Zn(L2)CI] (56%)
57 [Zn(L^)CI] (51%)
58 [Zn(Li)(OAc)] (77%)
59 [Zn(L2)(0Ac)] (55%)
(Scheme 10)
34

Attempted preparation of zinc(II) complexes supported by proligands 37-39
were not successfully. Attempts to modify the reaction conditions such as
temperature, solvents and the bases used were unsuccessful and no isolabe products
could be obtained.
Details of the preparation of complexes are described in the Experimental
Section. For paramagnetic copper(II) model complexes prepared in our work, their
molecular structures could only be determined by X-ray crystallography. For y
diamagnetic zinc(II) analogs, their structures have been characterized by NMR
spectroscopy, in addition to X-ray crystallographic analysis.
Besides, the generation and characterization of radical species have also been
carried out. Details of the experimental procedures are described in the following
sections.
35

2-C. Characterization
2-C-L Structural Studies
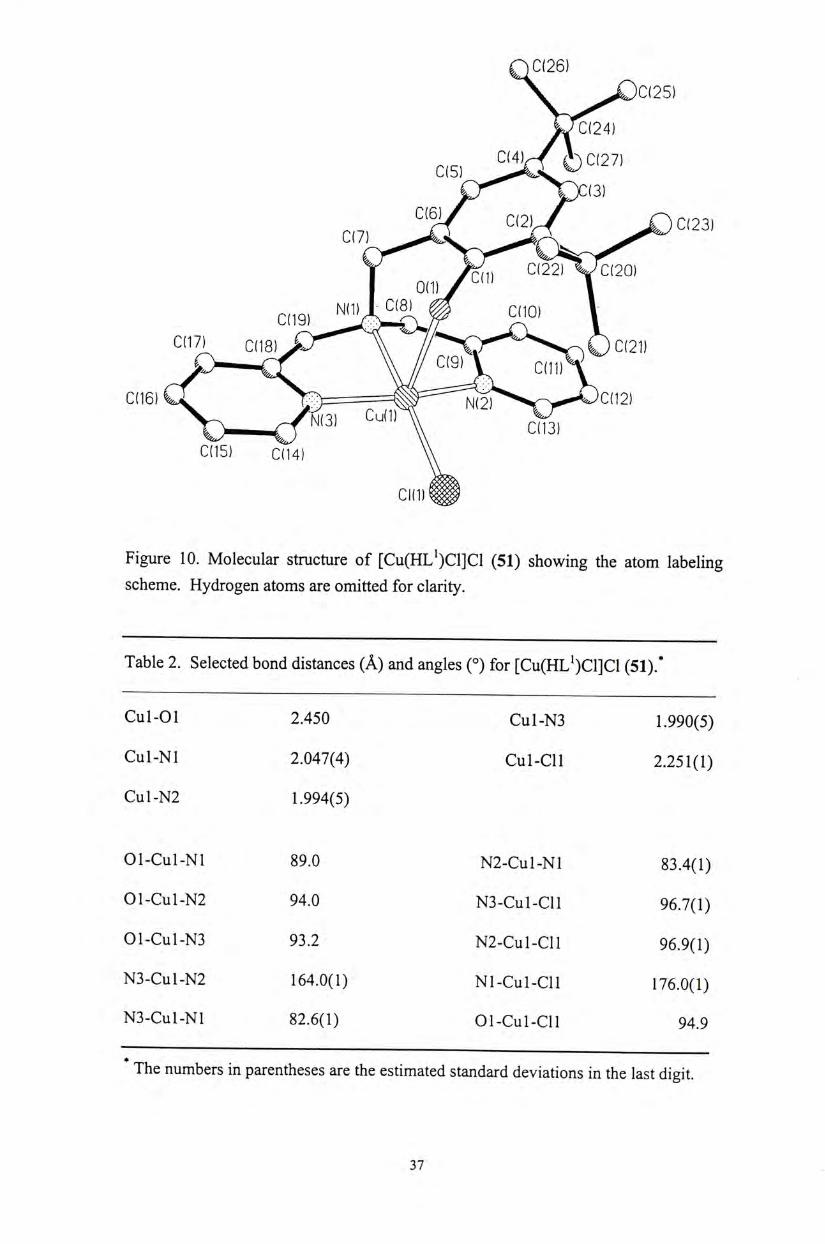
The molecular structures of the copper(II) phenoxyl complexes [Cu(HL")X]X
(51,52) (n = 1, 2, 3; X = CI, OAc), as well as copper(II) and zinc(II) phenolate
complexes [M(L")X] (54-59) (M = Cu, Zn; « = 1, 2, 3; X = CI, OAc) have been
characterized by X-ray crystallographic analyses. A l l of the metal complexes exhibit
a distorted square pyramidal coordination geometry on the metal centers (Figures 10-
16). The structure of these complexes is similar to that observed for the active site of
the native enzyme. ( Selected bond distances and angles for these metal complexes
are shown in Tables 2-9. The Cu-0(phenol) bond distances (2.437-2.450 A ) of the
copper(II) phenoxyl complexes 51 and 52 are similar to the observed Cu-0(Tyr 495)
bond distance (2.69 A ) for the native enzymes, although the phenoxyl protons are not
deprotonated in our current complexes. Upon deprotonation of the phenolate
pendant,the Cu-0 (phenolate) distances are comparatively shorter. For the
copper(II) phenolate complex 54,only the Cu-N bond distances (2.040-2.289 A ) are
comparable to the corresponding distances observed for the native enzyme」
36
Q C ( 2 6 )
� 9 ) J i l i l ^ / ^ C(IO) I
C(15) C(14) \\ c m )爆
Figure 10. Molecular structure of [Cu(HL^)Cl]Cl (51) showing the atom labeling scheme. Hydrogen atoms are omitted for clarity.
Table 2. Selected bond distances (A) and angles (。)for [Cu(HL^)Cl]Cl (51).*
Cul-01 2.450 Cul-N3 1.990(5)
Cul-Nl 2.047(4) Cul-Cll 2.251(1)
Cul-N2 1.994(5)
01-Cul-Nl 89.0 N2-Cul-Nl 83.4(1)
01-Cul-N2 94.0 N3-Cul-Cll 96.7(1)
01-Cul-N3 93.2 N2-CUL-CLL 96.9(1)
N3-Cu1-N2 164.0(1) Nl-Cul-Cl l 176.0(1)
N 3 - C U 1 - N 1 8 2 . 6 ( 1 ) 01-Cul-Cll 9 4 . 9
The numbers in parentheses are the estimated standard deviations in the last digit.
37

C(26)
c(27)
C ( 2 4 , ( ^ ^ C , 2 5 ) c(29)
CO)
Figure 11. Molecular structure of [Cu(HL^)(OAc)](OAc) (52) showing the atom labeling scheme. Hydrogen atoms are omitted for clarity.
Table 3. Selected bond distances ( A ) and angles (°) for [Cu(HL^)(OAc)](OAc) (52)广
Cul-01 2.437(7) Cul-N2 2.006(8)
Cul-02 1.905(7) Cul-N3 2.044(7)
Cul-Nl 1.989(8)
01-Cul-Nl 91.1(3) 02-Cul-Nl 98.5(3)
01-Cul-N2 92.8(3) 02-Cul-N2 96.7(4)
01-Cul-N3 88.0(3) 02-Cul-N3 173.8(3)
Nl-Cul-N2 164.5(4) Nl-Cul-N3 81.5(4)
01-Cul-02 85.7(3) N2-Cul-N3 83.6(4)
The numbers in parentheses are the estimated standard deviations in the last digit.
38
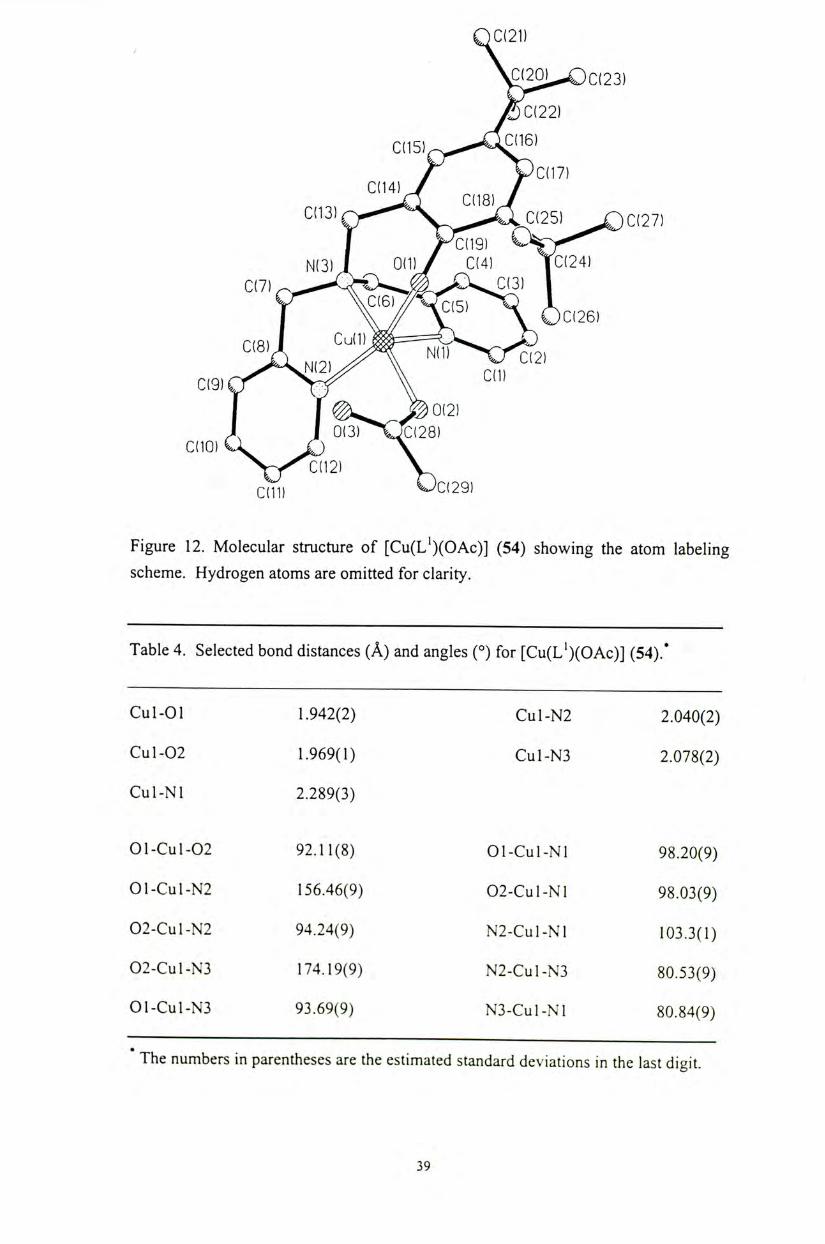
O C ( 2 1 )
V 2 0 ^ C ( 2 3 )
cn4) J
I yaiG) N(3) J 0(1)/ C(4) YC(24)
I � ^ ^ > 0 ( 2 1 A I 0(3) O:(28) CdO) Cl J j \ y C(ll) C(29) Figure 12. Molecular structure of [ C U ( L ' ) ( O A C ) ] (54) showing the atom labeling scheme. Hydrogen atoms are omitted for clarity.
Table 4 . Selected bond distances (A) and angles (。)for [C U ( L ' ) ( O A C ) ] ( 5 4 ) . '
Cul-01 1.942(2) Cul-N2 2.040(2)
Cul-02 1.969(1) Cul-N3 2.078(2)
Cul-Nl 2.289(3)
01-Cul-02 92.11(8) Ol-Cul-Nl 98.20(9)
01-Cul-N2 156.46(9) 02-Cul-Nl 98.03(9)
02-Cul-N2 94.24(9) N2-Cul-Nl 103.3(1)
02-Cul-N3 174.19(9) N2-Cul-N3 80.53(9)
01-Cul-N3 93.69(9) N3-Cul-Nl 80.84(9)
The numbers in parentheses are the est imated standard deviat ions in the last digit.
39

OC(251 广 1)
I C(22) /
C(23)
VI C )C (13 )
C(3) C(4) ⑶
C(11)
Figure 13. Molecular structure of [Zn(L^)Cl] (55) showing the atom labeling scheme. Hydrogen atoms are omitted for clarity.
Table 5. Selected bond distances ( A ) and angles (。)for [Zn(L^)Cl] (55)广
Znl -01 1.920(1) Znl-N2 2.134(2)
Znl -Cl l 2.294(1) Znl-N3 2.269⑵
Zn l -N l 2.078(2)
01-Znl-Nl 105.30(8) 01-Znl-Cl l 101.93(6)
01-Znl-N2 128.38(9) N l -Zn l -C l l 99.08(7)
Nl-Znl-N2 119.12(9) N2-Znl-Cl l 95.98(7)
01-Znl-N3 91.22(7) N2-Znl-N3 75.09(8)
Nl-Znl-N3 77.61(8) N3-Znl-Cl l 166.84(5)
• The numbers in parentheses are the estimated standard deviations in the last digit.
40

O C ( 2 1 )
C(17) I
l l b y ^ C(22) C c { 2 3 )
\ o ( 1 )
CO) a .
C(IO)
Figure 14. Molecular structure of [Zn(L^)Cl] (56) showing the atom labeling scheme. Hydrogen atoms are omitted for clarity.
Table 6. Selected bond distances (A) and angles (。)for [Zn(L^)Cl] (56). '
Z N L - 0 1 1.934(2) Zn l -N2 2.107(3)
Znl-Cll 2.321(1) Znl-N3 2.298(3) Znl-NM 2.134(3)
0 1 - Z n l - N l 125.20(1) O l - Z n l - C l l 99.97(8)
0 1 - Z n l - N 2 113.1(1) Nl -ZnI-CI l 98.71(8) Nl-Znl-N:! 113.8(1) N2-Znl-Cll 99.32(9)
N l - Z n l - N 3 75.4(1) N2-Znl-N3 77.0(1)
O l - Z n l - N 3 89.4(1) N3-ZnI-CIl 170.66(7) • The numbers in parentheses are the es t imated standard devia t ions in the last digit.
41

C ( 9 ) O Q C ( 1 0 )
I Vi”)
\ 1 C(13)
有 / 。"‘ Cld)
Figure 15. Molecular structure of [Zn(L^)Cl] (57) showing the atom labeling scheme. Hydrogen atoms are omitted for clarity.
Table 7. Selected bond distances ( A ) and angles (。)for [Zn(L^)Cl] (57).*
Znl-01 1.926(2) Znl-N2 2.104(2)
Znl-Cl l 2.3209(8) Znl-N3 2.269(2)
Znl -Nl 2.116(3)
01-Znl-Nl 116.9(1) 01-Znl-Cll 98.24(6)
01-Znl-N2 124.60(9) Nl -Znl -Cl l 100.54(8)
Nl-Znl-N2 112.5(1) N2-Znl-Cll 96.07(7)
Nl-Znl-N3 76.2(1) N2-Znl-N3 77.65(9)
01-Znl-N3 90.80(8) N3-Znl-Cll 170.89(6)
The numbers in parentheses are the estimated standard deviations in the last digit.
42
C(21) C(22)
尸 C ( 2 3 )
J k m y P c(26)
Njh^f^⑶ 5)
0(3)令 (2) C ( ^ c n i )
V C ( 2 8 )
O c ( 2 9 )
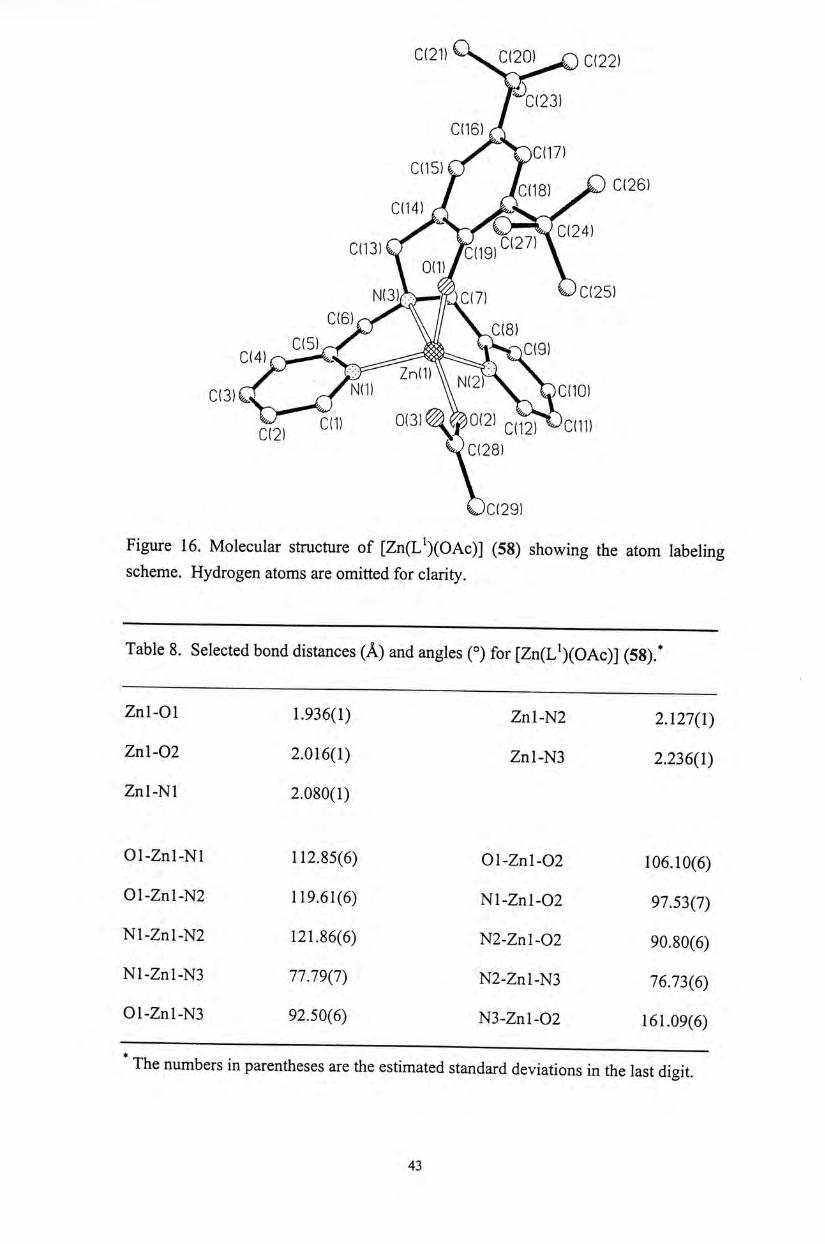
Figure 16. Molecular structure of [Zn(L^)(OAc)] (58) showing the atom labeling scheme. Hydrogen atoms are omitted for clarity.
Table 8. Selected bond distances ( A ) and angles (°) for [Zn(L^)(OAc)] (58)广
Znl-01 1.936(1) Znl-N2 2.127(1)
Znl-02 2.016(1) Znl-N3 2.236(1)
Znl-Nl 2.080(1)
01-Znl-Nl 112.85(6) Ol-Znl-02 106.10(6)
01 -Zn 1-N2 119.61(6) Nl-Znl-02 97.53(7)
Nl-Znl-N2 121.86(6) N2-Znl-02 90.80(6)
Nl-Znl -N3 77.79(7) N2-Zn 1-N3 76.73(6)
01-Znl-N3 92.50(6) N3-Znl-02 161.09(6)
The numbers in parentheses are the estimated standard deviations in the last digit.
43
; ; ^ I C ( 1 2 ) ^ ©CdO)
^^^ r .
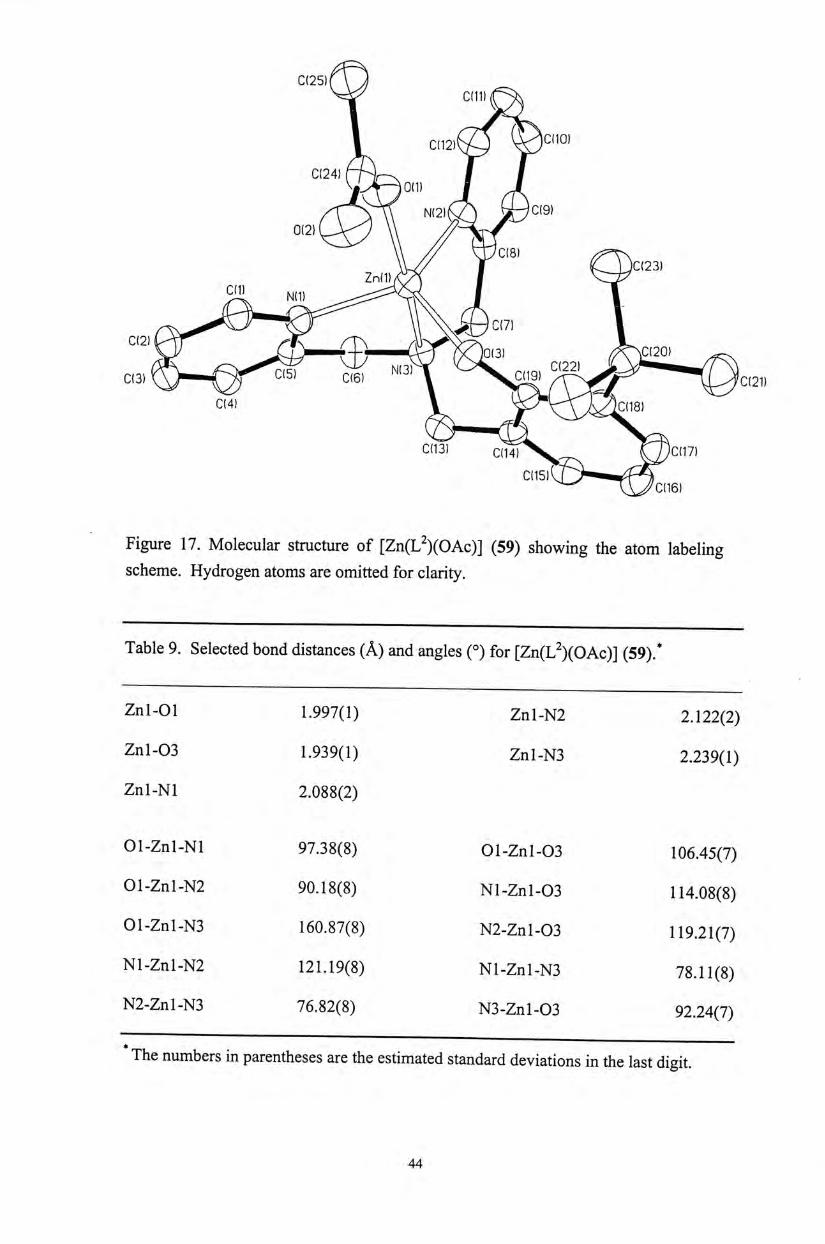
Figure 17. Molecular structure of [Zn(L^)(OAc)] (59) showing the atom labeling scheme. Hydrogen atoms are omitted for clarity.
Table 9. Selected bond distances ( A ) and angles (。)for [Zn(L^)(OAc)] (59).*
Znl-01 1.997(1) Znl-N2 2.122(2)
Znl-03 1.939⑴ Znl-N3 2.239(1)
Znl-Nl 2.088(2)
01-Znl-Nl 97.38(8) O l - Z n l - 0 3 106.45(7)
01-Znl-N2 90.18(8) N l - Z n l - 0 3 114.08(8)
01-Znl-N3 160.87(8) N 2 - Z N L - 0 3 119.21(7)
Nl-Znl-N2 121.19(8) Nl-Znl-N3 78.11(8)
N2-Znl-N3 76.82(8) N3-Znl-03 92.24(7)
The numbers in parentheses are the estimated standard deviations in the last digit.
44

2-C-II. Spectroscopic Studies
In this work, four copper(II) complexes, 51-54,were structurally characterized
by X-ray crystallography and elemental analyses. Copper(II) and zinc(II) phenolate
complexes derived from HL^ (34) were further studied and characterized in order to
correlate their solution structures with that of the solid state, and to understand the
electronic influences of the phenolate substituents.
The X-band electron paramagnetic resonance (EPR) spectra of all copper(II)
complexes of N3O ligands (51,52,53,54) taken at 77K exhibited axial signals with
g//-values in the range of 2.26 and 2.28 and g丄-values within 2.04 and 2.08. The
signals for copper(II) complexes are consistent with those of the reduced inactive
form of the native enzymes (g" = 2.28 and g丄=2.05).^ A l l zinc(II) complexes were
EPR silent.
The UV-Vis spectra of all zinc(II) complexes did not show any absorption band
within the range of 350 nm to 1100 nm. For the copper(II) acetate complex 54,a
sharp absorption band at 492nm was observed at room temperature which is assigned
to the PhO -»Cu(II) ligand-to-metal charge transfer (LMCT) transition.
Consequently, the complex acquires a deep blue coloration.⑴�
Al l complexes have been characterized by mass spectrometry. Their LSI or EI
mass spectra showed the corresponding isotopic ion distribution of [M]+ species
45

(Figures 18-22). However no parent peak signals have been observed in the LSI
mass spectra of the acetate complexes 54 and 58.
The 1h and ^ C NMR spectra of the N3O ligands 34-39 and the zinc(II)
complexes 55-59 have been measured. Upon complexation, the two methylene
protons located next to the pyridine ring are shown to be chemically non-equivalent.
They coupled to each other to give two doublet signals. For example, for complex
55,the two doublets appeared at (54.05 and 4.14 (Figure 23), while for complex 58,
they were resolved at (54.19 and 4.26 (Figure 24)。
46

P o s i t i i o n L — S I M S S p e c t r u r r x o i ' E C : A O a , i . ( u s i n s N B A a , m a t r i x )
.r, r- -f :二
二 汁 I . : - J r •二. I • V ... ;.>0 r-- ��� � 4000000 - ro 寸 uOLOf COCl - LT) LT) LO ltj u un LD
3 0 0 0 0 0 0 二
5 I 2 0 0 0 0 0 0 -
: , i j 1000000 - j j
,: juJvjLILLL ‘ I ‘ ‘ I ‘ ‘ ‘ ‘ 1 ‘ ‘ ‘ ‘ 1 1 1 ‘ ‘ 1 1 1 1 1 1~
50S 511 51S 521 52S m/z
Figure 18. The LSI mass spectrum of [Cu(HL^)Cl]Cl (51).
; f t ^ ^ r ^ l S 。 = l L " E n c t 〜 m Of ECA03:
4.Oe+07 一 T-.l 吁 r 产.
U.: ("O ^ ’一
3 . 5 e + 0 7 - — 一 一 — —
_ ‘ - lo CO r-w CO cr> - U-) LO to LT) LO
3 . 0 e - f 0 7 一
2.5e+07 一 :“ I
2 . O e + 0 7 —; |j ] I 1 . 5 e > 0 7 —
1 . O e + 0 7 - I I
: 丨 I : I I 1
5.0e-06 - I I ! !
。、。。: ^jjujUiJLj一 -5 .Qe+Q6 - ^ r~---r-
5 0 9 5 1 4 ‘ ‘ ‘ ‘ ~ _‘ ‘
二丄3 5 2 4 5 2 9 m / z
Figure 19. The LSI mass spectrum of [Cu(L^)Cl] (53).
47

S E u n d i n c a “ ~ A v e r a g e of 9.845 Co 9.94S tnin.: E C A 0 3 3 . D 240000 ^ 3 l 2
1 9 6
220000 -
200000 -
180000-
I S O O O O -
1 4 0 0 0 0 -
1 2 0 0 0 0 -
100000 - 幻
8 0 0 0 0 -
S O O O O - 1 119 198
40000- I … 58 40 7 425 20000. I 133 I J 219 267 306
0 • , , J l M . t t f i . , 1 , 1 . 1 , ,,,.I|Im, , J | I . , 〒 f , 、 , I „ i , l l , , 1 , 1 . ?357 3 7 9 … ! J 丨
n/z--> 40 S O a o 1 0 0 1 2 0 1 4 0 1 6 0 l a o 200 2 2 0 2 4 0 2 S 0 2 8 0 300 3 2 0 3 4 0 3 6 0 3 8 0 4 0 0 4 2 0
Figure 20. The EI mass spectrum of [Cu(L^)(OAc)] (54).
a.i. ” Po s'ltive ion L —SiMS Speo trtam of ECA03S ,\.siTis NBA as matrix ) 2 .0e+.07
“ �uo oo 寸 CT) LO r- • uo csi CO o r CO CO . " OD f CO to �� 1.5e+07 — uo '-JD r CO CD CO
‘ 一 一 一 _ _ C s l C ^ J 1 un Ln un LO Ln Ln un ; 观 V
1.0e-f07 -“
5.0e+06 - I I
: , I ! || I - i /働 ill , l| 0.0e.00 ‘ .—LU丄丄、�> a.丨wL«�ij丄L
‘ —I ‘ I ””‘ '1 r一—— r — ‘ - I — , r. •一 480 500 520 m/z
Figure 21. The LSI mass spectrum of [Zn(L^)Cl] (55).
48

a.i. - --'o. -:. li-^e i o n L … . S I U S S p e c: Lr 1.1 m o f E C A O S o (•-••si x ^ N B A a s m a t r i x )
:: S.0e>03,
“ : : - T : rj-)
•• r-v ‘ ― • — 一
_ 00 oc c o c o - T -sr
6.0e>08 - ^ : ^ “
S.Oe+03 -
4.0e+08 -
3.0e-^08 - 、.
2.0e+08 -
1.0e-f08 -
- ^ ^ _ _ _ _ _ _ O.Oe^OO ^ • ‘ •'"' —
1 ‘ • ‘ 1 ‘ ^‘ • • _•-, 1 ‘ ‘~ 490 540 590 m/z
Figure 22(a). The LSI mass spectrum of [Zn(L^)(OAc)] (58).
A b u n d a n c e A v e r a g e o t S . 1 2 2 Co 5 . 5 7 9 m i n .: E C A O S S . D .
9(3
4 0 0 0 0 - 3 1 9
3 5 0 0 0 -
261 425 3 0 0 0 0 -
58 4 3 2
2 5 0 0 0 -
2 0 0 0 0 - 1 9 8 3 8 6
119 丨 !|
I S O O O . 5 4 0
10_- , 232
。.J此 iiiiliLi iij J灿 k iiux 丄 ^ njUi 50 1 5 0 2 0 0 ^ ^ ^ ^ ^ 55o'‘
Figure 22(b). The EI mass spectrum of [Zn(L^)(OAc)] (58).
49

S
M K
P
I
•
.
丨
L
咖
饥 g
•
肌
iiti F工
f.^^
一…『L二
I
」:i
sm c-snsrs讯ssrssgss^s
!狀sSS
sa⑴q…i
Jo
lo
j il
5
『
^
Sri MMMMMMMMMMMMMMMMMMM
f—^
Ao^
a
1
-—_
KM^I^ ⑶
;”;= I
MMMM^ffi
) 认
-2
1
o
^ ——
maim
,2
X
,
J i
I-寬
L
:
L斤 ^
—-^Affl
棚
【【、
ul—
o
二『c
Myui
i ^
s
l^nl
m
p !4
V
J
ylu
-g
o
. ,5
嫩 K
fl^
印
!一『厂 ^
i
J ,6
H
i.么 nl
7
3.
1
vi
^
: p
”;〒
s
遣 丄I
3
i等
丄I
3
I
M
‘ I_
H
s- m Jy l\
.9
—9
A5S
~ 699,0、
ia
.
w n
;i!
^^
maa
li

2-D. Generation of Metal Phenoxyl Radical Species
Metal-radical array plays a very important role in many metalloproteins and
on
catalytic reactions.
Copper(II) and zinc(II) phenolate complexes (53, 54,58,59) could be oxidized
with one equivalent of eerie ammonium nitrate (CAN) in dry dichloromethane under
nitrogen at -80 °C to generate the corresponding metal phenoxyl radical species
(Scheme 11).
R, ^ J ^ '
^ ^ (剛C e > 0 3 ) 6 ] i CH2CI2 -80 °C y
0 亡
M = Cu, Zn X = CI, OAc
R = f-Bu; R' = t-Bu, H (1_1’2) R=H,. R' = t-Bu (l3) (Scheme 11)
Upon oxidation with CAN at -80。C,the solutions acquired a deep green
coloration immediately. The deep green intermediate species were stable only at low
temperatures (<-40 °C). However, for the copper(II) phenoxyl complex (51),no such
51

observable change was observed upon oxidation. Both EPR and UV-vis
spectroscopy were used to characterize the intermediate species in our work.
No obvious change has been observed in the EPR spectra of compound 51 both
before and after oxidation with CAN (Figure 25), suggesting that the compound was
inert toward oxidation and no evidence for the existence of any radical species has
been observed. On the other hand, both complexes 53 and 54 exhibited typical axial
Cu(II) signals before oxidation. Upon oxidation with CAN at -80 °C, the signal
amplitudes diminished substantially (Figures 26 and 27). These suggest the
formation of Cu(II) phenoxyl radical species. However, evidence on whether the
phenoxyl radical is tightly or loosely bound to the copper center is still missing in
these cases. Oxidation of the zinc(II) complex 58 gave a nearly isotropic signal at g
=2.00 (Figure 28),indicating the existence of the corresponding phenoxyl radical
species.
Further evidences on the existence of the radical intermediates could be
obtained from UV-Vis spectroscopy. Only complexes 54 and 58 were fully
characterized. The absorption band for the former complex 54 shifts from 491 nm to
413 nm and 662 nm upon oxidation with CAN (Figure 29). No absorption feature
has been observed in the range from 350-1050 nm for 58. Upon oxidation with
52

CAN, two absorption maxima at 406 nm and 753 nm were observed (Figure 30).
These absorption features suggest the existence o f phenoxyl radical species.^' ^
On the basis o f EPR and UV-vis spectroscopy,highly reactive phenoxyl radical
species have been successMly generated by chemical oxidation o f metal phenolate
complexes with C A N at very low temperatures.
& ^ “ I ——51 +CAN
2400 2600 2800 3000 3200 3400 3600 3800 4000 Magnetic Field (Gauss)
Figure 25. EPR spectra of [Cu(HL^)Cl]Cl (51) at 77K. Instrumental parameters: microwave power,0.2 microwatts; microwave frequency, 9.443 GHz; modulation amplitude, 2 G; modulation frequency, 100 kHz.
53
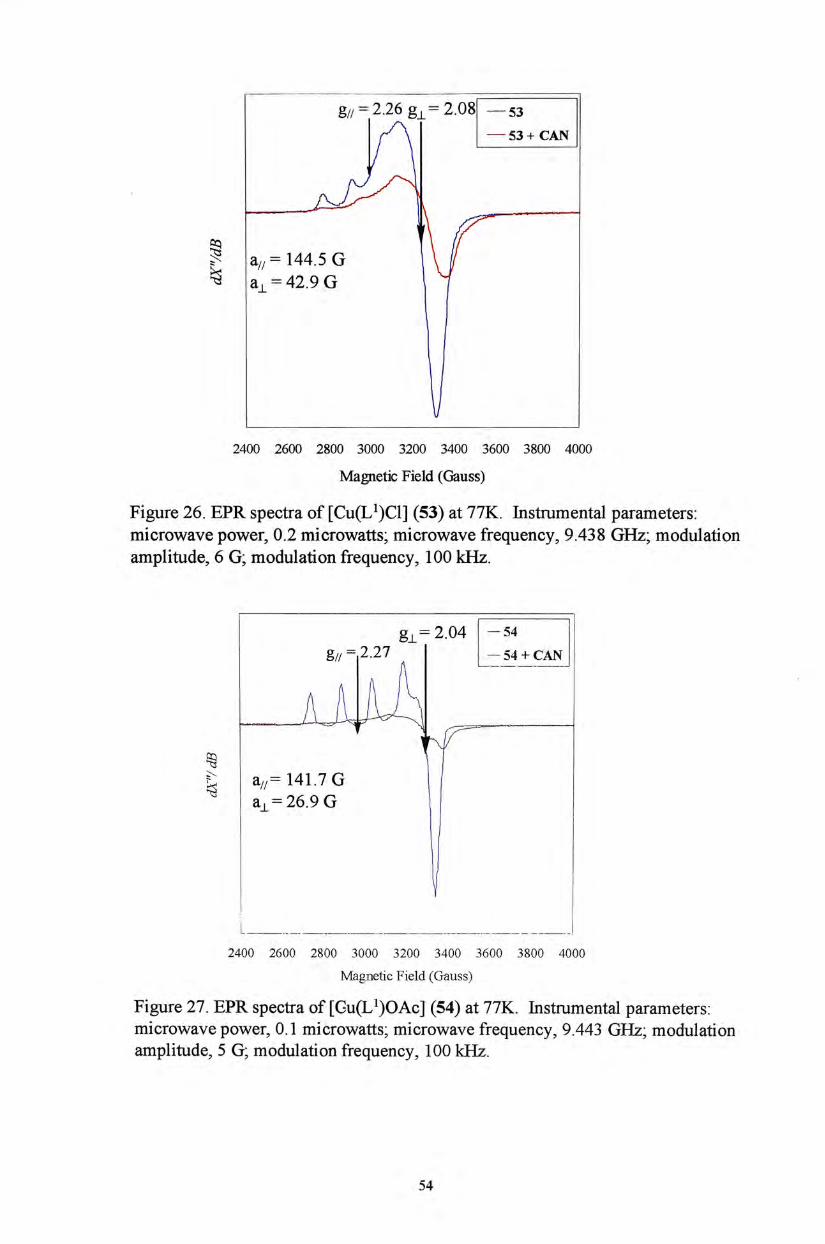
g// = 2.26 g丄=2.081 —53
—53 +CAN
_ _ _ _ _
, H I a / / -144.5 G \ ^ a丄=42.9 G ^
1/
2400 2600 2800 3000 3200 3400 3600 3800 4000
Magnetic Field (Gauss)
Figure 26. EPR spectra of [Cu(L^)Cl] (53) at 77K. Instrumental parameters: microwave power, 0.2 microwatts; microwave frequency, 9.438 GHz; modulation amplitude, 6 G; modulation frequency, 100 kHz.
I —
g 丄 = 2 . 0 4 — 5 4
g// = 2.27 —54 +CAN
^ J I a//= 141.7 G
a 丄 二 26.9 G
2400 2600 2800 3000 3200 3400 3600 3800 4000
Magnetic Field (Gauss) Figure 27. EPR spectra of [Gu(L^)OAc] (54) at 77K. Instrumental parameters: microwave power, 0.1 microwatts; microwave frequency, 9.443 GHz; modulation amplitude, 5 G; modulation frequency, 100 kHz.
54

—58 + CAN
宅 J ‘
^ I
2800 3000 3200 3400 3600 3800 4000
Magnetic Field (Gauss)
Figure 28. EPR spectrum of [Zn(Li)(OAc)] (58) at 77K. Instrumental parameters: microwave power, 0.2 microwatts; microwave frequency,9.438 GHz; modulation amplitude, 2 G; modulation frequency, 100 kHz.
3 r ——n ——54
2.5 ——54 + C A N
0 350 450 550 650 750 850 950 1050
wavelength (nm)
Figure 29. UV-vis spectra of [Cu(L^)(OAc)] (54) at room temperature.
55

0.7 [― 〒
— 5 8 0.6
— 5 8 + C A N 0.5
S 0.4 \f\ s 召 0.3 o
# 0.2 \
0.1 V “ 0 ^ “ — ~ —
-0.1
350 450 550 650 750 850 950 1050
wavelength (nm)
Figure 30. UV-vis spectra of [Zn(L^)(OAc)] (58) at room temperature.
56

2-E. Summary
In conclusion, a series of mononuclear copper(II) and zinc(II) complexes (51-
59) with tripodal tetradentate A/jO-type ligands were successfully synthesized as
mimics to the active site of galactose oxidase. X-Ray crystallography revealed a
distorted square pyramidal N3O coordination sphere around the metal center in these
complexes. Thus, these complexes serve as structural models for the reduced
inactive form of the active site of GAO. On the basis of EPR and UV-vis
spectroscopy, highly reactive phenoxyl radical species were successfiilly generated
by chemical oxidation of the corresponding metal phenolate complexes with cerium
ammonium nitrate.
The oxidation of primary alcohols to the corresponding aldehydes catalyzed by
these complexes have also been investigated. Unfortunately, none of the complexes
reported in this work showed catalytic properties towards oxidation of alcohols to
aldehydes. During optimization of the reaction conditions for this catalysis,
Yamauchi and co-workers have reported a series of copper(II) and zinc(II)
complexes and two of them were prepared by ligand HL^ (34) used in our studies. ^
However, the difference between our complexes (53-55, 58) and those complexes
reported by the Japanese research group is on the choice of counter anions. They
used mixed counter anions, CIO4" and CI" for the preparation of copper(II) complex,
57

but C r for the preparation of the zinc(II) complex supported by l A Besides, similar
approaches have been employed by both research group in the studies of the
phenoxyl radical species.
By using ligands 34-39, several copper(I) complexes were synthesized and their
chemistry has been found to be totally different from that of their copper(II)
counterparts, as wil l be described in Chapter 3.
58

2-F. References
1. Ito, N.; Philips, S. E. V.; Stevens, C.; Ogel, Z. B.; McPherson, M. J.; Keen, J. N.;
Yagaz, K. D. S.; Knowles, P. F. Nature 1991,350, 87-90.
2. Whittaker, M. M.; Whittaker, J. W. J. Biol Chem. 1988, 262, 6074-6080.
3. Whittaker, M. M.; Ballou, D. P.; Whittaker, J. W. Biochemistry 1998,37, 8426-
8436.
4. (a) Wang, Y.; Stack, T. D. P. J. Am, Chem. Soc. 1996, 13097-13098; (b) Wang,
Y.; BuBois, J. L.; Hedman, B.; Hodgson, K. O.; Stack, T. D. P. Science 1998,
279, 537-540.
5. (a) Chaudhuri, P.; Hess, M.; Florke, U.; Wieghardt, K. Angew. Chem. Int. Ed.
1998,37, 2217-2220; (b) Chaudhuri, P.; Hess, M.; Muller, J.; Hildenbrand, K.;
Bill, E.; Weyhermuller, T.; Wieghardt, K. J. Am. Chem. Soc. 1999,121, 9599-
9610.
6. (a) Sokolowski,A.; Lentbecher, H.; Weyhermuller, T.; Schnepf,R.; Bothe, E•;
Bill, E.; Hildebrandt, P.; Wieghardt, K. J. Biol Chem. 1997’ 2’ 444-453; (b)
Schnepf, R.; Sokolowski, A.; Muller, J.; Bachler, V.; Wieghardt, K.;
Hildebrandt, P. J. Am. Chem. Soc, 1998, 120, 2352-2364; (c) Muller, J.;
Weyhermuller, T.; Bill, E•; Hildebrandt, P.; Ould-Moussa, L.; Glaser, T.;
59

Wieghardt, K. Angew. Chem. Int. Ed. 1998,57, 616-619; (d) Bill, E.; Muller, J.;
Weyhermuller, T.; Wieghardt, K. Inorg. Chem. 1999, 38, 5795-5802.
7. Sokolowski, A.; Muller, J.; Weyhermuller, T.; Schnepf,R.; Hildebrandt P.;
Hildenbrand, K.; Bothe, E.; Wieghardt, K. J. Am. Chem. Soc. 1997,119, 8889-
8900.
8. Zurita, D.; Gautier-Luneau, L; Menage, S.; Pierre, J. L.; Saint-Aman, E. J. Biol.
Inorg. Chem. 1997, 2, 46-55.
9. Saint-Aman, E.; Menage, S.; Pierre, J. L.; Defrancq, E.; Gellon, G. New J .
Chem. 1998,393-394.
10. Halfen, J. A.; Jazadzewski, B. A.; Maphapatra, S.; Berreau, L. M.; Wilkinson, E.
C.; Que, L., Jr.; Tolman, W. B. J. Am. Chem. Soc. 1997,119, 8217-8227.
11. Halfen, J. A.; Young, V. G.,Jr.; Tolman, W. B. Angew. Chem. Int. Ed. Engl
1996, 55, 1687-1690.
12. Whittaker, M. M.; Duncan, W. R.; Whittaker, J. W. Inorg. Chem. 1996,35’ 382-
386.
13. (a) Adams, H•; Bailey, N. A.; Rodriguez de Barbarin, C. O.; Fenton, D. E.; He,
Q. Y. J. Chem. Soc” Dalton Trans. 1995, 2323-2331; (b) Adams, H.; Bailey, N.
A.; Campbell, I. K.; Fenton, D. E.; He, Q. Y. J. Chem. Soc., Dalton Trans. 1996,
2233-2237.
60

14. (a) Itoh, S.; Takayama, S.; Arakawa, R.; Furuta, A.; Komatsu, M.; Ishida, A.;
Takamuku, S.; Fukuzumi, S. Inorg. Chem. 1997,36, 1407-1416; (b) Itoh, S.;
Taki, M.; Takayama, S.; Nagatomo, S.; Kitagawa, T.; Sakurada, N.; Arakawa,
R.; Fukuzumi, S. Angew. Chem. Int. Ed. 1998,38, 2774-2776; (c) Itoh, S.; Taki,
M.; Kumei, H.; Takayama, S.; Nagatomo, S.; Kitagawa, T.; Sakurada, N.;
Arakawa, R.; Fukuzumi, S. Inorg. Chem. 2000, 39, 3708-3711.
15. Berends, H. P.; Stephan, D. W. Inorg. Chim. Acta 1984,93’ 173-178.
16. Whittaker, M. M.; Chuang, Y .Y.; Whittaker, J. W. J. Am. Chem. Soc. 1993,
115, 10029-10035.
17. Chaudhuri, P.; Hess, M.; Weyhermuller, T.; Wieghardt, K. Angew. Chem. Int.
Ed. 1999, 38, 1095-1098.
18. Shimazaki, Y.; Huth, S.; Odani, A.; Yamauchi, O. Angew. Chem. Int. Ed. 2000,
39, 1666-1669.
19. Halcrow, M. A.; Chia, L. M. L.; Liu, X.; Mclnnes, E. J. L.; Yellowlees, L. J.;
Mabbs, F. E:; Scowen, 1. J.; McPartlin, M.; Davies,J. E. J. Chem. Soc., Dalton
Trans., 1999,1753-1762.
20. Stubbe, J.; Donk, W. A. Chem. Rev. 1998,98, 705-762.
21. Shimazaki, Y.; Huth, S.; Hirota, S.; Yamauchi, O. Bull Chem. Soc. Jpn. 2000,
75,1187-1195.
61

CHAPTER 3
Copper(I) Complexes with N3O Tetradentate Ligands
3-A. Introduction
3-A-I. Studies on copper(I) complexes with oxygenation
Oxidation reactions play a very important role in various biological and
industrial processes. The use of dioxygen as the primary oxidant has attracted much
interest in the chemical industry.【Copper plays a crucial role in proteins involved in
electron transfer and dioxygen processing (Table 10). Besides, it has widespread
applications in both stoichiometric and catalytic oxidative transformation. The
interaction between copper(I) and dioxygen is a key component for both chemical
and biological processes. Thus, much interest has been attracted to the investigation
on the binding, interaction, and subsequent reactivity of dioxygen with copper ion
centers.
A number of dioxygen binding modes in copper complexes have been
^ 1
proposed. Figure 31 shows the six common ligation modes: end-on, r) -superoxo,
side-on, ri^-superoxo, |a-l,2-peroxo, |a-Ti :ri -peroxo, bis-)i-oxo and |i-hydroperoxo.
62

Table 10. Copper proteins involved in dioxygen processing.
Protein Biological Function Oxygen Carrier Hemocyanin* O2 transport
Copper Monooxygenases Tyrosinase tyrosine oxidation Dopamine p-monooxygenase (DpM) dopamine->norepinephrine Peptidylglycine a-amidating monooxygenase (PAM)* oxidative N-dealkylation Methane monooxygenase (MMO) methane—methanol Ammonia monooxygenase (AMO) ammonia->hydroxylamine Copper Dioxygenases Quercetinase quercetin oxidative cleavage
Copper Oxidases 'Blue,multi-copper oxidases (converting O2 to 2H2O) Laccase* phenol and diamine
oxidation Ascorbate oxidase* oxidation of L-ascorbate Ceruloplasmin* Fe(II)->Fe(III) and/or radical
scavenging
'Non-blue ‘ oxidases (converting O2 to 2H2O2) Amine oxidase* elastin, collagen formation
and alcohol oxidative deamination
Galactose oxidase* galactose oxidation alcohol—aldehyde
Glyoxal oxidase aldehyde->carboxylic acid Phenoxazinone synthase phenoxazinone formation
and oxidative coupling
Heme-copper oxidases terminal oxidases Cytochrome c oxidase (CcO)* proton translocation (pump) Quinol oxidase
Others Superoxide dismutase (SOD)* O2' dismutation
(detoxification) *X-ray structure(s) available.
63

- -11+ 1+ 「 -|2+
c u - \ H < 1 CU.....O _ _ L J 〇、-CLi
厂 l2+ r ^2+ ^ O� 「 J 1 +
广. . . -0- . . . / \ C u . 、 . , C u
Cu:、、人...-Cu Cu; :Cu 、〇••
O H - -I L J 匕 J
Figure 31. Dioxygen binding modes in copper complexes.
Most of the copper proteins in Table 10 have been introduced in Chapter 1. The
following section gives a brief review on the chemistry of copper complexes with
oxygen binding modes.
3-A-II. Studies on copper complexes with oxygen binding
Although copper-dioxygen complexes are important in many catalytic oxidation
reactions, only a few of the corresponding intermediates in these reactions have been
characterized, and the structural factors substantiating the catalytic effectiveness of
the copper complexes remain unclear. The reasons for the development of the
oxidation chemistry of copper-dioxygen complexes are: (1) providing the basis for
designing and developing new catalysts, and (2) being found to be common in highly
efficient biooxidation.
64

It has been suggested that copper-hydroperoxo species (Cu„-OOH; n = 1, 2) are
intermediates in many enzymatic reactions. These species have been known to be
key intermediates in the catalytic reactions of dopamine p-monooxygenase and
peptidylglycine a-hydroxylating monooxygenase. It was also proposed that these
species involve in the disproportionation of superoxide catalyzed by copper-zinc
superoxide dismutase (SOD), and the reduction of dioxygen to water by laccase and
multicopper oxidases.
Sorrell and co-workers have reported the dioxygen reactivities of dinuclear
copper-phenolate and -phenol complexes (60-64) with pyrazole and pyridine donors
(Schemes 12-14).4 In their reaction with dioxygen, the corresponding peroxo and
hydroperoxo adducts were generated at low temperature (Scheme 15).4
R
b ' " ' J n C I 八 — ^ 力 〜 叫 _ _ .
. a 亏 C I . Q Q . 乂 严 、 t h f 1 y ^ ^ ^ H
60 R = H
61 r = ch3 (Scheme 12)
65

p a y
62 (Scheme 13)
thf ,
63 R = H
64 r = ch3 (Scheme 14)
Dinucleating ligands 60 and 61 are unsymmetrical, whilst the 62-64 are
symmetrical. In these ligands, the para methyl substituent of the phenol group
prevents further substitution during the reactions. At -78 °C,the copper(I) phenolate
species 65,which showed no absorption in the UV-Vis region, reacted rapidly with
dioxygen to yield the copper(II)-peroxo adduct 70 (2:1 Cii:02),which was
characterized by an intense absorption at about 500 nm (a peroxide to Cu(II) charge-
transfer band), a smaller band at about 395 nm (a phenoxide to Cu(II) charge-transfer
band), and a shoulder at about 630 nm (a peroxide to Cu(II) charge-transfer band).
66
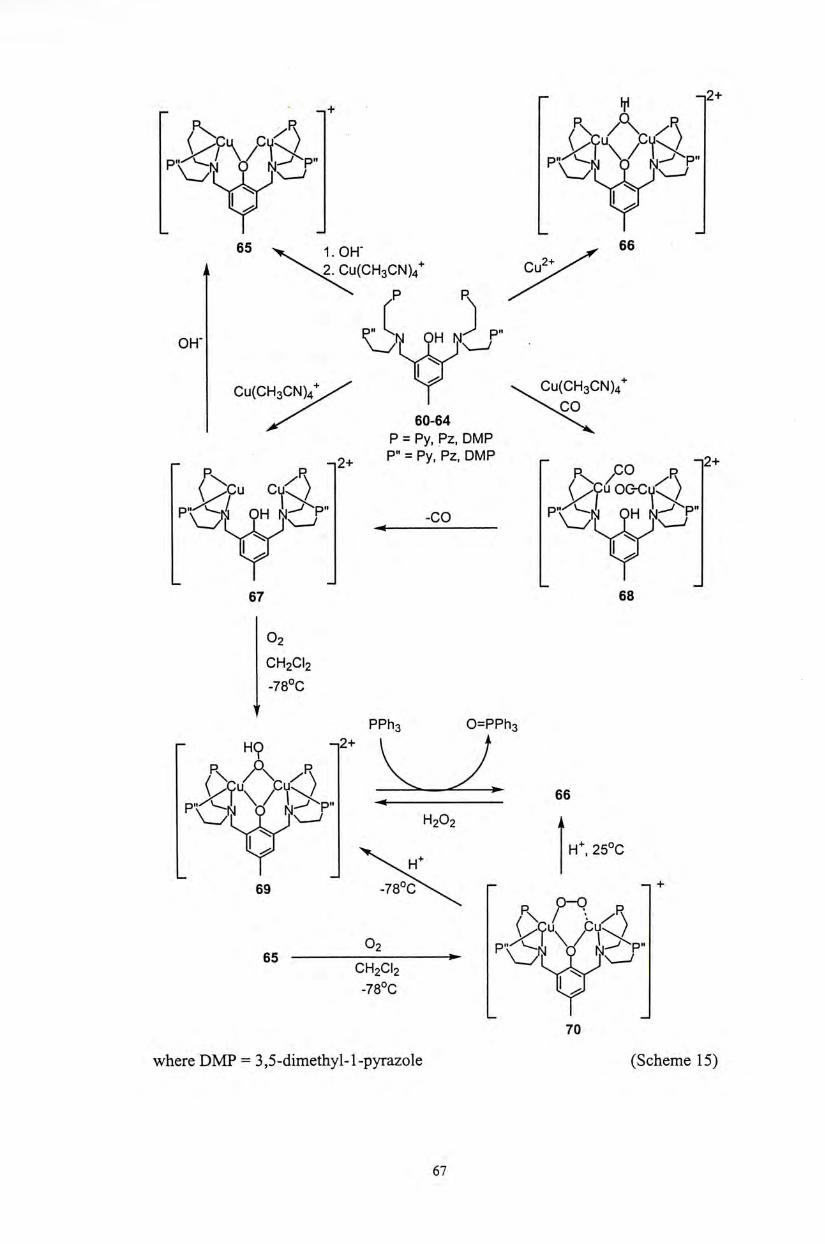
r 「 a T 彻会。..
y y 一 J 一
65 1.0H- Z 66 “ \s^CU(ch3CN)4+
/ P、
OH- d^X^ 丄。"
Cu(CH3CN)4>/ J \CU(CH3CN)/
60-64 P = Py, Pz, DMP P" = Py, Pz, DMP 2+
一 P P \ p CO p p p u C u Q p C u OC-CuCQ
乙 p " , -CO
y y 67 68
02 CH2CI2 -78。C
1 f PPha 0=PPh3
- H? n2+ \ f
举 : X X \ + H+, 25。C
- 丫 -69 -78°C\ . 「 "] +
65 " " " " P , ^ ; ^ 込 ” -78。C I L ^
70
where DMP = 3,5-dimethyl-l-pyrazole (Scheme 15)
67

The copper(I) phenol species 67 and 68 also reacted with dioxygen at very low
temperature to form a // -OOH copper(II) species, which were characterized by an
intense absorption at about 395 nm in the UV-Vis region. This species gave
triphenylphosphine oxide and the corresponding i i -hydroxo- j i -phenoxo copper(II)
dimeric species upon reaction with triphenylphosphine. It was reported that the
carbonyl groups in species 68 were removed under vacuum before its reaction with
dioxygen. The phenol species 67,without coordinated carbonyl groups, reacted
analogously with dioxygen and gave products with similar spectroscopic properties.
Karlin and co-workers have modified the binucleating ligands 60-64 and
prepared the imsymmetrical ligand 71 (Scheme 16).
N C t C L b p + 0 ^ 广 3 ~~EtOAc ‘
H iH3
L _ 4 . ( X ^ X ^ N H 2 MeOH/AcO” QL ^ Et2〇 N f ^ ^ J
H3C /
o 71
(Scheme 16)
68
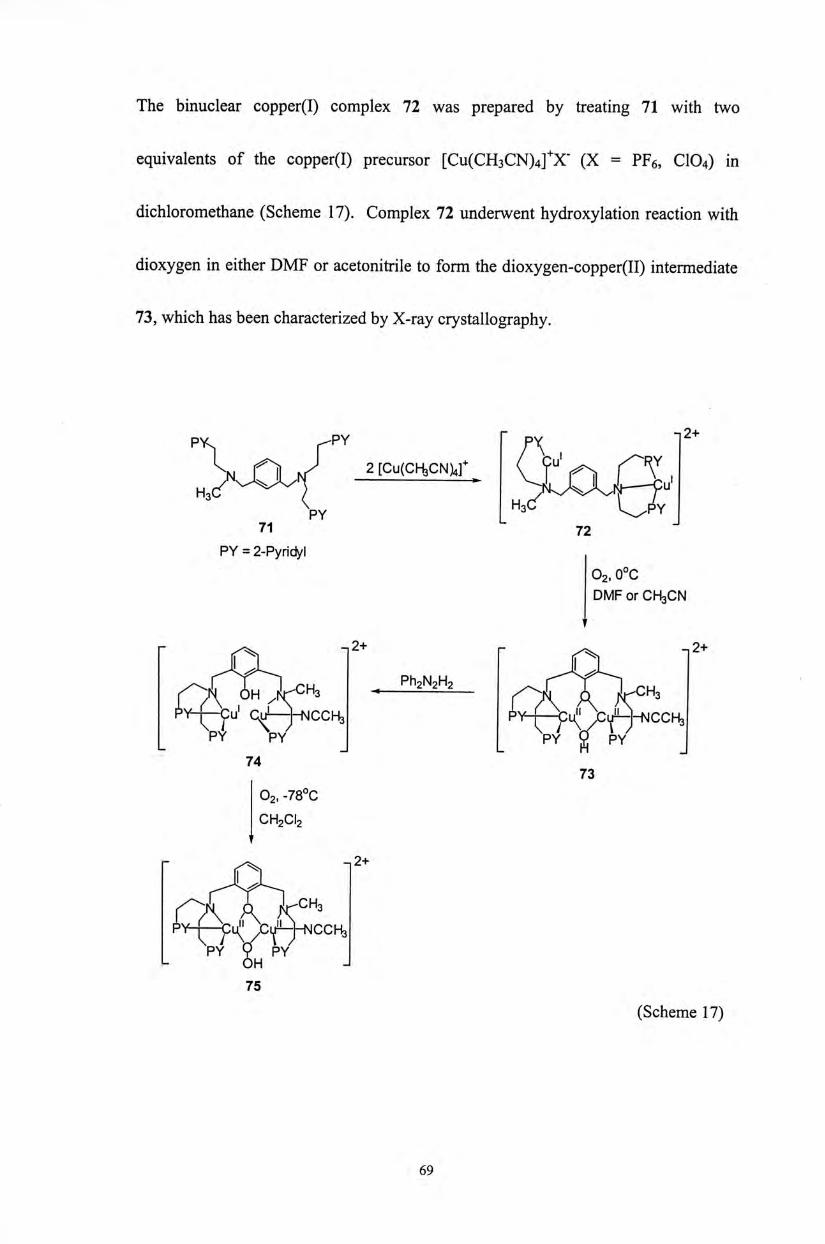
The binuclear copper(I) complex 72 was prepared by treating 71 with two
equivalents of the copper(I) precursor [Cu(CH3CN)4]+X- (X = PFe, CIO4) in
dichloromethane (Scheme 17). Complex 72 underwent hydroxylation reaction with
dioxygen in either DMF or acetonitrile to form the dioxygen-copper(II) intermediate
73,which has been characterized by X-ray crystallography.
py^ fPY 「 f \ 广
S g A hT 2 [Cu(CH3CN)4r (〒u' 广F Y H3 多 ( Y ^
71 72 」
PY = 2-Pyridyl O2, 0°C DMF or CH3CN
V V W Y S/ � L n _
74 73
O2, -78°C
CH2CI2
- ^ -|2+
OCX^CHs py-\~Cu;' /Ci/hhsicch3 V Y W
L 6H � 75
(Scheme 17)
69

The dicopper(I) complex 74 was prepared by reduction of dicopper(II) complex
73 with diphenylhydrazine. 74 has been characterized by two absorption bands in
the UV-Vis region at 312 nm and 366 nm. It reacted with dioxygen at -78 °C to give
(hydroperoxo)dicopper(II) complex 75, which has been characterized by a strong
absorption band at 390 nm (HOCT—Cii(II) LMCT transition), a shoulder at about
450 nm, and some weaker absorptions at about 630 nm and 1000 nm (d-d transition
for Cu(II)). Besides, complex 75 reacted with triphenylphosphine to yield
triphenylphosphine oxide (85%), a characteristic reaction for the hydroperoxo
complexes (Scheme 17).6a’ 6b
Karlin et al have reported a number of dinucleating ligands and the
corresponding dicopper-peroxo and -oxo complexes,; Some of them formed
(hydroperoxo)dicopper(II) complexes (76 and 77), converting triphenylphosphine to
triphenylphosphine oxide upon oxygenation,,6b’ 6d
- ^ 广 「 ^ 2+
V V Vy� V 9 V L OH 」 L OH J
76 77
PY = 2-Pyridyl PY = 2-Pyridyl
70

Apart from dicopper complexes, dicopper-peroxo, -oxo and -hydroperoxo
derived from copper complexes containing mononucleating ligands could also be
prepared.
Suzuki and co-workers have reported a bis( i i -oxo)dicopper(III) complex 79
which was prepared by the reaction of the monomeric copper(I) complex 78 with
dioxygen in acetone/methanol (10:1) at -78 °C. 79 has been characterized by X-ray
crystallography, UV-Vis, and resonance Raman spectroscopy.^^ It is noteworthy that
a reversible conversion of 79 to 78 could be achieved by bubbling nitrogen gas into a
dichloromethane solution of 79 at -80�C (Scheme 18)/®
A fS f| CH2=2_8Q。C 0 M \ y O
u 》 ^ 办 78 79 (Scheme 18)
The bis( // -oxo)dicopper(III) complex 79 exhibited an isotope-sensitive band at
590 cm" with (564 cm' with in its resonance Raman spectrum measured
in acetone at -80 °C with a laser excitation at 488 nm. These characteristic bands
71

were similar to those observed for the bis(}i-oxo)dicopper(in) complexes."’ The
UV-Vis spectrum of 79 in dichloromethane at -80。C showed an intense absorption at
378 nm, a shoulder at about 490 nm, and two more bands at 258 nm and ca. 300 nm.
The solid state structure of 79 has been determined by X-ray crystallography, which
showed an average Cu-0 and Cu-Cu bond lengths of 1.803 A and 2.758 A,
respectively. Al l of the data were comparable to those of bis( // -oxo)dicopper(III)
complexes."’ !之
Reaction of 79 with triphenylphosphine in acetone at -78。C under an argon
atmosphere gave dioxygen and the monomeric copper(I) complex 80 (Scheme 19).
\ \ / j f V ^ 形<!> : 1 ^^^ 02 + 广
r f ^ N f cr \ acetone. -78。C ( T ^ h Y
0 力 79 80
(Scheme 19)
72

Tolman and co-workers have reported a series of bis( i i -oxo)dicopper
complexes derived from monomeric copper(I) complexes (81-83) with macrocyclic
ligands (Scheme
r f x ^ R 1
~ V ^ R [CU(CH3CN)4]X I ^ J ^ 一
V n ^ X=SbFe-orCI04- 乂
k [ ^
R = Me, iPr, CHsPh 81 R = Me
82 R = iPr
83 R = CH2Ph (Scheme 20)
Upon oxygenation of complex 82 at - 7 8 � C in dichloromethane,
peroxo)-dicopper(II) species 84 was obtained. This species could be converted to
bis(|i-oxo)-dicopper(II) species 85 in tetrahydrofliran (Scheme 21)." At -75 °C,the
UV-Vis spectrum of 84 exhibited two absorption bands at 366 nm and 510 nm whilst
85 showed two absorption maxima at 324 nm and 448 nm. The resonance Raman
spectra of these two species have been measured as frozen solutions at -196 °C, with
laser excitation at 514 nm. Species 84 showed a signal at 722 cm'' and the bis(|u-
oxo)-dicopper(II) species 85 gave a signal at 600 cm' in their resonance Raman
spec 仕 a. 14
73
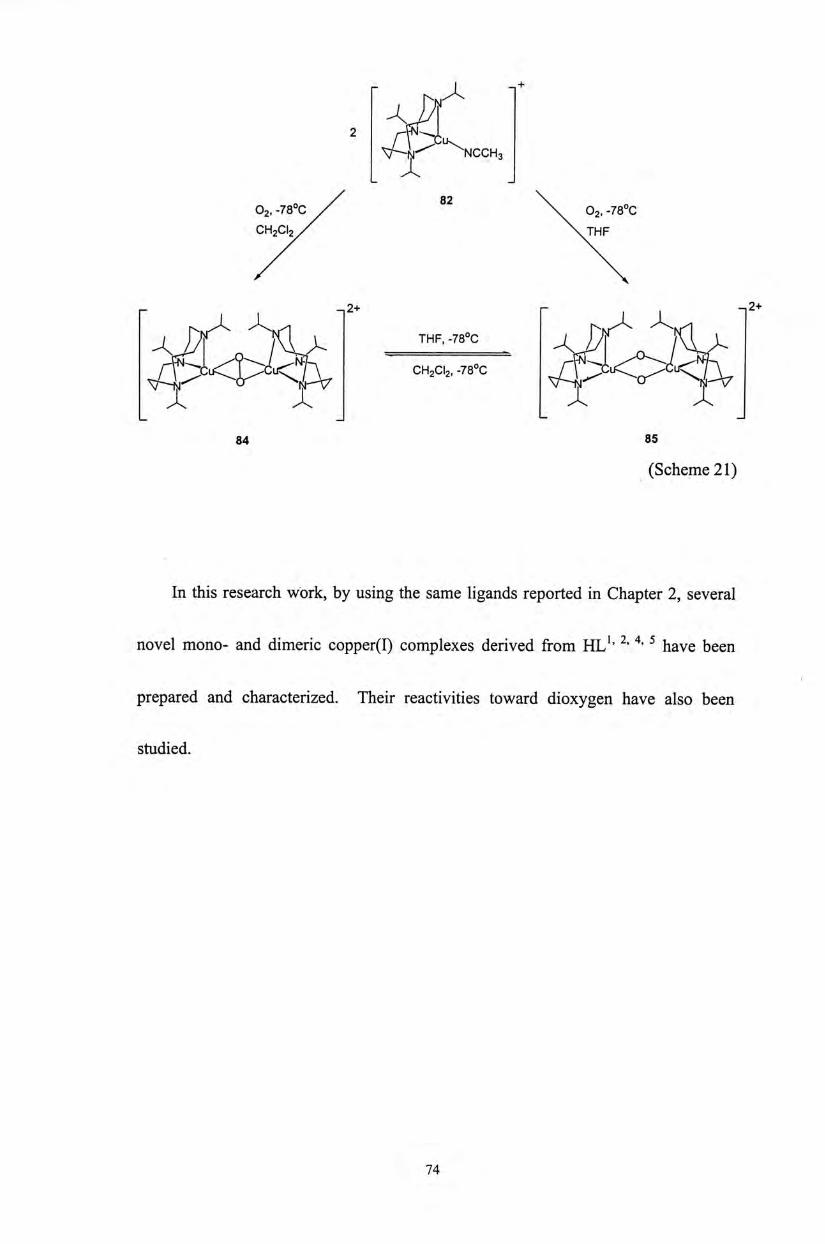
r 乂 "T
2 j i d L V^Z、CCH3 r
似 O2, -78°C
\ T H F
一 - 2 + - ^ ^ 一 2 +
— _j ‘― — 84 85
(Scheme 21)
In this research work, by using the same ligands reported in Chapter 2, several
novel mono- and dimeric copper(I) complexes derived from HL^' ^ have been
prepared and characterized. Their reactivities toward dioxygen have also been
studied.
74

Results and Discussion
3-B. Preparation of Copper(I) Complexes with N^O Tetradentate Ligands
In recent years, Karlin et al,、^^ and Tolman et al.^^' ^^ have studied the
oxygenation reaction of a series of monomeric and dimeric copper(I) complexes,
derived from similar or the same macrocyclic ligands as those they used to prepare
model complexes for galactose oxidase.
Copper(I) salts, such as [Cu(CH3CN)4]PF6 '''' ^^ [Cu(CH3CN)4](C104) 6b’ 6〜
[C u (CH3CN)4] (S03CF3)15 were commonly used for the preparation of monomeric
and dimeric copper(I) complexes. These copper(I) salts contain weakly coordinating
anions so that coordination to the metal centers during oxygenation reactions may be
suppressed. Reduction of copper(II) complexes may also be an alternative route to
copper(I) complexes.
In this part of our studies, a series of copper(I) complexes have been prepared
using proligands 34-39. In our study, Cul, [Cu(CH3CN)4]PF6 and
[Cu(CH3CN)4](C104) have been used as starting materials for the preparation of the
corresponding complexes. The former one, with a strong coordinating anion, was
stable to air and thus easy to be handled. The latter two, with weakly coordinating
anions, resulted in an intactable oil or disproportionation.
75

Treatment of copper(I) iodide with compounds 34 or 35 and triethylamine in
tetrahydrofliran gave the bisO-I) dicopper(I) complexes 86 and 87,respectively
(Scheme 22). However, attempted synthesis of the corresponding dicopper(I)
compound derived from 36 was unsuccessful. Only a small amount of
uncharacterizable solid was obtained after the reaction. Both compounds 86 and 87
have been structurally characterized by X-ray crystallography.
(f^ A (T H O ^ ' 2. cu., THF
34 R = R- = t-Bu (HLI) 86 [Cu(HL^)(l)]2 (80%)
35 R = t-Bu, R' = H (Hl2) g? [Cu(HL2)(|)]2 (77%)
(Scheme 22)
It is noteworthy that attempted deprotonation of the phenol pendant using
triethylamine was unsuccessful. When stronger bases such as sodium hydroxide and
«-butyllithium were used, only a deep reddish brown intractable oil was obtained.
Attempted preparations of monomeric copper(I) complexes supported by 34-36,
starting from [Cu(CH3CN)4]PF6 and [Cu(CH3CN)4](C104) as the starting materials
were also unsuccessful.
76

Although the benzimidazole-containing proligands 37-39 are structurally similar
to the pyridine-based proligands 34-36, the structures of the copper(I) complexes
derived from these ligands are totally different. As shown in Scheme 23, compounds
37-39 reacted with copper(I) iodide to give the mononuclear three-coordinate
copper(I) complexes 88 and 89. Attempted preparation of copper complex derived
from 39 was unsuccessful.
H O ^ 2.CU,.THF HN 么 广 丨 R
3 7 R = R ' -Bu (Hl4)
38R = f-Bu, R' = H (Hl5) 88 Cu(HL^)l (61%)
39 R=H. R' = f-Bu (HL®) 89 Cu(HL5)| (22%)
(Scheme 23)
Being similar to 86 and 87, attempted deprotonation of the phenol pendant on
37 and 38 with triethylamine was unsuccessful.
Details of the preparation of compounds 86,87,88 and 89 are described in
Chapter 4 Experimental Section. These complexes have been characterized by NMR
77

spectroscopy, mass spectroscopy, in addition to X-ray crystallography. Their
reactivities toward dioxygen have also been investigated.
In our work, four copper(I) complexes (86,87,88 and 89) have been
successfully synthesized by using copper(I) iodide as the staring materials.
Conceivably, the iodide ion reduces the potential problem of disproportionation of
the copper(I) compounds.
7S

3-C. Characterization
3-C-L Structural Studies
The molecular structures of two bis(}a-iodo)dicopper(I) complexes [Cu(HL")0-
I)]2 (86,n= 1;87, « = 2) and the monocopper(I) complex [Cu(HL'^)I] (88), have been
determined by X-ray crystallographic analyses. Figures 32-34 show the molecular
structures of 86,87, and 88. Selected bond distances (A) and angles (。)for these
complexes are listed in Tables 11-13. Both the binuclear complexes 86 and 87
exhibit a tetrahedral N2I2 coordination geometry, whereas 88 exhibits a trigonal
planar N2I coordination geometry around the metal center. The Cu-Cu separations
fall in the range of 2.9-3.0 人.The Cu-I bond distances of 〜2.6-2.8 人 in the bisO-
iodo)-dicopper(I) complexes (86 and 87) are longer than that of 2.5136(8) A in the
monocopper(I) complex 88. Besides, the observed Cu-N bond lengths of 2.043(7)-
2.094(7) A in the dimeric copper(I) compounds 86-87 are also longer than that of
1.986(2) - 2.009(2) A in the monomeric copper(I) complex 88. The Cu(l)-I( l )
[2.5136(8)入]distance in 88 is similar to the observed Cu-I distance of 2.511(1)-
2.5335(9)人 in [(CuI)3L] (L = l,3,5-tris[bis(pyridin-2-ylmethyl)aminomethyl]-2,4,6-
triethylbenzene) reported by Kim et al.}^
79

A common structural feature observed among the three copper(I) complexes,
86,87 and 92 is that the tertiary amino groups of the ligand are not participating in
coordination to the copper center. It is generally believed that copper(I) prefers
tetrahedral coordination. Trigonal planar geometry has also been observed.
It is noteworthy that a mononuclear three-coordinate copper(I) complex (88)
was obtaining by employing the benzimidazole-containing ligand 37. This is in
contrast to the pyridine-based ligands 34 and 35,which gave rise to dicopper
complexes 86 and 87. Plausible reasons for the difference may be: (1) the
benzimidazole-containing ligand 37 is sterically more bulky than the pyridine-based
derivatives 34 and 35; (2) an interaction between adjacent molecules as observed in
the unit of cell of 88 (Figure 35).
80
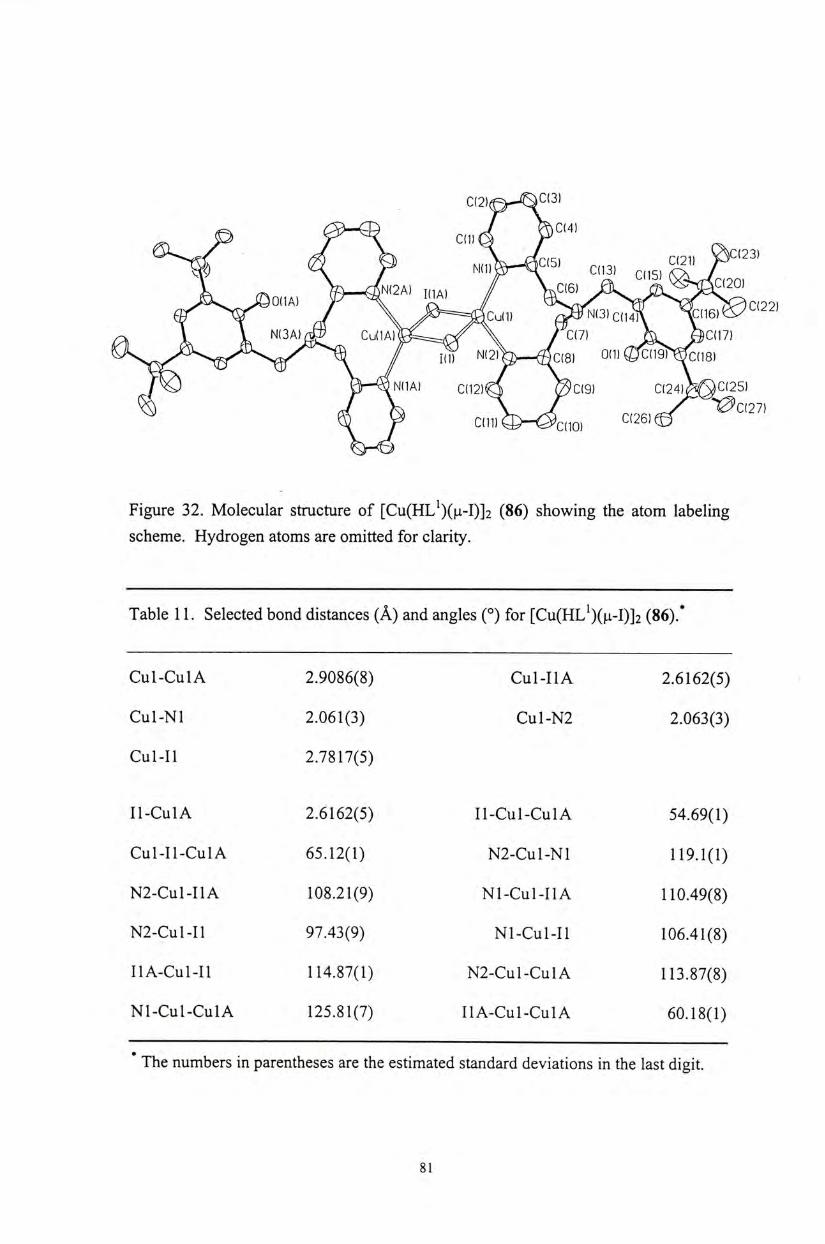
^ \ V N ( 3 A ) ^ / C(7) h ec(17)
p - ^ N d A ) C(12)Q >C(9) C(24^C(25)
Figure 32. Molecular structure of [Cu(HL^)(|a-I)]2 (86) showing the atom labeling scheme. Hydrogen atoms are omitted for clarity.
Table 1 1 . Selected bond distances ( A ) and angles (。)for [Cu(HL^)(^-I)]2 (86).丰
Cul-CulA 2.9086(8) Cu l - I IA 2.6162(5)
Cul -N l 2.061(3) Cul-N2 2.063(3)
Cu l - I l 2.7817(5)
I l -Cu lA 2.6162(5) I l -Cu l -Cu lA 54.69(1)
Cu l - I l -Cu lA 65.12(1) N2-Cul-Nl 119.1(1)
N2-Cul-I1A 108.21(9) N l -Cu l - I IA 110.49(8)
N2-Cul- I l 97.43(9) N l -Cu l - I l 106.41(8)
I IA -Cu l - I l 114.87(1) N2-Cul-CulA 113.87(8)
N l -Cu l -Cu lA 125.81(7) I lA-Cu l -Cu lA 60.18(1)
* The numbers in parentheses are the estimated standard deviations in the last digit.
81
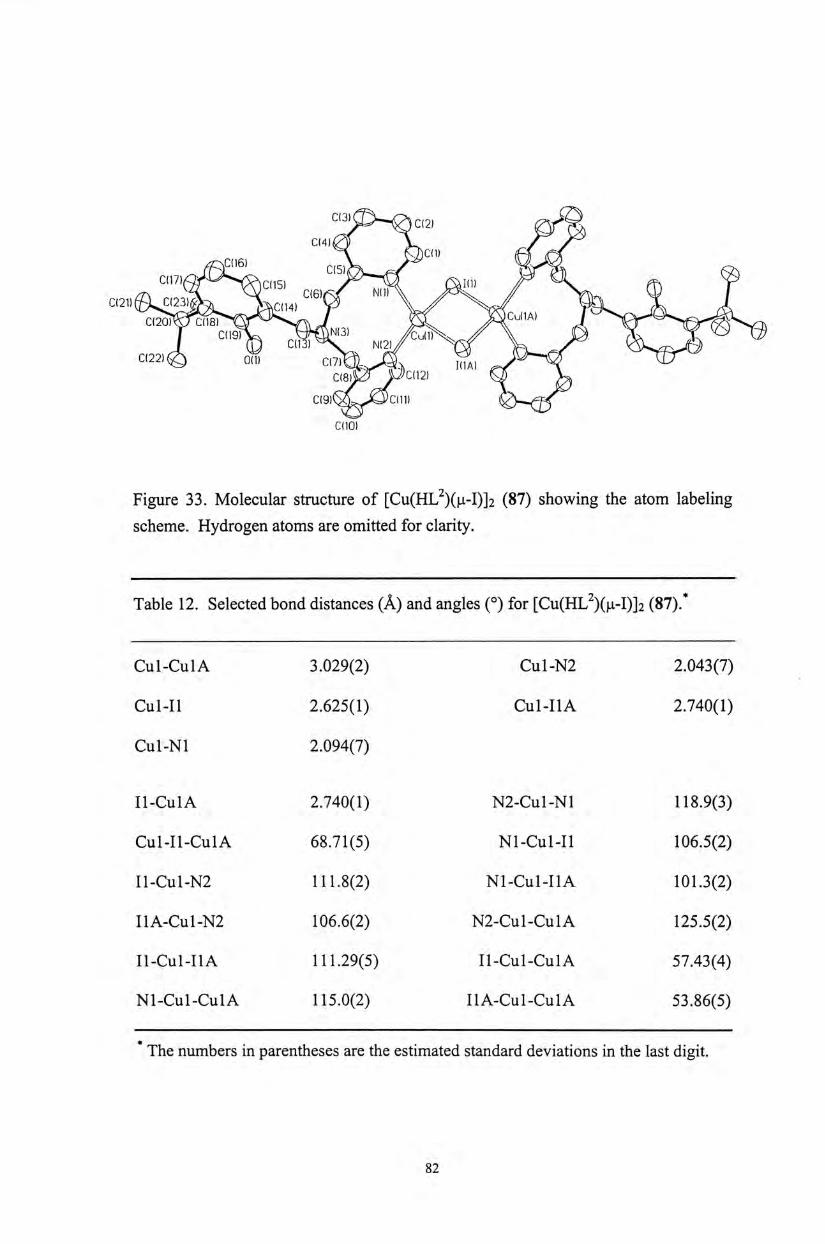
C(101
A
Figure 33. Molecular structure of [Cii(HL )(|i-I)]2 (87) showing the atom labeling scheme. Hydrogen atoms are omitted for clarity.
Table 12. Selected bond distances (A) and angles (。)for [Cu(HL^)(|a-I)]2 (87).*
Cul-CulA 3.029(2) Cul-N2 2.043(7)
Cu l - I l 2.625(1) Cu l - I IA 2.740(1)
Cul -N l 2.094(7)
I l -Cu lA 2.740(1) N2-Cul-Nl 118.9(3)
Cu l - I l -Cu lA 68.71(5) N l -Cu l - I l 106.5(2)
I l -Cul-N2 111.8(2) N l -Cu l - I IA 101.3(2)
I1A-Cul-N2 106.6(2) N2-Cul-CulA 125.5(2)
I l -Cu l - I IA 111.29(5) I l -Cu l -Cu lA 57.43(4)
N l -Cu l -Cu lA 115.0(2) I lA-Cu l -Cu lA 53.86(5)
* The numbers in parentheses are the estimated standard deviations in the last digit.
82

C(5)
V
Figure 34. Molecular structure of [ C u ( H L 4 ) I ] (88) showing the atom labeling scheme. Hydrogen atoms are omitted for clarity.
Table 13. Selected bond distances (A) and angles (。)for [ C u ( H L 4 ) I ] (88).*
Cul- I l 2.5136(8) Cul-N3 1.986(2)
Cul-Nl 2.009(2)
Nl-Cul-N3 116.58(9) N3-Cul-I l 125.17(7)
N l -Cu l - I l 117.39(6)
* The numbers in parentheses are the estimated standard deviations in the last digit.
83

禪 Figure 35. The interaction between two copper(I) complexes 88. Cu(lA)-N(2B): 3.468 A; I(1A)-N(2B): 3.575 A and Cu(lA)-N(2B)-I(lA): 66.8。.
84

3-C-II. Spectroscopic Studies
The copper(I) complexes 86, 87 and 88 have been flil ly characterized by
elemental analysis, mass spectrometry and NMR spectroscopy, in addition to single-
crystal X-ray crystallography. Complex 89 has been characterized by NMR
spectroscopy and LSI mass spectroscopy. However, correct elemental analysis of the
compound could not be obtained.
Both the mononuclear copper(I) compounds 88 and 89 have been characterized
by mass spectroscopy. The LSI spectrum of 88 showed the corresponding isotopic
ion distribution of the [M]+ species (Figure 36),whilst that of 89 showed
fragmentation peaks due to the [M-I]+ species (Figure 37).
With a dio electronic configuration for copper(I), all of the copper(I) complexes
are not suitable for characterization with electron paramagnetic resonance (EPR).
The ^H and ^ C NMR spectra of the four copper(I) complexes (86,87, 88 and 89)
were obtained. Upon complexation, the two methylene protons located adjacent to
the pyridine or benzimidazole moiety of the ligands 34,35, 37 and 38 are chemically
and magnetically equivalent as they did not couple to each other and hence a singlet
was observed.
85
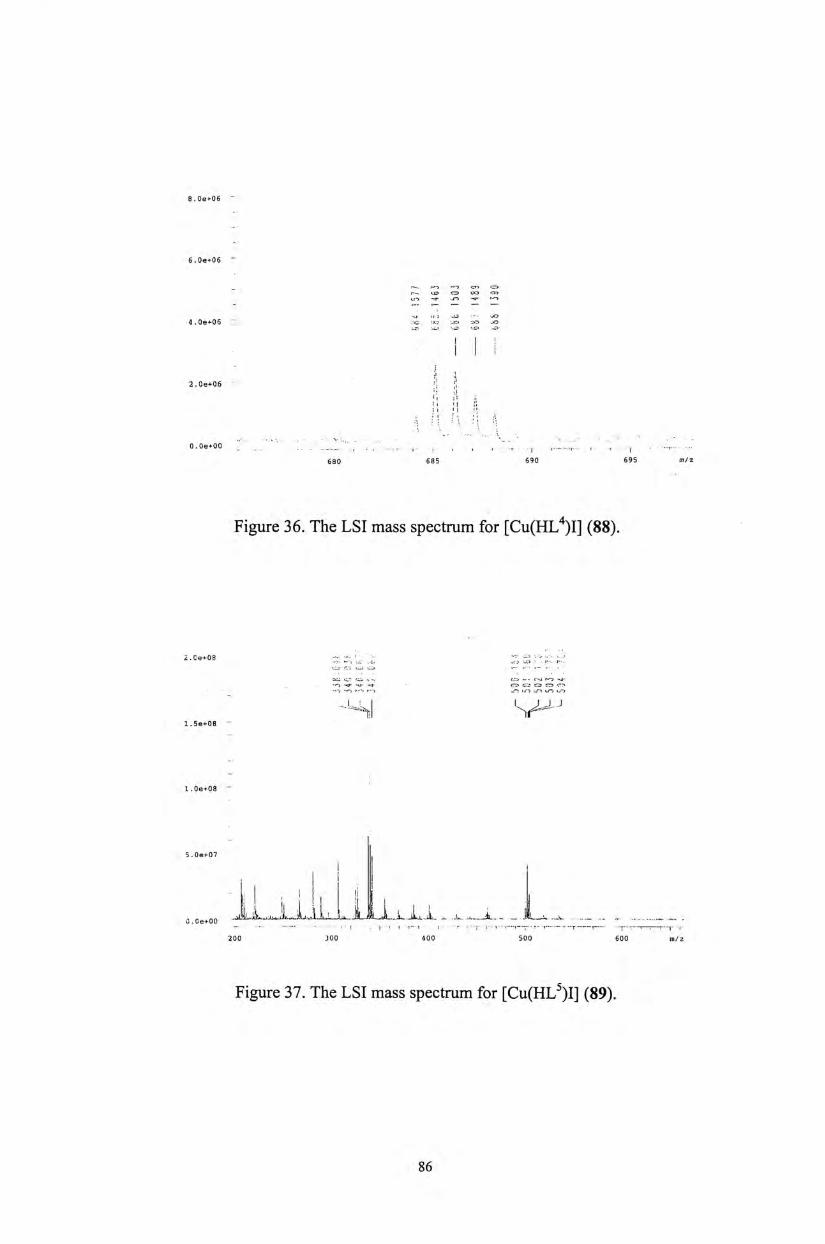
8 .Oe+06 -
6 . 0 e + 0 6 -
_� � I-O cr> cr> I—to CO o->
u-> 寸 ••O r-
•Kj- ».( }� 、0� -�.- JO 4 . 0e-»-06 •- fx? - c . •:、,
I.—.� ..“》 '.o '-O .o I !
I i _ :1� J�
2.0e+0;6 .. ;; ;1 ‘i� .�i�'1 ii ; 丨 I. : I ;i I I
A h h ;; i\ V • .•..、•. , - .. , ••• -、.:.... . . ... 、 ... ,、. - ‘ ..... . • . .、 ’、 •‘...
O . O e + 0 0 _ 一 … … . . . • . I : • • I , I • , , I I -—-- . - r - - ,. -1 1 , 1
6 8 0 6 8 5 6 9 0 6 9 5 m / z
Figure 36. The LSI mass spectrum for [Cu(HL4)I] (88).
• • I • - - . . I... .-.i
2 Ce+08 ‘‘� 、.•••' •� f-••; VC3 ;• ‘ • r— r -
'.:_二 .:二 «•_; ‘--• 、-.• - - - •
Cli ':-••‘ •;�V.� cr-v T-�c、ro� -•J- • "-} •-t.t- TTf c--> ctj 、‘.•> r-'-. U-J iJ") U-)
- 、 S f ^ j 1 . 5 e + 0 8 —
l . O e + 0 8 - I
5 . 0 e + 0 7 I
i I i I I
_I. I I ; i I 1 :i LL_LJiLJJLi...L.丄“1..‘.....L...丄―丄.‘ —1 一..丄
U.Ce+OU 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0 m / z
Figure 37. The LSI mass spectrum for [Cu(HL^)I] (89).
86
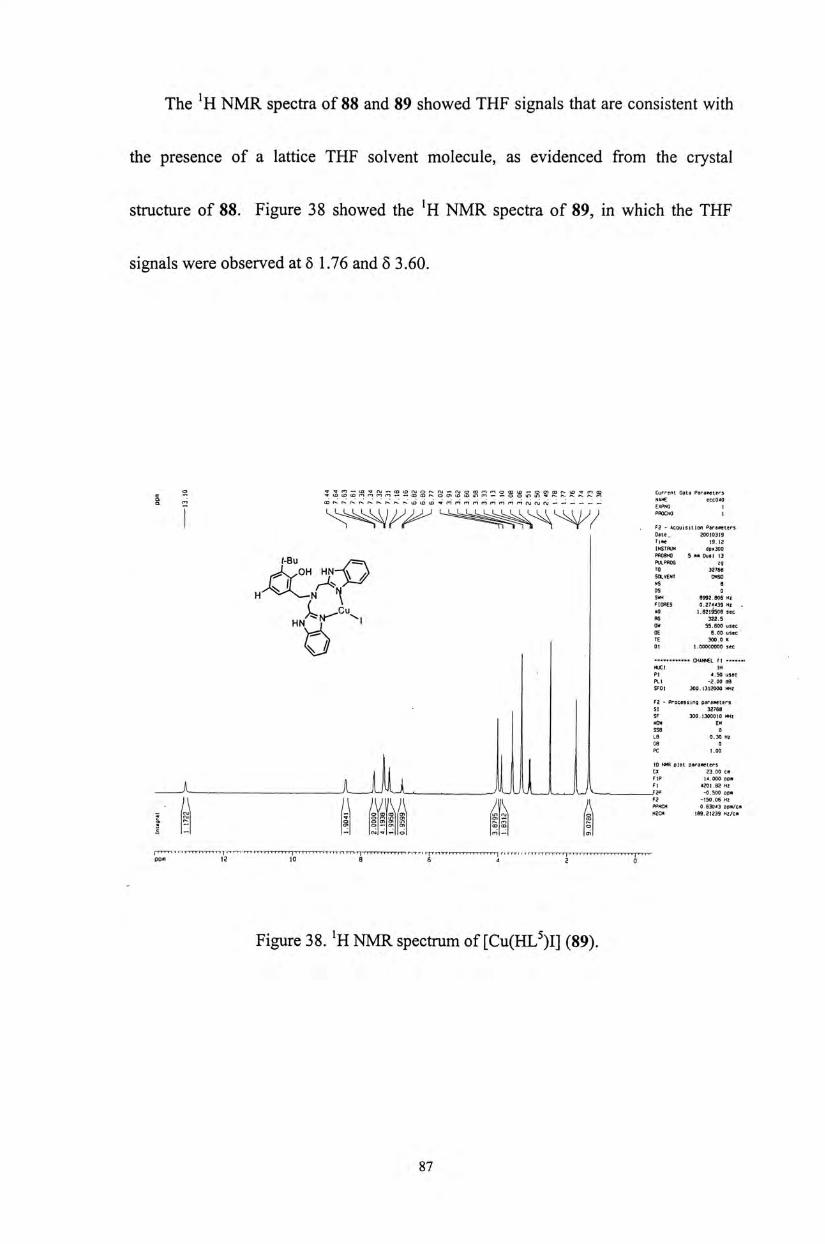
The NMR spectra of 88 and 89 showed THF signals that are consistent with
the presence of a lattice THF solvent molecule, as evidenced from the crystal
structure of 88. Figure 38 showed the NMR spectra of 89,in which the THF
signals were observed at 5 1.76 and 5 3.60.
E 2 Curr.ot Osta Parawters a • Nine eccO O
a f^ CD 卜 卜 j ^ 、 r v r • • 卜 卜 〜 — 一 一 一 一 一
丨 ^ ^ ^ ^ 广 厂 二 。 ‘ ’� I I 【 『� n� I�1�a� I T f . F2 - Acquisition Parairoteps
Oate_ 20010319 riM 19.12 INSTflUM dtu300 PROBHO 5 m» Dual 13
t-BU PULPROG zg
M OS 0 n ^―-N \ SWH 8993.806 Hz
/ ^ FIOTES 0.274439 Hz . L ^ C U AO sec
H N ^ 恥 进 niN J ow 55.600 usee
Oe 6.00 usee / )) TE 300.0 K (v J J 01 1.00000000 sec
.……••••• channel fl NUCl IH
‘ PI 4 . 5 0 u s e e PLl -2.00 06 SFOI 300.1312000 >*iz
F2 - Processing paraaeter, • SI 32768
SF 300.1300010 MHZ WOK EM ssa 0 LB 0.30 HZ C8 0 PC t.OO
to NMR pjot p»ra«ters I J CX 23.00 c«
. A l l 门P� 14.000 op»
L L__ J^ULm II___JUv Ts : .八 j\ 纖八 八 A 二 二二 c,
~� fy /oYm ml|oi| An /o\ HZCM 189.21239 Hz/cm
( T > 0 — C7101 (D (S o —� o j,” o� n�—. OT
I I I I I 11 r"r-i I ( I m r t i n j i~n~ri-n-r rrT i i i i i i > i | i i i i i i i i • -rn i n�ttvt•丁i�ti,i�i�i�i�i�i�i�i�titr�i�r pT n r • i ••••••• i • r'r r'T-|- ri i tti i r rTT.i丁ri,ri-’ r | n-rr-i • " i i • -i r' i't t i i i | r i i-i ppm 12 10 a 5 4 2 0
Figure 38. ^H NMR spectrum of[Cu(HL^)I] (89).
87

3-D. Reactivities of 86, 87 and 88 toward Dioxygen
The reactivities of copper(I) complexes toward dioxygen play an important role
in many biological processes」,、,Most of these processes involve dicopper systems.
In our studies, the reactivities of the binuclear copper(I) complex 86 toward dioxygen
have been studied. The oxygenation reaction was followed by an UV-Vis
spectrophotometer at -80 °C in order to identify the short-lived copper-dioxygen
intermediate. Figure 39 shows the changes of the UY-Vis spectra of an oxygenated
solution of 86 in dichloromethane from -80 °C to 10 °C. Absorption peaks at -320
nm, 420 nm and 600 nm were observed.
Upon oxygenation of 86 at -80 °C,a dark greenish brown solution was obtained,
which is believed to be due to a copper(II) species with absorption at 350 nm and
500 nm (Spectrum c, Figure 40). These absorptions are typical for copper(II) ion
with a d^ electronic configuration (Figure 40). Interestingly, this intermediate
species is stable at room temperature for about one day. Figure 40 summarizes the
spectra due to complex 86, an oxygenated intermediate, and the resultant reaction
product upon standing at room temperature for one day.
88

1.4『 8(fC
‘ ——-60'C 1.2 [ 50'C
丨 一-40^C -——30'C
1 - - 2 0 ' C I 15'C [ -lO'C
n ;;c
I * ^ 。.4 Vm
jo 37。 . 0 . 0
wavelength (ran)
Figure 39. Changes of UV-Vis spectra of an oxygenated solution of 86 in dichloromethane from -80 to 10
89

- 1
1.75 A
i 1.25 戟 8 1 — \\ \ I 0.75 i \ V
- —"‘ I I - 一 - • 一 ^ , • — • • . - . - — •
〇 一 • 、 ^ • • — • ^ 一 广 • 一 〜 — _ - — , , - , — —
1� “� I� ‘� ‘� ‘� ‘� I� ‘� “� ‘� ‘� I� ‘� “ I — I — I — I — — I — I — I — ] — 1 — I — I — 1 ~
^ 400 ^ 700 Wavelength (n
Figure 40. UV-Vis spectra of an oxygenated solution of 86 in dichloromethane. Spectrum a: -80。C; Spectrum b: 0 °C; Spectrum c: after standing at room temperature for 18 hours.
As shown in Figure 40,spectrum a represents the UV-Vis spectrum of the
reactive intermediate obtained upon oxygenation of 86 in dichloromethane at -80。C.
When the temperature of the solution was slowly raised, a red-shift of the absorption
spectrum was observed. Spectrum b shows the spectrum of the solution at 0 °C. The
absorption maxima at 330 nm and 350 nm on spectra a and b,respectively, were
assigned to the ligand-to-metal charge transfer (LMCT) bands. Presumably, the
absorptions at 410 nm and 600 nm might be due to a copper-hydroperoxo species. ' ^
90

u p o n standing at room temperature for 18 hours, both peaks at 410 nm and 600
nm disappeared (Spectrum c). A broad signal at 500 nm, however, was observed
which was assigned to be due to a copper(II) species.
Karlin and co-workers have reported that copper-hydroperoxo species could
oxidize triphenylphosphine to triphenylphosphine oxide. In order to prove the
possible formation of a hydroperoxo species upon oxygenation of 86,87 and 88,a
similar reaction has also been carried out.
Solutions of 86 and 87 in dichloromethane at -80 °C were bubbled with
dioxygen, followed by the addition of triphenylphosphine. After stirring 18 hours,
the resultant mixture was subjected to GC-MS analysis for triphenylphosphine oxide.
Interestingly, a quantitative conversion of triphenylphosphine to triphenylphosphine
oxide was observed. No oxo-transfer reaction was observed in control experiments,
in which no complex has been added. Complex 88,on the other hand, does not
facilitate the similar oxo-transfer reaction upon oxygenation with dioxygen. Results
of these experiments showed that a copper-hydroperoxo species may be the reactive
intermediate upon oxygenation of the binuclear complexes 86 and 87 at low
temperature.
Besides, the corresponding copper(II) species of both complexes in
dichloromethane after the reaction of 86 and 87 with triphenylphosphine were
91
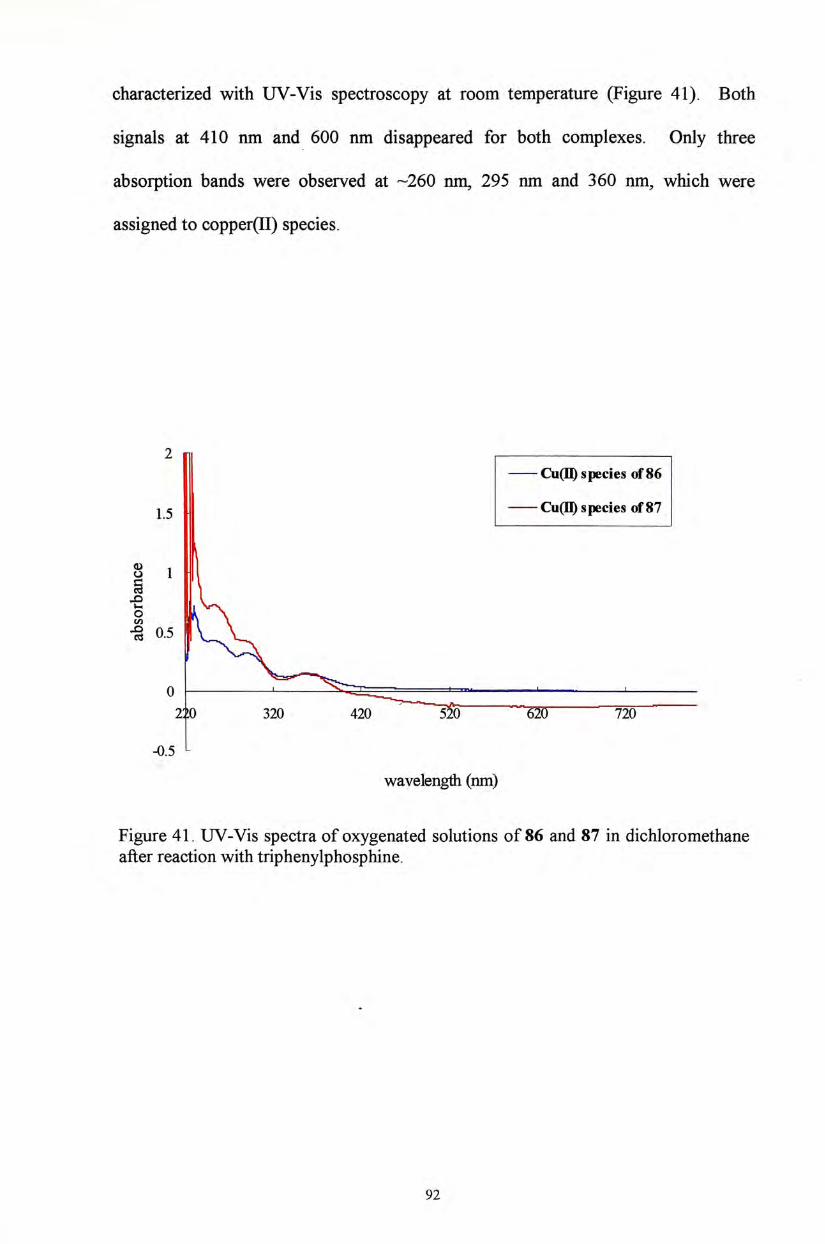
characterized wi th UV-Vis spectroscopy at room temperature (Figure 41). Both
signals at 410 nm and 600 nm disappeared for both complexes. Only three
absorption bands were observed at 〜260 nm, 295 nm and 360 nm,which were
assigned to copper(n) species.
2 Cu(II species of 86
I 5 Cu仰 species of 87
I i | 、
0� ‘� I� ‘� ‘�
2: to 320 420 ^ 720
-0.5 -wavelengtti (nm)
Figure 41. UV-Vis spectra of oxygenated solutions of 86 and 87 in dichloromethane after reaction with triphenylphosphine.
92

3-E. Summary
In conclusion, two bis( {i -I) dicopper(I) complexes 86 and 87 were synthesized
successfully with the pyridine-based tripodal tetradentate ligands 34 and 35. X-Ray
crystallography revealed that both complexes 86 and 87 are binuclear with
tetrahedrally coordinated copper(I) centers. With benzimidazole-based ligands 37
and 38,two mononuclear copper(I) complexes 88 and 89 were successfully prepared.
X-Ray crystallography revealed a trigonal planar coordination geometry around the
copper center in 88.
Oxygenation reactions of the bis( i i -iodo) dicopper(I) compounds 86 and 87
have been monitored by UV-Vis spectroscopy at low temperatures. The
corresponding copper hydroperoxo species have been suggested as the reactive
intermediates in these reactions, as evidenced by the conversion of
triphenylphosphine to triphenylphosphine oxide.
On the other hand, no oxo-transfer to triphenylphosphine has been observed
upon the oxygenation of a dichloromethane solution of the three-coordinate copper(I)
complex 88 at low temperature.
93

3-F. References
1. Schindler, S. Eur. J. Inorg. Chem. 2000, 2311-2326.
2. Kopf,M. -A.; Karlin, K. D. In Biomimetic Oxidations Catalyzed by Transition
Metal Complexes; Meunier, B., Ed.; Imperial College Press: London, 2000;
Chapter 7,pp 309-362.
3. Karlin, K. D.; Zuberbuhler, A. D. In Bioinorganic Catalysis: Second Edition,
Revised and Expanded., Reedijk, J., Bouwman, E.,Eds.; Marcel Dekker: New
York, 1999; pp 469-534.
4. Sorrell, T. N.; Vankai, V. A. Inorg. Chem, 1990, 29, 1689-1692.
5. Murthy, N. N.; Mahroof-Tahir, M.; Karlin, K. D. Inorg. Chem. 2001,40, 628-635.
6. (a) Mahroof-Tahir, M.; Murthy, N. N.; Karlin, K. D.; Blackburn, N. J.; Shaikh, S.
N.; Zubieta, J. Inorg. Chem. 1992, 31, 3001-3003; (b) Karlin, K. D.; Ghosh, P.;
Cruse, R. W.; Farooq, A.; Gultneh, Y.; Jacobson, R. R.; Blackburn, N. J.; Strange,
R. W.; Zubieta, J. J. Am. Chem. Soc. 1988, 110, 6769-6780; (c) Zhang, C. X.;
Liang, H. C.; Kim, E. I.; Gan, Q. F.; Tyeklar, Z.; Lam, K. C.; Rheingold, A. L.;
Kaderli, S.; Zuberbuhler, A. D.; Karlin, K. D. Chem. Commun. 2001, 631-632; (d)
Karlin, K. D.; Cruse, R. W.; Gultneh, Y.; Farooq, A.; Hayes, J. C.; Zubieta, J. J ,
Am. Chem. Soc. 1987,109’ 2668-2679.
94

7. Paul, P. P.; Tyeklar, Z.; Jacobson, R. R.; Karlin, K. D. J. Am. Chem. Soc. 1991,
113, 5322-5332.
8. Liang, H. C.; Karlin, K. D.; Dyson, R.; Kaderli, S.; Jung, B.; Zuberbuhler, A. D.
Inorg. Chem. 2000,39, 5884-5894.
9. (a) Jacobson, R. R.; Tyeklar, Z.; Farooq, A.; Karlin, K. D.; Liu, S.; Zubieta, J. J .
r
Am. Chem. Soc. 1988, 110, 3690-3692; (b) Schatz, M.; Becker, M.; Thaler, F.;
Hampel. F.; Schindler, S.; Jacobson, R. R.; Tyeklar, Z.; Murthy, N. N.; Ghosh, P.;
Chen, Q.; Zubieta, J.; Karlin, K. D. Inorg. Chem. 2001, 40, 2312-2322.
10. Hayashi, H.; Fujinami, S.; Nagatomo, S.; Ogo, S.; Suzuki, M.; Uehara, A.;
Watanabe, Y.; Kitagawa, T. J. Am. Chem. Soc. 2000,122, 2124-2125.
11. Mahapatra, S.; Halfen, J. A•; Wilkinson, E. C.; Pan, G.; Wang, X.; Young, V. G.,
Jr.; Cramer, C. J.; Que, L.,Jr.; Tolman, W. B. J. Am. Chem. Soc. 1996,118,
11555-11574.
12. Mahadevan, V.; Hou, Z.; Cole, A. P.; Root, D. E.; Lai, T. K.; Solomon, E. I.;
Stack, T. D. P. J. Am. Chem. Soc. 1997,119, 11996-11997.
13. Lam, B. M. T.; Halfen, J. A.; Young, V. G.,Jr.; Hagadom, J. R.; Holland, P. L.;
Lledos, A.; Cucurull-Sanchez, L.; Novoa, J. J.; Alvarez, S.; Tolman, W. B. Inorg.
Chem. 2000, 39, 4059-4072.
95

14. Halfen, J. A.; Mahapatra, S.; Wilkinson, E. C.; Kaderli, S.; Young, V. G.,Jr.;
Que, L.,Jr.; Zuberbuhler, A. D.; Tolman, W. B. Science 1996,271, 1397-1400.
15. He, C.; DuBois, J. L.; Hedman, B.; Hodgson, K. O.; Lippard, S. J. Angew. Chem.
Int. Ed. 2001, 40, 1484-1487.
16. Walsdorff, C.; Park, S.; Kim, J.; Heo. J.; Park, K. -M.; Oh, J•; Kim, K. J. Chem.
Soc” Dalton Trans., 1999, 923-929.
17. Kitajima, N.; Moro-oka, Y. Chem. Rev. 1994,94, 737-757.
96

CHAPTER 4
Experimental Section
4-A. General Preparations and Physical Measurements
4-A-I. General Preparations
Al l reactions were carried under a purified nitrogen atmosphere using Schlenk-
line techniques. Solvents were distilled under nitrogen from sodium benzophenone
(tetrahydrofliran, toluene, diethyl ether) or calcium hydride (dichloromethane,
acetonitrile), and degassed twice by freeze-thaw cycles before use. Triethylamine
was distilled over sodium under a nitrogen atmosphere.
Al l reagents, unless otherwise indicated, were of analytical grade and were
used without further purification. iV,A^-Bis(2-pyridylmethyl)amine [BPMA]^ N,N-
bis(2-benzimidazolylmethyl)amine [BBA]^, and 3,5-di-^err-butyl-2-hydroxybenzyl
bromide〗 were prepared according to literature procedures.
97

4-A-IL Physical Measurements
1h NMR (300 MHz) and ^ C NMR (75.4 MHz) spectra were obtained on a
Bruker DPX 300 spectrometer. LSI mass spectra were obtained on a Bruker APEX
47e Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer. EI mass
spectra were measured by a Hewlett-Packard 5989B Mass Engine spectrometer.
Elemental analyses were performed by MED AC Ltd., Brunei University, U. K.
Melting point was measured by Electrothermal 9100 digital melting point apparatus.
EPR analyses were performed by Bruker EMX-EPR spectrometer equipped with a
helium-flow cryostat (Oxford Instrument). UV-Vis spectra were obtained on a Gary
5E UV-Vis-NIR spectrophotometer or a Beckman DU-7500 photo-diode array
spectrophotometer. Low-temperature UV-Vis spectra were obtained on a Hewlett-
Packard 8453 UY-Vis spectrophotometer. GC-MS spectra were obtained by a
Hewlett-Packard 6890 GC - 5973 MSD system.
98

4-B. Compounds Described in Chapter 2
Preparation of Di-Substituted Amines
iV,iV-Bis(2-pyridylmethyl)amine [BPMA] (49). To a solution of 2-
pyridinecarboxaldehyde (0.11 g, 1.0 mmol) in methanol was added a solution of 2-
aminomethylpyridine (0.11 g, 1.0 mmol) in methanol. A dark brown mixture was
obtained immediately. After stirring overnight, sodium borohydride (0.076 g, 2.0
mmol) was added slowly to yield a pale yellow solution. Stirring was continued for
another 2 hours. Al l the volatiles were removed under reduced pressure and water
was added. The mixture was neutralized with concentrated hydrochloric acid
followed by extraction with dichloromethane. The combined extracts was dried over
anhydrous magnesium sulphate and rotary evaporated to give a yellow oily liquid.
Yield: 0.18 g (90 %). tf NMR (CDCI3): 5 8.50 (d,J= 4.8 Hz, 2 H, pyridyl), 7.58 (t,
J = 7.7 Hz, 2 H,pyridyl), 7.30 (d, •/= 7.5 Hz, 2 H, pyridyl), 7.10 (t, J = 7.7 Hz, 2 H,
pyridyl), 3.93 (s, 4 H, CH2), 3.21 (br, 1 H,NH).
7V,A^-Bis(2-benzimi(lazolylmethyl)amine [BBA] (50). A mixture of iminodiacetic
acid (6.66 g, 50 mmol) and o-phenylenediamine (10.8 g,100 mmol) was heated at
200。C in a capped round-bottom flask to give a dark purple solid. It was grounded
with a mortar and then extracted in refluxing methanol for about 18 hours. The
99

solution was filtered and the purple-red filtrate was concentrated. Recrystallization
of the crude product from diethyl ether gave 50 as a white solid. Yield: 7.0 g (50 %).
Hi NMR (CD3OD): 5 7.53 (m, 4 H, Ar), 7.22 (m, 4 H, Ar), 4.11 (s, 4 H, CH2).
Preparation of Substituted Benzaldehydes
3,5-Di-,忍"-butyI-2-hydroxybenzaldehyde (40). To a solution of 1>,5-6i-tert-
butylphenol (2.10 g, 10.0 mmol) and tributylamine (0.95 ml, 4.0 mmol) in toluene
was added SnCU (0.12 ml, 1.0 mmol) dropwise under nitrogen. A white fiime
appeared immediately during the addition and stirring was continued for 15 minutes
at room temperature. Paraformaldehyde (0.66 g, 22.2 mmol) was then added in a
single portion and the mixture was refluxed overnight. Water was added and the
residue was extracted with diethyl ether. The combined extracts were evaporated to
dryness. After purification by column chromatography, a pale yellow solid was
obtained. Yield: 1.90 g (80 %). ^H NMR (CDCI3): 6 9.87 (s, 1 H, CHO), 7.60 (d, J
=2 .4 Hz, 1 H, Ar), 7.35 (d, J = 2.4 Hz, 1 H, Ar), 1.43 (s, 9 H,/-Bu), 1.33 (s,9 H, t-
Bu).
3-r^i^-Butyl-2-hydroxybenzaldehyde (41). This was prepared analogously as for
compound 40, starting with using 3-^er^-butylphenol (1.50 g, 10.0 mmol). Yield:
100

1.30 g (72 %). ^H NMR (CDCI3): 5 9.87 (s, 1 H, CHO), 7.53 (dd,《/= 1.7, 7.7 Hz, 1
H, Ar), 7.39 (dd, J=L7, 7.7 Hz, 1 H, Ar), 6.94 (t, 7.7 Hz, 1 H, ArH), 1.42 (s, 9 H,
Bu).
5-^^#t-Butyl-2-hydroxybenzaldehyde (42). This was prepared analogously as for
compound 40,starting with 5-/err-butylphenol (1.50 g, 10.0 mmol). Yield: 1.24 g
(70 %). ^H NMR (CDCI3): 5 9.89 (s, 1 H,CHO), 7.59 (dd, J = 2.4,8.7 Hz, 1 H, Ar),
7.52 (d,J= 2.4 Hz, 1 H, Ar), 6.94 (d, J= 8.7 Hz, 1 H,^Bu), 1.33 (s, 9 H, /-Bu).
Preparation of Substituted Benzyl Alcohols
3,5-Di-r忍"-butyl-2-hydroxybenzyl alcohol (43). To a solution of compound 40
(0.23 g, 1.0 mmol) in methanol was slowly added sodium borohydride (0.08 g, 2.0
mmol). The pale yellow solution was stirred overnight and then evaporated to
dryness. Water was added to dissolve the residue. The mixture was neutralized with
hydrochloric acid and then extracted with dichloromethane. The combined extracts
was pumped to dryness to give a yellow oil which was used directly without further
purification. Yield: 0.23 g (95 %).
101

3-^^/t-Butyl-2-hydroxybenzyl alcohol (44). This was prepared analogously as for
compound 43,starting with compound 41 (0.18 g,1.0 mmol). Yield: 0.17 g (93 %).
5-^^r^-Butyl-2-hydroxybenzyl alcohol (45). This was prepared analogously as for
compound 43,starting with compound 42 (0.18 g, 1.0 mmol). Yield: 0.17 g (96 %).
Preparation of Benzyl Bromides
3,5-Di-,衫"-butyl-2-hydroxybenzyl bromide (46)。To a solution of compound 43
(0.24 g,1.0 mmol) in chloroform was added phosphorus tribromide (0.10 ml, 1.0
mmol). A white fume appeared during the addition and stirring was continued for an
hour. The resultant solution was quenched with cold water. The aqueous layer was
extracted with chloroform. The combined extracts was evaporated to dryness to give
a pale yellow oily liquid which was used directly without further purification. Yield:
0.29 g (95 %).
3-^^/t-Butyl-2-hydroxybenzyl bromide (47). This was prepared analogously as for
compound 46, starting with compound 44 (0.18 g, 1.0 mmol). Yield: 0.23 g (96 %).
102

5-,忍"-Butyl-2-hydroxybenzyl bromide (48). This was prepared analogously as for
compound 46’ starting with compound 45 (0.18 g,1.0 mmol). Yield: 0.23 g (96 %).
Preparation of Tetradentate Ligands
[A^-(3,5-Di-^^rr-butyl-2-hydroxybenzyl)-AyV-di-(2-pyridylmethyl)]ainine [HL^]
(34). A solution of 3,5-di-^er^-buty 1-2-hydroxylbenzy 1 bromide 46 (0.30 g, 1.0
mmol) in THF was added dropwise to a solution of BPMA 49 (0.20 g, 1.0 mmol) in
THF under nitrogen. After stirring for 5 min., triethylamine (0.15 mL,1.1 mmol)
was added. The mixture was refluxed overnight. After cooling down to room
temperature, the resulting yellow suspension was filtered. The filtrate was
concentrated under reduced pressure and purified by column chromatography using
ethyl acetate as eluent. The third band was collected. Al l the volatiles were removed
under reduced pressure to give a yellow solid. Yield: 0.35 g (84 %). M.p.: 81-82 °C.
1h N M R (CDCI3): (5 = 8.55 (d,J= 7.8 Hz, 2 H, pyridyl),7.61 (dt, J= 1.8, 7.8 Hz, 2
H, pyridyl), 7.37 (d, J = 7.8 Hz, 2 H, pyridyl), 7.22 (d, J= 2.4 Hz, 1 H, Ar), 7.11-
7.15 (m,2 H,pyridyl), 6.89 (d, J= 2A Hz, 1 H, Ar), 3.88 (s, 4 H, CH2), 3.81 (s, 2 H,
CH2), 1.47 (s, 9 H,^Bu), 1.27 (s, 9 H, t-Bu). NMR (CDCI3): 5 = 158.0,
153.7, 148.9, 140.2,136.5,135.4,124.4, 123.5, 123.0, 122.1, 121.6, 59.4,58.1,34.9,
34.0, 31.6,29.5. HRMS (LSI): m/z 418.2847 (calcd. for C27H35N3O [MH]+
103

418.2853).
[^-(3-^^ft-Butyl-2-hydroxybenzyl)-AVV-di-(2-pyridylmethyl)] amine [HL^] (35).
This was prepared analogously as for 34,starting with 3 -^er/-butyl-2-hydroxylbenzyl
bromide 47 (0.24 g, 1.0 mmol). Yield: 0.29 g (80 %). M.p.: 75-76 °C. ^H NMR
(CDCI3): 5 = 8.56 (d,J= 6.6 Hz, 2 H,pyridyl), 7.63 (dt, J = 1.2,7.7 Hz, 2 H,
pyridyl),7.36 (d,J= 7.4 Hz, 2 H, pyridyl), 7.14-7.21 (m,3 H, pyridyl and Ar), 6.92
(dd,J= 0.7,7.3 Hz, 1 H, Ar), 6.71 (t ,y=7.5 Hz, 1 H, pyridyl), 3.87 (s,4 H, CH2),
3.82 (s,2 H, CH2), 1.46 (s, 9H,/-Bu). NMR (CDCI3): (5 = 157.9,156.4,
148.8, 136.7, 128.0,126.2,123.5,122.6,122.2,118.2, 59.2,57.7, 34.7,29.5. HRMS
(LSI): m/z 362.2221 (calcd. for C23H28N3O [MH]+ 362.2227).
[A^-(5-r^rr-Butyl-2-hy(lroxylbenzyl)-iV^-di-(2-pyridylmethyl)] amine [HL^] (36).
This was prepared analogously as for 34,starting with 5-rer^butyl-2-hydroxylbenzyl
bromide 48 (0.24 g, 1.0 mmol). Yield: 0.28 g (78 %). M.p.: 76-78。C. ^H NMR
(CDCI3): 5 = 8.57 (d,J = 7.5 Hz, 2 H, pyridyl), 7.63 (dt, J= 1.8, 7.7 Hz, 2 H,
pyridyl), 7.37 (d,J= 7.8 Hz, 2 H,pyridyl), 7.14-7.21 (m, 3 H, pyridyl and Ar), 7.04
(d,J= 2.4 Hz, 1 H, At), 6.84 (1,7= 8.4 Hz, 1 H, pyridyl), 3.90 (s, 4 H, CH2), 3.79 (s,
2 H,CH2), 1.27 (s,9 H, t-Bu). NMR (CDCI3): 5 = 158.2, 155.0, 148.8,
141.1,136.8, 126.9,125.7, 123.2,122.2, 121.8,115.8, 59.1, 57.4,33.8, 31.5. HRMS
104

(LSI): m/z 362.2244 (calcd. for C23H28N3O [MH]+ 362.2227).
[AyV-Di-(2-benzimidazolylmethyl)-iV"(3,5-di-/忍"-butyl-2-hydroxylbenzyl)]amine
[HL4] (37). A solution of 3,5-di-^er^butyl-2-hydroxybenzyl bromide 46 (0.30 g, 1.0
mmol) in THF was added dropwise to a solution of BBA (50) (0.28 g, 1.0 mmol) in
THF under nitrogen. After stirring for 5 min.,triethylamine (0.15 mL, 1.1 mmol)
was added. The mixture was refluxed overnight. The resulting reddish brown
suspension was cooled down and then filtered. The filtrate was concentrated under
reduced pressure and purified by column chromatography using ethyl acetate as
eluent. The third band was collected and evaporated to give 37 as a pink solid.
Yield: 0.25 g (50 %). ^H NMR (DMSO-de): (5 = 12.41 (s,2 H,NH),10.73 (s,1 H,
OH), 7.61 (m,4H, Ar), 7.18 (m, 4 H,Ar), 7.10 (s,1 H, Ar) , 6.04 (s, 1 H, Ar) , 4.11 (s,
4H, CH2), 3.92 (s, 2 H, CH2), 1.40 (s,9 H, /-Bu), 1.21 (s, 9 H, t-Bu). NMR
(DMSO-de): 5 = 153.6, 151.6, 142.8,139.6,134.9,134.0,125.4,122.5, 122.2,
122.0,121.4,118.4, 111.4, 55.6,50.2, 34.7, 33.8, 31.5, 29.7. HRMS (LSI): m/z
496.3210 (calcd. for C31H37N5O [MH]+ 496.3595). Anal. Calcd. for
C31H37N5O • CH3COOCH2CH3: C, 72.00; H, 7.77; N, 12.00 %. Found: C, 72.62; H,
7.74; N, 12.630/0.
105

[iV^-Di-(2-benzimidazolylmethyl)-A^-(3-/^/t-butyl-2-hydroxylbenzyl)]amine
HL^] (38). This was prepared analogously to HL* 37, starting with 3-/er^butyl-2-
hydroxybenzyl bromide 47 (0.24 g, 1.0 mmol). Yield: 0.18 g (40 %). ^H NMR
(CD3OD): (5 = 7.54 (m, 4 H, Ar), 7.22 (m, 4 H, Ar), 7.05 (dd,J= 1.5, 7.8 Hz, 1 H,
Ar), 6.90 (dd ,y= 1.5, 7.4 Hz, 1 H,Ar), 6.64 (t, 7.8 Hz, 1 H,Ar), 4.04 (s, 4 H,
CH2), 3.83 (s, 2 H, CH2), 1.33 (s,9 H,f-Bu). NMR (CD3OD): 5 = 156.9,
152.4, 139.4, 137.8,129.2,127.4,123.7, 123.3, 120.0,115.8,58.8,52.8, 35.5,30.1.
HRMS (LSI): m/z 440.2454 (calcd. for C27H29N5O [MH]+ 440.5615). Anal. Calcd..
for C27H29N5O: C, 73.78; H,6.65; N, 15.93 %. Found: C, 73.36; H, 6.71; N, 15.44
%.
[AyV-Di-(2-benzimidazolylmethyl)-7V-(5-/^i^-butyl-2-hydroxylbenzyl)]amine
[ h i / ] (39). This was prepared analogously to H i / 37, starting with S-tert-hutyX-l-
hydroxybenzyl bromide 48 (0.24 g, 1.0 mmol). Yield: 0.16 g (36 %). ^H NMR
(CD3OD): 6 = 7.54 (m, 4 H, Ar), 7.23 (m, 4 H, Ar), 7.15 ( d , / = 2.4 Hz, 1 H, Ar),
7.09 (dd,y= 2.4,8.4 Hz, 1 H, Ar), 6.72 (d, •/= 8.4 Hz, 1 H, Ar), 4.04 (s, 4 H, CH2),
3.83 (s, 2 H,CH2), 1.21 (s, 9 H,/-Bu). NMR (CD3OD): (5 = 155.0,153.7,
143.3, 139.3,129.1, 126.9’ 123.5, 123.1,116.5,115.7,56.4,53.1, 34.7’ 31.9. HRMS
(LSI): m/z 440.2409 (calcd. for C27H29N5O [MH]+ 440.5615). Anal. Calcd. for
C27H29N5O • CH3COOCH2CH3: C, 70.56; H, 7.07; N,13.27 %. Found: C, 70.33; H,
106

6.90; N , 1 3 . 6 5 % .
Preparation of Copper(II) Complexes
[Cu(HL^)Cl]Cl (51). A solution of CuCl] (0.13 g, 1.0 mmol) in methanol was added
to HL^ (34) (0.42 g, 1.0 mmol) in the same solvent to afford a deep blue solution.
Triethylamine (0.14 mL, 1.0 mmol) was then added dropwise with stirring to give a
deep purple solution. The solution was refluxed for 2 hours and the resulting
suspension was cooled down and then filtered. Al l the volatiles were removed under
reduced pressure to give a deep purple solid. Recrystallization from
dichloromethane afforded blue crystals suitable for X-ray structural analysis. Yield:
0.44 g (80 %). M.P.: 213-214。C (dec.). HRMS (LSI): m/z 517.1734 (calcd. for
C27H36CIC11N3O [M-C1]+ 517.3528). Anal. Calcd. for C27H35CI2CUN3O • H2O: C,
56.88; H, 6.54; N, 7.37 %. Found: C, 56.24; H,6.60; N,7.17 %.
[CU(HL^)(CH3COO)1(CH3COO) (52) . Triethylamine (0.14 mL, 1.0 mmol) was
added dropwise to a solution of HL^ (34) (0.42 g,1.0 mmol) in methanol, followed
by a solution of Cu(CH3COO)2 • H2O (0.20 g, 1.0 mmol) in methanol. The resulting
deep purple solution was stirred overnight and then filtered. The filtrate was
evaporated under reduced pressure. Recrystallization from ethyl acetate afforded
107

blue needle-shaped crystals suitable for X-ray structural analysis. Yield: 0.30 g (50
%). M.p.: 96-97 °C (dec.). Anal. Calcd. for C31H41CUN3O5 • CH3COOCH2CH3: C,
61.14; H, 7.19; N,6.12 %. Found: C, 60.31; H, 6.87; N, 6.27 %.
Cu(L^)Cl (53). 1 M NaOH (1.0 mL, 1.0 mmol) in water was added to a solution of
HL^ ( 3 4 ) (0.420 g,1.0 mmol) in methanol to afford a yellow solution. A solution of
C11CI2 (0.14 g, 1.0 mmol) in methanol was then added drop wise. The solution was
stirred overnight and the resulting suspension was filtered. The filtrate was
evaporated under reduced pressure to give a purple solid. Yield: 0.38 g (73 %).
M.P.: 191-192 °C (dec.). HRMS (LSI): m/z 515.1759 (calcd. for C27H34N3OCUCI
[M]+ 515.2681). Anal. Calcd. for C27H34CIC11N3O • O.5H2O: C, 61.82; H, 6.73; N,
8.01 %. Found: C,61.84; H, 6.68; N, 7.94 %.
CU(L^)(CH3COO) (54). This was prepared analogously as for Cu(L^)Cl 53,starting
with Cu(CH3COO)2 • H2O (0.20 g,1.0 mmol). Recrystallization from acetone
afforded deep red block-shaped crystals suitable for X-ray structural analysis. Yield:
0.43 g (80 %). M.P.: 177-178 °C. Anal. Calcd. for C29H37CUN3O • H2O: C, 62.51;
H, 7.06; N,7.54 %. Found: C, 62.74; H,7.29; N, 7.54 %.
108

Preparation of Zinc(II) Complexes
Zn(L^)Cl (55) . 1 M NaOH (1.0 mL, 1.0 mmol) in water was added to a solution of
HLI (34) (0.42 g,1.0 mmol) in methanol to afford a yellow solution. A solution of
ZnCl2 (0.141 g,1.0 mmol) in methanol was then added dropwise. The solution was
stirred overnight and the resulting suspension was filtered. The filtrate was
evaporated under reduced pressure to give a yellow solid. Recrystallization from
acetonitrile afforded colorless block-shaped crystals suitable for X-ray structural
analysis. Yield: 0.37 g (71 %). M.p.: 199-200 °C (dec.). ^H NMR (CD3OD): d =
9.16 (d ,y= 4.8 Hz, 2 H, pyridyl), 7.98 (dt,《/= 1.8,7.6 Hz, 2 H, pyridyl), 7.54 (t, J =
6.9 Hz, 2 H, pyridyl), 7.46 (d,/= 7.8 Hz, 2 H, pyridyl), 7.07 (d, J= 2.7 Hz, 1 H, Ar),
6.92 (d, J= 2.7 Hz, 1 H, Ar), 4.14 (d, J= 16.5 Hz, 2 H, CH2), 4.06 (d, J= 16.5 Hz, 2
H, CH2), 3.76 (s, 2 H,CH2), 1.36 (s, 9 H, /-Bu), 1.21 (s,9 H, /-Bu). ^ C NMR
(CD3OD): (5 = 163.0,157.3,150.5, 141.7,140.3’ 138.1,126.9, 125.1,124.9,124.8,
60.3, 58.8, 36.0, 34.7, 32.2, 30.7. HRMS (LSI): m/z 518.1694 (calcd. for
C27H34ClN30Zn [MH]+ 518.1627). Anal. Calcd. for C27H34ClN30Zn: C, 62.68; H,
6.62; N, 8.12 %. Found: C, 62.54; H, 6.68; N,8.01 %.
Zn(L2)Cl (56) . This was prepared analogously to Zn(L^)Cl (55), starting with HL^
(35) (0.36 g, 1.0 mmol). Recrystallization from acetone afforded colorless needle-
109

shaped crystals suitable for X-ray structural analysis. Yield: 0.29 g (56 %). M.p.:
205-206 °C (dec.). ^H NMR (CDCI3): (5 =6.41 (d, J= 4.8 Hz, 2 H,pyridyl), 7.82 (t,
J= 7.6 Hz, 2 H, pyridyl), 7.41 ( t , 6 . 4 Hz, 2 H,pyridyl), 7.27 (d, J= 7.7 Hz, 2 H,
pyridyl), 7.11 (d,《/=7.6 Hz, 1 H, Ar), 6.84 ( d d , / = 1.3,7.2 Hz, 1 H, Ar), 6.38 (t,《/ =
7.4 Hz, 1 H, At), 4.09 (d, J= 15.0 Hz, 2 H,CH2),3.86 (d,J= 15.0 Hz, 2 H, CH2),
3.78 (s, 2 H, CH2), 1.42 (s, 9 H,fBu). ^ C NMR (CDCI3): (5 = 155.0, 150.5,140.5,
139.9,129.1,126.8, 124.2, 123.2, 122.7,112.8, 59.3, 57.7, 34.9,29.6. Anal. Calcd.
for C23H26ClN30Zn: C, 59.88; H, 5.68; N,9.10 %. Found: C,59.70; H,5.87; N,
8.80 % .
Zn(L^)Cl (57) . This was prepared analogously to Zn(L^)Cl (55),starting with HL^
(36) (0.36 g, 1.0 mmol). Recrystallization from dichloromethane afforded colorless
needle-shaped crystals suitable for X-ray structural analysis. Yield: 0.26 g (51 %).
M.P.: 203-205 °C (dec.). ^H NMR (CD3OD): (5 = 8.82 (s, 2 H, pyridyl), 7.99 (dt, J=
1.8,7.8 Hz, 2 H, pyridyl), 7.49 (t, 7.8 Hz, 4 H, pyridyl), 7.08 (s, 1 H, Ar), 6.98
(dd, J= in, 8.7 Hz, 1 H,Ar), 6.57 (d, J= 8.4 Hz, 1 H,Ar), 4.23 (d, J= 16.2 Hz, 2 H,
CH2), 4.15 (d, J = 16.2 Hz, 2 H, CH2),1.23 (s, 9 H, t-Bu). ^ C NMR (CD3OD): 5 =
157.1,149.9’ 141.9,140.1, 129.3,127.9,125.5, 125.3, 123.1, 120.4,60.2’ 59.1,34.5,
32.1. Anal. Calcd. for CzsHzeClNsOZn: C, 59.75; H, 5.89; N,9.08 %. Found: C,
110

59.46; H, 5.66; N, 8.95 %.
Zn(L^)(CH3COO) (58). 1 M NaOH (1.00 mL, 1.0 mmol) in water was added to a
solution of HL^ (34) (0.420 g, 1.0 mmol) in methanol to afford a yellow solution. A
solution of Zn(CH3COO)2 • 2H2O (0.22 g,1.0 mmol) in methanol was then added
dropwise. The solution was stirred overnight and the resulting suspension was
filtered. The filtrate was evaporated under reduced pressure to give a yellow solid.
Recrystallization from acetone afforded colorless block-shaped crystals suitable for
X-ray structural analysis. Yield: 0.42 g (77 %). M.p.: 216-217 °C (dec.). ^H NMR
(CD3OD): (5 = 8.66 (d, 5.1 Hz, 2 H, pyridyl), 7.92 (dt, J = 1.8, 7.6 Hz, 2 H,
pyridyl), 7.46 ( t , 6 . 6 Hz, 2 H, pyridyl), 7.38 (d, 7.8 Hz, 2 H,pyridyl), 6.97 (d,
J = 2.4 Hz, 1 H, Ar), 6.89 (d, J = 2.4 Hz, 1 H, Ar), 4.26 (d,•/= 16.5 Hz, 2 H,CH2)’
4.19 (d ,y= 16.5 Hz, 2 H, CH2), 3.73 (s,2 H, CH2),2.08 (s, 3 H,CH3)’ 1.34 (s, 9 H,
/-Bu), 1.18 (s,9 H, ^Bu). 13c NMR (CD3OD): 5 = 180.6,163.3, 157.1,149.5,
1 4 1 . 3 ’ 139.4,138.0’ 127.0,125.1’ 1 2 4 . 9 , 124.7,124.3,61.1,60.1,36.0, 34.6,32.2,
30.6. Anal. Calcd. for CsgHsvNsOZn • H2O : C, 62.31; H, 7.03; N, 7.51 %. Found:
C,63.14; H,6.86; N, 7.58 %.
I l l

Zn(L^)(CH3COO) (59). This was prepared analogously to Zn(L^)(CH3C00) (58),
starting with HL^ (35) (0.36 g, 1.0 mmol). Recrystallization from a mixed solvent of
chloroform and ethyl acetate (3:1) afforded colorless block-shaped crystals suitable
for X-ray structural analysis. Yield: 0.27 g (55 %). M.p.: 202-203 °C (dec.). ^H
NMR (CD3OD): 5 = 8.69 (d, J = 5.4 Hz, 2 H, pyridyl), 7.93 (dt, J = 1.8, 7.7 Hz, 2
H, pyridyl), 7.48 (t,J二 6.6 Hz, 2 H, pyridyl), 7.40 (d,J= 7.8 Hz, 2 H, pyridyl), 6.93
(dd, J= 1.8, 7.7 Hz, 1 H, Ar), 6.87 (dd, / = 1.8,7.5 Hz, 1 H,Ar), 6.32 (t, J = 7.5 Hz,
1 H, Ar), 4.26 (d, J = 16.5 Hz, 2 H,CH2), 4.19 (d, 16.5 Hz, 2 H, CH2),3.73 (s, 2
H,CH2), 2.10 (s, 3 H,CH3), 1.34 (s, 9 H, ^ C NMR (CD3OD): 5 = 180.6,
166.1, 157.1,149.5, 141.4,140.6,130.5, 128.0, 125.6, 125.1,124.4, 115.8, 60.6,
59.9,35.7,30.5,23.7. Anal. Calcd. for C25H29N30Zn • 2CH3OH : C, 59.02; H, 6.74;
N, 7.65 %. Found: C,58.51; H,6.12; N, 8.18 %.
112

4-C. Compounds Described in Chapter 3
Preparation of Dimeric Copper(I) Complexes
[Cu(Hl/)(//-!)】2 (86). Triethylamine (0.14 mL, 1.0 mmol) was added dropwise to
a solution of HL! (34) (0.42 g, 1.0 mmol) in dry THF at 0。C under nitrogen. The
resulting yellow solution was stirred for 15 min. and then added dropwise to a slurry
of Cul (0.191 g, 1.0 mmol) in dry THF at 0 °C to give a bright yellow mixture. The
mixture was stirred overnight and then filtered. The yellow filtrate was evaporated
in vacuo to give compound 86 as a yellow solid. Recrystallization of the crude
product from dry dichloromethane afforded colorless block-shaped crystals suitable
for X-ray structural analysis. Yield: 0.48 g (80 %). M.p.: 162-163 °C. ^H NMR
(C6D6): 5 = 10.46 (s,2 H, OH), 9.35 ( d , y = 3.0 Hz, 4 H, pyridyl), 7.54 (dt,J= 3.0
Hz, 2 H, Ar), 6.94 ( d t, 2 . 0 ’ 7.5 Hz, 4 H,pyridyl), 6.79 (m, 6 H, Ar and pyridyl),
6.59 ( t , y = 6.0 Hz, 4 H,pyridyl), 3.87 (s,8 H,CH2), 3.45 (s,4 H, CH2),1.75 (s, 18
H, /-Bu), 1.33 (s,18 H, t-Bu). '^C N M R (CeDg): (5 = 156.2, 154.8,151.9, 141.2,
137.0’ 136.3,125.9, 124.8,123.6, 123.1, 121.7,58.7,56.1,35.4,34.4, 32.0, 30.1.
Anal. Calcd. for C54H70I2N6O2CU2 • 2CH3OH: C, 52.54; H, 6.14; N,6.56 %. Found:
C, 52.42; H, 6.28;N, 6.17%.
113

[Cu(HL^)( n -I)]2 (87). This was prepared analogously to [Cu(HL^)(/z-I)]2 (86),
starting with HL 35 (0.36 g, 1.0 mmol). Recrystallization from dry dichloromethane
afforded colorless block-shaped crystals suitable for X-ray structural analysis. Yield:
0.85 g (77 %). M.P.: 167-169�C. ^H NMR (CsDe): d = 10.56 (s’ 2 H, OH), 9.29 (d,
J = 6.0 Hz, 4 H, pyridyl), 7.33 (dd,J= 1.5, 9.0 Hz, 2 H, Ar), 6.90 (dt,J二 1.5, 7.5
Hz, 4 H, pyridyl), 6.79 (d,•/= 7.5 Hz, 2 H, Ar), 6.74 ( d , J = 7.8 Hz, 4 H,pyridyl),
6.61 (dd, J 二 1.2,7.5 Hz, 2 H, Ar), 6.56 (t, 6.6 Hz, 4 H, pyridyl), 3.75 (s,8 H,
CH2), 3.36 (s,4 H, CH2),1.68 (s,18 H,PBu). ^ C NMR (CeDe): S = 157.5,156.4,
152.1, 137.5,137.3, 127.3,126.0, 123.4,122.6, 119.5, 58.4,56.4,35.4, 30.3. Anal.
Calcd. for C46H54I2N6O2CU2: C,50.05; H, 4.93; N,7.61 %. Found: C,49.61; H,
5.07; N, 6.99 %.
114

Preparation of Monomeric Copper(I) Complexes
Cu(HL4)I (88). Triethylamine (0.14 mL, 1.0 mmol) was added dropwise to a
solution of HL4 (37) (0.50 g, 1.0 mmol) in dry THF at 0 °C under nitrogen. The
resulting yellow solution was stirred overnight and then added dropwise to a slurry of
Cul (0.19 g, 1.0 mmol) suspended in dry THF at 0 °C to give an orange solution. The
mixture was stirred overnight and then filtered. The orange filtrate was concentrated
in vacuo to afford 88 as colorless crystals suitable for X-ray structural analysis.
Yield: 0.42 g, 61 %. M.p.: 230-232。C. ^H NMR (DMSO-de): 6 = 13.10 (s, 2 H,
NH), 9.26 (s,1 H, OH), 8.43 (s, 2 H, Ar), 7.60 (m, 2 H, Ar), 7.32 (m,4 H, Ar), 7.18
(d, J = 3.0 Hz, 1 H, Ar) , 7.01 { d , J = 3.0 Hz, 1 H,Ar), 4.04 (s, 4 H, CH2), 3.90 (s,2
H, CH2), 1.41 (s, 9 H, t-Bu). 13c NMR (DMSO-de): 5 = 153.4,150.1,141.6,140.6,
135.3, 133.2, 124.6,123.5,122.8, 122.3, 121.2,119.3, 112.0,67.0, 57.6,47.8,35.5,
33.9,31.5, 29.8, 25.1. HRMS (LSI): m/z 686.1503 (calcd. for C31H37IN5OC11 [M]+
686.1183). Anal. Calcd. for C31H37IN5OCU • THF: C,55.44; H, 5.98; N, 9.24 %.
Found: C, 55.47; H, 6.19; N, 9.35 %.
115

Cu(HL^)I (89). This was prepared analogously to Cu(HL^)I 88,starting with HL^
(38) (0.44 g,1.0 mmol). Recrystallization from THF afforded compound 89 as
colorless crystals suitable for X-ray structural analysis. Yield: 0.13 g (22 %). M.p.:
249-250 °C. 1h NMR (DMSO-de): (5 = 13.10 (s,1 H, OH), 8.44 (s,2 H, Ar), 7.63
(m, 2 H, Ar), 7.33 (m, 4 H, Ar), 7.17 (d,J= 6.0 Hz, 1 H, Ar), 6.80 (t, J= 7.5 Hz, 1
H, Ar) , 4.02 (s,4 H,CEb),3.91 (s,2 H, CH2), 1.38 (s,9 H,,-Bu). ^^C N M R
(DMS0-d6): (5 = 155.5, 150.2, 141.5, 135.3, 133.2,127.9,126.0,123.5,122.2,
119.3, 119.2, 112.0,67.0, 56.5, 47.8,34.3, 30.0,25.1. HRMS (LSI): m/z 502.1701
(calcd. for C27H29N5OCU [M-I]+ 502.8369). Anal. Calcd. for
C27H29IN5OCU • 4CH3OH: C,49.11; H, 5.98; N, 9.23 %. Found: C, 48.78; H, 5.45;
N,9.10 0/0.
116

4-D. Oxo-Transfer to Triphenylphosphine as Described in Chapter 3
Reactivity of [Cu(HL^)(//-!)]2 (86). Dioxygen was bubbled into a yellow solution
of[Cu(HL^)(/z-I)]2 86 (0.46 g, 0.38 mmol) in dichloromethane (15 mL) at -80 °C for
5 min. to give a deep brown solution. A slight excess of 1 equivalent of PPhs (0.10
g, 0.38 mmol) in dichloromethane (15 mL) was then added dropwise with stirring.
The solution was stirred at -80 °C for 18 hours. The resulting solution was wanned
to room temperature, and all the volatiles was removed under vacuum. The residue
was extracted with several portions of diethyl ether. GC-MS analysis of the ether
extracts showed a quantitative conversion of PPha to PhsPO.
Reactivity of [Cu(HL^)(/z-I)]2 (87). The reaction was studied under a similar
conditions and work-up procedures, starting from [Cu(HL^)( [i -I)]2 87 (0.32 g, 0.29
mmol). GC-MS analysis of the ether extracts showed a quantitative conversion of
PPhs to PhaPO.
Reactivity of [Cu(HL'')I] (88). Dioxygen was bubbled into a colorless solution of
[Cu(HL4)I] 92 (0.16 g, 0.23 mmol) in tetrahydrofliran (15 mL) at -80。C for 5 min. to
give a yellow solution. A slight excess of 1 equivalent of PPha (0.06 g, 0.24 mmol)
in tetrahydrofliran (15 mL) was added dropwise with stirring. The solution was
117

stirred at -80 °C for 18 hours. After the solution had been warmed to room
temperature, all the volatiles were removed under vacuum. The residue extracted
with diethyl ether. GC-MS analysis of the ether extracts showed the presence of
PPhs only, with no formation ofPhaPO.
118

4-E. References
1. Larsen, S.; Michelson,K.; Pedersen, E. Acta Chem. Scand A 1986, 40, 63-76.
2. Berends,H. P.; Stephan,D. W. Inorg. Chim. Acta 1984,93, 173-178.
3. Sokolowski, A.; Muller, J.; Weyheraiuller, T.; Schnepf, R.; Hilderbrand, P.;
Bothe, E.; Wieghardt, K. J. Am. Chem. Soc., 1997,119’ 8889-8900.
119

APPENDIX A H and NMR Spectra
A- l . ^H and NMR Spectra for Compounds Described in Chapter 2
A-1-1. 1h and NMR Spectra of H i / (37) 外 成 — r t� m O 一�r Current Data Parameters
r. 实另�2S:: 二�S� � ° - MAKE ecc033h,-d«0 ^ 2 rvr^r^h r^r^hU;� � V W tt m m m m� � EXPNO »
° — ^ v / / / I I \ \ l :。:=, t一一
"’灯「� II丨I Date. 20010109 Time 11.50 !NSinUM dD«300 PffOBHO 5 m« Dual 13 PULPROG zg TO 32760
H SOLVENT *ceton ( N ^ ^ FI ORES 0 .274439 »Z
AO 1.9219508 sec J V-f•日 U AG 101.6 OW 55.500 usee OE 6.00 usee
/ TE 300.0 K f - B U 01 1 .00000000 s e c
CHANNEL fl NUCI IH FM 4.50 usee PLl -2.00 d8 SFOl 300.1312000 KHZ
F2 - Processing parawters SI 32768 SF 300.1300012 MHZ NOH EH SSB 0 LB 0.30 HI GB 0 PC 100
10 NMR plot paraneiers , CX 23.00 cm A I FIP 12.000 DO"
MJu iLLJ V——I_1~JUl 三 r /V\ \ / l \ z" ,二二r
. s g[ig]s] gS s 2 _ I i s sss s s CO m o I o m .r" - o -
_ .0 T 5 ^ _. ] . . . .. ° •
Current Data Parameters NAHE ecc033C-c}niso EXPNO 1
\l \\\I\W/ ^ ^ ^ ^ ^ ^ ^ ^ TO 55536 SOLVENT CDC 13 NS 837 OS 0
jj |j SWH 22675.736 Hz A n n ^ ^ FI ORES 0.346004 HZ
^ ^ ^ 0€ 6.00 usee D,, TE 300.0 K
y - ^ l - D U D, 1.00000000 s e c n U ' ^ y ^ ^ / a n 0 .03000000 s e c
i D,f …………CHANNEL f 1……• f-SU HUC1 13C
PI 3.00 usee PU -6.00 (10 SFOl 75.47451U MHZ
. . . . . . . . . . . . C H A N N E L f2 CP0PRG2 waltz 16 NUC2 )H PCP02 100.00 usee Pia 120.00 d8 PU2 19.00 as ST02 300.1315007 MHz
F2 - Processing oarameters SI 65536 Sf 75 4677BOO MHz MOW EH SSB 0 La 3.00 HZ GB 0 PC I 40
10 NMR D】ol parameters CX 23.00 cn FlP ?00 000 Otjm F1 15093.55 Hz
I I I I F2P -?0.0Q0 oon 'I . J J ... . _ 」 • 、 . ,� .� f�..�rfg� -1509-35 HZ
* 納 , 一 Ml•"〜(-.1.�X.,,.•”II、...11 ":「••- '•‘ -fpp ^ 9 55522 Dom/cm HZCH 7?1 86572 Hz/cm
Dpm 180 150 140 120 100 80 60 40 20 0
120

胁
ssS
ii
」一
ri
s
i
? Errll
s
2二
ii
」s
一
一
rs邮
f
s
srrii
二 I:
J
g
i 。
,
Ii:二
ii
SJ^
r3二
=
咖
⑶
:
3
,
嫩舰•测二
Ja
w
2
80L
ow
J-
L
?
n
r
c
n
2
3
o .
wow
.w
a—
-
1
8
h
⑴
咖
丨
1
5
挪
叫
则
刚 丨
2
丨
10
5
S
S
晰
7
U
I
2
5;
5
“
“
t
加
则
S
拟
1
S
叩
-
-
1 02
, I
s
嶋
舰
g
S
汗 5
M
I
咖-r咖
E
朽
™
I
M
t 呢
•SStJiSS
5
H
脚
"s
Is
-
WB
N
p
p
w
w t
蛇视 8
2
H
赃
1
“
a Is
-
w
8
N
叱
5
lo
lo
3 1
T
5
HA^MAyn^
d s
o"
/ J
f ,
y
J-
c 2-
?
「冲
ST
1
i」」
s J
g.s
」
z.
S.E、
I,
w E5-1-——
IX
mam^^
i
N
{
彳
c J
hn/^n
w/^
A
n H
“乂
o
•一
w
MVD/U
^
d 5®.HI
X
H
.6
N/vh^
一
:
.H 悬、多
a
—”⑶
2. so.^u^
HN\vn
H
rt^
八。19:
is.gsl
——
I
^
A
o
J
丁
•
si
」//
s;
」//
- L
」
们S5;
」
g
m a
»
t
i
〔
p
eaa
n
L
D
1
PP®
I,
丨0,叫
ra丨
咖
‘ Q
。彳
Q 户
丨
. R
R
. S 千
_
�
f--
^
二;二
:
^r
�fi:
::
; ffi
^^129.104
\V7.O84
P f
—
=:12
5.876
cn
- \\V
-7.076
P
r�
.�123.504�
\V-6�73d
CI*
、123..130
[6.706
—
r 115.456
厂4�872�
^�
、丨�5.700�
⑴ O
m-
/ r 4.087
r"^^
‘:
1 p
jf
^ 3
‘ 1
|i I
r 56.3
96
> 00
°�
〉�—�
�.9.285
S
___
I;:丨丨丨
I
丨I
^^
34.67
4
W
、31.923
厂1.263�
p�r
t -21
5
—
一
^1.2
07
Q
、I.179�
V^�
o-
= °
sssgjv! 二�
=�A 丨
⑶
sssg室�ssi 旨
宝二�ss:�ssse
,„
〜,
。„
,。
” i
,…
"丨
--
j i
i H
fr-
PI
…s
i,『
!丨
…!
。|
p?
iiiiS
iP
i-.J
il
llll
M
kd
Ihii
il.i
iJk
il
y ii
i^ii
J -
。
L5
L
i
L.-
iii
..
Ij
If
Ir
--
‘ ^
‘至舌”
i
”i
丨『
y
! 一
…
Ipm
r ll
ri
—
•
•
9 2
“
u “
on
“

=
a
沖
议
£
m
【
二」-rr
s
srrli
I
=」-一
ri
一
r二
s
s
s srrll
ls"g。?ss£。二二咖g二 "Esgg
fsssosog
sggsgssig
SB丨
二二
= Sgo二§S§11
观sm
二
l
g
l
s
。
g
。
s
;咖sis^s
Ii
P>§
— SI
s
M
-
m r§
「二二二
II
pg
~
§§
i
"
!
"
I
i
pis
J
80
1
2
™
则
叫
g
们
=
™
~
丨
lo
5
e
£
咖
叫
e
邮
6
t
3
M
3
p
a
1
5
t
i
n
s
a
1
5 a
t
s
n
_
s
t
t
9
a
s
t
d
I
-
•
c
o
•
V
I
K
e
o
山
u
"
!
I
Z
I
l
o
g
"
o
a
m
o
p
州 s
y
w
例
。
•
你
阳
S
I
1
I
w
H
們
s
卿
删
=
例 m H
I
S
J
J
l
s
i
哪
-
“
9
m
p
p
raw
lo
6
nM- 5-1
mn^
Aso"
5-OE
1/
c 。浜二—-1
H
A
.
八
6
C
2
突-^
j
K 1
ss
々
:
1 2s
9£
L -2
9
二 Bt^-^
叩
n 二Z.0.
」
z 856•芝
A^ 1
-—-
」
s I
丄
口二、
沈
二二\
—
UAI
:
.g
ogvlv M
M®M
;
R 3
公 2
^ I
,——
.,5
B
⑴
I
:
XK&P
IT J
z^
-则
Q i
u
,s
”
”
?
咖
——L
% 頸I
SY
\l
:
:“::〔
J
,H Ij
c
xft
夂
§01
—mm^
• ;Jnfr
、
1
la^
nnn^s
18
I
1
4 ^
2E0-SI
M (!”『
- S6;
」/
A eoo-e
」
-9
s JJY
i
八 goo
g
m
. i
l
uidd
【卯
i a
LP

c
c
»
c
wc
s c
e
e
c
a
e
z
z
ffi
n
A
I
c
c
•
c
>
c
A
浓 S
S
K
S
S SSJ
S
S
SS£g£g
似
们
讯
r
i 咖
s
敝
!
咖
ss
晰
,
附
5
•
r r
^
二; ss
J;⑷二•"§
J
ssosog
"gispi
i2£ISOS§is
。ss
2…ggs
…s。g
。s
sggsgE
r pnl
gSS
®
§
I
“
“
“
II
s
I
他
-J
I—
晰
⑶
^
—
1
5
咖
叫
咖
=
p时刚
「
™
。咖
S
咖
g
叫
f
C
-
'
-
«
她
m
s
棚
S
“
“
3
二“隨 IJElei略SUSSES
f
ssS〔…i
IE
imiieLsi謹Esiesi
liLi
…⑶
…」1
lo
7 o
^^ I
i
八专
d T
™
n
z se.工
6I9>E——
丄
f 2E’?
;
o 13
K srE-^
000.?
-
-
-
e—^
\
to 墨
H
二”^
1
^
i.E——
1
MMnyw
ogs
、/
s A
/i.z
仪 "二
% ^
M i.
J
a
n
p. SB
+ r--
w u
§
a •
i
:
J.
I •
」_「7
I
卷
-
l
A
-
.
IH ")>
丄I
;
y
. i-7
t^Jj
L
1\
-so--/
M^
⑷
5 <
〜0000-g
「8
一
1
察•
L」 8t6.6M
^ j
<; 220-8」
I
i--
沒0V51
I
_叨
>9
i
1
印
i If,
【卿
i
】
LS

o 1
ppm
si
一。sg
「al
ppm
o .
w
:
w .
/-8.6s
V
SI
T
/^8.652
-
o J^
「7.g48
I
-f h
w T
‘
I 1
>
#
丨
S-
—157.…
1
V
A.要
H
;
V
^ lis
I
v.〜§
热H
g
/ \J
」Y^
v-25.089
《VL
vs.906
}
.Tl
?^cc/
^
g」
^
叩
f
HI
o \_JI
8
\-124.341
r\
n
N
w
n §
SJ
T
——4.906
c. 、4.2S
s
“
mT
小二:“
I
:IVT
二〕-
即
「61.066
^3.320
o
l「g-os T
K^
」/「-19.852 夕3.310
-_.
M
‘ —
」「49.569
El
一/「
S.2B5 \-3.2g
— J^
自-
u/
「 •二
S.717
T_
V- 48.434
/
r
I
w V
4P150
a^
-iv
飞
2.082
/(V
^Mn
— —--—
36.007
Mv 34.2B
c
^Y 32.227
^
,3P23 Tl
—一.341
2
L
M0V
n
1.181
s
_
1
丨
™
i
„
卿
咖 f
I
一
g
帅
啊
9
“
TO
Q
s
s
o
l
l
s
I
叫
SJ
1
咖
咖
仙
咖
g
l
a
n
d
!
旧
“
s 二
i
iie
«
s 9
!r二 li
二:iirj
g
e
z
s
二咖 r
二
二
HS^OCOEK
1
SKI
K
6 .o
o
1
.9
E
I n
gggy-osus!^;
^^^^
^
^ 训
盼
SSWSK 川
I
o 咖
Km w
Mres^e^^^
hk
;-SMg
进
Ks
二二二 “
!
=
二
;
2
;
二-二
二
S
I
二:二
S
S ?
rss
“「SS
E
5
D
/
3
•
3
•
3
3
B
V
W
W
2
I
a
=
3
3
a
D
&
3 们
“
„
们
J
3
“ 3
3

J
003
^ u Intel
;
XX
二 PI
1
: ;
0
i
出-
「8-S8
: -
y^B.570
一 >7.963
/r
t:
I
180.630
//r,s8
\
.「
:
」
w 个
I
viii
X
则」
>
——157.114
B,
ihwn
了
^
i
「「Izd
ru.
/, A
/
\/Y>
S
J/\.
彦 H
—
i)
.
!E
:
办
则
r
M
ZK
“ ,
」
i
c
p
^
I ./yVi
⑶一“
-
c
u
j
n
^^(CH
乂;•
:
营
\-J <124.3B0
1\广5.866
JM
T I
”二?
怕
\广6.322
rv
1
: 广
6.2s
M
SI
4.912
I P
9 /
V4.230
?sg/ ——
个,217
d
S-j
p」
g.l779
、「
9
/. /r
:—\^3-732
「60.593
riv
>3.321
/、59.870
议
9
一 -
=^
「49.851
1=—
"^3.310
o
o ,
49.567
\--3.304
J
j/y- S.2B3
£1
_—_
肿
V
4814B
/
^
^ /
_
UI^
〈2.102
I
o
妃—
9.25V
un
1
rs
V
!.
;^
ol
ss^ Iskii
s
=
对
二
i
j
l
" 二p
d
o
-
>
3
K-
3
0
n
£
>
K
n
•
韋
C
o
o
a
•
I'
B
o
a
j
"
s
•
u
- i
s
i
咖
o.
>
E
【
卯
sw
n
咖
赃
w
咖
i
s
'09
-
«
lislsr
〕 sorsasj
rggsss
mggs"
§n§g:!§IS。rs
二
::胍
is
二 丨SS
gorssss
sgsm"
gogisisrsisssgs
二
丨
柳
pi 二 g
. ™
S
j
I
二 ™
SSll
-rH
1
= 二二
5
I
s
1二
一
sil
-rH
3
们
似
似
w
们
则
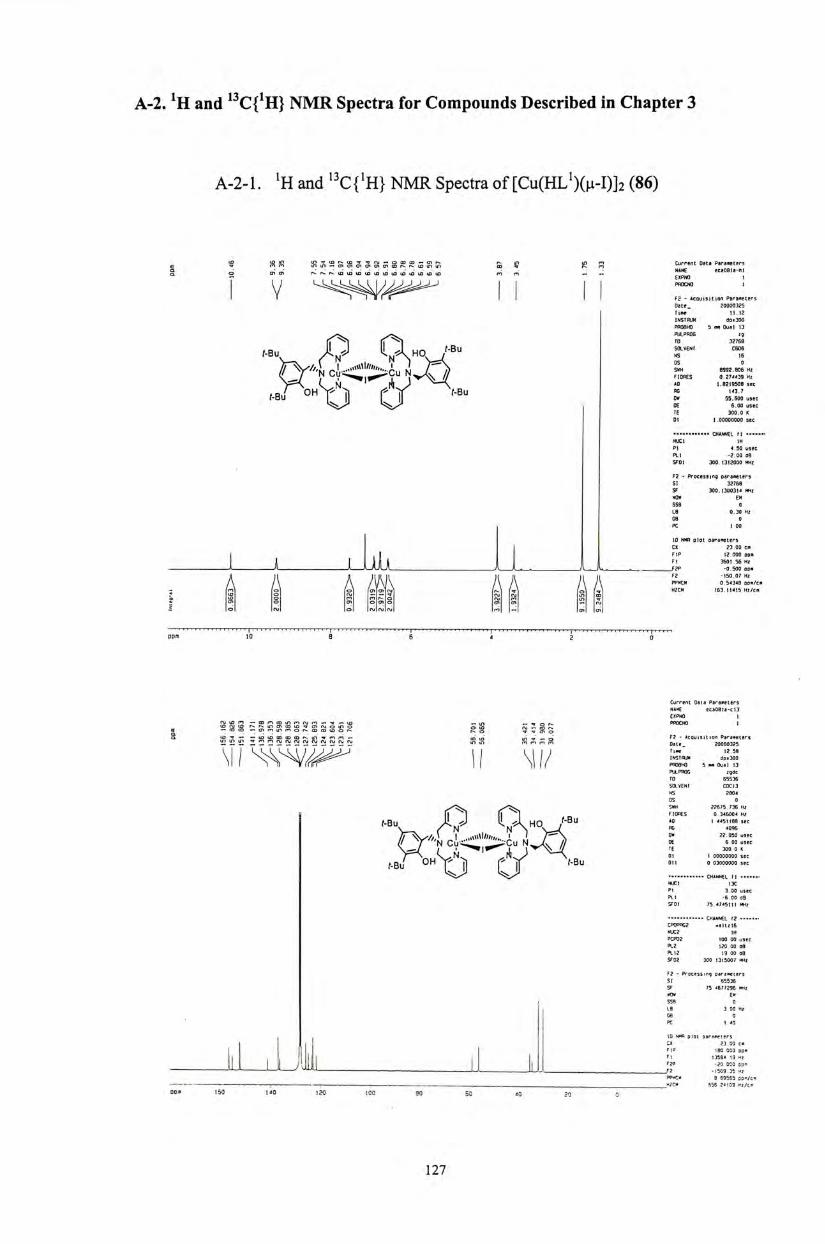
A-2. 1h and N M R Spectra for Compounds Described in Chapter 3
A-2-1. ^H and NMR Spectra of [Cu(HL^)(M)]2 (86)
6 专 浜�m� S ^ S & S S S S i m S S S S S S � £� ^ ^� Current�Data�Parameters�
S� o� oi�cn� (^r>>fN.u5iDU3tou3U5u3u3ujioioio� m� m — 二 朴明� ecaoeia-ni�
“ V ^^^^^^^^ 二 ‘ I� 1 I a r r� F2�-�ACQuisition�Parameters�
Oate^� 20000325�rime� 11.12�INSTRUM� dOK300�PflOeHO� 5�am�Dual�13�PULPROG iq
f T 》� f D� TO� 32768�f -Bu l ^ J / - B " r " ' ,
c i O c u I X j i L 二 fg� ‘� fll^J� *Q 1.8219509 sec
/-Bu C H I ^ 1 iT ^ ^ ^ DH 55.SCO usee ^ 6.00 usee re 300.0 K 01 丨.00000000�sec�............CHANNEL�fl … … , NUCl IH PI 4.50 usee
PLl� -2.00�OB�SFOl� 300�1312000�MHz�f2 -�Processing�parameters�SI� 32768�SF� 300.1300314�MHz�HOW� EM�SSB� 0�L8� 0.30�Hz�GB 0 PC 1 00 10 NHR plot Ddrameters CX 23.00 cm
I I FIP 12.000 pon I I H i 3601,56 Hi
A A n /LJULA /V A . > -O.soo pp*
A l\ A m l\ A 二 。’二 - fn] fo fol fcnloi f\il / 卜 、 a )� f l HZCM 163.11415 H"c« ?� u 3 o� rvj� �—.�� oj� r\j� i n c D�9� o� m m i ^ o� c\i� rn� In� ^�2� cr>� o� CT0010� OTOT� —� r\j�—� o� ru� o� f\j�ru�OJ� m� --� crioi�
I I 丨'_ I 丨• , I I t
卩 Jjm 10 9 6 4 2 0
Current�Data�Par«»eiers�NAME ecd0e]a-cl3
« ^ is SsSssgSssisS oS 5兰sg = ‘ a.� ua^v 一 一� u3io®a)®r«-inTrfn<n 一 m in iri a� f?�-�ACQuisil�ion�Paraoeters�
S� S� m�n�m� Date� 20000325�
\lI \l \\l/ S r � 丨 PULPROG ,90c TO 55536 SOLVENT CDC 13 MS 2B04 OS 0
/ : ^� SWH� 2?675�736�HI�fj� 〕� fl� FIWES� 0.346004 Hi
* D,, il ^J 11 yA u n N B U AO 1 4451188 sec
_ An N ^ V ^ I ^ -096 ^ \ „ 01 1 00000000 sec
f-Bu ^ 1] K II /-Bu on 0 03000000 sec
CHANNEL “ WXl 13C Pt 3.00 usee PU -5.00 d9 STOl 75 4745in MMr
CHANNEL r? CPOPPC? xaUi 16 MUC? ]H PCP02 100 00 usee 气2 】?0�00�OB�PL12� 19�00 OB STO?� 300�1315007�mw,�
F?�-�Processing oara*ters SI 65536 SF 75 <57 7296 mi tiOit EM
I SS9 0 La 3 00 Hi se 0 PC 1 40
ID plot oarareiers CX 23 00 cm
1 FtP 180 000 0D» } I ' ' I Pi 13564 19 H;
J L _J、i I ^ U-J� 1_y� /� LJ� L U J � F?� -1509.35�h?�
B�59555 po /c* ;� ^H^CM� 5S6 ?<109�
150� UO i?Q� 100 50� 50� 40 20� 0�
127

A-2-2. ^H and NMR Spectra of [Cu(HL^)(^-I)]2 (87)
in o o) mnj(Mioo)rv.r>.o>iDinfnf\>oj(Oi£Jfn r- in us� ①� Current�Data�Pararaeters�g� m�<M� m m n� — 卜 ( C i D i n m m� cm� tn NAME� ecc084�g� d� oi�o>� •�lOioiOiOiDioiDiDtoiOifito ,� m m� —� EXPNO� j�
\ / - Acqu is i t i on Parameters
Da te . 200104!0
Time 18.46
INSTPUH dp1300
PflOBHO 5 IBM Dual 13 PULPflOG zg
^ ^ TO 32768 f ] f i ^ SOLVENT C606
r 尋 , - 「 O H V ^ ' Y H oh 55.600 usee
f - B U OE 6.00 usee
k:^^� TE� 300.0�K�01 1.00000000 sec
. . . . . . . . . . . . C H A N N E L M
NUCl IH
PI 4.50 usee
PL! -2.00 dB
SFOJ 300.1312000 mz
F2 - Processing parameters
SI 32760
SF� 300.1300322�MHz�MOM EH
SS8 0
LB 0.30 Hz
68 0
PC 1.00
10 _ p lo t paramelers
CX 23 00 cm
I J FIP 12.000 Dpm I jl jJLMi : I , L I jj- • . F p p ^-O'mo :
A A / V A V A A A A 監 : 王 " rvj� o� o� r�m� �m�o>� to� o� m� m�
u� � IT)� o� oj�m�•-«�oi�ID� OT� � (D� cn�J� OT� 03� o� o�r�CM�OT�O)� n j r - m� r-c� o� —•� —�cu o r u o— 一� oo�
ppm 10 B 6 4 2 0
Current Oata Paraneters
NAME ecc084_cl3 exPNO 1
CM CD LO • ^ ' » c \ i ( \ j o a 3 n f n m m m m ru o i PflOCNO i ① ( D O o - * o a o t D m o > o o \ ^ in •»- i n i o g • rn 一 if)mr«.non<J>*J"ioin rr ” ru
g r.- oi rvj r^r^aidoir^ir^f^�� o;� cr>�ua� m o� F2�-�Acqujs.tion�Parameters� .�if> i n tn m m n j c g o j r u f v j r u o j - r ' m m m m Oate 300)0417
V I \| I I iHr 二 ? PULPflOG zgdc TO 6553B SOLVENT C0C13 NS 3693 OS 0 SHH 22575.736 H2 FIDOES 0.346004 Hz
H ii Q ^ H O /-Bu II usee "V- I ^N^^^V^ OE 5.00 usee I� .“、i、、l/""“� t� I� Te� 300.0�<�
//� C U < 1� N ^ v� //� ot� 1.00000000�
/� � � \� 《 01] 0.03000000 sec
I J ^Y H …•….•…••. PI 3.00 usee PU -6.00 dB SFOl 75.4745111 MH?
CHANNCL t2 • • • • • • , CPOPflG? » a l t r l 6 NUC2 IH PCPO? 100 00 usee PL2 IPO.OO flB PLl? 19.00 dB SF02 300 1315007 MHz
F2 ‘ Processing oarawte rs SI 65536 SF 75.^677053 MHl . HOW EM S5B 0 LB 3.00 HZ ca 0
PC 1.40
10 NHp olot D«f"a"Mters I CX 23.00 c«
FIP IBO 000 DP* n 135BM9 Hi
I I I F?P -20 000 00(»
‘ . , ^ HZCM 555 24091 Mz/Cff ODm 160 MO 120 100 80 50 -JO 20 0
128

A-2-2. ^H and NMR Spectra of [Cu(HL )( -I)]2 (87)
„ r*. T r g c v j o i r - i D Current 0«t« ParanetBrs g ti ry ^ l o i o m m m m m r u - ^ — ' o o O O T U J i n m m ^ ^ ecc071 a ^ o; a i r." r " r: r." r.' r - r - f-" f - f-' r^' ^ m m m m r ; £XPNO 1
I I WW KW// Date. 20010315
rime 13 27
INSTRUH dox300
PR06K0 5 Dual 13
NBu 舰麵 rt^rV/OH \ SOLVENT DHSO XX 0 l^QU N SHH� 8992.806 Hi
/ \ FIORES 0.27i439 Hz V C u AO 1.821950B sec / ^ M ^ TO 138
H N I 丨 OW 55.600 usee
OE 5.00 usee
TE 300.0 K
A j j 0� I.OOOOOOOO sec . . . . . . . . . . . . C H A N N E L f ! " " “ •
NUCt IH
PI 4.50 usee
PL! -2.00 dS
SFOI 300.1312000 MHz
F2 - Processing oarameters
SI 3276B
SF 300.1300015 MHZ
WW EM
ssa� 0�LB 0.30 HZ
GB 0 PC 1.00
10 _ o i o l parameters I CX 23.00 cm
丨 f IP 14.000 ppn
I A A 1 Al I JUjUl I lUul '二: . . . , , , , ,, 11 F2 -150.06 Hi
? N 3 5 12 g g] 2 S S £ 5 m o? r o o S r- C71 :丨 二 ? 二� o� —•� r\j� o�o� m卜� oilcn�
I�M�•� '�i�"�I�•�I�I�I�•�I�>�.�• ,,r-� '�I�"� '_..-.� I 丨� I� I�'-"�-ppm� .12� 10� 8� 6� 4� 2� 0�
Current Data Parameters
NAME� ecc014C-amso�EXPNO 1
c n — o i f - i n t D 臂 r n u j i o 卜 o o ) r«. l o o j 臂 r - » o i — n m r > « r ^ , o j o t n PBOCNO 1 o�in� inf^ioo»f\jotDO>aocDo� —� inrviinr-cnru^LDCD-^cDOoru�
I.� -� o� pj�.� p p t rs�
SS ? £ Date, jooioao? • n \ i \ r s \ w I = “ 。 H
pulPTOG zgdc TO 65536 SOLVENf CDC13 NS 2933 OS 0 5MH 22675.736 HZ
t P y FIOflES 0.346004 Hz I . / ^ AO 1.4451108 sec
- j ^ k ^ O H H N - \ \ y ) 呢� 9,98�r ^ if J� j i y� OH� 22.050�usee�J ^� 11� I — O E� 6.00�usee�
k O i l ISI 1 TE 300.0 K
f-D"� f� \� 01� r�00000000�sec�( C u Oi l 0.03000000 sec
UfsJ� 7 丨� ••……•…CHANNEL�fl……'�‘ 1 NUCJ 13C
P 3.00 usee
/ / / PU -6.00 dB
^ ^ ^ ! SFOI 75.4745111 KHz
. . . . . . . . . . . . C H A N N E L f2
CP0PRG2 wa lU ie NliC3 IH PCP02 100.00 usee n.2 120.00 dB PL12 19.00 08 SF02 300.1315007 MHz
F2 - Processing parameters I SI 65536
SF 75 4677830 MHz KOK EH SSB 0 LB 3.00 HZ G8 0 PC 1,40
10 NMfl p lot parameters CX 23.00 cm FtP leo 000 00*
Fl 13584,20 Hz
U | I / F2P -20 000 ppm
HZCH 655.2415日 H"cm
Dpm� 160 !�40� 120� 100� 80� 50� dO� 20� 0�
129

A-2-2. ^H and NMR Spectra of [Cu(HL )( -I)]2 (87)
o q*in”iD^^ru — ODiD(Mor-nj — r u o c o m m o c o l u — oCT>CDr-iD"«»ncD Current Qata Para»eie'"S p l O i D U j m m m m — '^aaajr^. o c n i o i o m m— Q NAME eCCO O Q m� a3r««r>-r»-r-«r»»r-r-•卜�r«-u3u3LD,r*"imf^nmnfnnnr\jf\jr\j 一� —�—�—� EXPNO� 】�
I 1 1 H iT ^ ! ^ 1� T [ F2 - Acquis i t ion Parameters Date. 20010319 Time 19.12 INSTRUM do*300
1 Q.. — PfiOBHO 5 im Dual 13 卜 tJU / = = \ PuLPnoG Jk OH HN—(v /) TO 32768
/ / / SOLVENT DMSO
n \ SMH 8992.806 H? ( p FIOBES 0.274439 H? k i ^ ^ ^ W 、 AO 1.8219508 sec
W M V ^ I TO 322.5 J W 55.600 usee
0£ 6.00 usee
I n TE .300.0 K
U 01 l.OOOOOOOO sec
CHANNEL f I
NUCl IH
PI 4,50 usee
PLl -2.00 06
SFOl 300. 1312000 MHz
F2 - Processing parameters
SI 32768
Sf 300.13000J0 mz
KOW EH
5Se� 0�L8 0.30 »Z
G8 0 PC I 00
to Nwn plot paratneters J CX 23 00 cn
I MP M.OOO pp.
k A JlRJ . 1,1,U . J.il P T 二 l\ i\ / \ / M / f \ A 二 二 二
- L ] H /OYCD ml ail |<n丨f^ c=,] H2CM 189,21239 H /cm « ‘ f\j| T (o m m oi ! oi 丨•“』 icD ^ irv o丨 [o cn cni m 〜丨rr ir-S" ‘ — cn o cn cn cd'od o -� •!� !�-�-I�-I •! •;� -1� I� .�三 一_� I»-•‘� I�rvi��I�11�o� m'—.� I�oi�I�
ppm 12 10 a 5 4 2
Current Data Parameters
MAME ecc040j:13
EXPNO 1
rx oj — ojocTiiD 〜o 对 ruu3 o r>.-«oiiflCDofM*jiDcoioiDruai PflOCNO \ o> •^nmtotDincDrxru uinrumr^oru*TiO(Oir)cncD"-*
g -T OJ If) mrvjcno'^fviry^o o m c D r - - m o m i n r u c p U 3 m o i o — g i n o ^ r.' u D r - i n d d c n o i O T m a s ^ o c r i i n F 2 - Acquis i t ion Parameters
m in T m m n j o j o j C M - - — lo i n - T r r T r - T m m m f n m m f n r u r u Date 20010320
M r \ r \ \ w I = 』 • PULPTOG jgdc TO 65536 SOLVENT OMSO NS 6066 t-Bu OS 0
I SHH 22575.735 Hz ^ ^ S ^ O H H N ' ^ V y ) FI ORES 0.34&004 HZ r I f j L ^ y AO 1.4451188 sec
Ji AG 9195.3 H 1 ON 22.050 usee
/ \ OE 6.00 usee L TE 300 0 K
\ , DI J.00000000 sec HN I I dll 0.03000000 sec 丨 … … … " C H A M N E L fl
( / / NUCl 13C
PI 3.00 usee
PLl -6.00 dS
SFOl 75.4745111 MHZ
. . . . . . . . . . . .CHANNEL f2
CP0PRG2 waltzlB NUC2 IH PCP02 100.00 usee PL2 120.00 d8 PL 12 19 .00 OB SF02 300.1315007 MHz
F2 - Processing parameters St 65336 Sf 75.467784丨 MHZ
MOW� EM�SSB 0
L8 3.00 HZ
GB 0 PC 1.40
10 NHR olot parameters CX 23 00 cm MP 160.000 Dpn
I I n 13564 20 HZ . I i F2P -20 000 ODO
HZCM 655 2415B Hz/cn I •… 厂 , … ' I I I T, r - ^ - r - . ~ ~ ~ ' ••‘~I … . ~ T ‘ ― ^ ' ' ' | '“''''''' ppm 160 140 120 100 80 60 40 20 0
130

APPE
NDIX
B C
rysta
llogr
aphic
Dat
a (C
ontin
ued)
B-1. X
-ray C
rystal
Struct
ure Da
ta for
Comp
lexes
(51,52,
54 and
55) D
escrib
ed in
Chapt
er 2
~
[Cu(HL»)C
l]C
l (51)
[Cu(HL»)(
OA
c)](
OA
c) (52)[CU(L')(OAC)] (54)
[Zn(L')
Cl] (55)
Empir
ical
Form
ula
C27
H35
CI2
CUN
3OCH
2CI2
O.5
H2O
C29
H37
CUN
3O3C
H3O
H2H
2O
C^^H
^^CIN
^O
^
Form
ula
Wei
ght
645.
95
656.
75
607.2
3 517.3
9 Cry
stal
Sys
tem
m
onocl
inic
tr
iclinic
tr
iclinic
ort
horh
om
bic
Sp
ace
Gro
up
C2c
PT
PT
Pna2(l
) a/
A
34.2
25(7
) 13
.237
(4)
8.6
502(1
7)
23.2
58(5
) b/A
12
.130
⑵
13.7
21(5
) 10.0
08(2
) 13.9
73(3
)
c/A
16.6
50⑶
20.9
26(7
) 20.4
36(4
) 8.2
841(1
7)
a/。
90
96.5
8(2
) 89.8
2(3
) 90
Q/o
110.2
5(3
) 100.3
8(2
) 84.0
8(3
) 90
- 90
108.8
1(2
) 64.5
2(3
) 90
—
Volu
me
/A^
6485(2
) 3477(2
) 1586.9
(5)
2692.2
(9)
Z
8
4
2
4
Densi
ty/M
gm
-^
1.3
23
1.2
54
1.2
71
1.2
77
Abso
rption
Coef
fici
ent/
mm
'
1030
0.67
8 0.7
32
1.03
4 F(O
OO
) 26
88
1394
64
6 10
88
Cry
stal
Siz
e/m
m
0.40
x 0
.35
x 0.
05
0.2
2x0.2
0x0.1
5 0.4
0 x
0.3
6 x
0.2
4 0.5
2 x
0.3
7 x
0.2
2 Te
mper
ature
/K
293(2
) 293(2
) 293(2
) 293(2
) G R
ange
for D
ata
Colle
ctio
n/^
1.79
-25.
00
1.67
-25.
00
2.0
1-2
4.0
0 2.2
8-2
5.4
8 In
dex
Ran
ges
0^
<40, -1
3<k<
12, -
19<1<
18
-1
5^
<1
4,0
^1
6,
-24<
1<24
-9
<h<
8, -
11
^0
, -2
3<
1^
3 -2
8<
h^
8, -1
6^
0,
-9<1<
9 N
o. of Ref
lect
ions
mea
sure
d
7373
63
30
4069
7778
N
o. of In
dep
enden
t Ref
lect
ions
4517
63
30
4069
4297
Goodne
ss-o
f-fit
on
F^
1-07
7 1.
102
1.06
4 1.
044
Final
R I
ndic
es [
I>2
ct(
I)]
R
= 0
.0824,
R^ =
0.2
132
R =
0.0
828,
RW
= 0
.1961
R =
0.0
710’
RW = 0
.1934
R =
0.0
672’ R^ =
0.1
730
R In
dic
es
(all d
ata
) R =
0.0
937, Rw
= 0
.2246
R =
0.1
455, Rw
= 0
.2320
R = 0
.0763, Rw
= 0
.1988
R =
0.0
699,
R^ =
0.1
760
R�= i
|FOI-|
fJ/
i|FOI. RW
= [Iw
( I Fo I -1 F
e I )'
/Sw I Fo I
w
= 4
Fo'/A\F
o').

APPE
NDIX
B Cr
ystallo
graph
ic Data
(Cont
inued)
B-1
. X-ra
y Crys
tal Str
ucture
Data f
or Co
mplex
es (56
- 59)
Descr
ibed i
n Chap
ter 2
[Zn(L
^)C
l]
(56)
[Zn(L
^)C
l]
(57)
[Zn(L
')(O
Ac)
] (5
8)
[Zn(L
^)(
OA
c)]
(59)
Empir
ical
Form
ula
C
^^
^C
IN^
O^
OS
lS^
^O
ZK
CH
^
C29H
37N
3O3Z
112H
2O
QA
^N
AZ
i^H
^
Form
ula
Wei
ght
461.
29
546.
21
577.0
2 52
0.91
Cry
stal
Sys
tem
m
onocl
inic
m
onocl
inic
tr
iclinic
tr丄
clin
ic
Space
Gro
up
P2(l
)/c
P2(l
)/c
Pi
Pl
a/A
16.4
60(3
) 19
.976
2(19
) 9.1
389(1
8)
9.0
580(5
) b/A
12
.827
(3)
10.9
377(1
1)
11.8
57(2
) 10.4
360(6
) c/
A
17.9
21 ⑷
11.9
829(1
1)
15.1
50(3
) 15.2
577(8
)
a/。�
90�90�
90.98(3)�
95.731(1)�
3/0
144.9
6(3
) 104.7
73(2
) 104.2
2(3
) 104.3
18(1
)
—
丨。
90
90
91.56(3)
112.238(1)
^
Volu
me/A
' 2172.6
(8)
2531.6
(4)
1590.3
(5)
1263.1
(1)
Z
4
4
2
2
Densi
ty/M
gm
-^
1-4
10
1.4
33
1.2
05
1.3
70
Abso
rption
Coef
fici
ent/
mm
'
1.27
2 1.
308
0.8
10
1.01
2 F(
OO
O)
960
1128
61
2 54
8 Cry
stal
Siz
e/m
m
0.6
0 x
0.30
x 0
.20
0.6
7x0.3
4x0.1
2 0.4
2 x
0.2
7 x
0.2
4 0
.48
x0
.36
x0
.11
Tem
per
ature
/K
293⑵
293(2
) 293(2
) 293(2
)
0 R
ange f
or D
ata
Collect
ion/^
2.0
2-2
4.0
0
2.1
1-2
5.0
0
2.2
4-2
4.0
0
1.4
1-2
8.0
2
Index
Ranges
-18让
《18,
-14<
k<
14, -1
8<1<20
-23:^
<23, -ll<
k<
13, -1
4<1<14
-1
0^
9,
-13<
k<
13, 0<1<17
-1
0^
11
, -1
3^
13
, -2
0<1<18
No. of Reflections
measu
red
5458
13695
4242
8917
No. of In
dependent Reflections
3133
4446
4242
5970
Goodne
ss-o
f-fit
on
1-03
4 0.
948
1.15
8 0.9
72
Final
R I
ndic
es [
I>2ct
(I)]
R = 0
.0506, Rw
= 0
.1339
R =
0.0
375,
Rw
= 0
.0972
R =
0.0
848, Rw
= 0
.2476
R =
0.0
429,
Rw
= 0
.0927
R In
dic
es
(all d
ata
) R =
0.0
565, R^ =
0.1
391
R =
0.0
573, Rw
= 0
.1095
R =
0.0
868,
Rw
= 0
.2510
R =
0.0
673, R^ =
0.1
028
R =
E|F
OI-|
FJ/I|
FO|.
Rw =
[Sw
(|Fol
-|Fj)2
/Iw|F
o|2f
2,w
= 4
Fo2
/cj2(F
o2).

APPE
NDIX
B Cr
ystallo
graph
ic Data
(Cont
inued)
B-2
. X-ra
y Crys
tal Str
ucture
Data f
or Co
mplex
es (86
, 87, 92)
Desc
ribed
in Ch
apter
3 [C
U(H
L')(^
-I)]2
(86)
[CU
(HL'
)(H-I)
]2 (8
7) [C
U(H
L')I]
(8
8)
Empir
ical
Form
ula
C54
H70
CU2I
2N6O
22CH2C
I2
C46
H54
CU2I
2N6O
22CH2C
I2
C31H
37CU
IN5O
2TH
F.O
.5CH
3O
H
Form
ula
Wei
ght
1385
.90
1273
.68
853.8
2 Cry
stal
Sys
tem
m
onocl
inic
m
onocl
inic
tr丄
clin
ic
Space
Gro
up
C2/
c ?
IJc
PT
a/A
38.6
58(2
) 18.4
17(4
) 10.0
48(3
)
b/A
9.2999(5)
10.202(2)
12.647(4)
c/A
18.7382(10)
14.137(3)
17.739(6)
a/。
90
90
100.9
84(7
)
p/。
110.7
820(1
0)
95.7
5(3
) 95.5
01(7
)
^
y/o
90
90
101.0
78(7
)
^
Volu
me/A
' 6298.4
(6)
2642.8
(9)
2151.0
(12)
Z
8
2
2
Densi
ty/M
gm
-3
1.4
62
1.6
01
1.3
18
Abso
rption
Coef
fici
ent/
mm
-'
1-86
7 2.
218
1.26
7 F(
OO
O)
2800
12
72
881
Cry
stal
Siz
e/m
m
0.71
x 0
.65
x 0.
54
0.35
x 0
.20
x 0.
18
0.5
6 x
0.4
8 x
0.4
4 Te
mper
ature
/K
293(2
) 293(2
) 293(2
) e
Ran
ge
for D
ata
Collection/。
2.1
9-2
8.0
2
2.2
2-2
5.0
0
1.6
8-2
4.0
0
Index
Ranges
-51^
<51, -1
0仏
12, -2
4<1<24
-21<
h<
21, -1
2^
0,
0<1<16
-11血
11,
-14^
4,-
19<
1<
19
No. of Reflections
measu
red
21369
4865
5070
No. of In
dependent
Reflections
7578
4654
5052
Goodne
ss-o
f-fit
on
F^
0.9
99
1.06
6 0.9
90
Final
R I
ndic
es [
I>2
ct⑴
] R =
0.0
393,
Rw
= 0
.1120
R =
0.0
648,
Rw
= 0
.1746
R =
0.0
591,R
, = 0
.1781
R In
dic
es
(all d
ata
) R =
0.0
545’ Rw
= 0
.1194
R =
0.1
304,
R^ =
0.2
086
R =
0.0
692, R
,. =
0.1
893
R =
L|Fo
l-|F
el/Z
|Fol
. Rw
= [2
W(|F。
|-|Fc
l)2/Z
W|F。
h"2
’W =
4F。
2/CJ2(
Fo2
).

APPENDIX C GC-MS Spectra
C- l . GC-MS Spectra for Standard Samples
C-1-1. GC-MS Spectra of Pure Triphenylphosphine (top) and Pure Triphenylphosphine oxide (bottom).
丨Abundance, ~ TiCrECXD" ! 2800000-i I 1 ! 2 6 0 0 0 0 0 - !
I ! j 2 4 0 0 0 0 0 j
I I 2200000
! i j 2000000 j
1800000 i
: !
I ‘ ; 1 6 0 0 0 0 0 j
! I
: 1 4 0 0 0 0 0 1
I 1200000] I I 1000000
i I 800000 I
! 600000j
I 4 0 0 0 0 0 I
! i 200000i
Time->。 6.00 8.00 10.00 12.00 14.00 16.00 18.00 20.00 22:00 24.00 26.00 28.00
•AbJHaiK^ nC:EC2.D ‘ I 1
1 3 0 0 0 0 0 ] I 1
. !
1200000j
(1100000 . I !
1000000j »
9 0 0 0 0 0 ,
1
8 0 0 0 0 0 . .;
.‘� I�
‘� •� j�
7 0 0 0 0 0 : I r ‘
i I 600000丨 I
: . I 5 0 0 0 0 0 ^ 丨
I :;
4 0 0 0 0 0 , ::
3 0 0 0 0 0 I
200000 *
1 0 0 0 0 0 J 丨 I
Time~> G j.OO 3.00 10.00 12.00 1.00 16.00 13.00 20.00 22.00 24.00 26.00 28,00_J
134

C-1-2. GC-MS Spectra of Pure HL^ 34 (top) and Pure HL^ 35 (bottom).
丨Abundance ""TTCTEUgTD 1 5 0 0 0 0
I 1 4 0 0 0 0
130000. l a 120000 iJ『胃
110000 Jf
100000.1 f
9 0 0 0 0 I
80000 • f •
I 7 0 0 0 0 I i f
! 6 0 0 0 0 •
5 0 0 0 0 • I
‘ 4 0 0 0 0 J f f l F
I : 1 jj\ , j r
! 竹 ; V� .�.�,� . , , 1 1 1� I�
i T i r n e — > . 6 . 0 0 3 . 0 0 1 0 : 0 0 . 1 2 . 0 0 . 1 4 : 0 0 16:00 1 8 . 0 0 2 0 . 0 0 2 2 . 0 0 2 4 . 0 0 2 6 . 0 0 2 8 . 0 0
,-n—^ nCTTCnXD ~ !
丨Abundance
110000] I ,
100000 |||fi|
9 0 0 0 0 • f '
if 80000-1 11 7 0 0 0 0 L j f
IH A/l / i _ V \ J I 4����_ I / 30000.
I 肩 iUr \
Time--> ^^^S.ho 8.00 10!00 ‘ 12: ‘ 14:00 16:00 18:00 20:00 22:00 24.00___2M9_J
135

C-2. GC-MS Spectra for the Reactions with Triphenylphosphine Described in
Chapter 3
C-2-1. GC-MS Spectra upon the reaction of [Cu(HL^)(|a-I)]2 86 with
triphenylphosphine (top), and upon the reaction of [Cu(HL XM)]2 87
with triphenylphosphine (bottom).
「Abundance rrcTEC-^D I I
; 2 0 0 0 0 0 0 ]
I
I 18000001 ‘ i ,1600000 I I I . I 14000001 I
I 1200000 i
: i
I 1000000;
1 i 800000
! 600000 I
4 0 0 0 0 0
! 200000J
i 1 i Iq j LI . 丨 . . ;
r r i m e - 〉 。 , I ‘ ' s . b o ‘ a.OO • I 0 I 0 O . U.OO ‘ ‘ 1 4 ; 0 0 isloo _ 1 8 : 0 0 2 0 : ^ 2 2 . 0 0 _ _ ^ _ _ ^ _ _ 2 8 - 0 0
bundance HC面:CT —
i 1 5 0 0 0 0 0
!
j 1 4 0 0 0 0 0 ]
1 1 3 0 0 0 0 0 j
1200000
j 1 1 0 0 0 0 0 .
1000000 9 0 0 0 0 0
800000 I
i 7 0 0 0 0 0
600000
500000
! 4 0 0 0 0 0
3 0 0 0 0 0
200000 i
i 100000 i V I !� I� j,� w——^�
' ' ' 6 . 0 0 ‘ I 8 . 0 0 ‘ ‘ i o ! o Q n l o o . ‘ U I Q O " ^ W . Q q ' ^ ^ I ^ Q “ 2 2 : 0 0 2 4 : 0 0 ‘ . 2 6 : 0 0
136

C-2-2. GC-MS Spectra upon the reaction of [Cu(HL'^)I] 88 with triphenylphosphine
(top), and the control experiment without addition of any metal complex
(bottom).
A b u n d a n c e ~ nCTEC'l 1. D ‘ 2e+07j
I 1
1.8e+07-j 1.6e+07 J
?
‘� 1�
1 1.4e+07-
‘1.2e+07 ,� I�.� 1�
] I
1e+07i !
8000000 J I . -I
: ! :6000000 J ! !
i 4000000
I I I 1�
:2000000 j
.Time-> ° ' ' ' 6.00 . ‘ 8.00 ‘ 10:00 , 12!o6 . u'.OO . ‘ 16:00 . ‘ 18:00 20:00 22:00 ‘ 24!00 26!qO 28:00
l A b u n d a n c e “ rrCTECTOTD “ 2.6e+07 2.4e+07 • 2.2e+07 2e+07
i.se+orj i
1.6e+07-1.4e+07 1.2e+Q7. 1e+07.
8000000
6000000
4000000 • 20000Q0 1
t / V - I . . . . I , .
r,me-> “ . ‘ 6.00 ‘ 3.00 10:00 12:00 u!oQ ‘ 16:00 . isloo ‘ 20!00 22.00 . 24:00 26:00 28:00
137

!
I "i i .
t�
V
^ •
I •;.
••
‘
5
\ 、
:
- 」
•
•‘
,I
[ -
r
;,•
.
:、
、 __「
)
-
:
—r�.�.�hiTiftre!?''.�

CUHK L i b r a r i e s
__illlllllll 0 D 3 f l 7 1 7 S l
Copyright © 2022 FDOKUMEN




















