
Syntaxin 1A is required for normal in utero development
6
Syntaxin 1A is required for normal in utero development John E. McRory a , Renata Rehak a , Brett Simms a , Clinton J. Doering a , Lina Chen a , Tamara Hermosilla a , Carlie Duke b , Richard Dyck b , Gerald W. Zamponi a, * a Department of Physiology and Biophysics, The Hotchkiss Brain Institute, University of Calgary, 3330 Hospital Dr. NW, Calgary, Canada T2N 4N1 b Department of Psychology, University of Calgary, 3330 Hospital Dr. NW, Calgary, Canada T2N 4N1 article info Article history: Received 25 July 2008 Available online 17 August 2008 Keywords: Syntaxin Knockout mouse Lethal Development PCR Southern blot abstract We have generated a syntaxin 1A knockout mouse by deletion of exons 3 through 6 and a concomitant insertion of a stop codon in exon 2. Heterozygous knockout animals were viable with no apparent phe- notype. In contrast, the vast majority of homozygous animals died in utero, with embryos examined at day E15 showing a drastic reduction in body size and development when compared to WT and hetero- zygous littermates. Surprisingly, out of a total of 204 offspring from heterozygous breeding pairs only four homozygous animals were born alive and viable. These animals exhibited reduced body weight, but showed only mild behavioral deficiencies. Taken together, our data indicate that syntaxin 1A is an important regulator of normal in utero development, but may not be essential for normal brain function later in life. Ó 2008 Elsevier Inc. All rights reserved. Syntaxin 1 is a critical element in calcium triggered exocytosis in both neuronal and non-neuronal tissues [1,2]. In vertebrate syn- apses, syntaxin 1 is known to interact tightly with both SNAP-25 and a synaptic protein interaction site found in both N-type and P/Q-type calcium channels [3–5]. This macromolecular protein complex allows for the docking of synaptic vesicles via interactions with synaptotagmin and synaptobrevin, thus localizing vesicles within calcium microdomains supported by channel activity—a key requirement for efficient calcium triggered neurotransmitter release (for review see [6,7]). In addition to its role in exocytosis, syntaxin 1 is known to directly inhibit calcium channel activity by reducing channel availability [8–13]. From a structural point of view, syntaxin 1 is organized as a four helix bundle that can un- dergo conformational changes, as well as a membrane insertion domain near the C-terminal which is located adjacent to the BotC cleavage site [14–16]. The mammalian nervous system expresses two syntaxin 1 isoforms, syntaxin 1A and 1B, which are about 85% homologous at the amino acid level, are both cleaved by BotC, and appear to be differentially distributed in the brain [12]. In addition, alternate splicing may generate additional syntaxin 1A variants [14–16]. A recent study reported on the functional knockout of syntaxin 1A in mice [17]. The authors reported that the mice were viable with apparent impairment in long term potentiation and condi- tioned fear memory, perhaps suggesting the possibility of compen- sation from syntaxin 1B. These authors targeted their gene deletion strategy towards exons 9 and 10, thus effectively removing the membrane insertion domain of syntaxin 1A, but sparing much of the syntaxin 1A molecule. The same group as well as others re- ported on the existence of a syntaxin variant (termed syntaxin 1C) that lacks the membrane insertion region as a result of alter- nate splicing and these truncated splice isoforms are also found in EST databases [18–20]. To determine if the lack of phenotype in the syntaxin 1A knockout animals may have been due to the fact that the authors effectively converted syntaxin 1A into a syntaxin 1C truncation protein, we created a syntaxin 1A null mouse in which we deleted exons three through six, and inserted an addi- tional stop codon into exon 2. Our findings show that the deletion of the syntaxin 1A gene resulted in reduced embryonic growth leading to death in utero. Hence, syntaxin 1A appears to may be a key requirement for appropriate embryonic development. Materials and methods To target and excise the endogenous syntaxin-1A gene a cDNA construct utilizing the pPNT vector was used. A segment encom- passing a portion of intron 2 to exon 10 was isolated from mouse DNA using PCR two oligos (ccaaagagcctcactgagc, acctccgtggttgatccc were used according to the manufacturer’s instructions, and the reaction product was analyzed on a 1.0% agarose gel; DNA bands were isolated, cloned into pGEM-T-easy (Stratagene), and se- quenced to confirm identity. DNA from the in house embryonic cell line was used to optimize targeting of the vector as substantial nucleotide differences do occur between mouse lines. The con- firmed DNA strand was cloned into the pPPNT vector for targeting 0006-291X/$ - see front matter Ó 2008 Elsevier Inc. All rights reserved. doi:10.1016/j.bbrc.2008.08.031 * Corresponding author. Fax: +1 403 210 8106. E-mail address: [email protected] (G.W. Zamponi). Biochemical and Biophysical Research Communications 375 (2008) 372–377 Contents lists available at ScienceDirect Biochemical and Biophysical Research Communications journal homepage: www.elsevier.com/locate/ybbrc
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Syntaxin 1A is required for normal in utero development

Biochemical and Biophysical Research Communications 375 (2008) 372–377
Contents lists available at ScienceDirect
Biochemical and Biophysical Research Communications
journal homepage: www.elsevier .com/locate /ybbrc
Syntaxin 1A is required for normal in utero development
John E. McRory a, Renata Rehak a, Brett Simms a, Clinton J. Doering a, Lina Chen a,Tamara Hermosilla a, Carlie Duke b, Richard Dyck b, Gerald W. Zamponi a,*
a Department of Physiology and Biophysics, The Hotchkiss Brain Institute, University of Calgary, 3330 Hospital Dr. NW, Calgary, Canada T2N 4N1b Department of Psychology, University of Calgary, 3330 Hospital Dr. NW, Calgary, Canada T2N 4N1
a r t i c l e i n f o a b s t r a c t
Article history:Received 25 July 2008Available online 17 August 2008
Keywords:SyntaxinKnockout mouseLethalDevelopmentPCRSouthern blot
0006-291X/$ - see front matter � 2008 Elsevier Inc. Adoi:10.1016/j.bbrc.2008.08.031
* Corresponding author. Fax: +1 403 210 8106.E-mail address: [email protected] (G.W. Zamp
We have generated a syntaxin 1A knockout mouse by deletion of exons 3 through 6 and a concomitantinsertion of a stop codon in exon 2. Heterozygous knockout animals were viable with no apparent phe-notype. In contrast, the vast majority of homozygous animals died in utero, with embryos examined atday E15 showing a drastic reduction in body size and development when compared to WT and hetero-zygous littermates. Surprisingly, out of a total of 204 offspring from heterozygous breeding pairs onlyfour homozygous animals were born alive and viable. These animals exhibited reduced body weight,but showed only mild behavioral deficiencies. Taken together, our data indicate that syntaxin 1A is animportant regulator of normal in utero development, but may not be essential for normal brain functionlater in life.
� 2008 Elsevier Inc. All rights reserved.
Syntaxin 1 is a critical element in calcium triggered exocytosis strategy towards exons 9 and 10, thus effectively removing the
in both neuronal and non-neuronal tissues [1,2]. In vertebrate syn-apses, syntaxin 1 is known to interact tightly with both SNAP-25and a synaptic protein interaction site found in both N-type andP/Q-type calcium channels [3–5]. This macromolecular proteincomplex allows for the docking of synaptic vesicles via interactionswith synaptotagmin and synaptobrevin, thus localizing vesicleswithin calcium microdomains supported by channel activity—akey requirement for efficient calcium triggered neurotransmitterrelease (for review see [6,7]). In addition to its role in exocytosis,syntaxin 1 is known to directly inhibit calcium channel activityby reducing channel availability [8–13]. From a structural pointof view, syntaxin 1 is organized as a four helix bundle that can un-dergo conformational changes, as well as a membrane insertiondomain near the C-terminal which is located adjacent to the BotCcleavage site [14–16]. The mammalian nervous system expressestwo syntaxin 1 isoforms, syntaxin 1A and 1B, which are about85% homologous at the amino acid level, are both cleaved by BotC,and appear to be differentially distributed in the brain [12]. Inaddition, alternate splicing may generate additional syntaxin 1Avariants [14–16].A recent study reported on the functional knockout of syntaxin1A in mice [17]. The authors reported that the mice were viablewith apparent impairment in long term potentiation and condi-tioned fear memory, perhaps suggesting the possibility of compen-sation from syntaxin 1B. These authors targeted their gene deletion
ll rights reserved.
oni).
membrane insertion domain of syntaxin 1A, but sparing much ofthe syntaxin 1A molecule. The same group as well as others re-ported on the existence of a syntaxin variant (termed syntaxin1C) that lacks the membrane insertion region as a result of alter-nate splicing and these truncated splice isoforms are also foundin EST databases [18–20]. To determine if the lack of phenotypein the syntaxin 1A knockout animals may have been due to the factthat the authors effectively converted syntaxin 1A into a syntaxin1C truncation protein, we created a syntaxin 1A null mouse inwhich we deleted exons three through six, and inserted an addi-tional stop codon into exon 2. Our findings show that the deletionof the syntaxin 1A gene resulted in reduced embryonic growthleading to death in utero. Hence, syntaxin 1A appears to may be akey requirement for appropriate embryonic development.
Materials and methods
To target and excise the endogenous syntaxin-1A gene a cDNAconstruct utilizing the pPNT vector was used. A segment encom-passing a portion of intron 2 to exon 10 was isolated from mouseDNA using PCR two oligos (ccaaagagcctcactgagc, acctccgtggttgatcccwere used according to the manufacturer’s instructions, and thereaction product was analyzed on a 1.0% agarose gel; DNA bandswere isolated, cloned into pGEM-T-easy (Stratagene), and se-quenced to confirm identity. DNA from the in house embryonic cellline was used to optimize targeting of the vector as substantialnucleotide differences do occur between mouse lines. The con-firmed DNA strand was cloned into the pPPNT vector for targeting
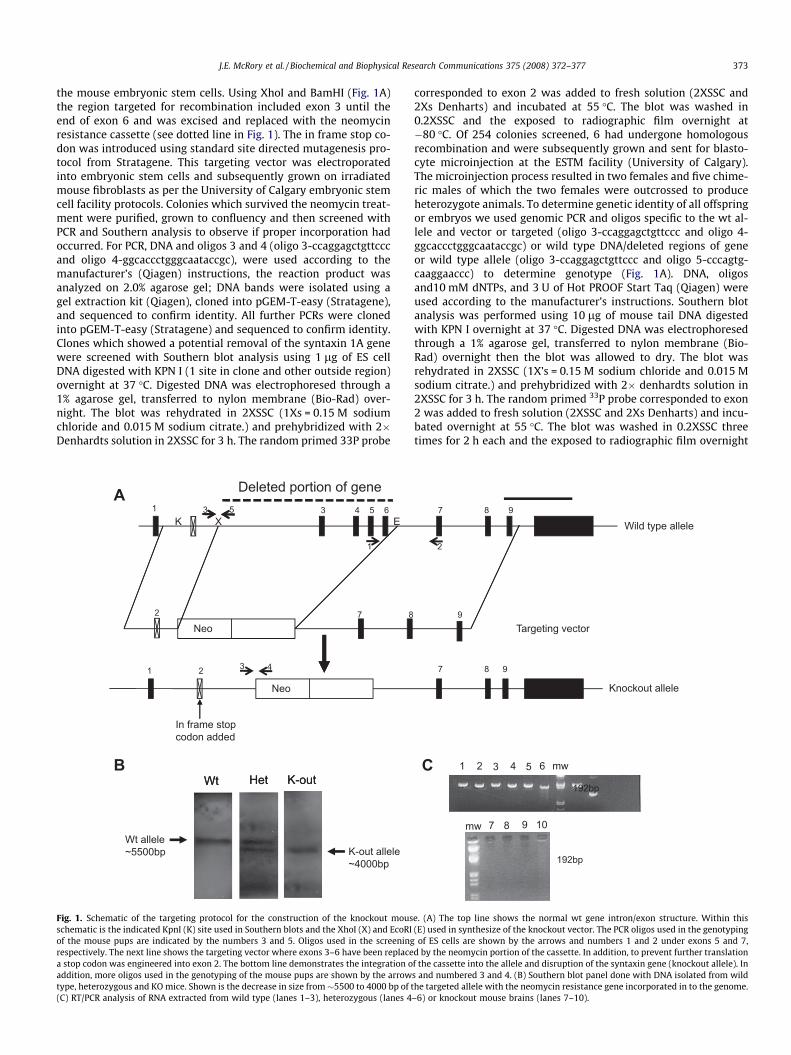
J.E. McRory et al. / Biochemical and Biophysical Research Communications 375 (2008) 372–377 373
the mouse embryonic stem cells. Using XhoI and BamHI (Fig. 1A)the region targeted for recombination included exon 3 until theend of exon 6 and was excised and replaced with the neomycinresistance cassette (see dotted line in Fig. 1). The in frame stop co-don was introduced using standard site directed mutagenesis pro-tocol from Stratagene. This targeting vector was electroporatedinto embryonic stem cells and subsequently grown on irradiatedmouse fibroblasts as per the University of Calgary embryonic stemcell facility protocols. Colonies which survived the neomycin treat-ment were purified, grown to confluency and then screened withPCR and Southern analysis to observe if proper incorporation hadoccurred. For PCR, DNA and oligos 3 and 4 (oligo 3-ccaggagctgttcccand oligo 4-ggcaccctgggcaataccgc), were used according to themanufacturer’s (Qiagen) instructions, the reaction product wasanalyzed on 2.0% agarose gel; DNA bands were isolated using agel extraction kit (Qiagen), cloned into pGEM-T-easy (Stratagene),and sequenced to confirm identity. All further PCRs were clonedinto pGEM-T-easy (Stratagene) and sequenced to confirm identity.Clones which showed a potential removal of the syntaxin 1A genewere screened with Southern blot analysis using 1 lg of ES cellDNA digested with KPN I (1 site in clone and other outside region)overnight at 37 �C. Digested DNA was electrophoresed through a1% agarose gel, transferred to nylon membrane (Bio-Rad) over-night. The blot was rehydrated in 2XSSC (1Xs = 0.15 M sodiumchloride and 0.015 M sodium citrate.) and prehybridized with 2�Denhardts solution in 2XSSC for 3 h. The random primed 33P probe
1 3 4 5 63
Deleted portion of gene5
EXK
1
Neo2 7 8
Neo
1 2 3 4
In frame stop codon added
Wt Het K-outWt Het K-out
Wt allele~5500bp K-out allele
~4000bp
Fig. 1. Schematic of the targeting protocol for the construction of the knockout mousschematic is the indicated KpnI (K) site used in Southern blots and the XhoI (X) and EcoRIof the mouse pups are indicated by the numbers 3 and 5. Oligos used in the screeningrespectively. The next line shows the targeting vector where exons 3–6 have been replacea stop codon was engineered into exon 2. The bottom line demonstrates the integration oaddition, more oligos used in the genotyping of the mouse pups are shown by the arrowtype, heterozygous and KO mice. Shown is the decrease in size from�5500 to 4000 bp of t(C) RT/PCR analysis of RNA extracted from wild type (lanes 1–3), heterozygous (lanes 4
corresponded to exon 2 was added to fresh solution (2XSSC and2Xs Denharts) and incubated at 55 �C. The blot was washed in0.2XSSC and the exposed to radiographic film overnight at�80 �C. Of 254 colonies screened, 6 had undergone homologousrecombination and were subsequently grown and sent for blasto-cyte microinjection at the ESTM facility (University of Calgary).The microinjection process resulted in two females and five chime-ric males of which the two females were outcrossed to produceheterozygote animals. To determine genetic identity of all offspringor embryos we used genomic PCR and oligos specific to the wt al-lele and vector or targeted (oligo 3-ccaggagctgttccc and oligo 4-ggcaccctgggcaataccgc) or wild type DNA/deleted regions of geneor wild type allele (oligo 3-ccaggagctgttccc and oligo 5-cccagtg-caaggaaccc) to determine genotype (Fig. 1A). DNA, oligosand10 mM dNTPs, and 3 U of Hot PROOF Start Taq (Qiagen) wereused according to the manufacturer’s instructions. Southern blotanalysis was performed using 10 lg of mouse tail DNA digestedwith KPN I overnight at 37 �C. Digested DNA was electrophoresedthrough a 1% agarose gel, transferred to nylon membrane (Bio-Rad) overnight then the blot was allowed to dry. The blot wasrehydrated in 2XSSC (1X’s = 0.15 M sodium chloride and 0.015 Msodium citrate.) and prehybridized with 2� denhardts solution in2XSSC for 3 h. The random primed 33P probe corresponded to exon2 was added to fresh solution (2XSSC and 2Xs Denharts) and incu-bated overnight at 55 �C. The blot was washed in 0.2XSSC threetimes for 2 h each and the exposed to radiographic film overnight
7 8 9
Wild type allele
2
Targeting vector9
7 8 9
Knockout allele
1 2 3 4 5 6 mw
7 8 9 10mw
192bp
192bp
e. (A) The top line shows the normal wt gene intron/exon structure. Within this(E) used in synthesize of the knockout vector. The PCR oligos used in the genotypingof ES cells are shown by the arrows and numbers 1 and 2 under exons 5 and 7,
d by the neomycin portion of the cassette. In addition, to prevent further translationf the cassette into the allele and disruption of the syntaxin gene (knockout allele). Ins and numbered 3 and 4. (B) Southern blot panel done with DNA isolated from wildhe targeted allele with the neomycin resistance gene incorporated in to the genome.–6) or knockout mouse brains (lanes 7–10).

374 J.E. McRory et al. / Biochemical and Biophysical Research Communications 375 (2008) 372–377
at �80 �C. Syntaxin-1A knockout mouse lines were then screened,backcrossed into non-related offspring and tested for eight gener-ations in the University of Calgary animal care facility. RT/PCR used10 lg RNA, 2 ll reverse oligo (oligo 2-tcactggtcgtggtccggc) and4.5 ll water, the mixture was heated at 70 �C for 5 min andquenched on ice. Added according in amount and order to manu-facturer’s instructions (Promega) to the denatured RNA mixturewere Superscript buffer, dNTPs, DTT, RNA Guard (Promega) and10 U of reverse transcriptase. The reaction was allowed to proceedfor 90 min followed by PCR. The PCR consisted DNA, oligos 1 and 2(oligo 1-gcattgagcagagcat and oligo 2-tcactggtcgtggtccggc) and10m dNTPs, and 3 U of Hot PROOF Start Taq (Qiagen) were usedaccording to the manufacturer’s instructions.
Several tests were employed to elucidate any behavioral differ-ences between Syntaxin 1A heterozygous(+/�) (n = 4), wild type(+/+)
(n = 5), and knockout(�/�) (n = 3) mice. The Irwin NeurologicalAssessment [21] tests basic neurological function. The assessmentincludes observational measures (e.g., locomotor activity, bodytone, spontaneous licking) and minor manipulations (e.g., pro-voked biting, corneal reflex, grip strength). Swimming behavior,including swim latency, limb-use asymmetry, episodes of forepawdisinhibition, and freezing behavior were assessed using the SwimAlley task [22]. The horizontal ladder [23] was used to measurefore- and hind-limb stepping, and inter-limb coordination. Thehanging wire test [23] measured limb strength. The aforemen-tioned tests were videotaped and analyzed using video replay.The Passive/Active avoidance tasks, used to assess associativelearning and memory, and the prepulse Inhibition task [24] weremeasured using equipment and software from Hamilton–KinderLLC; San Diego, CA. Spontaneous walking behavior was assessedin the open field test [22], and anxiety-like behavior in the elevatedplus maze. The Morris Water Maze [25] was used to test spatialand working memory. The open field, elevated plus and MorrisWater Maze tests were recorded and quantified using an auto-mated video tracking system (HVS Image, Hampton, UK). All testswere performed independently of each other and during the ani-mal’s light cycle. Mice were weighed every second day throughoutthe two-month testing period.
Human embryonic kidney tsA-201 cells were transfected withrat Cav2.2, rat b1b, rat a2-d1 and enhanced green fluorescent protein+/� rat syntaxin constructs, as described by us previously [12].Electrophysiological recordings were conducted using an Axopatch200B amplifier and pCLAMP9 software. The external solution con-tained (in mM) : BaCl2 20, MgCl2 1, Hepes 10, TEA-Cl 40,CsCl 65,Glucose 10; pH 7.2 adjusted with TEA-OH. The internal solutionwith resistances between 2 and 4 MX and contained (in mM):Cs-methanesulfonate 108, MgCl2 4, EGTA 9, Hepes 9; pH 7.2 ad-justed with Cs–OH. Half inactivation potentials were obtained byholding cells at various holding potentials for 5 s, prior to applyinga test pulse to +20 mV. Steady state inactivation curves were fittedusing the Boltzmann equation.
All data are presented as means ± standard errors. Statisticalsignificance was evaluated using one way ANOVA or t-tests asappropriate.
Results and discussion
A breeding program was implemented based after the success-ful generation of seven heterozygous syntaxin 1A mice. In numer-ous breeding rounds of heterozygous breeding pairs, the relativenumbers of wild type and heterozygous offspring was inconsistentwith Mendelian genetics (i.e., 41 L of Het/Het crosses resulted in 64(wild type)/156 (heteros)/4 (knockout). Since only four homozy-gous offspring were born (1 male, 3 female), the possibility of inutero death of homozygous animals was considered. To test this
possibility, we dissected pregnant mothers at day E15, and exam-ined the embryos. As shown in Fig. 2, homozygous knockout em-bryos (as verified by PCR) were substantially smaller than theirwild type or heterozygous litter mates, and lacked limbs, tail andfacial features, thus resembling what would normally be observedat an earlier developmental stage. Similar results were observed inthree different dissections, a total of 37 embryos were examined,with only three being homozygous knockout embryos (Fig. 2). Thisdistribution of WT and heterozygous animals is consistent with aMendelian heritance pattern, whereas the low numbers of homo-zygous embryos suggests reduced viability of homozygous knock-out animals. Altogether, these data indicate that syntaxin 1A mayhave a critical determinant of normal embryonic development.
Despite the aberrant development, four viable homozygoussyntaxin 1A knockout animals were born. These animals showeda significant reduction in body weight over a two month period(Fig. 3A), consistent with our observations with the knockout em-bryos. These four animals, as well as wild type and heterozygousmice, were subjected to a comprehensive behavioral analysis atage 60 days. These included a Morris Water Maze test to probefor alterations in memory, an elevated plus maze test to examineanxiety behavior, an open field test to determine spontaneouswalking behavior, passive and active avoidance tests to assess cog-nitive function, a swim alley task to assess swimming behavior, ahanging wire test to measure limb strength, a prepulse inhibitiontask, and a horizontal ladder task to examine limb placing andcoordination. The three groups of animals (wt, Syntaxin 1A+/�, Syn-taxin 1A�/�) differed, significantly, only during the horizontal lad-der test (Fig. 3B). The mice were tested over four differentconditions: (i) all of the rungs in place, (ii) 9 rungs removed, (iii)13 rungs removed, and (iv) 16 rungs removed. Foot faults, definedas a miss or slip (miss: limb fell through rung space, missing therung completely and causing a loss of balance; slip: limb wasplaced on the rung, but slipped off upon weight-bearing) weremeasured for each condition. The number of foot faults was signif-icantly different between heterozygote and wild type mice(t(9) = 8.12, p < 0.001), and heterozygotes and knockouts(t(9) = 5.41, p < 0.001) when 9 rungs were removed at random,but not between knockouts and wild type mice (t(9) = 1.81,p = 0.104). When 16 rungs were removed at random, significantdifferences were noted between heterozygotes and knockout mice(t(9) = 2.28, p = 0.048) and between wild type and knockout mice(t(9) = 2.32, p = 0.045) but not between wild type and heterozygousmice (t(9) = 0.07, p = 0.942). No significant differences were seenwhen all of the rungs were in place, nor, when 13 rungs were re-moved (p > 0.05). All other behavioral assessments failed to showany significant differences among the three groups of animals(p > 0.05). Overall, these data indicate that the absence of syntaxin1A result in only a mild phenotype in viable knockout animals.
We also attempted to breed the homozygous animals. This wassuccessful in one instance, however, the mother eviscerated thepups within hours after birth (interestingly, without devouringthem), and hence a further analysis of homozygous syntaxin 1Aknockout animals was precluded.
At this point, it is not clear if deletion of syntaxin 1A results in atruly embryonic lethal phenotype, or if incompletely developedmice are in fact born alive, but destroyed and eaten by the mother.The notion that only three homozygous embryos could be foundamong 35 embryos dissected from pregnant mothers is inconsis-tent with such a scenario, and suggests increased lethality or lackof development in utero. The fact that four animals did in fact sur-vive may be due to some form of some compensation that mayhave occurred in these four animals, thus resulting in their viabil-ity. Overall, these data suggest that although syntaxin 1A is impor-tant for in utero development, its presence may be less critical laterin life.
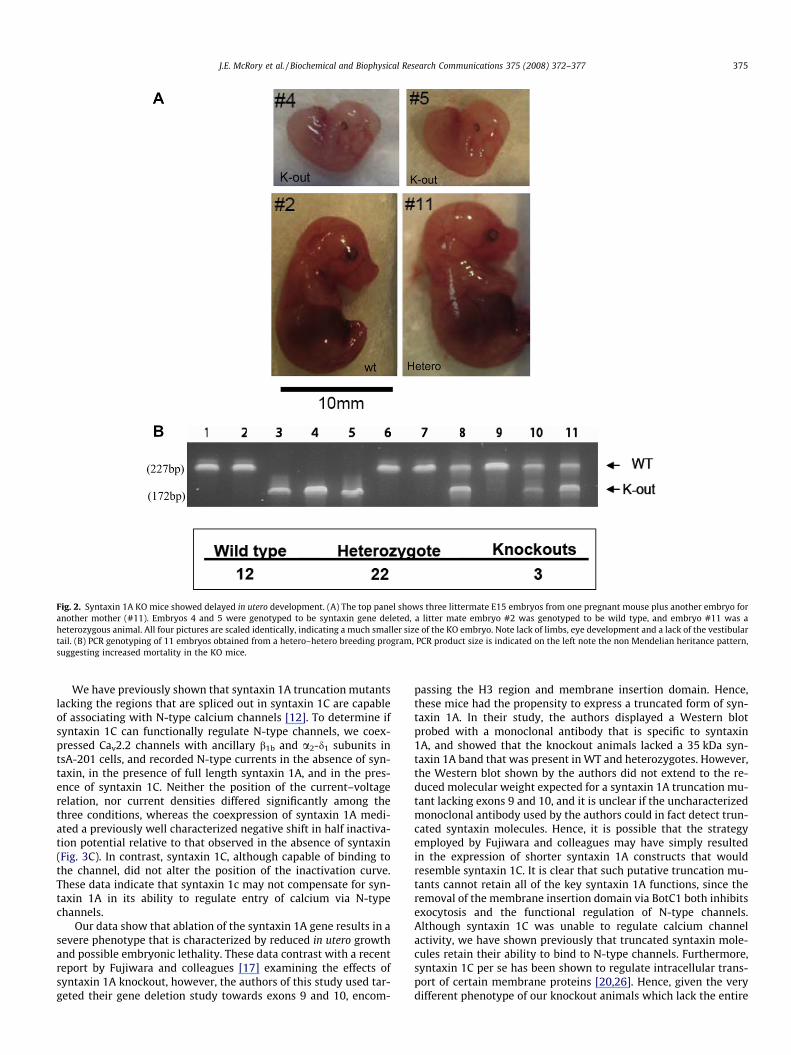
Fig. 2. Syntaxin 1A KO mice showed delayed in utero development. (A) The top panel shows three littermate E15 embryos from one pregnant mouse plus another embryo foranother mother (#11). Embryos 4 and 5 were genotyped to be syntaxin gene deleted, a litter mate embryo #2 was genotyped to be wild type, and embryo #11 was aheterozygous animal. All four pictures are scaled identically, indicating a much smaller size of the KO embryo. Note lack of limbs, eye development and a lack of the vestibulartail. (B) PCR genotyping of 11 embryos obtained from a hetero–hetero breeding program, PCR product size is indicated on the left note the non Mendelian heritance pattern,suggesting increased mortality in the KO mice.
J.E. McRory et al. / Biochemical and Biophysical Research Communications 375 (2008) 372–377 375
We have previously shown that syntaxin 1A truncation mutantslacking the regions that are spliced out in syntaxin 1C are capableof associating with N-type calcium channels [12]. To determine ifsyntaxin 1C can functionally regulate N-type channels, we coex-pressed Cav2.2 channels with ancillary b1b and a2-d1 subunits intsA-201 cells, and recorded N-type currents in the absence of syn-taxin, in the presence of full length syntaxin 1A, and in the pres-ence of syntaxin 1C. Neither the position of the current–voltagerelation, nor current densities differed significantly among thethree conditions, whereas the coexpression of syntaxin 1A medi-ated a previously well characterized negative shift in half inactiva-tion potential relative to that observed in the absence of syntaxin(Fig. 3C). In contrast, syntaxin 1C, although capable of binding tothe channel, did not alter the position of the inactivation curve.These data indicate that syntaxin 1c may not compensate for syn-taxin 1A in its ability to regulate entry of calcium via N-typechannels.
Our data show that ablation of the syntaxin 1A gene results in asevere phenotype that is characterized by reduced in utero growthand possible embryonic lethality. These data contrast with a recentreport by Fujiwara and colleagues [17] examining the effects ofsyntaxin 1A knockout, however, the authors of this study used tar-geted their gene deletion study towards exons 9 and 10, encom-
passing the H3 region and membrane insertion domain. Hence,these mice had the propensity to express a truncated form of syn-taxin 1A. In their study, the authors displayed a Western blotprobed with a monoclonal antibody that is specific to syntaxin1A, and showed that the knockout animals lacked a 35 kDa syn-taxin 1A band that was present in WT and heterozygotes. However,the Western blot shown by the authors did not extend to the re-duced molecular weight expected for a syntaxin 1A truncation mu-tant lacking exons 9 and 10, and it is unclear if the uncharacterizedmonoclonal antibody used by the authors could in fact detect trun-cated syntaxin molecules. Hence, it is possible that the strategyemployed by Fujiwara and colleagues may have simply resultedin the expression of shorter syntaxin 1A constructs that wouldresemble syntaxin 1C. It is clear that such putative truncation mu-tants cannot retain all of the key syntaxin 1A functions, since theremoval of the membrane insertion domain via BotC1 both inhibitsexocytosis and the functional regulation of N-type channels.Although syntaxin 1C was unable to regulate calcium channelactivity, we have shown previously that truncated syntaxin mole-cules retain their ability to bind to N-type channels. Furthermore,syntaxin 1C per se has been shown to regulate intracellular trans-port of certain membrane proteins [20,26]. Hence, given the verydifferent phenotype of our knockout animals which lack the entire
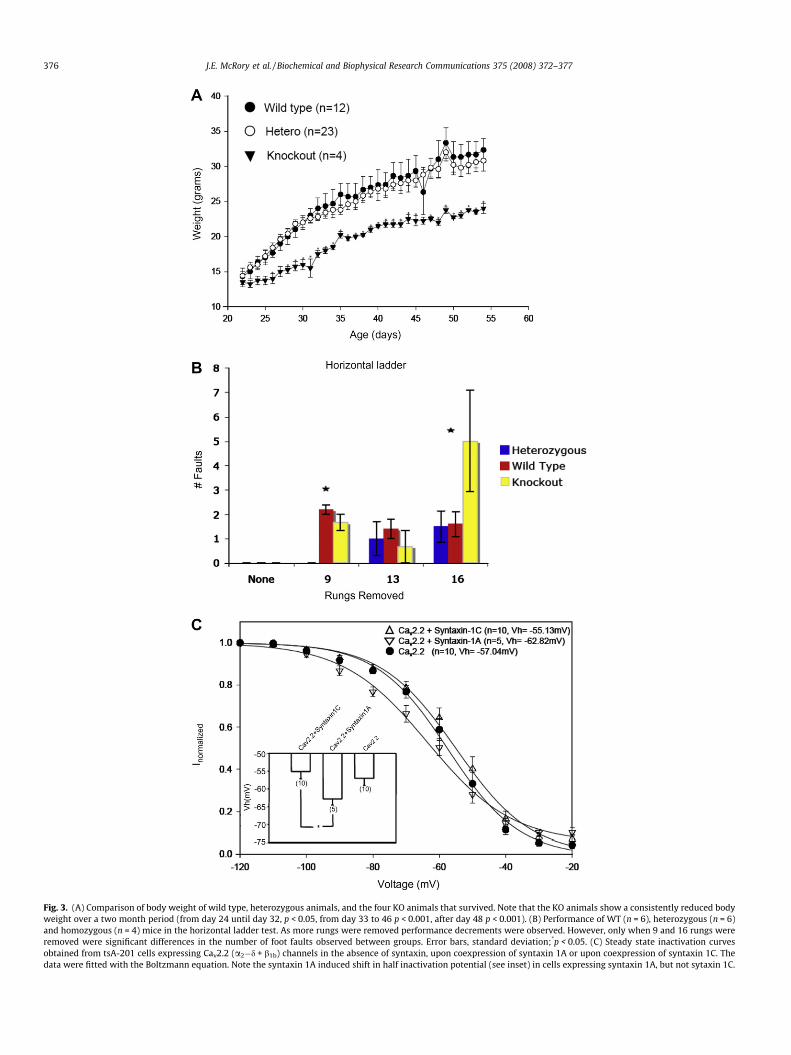
Fig. 3. (A) Comparison of body weight of wild type, heterozygous animals, and the four KO animals that survived. Note that the KO animals show a consistently reduced bodyweight over a two month period (from day 24 until day 32, p < 0.05, from day 33 to 46 p < 0.001, after day 48 p < 0.001). (B) Performance of WT (n = 6), heterozygous (n = 6)and homozygous (n = 4) mice in the horizontal ladder test. As more rungs were removed performance decrements were observed. However, only when 9 and 16 rungs wereremoved were significant differences in the number of foot faults observed between groups. Error bars, standard deviation;*p < 0.05. (C) Steady state inactivation curvesobtained from tsA-201 cells expressing Cav2.2 (a2�d + b1b) channels in the absence of syntaxin, upon coexpression of syntaxin 1A or upon coexpression of syntaxin 1C. Thedata were fitted with the Boltzmann equation. Note the syntaxin 1A induced shift in half inactivation potential (see inset) in cells expressing syntaxin 1A, but not sytaxin 1C.
376 J.E. McRory et al. / Biochemical and Biophysical Research Communications 375 (2008) 372–377

J.E. McRory et al. / Biochemical and Biophysical Research Communications 375 (2008) 372–377 377
syntaxin 1A gene, we speculate that residual syntaxin 1A frag-ments in the exon 9/10 deletion mutant mouse of Fujiwara andcolleagues may be sufficient to maintain some of the key syntaxin1A functions required for appropriate in utero development. Alter-natively, it is possible that other syntaxin isoforms such as syn-taxin 1B may have more effectively compensated in miceexpressing truncated syntaxin 1A proteins.
Overall, our data reveal an important function of syntaxin 1A indevelopment. The notion that vastly different phenotypes are ob-served in two separate knockout mouse studies underscores theimportance to ensure that knockout strategies do not result intruncated versions of a protein of interest.
Acknowledgments
This works was supported by a Grant from the Canadian Insti-tutes of Health Research (CIHR) to GWZ and the Novartis Chairfor Schizophrenia Research. GWZ is a Scientist of the Alberta Her-itage Foundation for Medical Research (AHFMR) and a Canada Re-search Chair. C.J.D. held a Studentship award from the AHFMR anda Canada Graduate Scholarship.
References
[1] M. Linial, Neurotoxins as tools in dissecting the exocytic machinery, SubcellBiochem. 34 (2000) 39–72.
[2] J. Pevsner, S.C. Hsu, J.E. Braun, N. Calakos, A.E. Ting, M.K. Bennett, R.H. Scheller,Specificity and regulation of a synaptic vesicle docking complex, Neuron 13 (2)(1994) 353–361.
[3] J. Rettig, C. Heinemann, U. Ashery, Z.H. Sheng, C.T. Yokoyama, W.A. Catterall, E.Neher, Alteration of Ca2+ dependence of neurotransmitter release bydisruption of Ca2+ channel/syntaxin interaction, J. Neurosci. 17 (17) (1997)6647–6656.
[4] Z.H. Sheng, J. Rettig, T. Cook, W.A. Catterall, Calcium-dependent interaction ofN-type calcium channels with the synaptic core complex, Nature 379 (6564)(1996) 451–454.
[5] Z.H. Sheng, J. Rettig, M. Takahashi, W.A. Catterall, Identification of a syntaxin-binding site on N-type calcium channels, Neuron 13 (6) (1994) 1303–1313.
[6] J.D. Spafford, G.W. Zamponi, Functional interactions between presynapticcalcium channels and the neurotransmitter release machinery, Curr. Opin.Neurobiol. 13 (3) (2003) 308–314.
[7] S. Mochida, Z.H. Sheng, C. Baker, H. Kobayashi, W.A. Catterall, Inhibition ofneurotransmission by peptides containing the synaptic protein interaction siteof N-type Ca2+ channels, Neuron 17 (4) (1996) 781–788.
[8] I. Bezprozvanny, R.H. Scheller, R.W. Tsien, Functional impact of syntaxin ongating of N-type and Q-type calcium channels, Nature 378 (6557) (1995) 623–626.
[9] I. Bezprozvanny, P. Zhong, R.H. Scheller, R.W. Tsien, Molecular determinants ofthe functional interaction between syntaxin and N-type Ca2+ channel gating,Proc. Natl. Acad. Sci. USA 97 (25) (2000) 13943–13948.
[10] S.E. Jarvis, W. Barr, Z.P. Feng, J. Hamid, G.W. Zamponi, Molecular determinantsof syntaxin 1 modulation of N-type calcium channels, J. Biol. Chem. 277 (46)(2002) 44399–44407.
[11] S.E. Jarvis, J.M. Magga, A.M. Beedle, J.E. Braun, G.W. Zamponi, G proteinmodulation of N-type calcium channels is facilitated by physical interactionsbetween syntaxin 1A and Gbetagamma, J. Biol. Chem. 275 (9) (2000) 6388–6394.
[12] S.E. Jarvis, G.W. Zamponi, Distinct molecular determinants govern syntaxin1A-mediated inactivation and G-protein inhibition of N-type calciumchannels, J. Neurosci. 21 (9) (2001) 2939–2948.
[13] E.F. Stanley, Syntaxin I modulation of presynaptic calcium channelinactivation revealed by botulinum toxin C1, Eur. J. Neurosci. 17 (6)(2003) 1303–1305.
[14] I. Dulubova, S. Sugita, S. Hill, M. Hosaka, I. Fernandez, T.C. Sudhof, J. Rizo, Aconformational switch in syntaxin during exocytosis: role of munc18, EMBO J.18 (16) (1999) 4372–4382.
[15] K.M. Misura, R.H. Scheller, W.I. Weis, Three-dimensional structure ofthe neuronal-Sec1-syntaxin 1a complex, Nature 404 (6776) (2000)355–362.
[16] M.A. Poirier, W. Xiao, J.C. Macosko, C. Chan, Y.K. Shin, M.K. Bennett, Thesynaptic SNARE complex is a parallel four-stranded helical bundle, Nat. Struct.Biol. 5 (9) (1998) 765–769.
[17] T. Fujiwara, T. Mishima, T. Kofuji, T. Chiba, K. Tanaka, A. Yamamoto, K.Akagawa, Analysis of knock-out mice to determine the role of HPC-1/syntaxin 1A in expressing synaptic plasticity, J. Neurosci. 26 (21) (2006)5767–5776.
[18] M.N. Jagadish, J.T. Tellam, S.L. Macaulay, K.H. Gough, D.E. James, C.W. Ward,Novel isoform of syntaxin 1 is expressed in mammalian cells, Biochem. J. 321(Pt 1) (1997) 151–156.
[19] T. Nakayama, K. Mikoshiba, T. Yamamori, K. Akagawa, Expression of syntaxin1C, an alternative splice variant of HPC-1/syntaxin 1A, is enhanced by phorbol-ester stimulation in astroglioma: participation of the PKC signaling pathway,FEBS Lett. 536 (1–3) (2003) 209–214.
[20] T. Nakayama, K. Mikoshiba, T. Yamamori, K. Akagawa, Activation of syntaxin1C, an alternative splice variant of HPC-1/syntaxin 1A, by phorbol 12-myristate 13-acetate (PMA) suppresses glucose transport into astrogliomacells via the glucose transporter-1 (GLUT-1), J. Biol. Chem. 279 (22) (2004)23728–23739.
[21] S. Irwin, Comprehensive observational assessment: Ia. A systematic,quantitative procedure for assessing the behavioral and physiologic state ofthe mouse, Psychopharmacologia 13 (3) (1968) 222–257.
[22] P. Sgado, L. Alberi, D. Gherbassi, S.L. Galasso, G.M. Ramakers, K.N. Alavian, M.P.Smidt, R.H. Dyck, H.H. Simon, Slow progressive degeneration of nigraldopaminergic neurons in postnatal engrailed mutant mice, Proc. Natl. Acad.Sci. USA 103 (41) (2006) 15242–15247.
[23] J.L. Tillerson, G.W. Miller, Grid performance test to measure behavioralimpairment in the MPTP-treated-mouse model of Parkinsonism, J. Neurosci.Methods 123 (2) (2003) 189–200.
[24] J.R. Ison, D.W. McAdam, G.R. Hammond, Latency and amplitude changes in theacoustic startle reflex of the rat produced by variation in auditoryprestimulation, Physiol. Behav. 10 (6) (1973) 1035–1039.
[25] R. Morris, Developments of a water-maze procedure for studying spatiallearning in the rat, J. Neurosci. Methods 11 (1) (1984) 47–60.
[26] K. Suga, T. Yamamori, K. Akagawa, Identification of the carboxyl-terminalmembrane-anchoring region of HPC-1/syntaxin 1A with the substituted-cysteine-accessibility method and monoclonal antibodies, J. Biochem. (Tokyo)133 (3) (2003) 325–334.




















