![Djelovanje don Šime Ljubića u Arheološkom odjelu Narodnog muzeja u Zagrebu [Don Šime Ljubić’s activity at the Department of Archaeology of the National Museum in Zagreb.]](https://static.fdokumen.com/doc/165x107/6343ffb3f474639c9b042bbc/djelovanje-don-sime-ljubica-u-arheoloskom-odjelu-narodnog-muzeja-u-zagrebu-don.jpg)
SVEUČILIŠTE U ZAGREBU FAKULTET KEMIJSKOG INŽENJERSTVA I TEHNOLOGIJE ZAVOD ZA ANALITIČKU KEMIJU
73
SVEUČILIŠTE U ZAGREBU FAKULTET KEMIJSKOG INŽENJERSTVA I TEHNOLOGIJE ZAVOD ZA ANALITIČKU KEMIJU A. J. M. Horvat, K. Margeta INSTRUMENTALNA ANALIZA Radni materijal za internu uporabu Zagreb, 2009. analitičar instrument ili proces korisničko sučelje opis (znanje upravljanje na nivou analitičara ugrađeno kontrola dijagnostika inteligentni procesor izlazni uređaj obrada podataka sirovi obrađeni podaci upravljanje računalo komande na razini
Transcript of SVEUČILIŠTE U ZAGREBU FAKULTET KEMIJSKOG INŽENJERSTVA I TEHNOLOGIJE ZAVOD ZA ANALITIČKU KEMIJU

SVEUČILIŠTE U ZAGREBU
FAKULTET KEMIJSKOG INŽENJERSTVA I TEHNOLOGIJE ZAVOD ZA ANALITIČKU KEMIJU
A. J. M. Horvat, K. Margeta
INSTRUMENTALNA ANALIZA
Radni materijal za internu uporabu
Zagreb, 2009.
analitičar
instrument ili proces
korisničko sučelje
opis (znanje
upravljanje na nivou analitičara
ugrađeno
kontrola dijagnostika
inteligentni procesor
izlazni uređaj
obrada podataka
sirovi
obrađeni podaciupravljanje
računalo
komande na razini

___________________________________________________________________________
___________________________________________________________________________

___________________________________________________________________________
___________________________________________________________________________
Radni materijal namijenjen je studentima slijedećih kolegija: INSTRUMENTALNA ANALITIČKA KEMIJA na dodiplomskim studiju Primijenjena kemija, PROCESNA I INSTRUMENTALNA ANALIZA na dodiplomskim studiju Kemijskog inženjerstva, KARAKTERIZACIJA MATERIJALA na dodiplomskim studiju Kemija i inženjerstvo materijala.

___________________________________________________________________________
___________________________________________________________________________

___________________________________________________________________________
___________________________________________________________________________
Sadržaj Predgovor Literatura 1. Uvod ..................................................................................................................................1 1.1. Vrste analitičkih signala...................................................................................................1 1.2. Instrumenti i uređaji .........................................................................................................2 1.3. Kalibracijski postupci .......................................................................................................3 2. Spektrometrije .................................................................................................................8 2.1. Atomske spektrometrije.................................................................................................13 2.1.1. Atomska apsorpcijska spektrometrija.........................................................................13 Laboratorijska vježba : Određivanje Cu ..................................................................14 2.1.2. Atomska emisijska spektrometrija ..............................................................................18 2.1.2.1. Plamena spektrometrija (Spektrometrija emisije EMZ) ..........................................18 Laboratorijska vježba : Određivanje Na+ i K+ iona plamenom spektrometrijom. ......19
2.2. Molekulske spektrometrije .............................................................................................23 2.2.1. Spektrometrija molekulske apsorpcije UV i/ili VID EMZ (spektrofotometrija) .............23 Laboratorijska vježba : Određivanje Fe(III), Ni(II), Cr(VI) ili NO3
- iona .....................24 2.2.2. Turbidimetrija i nefelometrija (Spektrometrija raspršenja EMZ) .................................28 Laboratorijska vježba : Određivanje sulfata u vodovodnoj vodi ...............................30 3. Elektroanalitičke metode ...............................................................................................32 3.1. Potenciometrija..............................................................................................................32 3.1.1. Potenciometrijska titracija...........................................................................................35 Laboratorijska vježba : Određivanje acetilsalicilne kiseline u tableti aspirina ..........35 3.1.2. Direktna potenciometrija.............................................................................................39 Laboratorijska vježba : Određivanje koncentracije iona u uzorku ion selektivnim elektrodama...................................................................... ..40 3.2. Konduktometrija ...........................................................................................................45 3.2.1. Konduktometrijska titracija ........................................................................................50 Laboratorijska vježba : Određivanje koncentracije slabe i jake kiseline u smjesi ....50 4. Instrumentalne metode separacije ..............................................................................52 4.1. Kromatografije ..............................................................................................................52 4.1.1. Plinska kromatografija ................................................................................................56 Laboratorijska vježba : Određivanje smjese eteričnih ulja .......................................57 5. Dodatak ..........................................................................................................................61 Upute za rad u laboratoriju ...................................................................................................61 Pisanje laboratorijskih izvještaja...........................................................................................62 Naslovna stranica laboratorijskog izvještaja.........................................................................63

___________________________________________________________________________________________
___________________________________________________________________________

___________________________________________________________________________________________
___________________________________________________________________________
Prije početka!
Ovaj pisani materijal namijenjen je studentima 2. i 3. godine studija na Fakultetu kemijskog inženjerstva i tehnologije i trebao bi biti pomoć pri izradi vježbi i spremanju kolokvija i ispita iz predmeta u kojima se obrađuju metode instrumentalne analize.
Preduvjet za uspješno savladavanje ovog gradiva su odslušana predavanja i završene vježbe iz Fizike, Analitičke kemije, Opće i anorganske kemije te Fizikalne kemije. Elektrokemija je također vrlo važna za razumijevanje, no ne predaje se na svim Studijima ovog Fakulteta.
Obzirom da se računanje i obrada podataka analize u analitičkoj kemiji ne može nikako izbjeći, Matematika je vrlo važan kolegij i određeno predznanje iz matematike je nužno. Također je i matematička statistika (Primijenjena matematika) alat koji je nezaobilazan za jednog analitičkog kemičara bez obzira kojom se metodom određivanja koristi.
I konačno u današnje vrijeme opće kompjuterizacije osnovna računalna pismenost uz znanje engleskog jezika nužan su preduvjet za uspješan rad. Naravno nešto od toga učit ćemo u hodu i nadam se savladati, a lavovski dio čeka vas na vašem budućem radnom mjestu i morat ćete se pošteno potruditi jer konkurencija je sve veća.
Instrumentalne metode osim toga nisu samo u funkciji određivanja sastava uzorka čime se bavi analitička kemija, nego i određivanja strukture npr. organskih spojeva čime se bave organski kemičari. Instrumentalnim metodama određuje se kinetika reakcija što je područje fizikalne kemije, određuju se konstante stabilnosti kompleksa što je područje tzv. koordinacijske kemije, itd. Instrumentalna analiza (Instrumentalna kemijska analiza), mogli bismo reći u užem smislu, obuhvaća dio instrumentalnih metoda koje se odnose na određivanje sastava i strukture.
Cilj ovog kolegija je dobivanje jednog općeg pregleda o metodama i postupcima koji će jednom studentu, budućem kemijskom inženjeru, sutra možda znanstveniku, nastavniku ili voditelju industrijskog laboratorija pomoći u određenim trenucima: kako riješiti problem koji je pred njega postavljen, koju literaturu konsultirati, koju metodu odabrati da bi konačna informacija, koju treba vjerojatno proslijediti dalje, bila što potpunija. Izbor metode ovisit će tada o znanju o osnovnim principima raznih metoda, o saznanjima o njihovim prednostima i ograničenjima.
Na samom početku odmah iza uvoda popis je literature dostupne na hrvatskom jeziku, prema poglavljima, kojom se možete poslužiti da bi dobili više informacija o metodama koje ćemo upoznati u praktikumu.
U ovom pisanom materijalu onaj dio teksta koji se odnosi na sva ili više područja uvijek prethodi područjima i metodama određivanja. Tako na samom početku par je riječi o građi instrumenata koliko se to odnosi generalno na sve instrumente a zatim slijede postupci kalibracije.
Nadam se da će vam ovaj pisani materijal, koji ću nadam se kroz godine koje slijede nadopunjavati, olakšati učenje za kolokvij ili ispit koji je pred vama, za ispit koji obuhvaća tako mnogo starih znanja, ali isto tako i dosta novoga te zahtijeva da se povežu sva ta znanja. Autori Zavod za analitičku kemiju U Zagrebu, listopada 2009.

LITERATURA
___________________________________________________________________________

LITERATURA
___________________________________________________________________________
LITERATURA
Popis literature koji slijedi je popis literature dostupne na hrvatskom jeziku o instrumentalnim metodama. Popis sadrži kako noviju tako i stariju literaturu. Literatura koju bi valjalo koristiti tokom praktikuma iz kolegija Procesna i instrumentalna analiza kao i za pripremu ispita označena je podebljanim slovima. I. UVOD U ANALITIČKU KEMIJU I STATISTIČKA OBRADA PODATAKA 1. M. Kaštelan-Macan, Kemijska analiza u sustavu kvalitete, Školska knjiga Zagreb
2003. 2. B. Petz, Osnovne statističke metode za nematematičare, Udžbenici Sveučilišta u
Zagrebu, 4. izdanje, Naklada Slap Jastrebarsko, 2002.
II. UVOD U INSTRUMENTALNU ANALIZU 1. V. Grdinić, Instrumentalne metode analitičke kemije, Kalibracija i standardizacija, u
Tehnička enciklopedija, Sv. 6, Leksikografski Zavod "Miroslav Krleža", Zagreb 1979., str. 495-496.
III. SPEKTROMETRIJSKE METODE 1. D. A. Skoog, D. M. West, F. J. Holler, Osnove analitičke kemije, 6. izdanje
(englesko), Školska knjiga, Zagreb 1999., str. 489-620. 2. S. H. Pine, Organska kemija, poglavlje Spektroskopske metode, Školska knjiga, Zagreb
1994., str. 1062-1132.
3. Da. Maljković, Spektrometrija, u Tehnička enciklopedija, Sv. 12, Leksikografski Zavod "Miroslav Krleža", Zagreb 1992., str. 150-178.
4. M. Laćan, M. Šuprina, Spektrometrijske metode u organskoj kemiji, Sveučilišna naklada, Zagreb 1976.
IV. ELEKTROANALITIČKE METODE 1. D. A. Skoog, D. M. West, F. J. Holler, Osnove analitičke kemije, 6. izdanje (englesko),
Školska knjiga, Zagreb 1999.
2. I. Piljac, Elektroanalitičke metode, RMC, Zagreb 1995. 3. Poglavlja I-VI u I. Filipović, P. Sabioncello (ur.). Laboratorijski priručnik, I dio - knjiga 2.,
Tehnička knjiga, Zagreb 1978.
4. Z. Štefanac, Instumentalne metode analitičke kemije, Elektrokemijske metode, u Tehnička enciklopedija, Sv. 6, Leksikografski Zavod "Miroslav Krleža", Zagreb 1979., str. 496-501.
V. INSTRUMENTALNE METODE SEPARACIJE – Kromatografija i elektroforeza 1. I. Piljac, Elektroforeza, Media Print, Zagreb 2006. 2. D. A. Skoog, D. M. West, F. J. Holler, Osnove analitičke kemije, 1. izd. (6. englesko
izdanje), Školska knjiga, Zagreb 1999., str. 645-716
3. Đ. Deur-Šiftar, D. Štefanović, Z. Šoljić, Kromatografija, Tehnička enciklopedija, Svezak 7., Leksikografski Zavod, Zagreb 1992., str. 387-395

INSTRUMENTALNA ANALIZA
___________________________________________________________________________
4. S. Turina, T. Bićan-Fišter, B. Starčević, Đ. Deur-Šiftar, Kromatografska analiza, poglavlje VII, u I. Filipović, P. Sabioncello (ur.). Laboratorijski priručnik, I dio - knjiga 2., Tehnička knjiga, Zagreb 1978., str. 485-601.
VI. OSTALE METODE 1. D. A. Skoog, D. M. West, F. J. Holler, Osnove analitičke kemije, 1. izd. (6. englesko
izdanje), Školska knjiga, Zagreb 1999.
2. Z. Štefanac, Instrumentalne metode analitičke kemije, Termokemijske metode, u Tehnička enciklopedija, Sv. 6., Leksikografski Zavod "Miroslav Krleža", Zagreb 1979., str. 506-509.
3. R. Despotović, M.J. Herak, M. Mirnik, P. Strohal, M. Vlarkovič. Radiometrijska analiza, poglavlje IX, u I. Filipović, P. Sabioncello (ur.). Laboratorijski priručnik, I dio - knjiga 2., Tehnička knjiga, Zagreb 1978., str. 649-713.
4. P. Sabioncello, Kalorimetrijska mjerenja, poglavlje X, u I. Filipović, P. Sabioncello (ur.). Laboratorijski priručnik, I dio - knjiga 2., Tehnička knjiga, Zagreb 1978., str. 714-728.
5. M. Vlatković, Radiokemija i radionuklidi, u Tehnička enciklopedija, Sv. 11., Leksikografski Zavod "Miroslav Krleža", Zagreb
VII. OSTALO 1. Vlatko Silobrčić, Kako sastaviti, objaviti i ocijeniti znanstveno djelo, 5. dopunjeno izdanje,
Medicinska naklada, Zagreb 2003.

1. UVOD
___________________________________________________________________________ 1
1. UVOD
Instrumentalne metode obuhvaćaju veliki broj vrlo raznih metoda i postupaka. Tu su prvenstveno spektrometrijske metode koje se baziraju na interakciji uzorka i energije a kao posljedicu interakcije mjerimo elektromagnetsko zračenje ili zračenje raznih čestica (elektrona, protona, iona), zatim elektroanalitičke metode kod kojih je ili signal pobude ili signal odziva (ili oba) električna veličina, radiokemijske metode, termičke (toplinske) metode (termometrijske), metode separacije koje obuhvaćaju kromatografije i elektroforeze.
Kao predmet u nastavi Instrumentalne metode trebaju biti okrenute ne samo razumijevanju građe pojedinog instrumenta nego i primjeni u razrješavanju kemijskih problema. Dobro razumijevanje instrumentalnih metoda zahtjeva poznavanje fizikalnih principa na kojima se osnivaju, njihova ograničenja kako bi se najbolje koristile u rješavanju zadanih problema.
Samo će takvim pristupom školovani kemičar ili kemijski inženjer znati odabrati metodu s kojom će na zadovoljavajući način riješiti zadatak koji je postavljen.
Prije nego se upustimo u avanturu otkrivanja već poznatog i ponešto nepoznatog trebalo bi ponoviti pojmove iz Osnova analitičke kemije i Analitičke kemije I i II kao što su: analit, uzorak, matrica, kvalitativna analiza, kvantitativna analiza, signal, slijepa proba, selektivni reagens, specifični reagens, standard, standardna otopina, preciznost, točnost, donja granica identifikacije, donja granica određivanja, validacija.
Cilj analitičke kemije je dobivanje informacija o kemijskom sastavu (kvantitativnom i kvalitativnom), strukturi i razdiobi ispitivanog materijala. Kemijska analiza proces je dobivanja tih informacija.
Kako informaciju o kemijskom sastavu nije moguće izravno mjeriti, mjeri se neko svojstvo, ponekad nazvano analitičkim svojstvom, koje je usko povezano sa strukturom ili sastavom ispitivanog uzorka.
1.1. VRSTE ANALITIČKIH SIGNALA
Kemijska analiza daje informaciju mjerenjem fizičkog svojstva koje je karakteristično u odnosu na analit. Takovo svojstvo pretvara se u signal i naziva se analitički signal. U tablici 1.1. navedeni su neki signali koji se mogu koristiti u analitičke svrhe.
Navedene analitička metoda na bazi mjerenog signala samo su dio metoda i postupaka koji su mogu naći u sve obimnijoj literaturi i svakim danom broj postupaka unutar pojedinih metoda se enormno povećava.
Instrumenti su alat današnje eksperimentalne kemije i s jednakim se uspjehom koriste u istraživanju, analizi i/ili nastavi. Instrumentalne metode jednako su važne u određivanju sastava i strukture kao i u istraživanju kinetike reakcija, ispitivanju površina ili u procesnoj kontroli.
Instrumentalna analiza obuhvaća onaj dio kemijske analize koji uz pomoć više ili manje sofisticiranih instrumenata daje podatke o kemijskom sastavu i strukturi tvari na temelju separacije, detekcije i mjerenja energetskih promjena što se događaju u atomnim jezgrama, atomnom elektronskom omotaču ili u molekulama.

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 2
Tablica 1.1. Neki analitički signali1
Signal Analitička metoda na bazi mjerenog signala masa gravimetrijska analiza volumen volumetrijska analiza emisija zračenja emisijske spektrometrije apsorpcija zračenja apsorpcijske spektrometrije raspršivanje zračenja turbidimetrija, nefelometrija, Ramanova spektrometrija refleksija zračenja refraktometrija, interferometrija difrakcija zračenja difrakcija rendgenskog zračenja, elektronska difrakcija polarizacija (zakretanje) zračenja
polarimetrija, optička zakretna disperzija
električni potencijal Potenciometrija, kronopotenciometrija električna struja polarografija, amperometrija, kulometrija električni otpor Konduktometrija odnos mase i naboja masena spektrometrija red reakcije kinetičke metode termička svojstva TGA, DTGA, DSC, termička vodljivost i određivanje entalpije
1.2. INSTRUMENTI I UREĐAJI
U najširem smislu uređaji za instrumentalnu analizu pretvaraju signal koji se uglavnom ne može direktno detektirati i nije razumljiv u signal koji to jest. Oni su veza između sustava koji se proučava i onoga (znanstvenika, inženjera u pogonu ili studenta) koji sustav proučava.
Slika 1.1. Komponente tipičnog instrumenta

1. UVOD
___________________________________________________________________________ 3
Bez obzira o kojem se instrumentu radi, temeljni sklop sadrži samo četiri osnovne
komponente (slika 1.1.): izvor signala, ulazni pretvornik ili detektor, procesor signala i izlazni pretvornik tj. jedinicu za očitavanje.
1
Izvor signala. Izvor signala daje analitički signal pojedinih sastojaka uzorka. Izvor signala može biti sam uzorak. Na primjer signal za analitičku vagu je sama masa uzorka, za pH-metar signal je aktivitet vodikovih iona u otopini. No kod mnogih instrumenata izvor signala nije tako jednostavan. Čine ga izvor energije i uzorak koji u interakciji daju analitički signal.
Ulazni pretvornik ili detektor. Pretvornik je uređaj koji pretvara jedan tip signala u drugi, na primjer termopar pretvara toplinski signal u električni signal. Najveći broj pretvornika koje ćemo spominjati pretvara analitički signal u električni. Pretvoreni analitički signal u električni ili mehanički ulazi dalje u procesor signala.
Procesori signala. Procesor signala modificira signal na taj način da se može jednostavnije i brže obrađivati u jedinici za zapis i eventualno obradu podataka. Najčešća modifikacija je pojačanje signala množenjem s veličinom većom od jedinice. Električni signal može se pojačati čak za faktor 106. Razne su druge modifikacije električnog signala moguće: integriranje, diferenciranje, oduzimanje ili dodavanje, itd.
Izlazni pretvornik ili jedinica za očitavanje. Moderni instrumenti danas kao jedinicu za očitavanje imaju računalo. Mi ćemo se tokom našeg rada u laboratoriju nažalost (ili na sreću) susresti s nizom uređaja starije generacije koji kao jedinicu za očitavanje imaju uređaj sa skalom (analogni prikaz), pisač s perom ili uređaj s digitalnim zapisom.
1.3. KALIBRACIJSKI POSTUPCI U svrhu kvantitativnog određivanja sastava uzorka metode instrumentalnih određivanja
koncentracije analita mogu biti apsolutne, tj. ne zahtijevaju kalibraciju da bi se dobio podatak o koncentraciji iz izlaznog signala instrumenta. Osjetljivost određivanja može se odrediti direktno iz teoretske ovisnosti signala i koncentracije analita. Takove su metode npr. kulometrija i kulometrijska titracija ili elektrogravimetrija.
Sve ostale instrumentalne metode su relativne i time zahtijevaju kalibraciju. Tri su postupka za određivanje koncentracije iz podataka izlaznog signala bilo kojeg instrumenta.
2
1.3.1. Metoda vanjskog standarda
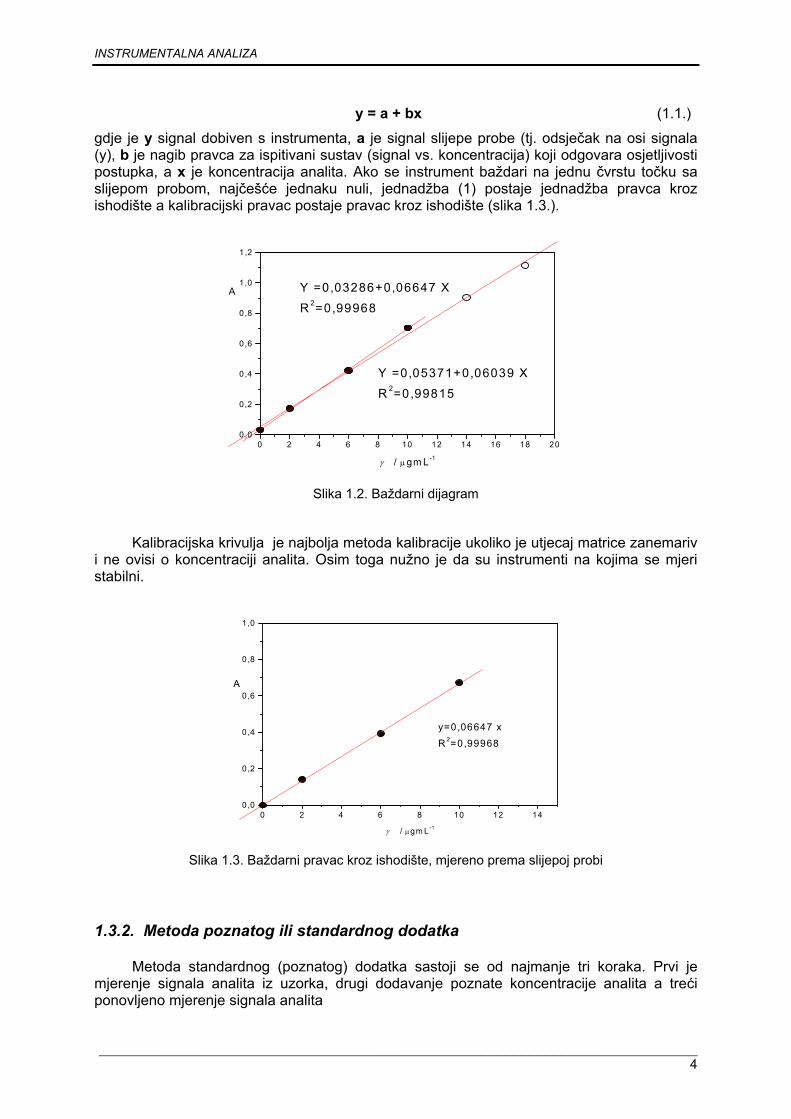
Kalibracijski, baždarni ili radni dijagram prikazuje ovisnost signala prema količini analita (npr. koncentraciji) dobiven mjerenjem signala serije standarda (standardnih otopina) pri točno određenim uvjetima.
Kalibracijski dijagrami nisu uvijek linearni, često su kod niskih ili visokih koncentracija krivulje a linearni je dio negdje u sredini takovog dijagrama (slika 1.2.). Takav linearni dio odnosa signala prema koncentraciji često nazivamo dinamičkim područjem. Dinamički dio kalibracijske krivulje može prikazati jednadžbom pravca.
1 D. A. Skoog, Principles of Instrumental Analysis, 3rd Ed., Saunders College Pub. Int. Ed.,Philadelphia 1985.
2 M. Kaštelan-Macan, Kemijska analiza u sustavu kvalitete, Kemijska analiza u sustavu kvalitete, Školska knjiga Zagreb 2003.
INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 4
y = a + bx (1.1.)
gdje je y signal dobiven s instrumenta, a je signal slijepe probe (tj. odsječak na osi signala (y), b je nagib pravca za ispitivani sustav (signal vs. koncentracija) koji odgovara osjetljivosti postupka, a x je koncentracija analita. Ako se instrument baždari na jednu čvrstu točku sa slijepom probom, najčešće jednaku nuli, jednadžba (1) postaje jednadžba pravca kroz ishodište a kalibracijski pravac postaje pravac kroz ishodište (slika 1.3.).
0 2 4 6 8 10 12 14 16 18 200,0
0,2
0,4
0,6
0,8
1,0
1,2
A
γ / μgm L-1
Y =0,05371+0,06039 X
R 2=0,99815
Y =0,03286+0,06647 X
R 2=0,99968
Slika 1.2. Baždarni dijagram
Kalibracijska krivulja je najbolja metoda kalibracije ukoliko je utjecaj matrice zanemariv i ne ovisi o koncentraciji analita. Osim toga nužno je da su instrumenti na kojima se mjeri stabilni.
0 2 4 6 8 10 12 140,0
0,2
0,4
0,6
0,8
1,0
y=0,06647 x
R 2=0,99968
A
γ / μgm L -1
Slika 1.3. Baždarni pravac kroz ishodište, mjereno prema slijepoj probi
1.3.2. Metoda poznatog ili standardnog dodatka
Metoda standardnog (poznatog) dodatka sastoji se od najmanje tri koraka. Prvi je mjerenje signala analita iz uzorka, drugi dodavanje poznate koncentracije analita a treći ponovljeno mjerenje signala analita

1. UVOD
___________________________________________________________________________ 5
Sa samo jednim dodatkom poznate koncentracije analita (drugi i treći korak) nužna je
pretpostavka da smo u linearnom dijelu odnosa signala i koncentracije analita. Slijedeći dodaci standardne otopine analita omogućuju veću preciznost i dokazuju ili opovrgavaju našu pretpostavku o linearnosti odziva prema koncentraciji.
Najprecizniji rezultati se dobiju ako je koncentracija prvog dodatka jednaka najmanje dvostrukoj koncentraciji analita u uzorku.
Metoda standardnog dodatka je posebno korisna kada je utjecaj matrice na određivanje analita znatan ili kada su koncentracije analita u uzorku vrlo blizu granice određivanja. Metodom standardnog dodatka utjecaj matrice znači postoji ali je konstantan za sva mjerenja. I kod ovog postupka kalibracije važna je stabilnost mjernog instrumenta.
Kalibracija metodom standardnog dodatka prikazana je na primjeru mjerenja apsorbancije (A) no na isti se način primjenjuje pri mjerenju bilo kojeg signala (npr. potencijala u direktnoj potenciometriji ili otklona galvanometra kod plamene spektrometrije).
Iz grafičkog prikaza (slika 1.4.) koncentracija analita u nepoznatom uzorku očita se kao apsolutna vrijednost na osi x u četvrtom kvadrantu (negativni dio osi x).
Tablica 1.2. Određivanje koncentracije analita u uzorku metodom standardnog dodatka
Volumen dodane SO mL
apsorbancija A
koncentracija dodane otopine μgmL -1
uzorak A uzorak
+ 1. dodatak V1 A1
γ 1= V SO SO
V ukupni1( ) ( )
( )⋅γ
(1.2.)
uzorak + 2. dodatak
V2 A2 γ 2 =
V SO SOV ukupni
2 ( ) ( )( )
⋅γ (1.3.)
SO = standardna otopina V1= 1. dodatak standardne otopine u mL V2= 2. dodatak standardne otopine u mL γ(SO) = masena koncentracija standardne otopine γ = masena koncentracija analita u nepoznatom uzorku γ 1 = masena koncentracija otopine za dodani volumen V1 γ 2 = masena koncentracija. otopine za dodani volumen V2 A = apsorbancija za nepoznatu otopinu uzorka A1= apsorbancija uz 1. dodatak standardne otopine analita A2= apsorbancija uz 2. dodatak standardne otopine analita
-3 -2 -1 0 1 2 3 4
0 ,2
0 ,4
0 ,6
γ 2γ 1γ (C u 2 + ) / μg m L -1
A
Slika 1.4. Grafički prikaz odnosa γ (Cu2+) μgmL-1 prema apsorbanciji za metodu standardnog dodatka

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 6
Metodom standardnog dodatka iz sličnosti trokuta moguće je izračunati koncentraciju
analita u nepoznatom uzorku:
tg α = A Aγ γ γ γ γ
= A+
1
1=
+2
2 (1.4.)
γ γ' =×−
1
1
AA A
γ γ' ' =×−
2
2
AA A
γγ γ
=+' ' '
2 (1.5.)
γ ' i γ '' = masene koncentracije analita nepoznate otopine uzorka iz dva mjerenja γ = masena koncentracija analita u nepoznatom uzorku (srednja vrijednost dva mjerenja uz
standardni dodatak) 1.3.3. Metoda unutarnjeg standarda
Unutarnji standard je tvar dodana uzorku, slijepoj probi i svim standardima (standardnim otopinama) a koncentracija unutarnjeg standarda je konstantna i poznata. Unutarnji standard je tvar potpuno različita od analita.
Ova metoda se koristi kada je osjetljivost instrumentalne metode promjenjiva s vremenom, kada je moguć gubitak uzorka za vrijeme određivanja i kod nestabilnog rada instrumenta. Uglavnom kada se količina uzorka i signal instrumenta mijenjaju od mjerenja do mjerenja iz nekog razloga koje je teško ili nemoguće kontrolirati. Često se upotrebljava u kromatografskim određivanjima, emisijskim spektrometrijama (spektrografiji, plamenoj spektro-metriji), rendgenskoj difrakciji.
Signal unutarnjeg standarda se mjeri istovremeno sa signalom analita. Priprema se poznata smjesa unutarnjeg standarda i analita za mjerenje relativnog signala detektora.
Slika 1.5. Kromatogram uz dodatak unutarnjeg standarda; A analit, B unutarnji standard
Na kromatogramu (slika 1.5.) površine ispod krivulja za tvari A (analit) i B (unutarnji standard) proporcionalne su koncentracijama. Detektor pak daje najčešće različit odziv za svaku komponentu. Primjerice ako je koncentracija obje tvari , analita i unutarnjeg standarda ista (5 µg/mL), površina ispod krivulja ne mora biti ista. Odnos površina ispod krivulja (ili bilo kojeg signala pri mjerenju) za iste koncentracije analita i unutarnjeg standarda naziva se omjer odziva (F):
F = signalA/signalB (1.6.)
vrijme (mim)
B
A

1. UVOD
___________________________________________________________________________ 7
odnosno uključujući koncentracije analita i unutarnjeg standarda:
signalA/signalB = F x [A]/[B] (1.7.)
odnosa signala analita prema signalu unutarnjeg standarda može se prikazti i grafički prema odnosu koncentracije analita i unutarnjeg standarda kao kod kalibracijske krivulje (slika 1.2.). Omjer odziva je u tom slučaju nagib pravca i daje osjetljivost postupka.
S istim unutarnjim standardom moguće je u jednom kromatografskom određivanju odrediti koncentraciju svih separiranih komponenti uz prethodno određivanje omjera odziva za svaku komponentu smjese.
0,1 0,2 0,3 0,4 0,5 0,6 0,70
2
4
6
y=-0,0621+11,213*x
omjer koncentracija [A]/[B]
omje
r sig
nala
(SA /
SB)
Slika 1.6. Kalibracija metodom unutarnjeg standarda
Ako je signal s detektora jednak za analit i unutarnji standard za istu koncentraciju oba, omjer odziva je 1.
Literatura 1. M. Kaštelan-Macan, Kemijska analiza u sustavu kvalitete, Školska knjiga, Zagreb 2003.,
str. 241-263.

INSTRUMENTALNA ANALIZA
_________________________________________________________________________________ 8
2. SPEKTROMETRIJE
Spektrometrije čine dio instrumentalnih metoda i postupaka kojima se mogu dobiti informacije o kemijskom sastavu i strukturi tvari na temelju separacije, detekcije i mjerenja energetskih promjena što se događaju u atomnim jezgrama, atomnom elektronskom omotaču ili u molekulama kao posljedica interakcije s energijom (Da. Maljković, [1
3]. Ta energija može biti
energija zračenja (elektrona, iona ili elektromagnetskog zračenja), toplinska, električna ili kemijska, a mi pratimo posljedicu te interakcije. Govoreći u najširim okvirima u pozadini svih spektrometrijskih tehnika, kao i svih instrumentalnih tehnika općenito, je interakcija energije i uzorka (slika 2.1.). Interakcija s uzorkom može se odvijati na atomnoj ili molekulskoj razini pa govorimo o atomskim odnosno molekulskim spektrometrijama.
Naziv spektrometrija može nositi svaki postupak mjerenja spektra tj. mjerenje intenziteta zračenja ovisno o energiji, valnoj duljini ili frekvenciji zračenja.
Povijest spektrometrije Tablica 2.1 Neke godine i ljudi važni za razvoj spektrometrija [1,2] prva polovica XVII st.
Marko Antonije de Dominis
Znanstveno objasnio nastajanje duge lomom sunčeve svjetlosti na kapljicama vode
1666 I. Newton zaključio da se bijela svijetlost sastoji od svjetlosnih zraka koje se razlikuju po boji a boja je u vezi s indeksom loma
1762 A.S. Marggraf prvi kemijski doprinos, zapaža da natrij i kalij različito boje plamen
1800 W. Herschel zaključuje da postoji nevidljivi dio spektra koji se nadovezuje na crveni dio vidljivog spektra (IC)
1801 J.W. Ritter otkriva UV zračenje na osnovi djelovanja svijetla na AgCl
J. Fraunhofer razvoj optičkih elemenata i proučavanje spektra
1825 W.H.F. Talbot povezuje prisutnost nekog spoja s pojavom linije u spektru
1838 W.A. Lampadius
objavio kolorimetrijsku metodu određivanja željeza i nikla u kobaltnoj rudi koja se osniva na usporedbi boja uzorka i standarda
sredina XIX st. otkrivena identičnost linija Sunčevog spektra i
emisijskih linija u plamenu
1852 A. Beer formulira zakon koji predstavlja temelj apsorpcijske spektrometrije
1853 A. Müller konstruirao prvi kolorimetar
1859 G.R Kirchhoff i R.W. Bunsen prvi upotrebljavaju spektroskop
1866 J.F. Bahr i R.W. Bunsen
apsorpcijsku spektrometriju prvi primijenili za kvantitativnu analizu
1866
otkriveni novi kemijski elementi pomoću spektroskopa, određene valne dužine karakterističnih linija elemenata u emisijskom spektru
1870 C. Vierordt prvi apsorpcijski spektrometar
3 Da. Maljković, Spektrometrije, Tehnička enciklopedija, Svezak 12, Leksikografski Zavod Miroslav Krleža, Zagreb 1992, str.150-178.
Koja je razlika između pojmova spektrometrija i spektroskopija? Pitanje postavila: Marica: [email protected] Može se uočiti da se pojam spektrometija i spektroskopija miješaju. Razlozi su u dobroj mjeri povijesni. Kao što je poznato, a o tome se može pročitati u poglavlju "povijest spektrometrije", prvi počeci ove grane znanosti imaju temelj u promatranju sunčevog spektra, te spektara različitih plamenova. Prvi instrument korišten u tu svrhu je spektroskop koji je konstruirao G. Kirchoff i upotrijebio u analizi sunčeve atmosfere. Od tog vremena ta grana znanosti se jako razvila, pa se danas većina informacija koje dobivamo proučavanjem spektara ne zasniva na direktnom vizuelnom promatranju. Instrumenti koji se koriste u tu svrhu nazivaju se spektrometri, npr. u laboratoriju imamo FT-IR spektrometar, te Moessbauerov spektrometar. Bilo bi vrlo pogrešno nazivati te instrumente spektro-skopom.Dakle, spektrometrija je širi pojam koji obuhvaća sve tehnike koje se bave proučavanjem i analizom nekakvih spektara. S druge strane, spektroskopija je grana fizikalne znanosti koja se bavi proučavanjem spektara na temelju promatranja. Usprkos ovim činjenicama spektroskopija se vrlo cesto koristi na mjestu gdje bi trebalo koristi izraz spektrometija, razlog tome je udomaćenost izraza "spectroscopy" u engleskoj literaturi
4.
Odgovorio: Goran Stefanic [email protected]
4 S. Kirin, G. Štefanić, e-škola – Spektrometrije, http://eskola.chem.pmf.hr/udzbenik/spektri/spektri_01.php3

2. SPEKTROMETRIJE
_________________________________________________________________________________ 9
nastavak tablice 2.1 874 J.N. Lockyer začetnik kvantitativne analize na temelju
emisijkih spektara 880 otografska registracija spektara
početak XX: st.
W.N. Hartley, A.G. Leonard, J.H. Pollok, A. DeGramont
azvijaju kvantitativnu analizu na bazi emisijskih spektara
923 Hadding spektrometrija emisije rendgenskog zračenja 929 H.G. Lundegardh uvodi fotoelektrično mjerenje intenziteta
Podjela spektrometrija Podjelu spektrometrija moguće je napraviti na osnovi raznih
postavki, npr. ovisno o nivou na kojem se događa interakcija (atomske ili molekulske spektrometrije), ovisno o energiji koja stupa u interakciju s uzorkom (spektrometrije potaknute toplinskom, kemijskom, električnom energijom ili zračenjem čestica), ovisno o posljedicama interakcije (apsorpcijske, emisijske spektrometrije, spektrometrije raspršenja, masena spektrometrija - omjer mase i naboja) i zračenju koje mjerimo nakon interakcije (zračenje čestica, toplinsko zračenje – fotoakustična spekrometrija). Najčešće spektrometrije se grupiraju ovisno o poslijedici interakcije energije i zračenja u grupama po vrsti zračenja koje se nakon interakcije mjeri.
Jednu od čini mi se dobrih podjela dao je profesor dr.sc. Darko Maljković u svojem opširnom poglavlju Spektromerija u Tehničkoj enciklopediji Leksikografskog Zavoda «Miroslav Krleža», Zagreb [3]. U okviru predavanja Instrumentalna analiza spektrometrije ćemo podijeliti prema slijedećoj shemi:
1 Atomske spektrometrije 1.1 Tehnike kod kojih mjerimo elektromagnetsko zračenje 1.1.1 Spektrometrije apsorpcije EMZ 1.1.1.1 Spektrometrija apsorpcije γ- i X-zračenja 1.1.1.2 Atomska apsorpcijska spektrometrija (UV-VID) 1.1.2 Spektrometrije emisije EMZ 1.1.2.1 Emisija γ-zračenja 1.1.2.2 Emisija X-zračenja 1.1.2.3 Emisija UV i VID zračenja 1.1.2.4 Atomska fluorescentna spektrometrija 1.2 Tehnike kod kojih mjerimo zračenje elektrona 1.2.1 Spektrometrije apsorpcije elektrona (odbijeni ili prolazni elektroni) 1.2.2 Spektrometrije emisije elektrona 1.3. Tehnike kod kojih mjerimo zračenje iona 1.3.1 Spektrometrija masa s ionizacijom u plazmi (ICP/MS - omjer mase i
naboja)
2 Molekulske spektrometrije 2.1 Tehnike kod kojih mjerimo elektromagnetsko zračenje 2.2.1 Spektrometrije apsorpcije i inducirane apsorpcije 2.2.1.1 Molekulska apsorpcija u UV i VID 2.2.1.2 IR spektrometrija 2.2.1.3 Spektrometrija apsorpcije mikrovalnog zračenja 2.2.1.4 Spektrometrija inducirane apsorpcije (NMR i ESR)
Sir Isaac Newton (1642-1727)
Joseph von Fraunhofer (1787-1826)
William Henry Fox Talbot (1800-1877)

INSTRUMENTALNA ANALIZA
_________________________________________________________________________________ 10
2.2.2 Spektrometrije emisije 2.2.2.1 Molekularna luminiscencija (fluorescencija i fosforescencija) 2.2.3 Spektrometrije raspršenja 2.2.3.1 Turbidimetrija i nefelometrija 2.2.3.2 Spektrometrija ramanovog raspršenja 2.2.4 Spekrometrije polarizacije 2.2.4.1 Spektropolarimetrija 2.2.4.2 Spektrometrija cirkularnog dikronizma 2.2. Tehnike kod kojih mjerimo zračenje iona 2.2.1 Spektrometrije raspršenih iona 2.2.2 Spektrometrija masa (omjer mase i nabija) 2.3 Tehnike kod kojih mjerimo toplinsko zračenje 2.3.2 Spektrometrija toplinskom lećom 2.3.1 Fotoakustična spektrometrija
Heinrich Lambert
(1728-1777)
Opći pojmovi
Svaka instrumentalna metoda, pa tako i spektrometrija, počiva na interakciji energije i uzorka. Ta energija može biti toplinska (plamena spektrometrija), električna (emisijska spektrometrija s pobudom u elektičnom luku), energija elektromagnetskog zračenja (AAS, molekularna apsorpcijska spektrometrija u UV ili VID), zračenja protona (PIXE), kemijska itd.
Zračenje je energija dobivena iz izvora koja putuje kroz neki materijal ili prostor. Svijetlost, toplina i zvuk su oblici zračenja, Ipak oblik zračenja o kojem će pretežno biti riječi u okviru spektrometrija je onaj oblik energije koji čestice (elektroni, protoni, ioni) ili elektromagnetski valovi usmjereno nose kroz prostor .
Signal pobude Signal odziva zračenje čestica (elektrona, iona) elektromagnetsko zračenje
zračenje nakon interakcije
toplinska energija kemijska reakcija iz
vor e
nerg
ije
električna energija
uzorak
promjena uzorka
Det
ekci
ja
Slika 2.2. Shematski prikaz interakcije energije i uzorka i posljedica te interakcije
Najveći broj spektrometrijskih tehnika prati elektromagnetsko zračenje nakon interakcije s uzorkom.
August Beer (1825 - 1863)
Bourguer

2. SPEKTROMETRIJE
_________________________________________________________________________________ 11
Elektromagnetsko zračenje (EMZ) je vrsta energije koja se prenosi
kroz prostor najvećom mogućom brzinom, a njegova svojstva okarakterizirana su valnom i korpuskularnom prirodom. Valni se karakter može opisati valnim parametrima: frekvencijom ν, brzinom širenja vala ν*, valnom duljinom λ, i valnim brojem ν. Korpuskularna svojstva opisuju elektromagnetsko zračenje kao česticu-foton. Količina energije koju prenosi foton, ovisi o frekvenciji zračenja, prikazana je izrazom:
E = h⋅ν = h⋅c/λ = h⋅c⋅ ν (2.1.)
gdje je h Planckova konstanta i iznosi 6,62x10-34 J s.
Intenzitet elektromagnetskog zračenja I0 smanjuje se prolazom kroz otopinu koja može apsorbirati zračenje. Smanjenje intenziteta ovisi o koncentraciji tvari (c) koja apsorbira zračenje, debljini sloja, tj. svjetlosnom putu kroz uzorak (b), molarnom apsorpcijskom koeficijentu (ε), specifičnom za svaku tvar, a mijenja se s valnom duljinom Odnos intenziteta prije i poslije prolaza kroz uzorak definirali su Lambert, Beer i Bourguer zakonom apsorpcije EMZ:
log I0/I = A = ε⋅c⋅b (2.2.)
Dio spektrometrija pri kojima se mjeri elektromagnetsko zračenja nakon interakcije energije i uzorka a koje obuhvaćaju ultraljubičasti (UV), vidljivi (VID) i infracrveni (IC) dio spektra u literaturi, pogotovo starijoj, nazivaju se i optičke spektrometrije. Vežu ih u tom nazivu klasični optički dijelovi instrumenata (prizme, leće, optičke mrežice).
Slika 2.4. Komponente i materijali kod spektrometrijska mjerenja
Slika 2.1. Shema atoma
Slika 2.3. Propusnost raznih materijala za zračenje čestica

INSTRUMENTALNA ANALIZA
_________________________________________________________________________________ 12
Slika 2.5. Blok sheme spektrometara: 1– jednozračni, 2- dvozračni sa signalom dijeljenim u vremenu, 3 - dvozračni sa signalom dijeljenim u prostoru,
4 – jednozračni s video kamerom, 5 – jednozračni s nizom dioda
Literatura
1. Da. Maljković, Spektrometrije, Tehnička enciklopedija, Svezak 12, Leksikografski Zavod Miroslav Krleža, Zagreb 1992, str.150-178.
2. S. Kirin, G. Štefanić, e-škola – Spektrometrije, http://eskola.chem.pmf.hr/udzbenik/spektri/spektri_01.php3
3. D.A. Skoog, D.M. West, F.J. Holler, Osnove analitičke kemije, 6. izdanje (1. izdanje hrvotsko), Školska knjiga, Zagreb 1999.

2. SPEKTROMETRIJE
_________________________________________________________________________________ 13
2.1. ATOMSKE SPEKTROMETRIJE 2.1.1. ATOMSKA APSORPCIJSKA SPEKTROMETRIJA
(SPEKTROMETRIJA ATOMSKE APSORPCIJE U UV/VIS PODRUČJU ELEKTROMAGNETSKOG ZRAČENJA)
Princip određivanja:
Fenomen atomske apsorpcije poznat je još od početka 19. stoljeća. Prva komercijalna oprema pojavila se u drugoj polovici 20. stoljeća (Walsh).
Atomska apsorpcijska spektrometrija (AAS) je tehnika koja daje informacije o atomima neovisno o molekulskim oblicima u kojima se nalaze u uzorku. AAS mjeri apsorpciju elektromagnetskog zračenja VIS ili UV područja, koje apsorbiraju atomi. Mjeri se razlika intenziteta upadne i izlazne zrake (smanjenje intenziteta), prije i poslije prolaza kroz atomizirani uzorak. Mjerenje se provodi tako da zraka pogodne valne duljine bude upućena u oblak atoma koji će apsorbirati jedan dio energije upadne zrake. Apsorbirano zračenje pobuđuje elektrone u vanjskim orbitalama atoma. Pobuđeni elektroni prelaze na različite više energetske nivoe.
Dio zračenja koji je apsorbiran u izravnom je odnosu s populacijom atoma u oblaku. To znači da možemo kvantitativno odrediti sadržaj analita (atoma) u oblaku atoma.
Elektromagnetsko zračenje određenog nepromjenjivog intenziteta iz izvora zračenja, šuplje katode, upućuje se u atomizirani oblak analita koji se dobiva uvođenjem otopine uzorka u plamen ili u grafitnu kivetu pri elektrotermalnoj atomizaciji (Slika 2.6.). U plamenu se uklanja otapalo, razgrađuje molekula i prevodi uzorak u atomizirano stanje. Kako samo nepobuđeni atomi mogu apsorbirati određenu energiju cilj je dobiti što veću atomizaciju uz što manju ionizaciju.
Slika 2.2. Otparavanje otapala, spaljivanje uzorka, atomizacija kod AAS s elektrotermalnom atomizacijom
Odnos apsorbirane energije i koncentracije je analogan Beer-ovom zakonu apsorpcije:
I=Io*exp (abc) log Io/I=A A=abc (2.3.)
Metoda se primjenjuje za određivanje koncentracija metalnih iona u različitim materijalima. Nemetalni analit može se odrediti samo posredno preko reakcije taloženja s otopinama metalnih soli.

INSTRUMENTALNA ANALIZA
_________________________________________________________________________________ 14
Laboratorijska vježba
Određivanje koncentracije Cu2+−iona atomskom apsorpcijskom Spektrometrijom
Zadatak:
1. Provesti kalibraciju metodom standardnog dodatka (odrediti koncentraciju γ (Cu2+) / μgmL-1 u uzorku grafički i računski iz kalibracije metodom standardnog dodatka,
2. Provesti kalibraciju direktno u masenoj koncentraciji (odrediti koncentraciju γ (Cu2+) / μgmL-1 u uzorku kalibracijom s tri (pet) standarda.
Instrument:
Spektrometar Perkin Elmer Model 3110 opremljen je za očitavanje apsorbancije, koncentracije ili intenziteta emisije zračenja uzorka unesenog u plamen. Očitanja se mogu provoditi u intervalima od 0,1 do 60 sekundi (slika 2.). Instrument se može opremiti dodatkom za bezplamenu atomsku apsorpcionu spektrometriju, zapisnim uređajem, štampačem koji otipkava rezultate mjerenja u toku identifikacije uzorka ili nekim drugim modelom za posebne namjene.
Slika 2.7. AA spektrofotometar Perkin Elmer 3110 s atomizacijom u plamenu
Atomski apsorpcijski spektrometar Perkin Elmer Model 3110 je dvozračni instrument s jednim detektorom koji prima energiju zračenja naizmjenično iz radne i referentne zrake (Slika 2.8.). Zračenje se dobiva iz izvora zračenja šuplje katode koja je načinjena od elementa koji se određuje (Slika 2.9.). Kod ovog modela postoji i mogućnost priključka deuterijske lampe za korekciju pozadinskog zračenja ("background corrector").

2. SPEKTROMETRIJE
_________________________________________________________________________________ 15
Slika 2.8. Shema dvozračnog atomskog apsorpcijskog spektrofotometra
Kod modela Perkin Elmer 603 moguće je priključiti deuterijsku lampu za korekciju pozadinskog zračenja i dodatak za bezplamenu atomizaciju (grafitna kiveta).
Slika 2.9. Šuplja katoda, linijski izvor elektromagnetskog zračenja, 1 ANODA pločica od volframa, 2 KATODA izrađena od materijala koji se određuje, 3 otvorena čašica (zaštita katode),
4 staklena cijev (ispunjena argonom ili neonom), 5 kvarcni prozor
Uvjeti mjerenja:
Tablica 2.1. Uvjeti mjerenja za određivanje bakra
Izvor zračenja: Cu šuplja katoda rezonantna linija: λ=324.75 nm radna struja katode 15 mA
Širina pukotine: d=0,7 mm
Smjesa plinova u plameniku: zrak i acetilen
protok: zraka 55 Lmin-1 acetilena 33 Lmin-1
Vrijeme integracije 2 s
detektor
šuplja katoda
plamenik
monokromator
čoper
zrcalo
5
1
argon ili neon3 4
2
1 3

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 16
Potrebne otopine:
- temeljna otopina bakra (pripremljena): γ (Cu2+) = 1 g/L (1000 mg/L, 1 mg/mL, 1000 µg/mL)
- standardna otopina (potrebno pripremiti): γ (Cu2+) = 10,0 mg/L
U odmjernim tikvicama od 25 mL pripremaju se otopine za kalibraciju. Potrebno je pripremiti otopine tako da koncentracija Cu2+ u otopinama ne prelazi 5 μg/mL.
Standardna otopina se dodaje iz mikro-birete od 10 mL. Nadopunite odmjerne tikvice deioniziranom vodom do oznake (25 mL), dobro promućkajte i izmjerite svaku otopinu. Pazite na faktor razrjeđenja.
Priprema otopina:
Pripremite standardne otopine razrjeđenjem temeljne standardne otopine (1,0 mg/mL). Pripremite otopine u rasponu koncentracije 0 - 5 μg/mL računato na Cu. Upotrebljavajte
samo čistu deioniziranu vodu za pranje staklenog posuđa i za sva razrjeđenja.
Temeljna standardna otopina je pripremljena ranije a radne standardne otopine mogu se pripremati zajednički za cijelu grupu.
Izmjerite i zapišite koncentracije nepoznatih uzoraka. Odredite ponovljivost mjerenjem barem tri (pet) puta. Između mjerenja aspirirajte deioniziranu vodu.
Pazite na faktor razrjeđenja.
Kalibracijski postupci:
Postavite instrument na NULU s deioniziranom vodom kao slijepom probom.
• Kalibrirajte prema uputsvu za rad s instrumentom5 s tri (šest) standarda.
Pripremite 3 (tri) od 6 (šest) otopina (sve koncentracije otopina trebaju ležati u linearnom području koncentracija) i provedite kalibraciju prema uputama u priručniku za instrument.
• Kalibrirajte metodom standardnog dodatka. Pripremiti 2 do 3 otopine uzorka sa standardnim dodatkom. Koncentracija analita u dodatku trebala bi biti u linearnom području odnosa A prema koncentraciji, što će se vidjeti iz grafa.
Priprema uzorka:
Uzorak izdaje voditelj laboratorija ili demonstrator u čistu odmjernu tikvicu od 100 mL. Nadopunite do oznake s deioniziranom vodom. Zbrinjavanje otpadnih kemikalija i pranje suđa i pribora:
Ovi eksperimenti ne proizvode opasan otpad. Sve otopine iz ove vježbe mogu se baciti u izljev nakon čega treba pustiti vodu. Stakleno posuđe i pribor na kraju rada dobro isperite s vodovodnom vodom i zatim nekoliko puta deioniziranom vodom. Rezultati i obrada podataka mjerenja:
• Skicirajte (blok shema) instrument na kojem ste mjerili,
5 Priručnik za rukovanje instrumentom Perkin-Elmer, Hardware

2. SPEKTROMETRIJE
___________________________________________________________________________ 17
• Nacrtajte odnos apsorbancije i koncentracije Cu2+ za metodu standardnog dodatka.
Odredite koncentraciju bakra u nepoznatom uzorku u μg/mL računski • Izračunajte i izrazite sadržaj bakra u nepoznatom uzorku u μg/mL iz grafičkog prikaza
za metodu standardnog dodatka, • Izrazite u μg/mL sadržaj bakra u nepoznatom uzorku direktnim očitanjem s AA
spektrometra nakon kalibracije s tri (pet) standarda
Literatura 3. D. Maljković, Spektrometrije, Tehnička enciklopedija, Svezak 12., Leksikografski zavod
Zagreb, 1985, str. 161

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 18
2.1.2. ATOMSKA EMISIJSKA SPEKTROMETRIJA
2.1.2.1. PLAMENA SPEKTROMETRIJA
(SPEKROMETRIJA EMISIJE U UV/VIS PODRUČJU ELEKTROMAGNETSKOG ZRAČENJA)
Princip određivanja:
Plamena spektrometrija (spektrofotometrija, fotometrija) je atomska emisijska metoda za rutinsko određivanje metalnih soli, uglavnom natrija, kalija, litija, kalcija, stroncija i barija (prva i druga grupa periodnog sustava). Kvantitativno određivanje tih iona provodi se mjerenjem emisije iz plamena otopina koje sadrže metalne soli. Ne-metalni elementi uglavnom ne daju izolirane neutralne atome u plamenu i ne mogu se odrediti plamenom spektrofotometrijom.
Otopine se nasisavaju u plamen. Plamen isparuje otapalo, atomizira metal i pobuđuje valentne elektrone na prelazak na viši energetski nivo. EMZ karakteristične valne duljine za svaki metal se emitira pri povratku elektrona u osnovno stanje. Interferencijski filtri ili monokromatori s optičkom mrežicom se koriste za odabir emisijskih valnih duljina analita. Iz vrijednosti intenziteta emisije zračenja standardnih otopina ili unutarnjeg standarda i analita određuje se kvantitativno metal u otopini uzorka.
Plamena fotometrija je jednostavna, relativno jeftina i brza metoda, koristi se u kliničkim i biološkim ispitivanjima, u analizi ruda i silikatnih materijala te u analizi okoliša.
Niska temperatura plamena (prirodni plin i zrak, acetilen i zrak) prema drugim postupcima pobude (električni luk, iskra, induktivno spregnuta plazma) ograničava metodu samo na metale s niskim pragom pobude. Kako temperatura nije dovoljna da pobudi prelazne metale metoda je selektivna za određivanje alkalija i zemnoalkalija. S druge strane, niska temperatura kriva je da je metoda osjetljiva na interferencije i stabilnost plamena te na uvjete aspiracije uzorka. Protok i čistoća gorivo i oksidansa, brzina nasisavanja uzorka, viskozitet otopina, onečišćenje uzorka (utjecaj matice) itd. ima znatnog utjecaja na određivanje. Zbog toga je vrlo važno mjeriti emisiju standarda i otopine nepoznatog uzorka pod istim uvjetima. Moguće je poboljšati mjerenje metodom kalibracije s unutarnjim standardom.

2. SPEKTROMETRIJE
___________________________________________________________________________ 19
Laboratorijska vježba
Spektrofotometrijsko određivanje koncentracije Na+- i/ili K+-iona
Zadatak: 1. Ispitati utjecaj natrija na intenzitet emisije kalija
2. Ispitati utjecaj viskoziteta otapala na intenzitet emisije natrija
3. Iz otopina poznatih koncentracija Na+-i/ili K+-iona izraditi baždarni dijagram, prikazati grafički odnos masene koncentracije (γ ) prema otklonu galvanometra (G) plamenog spektrometra
4. Iz baždarnog dijagrama odrediti koncentracije γ (Na+) i/ili γ (K+) u uzorku nepoznatog sastava, vodovodnoj vodi i onečišćenoj deioniziranoj vodi
5. Nacrtati shemu instrumenta.
Instrumenti: a) Plameni spektrofotometar Model III, Carl Zeiss, Jena jednozračni je fotometar. Opremljen
je apsorpcijskim i interferencijskim filterima za dobivanje grupa spektralnih linija karakterističnih za ispitivani element u uzorku. Filteri iz emisije izdvajaju samo jednu grupu spektralnih linija. Otopljeni uzorak koji se ispituje uvodi se u raspršivač gdje se raspršuje stlačenim zrakom te se nastala smjesa (aerosol) uvodi u komoru za miješanje s gorivim plinom. Ovako nastala smjesa uzorka, gorivog plina i zraka uvodi se u plamenik gdje dolazi do pobude uzorka i emisije elektromagnetskog zračenja. Emitirano zračenje iz uzorka optičkim sustavom se sakuplja i usmjerava na detektor. Detektor je selenska fotoćelija za vidljivi dio spektra, a cezijeva fotoćelija za UV. Izlazni signal iz detektora dolazi u galvanometar, čiji otklon kazaljke je proporcionalan intenzitetu zračenja odnosno koncentraciji ispitivane tvari u uzorku. Galvanometar ima dva područja osjetljivosti, očitanje na skali Gx1 odnosno Gx10.
Slika 2..10. Slika atomskog emisijskog spektrofotometra Zeiss III, Jena za plamenu spektrofotometriju
b) Atomski apsorpcijski spektrometar Perkin Elmer Model 603 opremljen je za očitavanje
apsorbancije, koncentracije ili intenziteta emisije zračenja uzorka unesenog u plamen. Model 603 je dvozračni instrument s jednim detektorom koji prima energiju zračenja naizmjenično iz radne i referentne zrake (slika 2.11.). Očitanja se mogu provoditi u intervalima od 0,2 do 60 sekundi. Instrument se može opremiti zapisnim uređajem, štampačem koji otipkava rezultate mjerenja.

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 20
Slika 2.11. Blok dijagram dvozračnog atomskog apsorpijskog spektrofotometra bez izvora EMZ kada se koristi kao plameni spektrofotometar.
Instrument je opremljen s dva monokromatora (optičke refleksijske mrežice), u radnom području od 180-900 nm (UV, VID i bliži IC dio spektra), te osjetljivim detektorom elektromagnetskog zračenja, fotomultiplikatorskom cijevi. Za atomizaciju uzorka služi plamenik PREMIX s komorom za predmiješanje uzorka i zraka, kojim se dobavlja oksidans zrak ili kisik, te gorivi plin acetilen. Komprimirani zrak dobavlja se komprimiran iz čeličnih boca ili iz kompresora bez podmazivanja klipa (teflon) koji na svom putu prije ulaska u jedinicu za regulaciju prolazi kroz suhi filter. Acetilen i kisik dobavljaju se iz čeličnih boca koje su opremljene odgovarajućim redukcijskim ventilima. Ugrađeni elektronički čitač s 6 digitalnih jedinica omogućuje očitanje apsorbancije, koncentracije ili intenziteta emisije s mogućnošću ekspandiranja skale 0,01 do 100 puta. Instrument ima automatsko postavljanje nule i automatsku kalibraciju s pet standardnih otopina. Interval integracije signala je 0,23 sekunde.
Uvjeti mjerenja:
• na spektrofotometru Zeiss Model III. selektor valne duljine: Na 59 I i K 77 I (interferencijski)
širine pukotina: ulazna 32 mm izlazna …… mm (pročitati na instrumentu) tlak zraka: …… kPa (u barima na manometru instrumenta)
tlak acetilena: …… kPa (u mm vodenog stupca na ploči instrumenta)
osjetljivost galvanometra: G x 10 (za Na) i G x 1 (za K)
• na spektrofotometru Perkin-Elmer Model 603 selektor valne duljine: optička mrežica
širina pukotine: ….… mm ((pročitati na instrumentu) tlak zraka: ….… kPa (u barima na manometru instrumenta)
tlak acetilena: ….… kPa (u mm vodenog stupca na ploči instrumenta)
detektor
šuplja katoda
plamenik
monokromator
čoper
zrcalo

2. SPEKTROMETRIJE
___________________________________________________________________________ 21
Potrebne otopine: Određivanje Na+- iona Određivanje K+- iona - TSO Na+-iona γ (Na+) = 1,0 mg/mL - TSO K+-iona γ (Ni2+) = 1,0 mg/mL Određivanje utjecaja Na+ na emisiju K+ - otopina Na+-iona γ (Na+) = 10 mg/mL Određivanje utjecaja viskoziteta na emisiju K+ etanol glicerol
TSO=temeljna standardna otopina
Postupak:
Ovaj postupak služi za određivanje natrija i kalija plamenom fotometrijom i pokazat će utjecaj čistoće i viskoziteta otopina na promatranu emisiju. Instrument je kalibriran serijom standardnih otopina koje pokrivaju područje koncentracija očekivanih u uzorku.
Pažljivo pročitajte upute koje ste dobili uz vježbu. Ukjučite instrument i dozvolite da se zagrije na radnu temperaturu. Aspirirajte deioniziranu vodu između uzoraka za čišćenje aspiratora. Natrij je sveprisutan. Vrlo je važno da se koristi pažljivo oprano posuđe da bi se dobili dobri rezultati.
Priprema otopina:
Pripremite standardne otopine razrjeđenjem temeljne standardne otopine (1,0 mg/mL).
Pripremite otopinu natrija 25 μg/mL u raznim otapalima: voda, etanol (w=10 %), etanol (w=50 %), glicerin (w=25 %).
Pripremite otopine za ispitivanje utjecaja natrija na intenzitet emisije kalija. U 5 (pet) odmjernih tikvica od 25 mL u koje ste najprije stavili određeni volumen otopine K+-iona (γ (K+)=25,00 μg/mL) dodajte otopinu natrija. Raspon koncentracija Na+ treba biti između 0-5 mg/mL. Tikvice nadopunite deioniziranom vodom.
Pripremite otopine slijedećih koncentracija: 5, 10, 25, 50, 75, i 100 μg/mL računato na Na i/ili K. Upotrebljavajte samo čistu deioniziranu vodu za pranje staklenog posuđa i za sva razrjeđenja. U standarde za kalibraciju kalija dodajte određenu koncentraciju natrija koju ste odredili u prethodnom ispitivanju.
Pripremite standarde u pažljivo očišćenom staklenom posuđu i prenesite otopine u plastične bočice. Staklo često sadrži visoki postotak natrija i može kontaminirati standarde ukoliko otopine ekstremno visokog ili niskog pH stoje dulje u staklenom posuđu.
Temeljna standardna otopina je pripremljena ranije a radne standardne otopine mogu se pripremati zajednički za cijelu grupu.
Priprema uzorka:
Uzorak izdaje voditelj laboratorija ili demonstrator u čistu odmjernu tikvicu od 50 mL. Uzorak vodovodne vode. Ne zaboravite faktor razređenja.

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 22
Kalibracijski postupak:
Postavite instrument na NULU s deioniziranom vodom kao slijepom probom. Postavite najviše očitanje prema uputstvu pomoću najkoncentriranije otopine (100 mg/mL). Izmjerite i zapišite intenzitete emisije svih preostalih standardnih otopina natrija i otopine nepoznatog uzorka (očitanje na skali galvanometra). Odredite točnost i ponovljivost mjerenjem standarnih otopina baren tri (pet) puta. Između mjerenja aspirirajte deioniziranu vodu.
Ispitivanje čistoće: Uronite dva prsta u čistu posudu s cca. 20 mL deionizirane vode. Izmjerite i zapišite
intenzitet emisije natrija i/ili kalija. Izmjerite intenzitet emisije vodovodne vode. Između mjerenja aspirirajte deioniziranu vodu.
Utjecaj promjene viskoziteta:
Izmjerite i zapišite intenzitet emisije svake otopne natrija (25 μg/mL) i/ili kalija u raznim otapalima: čista deionizirana voda, etanol (w=10 %), etanol (w=50 %), glicerol (w=25 %). Između mjerenja aspirirajte deioniziranu vodu.
Provjera ponovljivosti: Ponovno izmjerite intenzitete emisije za dvije ili tri standardne otopine. Ukoliko se javi
osjetna razlika ponovno podesite NULU i najveće očitanje i ponovno izmjerite intenzitete emisije standardnih otopina i nepoznatog uzorka.
Zbrinjavanje otpadnih kemikalija i pranje suđa i pribora: Ovi eksperimenti ne proizvode opasan otpad. Sve otopine iz ove vježbe mogu se baciti u
izljev nakon čega treba pustiti vodu. Stakleno posuđe i pribor na kraju rada dobro isperite s vodovodnom vodom i zatim
nekoliko puta deioniziranom vodom.
Rezultati i obrada podataka mjerenja:
• Skicirajte (blok shema) instrument na kojem ste mjerili
• Nacrtajte odnos otklona galvanometra (G) i koncentracije Na+ pri ispitivanju utjecaja natrija na emisiju kalija. Odredite potrebnu koncentraciji natrija,
• Nacrtajte odnos otklona na skali galvanometra (G) prema viskozitetu ispitivanih otopina kod određivanja K+,
• Nacrtajte odnos otklona galvanometra (G) i koncentracije μg/mL natrija i/ili kalija,
• Izračunajte i izrazite u μg/mL sadržaj natrija i/ili kalija u deioniziranoj vodi u koju ste uronili prste (kontaminacija otopina natrije i/ili kalijem),
• Izračunajte i izrazite u μg/mL sadržaj natrija i/ili kalija u nepoznatom uzorku.
Literatura
1. D. Maljković, Spektrometrije, Tehnička enciklopedija, Svezak 12., Leksikografski zavod Zagreb, 1985, str. 167-169

2. SPEKTROMETRIJE
___________________________________________________________________________ 23
2.2. MOLEKULSKE SPEKTROMETRIJE 2.2.1. SPEKTROMETRIJE MOLEKULSKE APSORPCIJE U UV / VIS PODRUČJU
ELEKTROMAGNETSKOG ZRAČENJA (SPEKTROFOTOMETRIJA) Princip određivanja:
Intenzitet elektromagnetskog zračenja I0 smanjuje se prolazom kroz otopinu koja može apsorbirati zračenje. Smanjenje intenziteta ovisi o koncentraciji tvari (c) koja apsorbira zračenje, debljini sloja, svjetlosnom putu kroz uzorak (b), molarnom apsorpcijskom koeficijentu, specifičnom za svaku tvar, a mijenja se s valnom duljinom (ε). Odnos intenziteta prije i poslije prolaza kroz uzorak definirali su Lambert, Beer i Bourguer zakonom apsorpcije EMZ:
log II 0 = A = ε⋅c⋅b (2.4.)
Molekularna apsorpcijska spektrometrija u UV i VID dijelu EMZ koristi se za kvantitativna određivanja organskih i anorganskih tvari. Često se primjenjuje za određivanje metala u otopinama (mogu biti u otopini prisutni kao kationske vrste, anionske ili metalni kompleksi).
Slijedeći primjeri opisuju spektrofotometrijsko određivanje iona u vodenim otopinama. Određivanja se osnivaju na reakcijama nastajanja kompleksnih vrsta između metalnih ona i anorganskih ili organskih liganada ili na apsorpciji EMZ samog iona (aniona).
Željezo u obliku Fe3+-iona reagira u kloridno kiselom mediju sa SCN–-ionom i stvara kompleksni ion [Fe(SCN)6]3– koji je crveno obojen. Nastali kompleksni ion ima maksimum apsorpcije elektromagnetskog zračenje kod valne duljine λ = 480 nm.
Krom(VI) reagira s difenilkarbazidom u kiseloj otopini dajući intenzivno obojeni kompleks. Nastali produkt ima apsorpcijski maksimum kod λ = 545 nm. Nitrati se određuju spektrofotometrijski u UV-području kod valne duljine λ=220 nm. Međutim ukoliko voda sadrži neka organska onečišćenja (npr. huminske kiseline) apsorbpcija kod 220 nm je suma apsorbancije nitrat-iona i organskih tvari. Stoga se određuje apsorbancija na dvije valne duljine, 220 nm i 275 nm. Pri valnoj duljini 275 nm apsorbiraju samo organske tvari a nitrati ne. Apsorbancija nitrata u tom slučaju odgovara razlici apsorbancije pri 220 i 275 nm. Ukoliko je uzorak vode zamućen, suspendirane čestice iz uzorka uklanjaju se filtriranjem, a zakiseljavanjem sa 1 M HCl uklanjaju se smetnje uzrokovane prisustvom anorganskih hidroksida ili karbonata. Određivanju nitrata ne smeta prisutnost klorida, ali smetaju nitriti i fosfati.

INSTREUMENTALNA ANALIZA
___________________________________________________________________________ 24
Laboratorijska vježba
Spektrofotometrijsko određivanje koncentracije Fe3+-, Cr(VI)- i/ili NO3
--iona
Zadatak:
1. Odrediti valnu duljinu maksimuma apsorpcije elektromagnetskog zračenja
(λmax) kompleksnih iona željeza, kroma i/ili nitrata
2. Iz otopina poznatih koncentracija Fe3+-, Cr(VI)- i NO3--iona izraditi
baždarni dijagram.
3. Iz baždarnog dijagrama odrediti koncentracije γ (Fe3+), γ [Cr(VI)] i/ili γ (NO3-)
u μgmL–1 u uzorku nepoznatog sastava.
InstrumentI:
a) Spektrofotometar Lambda 20, Perkin Elmer, dvozračni s monokromatorom optičkom refleksijskom mrežicom, valno područje od 190-1100 nm, dva izvora zračenja (halogeni za vidljivi dio spektra i deuterijski za ultraljubičasti dio spektra) i dva detektora. Ispis na zaslonu PC-a, pisač ili ploter.
b) Spektrofotometar Perkin Elmer 124, dvozračni s monokromatorom optičkom refleksijskom mrežicom. Radno područje spektrofotometra je od 190 nm do 800 nm. Opremljen je s dva izvora elektromagnetskog zračenja (volframov izvor za vidljivi dio spektra i deuterijev izvor za ultraljubičasti dio spektra) i dva detektora. Ispis ploter.
c) Spektrofotometar Lambda 1, Perkin Elmer, jednozračni s monokromatorom optičkom refleksijskom mrežicom, valno područje od 190-800 nm. Prikaz na zaslonu instrumenta, digitalni.

2. SPEKTROMETRIJE
___________________________________________________________________________ 25
d) Spektrofotometar MA 9525-SPEKOL 210, Iskra, Kranj, jednozračni s monokromatorom, optičkom refleksijskom mrežicom. Radno područje od 335-800 nm. Prikaz na zaslonu instrumenta, digitalni.
e) Spektrofotometar VSU-1 Carl Zeiss, Jena, jednozračni s monokromatorom, staklenom optičkom prizmom za vidljivi dio elektromagnetskog zračenja, 400-700 nm, i prizmom od taljenog SiO2 za ultraljubičasti dio elektromagnetskog zračenja, 200-400 nm. Prikaz skala instrumenta, analogno.
Slika 2.12. Spektrofotometri
Uvjeti mjerenja na spektrofotometru Perkin Elmer 124
- snimanje apsorpcijskog spektra u području valne duljine λ = 800-190nm - izvor zračenja; volframova ili deuterijska lampa - svjetlosni put radne i referentne kivete b = 1 cm - brzina zapisne sprave ; 10 mmmin–1
Uvjeti mjerenja na spektrofotometru Perkin Elmer Lambda 20
- snimanje apsorpcijskog spektra u području valne duljine λ = 190-800 nm - izvor zračenja; volframova i deuterijska lampa - svjetlosni put radne i referentne kivete b = 1 cm - zapis na zaslonu PC-a, printer ili ploter
Uvjeti mjerenja na spektrofotometru VSU-1 Carl Zeiss, Jena u UV području elektromagnetskog zračenja: - valna duljina λ=267 nm, TT=1016,7 (jedinice na skali noniusa na bubnju
monokromatora koje odgovaraju valnoj duljini prema tablici proizvođača) - prorez optičkog klina; p = 3.5 mm - širina pukotine; d = 0.2 mm - debljina sloja; b = 0.5 cm (kivete od taljenog SiO2 za UV zračenje)
Uvjeti mjerenja na spektrofotometru MA 9525 - SPECOL 21
- valna duljina λ=480 ili 545 nm - širina pukotine; d = 0.5 mm (oznaka na instrumentu) - svjetlosni put radne i referentne kivete; b = 1 cm
Uvjeti mjerenja na spektrofotometru Perkin Elmer Lambda 1
- valna duljina λ=480 ili 545 nm - širina pukotine; d = 0.5 mm (oznaka na instrumentu) - svjetlosni put radne i referentne kivete; b = 1 cm

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 26
Potrebne otopine: Određivanje Fe3+- iona Određivanje Cr(VI)- iona - otopina poznate koncentracije Fe3+- iona
γ (Fe3+) = 10 μgmL–1 - otopina H2O2; w (H2O2) = 3 % - otopina KSCN; w (KSCN) = 15 % - HCl konc.
- otopina poznate koncentracije Cr(VI)-iona γ (Cr) = 40 μgmL–1
- otopina H2SO4; w (H2SO4) = 2% - otopina difenilkarbazida; w (dfk) = 1%
Određivanje NO3--iona
- otopina poznate koncentracije NO3— - iona
γ (NO3-) = 44,3 μgmL–1
- otopina HCl, c (HCl)=1 M
Postupak:
Pažljivo pročitajte upute koje ste dobili uz vježbu. Ukopčajte instrument i dozvolite da se zagrije na radnu temperaturu. Vrlo je važno da se koristi pažljivo oprano posuđe da bi se dobili dobri rezultati. Upotrebljavajte samo čistu deioniziranu vodu za pranje staklenog posuđa i za sva razrjeđenja.
Priprema otopina za određivanje Fe3+, Cr(VI) i NO3—iona:
Pripremite standardne otopine razrjeđenjem temeljne standardne otopine.
Pripremite 5 otopina koncentracija: od 1 - 5 μg/mL računato na Fe, 1 - 5 μg/mL računato na Ni, 0,25 - 2 μg/mL računato na Cr te 1 - 8 μg/mL računato na nitrat-ion dodavajući u odmjerne tikvice od 25,00 mL iz birete potrebne volumene standardnih otopina.
Zatim za određivanje: Fe - u odmjerne tikvice s uzorkom dodajte do polovice volumena deioniziranu vodu i u sve
dodajte 1,0 mL HCl konc. i 2,5 mL otopine KSCN, w (KSCN) = 15 %. Ako je u uzorku prisutno i Fe(II) potrebno je dodati 0,5 mL H2O2 (w=3%) u svaku tikvicu prije dodatka reagensa.
Cr - u odmjerne tikvice s kromatom dodajte 2 mL 2%-tne otopine H2SO4 i 4 mL otopine difenilkarbazida u acetonu.
NO3- - u odmjerne tikvice s nitratom dodati 0,5 mL 1 M HCl.
Sve odmjerne tikvice nadopunite deioniziranom vodom do oznake (25 mL), dobro promućkajte i izmjerite apsorbancije za svaku otopinu.
Temeljne standardne otopine su pripremljene ranije a radne standardne otopine mogu se pripremati zajednički za cijelu grupu.
Priprema nepoznatog uzorka:
Nepoznati uzorak izdaje voditelj laboratorija ili demonstrator u čistu odmjernu tikvicu od 25,00 mL. Nadopunite do oznake deioniziranom vodom. Priprava otopine za određivanje valne duljine maksimuma apsorpcije:
Za snimanje spektara uzmite najkoncentriraniju otopinu iz niza otopina pripremljenih za izradu baždarnog dijagrama, snimite spektar i odredite valnu duljinu maksimuma apsorpcije.

2. SPEKTROMETRIJE
___________________________________________________________________________ 27
Kalibracijski postupak:
Postavite instrument na nulu sa slijepom probom. Izmjerite i zapišite vrijednosti apsorbancija svih standardnih otopina Ni, ili Fe ili Cr ili NO3
-i otopine nepoznatog uzorka. Odredite točnost i ponovljivost mjerenjem standardnih otopina barem tri (pet) puta. Mjerite u rastućem nizu koncentracija kod izrade baždarnog dijagrama.
Rezultati i obrada podataka mjerenja:
Nacrtajte blok sheme instrumenata na kojima ste mjerili. Priložite rezultatima snimljeni spektar (na slici 2.13. primjer je spektra Fe-tiocijanata),
Slika 2.13. Apsorpcijski spektar [Fe(SCN)6]3–-iona u vidljivom dijelu EMZ
• Nacrtajte i statistički obradite baždarni dijagram za izmjerene standardne otopine. Nacrtajte odnos apsorbancije prema masenoj koncentraciji g ( Fe, Cr i/ili NO3
-) u μg/mL,
• Izračunajte i izrazite u μg/mL sadržaj (Fe, Cr ili NO3-) u nepoznatom uzorku.
Zbrinjavanje otpadnih kemikalija i pranje suđa i pribora:
Ovi eksperimenti ne proizvode opasan otpad. Sve otopine iz ove vježbe mogu se baciti u izljev nakon čega je potrebno izljev isprati i vodom.
Literatura 1. Instrumentalna i procesna analiza, (Radni materijal za internu uporabu), Zavod za
analitičku kemiju FKIT, Zagreb, 2005.
2. D. Maljković, Spektrometrije, Tehnička enciklopedija, Svezak 12., Leksikografski zavod Zagreb, 1985, str. 150-178.
3. D.A. Skoog, D.M. West, F.J. Holler, Osnove analitičke kemije, 6. izdanje (1. izdanje hrvotsko), Školska knjiga, Zagreb 1999.
4. B. Petz, Osnovne statističke metode za nematematičare, Udžbenici Sveučilišta u Zagrebu, 4. izdanje, Naklada Slap Jastrebarsko, 2002.
5. M. Kaštelan-Macan, Kemijska analiza u sustavu kvalitete, Školska knjiga Zagreb 2003.
300 400 500 600 7000,00
0,05
0,10
0,15
0,20
0,25
0,30
A
λ / nm

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 28
2.2.2. TURBIDIMETRIJA I NEFELOMETRIJA
(SPEKTROMETRIJA raspršenja EMZ)
Princip određivanja:
Turbidimetrija i nefelometrija su metode kojima se određuje koncentracija čestica u suspenziji. Osniva se na elastičnom raspršivanju EM zračenja na suspendiranim česticama u otopini. Mjeri se smanjenje intenziteta prolaznog zračenja ili intenzitet raspršenog zračenja kao posljedicu sraza s česticama. Raspršivanje EMZ na suspendiranim česticama često se naziva i Tyndallov efekt. Raspršenje može biti Rayleighovog, Debyeovog ili Mieovog tipa, ovisno o veličini čestica.
Ako je dimenzija čestica reda veličine valne duljine upadnog zračenja ili manja, zračenje će se raspršivati no ako je veća doći će do refleksije. Optimalna veličina čestica da bi došlo do raspršenja je 100−1000 nm, dakle veličina koloida.
Kod turbidimetrijskih mjerenja pravocrtno usmjereno zračenje prolazi iz izvora kroz otopinu uzorka do detektora, a mjeri se smanjenje intenziteta prolaznog zračenja. Turbidimetrijski se određuje zamućenje vode u ekološkom okruženju ili koncentracija u sustavima u kojima reakcijom nastaje talog koji se teško filtrira zbog malih čestica ili želatinozne prirode taloga. Turbidimetrija često zamjenjuje dugotrajno gravimetrijsko određivanje.
U turbidimetriji mjeri se transmitancija primarne zrake:
TII
=0
(2.5.)
gdje je I0 intenzitet ulaznog zračenja nakon prolaska kroz slijepu probu, a I je intenzitet zračenja nakon prolaska kroz uzorak. Propušteno zračenje je proporcionalno koncentraciji suspendirane tvari prema izrazu koji je analogan Beerovom zakonu:
S II
kbc= =log 0 (2.6.)
gdje je S zamućenje, k je konstanta proporcionalnosti, koji puta nazvana koeficijentom zamućenja, b je duljina puta kroz uzorak a c je koncentracija.
Parametri koji utječu na određivanja su: a) koncentracija čestica, b) odnos indeksa loma čestice i okolnog medija, c) veličina, oblik i raspodjela čestica. d) valna duljina ulaznog zračenja.
Vrijednosti zamućenja ovise i o orijentaciji čestica u suspenziji obzirom da sve čestice nisu sferične.
Plavo svijetlo se raspršuje efikasnije nego crveno (zbog čega je i boja neba plava). Kroz obojene suspenzije propušta se zračenje iste boje, opet zbog smanjenja apsorpcije.
Instrumenti koji se koriste u turbidimetriji su vrlo slični spektrometrima za UV/VIS područje (spektrofotometri), a mogu se koristiti i obični spektrofotometri ili čak kolorimetri. Izvori zračenja su živin luk s filtrima za odabir samo jedne valne duljine ili volframova lampa u kombinaciji s monokromatorima ili filtrima. Monokromatsko svijetlo je nužno kod turbidimetrijskih određivanja da se smanji apsorpcija EMZ na česticama, te da je smanjenje intenziteta posljedica uglavnom raspršenja (prividna apsorpcija). Detektori su u turbidimetriji kao i u spektrofotometriji, fotoćelije, a fotomultiplikatori su potrebni kod nefelometrijskih instrumenata. Kivete za tekuće uzorke su u turbidimetriji iste kao i za spektrofotometrijska određivanja.

2. SPEKTROMETRIJE
___________________________________________________________________________ 29
Slika 2.14. Blok shema turbidimetra (fotometra s filtrerima i okruglim kivetama)
U nefelometriji mjeri se intenzitet elastično raspršenog zračenja na koloidnim česticama pod kutem, uglavnom od 90o, na smjer inicijalne zrake. Intenzitet raspršenog zračenja proporcionalan je zamućenju otopine. Instrumenti za nefelometrijska mjerenja su posebni instrumenti s vrlo stabilnim izvorima zračenja, kivete su ili okrugle gdje zračenje dolazi kroz dno a mjeri se raspršeno kroz stijenku cilindra, ili slične kao kod molekularne luminiscencije, a detektori su fotomultiplikatorske cijevi.
Slika 2.15. Blok shema nefelometra s okruglim kivetama
Nefelometrijska mjerenja su pogodna za analizu otopina slabog zamućenja.
Sustavi se, ukoliko se određuje zamućenje, kalibriraju sa suspenzijama formazina (polimerni spoj, primarni standard za kalibraciju) ili sa sekundarnim standardima koji mogu biti stakleni štapovi raznog stupnja neprozirnosti i na taj način simuliraju zamućene sustave. Suspenzije formazina su po boji slične mlijeku. Oprez! Za formazin postoji vjerojatnost da je kancerogen. Ako je mjerenje zamućenja u funkciji zamjene dugotrajnog gravimetrijskog određivanja, sustav se kalibrira kao i svako relativno instrumentalno određivanje jednim od naprijed objašnjenih postupaka kalibracije.
Rezultati mjerenja s ove dvije tehnike ne mogu se direktno uspoređivati kada se određuje zamućenje otopina. Jedinice u kojima se izražava zamućenje su NTU (nephelometric turbidity units), FTU (formazine turbidity units) ukoliko se mjeri nefelometrijski, a FAU (formazine attenuation units) ako se mjeri smanjenje intenziteta u prolaznom zračenju (turbidimetrijski). Svi propisi i standardi za vodene ekosustave traže nefelometrijska određivanja i rezultate daje u NTU jedinicama. U literaturi postoje podaci da 1 NTU odgovara ekvivalentu od 1 mg/mL suspendiranog SiO2.
Metode koje se osnivaju na raspršenju EMZ kao što je gore navedeno upotrebljavaju se najčešće za određivanje koncentracije u suspenzijama (turbidimetrija i nefelometrija), ili za određivanje stupnja zamućenja u ekosustavima ali primjena nije ograničena samo na ta određivanja. Raspršenjem EMZ može se odrediti veličina čestica, raspodjela i molekulska masa (posebno za polimerne čestice). Ta određivanja, iako se osnivaju na istim principima nisu tako jednostavna.

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 30
Laboratorijska vježba
Određivanje koncentracije SO42-− iona u vodi
Zadatak:
1. Izraditi baždarni dijagram za određivanje sulfat-iona
2. Iz baždarnog dijagrama odrediti koncentraciju SO42--iona u uzorku vode
Instrumenti:
a) fotometar MA 9502, MA 9507 Iskra; Kranj, Slovenija fotometar s obojenim filtrima
b) turbidimetar 2100A Hach, USA
Slika 2.16. Slika fotometra s okruglim kivetama i obojenim filterima /a) i nefelometra s okruglim kivetama (b)
Kivete Čistoća kiveta za mjerenje zamućenja vrlo je važna. Otisci prstiju, prljavština ili
ogrebotine smetaju pri određivanju jer mogu uzrokovati dodatno raspršenje ili mogu apsorbirati dio zračenja.
Uvjeti mjerenja: fotometar MA 9502, MA 9507 Iskra; Kranj Filtar: obojeni (plavi)
turbidimetar 2100A Hach, USA Skala: 1-100 NTU

2. SPEKTROMETRIJE
___________________________________________________________________________ 31
Potrebne otopine i reagensi: - otopine poznate koncentracije SO4
2-- iona (otopina Na2SO4): temeljna standardna otopina (TSO) − γ (SO4
2-)=1000,00 μg/mL standardna otopina − γ (SO4
2-)=200,00 μg/mL
- otopina za kondicioniranje: glicerol 50 mL, HCl konc. 30 mL, etanol 95 %-ni ili izo-propanol 100 mL, NaCl 75 g , H2O 300 mL; dobro promiješati
- BaCl2, p.a. Priprema otopina:
Pripremite radnu standardnu otopinu (200 μg/mL) razrjeđenjem temeljne standardne otopine (1,0 mg/mL).
Pripremite u odmjernim tikvicama od 25 mL otopine SO42— iona za izradu baždarnog
dijagrama tako da koncentracija bude između 0 – 180 μg/mL za mjerenja turbidimetrijski a 0-40 μg/mL za mjerenja nefelometrijski. U svaku odmjernu tikvicu dodajte 2,5 mL otopine za kondicioniranje, nadopunite do oznake (25 mL) deioniziranom vodom i dobro promućkajte.
Pripremite standarde u pažljivo očišćenom staklenom posuđu. Upotrebljavajte samo čistu deioniziranu vodu za ispiranje staklenog posuđa i za sva razrjeđenja.
Temeljna standardna otopina je pripremljena ranije a radne standardne otopine mogu se pripremati zajednički za cijelu grupu.
Priprema uzorka: Uzorak je vodovodna voda. Priprema se pod istim uvjetima kao i standardne otopine za
izradu baždarnog dijagrama. Ne zaboravite na faktor razređenja.
Kalibracijski postupak: Otopine sulfata za izradu baždarnog dijagrama staviti u Erlenmeyerovu tikvicu od 100
mL i dodati na vrhu spatule krutog BaCl2. Miješati na magnetskoj miješalici 60 sekundi. Na isti način pripremiti slijepu probu. Prenijeti slijepu probu i otopine sulfata u kivete, staviti u držač na fotometru i kad se kazaljka umiri, očitati otklon na skali apsorbancije fotometra pet puta (5). Očitane otklone kazaljke fotometra (intenzitet raspršenog zračenja u NTU ili prividna apsorpcija na skali fotometra E ili A) prema koncentraciji SO4
2-−iona u otopini i prikazati grafički.
Zbrinjavanje otpadnih kemikalija i pranje suđa i pribora Ovi eksperimenti ne proizvode opasan otpad. Sve otopine iz ove vježbe mogu se baciti u
izljev nakon čega treba pustiti vodu. Stakleno posuđe i pribor na kraju rada dobro isperite vodovodnom vodom i zatim
nekoliko puta deioniziranom vodom.
Rezultati i obrada podataka mjerenja: - Skicirajte (blok shema) instrument na kojem ste mjerili,
- Nacrtajte i statistički obradite baždarni dijagram za izmjerene standardne otopine, zamućenje (S) prema masenoj koncentraciji SO4
2—iona, i/ili intenzitet raspršenog zračenja (otklon na skali nefelometra) prema masenoj koncentraciji SO4
2—iona,
- Izračunajte i izrazite u μg/mL sadržaj SO42- -iona u vodovodnoj vodi.

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 32
3. ELEKTROANALITIČKE METODE 3.1. POTENCIOMETRIJA Princip određivanja:
Potenciometrijska određivanja se temelje na mjerenju razlike potencijala između dvije elektrode, indikatorske i referentne, uronjene u elektrokemijsku ćeliju s elektrolitom. Signal pobude je kemijska reakcija a signal odziva električna veličina (razlika potencijala).
Indikatorska elektroda reagira promjenom potencijala na promjenu aktiviteta specije koju određujemo u eksperimentu. Referentna elektroda ima konstantan potencijal bez obzira na promjenu aktiviteta bilo koje vrste uotopini.
Potencijal elektrokemijske ćelije dan je izrazom:
Ećelije=Eind-Eref+Epren (3.1.)
gdje je Ećelije=potencijal elektrokemijske ćelije (elektromotorna sila) Eind=potencijal indikatorske elektrode, Eref=potencijal referentne elektrode Epren=prenapon (liquid junction)
Prenapon nastaje na granici između dva elektrolita. Javlja se na kontaktu referentne elektrode i otopine u ćeliji. Potencijal indikatorske elektrode odgovor je na promjenu aktiviteta specije koja se određuje, mjeri se u sklopu s odabranom pogodnom referentnom elektrodom, i dan je Nernstovim izrazom:
)()(log303,2
)()(ln 00
redaoksa
zFRTE
redaoksa
zFRTEE +=+= (3.2.)
gdje je E potencijal, E0 je standardni redukcijski potencijal, R i F su termodinamičke konstante; T je apsolutna temperatura; a(oks) i a(red) su aktiviteti oksidiranog i reduciranog oblika iona koji se određuje u ispitivanoj otopini; z je naboj iona. Ion selektivne elektrode
Ion-selektivna elektroda (ISE) je elektrokemijski senzor koji stvara elektrokemijski potencijal prema Nernstovoj jednadžbi kada se uroni u otopinu koja sadrži slobodne ione određene vrste. Svaka ion-selektivna elektroda pokazuje relativno visoki stupanj selektivnosti za određeni tip ili vrstu iona u otopini.
Iako je materijal iz kojeg su konstruirane ion-selektivne elektrode različit teorija određivanja s ion-selektivnim elektrodama je razrađena i osnovni proces uspostavljanja razlike potencijala u suštini je poznat. To su procesi ionske izmjene, difuzije, odnosno prijenosa iona. Ion-selektivna elektroda poprima određeni potencijal kao odgovor na aktivitet iona u otopini na koji je elektroda selektivna.
Ion-selektivne elektrode imaju osjetljivu membranu koja odvaja ispitivanu otopinu od unutrašnje referentne otopine, tzv. elektrokemijsku membranu. Vrlo važno svojstvo elektrokemijskih membrana je formiranje razlike potencijala između dvije otopine. Kada se elektroda uroni u ispitivanu otopinu, momentalno se uspostavi tok čestica unutar membrane u smjeru otopine koja ima niži aktivitet pokretnih čestica. Dok se ne uspostavi ravnoteža traje prijenos mase i naboja (ako se radi o nabijenim česticama) koji rezultira razlikom potencijala i uspostavlja se ravnoteža. Ravnotežni potencijal je mjera razlike aktiviteta iona u dvije otopine i svaka promjena aktiviteta rezultirat će proporcionalnom promjenom potencijala:

3. ELEKTROANALITIČKE METODE
___________________________________________________________________________ 33
2
1'
2
1' log303,2lnaa
zFRTE
aa
zFRTEE +=+= (3.3.)
gdje je E potencijal, E' je konstantna vrijednost za određenu temperaturu i ovisi o izboru unutrašnje i vanjske referentne elektrode. R i F su termodinamičke konstante; T je apsolutna temperatura; a1 je aktivitet iona u ispitivanoj otopini, a2 u unutrašnjoj referentnoj otopini; z je naboj iona.
Kako je aktivitet ispitivanog iona u unutrašnjoj otopini elektrode konstantan, relacija (3.3.) se može pisati:
azF
RTEE o log303,2' += (3.4.)
Eo' je nova konstanta i ovisi o unutrašnjoj referentnoj otopini kao i ostalim faktorima spomenutim za E'. Jednadžba (3) pokazuje da je potencijal direktno proporcionalan apsolutnoj temperaturi.
Elektrode reagiraju samo na promjenu aktiviteta slobodnih, neasociranih iona, a ne reagiraju na ione koji su na bilo koji način vezani. ISE nisu, nažalost, specifične za određenu vrstu iona te reagiraju i na neke druge ione u otopini. U otopini koja osim ispitivanih iona sadrži i smetajući ion jednadžba (3) dobiva oblik:
[ ]xzaKazF
RTEE xpotAB
o /1' log303,2⋅++= (3.5.)
gdje je K ABpot konstanta odnosno koeficijent selektivnosti za smetajući ion; ax aktivitet
smetajućeg iona; zx naboj smetajućeg iona. Za sve smetajuće ione istog naboja izraz (4) postaje:
E E RTzF
a K a zABpot
x x= ± + ⋅⎡⎣⎢
⎤⎦⎥
∑0 12 303' /, log (3.6.)
gdje logaritamski dio dobiva predznak plus za katione, a minus za anione.
Pri niskim koncentracijama iona u otopini vrijedi Debye-Hückel-ova jednadžba: a = γ· c
log = - 0,5122 z I-γ ⋅ +z (3.7.)
I c zi i= ∑12
2
Ako ionsku jakost otopine održavamo konstantnom, aktivitet iona ovisiti će samo o njihovoj koncentraciji. Dakle, ako su ova tri uvjeta ispunjena, potencijal elektrode ovisit će samo o koncentraciji iona koji se određuje. Konstantna ionska jakost postiže se dodatkom, u velikom suvišku, elektrolita čiji ioni ne sudjeluju u promatranoj ravnoteži, tako da se ionska jakost ne mijenja bitno promjenom koncentracije ispitivanih iona.
Kako je vidljivo iz Nernst-ove jednadžbe, potencijal elektrode pada sa smanjenjem koncentracije aniona a raste sa smanjenjem koncentracije kationa. Pri ekstremno niskim koncentracijama (do 10−7 molL−1) promjena potencijala za istu razliku u koncentraciji počinje se smanjivati zbog pomanjkanja iona nosioca naboja (granica određivanja). Slika 3.3. prikazuje odnos promjene potencijala prema logaritmu koncentracije aniona. Odnos je u velikom rasponu koncentracija linearan da bi blizu granice određivanja dobio otklon prema osi x.

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 34
Tipovi ion selektivnih elektroda
Ion-selektivne elektrode se klasificiraju ovisno o tipu membrane koja je upotrebljena: elektrode s čvrstom membranom, elektrode s tekućom membranom.
Čvrste membrane ion-selektivnih elektroda mogu biti homogene, izrađene od stakla, monokristala ili prešanih kristala nekog spoja tzv. kristal-membranske elektrode, ili nehomogene. Nehomogene čvrste membrane se priređuju od smjese aktivne tvari u inertnom nosaču kao što je npr. silikonska guma, PVC ili pak nanošenjem aktivne tvari membrane na hidrofobizirani grafitni nosač, stvarajući tako membrane heterogene strukture.
Tekuće membrane za ion-selektivne elektrode pripremaju se iz ionskih izmjenjivača ekstrakcionom tehnologijom. Inertna membrana (filtar papir, sinterirano staklo, porozni teflon ili PVC, tekstilni materijal) može se učiniti selektivnom prema određenom ionu zasićivanjem s organskim sintetskim ionskim izmjenjivačem otopljenim u nepolarnom otapalu.
Kao referentna elektroda u sustavu za mjerenje može se upotrijebiti Ag/AgCl elektroda, zasićena kalomel-elektroda, zasićena Hg/Hg2SO4 elektroda ili npr. fluorid selektivna elektroda kod određivanja klorida. Referentna elektroda ne smije povećavati u ispitivanoj otopini koncentraciju iona koji se određuje.
Promjena potencijala registrira se na pION-metru, voltmetru visoke ulazne impedancije. Postupci određivanja su direktna potenciometrija i potenciometrijska titracija.
Rad s ion-selektivnim elektrodama ima apsolutnu prednost u pogledu brzine i raspona koncentracija određivanih iona.
Literatura 1. M. Metikoš-Huković, Elektrokemija, skripta, FKIT Zagreb, 2000.
2. I. Piljac, Elektroanalitičke metode, RMC, Zagreb 1995.
3. M. Mirnik, III. Potenciometrijska određivanja, u I. Filipović, P. Sabioncello (ur.), Laboratorijski priručnik, I. dio - knjiga 2., Tehnička knjiga, Zagreb 1978., str. 330-407

3. ELEKTROANALITIČKE METODE
___________________________________________________________________________ 35
3.1.1. POTENCIOMETRIJSKA TITRACIJA
Laboratorijska vježba
Određivanje masenog udjela acetilsalicilne kiseline u tableti aspirina potenciometrijskom titracijom
Zadatak:
1. Titrirati slabu acetilsalicilnu kiselinu jakom lužinom (NaOH) i odrediti točku
završetka reakcije: a) iz odnosa pH − V, b) iz odnosa ΔpH/ΔV − (Vi+Vi+1)/2, c) Granovom metodom
2. Odrediti maseni udio acetilsalicilne kiseline u uzorku tablete aspirina Instrumenti:
U potenciometrijskoj titraciji određuje se elektrokemijski točka završetka titracije. Slika 3.1 prikazuje uređaj za potenciometrijska određivanja.
Slika 3.1. Moderan uređaj za potenciometrijsku
titraciju firme Metrohm, Švicarska
- pH-metar MA 5705 Iskra Kranj, Slovenija
- pION metar, model E940 (Expandable Ion Analyser), Orion Research, USA
- kombinirana staklena elektroda HEC 0102; radno područje elektrode 2-10 pH (staklena i referentna Ag/AgCl elektroda u jednom tijelu), Iskra Kranj, Slovenija
- kombinirana staklena elektroda BlueLine, (staklena i referentna Ag/AgCl elektroda u jednom tijelu), radno područje pH 1-14, Schott, Njemačka
Uvjeti mjerenja:
- mjerno područje na pH-metru: 2-10 pH - temperatura: 25 oC - osjetljivost: 94 %

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 36
Princip određivanja acetilsalicilne kiseline u tableti aspirina: Aspirin se dobiva reakcijom OH-skupine salicilne kiseline s karboksilnom (-COOH) skupinom octene kiseline prema reakciji:
C OHO
CH3
OH
HOOC
+ CH3 C
O
O
HOOC
+ H2O
octena kiselina salicilna kiselina acetilsalicilna kiselina Acetilsalicilna kiselina je srednje slaba kiselina (Kk= 3,0*10-4). Reakcija neutralizacije teče prema jednadžbi:
+ + H2OOH-CH3 C
O
O
HOOC
CH3 C
O
O
-OOC
pri čemu jedan mol acetilsalicilne kiseline reagira s jednim molom lužine. Promjena koncentracije H+-iona u otopini mjeri se kombiniranom elektrodom (indikatorska staklena i referentna Ag/AgCl, u jednom tijelu), čiji je potencijal ovisan o aktivitetu H+-iona u otopini. Na pH-metru se nakon svakog dodatka volumena NaOH poznate koncentracije očita odgovarajuća pH vrijednost.
Ta ovisnost promjene pH vrijednosti otopine koja se titrira nije linearna (dobivena titracijska krivulja je karakteristična S-krivulja), pa se volumen u točki završetka titracije procjenjuje određivanjem točke infleksije krivulje. Točka infleksije krivulje može se odrediti pomoću tangenata ili prvom ili drugom derivacijom S-krivulje, pri čemu dolazi do izraza subjektivna procjena (posebno kod S-krivulje).
Granova metoda, koja određenim matematičkim transformacijama pretvara krivulju titracije u dva pravca koji se sijeku u točki završetka reakcije, je također metoda za određivanje točke završetka titracije. Metoda je vrlo jednostavna uz pretpostavku da su zadovoljeni neki uvjeti:
• analit i titrant ne sadrže karbonate, tj. drugi kiselo-bazični sustav, • analit ne taloži i ne otparava za vrijeme titracije, • pH je očitan tek nakon uspostavljanja ravnotežnog stanja u otopini, • nema oscilacija u temperaturi za vrijeme titracije, • ionska jakost otopine se bitno ne mijenja za vrijeme titracije i • pH-metar je ispravno kalibriran.
Nakon određivanja točke završetka reakcije iz volumena utrošene lužine za neutralizaciju odredi se masa acetilsalicilne kiseline. Iz određene mase acetilsalicilne kiseline i odvage tablete aspirina, izračuna se maseni udio acetilsalicilne kiseline u tableti aspirina.
Potrebne otopine:
- Standardna otopina NaOH:
c(NaOH)= 0,1 molL-1 (pripremljeno iz otopine c(NaOH) =1,0000 mol/L, Titrisol, E. Merck,
Darmstadt Njemačka) - Etanol, p.a. - pufer otopine (pH=5 i 9) za baždarenje kombinirane elektrode HEC 0102; radno
(3.8.)
(3.9.)

3. ELEKTROANALITIČKE METODE
___________________________________________________________________________ 37
područje elektrode je pH 2 do pH 10, Standard pufer Titrival, Kemika, Zagreb Hrvatska - tableta aspirina, acetilsalicilna kiselina (C9H5O4); M (C9H5O4)=180,2 gmol-1
Kalibracija instrumenta i elektrode: pH-metar i kombinirana elektroda HEC 0102 (2-10 pH) prije mjerenja se kalibrira/baždari
pufer otopinama poznate vrijednosti pH. Kombinirana elektroda prije kalibracije uronjena je u kiseli pufer ili po potrebi u otopini kloridne kiseline, c(HCl)=0,1 molL-1 najmanje 3 sata. Kod nekih tipova elektroda stakleni osjetljivi dio je zaštićen poklopcem od polimernog materijala koji održava vlažnim membranu elektrode. Određivanje nagiba pravca staklene elektrode:
Nagib pravca elektrode definiran je kao promjena E/mV kada se koncentracija vodikovih iona promijeni za deset puta, tj. za jednu pH jedinicu. Nagib pravca staklene elektrode određuje se s dva standardna pufera vrijednosti pH u području u kojem očekujemo promjenu kiselosti.
Kombiniranu elektrodu uroniti u otopinu pufera poznatog pH (1. pufer), na pH-metru dovesti čitač na tu vrijednost. Prebaciti skalu pH-metra na očitavanje potencijala u mV. Promijeniti pufer (2. pufer) i očitati novu vrijednost u mV (ne dirati regulator za standardizaciju). Izračunati nagib pravca za dva pufera poznatog pH.
Teoretska vrijednost nagiba je 59,1 mV. Ako dobivena vrijednost jako odstupa treba provjeriti sustav (pH-metar, elektrode, pufere). Ako i nakon provjere nagib nije u okviru dopuštenog odstupanja od teorerske vrijednosti promijeniti elektrodu.
Nakon kalibracije i provjere nagiba pravca elektroda se ispire destiliranom vodom i spremna je za praćenje promjene pH vrijednosti otopine koja se titrira. Kalibraciju instrumenta i elektrode obvezatno se provodi prije prvog mjerenja pH a po potrebi se kalibrira i tijekom radnog dana.
Priprema uzorka: Izvaganu tabletu aspirina staviti u čašu, smrviti staklenim štapićem, pipetom dodati 25
mL etanola (radi bolje topivosti acetilsalicilne kiseline) i 25 mL destilirane vode.
Titracija acetilsalicilne kiseline otopinom NaOH i praćenje promjene pH vrijednosti tijekom titracije
Uroniti kombiniranu elektrodu u otopinu tako da je razina otopine iznad (keramičkog) spoja elektrolita elektrode i vanjske otopine. U otopinu koja se titrira stavi se magnetni štapić, za intenzivno miješanje otopine na magnetskoj miješalici tijekom titracije.
Poslije stabilizacije elektrode očita se i zabilježi pH otopine (pH otopine acetilsalicilne kiseline u rastopljenoj tableti je oko 4). Zatim dodavati iz birete po 0,2 mL standardne otopine NaOH i svaki puta dozvoliti sustavu da se stabilizira te očitati vrijednost. Dodavanje standardne otopine nastaviti i poslije prelaza pH vrijednosti preko 7, sve dok se ne postigne vrijednost pH 10. Nacrtati krivulju titracije; na apscisu nanijeti volumen dodane NaOH, a na ordinatu odgovarajući pH.
Zbrinjavanje otpadnih kemikalija i pranje suđa i pribora: Sve otopine iz ove vježbe mogu se baciti u izljev nakon čega treba pustiti vodu.
Stakleno posuđe i pribor na kraju rada dobro isperite s vodovodnom vodom i zatim nekoliko puta deioniziranom vodom.

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 38
Rezultati i obrada podataka mjerenja:
1. dio vježbe:
• Skicirajte kombiniranu elektrodu s kojom ste mjerili, • Nacrtajte titracionu krivulju,
2. dio vježbe (u računalnoj učionici):
• Izračunajte i grafički prikažite prvu i drugu derivaciju S-krivulje, • Izračunajte i grafički prikažite Granovu metodu linearizacije S-krivulje, • Izračunajte i izrazite sadržaj acetilsalicilne kiseline u uzorku kao w (maseni udjeli) iz
grafičkih prikaza i prikažite u tablici. Objedinite rezultate u jednom laboratorijskom izvještaju

3. ELEKTROANALITIČKE METODE
___________________________________________________________________________ 39
3.1.2. DIREKTNA POTENCIOMETRIJA Princip određivanja:
Ion-selektivne elektrode (ISE) za određivanje klorida ili fluorida su prema sastavnim dijelovima iste kao i staklena elektroda selektivna na hidronium ion ali su materijali, posebno membrana, različiti. (slika 3.5.).
ISE za određivanje amonijevog iona je tzv. plinska ion selektivna elektroda koja se sastoji od vanjskog djela elektrode s polupropusnom membranom za plinoviti amonijak i unutarnje kombinirane staklene elektrode (slika 3.6.).
Slika 3.2. Shema potenciometrijskog mjerenja s ion selektivnom elektrodom
Slika 3.3. Karakteristična krivulja odnosa koncentracije ispitivanog iona prema potencijalu E/mV na log-lin dijagramu (os x logaritamska skala)
donja granica određivanja
keramička frita
unutarnja otopina
membrana
referentna elektroda ion-selektivna elektroda
Ag/AgCl elektrode

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 40
Laboratorijska vježba
Određivanje koncentracije iona u uzorku ion-selektivnim
elektrodama (ISE)
Zadatak: 1. Odredite nagib pravca ion selektivne elektrode (Cl-ISE, F-ISE, NH4-ISE), 2. Ispitajte utjecaj N03
− −iona na određivanje Cl−−iona, 3. Odredite linearno područje rada ISE, 4. Odredite koncentraciju klorida i/ili amonijevog iona u uzorku vode, 5. Odredite koncentraciju fluorida u pasti za zube, 6. Skicirajte ion-selektivnu elektrodu koju ste koristili
Instrumenti:
Digitalni pH/mV−metar, model 801/A, Orion Research, USA
pION metar, model E940 (Expandable Ion Analyser), Orion Research, USA

3. ELEKTROANALITIČKE METODE
___________________________________________________________________________ 41
3. pH metar MA 5736, Metrel, Horjul Slovenija
Slika 3.4. p-ION metri za potenciometrijska određivanja Elektrode: A. Ion selektivne elektrode: klorid selektivna elektroda, ORION model 94-17B fluorid selektivna elektroda, ORION model 94-09
amonij selektivna plinska elektroda, Metrohm, model 6.506.010
B. Referentne elektrode: Ag/AgCl elektroda, s dvije radne otopine (Duble Junction Reference Elektrode), model 90-02 Ag/AgCl elektroda, s jednom radnom otopinom (Single Junction Reference Elektrode), model 90-01
Slika 3.5. Shema ISE s krutom membranom

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 42
Slika 3.6. Elektroda za određivanje amonijaka i amonijevog iona (ISE osjetljiva na plinove)
Potrebne otopine:
− temeljna standardna otopina C1−−iona; otopina NaCI, c(NaC1) = 10−2 molL−1 (pripremljena)
− temeljna standardna otopina F−−iona: otopina NaF, g(NaF)=500 mg/L (pripremljena) − temeljna standardna otopina NH4
+−iona: otopina NH4Cl, γ (NH4C)=1000 mg/L (pripremljena)
− standardne otopine klorida, fluorida i amonijevog iona pripremaju se razrjeđenjem TSO (potrebno pripremiti)
− otopina ISA, otopina NaN03, c(NaN03) = 5 molL−1 − otopina TISAB II (Total Ionic Strenght Adjustment Buffer): u 500 mL destilirane vode
doda se 57 mL ledene octene kiseline, 58,0 g NaCl i 4 g CDTA (1,2 - cikloheksan-diamin-tetraoctena kiselina), zatim se polako dodaje NaOH, 5M, uz miješanje dok se ne postigne pH 5 -5,5. Ohladi se na sobnu temperaturu i nadopuni u tikvici do 1L (pripremljena)
− kruti NaOH p.a.
Priprema elektrode za mjerenje:
1. Ako je elektroda s krutom membranom (fluoridna ili kloridna) bila čuvana suha pripremi na slijedeći način: skine se kapica od polimernog materijala i pogleda površina ion osjetljivog elementa. Površina mora biti sjajna bez istaloženih drugih materijala. Ako je potrebno lagano i oprezno se površina izbrusi za to osiguranim brusnim papirom (asistent).
2. Ako je ISE osjetljive na plinove (amonijak) bila čuvana suha priprema se tako da se unutarnja elektroda (kombinirana staklena) ostavi stajati barem 6 sati (najbolje preko noći) u otopini pufera pH 7. Tako pripremljena unutarnja elektroda se zatim stavi u vanjsko tijelo elektrode, stavi se membrana propusna za plinove na vrh elektrode, zašarafi vrh i napuni s unutarnjom referentnom otopinom. Sada je elektroda spremna za mjerenje.
3. Elektrode (radna i referentna ili kombinirana) se zatim priključe se na instrument tako da se utikač referentne elektrode (igličasti utikač) i utikač osjetljive elektrode stave u za to predviđene utičnice na digitalnom pH/mV instrumentu ili nekom drugom specifičnom instrumentu. Instrumenti koji nisu ORION proizvodnje moraju imati specijalni adapter. Uvijek je preporučljivo konzultirati upute za rad i rukovanje instrumentom (asistent).

3. ELEKTROANALITIČKE METODE
___________________________________________________________________________ 43
Određivanje nagiba pravca ion selektivne elektrode:
Da bi se provjerilo stanje elektrode potrebno je odrediti nagib kalibracijskog pravca. Razlika potencijala za razliku koncentracije jedne dekade definira nagib pravca elektrode. Razlika će biti u području 54-60 mV/dekadi kada je temperatura pripremljenih otopina 25 °C.
Ako se ne postigne razlika potencijala u predviđenom rasponu pogledati u originalne upute koja daje proizvođač za elektrodu.
Određivanje linearnog područja (dinamičko područje) kalibracijske krivulje:
U 7 tikvica od 100 mL stavite određeni volumen ISE (kloridi) ili TISAB-a IV (fluoridi), volumen standardne otopine da koncentracija iona koji se određuje bude u rasponu od 1*10-6 M do 1*10-2 M i deionizirane vode do 100 mL. Prelijte otopine u čaše, uronite elektrode i pričekajte da se sustav stabilizira te očitajte potencijal.
Rezultate prikažite grafički za cijelo područje koncentracija. Očitajte dinamičko područje kalibracijske krivulje.
Određivanje utjecaja N03
−−iona na određivanje C1−−iona:
U niz odmjernih tikvica volumena 100 mL dodajte odabrani volumen otopine NaC1, c(NaCI)=10−2 molL−1 i volumene otopine N03
−−iona, NaN03, c(NaN03)=5 molL−1 u rasponu koncentracija N03
−−iona u otopini od 0,1 do 2 molL−1. Nakon stabilizacije napona zabilježite dobivenu vrijednost.
Rezultate unesite u tablicu 1 i nacrtajte promjenu potencijala s porastom koncentracije nitrata u mjernim otopinama. Veličinu razdiobe u grafičkom prikazu za os y (E/mV) ujednačite s prikazom za baždarni dijagram.
Kalibracija postupka za određivanje klorida i/ili fluorida:
Rezultate unesite u tablicu. Nacrtajte odnos potencijala prema koncentraciji klorid/fluorid-iona ili prema logaritmu koncentracije prema vlastitom izboru. Ako crtate odnos prema koncentraciji koristite lin-log papir.
Digitalni instrument pION metar, model E940 (Expandable Ion Analyser), Orion Research, USA omogućuje direktnu kalibraciju i očitavanje koncentracije klorida i/ili fluorida u nepoznatom uzorku.
Kloridi:
U odmjernim tikvicama od 100 mL pripremite otopine C1−−iona za izradu baždarnog dijagrama. Raspon koncentracija klorida određen je u eksperimentu određivanja dinamičkog područja odnosa potencijala prema negativnom logaritmu koncentracije (-log c). U svaku tikvicu dodaje volumen otopine NaN03 određen u eksperimentu ispitivanja utjecaja N03
−−iona na određivanje klorida i nadopunite do markice deioniziranom vodom.
Fluoridi:
U odmjerne tikvice od 50 mL pipetiraju se potrebni volumeni otopina F−−iona i u svaku dodaje 2 mL otopine TISAB-a II te nadopunite do markice deioniziranom vodom.. Raspon koncentracija fluorida određen je u prvom eksperimentu određivanja dinamičkog područja odnosa potencijala prema negativnom logaritmu koncentracije (-log c).

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 44
Kalibracija postupka za određivanje fluorida, fluorid-ionselektivnom elektrodom provodi
se na instrumentu s mogućnošću kalibracije s pet standarda pripremljenih razrjeđenjem temeljne standardne otopine prema propisu za određivanje fluorida.
Amonijev ion:
U odmjerne tikvice od 100 mL pipetira se potrebni volumen otopina NH4+−iona i te
nadopunite do markice deioniziranom vodom.. Raspon koncentracija amonijevog iona određen je u prvom eksperimentu određivanja dinamičkog područja odnosa potencijala prema negativnom logaritmu koncentracije (-log c). Standardna otopina se prenese u čašu u kojoj se mjeri te se uz konstantno miješanje doda 1 peleta krutog NaOH p.a. Otopina mora imati pH 12.
Priprema uzoraka:
Pripremite uzorak vode nepoznate koncentracije klorida i/ili amonijevog iona. Uzorci se pripremaju u tikvicama od 100 mL na isti način kao za mjerenja pri kalibraciji.
Pripremite uzorak paste za zube odvagom 500 ±0,1 mg paste u čašu od 250 mL Dodajte cca 40 mL destilirane vode, zagrijte na električnoj ploči i oprezno kuhajte 5 minuta. Ohladite, prenesite u odmjernu tikvicu od 50 mL, nadopunite do oznake destiliranom vodom. Alikvot od 10,0 mL prenesite u odmjernu tikvicu od 50 mL dodajte 2 mL TISAB-a nadopunite do oznake, prenesite u čašu i uz stalno miješanje očitajte kad se potencijal stabilizira. Zbrinjavanje otpadnih kemikalija i pranje suđa i pribora:
Ovi eksperimenti ne proizvode opasan otpad. Sve otopine iz ove vježbe mogu se baciti u izljev nakon čega treba pustiti vodu.
Stakleno posuđe i pribor na kraju rada dobro isperite s vodovodnom vodom i zatim nekoliko puta deioniziranom vodom.
Rezultati i obrada podataka mjerenja:
− skicirajte ISE s kojom ste mjerili, − odredite linearno područje kalibracijske krivulje za kloride, fluoride i /ili amonijev ion, − odredite potrebnu koncentraciju nitrata kod određivanja klorida, − nacrtajte odnos potencijala i koncentracije iona. Odredite koncentraciju iona u
nepoznatom uzorku, − odredite koncentraciju fluorid-iona u nepoznatom uzorku na temelju kalibracije s pet
standard

3. ELEKTROANALITIČKE METODE
___________________________________________________________________________ 45
3.2. KONDUKTOMETRIJA
Princip određivanja:
Električnu struju kroz otopinu prenose ioni. Pozitivni ioni kreću se prema negativnoj elektrodi, a negativni prema pozitivnoj elektrodi. Pri mjerenju provodnosti elektrolita koristi se izmjenična struja frekvencije 1000 Hz između elektroda dovoljno velike površine. Istosmjerna struja se ne može koristiti jer bi došlo do elektrolize, a osim toga javila bi se i polarizacija suprotnog smjera upotrebljenom članku. Za mjerenje provodnosti koristi se aparatura čija shema je prikazana na slici 3.7.
Električni otpor otopine elektrolita ovisi o: • broju prisutnih iona • naboju iona • sposobnosti ionske vrste da provodi struju • efektivnoj površini elektrode • udaljenosti između elektroda i • temperaturi otopine.
Tok struje prouzrokuje stvaranje veće ili manje količine topline, što ovisi o električnom otporu sustava. Kod otopina elektrolita povišenje temperature povećava provodnost električne struje, jer se smanjuje viskoznost i solvatizacija iona, a povećava disocijacija.
Mjera za provodnost elektrolita je recipročna vrijednost otpora 1/R i izražava se u simensima odnosno u "recipročnim omima" [ohm-1 = siemens (S)].
Slika 3.7. Shema uređaja za konduktometrijsku titraciju
Specifična, ekvivalentna i ekvivalentna provodnost kod beskonačnog razrjeđenja
Provodnost otopina G je recipročna vrijednost električnom otporu; jedinica je ohm-1 ili S.
GR
=1
(3.10.)
Ako je površina elektroda 1 cm2, a elektrode su na udaljenosti 1 cm, provodnost otopine, G, naziva se specifična provodnost, κ. Specifična provodnost, κ, je dakle provodnost kocke tekućine s bridom od 1 cm; jedinica je ohm-1 cm-1 ili S cm-1.
G Pl
= κ (3.11.)
Ekvivalentna provodnost, Λ, je definirana kao provodnost 1 mola otopljene tvari koja se nalazi između elektroda koje su udaljene 1 cm. Volumen i površina elektroda nisu

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 46
definirani. Direktno mjerenje ekvivalentne provodnosti provodi se vrlo rijetko, već se određuje indirektno iz vrijednosti izmjerenih specifičnih provodnosti.
Volumen otopine koji sadrži 1 mol otopljene tvari definiran je u cm3 kada je koncentracija c definirana u molovima po litri, odnosno volumen se može definirati i iz dimenzija ćelije pa onda imamo:
Vc
V l P= = ⋅1000 odnosno (3.12.)
Ako su elektrode na udaljenosti od 1 cm dobivamo da je ekvivalentna provodnost jednaka:
Λ =1000
cκ
(3.13.)
Dobiveni izraz dozvoljava izračunavanje ekvivalentne provodnosti iz eksperimentalnih vrijednosti κ za otopinu poznate koncentracije.
Ekvivalentna provodnost raste s razrjeđenjem. Za jake elektrolite, ako se odnos ekvivalentne provodnosti i drugog korijena iz koncentracije prikaže grafički, dobije se ekstrapolacijom tog odnosa na koncentraciju jednaku nula vrijednost ekvivalentne provodnosti kod beskonačnog razrjeđenja, Λo (tablica 3.1).
Tablica 3.1. Ekvivalentna provodnost Λ za otopinu NaCl c (NaCl) / mol L-1 Λ / Scm2mol-1 0,1 106,7
0,01 118,5
0,01 123,7
kod beskonačnog razrjeđenja 126,4 (Λo)
Kod beskonačnog razrjeđenja teoretski nema interakcije među ionima, te provodnost otopine ovisi samo o ionskoj provodnosti i jednaka je:
Λ00 0= ++ −λ λ (3.14.)
Vrijednosti za brojne katione i anione eksperimentalno su određene i mogu se naći u
priručnicima (tablica 3.2). Poznate vrijednosti ionske provodnosti omogućuju da se predvidi relativna provodnost otopina raznih tvari, a time je dana i mogućnost da se predvidi tok konduktometrijske titracije.
Tablica 3.2. Ionske provodnosti kod beskonačnog razrjeđenja kod 25 oC
Kationi λ+0 anioni λ−
0 H+ 349,8 OH− 198 Na+ 50,1 Cl− 76,3 K+ 73,5 NO3
− 71,4 Li+ 38,7 ClO4
− 68,0 NH4
+ 73,4 CH3COO− 40,9 Mg2+ 53,1 SO4
2− 79,8 Fe3+ 68 C2O4
2− 24

3. ELEKTROANALITIČKE METODE
___________________________________________________________________________ 47
Primjena direktnog mjerenja provodnosti u kvalitativnoj i kvantitativnoj analizi je
ograničena. zbog ne selektivnosti. Koristi se za praćenje ukupne minealizacije otopine (kvaliteta demineralizirane ili destilirane vode), kao detektor u kromatografijama te za za određivanje točke završetka titracije. Za razliku od potenciometrijske titracije gdje se koriste elektrode koje reagiraju samo na titrirani sustav, kod konduktometrijskie titracije svi prisutni ioni sudjeluju u provođenju naboja kroz otopinu. Kod konduktometrijske titracije mjeri se specifična provodnost otopine nakon dodavanja standardne otopine, a dobivene vrijednosti se unose u dijagram prema volumenu potrošene standardne otopine. U pravilu se na taj način dobivaju pravci koji se sijeku u točki ekvivalencije.
Ćelije za mjerenje provodnosti Ćelije za mjerenje provodnosti moraju biti od netopljivog materijala. Elektrode su
najčešće od platine ili legure platine i iridija. Ako otopine nisu jako kisele i nije prisutna dušična kiselina elektrode mogu biti i od srebra. U alkalnim otopinama se upotrebljavaju V2A čelici, te ugljen i grafit. Ipak elektrode od platine su najčešće u upotrebi. To su obično platinirane platinske elektrode da bi se povećala efektivna površina i minimalizirali kapacitivni efekti.
Ćelije za mjerenje provodnosti otopina (slika 3.8.) moraju osim toga imati elektrode sa stalnom geometrijom .
Slika 3.8. Različiti oblici ćelija za mjerenje provodnosti
Određivanje konstante ćelije
Kod konduktometrijskih mjerenja najčešće je relativna provodnost κ vrijednost koja nam je potrebna. Vrijednost κ je proporcionalna provodnosti otopine, a ovisi o površini elektroda i njihovoj međusobnoj udaljenosti. Taj odnos, konstantan za svaku konduktometrijsku ćeliju, naziva se konstanta ćelije.
⋅==CC
GCCl
P κ 1 (3.15.)

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 48
Tablica 3.3. Specifične provodnosti otopina KCl kod 25 oC
m (KCl)/1000 g otopine κ (25 oC) S cm-1
71,1352 0,111342
7,41913 0,0128560
0,745263 0,00140877 Konstanta ćelije (CC) određuje se eksperimentalno pomoću otopina poznate
koncentracije pri određenoj temperaturi čija je specifična provodnost poznata iz literature. Za tu svrhu najčešće se koristi otopina KCl (tablica 3.3).
1. Kiselo-bazne titracije Kod reakcija neutralizacije moguće je uspješno koristiti konduktometrijsko određivanje
točke završetka titracije zbog visoke provodnosti hidronium i hidroksidnih iona u odnosu prema provodnosti produkata reakcije. Konduktometrijski je moguće odrediti točku završetka titracije kod titracije jakih kiselina (slika 3.9.a) ili baza, titracije slabih kiselina (slika 3.9.c) ili baza, titracija dvoprotonskih kiselina (slika 3.9.d i 3.9.e) i titracije soli slabih kiselina ili baza.
Također je moguće odrediti sastav smjese dviju kiselina različite jakosti. Konduktometrijska titracija takove smjese najčešće je mnogo točnija od potenciometrijske titracije.
volumen jake baze / mL
prov
odno
st / μS
volumen slabe baze / mL
prov
odno
st / μS
volumen jake baze / mL
prov
odno
st / μS
3
21
volumen slabe baze / mL
prov
odno
st / μS
3
21
volumen slabe baze / mL
prov
odno
st / μS
Slika 3.9. Titracije otopina kiselina: a) jaka kiselina s jakom bazom, b) jaka kiselina sa slabom bazom,
c) slaba kiselina s jakom bazom, d) dvoprotonska kiselina s jakom bazom, e) dvoprotonska kiselina sa slabom bazom.
Za razliku od potenciometrijskih titracija gdje se gotovo isključivo kao standardna
otopina s kojom se titrira koristi jaka kiselina ili jaka baza, kod titracija s konduktometrijskim određivanjem točke završetka titracije mogu se koristiti i slabe baze (slika 5.4b i 5.4e) odnosno slabe kiseline.
2. Taložne titracije i titracije nastajanja kompleksa Konduktometrijska titracija može se primijeniti kod taložnih titracija i titracija u kojima
nastaju kompleksi. Međutim, postoji čitav niz ograničenja obzirom na komplikacije koje mogu
A B C
D E
1 2 1
12
2

3. ELEKTROANALITIČKE METODE
___________________________________________________________________________ 49
nastati tokom reakcije: adsorpcija na talogu, taloženje na elektrodi, nestabilnost kompleksa, sporost taložnih reakcija itd.
Ipak, u taložnim reakcijama i u reakcijama gdje nastaju kompleksi, primjenjuje se konduktometrijska titracija. To su uglavnom titracije sa srebrovim i olovnim nitratom, barijevim acetatom ili kloridom, te otopinama litijeva sulfata ili oksalata, kalijeva ferocijanida i kalijeva fericijanida, barijevog hidroksida.
Kontrola temperature Temperaturni koeficijent kod konduktometrijskih mjerenja je oko 2% po promjeni
temperature od 1 oC. Zbog toga je kod konduktometrijskih titracija potrebna kontrola temperature. Najčešće je dovoljna primjena vodene ili uljne kupelji na temperaturi oko sobne.
Kontrola volumena Za vrijeme titracije povećava se volumen otopine. Zbog toga je potrebno korigirati
volumen jer se u protivnom dobiju ne linearne titracione krivulje. Grešku zbog promjene volumena moguće je smanjiti i tako da se koristi standardna otopina čija je koncentracija cca 10 puta veća od koncentracije otopine koja se titrira.
Krivulje titracije Da bi se odredila točka ekvivalencije kod konduktometrijskih titracija potrebno je
odrediti nekoliko točaka prije i nekoliko točaka nakon točke ekvivalencije. Broj određivanja je relativno mali. Nakon korekcije volumena vrijednosti provodnosti se nanose u dijagram kao funkcija volumena dodatka standardne otopine. Dva linearna dijela dijagrama se ekstrapoliraju, a sjecište produženih pravaca odgovara točki ekvivalencije. Oko točke ekvivalencije dobivene vrijednosti odstupaju od pravca, te se stoga koriste mjerene vrijednosti koje su dovoljno daleko od točke ekvivalencije.
Smanjenje pogreške kod konduktometrijskih titracija Da bi se smanjila pogreška pri konduktometrijskim tiracijama potrebno je dobro
odabrati otopinu za titraciju. Točka završetka titracije će biti određena to točnije što je manji kut koji zatvaraju pravci na dijagramu provodnost − volumen standardne otopine. To se može postići odabirom iona pogodne gibljivosti.
Kod taložnih titracija potrebno je smanjiti topljivost taloga što se često postiže dodatkom 30-40% alkohola. Ako talog nastaje polagano može se otopina cijepiti kristalima taloga itd. Neke pogreške gotovo je nemoguće izbjeći, kao na pr. uslijed okluzije pa se te taložne reakcije ne mogu koristiti kod konduktometrijskih titracija.

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 50
3.2.1. KONDUKTOMETRIJSKA TITRACIJA
Laboratorijska vježba
Određivanje koncentracije slabe i jake kiseline u smjesi mjerenjem
specifične provodnosti otopine pri titraciji s jakom lužinom
Zadatak:
1. Odrediti konstantu ćelije
2. Titrirati otopinu HCl, HAc i smjesu HCl i HAc otopinom NaOH, te iz grafičkog prikaza ovisnosti specifične provodnosti o volumenu standardne otopine odrediti točke završetka titracija.
3. Izračunati γ (HCl) / mg mL-1 i γ (HAc) / mg mL-1 u smjesi Potrebni reagensi:
− otopina KCl poznate koncentracije za određivanje konstante ćelije − standardna otopina NaOH; c(NaOH)=0,1 molL-1; f=1,0000 (Titrival, Merck
Darmstadt, Njemačka)
Instrumenti:
• Mikroprocesorski laboratorijski konduktometar MA 5964 Iskra Kranj, Slovenija
• ćelija za mjerenje vodljivosti (za uranjanje)
Slika 3.10. Mikroprocesorski laboratorijski konduktometar
Uvjeti mjerenja:
− Vukupno 200 mL − temperatura: 25 oC − miješanje
Određivanje konstante ćelije: Otopinu KCl poznate koncentracije (termostatiranu na zadanu temperaturu) stavi u
čašu s dvostrukim stjenkama. Uključi termostat i otvori protok vode iz termostata kroz dvostruke stjenke. Uključi magnetsku miješalicu ispod čaše.
Pronađi u tablicama vrijednost za specifičnu vodljivost κ otopine i prema uputstvu za određivanje konstante ćelije (CC) unesi specifičnu vodljivost u program konduktometra i očitaj konstantu ćelije.
Sva daljnja mjerenja se provode uz tu određenu konstantu ćelije.

3. ELEKTROANALITIČKE METODE
___________________________________________________________________________ 51
Titracija jake kiseline, slabe kiseline i smjese slabe i jake kiseline:
Alikvote otopina nepoznate koncentracije titrira se standardnom otopinom NaOH.
Iz volumena dodanog NaOH u točkama završetka titracije odredite koncentraciju slabe i jake kiseline u smjesi. Zbrinjavanje otpadnih kemikalija i pranje suđa i pribora:
Ovi eksperimenti ne proizvode opasan otpad. Sve otopine iz ove vježbe mogu se baciti u izljev nakon čega treba pustiti vodu.
Stakleno posuđe i pribor na kraju rada dobro isperite s vodovodnom vodom i zatim nekoliko puta deioniziranom vodom.
Rezultati i obrada podataka mjerenja:
• Skicirajte (blok shema) instrument na kojem ste mjerili,
• Nacrtajte titracione krivulje za sve ispitivane otopine,
• Izračunajte sadržaj jake i slabe kiseline u nepoznatom uzorku iz grafičkog prikaza.
Literatura 4. I. Piljac, Elektroanalitičke metode, RMC, Zagreb 1995
5. S. Ašperger, IV. Konduktometrijska analiza, u I. Filipović, P. Sabioncello (ur.), Laboratorijski priručnik, I. dio - knjiga 2., Tehnička knjiga, Zagreb 1978, str. 408-451

INSTRUMENTALNA ANALIZA
____________________________________________________________________________________ 52
4. INSTRUMENTALNE METODE SEPARACIJE
Instrumentalne metode separacije obuhvaćaju kromatografije: plinsku, tekućinsku (tekućinska visoke djelotvornosti, ionska, isključenjem), kromatografiju s fluidom pri superkritičnim uvjetima, elektroforezu a tu bi se svakako trebala uvrstiti i masena spektrometrija.
4.1. KROMATOGRAFIJE Opći pojmovi
Kromatografija je postala jedna od najvažnijih metoda analitičke kemije kojom se sastojci smjese mogu razdvojiti te u tekućoj ili plinovitoj fazi ili u fluidu pri superkritičnim uvjetima identificirati i kvantificirati.
Kromatografski sustav čine pokretna ili pokretna faza, nepokretna ili nepokretna faza i uzorak koji treba razdvojiti. Po IUPAC-ovoj (Union of Pure and Applied Chemistry) definiciji kromatografija je: « fizikalna metoda separacije u kojoj se sastojci koje treba razdvojiti raspodjeljuju između dvije faze, jedne koja je nepokretna dok se druga kreće (pokretna faza ili eluens) u određenom smjeru «(slika 4.1).
6
Prvi objašnjeni rad u kojem je opisan postupak odjeljivanja sastojaka smjese koja prolazi kroz stupac adsorbensa, nazvan kromatografijom, objavio je M. Cvet 1906. (slika 4.2). Tek od 1931. godine kada su R. Kuhn i E. Lederer opisali odjeljivanje karotena, kromatografski se postupci počinju šire primjenjivati.
A.J.P. Martin i R.L.M. Synge (1941.) prvi su upotrijebili tekućinu kao nepokretnu fazu nanijetu u tankom sloju na podlogu velike površine uz tekućinu kao mobilnu fazu.. Uveli su i objasnili pojam razdjelne kromatografije, uveli teoriju tavana u kromatografiju i 1952. za teoriju razdjelne kromatografije dobili Nobelovu nagradu.
7
Primjenom plina kao pokretne faze i tekućine kao nepokretne faze otvorili su A.J.P. Martin i A.T. James 1951. godine novo područje - plinsku kromatografiju.
7
E.Stahl je 1958. opisao mogućnost kromatografskog razdvajanja u kojem je adsorbens nanesen u tankom sloju na staklenu ploču - tankoslojnu kromatografiju.
Prva knjiga o kromatografiji objavljena je 1936., prvi plinski kromatograf proizveden je 1952. a instrumentalna tekućinska kromatografija počela se raditi oko 1965.
Zahvaljujući svojoj širokoj primjenjivosti i jednostavnosti, kromatografija je danas postala rutinska metoda i ima najširu primjenu: analiza okoliša, biomedicinska ispitivanja, forenzička analiza, farmaceutska ispitivanja čistoće lijekova, praćenje reaktanata i produkata sinteze u organskoj sintezi itd. Kromatografijom se mogu izolirati i pročišćavati najrazličitiji uzorci.
6 Kromatografsko nazivlje, IUPAC preporuke 1993. i 1998., HINUS i Sekcija za
kromatografiju HDKI, Zagreb 1999 7 L.S. Ettre, Milestones in chromatography – The birth of patrtition chromatography,
LC-GC 19(5), (2001), http://www.lcgcmag.com/lcgc/data/articlestandard/lcgc/462001/2090/article.pdf
Slika 4.1. Raspodjela između
dviju faza1: a) dinamička ravnoteža molekula između mirujućih faza, b) model kromatografskog procesa
Mihovil Semjonovič Cvet (1872-1919)

4. INSTRUMENTALNE METODE SEPARACIJE
____________________________________________________________________________________ 53
Princip se osniva na ravnotežnoj razdiobi između dviju faza;
nepokretne imobilizirane u koloni ili na ravnoj podlozi i pokretne koja nosi sastojke smjese kroz sustav (slika 4.1). Različita brzina putovanja komponenata dovodi do njihove separacije. Dvije su teorije kojima se tumači separacija u kromatografiji: teorija tavana i kinetička teorija. Teorija tavana tumači da je separacija definirana, kao prolaz otapala kroz imaginarni odsječak i posljedica je afiniteta otopljene tvari, tavani su definirani visinom i brojem (mjera efikasnosti separacije):
NLH = (4.1.)
gdje je H (ili HEPT) visina teoretskog tavana, N je broj tavana a L je duljina puta.
Prema fizičko-kemijskim fenomenima na kojima se osnivaju, kromatografije mogu biti adsorpcijske (nepokretna faza krutina), razdjelne (nepokretna i/ili pokretna faza tekućina), iono-izmjenjivačke (nepokretna faza ionski izmjenjivač) ili kromatografje isključenjem po veličini čestica (nepokretna faza gel, molekularna sita), no razdvajanje je najčešće kompeticija više mehanizama i ne može se u potpunosti opisati jednim matematičkim modelom već su algoritmi koji opisuju razdvajanje uglavnom empirijski. Ispitivani uzorak (smjesa tvari) uvodi se sa strujom plina, tekućine ili fluida pod superkritičnim uvjetima u kromatografsku kolonu ispunjenu krutom ili tekućom stacionarnom fazom, ili se nanosi na ravni sloj.
Kromatografija na koloni
Kod kromatografija na koloni duljine puta za sve sastojke smjese su iste (duljina kolone) i definirane odabirom kolone, a vrijeme zadržavanja na koloni je, kad je ostvarena dobra separacija, karakteristična veličina sastojke smjese i za svaki pojedini sastojak je različito.
Odijeljeni sastojke na izlazu iz kolone pomiješane samo s mobilnom fazom ulaze u detektor. Izlazni signal iz pojačala je ulazni signal za zapisni uređaj (pisač).
Vrijeme zadržavanja tR je vrijeme koje prođe od trenutka ubacivanja uzorka u injektor do vremena kada se zapisuju maksimalne vrijednosti kromatografskog maksimuma a ovisi o vrsti punila u koloni kao i o drugim radnim uvjetima; temperaturi kolone, protoku pokretne faze. Vrijeme tM je tzv. Mrtvo vrijeme, tj. Vrijeme od trenutka injektiranja potrebno da sama pokretna faza prođe kroz kolonu. Visina kromatografskog maksimuma je udaljenost između vrha kromatografskog maksimuma i osnovne linije, a udaljenost između sjecišta osnovne linije s tangentama povučenim u točkama infleksije označava se s w i predstavlja širinu kromatografskog maksimuma (slika 4.3.).
Vrijeme zadržavanja odijeljenog sastojka u koloni karakteristično je za svaki sastojak, a visina kromatografskog maksimuma i/ili površina ispod njega proporcionalna je količini sastojka.
Izgled idealnog kromatograma je isti kao normalna raspodjela slučajne pogreške (Gausova raspodjela), dok kod realnih kromatograma pikovi često imaju izgled ne-Gausove raspodjele (slika 4.4.).
Za kvantificiranje separacije između dva kromatografska maksimuma upotrebljava se faktor razdvajanja koji se može izračunati iz kromatograma prema izrazu:
Slika 4.3. Prikaz vremena
zadržavanja tM i tR u koloni
Slika 4.4 Izgled
kromatograma kod adsorpcijske raspodjele: a-Gaussova raspodjela, b,c,d-ne Gaussove raspodjele
a
b
d
c

INSTRUMENTALNA ANALIZA
____________________________________________________________________________________ 54
21
122wwtt
Rs RR
+
−= (4.2.)
Faktor zadržavanja daje odnos ukupne mase tvari razdijeljene na dvije vrijednosti; mase u mobilnoj fazi mM i mase u stacionarnoj fazi mS. Faktor je neovisan o duljini kolone i brzini protoka plina ali varira ovisno o uvjetima eksperimenta i najvažniji je parametar u kromatografiji za određivanje ponašanja kolone. Ne smije biti preveliki jer je u tom slučaju vrijeme trajanja separacije jako produženo.
k mm
K VV
S
M
S
M
= = (4.3.)
Faktor separacije dozvoljava usporedbu između dvije otopljene tvari na istom kromatogramu i jednak je odnosu faktora zadržavanja dviju sastojakai na istom kromatogramu.
α = =tt
kk
R
R
''
2
1
2
1
(4.4.)
Plošna kromatografija Plošne kromatografije pripadaju u grupu tekućinskih kromatografija,
pokretna faza je tekućina a nepokretna faza je papir ili tanki sloj sorbrnsa nanesen na podlogu koja je najčešće staklo ali može biti i metalna ili polimerna folija. Ime se odvodi od sorbensa (nepokretna faza): papirna, tankoslojna, gel kromatografija.
Kod plošnih kromatografija vrijeme potrebno za razvijanje kromatograma je isto za sve sastojke smjese a duljine puta koje prođu pojedini sastojci su, kad je ostvarena dobra separacija, karakteristične veličine i različiti za svaki sastojak smjese.
Faktor razdvajanja u plošnoj kromatografiji je omjer puteva koje su prošli sastojci smjese i puta koje je prošlo otapalo a označava se s RF, lA je put koji je prošao sastojak smjese a L duljina puta otapala od linije starta do fronte (slika 4.5):
Ll
R AF = (4.5.)
Za procjenu uspješnosti separacije također se može upotrebiti faktor razdvajanja (Rs) koji se može izračunati između dvije mrlje na kromatografskoj pločici ili na papiru iz kromatograma prema izrazu:
21
122wwRR
Rs FF
+
−= (4.6.)
Literatura 4. M. Kaštelan-Macan, Kemijska analiza u sustavu kvalitete, Školska
knjiga, Zagreb 2003., str. 217-237.
5. Đ. Deur-Šiftar, D. Štefanović, Z. Šoljić, Tehnička enciklopedija, Svezak 7., Leksikografski Zavod, Zagreb 1992., str. 387-395.
Slika 4.5. Razdvajanje u plošnoj kromatografiji
Slika 4.6. Uređaj za automatsko nanošenje uzoraka u tankoslojnoj kromatografiji

4. INSTRUMENTALNE METODE SEPARACIJE
____________________________________________________________________________________ 55
3. S. Turina, Tankoslojna kromatografija, SKTH/Kemija u industriji, Zagreb, 1984
4. S. Turina, Kromatografska analiza, u I. Filipović, P. Sabioncello (ur.), Laboratorijski priručnik, I. dio-knjiga 2., Tehnička knjiga, Zagreb 1978, str.485-498.
5. Đ. Deur-Šiftar, Plinska kromatografija, u I. Filipović, P. Sabioncello (ur.), Laboratorijski priručnik, I. dio-knjiga 2., Tehnička knjiga, Zagreb 1978, str.557-601.
6. T. Bićan-Fišter, Metode plošne kromatografije, u I. Filipović, P. Sabioncello (ur.), Laboratorijski priručnik, I. dio-knjiga 2., Tehnička knjiga, Zagreb 1978, str.499-537.

INSTRUMENTALNA ANALIZA
____________________________________________________________________________________ 56
4.1.1. PLINSKA KROMATOGRAFIJA Princip određivanja:
Plinska kromatografija je kromatografija kod koje je pokretna faza plin, a nepokretna faza tekućina ili krutina u koloni. Prema fizičko-kemijskim fenomenima na kojima se osnivaju kromatografije, plinska kromatografija je adsorpcijska (plin-krutina) ili razdjelna (plin-tekućina) kromatografija.
Slika 4.7. Moderni plinski kromatograf s automatskim dodavačem uzoraka firme Shimadzu, Japan.
Ispitivani uzorak (smjesa tvari) uvodi se sa strujom inertnog plina nosioca u kromatografsku
kolonu ispunjenu krutom ili tekućom stacionarnom fazom. Tekuća stacionarna faza može biti adsorbirana na inertnom čvrstom nosaču-diatomejskoj zemlji ili može biti imobilizirana na stijenke kapilare (kapilarne kolone).
Ako je stacionarna faza tekućina prolazom kroz kolonu smjesa tvari se razdjeljuje između nepokretne tekuće faze i pokretne faze plina nosioca. Odjeljivanje smjese tvari provest će se pod uvjetom da imaju različitu topljivost u tekućoj stacionarnoj fazi pa će i eluiranje teći prema njihovoj topljivosti. Prva komponenta koja izlazi iz kolone je najslabije topljiva u stacionarnoj fazi.
Odjeljene komponente na izlazu iz kolone pomješane su samo s inertnim plinom (mobilnom fazom) i ulaze u detektor. Detektor može biti plameno ionizacijski (slika 4.14.), detektor termičke vodljivosti, foto-ionizacijski, maseni spektrometar itd. Izlazni signal iz pojačala je ulazni signal za zapisni uređaj (pisač). Blok shema plinskog kromatografa prikazana je na slici 4.8. gdje je t’Ri vrijeme zadržavanja, a wi je širina pika.

4. INSTRUMENTALNE METODE SEPARACIJE
____________________________________________________________________________________ 57
Laboratorijska vježba
Analiza smjese organskih tvari plinskom kromatografijom
Zadatak: 1. Snimiti kromatogram,
2. Iz kromatograma odrediti vrijeme zadržavanja tR za pojedine komponente,
3. Iz kromatograma odrediti faktore rezolucije Rs,
4. Iz visine pika ili površine pod pikom odrediti metodom unutarnjeg standarda koncentraciju pojedinih komponenata smjese.
Instrument:
Plinski kromatograf PYE UNICAM GCV. Blok shema plinskog kromatografa prikazana je na slici 4.8 a sliku plinskog kromatografa prikazuje slika 4.9.
Slika 4.8. Blok dijagram plinskog kromatografa PYE UNICAM GCV, Cambridge,UK
Slika 4.9. Slika plinskog kromatografa PYE UNICAM GCV, Cambridge,UK
PLIN NOSILAC INJEKCIJSKI
BLOK KOLON
A
DETEKTOR (plameno ionizacijski)
UZORAK PISAČ
H2 O2 ili zrak
termostatirano -80 do 450 oC

INSTRUMENTALNA ANALIZA
____________________________________________________________________________________ 58
Slika 4.10. Shema injektora (lijevo) i načina propreme alikvota uzorka za GC (desno)
Slika 4.11. Petlja za ručno unošenje uzorka na kolonu
Slika 4.12. Utjecaj brzine aplikacije uzorka na kolonu na separaciju komponentu smjese
uzorak uzorak
ventil ventil
plin nosilac plin nosilac
na kolonu na kolonu
sporo brzo

4. INSTRUMENTALNE METODE SEPARACIJE
____________________________________________________________________________________ 59
Slika 4.13. Kolone za plinsku kromatografiju: punjena kolona (lijevo) i kapilarna kolona (desno)
Slika 4.14. Shema plameno ionizacijskog detektora
Uvjeti rada:
Kolona A: OV-17 943 H (1248-21 Q-3) za odjeljivanje eteričnih ulja Kolona B: E 30 02921 ( 12428-20-3) za odjeljivanje ugljikovodika Položaj indikatora na rotametru i manometrima (dimenzija jedinice pritiska u kolonama na
instrumentu je bar) preračunajte iz Pa u bar da možete zadati odgovarajuće pritiske instrumentu.
.................................................................................................... N2 / bar H2 / bar zrak / bar rotametar manometar manometar
................................................................................................... Kolona A 1,5 0,35 0,40 Kolona B 1,6 0,40 0,72
.................................................................................................... Temperatura injektora: 240 °C Temperatura kolone: početna 90 °C, konačna 160 °C Temperatura detektora: 170 °C Volumen (uzorka) = 0,15 μL Brzina (pisača) = 600 mm / sat = 10 mm / min Osjetljivost = 10 mV (pikovi moraju po mogućnosti ostati unutar papira) Pojačalo postavljeno na 2102x a birač Back off na +1 Temperaturni program kolone: 90 °C / 2 min → 10 °C / min do 160 °C → 160 °C / 10 min → hlađenje
sakupljačka elektroda
zrak vodik H2, N2
ili A
kolona
ampermetar

INSTRUMENTALNA ANALIZA
____________________________________________________________________________________ 60
Kalibracijski postupak:
Metoda unutarnjeg standarda.
Rezultati i obrada podataka mjerenja:
• Skicirajte (blok shema) instrument na kojem ste mjerili.
• Nacrtajte baždarni dijagram za izmjerene standardne otopine.
• Nacrtajte dijagram za metodu unutarnjeg standarda i usporedite s baždarnim dijagramon iz čistih standardnih otopina.
• Izračunajte i izrazite u μg/mL sadržaj ispitivanih komponenti uzorku.

5.DODATAK
___________________________________________________________________________ 61
5. DODATAK UPUTE ZA RAD U LABORATORIJU Laboratorijski izvještaj i ocjenjivanje Za svaku vježbu potrebno je pisati laboratorijski izvještaj. Laboratorijski izvještaj piše se prema dobivenom uputstvu za pisanje izvještaja. Svi laboratorijski izvještaji se predaju nakon završenog eksperimenata a prije početka slijedeće vježbe. Laboratorijske izvještaje pregledava i ocjenjuje asistent, voditelj vježbi. Svako kašnjenje u predaji izvještaja ima za posljedicu zabranu pristupa sljedećoj vježbi i smanjenje ocjenu 10% po danu kašnjenja. Laboratorijske izvještaje treba pisati na predlošku koji se dobije od asistenta uz svaku vježbu, piše se tintom, izvještaji pisan olovkom bit će vraćeni studentu. Laboratorijska bilježnica i ostali pribor Za rad na vježbama potrebna je laboratorijska bilježnica u koju se upisuju sva mjerenja, opažanja, bilješke potrebne za izradu laboratorijskog izvještaja. Osim laboratorijske bilježnice svaki student treba imati kutu i pribor za pisanje. Grupe Polaznici vježbi podijeljeni su u grupe po 6-10 studenata prema rasporedu. Prisustvovanje vježbama je obavezno u terminima grupa i svaka promjena se mora unaprijed dogovoriti s asistentom voditeljem vježbi. Korištenje mobilnih uređaja kao i konzumiranje hrane i pića nije dozvoljeno u laboratoriju.

INSTRUMENTALNA ANALIZA
___________________________________________________________________________ 62
PISANJE LABORATORIJSKIH IZVJEŠTAJA Laboratorijski izvještaj treba biti napisan jasno, sažeto i treba sadržavati:
1. Sažetak rada 2. Teorijski uvod 3. Eksperimentalni dio 4. Rezultate i raspravu 5. Popis literature korištene pri izradi izvještaja
1 Sažetak rada
• Sažetak treba biti napisan sažeto i jasno. U tri do četiri rečenice sažetak bi trebao navesti cilj ispitivanja, alate s kojima se dolazi do cilja (metoda) i rezultat dobiven ispitivanjem.
2 Teorijski uvod
• Sastoji se iz kratkog uvoda s teorijskim osnovama. Teorijske činjenice potkrepljene su literaturnim navodom u uglatim zagradama [1].
3 Eksperimentalni dio Mora sadržavati
• popis aparatura i svih kemikalija: standardne otopine, reagensi, otapala, • opis i blok shemu instrumenta, • pripremu otopina i uzoraka, • kratki opis eksperimenta.
4 Rezultati Uključuju grafove, slike i tablice. Sirove podatke mjerenja i izračunate rezultate dobivene iz podataka mjerenja, te raspravu o dobivenim rezultatima.
5 Literatura Literaturne reference trebaju biti označene brojevima redoslijedom kako se javljaju u radu.
- refrenca iz časopisa:
1. A. Novak, S. Danko i B. Utorak, Određivanje željeza u uzorcima tla s područja sjevero-zapadne Hrvatske, Kemija u industriji 23 (3), 101-105 (1995)
- refrenca iz knjige:
2. A. Novak, Instrumentalna analiza, Školska knjiga Zagreb, 1999., str. 459-470
- za svaku drugu referencu pitaj asistenta ili voditelja laboratorija.
Literatura
2. Vlatko Silobrčić, Kako sastaviti, objaviti i ocijeniti znanstveno djelo, 5. dopunjeno izdanje, Medicinska naklada, Zagreb 2003

Sveučilište u Zagrebu Fakultet kemijskog inženjerstva i tehnologije ZAVOD ZA ANALITIČKU KEMIJU Zagreb, Marulićev trg 20
Akademska godina: 2008. / 2009. Vježba broj: ______________
PROCESNA I INSTRUMENTALNA ANALIZA Laboratorijski izvještaj
Naslov vježbe: ______________________________________________________________
Ime i prezime: ______________________________________ Grupa:_________________ Referat predan dne: _________________________________ Imena ostalih studenata u grupi: _______________________ _______________________ _______________________ _______________________ _______________________ _______________________ Potpis: ____________________________
(ne ispunjavati ispod crte) ___________________________________________________________________________ Referat pregledala/o: ___________________________ datum: ________________________ Ocjena referata: _______________________________ Bodovna lista: ________________ / ( 3 ) Sažetak ________________ / ( 3 ) Rezultati ________________ / ( 2 ) Statistička obrada rezultata (ANOVA)
(samo za neke vježbe) ________________ / ( 1 ) Literatura ________________ / ( 1 ) Opći dojam ( stil, urednost) ________________ / ( 10 ili 8 ) Ukupno




















