
Studies on peptide fragments of prion proteins
31
STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS BY FABRIZIO TAGLIAVINI, GIANLUIGI FORLONI,* PASQUALINA D’URSI,* ORSO BUGIANI, AND MARIO SALMONA* Istituto Nazionale Neurologico Carlo Besta and *Istituto di Ricerche Farmacologiche Mario Negri, Milano, Italy I. Introduction. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 171 II. The Amyloid Peptides of Gerstmann-Sträussler-Scheinker Disease . . . . . . . . . 174 III. Unraveling the Conformational Conversion of PrP C to PrP Sc Using Synthetic Peptides . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 175 A. The Flexible N-terminal Domain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 177 B. The Globular Domain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 182 IV. Unraveling the Pathogenesis of Prion Diseases Using Synthetic Peptides . . . 185 A. Interaction of PrP Peptides with Cell Membranes . . . . . . . . . . . . . . . . . . . . 185 B. Effects of PrP Peptides on Neuronal Cultures . . . . . . . . . . . . . . . . . . . . . . . 187 C. Effects of PrP Peptides on Astroglial and Microglial Cultures . . . . . . . . . . 194 V. Concluding Remarks . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 196 References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 196 I. INTRODUCTION The prion diseases are neurodegenerative disorders of humans and animals that are sporadic or inherited in origin and can be transmitted (Prusiner, 1991). Despite remarkable differences in phenotypic expres- sion, these disorders share a similar pathogenic mechanism, that is, a posttranslational modification of the prion protein from a normal cel- lular isoform (PrP C ) to disease-specific species (PrP Sc ). Nuclear mag- netic resonance (NMR) studies of recombinant murine, hamster, bovine, and human PrP indicate that the normal protein is composed of two structurally distinct moieties: an extended N-terminal segment (residues 23–125) with features of a flexibly disordered polypeptide chain, and a well-defined globular domain (residues 126–231) with three α helices and two-stranded antiparallel β sheet (Fig. 1) (Riek et al., 1996; Riek et al., 1997; Donne et al., 1997; Lopez Garcia et al., 2000; Zahn et al., 2000). The PrP C >PrP Sc transition involves a profound con- formational change with decrease in α-helical secondary structure (~40% > 30%) and striking increase in β-sheet content (~3%>40%) (Caughey et al., 1991; Pan et al., 1993; Safar et al., 1993). This rearrange- ment is accompanied by the acquisition of abnormal physicochemical properties, including insolubility in nondenaturing detergents and 171 Copyright © 2001 by Academic Press. All rights of reproduction in any form reserved. 0065-3233/01 $35.00 ADVANCES IN PROTEIN CHEMISTRY, Vol. 57
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Studies on peptide fragments of prion proteins

STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS
BY FABRIZIO TAGLIAVINI, GIANLUIGI FORLONI,* PASQUALINA D’URSI,*
ORSO BUGIANI, AND MARIO SALMONA*
Istituto Nazionale Neurologico Carlo Besta and *Istituto di Ricerche Farmacologiche Mario Negri, Milano, Italy
I. Introduction. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 171II. The Amyloid Peptides of Gerstmann-Sträussler-Scheinker Disease . . . . . . . . . 174
III. Unraveling the Conformational Conversion of PrPC to PrPSc
Using Synthetic Peptides . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 175A. The Flexible N-terminal Domain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 177B. The Globular Domain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 182
IV. Unraveling the Pathogenesis of Prion Diseases Using Synthetic Peptides . . . 185A. Interaction of PrP Peptides with Cell Membranes . . . . . . . . . . . . . . . . . . . . 185B. Effects of PrP Peptides on Neuronal Cultures . . . . . . . . . . . . . . . . . . . . . . . 187C. Effects of PrP Peptides on Astroglial and Microglial Cultures . . . . . . . . . . 194
V. Concluding Remarks . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 196References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 196
I. INTRODUCTION
The prion diseases are neurodegenerative disorders of humans andanimals that are sporadic or inherited in origin and can be transmitted(Prusiner, 1991). Despite remarkable differences in phenotypic expres-sion, these disorders share a similar pathogenic mechanism, that is, aposttranslational modification of the prion protein from a normal cel-lular isoform (PrPC) to disease-specific species (PrPSc). Nuclear mag-netic resonance (NMR) studies of recombinant murine, hamster,bovine, and human PrP indicate that the normal protein is composedof two structurally distinct moieties: an extended N-terminal segment(residues 23–125) with features of a flexibly disordered polypeptidechain, and a well-defined globular domain (residues 126–231) withthree α helices and two-stranded antiparallel β sheet (Fig. 1) (Riek etal., 1996; Riek et al., 1997; Donne et al., 1997; Lopez Garcia et al., 2000;Zahn et al., 2000). The PrPC>PrPSc transition involves a profound con-formational change with decrease in α-helical secondary structure(~40% > 30%) and striking increase in β-sheet content (~3%>40%)(Caughey et al., 1991; Pan et al., 1993; Safar et al., 1993). This rearrange-ment is accompanied by the acquisition of abnormal physicochemicalproperties, including insolubility in nondenaturing detergents and
171Copyright © 2001 by Academic Press.
All rights of reproduction in any form reserved.0065-3233/01 $35.00
ADVANCES INPROTEIN CHEMISTRY, Vol. 57

partial resistance to proteinase K digestion (Prusiner, 1991). In thepresence of detergents, the protease-resistant core of PrPSc, which is N-terminally truncated near residue 90, assembles into insoluble fibrillarystructures that exhibit the tinctorial and ultrastructural properties ofamyloid (Prusiner et al., 1983). Thus, prion diseases share a basic mole-cular mechanism with amyloid diseases in that they both involve theconversion of a normally soluble form of a protein into an insolublequaternary structure having an extensive β-sheet conformation.
PrP amyloid formation occurs consistently and to the highest degreein two human disorders, (i.e., Gerstmann-Sträussler-Scheinker disease[GSS] and PrP-cerebral amyloid angiopathy [PrP-CAA]) (Ghetti et al.,1996a). In the former, amyloid deposits are essentially parenchymal,whereas in the latter, they are predominantly vascular. Deposition ofPrP amyloid in the neuropil is also a consistent feature of the new vari-
172 FABRIZIO TAGLIAVINI ET AL.
FIG. 1. Schematic representation of PrPC, of amyloid peptides isolated from GSSbrains, and a partially deleted PrP sequence that supports prion propagation. The topdiagram illustrates the polypeptide chain of mature human PrP with the ocatapeptiderepeat region spanning residues 51–90 and the common M/V polymorphism at codon129. The normal protein is composed of two structurally distinct moieties, that is, a flex-ible N-terminal segment (residues 23–125) and a globular domain (residues 126–231)with three α helices (black areas) and two stranded antiparallel β sheets (arrows). Thesecond and third diagrams correspond to amyloid peptides isolated from GSS patientscarrying A117V, F198S, or Q217R mutation in coupling phase with valine at codon 129.The major N- and C-terminal signals obtained from protein sequence analysis and theamino acid residue at position 129 are indicated. The amyloid peptides originated frommutant molecules, because only valine was found at position 129, although the patientswere M/V heterozygous at that codon. The last diagram illustrates a redacted version ofPrP lacking residues 23–88 and 141–176, which is sufficient to support prion disease.

ant of Creutzfeldt-Jakob disease (CJD), which seems to be causallylinked to bovine spongiform encephalopathy (Will et al., 1996; Bruce etal., 1997). In all these conditions, amyloid fibrils are associated with PrPaggregates that do not exhibit the tinctorial, optical, and ultrastructuralproperties of amyloid. The latter are the only or the prevalent form ofPrP deposits in sporadic and familial CJD, fatal familial insomnia (FFI),and natural prion disease of animals (e.g., scrapie of sheep and goatand spongiform encephalopathy of cattle). Whether these nonfibrillardeposits possess an underlying regular molecular structure is unknown.
Co-occurrence of nonfibrillar and fibrillar amyloid deposits has beenobserved in many other conditions such as amyloid-β (Aβ) andimmunoglobulin light chain aggregation disorders (Bugiani et al.,1989; Kaplan et al., 1997; Miravalle et al., 2000). Since at least in someinstances protein aggregation in vivo is based on self-association of par-tially folded intermediates, different sequential or divergent foldingintermediates might be the building blocks for nonfibrillar and fibrillarPrP aggregates. The existence of different abnormal conformers ofPrPSc has been inferred by the observation that distinct protease-resis-tant fragments of PrPSc are found in vivo and can be generated in vitroby limited proteolysis with proteinase K (Bessen and Marsh, 1992;Bessen and Marsh, 1994; Parchi et al., 1996; Piccardo et al., 1996; Pic-cardo et al., 1998). It is noteworthy that these PrPSc types and fragmentsthereof are linked to distinct neuropathological profiles. In particular,amyloid formation is consistently present in conditions, such as GSSand PrP-CAA, in which PrPSc undergoes a specific proteolytic process-ing that originates low-molecular-weight, N- and C-terminal truncatedpeptides (Tagliavini et al., 1991; Tagliavini et al., 1994; Piccardo et al.,1996; Piccardo et al., 1998). On the other hand, nonfibrillar proteinaggregates, which are the prevalent form of PrPSc deposition in CJD,FFI and natural prion diseases of animals, are associated with full-length PrPSc and N-terminal truncated peptides.
The recognition of the protein domains involved in the conforma-tional change of PrPC into PrPSc and the structure of PrPSc conformershas been limited by the insolubility of these molecules. To gain insightsinto this issue, physicochemical studies have been carried out using syn-thetic or recombinant fragments of PrP corresponding to regions ofputative α-helical structure as deduced by structural predictions. Analternative approach was to design synthetic peptides homologous tothe PrP region essential for amyloid fibril formation, as amyloid pro-teins have a high content of β-sheet secondary structure—the centralfeature that distinguishes PrPSc from PrPC—and are often small frag-ments of a precursor protein.
STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS 173

II. THE AMYLOID PEPTIDES IN GERSTMANN-STRÄUSSLER-SCHEINKER DISEASE
GSS disease is an autosomal dominant disorder linked to variantPRNP genotypes resulting from the combination of a pathogenic muta-tion (P102L, P105L, A117V, F198S, D202N, Q212P, Q217R, 192 bpinsertion corresponding to 8 extra copies of the octapeptide repeat)with a common polymorphism at codon 129 (M/V) (Young et al.,1999). In most instances, specific GSS genotypes are associated with dis-tinct clinicopathological phenotypes, a circumstance that is particularlyrelevant for unraveling the ground of phenotypic variability (Ghetti etal., 1996a).
The biochemical composition of PrP amyloid was first determined inpatients of the Indiana kindred of GSS, carrying a F>S substitution atPrP residue 198 in coupling phase with V129. Protein fractionsextracted from amyloid cores isolated from cerebral cortex containedtwo major peptides of ~11 and ~7 kDa, spanning residues ~58–150 and~81–150 of PrP, respectively (Fig. 1) (Tagliavini et al., 1991; Tagliavini etal., 1994). To verify whether these peptides originated from mutant PrPor from both mutant and wild-type molecules, M/V heterozygotes atcodon 129 were selected for study, and V129 was used as a marker of themutant allele. Amino acid sequencing and mass spectrometry showedthat the purified amyloid fractions contained only peptides with V129,suggesting that only mutant PrP was involved in amyloid formation(Tagliavini et al., 1994). The analysis was subsequently extended to GSSpatients with other PRNP mutations (i.e., A117V and Q217R). In allinstances the major amyloid subunit was a ~7 kDa N- and C-terminaltruncated fragment of PrP of similar size and sequence, which wasderived from the mutant allele (Tagliavini et al., 1994; Tagliavini et al.,2001); in GSS A117V, the amyloid protein contained the mutant V117.
The finding that the GSS amyloid protein is an internal fragment ofPrP was verified by immunostaining brain sections with antisera to syn-thetic peptides homologous to different PrP domains (Giaccone et al.,1992). The amyloid cores were strongly immunoreactive with antiserato the mid-region of the molecule, and the periphery of the cores wasimmunostained by antibodies to N- or C-terminal domains. In addition,the antisera to PrP labeled large areas of the neuropil that did not showthe tinctorial and optical properties of amyloid, suggesting that amyloiddeposition in GSS is accompanied by accumulation of PrP peptides,which are not assembled into amyloid fibrils.
Evidence suggests that N- and C-terminal cleavage of abnormal PrPisoforms that generate amyloid peptides occurs before, rather thanafter, fibril formation. Western blot analysis of total brain extracts from
174 FABRIZIO TAGLIAVINI ET AL.

F198S patients has revealed three major protease-resistant PrP frag-ments of 27–29, 18–19, and 8 kDa. The 18–19 and 8 kDa peptides are N-and C-terminal truncated, as deduced by their antigenic profile, likelyrepresenting amyloid protein precursors (Piccardo et al., 1996). Theselow-molecular-weight PrP fragments are also present in areas withoutamyloid deposits, suggesting that brain regional factors play a key rolein amyloidogenesis. Identical PrP peptides are found in patients withD202N and Q217R mutations, whose clinicopathological profile is simi-lar to that of GSS F198S (Ghetti et al., 1996a; Lievens et al., 1998; Pic-cardo et al., 1998). N- and C-terminal truncated fragments are also aconsistent feature of GSS P102L, A117V, and Q212P, although they canbe distinguished from those of GSS F198S, D202N, and Q217R by slightdifferences in migration pattern (Parchi et al., 1998; Piccardo et al.,1998). This suggests structural variations of the relevant precursor pro-tein, which could account for the differences in phenotypic expressionobserved in these conditions.
The identification of the minimal PrP segment involved in amyloidformation in GSS (i.e., residues ~81 to ~150) along with the observa-tions that truncation of the N-terminus (residues 23–88) and deletionof residues 141–176 do not prevent the conversion of PrPC to PrPSc
(Muramoto et al., 1996; Supattapone et al., 1999) suggest that, at least insome instances, the region comprising residues ~90–140 plays a majorrole in the conformational change of PrP. This hypothesis is supportedby in vitro studies with synthetic peptides.
III. UNRAVELING THE CONFORMATIONAL CONVERSION OF PrPC TO PrPSC
USING SYNTHETIC PEPTIDES
Initial studies with synthetic PrP peptides focused on the identificationof protein domains that could be involved in the conformational conver-sion of PrPC into disease-associated species. To test the hypothesis thatthis conversion involves the transition of α helices into β sheet, Gasset etal. (1992) examined the physicochemical properties of peptides homolo-gous to residues 109–122, 129–141, 178–191, and 202–218 of Syrian ham-ster PrP, which were predicted to correspond to α-helical regions(designated H1, H2, H3, and H4, respectively) by computational studies.At variance with the theoretical predictions, peptides H1, H3, and H4exhibited extensive β-sheet structure in the solid state and in aqueoussolution and assembled into fibrils showing ultrastuctural and tinctorialproperties of amyloid, as described in detail later. Multidimensional het-eronuclear NMR has revealed that the prediction of the two C-terminal
STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS 175

helices was largely correct, because the recombinant Syrian hamster PrPcontains three helical domains spanning residues 144–156, 172–193, and200–227. Conversely, the predicted H1 region, which showed the highestpropensity to adopt extended β-sheet conformation when synthesized aspeptide, does not form part of any stable secondary or tertiary fold in theentire protein and is highly flexible (Donne et al., 1997). This is consis-tent with the observation that PrPC to PrPSc transition is accompanied byremarkable increase in β-sheet structure and only slight decrease in α-helical content, suggesting that the conformational rearrangementoccurs primarily in unstructured regions.
A different strategy for identifying PrP regions involved in the confor-mational conversion of PrPC into PrPSc was based on physicochemicalcharacterization of synthetic peptides homologous to consecutive seg-ments of the GSS amyloid protein. This approach issued from the obser-vation that the amyloid subunit was a relatively small fragment of PrP(residues ~81 to ~150) with high intrinsic ability to form insoluble qua-ternary structures with extensive β-sheet conformation. Furthermore,such peptides could allow in vitro experiments to investigate the molecu-lar basis of neuronal degeneration and glial activation associated withextracellular deposition of PrP amyloid. These studies showed that thesequence spanning residues 106–147 is central to amyloid fibril forma-tion (Tagliavini et al., 1993). In particular, a peptide homologous toresidues 106–126 of human PrP (PrP106–126) exhibited high propen-sity to adopt stable β-sheet secondary structure and to assemble intostraight, unbranched, ~8-nm-diameter filaments, ultrastructurally simi-lar to those observed in GSS patients (Fig. 2). The fibrillary assemblieswere partially resistant to protease digestion, displayed tinctorial proper-ties of in situ amyloid (i.e, bi-refringence under polarized light afterCongo red staining and yellow fluorescence after thioflavine S treat-ment) and showed x-ray diffraction patterns consistent with that ofnative amyloid fibrils (Fig. 2) (Selvaggini et al., 1993; Tagliavini et al.,1993). Most interestingly, PrP106–126 was toxic to neurons in culture,whereas it induced activation of glial cells (Forloni et al., 1993; Forloni etal., 1994). These data suggested that the distal part of the flexible N-ter-minal domain of PrP may be central to both the process of PrPC>PrPSc
transition and the pathogenic properties of abnormal PrP isoforms.These observations and the finding that amino acid substitutions
associated with genetic forms of CJD, such as D178N and E200K,increase the fibrillogenic properties of PrP fragments (Goldfarb et al.,1993) promoted a variety of studies with synthetic peptides homolo-gous to segments of most PrP regions (Fig. 3, see color insert). The fol-lowing sections review structural studies with peptides derived from the
176 FABRIZIO TAGLIAVINI ET AL.

flexible N-terminal domain and the globular C-terminal domain of PrP,and the subsequent chapter focuses on the biological properties of PrPpeptides in vitro.
A. The Flexible N-terminal Domain
1. The Octapeptide Repeat Region (Residues 51–90)
This region, which contains a series of unusual glycine-rich repeatswithout significant homology to other known protein sequences, isamong the most conserved parts of PrP in mammals, and analogoussequences are present in other prion proteins (e.g., hexameric repeatsin chicken PrP) (Harris et al., 1991; Gabriel et al., 1992). In humans, therepeat region is composed of one nonapeptide (termed R1) followedby four octapeptides (termed R2, R2, R3, R4) that have the same aminoacid sequence (PHGGGWGQ) but can be distinguished by variations inthe DNA sequence (Fig. 3A). Deletion of a single octarepeat (R2 or R3)is present in about 1% of the population and does not have deleteriousconsequences. By contrast, the insertion of one, two, four, five, six, seven,eight, or nine extra octapeptides is associated with inherited prion dis-ease (for review see Young et al., 1999).
Based on the observation that the octapeptide repeats share thesequence PHG with the histidine-rich glycoprotein, which is thought tobe involved in plasma copper transport, Hornshaw et al. (1995a) hypoth-
STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS 177
FIG. 2. Structural features of PrP106–126 assemblies. (A) Electron micrograph of fib-rils generated by the peptide in physiological conditions. Magnification bar = 100 nm.(B) X-ray diffraction pattern of PrP106–126, with reflections corresponding to H-bond-ing between antiparallel β sheets (arrow).

esized that this region could be a metal-binding domain. To test thishypothesis, they generated synthetic peptides containing three or fourcopies of the octarepeat and tested their ability to bind metal ions usingmass spectrometry and size exclusion chromatography. They found thatthe octapeptide repeat sequence preferentially binds copper over otherdivalent ions such as zinc, manganese, cobalt, nickel, iron, calcium, andmagnesium. This finding is consistent with the observation that full-length PrPC, but not N-terminal-truncated PrPC, can be purified by Cu2+
chelate chromatography (Pan et al., 1992). However, whether PrP has asignificant role in copper binding and cuproenzyme activity—e.g.,Cu/Zn superoxide dismutase—in the brain is unclear (Brown et al.,1997; Waggoner et al., 2000).
Subsequently, Hornshaw et al. (1995b) investigated the secondarystructure of a synthetic peptide containing four tandem repeats (PrPresidues 60–91). Circular dichrosim (CD) spectroscopy revealed a ran-dom coil conformation that was independent from absence or pres-ence of Cu2+. Miura et al. (1996) did not detect any regular structure ina single octapeptide unit in the absence of divalent metal ions usingRaman spectroscopy. However, Cu2+ binding to the HGGG motif of theoctapeptide induced a conformational transition in the adjacent C-ter-minal portion from an irregular loop to an α helix. The analysis of alonger segment comprising an octapeptide and the succeeding 12amino acids (PrP residues 84–103) showed that the α-helical structurenucleated by Cu2+ binding in the octapeptide extended over the C-ter-minal sequence.
Stöckel et al. (1998) investigated the metal-binding properties of anearly full-length recombinant hamster PrP (residues 29–231). Afterpurification, the polypeptide chain was refolded into a predominantlyα-helical structure resembling that of PrPC isolated from hamster brain,as deduced by near-UV CD spectroscopy. On heating, the recombinantprotein could be converted into a β-sheet-rich form. CD and trypto-phan fluorescence spectroscopy revealed that the addition of Cu2+
induced a structural change in hamster PrP29–231. Furthermore, thecopper-complexed PrP was more prone to shift from a soluble α-helicalstructure to a β-sheet aggregate on heat denaturation. The study alsoconfirmed that the metal-binding sites of PrP are highly specific forCu2+, as no change in tryptophan fluorescence emission was observedwith other divalent cations, and pointed to a role for histidine as achelating ligand on the basis of pH titration of copper binding. Equi-librium dialysis experiments indicated a binding stoichiometry of twocopper molecules per PrP molecule at physiologically relevant concen-trations, with a dissociation constant of 14 µM. This finding was at vari-
178 FABRIZIO TAGLIAVINI ET AL.

ance with previous data suggesting 5.6 Cu2+ binding sites per PrP mole-cule (Brown et al., 1997).
To further investigate the site and mode of binding of Cu2+ to PrP,Viles et al. (1999) analyzed a series of peptides containing two, three,and four octarepeats using various spectroscopic techniques includingCD, NMR, absorption spectroscopy, and electron spin resonance spec-troscopy. They found that two octapeptides bind a single Cu2+ ion witha Kd of 6 µM, whereas four octapeptides cooperatively bind four Cu2+
ions. In the absence of Cu2+, all peptides were unstructured in solution;however, a distinctive structuring consistent with turns and structuredloops, without indication of α helix or β sheet, was observed on addi-tion of Cu2+. The authors suggested that the discrepancies betweenthese and previous data could be due to experimental conditions, inparticular pH below 7.4, which is the optimum for copper binding, anduse of Tris buffer, which competes for copper with the octarepeat pep-tides at pH 7.4.
Based on spectroscopic data, Viles et al. (1999) proposed a model inwhich the four histidines in successive octarepeats form a bridged cop-per complex, with the Nε2 and Nδ1 imidazole nitrogens from each histi-dine coordinating two adjacent copper ions. This model would implythat, although the octarepeat region is unstructured in the absence ofCu2+, it could be constrained by the four copper-coordinating histidinesinto a rather compact structure at physiological Cu2+ concentrations.Based on Raman and absorption spectroscopic analysis of peptides con-taining one, two, and four octarepeats, Miura et al. (1999) suggested adifferent copper-binding mode. In this model, at neutral or basic pH,each octapeptide unit binds a Cu2+ ion via the Nπ atom of the histidineside chain and two deprotonated amide nitrogens in the adjacenttriglycine segment. However, at weakly acidic pH, the Cu2+-amide– bondsdissociate and only the histidine residues provide metal coordinationsites, resulting in decreased affinity of the protein to Cu2+. Because cop-per stimulates endocytosis of PrPC from the cell surface (Pauly and Har-ris, 1998), the pH-dependent change of binding mode may play a role ininternalizing Cu2+ ions from extracellular to endocytic compartments.Furthermore, this change may allow formation of His-Cu-His bridgesbetween different peptide chains, resulting in peptide aggregation.
The octapeptide repeat region is not required for the conversion ofPrPC into PrPSc and propagation of the disease process. Nevertheless,extra-repeat insertional mutations in one PRNP allele trigger disease. Itis conceivable that disorder of copper binding in these extended pro-tein species might feature in the formation of altered PrP isoformsand/or PrP aggregation. In this regard, it is noteworthy that PrPSc is
STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS 179

extracted from brain tissue of subjects with prion disease in a metal-ion-occupied form and can undergo conformational change as a result ofbinding metal ions (Wadsworth et al., 1999).
2. The Distal Part of the N-terminal Domain (Residues 90–125)
This region corresponds to the N-terminal part of the protease-resis-tant core of PrPSc (Prusiner et al., 1984). Protection against proteinaseK digestion suggests that it undergoes a conspicuous structuralrearrangement in PrPC to PrPSc conversion. On the other hand, mutantPrP lacking parts of this sequence (i.e., residues 95–107, 108–121, or114–121) is not converted into a protease-resistant form in scrapie-infected mouse neuroblastoma cells (Muramoto et al., 1996; Hölscheret al., 1998). Further, overexpression of the deletion mutantPrP∆114–121 inhibits in a trans-dominant fashion the accumulation ofendogenous PrPSc in these cells (Hölscher et al., 1998).
Structural analysis has shown that peptides derived from the C-termi-nal half of this region, which is the most conserved PrP sequence acrossall species, are able to adopt different conformations in distinct envi-ronment, although intrinsically prefer a β-sheet structure (Fig. 3B).The first peptide to be investigated corresponded to residues 109–122of Syrian hamster PrP (Gasset et al., 1992). Fourier transform IR (FTIR)spectra of hydrated peptide films were marked by an amide I band withmaximum absorbance at 1621 cm–1, which is typical of a β-pleated sheetstructure maintained by strong hydrogen bonds. The presence of inter-molecular interactions was confirmed by the residual amide II bandafter 2 hours of hydrogen/deuterium exchange. Within the sequence109–122, the hydrophobic segment AGAAAAGA (i.e., residues113–120) was found to be central to β-sheet association. On the otherhand, the α-helix promoting solvent hexafluoroisopropanol inducedPrP109–122 to adopt α-helical conformation. The extension of thissequence toward the N-terminus to include the lysine residues 106 and104 (i.e., peptides PrP106–122, 105–122, and 104–122) resulted in anincrease in α-helix/random coil structure in aqueous solutions at phys-iological pH (Nguyen et al., 1995a).
Solid-state NMR confirmed that peptide PrP109–122 can exist in atleast two different conformations, depending on the solvent fromwhich the final preparation is obtained (Heller et al., 1996). Althoughpeptide lyophilized from acetonitrile/water solution showed β-sheetstructure in the segment spanning residues 112–121, the samplelyophilized from hexafluoroisopropanol had a metastable α-helicalconformation in the segment comprising residues 113–117. The expo-
180 FABRIZIO TAGLIAVINI ET AL.

sure of lyophilized PrP109–122 to water vapor caused a complete con-version of the secondary structure to β sheet within a few hours.
Evidence indicates that the physicochemical environment primarilyaffects the structure of the N-terminal segment of PrP109–122. X-raydiffraction of samples lyophilized from water showed that the β sheetswere composed of small side chain, suggesting a single β strand in theC-terminal segment. Conversely, specimens dehydrated after acetoni-trile solubilization exhibited two types of β sheets, one with larger andone with smaller side chains; this suggested the presence of two βstrands corresponding to residues 109–113 and 116–122, respectively,with a turn creating an intrachain intersheet interaction (Inouye andKirschner, 1998). It was advanced that the ionization state of His111may play an important role in these structural modifications, as it facesthe hydrophobic segment 116–122. When this residue becomes neutralby being deprotonated, the structure of the peptide may be stabilized;however, when His111 is positively charged at acidic pH, the side chainis unlikely to remain in such a nonpolar environment.
Physicochemical studies of the synthetic peptide spanning residues106–126 of human PrP led to similar conclusions. PrP106–126 consistsof an N-terminal polar head (KTNMKHM-) followed by a longhydrophobic tail (–AGAAAAGAVVGGLG); and its structural featuresare markedly influenced by solvent composition, ionic strength, andpH. CD spectroscopy showed that the peptide adopts a random coilconformation in deionized water, a combination of random coil and βsheet in phosphate buffer pH 7.0, a predominantly β-sheet structure inphosphate buffer pH 5.0, and an α-helical structure in trifluoroethanolor in the presence of micelles formed by a 5% SDS solution. Notably,the β-sheet conformation is extremely stable, because it is not affectedby trifluoroethanol if the peptide was previously suspended in phos-phate buffer pH 5.0 (De Gioia et al., 1994).
Studies with mutated analogs of PrP106–126 support the hypothesisthat the conformational plasticity of the peptide is largely due toHis111, which is located between the hydrophilic and hydrophobic seg-ments (Salmona et al., 1999). Replacement of L-His111 with D-His111abolished the pH-dependent conformational polymorphism andcaused loss of β-sheet structure. Conceivably, the orientation of the imi-dazo moiety of D-His produces a steric hindrance with the neighboringLys110 and Met112, resulting in the formation of a random coil struc-ture. Substitution of His111 with the hydrophobic, nonionizable aminoacid alanine, remarkably decreased the solubility of the peptide, whichinstantly formed amorphous aggregates in aqueous solutions, suggest-ing that hydrophobic forces prevailed over the kinetics of fibril assem-
STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS 181

bly. Unexpectedly, substitution of His111 with lysine, which is alwaysionized in the pH range 5–7, did not abolish the pH dependent confor-mational polymorphism and fibrillogenic ability of the peptide. Thisfinding is likely related to the intramolecular environment of residue111, which is flanked by hydrophilic and hydrophobic regions thatinfluence the pKa of lysine (Salmona et al., 1999).
Determination of solution structure by NMR spectroscopy showedthat PrP106–126 adopts an α-helical conformation in the hydrophobicC-terminal segment (residues 112–125) when dissolved in deionizedwater at pH 3.5, the most populated helical region corresponding toAla115-Ala118. In water/trifluoroethanol, the α helix increased in pop-ulation, particularly in the Gly119-Val122 tract (Fig. 4A). Conversely, indimethyl sulfoxide, the region corresponding to the hydrophobic clus-ter Ala113-Ala120 adopted a prevalently extended conformation (Fig.4B). Under all experimental conditions, the N-terminal segment span-ning residues Asn108-Met112 showed a “turn-like” conformation. Theanalysis confirmed that the structure of PrP106–126 is remarkably sen-sitive to pH, supporting the view that the pH-dependent ionizable sidechain of His111 may be important in modulating the conformationalmobility of the peptide (Ragg et al., 1999).
B. The Globular Domain
Evidence suggests that the proximal part of the gobular domainextending from residue 125 to the origin of the first α helix is involvedin the conformational transition of PrPC into disease-specific species.This region, which includes a short β strand, corresponds to the C-ter-minal segment of the amyloid protein purified from GSS brains (Fig.3B). A synthetic peptide homologous to residues 127–147 of humanPrP was found to assemble into fibrils having ultrastructural features,tinctorial properties, and x-ray diffraction patterns of amyloid (Tagli-avini et al., 1993). However, the intrinsic potential of this peptide toamyloid formation appeared to be significantly lower than that ofPrP106–126 and required the peptide to be intact, as shorter peptidesspanning residues 127–135 or 135–147 were nonfibrillogenic. Further,the fibrils showed a distinct morphology and the propensity to helicallywind around each other with a well-defined periodicity, suggesting thatthe sequence comprising residues 127–147 may contribute to the mor-phology of scrapie-associated fibrils extracted from brains of humanand animals with prion diseases (Merz et al., 1981; Merz et al., 1983).
Gasset et al. (1992) examined the physical characteristics of a peptidespanning residues 129–141 of hamster PrP by FTIR. The spectrum of
182 FABRIZIO TAGLIAVINI ET AL.

the amide I region of hydrated peptide films was marked by maximumabsorbance at 1655 cm–1, consistent with multiple-ordered structureswith little indication of any intermolecular interaction. Nguyen et al.(1995a) found that PrP129–141 is soluble and has coil or α-helicalstructures in solution; however, this peptide formed β-sheet-rich, insol-uble aggregates on addition of a peptide corresponding to residues109–122 of Syrian hamster PrP. This remarkable effect appeared to bespecific, as it was not observed using unrelated amyloid peptides (e.g.,Aβ11–25 and Aβ25–35) instead of PrP109–122; furthermore, the con-version process was significantly less efficient using a peptide homolo-gous to the mouse PrP sequence, which differs from Syrian hamster PrPat residues 109 and 122 (Nguyen et al., 1995a). These findings sug-gested that the high propensity for β structure and fibril formationenables PrP109–122 to convert peptides having an intrinsically lowamylidogenic potential from a soluble coiled state to an insoluble β-sheet-rich form.
Overall these data pointed to the region comprising the distal part ofthe flexible N-terminal domain and the proximal part of the globulardomain as a possible nidus at which the conformational change is initi-ated in PrPC to PrPSc conversion. On this ground, larger peptides cor-responding to residues 109–141 and 90–145 of Syrian hamster PrP wereanalyzed (Zhang et al., 1995). The latter is of particular interest because
STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS 183
FIG. 4. Solution conformations of PrP106–126 as determined by NMR and restrainedmolecular dynamics. Representative structures of the peptide in trifluroethanol/watersolution (A) and dimethyl sulfoxide (B).

residue 90 is the N terminus of the protease-resistant core of PrPSc, andresidue 145 coincides with a stop codon in the PRNP gene, which isassociated with a cerebrovascular form of PrP amyloidosis (Kitamoto etal., 1993; Ghetti et al., 1996b). CD and FTIR spectroscopy showed thatPrP90–145 adopts α-helical structure in aqueous solutions containingorganic solvents such as trifluoroethanol and hexafluoroisopropanol,or detergents such as sodium dodecyl sulfate and dodecyl phospho-choline. Sodium chloride at physiological concentration or acetonitrileinduced the formation of β sheets, whose intermolecular nature wasevident from the presence of rod–shaped polymers as revealed by elec-tron microscopy (Zhang et al., 1995). X-ray diffraction analysis con-firmed the three-dimensional organization of these polymers, whichcontained β-sheet structures with intermolecular β sheets. The hydro-gen-bonding distances within the β sheets were similar to thoseobserved for hydrated prion rods, suggesting that the amyloidogeniccore of PrPSc is closely modeled by the peptide PrP90–145 (Nguyen etal., 1995b).
Recently, Kaneko et al. (2000) addressed the question as to whether asimilar synthetic peptide is able to initiate prion disease. In particular, theyused a peptide homologous to mouse PrP residues 89–143 (i.e., humanresidues 90–144) with the P101L substitution corresponding to GSS-linked P102L mutation. Intracerebral inoculation of a β-sheet-rich form ofthis peptide into transgenic mice expressing low levels of the P101L muta-tion resulted in neurological dysfunction and neuropathological changesconsistent with GSS. Conversely, larger doses of a non β form of the samepeptide failed to induce these changes, emphasizing the importance ofprotein conformation in disease initiation and propagation.
NMR spectroscopy of recombinant murine PrP showed that helix 1corresponding to residues 144–154 is located in the outer shell of theglobular domain. Six of the 11 amino acid residues are able to changetheir ionization state in dependence of pH variations (Riek et al., 1996).The N-terminal half of this helix is part of the epitope of the PrPSc-spe-cific monoclonal antibody 15B3 and is the epitope of the antibody 6H4,which recognizes both recombinant PrP and PrPC (Korth et al., 1997).On this ground, it was advanced that helix 1 might be involved in con-formational changes leading to PrPSc formation. To test this hypothesis,Liu et al. (1999) examined a peptide corresponding to residues143–158 of mouse PrP and found that it has an extraordinary high helixpropensity. Comparison of NMR structure of this peptide and recombi-nant mouse PrP revealed striking similarities, as both formed apincette-like motif in the N-terminal part of the helix.
184 FABRIZIO TAGLIAVINI ET AL.

Gasset et al. (1992) investigated the secondary structure of peptidesspanning residues 178–191 and 202–218 of Syrian hamster PrP (Fig.3C). These peptides were predicted to correspond to helical regions,and indeed are part of helix 2 and helix 3, respectively, as determinedby NMR (Donne et al., 1997). Unexpectedly, FTIR spectroscopy showedthat hydrated PrP178–191 contained a mixture of β sheet and turns,whereas PrP202–218 had a predominantly β-sheet structure.
IV. UNRAVELING THE PATHOGENESIS OF PRION DISEASES
USING SYNTHETIC PEPTIDES
A. Interaction of PrP Peptides with Cell Membranes
A variety of data argue that plasma membranes are central to thepathogenesis of prion disease. Myoclonus, periodic electroencephalo-graphic (EEG) changes, and spongiform degeneration of neurons,which are typical features of CJD, have been regarded as a consequenceof nerve cell damage following plasma membrane abnormalities (Bass etal., 1974; Chou et al., 1980; Traub and Pedley, 1981). This view was con-sistent with the observation that rats injected intracortically with a mem-brane-bound ATPase inhibitor developed periodic EEG waves andcortical spongiosis (Bignami and Palladini, 1966). The membranehypothesis was further supported by the demonstration that scrapieinfectivity is associated with the brain membrane fraction (Millson et al.,1971; Semancik et al., 1976; Marsh et al., 1984; McKinley et al., 1991).Subsequent studies showed that cultured neurons exposed to PrPSc
exhibit alteration of membrane protein, increased membrane microvis-cosity, and abnormal receptor-mediated calcium ion responses in theabsence of morphological changes, suggesting that PrPSc affects theplasma membrane early (Kristensson et al., 1993). Caveolae-likedomains purified from scrapie-infected cells and scrapie-infected brainscontain both PrPC and PrPSc (Vey et al., 1996). Notably, the conversionof PrPC into PrPSc is inhibited by lovastatin, a drug that reduces mem-brane cholesterol levels, a critical determinant of the architecture ofcaveolae-like domains (Taraboulos et al., 1995, Murata et al., 1995).Accordingly, this membrane compartment has been regarded as a puta-tive site of PrPC>PrPSc conversion. Harris and co-workers reported thatmutant PrP generated by transfected CHO cells and PrPSc produced byscrapie-infected neuroblastoma cells remain associated with the plasmamembrane after digestion with a specific bacterial phospholipase that
STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS 185

cleaves the glycosylphosphatidylinositol (GPI) anchor. However, theanchor becomes susceptible to the enzyme when membrane proteinsare denatured with SDS, suggesting that the GPI of mutant PrP andPrPSc is physically shielded from the phospholipase, either by aggrega-tion of the protein or by intrinsic conformational features of thepolypeptide chain (Lehmann and Harris, 1995; Narwa and Harris,1999). Studies with cell-free translation systems containing ER-derivedmicrosomal membranes have revealed that PrP may exist in differenttopological forms, including two transmembrane forms with oppositemembrane orientation. A specific transmembrane form was found to beincreased in transgenic mice expressing the A117V mutation and wasdetected in brain tissue of patients with GSS A117V (Hedge et al., 1998).
The membrane-spanning domain of the transmembrane forms ofPrP corresponds to residues 113–135. Noteworthy, synthetic peptidesrelevant to this domain interact with artificial and natural membranesin vitro, causing profound changes. The incubation of peptidePrP106–126 with liposomes resulted in a remarkable increase in micro-viscosity, as determined by steady-state fluorescence spectroscopy usingthe lipophylic probe 1,6-diphenyl-1,3,5-hexatriene (DPH), which incor-porates into the lipid bilayer. This effect was also observed using aPrP106–126 analog with amidated C terminus (PrP106–126 NH2), amodification that substantially decreases peptide fibrillogenic ability.Conversely, a scrambled sequence of PrP106–126 and peptidePrP127–147 was not effective (Salmona et al., 1997).
Similar changes in membrane microviscosity following PrP106–126treatment were observed in several cell lines as well as in primary nerveand glial cell cultures. These changes occurred in the first few minutesof exposure and remained stable for at least 60 minutes. The increasein microviscosity was unrelated to membrane protein and lipid compo-sition or phase transition of membrane lipids in the range of biologi-cally significant temperatures (Salmona et al., 1997).
To investigate whether this effect was due to insertion of PrP106–126in the membrane bilayer, the peptide and its scrambled congener werecoupled to a fluorescent dye and incubated for 60 minutes with murinefibroblasts. Fluorescence microscopy showed that PrP106–126 labelingwas punctuated and confined to the cell surface, whereas scrambledPrP106–126 was homogeneously distributed over the cell body (Fig.5A). When the incubation was prolonged for 24 hours, PrP106–126 waslargely internalized from the cell surface to cytoplasmic organelles (Fig.5B). These observations suggest that PrP106–126 has a high propensityto embed itself into the lipid bilayer, increasing the lipid density and
186 FABRIZIO TAGLIAVINI ET AL.

membrane microviscosity. This propensity seems to be related to theprimary structure rather than the aggregation properties of the pep-tide, because the poorly fibrillogenic PrP106–126 NH2, but not the fib-rillogenic PrP127–147, induced similar changes. It is conceivable thatthe membrane modifications induced by PrP106–126 are at the basis ofreceptor and channel dysfunction of nerve and glial cells, and accountfor cell responses to the peptide in vitro.
The membrane effects of PrP 106–126 were also investigated by Linet al. (1997) using a model of planar lipid bilayer membranes. Thestudy showed that ion-permeable channels are rapidly formed on theaddition of peptide at concentrations comparable to those required forneurotoxicity. These channels were permeable to all ions and irre-versibly associated with the membranes, disrupting the selectivity of cellpermeability. The effects were more pronounced using an “aged”PrP106–126 and were enhanced by acidic pH, suggesting that theaggregation state of the peptide influenced channel formation.
Based on the observation that a C-terminal fragment of Aβ compris-ing residues 29–42 is highly fusogenic (Pillot et al., 1996) and that thisfragment has sequence homology with residues 118–135 of PrP, Pillotet al. (1997) investigated the interaction of peptides PrP118–135 andPrP120–133 with artificial membranes. The conformation of thesepeptides at the lipid/water interface was calculated by the energy min-imization approach (Brasseur, 1991). PrP118–135 and PrP120–113inserted into the lipid bilayer in an oblique way, although with differ-ent angles (25° and 45°, respectively). PrP118–135 penetrated the lipidphase through its more hydrophobic N-terminal segment, which cor-responds to the hydrophobic domain of PrP106–126. Both peptidesresulted highly fusogenic when exposed to unilamellar lipid vesiclesand caused the leakage of entrapped calcein as well (Pillot et al., 1997).The high propensity of these peptides to insert stably into cell mem-branes, resulting in membrane perturbation, has recently beenpropsed as a mechanism of neurotoxicity for PrP peptides, whichseems to be independent from the expression of endogenous PrPC
(Haïk et al., 2000).
B. Effects of PrP Peptides on Neuronal Cultures
1. PrP 106–126
The accumulation of PrPSc and PrP amyloid in the central nervoussystem is accompanied by activation of microglial cells, hypertrophy,and proliferation of astrocytes and degeneration of neurons leading to
STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS 187

variable degrees of atrophy of the target regions. The temporal andtopographical relationships between PrP deposition and brain changes(DeArmond et al., 1988; Bruce et al., 1989; Williams et al., 1994) suggestthat altered forms of the protein may be responsible for nerve celldegeneration and glial cell reaction. To test this hypothesis, severalstudies have been carried out to assess the effects of synthetic PrP pep-tides on neurons and glial cells in culture. The first peptides to be usedcorresponded to consecutive segments of the GSS amyloid protein(residues 81–88, 89–106, 106–126, and 127–147). Exposure of primaryrat hippocampal neurons to these peptide for 24 hours did not affectcell survival. Conversely, there was great neuronal loss in cultures after7-day treatment with micromolar concentrations of PrP106–126. Under
188 FABRIZIO TAGLIAVINI ET AL.
FIG. 5. Fluorescence pattern of murine fibroblasts after short or prolonged incuba-tion with PrP106–126 coupled to a fluorescent dye. After one hour incubation, the fluo-rescent peptide is confined to the cell surface (A) while after 24 hours is internalized,the fluorescence being associated with cytoplamic organelles (B).

the same conditions, the other PrP peptides and a scrambled sequenceof PrP106–126 did not significantly reduce cell viability (Fig. 6A and B).The neurotoxicity of PrP106–126 was dose-dependent; the toxicresponse was first detected at a concentration of 10 µM, was statisticallysignificant at 25 µM, and resulted in virtually complete neuronal loss at50 µM. Fluorescence microscopy following culture treatment withDNA-binding fluorochromes (e.g., Hoechst 33258), as well as electronmicroscopy revealed that neurons exposed to PrP106–126 exhibitedapoptotic changes such as condensation of the chromatin and frag-mentation of the nucleus. Accordingly, agarose gel electrophoresis ofDNA extracted from cultured cells after 7-day treatment with the pep-tide showed an apoptotic pattern of DNA fragmentation, resulting fromcleavage of nuclear DNA in internucleosomal regions (Forloni et al.,1993). A subsequent study revealed that the neurotoxic effect of thepeptide involves calcium entry through L-type voltage-sensitive calciumchannels (Florio et al., 1998). Exposure of GH3 cells—a clone derivedfrom a GH-secreting rat pituitary adenoma—to PrP106–126 resulted inapoptosis and dose-dependent inactivation of L-type calcium channels,as determined by elctrophysiological measurements.
The apoptotic pathway induced by PrP106–126 is not associated withincreased expression of classic oncogenes such as C-fos, C-jun, and C-mycbut is accompanied by a reduction of Bcl-2 expression (Forloni et al.,1996). Recently, we have evaluated the role of caspases in peptide neu-rotoxicity by measuring the activity of caspase-3 (CPP32), a major apop-tosis effector enzyme, in primary cortical neurons. Culture treatmentwith 2.5 to 50 µM PrP106–126 resulted in dose-dependent activation ofCPP32, which was 6-fold increased at the maximal dosage. Co-treat-ment of cultures with a nonselective caspase inhibitor (z-VAD-fmk) or aCPP32-specific inhibitor (DEVD-CHO) abolished CPP32 activation(Fig. 7A). Although z-VAD-fmk also reduced PrP106–126 neurotoxicity,co-treatment of cultures with DEVD-CHO did not prevent neuronaldeath, suggesting that the neurotoxic effect of the peptide was notdependent on caspase-3 activation (Fig. 7B). This observation is consis-tent with the immunohistochemical finding of a large disproportionbetween activated caspase-3 immunoreactive cells and apoptotic cells inthe cerebellum of GSS patients (Migheli et al., 2000).
Brown et al. (1994) reported that PrP106–126 neurotoxicity is depen-dent on the expression of endogenous PrPC. This finding is consistentwith in vivo observations that PrP0/0 mice are resistant to scrapie infec-tion (Büeler et al., 1993), and the presence of PrPC is essential for thedevelopment of PrPSc-related neurodegenerative changes (Brandner etal., 1996). The relationship between peptide toxicity and PrPC expres-
STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS 189
sion was further analyzed by Hope et al. (1996) who found that cell lineswith low expression levels of PrPC are less susceptible to PrP106–126than cells with high PrPC expression. Using cerebellar cultures obtainedfrom different lines of PrP transgenic mice, Brown (1998) observedthat granule cells from Tg35 mice having 8 to 10 times PrPC overex-
190 FABRIZIO TAGLIAVINI ET AL.
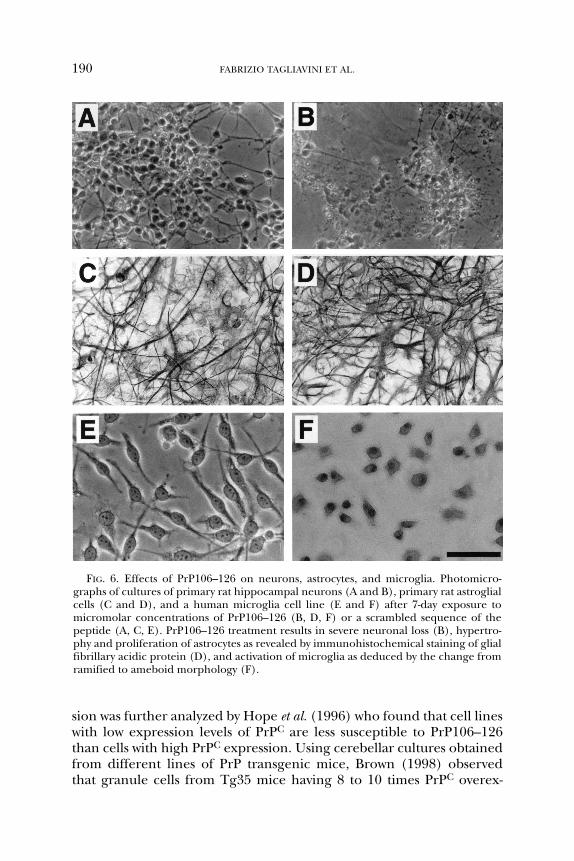
FIG. 6. Effects of PrP106–126 on neurons, astrocytes, and microglia. Photomicro-graphs of cultures of primary rat hippocampal neurons (A and B), primary rat astroglialcells (C and D), and a human microglia cell line (E and F) after 7-day exposure tomicromolar concentrations of PrP106–126 (B, D, F) or a scrambled sequence of thepeptide (A, C, E). PrP106–126 treatment results in severe neuronal loss (B), hypertro-phy and proliferation of astrocytes as revealed by immunohistochemical staining of glialfibrillary acidic protein (D), and activation of microglia as deduced by the change fromramified to ameboid morphology (F).

pression were more susceptible to PrP106–126 neurotoxicity that thosederived from normal mice. Unexpectedly, no significant differenceswere detected between granule cells from Tg20 transgenic mice having12 to 14 times PrPC overexpression and neurons from normal mice.
STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS 191
FIG. 7. Relationship between caspase activation and neurotoxicity of PrP106–126.(A) The activity of caspase-3 (CPP32) was determined after 5-day exposure of corticalneurons to 50 µM PrP106–126 or scrambled PrP106–126 (Control). The remarkableincrease in CPP32 activity induced by PrP106–126 was abolished by cotreatment of cul-tures with a specific (DEVD-CHO) or a nonselective (z-VAD-fmk) caspase inhibitor. (B)Viability of cortical neurons under the conditions described in (A). The neurotoxiceffect of PrP106–126 was partially reduced by cotreatment of cultures with z-VAD-fmk,whereas DEVD-CHO was ineffective. Data are the mean ± SE of 6 determinations; *P <0.01 versus control group (Tukey’s test).

This discrepancy was explained by a different behavior of Tg35 andTg20 microglial cells present in culture.
The role of microglia in PrP106–126 neurotoxicity was first investi-gated by Brown et al. (1996). These authors showed that the toxic effectof the peptide to mouse cerebellar granule neurons was related to thepresence of microglial cells, which responded to PrP106–126 by increas-ing their oxygen radical production. The neurotoxicity was remarkablyreduced by treatment of cultures with L-leucine methylester (LLME),which is toxic to microglia, and was restored by addition of microglialcells. Similar results were observed using phaeochromocytoma PC12 cells(Brown et al., 1997). Treatment of undifferentiated or NGF-differentiatedPC12 with 80 µM PrP106–126 did not reduce cell viability; however, whenPC12 were co-cultured with microglia, the peptide induced cell degener-ation, that was more pronounced after differentiation with NGF.
Based on the finding that selective expression of PrPC in astrocytes issufficient to restore susceptibility of PrP0/0 mice to scrapie infection, itwas advanced that astroglial cells feature in prion disease pathogenesis,possibly by an indirect toxic effect on neurons (Raeber et al., 1997).This hypothesis is supported by an in vitro study where PrP0/0 neuronsbecame susceptible to PrP106–126 neurotoxicity when co-cultured withPrP+/+ astrocytes (Brown, 1999).
Our studies do not support the view that PrP106–126 neurotoxicityrequires microglia or astrocytes. On the contrary, the presence of astro-cytes in neuronal cultures may favor the neuroprotective effect of antioxi-dants. This was deduced by the observation that although primaryneurons cultured with or without 10% fetal calf serum (FCS) are equallysensitive to the toxic effect of the peptide, only neurons cultured withFCS, a condition that markedly increases astrocyte contamination, areprotected by antioxidants even in the absence of microglia (Angeretti etal., in press). The view that PrP106–126 has a direct effect on neurons issupported by the finding of peptide-induced toxicity in glia-free, neuronalcell lines such as PC12 and NB41A3 (Hope et al., 1996).
The neurotoxicity of PrP106–126 has been used as a tool to identifyneuroprotective compounds. Flupirtine and sulfated glycosaminogly-cans have been found to prevent the toxic effects of the peptide on pri-mary rat cortical neurons and human neuroblastoma cell lines,respectively (Perovic et al., 1995; Perez et al., 1998). Although flupirtinehas been regarded as an antiapoptotic agent (Perovic et al., 1998), theeffect of sulfated glycosaminoglycans was associated with the ability ofthese molecules to inhibit peptide aggregation and amyloid formation.However, evidence suggests that the neurotoxicity of PrP106–126 is notstrictly related to the fibrillogenic properties of the peptide. Based on
192 FABRIZIO TAGLIAVINI ET AL.

conformational studies on Aβ 25–35 (Terzi et al., 1994), we synthesized aPrP106–126 analog with amidated C-terminus (PrP106–126 NH2).Although this modification strongly reduced the ability of the peptide toform amyloid fibrils, it did not affect its neurotoxicity (Salmona et al.,1999). On the other hand, PrP106–126 NH2 lost the capability to induceastroglial proliferation, suggesting that this effect is related to aggrega-tion state of the peptide (Rizzardini et al., 1997, Salmona et al., 1999).
Jobling et al. (1999) investigated the role of the hydrophobic coresequence AGAAAGA in PrP106–126 neurotoxicity. The substitution oftwo or more hydrophobic residues with hydrophilic serine residues abol-ished the toxic effect of the peptide on mouse cerebellar neuronal cul-tures. This correlated with a decrease in β-sheet structure that altered theaggregation and fibrillogenic properties of the peptide. The discrepancybetween this observation and our results with PrP106–126 NH2 could bedue to substantial differences in the nature of the changes introduced inthe peptides (i.e., alteration of the primary structure and disruption ofthe hydrophobic core versus removal of the C-terminus electric charge byamidation without sequence modification). The view that fibrillogenesisis not essential for neurotoxicity is supported by studies with syntheticpeptides homologous to Alzheimer’s disease Aβ protein (Pike et al., 1991;Forloni et al., 1997; Johnson and Gibbs, 1998; Kshet et al., 1999).
A recently published article did not confirm the neurotoxic effect ofPrP106–126 and indicated that the possible source of the neurotoxicactivity could be an high-performance liquid chromatography (HPLC)contaminant (Kunz et al., 1999). In this regard, it is noteworthy that theneurotoxicity of PrP106–126 has been successfully replicated in manylaboratories throughout the world. Furthermore, PrP 106–126 wasobtained from several different sources, both industrial and academic,and the possibility that a chemical contaminant with similar propertieswas systematically and exclusively associated with PrP106–126 is unreal-istic (Forloni et al., 2000).
2. PrP Peptides Carrying Mutations Associated with Human Prion Diseases
We have analyzed the influence of point mutations linked to CJD(D178N, E200K, I210V) and GSS (P102L, P105L, A117V, F198S, Q217R)on physicochemical properties and biological activity of PrP peptides(Foloni et al., 1999). For the study, wild-type and mutated PrP fragmentscorresponding to residues 89–106 (P102L and P105L variants),106–126 (A117V), 169–185 (D178N), 195–213 (F198S, E200K andI210V variants), and 201–220 (Q217R) were synthesized. Nerve andglial cell viability was determined after prolonged exposure of rat pri-mary neuronal and astroglial cultures to micromolar concentrations of
STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS 193

peptides. Although mutations P102L and D178N strongly increased theneurotoxic activity of the native sequence, the other substitutions hadno effect. The neurotoxicity of PrP89–106 carrying the P102L mutationwas not related to changes in aggregation properties, because the pep-tide was not able to form amyloid fibrils, similar to the wild-typesequence. Conversely, the D187N substitution resulted in strikingincrease in fibrillogenic capacity. Furthermore, PrP169–185 withD187N mutation was able to induce astroglial proliferation, at variancewith the wild-type sequence. These data suggest that PrP sequences inthe mutated form may be neurotoxic and further emphasize that neu-rotoxicity may be independent of amyloidogenesis.
C. Effects of PrP Peptides on Astroglial and Microglial Cultures
Based on the observation that glial cell activation occurs early in thecourse of experimental prion disease and is related to PrP deposits(Williams et al., 1994), several studies focused on the effects of syntheticPrP peptides on astrocytes and microglia in vitro. Prolonged exposure ofprimary astroglial cultures to peptide PrP106–126 resulted in a remark-able increase in size and density of astroglial processes (Forloni et al.,1994, Fig. 4C and D). The hypertrophy of astrocytes was associated with astriking increase in glial fibrillary acidic protein (GFAP) transcripts asrevealed by Northern blot analysis. Densitometric quantification of GFAPmRNA showed that this increment was dependent on peptide concentra-tion, being significant at 10 µM and resulting in 3- and 5-fold increaseabove control values at 25 µM and 50 µM, respectively. The rise of GFAPtranscripts was accompanied by a substantial increase in GFAP, as deter-mined by Western blot analysis (Forloni et al., 1994). Furthermore, astro-cytes exposed to PrP106–126 showed increased expression ofapolipoprotein J (ApoJ). This effect was maximal after 7 days and was con-centration-related, being significant at 25 µM and resulting in 3-fold incre-ment at 50 µM (Chiesa et al., 1996). It is noteworthy that a strikingincrease in ApoJ mRNA has been found in experimental scrapie and ApoJimmunoreactivity is associated with PrPSc and PrP amyloid in CJD andGSS brains. Furthermore, PrP106–126 was able to enhance proliferationof astrocytes. This effect was remarkable when cultures were kept inserum-free medium. Under these conditions, the uptake of thymidineafter 9-day treatment with 50 µM peptide was 50 times higher than thebasal values (Florio et al., 1996) The proliferative effect of PrP106–126 wasabolished by cotreatment of astroglial cultures with nicardipine, (i.e., ablocker of L-type voltage-sensitive calcium channels). Microfluorimetricanalysis of intracellular calcium levels in single astrocytes showed that
194 FABRIZIO TAGLIAVINI ET AL.

PrP106–126 induced a rapid increase in cytosolic calcium concentrations,followed by a slow return to basal levels. This effect was not observed withthe scrambled peptide; it was absent when calcium was removed from themedium and was prevented by preincubation of cultures with nicardipine.These data suggest that PrP106–126 stimulates astroglial proliferation viaan increase in intracellular calcium concentration, through the activationof L-type voltage-sensitive calcium channels. Brown (1999) has proposedthat PrP106–126 neurotoxicity may be partly mediated by the ability of thepeptide to inhibit the glutamate uptake by astrocytes. Further, it has beenadvanced that this inhibition may occur through the interaction of thepeptide with PrPC of astrocytes based on the finding that a lower rate ofglutamate uptake was related to PrPC expression (Brown and Mohn,1999). The involvement of glutamate-mediated neurotoxicity in prion dis-ease pathogenesis has been hypothesized by other authors (Müller et al.,1993; Scallet and Ye, 1997); however, the supposed role of PrPC in gluta-mate uptake needs to be further investigated.
The prolonged exposure of primary mouse microglial cultures tomicromolar concentrations of PrP106–126 resulted in enhanced cell pro-liferation and increased oxygen radical production (Brown et al., 1996).These effects were not dependent on the expression of endogenousPrPC. Treatment of a pure culture of human resting microglia withPrP106–126 induced morphological changes with a shift from ramifiedto ameboid morphology, enhanced phagocitic activity, and cell prolifera-tion (Fig. 6E and F). These effects were associated with calcium entrythrough voltage-sensitive calcium channels and were blocked by specificinhibitors of these channels (Silei et al., 1999). Studies with neuron-microglia co-cultures and mixed glia cultures indicate that the activationof microglia increases nerve cell death and astrocyte proliferation afterpeptide treatment. Nevertheless, the observations that PrP106–126 istoxic to microglia-free neuronal cell lines and induces hypertrophy andproliferation of highly purified astrocytes (Florio et al., 1996; Flemingerand Curtis, 1997) point to a direct effect of the peptide on nerve andastroglial cells, in addition to a microglia-mediated effect.
Peyrin et al. (1999) found that microglial activation by PrP106–126was associated with 5-fold overexpression of proinflammatory cytokineIL-1β and IL-6, but TNFα and anti-inflammatory cytokines IL-10 andTGF(β1) were unchanged. These effects were dependent on the abilityof the peptide to form fibrils, because PrP106–126 NH2 was ineffective.These findings are in agreement with microglial activation andcytokines production observed in experimental scrapie (Williams et al.,1994; Kim et al., 1999; Baker et al., 1999) and suggests that proinflam-matory cytokines may contribute to prion disease pathogenesis.
STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS 195

V. CONCLUDING REMARKS
The hypothesis that PrPC misfolding leads to a neurodegenerativeprocess that can be transmitted by abnormal PrPSc conformers urged atthe recognition of polypeptide segments that are central to PrPC-PrPSc
interaction and PrPC>PrPSc conversion. Molecular modeling and NMRstudies have suggested putative critical regions. Noteworthy, some PrPfragments from these regions were found to possess both an unusualstructural polymorphism and the ability to reproduce pathological hall-marks of the disease process in vitro. Accordingly, PrP peptides can beregarded as a prime tool for investigating the molecular basis of proteinmisfolding and disease pathogenesis.
The need to develop therapies for prion diseases has remarkablyincreased following the emergence of the new variant CJD. Althoughseveral compounds have been found to antagonize prion propagationin vitro or in vivo, the most suitable target for pharmacological therapyhas not yet been identified. One possible strategy is to recognize com-pounds able to interfere with PrP domains involved in the interactionof PrPC with PrPSc or other macromolecules that feature in the conver-sion process. An alternative approach is to develop compounds capableof binding PrPSc selectively and of destabilizing the misfolded structureof the protein. Evidence suggests that short synthetic peptides homolo-gous to PrP domains implicated in abnormal folding and bearingamino acid residues that destabilize β-conformation may possess theseproperties (Chabry et al., 1998; Soto et al., 2000). Furthermore, syn-thetic PrP amyloid and large PrP peptides that can be folded into dif-ferent conformers can be conveniently used for large-scale screening ofcompounds in vitro.
ACKNOWLEDGMENTS
This study was supported by the Italian Ministry of Health, Department of Social Services(RF99–38), the European Community (BMH4 CT98-6011, CT98-6051) and Telethon Italia(E574). We are grateful to Dr. Thomas Stockner for the critical comments given in the prepara-tion of the manuscript.
REFERENCES
Angeretti, N., Rodrigues Martin, T., Peressini, E., Lucca, E., Galbeten, J. L., Bugiani, O.,Tagliavini, F., Salmona, M., and Forloni, G. (2001). Neuroscience, in press.
Baker, C.A., Lu, Z.Y., Zaitsev, I., and Manuelidis, L. (1999). J. Virol. 73, 5089–5097.Bass, N.H., Hess, H.H., and Pope, A. (1974). Arch. Neurol. 31, 174–182.
196 FABRIZIO TAGLIAVINI ET AL.

Bessen, R.A., and Marsh, R.F. (1992). J. Virol. 66, 2096–2101.Bessen, R.A., and Marsh, R.F. (1994). J. Virol. 68, 7859–7868.Brandner, S., Isenmann, S., Raeber, A., Fischer, M., Sailer, A., Kobayashi, Y., Marino, S.,
Weissmann, C., and Aguzzi, A. (1996). Nature 379, 339–343.Brasseur, R. (1991). J. Biol. Chem. 266, 16120–16127.Brown, D.R., Herms, J., and Kretzschmar, H.A. (1994). Neuroreport 5, 2057–2060.Brown, D.R., Schmidt, B., and Kretzschmar, H.A. (1996). Nature 380, 345–347.Brown, D.R., Qin, K., Herms, J.W., Madlung, A., Manson, J., Strome, R., Fraser, P.E.,
Kruck, T., von Bohlen, A., Schulz-Schaeffer, W., Giese, A., Westaway, D., and Kret-zschmar, H.A. (1997). Nature 390, 684–695.
Brown, D.R. (1998). J. Neurosci. Res. 54, 331–340.Brown, D.R. (1999). J. Neurochem. 73, 1105–1113.Brown, D.R., and Mohn, C.M. (1999). Glia 25, 282–292.Bruce, M.E., McBride, P.A., and Farquhar, C.F. (1989). Neurosci. Lett. 102, 1–6.Bruce, M.E., Will, R.G., Ironside, J.W., McConnell, I., Drummond, D., Suttle, A., McCar-
dle, L., Chree, A., Hope, J., Birkett, C., Cousens, S., Fraser, H., and Bostock, C.J.(1997). Nature 389, 498–501.
Büeler, H., Aguzzi, A., Sailer, A., Aguet, M., and Weissmann, C. (1993). Cell 73, 1339–1347.Bugiani, O., Giaccone, G., Frangione, B., Ghetti, B., and Tagliavini, F. (1989). Neursci.
Lett. 103, 263–268.Caughey, B.W., Dong, A., Bhat, K.S., Ernst, D., Hayes, S.F., and Caughey, W.S. (1991).
Biochemistry 30, 7672–7680.Chabry, J., Caughey, B., and Chesebro, B. (1998). J. Biol. Chem. 273, 13203–13207.Chiesa, R., Angeretti, N., Lucca, E., Salmona, M., Tagliavini, F., Bugiani, O., and Forloni,
G. (1996). Eur. J. Neurosci. 8, 589–597.Chou, S.M., Payne, W.N., Gibbs, C.J., Jr., and Gajdusek, D.C. (1980). Brain 103, 885–904.De Gioia, L., Cantù, L., Ghibaudi, E., Diomede, L., Passerini, F., Forloni, G., Orso, O.,
Tagliavini, F., and Salmona, M. (1994). J. Biol. Chem. 269, 563–570.DeArmond, S.J., Gonzales, M., Mobley, W.C., Kon, A.A., Sern, H., and Prusiner, S.B.
(1988). In “Alzheimer’s Disease and Related Disorders. Progress in Clinical BiologyResearch” (K. Ikbal, H.M. Wisniewsky, and B. Wimblad, eds.) Vol. 317 pp. 601–618.Alan R. Liss, New York.
Donne, D.G., Viles, J.H., Groth, D., Mehlhorn, I., James, T.L., Cohen, F.E., Prusiner, S.B.,Wright, P.E., and Dyson, H.J. (1997). Proc. Natl. Acad. Sci. U.S.A. 94, 13452–13457.
Fleminger, S., and Curtis, D. (1997). Br. J. Psychiatry 170, 103–105.Florio, T., Grimaldi, M., Scorziello, A., Salmona, M., Bugiani, O., Tagliavini, F., Forloni,
G., and Schettini, G. (1996). Biochem. Biophys. Res. Commun. 228, 397–405.Florio, T., Thellung, S., Amico, C., Robello, M., Salmona, M., Bugiani, O., Tagliavini, F.,
Forloni, G., and Schettini, G. (1998). J. Neurosci. Res. 54, 341–352.Forloni, G., Angeretti, N., Chiesa, R., Monzani, E., Salmona, M., Bugiani, O., and Tagli-
avini, F. (1993). Nature 362, 543–545.Forloni, G., Del Bo, B.R., Angeretti, N., Chiesa, R., Smiroldo, S., Doni, R., Ghibaudi, E.,
Salmona, M., Porro, M., Verga, L., Giaccone, G., Bugiani, O., and Tagliavini, F.(1994). Eur. J. Neurosci. 6, 1415–1422.
Forloni, G., Bugiani, O., Tagliavini, F., and Salmona, M. (1996). Mol. Chem. Neuropathol.28, 163–171.
Forloni, G., Lucca, E., Angeretti, N., Della Torre, P., and Salmona, M. (1997). J. Neu-rochem. 69, 2048–2056.
Forloni, G., Angeretti, N., Malesani, P., Peressini, E., Martin, R., Della Torre, P., andSalmona, M. (1999). Ann. Neurol. 45, 489–494.
STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS 197

Forloni, G., Salmona, M., Bugiani, O., and Tagliavini, F. (2000). FEBS Lett. 466, 205–206.Gabriel, J.M., Oesch, B., Kretzschmar, H., Scott, M., and Prusiner, S.B. (1992). Proc. Natl.
Acad. Sci. U.S.A. 89, 9097–9101.Gasset, M., Baldwin, M.A., Lloyd, D.H., Gabriel, J.M., Holtzman, D.M., Cohen, F., Flett-
erick, R., and Prusiner, S.B. (1992). Proc. Natl. Acad. Sci. U.S.A. 89, 10940–10944.Ghetti, B., Piccardo, P., Frangione, B., Bugiani, O., Giaccone, G., Young, K., Prelli, F.,
Farlow, M.R., Dlouhy, S.R., and Tagliavini, F. (1996a). Brain Pathol. 6, 127–145.Ghetti, B., Piccardo, P., Spillantini, M.G., Ichimiya, Y., Porro, M., Perini, F., Kitamoto, T.,
Tateishi, J., Seiler, C., Frangione, B., Bugiani, O., Giaccone, G., Prelli, F., Goedert, M.,Dlouhy, S.R., and Tagliavini, F. (1996b). Proc. Natl. Acad. Sci. U.S.A. 93, 744–748.
Giaccone, G., Verga, L., Bugiani, O., Frangione, B., Serban, D., Prusiner, S.B., Farlow,M.R., Ghetti, B., and Tagliavini, F. (1992). Proc. Natl. Acad. Sci. U.S.A. 89, 9349–9353.
Goldfarb, L.G., Brown, P., Haltia, M., Ghiso, G., and Frangione, B. (1993). Proc. Natl.Acad. Sci. U.S.A. 90, 4451–4454.
Haïk, S., Peyrin, J.M., Lins, L., Rosseneu, M.Y., Brasseur, R., Langeveld, J.P., Tagliavini, F.,Deslys, J.P., Lasmézas, C., and Dormond, D. (2000). Neurobiol. Dis. 7, 644–656.
Harris, D.A., Falls, D.L., Johnson, F.A., and Fischbach, G.D. (1991). Proc. Natl. Acad. Sci.U.S.A. 88, 7664–7668.
Hedge, R.S., Mastrianni, J.A., Scott, M.R., De Fea, K.A., Tremblay, P., Torchia, M., DeAr-mond, S.J., Prusiner, S.B., and Lingappa, V.R. (1998). Science 279, 827–834.
Heller, J., Kolbert, A.C., Larsen, R., Ernst, M., Bekker, T., Baldwin, M., Prusiner, S.B.,Pines, A., and Wemmer D.E. (1996). Protein Sci. 5, 1655–1661.
Hölscher, C., Delius, H., and Burkle, A. (1998). J. Virol. 72, 1153–1159.Hope, J., Shearman, M.S., Baxter, H.C., Chong, A., Kelly, S.M., and Price, M.C. (1996).
Neurodegeneration 5, 1–11.Hornshaw, M.P., McDermott, J.R., and Candy, J.M. (1995a). Biochem. Biophys. Res. Com-
mun. 207, 621–629.Hornshaw, M.P., McDermott, J.R., Candy, J.M., and Lakey, J.H. (1995b). Biochem. Biophys.
Res. Commun. 214, 993–999.Inouye, H., and Kirschner, D.A. (1998). J. Struct. Biol. 122, 247–255.Jobling, M.F., Stewart, L.R., White, A.R., McLean, C., Friedhuber, Maher, F., Beyreuther,
K., Masters, C.L., Barrow, C.J., Collins, S.J., and Cappai, R. (1999). J. Neurochem. 73,1557–1565.
Johnson, R.T., and Gibbs, C.J. (1998). N. Engl. J. Med. 339, 1994–2004.Kaneko, K., Ball, H.L., Wille, H., Zhang, H., Groth, D., Torchia, M., Tremblay, P., Safar, J.,
Prusiner, S.B., DeArmond, S.J., Baldwin, M.A., and Cohen, F.E (2000). J. Mol. Biol.295, 997–1007.
Kaplan, B., Vidal, R., Kumar, A., Ghiso, J., Frangione, B., and Gallo, G. (1997). Clin. Exp.Immunol. 110, 472–478.
Kim, J.I., Ju, W.K., Choi, J.H., Choi, E., Carp, R.I., Wisniewski, H.M., and Kim, Y.S.(1999). Brain Res. 73, 17–27.
Kitamoto, T., Iizuka, R., and Tateishi, J. (1993). Biochem. Biophys. Res. Commun. (1993).192, 525–531.
Korth, C., Stierli, B., Streit, P., Moser, M., Schaller, O., Fischer, R., Schulz-Schaeffer, W.,Kretzschmar, H., Raeber, A., Braun, U., Ehrensperger, F., Hornemann, S., Glockshu-ber, R., Riek, R., Billeter, M., Wüthrich, K., and Oesch, B. (1997). Nature 390, 74–77.
Kristensson, K., Feuerstein, B., Taraboulos, S., Hyun, W.C., Prusiner, S.B., and DeArmond, S.J. (1993). Neurology 43, 2335–2341.
Kshet, I., Ovadia, H., Taraboulos, A., and Gabizon, R. (1999). Neurochemistry 72, 1224–1231.Kunz, B., Sandmeier, E., and Christen, P. (1999). FEBS Lett. 458, 65–68.
198 FABRIZIO TAGLIAVINI ET AL.

Lehmann, S., and Harris, D.A. (1995). J. Biol. Chem. 270, 24589–24597.Lin, M.C., Mirzabekov, T., and Kagen, B.L. (1997). J. Biol. Chem. 272, 44–47.Liu, A., Riek, R., Zahn, R., Hornemann, S., Glockshuber, R., and Wüthrich, K. (1999).
Biopolymers 51, 145–152.Lievens, P.M.J., Quaglio, E., Piccardo, P., Tranchant, C., Warter, J.M., Bird, T., Bugiani,
O., Ghetti, B., and Tagliavini F. (1998). J. Neuropathol. Exp. Neurol. 57, 517.Lopez Garcia, F., Zahn, R., Riek, R., and Wüthrich, K. (2000). Proc. Natl. Acad. Sci. U.S.A.
97, 8334–8339.Marsh, R.F., Dees, C., Castle, B.E., Wade, W.F., and German, T.L. (1984). J. Gen. Virol. 65,
415–421.McKinley, M.P., Taraboulos, A., Kenaga, L., Serban, D., Stieber, A., De Armond, S.J.,
Prusiner, S.B., and Gonatas, N. (1991). Lab. Invest. 65, 622–630.Merz, P.A., Somerville, R.A., Wisniewski, H.M., and Iqbal, K. (1981). Acta Neuropathol. 54,
63–74.Merz, P.A., Somerville, R.A., Wisniewski, H.M., Manuelidis, L., and Manuelidis, E.E.
(1983). Nature 306, 474–476.Migheli, A., Atzori, M., Srinivasan, A.N., and Ghetti, B. (2000). Neurobiol. Aging 21, S266.Millson, G., Hunter, G.D., and Kimberlin, R.H. (1971). J. Comp. Pathol. 81, 255–265.Miravalle, L., Tokuda, T., Chiarle, R., Giaccone, G., Bugiani, O., Tagliavini, F., Frangione,
B., and Ghiso, J. (2000). J. Biol. Chem. 275, 27110–27116.Miura, T., Hori-i, A., and Takeuchi, H. (1996). FEBS Lett. 396, 248–252.Miura, T., Hori-i, A., Mototani, H., and Takeuchi, H. (1999). Biochemistry 38,
11560–11569.Müller, W.E., Ushijima, H., Schroder, H.C., Forrest, J.M., Schatton, W.F., Rytik, P.G., and
Heffner-Lauc, M. (1993). Eur. J. Pharmacol. 246, 261–267.Muramoto, T., Scott, M., Cohen, F.E., and Prusiner, S.B. (1996). Proc. Natl. Acad. Sci.
U.S.A. 93, 15457–15462.Murata, M., Peranen, J., Schreiner, R., Wieland, F., Kurzhalla, T.V., and Simons, K.
(1995). Proc. Natl. Acad. Sci. U.S.A. 92, 10339–10343.Narwa, R., and Harris, D.A. (1999). Biochemistry 38, 8770–8777.Nguyen, J.T., Baldwin, M.A., Cohen, F.E., and Prusiner, S.B. (1995a). Biochemistry 34,
4186–4192.Nguyen, J.T., Inouye, H., Baldwin, M.A., Fletterick, R.J., Cohen, F.E., Prusiner, S.B., and
Kirschner, D.A. (1995b). J. Mol. Biol. 252, 412–422.Pan, K.M., Stahl, N., and Prusiner, S.B. (1992). Protein Sci. 1, 1343–1352.Pan, K.M., Baldwin, M.A., Nguyen, J.T., Gasset, M., Serban, A., Groth, D., Mehlhorn, I.,
Huang, Z., Fletterick, R.J., Cohen, F.E., and Prusiner, S.B. (1993). Proc. Natl. Acad. Sci.U.S.A. 90, 10962–10966.
Parchi, P., Castellani, R., Capellari, S., Ghetti, B., Young, K., Chen, S.G., Farlow, M., Dick-son, D.W., Sima, A.A.F., Trojanowski, J.Q., Petersen, R.B., and Gambetti, P. (1996).Ann. Neurol. 39, 767–778.
Parchi, P., Chen, S.G., Brown, P., Zou, W., Cappellari, S., Budka, H., Hainfellner, J.,Reyes, P.F., Golden, G.T., Hauw, J.J., Gajdusek, D.C., and Gambetti, P. (1998). Proc.Natl. Acad. Sci. U.S.A. 95, 8322–8327.
Pauly, P.C., and Harris, D.A. (1998). J. Biol. Chem. 273, 33107–33110.Perez, M., Wandosell, F., Colaco, C., and Avila, J. (1998). Biochem. J. 335, 369–374.Perovic, S., Pergande, G., Ushijima, H., Kelve, M., Forrest, J., and Muller, W.E. (1995).
Neurodegeneration 4, 369–374.Perovic, S., Bohm, M., Meesters, E., Meinhardt, A., Pergrande, G., and Muller, W.E.
(1998). Mech. Ageing Dev. 101, 1–19.
STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS 199

Peyrin, J.M., Lasmézas, C.I., Haïk, S., Tagliavini, F., Salmona, M., Williams, A., Richie, D.,Deslys, J.P., and Dormond, D. (1999). Neuroreport 10, 723–729.
Piccardo, P., Seiler, C., Dlouhy, S.R., Young, K., Farlow, M.R., Prelli, F., Frangione, B.,Bugiani, O., Tagliavini, F., and Ghetti, B. (1996). J. Neuropathol. Exp. Neurol. 55,1157–1163.
Piccardo, P., Dlouhy, S.R., Lievens, P.M.J., Young, K., Bird, T.D., Nochlin, D., Dickson,D.W., Vinters, H.V., Zimmerman, T.R., Mackenzie, I.R., Kish, S.J., Ang, L.C., De Carli,C., Pocchiari, M., Brown, P., Gibbs, C.J.Jr., Gajdusek, D.C., Bugiani, O., Ironside, J.,Tagliavini, F., and Ghetti, B. (1998). J. Neuropathol. Exp. Neurol. 57, 979–988.
Pike, C.J., Walencewicz, A.J., Glabe, C.G., and Cotman, C.W. (1991). Eur. J. Pharmacol.207, 367–368.
Pillot, T., Goethals, M., Vanlao, B., Talussot, C., Brasseur, R., Rosseneu, M., Vandekerck-hove, J., and Lins, L. (1996). J. Biol. Chem. 271, 28757–28765.
Pillot, T., Lins, L., Goethals, M., Vanlao, B., Baest, J., Vandekerckhove, J., Rosseneu, M.,and Brasseur, R. (1997). J. Mol. Biol. 274, 381–393.
Prusiner, S.B., McKinley, M.P., Bowman, K.A., Bolton, D.C., Bendheim, P.E., Groth, D.F.,and Glenner, G.G. (1983). Cell 35, 349–358.
Prusiner, S.B., Groth, D.F., Bolton, D.C., Kent, S.B., and Hood, L.E. (1984). Cell 38,127–134.
Prusiner, S.B. (1991). Science 252, 1515–1522.Raeber, A.J., Race, R.E., Brandner, S., Priola, S.A., Sailer, A., Bessen, R.A., Mucke, L.,
Manson, J., Aguzzi, A., Oldstone, M.B.A., Weissmann, C., and Chesebro, B. (1997).EMBO J. 16, 6057–6065.
Ragg, E., Tagliavini, F., Malesani, P., Monticelli, L., Bugiani, Forloni, G., and Salmona, M.(1999). Eur. J. Biochem. 266, 1192–1201.
Riek, R., Hornemann, S., Wider, G., Billeter, M., Glockshuber, R., and Wüthrich, K.(1996). Nature 382, 180–182.
Riek, R., Hornemann, S., Wider, G., Glockshuber, R., and Wüthrich, K. (1997). FEBS Lett.413, 282–288.
Rizzardini, M., Chiesa, R., Angeretti, N., Lucca, E., Salmona, M., Forloni, G., and Can-toni, L. (1997). J. Neurochem. 68, 715–720.
Safar, J., Roller, P.P., Gajdusek, D.C., and Gibbs, C.J.Jr. (1993). J. Biol. Chem. 268,20276–20284.
Salmona, M., Forloni, G., Diomede, L., Algeri, M., De Gioia, L., Angeretti, N., Giaccone,G., Tagliavini, F., and Bugiani, O. (1997). Neurobiol. Dis. 4, 47–57.
Salmona, M., Malesani, P., De Gioia, L., Gorla, S., Bruschi, M., Molinari, A., DellaVedova, F., Pedrotti, B., Marrari, M.A., Awan, T., Bugiani, O., Forloni, G., and Tagli-avini, F. (1999). Biochem. J. 342, 207–214.
Scallet, A.C., and Ye, X. (1997). Ann. N.Y. Acad. Sci. 825, 194–205.Selvaggini, C., De Gioia, L., Cantù, L., Ghibaudi, E., Diomede, L., Passerini, F., Forloni,
G., Bugiani, O., Tagliavini, F., and Salmona, M. (1993). Biochem. Biophys. Res. Commun.194, 1380–1386.
Semancik, J.S., Marsh, R.F., Geelen, J.L., and Hanson, R.P. (1976). J. Virol. 18, 693–700.Silei, V., Fabrizi, C., Venturini, G., Salmona, M., Bugiani, O., Tagliavini, F., and Lauro,
G.M. (1999). Brain Res. 818, 168–170.Soto, C., Kascsak, R.J., Saborio, G.P., Aucouturier, P., Wisniewski, T., Prelli, F., Kascsak, R.,
Mendez, E., Harris, D.A., Ironside, J., Tagliavini, F., Carp, R.I., and Frangione, B.(2000) Lancet 355, 192–197.
Stöckel, J., Safar, J., Wallace, A.C., Cohen, F.E., and Prusiner S.B. (1998). Biochemistry 37,7185–7193.
200 FABRIZIO TAGLIAVINI ET AL.

Supattapone, S., Bosque, P., Muramoto, T., Wille, H., Aagaard, C., Peretz, D., Nguyen,H.O., Heinrich, C., Torchia, M., Safar, J., Cohen, F.E., DeArmond, S.J., Prusiner, S.B.,and Scott, M. (1999). Cell 96, 869–878.
Tagliavini, F., Prelli, F., Ghiso, J., Bugiani, O., Serban, D., Prusiner, S.B., Farlow, M.R.,Ghetti, B., and Frangione, B. (1991). EMBO J. 10, 513–519.
Tagliavini, F., Prelli, F., Verga, L., Giaccone, G., Sarma, R., Gorevic, P., Ghetti, B.,Passerini, F., Ghibaudi, E., Forloni, G., Salmona, M., Bugiani, O., and Frangione, B.(1993). Proc. Natl. Acad. Sci. U.S.A. 90, 9678–9682.
Tagliavini, F., Prelli, F., Porro, M., Rossi, G., Giaccone, G., Farlow, M.R., Dlouhy, S.R.,Ghetti, B., Bugiani, O., and Frangione, B. (1994). Cell 79, 695–703.
Tagliavini, F., Lievens, P. M.-J., Tranchant, C., Warter, J.-M., Mohr, M., Giaccone, G.,Perini, F., Rossi, G., Salmona, M., Piccardo, P., Ghetti, B., Beavis, R.C., Bugiani, O.,Frangione, B., and Prelli, F. (2001). J. Biol. Chem. 276, 6009–6015.
Taraboulos, A., Scott, M., Semenov, A., Avrahami, D., Laszlo, L., Prusiner, S.B., and Avra-ham D. (1995). J. Cell Biol. 129, 121–132.
Terzi, E., Holzemann, G., and Seelig, J. (1994). Biochemistry 33, 1345–1350.Traub, R.D., and Pedley, T.A. (1981). Ann. Neurol. 10, 405–410.Vey, M., Pilkuhn, S., Will, H., Nixon, R., DeArmoud, S.J., Smart, E.J., Anderson, R.G.W.,
Taraboulos, A., and Prusiner, S.B. (1996). Proc. Nat. Acad. Sci. U.S.A. 93, 14945–14949.Viles, J.H., Cohen, F.E., Prusiner, S.B., Goodin, D.B., Wright, P.E., and Dyson, H.J.
(1999). Proc. Natl. Acad. Sci. U.S.A. 96, 2042–2047.Wadsworth, J.D.F., Hill, A.F., Joiner, S., Jackson, G.S., Clarke, A.R., and Collinge, J.
(1999). Nature Cell Biol. 1, 55–59.Waggoner, D.J., Drisaldi, B., Bartnikas, T.B., Casareno, R.L.B., Prohaska, J.R., Gitlin, J.D.,
and Harris, D.A. (2000). J. Biol. Chem. 725, 7455–7458.Will, R.G., Ironside, J.W., Zeidler, M., Cousens, S.N., Estibeiro, K., Alperovitch, A., Poser,
S., Pocchiari, M., Hofman, A., and Smith, P.G. (1996). Lancet 347, 921–925.Williams, A.E., van Dam, A.M., Man-A-Hing, W.K., Berkenbosch, F., Eikelenboom, P.,
and Fraser, H. (1994). Brain Res. 654, 200–206.Young, K., Piccardo, P., Dlouhy, S., Bugiani, O., Tagliavini, F., and Ghetti, B. (1999). In
“Prions: Molecular and Cellular Biology” (D.A. Harris, Ed.), pp. 139–175. HorizonScientific Press, Wymondham, UK.
Zahn, R., Liu, A., Luhrs, T., Riek, R., von Schroetter, C., Lopez Garcia, F., Billeter, M.,Calzolai, L., Wider, G., and Wüthrich, K. (2000). Proc. Natl. Acad. Sci. U.S.A. 97,145–150.
Zhang, H., Kanelo, K., Nguyen, J., Livshits, T., Baldwin, M.A., Cohen, F.E., James, T.L.,and Prusiner, S.B. (1995). J. Mol. Biol. 250, 514–526.
STUDIES ON PEPTIDE FRAGMENTS OF PRION PROTEINS 201




















