
Galacto-oligosaccharides attenuate renal injury with microbiota modification
Upload
independentCategory
view
0download
0
ELSEVIER Carbohydrate Research 266 (1995) 237-261
CARBOHYDRATE RESEARCH
Structural studies of the O-antigen oligosaccharides from two strains of Moraxella catarrhalis
serotype C
Per Edebrink a, Per-Erik Jansson a,*, M. Mahbubur Rahman a, G6ran Widmalm ~ Tord Holme b Motiur Rahman b
"Department of Organic Chemistry, Arrhenius Laboratory, Stockholm University, S-106 91 Stockholm, Sweden b Microbiology and Tumor Biology Centre, Division of Bacteriology, Karolinska Institute, S-171 77 Stockholm,
Sweden
Received 19 May 1994; accepted 12 August 1994
Abstract
The oligosaccharide parts from Moraxella (Branhamella) catarrhalis serotype C lipooligosac- charities were isolated by mild acid hydrolysis followed by gel permeation chromatography. Four different oligosaccharides could be identified from strain RS26 and two from strain RSI0. The structures of the O-oligosaccharides were established by methylation analyses, mass spectrometry, and NMR spectroscopy. It is concluded that the oligosaccharide O-antigens from RS26 are a mixture of octa-, deca-, and undeca-saccharides, and most likely a heptasaccharide. Strain RSI0 contains the deca- and the undeca-saccharide only. The structures for the oligosaccharides are shown below.
(x-D-GIcpNAc-(l-.~2)-l~-D-Glcp I J, 4
a-D-GIcp-( I..~2)-[~-D-GIcp-(l-a6)-Ot-D-Glcp-( I.-~5)-Kdo 3 1' I
{3.o-GIcp
OS(7)
* Corresponding author.
0008-6215/95/$09.50 © 1995 Elsevier Science B.V. All rights reserved SSDI0008-6215 (94) 00276-2

238 P. Edebrink et at. / Carbohydrate Research 266 (1995) 237-261
[3-O-Galp-( I --v4)-ct-D-GlcpNAc-( I --->2)-~-D-Glcp I $ 4
ot-D-Glcp-( I --,'.2)-~D-Glcp-( I .---.~ )-ct-D-Glcp-( I ---~5)-Kdo 3 1" I
~O-Glcp
OS(8)
~]-D-Galp-( I ---~)-ot-D-GlcpNAc-( I --*2)-~-D-Glcp I ,L 4
ta-D-Galp-( I -¢4)-~-D-Galp-( t ---~4).ct-D-Glcp-( I-~2)-I~-D-Glcp-( I --¢6)-ot-D-Glcp-( I ---¢5)-Kdo 3 1" I
OS(10)
ot-OoGalp-( I ~)4)ol3-D-Galp-( I --~)4x-O-GlcpNAc-( I -~2)-13-O-Glcp I
4 ~t,OoGalp.( I -,)4)°~-OoOalpo( I -~4)-ot-O°Glcp-( I -*2)o0-DoOlcpo( I .~6)-mD-Glcp°( I .-)5) Kdo
3
I ~.D.olcv
OS(11) Methylation analysis of the intact lipooligosaccharides showed that two Kdo residues were present,
one terminal and one 4,5-substituted residue, it also showed that they consisted of a lipid A portion with 6-substituted glucosamine residues.
Keywords: Mortvcella catarrhalis; Branhameila; Lipooligosaccharide; NMR
1. lu~v¢~uction
Moraxella catarrhalis causes, inter alia, respiratory diseases in patients with chronic bronchitis and in immunocompromized hosts [ 1 ], childhood otitis media, and maxillary siausitis [2]. The lipooligosaccharide (LOS) of M. catarrhalis lacks the extended O- antigenic side chains characteristic of enteric pathogens, thus being similar in general structure to the LOS of Neisseria, Haemophilus, and Bordetella [3-6]. We have in this paper adopted the term lipooligosaccharide in order to conform with recent carbohydrate nomenclature rules [7]. Also, consequently, the product after hydrolysis is an oligosac- charide. Serological typing of M. catarrhalis based on LOS antigens has been described

P. Edebrink et al. / Carbohydrate Research 266 (1995) 237-261 239
[8]. Three major antigenic types, A, B, and C, were distinguished, accounting for 95% of strains isolated from different sources. It was shown that D-glucose, D-galactose, D-giucos- amine, and Kdo (but not heptose) were present [5,9,10]. The qualitative composition of the different sugar constituents was similar in all strains tested, but differences in their relative amounts were recorded.
We have recently studied the serotype A LOS (strain ATCC 25238) and described structure I for the oligosaccharide part [ 11 ]. A second Kdo residue could be shown to be linked to the Kdo shown in 1.
ot-D..GlcpNAc-(I.--~2)-~D-Glcp i ,1, 4
~-D-Oalp-( l--~)-I]=D=Galp-( I ---~4)-ct-D-Glep-(I--~2)-~-D-Glcp-(! --~6)-ot-l>-Olcp-(I.-~5)-Kdo 3
I ~ctcp
1 The same structure was derived by another group working on the same LOS [ 12]. In
this paper we report the structure of the oligosaccharide part of the LOS from two strains of M. catarrhalis serotype C.
2. Results and discussion
Two strains of Moraxella catarrhalis serotype C, RSI0 and RS26, were used, both obtained from clinical isolates from Roslagstulls Sjukhus in Stockholm, Sweden. Cells of M, c¢aat~'halis were grown in brain-heart infusion broth containing yeast extract and the bacterial pellet was ist~lated by ccntrifllgation. Extraction of the freeTe-dried pellet with phenol--chlorotbrm°-petroleum ether yielded a purified LOS. A low UV absorption indicated the absence of nucleic acids and Lowry tests showed that the content of protein also was low. Phospholipids were present as evident from fatty acid analysis and TLC, but were not removed.
SDS-polyacrylamide gel electrophoresis of the LOS showed a single band upon silver staining (data not shown). The average molecular mass was estimated by comparison of migration distance with those from Salmonella LOS of known molecular mass, and was approximately 4200 Daltons. These results were in agreement with those for M. catarrhalis serotype A.
Treatment of the LOS from RS10 or RS26 in 0.1 M ~cetate buffer of pH 4.4 containing 0.1% SDS for 2 h at 100°C, yielded the oligosaccharid~s OS(10) and OS( 11 ) for strain RSI0, and OS(8), OS(10), and small amounts of OS(7) and OS( 1 i) for strain RS26, the number in parentheses referring to the number of sugar residues in the oligosaccharide. Subsequent work-up gave lipid-free solutions of the oligosaccharides which were further purified by gel permeation chromatography. The oligosaccharides from RS26 were eluted in the region for large oligosaccharides, OS(7) and OS(8) as one peak partially overlapping the peak of the unresolved OS(10) and OS(11). The oligosaccharides from strain RS10.

240 Po Edebrink et al. / Carbohydrate Research 266 (1995) 237-261
(a)
PS(IO)
PS( l l )
t I I I I I 0 $ 10 15 20 23 min
PS(tO)
(hi
PS(8 )
PS(t|)
I I I I I I (I '~ I0 I ~ ~1 2:1 mr .
Fig, l, Pionex ch|omatogram of the oligo~echaridcs from the lipooligosaccharides of M, catarrhalis serotype C, RSI0 (a) and RS26 (b), The samples were elutcd with a gradient starting from 85:15 100 mM NaOH (eluent A)~100 mM NaOH and 500 mM NaOAc (eluent B) and ending at 75:25 after 30 rain.
OS(10), and OS(II) were eluted as one peak in the region for large oligosaccharides. Chromatograms from high-pH high-performance anion-exchange chromatography (Dionex) of the oligosaccharides from the lipooligosaccharides of RSI0 and RS26 are shown in Fig, 1. Separation of OS(10) and OS( 11 ) from RSI0 was performed using the Dionex system, Subsequent gel permeation chromatography yielded the desalted oligosac- charides. Repeated gel permeation chromatography of the oligosaccharides obtained from RS26 yielded one fraction containing mainly OS(8) and some OS(7); another fraction contained OS (10) and a smaller proportion of OS ( 11 ), The total yield of oligosaccharides was low, ca. 10%, but corresponded to the content of hexoses and hexosamines in the LOS, as indicated by quantitative sugar analyses which showed 9% carbohydrate. These low yields can be explained by the presence of phospholipids (see above). Sugar analysis of the oligosaccharides gave glucose, galactose, and glucosamine (Table 1 ). GLC analysis of the LOS showed the same sugars but the presence of Lipid A increased the proportion of glucosamine, The absolute configurations of the hexoses and hexosamines were shown all

P. Edebrink et aL / Carbohydrate Research 266 (1995) 237-261 241
Table 1 Sugar analysis of the lipooligosaccharides and the oligosaccharides from M. catarrhalis serotype C (RS10 and RS26)
Sugars a Detector response (%)
RS10 RS26
LOS OS(10) OS(11) LOS OS(8) OS(IO)
D-GIc 48 58 55 50 62 53 D-Oal 33 35 39 23 22 33 D-GleN 20 8 16 27 16 14
"Sugars were analysed as alditol acetates on an HP-5 capillary column using the temperature program 180°C (1 min) to 250°C at 3°C/min.
to be I) when analysed as their respective trimethylsilylated (S) -2-butyl glycosides [ 13,14]. Analysis of the molecular masses of the oligosaccharides by positive-ion FABMS, using
glycerol or glycerol and thioglycerol (1:1) as matrix, showed pseudomolecular ions at m/z values 1252.4,1414.5, 1738.4, and 1900.5 corresponding to hepta-, octa-, deca-, and undeca- saccharides, respectively, with the respective formulas HexsHexNAcKdo+ H, Hex6HexNAcKdo + H, HexsHexNAcKdo + H, and Hex9HexNAcKdo + H. On the addition of sodium ions, mono- and some di-sodiated forms were observed.
Methylation analysis of the oligosac, harides gave the methyl ethers listed in Table 2. The results indicated that OS(8) contains two residues of terminal glucose, one residue of 3,4,6-substitmed glucose, two residues of 2-substituted glucose, and one residue each of terminal galactose and 4-substituted glucosamine. Some terminal glucosamine was also
Table 2 Methylatton analysis o1' lipooligosaccharides and oligosaceharidcs from M. catarrhalis scrolypc C (strains RSI0 and RS26)
Sugar T" Detector response (%)
RS10 RS26
LOS OS(10) OS( 11 ) LOS OS(8) OS(10)
2,3,4/ GIc b 1.00 12 12 10 8 29 12 2,3,4,6-Gal 1.04 12 18 15 12 10 21 3,4,6-G1c 1.20 23 23 22 17 27 20 2,3,6-Gal 1.22 12 12 16 13 13 2,3,6-G1c 1.24 15 14 14 13 13 2-GIc 1.77 16 15 17 21 21 13 2,3,4,6-GIcNAc 1.86 + 6 2,3,6-GIcNAc 2.13 5 7 5 10 7 8 2,3,4-GIcNAc 2.32 4 7
"Retention time relative to 1,5-di-O-acetyl-2,3,4,5-tetra-O-methylglucitol (1.00) and hexa-O-acetylglucitol r2.00) on an HP-5 capillary column using the temperature program 180°C ( 1 min) to 250°C at 3°C/min. b 2,3,4,6-G1c ffi 2,3,4,6-tetra-O-methylglucose.

242 P. Edebrink et al. / Carbohydrate Research 266 (1995) 237-261
present and the reason for this is discussed below. OS(10) contains one residue each of terminal, 4-substituted and 3,4,6-substituted glucose, two residues of 2-substituted glucose, two residues of terminal galactose, one residue of 4-substituted galactose, and one residue of 4-substituted glucosamine. These data differ from serotype A in that the glucosamine residue is substituted and no longer terminal, and that one additional terminal galaetose is observed.
Finally, OS(11) contains the same residues as OS(10) but with an increase in the amount of 4-substituted galactose.
The Kdo residue in OS(8) and OS(10) could be shown by blMR spectroscopy to be 5-substituted, see below. The oligosaccharides OS(7) and OS(11) were not investigated in this respect but the Kdo residues must be assumed to be substituted in the same position. Additional evidence about the Kdo region of the LOSs was obtained through methylation of the LOS followed by methanolysis and analysis of the Kdo methyl ester methyl glyco- sides. The results, albeit with poor stoichiometry, indicate that the LOSs have two Kdo residues, one terminal and one 4,5-substituted as shown by comparison with authentic standards [ 15,16]. The terminal Kdo residue should thus substitute the 4-position of the 4,5-substituted branch-point Kdo. In addition, methylation analyses of the intact lipooli- gosaccharides suggest that the lipid A portion consists of 6-substituted glucosamine residues.
The structure of OS( 10).--NMR spectra for the anomeric region of D20 solutions of OS(10) isolated from RSL0 and RS26 (Figs. 2a and 2b, respectively), as well as the molecular masses and the methylation analysis data (Table 2), suggested an identical structu,+e for the isolated decasaccharides. Thus, only OS(10) from RS26 was further investigated by NMR spectroscopy, as it was present in a larger amount. The ;H NMR spectrum of OS (10) (;cgicn 1 ~ ppm is shown in Fig. 3) demonstrated that the glueosamine residue was Noacetylated, evident from a singlet at 2.18 ppm. In some preparations one or both of the protons in the 3-deoxy group of the reducing Kdo residue were exchanged for deuterium, although the pD was neutral, as had been observed in previous work [11,17]. This incorporation of deuterium atoms also accounts for an observed increase of the molec- ular masses by two mass units when recording FABMS spectra of samples that had been dissolved in D:O, used for NMR, lyophilised, and redissolved in H20, compared to samples that had only been dissolved in H~O. This demonstrates that the Kdo residue makes up the reducing end of the oligosaccharide+ The chemical shifts at 2.02 ~nd 1.88 ppm demonstrate that it has predominantly the ot configuration [ 18].
The anomeric region of the ZH NMR spectrum (Figs. 2a and 2b) of OS(10) contained signals for twelve protons. Three of these were later shown to be the signals for H-5 (8 4.36) of the terminal a-D-galactose residue, and H-3 (8 4.55) and H-5 (8 4.66) of the 3,4,6-substituted ¢z-D-glucose residue. These three protons were also observed in the spec- trum of the serotype A oligosaccharide with similar chemical shifts. At low field, signals for four c-linked residues were observed as indicated by the Jl~-m,H-... values of ca. 4 Hz. Signals for five/]-linked residues were detected, as evident t'rom their JH.~.H-2 values of 7--8 Hz, two of these at the unusual downfield chemical shifts of 5.21 and 5.08 ppm. Them, signals were also present in the t H NMR spectrum of the oligosaccharide from M. catarrhali~ serotype A for which they were identified as 2-substituted and terminal/]-glucose, respec- tively. The other three signals derived from/3-1inked residues were observed at more moderate chemical shifts, 4.60, 4.52, and 4.48 ppm.
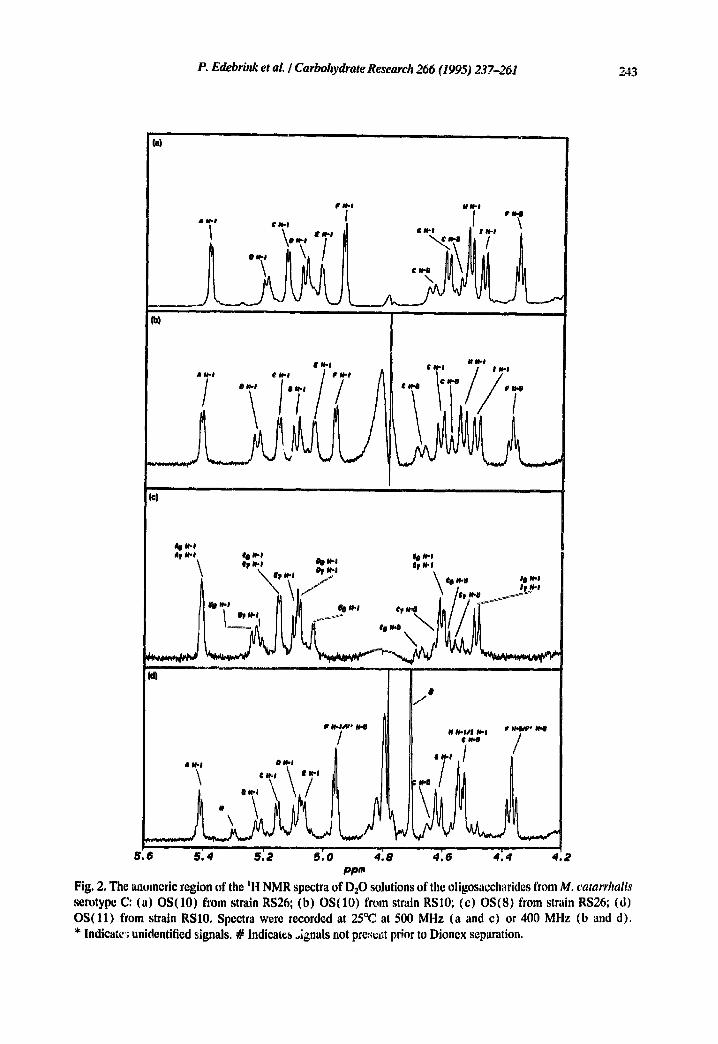
P. Edebrink et al. I Carbohydrate Research 266 (1995) 237-261 243
P N. I # N ' l
} " "" "'k~ 'r .,. / . .
/ I
(©)
\ fe ~-I 0, Nq er ~J \ \ wp~.l *rM-i
ii " ~ i i / \ -/ #rN'I " a / " Y ~
t , I # /
I~ N-# ,~ e* N Iv ~ ' O ~ ' / ..-.,..:. / | N - O & i~l • N.I /
i i i i v 1
5 .e 5 .4 5 .2 5 . 0 ~ .8 4 . 6 4 .4 ,t .2 ppm
Fig. 2, The anomeric region of the tl~l NMR spectra of D:O solutions of tile oligosaccharides from M. catarrhalis serotype C: (a) OS(10) from strain RS26; (b) OS(10) from strain RSI0; (c) OS(8) from strain RS26; (d) OS(l l ) from strain RS10. Spectra were recorded at 25°C at 500 MHz (a and c) or 400 MHz (b and d). * Indicate~; unidentified signals. # Indicates oignals not pres.,it prior to Dionex separation.
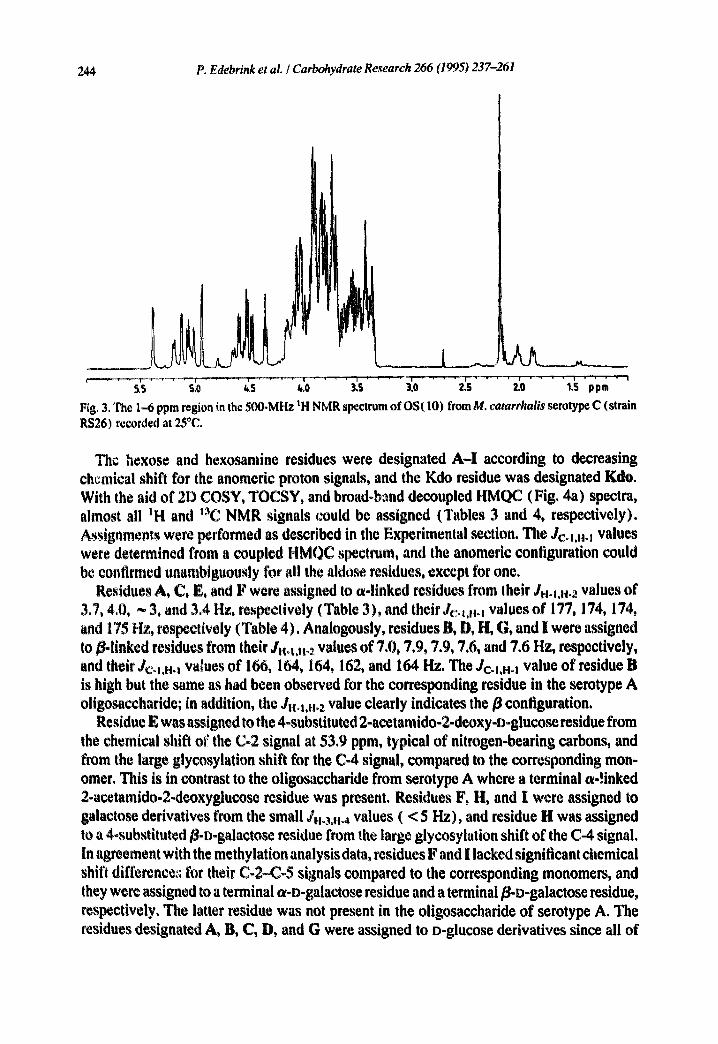
244 P. Edebrink et aL / Carbohydrate Research 266 (1995) 237-261
. . . . . . s,o ~,s 4.0 3'.s 3.o . . . . i s io ~.~ pp~
F~g. 3. The 1.-6 ppm region in the 500-MHz ~H NMR spectrum of OS(10) from M. catarrhalis serotype C (strain RS26) recorded at 25°C.
The hexose and hexosamine residues were designated A-I according to decreasing chemical shift for the anomeric proton signals, and the Kdo residue was designated Kdo. With the aid of 2D COSY, TOCSY, arid broad-band decoupled HMQC (Fig. 4a) spectra, almost all IH and 13C NMR signals could be assigned (Tables 3 and 4, respectively). Assig, mcnts were performed as described in the Experimental section. The Jc°~,..i values were determined from a coupled HMQC spectrum, and the anomeric configuration could be confirmed unambiguously for all the aldose residues, except for one,
Residues A, C, E, and F were assigned to ¢~-linked residues from their J.o1,.o~ values of 3.7, 4,0, ~ 3, and 3.4 Hz, respectively (Table 3), and their Jc~1,.°i values of 177, 174, 174, and 175 I~, respectively (Table 4). Analogously, residues B, D, H, G, and I were assigned to B-linked residues from their J..t.tt~ values of 7.0, 7.9, 7.9, 7.6, and 7.6 Hz, respectively, and their Jco1,..i values of 166, 164, 164, 162, and 164. Hz. The Jc-1,H-t value of residue B is high but the same as had been observed for the corresponding residue in the serotype A oligosaccharide; in addition, the J.a..~2 value clearly indicates the B configuration.
Residue E was assigned to the 4-substituted 2-acetamido-2-deoxy-D-glucose residue from the chemical shift of the C-2 signal at 53.9 ppm, typical of nitrogen-bearing carbons, and from the large glycosylation shift for the C-4 signal, compared to the corresponding mon- omer. This is in contrast to the oligosaccharide from serotype A where a terminal a-linked 2-acetamido-2-deoxyglucose residue was present, Residues F, II, and I were assigned to galactose derivatives from the small J..3,.~ values ( < 5 Hz), and residue II was assigned to a 4~substituted B-D-galactose residue from the large glycosylation shift of the C-4 signal. In agreement with the methylation analysis data, residues F and I lacked significant chemical shift difference:; for their C-2-4~-5 signals compared to the corresponding monomers, and they were assigned to a terminal wD-galactose residue and a terminal/~.n-galactose residue, respectively, The latter residue was rmt present in the oligosaccharide of serotype A. The residues designated A, B, C, D, and G were assigned to D-glucose derivatives since all of

F2
] (a
) (D
Pm~
3.4~
3.6~
-t
3.8~
4.0~
4.2~
4.44
4.6
i
l 8
5.0~
5.2-
~
5.4-
5.6
,5.8
-
8 0 6 &
Q
6
6
®
6
t05
t00
95
90
85
80
75
70
65
60
55
50
F1
(pgm
)
.-,.
O~
Fig.
4. B
road
-ban
d dec
oup!
ed H
MQ
C sp
ectra
of I)
20 so
lutio
ns of
the o
ligos
acch
arid
es fr
om M
. ca
tarr
hal[
s se
roty
pe C
: (a)
OS(
10) f
rom
stra
in R
S26;
(b)
0S(8
) fr
om
strai
n RS2
6. S
pect
ra w
ere r
ecor
ded a
t 600
MH
z at 2
5°C.
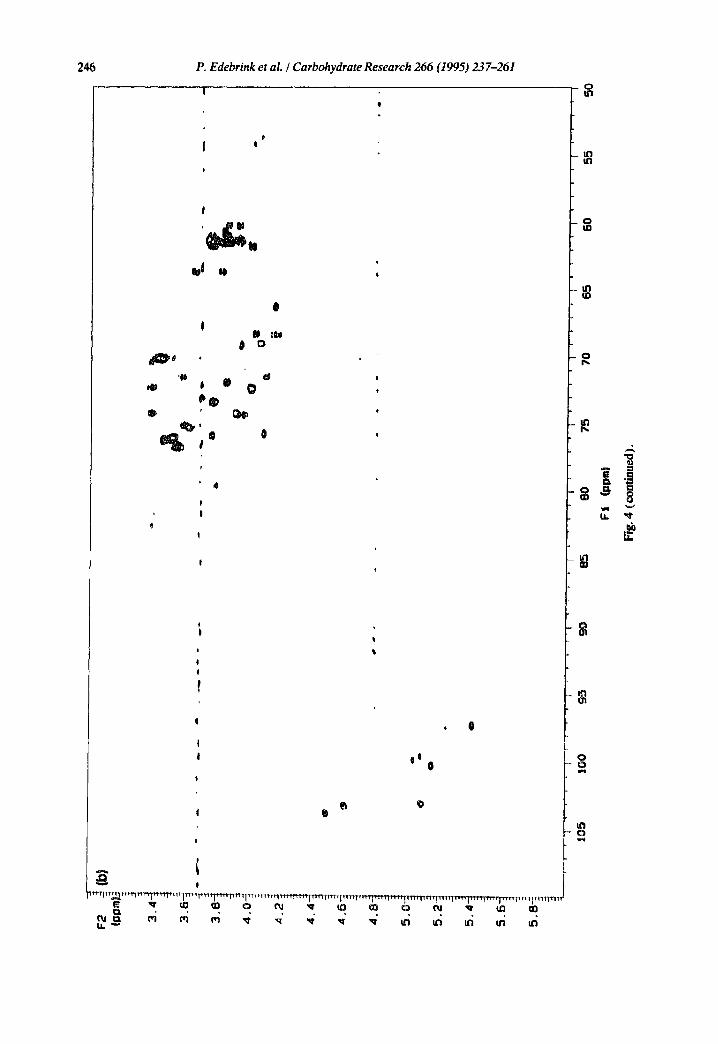
246 P. Edebrink et al. / Carbohydrate Research 266 (1995) 237-261 . . . . . |
Q
I
'~
$$
° 4
! !
J
N o0~ # o
eJ
| Q
ii I 0
, O
o I f l
111 111
0
I
~ " ~ I j ' ~ l ~ ~ r ~ - ~ ~ ~ ~ i~ ~ p ~ , t~ I ~ ~ ~ ~ ~ I ' ~ r ~ ~ i ' m ~ m ~ q ' m ~ " ~ ~ ~ "~-- ~,,, ' I ' ' n r rmJ
l . a . ~
0 p~
II1 p,.
$
o
I"
o o q ~
$
P, Edebrink et al. I Carbohydrate Research 266 (1995) 237-261
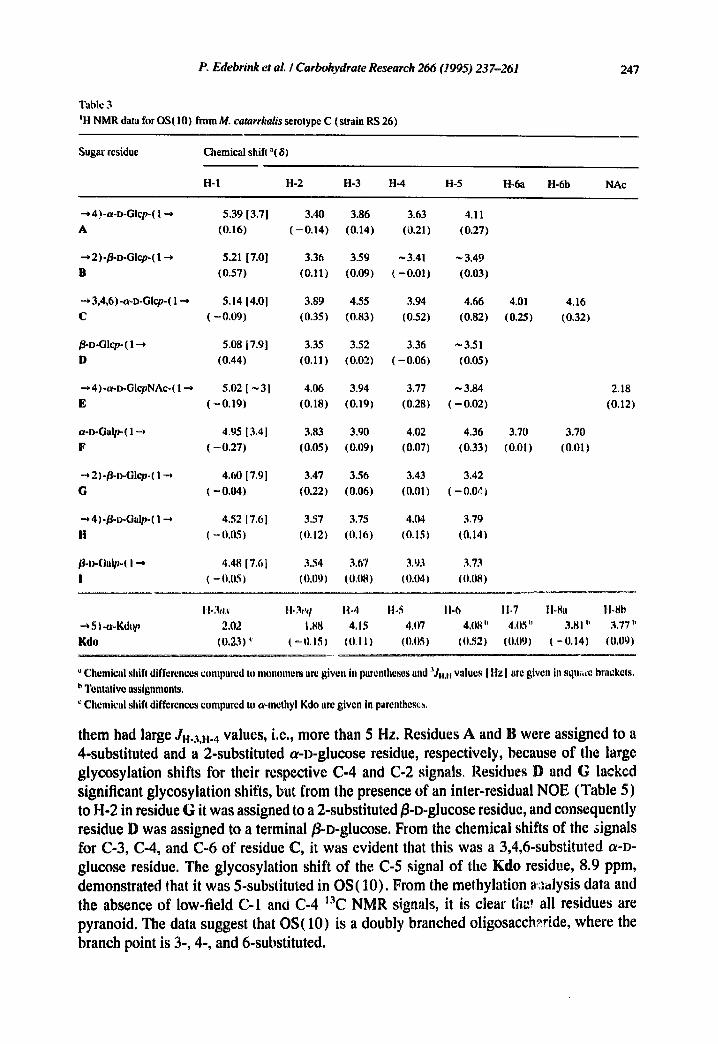
Table 3 JH NMR data for OS(10) from M. catarrhalis setotype C (strain RS 26)
247
Sugar residue Chemical shift "( 61
H-1 H-2 H-3 H-4 H-5 H-6a H-6b NAc
~4)-a-D-GIcp-(l~ 5.39[3.7] 3.40 3.86 3.63 4.11 A (0.16) (-0.14) (0.14) (0.21) (0.27)
~2)-/]-o-Glcp-(l~ 5.21 [7.0] 3.36 3.59 -3.41 ~3.49 B (0.57) (0.11) (0.09) (-0.01) (0.03)
--',3,~6)-a-D-Glcp-(l--* 5.14 [4.0] 3.89 4.55 3.94 4.66
C (-0,09) (0.35) (0.83) (0.52) (0.82)
~.D-Glcp-(l~ 5.08 [7.9] 3.35 3.52 3.36 ~3.51 D (0.44) (0.11) (0.02) (-0.06) (0.05)
~4)-a-D-GlcpNAc-(l-.-, 5.02[ ~3] 4.06 3,94 3.77 ~3.84 E ( - 0 . 1 9 ) (0.18) (0.191 (0.28) (-0.02)
a-D-Oa~ql-~ 4.95 [3.4] 3.83 3.90 4.02 4.36 F (-0.27) (0.05) (0.09) (0.07) (0.33)
-~2)-~-D-Glcp-(I-~ 4.60 [7.9] 3.47 3.56 3.43 3.42 G (-0.04) (0.22) (0.06) (0.011 (-0.0~)
• "~ 4)°~-D-Ga~-( 1 -~ 4.52 [7,6] 3.57 3.75 4.04 3.79 H ( ~ ().05) (0 ,12) (0 ,16) (0.15) (0 ,14)
~-l)-Ga~-( I ~ 4.4817.~I 3.54 3.67 3.93 333
I ( = 11,05 ) (0,11~) (0,tl81 (0.041 (0.08)
4.01 4.16 (0.25) (0.32)
3.70 3.70 (0.01) (0.01)
2.18 (0.12)
11-3aa 11o3eq 11.4 1t-5 t l .6 11-7 11-8. tbSb 5 ) -e~Kdop 2.02 1,88 4,15 4,07 4,1)8" 4,05" 3.81 b 3,77"
Kdo ((I.23) ~' ( - 0.1~) (0,11 ) (0,051 (0,52) (0,09) ( -(I.141 (0,09)
Chemical shift differences compared to monomers ar~ given in parentheses and '~J... values [ Hz] are given in squ~re brackets. t, Tentative assignments. ~' Chemical shift differences compared to a-methyl Kdo arc given in parentheses.
them had large JH-3,}l.4 values, i.e., more than 5 Hz. Residues A and B were assigned to a 4-substituted and a 2-substituted a-D-glucose residue, respectively, because of the large glycosylation shifts for their respective C-4 and C-2 signals. Residues D and G lacked significant glycosylation shifts, but from the presence of an inter-residual NOE (Table 5) to H-2 in residue G it was assigned to a 2-substituted g-D-glucose residue, and consequently residue D was assigned to a terminal fl-D-glucose. From the chemical shifts of the signals for C-3, C-4, and C-6 of residue C, it was evident that this was a 3,4,6-substituted a-D- glucose residue. The glycosylation shift of the C-5 signal of the Kdo residue, 8.9 ppm, demonstrated that it was 5-substituted in OS(10). From the methylation ~.:,~lysis data and the absence of low-field C-1 anO C-4 13C NMR signals, it is clear tha! all residues are pyranoid. The data suggest that OS(10) is a doubly branched oligosacch~ride, where the branch point is 3-, 4-, and 6-substituted.
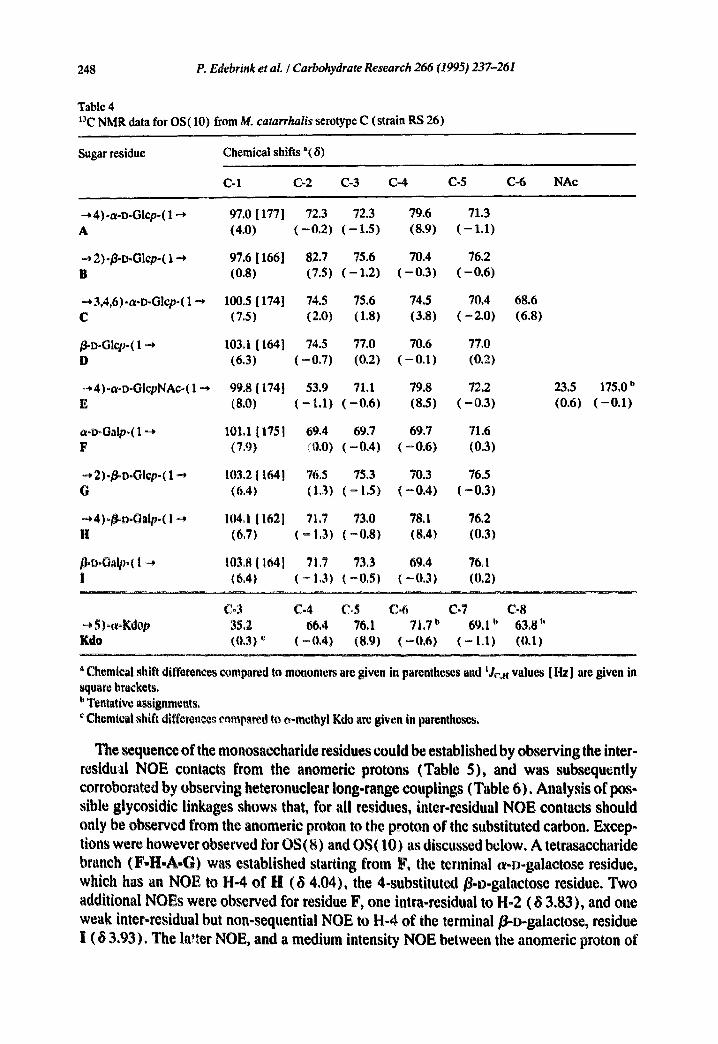
248 P. Edebrink et al. / Carbohydrate Research 266 (1995) 237-261
Table 4 t'~C NMR data for OS(10) from M. catarrhalis serotype C (strain RS 26)
Sugar residue Chemical shifts "(8)
C-1 C-2 (2-3 C-4 C-5 C-6 NAc
--* 4)-a-D-Glcp-( 1 --+ 97.0 [ 177 ] 72.3 72.3 79.6 71.3 A (4.0) ( - 0 . 2 ) ( - 1.5) (8.9) ( - 1.1)
--* 2)-~D-Glcp-( 1 -* 97.6 [ 166] 82.7 75.6 70.4 76.2 B (0.8) (7.5) ( - 1 . 2 ) ( - 0 . 3 ) ( - 0 . 6 )
• -* 3,4,6)-a-D-Glcp-( 1 -.* 100.5 [ 174] 74.5 75.6 74,5 70.4 68.6 C (7.5) (2.0) (1.8) (3.8) ( - 2 , 0 ) (6.8)
/~D-Glcp-( 1 ~ 103.1 [ 164] 74.5 77.0 70.6 77.0 D (6.3) ( - 0 . 7 ) (0.2) ( - 0 . 1 ) (0.2)
~,4)-a-o-GIcpNAc-( 1 -* 99.8 [ 174] 53.9 71.1 79.8 72,2 23.5 E (8.0) ( - 1.1) ( - 0 . 6 ) (8.5) ( - 0 , 3 ) (0.6)
a-D-Galp-(1 --* 101.1 [175] 69.4 69.7 69.7 71.6 F (7,9) (0.0) ( - 0 . 4 ) ( - 0 . 6 ) (0.3)
-*2)-~D-Glcp-( 1 ~ 103,2 [ 164] 76.5 75.3 70.3 76.5 G (6,4) (1.3) ( - 1.5) ( - 0 . 4 ) ( - 0 . 3 )
=,4)o/~a.Oalpo( I ~* 104.1 [ 162l 7[3 73.0 78.1 76.2 H (63) ( ~ 1,3) (=0,8) (8.4) (0,3)
/]+l~oO,lpo( I ~ 103.8 [ 164] 71.7 73.3 69.4 76.1 I (6,4) (- 1.3) (-0,$) (-0,3) (0,2)
175.0 b (-0.1)
Co3 C-4 Co5 C-6 C-7 C-8 ~* $)*wKdop 35.2 66.4 76.1 71.7 u 69,1 a 63.8 h Kdo (0.3) ~ (-0.4) (8,9) (-0.6) ( - I.I) (0.I)
' Chemical shift differences compared to monomers arc given in parentheses and 'Jr,. values [Hz] are given in square brackets. I' Tentative assignments,
Chemical shift diffcrence~ eampared to o-methyl Kdo are given in parentheses.
The sequence of the monosaccharide residues could be established by observing the inter- residufl NOE contacts from the anomeric protons (Table 5), and was subsequently corroborated by observing heteronuclear long-range couplings (Table 6). Analysis of pos- sible glycosidic linkages shows that, for all residues, inter-residual NeE contacts should only be observed from the anomeric proton to the proton of the substituted carbon. Excep- tions were however observed for OS(8) and OS(10) as discussed below. A tetrasaccharide branch (F-H-A-G) was established starting from F, the terminal o~-o-galactose residue, which has an NeE to H-4 of H (~ 4.04), the 4-substituted/3-o-galactose residue. Two additional NOEs were observed for residue F, one intra-residual to H-2 ( 6 3.83), and one weak inter-residual but non-sequential NeE to H-4 of the terminal/3-D-galactose, residue l (6 3.93), The In*tar NeE, and a medium intensity NeE between the anomeric proton of

P. Edebrink et al. I Carbohydrate Research 266 (1995) 237-261 249
Table 5 NOE data for OS(10) from M. catarrhafis serotyp~ C (strain RS 26). Data were obtained at 500 MHz, using a mixing time of 350 ms
Anomeric proton
Residue
NOE contact to proton
8 8 Intensity ° Residue, atom
--* 4)-a-D-Glcp-( 1 ~ 5.39 3.40 s A, H-2 A 3.47 s G, H-2
3.94 m C, H-4 4.60 vw G, H-1
2)-~D-Glcp-( 1 -* 5.21 3.36 w B, H-2 B 3.49 s B, H-5
3.59 s B, H-3 3.94 w C, H-4 4.55 s C, H-3
~ 3,4,6)-Or-D-Glcp-( 1 --* 5.14 3.89 s C, H-2 C 4.07 s Kdo, H-5 /~-D-Glcp-(1 -* 5.08 3.35 m D, H-2 D 3.52 s D, H-3
3.89 m C, H-2 4.55 s C, H-3
~ 4)-a-D-GlcpNAe-( 1 -~ 5.02 3.36 s B, H*2 E 4.06 s E, H-2 a-D-Galp-(1 -~ 4.95 3.83 s F, H-2 F 3.93 w 1, I~-4
4.04 m H, H-4
-~ 2)-~-D-Glep-( 1 ~ ' 4.60 3.42 s G, H-5 G 3.47 in G, H-2
3.56 s G, H-3 3.94 w C, H-4 4.01 s C, H-6a 4.16 s C, H-6b
~, 4)-goDoGalpo( 1 ~ 4.52 3.57 w H, H-2 H 3.63 s A, H-4
3.75 s I i , H-3 3.79 m H, H-5
~-D-Galp-( I ~ 4.48 3.67 m I, H-3 1 3.73 s 1, H-5
3.77 s E, H-4 3.90 m F, H-3
and/orD, H-5
' The intensities are estimated and given as, s ~ strong, m m medium, w ~ weak, and vw ~ very weak.
residue I and the H-3 of residue F (8 3.90), suggested that the two terminal D-galactose residues are close to each other. Residue H has an NOE to H-4 of A (8 3.63), the 4-substituted a-D-glucose residue, in addition to the internal NOEs to H-2, H-3. and H-5 (8 3.57, 3.75, and 3°79, respectively). Residue A has one strong internal NOE to H-2 ( 8 3.40), as well as inter-residual NOEs to H-2 (8 3.47 strong) and H-1 (8 4.60, weak) of G, a 2-substituted ~-D-glucose residue. The latter NOE is to an axial proton and is normally not observed. A non-sequential NOE of medium intensity between the anomeric proton of
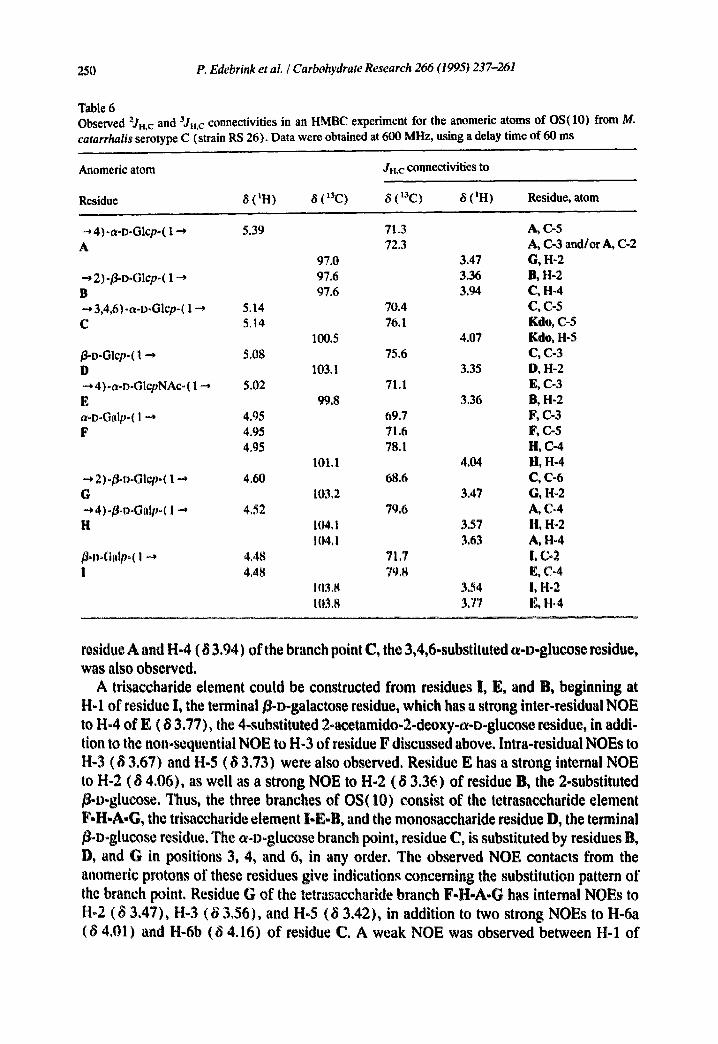
250 P. Edebrink et aL / Carbohydrate Research 266 (1995) 237-261
Table 6 Observed 'JH,c and 3Jn.c connectivities in an HMBC experiment for the anomeric atoms of OS(10) from M. catarrhalis serotype C (strain RS 26). Data were obtained at 600 MHz, using a delay time of 60 ms
Anomeric atom Ju,c conneetivities to
Residue 8 (tH) 8 (13C) 8 (13C) 8 (IH) Residue, atom
--,4)-a-D-Glep-( 1 ~ 5.39 71.3 A, {2-5 A 72.3 A, C-3 and/or A, C-2
97.0 3.47 G, H-2 2)-~D-Glcp-( 1 --, 97.6 3.36 B, H-2
B 97.6 3.94 C, H-4 • -* 3,4,6)-a-D-Glcp-( 1 ~ 5.14 70.4 C, C-5 C 5.14 76.1 Kale, C-5
100.5 4.07 Kale, H-5 ~o-Glcp-( 1 --* 5.08 75.6 C, C-3 D 103.1 3.35 D, H-2 ~, 4)-a-D-GlepNAc-( I -~ 5.02 71.1 E, C-3 E 99.8 3.36 B, H-2 a-D-Galp-( 1 ~, 4.95 69.7 It, {2-3 F 4.95 71.6 F, C-5
4.95 78.1 H, C-4 101.1 4.04 li, H-4
"~, 2)-/~,o-Glcp-( 1 ~ 4.60 68.6 C, C-6 G 103.2 3.47 G, H-2 ~, 4)-/~l~oGalp-( 1 ~, 4,52 79,6 A, C-4 H 104,1 3.57 H, H-2
104. I 3.63 A, H*4 ~°D-Galp-( I ~ 4.48 71,7 !, C-2 i 4,48 79,8 E, C°4
103,8 3.54 I, H-2 11)3.8 337 E, H.4
residue A and H-4 ( 8 3.94) of the branch point C, the 3,4,6-substituted a-D-glucose residue, was also observed.
A trisaccharide element could be constructed from residues I, E, and B, beginning at H-1 of residue l, the terminal ~-D-galactose residue, which has a strong inter-residual NeE to H-4 of E ( 8 3.77), the 4-substituted 2-acetamido-2-deoxy-a-D-glucose residue, in addi- tion to the non-sequential NeE to H-3 of residue F discussed above. Intra-residual NOEs to H-3 (8 3.67) and H-5 (8 3.73) were also observed. Residue E has a strong internal NeE to H-2 ( 8 4.06), as well as a strong NeE to H-2 (8 3.36) of residue B, the 2-substituted E-D-glucose. Thus, the three branches of OS(10) consist of the tetrasaccharide element F.H-A-G, the trisaccharide element I-E-B, and the monosaccharide residue D, the terminal /I-D-glucose residue. The o~-D-glucose branch point, residue C, is substituted by residues B, D, and G in positions 3, 4, and 6, in any order. The observed NeE contacts from the anomeric protons of these residues give indications concerning the substitution pattern of the branch point. Residue G of the tetrasaccharide branch F-H-A-G has internal NOEs to H-2 (8 3.47), H-3 (6 3.56), and H-5 (6 3.42), in addition to two strong NOEs to H-6a (8 4.01) and H-6b (6 4.16) of residue C. A weak NeE was observed between H-1 of

P. Edebrink et al. / Carbohydrate Research 266 (1995) 237-261 251
residue G and H-4 ( 6 3.94) of residue C; this NOE was also observed for the oligosaccharide of serotype A. Residue B of the trisaccharide branch I-E-B, has an internal NOE to H-2 (B 3.36), H-3 (6 3.59), and H-5 (6 3.4 9), which further corroborates the/3 configuration. Similarly to serotype A, two inter-residual NOEs to residue C were also observed, one strong to H-3 (6 4.55), and one weak to H-4 (6 3.94). Residue D has in addition to an internal NOE to H-2 (6 3.35) and the overlapping signals of H-3 (6 3.52) and H-5 ( 6 3.51), two inter-residual NOEs to residue C, one strong to H-3 ( 6 4.55), and one medium intensity to tL2 (6 3.89). The latter was not observed for serotype A. These data suggest that the branch-point residue is substituted by the/3-D-glucose group in the 3-position, by the trisaccharide branch I-E-B in the 4-position, analogously to type A, and by the tetrasac- charide branch F-H-A-G in the 6-position. The branch-point residue has an internal NOE to H-2 (B 3.89), and an inter-residual NOE to H-5 (6 4.07) of the Kdo residue. All of these NOEs indicated a substitution pattern of the branch-point glucose residue identical to that of the corresponding residue in the oligosaccharide from M. catarrhalis serotype A.
Further evidence for the substitution pattern of residue C was derived from an HMBC experiment (60-ms delay time), which should, inter alia, show 3Jr. • cross-peaks from anomeric protons and carbons to substitution carbons or the protons of the linkage carbons, respectively. Less diagnostic correlations due to intra-residual two- and three-bond hetero- nuclear couplings should also be observed (Table 6). Not all of the expected cross-peaks were observed in this single experiment, since the intensity of a cross-peak depends on the size of the particular long-range coupling constant, as well as on the length of the delay time used for transfer of the multiple bond coherence [ 19]. Inter glycosidic correlations confirming structures of the branches, F-H-A-G and I-E-B, were observed from the anom- eric carbons to the protons of the substitution carbons. In addition, cross-peaks were observed from the anomeric protons of residues F, H, and 1 to the linkage carbons of residues H, A, and E, respectively. Residue C, the branch point, has couplings to H-5 and C-5 of the Kdo residue from its C-I and H-I, respectively. Correlations were observed to C-3, H-4, and C-6 of the branch point from H-1 of residue D, C-I of residue B, and the H- 1 of residue G, respectively. Thus, from the combined evidence of the NOESY and the HMBC experiments, structure 2 is proposed for OS(10), the decasaccharide isolated from the LOS from M. catarrhalis serotype C. Data from the modified methylation analysis indicate that a terminal Kdo residue substitutes the S-substituted Kdo residue of structure 2 in its 4-position.
i E B I}-D-Galp-( I --)4)-Ot-D-GlcpNAc-( l-+2)-I}-D-Glcp
I J,
F H A G C 4 Kdo ¢~-I~Gall)-( I --)4)-I}-D-Galp-( I ---)4)-ot-I>Glcp-( 1 --~2)-I}-D-Glcp-( 1 --q.b)-0t-I)-Glcp-( I -.~5)-Kdop
3 T
D I [3-1) Glcp
2 The structure of OS(8) .--A comparison of the methylation analysis data (Table 2) for
OS(10) and OS(8) showed a significantly increased relative amount of terminal glucose

252 P. Edebrink et al. / Carbohydrate Research 266 (1995) 237-261
Table 7 tH NMR data for the mixture of 0S(8) and OS(7) from M. catarrhalis serotype C (strain RS 26)
S,Jgar residue ' Chemical shifts b (8)
H-I H-2 H-3 H,-4 H-5 H-6a H-6b NAc
wD-Glcp-( 1-4 5.41 |3.5] 3.36 3.75 3.39 4.00 A (0.18) ( -0 .18 ) (0.03) ( -0 .03 ) (0.16)
~* 2)..~v-Olcp-( ! -4 5.23 [ ~8] 3.38 3.61 3.43 3.51 B (0.59) (0.13) (0.11) (0.01) (0.05)
-* 2)./~D-Glcp-( 1 --* 5.21 [n.d.] 3.39 3.60 3.43 B, (0.57) (0.14) (0.11) (0.01)
-4 3,4.6)-wD-Glcp-( 1 ~ 5.15 [ ~4] 3.91 4.57 3.96 4.68 C ( -0 ,08) (0.36) (0.85) (0.54) (0.84)
=* 3,4,6)-a-o-Glcp-( t ~ 5.15 [n.d.] 3.91 4.53 3.96 4.62 C~ ( -0 .08) (0.37) (0.81) (0.54) (0.78)
~-o,Glcp-( I -. 5,09 [8] 3,37 3,54 3.35 ",3.52 D (0,45) (0,12) (0.04) (-0.07) (0.06)
~D.GI~-( 1 ~ 5.09 [ ~ 7l 3.36 3.55 3.36 I~ (0,45) (OAt) (0,05) ( -0 .06)
~4)oWDoGIcpNAc.( t ~* $.04 [ ,- 3] 4.0? 3.96 3.78 3.86 '~
E (~0.17) (0,19) (0,21) (0.29) (0,00)
wt~oOl~NAco( t ~ L08 [ ,,, 41 4,02 3,74 3,r~! i~ (~|).13) (0,14} (=0 ,0 t ) (0 . t l )
~* ~)o~p431¢.#~( I ~* 4,f~0 J7,$] 3,49 3.57 3,42 ~- 3,44 G (~0,04) (0,24) (0,07) (0.00) ( -o .o2)
~t)~Oalp-( 1 ~ 4,49 [8.0] 3,,~5 3,68 3.94 3,75 ~ l ( ~ 0,04 ) (0,11) (0.09) (0.05) (0.10)
4.03 4.15 (0.27) (0.31)
2 . ~
(0.14)
H.3~tz H-3eq tl-4 H-5 H-6 H-7 H-Sa H-gb
~5)-wKdop 2.03 i.88 4.15 4.08 4.10 ~ 4.06 ~ 3.81 e 3.64 ~
Kale (0.24) '~ ( -0 .15) (0.11) (0.06) (0.54) (0.10) ( -0 .14) ( -0 .04)
a Bold I©tte~ (B) indicate an ~tasaccharide residue; bold lettem with index 7 (B~) indicate a heptasaccharide residue. b Chemical shift differences compared to monomers are given in parentheses and ~J.,,, values [ Hz ] are given in .square brackets; in,d,] indicates that the coupling constant was not measured.
Tentative assignments, d Chemical shift differences compared to •-methyl Kdo ~re given in parenthe.~s.
in the latter, The 4-substituted glucose and galactose residues were absent in the analysis of OS(8). This suggests that OS(8) lacks the F-H disaccharide element, a-D-Galp-(l --* 4)- /~D-Galp-( 1 .-,, which is substituting the 4-position of residue A, a-D-Glc in OS(I0). The glucosamine of OS(8) was N-acetylated as evident from a singlet at 2.20 ppm in the tH NMR spectrum of the oligosaccharide in a D20 solution. It was concluded that the Kdo

P. Edebrink et al. I Carbohydrate Research 266 (1995) 237-261
Table 8 ISC NMR data for the mixture of OS(8) and OS(7) from M. catarrhalis serotype C (strain RS 26)
~ 3
Sugar residue ~ Chemical shift b (8)
C-1 C-2 C-3 C-4 C-5 C-6 NAc
a-D-Glcp-( 1 --* 97.2 72.4 73.4 70.4 72.4 A (4.3) ( - 0 . 1 ) ( - 0 . 4 ) ( - 0 . 3 ) (0.0)
--* 2)-~D-Glcp-( 1 ~ 97.5 82.7 75.4 70.2 76.2
B (0.7) (7.5) ( - 1.4) ( - 0 . 5 ) ( - 0 . 6 )
--* 2)-~-D-Glcp- ( 1 ~ 98.0 81.8 75.4 70.2 B, (1.2) (6.6) ( - 1.4) ( - 0 . 5 )
-* 3,4,6)-a-D-Glcp-( 1 ~ 100.3 74.4 75.5 74.4 70.2 68.4 C (7.3) (1,9) (1.7) (3.7) ( - 2.2) (6.6)
3,4,6)-a-D-Glcp-( 1 --* 100.3 74.4 75.5 74.4 70.4 C~ (7.3) (1.9) (1.7) (3.7) ( - 2 . 0 )
/~-D-Giep-( 1 ~ 103.0 74.3 76.8 70.6 76.9 D (6.2) ( - 0 . 9 ) (0.0) ( - 0 . 1 ) (0.1)
-* 4)-a-D-GlcpNAc- ( 1 -* 99.8 53.7 71.0 79.7 72.0 ~ E (8.0) ( - 1 . 3 ) ( - 0 . 7 ) (8.4) ( - 0 . 5 )
a-D-GicpNAc-( l -~ 99.6 54.2 72.6 70.7 E7 (7.8) ( - 0 . 8 ) (0.9) t -0 .6)
-~ 2)-/~-D-Glcp-( 1 ~ 103.2 76.3 75.2 702 76.3 G (6.4) ( I . I ) ( - ! . 6 ) ( - 0 . 5 ) (~0 .5 )
oO-D-Galpo ( 1 ~* 103.8 71,6 73.2 69,2 76,0 ~' I (6.4) ( - 1 . 4 ) ( - 0 . 6 ) ( - 0 . 5 ) (0.1)
23.5 (0.6)
C-4 C-5 C-6 C-7 C-8 --* 5)-a-Kdop 66.3 75.8 71.5 ~ 69.1 c 63.7 ~ Kdo ( - 0 . 5 ) d (8.6) ( - 0 . 8 ) ( - 1 . 1 ) (0.0)
Bold letters (B) indicates an octasaccharide residue; letters with index 7 (B~) indicate a heptasaccharide residue. b Chemical shift differences compared to monomers are given in parentheses. c Tentative assignments. ' d Chemical shift differences compared to a-methyl Kdo :,re given in parentheses.
residue was the reducing end of the oligosaccharide, since the 3-deoxy protons of the Kdo residue were exchangeable. The anomeric configuration of the Kdo residue was predomi- nantly o~, as the signals for the axial and equatorial 3-deoxy protons were observed at 8 2.03 and 1.88, respectively.
The anomeric region of the 1H spectrum of OS(8) (Fig. 2c) displayed heterogeneity of the material. However, the spectrum for the m::!or component of the 08(8) preparation from RS26 contains signals for nine proto~, ~,o of which later were shown to be H-3

254 P. Edebrink et al. / Carbohydrate Research 266 (1995) 237-261
(8 4.57) and H-5 (8 4.68) of the 3,4,6-substituted glucose residue. Three anomeric signals for a-linked residues were observed at 8 5.41, 5.15, and 5.04, as indicated by the small JH-I.H-2 values of 3-4 Hz, and four signals at 6 5.23, 5.09, 4.60, and 4.49 were observed for anomeric protons of jS-linked residues, as indicated by the large JH-~,H-2 values of ~ 8 Hz. The chemical shifts for these anomeric signals are close to those of residues A, B, C, D, E, G, and I of OS(10), the chemical shift differences being no larger than 0.02 ppm. Hence, the hexose and hexosamine residues were designated as A, B, C, D, E, G, and I in order of decreasing chemical shift values, and the Kdo residue was designated as Kdo. In agreement with methylation analysis data, the 1D ~H NMR spectrum of OS(8) lacked the signals which for OS(10) corresponded to H-I (8 4.95) and H-5 (6 4.36) of residue F (terminal a-D-galactose) and H-1 (8 4.52) of residue H (4-substituted fi-D-galactose).
Almost all 1H and ~3C signals could be assigned (Tables 7 and 8, respectively), inter alia aided by an HMQC spectrum (Fig. 4b). It was however not possible to obtain the Jc-~.H.~ values from a coupled HMQC experiment because of the heterogeneity and the small amount of material. Residue A was assigned to terminal a-linked v-glucose as the tH and ~3C chemical shifts were in agreement with those for monomeric c~-v-glucose. The other spin systems were assigned essentially as described for OS(10). Thus, residues C and E were assigned to 3,4,6-substituted a-v-glucose and 4-substituted 2-acetamido-2- deoxy-a-D-glucose, respectively. Residues B, D, G, and I were assigned to 2-substituted fi-D-glucose, terminal/]-D-glucose, 2-substituted/]-D-glucose, and terminal ~-v-galactose, respectively. The tH and t3C chemical shifts of the Kdo residue indicated that it was 5- substituted.
Sequential information was obtained by observing inter-residual NOEs from anomeric protons (Table 9). The disaccharide branch A-G was established starting from A, the terminal c-D-glucose residue, which has an NOB to H-2 ( 6 3.49 ) of residue G, 2-substituted ~-Doglucose. The trisaceharide branch I-E-B was established starting from I, the terminal ~-D-galactose residue, which, imer alia, has an NOB to H-4 (/~ 3.78) of residue E, the 4-substituted 2-acvtamido-2°deoxy°ot-D-glucose residue, which has a sequential NOE to Ho2 of residue B (/~ 3.38), a 2-substituted/I-D-glucose residue. Residue C, the 3,4,6- substituted e-D-glucose branch-point residue, has an inter-residual NOE to H-5 of Kdo (~ 4,08). Inter-residual NOEs were observed from H-6a (8 4.03, medium), H-6b ( 8 4.15, medium), and H-4 (8 3.96, weak) of residue C to the anomeric proton of residue G. The anomeric proton of residue B has a weak NOE to H-3(8 4.57) of residue C, and the anomeric proton of residue D has a medium intensity NOE to H-3 ( 8 4.57) and a very weak contact to H-2 (~ 3.91 ) of residue C. These NOEs to the ring protons of the branch-point residue are in agreement with those observed for OS(10), and it is concluded that OS(8) has structure 3.
I E B
I
A G C 4 K d o (~-l)-Glcp-i I ~2)ol3-o-Glcp-( I ~6)-cz-I)-Glcp-( I-*5)-Kdop
3 T
D I l~-o-Olcp
3

P. Edebrink et al. / Carbohydrate Research 266 (1995) 237-261 255
For the minor component of OS(8) from RS26, the spin systems for terminal O~-D- GIcNAc (E~,) and 2-substituted fl-D-glucose (BT) could, inter alia, be identified (Tables 7 and 8). The chemical shifts for the signals assigned were in agreement with those for the disaccharide element n,-D-GlcpNAc-( 1 --, 2)-//-D-Glcp-( 1 -* of the nonasaccharide from the LOS of Moraxella catarrhalis serotype A. Some 2-deoxy-3,4,6-tri-O-methyl-2-(N- methylacetamido)glucose was detected in the methylation analysis of OS(8) from RS26 (Table 2). These indications together with the observation of a pseudomolecular ion at m/z 1252.4 in the positive-ion FABMS spectrum of the oligosaccharide preparation suggest the presence of a small amount of a heptasaccharide with structure 4.
E7 B7 ~-D-GlcpNAc-(I--~2)-~-D-Glcp
I
C? 4 ct-D-Glcp-( i--*2)-[3-o-Gicp-( I--~6)-ot-a-Glcp-(l-.~5)-Kdop
3 1"
D7 1 [~-D-Glcp
4
The structure of OS(ll).--Only a small amount of OS(11) was purified and therefore no 2D NMR experiments were performed. The 1D 1H NMR spectrum ofOS(11) displayed similarities to that of OS(10), but in the anomeric region (Fig. 2d), slight differences were also observed and two additional signals were present. The two new signals, at 8 4.96 and 8 4.37, overlap the signals which for OS(10) were assigned to H-I (8 4.96) and H-5 (8 4.37) of residue F, the terminal O~-D-galactose. The signals for the anomeric protons of re:~,idues I (8 4.48) and E (8 5.02) of OS(10) were not observed at those chemical shifts but instead two signals at 8 4.54 and 8 5.07 were observed, which are assigned to I and E, respectively, and which consequently are shifted 0.06 and 0.05 ppm downfield. This indi- cates that the trisaccharide branch I-E-B of OS(10) is further substituted by a terminal O~-D-galactose residue in OS(11). The chemical shifts of the signals from the remaining anomeric protons were observed at 8 5.41 (A), 5.22 (B), 5.15 (C), 5.09 (D), 5.06 (E), 4.96, 4.95 ( F/It' ), 4.61 (G), and 4.54 ( H and I). The 3Jn.n values for the anomeric protons of residues A, B, C, D, E, and G were 3.7, 7.7, 4.4, 8.0, 4.4, and 8.0 Hz, respectively. The 3JH. a values for the anomeric proton signals of residues 1¢ and F' were ca. 3 Hz, and for H and I ca. 8 Hz. A comparison between the methylation analyses of OS(10) and OS( 11 ) (Table 2) showed that for OS( l l ) the proportion of 2,3,6-tri.O-methyl-galactose is increased and that of 2,3,4,6-tetra-O-methyl-galactose is approximately the same. This suggested that one of the terminal galactose residues of OS(10) was substituted at position 4 by another galactose in OS( 11 ). In order to establish which of the branches of OS(10) had b~.en elongated by a galactose residue in order to form OS(11), positive-ion FABMS B/E-linked scans were recorded for both oligosaccharides. The most intense fragment ions were observed at m/z 365.8 for OS(10) and at m/z 528.1 for OS(11). The fragment at m/z 365.8 corresponds to Hex-HexNAc, and the fragment at m/z 528.1 corresponds to a Hex-Hex-HexNAc fragment, both fragments are formed via pathway A [20]. Other less
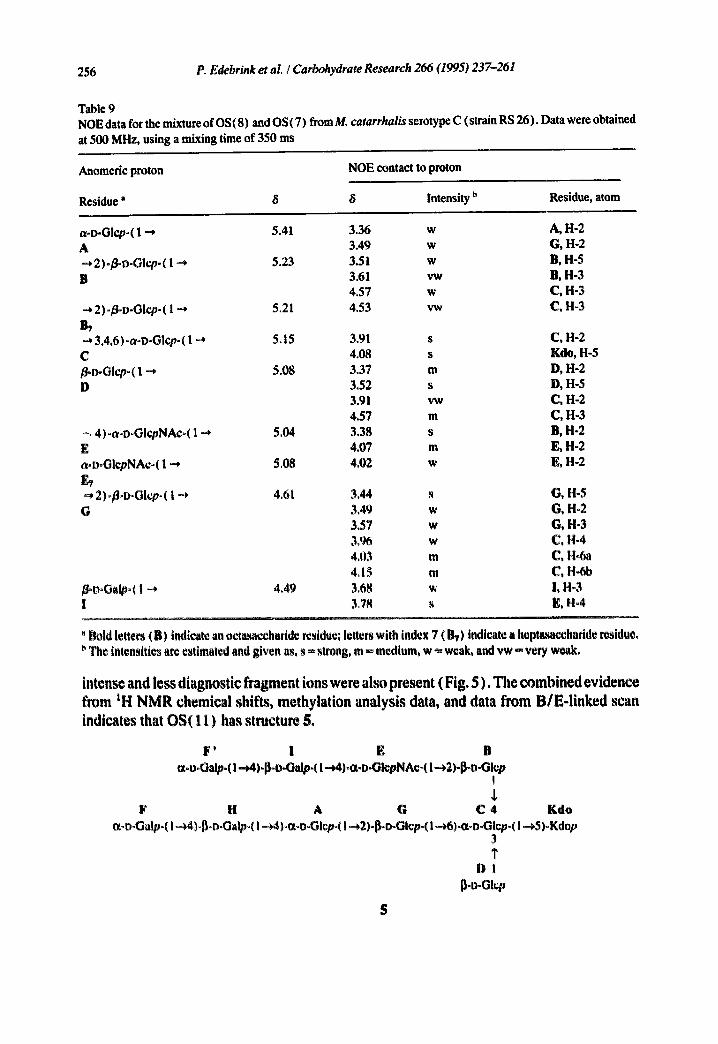
256 P. Edebrink er al. / Carbohydrate Research 266 (1995) 237-261
Table 9 NOE data for the mixture of OS(8) and OS(7) from M. catarrhalis serotype C (strain RS 26). Datawere obtained at 500 MHz, using a mixing time of 350 ms
Anomedc proton NOE contact to proton
Residue" 8 8 Intensity b Residue, atom
a-D-Glcp-( 1 --* 5.41 3.36 w A 3.49 w -* 2)-/~-Glcp-( 1-4 5.23 3.51 w B 3.61 vw
4.57 w .~, 2)-~D-Glcpo ( 1 "', 5.21 4.53 vw B,
3.4,6)-a-D-Glcp-( 1 ~ 5.15 C /~o-Glcp-( 1 -* 5.08 I)
-, 4)-a-D-GlcpNAc-( 1-4 5.04 E a-D-OlcpNAc-( 1 ~* 5.08
2)-/~D-Glcp- ( 1 -4 4.61 G
~°o°Galp°( 1 -4 4.4~ I
A, H-2 G, !-I-2 B, H-5 B, H-3 C, H-3 C, H-3
3.91 s C, H-2 4.08 s Kdo, H-5 3.37 m D, H-2 3.52 s D, H-5 3.91 vw C, H-2 4.57 m C, H-3 3.38 s B, H-2 4.07 m E, H-2 4.02 w E, H-2
3.44 s G, H-5 3,49 w G, H-2 3.57 w G, H-3 3,% w C, H-4 4,03 m C, H-Sa 4, I ~ m C, H-6b 3.68 ~, !, H°3 3,78 s E, H~4
' Bold letters (B) indicate an octa~ccharide residue; letters with index 7 (1~) indicate a heptasacchadde residue. The intensities are estimated and given as, s ~ strong, m ~, medium, w ~ weak, and vw ~ very weak.
intense and less diagnost ic f r agmen t ions were also p resen t (F ig . 5 ) . T h e c o m b i n e d ev id en ce
f rom tH N M R chemica l shifts , me thy la t ion analys is data, and data f r o m B / E - l i n k e d scan
indicates that O S ( 1 1 ) has s t ructure 5.
F' i E B ¢x-D-Oalp-( I -~4)-~-o-Galp-( I --J4)-a-I>-GlepNA¢~( I-*2)-p-D-Glep
!
F 14 A G C 4 Kdo ~X-boGalp~( l~4)ol~DoGalp~( I ~4)oo~-u-Glcpo( I -*2)-13-o-Gicp-( l--~6)-a-DoGlcp-( I -~,5)-Kdop
3 I"
D l p.o-Gl,~p
5

P, Edebrink et aLI Carbohydrate Research 266 (1995) 237-261 257
1 0 8 -
90
oo,
7o,
so.
50-
40
30
2is
IO
(el
il
3GS, O
S~O, I
! ! - i i
, , , 4 I ~ * < ' I ~ ' ~ ' ~ " ~ ' < ' F ' ~ ' ~ ' i ~ ' ~ ' : ~ "
' I ' ~ I ' ' '+ i ' "+ I '+ ' - , ' , ~
MW 1737.4
173~.4
I I 1'4 ISP6,0
llm8 15i~ 151/ '~
Im~ (b)
g6,
go
~o
6Q
S~I.I
3G~,O
i
4
T !
MW 1899.5
1809, '~
69O. I
i " ! i •
5 loam I~,ee m / s ,
Fig. 5. Positive-ion FAL]MS B/E-linked scan spectra of the oligos',,ccharides from M. c , tarrhalis scrotyi~ C strain RSI0: < a) OS( I0); (b) OS(I 1 ). Experimental ,,/z-values for the fragment ions are given in the spectra and ~Iculated values are given in the st1"1]cturc~.. M in the spectra indicates signals originating from the matrix.

258 P Edebrtnk et aL / Carbohydrate Research 266 (1995) 237-261
3. Experimental
General methods.---Concentrations were performed under diminished pressure at < 40°C or by flushing with air at room temperature. For GLC, a Hewlett-Packard 5830 instrument fitted with a flame-ionisation detector was used. GLC-MS (El) was performed on a Hewlett-Packard 5970 MSD. Fractionations of alditol acetates and partially methylated aiditol acetates were performed on an HP-5 capillary column (25 mxO.20 ram), using a temperature program 180°(2 ( 1 rain) ~ 250°C at 3°C/min. GPC was performed on an FPLC system using a column of Superdex 30 (Pharmacia AB), with aqueous 0.07 M pyridinium acetate buffer (pH 5.4) as irdgant, and was monitored with a differential refractometer.
NMR spectroscopy.--The tH and 13C NMR spectra were recorded with a Jeol Alpha 400, a Varian Unity 500, or a Varian Unity 600 spectrometer using standard pulse sequences. NMR data were processed using standard Jeol and Varian software or using Felix V2.05 (Hare Research Inc.). Spectra of D20 solutions were recorded at 25°C. Samples in D20 were iyophilised twice with D20. t~hemical shifts are reported in ppm, using sodium 3-trimethylsilylpropanoate-d4 (~t 0.00) or acetone (~c 31.00) as internal references. Chemical shifts were obtained from 1D spectra when possible, or from proton-proton correlated 2D (COSY) or total proton-proton correlated (TOCSY) spectra; 33,,. coupling constants were obtained from ID spectra or from COSY spectra, and IJc., values were determined from the coupled HMOC spectrum. The TOCSY experiments were recorded using mixing times of 30, 60, and 90 ms. NOESY experiments were performed using mixing times of 200 (at 400 MHz) and 350 ms (at 500 MHz). Proton-.carbon correlated spectra, HMOC, were obtained with or without decoupling and the long-range proton-carbon cor- related spectra, HMBC, were performed using a delay time of 60 ms,
Fast atom bombardment mass spectrometrv.--A Jeol SXI02 instrument was used for the FABMS experiments. Ions were produced by a beam of Xe atoms (6 keV), using matrices of glycerol or 1:1 mixtures of glycerol and thioglycerol. The instrument was calibrated with Csl for ordinary scans and with a 3:1 mixture of Nal and Csl for linked scans. Helium was used as collision gas in the first field-tree region for the linked scan experiments. The gas pressure was increased until ca. 90% of the selected mother ion had collapsed, or until the ion source pressure reached 1.5 × 10-4 torr.
Bacteria and caltivation,~Moraxeila catarrhalis serotype C strains, RS10 and RS26, obtained from clinical isolates at Roslagstuils Sjukhus in Stockholm (Sweden), were used in this study. Bacteria were grown overnight in horse-blood agar plates at 37*C. Bacterial cultures used for the production of lipooligosaccharides were grown in brain-heart infusion broth ( Difco laboratories, Detroit, MI, USA) supplemen'ed with 0.5% (w/v) yeast extract. A 50-L fermentor containing 30 L of medium was inoculated with 5 L of late logarithmic phase culture, The bacteria were grown at constant pH of 7.2 to late logarithmic or early stationary phase, treated with formaldehyde to a final concentration of 1%, and kept over- night at 4°C, The cells were harvested by centrifugation, washed once with PBS, suspended in HzO, and lyophilised, Average yield was 16.6 g /L (dry weight).
Preparation of LOS,--Lipooligosaccharides were extracted from iyophilised bacteria, using phenol-CHCI3-petroleum ether, as described by Galanos et al. [ 21] with the follow- ing modification, Lipooligosaccharides were precipitated by 6 vol of 1:5 diethyl ether-

P. Edebrink et al. / Carbohydrate Research 266 (1995) 237-261 259
acetone [ 22]. No detectable amount of protein or nucleic acid was found in LOS. The yield was 1.3 mg/g (dry weight).
Sugar analysis.--The solutions of LOS and US (0.1 mg each) in 2 M CF3CO2H (0.5 mL) were kept in closed vials at 120°C for 3 h. The sugars in the hydrolysates were then converted into alditoi acetates by conventional methods.
Methylation analysis.--Methylation analyses were performed using a modification of the NaOH method [23,24]. Samples ( 1.0 mg of LOS and 0.2 mg of US) were dis,~olved in dimethyl sulfoxide (50/~L) and methylated by sequential addition of an NaOH slurry in dimethyl sulfoxide (120 mg/mL, 50/.~L; 20 min) and three additions of Mel ( 10, 10, and 20 pL) at 10-rain intervals. The methylated glycans were recovered in the organic phase after addition of chloroform (0.5 mL x 3) and M Na2S203 ( 1 mL). The permethylated products were further purified by reversed phase-chromatography on a Sep-Pak C18 car- tridge [ 25 ].
Identification ofKdo linkages.--A LOS sample (2 mg) was dried in a desiccator, and was methylated as described above and purified by reversed-phase chromatography on a Sep-Pak C18 cartridge. The dried permethylate0 LU• was subjected to methanolysis with M HCI in MeOH (0.1 mL) for 16 h at 85°C. Methanol was removed by flushing with N2. The product was then acetylated with 1:1 Ac20-pyridine for 30 min at 120°C.
SDS-polyacrylamide gel electrophoresis.--LOS samples (0.1 mg/mL) were suspended in sample buffer containing 0.1 M Tris. HC! (pH 6.8), 2% w/v SDS, 20% w/v sucrose, and 0.01% Bromophenol Blue in 1% dithiothreitol to a concentration of 1 pg/pL, and heated at 95°C for 5 rain. LOS in sample buffer was fractionated on a gel containing acrylamide (4%) in the stacking gel and acrylamide (18%) and 4 M urea in the separating gel. Each well was loaded with 10 btg of LOS preparation. SDS-PAGE was carried out at 100 V, 54 mA for the first 15 min, then 200 V, 54 mA until the front line reached the end of the gel. Bio-Rad mini protein electrophoresis equipment was used. Silver staining was done as described [261 with the following modifications. The gel was oxidised with 1% HIO4 in aq 40% EtOH containing AcOIt (5%) Ibr 25 mill. Staining with AgNO:~ was done for 35 rain. The R-type lipooligosaccharides from Salmonella typhimurimn rough mutants were prepared as described by Galanos et al. [ 21 ].
High-pH anion.exchatlge chromatograph), ( Dionex ).~High-pH anion-exchange chro- matography was performed on a Dionex system fitted with a pulsed amperometric detector (PAD) and using a Carbo Pac PAl column. The samples were eluted with a gradient of a 100 mM NaOH solution (eluent A) and a 100 mM NaOH and 500 mM NaOAc solution (eluent B) starting at an 85:15 ratio and ending at a 75:25 ratio after 30 min. The fractions were collected and desalted on Sephadex G-50 and Superdex 30 columns.
l,~olation of OS.~The iipooligosaccharides of RSI0 and RS26 (100 mg each) were suspended separately in aq 0.1 M NaOAc (20 mL) containing 0.1% SDS and stirred for 5 rain. Glacial AcOH was added to pH 4.4 and the solutions were kept at 100°C for 2 h [27,28l. After lyophilisation, the material was washed three times with EtOH, suspended in water, and centrifuged. The supernatant solutions were then applied to a Sep-Pak C18 cartridge and were eluted with water. The eluates were lyophilised and purified by GPC using the FPLC system and a Superdex 30 column. The LOS from strain RS10 (50 rag) yielded two oligosaccharides 0S(10) and OS(II ) co.eluting as one peak at 1.9 void volumes. This fraction was further purified on a Carbo Pac PAl column using the Dionex

2611 P. Edebrink et aL / Carbohydrate Research 266 (1995) 237-261
system, and, after desalting on Sephadex 50 and Superdex 30 columns, OS(10) and OS(11) were obtained in 1.2 and 0.5 mg yield, respectively. The LOS from strain RS26 yielded four oligosaccharides which eluted as two partially overlapping peaks at 1.9 void volumes, including OS(10) and OS(11), and at 2.0 void volumes, including OS(7) and OS(8). Repeated GPC of the fractions on a Superdex 30 column yielded OS(8) ( 1.5 rag), con- taining small amounts of OS(7), and OS(10) (3.0 mg), containing a small amount of OS(11).
NMR assignments,~The chemical shifts for the monomeric hexoses and hexosamine [ 29] and for Kdo-OMe [30] were obtained from the literature. The hexose and hexosamine residues are designated A-I, The tH NMR chemical shifts of the residues for OS(10) and OS(8) are listed in Tables 3 and 7, respectively, and the signals were assigned as described below. The tH NMR signals were assigned as far as possible using double quantum filter 2D COSY and 2D TOCSY (30, 60, and 90 ms mixing time, including trim-pulses) exper- iments in the phase sensitive mode (TPPI). The assignment of signals for H-5 of/i-linked residues was assisted or corroborated by phase sensitive 2D NOESY spectra (350 ms mixing time), The protons substituting the primary carbon of residue C had small 3,]H, H values and COSY cross-peaks to H-5 were not observed, but strong intra-residual NOEs from H-5 aided the assignments. Proton-detected broad-band-decoupled HMQC experiments in the phase sensitive mode (i'PPl) facilitated the assignment of the t3C signals. The t3C NMR chemical shifts for OS(10) and OS(8) are listed in Tables 4 and 8, respectively. The tH and t~C NMR signals that could not be assigned according to the scheme indicated above were assigned tentatively (as indicated in the tables), where the experimental tH and t~C chemical shifts were assigned to atoms having chemical shifts closest to those of the corresponding monosaccharides~ It was indicated from the J , . . coupling constants that all hexose rings were maintained in their normal 4C, conformation,
Acknowledgements
This work was supported by grants from the Swedish National Board for Technical and Industrial Development, the Swedish Research Council for Engineering Sciences, the Swed- ish Natural Science Research Council, and the Swedish Medical Research Council. We thank the Swedish Institute and the Wenner-Gren Center foundation for fellowships to members of the group (MMR and MR), and the Swedish NMR Centre for putting 500- and 600-NMR facilities at our disposal. Mrs. Olga Hegedtis is thanked for performing the Dionex separatiors,
References
[ 11 G,V, Doom, Diagn, Microbiol, le~f¢ct, DL~,, 4 (198f~) 191-201, [ 21 G~F, van Hare, P,A, Shurin, C.D~ Marchaut, N,A, Cartclli, C.E, Johnson, D, Fulton, S, Carlin, and C.H. Kim,
Ret,, Infect, Dis,, 9 (1987) 16-27, 131 P,J, Hitchcock, L Loire, P,R, M~ike|~i, E,T, Rietschel, W. Strittmatter, and D.C. Morrison, J. Bacteriol., 166
(1986) 699-705, 14] K,G, Johnson, l,J, McDonald, and M.B. Perry, Can. $. MicrobioL, 22 (1976) 460-,A67~

P. Edebrink et al. / Carbohydrate Research 266 {1995) 237-261 261
[5] T.F. Murphy, Pediatr. Infect. Dis. J., 8 (1989) $75--$77. [6] T.F. Murphy and L.C. Bartos, Infect. lmmm~., 57 (1989) 2938-2941. [7] IUPAC-IUB Joint Commision on Biochemical Nomeclature, Nomenclature of Carbohydrates, JCBN 24.8. [8] IVy. Vaneechoutle, G. Verschraegen, G. Claeys, and A.van den Abeele, J. Clin. Microbiol., 28 (1990) 182-
187. [9] (;~.A. Adams, T.G. Tornabene, and M. Yaguchi, Can. J. Microbiol., 15 (1969) 365-374.
[ 10] J.S. Fomsgaard, A. Fomsgaard, B. Bruun, N. Hfiby, and C. Galanos, Infect. immun., 59 (1991) 3346-3349. [ 11 ] P. Edebrink, P.-E. Jansson, M.M. Rahman, G. Widmalm, T. Holme, M. Rahman, and A. Weintraub,
Carbohydr. Res., 257 (1994) 269-284. [ 12] H. Masoud, M.B. Perry, J.-R. Brisson, D. Uhrin, and J.C. Richards, Can. J. Chem., (1994) in press. [ 13] G.J. Gerwig, J.P. Kamerling, and J.F.G. Vliegenthart, Carbohydr. Res., 62 (1978) 349-357. [ 14] GJ. Gerwig, J.P. Kamerling, and J.F.G. Vliegenthart, Carbohydr. Res., 77 (1979) 1-7. [ 15] H. Brade, U. Ziihringer, and E.T. Rietschel, Carbohydr. Res., 134 (1984) 157-166. [ 16] H. Brade and E.T. Rietschel, Eur. J. Biochem., 145 (1984) 231-236, [ 17] H. Schmidt and H. Friebolin, J. Carbohydr. Chem., 2 (1983) 405-413. [ 18] F. Unger, Adv. Carbohydr. Chem. Biochem., 38 (1982) 323-388. [ 19] A. Bax and M.F. Summers, J. Am. Chem. Soc., 108 (1986) 2093-2094. [20] A. Dell, Adv. Carbohydr. Chem. Biochem., 45 (1987) 19-72. [21 ] C. Galanos, O. Liideritz, and O. Westphal, Eur. J. Biochem., 9 (1969) 245-249. [22] N, Qureshi, K. Takayama, and E. Ribi, J. Biol. Chem., 19 (1982) 11808-11815. [23] I. Ciucanu and F. Kerck, Carbohydr. Res., 131 (1984) 209-217. [24] MJ. McConville, S.W. Homans, J.E. Thomas-Oates, A. Dell, and A. Bacic, J. Biol. Chem., 265 (1990)
7385-7390. [23] TJ. Waeghe, A.G. Darviil, M. McNeil, and P. Albersheim, Carbohydr. Res., 123 (1983) 281-304. [26] C. Tsai and C.E. Frasch, Anal. Biochem., 119 (1982) 115-119. [27] M. Caroff, A. Tacken, and L. Szab6, Carbohydr. Res., 175 (1988) 273-282. 128l O. Hoist, E. Rfhrscheidt-Andrzejcwski, H.-P. Cordes, and H. Brade, Carbohydr. Re,~., 188 (1989) 213-
218. 1291 P,°E. Jalnsson, L. Kenm~, and G. Widmahn, Carbohydr. Re.~'., 188 (1989) 1fi9-o191. 1301 K, Luthm~m, A. Claesson, L. Kcnne, and I. Csilregh, Cm'btJhydr. Re.~'., 171) (1987) 1(~7~179.
Copyright © 2022 FDOKUMEN




















