
Structural, electronic and thermodynamic properties of Al- and Si-doped α-, γ-, and β-MgH2:...
7
Structural, electronic and thermodynamic properties of britholites Ca 10x La x (PO 4 ) 6x (SiO 4 ) x F 2 (0 x 6): Experiment and theory H. Njema a , M. Debbichi b, *, K. Boughzala a , M. Said b , K. Bouzouita a a Laboratoire de Chimie Industrielle, Ecole Nationale d’Ingnieurs de Sfax, BP1173 Sfax, Tunisia b Laboratoire de la matie `re condense ´e et nanosciences, De ´partement de Physique, Faculte ´ des Sciences de Monastir, 5019 Monastir, Tunisia 1. Introduction The hydroxyapatite (Ca 10 (PO 4 ) 6 OH 2 , HA) and to a lesser extent the fluorapatite (Ca 10 (PO 4 ) 6 F 2 , FA) have been extensively studied because of to their physicochemical and biological properties and for their applications as biomaterials [1,2]. Furthermore, the ability of their structure to accommodate a great number of substitutions, both cationic and anionic substitutions, has widely expanded their field of application [3–5]. For example, the simultaneous substitutions of rare earth elements (Ln 3+ , La 3+ , Nd 3+ , etc.) and SiO 4 4 for Ca 2+ and PO 3 4 , result in a new family of compounds called britholites [6]. The discovery of the natural nuclear reactor of Oklo (Gabon) [7] has demonstrated that these compounds can retain minor actinides and long-lived fission products over several million years without suffering major damage [8–11], suggesting that they can be very suitable for the confinement of the nuclear waste [12]. Compared to FA, it is well known that the chemical and physical properties of the substituted counterparts are in direct correlation with the atoms in substitution. Therefore, britholites have been the aim of several experimental studies, using X-ray diffraction (XRD), infrared (IR) and Raman spectroscopy, nuclear magnetic resonance (NMR) and so on, in order to determine their thermal, chemical, physical and structural properties [13,14]. However, the interpretation of some experimental results is still somewhat difficult, such as the evolution of their stability with the substitution. Nowadays, ab initio calculations have proved to be an effective way to analyze and model the structure of apatites at the atomic level in order to predict their physical and chemical properties. Several theoretical studies on the apatites have been reported for the interpretation of the electronic structure, charge distribution and atomic configuration using ab initio Hartree-Fock method [15], molecular dynamic simulation [16], and density functional theory [17–19]. Katsuyuki et al. [20,21] have proposed the thermody- namic treatment of total energies to analyze the defect stability under chemical equilibrium between the solid and aqueous solution. Rabone and de Leeuw have studied Sr 2+ substitution for Ca 2+ in fluorapatite and hydroxyapatite using classical potentials [22]. Other studies have focused on the position and orientation of the common anionic groups along the c-axis channel in apatites [19,23,24]. In addition, Meis et al. [25] have investigated the possibility of incorporating selectively plutonium and cesium in a mono-silicate neodymium-fluoroapatite using analytical potentials. These previous studies have established that the atomic scale simulation is useful and reliable for the study of apatites. According to the properties of natural silicate-apatites and the strong bonds between lanthanides and fluorine anions as well as the rigidity of the PO 4 groups [6], the possibility of obtaining synthetic apatites with a definite composition allows the repara- tion of materials suitable for use as high performance matrices for long-lived radioactive wastes. The goal of this paper is to undertake Materials Research Bulletin 51 (2014) 210–216 A R T I C L E I N F O Article history: Received 25 July 2013 Received in revised form 2 December 2013 Accepted 14 December 2013 Available online 21 December 2013 Keywords: Ceramics Chemical synthesis X-ray diffraction Inorganic compounds Crystal structure A B S T R A C T The apatite-type compounds Ca 10x La x (PO 4 ) 6x (SiO 4 ) x F 2 with 0 x 6 were prepared using a high temperature solid state reaction and were characterized by X-ray diffraction. The crystal structure, chemical bonding, electronic structure and formation energy of all relaxed structures were analyzed by density functional theory (DFT). The calculated results show that the predicted geometry can well reproduce the structural parameters. The incorporation of La 3+ into the fluorapatite (FA) structure induced especially at the level of the S(2) sites a certain disorder which is responsible for the weakening in the stability with x. Excellent agreement were obtained between the calculated and experimental results. Moreover, the band structure indicates that despite the reduction of the band gap with x content all materials remain insulating. ß 2013 Elsevier Ltd. All rights reserved. * Corresponding author. Tel.: þ216 73 500276; fax: þ216 73 500 278. E-mail address: [email protected] (M. Debbichi). Contents lists available at ScienceDirect Materials Research Bulletin jo u rn al h om ep age: ww w.els evier.c o m/lo c ate/mat res b u 0025-5408/$ – see front matter ß 2013 Elsevier Ltd. All rights reserved. http://dx.doi.org/10.1016/j.materresbull.2013.12.022
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Structural, electronic and thermodynamic properties of Al- and Si-doped α-, γ-, and β-MgH2:...

Materials Research Bulletin 51 (2014) 210–216
Structural, electronic and thermodynamic properties of britholitesCa10�xLax(PO4)6�x(SiO4)xF2 (0 � x � 6): Experiment and theory
H. Njema a, M. Debbichi b,*, K. Boughzala a, M. Said b, K. Bouzouita a
a Laboratoire de Chimie Industrielle, Ecole Nationale d’Ingnieurs de Sfax, BP1173 Sfax, Tunisiab Laboratoire de la matiere condensee et nanosciences, Departement de Physique, Faculte des Sciences de Monastir, 5019 Monastir, Tunisia
A R T I C L E I N F O
Article history:
Received 25 July 2013
Received in revised form 2 December 2013
Accepted 14 December 2013
Available online 21 December 2013
Keywords:
Ceramics
Chemical synthesis
X-ray diffraction
Inorganic compounds
Crystal structure
A B S T R A C T
The apatite-type compounds Ca10�xLax(PO4)6�x(SiO4)xF2 with 0 � x � 6 were prepared using a high
temperature solid state reaction and were characterized by X-ray diffraction. The crystal structure,
chemical bonding, electronic structure and formation energy of all relaxed structures were analyzed by
density functional theory (DFT). The calculated results show that the predicted geometry can well
reproduce the structural parameters. The incorporation of La3+ into the fluorapatite (FA) structure
induced especially at the level of the S(2) sites a certain disorder which is responsible for the weakening
in the stability with x. Excellent agreement were obtained between the calculated and experimental
results. Moreover, the band structure indicates that despite the reduction of the band gap with x content
all materials remain insulating.
� 2013 Elsevier Ltd. All rights reserved.
Contents lists available at ScienceDirect
Materials Research Bulletin
jo u rn al h om ep age: ww w.els evier .c o m/lo c ate /mat res b u
1. Introduction
The hydroxyapatite (Ca10(PO4)6OH2, HA) and to a lesser extentthe fluorapatite (Ca10(PO4)6F2, FA) have been extensively studiedbecause of to their physicochemical and biological properties andfor their applications as biomaterials [1,2]. Furthermore, the abilityof their structure to accommodate a great number of substitutions,both cationic and anionic substitutions, has widely expanded theirfield of application [3–5]. For example, the simultaneoussubstitutions of rare earth elements (Ln3+, La3+, Nd3+, etc.) andSiO4�
4 for Ca2+ and PO3�4 , result in a new family of compounds called
britholites [6]. The discovery of the natural nuclear reactor of Oklo(Gabon) [7] has demonstrated that these compounds can retainminor actinides and long-lived fission products over severalmillion years without suffering major damage [8–11], suggestingthat they can be very suitable for the confinement of the nuclearwaste [12].
Compared to FA, it is well known that the chemical andphysical properties of the substituted counterparts are in directcorrelation with the atoms in substitution. Therefore, britholiteshave been the aim of several experimental studies, using X-raydiffraction (XRD), infrared (IR) and Raman spectroscopy, nuclearmagnetic resonance (NMR) and so on, in order to determine theirthermal, chemical, physical and structural properties [13,14].
* Corresponding author. Tel.: þ216 73 500276; fax: þ216 73 500 278.
E-mail address: [email protected] (M. Debbichi).
0025-5408/$ – see front matter � 2013 Elsevier Ltd. All rights reserved.
http://dx.doi.org/10.1016/j.materresbull.2013.12.022
However, the interpretation of some experimental results is stillsomewhat difficult, such as the evolution of their stability with thesubstitution.
Nowadays, ab initio calculations have proved to be an effectiveway to analyze and model the structure of apatites at the atomiclevel in order to predict their physical and chemical properties.Several theoretical studies on the apatites have been reported forthe interpretation of the electronic structure, charge distributionand atomic configuration using ab initio Hartree-Fock method [15],molecular dynamic simulation [16], and density functional theory[17–19]. Katsuyuki et al. [20,21] have proposed the thermody-namic treatment of total energies to analyze the defect stabilityunder chemical equilibrium between the solid and aqueoussolution. Rabone and de Leeuw have studied Sr2+ substitutionfor Ca2+ in fluorapatite and hydroxyapatite using classicalpotentials [22]. Other studies have focused on the position andorientation of the common anionic groups along the c-axis channelin apatites [19,23,24]. In addition, Meis et al. [25] have investigatedthe possibility of incorporating selectively plutonium and cesiumin a mono-silicate neodymium-fluoroapatite using analyticalpotentials. These previous studies have established that the atomicscale simulation is useful and reliable for the study of apatites.
According to the properties of natural silicate-apatites and thestrong bonds between lanthanides and fluorine anions as well asthe rigidity of the PO4 groups [6], the possibility of obtainingsynthetic apatites with a definite composition allows the repara-tion of materials suitable for use as high performance matrices forlong-lived radioactive wastes. The goal of this paper is to undertake

Fig. 1. Primitive unit cell of the fluoroapatite Ca10(PO4)6F2 with atoms labeled
according to element and symmetric type.
Fig. 2. XRD patterns of Ca10�xLax(PO4)6�x(SiO4)xF2 sample, with 0 � x � 6.
H. Njema et al. / Materials Research Bulletin 51 (2014) 210–216 211
a systematical analysis of the structural and electronic propertiesof the Ca10�xLax(PO4)6�x(SiO4)xF2 (0 � x � 6) in order to evaluatethe influence of the substitutional atoms at the atomic level inorder to help understand thermodynamic data, using first-principles DFT calculations. The obtained results will be comparedto those experimentally determined.
2. Materials and methods
2.1. Sample preparation and characterization
The samples were prepared by utilizing a solid-state reactionfrom a stoichiometric mixture of CaCO3, CaF2, La2O3, SiO2, andCa2P2O7 according to the following reaction:
3CaCO3þ 6�x2
� �Ca2P2O7þCaF2þxSiO2þxLa2O3 ! Ca10�xLax(-
(PO4)6�x(SiO4)xF2þ3CO2
The calcium diphosphate was obtained by heating a mixture ofcalcium carbonate and diammonium hydrogen phosphate at900 8C for 10 h. Before use, the lanthanum oxide was calcined at1000 8C for 12 h.
The starting reagents were thoroughly ground in an agatemortar. Then, they were pressed into pellets and heat-treatedunder an argon flow at 900 8C for 12 h. Thereafter, the pellets weremanually crushed and homogenized. The resulting powders wereagain uniaxially pressed and heat-treated between 1200 and1400 8C according to their SiO2 content. The heating rate was of10 8C/min. Finally, the samples were cooled naturally to roomtemperature. Samples were characterized by X-ray diffraction(XRD) using a Bruker D8 ADVANCE diffractometer operating withCu-Ka radiation for a 2u range from 98 to 808, with a step size of0.028 and a counting time of 10 s per step.
2.2. Density functional theory based calculations
The calculations have been performed using a plan-wave basisset and ultrasoft pseudopotentials as implemented in theQUANTUM-ESPRESSO package [26]. The wave functions areexpanded in plane waves with an energy cutoff of 110 Ry. Theexchange-correlation potential was treated in the framework ofthe generalized-gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) [27]. The Brillouin-Zone (BZ) integrations havebeen carried out using sets of special points corresponding to a4�4�2 Monkhorst-Pack mesh [28]. Each eigenvalue was convo-luted with a Gaussian with a full width at half maximum ofs = 0.02 Ry. All structures were optimized by simultaneouslyrelaxing the atomic basis vectors and the atomic positions insidethe unit cells using Broyden–Fletcher–Goldfarb–Shanno (BFGS)algorithm [29]. The relaxation was considered to be completewhen the atomic forces were less than 1.0 mRy/Bohr, and the totalenergy during the structural optimization iterative process wasconverged to less than 0.1 mRy. Unit-cell and initial positionalparameters used in the optimization were referred from theRietveld refinement results.
2.3. Crystal structure
Fluorapatite (FA) crystallizes in a hexagonal crystal structurewith a P63/m space group. The unit cell contains 42 atoms. In thisstructure, PO3�
4 tetrahedra form the network with two types ofchannels where the Ca atoms are located. The tunnels of the firstkind are filled with four Ca(1), noted S(1) sites (4f position), along athree-fold axis. Each of these Ca atoms, which are aligned incolumns parallel to the c-axis, is surrounded by nine oxygenatoms: three O(1), three O(2) and three O(3), forming aCaO9-polyhedron with six short and three long Ca–O bonds. The
second-type-tunnel, which is the largest, is lined by oxygen atomsand the six other Ca, noted S(2) sites (6h position), along a six-foldaxis. These Ca coordinated by six oxygen atoms (O(1), O(2), fourO(3), and a fluorine atom form an irregular CaO6F-polyhedron.Otherwise, Ca(2) atoms are located at the vertices of two alternatedequilateral triangles at z = 1/4 and 3/4, which are centered on thesix-fold axis, where the fluorine ions are located at 2a positions. Amodel for FA structure is given as a reference in Fig. 1.
3. Results and discussion
3.1. Lattice parameters and stability
The XRD patterns of the samples are shown in Fig. 2. All the XRDpatterns showed reflections corresponding to an apatite phase,matching the ICDD standard (JCPDS 00-071-0880) for FA. Nosecondary phases were detected in any of the patterns.
Prior to the simulation, the XRD data were analyzed via theRietveld method using the Fullprof program [30]. The computedtogether with experimental lattice parameters of the samples aregiven in Table 1 and are approximately 0.1–0.2 A larger than theexperimental ones. This overestimation is typical of the GGAfunctional. We notice that the increase of both a and c parameterswith the simultaneous incorporation of La3+ and SiO4�
4 has abasically linear evolution in accordance with Vegard’s law. This
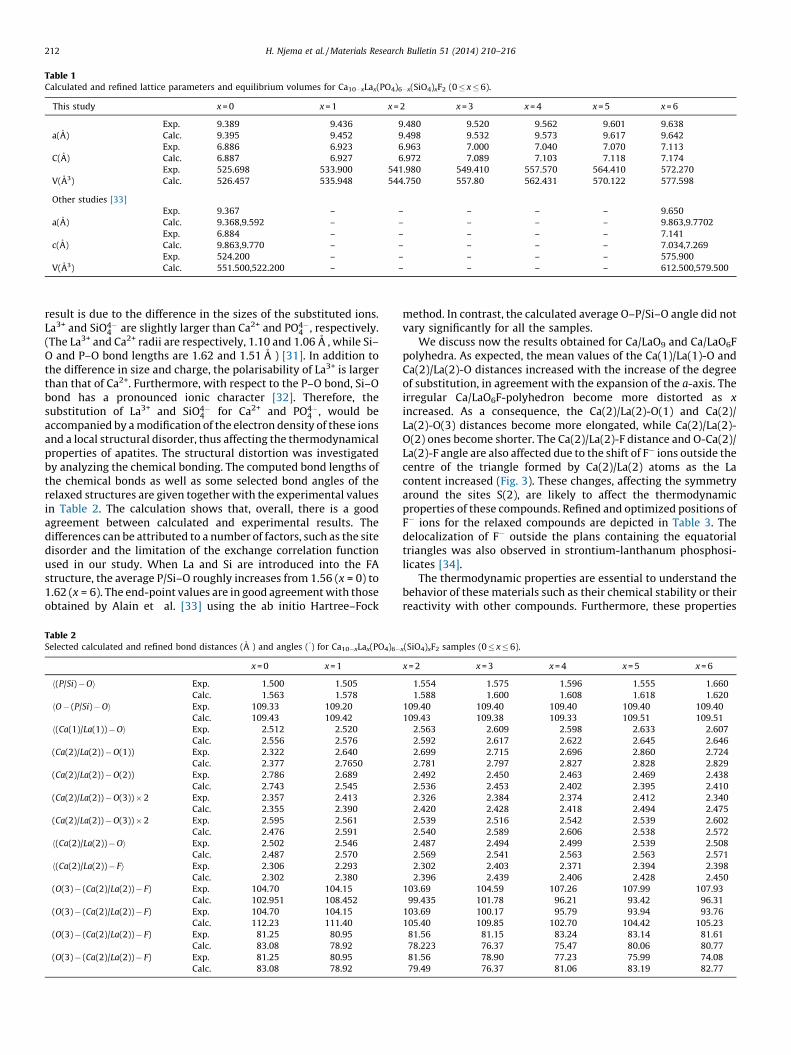
Table 1Calculated and refined lattice parameters and equilibrium volumes for Ca10�xLax(PO4)6�x(SiO4)xF2 (0 � x � 6).
This study x = 0 x = 1 x = 2 x = 3 x = 4 x = 5 x = 6
Exp. 9.389 9.436 9.480 9.520 9.562 9.601 9.638
a(A) Calc. 9.395 9.452 9.498 9.532 9.573 9.617 9.642
Exp. 6.886 6.923 6.963 7.000 7.040 7.070 7.113
C(A) Calc. 6.887 6.927 6.972 7.089 7.103 7.118 7.174
Exp. 525.698 533.900 541.980 549.410 557.570 564.410 572.270
V(A3) Calc. 526.457 535.948 544.750 557.80 562.431 570.122 577.598
Other studies [33]
Exp. 9.367 – – – – – 9.650
a(A) Calc. 9.368,9.592 – – – – – 9.863,9.7702
Exp. 6.884 – – – – – 7.141
c(A) Calc. 9.863,9.770 – – – – – 7.034,7.269
Exp. 524.200 – – – – – 575.900
V(A3) Calc. 551.500,522.200 – – – – – 612.500,579.500
H. Njema et al. / Materials Research Bulletin 51 (2014) 210–216212
result is due to the difference in the sizes of the substituted ions.La3+ and SiO4�
4 are slightly larger than Ca2+ and PO4�4 , respectively.
(The La3+ and Ca2+ radii are respectively, 1.10 and 1.06 A , while Si–O and P–O bond lengths are 1.62 and 1.51 A ) [31]. In addition tothe difference in size and charge, the polarisability of La3+ is largerthan that of Ca2+. Furthermore, with respect to the P–O bond, Si–Obond has a pronounced ionic character [32]. Therefore, thesubstitution of La3+ and SiO4�
4 for Ca2+ and PO4�4 , would be
accompanied by a modification of the electron density of these ionsand a local structural disorder, thus affecting the thermodynamicalproperties of apatites. The structural distortion was investigatedby analyzing the chemical bonding. The computed bond lengths ofthe chemical bonds as well as some selected bond angles of therelaxed structures are given together with the experimental valuesin Table 2. The calculation shows that, overall, there is a goodagreement between calculated and experimental results. Thedifferences can be attributed to a number of factors, such as the sitedisorder and the limitation of the exchange correlation functionused in our study. When La and Si are introduced into the FAstructure, the average P/Si–O roughly increases from 1.56 (x = 0) to1.62 (x = 6). The end-point values are in good agreement with thoseobtained by Alain et al. [33] using the ab initio Hartree–Fock
Table 2Selected calculated and refined bond distances (A ) and angles (8) for Ca10�xLax(PO4)6�
x = 0 x = 1
h(P/Si) � Oi Exp. 1.500 1.505
Calc. 1.563 1.578
hO � (P/Si) � Oi Exp. 109.33 109.20
Calc. 109.43 109.42
h(Ca(1)/La(1)) � Oi Exp. 2.512 2.520
Calc. 2.556 2.576
(Ca(2)/La(2)) � O(1)) Exp. 2.322 2.640
Calc. 2.377 2.7650
(Ca(2)/La(2)) � O(2)) Exp. 2.786 2.689
Calc. 2.743 2.545
(Ca(2)/La(2)) � O(3)) � 2 Exp. 2.357 2.413
Calc. 2.355 2.390
(Ca(2)/La(2)) � O(3)) � 2 Exp. 2.595 2.561
Calc. 2.476 2.591
h(Ca(2)/La(2)) � Oi Exp. 2.502 2.546
Calc. 2.487 2.570
h(Ca(2)/La(2)) � Fi Exp. 2.306 2.293
Calc. 2.302 2.380
(O(3) � (Ca(2)/La(2)) � F) Exp. 104.70 104.15
Calc. 102.951 108.452
(O(3) � (Ca(2)/La(2)) � F) Exp. 104.70 104.15
Calc. 112.23 111.40
(O(3) � (Ca(2)/La(2)) � F) Exp. 81.25 80.95
Calc. 83.08 78.92
(O(3) � (Ca(2)/La(2)) � F) Exp. 81.25 80.95
Calc. 83.08 78.92
method. In contrast, the calculated average O–P/Si–O angle did notvary significantly for all the samples.
We discuss now the results obtained for Ca/LaO9 and Ca/LaO6Fpolyhedra. As expected, the mean values of the Ca(1)/La(1)-O andCa(2)/La(2)-O distances increased with the increase of the degreeof substitution, in agreement with the expansion of the a-axis. Theirregular Ca/LaO6F-polyhedron become more distorted as x
increased. As a consequence, the Ca(2)/La(2)-O(1) and Ca(2)/La(2)-O(3) distances become more elongated, while Ca(2)/La(2)-O(2) ones become shorter. The Ca(2)/La(2)-F distance and O-Ca(2)/La(2)-F angle are also affected due to the shift of F� ions outside thecentre of the triangle formed by Ca(2)/La(2) atoms as the Lacontent increased (Fig. 3). These changes, affecting the symmetryaround the sites S(2), are likely to affect the thermodynamicproperties of these compounds. Refined and optimized positions ofF� ions for the relaxed compounds are depicted in Table 3. Thedelocalization of F� outside the plans containing the equatorialtriangles was also observed in strontium-lanthanum phosphosi-licates [34].
The thermodynamic properties are essential to understand thebehavior of these materials such as their chemical stability or theirreactivity with other compounds. Furthermore, these properties
x(SiO4)xF2 samples (0 � x � 6).
x = 2 x = 3 x = 4 x = 5 x = 6
1.554 1.575 1.596 1.555 1.660
1.588 1.600 1.608 1.618 1.620
109.40 109.40 109.40 109.40 109.40
109.43 109.38 109.33 109.51 109.51
2.563 2.609 2.598 2.633 2.607
2.592 2.617 2.622 2.645 2.646
2.699 2.715 2.696 2.860 2.724
2.781 2.797 2.827 2.828 2.829
2.492 2.450 2.463 2.469 2.438
2.536 2.453 2.402 2.395 2.410
2.326 2.384 2.374 2.412 2.340
2.420 2.428 2.418 2.494 2.475
2.539 2.516 2.542 2.539 2.602
2.540 2.589 2.606 2.538 2.572
2.487 2.494 2.499 2.539 2.508
2.569 2.541 2.563 2.563 2.571
2.302 2.403 2.371 2.394 2.398
2.396 2.439 2.406 2.428 2.450
103.69 104.59 107.26 107.99 107.93
99.435 101.78 96.21 93.42 96.31
103.69 100.17 95.79 93.94 93.76
105.40 109.85 102.70 104.42 105.23
81.56 81.15 83.24 83.14 81.61
78.223 76.37 75.47 80.06 80.77
81.56 78.90 77.23 75.99 74.08
79.49 76.37 81.06 83.19 82.77
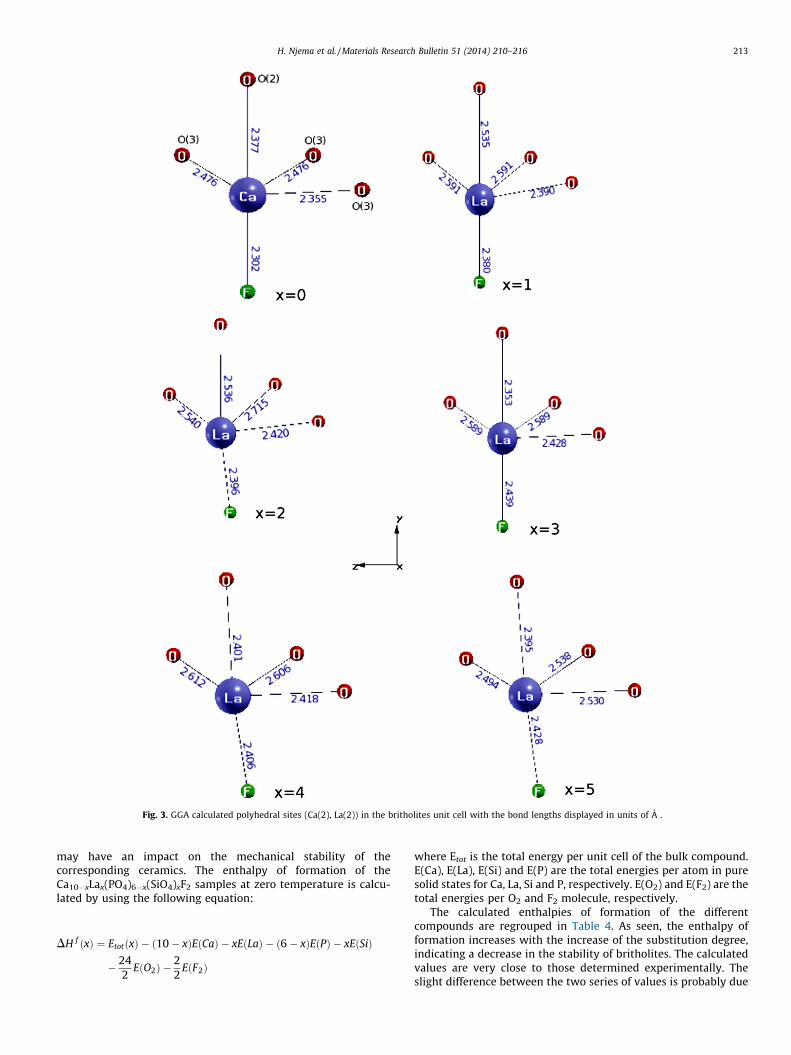
Fig. 3. GGA calculated polyhedral sites (Ca(2), La(2)) in the britholites unit cell with the bond lengths displayed in units of A .
H. Njema et al. / Materials Research Bulletin 51 (2014) 210–216 213
may have an impact on the mechanical stability of thecorresponding ceramics. The enthalpy of formation of theCa10�xLax(PO4)6�x(SiO4)xF2 samples at zero temperature is calcu-lated by using the following equation:
DH f ðxÞ ¼ EtotðxÞ � ð10 � xÞEðCaÞ � xEðLaÞ � ð6 � xÞEðPÞ � xEðSiÞ
� 24
2EðO2Þ � 2
2EðF2Þ
where Etot is the total energy per unit cell of the bulk compound.E(Ca), E(La), E(Si) and E(P) are the total energies per atom in puresolid states for Ca, La, Si and P, respectively. E(O2) and E(F2) are thetotal energies per O2 and F2 molecule, respectively.
The calculated enthalpies of formation of the differentcompounds are regrouped in Table 4. As seen, the enthalpy offormation increases with the increase of the substitution degree,indicating a decrease in the stability of britholites. The calculatedvalues are very close to those determined experimentally. Theslight difference between the two series of values is probably due
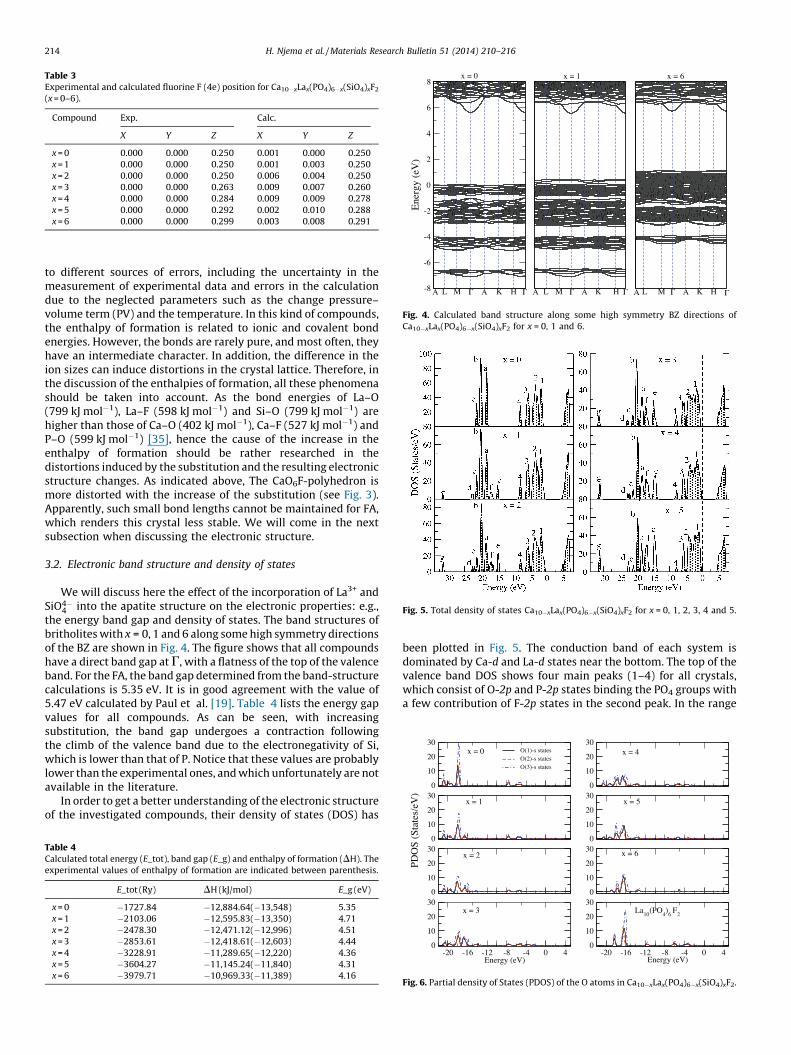
Table 3Experimental and calculated fluorine F (4e) position for Ca10�xLax(PO4)6�x(SiO4)xF2
(x = 0–6).
Compound Exp. Calc.
X Y Z X Y Z
x = 0 0.000 0.000 0.250 0.001 0.000 0.250
x = 1 0.000 0.000 0.250 0.001 0.003 0.250
x = 2 0.000 0.000 0.250 0.006 0.004 0.250
x = 3 0.000 0.000 0.263 0.009 0.007 0.260
x = 4 0.000 0.000 0.284 0.009 0.009 0.278
x = 5 0.000 0.000 0.292 0.002 0.010 0.288
x = 6 0.000 0.000 0.299 0.003 0.008 0.291
-8
-6
-4
-2
0
2
4
6
8
Ene
rgy
(eV
)
ΓΓMLA HK AA A A ALL MM ΓΓ Γ ΓKK HH
x = 0 x = 1 x = 6
Fig. 4. Calculated band structure along some high symmetry BZ directions of
Ca10�xLax(PO4)6�x(SiO4)xF2 for x = 0, 1 and 6.
Fig. 5. Total density of states Ca10�xLax(PO4)6�x(SiO4)xF2 for x = 0, 1, 2, 3, 4 and 5.
0
10
20
30
0
10
20
30
20
30
20
30
O(1)-s state sO(2)-s state sO(3)-s state s
tes/
eV)
x = 0
x = 1
x = 4
x = 5
H. Njema et al. / Materials Research Bulletin 51 (2014) 210–216214
to different sources of errors, including the uncertainty in themeasurement of experimental data and errors in the calculationdue to the neglected parameters such as the change pressure–volume term (PV) and the temperature. In this kind of compounds,the enthalpy of formation is related to ionic and covalent bondenergies. However, the bonds are rarely pure, and most often, theyhave an intermediate character. In addition, the difference in theion sizes can induce distortions in the crystal lattice. Therefore, inthe discussion of the enthalpies of formation, all these phenomenashould be taken into account. As the bond energies of La–O(799 kJ mol�1), La–F (598 kJ mol�1) and Si–O (799 kJ mol�1) arehigher than those of Ca–O (402 kJ mol�1), Ca–F (527 kJ mol�1) andP–O (599 kJ mol�1) [35], hence the cause of the increase in theenthalpy of formation should be rather researched in thedistortions induced by the substitution and the resulting electronicstructure changes. As indicated above, The CaO6F-polyhedron ismore distorted with the increase of the substitution (see Fig. 3).Apparently, such small bond lengths cannot be maintained for FA,which renders this crystal less stable. We will come in the nextsubsection when discussing the electronic structure.
3.2. Electronic band structure and density of states
We will discuss here the effect of the incorporation of La3+ andSiO4�
4 into the apatite structure on the electronic properties: e.g.,the energy band gap and density of states. The band structures ofbritholites with x = 0, 1 and 6 along some high symmetry directionsof the BZ are shown in Fig. 4. The figure shows that all compoundshave a direct band gap at G, with a flatness of the top of the valenceband. For the FA, the band gap determined from the band-structurecalculations is 5.35 eV. It is in good agreement with the value of5.47 eV calculated by Paul et al. [19]. Table 4 lists the energy gapvalues for all compounds. As can be seen, with increasingsubstitution, the band gap undergoes a contraction followingthe climb of the valence band due to the electronegativity of Si,which is lower than that of P. Notice that these values are probablylower than the experimental ones, and which unfortunately are notavailable in the literature.
In order to get a better understanding of the electronic structureof the investigated compounds, their density of states (DOS) has
Table 4Calculated total energy (E_tot), band gap (E_g) and enthalpy of formation (DH). The
experimental values of enthalpy of formation are indicated between parenthesis.
E_tot (Ry) DH (kJ/mol) E_g (eV)
x = 0 �1727.84 �12,884.64(�13,548) 5.35
x = 1 �2103.06 �12,595.83(�13,350) 4.71
x = 2 �2478.30 �12,471.12(�12,996) 4.51
x = 3 �2853.61 �12,418.61(�12,603) 4.44
x = 4 �3228.91 �11,289.65(�12,220) 4.36
x = 5 �3604.27 �11,145.24(�11,840) 4.31
x = 6 �3979.71 �10,969.33(�11,389) 4.16
been plotted in Fig. 5. The conduction band of each system isdominated by Ca-d and La-d states near the bottom. The top of thevalence band DOS shows four main peaks (1–4) for all crystals,which consist of O-2p and P-2p states binding the PO4 groups witha few contribution of F-2p states in the second peak. In the range
0
10
0
10
0
10
20
30
0
10
20
30
-20 -16 -12 -8 -4 400
10
20
30
-20 -16 -12 -8 -4 400
10
20
30
PDO
S (S
ta
Energy (eV) Energy (eV)
x = 2
x = 3
x = 6
La10
(PO4)6 F
2
Fig. 6. Partial density of States (PDOS) of the O atoms in Ca10�xLax(PO4)6�x(SiO4)xF2.
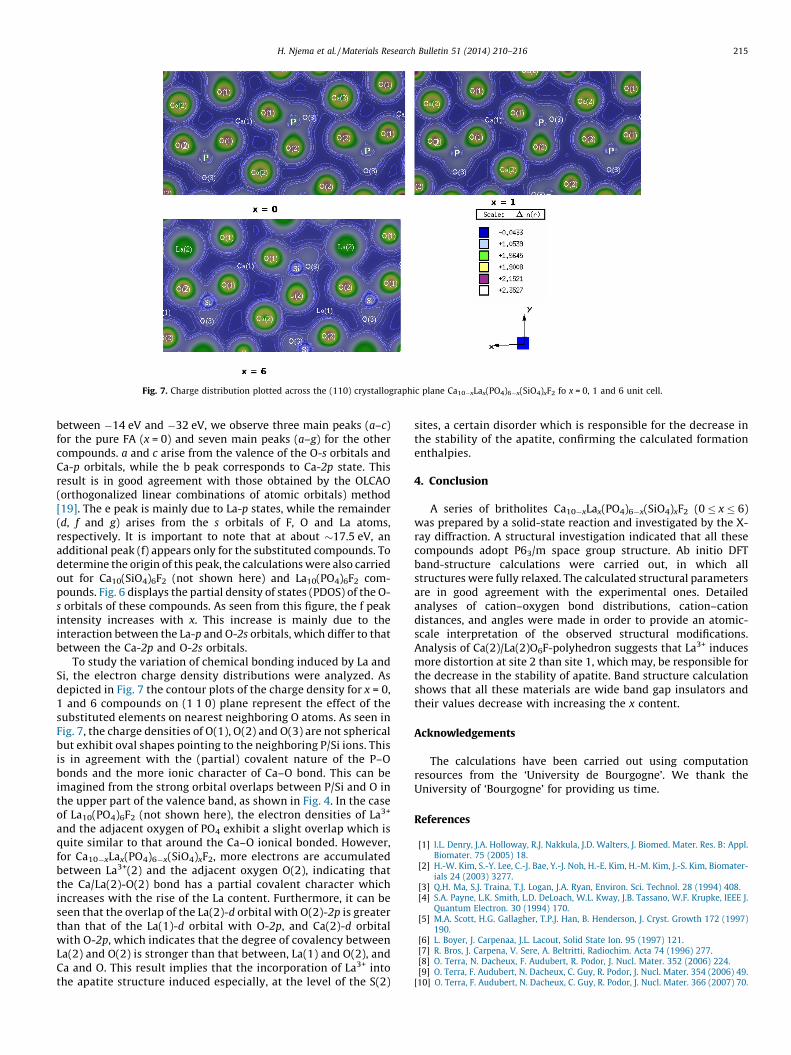
Fig. 7. Charge distribution plotted across the (110) crystallographic plane Ca10�xLax(PO4)6�x(SiO4)xF2 fo x = 0, 1 and 6 unit cell.
H. Njema et al. / Materials Research Bulletin 51 (2014) 210–216 215
between �14 eV and �32 eV, we observe three main peaks (a–c)for the pure FA (x = 0) and seven main peaks (a–g) for the othercompounds. a and c arise from the valence of the O-s orbitals andCa-p orbitals, while the b peak corresponds to Ca-2p state. Thisresult is in good agreement with those obtained by the OLCAO(orthogonalized linear combinations of atomic orbitals) method[19]. The e peak is mainly due to La-p states, while the remainder(d, f and g) arises from the s orbitals of F, O and La atoms,respectively. It is important to note that at about �17.5 eV, anadditional peak (f) appears only for the substituted compounds. Todetermine the origin of this peak, the calculations were also carriedout for Ca10(SiO4)6F2 (not shown here) and La10(PO4)6F2 com-pounds. Fig. 6 displays the partial density of states (PDOS) of the O-s orbitals of these compounds. As seen from this figure, the f peakintensity increases with x. This increase is mainly due to theinteraction between the La-p and O-2s orbitals, which differ to thatbetween the Ca-2p and O-2s orbitals.
To study the variation of chemical bonding induced by La andSi, the electron charge density distributions were analyzed. Asdepicted in Fig. 7 the contour plots of the charge density for x = 0,1 and 6 compounds on (1 1 0) plane represent the effect of thesubstituted elements on nearest neighboring O atoms. As seen inFig. 7, the charge densities of O(1), O(2) and O(3) are not sphericalbut exhibit oval shapes pointing to the neighboring P/Si ions. Thisis in agreement with the (partial) covalent nature of the P–Obonds and the more ionic character of Ca–O bond. This can beimagined from the strong orbital overlaps between P/Si and O inthe upper part of the valence band, as shown in Fig. 4. In the caseof La10(PO4)6F2 (not shown here), the electron densities of La3+
and the adjacent oxygen of PO4 exhibit a slight overlap which isquite similar to that around the Ca–O ionical bonded. However,for Ca10�xLax(PO4)6�x(SiO4)xF2, more electrons are accumulatedbetween La3+(2) and the adjacent oxygen O(2), indicating thatthe Ca/La(2)-O(2) bond has a partial covalent character whichincreases with the rise of the La content. Furthermore, it can beseen that the overlap of the La(2)-d orbital with O(2)-2p is greaterthan that of the La(1)-d orbital with O-2p, and Ca(2)-d orbitalwith O-2p, which indicates that the degree of covalency betweenLa(2) and O(2) is stronger than that between, La(1) and O(2), andCa and O. This result implies that the incorporation of La3+ intothe apatite structure induced especially, at the level of the S(2)
sites, a certain disorder which is responsible for the decrease inthe stability of the apatite, confirming the calculated formationenthalpies.
4. Conclusion
A series of britholites Ca10�xLax(PO4)6�x(SiO4)xF2 (0 � x � 6)was prepared by a solid-state reaction and investigated by the X-ray diffraction. A structural investigation indicated that all thesecompounds adopt P63/m space group structure. Ab initio DFTband-structure calculations were carried out, in which allstructures were fully relaxed. The calculated structural parametersare in good agreement with the experimental ones. Detailedanalyses of cation–oxygen bond distributions, cation–cationdistances, and angles were made in order to provide an atomic-scale interpretation of the observed structural modifications.Analysis of Ca(2)/La(2)O6F-polyhedron suggests that La3+ inducesmore distortion at site 2 than site 1, which may, be responsible forthe decrease in the stability of apatite. Band structure calculationshows that all these materials are wide band gap insulators andtheir values decrease with increasing the x content.
Acknowledgements
The calculations have been carried out using computationresources from the ‘University de Bourgogne’. We thank theUniversity of ‘Bourgogne’ for providing us time.
References
[1] I.L. Denry, J.A. Holloway, R.J. Nakkula, J.D. Walters, J. Biomed. Mater. Res. B: Appl.Biomater. 75 (2005) 18.
[2] H.-W. Kim, S.-Y. Lee, C.-J. Bae, Y.-J. Noh, H.-E. Kim, H.-M. Kim, J.-S. Kim, Biomater-ials 24 (2003) 3277.
[3] Q.H. Ma, S.J. Traina, T.J. Logan, J.A. Ryan, Environ. Sci. Technol. 28 (1994) 408.[4] S.A. Payne, L.K. Smith, L.D. DeLoach, W.L. Kway, J.B. Tassano, W.F. Krupke, IEEE J.
Quantum Electron. 30 (1994) 170.[5] M.A. Scott, H.G. Gallagher, T.P.J. Han, B. Henderson, J. Cryst. Growth 172 (1997)
190.[6] L. Boyer, J. Carpenaa, J.L. Lacout, Solid State Ion. 95 (1997) 121.[7] R. Bros, J. Carpena, V. Sere, A. Beltritti, Radiochim. Acta 74 (1996) 277.[8] O. Terra, N. Dacheux, F. Audubert, R. Podor, J. Nucl. Mater. 352 (2006) 224.[9] O. Terra, F. Audubert, N. Dacheux, C. Guy, R. Podor, J. Nucl. Mater. 354 (2006) 49.
[10] O. Terra, F. Audubert, N. Dacheux, C. Guy, R. Podor, J. Nucl. Mater. 366 (2007) 70.

H. Njema et al. / Materials Research Bulletin 51 (2014) 210–216216
[11] N. Dacheux, N. Clavier, A.C. Robisson, O. Terra, F. Audubert, J.E. Lartigue, C. Guy, C.R. Chimie 7 (2004) 1141.
[12] J. Rakovan, R.J. Reeder, E.J. Elzinga, D.J. Cherniak, C.D. Tait, D.E. Morris, Environ. Sci.Technol. 36 (2002) 3114.
[13] A. Meldrum, L.M. Wang, R.C. Ewing, Nucl. Instrum. Methods Phys. Res. B 116(1996) 220.
[14] W.J. Weber, L.M. Wang, Nucl. Instrum. Methods Phys. Res. B 91 (1994) 63.[15] A. Peeters, E.A.P. De Maeyer, C. Van Alsenoy, R.M.H. Verbeeck, J. Phys. Chem. B 101
(1997) 3995.[16] D. Zahn, O. Hochrein, Phys. Chem. Chem. Phys. 5 (2003) 4004.[17] R. Astala, M.J. Stott, Chem. Mater. 17 (2005) 4125.[18] V.L. Achille, L. De Windt, M. Defranceschi, Comput. Mater. Sci. 10 (1998) 346.[19] P. Rulis, L. Ouyang, W.Y. Ching, Phys. Rev. B 70 (2004) 155104.[20] K. Matsunaga, A. Kuwabara, Phys. Rev. B 75 (2007) 014102.[21] K. Matsunaga, J. Chem. Phys. 128 (2008) 245101.[22] J.A.L. Rabone, N.H. de Leeuw, J. Comput. Chem. 27 (2006) 253.
[23] N.H. de Leeuw, Phys. Chem. Chem. Phys. 4 (2002) 3865.[24] L. Calderın, M.J. Stott, A. Rubio, Phys. Rev. B 67 (2003) 134106.[25] C. Meis, J.D. Gale, L. Boyer, J. Carpena, D. Gosset, J. Phys. Chem. A 104 (2000) 5380.[26] P. Giannozzi, et al. J. Phys.: Condens. Matter 21 (2009) 395502.[27] J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865.[28] H.J. Monkhorst, J.D. Pack, Phys. Rev. B 13 (1976) 5188.[29] S.R. Billeter, A. Curioni, W. Andreoni, Comput. Mater. Sci. 27 (2003) 437.[30] J. R. Carvajal, Satellite Meeting on Powder Diffraction of the XV Congress of the
IUCr. Book of Abstracts, Toulouse, France, 1990, p. 127.[31] R.D. Shannon, Acta Crystallogr. A 32 (1976) 751.[32] L. Pauling, Am. Mineral. 65 (1980) 321.[33] A. Chartier, C. Meis, J.D. Gale, Phys. Rev. B 64 (2001) 085110.[34] K. Boughzala, E. Ben Salem, F. Kooli, P. Gravereau, K. Bouzouita, J. Rare Earth 26
(2008) 483.[35] D.R. Lide, Handbook of Chemistry and Physics, 85, CRC Press, Boca Raton (FL),
London, New York, 2004p. 52.




















