
Electronic structure of copper phthalocyanine: A comparative density functional theory study
Upload
independentCategory
view
0download
0
ANALYSIS OF VIBRATIONAL, STRUCTURAL, AND ELECTRONICPROPERTIES OF RIVASTIGMINE BY DENSITY FUNCTIONAL THEORY
O. Prasad,* L. Sinha, N. Misra, V. Narayan, N. Kumar, and A. Kumar
UDC 539.19
The present work deals with the structural, electronic, and vibrational analysis of rivastigmine. Rivastigmine,an antidementia medicament, is credited with significant therapeutic effects on the cognitive, functional, andbehavioural problems that are commonly associated with Alzheimer’s dementia. For rivastigmine, a number ofminimum energy conformations are possible. The geometry of twelve possible conformers has been analyzedand the most stable conformer was further optimized at a higher basis set. The electronic properties and vi-brational frequencies were then calculated using a density functional theory at the B3LYP level with the 6-311+G(d, p) basis set. The different molecular surfaces have also been drawn to understand the activity ofthe molecule. A narrower frontier orbital energy gap in rivastigmine makes it softer and more reactive thanwater and dimethylfuran. The calculated value of the dipole moment is 2.58 debye.
Keywords: normal modes, frontier orbital energy, molecular electrostatic potential surface.
Introduction. Alzheimer’s disease is a progressive neurodegenerative disease and the most prevalent cause ofdementia with aging [1]. Symptomatic pharmacological treatment of AlzheimerÒs disease (AD) is mainly based on theuse of acetylcholinesterase (AchE) inhibitors. Rivastigmine, an antidementia medicament, has beneficial effects on cog-nitive, functional, and behavioral symptoms of the disease [2, 3]. These inhibitors have now become one of the mostactively investigated classes of compounds in the search for an effective treatment of AD [4–14]. Rivastigmine has anumber of minimum energy conformers, say, by the location of methyl and ethyl groups at the N site as regard to theposition of the other N site of a dimethylamine group. Therefore it becomes essential to explore and analyze all theexpected conformers and to carry out the vibrational study of rivastigmine as such. The vibrational analysis also yieldsdetailed information about intramolecular interactions in the molecular functional and fingerprint regions. The aim ofthe present theoretical study is to investigate the structural, electronic, and vibrational properties of rivastigmine due toits biological and medical importance. Structures of twelve possible conformers were first analyzed and compared atthe DFT/6-311+G(d) level. The conformer with the least ground state energy was further analyzed at the higher basisset 6-311+G(d, p), and harmonic frequencies were then calculated. The reported geometry and molecular propertiessuch as equilibrium energy, highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital(LUMO) band gaps, a molecular electrostatic potential energy map, a dipole moment, and vibrational frequencies havealso been used to understand the properties and activity of the molecule under investigation.
Computational Details. All the twelve possible conformers of rivastigmine have been analyzed with the den-sity functional theory (DFT) employing Becke’s three-parameter hybrid exchange functionals [15, 16] with Lee-Yang-Parr functionals (B3LYP) [17, 18] using the Gaussian 03 program package [19]. The vibrational frequencies have alsobeen calculated and scaled down by a factor of 0.9679 [20]. By combining the results of the GaussView programpackage [21] with symmetry considerations, vibrational frequency assignments were made with a high degree of accu-racy. For the vibrational assignments to be precise, the normal modes have also been analyzed using the VEDA 4 pro-gram for vibrational energy distribution analysis [22].
Physics Department, University of Lucknow, Lucknow, 226007, India; e-mail: [email protected],[email protected] Published in Zhurnal Prikladnoi Spektroskopii, Vol. 77, No. 4, pp. 507–516, July–August,2010. Original article submitted October 23, 2010.
Journal of Applied Spectroscopy, Vol. 77, No. 4, 2010
0021-9037/10/7704-0468 ©2010 Springer Science+Business Media, Inc.468
∗To whom correspondence should be addressed.

Results and Discussion. Molecular geometry optimization scheme and energies. For rivastigmine, a number ofminimum energy conformations are expected. These conformers differ from one another by the dispositions of methyland ethyl groups at the N atom site (atom number 4, defined as N′) relative to their positions at the other N atom site(atom number 3) of the dimethylamine group. The twelve possible conformations of rivastigmine corresponding to thecis- or trans-, or nearly perpendicular (gauche-) orientations of different methyl groups, have been analyzed and com-pared. The geometries of these possible conformers are shown in Fig. 1. The analysis of each of the possible twelveconformers was first carried out using the density functional theory and employing the 6-311+G(d) basis set. Theground state energies of the twelve possible conformers (A to L) are given in Table 1. The most stable conformer (L)has energy differing from that of the next stable conformer (J) by a value of 6230 cm–1/0.7728 eV. The most stableconformer (L) was further optimized at the same level of the theory but at the higher basis sets 6-311+G(d, p), andthe normal mode frequencies were also calculated. An optimized molecular structure of the most stable conformer to-gether with the numbering scheme of atoms is shown in Fig. 2, and the optimized parameters are given in Table 2.The skeleton of the optimized molecule is nonplanar. Since the diffuse functions provide an accurate description ofmolecules with unshared pairs of electrons, the choice of the basis set was based on the fact that rivastigmine also car-ries electron-pair rich atoms like oxygen and nitrogen. The Mulliken charge analysis indicates that while both the oxy-gen atoms and one of the nitrogen atoms N4 are negatively charged, the other nitrogen atom N3 at the dimethylaminesite is positively charged (Table 3). As the calculated vibrational spectra have no imaginary frequency, the optimizedgeometry is confirmed to be located at the local minima on a potential energy surface. The ring C–C bond distancesare between 1.39 and 1.40 A° , and in other parts of the molecule the C–C bond distances vary from 1.53 to 1.54 A° .The calculated C–N bond lengths are found to be shorter than those for C–C and lie between 1.46 and 1.47 A° . Theoptimized ring C–H bond lengths are 1.08 A° . The C–O bond lengths vary between 1.38 and 1.40 A° , and the valuefor C=O is 1.21 A° (refer to Table 4). Most of the bond distances are very similar to the standard bond lengths [23].The angle between the plane of the benzene ring and the plane containing the C5 and C6 carbon atoms and the N3nitrogen atom is τN3–C5–C6–C8 = 135.57o, while the angle between the planes of the benzene ring and carbonyl is con-siderably smaller (τC8–C12–C5–N3 = 62.00o). Owing to the smaller interatomic distance, calculated to be 2.32 A° , there ex-ists a possibility of a strong hydrogen bond between the partially negative oxygen atom O2 of the carbonyl group andthe hydrogen atom H37 of the methyl group attached to nitrogen N4. The five-member ring resulting from the forma-tion of intramolecular hydrogen bonding may also contribute a little towards the stability of the whole molecule as
TABLE 1. Theoretically Computed Ground State Energy of Possible Conformers of Rivastigmine at B3LYP/6-311+G(d)
ConformersGround state
energy in hartree
Ground stateenergy in cm–1,
relative to conformer LConformers
Ground stateenergy in hartree
Ground state energyin cm–1, relative to conformer L
A –806.8079 9810 G –806.8062 10,180B –806.8095 9460 H –806.8057 10,290C –806.8088 9610 I –806.8225 6610D –806.8083 9720 J –806.8242 6230E –806.8053 10,380 K –806.8236 6360F –806.8070 10,010 L –806.8526 0
TABLE 2. Theoretically Computed Ground State Optimized Parameters of Rivastigmine
Parameters B3LYP/6-311+G(d, p)
Energy (in hartree) –806.9223HOMO (in eV) –5.9899LUMO (in eV) –0.4161
Frontier orbital energy gap (in eV) 5.5738Dipole moment (in debye) 2.58
469

Fig. 2. Optimized structure of the most stable conformer of rivastigmine.
Fig. 3. Frontier orbitals and surfaces of rivastigmine.
Fig. 1. Different conformers of rivastigmine.
470

such. On the basis of compiled data for a large number of hydrogen binding C–H...O sites, Desiraju et al. [24] haveconcluded that even sites that are as far out as 3.0 A° can be safely viewed as "weak" hydrogen-bonded ones, with agreater contribution to packing forces than the simple van der Waals attractions they appear to be.
Electronic properties. The electronic structure of the most stable conformer in the gas phase has been calcu-lated with DFT using the 6-311+G(d, p) basis set with an exchange-correlation functional (B3LYP). The basic elec-tronic parameters related to the orbitals in a molecule are the HOMO and LUMO and their resulting energy gap.These orbitals not only determine the way the molecule interacts with other species, but their energy gap (frontier or-bital gap) helps to characterize the chemical reactivity and kinetic stability of the molecule. The plots of the HOMO,LUMO, total electron density (TED) for rivastigmine as well as that of the electrostatic potential (ESP) mapped on anisodensity surface are shown in Fig. 3. It is seen from the figure that both the HOMO and LUMO have nodes, and,moreover, the nodes in the orbitals are placed symmetrically. The HOMO is found to be concentrated mainly over theN3 site and around its surrounding groups, but the LUMO lies mainly over the benzene ring. The calculated value ofthe frontier orbital energy gap, 5.5738 eV, in the case of rivastigmine (refer to Table 2) makes of it a soft and morepolarizable molecule as compared to water and dimethylfuran (DMF). These have been taken as reference molecules,as both are eminently polar solvents due to their high dielectric constant and feature the electron donor property andability to form complexes. Their frontier orbital energy gaps (9.3088 and 6.8054 eV respectively) have been calculatedat the same level of theory using the same basis sets. The low frontier orbital gap is also associated with a highchemical reactivity and low kinetic stability [25].
The ESP is a physical property of a molecule related to how a molecule is first "seen" or "felt" by anotherapproaching species. A portion of a molecule that has a negative electrostatic potential is susceptible to an electrophilicattack — the more negative the better. The ESP, which is related to the electronegativity and the partial charges onthe different atoms of the molecule, when plotted on the isodensity surface of the molecule is termed the MEP. TheMEP and the TED are important parameters, and their study leads to a better understanding of complex biologicalprocesses involving the charge-dipole, dipole-dipole, and quadrupole-dipole interactions. As seen from Fig. 3d, theTED surface of rivastigmine depicts a uniform distribution. Red and blue areas in the MEP map refer to the regions
TABLE 3. Optimized Atomic Partial Charges in Rivastigmine at B3LYP/6-311+G(d, p) Level
Atom Atomic charge Atom Atomic charge
O1 –0.01 H21 0.17O2 –0.28 H22 0.15N3 0.13 H23 0.13N4 –0.11 H24 0.14C5 0.16 H25 0.16C6 0.80 H26 0.14C7 –0.75 H27 0.12C8 0.17 H28 0.14C9 –0.28 H29 0.13C10 –0.40 H30 0.12C11 –0.27 H31 0.13C12 –1.14 H32 0.12C13 –0.42 H33 0.14C14 0.30 H34 0.18C15 –0.30 H35 0.14C16 –0.11 H36 0.15C17 –0.25 H37 0.18C18 –0.34 H38 0.14H19 0.10 H39 0.13H20 0.16 H40 0.15
471

TABLE 4. Optimized Parameters of Rivastigmine at B3LYP/6-311+G(d, p) Level
Parameter Value Parameter Value Parameter Value
R(O1,C12) 1.40 A(C6,C9,H24) 119.10 D(C10,N3,C11,H30) –66.99R(O1,C16) 1.38 A(C13,C9,H24) 120.51 D(C16,N4,C15,C18) 87.36R(O2,C16) 1.21 A(N3,C10,H25) 110.45 D(C16,N4,C15,H33) –151.14R(N3,C5) 1.47 A(N3,C10,H26) 109.47 D(C16,N4,C15,H34) –35.73
R(N3,C10) 1.46 A(N3,C10,H27) 112.44 D(C17,N4,C15,C18) –87.14R(N3,C11) 1.46 A(H25,C10,H26) 108.16 D(C17,N4,C15,H33) 34.36R(N4,C15) 1.47 A(H25,C10,H27) 108.12 D(C17,N4,C15,H34) 149.77R(N4,C16) 1.36 A(H26,C10,H27) 108.07 D(C15,N4,C16,O1) 2.91R(N4,C17) 1.46 A(N3,C11,H28) 111.16 D(C15,N4,C16,O2) –177.55R(C5,C6) 1.53 A(N3,C11,H29) 108.83 D(C17,N4,C16,O1) 177.36R(C5,C7) 1.54 A(N3,C11,H30) 112.93 D(C17,N4,C16,O2) –3.10
R(C5,H19) 1.11 A(H28,C11,H29) 107.56 D(C15,N4,C17,H35) 58.81R(C6,C8) 1.40 A(H28,C11,H30) 108.42 D(C15,N4,C17,H36) –61.46R(C6,C9) 1.40 A(H29,C11,H30) 107.75 D(C15,N4,C17,H37) 178.75
R(C7,H20) 1.09 A(O1,C12,C8) 121.17 D(C16,N4,C17,H35) –115.98R(C7,H21) 1.09 A(C8,C12,C14) 121.27 D(C16,N4,C17,H36) 123.75R(C7,H22) 1.09 A(C9,C13,C14) 120.53 D(C16,N4,C17,H37) 3.96R(C8,C12) 1.39 A(C9,C13,H31) 119.95 D(N3,C5,C6,C8) 135.57R(C8,H23) 1.08 A(C14,C13,H31) 119.52 D(N3,C5,C6,C9) –47.71R(C9,C13) 1.39 A(C12,C14,C13) 118.78 D(C7,C5,C6,C8) –100.10R(C9,H24) 1.08 A(C12,C14,H32) 119.63 D(C7,C5,C6,C9) 76.63R(C10,H25) 1.09 A(C13,C14,H32) 121.60 D(H19,C5,C6,C8) 15.85R(C10,H26) 1.09 A(N4,C15,C18) 113.70 D(H19,C5,C6,C9) –167.43R(C10,H27) 1.11 A(N4,C15,H33) 106.63 D(N3,C5,C7,H20) 172.55R(C11,H28) 1.09 A(N4,C15,H34) 108.73 D(N3,C5,C7,H21) 53.59R(C11,H29) 1.09 A(C18,C15,H33) 110.09 D(N3,C5,C7,H22) –68.06R(C11,H30) 1.11 A(C18,C15,H34) 110.14 D(C6,C5,C7,H20) 49.12R(C12,C14) 1.39 A(O1,C16,O2) 1.40 D(C6,C5,C7,H21) –69.84R(C13,C14) 1.40 A(O1,C16,N4) 1.38 D(C6,C5,C7,H22) 168.51R(C13,H31) 1.08 A(O2,C16,N4) 1.21 D(H19,C5,C7,H20) –66.84R(C14,H32) 1.08 A(N4,C17,H35) 1.47 D(H19,C5,C7,H21) 174.20R(C15,C18) 1.53 A(N4,C17,H36) 1.46 D(H19,C5,C7,H22) 52.55R(C15,H33) 1.09 A(O1,C12,C14) 117.41 D(C5,C6,C8,C12) 175.93R(C15,H34) 1.09 A(H33,C15,H34) 107.31 D(C5,C6,C8,H23) –3.21R(C17,H35) 1.10 A(N4,C17,H37) 109.00 D(C9,C6,C8,C12) –0.86R(C17,H36) 1.10 A(H35,C17,H36) 108.57 D(C9,C6,C8,H23) –180.00R(C17,H37) 1.09 A(H35,C17,H37) 108.92 D(C5,C6,C9,C13) –176.50R(C18,H38) 1.09 A(H36,C17,H37) 109.17 D(C5,C6,C9,H24) 4.48R(C18,H39) 1.09 A(C15,C18,H38) 110.11 D(C8,C6,C9,C13) 0.26R(C18,H40) 1.09 A(C15,C18,H39) 111.52 D(C8,C6,C9,H24) –178.77
A(C12,O1,C16) 118.89 A(C15,C18,H40) 110.66 D(C6,C8,C12,O1) 176.22A(C5,N3,C10) 111.45 A(H38,C18,H39) 107.83 D(C6,C8,C12,C14) 0.76A(C5,N3,C11) 113.75 A(H38,C18,H40) 108.14 D(H23,C8,C12,O1) –4.63
A(C10,N3,C11) 109.91 A(H39,C18,H40) 108.47 D(H23,C8,C12,C14) 179.91A(C15,N4,C16) 123.63 D(C16,O1,C12,C8) 62.00 D(C6,C9,C13,C14) 0.46
472

of negative and positive potentials and correspond to the electron-rich and electron-poor regions, respectively, whereasthe green color signifies the neutral electrostatic potential. The MEP surface provides necessary information about thereactive sites. The MEP of rivastigmine reveals some interesting features. The region around the oxygen atom in rivas-tigmine reflects the large concentration of electrons and is responsible for electrophillic interactions/activity, whereasthe other regions seem to present almost neutral potential as represented by the green region.
Rivastigmine is a polar molecule, and its calculated dipole moment value is 2.58 debye. Comparison of thecalculated dipole moment of rivastigmine with those of water and DMF shows that it is slightly higher than the formerbut relatively smaller than the latter. (Dipole moment values for DMF and water are 4.24 and 2.16 debye, respectively,at the same level of calculations i.e., B3LYP/6-311G+(d, p).)
Thermal properties. Standard thermodynamic functions such as free energy, constant volume heat capacity CV,and entropy S have also been calculated and are given in Table 5. All the thermodynamic parameters reported in thispaper correspond to the 1 atm and 298 K reference state. These functions can provide helpful information for furtherthermal study of the title molecule.
Vibrational spectra. The optimized molecular structure of rivastigmine belongs to the C1 point group as itdoes not display any special symmetries. As a result, all the normal modes are both infrared and Raman active. Atotal of 114 (3-N6) normal modes of vibrations have been calculated theoretically as the title molecule has 40 atoms.The vibrational frequencies were computed and the spectra plotted as a prediction (refer to Table 6 and Fig. 4).
It is well known that ab initio and DFT methods systematically overestimate the vibrational wavenumbers.These discrepancies can be corrected by computing anharmonic correlations explicitly, by introducing a scaled field[26], or even by direct scaling the calculated wavenumbers with a proper factor [20]. The vibrational wavenumberswere calibrated accordingly with scaling by 0.9679 for DFT at B3LYP. The scaled vibrational frequencies are listed in
TABLE 4. (Continued)
Parameter Value Parameter Value Parameter Value
A(C15,N4,C17) 117.58 D(C16,O1,C12,C14) –122.37 D(C6,C9,C13,H31) –179.76A(C16,N4,C17) 118.57 D(C12,O1,C16,O2) –0.10 D(H24,C9,C13,C14) 179.47A(N3,C5,C6) 110.96 D(C12,O1,C16,N4) 179.46 D(H24,C9,C13,H31) –0.74A(N3,C5,C7) 112.49 D(C10,N3,C5,C6) –176.69 D(O1,C12,C14,C13) –175.67
A(N3,C5,H19) 109.57 D(C10,N3,C5,C7) 174.57 D(O1,C12,C14,H32) 3.98A(C6,C5,C7) 108.90 D(C10,N3,C5,H19) 55.25 D(C8,C12,C14,C13) –0.04
A(C6,C5,H19) 107.38 D(C11,N3,C5,C6) 171.92 D(C8,C12,C14,H32) 179.60A(C7,C5,H19) 107.35 D(C11,N3,C5,C7) 49.64 D(C9,C13,C14,C12) –0.56A(C5,C6,C8) 119.90 D(C11,N3,C5,H19) –69.68 D(C9,C13,C14,H32) 179.80A(C5,C6,C9) 120.92 D(C5,N3,C10,H25) 56.56 D(H31,C13,C14,C12) 179.65A(C8,C6,C9) 119.10 D(C5,N3,C10,H26) 175.55 D(H31,C13,C14,H32) 0.01
A(C5,C7,H20) 109.32 D(C5,N3,C10,H27) –64.31 D(N4,C15,C18,H38) 177.34A(C5,C7,H21) 110.61 D(C11,N3,C10,H25) –176.39 D(N4,C15,C18,H39) 57.67A(C5,C7,H22) 112.08 D(C11,N3,C10,H26) –57.40 D(N4,C15,C18,H40) –63.17
A(H20,C7,H21) 108.14 D(C11,N3,C10,H27) 62.74 D(H33,C15,C18,H38) 57.79A(H20,C7,H22) 107.75 D(C5,N3,C11,H28) –63.36 D(H33,C15,C18,H39) –61.88A(H21,C7,H22) 108.83 D(C5,N3,C11,H29) 178.36 D(H33,C15,C18,H40) 177.28A(C6,C8,C12) 119.93 D(C5,N3,C11,H30) 58.76 D(H34,C15,C18,H38) –60.35A(C6,C8,H23) 120.45 D(C10,N3,C11,H28) 170.89 D(H34,C15,C18,H39) 179.99A(C12,C8,H23) 119.62 D(C10,N3,C11,H29) 52.61 D(H34,C15,C18,H40) 59.15A(C6,C9,C13) 120.38
Note. Bond lengths (R) are in A° , bond angles (A) and dihedrals (D) are in degrees.
473

TABLE 6. Theoretical Unscaled and Scaled Wavenumbers (in cm–1) of Rivastigmine
Calculated(unscaled)
wavenumber
Scaled wavenumber
Description of dominant modes in order of decreasing frequency
3196 w 3093 νs (CH)R
3190 vw 3088 νas (CH)R
3189 vw 3087 νas (CH) R
3169 vw 3067 νas (CH) R
3156 vw 3055 νas (CH) {methyl}4
3124 w 3024 νas (CH){ethyl}
3120 w 3019 νas (CH) {methyl}1,3
3116 w 3016 νas (CH) {methyl}2
3109 vw 3009 νas (CH) {methyl}1,3
3105 w 3005 νas (CH) {ethyl}
3102 w 3002 νas (CH) {methyl}1,3
3089 vw 2989 νas (CH) {ethyl}
3070 m 2971 νas (CH) {methyl}2,3
3065 w 2967 νas (CH) {methyl}2,3
3060 m 2962 νas (CH) {methyl}4
3043 m 2945 νas (CH) {ethyl}
3034 w 2937 νs (CH) {methyl}1
3031 w 2934 νs (CH) {ethyl}
3015 m 2918 νs (CH) {methyl}4
2914 s 2820 νs (CH) {methyl}2,3 + ν (CH)adjR
2902 s 2809 νas (CH) {methyl}2,3
2883 w 2790 ν (CH)adjR1777 vs 1720 ν (C=O)1644 vw 1591 ν(C–C)R + Φ(H–C–C)R1630 w 1578 ν(C–C)R + Φ(H–C–C)R + Φ(H–C–C)adjR1524 w 1475 {methyl}4 deformation + Φ(H–C–C)R1519 vw 1470 {methyl}1,2,3 bending1516 w 1468 Φ(H–C–C)R + {methyl}1,2,3,4 deformation1509 vw 1461 {methyl}1,2,3 deformation1506 vw 1458 {methyl}4 deformation + {ethyl} deformation1502 vw 1456 {ethyl} deformation + {methyl}4 deformation1501 vw 1453 {methyl}1,2,3 deformation1500 vw 1452 {methyl}1,2,3 deformation1494 vw 1446 {ethyl} deformation + {methyl}4 deformation1493 vw 1445 {ethyl} deformation + {methyl}1,2,3,4 deformation1492 vw 1444 {methyl}1,2,3,4 deformation + {ethyl} deformation1488 vw 1440 {methyl}1,2,3 deformation1473 vw 1426 {methyl}2,3 umbrella bending1468 vw 1421 {methyl}2,3 umbrella bending + Φ(H–C–C)R + ν(C–C)R1448 s 1402 {methyl}4 umbrella bending
474

TABLE 6. (Continued)
Calculated(unscaled)
wavenumber
Scaled wavenumber
Description of dominant modes in order of decreasing frequency
1441 vw 1395 {methyl}2,3 umbrella bending1432 vs 1386 ν(C–N′) + γ(CH2)ethyl + {methyl}4 umbrella bending1411 vw 1366 ω(CH2)ethyl + ethyl(CH3) umbrella bending1405 vw 1360 {methyl}1 umbrella bending1393 vw 1348 ω(CH2)ethyl + ethyl(CH3) umbrella bending1366 vw 1322 ω(C–H) adj R + Φ(H–C–C)R + ν(C–C)R1352 w 1309 ω(C–H) adj R1340 vw 1297 Φ(H–C–C)R + ν(C–C)R + ω(C–H) adj R1311 s 1269 γ(CH2)ethyl + ν(C–N′)1304 vs 1262 Φ(H–C–C)R1291 vw 1250 r{methyl}1,2,3 + ν(C–N)1272 w 1231 ν(C–N′) + Φ(C–C–C)R + Φ(H–C–C)R + ω(C–H) adj R + r{methyl}4
1251 vs 1211 Ring deformation + ω(C–H) adj R + ν(C–O) + γ(CH2)ethyl1235 w 1195 Ring deformation + r {methyl}1,2,3 + Φ(C–N–C)1183 vs 1145 Φ(H–C–C)R1177 w 1139 Φ(H–C–C)R + r {methyl}2,3
1172 vs 1134 Φ(H–C–C)R + ν(C–O) + r {methyl}4 + ethyl deformation1162 vs 1125 Φ(H–C–C)R + r{methyl}4 + ethyl (CH2 rocking) + Φ(O–C–O) + Φ(O–C–N)1150 s 1113 r {methyl}4
1118 vw 1082 r {methyl}2,3
1105 vw 1070 r {methyl}1,2,4 + r ethyl + Φ(H–C–C)R1104 w 1069 r {methyl}4 + r {ethyl}+ Φ(H–C–C)R1099 w 1064 r {methyl}1,2,3 + Φ(H–C–C)add R + Φ(H–C–C)R1090 vw 1055 r{methyl}4 + r ethyl1084 vw 1049 Φ(H–C–C) R + r {methyl}1,2,31059 vw 1025 r {methyl}2,3
1038 w 1005 r {ethyl} + r {methyl}4 + Φ(H–C–C)R1028 w 995 r {methyl}1,2,3 + Ψ (H–C–N)1016 vw 983 Φ(C–C–C) R993 vw 961 C–H out-of-plane Ring wag974 vw 943 r {methyl}1,2,3956 w 925 r {ethyl} + r {methyl}4928 w 898 C–H out-of-plane Ring wag916 w 887 C–H out-of-plane Ring wag + r{methyl}1
902 vw 873 C–H out-of-plane Ring wag834 w 807 C–H out-of-plane Ring wag + r {ethyl}812 w 786 C–H out-of-plane Ring wag+ r {methyl}1 + Ψ (H–C–N)799 w 773 r {ethyl} + C–H out-of-plane Ring wag
789 vw 764 C–H out-of-plane Ring wag + r {ethyl}761 w 737 Ψ (O=C–O) + Ψ (N–C=O) + Ψ (C–N–C) + Φ(C–C–C) R
758 vw 734 Ψ (O=C–O) + Ψ (N–C=O) + Ψ (C–N–C) + C–H out-of-plane ring wag713 w 690 C–H out-of-plane ring wag
475

Table 6. The description of vibrational modes has been done based on relative intensities, line shapes, the VEDA 4program, and the animation option of the GaussView 3.07.
CH3 Group Modes. Four methyl groups are present in rivastigmine, out of which two groups are attached tothe first nitrogen atom (N), one to the second nitrogen atom (N′), and the fourth to the carbon atom (C7) to whichdimethylamine is also connected. The methyl group shows several characteristic fundamental vibrations correspondingto asymmetric and symmetric stretches and to bending, rocking, and torsion modes. The CH3 antisymmetric stretchingvibrations are generally observed in the region of 2940–2980 cm–1, while the symmetric stretching vibrations usuallyappear between 2850 and 2890 cm–1. In rivastigmine the frequency range for antisymmetric CH3 modes is found to be3060–2960 cm–1, while the frequencies of symmetric stretching vibrations are calculated to lie in the range of 2940–2810 cm–1. Methyl deformation modes appear as mixed modes in the fingerprint region. The methyl bending modescalculated for the present case are in the frequency range of 1480–1440 cm–1. These bending modes are in generalmixed with ethyl bending and ring (H–C–C) bending modes (Table 6). The methyl umbrella bending is calculated tobe in the frequency region below 1430 cm–1, while methyl rocking modes lie below 1260 cm–1.
C=O and C–N Stretches. The appearance of a strong band in the IR spectra around 1700 cm–1 shows thepresence of the carbonyl group in the molecule. This is due to the C=O stretch. The frequency of the stretch due tothe carbonyl group depends mainly on the force constant, which in turn depends upon inductive, conjugative, field,and steric effects. The effect of electron-withdrawing oxygen and nitrogen atoms attached to the carbonyl group is toincrease the strength of the C=O bond, hence in the case of rivastigmine the vibration occurs at a relatively higherfrequency. The calculated mode at 1720 cm–1 is correctly identified as the pure C=O stretching mode. On the otherhand, the modes calculated to be in the fingerprint region at 1211 and 1134 cm–1 represent mixed modes having asubstantial contribution from the C–O stretch The identification of C–N stretching vibrations is not an easy task dueto possible mixing with other vibrations. As expected, the C–N stretching modes in the case of rivastigmine are cal-culated to appear at 1386, 1269, and 1250 cm–1 and found to be higher in frequency as compared to the C–O ones.
Ring Modes. The C–H stretching in the benzene ring is assigned at 3093, 3088, 3087, and 3067 cm–1. Thecalculated modes involving a major contribution from C–H in-plane bending are at 1591, 1578, 1468, 1297, 1262, and1145 cm–1. The calculated wavenumbers of 898, 873, and 690 cm–1 are identified with the pure C–H out-of-planewagging modes. The modes calculated at 983, 618, and 593 cm–1 arise from the C–C–C in-plane bending. The modes
TABLE 6. (Continued)
Calculated(unscaled)
wavenumber
Scaled wavenumber
Description of dominant modes in order of decreasing frequency
657 vw 636 Ring torsion mode + r {ethyl}638 vw 618 Φ(C–C–C)R + r {methyl}1
613 vw 593 Φ(C–C–C)R + Φ(N′–C–O)599 vw 580 Ring torsion + Φ(C–C–N′)514 vw 498 Ψ(H–C–N) + Ψ(H–C–C) {methyl+ethyl}480 vw 465 Ring torsion457 vw 442 τ(C–N′) + τ(C–C)ethyl448 vw 434 Ring torsion + τ (C–N) + τ(C–C)430 vw 416 τ(C–N) + τ(C–N′) + τ(C–C) + τ(C–O)413 vw 400 τ(C–N) + τ(C–N′) + τ(C–C) + τ(C–O) + τ(C–C)ethyl
Note. Abbreviations used here have following meanings: ν — stretching; νs — symmetric stretching; νas — asymmetric stretching;Φ — in-plane bending; Ψ — out-of-plane bending; τ — torsion; ω — wagging; γ — twisting; R — benzene ring; adj R — carbonatom adjacent to ring; {methyl}1,2,3,4 — four methyl groups involving carbon atoms numbered 7, 10, 11 and 17 respectively; N′— nitrogen atom attached to C=O group.
476
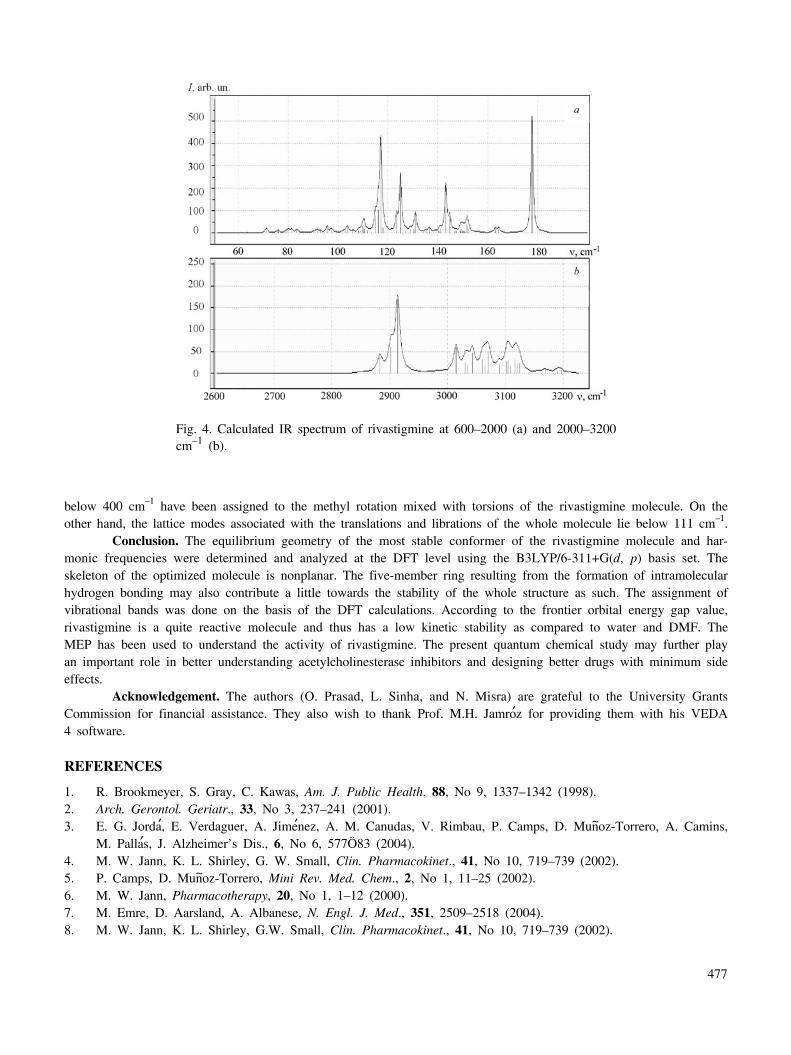
below 400 cm–1 have been assigned to the methyl rotation mixed with torsions of the rivastigmine molecule. On theother hand, the lattice modes associated with the translations and librations of the whole molecule lie below 111 cm–1.
Conclusion. The equilibrium geometry of the most stable conformer of the rivastigmine molecule and har-monic frequencies were determined and analyzed at the DFT level using the B3LYP/6-311+G(d, p) basis set. Theskeleton of the optimized molecule is nonplanar. The five-member ring resulting from the formation of intramolecularhydrogen bonding may also contribute a little towards the stability of the whole structure as such. The assignment ofvibrational bands was done on the basis of the DFT calculations. According to the frontier orbital energy gap value,rivastigmine is a quite reactive molecule and thus has a low kinetic stability as compared to water and DMF. TheMEP has been used to understand the activity of rivastigmine. The present quantum chemical study may further playan important role in better understanding acetylcholinesterase inhibitors and designing better drugs with minimum sideeffects.
Acknowledgement. The authors (O. Prasad, L. Sinha, and N. Misra) are grateful to the University GrantsCommission for financial assistance. They also wish to thank Prof. M.H. Jamro′z for providing them with his VEDA4 software.
REFERENCES
1. R. Brookmeyer, S. Gray, C. Kawas, Am. J. Public Health, 88, No 9, 1337–1342 (1998).2. Arch. Gerontol. Geriatr., 33, No 3, 237–241 (2001).3. E. G. Jorda′, E. Verdaguer, A. Jime′nez, A. M. Canudas, V. Rimbau, P. Camps, D. Mun~oz-Torrero, A. Camins,
M. Palla′s, J. Alzheimer’s Dis., 6, No 6, 577Ö83 (2004).4. M. W. Jann, K. L. Shirley, G. W. Small, Clin. Pharmacokinet., 41, No 10, 719–739 (2002).5. P. Camps, D. Mun~oz-Torrero, Mini Rev. Med. Chem., 2, No 1, 11–25 (2002).6. M. W. Jann, Pharmacotherapy, 20, No 1, 1–12 (2000).7. M. Emre, D. Aarsland, A. Albanese, N. Engl. J. Med., 351, 2509–2518 (2004).8. M. W. Jann, K. L. Shirley, G.W. Small, Clin. Pharmacokinet., 41, No 10, 719–739 (2002).
Fig. 4. Calculated IR spectrum of rivastigmine at 600–2000 (a) and 2000–3200cm–1 (b).
477

9. J. Corey-Bloom, R. Anand, J. Veach, Int. J. Geriatr Psychopharmacol., 1, 55–65 (1998).10. M. Ro
..sler, R. Anand, A. Cicin-Sain, Br. Med. J., 318, 633–640 (1999).
11. S. I. Finkel, Clin. Ther., 26, 980–990 (2004).12. M. Ro
..sler, W. Retz, P. Retz-Junginger, H. J. Dennler, Behav. Neurol., 11, No 4, 211–216 (1998).
13. A. A. N. de. Paula, J. B. L. Martins, R. Gargano, M. L. dos Santos, L. A. S. Romeiro, Chem. Phys. Lett., 446,304–308 (2007).
14. A. A. N. de Paula, J. B. L. Martins, M. L. dos Santos, L. de C. Nascente, R. Gargano, L. A. S. Romeiro, T. F.M. A. Areas, K. S. T. Vieira, N. F. Gamboa, N. G. Castro, R. Gargano, Eur. J. Med. Chem., 44, 3754–3759(2009).
15. W. Kohn, L. J. Sham, Phys. Rev., 140, A1133–A1138 (1965).16. A. D. Becke, J. Chem. Phys., 98, 5648–5652 (1993).17. C. Lee, W. Yang, R.G. Parr, Phys. Rev., B 37, 785 (1988).18. B. Miehlich, A. Savin, H. Stoll, H. Preuss, Chem. Phys. Lett., 157, 200–206 (1989).19. M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery
Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci,M. Cossi, G. Scalmani, N. Rega, G.A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda,J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox,H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev,A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J.Dannenberg, V. G. Zakrzewski, S. Dapprich, A.D. Daniels, M. C. Strain, O. Farkas, D. K. Malick,A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski,B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, Gaussian 03, Revision C01 (2003).
20. A. P. Scott, L. Random, J. Phys. Chem. US, 100, 16502–16513 (1996).21. R. Denington II, T. Keith, J. Millam, K. Eppinnett, W. L. Hovell, R. Gilliland, GaussView, Version 3.07,
Semichem Inc., Shawnee Mission, KS (2003).22. M. H. Jamro′z, Vibrational Energy Distribution Analysis: VEDA 4 program, Warsaw (2004).23. Introduction to Physical Chemistry, Ed. Marcus Frederick Charles Ladd, 3 rd ed., Cambridge University Press,
Chapter 6, 251 (1998).24. G. R. Desiraju, T. Steiner. The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University
Press, New York, 13 (1999).25. I. Fleming, Frontier Orbitals and Organic Chemical Reactions, John Wiley and Sons, New York (1976).26. P. Pulay, G. Fogarasi, G. Pongor, J. E. Boggs, A. Vargha, J. Am. Chem. Soc., 105, 7037–7047 (1983).
478
Copyright © 2022 FDOKUMEN


![Density functional theory study of electronic structure and spectra of tetraoxa[8]circulenes](https://static.fdokumen.com/doc/165x107/6335e140379741109e00d238/density-functional-theory-study-of-electronic-structure-and-spectra-of-tetraoxa8circulenes.jpg)

















