
ß ¤¤ ¥¡ ¤j
195
國 立 中 央 大 學 化學工程研究所 博 士 論 文 環氧樹脂/聚氧化二甲苯摻合體反應性、 相行為及機械性質之研究 指導教授:徐新興 博士 研 究 生:吳紹榮 中華民國八十九年六月
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of ß ¤¤ ¥¡ ¤j

國 立 中 央 大 學
化學工程研究所
博 士 論 文
環氧樹脂/聚氧化二甲苯摻合體反應性、
相行為及機械性質之研究
指導教授:徐新興 博士
研 究 生:吳紹榮
中華民國八十九年六月

中文摘要
關鍵詞:環氧樹脂;聚氧化二甲苯;摻合;相分離;反應動力;複合
材料
本研究第一部分是以熱熔融法摻合聚氧化二甲苯 (PPO) 於氰酸
酯硬化之環氧樹脂中來探討摻合體熱性質、機械性質與相行為。由
DSC實驗得知,摻合體的反應起始溫度隨著 PPO的含量增加而降低,
且反應之總放熱量卻隨著 PPO 增加而下降。環氧樹脂/PPO系統之硬
化遵守著自動催化反應動力模式,但是在此混合系統中之硬化反應速
率高於純環氧樹脂系統。環氧樹脂/PPO 系統中有較高的反應速率原
因是由於聚氧化二甲苯的末端 OH 基會催化環氧樹脂與氰酸酯的硬
化反應。FTIR 與 NMR分析顯示特性官能基(如環氧基、氰酸酯基)
的反應隨 PPO 增加而加速並且有 coreaction 產生,例如 cyanate-
hydroxyl addition (imidocarbonate, carbamate),epoxy-cyanate addition
(oxazolidinone)等。由 SEM與 DMA發現摻合體相型態與聚氧化二甲
苯的含量有關。當 PPO 含量低時,相分離是經由 nucleation and growth
(NG) 進行。PPO含量高時,相分離是經由 spinodal相分離模式。當
PPO 含量大於 20 phr時,系統的相反轉開使產生。當 PPO含量到達
50 phr 時,形成 PPO 為基材連續相,環氧樹脂為分散相。摻合體的
抗張強度及模數僅有些許變化,但是韌性卻大幅提升,由其是在系統
開使相反轉現象時特別明顯,原因是 PPO 的延展性及塑性形變造成
的。環氧樹脂的介電常數也隨 PPO 的增加而下降。此外添加反應性
單體三丙烯基三聚氰酸鹽 (Triallylisocyanurate, TAIC) 可改善摻合體
的相容性和抗溶劑性。
第二部分是以分散法摻合 Dicyandiamide (DICY) 硬化之環氧

樹脂和 PPO,來探討其韌性、熱性質、動態黏彈、介電常數和相形態
等。由分析中得知摻合 PPO 提升了摻合體的韌性與絕緣性質,而且
抗張強度及模數並未因 PPO 之摻合而改變;PPO 添加量低的摻合體
中,PPO粒子有聚集發生;不過在 PPO 添加量高的摻合體中,粒子
間皆有互相連結情形發生,形成 PPO 與環氧樹脂的共連續相。由動
態機械分析 (DMA) 得知摻合體會隨著 PPO 添加量的增加,其兩相
(環氧樹脂相和 PPO 相) 的玻璃轉化溫度有向內轉移的現象,產生此
現象的原因可能是摻合體在混合及硬化過程中,環氧樹脂比起硬化劑
dicy更容易溶於 PPO相,結果是較多的環氧樹脂、較少的 dicy溶於
PPO 相,導致 PPO相的塑化作用。PPO 相的 Tg大幅下降可證實這個
觀點,同時 dicy 濃度在兩相中分配不均勻,使得環氧樹脂相的 dicy
濃度較高,造成環氧樹脂相的 Tg點提升。此外,我們添加三丙基烯
基三聚氰酸鹽 (TAIC) 於摻合體中,可改善摻合體之相容性及抗溶劑
性。在破裂能量方面,隨著TAIC含量的增加而提升。在絕緣電性方
面,隨著 TAIC 含量增加,其介電常數明顯下降。在 TGA 分析中,
在最大裂解速率溫度隨著 TAIC 含量增加而提高。
第三部分是環氧樹脂/PPO 摻合體與高功能克維拉纖維製成複合
材料,其相分離的模式不同於純摻合體樹脂。當有纖維存在時,相分
離過程中環氧樹脂會往極性的纖維表面移動,形成 epoxy-coated fiber
的相形態,所以 microbond 的界面性質並不會因 PPO的存在而下降。
在複合材料積層板中,環氧樹脂會大量聚集在富纖維區域,形成
epoxy-coated fiber分佈於 PPO相中,此種相形態對於複合材料的破壞
韌性、機械性質有正面的影響。

ABSTRACT
Cure behavior, miscibility and phase separation have been studied in
blends of polyphenylene oxide (PPO) with diglycidyl ether of bisphenol
A (DGEBA) resin and cyanate ester hardener. An autocatalytic
mechanism is observed for the epoxy/PPO blends and the neat epoxy. It is
also found that the epoxy/PPO blends react faster than the neat epoxy.
The effects of PPO content on the cure behavior in the cyanate ester
cured epoxy were investigated with FTIR. FTIR analysis reveals that the
cyanate functional group reactions are accelerated by adding PPO and
indicates that several coreactions have occurred, such as cyanate-
hydroxyl addition and epoxy-cyanate addition. This is caused by the
reaction of cyanate ester with PPO phenolic end-group and water yield ing
imidocarbonate and carbamate intermediate which can react with cyanate
ester to form cyanurate. Then the cyanurate can further react with epoxy
resin. During cure, the epoxy resin is polymerized and the reaction-
induced phase separation is accompanied by phase inversion upon the
concentration of PPO greater than 50 phr. At low PPO content, the phase
separation takes place via nucleation and growth (NG). At high PPO
content, the phase separation takes place via spinodal decomposition
(SD). The dynamic mechanical measurements indicate that the two-phase
character and partial mixing existed in all the mixtures. The fracture
toughness (GIC) and thermal mechanical property are improved by PPO
content. However, the two-phase particulate morphology is not uniform
especially at a low PPO content. In order to improve the uniformity and
miscibility, triallylisocyanurate (TAIC) is evaluated as an in situ
compatibilizer for epoxy/PPO blends. TAIC is miscible in epoxy and the
PPO chains are bound to TAIC network. SEM observations show that

adding TAIC improve the miscibility and solvent resistance of the
epoxy/PPO blends.
A series of blends has been prepared by adding a polyphenylene
oxide, in varying proportions, to an epoxy resin cured with dicyandiamide.
All the materials show two-phase morphology when characterized by
SEM and DMA. The SEM and DMA indicate that partial mixing exists in
all the blends especially in high PPO content. It implies that the epoxy
oligomer or low crosslinking density epoxy exists in PPO phase after
curing. The tensile strength and modulus of these blends are nearly
independent of PPO content. While the fracture toughness (GIC) is
improved by PPO content. Furthermore, the dielectric constant decreases
with increasing PPO content in a linear fashion. However, two-phase
particulate morphology is not uniform. In order to improve the uniformity
and miscibility, triallylisocyanurate (TAIC) has been used as an in situ
compatibilizer for the polymer blends of epoxy and PPO. SEM and DMA
reveal the improvement of miscibility and solvent resistant in this system.
The fracture toughness and dielectric constant of these TAIC-modified
systems are also improved by adding TAIC (0-20 phr).
The morphology of the fiber-rich areas in the composite is different
from that of the epoxy/PPO blend without Kevlar fiber. In the pure
polymer blends for high PPO content (30 and 50 phr), phase separation
and phase inversion are observed. In the composites, the majority of the
epoxy resin migrates to the polar fiber surface resulting in the epoxy-
coated fibers. So the interfacial shear strength (IFSS) between Kevlar
fiber and epoxy/PPO blends is almost the same as that between Kevlar
fiber and neat epoxy. The presence of PPO does not affect the interfacial
property in the epoxy/PPO/fiber composite. So the interlaminar shear
strength (ILSS) increase with the PPO content is due to an increase in the
composite’s ductility or toughness.

目 錄
中文摘要 … … … … … … … … … … … … … … … … … … … … … … … … … . I
英文摘要 … … … … … … … … … … … … … … … … … … … … … … … … ..III
圖表索引 … … … … … … … … … … … … … … … … … … … … … … … … ..IX
第一章 緒論 … … … … … … … … … … … … … … … … … … … … … … … 1
第二章 文獻回顧 … … … … … … … … … … … … … … … … … … … … … 4
2-1 環氧樹脂的配方組成 … … … … … … … … … … … … … … … … … … . 4
2-2 環氧樹脂的增韌改質劑 … … … … … … … … … … … … … … … … … . 5
2-3 增韌機構 … … … … … … … … … … … … … … … … … … … … … … … . 8
2-4 兩相形態的形成 … … … … … … … … … … … … … … … … … … … … . 9
2-4-1 反應誘導相分離 … … … … … … … … … … … … … … … … … … … . 9
2-4-2 分散第二相於熱固性高分子的單體中 … … … … … … … … … … 11
2-5 反應誘導相分離與分散法的比較 … … … … … … … … … … … … ...16
2-6 相分離過程的熱力學描述 … … … … … … … … … … … … … … … ...16
2-6-1 摻合體的相容性… … … … … … … … … … … … … … … … … … ...17
2-6-2 Binodal Curve … … … … … … … … … … … … … … … … … … … … .17
2-6-3 Spinodal Curve … … … … … … … … … … … … … … … … … … … ...18
2-6-4 Critical Point … … … … … … … … … … … … … … … … … … … … ...18
2-7 硬化反應動力學 … … … … … … … … … … … … … … … … … … … ...18
2-7-1 微差掃瞄熱卡計… … … … … … … … … … … … … … … … … … ...19
2-7-2 霍氏轉換紅外光譜儀 … … … … … … … … … … … … … … … … ...21
2-8 環氧樹脂硬化的凝膠效應 … … … … … … … … … … … … … … … ...21
2-9 聚醯胺纖維與複合材料 … … … … … … … … … … … … … … … … ...23
第三章 氰酸酯硬化之環氧樹脂與聚氧化二甲苯摻合體性質之研
究 … … … … … … … … … … … … … … … … … … … … … … … ..30

3-1 前言 … … … … … … … … … … … … … … … … … … … … … … … … ...30
3-2 實驗 … … … … … … … … … … … … … … … … … … … … … … … … ...31
3-2-1 材料與藥品 … … … … … … … … … … … … … … … … … … … … ...31
3-2-2 儀器設備 … … … … … … … … … … … … … … … … … … … … … ...32
3-2-3 樣品製備 … … … … … … … … … … … … … … … … … … … … … ...33
3-2-4 實驗步驟 … … … … … … … … … … … … … … … … … … … … … ...34
3-3 結果與討論 … … … … … … … … … … … … … … … … … … … … … ...37
3-3-1 氰酸酯硬化動力學 … … … … … … … … … … … … … … … … … ...37
3-3-2 氰酸酯硬化環氧樹脂/聚氧化二甲苯摻合體反應性分析 … … ..38
3-3-3 摻合體反應過程之 FTIR 分析 … … … … … … … … … … … … … .64
3-3-4 摻合體硬化過程相分離行為及摻合體硬化過後的相形態之探
討 … … … … … … … … … … … … … … … … … … … … … … … … ....83
3-3-5 摻合體的動態機械分析 … … … … … … … … … … … … … … … ...96
3-3-6 摻合體的機械性質及介電性質之探討 … … … … … … … … … ...99
3-3-7 摻合體熱性質分析 … … … … … … … … … … … … … … … … … .103
3-4 結論 … … … … … … … … … … … … … … … … … … … … … … … … .107
第四章 PPO填充粒子對環氧樹脂熱性質與機械性質之研究 … … .111
4-1 前言 … … … … … … … … … … … … … … … … … … … … … … … … .111
4-2 實驗 … … … … … … … … … … … … … … … … … … … … … … … … .111
4-2-1 材料與藥品 … … … … … … … … … … … … … … … … … … … … ..111
4-2-2 儀器設備 … … … … … … … … … … … … … … … … … … … … … ..112
4-2-3 樣品製備 … … … … … … … … … … … … … … … … … … … … … ..112
4-2-4 實驗步驟 … … … … … … … … … … … … … … … … … … … … … ..113
4-3 結果與討論 … … … … … … … … … … … … … … … … … … … … … .114
4-3-1 Dicy硬化環氧樹脂/PPO摻合體加工條件之訂定 … … … … … .114

4-3-2 環氧樹脂與 PPO摻合體相形態之探討 … … … … … … … … … .115
4-3-3 TAIC改質環氧樹脂與 PPO摻合體相形態之探討 … … … … … 118
4-3-4 環氧樹脂與 PPO摻合體機械性質之探討… … … … … … … … .118
4-3-5 環氧樹脂與PPO摻合體動態機械性質之探討 … … … … … … .120
4-3-6 環氧樹脂與PPO摻合體熱穩定性之探討… … … … … … … … .131
4-3-7 環氧樹脂與PPO摻合體介電性質之探討… … … … … … … … .134
4-4 結論 … … … … … … … … … … … … … … … … … … … … … … … … .139
第五章 氰酸酯硬化環氧樹脂/聚氧化二甲苯摻合體為基材之克維拉
纖維複合材料性質之研究 … … … … … … … … … … … … … 141
5-1 前言 … … … … … … … … … … … … … … … … … … … … … … … … .141
5-2 實驗 … … … … … … … … … … … … … … … … … … … … … … … … .143
5-2-1 材料與藥品 … … … … … … … … … … … … … … … … … … … … .143
5-2-2 儀器與設備 … … … … … … … … … … … … … … … … … … … … .143
5-2-3 纖維的前處理 … … … … … … … … … … … … … … … … … … … .144
5-2-4 纖維表面形態的觀察 … … … … … … … … … … … … … … … … .144
5-2-5 Microbond試件製作及測量 … … … … … … … … … … … … … … 144
5-2-6 Kevlar纖維/環氧樹脂複合材料試片之製作程序及測試 … … ..145
5-2-7 SEM試件之製備與觀察 … … … … … … … … … … … … … … … ..146
5-3 結果與討論 … … … … … … … … … … … … … … … … … … … … … .148
5-3-1 以微粒拉出法研究 PPO對 Kevlar纖維/環氧樹脂界面性質的影
響 … … … … … … … … … … … … … … … … … … … … … … … … ..148
5-3-2 複合材料的破壞面 … … … … … … … … … … … … … … … … … .152
5-3-3 複合材料機械性質之探討 … … … … … … … … … … … … … … .159
5-3-4 複合材料熱性質測試 … … … … … … … … … … … … … … … … .163
5-3-5 Kevlar纖維表面處理對於複合材料機械性質的影響 … … … ...168

5-4 結論 … … … … … … … … … … … … … … … … … … … … … … … … .168
第六章 總結 … … … … … … … … … … … … … … … … … … … … … … 169
參考文獻 … … … … … … … … … … … … … … … … … … … … … … … … 172
簡歷 … … … … … … … … … … … … … … … … … … … … … … … … … … 178

圖表索引
Table caption
Table 3-1 Heats of reaction and autocatalytic model constants of neat
cyanate ester and mixtures of cyanate ester with various PPO contents..43
Table 3-2 Dynamic DSC measurement for cyanate ester cured epoxy/PPO
blends with various PPO contents … … … … … … … … … … … … … … … 5 0
Table 3-3 Isothermal cure reaction for cyanate ester cured epoxy/ PPO
blends with various PPO contents … … … … … … … … … … … … … … … 5 9
Table 3-4 Autocatalytic model constants for cyanate ester cured
epoxy/PPO blends system … … … … … … … … … … … … … … … … … … 6 5
Table 3-5 The most rapid decomposition rate temperatures of epoxy/PPO
blends … … … … … … … … … … … … … … … … … … … … … … … … … ..108
Table 3-6 The coefficient of thermal expansion of cured resin blends...109
Table 4-1 Glass transition temperatures of epoxy and PPO phase of
epoxy/PPO blends in cured states … … … … … … … … … … … … … … ...129
Table 5-1 Glass transition temperature Tg of cured resin blends and
composites … … … … … … … … … … … … … … … … … … … … … … … ..166
Table 5-2 The result of TGA analysis for Kevlar fiber composites … ...167
Figure caption
Figure 2-1 Evolution of the miscibility gap with conversion (p) for a
modified- thermosetting polymer showing an upper-critical-solution-
temperature behavior, UCST (φM0 = initial volume fractionof modifier, pcp
= cloud-point conversion) … … … … … … … … … … … … … … … … … … . 1 0
Figure 2-2 Evolution of the miscibility gap with conversion (p) for a
modified- thermosetting polymer showing an lower-critical-solution-
temperature behavior, LCST (φM0 = initial volume fractionof modifier, pcp
= cloud-point conversion) … … … … … … … … … … … … … … … … … … . 1 2

Figure 2-3 Miscibility gap in conversion-modifier concentration
coordinates, in the pre-gel region at a constant temperature (φM0 = initial
volume fractionof modifier, pcp = cloud-point conversion) … … … … … ..13
Figure 2-4 Shift of the miscibility gap with temperature: (a) for UCST; (b)
for LCST (only the pre-gel region is shown) … … … … … … … … … … ...14
Figure 2-5 Time temperature transformation (TTT) diagram … … … … ..22
Figure 2-6 Structure of the PPTA polymer … … … … … … … … … … … ...25
Figure 2-7 Models of Kevlar structure according to: (a) Dobb et al., (b)
Morgan et al., (c) Panar et al., (d) Li et al … … … … … … … … … … … … 2 9
Figure 3-1 Plots of the reaction rate versus time for neat cyanate ester and
10, 30, 50 phr PPO/cyanate ester system at three different temperatures:
(a) 0 phr; (b) 10 phr; (c) 30 phr; (d) 50 phr … … … … … … … … … … … ..39
Figure 3-2 Comparison between the autocatalytic model and data for neat
cyanate ester and 10, 30, 50 phr PPO/cyanate ester system at three
different temperatures: (a) 0 phr; (b) 10 phr; (c) 30 phr; (d) 50 phr … … .44
Figure 3-3 Dynamic DSC thermograms of cyanate ester cured epoxy/PPO
mixture samples … … … … … … … … … … … … … … … … … … … … … … 4 9
Figure 3-4 Isothermal DSC thermograms of cyanate ester cured
epoxy/PPO mixture samples … … … … … … … … … … … … … … … … … . 5 1
Figure 3-5 Plots of the conversion versus time for cyanate ester cured
epoxy/PPO mixture samples at four different temperatures: (a) 180; (b)
190; (c) 200; (d) 210oC … … … … … … … … … … … … … … … … … … … .55
Figure 3-6 Plots of the reaction rate versus time for cyanate ester cured
epoxy/PPO blends with various PPO contents at three different
temperatures: (a) 180; (b) 190; (c) 200; (d) 210oC … … … … … … … … ...60
Figure 3-7 Changes in the FTIR spectrum with curing at 180oC in the
epoxy/PPO system without cyanate ester … … … … … … … … … … … … .67
Figure 3-8 Changes in the FTIR spectrum with curing at 180oC in the
epoxy/cyanate ester system … … … … … … … … … … … … … … … … … ..68

Figure 3-9 Reaction scheme proposed between epoxy and cyanate
ester … … … … … … … … … … … … … … … … … … … … … … … … … … ...70
Figure 3-10 Solid-state 13C NMR for the epoxy/cyanate ester system
heated at 180oC for 35 min … … … … … … … … … … … … … … … … … ...71
Figure 3-11 Changes in the FTIR spectrum with curing at 180oC in the
cyanate ester system … … … … … … … … … … … … … … … … … … … … .72
Figure 3-12 Changes in the FTIR spectrum with curing at 180oC in the
cyanate ester/PPO (30 phr) system … … … … … … … … … … … … … … ...73
Figure 3-13 Changes in the FTIR spectrum with curing at 180oC in the
cyanate ester/PPO (100 phr) system … … … … … … … … … … … … … … .74
Figure 3-14(a) Relative decrease of cyanate group obtained by FTIR with
various PPO contents … … … … … … … … … … … … … … … … … … … … 7 5
Figure 3-14(b) Relative increase of triazine group obtained by FTIR with
various PPO contents … … … … … … … … … … … … … … … … … … … … 7 6
Figure 3-15 Changes in the FTIR spectrum with curing at 180oC in the
cyanate ester cured epoxy/PPO (50 phr) system … … … … … … … … … ..78
Figure 3-16 Plots of the oxazolidinone group peak intensity versus time
for cyanate ester cured epoxy/PPO blends system at curing temperature
180oC … … … … … … … … … … … … … … … … … … … … … … … … … … 7 9
Figure 3-17 Relative decrease of cyanate group obtained by FTIR with
various PPO contents … … … … … … … … … … … … … … … … … … … … 8 0
Figure 3-18 Solution-state 13C NMR for the epoxy/cyanate ester system at
uncured state … … … … … … … … … … … … … … … … … … … … … … … .82
Figure 3-19 Scanning electron micrographs of cyanate ester cured
epoxy/PPO (50 phr) blend specimens cured for different times at 180oC :
(a) 30 s; (b) 5 min; (c) 10 min; (d) 20 min … … … … … … … … … … … ...84
Figure 3-20 Phase diagram of cyanate ester cured epoxy/PPO (50 phr)
blend … … … … … … … … … … … … … … … … … … … … … … … … … … .85
Figure 3-21 Optical micrographs of cyanate ester cured epoxy/PPO (10

phr) cured for different times at 180oC: (a) 5 min; (b) 10 min; (c) 20
min … … … … … … … … … … … … … … … … … … … … … … … … … … … 8 7
Figure 3-22 Optical micrographs of cyanate ester cured epoxy/PPO (30
phr) cured for different times at 180oC: (a) 1 min; (b) 2 min; (c) 5 min; (d)
10 min; (e) 15 min; (f) 20 min … … … … … … … … … … … … … … … … ..88
Figure 3-23 Possible trajectories in the metastable region of the
conversion vs. composition phase diagram; a’ and b’ represent the
compositions of the dispersed phase corresponding to trajectories a and b,
respectively (K = phase separation rate/cure reaction rate, trajectory a is
associated to an NG mechanism while trajectory b leads to SD) … … … .90
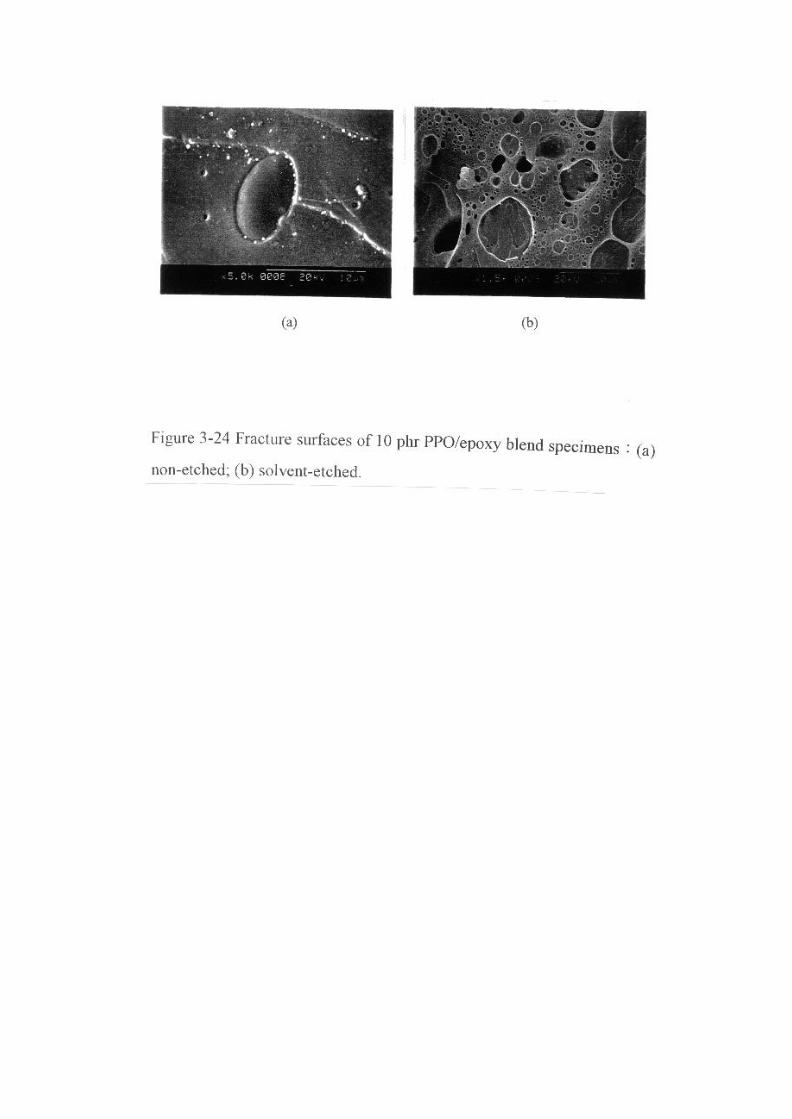
Figure 3-24 Fracture surfaces of 10 phr PPO/epoxy blend specimens:(a)
non-etched; (b) solvent-etched … … … … … … … … … … … … … … … … .92
Figure 3-25 Fracture surfaces of 50 phr PPO/epoxy blend specimens:(a)
non-etched; (b) solvent-etched … … … … … … … … … … … … … … … … .93
Figure 3-26 Fracture surfaces of 30 phr PPO/epoxy blend specimens:(a)
non-etched; (b) solvent-etched … … … … … … … … … … … … … … … … .94
Figure 3-27 Scanning electron micrographs of 30 phr PPO/epoxy blends
with various TAIC content:(a) 5 phr TAIC; (b) 5 phr TAIC (etched); (c)
10 phr TAIC; (d) 10 phr TAIC (etched); (e) 20 phr TAIC; (f) 20 phr TAIC
(etched)… … … … … … … … … … … … … … … … … … … … … … … … … .95
Figure 3-28 Dynamic storage modulus (E’) in the temperature range of
50°C-250°C for the epoxy/PPO blends … … … … … … … … … … … … … 9 7
Figure 3-29 Relaxation peaks (tan δ) in the temperature range of 50°C-
250°C for the epoxy/PPO blends … … … … … … … … … … … … … … … ..98
Figure 3-30 Relaxation peaks (tan δ) of epoxy/PPO (30 phr) blend and of
epoxy/PPO (30 phr) blend with 20 phr TAIC … … … … … … … … … … .100
Figure 3-31 The tensile modulus and strength as a function of blend
composition for the epoxy/PPO system … … … … … … … … … … … … .101

Figure 3-32 The fracture energy (GIC) as a function of blend composition
for the epoxy/PPO system … … … … … … … … … … … … … … … … … .102
Figure 3-33 Dielectric constant behavior of cyanate ester cured
epoxy/PPO system in high frequency regions … … … … … … … … … … 104
Figure 3-34 Dielectric constant as a function of blend composition for the
cyanate ester cured epoxy/PPO system … … … … … … … … … … … … ..105
Figure 3-35 TGA curves for the systems with various PPO content … ..106
Figure 4-1 Scanning electron micrographs of epoxy/PPO blend samples
for different PPO content : (a) 0 phr; (b) 20 phr; (c) 60 phr; (d) 100
phr … … … … … … … … … … … … … … … … … … … … … … … … … … ...116
Figure 4-2 Scanning electron micrographs of epoxy/PPO blend samples
for different PPO content (solvent-etched): (a) 0 phr; (b) 20 phr; (c) 60
phr; (d) 100 phr … … … … … … … … … … … … … … … … … … … … … ...117
Figure 4-3 Scanning electron micrographs of 30 phr PPO/epoxy blends
with various TAIC contents: (a) 0 phr TAIC; (b) 0 phr TAIC (etched); (c)
10 phr TAIC; (d) 10 phr TAIC (etched); (e) 20 phr TAIC; (f) 20 phr TAIC
(etched) … … … … … … … … … … … … … … … … … … … … … … … … ...119
Figure 4-4 Results of tensile strength and modulus for the epoxy/PPO
blends … … … … … … … … … … … … … … … … … … … … … … … … … ..121
Figure 4-5 Results of fracture energy (GIC) for the epoxy/PPO blends..122
Figure 4-6 Results of tensile strength and modulus for the epoxy/PPO (30
phr) blends with various TAIC contents … … … … … … … … … … … … .123
Figure 4-7 Results of fracture energy (GI C) for the epoxy/PPO (30 phr)
blends with various TAIC contents … … … … … … … … … … … … … … 124
Figure 4-8 Dynamic DSC thermograms of dicy cured epoxy/PPO (30
phr)/TAIC mixture samples: (a) 0 phr TAIC (first scan), (b) 0 phr TAIC
(second scan), (c) 10 phr TAIC (first scan), (d) 10 phr TAIC (second
scan) … … … … … … … … … … … … … … … … … … … … … … … … … … 125
Figure 4-9 Dynamic DSC thermograms of dicy cured epoxy/PPO (30

phr)/TAIC mixture samples: (a) 20 phr TAIC (first scan), (b) 20 phr TAIC
(second scan), (c) 30 phr TAIC (first scan), (d) 30 phr TAIC (second
scan) … … … … … … … … … … … … … … … … … … … … … … … … … … 126
Figure 4-10 Relaxation peaks (tan δ) for the epoxy/PPO blends … … ...128
Figure 4-11 Relaxation peaks (tan δ) of epoxy/PPO (30 phr) blends with
various TAIC contents … … … … … … … … … … … … … … … … … … … 130
Figure 4-12 TGA curves for the systems with various PPO contents … 132
Figure 4-13 TGA curves for the systems with various PPO contents (scale
up) … … … … … … … … … … … … … … … … … … … … … … … … … … ...133
Figure 4-14 TGA curves for the epoxy/PPO systems with various TAIC
contents … … … … … … … … … … … … … … … … … … … … … … … … ...135
Figure 4-15 Plots of the most rapid decomposition rate temperatures
versus time for dicy cured epoxy/PPO blends system … … … … … … … 136
Figure 4-16 Dielectric constant as a function of blend composition for the
dicy cured epoxy/PPO system … … … … … … … … … … … … … … … … 137
Figure 4-17 Dielectric constant as a function of blend composition for the
dicy cured epoxy/PPO system with various TAIC contents … … … … ...138
Figure 5-1 Typical load-displacement curve of short beam test … … … .147
Figure 5-2 Effect of the plasma treatment on IFSS of the Kevlar 49 fiber-
cyanate ester cured epoxy system … … … … … … … … … … … … … … ...149
Figure 5-3 SEM micrographs of fracture surfaces of the microbond
specimens: (a) Control, (b) 10 phr PPO, (c) 30 phr PPO, (d) 50 phr PPO,
(e) neat PPO … … … … … … … … … … … … … … … … … … … … … … … 150
Figure 5-4 The interfacial shear strength of microbond test with various
PPO contents … … … … … … … … … … … … … … … … … … … … … … ..153
Figure 5-5 Fracture surfaces of the cured epoxy/PPO blends: (a) 30 phr
PPO, (b) 50 phr PPO … … … … … … … … … … … … … … … … … … … ..154
Figure 5-6 Fracture surfaces of the cured epoxy/PPO blends (etched): (a)
30 phr PPO, (b) 50 phr PPO … … … … … … … … … … … … … … … … ...155

Figure 5-7 Fracture surfaces of the composite: (a) neat epoxy, (b) 30 phr
PPO … … … … … … … … … … … … … … … … … … … … … … … … … … .156
Figure 5-8 Fracture surfaces of the composite (etched): (a) neat epoxy, (b)
30 phr PPO … … … … … … … … … … … … … … … … … … … … … … … .157
Figure 5-9 SEMs of the polished cross-section of the epoxy/PPO-base (30
phr PPO) Kevlar fiber reinforced composite: (a) non-etched, (b) solvent-
etched … … … … … … … … … … … … … … … … … … … … … … … … … ..158
Figure 5-10 Schematic representation of phase separation of epoxy/PPO
blend in microbond sample … … … … … … … … … … … … … … … … … 160
Figure 5-11 Results of fracture energy (GI C) for the epoxy/PPO/Kevlar
fiber composite … … … … … … … … … … … … … … … … … … … … … ...161
Figure 5-12 The interlaminar shear strength of composites with various
PPO contents … … … … … … … … … … … … … … … … … … … … … … ..162
Figure 5-13 The T-peel strength of composites with various PPO
contents … … … … … … … … … … … … … … … … … … … … … … … … ...164
Figure 5-14 Fracture surfaces of the composites with various PPO
contents after T-peel test: (a) 0 phr, (b) 30 phr, (c) 50 phr … … … … … .165

第一章 緒論
高分子材料近年來在電子構裝技術的各種製程,例如銅箔基板材
料,層間絕緣及半導體元件的封裝材料等技術中扮演重要的角色,應
用於各項構裝製程的樹脂材料,其耐熱、電氣及機械等特性必須進一
步提升。目前在電子工業應用最廣的樹脂是環氧樹脂,其使用上仍有
許多限制,例如在高溫使用時尺寸安定性不佳,在高頻使用範圍中介
電常數過高,所以傳統的環氧樹脂對今日高性能需求的電子工業已無
法應付技術上的要求,因此希望能藉由摻合高性能的聚氧化二甲苯
(Polyphenylene Oxide,PPO)來改善此問題。
聚氧化二甲苯主幹的結構是在苯環上 1,4 的位置藉由氧原子來
鍵結,由於此結構分子鏈有良好的耐熱性、低介電常數、低電氣損耗
因子和高的玻璃轉換溫度等,此外聚氧化二甲苯尚有低吸水性、抗溶
劑性、耐衝擊性、耐燃性及對銅箔優良的剝離強度等優點[1]。但是
聚氧化二甲苯主要缺點在於加工性不佳,由於非極性之熱塑性線性高
分子,與極性的環氧樹脂相容性不佳,如果直接摻混,並不能得到最
佳的效果。
在文獻中有許多改質環氧樹脂的方法,有添加無機物如玻璃珠、
氧化矽等物質;橡膠物質如壓克力彈性體、矽橡膠、核殼式橡膠和液
態橡膠等;或是添加熱塑性高分子等方法。但是以無機物添加入環氧
樹脂系統中,填充劑與環氧樹脂界面作用力差,降低了系統的機械性
質。在以橡膠改質熱固性環氧樹脂系統中,當環氧樹脂交聯密度增加
時,由於相分離後之橡膠被高交聯密度環氧樹脂的網狀結構所包圍
著,所以橡膠顆粒無法造成基材的剪切降服或空洞形成,因此缺乏增
韌效果。此外,大部份的橡膠增韌熱固性樹脂系統中,由於會有少量
的橡膠無法從環氧樹脂中完全分離出來,導致硬化後的樹脂之玻璃轉

移溫度降低。所以選擇具有高玻璃轉移溫度的熱塑性樹脂為改質劑,
乃是現今改質環氧樹脂的主流。熱塑性改質劑不僅可增強環氧樹脂系
統的韌性,更不會犧牲熱固性所具備的玻璃轉移溫度、硬度、強度或
其他特有的物性。
氰酸酯 (cyanate ester) 硬化環氧樹脂系統與胺類 (amine) 硬化
環氧樹脂系統比較起來,算是較新的反應系統,所以較少有文獻提及
環氧樹脂與氰酸酯之間的反應[2, 3]。以氰酸酯硬化後的環氧樹脂與
傳統的環氧樹脂比較起來有較好的電氣性質、較佳的韌性、耐熱性及
提升耐燃性與降低發煙性,此外氰酸酯硬化環氧樹脂的反應溫度較
高,有利於摻合加工過程。氰酸酯與環氧樹脂之硬化過程的化學反應
非常複雜,因此不容易精確的描述其反應機構,所以在文獻中有利用
模式化合物 (model compounds) 來探討其反應機構[2]。
本研究第一部分探討以高玻璃轉移溫度的熱塑性高分子
Polyphenylene Oxide 改質氰酸酯硬化環氧樹脂的硬化反應動力和反
應機構;此外,也觀察硬化過程中熱固性與熱塑性高分子之相變化。
硬化之後的摻合體的熱性質與機械性質也加以探討。為了改善摻合體
的相容性,利用添加多丙烯基反應性單體於聚氧化二甲苯/環氧樹脂
摻合體中,利用含丙烯基單體 (triallylisocyanurate, TAIC) 所產生的自
由基與聚氧化二甲苯產生反應,形成 PPO-TAIC copolymer,與環氧
樹脂摻合時,可增加摻合體的相容性與抗溶劑性,並針對此摻合系統
之物理性質作一番探討。第二部分是聚氧化二甲苯以分散法與環氧樹
脂-Dicyandiamide硬化系統混合,來探討韌性、熱性質、動態黏彈、
介電常數和相型態等,摻合系統也將添加 TAIC來改善其相容性及機
械性質等。
此外在摻合體與纖維複合材料的性質中,以界面性質最為重要。

因為電子工業中之印刷電路板,就是以纖維補強樹脂製成的複合材
料,它必須通過嚴格的漂鍚實驗 (solder resistance),否則就有爆板之
虞。所以除了基材穩定性要好之外,複合材料層間介面強度是重要因
素。所以本研究將探討環氧樹脂 /PPO 摻合體與克維拉纖維間的界面
性質,以了解 PPO 的存在對複合材料界面性質的影響。接著再對此
複合材料進行一系列的測試及分析,探討其界面接著、破壞機構、機
械強度及物理性質。

第二章 文獻回顧
2-1 環氧樹脂的配方組成
環氧樹脂的配方組成主要是由樹脂與硬化劑所構成,環氧樹脂的
種類很多,最常用的是雙官能基的 Diglycidyl Ether of Bisphenol A
(DGEBA) 環氧樹脂,1947年工業生產以來,因其優良的性能而用於
塗料、接著劑、鑄模品、積層品、複合材料、電子封裝材料,甚至航
太材料,它都佔有十分重要的地位。此類型環氧樹脂是在鹼的存在
下,使雙酚A (Bisphenol A) 與環氧-[1,2]-氯-[3]丙烷 (epichloro hydrin)
反應而製造,其結構如下:
n值可因反應條件而變化,從 n值為 1以下的液態樹脂到 n值約
15 的固態樹脂,有各種不同分子量的市售產品。由環氧樹脂的分子
結構觀之,Diglycidyl 部份愈長,則樹脂的柔軟性愈佳。所以環氧樹
脂的分子結構影響環氧樹脂的性能甚巨。
其次,環氧樹脂所使用的硬化劑有很多種類,硬化後樹脂成品的
性質、樹脂與硬化劑的混合物可用的硬化溫度、硬化時間、加工條件
等依硬化劑的種類而有所不同,所以硬化劑的選擇非常重要。環氧樹
脂的硬化劑依反應機構、硬化溫度、化學構造而分類如下:
1. 依反應機構分類
(1) 觸媒作用者:如三級胺類、三氟化硼-胺複合物。
(2) 與環氧樹脂官能基反應者:如胺類、酸酐、氰酸酯等。
CH2 CH2CH2CH
O- -O C
CH3
CH3
O CH2 CH
OH
O
CH3
CH3
C CH2O
O
CHCH2

2. 依硬化反應溫度分類
(1) 常溫硬化:如聚醯胺樹脂、diethylene triamine等。
(2) 中溫硬化:如 diethyl amino propyl amine等。
(3) 高溫硬化:如雙氰胺 (dicyamdiamide)、氰酸酯 (cyanate ester)、
無水苯二甲酸等。
3. 依化學構造分類
(1) 胺類:如脂肪族胺類、芳香族胺類、二級及三級胺等。
(2) 酸酐
(3) 聚醯胺樹脂
(4) 三氟化硼-胺複合物
(5) 雙氰胺
(6) 氰酸酯
本研究所選用的硬化劑為氰酸酯,其可與環氧樹脂反應生成一個
穩定的五環結構 (oxazoline) [3]。氰酸酯可以改善環氧樹脂的耐熱性
(temperature resistance)、耐燃性 (fire resistance)、降低介電常數、提
升模數 (modulus) 及硬度 (hardness),而且在硬化過程中無揮發物產
生及較低的硬化收縮率;氰酸酯對環氧樹脂的硬化反應溫度較高,所
以有利於高溫下加工的進行。
2-2 環氧樹脂的增韌改質劑
環氧樹脂自 1950 年代即被廣泛使用為接著劑,因其具有某些優
越的性質。不過當環氧樹脂硬化後其交聯密度 (degree of crosslinking)
增加,使環氧樹脂成為一易脆的材料,會減弱環氧樹脂的韌性,而限
制了其應用。這個缺點會造成鑄模品在成型時易產生裂紋;在作黏著
劑時容易有剝離的現象;而用於複合材料的基材時,內部的微小裂紋

容易沿著纖維方向傳遞擴大。針對這些缺點,環氧樹脂的韌性
(toughness)之改良已有許多研究探討。一般而言在改質環氧樹脂方
面有如下的方法:
(1) 化學方法:經由共聚合,共縮合,接枝聚合,塊聚合以及架橋反
應等化學反應而進行改質。
(2) 物理方法:摻混填充劑,添加劑,改質劑,增韌劑等而進行改質。
1968年麻省理工學院的McGarry及Willner等人利用具有反應性
末端基的液態橡膠來改質環氧樹脂,雖然熱固性環氧樹脂的韌性增加
了,但是其彈性模數 (elastic modulus)、降伏強度 (yield strength) 、
蠕變阻抗 (creep resistance) 和玻璃轉移溫度也因此而變小了,此結果
相對的限制了增韌後樹脂的用途,且研究中指出利用橡膠增韌之效果
只對低交聯密度之環氧樹脂有較佳的效果,對高度交聯者效果不佳,
而高強度、高硬度、高玻璃轉移溫度之熱固性樹脂多屬於是高度交聯
之熱固性樹脂,但也因為其高度交聯而變脆(brittle),因此在增韌的
課題上就顯得相當重要[4]。幸運的是一種新的環氧樹脂增韌方法可
避開傳統橡膠增韌所遭遇的缺點,那就是使用堅硬的熱塑性塑膠。利
用熱塑性塑膠增韌的技術源自於 1980 年代初期,Bucknall等人首先
發表此類論文,可惜的是這些研究結果對於樹脂的韌性只有微幅提升
[5]。然而發現熱塑性塑膠增韌環氧樹脂有重大進展的是在維吉尼亞
高分子技術學院 (Virginia Polytechnic Institute) 的研究人員所提出,
他們是利用合成 polysulfone (PSF) 寡聚物來當作環氧樹脂的增韌劑
[6, 7]。此外,有些研究者使用 polyethersulphones (PES) 來改質環氧
樹脂,會產生相分離及良好的界面黏著性,得到不錯的增韌效果[8]。
還有一些研究發現,利用 polyetherimide (PEI) 改質環氧樹脂的效果
比 PES好[9, 10]。另外改質環氧樹脂的熱塑性塑膠還有 polycarbonate

[11, 12]、或是結合了橡膠和熱塑性高分子等[13],熱塑性塑膠改質劑
不僅可以增加環氧樹脂系統的破壞韌性,更不會犧牲熱固性樹脂所具
備的玻璃轉移溫度、強度、硬度等。熱塑性高分子在添加入環氧樹脂
後,不僅會改變網狀結構形態而且也會影響到固化動力。如果熱塑性
高分子和交聯之環氧樹脂在固化後不相溶,則系統會發生相分離並發
展成一不均勻相之形態。當直鏈型熱塑性高分子混合熱固性樹脂系統
時,溶解程度會影響到系統之形態、物理和機械上的性質,所以在經
過細心的設計混合系統組成時,就可真正的使直鏈型熱塑性高分子達
到改質熱固性環氧樹脂的目的。
本研究所選用的熱塑性改質劑為聚氧化二甲苯 (PPO),PPO為一
種重要的熱塑型工程塑膠,具有很高的玻璃轉移溫度 (205-220℃),
同時在 200℃之下仍可保有足夠的耐衝擊強度,它的介電常數及耗損
因子均低,在不同的溫度、頻率、濕度下改變量極小。另外它的機械
性質優異,是一種堅硬且強韌的材料,其熱變形溫度可高達 179℃,
此外 PPO 尚有低吸水性、耐衝擊性和對銅箔有優良的剝離強度。雖
然 PPO 具有這麼多優點,但是因為它的耐化學藥品性差,以及它有
很高的熔融溫度及黏度,增加了在加工上的困難,因而阻礙 PP0商業
化的腳步。幸運的是,PPO可與 PS任何比例混合完全互溶,大大提
高了它的加工性,使得 PPO 的商業化變成可能,同時因為它在線型
分子鏈的末端基上帶有-OH基,使得它也可與酸酐或環氧基等官能基
進行反應,得以與他種聚合物摻混而成可用的合膠。
對於聚氧化二甲苯與環氧樹脂摻合系統的相關文獻有 1993年 R.
A. Pearson等人在PPO/Epoxy摻合系統中,添加不同組成比例的 SMA
(styrene-co-maleic anhydride) 共聚物為相容劑,研究發現相容劑的添
加會使 PPO 分散相的粒徑減小,減少 PPO 粒子聚集的產生,增加

Epoxy與 PPO的相容性[14]。R. A. Pearson等人在另一篇文獻指出,
PPO/Epoxy 摻合體的韌性隨 PPO 的含量而線性增加,並以不同的破
壞機構說明 PPO 增韌的原因;此外該研究也指出相容劑的添加可增
加摻合體的相容性及機械性質[15]。1994年 R. W. Venderbosch等人在
PPO/Epoxy摻合系統的研究中指出,混合系統在硬化過程中相分離始
終是進行著,且 PPO 的含量多寡會影響到硬化後摻合體的相形態。
此外該研究也指出高溫的硬化條件會使 PPO 末端的-OH 基與環氧基
產生醚化反應,進而提升相容性,增強機械性質[16]。1995年 R. W.
Venderbosch等人利用纖維補強 PPO/Epoxy系統,研究中發現纖維補
強對於材料的機械性質有明顯的提升,其原因是經由相分離後,環氧
樹脂會附著在纖維表面,與纖維有良好的接著力[17]。此外在 1995
年 R. W. Venderbosch 等人在 PPO/Epoxy 摻合系統中添加聚丙醚
(polypropylene oxide)發現,當相反轉發生後,環氧樹脂分散相的 Tg
會降低而形成橡膠態,增加系統的韌性[18]。1998年張國輝等人研究
組成與硬化溫度對 PPO-DER332/DETDA摻合物的相形態、分相行為
與機械性質的影響[19]。
2-3 增韌機構
熱固性高分子材料具有硬脆特性,對於裂縫起始與裂縫成長的
抵抗能力較弱,所以必須添加改質劑形成第二相結構,增加基材對裂
縫起始及成長的抑制能力,本研究希望藉由添加熱塑性 PPO 來增加
環氧樹脂的韌性。許多不同的機構已被提出解釋熱固性樹脂在含一散
佈相熱塑性塑膠粒子的微結構下會使其破壞能量改良之原因,研究指
出分散粒子可能產生以下之增韌機構:
(1) 大頭針形狀之裂痕 (crack pinning) [20]。

(2) 粒子架橋作用 (particle bridging) [21]。
(3) 裂縫繞道 (crack path deflection) [22]。
(4) 粒子降伏 (particle yielding) [23]。
(5) 剪切降伏帶 (particle-yielding-induced shear banding) [24]。
(6) 微裂縫 (microcracking) [25]。
2-4 兩相形態的形成
在熱固性樹脂改質系統中,形成兩相形態可由下列兩種方法:
(a) 最初是個均勻溶液,經由熱固性樹脂的聚合反應產生相分離。(反
應-誘導相分離,reaction-induced phase separation)
(b) 分散第二相於最初的熱固性單體中。
2-4-1 反應誘導相分離
MaGarry於 1960年末期至 1970年初期發表了橡膠改質熱固性環
氧樹脂的方法 [4, 25, 26],他們是利用添加低分子量的 carboxyl-
terminated poly(butadiene-acrylonitrile) copolymers (CTBN)來增韌
diglycidyl ether of bisphenol A (DGEBA),使用的硬化劑為二胺
(diamines)。橡膠區域會經由熱固性樹脂硬化反應而沉積出來,進而
增韌環氧樹脂。1980 年代開始利用熱塑性塑膠來取代橡膠來增韌環
氧樹脂。大部分的改質劑與環氧樹脂會呈現出上臨界溶解溫度行為
(UCST),也就是說相容性隨溫度而增加。像 Figure 2.1所示,一組成
包含了一最初體積分率為φM0的改質劑,在反應溫度 Tr的初期時與熱
固性樹脂是相容的。當反應進行時,改質劑與樹脂的相容性會變差。
若 miscibility gap 到達了點 (φM0, Tr) 的位置,相分離就開始發

Figure 2-1 Evolution of the miscibility gap with conversion (p) for a
modified- thermosetting polymer showing an upper-critical-solution-
temperature behavior, UCST (φM0 = initial volume fractionof modifier, pcp
= cloud-point conversion).

生,此時轉化率定義為霧點轉化率 (cloud-point conversion, pcp)。
相似的情況也發生在改質劑為 poly(ether sulfone) (PES)[8, 27,
28],如 Figure 2.2,其所呈現的是下臨界溶解溫度行為。在此系統中,
反應溫度 Tr是低於反應初期的 miscibility gap。
其實上述兩種行為皆可利用轉化率對改質劑濃度的座標來描述
[29, 30],如 Figure 2.3,此圖的溫度是固定的。若改變了溫度其
miscibility gap 也會移動,如Figure 2.4所示。在Figure 2.3與 Figure 2.4
中所描述的是在摻合體凝膠前才可繪出兩相區域。雖然有一些文獻證
明熱固性高分子在凝膠之後仍有繼續相分離的行為,但是大多數的相
分離是發生在凝膠之前[31-36]。
有效地使用反應誘導相分離的程序來改質熱固性高分子需要了
解控制相形態形成的因素及相形態與材料性質之間的關係。反應誘導
相分離的過程是由熱力學及動力學因素來決定。在轉化率-組成的座
標中,由熱力學分析能決定系統的穩態區 (無相分離產生)、介穩態
區 (相分離可能產生) 及非穩態區 (相分離產生)。當系統狀態進入介
穩態區,開始發生相分離的可能性是由本身的相分離速率與反應速率
的競爭來決定。假使一系統的相分離速率遠大於反應速率,新的相將
會形成,這可利用熱力學分析來預測。若反應速率遠大於相分離速
率,代表系統的反應速率非常快,系統一直到非穩定區才產生 spinodal
相分離。本研究的第三章是以反應誘導相分離的方式來製備兩相形態
的高分子,並且討論其反應性、相形態、機械性質等與熱塑性 PPO
的關係。
2-4-2 分散第二相於熱固性高分子的單體中

Figure 2-2 Evolution of the miscibility gap with conversion (p) for a
modified- thermosetting polymer showing an lower-critical-solution-
temperature behavior, LCST (φM0 = initial volume fractionof modifier, pcp
= cloud-point conversion).

Figure 2-3 Miscibility gap in conversion-modifier concentration
coordinates, in the pre-gel region at a constant temperature (φM0 = initial
volume fractionof modifier, pcp = cloud-point conversion).

Figure 2-4 Shift of the miscibility gap with temperature: (a) for UCST; (b)
for LCST (only the pre-gel region is shown).

Figure 2-4 Shift of the miscibility gap with temperature: (a) for UCST; (b)
for LCST (only the pre-gel region is shown).

此方法是利用在最初的組成中放入已成形的粒子來改質熱固性
高分子,此方法也常見於改質熱塑性高分子。文獻中有利用分散壓克
力系彈性體 [37-39],也有利用殼-核粒子來改質熱固性高分子 [40-
43]。殼-核粒子是由橡膠中心核包埋在可相容的熱塑性殼層中,殼-
核粒子分散的穩定性可利用調整殼層的組成來改善[40]。本研究的第
四章將加以探討此製備法所得的摻合體性質。
2-5 反應誘導相分離與分散法的比較
根據應用性,此二種程序來得到兩相形態之改質過的熱固性高
分子各有其優缺點。關於增韌熱固性網狀結構,利用反應誘導相分離
的優點是在最初的均勻相溶液的黏度是較低的且較穩定的,利用此法
也可得到各種不同硬化後的相形態。缺點是需要建立一套在硬化條
件、相形態與材料性質的關係式;另一缺點是當反應完成後,仍然可
能有部分的改質劑殘留於熱固性樹脂中,如果改質劑的 Tg點較低,
就會造成整個材料的Tg點下降,也就是說造成整個材料的塑性效應。
若是使用高 Tg的熱塑性樹脂,則殘餘的改質劑不會造成這個問題。
關於分散第二相改質劑於熱固性單體中的方式,其最大的優點
是能控制分散相的體積分率、粒徑分布及組成,也可能完成較精確的
研究關於界面黏著力[40]或是粒徑分布[44]對材料性質的影響。此方
法缺點是加入分散相的最初黏度相當高,加工不易,有可能造成分散
相粒子的聚集,甚至在儲存或是加工過程會有巨觀的相分離。此外有
些添加無機填充劑的系統會降低材料的性質[45]。
2-6 相分離過程的熱力學描述
2-6-1 摻合體的相容性

在熱力學平衡上,在一定溫度及壓力下,惟有混合的自由能
(∆GM) 為零或負值,不同種類的高分子才可互溶。而混合自由能可表
示為:
∆GM = ∆HM - T∆SM
∆HM = V (δ1 - δ2)2 φ1φ2
∆SM = -R (n1 ln φ1 + n2 ln φ2)
∆GM = V (δ1 - δ2)2 φ1φ2 + RT (n1 ln φ1 + n2 ln φ2)
其中∆HM與∆SM分別為系統的混合焓與混合熵,T為混合時的溫度,
V為混合物的莫耳體積,n1與 n2為改質劑與樹脂的莫耳分率,φ1與φ2
為改質劑與樹脂的體積分率,δ1與δ2為改質劑與樹脂的溶解度參數。
在混合高分子過程中其分子的不同種類排列數目大大地減少,所以其
∆SM非常小,所以要使摻合體的相容性佳,在混合過程∆HM必須甚小
或為負值。對於造成∆HM值變小或為負值的原因很多,例如分子間的
氫鍵、質子轉移、電子轉移、過渡金屬複合物及高分子間之官能基產
生化學交互作用力等,例如增加橡膠的極性,會增加與環氧樹脂的相
容性。此外,當樹脂開始進行硬化反應,樹脂與樹脂間的聚合反應及
樹脂與改質劑間的反應,莫耳體積 V 會急速的增大,導致∆GM變成
正值,改質劑便逐漸地從樹脂中分離出來,這就是分散相與基材相形
成的原因。
2-6-2 Binodal Curve
Binodal curve是熱力學上的穩態與介穩態的邊界。除了熱力學的
計算外,通常可利用霧點曲線 (cloud-point curve, CPC) 實驗來決定此
邊界。系統的兩相組成在平衡時,此平衡狀態可由 binodal 來描述,
各成份的化勢 (chemical potential) 在兩相中應相等。化勢定義為:

系統平衡狀態 (binodal curve) 可由下列式子表示:
∆µ1α = ∆µ1
β
∆µ2α = ∆µ2
β
其中 α 與 β 表示在平衡時的兩相。當固定溫度 T與φ2α (φ1
α = 1 - φ2α)
時,利用上述平衡式可求得轉化率 p與φ2β (φ1
β = 1 - φ2β),即可求出轉
化率與組成座標圖中的 binodal curve。
2-6-3 Spinodal Curve
Spinodal curve 是熱力學上介穩態與非穩態區域的邊界。系統在
此邊界上,必須滿足 (∂ ∆µ i / ∂ φ j)T, P = 0 的式子。由熱力學的方法也
可解出 spinodal curve。在實驗中求 spinodal curve最常使用的儀器為
Pulse Induced Critical Scattering (PICS)[46]。
2-6-4 Critical Point
當 binodal curve與 spinodal curve的交接點稱為臨界點,所以此
點為化勢對組成的一次微分與二次微分皆為零。若改質劑的含量超過
臨界點,摻合體就會產生相反轉的現象。
2-7 硬化反應動力學
熱塑性高分子 PPO摻合到環氧樹脂中,不僅會改變硬化後相形
態,而且會影響系統的硬化行為,此為本研究的一個重要課題。研究
i
Mi
nG
∂∆∂
=∆µ

熱固性樹脂硬化動力學的方法很多,而最常使用的儀器為下列數種:
(1) 微差掃瞄熱卡計 (Differential Scanning Calorimeter, DSC)
(2) 霍氏轉換紅外光譜儀 (Fourier Transform Infrared Spectroscopy,
FTIR)
(3) 介電測試儀 (Dielectric Measurements)
(4) 流變動力測試儀 (Rheokinetic measurements)
2-7-1 微差掃瞄熱卡計 (Differential Scanning Calorimeter,
DSC)
微差掃瞄熱卡計最常用在決定熱固性樹脂硬化反應的動力學參
數與動力學方程式。在 DSC測試動力學方法的基本假設為反應熱(Q)
的改變正比於反應程度(α)的改變,如下式表示。
或是
其中 Qo 是總反應熱,它可由非等溫掃瞄程序來獲得;由時間對上式
積分可求得反應轉化率。
利用 DSC 來偵測熱固性樹脂的方法有兩種,一種是非恆溫掃瞄
測試 (non-isothermal scanning tests),另一種是恆溫測試 (isothermal
tests)。在非恆溫測試中最常使用的是 Borchardt and Daniels Method
[47],此法可由單一的 DSC 掃瞄就可記算反應活化能、指數前因子
dt
d
dt
dQ α∝
dt
d
dt
dQ
Qo
α=
1

(pre-exponential)、反應熱、反應級數及速率常數,其計算如下:
The Calculations of Borchardt and Daniels Method
dα/dt = k(T) (1-α)n
k(T) = A e-E/RT
⇒ dα/dt = A e-E/RT (1-α)n
⇒ ln(dα/dt) = lnA - E/RT + n ln(1-α)
Borchardt and Daniels Method有幾項假設:(1)反應必須遵循 n階反應
動力;(2)反應速率必須符合阿瑞尼士方程式 (Arrhenius expression);
(3)反應只能有一種反應機構;(4)擴散效應必須忽略。Khanna et al.利
用非等溫 DSC 測試決定 phenolic triazine 系統的動力學[48]。Ng 和
Manas-Zloczower也利用非等溫DSC來求不飽和聚酯的硬化動力學,
得到不錯的效果[49]。
在恆溫測試中,主要的假設是總反應熱能準確地求得和所有反應
具有相同的焓 (enthalpy)。恆溫反應同時適用於 n階反應 (nth order)
與自催化反應 (autocatalyed) 模式。恆溫測試也可除去熱遲緩效應
(thermal lag effect) 和降低熱裂解的干擾。Lee 利用恆溫測試
polyetherstyrene 系統來決定抑制劑與起使劑對動力學參數和速率常
數的影響[50]。Sourour 和 Kamal 研究指出環氧樹脂與芳香族胺類的
恆溫反應動力學數據符合所假設的自催化反應模式[51],本研究是選
用 Kamal的自催化反應模式來作氰酸酯硬化系統的動力學分析。
2-7-2 霍氏轉換紅外光譜儀 (Fourier Transform Infrared
Spectroscopy, FTIR)
FTIR 用來偵測熱固性樹脂的硬化反應主要是分析反應過程中官

能基光譜吸收峰高度的變化。FTIR 分析可提供硬化過程組成的變
化,可獲得更詳細的訊息。St John et al.利用 FTIR導出動力學模式,
以了解不純物對 TGDDM/DDS 系統之硬化動力的影響[52]。Yang 和
Lee利用 FTIR了解聚胺基甲酸酯 (PU) 與聚酯 (polyester) 之 IPN系
統的多種反應機構,這些機構是無法利用 DSC求得[53]。Simon等人
同時利用 DSC與 FTIR 來探討雙官能基氰酸酯的硬化動力學,並且經
由實驗求出 TTT 圖[54]。
2-8 環氧樹脂硬化的凝膠效應
凝膠點 (gel point) 的定義為某一時間或溫度下,分子間的共價
鍵連接成網路,此時無窮大的網路形成。到達凝膠點時硬化反應仍繼
續進行,然而系統黏度加大導致擴散速率降低,可能會抑制交聯反
應。反應系統若是到達玻璃化點 (vitrification),更進一步的反應將被
抑止,Wisanrakkit和 Gillham研究指出在玻璃態時只可能發生非常緩
慢擴散控制 (diffusion control) 的反應[55]。所以在玻璃態的 Tg點與
反應溫度相同原因是形成了堅硬的玻璃狀。恆溫反應的熱固性系統之
凝膠效應可以利用 Time-Temperature-Transformation (TTT) 圖加以描
述[56]。TTT圖 (Figure 2-5) 中有七個明顯的區域;液體 (liquid),溶
膠-凝膠橡膠 (sol-gel rubber),凝膠-橡膠 (gel-rubber),溶膠-凝膠玻璃
(sol-gel glass),凝膠-玻璃 (gel-glass),溶膠-玻璃 (sol-glass) 及焦炭
(char)。Tgo是指沒有硬化反應的玻璃轉移溫度,gel-Tg是指凝膠與玻
璃化同時發生的溫度,Tg∞是指完全硬化後的玻

Figure 2-5 Time temperature transformation (TTT) diagram.

璃轉移溫度。
若恆溫加熱樣品,我們可在 TTT 圖中以一水平線來表示,樣品
根據最初的加熱溫度來決定其所經過的區域。當硬化反應的溫度低於
Tgo 時,此時的溫度所提供的動能不夠大,造成分子鏈無法移動或滑
動的溶膠-玻璃區,故硬化反應無法進行。若硬化反應的溫度在 Tgo
和 gel-Tg之間,則硬化反應在到達凝膠化之前就進行到溶膠-玻璃區
域時,硬化反應便停止下來。若把硬化反應溫度升高,則硬化反應因
分子鏈可以移動或滑動而繼續進行,直到另一個溶膠-玻璃區域後,
硬化反應又停止了。若反應溫度是介於 gel-Tg與 Tg∞之間,系統最初
是液體狀,隨反應時間增加則凝膠產生,最後系統玻璃化。若反應溫
度為 Tg∞,系統會得到最大 Tg。若硬化溫度大於 Tg∞,系統將會凝膠
而不會玻璃化,最後變成焦炭。
環氧樹脂硬化過程分成幾個步驟,未反應前是小分子結構,稱為
A-stage。在反應初期分子受熱,分子運動增強而黏度下降,而隨著時
間 (溫度) 的進行,小分子便相互結合形成線性及部份分枝的結構,
此時稱為 B-stage,此時材料尚未到達凝膠點。在凝膠點附近,黏度
急速增加,在凝膠點時,材料已失去流動性,無窮大的分枝狀的大分
子突然形成。在凝膠之後,硬化反應仍繼續進行,交聯密度逐漸增加,
直到完全硬化為止,此時為 C-stage,由於分子鏈受到網狀結構的限
制而無法移動,所以反應驟然停止或是反應速率極為緩慢,此現象稱
之為玻璃化,硬化反應也從 reaction control 變成 diffusion control
[57]。本研究將探討環氧樹脂的硬化反應,接著再探討添加 PPO 後對
環氧樹脂硬化系統反應性的影響。
2-9 聚醯胺纖維與複合材料

複合材料的組成成份主要是高分子基材 (matrix) 與纖維補強材
(reinforcements)。過去幾十年來,由於複合材料的快速發展,各種高
性能的合成纖維相繼推出,這些纖維通常具有高度的結晶性[58],其
機械性能方面也幾乎能達到理論值的上限。在高性能的有機纖維中,
尤以芳香族的高分子所合成之纖維性能為最佳,克維拉 (Kevlar) 纖
維是種有機纖維具有高強度、高模數 (modulus) 及耐熱性,Kevlar 纖
維主要是應用於有韌性及耐衝擊性需求的複合材料上,其結構可由化
學分析 (chemical analysis)、X 光射線檢晶儀 (X-ray crystallography)
和紅外光光譜儀 (infrared spectroscopy) 測定。Kevlar 纖維是由具有
高度方向性的柱狀 (rod-like) 分子所構成,直徑大小約為 12μm。醯
胺的單體是 para-phenylene terephthalamide (PPTA),如 Figure 2-6所
示,堅硬的線性高分子鏈的排列方向和纖維縱軸的方向是一致的,而
纖維橫向則靠分子間所形成的氫鍵所維繫;所以 Kevlar 纖維具有很
高的縱向的 (longitudinal) 強度,而橫向的 (transverse) 強度則較
低。PPTA 的製造是以適當化學當量的 p-phenylenediamine 和
terephthaloyl chloride 在低溫下經由溶液縮合聚合而成,其反應如
下:
NH2H2N C Cl
O
CCl
O
+ nn
N C
O
C
O
N
HH
+ 2n HCl( )n
聚合過程中所產生 HCl 則以 NaOH 中和並加以水洗。然後將
PPTA 溶於 100%濃硫酸 (∼20 wt%PPTA) 形成低黏度的PPTA液晶
態,溫度在 80℃下經由紡積器 (spinneret) 進行擠壓抽絲成型,再以
NaOH中和,並以清水洗去中和過程形成的 Na2SO4,再經過乾燥、


沿伸和拉長等處理,以增加纖維的勁度 (stiffness) 和強度[59]。
Kevlar 纖維針對不同的需要有四種型態[59]:
(1) 帶狀克維拉 (tyre core kevlar)。
(2) 用於繩索 (ropes) 和織布 (fabrics) 的中度模數之 Kevlar 29。
(3) 用於複合材料強化的纖維,具有較高模數之 Kevlar 49。
(4) 較 Kevlar 49高 40 % 模數的新產品-Kevlar 149。
其他聚醯胺纖維尚包括 Enka 生產的“Twaron”及 Teijin 生產的
“Technora”。
Kevlar 纖維的耐熱性質亦相當好,根據 Keinath 和 Morgan [60]
的研究,在氮氣的環境下,Kevlar 49和 Kevlar 149纖維的最大熱分
解 (thermal decomposition) 溫度高達 600oC。除此之外,Kevlar 纖維
本身的吸濕性也相當低。貯存在 97 %RH中一個星期後,經過熱分析
(TGA) 的結果,Kevlar 49 纖維的重量損失為 7.96%,而 Kevlar 149
纖維則祇有 2.86 %;貯存在 75%RH 後,Kevlar 49纖維中的水份含
量為 5.34 %,而 Kevlar 149纖維則更低至 1.19 % [60]。另外Kevlar 纖
維若暴露在太陽光及紫外光下將使纖維產生去色 (discoloration)、劣
化 (degradation) 以及抗張強度明顯下降的現象。在製造複合材料時
可添加吸收紫外光的填充物,降低此問題的影響性[61]。
關於 Kevlar 纖維的立體結構,多位學者[62-68]曾做過詳細的研
究,並且提出多種的結構模型:
(1) 摺曲結構 (pleat structure):
Dobb等人[64]以 electron diffraction及 electron microscope dark-
field image的技術對 Kevlar 49纖維研究結果顯示纖維為沿著軸向打
褶的平板狀的結構,如 Figure 2-7(a) 所示,褶曲主要有 500 nm和 250
nm兩種大小;而打褶的平板結構 (pleated sheet structure) 對纖維的機

械性質影響很大,特別是纖維的彈性模數 (elastic modulus)。
(2) 鏈-末端分佈結構 (chain-end distribution structure):
Morgan等人[62]根據 PPTA纖維的變形及破壞機構研究結果,提
出了 chain-end distribution模型,如 Figure 2-7(b) 所示。纖維表層的
chain-end的分佈是不規則的,而較內層的結構則是由直徑∼60 nm的
柱狀晶體規則及週期地排列而成,因此導致週期性的橫向弱平面
(periodic transverse weak plane),其間距約為 220 nm。
(3) 表層-核心結構 (skin-core structure):
Panar等人[65]利用 TEM,SEM,small-angle X-ray scattering以
及 wide-angle X-ray diffraction 等研究 PPTA 纖維,提出了 skin-core
模型,如 Figure 2-7(c) 所示。纖維的表層 (skin) 和核心 (core) 的結
構不同,skin的鬚狀結構沿著纖維的軸向排列得很規則,而 core的鬚
狀結構排列的次序則不規則;其中的鬚狀結構大約 600 nm。此外,
纖維的結構中有缺陷層 (defect layer) 存在,defect spacing大約是 35
nm。
(4) 結晶子層結構 (crystallinity layer structure):
Li 等人 [63]以 TEM, secondary electron imaging 和 electron
diffraction 等技術對 Kevlar 進行研究,提出了一個芳香族的聚醯胺
纖維模型,如 Figure 2-7(d) 所示。纖維的核心 (core) 是由層狀的結
構 (layered structure) 垂直於纖維軸的方向堆疊而成,層狀組織是由
直徑∼50 nm的棒狀的 (rod-sphaped) 晶體緊密排列而成;晶體的長
短則隨材料的分子量提高而增長,對於 Kevlar 49纖維而言,晶體長
度約為 210 nm,而且晶體的長度會影響纖維軸向的強度。纖維軸向
的鍵結力主要為化學鍵,而徑向的鍵結力則以氫鍵為主。
除了上述各種不同的聚醯胺纖維的結構模型的提出之外,另外有

一些學者[66-68]針對聚醯胺纖維的微結構和其機械性質做了詳細的
研究。如:Young 等人[66]研究的結果發現纖維的機械性質由其結構
所控制,整體分子排列的方向性及整齊度,分別決定纖維的強度及模
數。而 Krause等人[67]的研究指出:Kevlar 149纖維結構中晶體排列
的方向性及晶體的大小都大於 Kevlar 49纖維,因此 Kevlar 149纖維
的模數比 Kevlar 49纖維高出 35 %。此外,Dobb和 Robson [68]也以
electron microscope 和 X-ray diffraction針對多種不同的 Kevlar 纖維
的結構和機械性質加以研究。本研究第五章將以 Kevlar 49纖維來補
強基材,並探討纖維對複合材料之相形態及機械性質的影響。


第三章 氰酸酯硬化之環氧樹脂與聚氧化二甲苯
摻合體性質之研究
3-1 前言
利用橡膠改質環氧樹脂以增加其韌性已經有很長一段的時間,
然而對於高交聯密度的環氧樹脂而言,橡膠被高交聯密度的環氧樹脂
所包圍,所以橡膠顆粒無法有效地造成環氧樹脂基材的剪切降服
(shear yielding) 或空穴形成 (caviation),因此沒有增韌的效果。此外,
在相分離的過程中,會有少量的橡膠無法完全從環氧樹脂中分離出
來,導致環氧樹脂的模數、強度及 Tg點下降。所幸地是,1980年代
開始,一種新的增韌方法可以增加熱固性樹脂的韌性而不破壞其他的
機械性質,此法是利用高模數、高 Tg的熱塑性樹脂來增韌熱固性樹
脂。
聚氧化二甲苯 (PPO) 是由 GE公司所發展出來的熱塑性工程塑
膠,1957年 GE公司的 A. S. Hay以氧化偶合方法,製得高分子量的
2,6取代基的 PPO,1965年 GE公司利用此製程製造耐熱性 190oC的
工程塑膠。1967 年開始工業化,生產稍微犧牲其耐熱性而改善其加
工性的變性 PPO,商品名稱為NORYL。PPO為無定形 (amorphous) 的
熱塑性高分子,具有低介電常數、低消散因子、高玻璃轉移溫度、低
吸水性、耐衝擊性及對銅箔優良的黏著性,很少有工程塑膠同時具備
這些優點。但是 PPO 為非極性高分子,與環氧樹脂的摻混效果不是
很好,所以改善其相容性為一重要課題。
本研究是利用熱塑性 PPO 來改質環氧樹脂,除了增加韌性之
外,更可提升其他如電氣性質及耐熱性質等。本章節先探討 PPO 對
氰酸酯反應性的影響,之後再對於氰酸酯硬化環氧樹脂與 PPO 摻合

體的硬化反應動力學以及摻合體相分離及硬化最終的相形態也會加
以討論。此外,本研究也要探討添加反應性單體 (triallylisocyanurate,
TAIC) 後摻合體相形態的改變情況,這是一種新的方法來改善熱塑性
塑膠與環氧樹脂的相容性。
3-2 實驗
3-2-1 材料與藥品
1. 環氧樹脂,DGEBA (Diglycidyl Ether of Bisphenol-A),Shell出品,
商業名稱為 Epon 828。
2. 氰酸酯 (cyanate ester, PT-30),MW為 320到 420,Ciba-Geigy出品。
3. 聚氧化二甲苯 (polyphenylene oxide, PPO),本性黏度為 0.4 dl/g於
25oC氯仿,G.E. 出品。
HH
CH3
CH3
O( )n
OCN
CH2 CH2
OCN OCN
n
CH2 CH2CH2CH
O
- -O C
CH3
CH3
O CH2 CH
OH
O
CH3
CH3
C CH2O
O
CHCH2

4. 三丙烯基三聚氰酸鹽 (Triallylisocyanurate, TAIC),ACROS出品。
N
NN
O
O O
CH2CH CH2
CH2CH2CH
CH2 CHCH2
3-2-2 儀器設備
1. 微差掃瞄熱卡計 (Differential Scanning Calorimeter, DSC)
為 Perkin-Elmer DSC 7型,實驗前先利用銦 (In) 與鋅 (Zn) 標準
品測試校正溫度及熔融熱,所有樣品的測試皆在氮氣下進行。
2. 動態機械分析儀 (Dynamic mechanical analyzer)
DMA的型號為 Du Pont, model 2980,樣品大小約 10×5×1 mm,
測試方法為單懸臂彎曲方式,振幅為 30 µm,測試頻率為 1Hz,升溫
速率為 5oC/min,範圍為 50 ~ 250 oC。
3. 熱重分析儀 (Thermogravimetric analyzer)
TGA的型號為 Perkin Elmer TGA 7,樣品重量約為 10 mg,測試
條件為升溫速率 20oC/min,溫度範圍為 100 ~800 oC。
4. 熱機械分析儀 (Thermomechanical analyzer)
TMA的型號為 Perkin Elmer TMA 7,樣品大小約為 5×5×1 mm,
測試條件為升溫速率 10oC/min,溫度範圍為 50~250 oC。
5. 傅立葉轉換紅外光譜儀 (Fourier transform infrared spectrometer)

FTIR的型號為 BIO RAD FTS-40,測試條件為解析度 8 cm-1,圖
譜掃描次數為 16次,光譜的記錄範圍為 400~4000 cm-1。
6. 掃描式電子顯微鏡 (Scanning electron microscopy)
儀器型號為 Hitachi S-2300,實驗前先將樣品鍍金,實驗所使用
的加速電壓為 15Kv。
7. 萬能拉力機 (Tensometer)
儀器型號為 Instron 1302,拉伸測試是遵循 ASTM D638-64T,臨
界應變能釋放率GIC是遵循Mostovoy所設計的方法[70]。
8. 光學顯微鏡
儀器型號為 Nikon Model SN, Japan。
9. 介電常數測試儀
儀器型號為 HP-4291A。
3-2-3 樣品製備
1. 熱塑性高分子 PPO與環氧樹脂以熱熔融法混合
環氧樹脂在使用前皆經過除水過程。PPO先於溶於二氯甲烷,再
將溶液倒入甲醇溶液中,使 PPO在 poor solution中析出,然後過濾烘
乾,在 130oC下置於真空烘箱內烘乾四小時,取出後再與環氧樹脂於
170oC下抽真空攪拌混合,待 PPO與環氧樹脂完全混合後得到一透明
橙色的黏稠溶液,之後再加入 20 phr 的氰酸酯硬化劑,攪拌約二分
鐘,最後得到一透明琥珀色的三成份的溶液。因受限於黏度因素,PPO
的組成在 50 phr範圍內均可得到一混合均勻的三成份系統。(phr:相

對於每 100克環氧樹脂所添加的量)。
2. 添加 TAIC於摻合系統
本實驗選用 30 phr PPO的添加量來研究TAIC對摻合體相容性的
影響。由於在文獻中得知 TAIC 與 PPO會形成共聚物,且 TAIC 與環
氧樹脂相容性佳[69]。當環氧樹脂、PPO混合均勻後,接著加入氰酸
酯與 TAIC攪拌約二分鐘。最後得到一混合澄清液體。
3. 摻合體試片之製作
當熱熔融法混合均勻後,將混合物倒入預熱的模具中進行硬化反
應,硬化條件為 180 oC四小時,220 oC四小時。當硬化完成後,樹脂
必須經過緩慢的降溫退火過程才可從烘箱中取出。
3-2-4 實驗步驟
1. 熱分析實驗
本實驗採用 Perkin-Elmer DSC 7 型,將配製好的環氧樹脂摻合
體,取約 5-10 mg於 DSC鋁盤中,以 10 oC/min的加熱速率測試樣品
的放熱曲線,加熱溫度範圍為 50-350 oC,可得到動態掃瞄反應熱∆HT
(J/g)。
為了研究熱塑性高分子 PPO 對環氧樹脂的硬化反應動力的影
響,我們選擇恆溫的硬化溫度為 180、190、200及 210 oC,在實驗中
等溫反應的反應熱為∆HI,而反應的總放熱量為動態掃瞄所得到的反
應熱∆HT。所以在時間 t時的等溫轉化率為
áI (t) = ∆HI (t) / ∆HT
其中∆HI (t)為樣品在等溫反應時間 t時的反應放熱量。

2. 霍氏轉換紅外光譜實驗
本實驗採用霍氏轉換紅外光譜儀為 BIO-RAD FTS-40,測試條件
為光譜解析度 8 cm-1,掃瞄次數為 16次。記錄的波數範圍為 400-4000
cm-1。在實驗中使用成膜法來製備不同組成的環氧樹脂/PPO摻合體,
首先把各組成份溶解於二氯甲烷中配成 2.5 wt %的溶液,再成膜於溴
化鉀錠上,並於 50oC的真空烘箱中抽真空 12小時,之後隨著 180 oC
的恆溫反應做一系列的測試。
3. 核磁共振光譜實驗:
各種組成的環氧樹脂/PPO 系統在恆溫 180oC 時以不同的時間作
熱處理,再將這些樣品溶解於 CDCl3中作液態 C13-NMR 及固態 C13-
NMR鑑定反應物可能的反應機構。
4. 掃瞄式電子顯微鏡(SEM)實驗:
將樣品以雙面膠固定於 SEM 試件平台上,經過鍍金後,放大觀
察摻合體的破裂表面形態。
5. 動態機械分析實驗:
本實驗所使用的 DMA為 Du Pont, model 2980,將硬化完畢的摻
合體試片裁成大小約 10×5×1 mm,測試方法為單懸臂彎曲方式,振幅
為 30 µm,測試頻率為1 Hz,升溫速率為 5oC/min,範圍為 50 ~ 250 oC。
本實驗由動態機械分析所得到的資料包括:(1)玻璃轉移溫度,(2)高
分子摻合物的相容性,(3)兩相系統的相轉移,(4)分子運動。

6. 熱重分析實驗:
樣品為不同配方已硬化完畢的樹脂,樣品重量約為 10 mg,測試
條件為升溫速率 20oC/min,通入氮氣,溫度範圍為 100 ~800 oC。由
TGA圖形分析可得起使熱裂解溫度及最大熱裂解速率溫度,以了解
材料的熱穩定性。
7. 熱機械分析實驗:
將各種不同組成已硬化的樹脂置入 TMA 中,樣品大小約為
5×5×1 mm,測試條件為升溫速率 10oC/min,溫度範圍為 50~250 oC。
由 TMA圖形中可測得樹脂的熱膨脹係數以及概略的 Tg點,可了解樹
脂的熱穩定性。
8. 光學顯微鏡實驗:
以不同重量比之環氧樹脂/PPO 以二氯甲烷當溶劑,在載玻片上
成膜,並於控溫加熱台中等溫處理各組成樣品,以光學顯微鏡觀察摻
合體的相形態。
9. 機械性質測試:
硬化後樣品的抗張強度及模數測試是根據 ASTM 638。側面拋光
的樣品長度為 180 mm,厚度為 2 mm,中間狹窄的部分寬度為 13
mm。測試條件為拉伸速率為 1 mm/min,環境為 25oC。對於臨界應
變能釋放率 GIC測試方法為 tapered double cantilever beam (TDCB)方
法[70]。

10. 介電常數測試:
將硬化過後的摻合體裁切為 20×20×2 mm的大小,測試條件為 1
MHz,溫度為 25oC。
3-3 結果與討論
3-3-1 氰酸酯硬化動力學
在研究氰酸酯硬化環氧樹脂的硬化動力學之前,我們先探討純氰
酸酯的硬化行為。氰酸酯的硬化動力學已有許多學者提出[71-85],大
多數的研究是使用 2, 2’-bis(4-cyanatophenyl) propane (dicyanate of
bisphenol A, DCBA)[71-82],也有研究使用其他類型的雙官能基及單
官能基的氰酸酯[71, 82, 85]。這些氰酸酯的硬化反應有利用 bulk模式
[71-73, 77, 79, 80, 82, 83, 85],或是在溶液中進行[74-78, 81],硬化反
應無使用觸媒者[71-73, 79, 80, 82, 83],有使用觸媒者[78, 80, 81,
85]。雖然這些反應機構有使用觸媒及反應條件的差異,但是有一些
共通的現象如下所述:
1. 氰酸酯反應成 cyanurates 的反應熱∆H 約為 -105 KJ/mol OCN-
group。
2. 在極高溫度下,官能基的轉化率幾乎可達完全。
3. 在反應過程中可生成活性中間體,形成自催化反應的特性。
4. 反應速率對於少量不純物或觸媒非常敏感。
5. 整體的動力模式可用簡單的式子表示,無關觸媒的存在與否。
實驗中我們將氰酸酯混合 10, 30, 50 phr的 PPO在三種不同溫度
(200,210 和 220 oC) 中進行硬化反應。在實驗數據分析使用 Kamal
的自催化反應模式[86]。三種不同溫度 200,210和 220 oC 之反應速
率曲線表示於 Figure 3-1,在圖中這些速率曲線很明顯地具有自催化

反應特性,並且隨著反應溫度的提高及 PPO 含量的增加,最大反應
速率出現的時間有往前移的趨勢,所以 PPO 在氰酸酯硬化系統中有
催化反應的特性。此外混合系統中最大反應速率也會隨著 PPO 含量
的增加而加大,因此可知 PPO 的存在很明顯的加速了氰酸酯反應速
率。
所有詳細的等溫 (∆HI) 和掃瞄 (∆HT) 反應放熱和轉化率 (α)
皆列於 Table 3-1。氰酸酯/PPO混合系統的總反應熱會低於純氰酸酯
反應放熱,結合反應速率的改變,我們推論系統中 PPO 的存在可能
會改變混合系統的反應機構。在 Table 3-1中當反應溫度由 200 oC增
至 220 oC,則最終轉化率也會增加,這代表著在較高反應溫度下反應
系統達到擴散控制區 (玻璃態) 將會在較高的轉化率情況時發生。氰
酸酯的硬化反應系統之數據分析以 Kamal 所建議的自催化模式來分
析,同時參考 Simon和 Gilham所導的自催化反應方程式[87, 88],設
定反應級數 n = 2:
r = dα/dt = (k1 + k2 αm) (1-α)n
式子中的 α 為 OCN官能基的轉化率,k1及 k2為速率常數,m 及 n
為反應級數。此自催化模式可將氰酸酯的硬化反應在進入玻璃化前的
實驗數據描述的很好,如 Figure 3-2所示。由實驗結果得知自催化動
力模式很適合用在氰酸酯/PPO 摻合體之硬化系統,但是在玻璃態時
的擴散控制效應限制了氰酸酯硬化反應的進行,在玻璃化後氰酸酯系
統的硬化反應幾乎已經停止,導致系統的轉化率受到限制,所以反應
最後階段受到玻璃化之擴散控制效應影響甚巨。
3-3-2 氰酸酯硬化環氧樹脂/聚氧化二甲苯摻合體反應性分析

0 20 40 60 80 100 120 140
0.0000
0.0001
0.0002
0.0003
0.0004
0.0005
0.0006
experimental
autocatalytic model
Ra
te(1
/s)
Time(min)
0 20 40 60 80 100 120 140
0.0000
0.0002
0.0004
0.0006
0.0008
0.0010
0.0012
experimental
autocatalytic model
Ra
te(1
/s)
Time(min)
0 20 40 60 80 100 120 140
-0.0002
0.0000
0.0002
0.0004
0.0006
0.0008
0.0010
0.0012
0.0014
experimental
autocatalytic model
Ra
te(1
/s)
Time(min)

0 20 40 60 80 100 120 140
-0.0001
0.0000
0.0001
0.0002
0.0003
0.0004
0.0005
0.0006
0.0007
experimental
autocatalytic model
Ra
te(1
/s)
Time(min)
0 20 40 60 80 100 120 140
0.0000
0.0002
0.0004
0.0006
0.0008
0.0010
0.0012
experimental
autocatalytic modelRa
te(1
/s)
Time(min)
0 20 40 60 80 100 120 140
0.0000
0.0003
0.0006
0.0009
0.0012
0.0015
experimental
autocatalytic model
Ra
te(1
/s)
Time(min)

0 20 40 60 80 100 120 140
0.0000
0.0002
0.0004
0.0006
0.0008
0.0010
experimental autocatalytic model
Ra
te(
1/
Time(min)
0 20 40 60 80 100 120 140
0.0000
0.0003
0.0006
0.0009
0.0012
0.0015
experimental
autocatalytic modelRa
te(1
/s)
Time(min)
0 20 40 60 80 100 120 140
0.0000
0.0005
0.0010
0.0015
0.0020
0.0025
0.0030
experimental
autocatalytic model
Ra
te(1
/s)
Time(min)

0 20 40 60 80 100 120 140
0.0000
0.0002
0.0004
0.0006
0.0008
0.0010
experimental
autocatalytic model
Ra
te(1
/s)
Time(min)
0 20 40 60 80 100 120 140
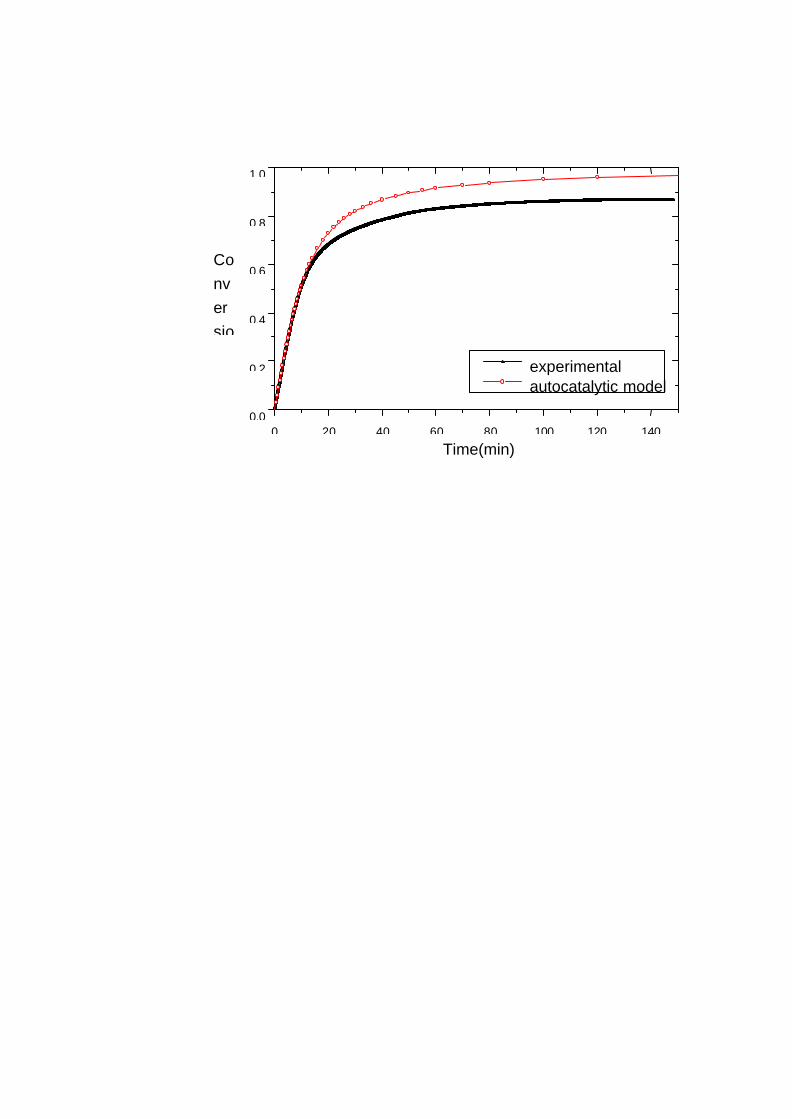
0.0000
0.0003
0.0006
0.0009
0.0012
0.0015
0.0018
0.0021
experimental
autocatalytic model
Ra
te(1
/s)
Time(min)
0 20 40 60 80 100 120 140
0.0000
0.0005
0.0010
0.0015
0.0020
0.0025
experimental
autocatalytic model
Ra
te(1
/s)
Time(min)

Table 3-1 Heats of reaction and autocatalytic model constants of neatcyanate ester and mixtures of cyanate ester with various PPO contents.Isothermal reaction
temperature (oC)∆HI (J/g) ∆HT (J/g) Conversion (α)
Neat cyanate ester200 593 89.2210 628 665 94.4220 637 95.8
10 phr PPO200 552 83.4210 598 662 90.3220 610 92.1
30 phr PPO200 516 85.0210 532 607 87.6220 570 93.9
50 phr PPO200 503 86.7210 521 580 89.8220 528 91.0
T (oC) m n k1 k2
Neat cyanate ester
200 1.60000 2 0.00025 0.00533
210 1.60000 2 0.00037 0.01092
220 1.80004 2 0.00053 0.01589
Cyanate ester/PPO (10 phr)
200 1.05710 2 0.00037 0.00322
210 1.25723 2 0.00063 0.00815
220 1.17506 2 0.00084 0.00968
Cyanate ester/PPO (30 phr)
200 0.93574 2 0.00083 0.00305
210 1.09790 2 0.00139 0.00546
210 1.19746 2 0.00229 0.01254
Cyanate ester/PPO (50 phr)
200 0.98688 2 0.00097 0.00277
210 1.00956 2 0.00175 0.00593
220 1.12361 2 0.00245 0.00846
0 20 40 60 80 100 120 1400.0
0.2
0.4
0.6
0.8
1.0
experimental autocatalytic model
Co
nv
er
sio
Time(min)

0 20 40 60 80 100 120 140
0.0
0.2
0.4
0.6
0.8
1.0
experimental
autocatalytic model
Co
nve
rsio
n
Time(min)
0 20 40 60 80 100 120 140
0.0
0.2
0.4
0.6
0.8
1.0
experimental
autocatalytic model
Co
nve
rsio
n
Time(min)
0 20 40 60 80 100 120 140
0.0
0.2
0.4
0.6
0.8
1.0
experimental
autocatalytic model
Co
nve
rsio
n
Time(min)

0 20 40 60 80 100 120 140
0.0
0.2
0.4
0.6
0.8
1.0
experimental
autocatalytic model
Co
nve
rsio
n
Time(min)
0 20 40 60 80 100 120 140
0.0
0.2
0.4
0.6
0.8
1.0
experimental
autocatalytic model
Co
nve
rsio
n
Time(min)
0 20 40 60 80 100 120 140
0.0
0.2
0.4
0.6
0.8
1.0
experimental
autocatalytic model
Co
nve
rsio
n
Time(min)

0 20 40 60 80 100 120 140
0.0
0.2
0.4
0.6
0.8
1.0
experimental
autocatalytic model
Co
nve
rsio
n
Time(min)
0 20 40 60 80 100 120 140
0.0
0.2
0.4
0.6
0.8
1.0
experimental
autocatalytic model
Co
nve
rsio
n
Time(min)
0 20 40 60 80 100 120 140
0.0
0.2
0.4
0.6
0.8
1.0
experimental
autocatalytic model
Co
nve
rsio
n
Time(min)

0 20 40 60 80 100 120 140
0.0
0.2
0.4
0.6
0.8
1.0
experimental
autocatalytic model
Co
nve
rsio
n
Time(min)
0 20 40 60 80 100 120 140
0.0
0.2
0.4
0.6
0.8
1.0
experimental
autocatalytic model
Co
nve
rsio
n
Time(min)
0 20 40 60 80 100 120 140
0.0
0.2
0.4
0.6
0.8
1.0
experimental
autocatalytic model
Co
nve
rsio
n
Time(min)

環氧樹脂 (DGEBA) 添加 20 phr氰酸酯和混合 10、30及 50 phr
的 PPO進行動態 DSC掃瞄,升溫速率是 10oC/min,反應熱的計算是
扣除掉摻合體中 PPO的重,根據環氧樹脂/氰酸酯的淨重計算而得。
Figure 3-3所示動態DSC熱分析,發現只有一個放熱峰在溫度200-300
oC的範圍內。總反應放熱量隨 PPO含量增加而下降,此現象可能的
原因是 PPO 含量的增加,會使環氧樹脂在硬化過程中會有交聯的立
體障礙;PPO的 Tg點高,會造成反應系統中擴散的限制。此外由 DSC
圖可知起始反應溫度 (Ti) 及最大放熱峰溫度 (Tp) 隨 PPO 的增加向
低溫處移動,起使反應溫度隨 PPO量增加 (0 phr→50 phr) 降低了 25
oC,最大放熱峰溫度降低了 24 oC,這意味著 PPO 會使系統的反應提
前在低溫進行,提升了硬化早期的反應速率。這些實驗結果列於 Table
3-2。在恆溫反應方面,我們選擇 180、190、200、210 oC進行硬化反
應,如 Figure 3-4 所示。特別的是在 180 oC 的恆溫反應中,未添加
PPO及添加10 phr PPO的系統中,動態DSC出現了兩個反應放熱峰,
原因是氰酸酯反應官能基空間位置不同,出現反應立體障礙效應,但
隨著PPO添加量的增加及反應溫度的提高,反應放熱峰又變成一個,
這可能是 PPO 的催化效應及高反應溫度會降低此效應的影響。由反
應轉化率對時間作圖 (Figure 3-5) 可知添加 PPO 其系統的總轉化率
下降,在玻璃化之後環氧樹脂的硬化反應幾乎停止,此外也與 PPO
的存在造成黏度的增加導致最終轉化率的下降。但是PPO的量愈多,
反應的起始時間會往前移,其結果列於 Table 3-3。下章節的 FTIR 圖
譜對系統反應性會做進一步的探討。由 Figure 3-6中顯示,系統的反
應速率必須反應一段時間後才會到達最大值,此結果符合自催化
反應模式,原因是系統中水氣及 PPO 的 -OH 基會容易與氰酸
酯的 -OCN起反應,產生催化效果,


Table 3-2 Dynamic DSC measurement for Epoxy/PPO blends withvarious PPO contents.
PPO content Ti (°C) Tp (°C) ∆HT (J/g)0 phr 235 264 30610 phr 234 263 30320 phr 224 250 28030 phr 218 245 27750 phr 210 240 274
100 phr 186 221 270Ramp : 10°C/min from 50°C to 350°C in 40 ml/min N2
Ti : the onset temperature of heat of reactionTp : the maximum peak temperature of heat of reaction∆HT : heat of reaction

Figure 3-4 Isothermal DSC thermograms of cyanate ester cured epoxy/PPO mixture samples.
Epoxy
210 oC
200 oC
190 oC
180 oC
Hea
t flo
w

Figure 3-4 Isothermal DSC thermograms of cyanate ester cured epoxy/PPO mixture samples.
210 oC
200oC
190oC
180 oC
PPO 10 phr
Hea
t flo
w

0 20 40 60 80 100 120
Figure 3-4 Isothermal DSC thermograms of cyanate ester cured epoxy/PPO mixture samples.
210 oC
200 oC
190 oC
180 oC
PPO 30 phr
He
at
flow
time (min)

0 20 40 60 80 100 120
Figure 3-4 Isothermal DSC thermograms of cyanate ester cured epoxy/PPO mixture samples.
210oC
200 oC
190 oC
180 oC
PPO 50 phr
Hea
t flo
w
time (min)

0 20 40 60 80 100 1200.0
0.2
0.4
0.6
0.8
1.0
Figure 3-5 Plots of the conversion versus time for cyanate ester cured epoxy/PPO mixture
samples at four different temperatures: (a) 180; (b) 190; (c) 200; (d) 210oC.
(a) Cure temperature: 180oC
0phr
10phr30phr 50phr
Co
nve
rsio
n (%
)
time (min)

0 20 40 60 80 100 1200.0
0.2
0.4
0.6
0.8
1.0
Figure 3-5 Plots of the conversion versus time for cyanate ester cured epoxy/PPO mixture
samples at four different temperatures: (a) 180; (b) 190; (c) 200; (d) 210oC.
(b) Cure temperature: 190oC
0phr10phr30phr50phr
Co
nve
rsio
n (%
)
time (min)

0 20 40 60 80 100 1200.0
0.2
0.4
0.6
0.8
1.0
Figure 3-5 Plots of the conversion versus time for cyanate ester cured epoxy/PPO mixture
samples at four different temperatures: (a) 180; (b) 190; (c) 200; (d) 210oC.
(c) Cure temperature: 200oC
0phr10phr30phr50phr
Co
nve
rsio
n (%
)
time (min)

0 20 40 60 80 100 1200.0
0.2
0.4
0.6
0.8
1.0
Figure 3-5 Plots of the conversion versus time for cyanate ester cured epoxy/PPO mixture
samples at four different temperatures: (a) 180; (b) 190; (c) 200; (d) 210oC.
(d) Cure temperature: 210oC
0phr
10phr30phr50phrC
on
vers
ion
(%)
time (min)

Table 3-3 Isothermal cure reaction for Epoxy/PPO blends with variousPPO contents.PPO content ti (min) tp (min) ∆HI (J/g) α (%)Isothermal
180°C0 phr 17.9 38.7 253 82.710 phr 13.2 30.3 242 79.930 phr 9.7 24.8 215 77.650 phr 6.2 19.2 208 75.9
Isothermal190°C0 phr 11.4 28.1 296 96.710 phr 8.0 24.4 267 88.130 phr 6.4 17.1 218 78.750 phr 4.5 14.5 212 77.3
Isothermal200°C0 phr 7.1 19.4 304 99.310 phr 5.8 19.7 292 96.430 phr 3.4 12.3 220 79.450 phr 3.5 10.1 217 79.2
ti : the exothermic initial timestp : the exothermic peak times∆HI : heat of reaction at isothermal temperature
0 20 40 60 80 100 1200.000
0.005
0.010
0.015
0.020
0.025
0.030
0.035
0.040
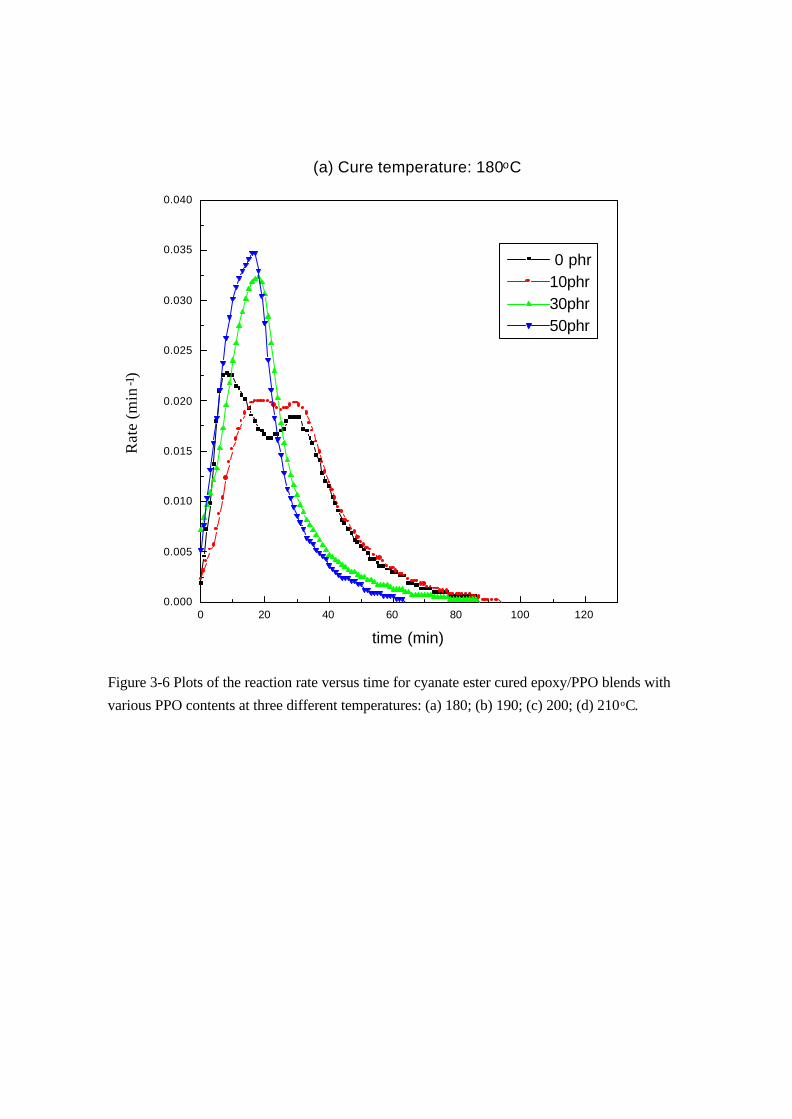
Figure 3-6 Plots of the reaction rate versus time for cyanate ester cured epoxy/PPO blends with
various PPO contents at three different temperatures: (a) 180; (b) 190; (c) 200; (d) 210oC.
(a) Cure temperature: 180oC
0 phr10phr30phr50phr
Rat
e (m
in -
1 )
time (min)

0 20 40 60 80 100 1200.00
0.01
0.02
0.03
0.04
0.05
Figure 3-6 Plots of the reaction rate versus time for cyanate ester cured epoxy/PPO blends with
various PPO contents at three different temperatures: (a) 180; (b) 190; (c) 200; (d) 210oC.
(b) Cure temperature: 190oC
0phr
10phr30phr50phr
Rat
e (m
in -
1)
time (min)

0 20 40 60 80 100 1200.00
0.02
0.04
0.06
0.08
Figure 3-6 Plots of the reaction rate versus time for cyanate ester cured epoxy/PPO blends with
various PPO contents at three different temperatures: (a) 180; (b) 190; (c) 200; (d) 210oC.
(c) Cure temperature: 200oC
0phr10phr30phr
50phr
Ra
te (
min
-1)
time (min)

0 20 40 60 80 100 1200.00
0.02
0.04
0.06
0.08
0.10
Figure 3-6 Plots of the reaction rate versus time for cyanate ester cured epoxy/PPO blends with
various PPO contents at three different temperatures: (a) 180; (b) 190; (c) 200; (d) 210oC.
(d) Cure temperature: 210oC
0phr10phr
30phr50phr
Ra
te (
min
-1)
time (min)

所以本系統選擇自催化機構來分析反應動力常數,自催化反應動力學
方程式如下[86]:
r = dα/dt = (k1 + k2 αm) (1-α)n
式中轉化率α(t)可從相同溫度下,不同時間所累積的放熱量∆H(t)除以
反應總放熱量∆HT求得。以下步驟求出各個常數及級數。
步驟一:At α = 0 ⇒ dα/dt = k1
步驟二:ln (dα/dt) = ln (k1 + k2 αm) + n ln (1-α)
Plot of ln (dα/dt) vs. ln (1-α)
⇒ n
步驟三:ln [(dα/dt)/(1-α)n - k1] = ln k2 + m ln α
Plot of ln [(dα/dt)/(1-α)n - k1] vs. ln α
⇒ m、k2
疊代法:將所求得的 m、k1、k2值代入步驟二,可得新的 n值,對步
驟二及步驟三做一系列的疊代,直到 m、n值收斂為止。
本實驗所測得的系統動力學參數列於 Table 3-4,由分析得知純的
環氧樹脂與添加 10 phr PPO 的摻合系統其反應級數 m 值約為一點
多,n值約為 0.7。而添加量為 30 phr與 50 phr時,其反應級數 m值
約為三左右,n值約為一點多,彼此接近但與純環氧樹脂系統大大不
同。由此可知 PPO 添加量增加時會改變系統的反應機構。在相同溫
度下,PPO 量愈多,其反應速率常數 k1與 k2皆變大。所以添加 PPO
於反應系統中會加速環氧樹脂的硬化反應,可能是 PPO 末端的 OH
基會催化此反應系統。
3-3-3 摻合體反應過程之 FTIR 分析
在以 FTIR 量測摻合體反應性之前,首先將環氧樹脂塗佈在 KBr

Table 3-4 Autocatalytic model constants for cyanate ester cured
epoxy/PPO blends system.
T (oC) m n k1 k2
Neat epoxy
190 1.3719 0.732 0.002395 0.1705
200 1.1707 0.639 0.003781 0.1977
210 1.1636 0.501 0.007597 0.1966
Epoxy/PPO (10 phr)
190 1.6677 0.7073 0.002220 0.1286
200 1.5726 0.7805 0.002591 0.2771
210 1.4995 0.7841 0.009551 0.3109
Epoxy/PPO (30 phr)
180 3.2885 1.2654 0.007166 0.4504
190 3.0798 1.1562 0.007889 0.5130
200 3.0937 1.1761 0.015263 0.7921
210 3.2679 1.1223 0.015263 1.0059
Epoxy/PPO (50 phr)
180 2.9863 1.0330 0.005183 0.3554
190 3.3983 1.1378 0.012734 0.5398
200 2.8353 1.0903 0.024148 0.4797
210 3.4884 1.1229 0.025907 1.1852

鹽片上,加熱並利用 FTIR 監測熱處理過程中環氧樹脂各吸收官能基
的圖譜變化,以了解環氧樹脂的本質。Figure 3-7 為純環氧樹脂
DGEBA在 180 oC做不同時間熱處理的 FTIR 圖譜,在圖譜中環氧基
的主要特性吸收峰在波數 900 cm-1,而氫氧基吸收範圍為 3200-3700
cm-1之間。在圖中環氧基的 IR吸收峰在熱處理 35分鐘前幾乎沒有變
化,之後隨加熱時間增加而稍為減小;而氫氧基隨環氧基吸收減少而
增加。有許多文獻顯示環氧基與氫氧基之間在高溫時會有鏈成長反
應,所以此反應可能是環境中的水氣及 PPO末端氫氧基所造成的。
完全純化的 cyanate ester在加熱後幾乎沒有反應,而少量的不純
物,如親核之活性氫物種 H2O、Ar′OH是起始反應所必須的催化劑。
氰酸酯的 C≡N 與系統中些微的水氣產生中間產物,最後生成穩態的
carbamate 結構,不過中間產物與 carbamate 也可形成氮雜環 triazine
結構 [89]。此外氰酸酯的 C≡N 也會與芳香族酚類反應產生
imidocarbonate,此為一平衡反應,若增加 triazine濃度或是降低溫度,
反應往 imidocarbonate 移動,一個 imidocarbonate 會繼續與兩個氰酸
酯反應形成芳香族三聚氰酸鹽 (aryl cyanurate),並放出一個酚[89,
90]。在環氧樹脂與氰酸酯的反應系統有文獻提出是利用模式反應物
來探討其可能的反應機構[3],研究中指出有三種反應出現在環氧樹
脂與氰酸酯的反應系統中,一是氰酸酯的環化反應,另一是環氧樹脂
與氫氧基的加成反應 (醚化反應),此外尚有C≡N官能基和環氧基反
應形成 oxazoline的五環結構。
本研究發現在環氧樹脂與氰酸酯的反應中 (Figure 3-8),除了少
量的 C≡N會直接與環氧基反應形成 oxazoline,其特性吸收峰的位置
為 1650 cm-1。大部分的氰酸酯中C≡N會與水氣反應產生 carbamate,



接著反應產生三聚氰酸鹽,三聚氰酸鹽之特性官能基的吸收峰為1566
cm-1 (ν C=N−C) 及 1369 cm-1 (ν N−C−O),所以C≡N (2270及 2230 cm-1)
的吸收峰強度隨著反應時間下降。接著環氧基經由插入反應進入
cyanurate環而形成 alkylcyanurate,alkylcyanurate 再進行異構化作用
生成 alkylisocyanurate,其特性吸收峰為 1696 cm-1及 1457 cm-1。最後
環氧基與 alkylisocyanurate 反應生成 oxazolidinone,其特性吸收峰位
置為 1753 cm-1,其相關反應式已有一些學者利用模式化合物證明出
來[91-93],其反應機構如 Figure 3-9所示。此外環氧樹脂/氰酸酯系統
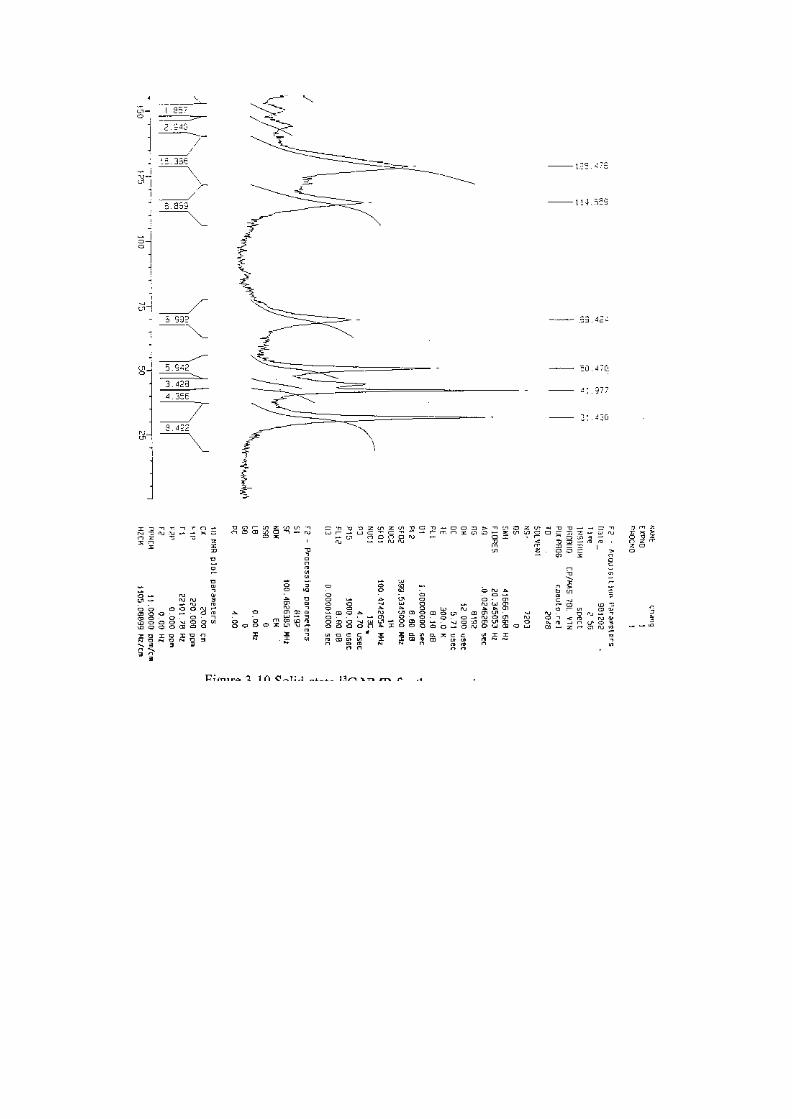
反應 35分鐘的 NMR圖譜 (Figure 3-10) 中也發現 triazine結構 (173
ppm)、isocyanurate的 C=O結構 (149 ppm) 及 oxazolidinone的 C=O
結構 (155 ppm),這時氰酸酯 C≡N (108.9 ppm) 訊號已經消失。綜合
FTIR 所觀察的結果,氰酸酯的C≡N與環氧樹脂的環氧基的吸收強度
隨反應時間增長而下降;而 oxazolidinone和 isocyanurate的吸收峰隨
反應時間增長而增強;此外 cyanurate的異構化結果,cyanurate的吸
收峰先隨反應時間增加而增強,之後又些微下降。
在實驗中發現環氧樹脂與氰酸酯系統中添加了 PPO之後,其反
應速率會隨著 PPO所加入的量增加而加速,所以我們先探討氰酸酯
與 PPO間的反應關係。由 Figure 3-11至 Figure 3-13可知隨著 PPO
含量增加,會加速 C≡N的反應,Figure 3-14(a)為 C≡N官能基的相對
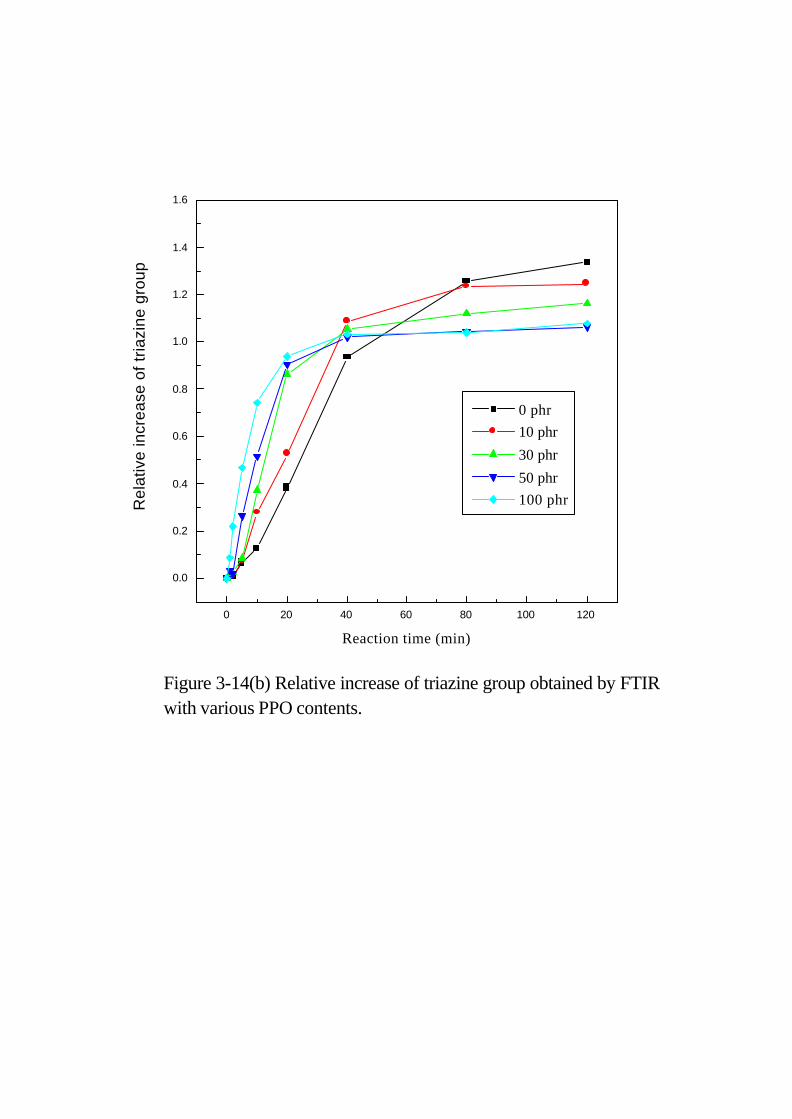
遞減圖,我們可更清楚的看出C≡N的反應速率。Triazine ring (1566
cm –1) 與 cyanurate (1369 cm –1) 吸收峰變大,但隨著 PPO量增加,
此兩結構的吸收峰強度卻減少,原因是 PPO的加入會阻礙網狀結構
形成,如 Figure 3-14(b)所示。中間產物 carbamate與 imidocarbonate,
出現在約 1680 cm –1,但並不明顯。當 PPO量為 0 phr、10 phr時,
carbamate與 imidocarbonate的吸收峰隨著時間增

Figure 3-9 Reaction scheme proposed between epoxy and cyanate ester.




0 20 40 60 80 100 120
-0.6
-0.5
-0.4
-0.3
-0.2
-0.1
0.0
Figure 3-14(a) Relative decrease of cyanate group obtained by FTIR with various PPO contents.
0phr
10ph r
30ph r
50ph r
100phr
Cya
nate
(Ai-
Ao)
/Ao
Reaction time (min)
0 20 40 60 80 100 120
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
Figure 3-14(b) Relative increase of triazine group obtained by FTIR with various PPO contents.
0 phr
10 phr
30 phr
50 phr
100 phrRe
lativ
e in
cre
ase
of
tria
zin
e g
rou
p
Reaction time (min)

加而增加之後又下降;但當 PPO量多至 50 phr時,吸收峰只有增加
而不會再下降。原因是少量 PPO 時, imidocarbonate 會繼續與
cyanate ester反應形成 triazine ring;但 PPO量多時,此反應則會受
到阻礙,使得反應停留在小分子而無法形成網狀結構。
由 FTIR 及 NMR 觀察得知,存在系統中之些微的水氣及 PPO
末端 OH基會催化此反應系統,產生了新的反應機制。在添加 50 phr
PPO 的 FTIR圖 (Figure 3-15) 中,反應初期氰酸酯中的 C≡N會與水
氣與 PPO-OH反應產生 carbamate及 imidocarbonate,接著反應產生
三聚氰酸鹽,此系統中C≡N的消失速率比未添加 PPO的系統還快,
但是由 FTIR圖中發現 isocyanurate和 oxazolidinone的吸收峰並不明
顯,原因可能是 C≡N 雖然可快速地與 PPO-OH 反應,其所產生的
cyanurate環端會接著分子量很大的 PPO部份,所以不容易再與環氧
基反應生成 isocyanurate與 oxazolidinone;在 NMR所作的吸收峰強
度變化趨勢圖譜中,在PPO含量高時oxazolidinone的C=O結構 (155
ppm) 在反應後期已無明顯的變化,未添加及低 PPO 含量時其
oxazolidinone 峰的強度有繼續增加的趨勢 (Figure 3-16)。所以 PPO
的存在雖然會加速與 C≡N 的反應,但是卻會阻礙 cyanurate 與環氧
基的反應,由 DSC 中也發現此現象,隨 PPO 含量增加,系統起始
反應加快,但是最終轉化率降低。由此可知混合系統並非只有單純
的環氧基與氰酸酯間的加成反應,當系統含有 H2O與 ArOH時,則
有其他反應發生,也改變了反應機構及反應速率。
為暸解其官能基反應的速率,我們可藉由 FTIR儀器,量測其反
應官能基隨時間變化之相對消長情形 (如 Figure 3-17所示)。在 FTIR
圖譜分析中我們知道存在環氧樹脂及PPO中之甲基是不隨著反應而
改變,其波數 (wavenumber) 是出現在 2975 cm-1,而氰酸酯

30 40 50 60 70 80 90 100 110 120 130
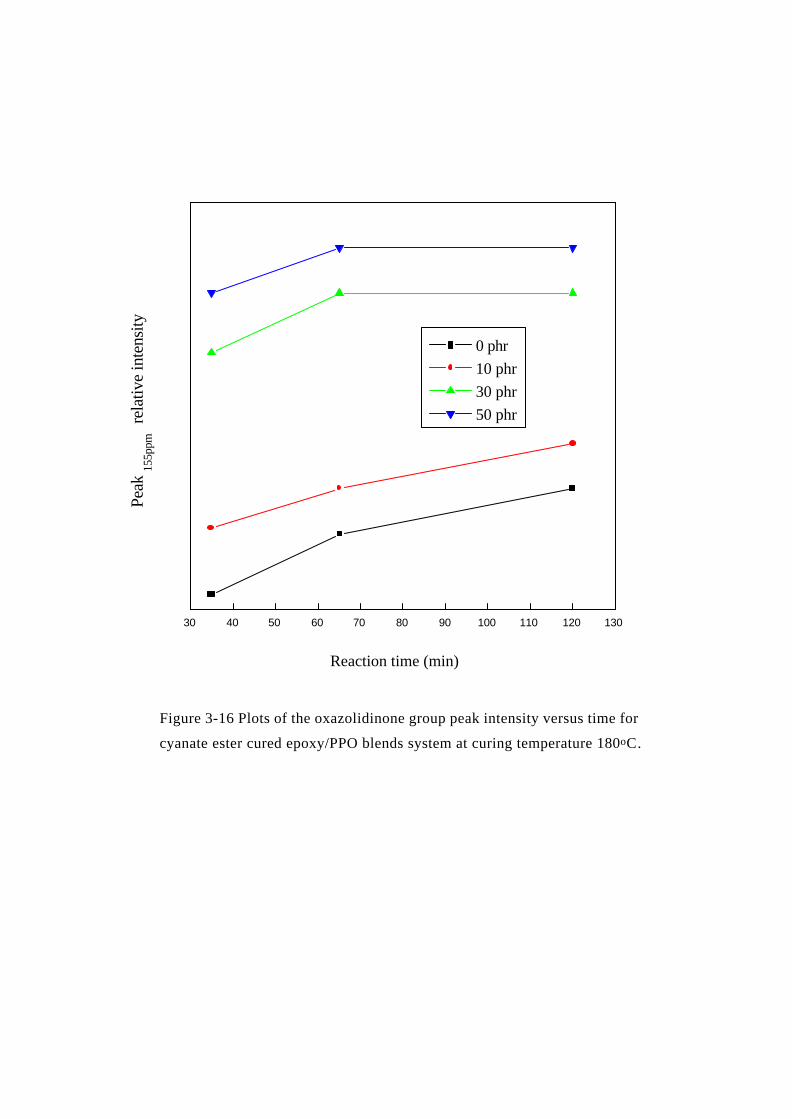
Figure 3-16 Plots of the oxazolidinone group peak intensity versus time for
cyanate ester cured epoxy/PPO blends system at curing temperature 180oC.
0 phr
10 phr
30 phr
50 phr
Peak
155p
pm r
elat
ive
inte
nsity
Reaction time (min)

0 5 10 15 20
-1.0
-0.8
-0.6
-0.4
-0.2
0.0
Figure 3-17 Relative decrease of cyanate group obtained by FTIR with various PPO contents
0 phr PPO
10 phr PPO
30 phr PPO
50 phr PPO
Rel
ativ
e de
crea
se o
f O
CN
gro
up
time (min)

之 C≡N反應官能基,其波數是出現在2220 cm-1及 2270 cm-1的位置,
我們可藉由圖形上 C≡N 的特性峰高度,相對於甲基特性峰高度的
變化作圖,其相對關係式如下:
t = 0,1,2,5,10,20 min
藉由此關係式可得到 C≡N 及環氧基其相對強度的消失速率對時
間作圖,我們發現 C≡N反應隨 PPO的增加而加速,證實了 PPO 的存
在會對系統造成影響。
由 FTIR 中 C≡N 官能基消失速率的觀察得到,在反應過了 20
分鐘後,即快完全消失;而 cyanurate的吸收峰先隨反應時間增加而
增強,之後又些微下降;。藉由 Solid NMR及 liquid NMR的分析,
我們可以由 liquid NMR 圖譜中發現,在反應前段仍可以發現 C≡N
官能基的碳,出現在 108 ppm (如 Figure 3-18所示),而在反應經過
35分鐘後的Solid NMR圖譜中已找不出此官能基 (如Figure 3-10所
示)。
在反應經過 35分鐘後的 Solid NMR圖譜中我們也由 (如Figure
3-10所示) 中 173 ppm處發現存在於氮環 (triazine) 上的碳,此氮環
的出現和我們先前所提及之反應機構是相符合的。而在 Solid NMR
反應過程中 oxazolidinone 上 C=O 官能基 (155 ppm) 的變化 (如
Figure 3-16所示),我們發現 PPO 含量為 0 phr與 10 phr的反應
t
t
group] methyl of[function decrease][function
=tA
0
0t
AA - A
=tR


趨勢是相近的;而 30 phr與 50 phr的反應趨勢也是相近的,且由反
應時間 35 min到 65 min的趨勢圖中,亦可發現 PPO含量為 30及 50
phr之 oxazolidinone上 C=O的訊號強度在反應後期已無明顯變化,
這點亦符合先前的推論。
3-3-4 摻合體硬化過程相分離行為及摻合體硬化過後的相型
態之探討
環氧樹脂/PPO 系統在混合後呈現一透明橙黃色的均勻相,相分
離在硬化過程中自始至終都在進行,並且在硬化過程中相區域及相大
小都有明顯的改變;在文獻中環氧樹脂/PPO 混合物是遵循上臨界溶
液溫度 (upper critical solution temperature, UCST) 行為[16]。實驗中
利用 SEM對 180 oC下 50 phr的摻合物樣品之相形態隨硬化時間的變
化做觀察。由於低 PPO 含量時樣品呈現黏稠液態狀,所以無法使用
SEM偵測。Figure 3-19為在等溫 180 oC不同的硬化時間 (30 sec、5、
10和 20 min) 時環氧樹脂/PPO (50 phr) 之混合物樣品之 SEM圖。在
經過 30 sec 的硬化時間,顯示混合物在反應初期是均勻狀的。硬化時
間到了 5 min,摻合體的破壞面呈現一種富環氧樹脂與富 PPO區域交
互分散著並沒有明顯的界面,這時無法分辨出連續相及非連續相。當
硬化持續到 10 min,富 PPO區域清楚地形成網狀結構且互相交錯連
接著,形成一連續相。由於環氧樹脂分子量增加,UCST圖便往上移,
所以此系統就經由 spinodal decomposition (SD) 產生相分離,如Figure
3-20。如同 SEM 所觀察到的,硬化過後的相形態是固定在 SD 的最
後階段,圓球狀環氧樹脂均勻分佈在 PPO的連續相中。
此外本實驗也利用光學顯微鏡觀察添加 10 phr及 30 phr PPO之


0 10 20 30 40 50 60
Figure 3-20 Phase diagram of cyanate ester cured epoxy/PPO (50 phr) blend.
PPO content (phr)
phase seperation occur
conversionp
p = 02 phases

摻合體。在 10 phr PPO 系統中,反應初期是呈現一均勻狀,以 180 oC
反應至 5 min,從原本均勻的混合物中分離出約 1µm的微小顆粒,
隨著反應時間的增加微小顆粒漸漸變大,此相分離方式是成核模式
(nucleation and growth mechanism, NG),所以在 PPO含量較低時,混
合系統是以 NG模式進行相分離,如Figure 3-21。而在 PPO含量 30 phr
時的相分離形式就與 10 phr的相分離形式大大不同。Figure 3-22為 30
phr PPO的摻合體以 180 oC反應不同時間的相圖,圖中顯示系統在反
應至 1 min時由均勻相開始轉變為極微小相互連接的結構,隨著反應
時間的增加,這些網狀微結構有變粗的現象。反應至 10 min,相分離
開始時之網狀結構的連續性被界面張力所打斷,反應至 20 min 後,
摻合體相形態形成一分散的粒滴形態。
當系統進入到介穩態區域 (metastable region) 時,相分離機構就
會被兩種因素控制,一為相分離的速率,另一為硬化反應速率[94]。
K = phase separation rate/cure reaction rate
Figure 3-23根據 K 值顯示在介穩態區域中可能的相分離趨勢走
向。當 K 值趨近於無窮大時,平衡會迅速地到達且系統進行的方向
沿著 binodal曲線 (a軌跡),分離相的組成會沿著另一邊的 binodal曲
線 (a’軌跡)。假使 K 值趨近於零,反應速率很快,沿軌跡 b 到達
spinodal曲線才有相分離產生,此時的相分離是藉著從低濃度擴散到
高濃度的區域,擴散係數為負值。Spinodal相分離所形成的相形態通
常是相互連接性的結構 (bicontinuous structure)。
改質劑最初的體積組成分率 (φMo) 也會影響相分離的反應機
構。若φMo接近臨界體積組成分率φMcrit,SD較容易產生;若φMo遠離

(a)
(b)
(c)
Figure 3-21 Optical micrographs of cyanate ester cured epoxy/PPO (10
phr) cured for different times at 180oC: (a) 5 min; (b) 10 min; (c) 20 min.

(a)
(b)
(c)
Figure 3-22 Optical micrographs of cyanate ester cured epoxy/PPO (30
phr) cured for different times at 180oC: (a) 1 min; (b) 2 min; (c) 5 min; (d)
10 min; (e) 15 min; (f) 20 min.

(d)
(e)
(f)
Figure 3-22 Optical micrographs of cyanate ester cured epoxy/PPO (30
phr) cured for different times at 180oC: (a) 1 min; (b) 2 min; (c) 5 min; (d)
10 min; (e) 15 min; (f) 20 min.

Figure 3-23 Possible trajectories in the metastable region of the conversion vs. composition phase diagram; a' and b' represent the compositions of the dispersed phase corresponding to trajectories a and b, respectively(K = phase separation rate/cure reaction rate, trajectory a isassociated to an NG mechanism while trajectory b leads to SD).
b'ba'a
K ∞0K
Ext
ent o
f rea
ctio
n, p
PPO content (phr)

φMcrit,則較容易產生 NG。由Monte Carlo 模擬熱塑性塑膠改質環氧樹
脂之相分離過程顯示[95],SD出現在較接近臨界點的位置時與硬化
反應速率無關;若改質劑組成遠離臨界點時,隨硬化反應速率增加則
相分離模式會由 NG 變成 SD。由此可知在環氧樹脂的改質系統中,
改質劑的濃度遠低於或大於臨界濃度,則相分離是以 NG模式進行。
SD 通常是發生在使用高分子量的熱塑性改質劑或是改質劑的濃度接
近臨界濃度,特別在高硬化反應速率或是兩相界面張力高時更容易發
生[96-101]。
在等溫硬化過程摻合體之形態改變不僅是硬化時間的函數,同時
也受系統組成的影響。由 Figure 3-24顯示摻合體的破壞面在 PPO含
量為 10 phr 時,PPO 形成顆粒狀分佈於環氧樹脂連續相中,破壞面
經由二氯甲烷蝕刻後,PPO相被溶解掉呈現坑洞狀散佈於環氧樹脂連
續相中。當摻合體 PPO 含量為 50 phr時,此時環氧樹脂形成顆粒狀
分佈於 PPO 連續相中,破壞面經由二氯甲烷蝕刻後呈現環氧樹脂顆
粒狀的結構,如 Figure 3-25。值得注意的是假使 PPO 與環氧樹脂間
沒有良好的鍵結,當溶劑溶蝕 PPO 後應呈現出光滑的環氧樹脂顆粒
球面。但是圖中顯示仍存有 PPO 被覆在環氧樹脂顆粒上,這表示兩
相界面間有良好的化學鍵結。當 PPO含量為 30 phr時,則摻合體硬
化後的相形態表現出一混合形態,此混合形態包含了一相類似環氧樹
脂摻合較少 PPO 時的 PPO 顆粒分散於環氧樹脂連續相之相形態,而
另一相為類似 PPO含量較高時環氧樹脂顆粒分散於 PPO連續相之相
形態,此結構也可稱之部分相反轉,如 Figure 3-26。
為了改善摻合體的相容性,我們使用 triallylisocyanurate (TAIC)
為相容改質劑。Figure 2-27顯示添加不同含量 TAIC 之 30 phr PPO摻
合體的破壞面;在富環氧樹脂的區域中,經由二氯甲烷蝕刻 PPO


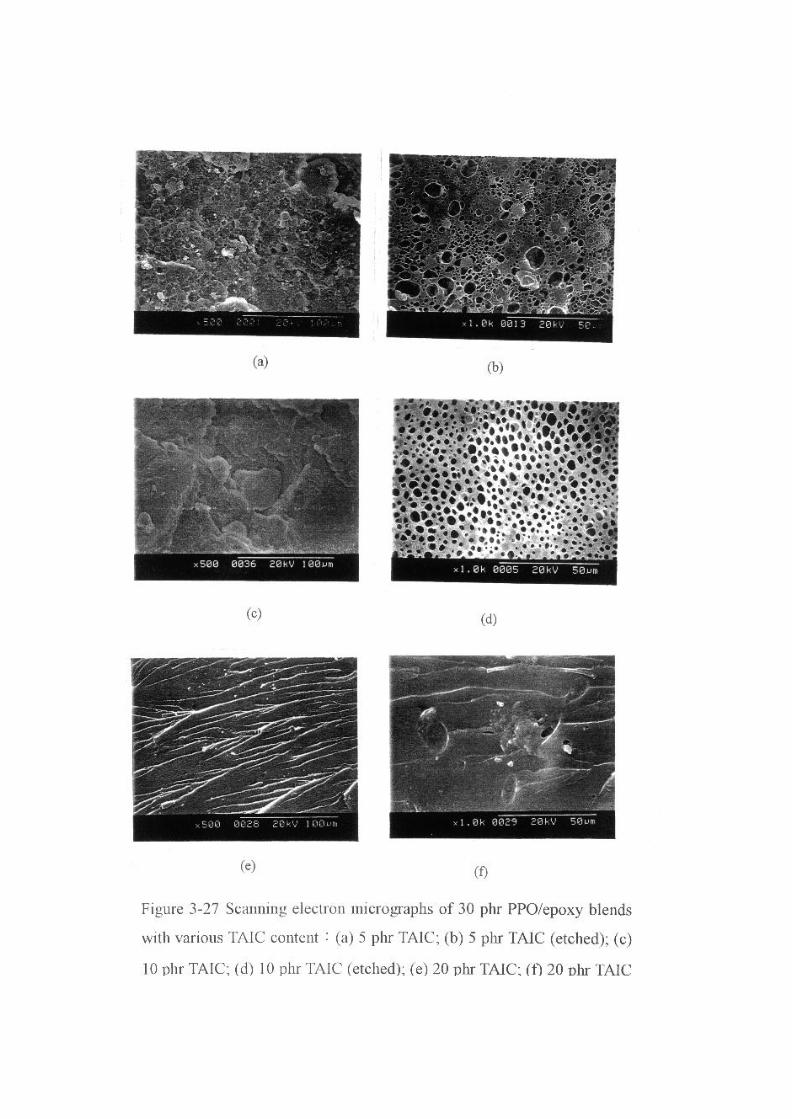

後留下孔洞,而未添加 TAIC 的破壞面之孔洞最大,這表示 PPO 聚
集的情況較嚴重。但隨著 TAIC的添加量增加,PPO相的區域漸漸
縮小,當 TAIC 添加量為 20 phr時,摻合體的破壞面變得非常均勻,
原因可能是環氧樹脂與 TAIC的相容性佳且 PPO分子鏈與 TAIC產生
鍵結。另一可能造成相容性增加的原因是產生接枝共聚物阻礙了 SD
相分離的進行,TAIC/PPO接枝共聚物的產生是由於 PPO 上的甲基很
容易受到 TAIC 自由基的攻擊[69],鍵結過後的 PPO 就不易受到二氯
甲烷的萃取,摻合體之抗溶劑性質提升。
3-3-5 摻合體的動態機械分析
由動態機械分析 (DMA) 得知氰酸酯硬化環氧樹脂與 PPO 摻合
呈現出兩相的特性和些微互溶的情況。Figure 2-28顯示純 PPO的 Tg
點為 220oC,當溫度高於 Tg點後,熱塑性 PPO的儲存模數 E’大幅度
的下降如同材料從塑性到黏彈性產生軟化,tanδ 也顯示很陡峭的阻
尼峰,這表示 PPO分子出現長距離的分子運動。但是 PPO與環氧樹
脂混合後,溫度在 PPO 相的 Tg之上,儲存模數下降的速率緩慢且橡
膠高原區的模數維持在一高水平,此現象意味著是 PPO 分子鏈與環
氧樹脂網狀結構相互纏繞導致分子運動受到限制。當 PPO 的含量增
加,摻合體的相形態變成為環氧樹脂顆粒分散在 PPO 連續相中,儲
存模數也顯示只有一個比較明顯的玻璃轉移區在靠近 PPO 相的轉移
區。所以在兩相玻璃轉移區之間的儲存模數大小是根據摻合物組成比
例與相之連續性或分散性而定。
摻合過後的環氧樹脂相與 PPO相的 tanδ 峰高度比純環氧樹脂相
與純 PPO 相的高度還低,如 Figure 3-29所示,原因是環氧樹脂與 PPO
分子相互纏繞的效應。tanδ 峰的高度變化也與相的連續性有
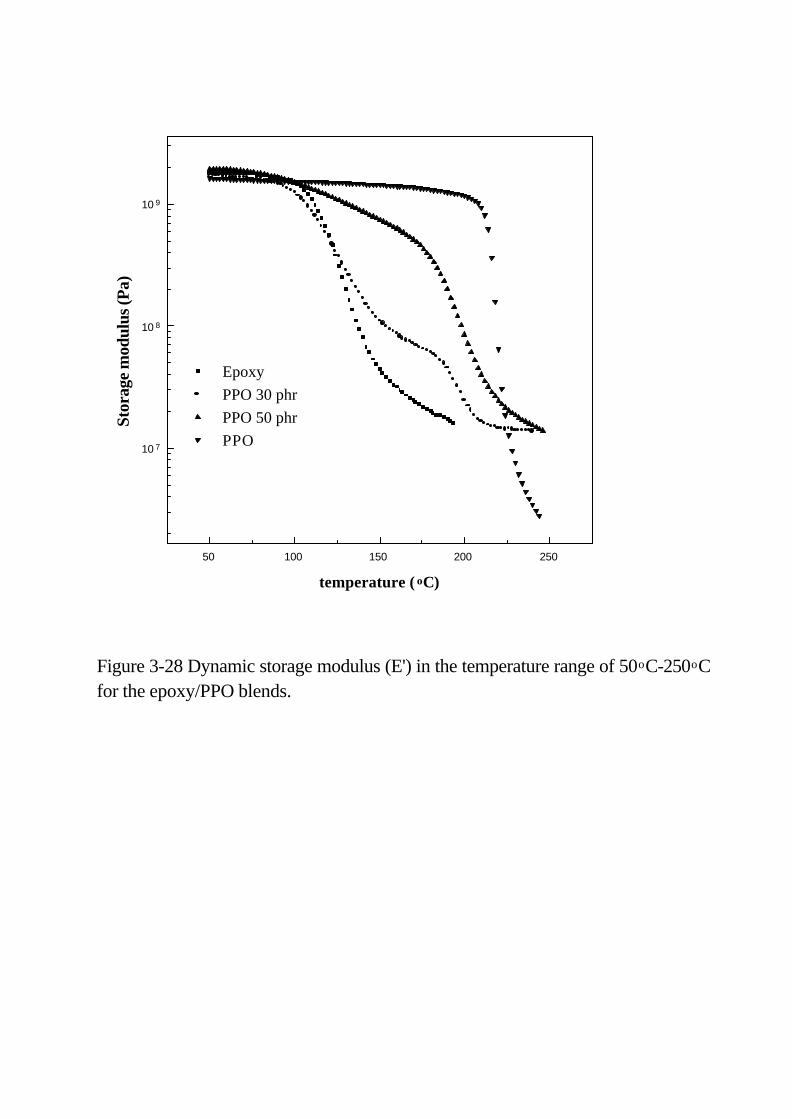
50 100 150 200 250
10 7
10 8
10 9
Figure 3-28 Dynamic storage modulus (E') in the temperature range of 50oC-250oC for the epoxy/PPO blends.
Epoxy
PPO 30 phr
PPO 50 phr
PPO
Sto
rage
mod
ulu
s (P
a)
temperature ( oC)

50 100 150 200 2500.01
0.1
1
Figure 3-29 Relaxation peaks (tan d) in the temperature range of 50oC-250oC for the epoxy/PPO blends.
Epoxy
PPO 30 phr
PPO 50 phr
PPO
tanδ
temperature (oC)

關,當 PPO添加量增加,PPO之 tanδ 峰的高度增加且環氧樹脂之 tanδ
峰的高度漸漸下降,當 PPO 變成連續相時,環氧樹脂之阻尼幾乎消
失,所以摻合體的性質被 PPO 相的阻尼所主導。由此可知摻合體的
相形態也可利用阻尼的特性來判斷[102-104]。
Figure 3-30為環氧樹脂/PPO (30 phr) 摻合 20 phr TAIC的 tanδ 對
溫度的作圖。未添加 TAIC之摻合體的 PPO相之 Tg約為 200 oC,但
隨著 TAIC的添加而 PPO相之 Tg下降。當 TAIC 添加量為 20 phr時,
兩相的 Tg往內移,這表示摻合體相容性的增加,最後形成一個形狀
寬廣的阻尼峰。造成阻尼峰寬廣的原因是 PPO鍵結於 TAIC的網狀結
構,形成摻合體複雜的結構。
3-3-6 摻合體的機械性質及介電性質之探討
Figure 3-31為摻合體在室溫下 PPO 含量與抗張強度和模數的圖
形。隨著 PPO含量增加,抗張強度從 68 Mpa增加至 72 MPa (添加量
為 50 phr PPO),抗張模數從 4.0 Gpa降至 3.7 GPa。雖然 PPO含量不
同會有不同的相形態及相分離程度,但是抗張強度和模數的變化並不
是很大,所以添加 PPO 並不會造成機械性質的負面影響。在破裂韌
性方面,有文獻指出熱塑性塑膠在裂縫成長時彼此粒子間產生裂縫,
較難貫穿粒子表面,所以表面能吸收裂縫能量而達成增韌效果。本研
究摻合體臨界應變能釋放速率隨著 PPO 含量增加而有小幅度上升,
直到 PPO 含量超過 20 phr時上升的幅度特別明顯,原因是高 PPO 含
量會造成摻合體的相反轉,熱塑性 PPO 變成連續相,所以主要的增
韌機構為 PPO的塑性形變。此外利用TAIC改質系統對摻合體的韌性
也有提升,如 Figure 3-32。
單位電場中,單位體積儲存的能量稱為介電容量。物質的介電

50 100 150 200 250
0.00
0.05
0.10
0.15
0.20
0.25
0.30
Figure 3-30 Relaxation peaks (tan d) of epoxy/PPO (30 phr) blend and of epoxy/PPO (30 phr) blend with 20 phr TAIC.
PPO 30 ph r
PPO 30phr/TAIC 20 phr
tanδ
temperature (oC)

0 10 20 30 40 5050
55
60
65
70
75
80
PPO content (phr)
Ten
sile
str
engt
h (M
Pa)
Tensile strength
0
2
4
6
8
Figure 3-31 The tensile modulus and strength as a function of blend composition for the epoxy/PPO system.
Ten
sile mod
ulu
s (GP
a)
Tensile modulus

0 10 20 30 40 50
50
100
150
200
250
300
Figure 3-32 The fracture energy (GIC
) as a function of blend composition
for the epoxy/PPO system.
Epoxy/PPO(30 phr)/TAIC(10 phr)
Fra
ctur
e en
ergy
(Jm
-2 )
PPO content (phr)

容量與真空的介電容量之比,稱為介電常數。在介電分析方面,PPO
具有優越的介電性質,在 1 MHz下介電常數為 2.45。綜觀整個產業
變化與國際科技趨勢,可以發現使用頻率越來越高,且頻寬精準性要
求也越高,在 1998年 2月 19日工研院 ITIS 計畫調查指出,基板材
料將朝低介電性、低熱膨脹、薄形多層化及高耐熱化等方向發展,樹
脂介電常數越低,會有較佳的電氣絕緣性及低逸散因子 (dissipation
factor),此外,對於不同頻率使用下亦要求樹脂材料能提供較佳的介
電穩度性。由 Figure 3-33中可以得知環氧樹脂/PPO (30 phr) 摻合體
在不同頻率下依然仍能維持相當穩定的介電常數,約 3.5左右,尤其
是當頻率在高頻 (1GHz) 下,介電常數仍能在高頻下穩定不變,使摻
合體對於高頻率的電子器材 (電腦中央處理器,通訊器材) 使用上會
具有較大的潛能。所以摻合PPO於環氧樹脂中可有效改進介電性質,
介電常數隨著 PPO含量增加有一線性下降的趨勢,如 Figure 3-34,
此性質與組成有關,與相形態無關。
3-3-7 摻合體熱性質分析
判斷一物質的耐熱性一般是藉由 Thermogravimetric analyzer
(TGA) 來評估,從 TGA 中可得到的資訊有材料的起始裂解溫度
(Td)、最大裂解速率溫度 (Tp) 及殘餘焦炭 (char residual) 之多寡。
Figure 3-35為硬化後環氧樹脂之 T-G 曲線圖,由 TGA得知環氧樹脂
在一大氣壓下氮氣中可以穩定到約 300oC (Td) 左右。然而環氧樹脂
/PPO 摻合體之起始裂解溫度 (Td) 比起純的環氧樹脂有些許
下降,可能的原因是添加直鏈型高分子 PPO 於中環氧樹脂
會阻礙環氧樹脂之交聯程度,另一原因可能是氰酸酯之 C≡N
會與 PPO末端 OH基產生反應生成 imidocarbonate,再與氰酸酯生

100000 1000000 1E7 1E8 1E9 1E10 1E110
1
2
3
4
5
Figure 3-33 Dielectric constant behavior of cyanate ester cured epoxy/PPO system in high frequency regions.
Epoxy
Epoxy/PPO(30 phr)
Die
lect
ric
cons
tant
Frequency (Hz)

0.0 0.2 0.4 0.6 0.8 1.0
2.5
3.0
3.5
4.0
Figure 3-34 Dielectric constant as a function of blend composition for the cyanate ester cured epoxy/PPO system.
Die
lect
ric
con
stan
t
Weight fraction of PPO

100 200 300 400 500 600 700 8000
20
40
60
80
100
Figure 3-35 TGA curves for the systems with various PPO content.
Wei
ght
(Wt
%)
Temperature ( oC)
0 phr PPO
20 phr PPO
40 phr PPO
PPO

成 cyanurate,但其環端接著分子量很大的 PPO 部分,所
以 不 易 再 與 環 氧 樹 脂 反 應 產 生 i s o c y a n u r a t e 及
oxazolidinone,導致部分 環氧樹脂反應不完全,降低了交聯密
度 ; 從 TGA 分析圖形中分析得知最大裂解速率溫度 (peak
temperature) 與 PPO 含量之關係 (如 Table 3-5所示) ,最大裂解速率
溫度亦隨著 PPO 的含量增加而大大的提高,而每增加 10 phr的 PPO
含量,其 peak溫度即提高 3.69oC,原因是摻合體中 PPO 含有許
多苯環所造成的。因此可知,由 PPO摻入環氧樹脂之中,PPO之
含量會對環氧樹脂的摻合體提升熱穩定性,也由 PPO 含量提升其扮
演熱穩定性角色相對重要。
硬化後樹脂的熱膨脹係數是以熱機械分析法 (TMA) 來測定,一
般而言大部份材料之尺寸會隨著溫度之升高而增加,而高分子材料之
熱膨脹係數約為金屬的十倍,因此要將兩材料結合,則必須考慮到此
複合系統之熱膨脹係數差異,降低高分子材料的熱膨脹係數,使其尺
寸安定,即為此一高分子是否能更廣泛的被應用有很大的關連,在此
摻合體系統中,無論是低溫或高溫時的熱膨脹係數皆會隨著 PPO 含
量的增加而降低 (如 Table 3-6),在高 PPO含量時 (> 20 phr) 摻合體
開始相反轉,PPO 成為連續相時特別明顯,此結果大幅提升了此材料
的應用性。
3-4 結論
聚氧化二甲苯 (Polyphenylene oxide, PPO) 以熱融法與 DGEBA
環氧樹脂-氰酸酯硬化系統混合,來探討硬化反應、相容性及相形態。
由DSC發現環氧樹脂/PPO摻合體符合自催化反應模式且摻合體中環
氧樹脂的硬化反應速率大於純的環氧樹脂,而反應之總放熱量卻

Table 3-5 The most rapid decomposition rate temperatures of epoxy/PPOblends.
PPO content Tp (°C)0 phr 45210 phr 45330 phr 46250 phr 470
Neat PPO 489Kevlar fiber 644
Ramp : 20°C/min from 100°C to 800°C in 40 ml/min N2
Tp : the most rapid decomposition rate temperatures

Table 3-6 The coefficient of thermal expansion of cured resin blends.
PPO (phr) α60-100oC (10-6/oC) α205-245oC (10-6/oC)
0 61 193
10 57 175
30 53 152
50 50 110
TMA measurement, ramp: 10oC/min in N2 100 ml/min, 0.05 N load.

隨著 PPO 增加而下降。FTIR 及 NMR分析顯示特性官能基的反應隨
PPO 增加而加速並且有 coreaction 產生,例如 cyanate-hydroxyl
addition (imidocarbonate, carbamate) , epoxy-cyanate addition
(oxazolidinone) 等。此外在氰酸酯摻合 PPO 系統也呈現自催化反應
模式。在硬化過程中,環氧樹脂開始聚合且同時進行著反應誘導式相
分離行為。當 PPO 含量低時,混合系統是以 NG 形式進行相分離,
當 PPO 含量高時,則以 SD 形式進行相分離。PPO 的含量增加時,
摻合體開始有部分相反轉的現象。利用 DMA可得知摻合體中兩相的
特性及部分相容的情況。然而摻合體兩相的形態並不均勻特別是在聚
氧化二甲苯含量較低時,所以添加 TAIC以改善摻合體之均勻性及相
容性。TAIC 與環氧樹脂的相容性佳且與聚氧化二甲苯形成接枝共聚
物。由 SEM 觀察可知添加 TAIC 後的摻合體之相容性大幅增加且抗
溶劑性提高。此外抗張強度隨著 PPO 增加而微幅上升,抗張模數卻
微幅下降;在破裂能量方面,當 PPO 變成連續相時則大幅增加,原
因是撕裂及延展 PPO需大幅能量。在熱性質方面,隨著 PPO含量增
加則摻合體之最大熱裂解速率溫度增加,但起始裂解溫度有些微下降
趨勢。熱膨脹係數也隨著 PPO 含量增加而降低,在摻合體相反轉時
特別明顯。介電常數隨 PPO含量的增加有線性下降的趨勢。

第四章 PPO 填充粒子對環氧樹脂熱性質與機械
性質之研究
4-1 前言
環氧樹脂已廣泛地使用在銅箔基板以及半導體封裝材料上,環氧
樹脂受歡迎的原因是它容易加工,極佳的抗化學性,而且有相當程度
的熱性質與機械性質。然而高交聯密度的網狀結構使產品性質變得硬
脆,在高頻範圍時的介電常數偏高,所以可藉著摻合 PPO 來改善這
些性質。
PPO 已經廣泛地使用在一些先進的複合材料上[17],主要的原因
是 PPO 的韌性、高玻璃轉移溫度、低介電常數可提升複合材料的性
質。一些學者在做熱固性與熱塑性樹脂摻合時常是利用熱熔融程序或
是添加溶劑的方法來混合摻合物,但是熱熔融法是在高溫下進行,可
能會使高分子裂解而且不容易控制;使用溶劑來摻合高分子會造成環
境的污染且硬化過後的產品容易留下孔洞 (void)。在此章節中,我們
將利用分散程序來摻合環氧樹脂及 PPO,也就是於反應初期時 PPO
就以第二相分散於環氧樹脂單體中。本研究將探討在環氧樹脂 -dicy
的硬化系統中加入 PPO對材料的相形態、熱性質及機械性質的影響。
由於 PPO 為非極性高分子,所以與環氧樹脂的相容性不佳,為了改
善系統的相容性,我們選擇 triallylisocyanurate (TAIC)為相容改質劑,
添加 TAIC後對於摻合體的相容性及熱機械性質的影響將加以討論。
4-2 實驗
4-2-1 材料與藥品
1. Dicyandiamide (Dicy),東京化成出品。

C NH
NH
CNH2N
2. 2-Methlyimidazole (2-MI),東京化成出品。
NN :H
CH3
其他的材料與藥品已於第三章所述。
4-2-2 儀器設備
儀器設備與第三章相同。
4-2-3 樣品製備
1. 熱塑性高分子 PPO與環氧樹脂以分散法混合
首先將環氧樹脂加熱到 100 oC左右,加入 DICY來當硬化劑,並
以均質機 24000 rpm均質 10 分鐘,之後再抽真空並以 400 rpm的機
械攪拌 30分鐘,接著把混合物溫度降到 80 oC,一邊再緩緩加入PPO,
一邊機械攪拌,真空攪拌 3 小時;之後再加入促進劑 2-MI,並持續
抽真空,攪拌約 30分鐘,得到一混合溶液。
2. 添加 TAIC於摻合系統
當環氧樹脂、DICY與 PPO 混合後,此時停止加熱抽氣,但仍持
續機械攪拌,再加入 TAIC,如此攪拌十分鐘後,然後取出混合物,

灌入已預熱的模版中取出。
3. 摻合體試片之製作
當分散混合完畢後,將混合物倒入預熱的模具中進行硬化反應,
硬化條件為 200 oC二小時。當硬化完成後,樹脂必須經過緩慢的降溫
退火過程才可從烘箱中取出。
4-2-4 實驗步驟
1. 熱分析實驗
利用 DSC可量測得知環氧樹脂與 dicy所需的硬化溫度與時間以
及添加 PPO後對系統反應性的影響,但硬化後的成品無法利用 DSC
量測得 Tg點,於是選擇了 DMA來分析。對於摻合體之熱穩定性質,
本研究選擇 TGA來進行分析,從 TGA分析中可得知最大裂解速率溫
度及起始裂解溫度,由此得知材料的抗熱裂解程度。
2. 動態機械性質之量測
動態黏彈測量對於聚合物的轉變,結晶型態改變,及構造上的變
化特別靈敏,阻尼峰與不同種類的分子運動和玻璃轉變,二級玻璃轉
變有關係,有時候也和結晶型態的轉變有關。本研究可利用 DMA測
試摻合體的玻璃轉移溫度與玻璃轉變強度、聚合體混合物的相容性及
聚合體混合物及其他兩相系統的相轉移。
3. 機械強度之量測
在拉力測試實驗中,所獲得 PPO 含量不同之摻合體的抗張模數
(Tensile modules)和抗張強度(Tensile strength),來判定材料是否會因
PPO 的摻入而破壞其機械性質。此外對於摻合體在不同 PPO 含量之

下,材料的破裂能量變化情形為何?這些需要依靠萬能拉力試驗機
(Instron)來測試。測試方法為 tapered double cantilever beam (TDCB)
方法[70]
4. 介電常數之量測
材料的介電性質的改進也是本論文主要目的之一,我們知道物質
的介電常數下降時,其絕緣效果增強,可廣泛使用於通訊器材、印刷
電路板及 IC封裝業等,其試片大小為 20×20×2 mm。
5. 相形態與破壞撕裂面的觀察
以掃描式電子顯微鏡 (Scanning Electron Microscopy, SEM)分
析,可由不同的倍率觀察得知環氧樹脂與 PPO 摻合的情形;掃描面
為樣品之破裂面,所以其破壞情形可加以分析,並與機械性質作一歸
納與討論。此外以二氯甲烷蝕刻摻合體中的 PPO 後再去做 SEM 分
析,我們就可以很清楚看到環氧樹脂與 PPO 分佈情形甚至可觀察出
基材中的粒徑分佈,或者是否有相反轉的產生,而相反轉情形可與
DMA分析相互佐證。
4-3 結果與討論
4-3-1 Dicy硬化環氧樹脂/PPO摻合體加工條件之訂定
使用 DSC儀器來決定加工條件,本研究中選用的樹脂為 DGEBA
(Diglycidyl Ether of Bispherol-A)型環氧樹脂,硬化劑為隱性硬化劑
dicyandmide (dicy),系統中並添加 0.07 phr的 2-MI (2-methyl imidazole)
作為硬化促進劑;由於 dicy系統添加了 2-MI之後,其加工溫度可提
前至 110oC開始硬化。環氧樹脂與 PPO摻合過程會產生許多的氣泡,

所以必須經過較長時間抽氣,否則溶液因溫度和時間效應作用下導致
黏度提高,這表示環氧樹脂已開始聚合,抽氣泡的過程會更加困難;
在多次嘗試之後,選用加入 dicy之加工條件就選在 100oC/30 min 為
最佳,而加入 PPO之加工條件為 80 oC /2 hr為最佳,以避免加工黏性
過大。至於 TAIC因屬於液體,加工時溫度就不再加熱,只要其與環
氧樹脂和 PPO的溶液相互混合即可。
4-3-2 環氧樹脂與 PPO摻合體相形態之探討
以掃描式電子顯微鏡 (SEM) 分析其破裂面,得知摻合體的撕裂
情況,再利用二氯甲烷溶解摻合體中的 PPO,如此我們就可以很清楚
看到環氧樹脂與 PPO分佈情形。在 Figure 4-1與 Figure 4-2 中,我們
可看到純環氧樹脂的破裂面非常平滑,經由溶劑蝕刻後破裂面幾乎沒
有改變。在 PPO 添加量為 20 phr時,PPO會彼此聚集形成一不規則
球形區域,其直徑約 5-50 µm,不均勻地分佈在基材中。經由蝕刻程
序移去 PPO後留下的孔洞更可看出 PPO 所分佈的區域。在 PPO 添加
量為 60 phr時,摻合體的破壞面變得更複雜,除了上述的 PPO 所分
佈的不規則球形區域之外,另有 PPO 不規則島狀的區域,此時環氧
樹脂仍是連續相;這些熱塑性島狀結構並非完成是 PPO 成份,在這
些區域內,我們發現相反轉的現象,微小的環氧樹脂粒子分佈在其
中。經由溶劑蝕刻後,可觀察到這些區域中有部分成份不被溶劑溶
解,有趣的是我們觀察到的環氧樹脂顆粒的表面被一連續性的表層所
覆蓋,這表示環氧樹脂與 PPO 之間應該有化學鍵結的存在導致 PPO
無法完全被二氯甲烷溶解。當 PPO添加量為 100 phr時,其破裂面變
得粗糙崎嶇,是屬不規則的撕裂面,所以當裂縫成



長時會受到 PPO 相所阻礙,導致破裂能量沿著撕裂面四處分散,達
成增韌效果。經由溶劑蝕刻後,環氧樹脂組成呈現一粗糙且相互連接
的結構,此現象相似於共連續摻合體之相形態。以上的摻合體相形態
之分析可用來解釋摻合體的破裂行為;當 PPO 為分散相時,系統的
增韌效應為 PPO 粒子的塑性形變,此外撕起 PPO粒子時,摻合系統
提供了充分的相界面強度,經由溶劑蝕刻便可證明 PPO 與環氧樹脂
間有化學交互作用。當 PPO 與環氧樹脂形成共連續相時,主要的增
韌機構變成是熱塑性連續相的塑性形變。
4-3-3 TAIC改質環氧樹脂與 PPO摻合體相形態之探討
Figure 4-3顯示 30 phr PPO 之摻合體中添加不同含量 TAIC的破
裂面形態,可看出未添加 TAIC 時的 PPO粒子聚集情形嚴重,經過溶
劑蝕刻後,留下 PPO 被溶解的孔洞分佈於環氧樹脂連續相中。這些
孔洞隨著 TAIC 添加量增加而縮小,當 TAIC添加量由 0 phr增至 20
phr,PPO溶解後的孔洞由 50 µm減至 5 µm,而且孔洞的數目增加。
當 TAIC 添加量至 20 phr時,PPO顆粒就不再聚集在一起,形成一較
均勻的分散情況。此外在高添加量 TAIC的摻合體經二氯甲烷蝕刻之
後,PPO孔洞卻是越來越小,由此可知 TAIC 是可以增加 PPO 之抗
溶劑性。由 SEM 顯示當 TAIC 含量高時,摻合體相容性有改善的趨
勢,原因是 TAIC與環氧樹脂的相容性極佳,而且 TAIC與 PPO 間可
產生鍵結,因為 PPO的甲基很容易受到 TAIC 的自由基攻擊,產生接
枝共聚合物,所以鍵結的 PPO 分子較不容易被溶劑萃取出來。
4-3-4 環氧樹脂與 PPO摻合體機械性質之探討
在拉力試驗中所測得不同 PPO 含量下摻合體之抗張模數


(Tensile modulus) 和抗張強度 (Tensile strength),由 Figure 4-4可看出
摻合體之模數和強度稍微地隨著 PPO 含量增加而提升,其提升趨勢
並不十分明顯,但最起碼摻合體不會因 PPO 的加入而破壞其機械性
質。Figure 4-5為不同 PPO 含量之摻合體的破裂能量圖形。PPO含量
低於 30 phr時,其破裂能量提升的幅度不大,但是 PPO 含量大於 30
phr 後,破裂能量的提升速率也隨著 PPO 含量增加而增加,當 PPO
含量增加至 100 phr時,其材料的破裂能量已提升 16倍之多。
Figure 4-6為 30 phr PPO 摻合體加入不同含量 TAIC之抗張模數
和抗張強度的圖形,由圖中可知當 TAIC添加至 15 phr時,其變化不
大,添加超過 20 phr時,有些許下降的趨勢,但整體而言,所有樣品
的抗張模數和抗張強度並無太明顯的改變。Figure 4-7為 30 phr PPO
摻合體添加不同含量 TAIC所做的破裂能量分析圖,摻合體之破裂能
量隨 TAIC含量大幅提升,由文獻得知 TAIC所產生的自由基會攻擊
PPO 的甲基,形成PPO/TAIC的共聚合物[69],而且環氧樹脂與 TAIC
的相容性佳,所以我們可推測摻合體中的 PPO 多了許多支鏈,連結
於 TAIC 網路上,增加了與基材的相容性,導致摻合體破裂能量的提
升;但是超過 20 phr TAIC時,摻合體破裂能量就有明顯下降,這種
現象就必須與其 DSC圖一起來探討 (如 Figure 4-8及 Figure 4-9),我
們可以知道添加量超過 20 phr TAIC 之摻合體,其內可能仍有少許無
法反應完全的 TAIC單體,產生一些微觀的缺陷,導致摻合體的破裂
能量下降。
4-3-5 環氧樹脂與 PPO摻合體動態機械性質之探討
高分子的機械性質可由黏性 (viscosity) 及彈性 (elasticity) 共同
表現出來,通常我們利用動態黏彈性分析來看材料中各種分子鏈的

0 20 40 60 80 10050
52
54
56
58
60
62
64
66
68
70
72
74
PPO content (phr)
Ten
sile
str
engt
h (M
Pa)
Tensile strength
3.5
4.0
4.5
5.0
Figure 4-4 Results of tensile strength and modulus for the epoxy/PPO blends.
Tensile m
odulus (GPa)
Tensile modulus

0 20 40 60 80 1000
200
400
600
800
Figure 4-5 Results of fracture energy (GIC
) for the epoxy/PPO blends.
Frac
ture
ene
rgy
(J/m
2)
PPO content (phr)

-5 0 5 10 15 20 25 30 3550
55
60
65
70
75
80
PPO content (phr)
Ten
sile
str
engt
h (M
Pa)
Tensile strength
0
2
4
6
8
Figure 4-6 Results of tensile strength and modulus for the epoxy/PPO (30 phr) blends with various TAIC contents.
Ten
sile mod
ulu
s (GP
a)
Tensile modulus

0 5 10 15 20 25 3050
100
150
200
250
300
Figure 4-7 Results of fracture energy (GIC
) for the epoxy/PPO (30 phr)
blends with various TAIC contents.
Frac
ture
ene
rgy
(J/m
2 )
TAIC content (phr)



運動,其中是可用來觀察摻合系統中各成分的玻璃轉移溫度 (Tg);將
tan δ 對溫度作圖時,tan δ 的峰通常可定為 Tg點 (α transition),當
二元摻合系統之相容性增加時,兩單獨成分的Tg點會向彼此間移動,
移動的程度可視為相容的程度,再參照 SEM便可知摻合體相容的情
形。在 DMA中顯示 (Figure 4-10),純環氧樹脂的 Tg點約在 122oC,
純 PPO的 Tg約為 220 oC。環氧樹脂/PPO摻合體有兩個玻璃轉化溫度
及部分互容情形;在環氧樹脂相中,隨著 PPO量的增加,其 Tg稍微
提昇;在 PPO相中,隨著 PPO量的增加,其 Tg卻明顯的下降;我們
從 Table 4-1 中可以清楚得知,摻合體的環氧樹脂相與 PPO 相的 Tg
溫度差距中,PPO 添加量由 0 phr至 50 phr時,其 Tg的差距由 89 oC
降至 46 oC,也就是說摻合體隨著 PPO 量的增加,兩相的 Tg便有向內
移的傾向,產生此現象的原因可能是摻合體在混合及硬化過程中,環
氧樹脂比起硬化劑 dicy更容易溶於PPO相,結果是較多的環氧樹脂、
較少的 dicy溶於 PPO相,導致 PPO 相的塑化作用。PPO相的 Tg大
幅下降可證實這個觀點,同時 dicy 濃度在兩相中分配不均勻,使得
環氧樹脂相的 dicy濃度較高,造成環氧樹脂相的 Tg點提升。
Figure 4-11是 30 phr PPO的摻合體添加不同含量 TAIC進行相容
改質, 當加入 5至 20 phr的 TAIC時,環氧樹脂相的 Tg隨著 TAIC
量增加而提升,由 127 oC提升到 141 oC;在 PPO相中,其 Tg隨著 TAIC
增加而下降;摻合體的環氧樹脂相與 PPO相的 Tg差距也有縮小的趨
勢,這表示 TAIC有效的改善環氧樹脂與 PPO的相容性。若 TAIC含
量超過 20 phr,則會有殘存的不易完全反應的 TAIC,導致環氧樹脂
相的Tg大幅下降。這現象我們也可從DSC圖 (Figure 4-8及Figure 4-9)
中得知,在加入低於 20 phr TAIC 含量之摻合體去做 DSC 分析時
(DSC設定條件溫度 50~300 oC,20 oC/min),同樣樣品的第一次
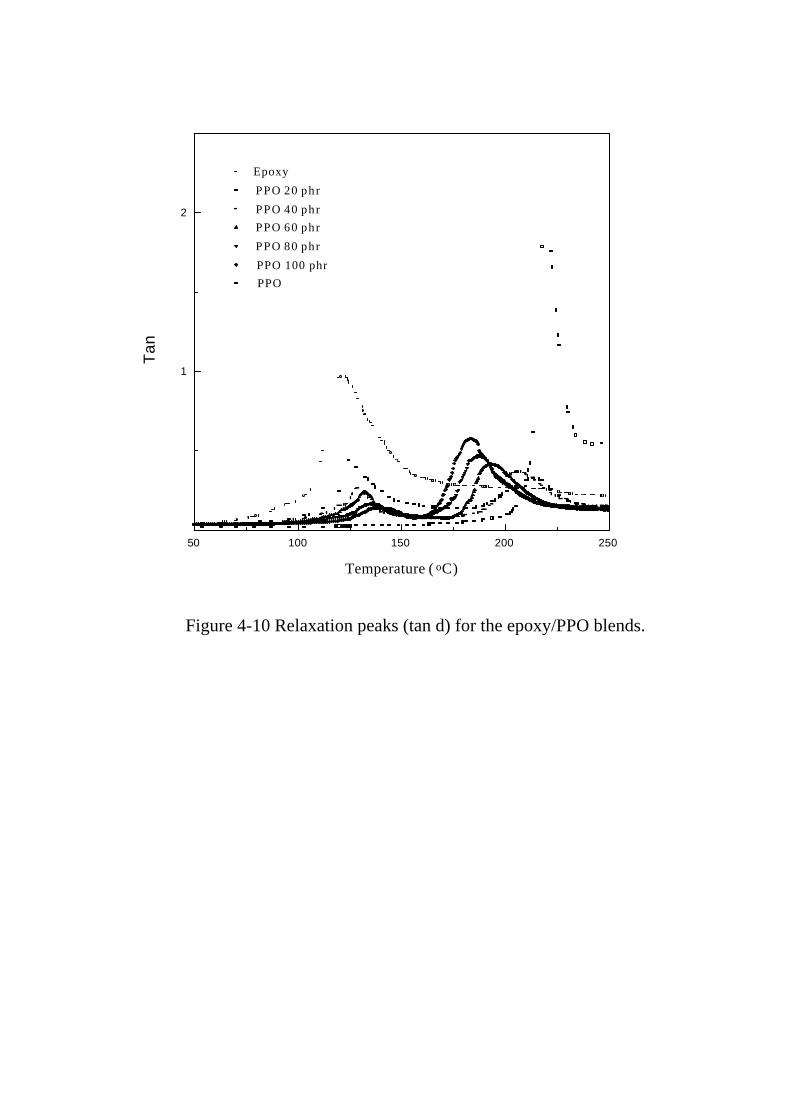
50 100 150 200 250
1
2
Figure 4-10 Relaxation peaks (tan d) for the epoxy/PPO blends.
Epoxy
PPO 20 phr
PPO 40 phr
PPO 60 phr
PPO 80 phr
PPO 100 phr
PPO
Ta
nδ
Temperature ( oC)

Table 4-1 Glass transition temperatures of epoxy and PPO phase ofepoxy/PPO blends in cured states.
PPO Tg of cured blend (oC)
(phr) PPO phase Epoxy phase
0 --- 122
20 215 125
40 207 129
60 193 132
80 188 136
100 184 138
Tg of PPO: 220oC; DMA measurement, ramp: 5oC/min from 50 to 250oC
at 1 Hz.

50 100 150 200 250
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
Figure 4-11 Relaxation peaks (tan d) of epoxy/PPO (30 phr) blends with various TAIC contents.
0 phr TAIC
5 phr TAIC
15 phr TAIC
20 phr TAIC
tanδ
Temperature ( oC)

熱掃描和第二次熱掃描作分析,可以很清楚得知並無太大的改變,但
是加入超過 20 phr TAIC之摻合體時,同樣樣品第一次與第二次熱掃
描就有明顯的改變;第一次熱掃描在 200~220 oC有明顯吸熱出現,這
可能有一些 TAIC單體未反應導致揮發之結果。所以在環氧樹脂/PPO
(30 phr) 的摻合系統中摻入 TAIC的最佳含量為 20 phr。
4-3-6 環氧樹脂與 PPO摻合體熱穩定性之探討
Figure 4-12顯示環氧樹脂/PPO 摻合體均為二階段的裂解行為;
在第一階段裂解範圍中,可觀察得知純環氧樹脂會在 300 oC之後重量
才迅速往下掉,而在高含量 PPO 之摻合體中,尚未到達純環氧樹脂
裂解溫度之前就出現緩緩裂解的現象 (Figure 4-13),由此可見摻合過
程中環氧樹脂會進入 PPO相,導致 PPO 內 dicy濃度較低,硬化後仍
有一些交聯不完全的環氧樹脂存留在 PPO相。此外,對 TGA微分得
到裂解速率對溫度的圖形中可得到一個主要的峰,這些峰的位置表示
最大裂解速率出現的溫度。由微分圖得知最大裂解速率溫度隨著 PPO
含量增加而提高,從純環氧樹脂的 441 oC提升到添加 100 phr PPO的
478 oC。在 TGA圖中,摻合體最後留下的焦炭重量比純環氧樹脂高,
純環氧樹脂在 800 oC 時幾乎已裂解完畢。PPO 摻入環氧樹脂中,雖
然會造成一些反應不完全的環氧樹脂存在於 PPO 中,使得起始裂解
溫度些微下降,但是對於摻合體的最大裂解速率溫度之提升,及最終
焦炭殘留之增加,也可知 PPO 對於摻合體熱穩定性的影響。PPO 含
量之提高也對摻合體提升了熱穩定性,在文獻中曾解釋環氧樹脂與
PPO 的高溫 (200oC) 混合加工系統中,PPO 會妨礙環氧樹脂的聚合
反應,因而降低環氧樹脂的聚合度;不過在熱重分析圖 (TGA) 中,
可知在純的環氧樹脂會在較高溫度才迅速往下掉,

0 200 400 600 8000
20
40
60
80
100
Figure 4-12 TGA curves for the systems with various PPO contents.
0 phr
30 phr
50 phr
80 phr
Wei
ght (
Wt %
)
Temperature ( oC)

50 100 150 200 250 300 350 40095
100
Figure 4-13 TGA curves for the systems with various PPO contents (scale up).
0 phr
30 phr
50 phr
80 phr
Wei
ght (
Wt %
)
Temperature ( oC)

但在 PPO 高含量之摻合體中,會在較低溫度才和緩往下掉,由此可
見環氧樹脂會進入 PPO 顆粒內,導致摻合體內含有未完成反應的環
氧樹脂單體。
TAIC會與 PPO產生反應,且可溶於環氧樹脂中,因此選用 TAIC
來改進環氧樹脂與 PPO的不相容情形;在環氧樹脂/PPO(30 phr)/TAIC
的熱重分析中得知它仍為二階段裂解,如 Figure 4-14所示。添加 TAIC
之摻合體與未添加 TAIC摻合體相比,其第一階段裂解會出現在較低
溫度,且進行緩慢;由此可見摻合體內含有反應不完全的環氧樹脂與
TAIC的單體。由 TAIC的含量對最大裂解速率溫度溫度作圖 (Figure
4-15),顯示最大裂解速率溫度會隨著 TAIC含量增加而提高,這表示
TAIC的網狀結構耐熱性佳。
4-3-7 環氧樹脂與 PPO摻合體介電性質之探討
非極性高分子置於電場中,電極間儲存電荷的能力小,其介電常
數較低。PPO 是屬於一非極性高分子,故其介電常數只有 2.45。Figure
4-16 為不同PPO 含量摻合體所測出的介電常數之圖形,我們可看出
摻合體隨著 PPO 的含量增加,其介電常數即大大下降,平均每增加
10 phr 的PPO 即下降 2.45% 左右,這些結果可提供工業界另一種低
介電常數的材料。Figure 4-17 為30 phr PPO 之摻合體加入不同含量
的TAIC 所做的介電常數分析圖,在圖中可以清楚的看出,當加入至
20 phr TAIC 時,其介電常數有明顯下降;這原因可能是相容性提升
所造成,當 TAIC 加至 30 phr 時,其介電常數卻升高,這原因是 30
phr TAIC 之摻合體含有一些未反應完全之 TAIC 所致,由於 TAIC
單體本身含有三個丙烯基的雙鍵,這些雙鍵容易導電,因此導致摻合
體介電常數升高。

0 200 400 600 800
0
20
40
60
80
100
Figure 4-14 TGA curves for the epoxy/PPO systems with various TAIC contents.
PPO 30 phr
PPO(30 phr) /TAIC(5 phr)
PPO(30 phr)/TAIC(10 phr)
PPO(30 phr)/TAIC(15 phr)
PPO(30 phr)/TAIC(20 phr)
Wei
ght (
Wt %
)
Temperature ( oC)

0 5 10 15 20460
470
480
490
500
510
520
530
Figure 4-15 Plots of the most rapid decomposition rate temperatures versus time for dicy cured epoxy/PPO blends system.
TAIC
Te
mp
era
ture
(o C
)
TAIC content (phr)

0 20 40 60 80 1002.4
2.6
2.8
3.0
3.2
3.4
3.6
3.8
4.0
4.2
Figure 4-16 Dielectric constant as a function of blend composition for the dicy cured epoxy/PPO system.
Neat PPO
Die
lect
ric
cons
tant
PPO content (phr)

0 5 10 15 203.0
3.1
3.2
3.3
3.4
3.5
3.6
3.7
3.8
Figure 4-17 Dielectric constant as a function of blend composition for the dicy cured epoxy/PPO system with various TAIC contents.
Die
lecr
ic c
onst
ant
TAIC content (phr)

4-4 結論
本研究是探討添加熱塑性 PPO 粒子對熱固性環氧樹脂的影響。
結果顯示所有組成的摻合體皆為兩相形態,且它們的相形態與 PPO
含量有關。在低 PPO含量下,呈現出 PPO顆粒分散在環氧樹脂的基
材相中,PPO粒子有聚集的現象。在高 PPO 含量下,相形態就顯得
較複雜,例如在 60 phr PPO含量的摻合體中之 PPO相有環氧樹脂溶
入其中;100 phr PPO 含量時則有共連續相的產生。在 DMA分析中,
所有組成都有部分互溶的情形,PPO相 Tg下降的原因是由於有交聯
不完全的環氧樹脂存在 PPO 相。在抗張強度及抗張模數方面,PPO
的添加對摻合體的影響並不大。在破裂能量方面,隨著 PPO 含量的
增加而大幅提升,在 PPO 含量高時特別明顯,此與相形態有密切的
關係。在 TGA分析中,PPO會妨礙環氧樹脂的交聯反應,因而降低
環氧樹脂的交聯程度;不過在熱重分析圖 (TGA) 中,可知在純的環
氧樹脂會在較高溫度才迅速往下掉,但在 PPO 高含量之摻合體中,
起始反應溫度有些微下降的現象,由此可知除了 PPO 會阻礙環氧樹
脂交聯之外,環氧樹脂還會進入 PPO 顆粒內,導致摻合體內含有未
完成反應的環氧樹脂。此外,摻合體隨著 PPO 含量增加其最大裂解
速率溫度大幅提升。在介電性質方面,隨著 PPO 的含量增加,其介
電常數大幅下降。
在環氧樹脂/PPO (30 phr)摻合系統中添加 TAIC之後,摻合體中
PPO 顆粒聚集現象有明顯的改善。由 DMA及 SEM可看出添加 TAIC
後摻合體的相容性增加,破裂面經由二氯甲烷蝕刻後,顯示出系統的
抗溶劑性質增加。TAIC 的添加並未造成摻合體的抗張強度及模數的
下降。在破裂能量方面,隨著 TAIC含量的增加而上升,20 phr是最
佳添加量。在 TGA分析中,最大裂解速率溫度隨著 TAIC 含量增加

而升高。介電性質方面,隨著 TAIC含量增加,摻合體的介電常數也
明顯下降,超過 20 phr有上升趨勢。

第五章 氰酸酯硬化之環氧樹脂/聚氧化二甲苯
摻合體為基材之克維拉纖維複合材料性
質之研究
5-1 前言
纖維補強的高分子複合材料 (Fiber Reinforced Plastics),因為技
術知識不斷地更新進步,使得此複合材料的應用愈來愈廣,其用途除
了運動器材外,最近又擴展至工業材料或作為飛機的初級結構性材料
[105]。一般纖維強化熱固性塑膠是指以不飽和聚酯、環氧樹脂、乙
烯基酯、酚醛樹脂等熱固性樹脂為基材,與玻纖、碳纖、硼纖、聚醯
胺纖維等為補強材料,經手工積層法、噴佈法、纏繞成型法、拉擠成
型法、樹脂轉強法、SMC、BMC、預浸材等加工步驟複合而成。纖
維強化塑膠產品的主要特點為質輕、強度大,並可以防止腐蝕;此種
複合材料比金屬材料有較高的比模數 (specific modulus) 和比強度
(specific strength)。故使得複合材料逐漸取代許多傳統的材料,特別
是金屬材料。
複合材料所使用的纖維有很多種[106],Aramid 纖維是所有芳香
族聚醯胺 (aromatic polyamide) 纖維的總稱。早在 1950年化學家即指
出如能合成芳香族型之聚合體,則材料之性能會更優越。1964年 H.
Mark博士證實以脂肪族或芳香族環狀物取代次甲基鏈節 -(CH2)n- 將
可提高聚合體鏈結之分子剛性,有助於聚合體整體性質之提升。美國
杜邦 (du Pont) 公司於 1955年至 1965年從事此方面之研究,但一直
不能掌握對位型全芳香族聚醯胺在製程及加工上的突破,且又無適當
的溶劑能予以抽絲,直至一名研究員發現 PPTA (即 Kevlar) 大部份可
溶於濃硫酸。1966年 du Pont申請首篇 Kevlar高強力、高係數纖維製

造專利;1970年建廠;1971年量產。Kevlar 纖維因為是由液晶相溶
液抽絲紡紗而成,及 6 m/s之抽絲速率提高分子在纖維軸方向的剪變
率,因而使得 PPTA具有高度的順向性,堅硬的線性高分子鏈排列方
向和纖維縱軸的方向是一致的,而纖維橫向則靠分子間所形成的氫鍵
來維繫,因此 Kevlar纖維具有很高的縱向 (longitudinal) 強度,而徑
向 (radial) 強度則較低。
對於纖維強化複合材料而言,如何選用適當的樹脂基材及補強纖
維材料,對於複合材料製品之品質影響甚鉅。就複合材料中基材而
言,其在複合材料中負擔著三種任務。
(1)使纖維定位並做為纖維與纖維間之橋樑
(2)傳遞力量之介質。
(3)保護纖維,使不受外界之腐蝕或損壞。
而纖維本身則擔任承受力量的補強擔體。引此纖維和樹脂複合材料的
好壞,除了取決於材料本身的強度外,適當的介面黏著亦是非常重
要,因為基材如能將所承受到的力量充分地傳送到纖維上,則可使複
合材料的性質大幅地提升。
目前用於複合材料之基材以環氧樹脂的使用量最大。但是環氧樹
脂在高頻使用時介電常數過高,環氧樹脂硬化後產生極高的交聯密
度,在硬化後其機械性質變得非常的脆,所以本研究選擇高 Tg、高
韌性、低介電常數的熱塑性塑膠 PPO 來改質環氧樹脂。已有許多熱
塑性塑膠改質熱固性樹脂的預浸材料應用在航空器材上[107-109],目
前的研究是朝向使用熱塑性塑膠改質劑來對基材產生增韌效果,以提
升複合材料的破壞容忍度。本研究目的主要是探討摻合 PPO 後的基
材對克維拉複合材料介面性質的影響。以克維拉複合材料在纖維軸向

性質相當良好,但是其非軸向性質可能會因纖維與非極性 PPO 間的
介面黏著性不佳而下降,因此如何控制良好的介面性質是達成複合材
料優良性質的基本要求。
5-2 實驗
5-2-1 材料與藥品
1. Kevlar 49 fabric, 1140 denier, style 281, E. I. du Pont de Nemours &
Co. (USA)。
2. 環氧樹脂,Epon 828 (diglycidyl ether of bisphenol A, DGEBA), Shell
公司產品。
3. 氰酸酯 (cyanate ester, PT-30),MW為 320到 420,Ciba-Geigy出品。
4. 聚氧化二甲苯 (polyphenylene oxide, PPO),本性黏度為 0.4 dl/g於
25oC氯仿,G. E. 出品。
5. 1, 2-dichloroethane, Merck, synthesis。
6. Methanol, Alps Chem. Co., Ltd., L. C. grade。
7. 去離子水。
5-2-2 儀器與設備
1. 萬能拉力機,Istron, Model 4302。
2. 掃瞄式電子顯微鏡,Hitachi S-570 及 S-2300 Scanning Electron
Microscope (SEM)。
3. 光學顯微鏡,儀器型號為 Nikon Model SN, Japan。
4. 長焦距立體顯微鏡,Nikon, Type 102。
5. 真空烘箱,EYELA vacuum oven, VOS-300SD。
6. 熱壓機,CARVER Laboratory Press, Model M。

7. 砂輪切割機。
5-2-3 纖維的前處理
購得之克維拉纖維於進行各種表面處理之前,先後分別以 1, 2-
二氯乙烷、甲醇及去離子水浸泡清洗後,再將纖維置於 100 oC烘箱中
真空乾燥 12小時。清洗乾燥後之纖維貯存於電子乾燥櫃中作為進一
步表面處理之用。
5-2-4 纖維表面型態的觀察
將纖維或纖維布剪下一小段,用雙面膠黏於 SEM圓台上,抽真
空至 10-2 torr後鍍金。送入 SEM儀器中,以 400~6000倍率進行纖維
表面及破壞型態之觀察。
5-2-5 Microbond試件製作及測量
以鑷子挑取單根纖維,用 AB膠固定於中空折好的錫箔中,將環
氧樹脂/PPO 摻合體用細針挑起少量的樣品固定於纖維上送入烘箱
中,以 180 oC/4 hr及 220 oC/ 4 hr進行硬化反應。硬化後之Microbond
試件以 200倍光學顯微鏡放大觀察摻合體樹脂粒的大小,並量取纖維
埋於樹脂中之長度 (ambedded length)。接著將試件固定於 Instron材
料試驗機上之特製小型夾具上,環氧樹脂/PPO 樹脂粒則位於微形虎
鉗 (microvise) 的間隙中,微形虎鉗間隙的大小則在長焦距立體顯微
鏡的監視下加以調整,以期能卡住樹脂球粒,而不致卡斷纖維。測試
時,夾頭移動速度為 0.5 mm/min,由儀器上可讀取拉動 microbond 所
需的負荷大小,以及microbond脫落後的摩擦力大小。測試過程中的
負荷大小變化對位移的關係圖可由連接 Instron 的繪圖機記錄。測試

方法參考 B. Miller等人文獻[110-112]。
5-2-6 Kevlar纖維/環氧樹脂複合材料試片之製作程序及測試
1. Mode I測試法 (Double Cantilever Beam)
本實驗採用 Berry [113]實驗模式來決定GIC值,實驗所需的負載
-卸載過程的負荷與位移的關係圖。試片兩測面塗以立可白修正液以
助裂縫成長的觀察,測試前將試片預裂 1.5 mm的長度,以鋁箔紙做
一預裂縫口,當施以負荷時,使裂縫延預埋方向生長至比鋁箔紙長
2-3 mm,維持該位移達到 V2,並觀察裂縫長度 a1,使負荷及位移回
到零,然後再施加負荷直到位移 V2,並記錄裂縫長度 a2,隨後重新
卸載-負載的程序。
實驗完成後,由負荷-位移圖讀取最大臨界點的負荷、位移及觀
察的裂縫成長值,將各實驗數據代入下式:
GIC=Ba
PcVc
2
3
其中 Pc為臨界點的負荷,a為裂縫長度,Vc為位移值,B為試片寬度。
即可求得該條件複合材料的韌性值GIC。
2. 層間剪切應力 (Interlaminar Shear Strength, ILSS) 試片
環氧樹脂先置於 100oC 烘箱中加熱並抽真空 30 分鐘以除去水
份,等冷卻後再與氰酸酯硬化劑 (20 phr)、PPO (0-50 phr) 和二氯甲
烷混合,配成預浸材溶液,固含量約 20 %。然後將纖維布含浸其中,
取出後先於室溫下置放 30分鐘,再置於 180oC烘箱中 5分鐘,將製
好的預浸材置於鋁製凹型模具內,依照所需的疊層順序 (0 度) 及層
數 (16層) 依次疊好,然後將此模具放置於熱壓機上,先以溫度 180

oC、壓力為 1.01×106 Pa熱壓四小時,再昇溫至 220oC、壓力 1.01×106
Pa 熱壓四小時,進行硬化。硬化完畢後將溫度緩慢降至室溫後再洩
壓。ILSS試片是將複合材料以砂輪切割機切割成 16 mm×11 mm的
大小。其測試方法是參考 ASTM D2334的建議,以零度、16層的積
層板短樑做三點彎曲的測試,採用三點荷重的方式,並以X-Y記錄
器記錄荷重與夾具位移的關係,如 Figure 5-1。
實驗完成後可得一負荷、位移關係圖,求其負荷的最高點 P,將
P代入下式:
τ= 34
PBh
其中 B為試片寬度,h為試片厚度,P為最大負荷。即可求得該
複合材料的臨界層間剪切力強度τ。
3. T-peel strength試片
其法也與製 ILSS試片相同,不同處為疊層數為兩層,需預裂縫。
T-peel strength的試片是將複合材料切割成 15 mm×90 mm的大小。
其測試方法參考 ASTM D1876,以零度、二層的積層做 T-peel strength
的測試,使用 Instron-Model 4302及 X−Y記錄器,測出層間剝離力。
以上所製的試片其環氧樹脂的含量約為 40 wt % 。
5-2-7 SEM 試件之製備與觀察
克維拉纖維以雙面膠固定於 SEM 試件平台上,經過鍍金後,放
大觀察纖維的表面形態。纖維的直徑大小則是以纖維橫截面的放大
SEM 照片經由游標卡尺測量換算比例後求得,取 10根纖維直徑的平
均值作為平均的纖維直徑。此外,經過測試後的 T-peel test試件及拋
光後複合材料橫切面也以 SEM 放大來觀察纖維與樹脂兩者
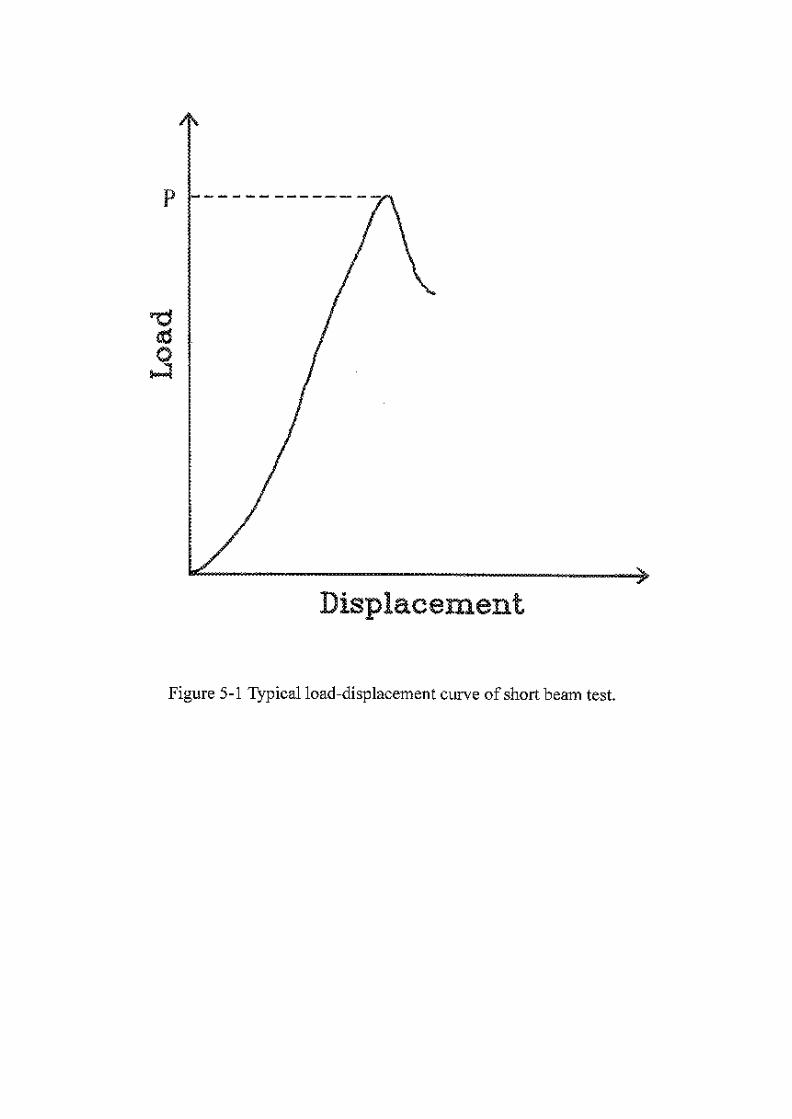

間界面黏著的情形。蝕刻後的破壞面及拋光之複合材料橫切面也以
SEM 放大來觀察。
5-3 結果與討論
5-3-1 以微粒拉出法研究 PPO 對 Kevlar 纖維/環氧樹脂界面
性質的影響
界面剪切應力 (Interfacial Shear Strength, IFSS) 的來源包括了兩
個部份,第一個為樹脂與纖維之間的摩擦剪應力,第二個為物理及化
學的鍵結力。若單體與纖維間有反應官能基的存在,則彼此間可產生
化學鍵結。像氰酸酯硬化之環氧樹脂與克維拉纖維之間即可形成鍵
結,克維拉纖維上的胺基可與環氧樹脂產生開環反應,此外,胺基會
與氰酸酯上的三鍵來反應,所以可與氰酸酯反應產生 iminocarbonate
[114]。所以在氨氣電漿處理完畢之克維拉纖維與氰酸酯硬化之環氧樹
脂製成複合材料後,在微粒拉出實驗時所得的界面剪切應力有上升的
趨勢,如 Figure 5-2所示。
Kevlar fiber/Epoxy/PPO複合材料微粒試件在測試後,以 SEM觀
測其界面破壞型態。一般而言,當纖維從樹脂球粒中拉出,破壞面有
可能發生在纖維與樹脂間的界面,或是發生在樹脂的破裂,或是纖維
的撕裂。Figure 5-3(a)為 Kevlar fiber/epoxy微粒試件的破壞面型態,
由圖中可得知纖維表面有殘留環氧樹脂附著,破壞面是發生在樹脂,
這表示氰酸酯硬化之環氧樹脂與克維拉纖維間有良好的接著力。而
Figure 5-3(b)-(d)為 Kevlar fiber/Epoxy/PPO微粒試件測試完畢的纖維
表面型態,由圖可得知雖然添加 PPO 於基材中,其破壞型態與純環
氧樹脂相近,皆有殘留樹脂於纖維上,代表摻合體與克維拉纖維間仍
有良好的接著力。但是純 PPO 卻與克維拉纖維間的界面黏

0 10 20 30 40
Figure 5-2 Effect of the plasma treatment on IFSS of the Kevlar 49 fiber-cyanate ester cured epoxy system.
Control
30W 5 min NH3-plasma
Interfacial shear strength (MPa)



著力不佳,由 Figure 5-3(e)可看出微粒拉出後的纖維表面相當光滑,
其破壞面是發生在 PPO 與克維拉纖維的界面。理論上摻合 PPO會造
成基材與克維拉纖維間的界面黏著力下降,但是由 IFSS 值測得結果
卻不然,IFSS幾乎不受到 PPO的加入而改變,如 Figure 5-4所示,
其原因將在 5-3-2節討論。
5-3-2 複合材料的破壞面
Figure 5-5 顯示未含纖維時環氧樹脂/PPO 摻合體的破壞面,在
PPO 含量 30 phr和 50 phr時,摻合體硬化完成後有相分離及相反轉
的現象,也就是說環氧樹脂顆粒分佈在 PPO 連續相中。為了瞭解顆
粒狀環氧樹脂的表面特性,摻合體的破壞面可利用二氯甲烷蝕刻。熱
塑性 PPO 區域,假使與交聯後的環氧樹脂間沒有充分的鍵結,則 PPO
會被二氯甲烷萃取出來,而顯露出光滑的環氧樹脂球粒表面。但是破
壞面經由二氯甲烷蝕刻後,發現在環氧樹脂球粒表面上仍有 PPO 附
著在其上,所以Figure 5-6顯示出 30 phr及 50 phr PPO摻合體經由二
氯甲烷後仍然非常粗糙。這個現象證實了環氧樹脂與 PPO 的界面間
有化學鍵結,彼此間有良好的界面黏著力。但是在環氧樹脂/PPO/克
維拉纖維複合材料的破壞面中,並未發現環氧樹脂顆粒分佈在 PPO
連續相中,如 Figure 5-7所示。為了進一步探討其相形態形成的原因,
我們將破壞面利用二氯甲烷蝕刻,觀察 PPO溶解的程度。由Figure 5-8
中我們發現 PPO 相被溶解掉,並未發現環氧樹脂顆粒顯露出來,而
發現克維拉纖維表面包覆著環氧樹脂。同時我們利用拋光複合材料的
橫切面來觀察其相形態,很明顯的分辨出纖維與基材所分佈的區域,
但是對於基材中環氧樹脂與 PPO相卻很難分辨出來,如 Figure 5-9(a)
所示。拋光後橫切面經由二氯甲烷蝕刻後,

0 10 20 30 40 5015
20
25
30
35
Figure 5-4 The interfacial shear strength of microbond test with various PPO contents.
PPO content (phr)
Neat PPO
Inte
rfac
ial s
hear
str
engt
h (M
Pa)
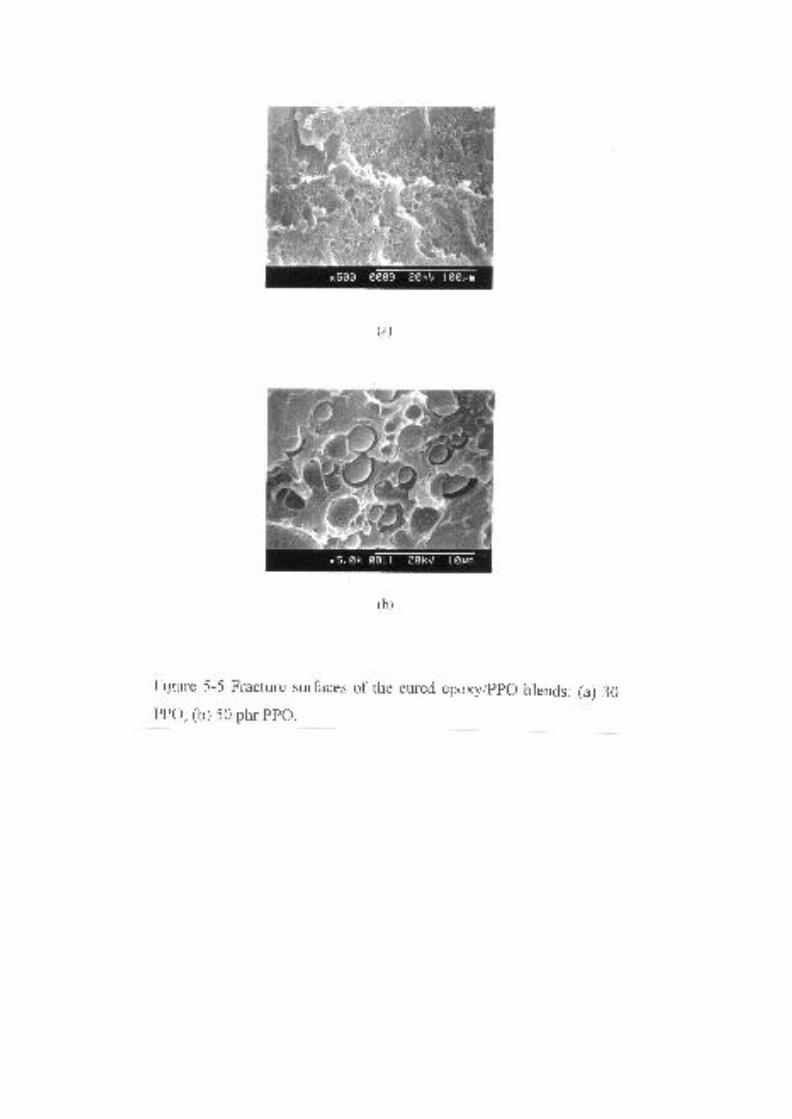





我們發現環氧樹脂會集中在富克維拉纖維 (Kevlar fiber-rich) 的區
域,如 Figure 5-9(b) 所示,原因可能是環氧樹脂的極性比 PPO高,
克維拉纖維因為具有高極性的醯胺基,所以在硬化過程中環氧樹脂較
易往纖維的極性表面移動,因此形成了環氧樹脂包覆克維拉纖維
(epoxy-coated Kevlar fiber) 的相形態,這種 epoxy-coated fiber之相形
態也出現在 Epoxy/PEI複合材料系統[115]。這也可解釋在 microbond
測試中摻合 PPO 後的 IFSS值變化不大的原因,如 Figure 5-10所示。
所以在複合材料中基材的相形態相異於未含纖維之摻合體的相形
態。
5-3-3 複合材料機械性質之探討
複合材料的臨界應變能釋放率 (GIC) 乃是剝離層間樹脂開口式的破
壞行為所需的能量。除了樹脂的破壞外,纖維的聚集 (nesting)、纖維
的架橋 (bridging)、纖維的拉出及斷裂等現象都會提高 GIC 值。以
Mode-I DCB法測GIC值,實驗時所需之循環負載-卸載過程,由 X-
Y記錄器繪製負載-位移 (loading-displacement) 圖。不同 PPO 含量
的Kevlar fiber/Epoxy/PPO複合材料之臨界應變能釋放率GIC值影響如
Figure 5-11所示,隨 PPO含量增加 GIC值有上升的趨勢,所以複合材
料破裂能量增加的主要因素為樹脂基材破壞強度的大小[116, 117]。在
層間剪切應力測試中,我們採用 ASTM D2334的方法,以 0度-16層
的積層材做三點彎曲測試,結果列於 Figure 5-12。複合材料的 ILSS
剪切力與破壞行為彼此相互關連。對於韌性基材而言,在剪切作用下
基材會有明顯的降伏及塑性變形,進而形成許多小裂紋。隨著力量增
加,小裂縫間互相聯結,最後造成破裂[118]。由以上可知,假使基
材的韌性愈大時,其抵抗裂縫的起始能力愈強,所需的

Figure 5-10 Schematic representation of phase separation of epoxy/PPO
blend in microbond sample.

0 10 20 30 40 50100
150
200
250
300
350
400
450
500
Figure 5-11 Results of fracture energy (GIC
) for the epoxy/PPO/Kevlar
fiber composite.
Fra
ctur
e en
ergy
(Jm
-2 )
PPO content (phr)

0 10 20 30 40 5025
30
35
40
45
Figure 5-12 The interlaminar shear strength of composites with various PPO contents.
Inte
rlam
inar
she
ar s
tren
gth
(MPa
)
PPO content (phr)

剪應力愈大,相對地破壞能量也較高。本實驗之複合材料的相形態
中,環氧樹脂會集中在纖維區域,形成 epoxy-coated fiber區域,此種
相形態不會因為 PPO 的添加而降低了纖維與基材間的界面黏著力,
所以摻合體之 IFSS值與純環氧樹脂之 IFSS值相當。但是複合材料的
ILSS值卻隨 PPO的添加量增加而大幅上升,原因是摻合熱塑性 PPO
會使複合材料的延展性與韌性大幅提升。在 T型剝離力與 PPO含量
的關係圖中 (Figure 5-13),我們也發現 T型剝離力也隨著 PPO 含量
增加而上升。由 SEM觀察發現 (Figure 5-14),纖維被破壞的程度並
未有明顯的差異,而在基材處則發現隨 PPO 增加而出現應力反白的
裂紋,這表示 T型剝離力的增加也是由於基材韌性增加所導致。
5-3-4 複合材料熱性質測試
Table 5-1顯示 PPO摻合量增加會造成環氧樹脂相的 Tg下降,尤
其是 PPO 含量大於 30 phr時特別明顯,這種現象同時發生在無纖維
補強的樹脂及纖維補強的複合材料之基材相中。會造成環氧樹脂相 Tg
下降的原因是 PPO的存在會阻礙環氧樹脂交聯,同時經由 FTIR 分析
得知 PPO 也會改變硬化反應機構。由純摻合體及含有纖維的基材中
之環氧樹脂與 PPO兩相 Tg的溫度差 (∆T) 得知,複合材料中兩相 Tg
的溫度差遠大於摻合體的 Tg溫度差,這意味著含有纖維時的相分離
程度遠大於純樹脂的相分離程度。
在 TGA分析中,複合材料的起始裂解溫度 (Td) 約在 300oC,其
最大裂解速率溫度 (Tp) 隨 PPO 的增加而提高,趨勢與未含纖維的摻
合體相同。在裂解完畢後所剩的焦炭,大約剩下 20 %,這應該是克
維拉纖維所留下的焦炭部分,如 Table 5-2所示。

0 10 20 30 40 50400
500
600
700
800
900
1000
1100
Figure 5-13 The T-peel strength of composites with various PPO contents.
T-p
eel s
tren
gth
(g/c
m)
PPO content (phr)


Figure 5-14 Fracture surfaces of the composites with various PPO
contents after T-peel test: (a) 0 phr, (b) 30 phr, (c) 50 phr.

Table 5-1 Glass transition temperature Tg of cured resin blends and
composites.
PPO Tg of cured blend (oC) Tg of composite (oC)
(phr) PPO phase Epoxy phase PPO phase Epoxy phase
0 --- 134 --- 126
10 194 134 212 126
30 198 132 202 122
50 200 --- 200 118
Tg of PPO: 220oC; DMA measurement, ramp: 5oC/min from 50 to 250oC
at 1 Hz; Cure condition: 4h at 180oC followed by 4h at 220oC.

Table 5-2 The result of TGA analysis for Kevlar fiber composites.
PPO content Td (oC) Tp (
oC) Char residual (wt %)0 phr 350 450 2210 phr 350 453 2130 phr 346 460 2050 phr 344 468 20
Heating rate: 20 oC/minChar residual at 800 oC in nitrogen.

5-3-5 克維拉纖維表面處理對於複合材料機械性質的影響
在 DDM硬化環氧樹脂系統中,利用電漿及氯磺酸進行克維拉纖
維表面處理可大幅增加複合材料的界面黏著力[117],所以我們也嘗試
以此種方式來增進氰酸酯硬化系統的複材界面黏著力。克維拉纖維
經由 NH3-plasma 30 W 5 min的處理後,複合材料的層間剪切應力與 T
型剝離力均上升,原因是纖維表面接上胺基後可與環氧樹脂之環氧基
及氰酸酯之 C≡N 反應,彼此間形成化學鍵結,增加了界面黏著力。
可是在氯磺酸纖維表面處理後,其層間剪切應力及 T 型剝離力卻下
降,其原因仍有待探討。
5-4 結論
複合材料中基材的相分離模式與純樹脂不同,純樹脂的相形態如
第三章所述,有分散相與連續相的分別;當有極性的克維拉纖維存在
時,在硬化過程中環氧樹脂會累積在纖維表面,在纖維上形成環氧樹
脂層。環氧樹脂層能提供良好的界面接著力,此界面力是純 PPO 所
無法達成的。所以摻合體/Kevlar fiber的界面剪切應力 (IFSS) 與純環
氧樹脂/Kevlar fiber 的 IFSS 的值接近。若纖維含量高時,則會形成
epoxy-coated fiber 在 PPO 基材相中,此種相形態能提供複合材料良
好的機械性質,例如破裂能量、層間剪應力 (ILSS)、T型剝離力等。
而複合材料熱性質與純樹脂相似,不會受到纖維存在的影響。

第六章 結論
本研究中氰酸酯硬化環氧樹脂/PPO混合系統與氰酸酯/PPO混合
系統的硬化行為是一自催化反應機構。隨著 PPO 添加量增加,氰酸
酯硬化環氧樹脂/PPO 混合系統的反應機構會改變且起始反應速率會
加快,原因是 PPO 末端 OH 基的催化作用所導致。但是隨反應進行
到凝膠態時,反應就受到擴散效應的控制,在高 PPO 含量時轉化率
有降低的趨勢,所以反應熱也隨 PPO 添加量增加而下降。FTIR 及
NMR 分析顯示特性官能基的反應隨 PPO 增加而加速並且有
coreaction 產生,例如 cyanate-hydroxyl addition (imidocarbonate,
carbamate),epoxy-cyanate addition (oxazolidinone) 等。氰酸酯硬化環
氧樹脂/PPO 混合系統當反應進行時其相形態會發生變化,在低 PPO
含量時 (10 phr),相分離是以成核模式 (NG) 進行;在高 PPO 含量
時 (30 和 50 phr),相分離是以 spinodal decomposition (SD) 模式進
行,原因是 PPO 的添加會造成系統硬化反應速率加快,轉化率提高
且迅速地穿越 binodal curve到達 spinodal curve,產生相分離。此外在
臨界點附近也容易形成 SD相分離。摻合體最終相形態是有規律且隨
PPO 含量而變化,在 10 phr PPO時,是形成 PPO為顆粒分散相分佈
在環氧樹脂連續相中;當 PPO 為 50 phr時,摻合體的相形態轉變為
環氧樹脂為分散相,PPO 為連續相,稱之為相反轉;而 PPO為 30 phr
時,形成一部分相反轉的相形態。這些相形態常見於熱塑性塑膠改質
熱固性系統中。此外我們也可利用 DMA可得知摻合體中兩相的特性
及部分相容的情況。然而摻合體兩相的形態並不均勻特別是在聚氧化
二甲苯含量較低時,所以添加 TAIC 以改善摻合體之均勻性及相容
性。TAIC 與環氧樹脂的相容性佳且與聚氧化二甲苯形成接枝共聚
物。由 SEM 觀察可知添加 TAIC 後的摻合體之相容性大幅增加且抗

溶劑性提高。此外抗張強度隨著 PPO 增加而微幅上升,抗張模數卻
微幅下降;在破裂能量方面,當 PPO 變成連續相時則大幅增加,原
因是撕裂及延展 PPO需大幅能量。在熱性質方面,隨著 PPO含量增
加則摻合體之最大熱裂解速率溫度增加,但起始裂解溫度有些微下降
趨勢。熱膨脹係數也隨著 PPO 含量增加而降低,在摻合體相反轉時
特別明顯。介電常數隨 PPO含量的增加有線性下降的趨勢。
另外我們以分散法製備 dicy硬化環氧樹脂/PPO摻合體,探討添
加熱塑性 PPO 粒子對熱固性環氧樹脂的影響。結果顯示所有組成的
摻合體皆為兩相形態,且它們的相形態與 PPO 含量有關。在低 PPO
含量下,呈現出 PPO 顆粒分散在環氧樹脂的基材相中,PPO 粒子有
聚集的現象。在高 PPO 含量下,相形態就顯得較複雜,例如是相反
轉及共連續相的產生。在 DMA分析中,所有組成都有部分互溶的情
形,PPO相 Tg下降的原因是由於有交聯不完全的環氧樹脂存在 PPO
相。在抗張強度及抗張模數方面,PPO 的添加對摻合體的影響並不
大。在破裂能量方面,隨著 PPO含量的增加而大幅提升,在 PPO含
量高時特別明顯,此與相形態有密切的關係。在 TGA 分析中,PPO
會妨礙環氧樹脂的交聯反應,因而降低環氧樹脂的交聯程度;不過在
熱重分析圖 (TGA) 中,可知在純的環氧樹脂會在較高溫度才迅速往
下掉,但在 PPO 高含量之摻合體中,起始反應溫度有些微下降的現
象,由此可知除了 PPO 會阻礙環氧樹脂交聯之外,環氧樹脂還會進
入 PPO 顆粒內,導致摻合體內含有未完成反應的環氧樹脂。此外,
摻合體隨著 PPO 含量增加其最大裂解速率溫度大幅提升。在介電性
質方面,隨著 PPO 的含量增加,其介電常數大幅下降。在環氧樹脂
/PPO (30 phr)摻合系統中添加 TAIC 之後,摻合體中 PPO顆粒聚集現
象有明顯的改善。由 DMA及 SEM可看出添加 TAIC後摻合體的相容

性增加,破裂面經由二氯甲烷蝕刻後,顯示出系統的抗溶劑性質增
加。TAIC 的添加並未造成摻合體的抗張強度及模數的下降。在破裂
能量方面,隨著 TAIC 含量的增加而上升,20 phr是最佳添加量。在
TGA分析中,最大裂解速率溫度隨著 TAIC 含量增加而升高。介電性
質方面,隨著 TAIC含量增加,摻合體的介電常數也明顯下降,超過
20 phr有上升趨勢。
最後在氰酸酯硬化環氧樹脂/PPO/Kevlar fiber複合材料方面,我
們發現複合材料的相形態與純樹脂的相形態不同,在基材相分離的過
程中,環氧樹脂會往極性的Kevlar fiber表面移動,形成一 epoxy-coated
fiber的相形態,所以在 microbond測試中其 IFSS值不受到 PPO添加
的影響而下降。在複合材料積層板中,環氧樹脂會大量聚集在纖維表
面,形成 epoxy-coated fiber 分佈於 PPO相中,此種相形態對於複合
材料的破壞韌性、機械性質有正面的影響。

參考文獻
1. Y. Ishii, T. Arai, S. Kinoshita and T. Katayose, PCWC VII, Basel 96, I-3-1 (1996).
2. M. F. Grenier-Loustalot, C. Lartigau and P. Grenier, Europ. Polym. J. 31, 1139
(1995).
3. D. A. Shimp, F. A. Hudock and S. J. Ising, Int. SAMPE symp. Exhib. 33, 754; Ch.
Abs. 110, 96323 (1988).
4. F. J. McGarry and A. M. Willner, Org. Coat Plast. Chem. (ACS) 28, 512 (1968).
5. C. B. Bucknall and I. K. Partridge, Polym. Eng. Sci. 26, 54 (1986).
6. J. L. Hedrick, I. Yilgor, G. L. Wilkes and J. E. McGrath, Polym. Bull. 13, 201
(1985).
7. J. A. Cecere and J. E. McGrath, Polym. Prepr. 27, 299 (1986).
8. C. B. Bucknall, C. M. Gomez and I. Quintard, Polymer 35, 353 (1994).
9. C. B. Bucknall and A. H. Gilbert, Polymer 30, 213 (1989).
10. D. J. Hourston and J. M. Lane, Polymer 33, 1379 (1992).
11. M. C. Chen, D. J. Hourston and W. B. Sun, Europ. Polym. J. 28, 1471 (1992).
12. T. Ohsako, K. Nagura and I. Nozue, Polymer 34, 5080 (1993).
13. E. M. Woo, L. D. Bravence and J. C. Seferis, Polym. Eng. Sci. 34, 1664 (1994).
14. R. A. Pearson and A. F. Yee, Polymer 34, 3658 (1993).
15. R. A. Pearson and A. F. Yee, J. Appl. Polym. Sci. 48, 1051 (1993).
16. R. W. Venderbosch, H. E. H. Meijer and P. J. Lemstra, Polymer 35, 4349 (1994).
17. R. W. Venderbosch, H. E. H. Meijer and P. J. Lemstra, Polymer 36, 1167 (1995).
18. R. W. Venderbosch, H. E. H. Meijer and P. J. Lemstra, Polymer 36, 2903 (1995).
19. 張國輝, 聚苯醚-環氧樹脂摻合物芳香族胺類固化系統之研究 , 國立交通大
學應用化學研究所碩士論文 (1998).
20. Z. Fu and Y. Sun, Polym. Prepr. 29, 177 (1988).
21. Z. B. Ahmad, M. F. Ashby and P. W. R. Beaumont, Scr. Metall. Mater. 20, 843
(1986).
22. K. T. Farber and A. G. Evans, Acta Mettal. 31, 565 (1983).
23. J. Kim and R. Robertson, Polym. Mater. Sci. Eng. (ACS) 63, 301 (1990).
24. H. J. Sue, R. A. Pearson and A. F. Yee, Polym. Eng. Sci. 31, 793 (1991).
25. C. B. Bucknall, In: Toughened Plastics, Applied Science, Barking Essex. (1997).
26. J. N. Sultan, R. C. Laible and F. J. McGarry, J. Appl. Polym. Sci. 6, 127 (1971).

27. B. S. Kim, T. Chiba and T. Inoue, Polymer 34, 2809 (1993).
28. J. P. Pascault, J. Galy and F. Mechin, In: I. Hamerton (ed), Chemistry and
Technology of Cyanate Ester Resins, Chapman & Hall, London, 112 (1994).
29. R. J. J. Williams, J. Borrajo, H. E. Adabbo and A. J. Rojas, In: C. K. Riew and J.
K. Gillham (eds), Rubber-Modified Thermoset Resins, Adv. Chem. Ser. 208
Washington DC; Am. Chem. Soc., 195 (1984).
30. A. Vazquez, A. J. Rojas, H. E. Adabbo, J. Borrajo and R. J. J. Williams, Polymer
28, 1156 (1987).
31. S. Viscont and R. H. Marchessault, Macromolecules 7, 913 (1974).
32. T. T. Wang and H. M. Zupko, J. Appl. Polym. Sci. 26, 2391 (1981).
33. L. C. Chan, J. K. Gillham, A. J. Kinloch and S. J. Shaw, In: C. K. Riew and J. K.
Gillham (eds), Rubber-Modified Thermoset Resins, Adv. Chem. Ser. 208
Washington DC; Am. Chem. Soc., 235 (1984).
34. A. C. Grillet, J. Galy and J. P. Pascault, Polymer 33, 34 (1992).
35. D. Chen, J. P. Pascault, H. Sautereau and G. Vigier, Polym. Int. 32, 369 (1993).
36. C. G. Delides, D. Hayward, R. A. Pethrick and A. S. Vatalis, J. Appl. Polym. Sci.
47, 2037 (1993).
37. D. K. Hoffman and C. B. Arends, US Patent 4, 708, 996 (1987).
38. D. K. Hoffman, C. Ortiz, D. L. Hunston and W. McDonough, Polym. Mat. Sci.
Eng. (ACS) 70, 7 (1994).
39. C. Ortiz, W. McDonough, D. L. Hunston and D. K. Hoffman, Polym. Mat. Sci.
Eng. (ACS) 70, 9 (1994).
40. H. J. Sue, E. I. Garcia-Meitin, D. M. Pickelman and P. C. Yang, In: Toughened
Plastics 1: Science and Engineering, Adv. Chem. Ser. 233 Washington DC; Am.
Chem. Soc., 259 (1993).
41. C. K. Riew, A. R. Siebert, R. W. Smith, M. Fernando and A. J. Kinloch, Polym.
Mat. Sci. Eng. (ACS) 70, 5 (1994).
42. J. Y. Qian, R. A. Pearson, V. L. Dimonie and M. S. El-Aasser, Polym. Mat. Sci.
Eng. (ACS) 70, 17 (1994).
43. A. Maaazouz, H. Sauteereau and J. F. Gérard, Polym. Bull. 33, 67 (1994).
44. G. Levita, A. Marchetti and A. Lazzeri, Makromol. Chem., Macromol. Symp. 41,
179 (1991).
45. R. Mülhaupt and U. Buchholz, Polym. Mat. Sci. Eng. (ACS) 70, 4 (1994).
46. M. Gordon, J. Goldsbrough, B. Ready and K. W. Derham, In: Industrial

Polymers, Characterization by Molecular Weight, Transcripta Books, London, 45
(1973).
47. H. J. Borchardt and F. Daniels, J. Am. Chem. Soci. 79, 41 (1957).
48. Y. P. Khanna, R. Kumar and S. Das, Polym. Eng. Sci. 30, 1171 (1990).
49. H. Ng and I. Manas-Zloczower, Polym. Eng. Sci. 29, 1097 (1989).
50. L. J. Lee, Polym. Eng. Sci. 21, 483 (1981).
51. S. Sourour and M. R. Kamal, Thermochimica Acta. 14, 41 (1976).
52. N. A. St John, G. A. Georage, P. A. Cole-Clarke, M. E. Mackay and P. J. Halley,
High Perform. Polym. 5, 21 (1993).
53. Y. S. Yang and L. J. Lee, SPE ANTEC Tech. Papers 32, 419 (1986).
54. S. L. Simon and J. K. Gillham, J. Appl. Polym. Sci. 47, 461 (1993).
55. G. Wisanrakkit and J. K. Gillham, J. Coat. Technol. 62, 35 (1990).
56. M. T. Aronhime and J. K. Gillham, Adv. Polym. Sci. 78, 84 (1986).
57. J. K. Gillham and J. B. Enns, J. Appl. Polym. Sci. 28, 2567 (1983).
58. J. W. S. Hearle and C. Y. Zhou, Textile Res. J. 57, 7 (1987).
59. J. Kalantra and L. T. Drzal, J. Mater. Sci. 25, 4186 (1990).
60. S. E. Keinath and R. J. Morgan, Thermochimica Acta 166, 17 (1990).
61. L. S. Penn and F. Larsen, J. Appl. Polym. Sci. 23, 59 (1979).
62. R. J. Morgan, C. O. Pruneda, and W. J. Steele, J. Polym. Sci.: Polym. Phys. Ed. 2,
1757 (1983).
63. L. S. Li, L. F. Allard, and W. C. Bigelow, J. Macromol. Sci. Phys. B22, 269
(1983).
64. M. G. Dobb, D. J. Johnson, and B. P. Saville, J. Polym. Sci.: Polym. Phys. Ed. 15,
2201 (1977).
65. M. Panar, D. Avakian, R. C. Blume and K. H. Gardner, J. Polym. Sci.: Polym.
Phys. Ed. 21, 1955 (1983).
66. R. J. Young, D. Lu, R. J. Day, W. F. Knoff and H. A. Davis, J. Mater. Sci. 27,
5431 (1992).
67. S. J. Krause, D. L. Vezie and W. W. Adams, Polym. Commun. 30, 19 (1989).
68. M. G. Dobb and R. M. Robson, J. Mater. Sci. 25, 459 (1990).
69. H. Fujiwara, B. S. Kim and T. Inoue, Polym. Eng. Sci. 36, 1541 (1996).
70. S. Mostovoy, P. B. Crosley and E. J. Ripling, J. Mater. 2, 661 (1967).
71. J. Bauer, Thesis, B. Academy of Sciences of the GDR, Berlin (1990).
72. M. Bauer, J. Bauer and B. Garske, Acta Polym. 37, 604 (1986).

73. M. Bauer, J. Bauer and G. Kühn, Acta Polym. 37, 604 (1986).
74. A. K. Bonezkaya, M. A. Kravtschenko, V. V. Korshak, Z. M. Frenkel, V. A.
Pankratov and S. V. Vinogradova, Vysokomol. Soed. B17, 282 (1975).
75. A. K. Bonezkaya, M. A. Kravtschenko, Z. M. Frenkel, V. A. Pankratov, S. V.
Vinogradova and V. V. Korshak, Vysokomol. Soed. A19, 1042 (1977).
76. A. K. Bonezkaya, V. V. Ivanov, M. A. Kravtschenko, V. A. Pankratov, Z. M.
Frenkel, V. V. Korshak and S. V. Vinogradova, Vysokomol. Soed. A22, 766 (1980).
77. A. K. Bonezkaya, M. A. Kravtschenko, Z. M. Frenkel, V. A. Pankratov, V. V.
Korshak and S. V. Vinogradova, Vysokomol. Soed. A23, 1494 (1981).
78. Z. M. Frenkel, V. V. Korshak, A. K. Bonezkaya, V. A. Pankratov, S. V.
Vinogradova and M. A. Kravtschenko, J. Prakt. Chem. 318, 923 (1976).
79. O. Georjon, J. Galy and J. P. Pascault, J. Appl. Polym. Sci. 49, 1441 (1993).
80. A. M. Gupta and C. W. Macosko, Makromol. Chem. Macromol. Symp. 45, 105
(1991).
81. V. V. Korshak, A. K. Bonezkaya, M. A. Kravtschenko, Z. M. Frenkel, V. A.
Pankratov and S. V. Vinogradova, Izv. Akad. Nauk SSSR, Ser. Chim. 1976, 923
(1976).
82. V. Micro, Z. Q. Cao, F. Mechin and J. P. Pascault, Proc. ACS, Div. Polym. Mat.
Sci. Eng. 66, 451 (1992).
83. Y. P. Khanna, R. Kumar and S. Das, Polym. Eng. Sci. 30, 1171 (1990).
84. R. H. Lin, J. L. Hong and A. C. Su, Proc. ACS, Div. Polym. Mat. Sci. Eng. 66,
464 (1992).
85. S. V. Vinogradova, M. M. Pazuriya, V. A. Pankratov, V. V. Korshak, L. I.
Makarova and K. A. Andrianov, Vysokomol. Soed. A19, 80 (1977).
86. M. R. Kamal, Polym. Eng. Sci. 14, 231 (1974).
87. S. L. Simon and J. K. Gillham, Proc. ACS, Div. Polym. Mat. Sci. Eng. 32, 182
(1991).
88. S. L. Simon and J. K. Gillham, Proc. ACS, Div. Polym. Mat. Sci. Eng. 66, 453
(1992).
89. E. Grigat and R. Putter, Angew. Chem. Internat. 6, 206 (1967).
90. D. A. Shimp, S. J. Ising and J. R. Christenson, Int. SAMPE symp. Exhib. 34, 222
(1989).
91. M. Bauer, W. Tänzer, H. Much and R. Ruhman, Acta Polym. 40, 335 (1989).
92. M. Bauer, J. Bauer, R. Ruhman and G. Kühn, Acta Polym. 40, 397 (1989).

93. D. A. Shimp and J. E. Wentworth, SAMPE, Los Angeles, 293 (1992).
94. D. Verchère, H. Sautereau, J. P. Pascault, S. M. Moschiar, C. C. Riccardi and R. J.
J. Williams, In: C. K. Riew and A. J. Kinloch (eds), In: Toughened Plastics 1:
Science and Engineering, Adv. Chem. Ser. 233 Washington DC; Am. Chem. Soc.,
335 (1993).
95. W. H. Jo and M. B. Ko, Macromolecules 27, 7815 (1994).
96. K. Yamanaka and T. Inoue, Polymer 30, 662 (1989).
97. K. Yamanaka, V. Takagi and T. Inoue, Polymer 30, 1839 (1989).
98. K. Yamanaka and T. Inoue, J. Mater. Sci. 25, 241 (1990).
99. H. S. Y. Hsich, J. Mater. Sci. 25, 1568 (1990).
100. H. S. Y. Hsich, Polym. Eng. Sci. 30, 493 (1990).
101. T. Inoue, Prog. Polym. Sci. 20, 119 (1995).
102. T. Miyamoto, K. Kodama and K. Shibayama, J. Polym. Sci. A2, 8, 2095 (1970).
103. R. Buchdahl and L. E. Nielsen, J. Appl. Phys. 21, 482 (1950).
104. L. E. Nielsen, Appl. Polym. Symp. 12, 249 (1969).
105. D. Hull, In: An Introduction to Composite Materials, Cambridge University
Press, Ch. 1 (1981).
106. S. Kumar, In: Structure and Properties of High Performance Polymeric and
Carbon Fiber, An Overview, SAMPE Quarterly, Jan., 3 (1986).
107. C. B. Bucknall and I. K. Partridge, Polymer 24, 639 (1983).
108. J. C. Hedrick, J. L. Hedriclc, J. A. Cecere, S.C. Liptak and J .E. McGrath, In:
Proc. ACS Division of Polymeric Materials: Science and Engineering, ACS Books
and Journal Division, Washington DC, 26-31, 190-194 (1990).
109. H. D. Stenzenberger, W. Römer, M. Herzog and P. König, In: Proc. 33rd Int.
SAMPE Symp., ed. G. Carrillo, E. D. Newell, W. D. Brown and P. Phelan, SAMPE,
Anaheim, CA, 1546 (1988).
110. B. Miller, P. Muri and L. Rebenfeld, Composites Sci. and Technol. 28, 17 (1987).
111. P. J. Merrera-Franco and L. T. Drzal, Composites 23, 2 (1992).
112. G. S. Sheu, T. K. Lin, S. S. Shyu and J.Y. Lai, J. Adhesion Sci. Technol. 8, 511
(1994).
113. J. P. Berry, J. Appl. Phys. 34, 62 (1963).
114. M. F. Grenier-Loustalot, C. Lartigau, F. Metras and P. Grenier, J. Polym. Sci.:
Polym. Chem. 34, 2955 (1996).
115. D. J.-P. Turmel and I. K. Partridge, Composites Sci. Technol. 57, 1001 (1997).

116. J. R. Brown, P. J. C. Chappell and Z. Mathys, J. Mater. Sci. 26, 4172 (1991).
117. S. R. Wu, G. S. Shyu and S. S. Shyu, J. Appl. Polym. Sci. 62, 1347 (1996).
118. D. C. Leach and D. R. Moore, Composites Sci. Technol. 23, 131 (1985).









![E=F9;mklge]j ZYjge]l]j L`ak lae] alÌk h]jkgfYd2 ^jge [gfkme]j lg [g%[j]Ylgj](https://static.fdokumen.com/doc/165x107/631789cb7451843eec0ab6f2/ef9mklgej-zyjgelj-lak-lae-alik-hjkgfyd2-jge-gfkmej-lg-gjylgj.jpg)










