
RuO 4 -mediated oxidation of N -benzylated tertiary amines. Are amine N -oxides and iminium cations...
21
DOI: 10.2478/s11532-006-0039-8 Research article CEJC 4(4) 2006 674–694 RuO 4 -Mediated Oxidation of N -Benzylated Tertiary Amines. 3. Behavior of 1,4-Dibenzylpiperazine and Its Oxygenated Derivatives Horia Petride ∗ , Constantin Dr˘ aghici, Cristina Florea, Aurica Petride “Costin D. Nenitzescu” Center of Organic Chemistry, 050461 Bucharest, P.O. 35, Box 108, Romania Received 28 April 2006; accepted 2 July 2006 Abstract: 1,4-Dibenzylpiperazine (1), -2-piperazinone (7), -2,6-piperazinedione (9), and 1-benzoyl-4- benzylpiperazine (30) were oxidized by RuO 4 (generated in situ ) by attack at their endocyclic and exocyclic (i.e., benzylic) aminic N -α-C-H bonds to afford various oxygenated derivatives, including acyclic diformamides, benzaldehyde, and benzoic acid. The reaction outcome was complicated by (i) the hydrolysis of diformamides, occurred during the work-up, and (ii) the reaction of benzaldehyde with the hydrolysis-derived amines giving imidazolidines and/or Schiff bases. Benzoic acid resulted from benzaldehyde only. Compounds 7, 30, and 1-benzylpiperazine, but not 9, were transiently formed during the oxidation of 1. In the same reaction conditions, 1,4-dibenzyl-2,3-(or 2,5)-piperazinedione, 1,4-dibenzyl-2,3,6-piperazinetrione, 4-benzoyl-1-benzyl-2-piperazinone, and 1,4-dibenzoylpiperazine were inert. The proposed oxidation mechanism involves the formation of endocyclic and exocyclic iminium cations, as well as of cyclic enamines. The latter intermediates probably result by base-induced deprotonation of the iminium cations, provided an N + −β-proton is available. In the case of 1, the cations were trapped with NaCN as the corresponding α-aminonitriles. The statistically corrected regioselectivity (endocyclic/exocyclic) of the RuO 4 -induced oxidation reaction of 1, 7, and 30 was 1.2-1.3. c Versita Warsaw and Springer-Verlag Berlin Heidelberg. All rights reserved. Keywords: Oxidation, ruthenium tetraoxide, amines, iminium cations, regioselectivity 1 Introduction It is known that the powerful oxidant ruthenium tetraoxide (RuO 4 ) functionalizes ter- tiary aliphatic amines at their N -α-positions to afford amides and/or dioxygenated com- pounds [1–6]. For instance, earlier studies indicated 4-benzoylmorpholine and 4-benzyl-3- ∗ E-mail: [email protected]
Transcript of RuO 4 -mediated oxidation of N -benzylated tertiary amines. Are amine N -oxides and iminium cations...

DOI: 10.2478/s11532-006-0039-8Research article
CEJC 4(4) 2006 674–694
RuO4-Mediated Oxidation of N -Benzylated TertiaryAmines. 3. Behavior of 1,4-Dibenzylpiperazine
and Its Oxygenated Derivatives
Horia Petride∗, Constantin Draghici, Cristina Florea, Aurica Petride
“Costin D. Nenitzescu” Center of Organic Chemistry,050461 Bucharest, P.O. 35, Box 108, Romania
Received 28 April 2006; accepted 2 July 2006
Abstract: 1,4-Dibenzylpiperazine (1), -2-piperazinone (7), -2,6-piperazinedione (9), and 1-benzoyl-4-benzylpiperazine (30) were oxidized by RuO4 (generated in situ) by attack at their endocyclic andexocyclic (i.e., benzylic) aminic N -α-C-H bonds to afford various oxygenated derivatives, includingacyclic diformamides, benzaldehyde, and benzoic acid. The reaction outcome was complicated by (i)the hydrolysis of diformamides, occurred during the work-up, and (ii) the reaction of benzaldehydewith the hydrolysis-derived amines giving imidazolidines and/or Schiff bases. Benzoic acid resultedfrom benzaldehyde only. Compounds 7, 30, and 1-benzylpiperazine, but not 9, were transiently formedduring the oxidation of 1. In the same reaction conditions, 1,4-dibenzyl-2,3-(or 2,5)-piperazinedione,1,4-dibenzyl-2,3,6-piperazinetrione, 4-benzoyl-1-benzyl-2-piperazinone, and 1,4-dibenzoylpiperazine wereinert. The proposed oxidation mechanism involves the formation of endocyclic and exocyclic iminiumcations, as well as of cyclic enamines. The latter intermediates probably result by base-induceddeprotonation of the iminium cations, provided an N +−β-proton is available. In the case of 1, the cationswere trapped with NaCN as the corresponding α-aminonitriles. The statistically corrected regioselectivity(endocyclic/exocyclic) of the RuO4-induced oxidation reaction of 1, 7, and 30 was 1.2-1.3.c© Versita Warsaw and Springer-Verlag Berlin Heidelberg. All rights reserved.
Keywords: Oxidation, ruthenium tetraoxide, amines, iminium cations, regioselectivity
1 Introduction
It is known that the powerful oxidant ruthenium tetraoxide (RuO4) functionalizes ter-
tiary aliphatic amines at their N -α-positions to afford amides and/or dioxygenated com-
pounds [1–6]. For instance, earlier studies indicated 4-benzoylmorpholine and 4-benzyl-3-
∗ E-mail: [email protected]

H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694 675
morpholinone as reaction products obtained from 4-benzylmorpholine [2], meaning that
both types of N -α-sites (i.e., exocyclic and endocyclic, respectively) were attacked. In
the same reaction conditions, 1-benzylpiperidine yielded only products of endocyclic
attack [3]. Same exclusive endocyclic functionalization showed also some piperazine
derivatives (Scheme 1). Thus, starting from 1,4-dibenzylpiperazine (1), Tortorella and
his co-workers [6] have isolated two reaction products, namely N,N’ -dibenzyl-N,N’ -1,2-
ethanediylbisformamide (2) and 1,4-dibenzyl-2,3-piperazinedione (3), in 65 and 5% yield,
respectively. Similar reaction of 1-benzylpiperazine (4) afforded only the corresponding
acyclic diformamide 5, but not also 6. The same piperazinedione 3 was obtained when
starting from piperazinone 7 [6]; the analogous possible reaction 8→6 has not been stud-
ied yet. In the same paper [6], the authors claimed also that only 10 has resulted from
2,6-piperazinedione 9. Besides the endocyclic functionalization in all these cases, a simul-
taneous oxidative attack at both N1-CH2-CH2-N2 methylene groups of 1 and 4 seems to
have been in action, unlike the behavior of 1-benzylpiperidine [3].
Scheme 1 Oxidation by RuO4 of piperazine derivatives (literature data).
At variance with some of these data, our previous works, devoted to the RuO4-
oxidation of tertiary amines of general structure Ph-CH2-NR2 (NR2= 1-pyrrolidinyl, 1-
piperidinyl, 4-morpholinyl [7]; R = alkyl [8]), have indicated that both types of N -α-sites
were attacked, even in the case of 1-benzylpiperidine. The reactions were interpreted as
occurring through the corresponding iminium cations, as well as through enamines, gen-
erated by N +−β-deprotonation of the non-benzylic cations (R �= CH3). The cations were
trapped with NaCN as the respective nitriles. Oxidation of the C=C bond of enamines

676 H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694
was the source of some dioxygenated reaction products.
Taking into account all these facts, we considered it interesting to extend our research
on the RuO4-oxidation reactions of piperazine 1 and of some of its mono- and dioxy-
genated derivatives. Do they really react at the endocyclic sites exclusively, as claimed
in the literature? At the same time, we questioned about the route(s) giving 2 and 3.
Do these compounds result by a simultaneous introduction of two oxygen atoms or by
two successive monooxidation steps? Piperazine 1 might be viewed as a 4-aza analog of
1-benzylpiperidine. Could the formation of 2 and 3 be attributable just to the presence
of the additional nitrogen atom? This paper aims to clarify all these questions. As shown
below, the oxidative patterns of 1, 7, or 9 were more complicated than those suggested
in the literature [6]. Generally speaking, the respective reactions followed the same path-
ways as those already reported by us for other substrates [7, 8]. Even more interesting
was the behavior of 4 and of its oxygenated derivatives; the respective results will be
detailed in a coming paper [9].
In order to distinguish between the two nitrogen atoms in 1-10, they were labeled by
1 and 2 superscripts in Scheme 1. The same type of labeling was followed in Schemes 2-6.
For the same reason, the yields of the various reaction products were written in the main
text as {compound number}, followed by i or exp subscripts for the initially formed
and the experimentally found amounts, respectively.
2 Results and discussion
Oxidation was performed in the same conditions as above [2, 3, 7, 8], that is with cat-
alytic amounts of RuO4 (generated in situ from catalytic RuO2 and excess NaIO4), in
CCl4/water heterogeneous mixtures and at room temperature. The reaction mixtures
were worked-up in two ways (A or B ; see Section 3.8), differing by the order of opera-
tions. Some oxidation products were unstable in alkaline media (see below); they could
be seen only following the work-up A. This information was correlated with the already
reported data [10] obtained on the hydrolysis of pure compounds.
The oxidation results are discussed in Section 2.1 and summarized in Table 1. All
yields quoted in Table 1 are of exp type. It is worth mentioning that the yield of benzalde-
hyde (BzH) actually corresponds to that of the benzaldehyde+benzoic acid sum. This is
entirely justified by the fact that benzoic acid is derived by oxidation from benzaldehyde.
As presented below, other possible sources of benzoic acid (i.e., by hydrolysis of N-COPh
functions) could be safely discharged. Some of the yields in Table 1 are quoted as sums;
the reason will be explained later. The last column of Table 1 contains the values of
regioselectivity (endocyclic/exocyclic), abbreviated as Sel ; the method of calculation will
be detailed in Section 2.2. The reaction products were identified by GLC and high-field
NMR spectroscopy, by adding small amounts of unambiguously synthesized compounds
into the analyzed samples. Pertinent NMR data are presented in Table 2 (Section 4.7).
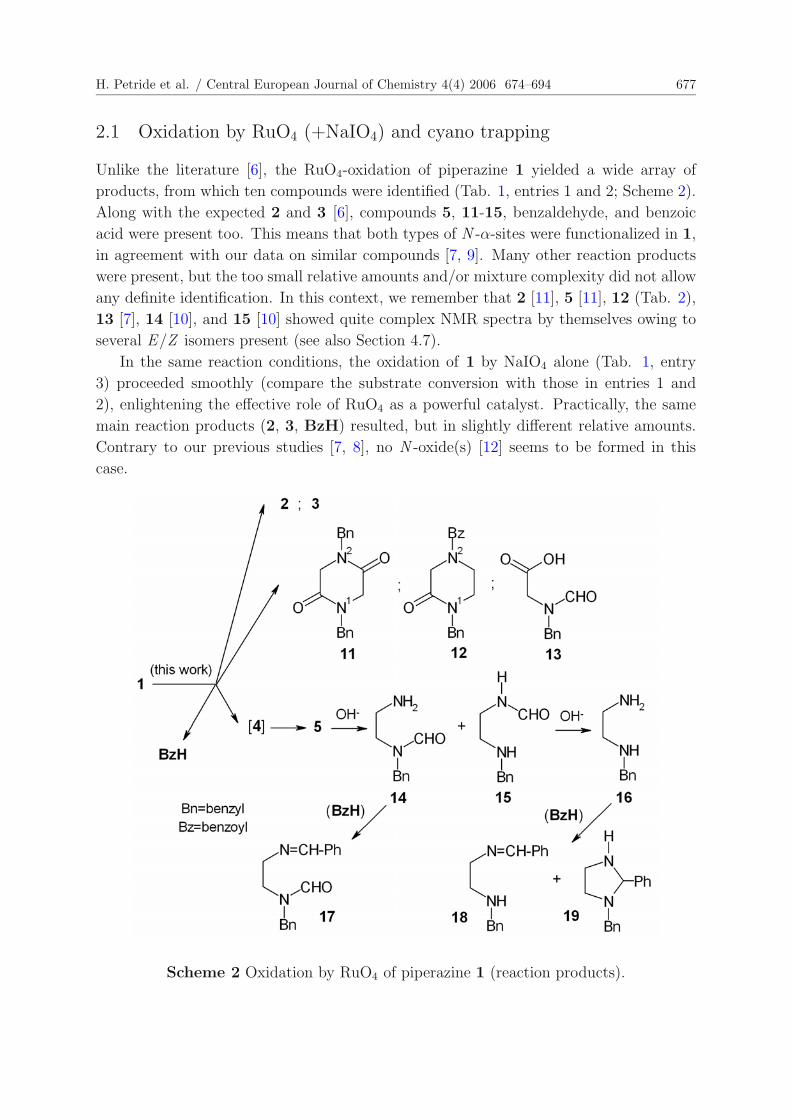
H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694 677
2.1 Oxidation by RuO4 (+NaIO4) and cyano trapping
Unlike the literature [6], the RuO4-oxidation of piperazine 1 yielded a wide array of
products, from which ten compounds were identified (Tab. 1, entries 1 and 2; Scheme 2).
Along with the expected 2 and 3 [6], compounds 5, 11-15, benzaldehyde, and benzoic
acid were present too. This means that both types of N -α-sites were functionalized in 1,
in agreement with our data on similar compounds [7, 9]. Many other reaction products
were present, but the too small relative amounts and/or mixture complexity did not allow
any definite identification. In this context, we remember that 2 [11], 5 [11], 12 (Tab. 2),
13 [7], 14 [10], and 15 [10] showed quite complex NMR spectra by themselves owing to
several E/Z isomers present (see also Section 4.7).
In the same reaction conditions, the oxidation of 1 by NaIO4 alone (Tab. 1, entry
3) proceeded smoothly (compare the substrate conversion with those in entries 1 and
2), enlightening the effective role of RuO4 as a powerful catalyst. Practically, the same
main reaction products (2, 3, BzH) resulted, but in slightly different relative amounts.
Contrary to our previous studies [7, 8], no N -oxide(s) [12] seems to be formed in this
case.
Scheme 2 Oxidation by RuO4 of piperazine 1 (reaction products).

678 H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694
Table 1 Oxidation of piperazine derivatives with the RuO4/NaIO4 system.
Entry Substrate[a] Conds. Identified reaction products[e] (yields, %)[b,f ] Sel(conv.)[b,c] [d] [g]
1. 1 (100) A 2 (43.5), 3 (16), 5 (9.5), 11 (1.5), 12 (2.5), 13 (tr),14 (3.5), 15 (2), BzH (28±2)
2. 1 (100) B 2 (43), 3 (16), 5 (6.5), 11 (1.5), 12 (2.5), 13 (tr),14 (5±1), 15 (3), BzH (28±2)
∼ 1.2
3. 1 (15) C 2 (37), 3 (10), BzH (30±2)
4. 7 (100) A 3 (58), 6 (2.5), 11 (4.5), 12 (4.5), 20[h] (5), 21 (5),23 (1), 24 (2), BzH (23.5±1)
5. 7 (100) B 3 (57), 6 (2.5), 11 (5), 12 (5), 21 (10), 23 (1), 24(2), BzH (23.5±1)
∼ 1.3
6. 9 (65) A 10 (70), BzH (25)[i]
7. 30 (85) A 12 (22.5), 31 (38), 32 (11.5), 33 (8), 34 (5), 35(6), 36 (2), 37 (tr), 39 (5), BzH (22.5)
8. 30 (84) B 12 (22), 31+32 (2+48=50)[j], 33 (8),34+35+36+37 (0+6+6+1=13)[j], 39 (4.5),BzH+37 (22+1=23)[j]
1.3
9. 1 (57) D 2 (3), 4 (7.5), 7 (1.5), 30 (2), 49 (66.5), 50 (19),BzH (8)
1.2
[a] Compounds 2, 3, 5, 6, 10-13, 31, 34, 38, and 39 were all stable against RuO4-oxidation.[b] Mean values (±0.5, unless otherwise noted) of two identical runs. All significant figures were corrected for
the work-up loss.[c] Calculated (%) with respect to the initial substrate amount.[d] Reaction conditions (substrate = 1 mmol): A – RuO2/ NaIO4/ CCl4/ H2O = 10 / 4 / 10 / 10
(mg/mmol/mL/mL), initial neutral work-up; B – as in A, but initial alkaline work-up; C – as in A,but without RuO2; D – as in A, but NaCN (4 mmol) in 10 mL of water was added too.
[e] BzH means benzaldehyde; its yield includes that of benzoic acid.[f ] Calculated with respect to the reacted substrate, by assuming a 1/1 stoichiometry; tr means traces (< 0.2%).[g] For the calculation of regioSelectivity (endocyclic/exocyclic), see text.[h] Tentative assignment.[i] Other reaction products are present too.[j] Variable single values (±2), but constant (±0.5) sum (see text).
As anticipated before, some of the reaction products listed in entries 1 and 2 of
Table 1 might result by subsequent reactions of various intermediates. For instance, as
2 resulted from 1, the presence of 5 might be due to the analogous oxidation of 4 [6].
Our preliminary investigations on the RuO4-oxidation of 4 confirmed the presence of 5
in the respective reaction mixtures, together with 13-19 (formulae in Scheme 2), BzH,
and other compounds [9]. Monoformamides 14 and 15 resulted from 5, by hydrolysis
during the work-up, as our study [10] on the mild hydrolysis of pure 5 [11] indicated.
In this last case, diamine 16 resulted also, by subsequent hydrolysis of both 14 and 15.
Diamine 16 was not identified in the 1-oxidation mixtures, but this does not mean that
it was not formed at all. In fact, 16 might be (partially or totally) hidden as a mixture

H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694 679
18+19 [13], by reaction with BzH. Analogously, some of the amine 14 could afford the
Schiff base 17. We verified the behavior of 14 and 16 towards BzH and really found
17 and 18+19 (18/19=1/2, molar) as reaction products, respectively [10]. At the same
time, we have noticed that pure 2 [11] is remarkably stable in the same mild hydrolysis
conditions [10].
It results that 5, 13-15, and some of BzH derived all from 4, transiently formed
during the oxidation of 1. Therefore, on one hand, 5 is not an initial oxidation product
of 1 and, on the other hand, 13, 14, and 15 are not true oxidation products of 1 at all.
To understand the origin of 3, 4, 11, and 12, additional oxidation experiments were per-
formed starting from possible reaction intermediates. As promising candidates we chose
1,4-dibenzyl-2-piperazinone (7) and 1-benzoyl-4-benzylpiperazine (30); the respective re-
sults are presented below.
Oxidation of piperazinone 7 gave complex reaction mixtures in which 3, 6, 11, 12,
21, 23, 24, benzaldehyde, and benzoic acid were identified (Tab. 1, entries 4 and 5;
Scheme 3). Only 3 has been isolated previously by Tortorella’s group, in a 65% crude
yield [6]. By analogy with the sequence 5→14+15→16 (Scheme 2), monoformamide 21
might be the hydrolysis product of diformamide 20. Unfortunately, attempts to prepare
20 failed, but the transformation 20→21 is supported by our competitive hydrolysis
study performed on similar substrates [10]. In fact, the study suggested that, from the
three types of amidic N-CO bonds existing in 20, the most reactive might be N1-CHO.
At the same time, both N1-CO and N2-CHO amide bonds in 21 could be resistant to
mild hydrolysis. Indeed, the hydrolysis of 21 to yield 22 did not occur in the conditions
employed (see Section 4.6). Following the work-up A, the NMR spectra of crude reaction
mixture contained signals that could be tentatively ascribed to the diformamide 20 [14].
They practically disappeared when the work-up B was adopted. To the extent of which
21 is the sole hydrolysis product of 20, the sum {20}exp+{21}exp should remain constant
in entries 4 and 5, which actually has been observed.
By analogy with our previous papers [7, 8], we imagined the oxidation course of 7
as depicted in Scheme 3. Piperazinone 7 contains two kinds of nitrogen atoms, namely
of amide (N1) and amine type (N2). To each of them correspond two kinds of N -α-C-H
bonds, that is of endocyclic and exocyclic (i.e., benzylic) type. Actually, as two different
endocyclic N2-CH2 methylene groups are present, there would be five types of N -α-C-H
bonds prone to be oxidized, at least in principle. From these, only the three types of
N2-C-H bonds would be much more susceptible to undergo oxidation. This derives from
our earlier observations substantiating the higher reactivity of amines vs. that of amides
during their RuO4-oxidation [7, 8]; we will return to this argument.
Accordingly, the first step of the oxidative attack on 7 yields three iminium cations:
two endocyclic (25, 26) and one exocyclic (27). Nucleophilic water molecule can trap
these cations to give the corresponding aminoalcohols, as indicated for 27→28. Their
subsequent oxidation at the OH level would yield the piperazinediones 3 and 11, as
well as the benzoyl derivative 12, respectively. It is well-known that alcohols are easily
converted by RuO4 into the corresponding carbonyl derivatives [15–19]. At the same

680 H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694
Scheme 3 Oxidation by RuO4 of piperazinone 7 ([ox] means oxidation).
time, alternative pathways are possible for 26 and 28. Thus, 26 might be N + − β-
deprotonated by a base and the resulted cyclic enamine 29 could be oxidized further in
order to form 20. This last step has many precedents in the literature [7, 8, 20]. Besides
oxidation to 12, the hemiaminal 28 could also give an equimolecular mixture of BzH
and 8. Similar steps have been invoked several times in the past [7, 8, 21]. Benzoic acid
originated from benzaldehyde only. In fact, 12 was inert during mild hydrolysis attempts
(see Section 4.6).
Besides the trivial oxidation of benzaldehyde to benzoic acid, control experiments
showed that, unlike 3, 11, and 12 (and probably also 20 [22]), 1-benzyl-2-piperazinone
(8) is the unique reaction product susceptible to undergo further oxidation. Indeed,
starting from pure 8, mixtures of piperazinedione 6, two pyrazinones (23 and 24), and

H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694 681
some BzH were obtained [9]. In this way the formation of all identified reaction products
listed in entries 4 and 5 of Table 1 finds a reasonable explanation. Obviously, {BzH}exp
includes the small contribution of the 8-oxidation.
If the two types of N1-C-H bonds of 7 would be attacked, some BzH and the 2,6-
piperazinedione 9 should be formed [2, 7]. Submission of 9 to the RuO4-oxidation gave
BzH and piperazinetrione 10 as main reaction products (Tab. 1, entry 6; Scheme 3).
We remember that only 10 has been isolated by the Italian team, in “good yield” [6].
Like 2, 3, 5, or 11, piperazinetrione 10 was inert towards further oxidation, this time in
accord with the literature [6]. Neither 9 nor 10 were identified in the oxidation mixture
derived from 7. By way of consequence, we did not study more in detail the oxidation of
9. These facts suggest that the reactions 7→9, 3→10, and 11→10 did not occur in the
conditions employed, enforcing the aforementioned “amide � amine” reactivity order.
According to Scheme 3, the derivatives 3 and 11+20+21 result from the cations
25 and 26, respectively. Taking into account the respective yields (Tab. 1, entry 4
or 5), it emerges that the initial {25}i/{26}i ratio could be about 4/1. Since 25 is
presumably less stable than 26 [23], this value suggests that their formation might be
kinetically controlled. In other words, the rate-determining step seems not to be the
iminium cations’ generation.
Eleven reaction products were identified in the oxidation mixtures of 1-benzoyl-4-
benzylpiperazine (30) (Tab. 1, entries 7 and 8; Scheme 4). Excepting benzaldehyde and
benzoic acid, all compounds contain two nitrogen atoms in their molecules. Like in 30, no
hydrogen atoms are attached to N1 or N2 atoms in 12, 31, and 39; they clearly derived
from 30. On the contrary, the remaining nitrogen-containing reaction products have
N2H (32), N1H (33-36), or N=C functions (37) in their structures, indicating complex
reactions, presumed to be those depicted in Scheme 4. Analogously to the previously
discussed case of 7, we considered that the aminic N1-CH2 sites of 30 are active only.
Their oxidation yields the corresponding endocyclic (40) and exocyclic (41) iminium
cations. Water trapping (like 41→42) and subsequent OH oxidation give 12 and 39,
respectively. N + − β-Deprotonation of 40 yields the enamine 43, oxidation of which
is the source of the diformamide 31. Analogously to 28, hemiaminal 42 splits into an
equimolecular mixture of BzH and 33.
Control experiments showed that, unlikely to 12, 31, or 39, monobenzoylpiperazine
(33) reacts further with RuO4 [9] to give initially 34 and also 38. Mild hydrolysis of 31
and 34, occurring during the work-up, was the source of 32 and of 35+36, respectively,
as indicated by our hydrolysis study [10] performed on unambiguously synthesized 31 and
34 [11]. The same study [10] put in evidence the inertness of 32 and the easy 35→36
hydrolysis, as well as the lacking of benzoic acid in all these hydrolysis reactions. The
experimentally found higher reactivity of 34 vs. that of 31 [10] explains their relative
yields in entries 7 and 8. Finally, the primary amine 36, generated from 30 via 33, with
benzaldehyde really gives the Schiff base 37 [10]. All benzoylated derivatives 12 and
31-39 did not give any benzoic acid on mild hydrolysis (see Section 4.6).
Some of the yields appear in entry 8 as sums. The single values varied in an un-
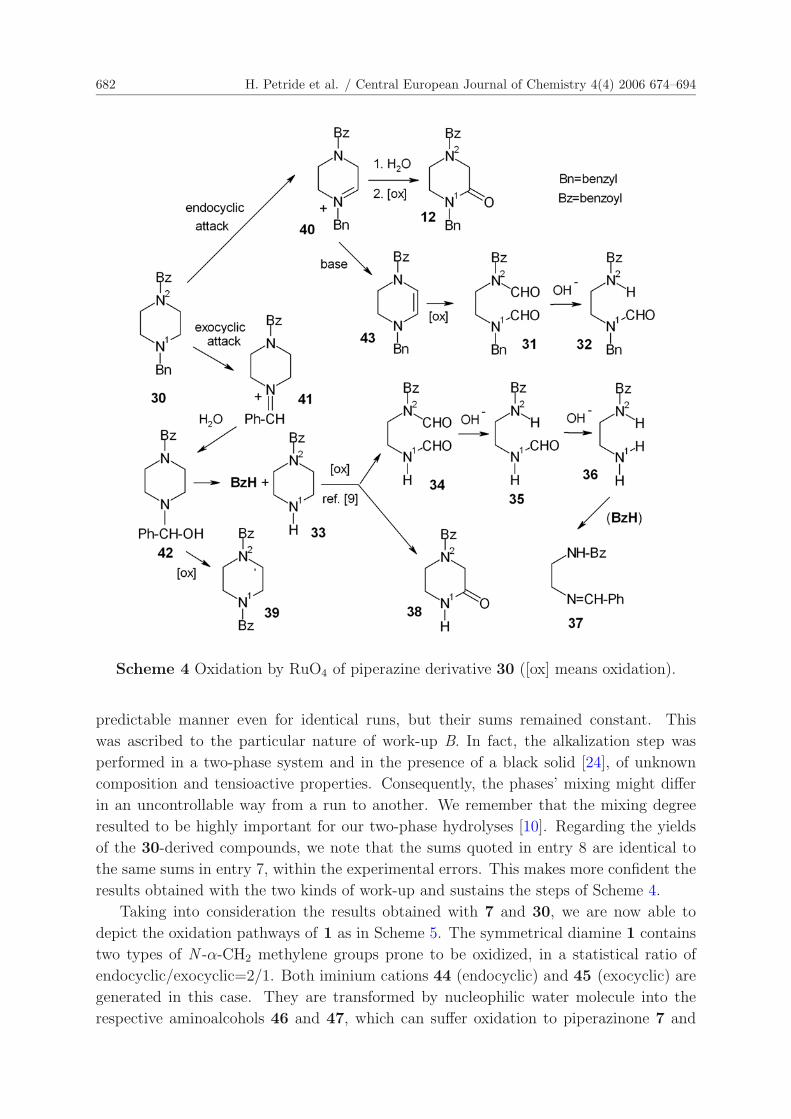
682 H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694
Scheme 4 Oxidation by RuO4 of piperazine derivative 30 ([ox] means oxidation).
predictable manner even for identical runs, but their sums remained constant. This
was ascribed to the particular nature of work-up B. In fact, the alkalization step was
performed in a two-phase system and in the presence of a black solid [24], of unknown
composition and tensioactive properties. Consequently, the phases’ mixing might differ
in an uncontrollable way from a run to another. We remember that the mixing degree
resulted to be highly important for our two-phase hydrolyses [10]. Regarding the yields
of the 30-derived compounds, we note that the sums quoted in entry 8 are identical to
the same sums in entry 7, within the experimental errors. This makes more confident the
results obtained with the two kinds of work-up and sustains the steps of Scheme 4.
Taking into consideration the results obtained with 7 and 30, we are now able to
depict the oxidation pathways of 1 as in Scheme 5. The symmetrical diamine 1 contains
two types of N -α-CH2 methylene groups prone to be oxidized, in a statistical ratio of
endocyclic/exocyclic=2/1. Both iminium cations 44 (endocyclic) and 45 (exocyclic) are
generated in this case. They are transformed by nucleophilic water molecule into the
respective aminoalcohols 46 and 47, which can suffer oxidation to piperazinone 7 and

H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694 683
Scheme 5 Reaction pathways followed during the RuO4-oxidation of 1 ([ox] means oxi-
dation).
benzoyl derivative 30, respectively. Alternatively, cation 44 may be deprotonated to the
enamine 48; oxidation of the C=C double bond in 48 gives diformamide 2 [15, 25, 26].
Like 28 or 42, hemiaminal 47 may be also split into an equimolecular mixture of BzH
and 1-benzylpiperazine (4). Subsequent oxidation of 4 [9], 7 (Scheme 3), and 30 (Scheme
4) yields the respective compounds listed in Scheme 5. As pointed out before, the unique
source of benzoic acid was the oxidation of benzaldehyde.
According to Scheme 5, the dioxygenated compound 3 was formed by sequential
monooxidation of the appropriate substrates (i.e., 1→7→3) and not by a simultaneous
introduction of two oxygen atoms in the molecule of 1. The latter alternative seems to
have been followed to generate 2 (via 48). However, working with N -benzylpiperidine
or -morpholine [7], we have found in the past that an enamine with a 48-like structure
yields both 2- and 3-like derivatives, in a 42/1-45/1 molar ratio. If this is true also for
the 1-oxidation case, {3}exp=16% of entry 2 would be the sum of yields due to both 7→3
and 48→3 reactions. Quantitatively, the contribution of the 48→3 step could represent
about 43/(42÷45)∼1% of the reacted 1. Accordingly, the remaining 15% of {3} would
be due to the monooxidation sequence. This implies that 3 and 11 should result from
transient 7 in a 15/1.5=10/1 ratio. This value is slightly smaller than that obtained in
entry 5 (i.e., 57/5∼11), suggesting that all 3 in entry 2 would be formed from 7 only,

684 H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694
within the experimental errors. This clarifies the corresponding question presented in the
Introduction.
A decisive proof favoring the proposed mechanism came from the 1-oxidation exper-
iment performed in the presence of NaCN (Tab. 1, entry 9). In this particular case,
the main reaction products were two α-aminonitriles (49 and 50 in Scheme 5), counting
for about 86% of the reacted 1. They clearly resulted from the iminium cations 44 and
45, respectively, by cyano trapping [7, 8, 27–29]. Excepting 4, the other reaction prod-
ucts (i.e., 2+7 and 30+BzH) were oxygen-containing compounds, clearly originating
from the same iminium cations, escaped from the cyanide anion trapping (i.e., captured
by water). Interestingly, benzoic acid was found only in trace amounts. Moreover, no
products of further oxidation of 4, 7, and 30 were present. These facts, correlated also
with the lower substrate conversion (compare entries 1 or 2 with 9), suggest that RuO4
and/or NaIO4 were rapidly consumed in other (unknown) reactions. Similar results were
obtained previously [7, 8]. It is worth mentioning that the amounts of 4 and BzH are
equal in entry 9, within the experimental errors, supporting the intermediacy of hemi-
aminal 47. At the same time, the transient existence of 4, 7, and 30, only presumed in
the absence of NaCN, finds now full confirmation.
2.2 Regioselectivity
Regioselectivity of the RuO4-oxidation reactions could be apprised if the amounts of
the endocyclic and exocyclic iminium cations would be separately known. As presented
below, the required amounts can be really calculated from those of the corresponding,
directly formed reaction products. The calculated values of regioselectivity, abbreviated
as Sel, are written in the last column of Table 1. In the following considerations, the
more reliable results of work-up B were manipulated only, excepting the cyano trapping
experiment.
Let us begin with the oxidation of 7. According to Scheme 3, the iminium cations 25,
26, and 27 give initially 3, 11+20, and 8+12, respectively, where {BzH}i={8}i. Taking
into accounts their previously discussed behavior towards RuO4 or during the work-up,
we can write {3}i={3}exp, {11}i={11}exp, {20}i={20}exp+{21}exp, and {12}i={12}exp,
where the experimental amounts are those listed in Table 1 (entry 5). Therefore, in order
to calculate Sel for the 7-oxidation we need to know {8}i or {BzH}i only. Calculation
of {8}i from {6}exp, {23}exp, and {24}exp is simpler. Thus, starting from 7 or pure 8,
compounds 6+23+24 resulted in 5.5% (entry 5) or 24.5% [9] cumulated yields, respec-
tively. Accordingly, a value of 5.5/0.245∼22% can be advanced for {8}i in the conditions
of entry 5. It emerges that the endocyclic and exocyclic routes in Scheme 3 correspond to
{3}exp+{11}exp+{20}exp+{21}exp=57+5+0+10=72% and {8}exp+{12}i ∼22+5=27%
of the reacted 7, respectively. Their sum matches well the theoretical value of 100%.
Consequently, the statistically corrected regioselectivity (endocyclic/exocyclic) results to
be around 72/(2∗27)=1.3.
More accurate Sel calculation can be made for the 30-oxidation, because all needed

H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694 685
yields are known. In fact, looking at the data of Table 1 (entry 8) and at Scheme 4, we
note that the endocyclic oxidative attack interested {12}exp+{31}exp+{32}exp=72% of
30. Analogously, the products of exocyclic attack (i.e., {39}exp and {BzH}i={BzH}exp
+{37}exp) counted for a 27.5%-consumption of 30. Consequently, we can calculate the
regioselectivity of the oxidation of 30 as being 72/(2∗27.5)=1.3.
The Sel calculation for the 1-oxidation in the absence of NaCN resulted to be more
complicated. According to Scheme 5, we need to know, on one hand, {2}i and {7}i, and,
on the other hand, {30}i and {BzH}i={4}i. At this moment, only {2}i={2}exp=43% is
known. From the identified reaction products (Tab. 1, entry 2), 2+3+11 and 5+13+14+
15 resulted from one route only, namely endocyclic and exocyclic, respectively, but 12
and BzH came from both pathways. As shown in the following, {7}i and {30}i can
be really calculated. In fact, starting from 7 or 1, the cumulated yield of 3+11 was
62% (entry 5) or 17.5% (entry 2), respectively. Accordingly, the yield of 7, initially
formed from 1, could be {7}i=17.5/0.62∼28%. Compounds 2 and 7 are the sole initial
1-oxidation products originating from the endocyclic route. Consequently, this pathway
interested about 43+28=71% of 1. Obviously, the remaining 29% should correspond to
the exocyclic route, viz. to the sum {30}i+{BzH}i. Although judging on small values
could be misleading, {30}i may be tentatively calculated from {12}exp=2.5% in entry 2.
In fact, 12 resulted from 1 via 7 and 30. Knowing that 5% of 7 is transformed into 12
(entry 5), the amount of 12 formed from {7}i in entry 2 could be about 28∗0.05=1.4%.
Therefore, the contribution of the 30→12 reaction in entry 2 would be 2.5-1.4∼1%.
Since this last value represents 22% of the starting {30}i (entry 8), it emerges that
{30}i=1/0.22=4.5% in the conditions of entry 2. Consequently, {BzH}i should be 29-
4.5=24.5% with respect to the reacted 1. Unfortunately, we could not verify this last
value. In fact, benzaldehyde resulted from 1 by both endocyclic (from 7) and exocyclic
routes (from 4, 30, and 47). Although the contributions in {BzH}exp due to 4-, 7-, and
30-oxidations might be calculated using the same method as before, the total amount of
benzaldehyde formed from 1 remains still unknown. We remember that some of BzH
could be used to form various Schiff bases and imidazolidine 19 [9, 10], but we do not
know their yields. This explains why {BzH}exp varied so much (28±2%), but, at the same
time, it makes impossible the calculation of {BzH}i. Anyway, as anticipated before, it
seems that the regioselectivity of the 1-oxidation in the absence of NaCN might have a
value of about 71/(2∗29)=1.2.
This value is confirmed when the data of 1-oxidation in the presence of NaCN (Tab. 1,
entry 9; Scheme 5) are taken into account. In this case, 2, 7, and 49 derived all from the
endocyclic cation 44. Analogously, 30, 50, and BzH (and 4) were formed from the exo-
cyclic cation 45. By way of consequence, Sel would be (3+1.5+66.5)/[2∗(2+19+8)]=1.2.
Summing-up, it results that 1, 7, and 30 underwent RuO4-oxidation with the same
poor endocyclic/exocyclic regioselectivity of 1.2-1.3. This value is intermediate between
those experienced [7] by N -benzylpiperidine and -morpholine (i.e., 2.1 and 0.8, respec-
tively), but more close to the latter. Compounds 1, 7, and 30, could be viewed as N -
benzylpiperidine derivatives bearing N -γ-electron withdrawing groups in their aliphatic

686 H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694
cycles. Consequently, 1, 7, and 30 are rather similar to N -benzylmorpholine than to N -
benzylpiperidine and their Sel ’s seem to feel this structural resemblance accordingly. This
suggests that, among other factors, the RuO4-catalyzed oxidation could be susceptible to
electronic effects exerted by remote N -substituents in the starting tertiary amines.
3 Conclusions
RuO4-Catalyzed oxidation of 1,4-dibenzylpiperazine, -2-piperazinone, and 1-benzoyl-4-
benzylpiperazine (1, 7, and 30, respectively) gave wide arrays of reaction products, in-
cluding acyclic diformamides and benzaldehyde. 1,4-Dibenzyl-2,6-piperazinedione (9)
was not involved in the 7- or 1-oxidation reactions. Control experiments showed that 1-
benzylpiperazine (4), 7, and 30 are all transiently formed during the oxidation of 1. Their
subsequent oxidation complicated the initial outcome of 1. Moreover, several acyclic
monoformamides and diamines with an ethylenediamine skeleton were also found in the
respective reaction mixtures. These false oxidation products derived from the acyclic
diformamides, by hydrolysis during the work-up. The RuO4-mediated oxidation was in-
terpreted as involving iminium cations and cyclic enamines as reactive intermediates.
Two kinds of cations resulted, depending on the attacked N -α-CH2 methylene group:
endocyclic or exocyclic (i.e., benzylic). Only the aminic N -α-sites were functionalized in
7 and 30. When structurally possible, N + − β-deprotonation of the endocyclic cations
gave the corresponding cyclic enamines, which were the sources of acyclic diformamides.
In the case of 1, the iminium cations were captured with NaCN as the corresponding
α-aminonitriles. The oxidation regioselectivity (endocyclic/exocyclic) had a statistically
corrected value of about 1.2-1.3. Comparison with the values previously found for N -
benzylpiperidine and -morpholine suggested that the RuO4-catalyzed oxidation could be
susceptible to electronic effects in the starting tertiary amines.
4 Experimental section
4.1 General remarks
Melting points were taken with a Boetius hot plate and are uncorrected. The GLC and
NMR apparatuses and procedures were already described [7]. IR spectra were recorded
on an UR-20 Carl Zeiss Jena spectrophotometer.
4.2 Materials
Hydrated ruthenium dioxide (Aldrich), sodium periodate (Merck), and N -benzylethy-
lenediamine (16; Aldrich) were used as purchased. Carbon tetrachloride (Chimopar,
Romania) was stored over anhydrous Na2CO3 and filtered prior to use. Compounds 1
[30], 2 [11], 4 [30], 5 [11], 7 [30], 11 [30], 13 [7], 14 [10], 15 [10], 17-19 [10], 31 [11], 32
[10], 34 [11], 35 [10], 36 [10], 37 [10], 49 [31], and 50 [31] were from our previous works.

H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694 687
Synthesis of 8, 23, 24, and 38 will be detailed in a future paper [9]. Derivatives 3 [6],
9 [6, 32], 10 [6], 12 [33], 30 [34], 33 [35], and 39 [36] are all known from the literature
and were synthesized according to the indicated procedures. All compounds presented
mp’s (or bp’s) in accord with the literature values, except 3 which melted at 201-202◦Cafter recrystallization from acetone (lit. [6] mp 188-190◦C). Elemental analyses were in
agreement with their molecular formulae. All compounds were characterized by IR and
NMR spectroscopy (see below).
4.3 1-Benzyl-2,3-piperazinedione (6)
A stirred mixture of 16 (3.75 g; 25 mmol) and diethyl oxalate (3.4 mL; 25 mmol) was
gradually heated on an oil bath. After all formed ethanol distilled off, the mixture was
heated at 180◦C for 30 minutes and allowed to cool at room temperature. The resulted
solid was triturated with acetone and then recrystallized from ethanol to leave colorless
crystals of 6 (3.45 g; yield 69%), melting at 229-231◦C. Elemental analysis gave (%): C
(64.75), H (5.82), N (13.45). Theoretical for C11H12N2O2 (%): C (64.69), H (5.92), N
(13.72). Its NMR features are given in Table 2.
IR spectrum (CH2Cl2, cm−1): 1680+1700 (large and strong, νCO), 3400 (medium,
νNH).
4.4 N -Benzyl-2-(benzylamino)acetamide (22)
A magnetically stirred solution of N -benzyl-2-(benzylideneamino)acetamide (1.36 g; 5.4
mmol) [9] in methanol (30 mL) was treated portionwise with a solution of NaBH4 (0.6 g;
15.4 mmol) in methanol (20 mL) and the stirring was continued overnight. The solution
was evaporated in vacuo, the residue taken in chloroform and washed with alkaline water.
The organic layer was anhydrized (Na2SO4) and evaporated to dryness to leave 1.3 g (yield
95%) of solid 22. It was purified through its hydrochloride, mp 243-244◦C (lit. mp 244-
245◦C [37]; 203◦C [38]). Elemental analysis gave (%): C (65.81), H (6.71), N (9.45), Cl
(12.36). Theoretical for C16H18N2O.HCl (%): C (66.09), H (6.59), N (9.63) Cl (12.19).
The NMR characteristics of free base 22 are presented in Table 2.
4.5 N -Benzyl-2-(N -benzyl-N -formylamino)acetamide (21)
Amine 22 (1.25 g; 4.9 mmol) was formylated with excess ethyl formate (4 mL; 0.05 mol)
by heating their mixture at reflux for 10 hours. The mixture was evaporated to dryness,
the residue triturated with ether and recrystallized from benzene/ether to leave colorless
crystals of 21 (1.05 g; yield 76%), melting at 95-97◦C. Elemental analysis gave (%): C
(72.02), H (6.22), N (10.18). Theoretical for C17H18N2O2 (%): C (72.32), H (6.43), N
(9.92). Its NMR characteristics are given in Table 2.
IR spectrum (CH2Cl2, cm−1): 1670+1690 (large and strong, νCO), 3430 (medium,
νNH).

688 H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694
Table 2 1H- and 13C-NMR data of some compounds of interest.
Compd. Chemical shifts[a] (δ, ppm, J in Hz, CDCl3) and assignments[b]
1 δH : 2.48 (s, 8H, 4∗CH2), 3.51 (s, 4H, 2∗Bn);δC : 53.2 (CH2), 63.2 (Bn), 127.1, 128.3, 129.3, 138.3.
3 δH : 3.32 (s, 4H, N1-CH2-CH2-N2), 4.62 (s, 4H, 2∗Bn), 7.20-7.33 (m, 10H, 2∗Ph);δC : 43.4 (N1-CH2-CH2-N2), 50.4 (Bn), 127.9 (Cpara), 128.2, 128.7, 135.4, 157.4(CO).
6 δH : 3.41-3.53 (m, 4H, N1-CH2-CH2-N2), 4.70 (s, 2H, Bn), 7.26-7.40 (m, 5H, Ph),7.80[c] (br, 1H, N2H);δC : 39.1 (N2-CH2), 44.6 (N1-CH2), 50.7 (Bn), 128.2, 128.4, 129.0, 135.6, 157.8(N2-CO), 159.1 (N1-CO).
7 δH : 2.58 (t, 3J=5.5, 2H, CH2-CH 2-N2), 3.14 (t, 3J=5.5, 2H, N1-CH2), 3.18 (s,2H, N2-CH2-CO), 3.48 (s, 2H, N2-Bn), 4.52 (s, 2H, N1-Bn), 7.23-7.37 (m, 10H,2∗Ph);δC : 46.1 (N1-CH2), 49.3 (CH2-CH2-N2), 49.5 (N1-Bn), 57.9 (N2-CH2-CO), 62.9(N2-Bn), 127.6, 128.2, 128.5, 128.7, 129.1, 136.5 (of Ph-CH2-N1), 137.5 (of Ph-CH2-N2), 167.2 (CO).
9 δH : 3.35 (s, 4H, CH2-N2-CH2), 3.53 (s, 2H, N2-Bn), 4.86 (s, 2H, N1-Bn), 7.1-7.4(m, 10H, 2∗Ph);δC : 42.3 (N1-Bn), 56.3 (CH2-N2-CH2), 60.7 (N2-Bn), 127.6+128.1 (Cpara),128.5+128.7+128.9+129.1 (Cmeta+Cortho), 135.2 (of Ph-CH2-N2), 136.6 (of Ph-CH2-N1), 169.8 (CO).
10 δH : 4.10 (s, 2H, N2-CH2), 4.56 (s, 2H, N2-Bn), 4.91 (s, 2H, N1-Bn);δC : 44.2 (N1-Bn), 49.7 (N2-CH2), 50.4 (N2-Bn), 133.6 (of N2-CH2-Ph), 135.0 (ofN1-CH2-Ph), 152.3 (N2-CO), 156.5 (N1-CO-CO), 164.8 (N1-CO-CH2).
11 δH : 3.93 (s, 4H, 2∗CH2), 4.58 (s, 4H, 2∗Bn), 7.24-7.30+7.30-7.39 (m+m, 4H+6H,2∗Ph);δC : 49.3 (CH2), 49.4 (Bn), 128.1, 128.6, 129.0, 134.3, 163.3 (CO).
12[d] δH : 3.32 (br, 2H, CH2-N1), 3.50-4.05 (br, 2H, CH2-CH 2-N2), 4.15-4.54 (br, 2H,N2-CH2-CO), 4.62 (s, 2H, Bn), 7.25-7.37 (m, 5H, Ph-CH2), 7.42 (s, 5H, Ph-CO);δC : 39.8 (br, CH2-CH2-N2), 45.0 (CH2-N1), 50.0 (Bn), 51.6 (br, N2-CH2-CO), 127.3(br)+128.7+130.5(br)+134.3 (Ph-CO), 127.9+128.3+128.8+135.9(Ph-CH2), 164.8 (br, CO-N1), 170.1 (CO-N2).δ[e,f ]H : 3.39+3.51 (br+br, 1.08+0.92H, CH2-N1), 3.73+4.05 (br+br, 1.08+0.92H,
CH2-CH 2-N2), 4.43+4.66 (br+br, 0.92+1.08H, N2-CH2-CO), 4.71 (br, 2H, Bn);δ[e,f ]C : 40.1+44.5 (CH2-CH2-N2), 45.1+45.9 (CH2-N1), 46.4+50.8 (N2-CH2-CO),
51.2 (Bn), 166.8 (N2-CO), 167.4 (N1-CO).21[e] δH : 3.79+3.88 (s+s, 0.5+1.5H, N2-CH2), 4.36+4.37 (d[g]+d[g], 3J=6, 2H, N1-Bn),
4.52+4.55 (s+s, 1.5+0.5H, N2-Bn), 6.16[c]+6.41[c] (t+t, 3J=6, 0.25+0.75H, N1H),7.10-7.30 (m, 10H, 2∗Ph), 8.16+8.32 (s+s, 0.25+0.75H, CHO);δC : 43.6+43.8 (N1-Bn), 46.4+50.2 (N2-CH2), 47.1+52.5 (N2-Bn), 127.6, 127.7,127.8, 127.96, 128.00, 128.16, 128.4, 128.5, 128.6, 128.8, 128.9, 129.8, 135.0+135.6(of Ph-CH2-N2), 137.8+137.9 (of Ph-CH2-N1), 163.4+163.6 (CHO), 167.6+167.7(CO).

H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694 689
Table 2 (continued) 1H- and 13C-NMR data of some compounds of interest.
Compd. Chemical shifts[a] (δ, ppm, J in Hz, CDCl3) and assignments[b]
22 δH : 2.53[c] (br, 1H, N2H), 3.29 (s, 2H, N2-CH2), 3.71 (s, 2H, N2-Bn), 4.42 (d[g],3J=6.0, 2H, N1-Bn), 7.18-7.40 (m, 10H, 2∗Ph), 7.60[c] (t, 3J=6.0, 1H, N1H);δC : 42.9 (N1-Bn), 51.7 (N2-CH2), 53.8 (N2-Bn), 127.2, 127.61, 127.71, 128.1, 128.5,129.1, 139.2 (of Ph-CH2-N2), 138.3 (of Ph-CH2-N1), 171.7 (CO).
23 δH : 3.29 (t, 3J=6.4, 2H, N1-CH2), 3.75 (td, 3J=6.4, 4J=2.4, 2H, N2-CH2), 4.60(s, 2H, Bn), 7.2-7.35 (m, 5H, Ph), 7.78 (t, 4J=2.4, 1H, N2=CH);δC : 42.5 (N1-CH2), 47.9 (N2-CH2), 49.5 (Bn), 127.7 (Cpara), 128.1 (Cortho), 128.7(Cmeta), 135.7, 155.9 (CO), 156.9 (N2=CH).
24 δH : 5.07 (s, 2H, Bn), 7.07 (d, 3J=4.4, 1H, CH-N1), 7.2-7.35 [m, 6H, Ph+(CH=CH -N2)[ni]], 8.17 (s, 1H, CO-CH);δC : 51.5 (Bn), 123.8 (CH=CH-N2), 127.9 (Cpara), 128.4+128.6 [Cortho+(CH-N1)],129.0 (Cmeta), 134.6, 149.6 (CO-CH), 156.0 (CO).
30[d] δH : 2.39 (br, 2H, N1-CH2 syn to CO), 2.55 (br, 2H, N1-CH2 anti to CO), 3.44(br, 2H, N2-CH2 anti to CO), 3.55 (s, 2H, Bn), 3.81 (br, 2H, N2-CH2 syn to CO);δC : 42.1 (N2-CH2 syn to CO), 47.7 (N2-CH2 anti to CO), 52.8 (N1-CH2 anti toCO), 53.3 (N1-CH2 syn to CO), 62.9 (Bn), 127.0, 127.3, 128.3, 128.4, 129.1, 129.6,135.9 (of Ph-CO), 137.6 (of Ph-CH2), 170.2 (CO).
33[d] δH : 1.73[c] (s, 1H, N1H), 2.82 (br, 2H, N1-CH2 anti to CO), 2.92 (br, 2H, N1-CH2
syn to CO), 3.40 (br, 2H, N2-CH2 anti to CO), 3.75 (br, 2H, N2-CH2 syn to CO),7.40 (s, 5H, Ph);δC : 43.3 (br, N2-CH2 syn to CO), 49.1 (br, N2-CH2 anti to CO), 46.1 (br, N1-CH2 syn to CO), 46.6 (br, N1-CH2 anti to CO), 127.1, 128.5, 129.6, 136.0, 170.5(CO).
39[d] δH : 3.2-3.9 (br, 8H, 4∗CH2), 7.40 (s, 10H, 2∗Ph);δC : 42.4+47.6 (br+br, CH2-CH2), 127.0, 128.6, 130.1, 135.1, 170.6 (CO);δ[e,f ]H : 3.52+3.68 (s+s, 1.6+2.4H, 2∗CH2 anti to CO), 3.86+4.01 (s+s, 2.4+1.6H,
2∗CH2 syn to CO), 7.35-7.60 (m, 10H, 2∗Ph);δ[e,f ]C : 42.6+43.3 (CH2 syn to CO), 47.6+48.2 (CH2 anti to CO), 127.1+129.2
(Cortho+Cmeta), 131.6sh+131.7 (Cpara), 132.1+132.2 sh, 173.4 (CO).[a] Data useful in product identificati on are listed only. Formulae as in Schemes 1-6.[b] Bn stands for benzylic protons or carbons. The value of aromatic Cipso is given in italics.[c] Vanishes upon addition of D2O.[d] Broadening and lack of multiplicity are due to slow rotation(s) about the N-COPh bond(s). For the meaning
of syn and anti see text and Scheme 6.[e] Two unequally populated E/Z isomers are present; the values belonging to the major one are underlined.[f ] In a CDCl3/trifluoroacetic acid mixture.[g] Collapses to a singlet upon addition of D2O.[i] According to 1H-1H and long-range 1H-13C correlation, the CH=CH -N2 proton could lie at about 7.28 ppm.
4.6 Mild Hydrolysis of 12, 21, 30, 33, 37, 38, and 39
Hydrolysis was checked in the same mild conditions as for 2, 5, 14, 15, 31, 32, or 34-36
[10], in CH2Cl2/aqueous NaOH mixtures and in the presence or not of benzyltriethy-
lammonium bromide (Aldrich) as a surfactant (substrate/NaOH/surfactant=1/10/0.1,

690 H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694
molar). No reaction occurred within six hours at room temperature, except for 37 which
partially yielded a 36+BzH mixture.
4.7 NMR Spectra
The 1H- and 13C-NMR characteristics of all compounds of interest, useful in product
identification, are collected in Table 2, unless those already reported belonging to 2 [11],
5 [11], 13 [7], 14-19 [10], 31 [11], 32 [10], 34 [11], 35-37 [10], 49 [31], and 50 [31]. Equally
missing are the NMR data of 4, 8, and 38; they will be found in a coming paper [9]. The
NMR characteristics of benzaldehyde [39]a, and benzoic acid [39]b are lacking too in Table
2, as being easily available. Even reported previously by us [30], the 1H-NMR features
of 1, 7, and 11 were instead maintained in Table 2, in order to have on hand a complete
view about their NMR spectral properties. We remember that the spectra of 2, 5, 13-15,
17, 31, 32, 34, 35, and 38 are complicated by the presence of several stereoisomers, due
to hindered rotation(s) about the N-CO bond(s). For instance, all theoretically possible
E/Z isomers of 2 and 5 were seen at room temperature, in 48/45/7 and 5.5/58.5/3/33%
ratios, respectively [11]. Only one species was instead observed for 18, 36, and 37.
The complex aliphatic part of the 1H-NMR spectrum of 49 was rationalized with full
assignment; the CN group resulted to be mainly axially located [31].
Scheme 6 Isomerism of benzoylated derivatives 12 and 39.
The 1H- and 13C-NMR chemical shifts in Table 2 are quoted with respect to internal
Si(CH 3)4 (0 ppm) and CDCl3 (77.16 ppm [40]), respectively. The assignments were made
by two-dimensional NMR experiments, including the long-range 1H-13C heteronuclear
correlation. Our 1H-NMR features of 3 and 11 matched well those found in the literature
[6]. On the contrary, partial (for 7 and 10) or total disagreement (for 9) was found
between our 1H-NMR data (and/or assignments) and Tortorella’s [6]. Two unequally
populated stereoisomers were observed for 12, 39, or 21, due to the slow rotation(s)
about the N-COPh or N-CHO bond(s), respectively. Structural assignment of 12- and
39-isomers (Scheme 6) was made according to the well-known effect of carbonyl bond
anisotropy: the protons in a syn relationship with the carbonyl oxygen are deshielded

H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694 691
with respect to those in an anti position. The same type of reasoning was followed in the
case of N2-CH2 of 30 and 33.
4.8 Oxidation by RuO4/NaIO4
Aqueous NaIO4solution (0.4M; 10 mL) was added to the substrate (1 mmol) dissolved in
CCl4 (10 mL). Solid RuO2 (10 mg) was added and the whole mixture was magnetically
stirred at room temperature for 4-7 hours. The final mixture contained a black solid.
The heterogeneous reaction mixture was worked-up in two different ways (A or B). In
the first case (A), the black amorphous solid was separated by filtration and the two-phase
filtrate retained. The solid was well triturated with fresh CCl4, the filtration repeated,
and the combined filtrates were separated into organic (Ia) and aqueous solutions (IIa).
The solid was triturated with CH2Cl2 and water, and the same operations as before gave
new organic (Ib) and aqueous (IIb) layers. The combined aqueous solutions (IIa+IIb)
were stirred for one hour with a NaOH solution [200 mg (5 mmol) in 2 mL of water]
to give the alkaline solution IIc. Alternatively (B), the alkalization step was made on
the genuine reaction mixture, all other operations being identical. In both cases, the
alkaline aqueous solution IIc was continuously extracted with CH2Cl2 and the organic
layer (Ic) separated. The remaining aqueous layer was rendered acidic with HCl, the
continuous CH2Cl2-extraction repeated, and the organic layer (Id) separated. The four
organic extracts [one in CCl4 (Ia), three in CH2Cl2 (Ib-Id)] were anhydrized (Na2SO4)
and analyzed as such and after solvent evaporation (residues Ia-Id). Some differences
were seen between the relative amounts determined on solutions and those on residues,
especially for benzaldehyde and the Schiff bases. The yields quoted in Table 1 refer to
the composition of the respective solutions only.
Identification of the various reaction products was performed by GLC and NMR anal-
yses [7, 8]. For this purpose, small amounts of unambiguously synthesized compounds
were added into the analyzed samples. In preliminary experiments, separation in basic
(amines) and neutral constituents (amides) by treatment of residues Ia-Ic with aqueous
HCl [8] was sometimes useful. The quantification was made especially by 1H-NMR, by
comparison of the integrals with that of a known amount of added cyclohexane. Ben-
zophenone and acetophenone were used as internal standards for GLC. Synthetic mixtures
were worked-up as before (blank experiments) and the results used to correct the NMR-
or GLC-derived amounts.
4.9 Cyano Trapping
The oxidation was performed as before, but NaCN (200 mg; 4 mmol) dissolved in water
(10 mL) was added just before adding RuO2. The followed work-up was of type A.
Caution! Sodium cyanide is highly toxic. Direct contact of the chemical or its solutions
with the skin or eyes should be strictly avoided. The acidification and following operations
should be performed in a good hood.

692 H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694
Acknowledgment
This work was supported by the Romanian Academy and by the Romanian Ministry of
Education and Research (grant n. 4-232/2004).
References
[1] J.C. Sheehan and R.W. Tulis: “Oxidation of Cyclic Amines with Ruthenium Tetrox-
ide”, J. Org. Chem., Vol. 39, (1974), pp. 2264–2267.
[2] R. Perrone, G. Bettoni and V. Tortorella: “Oxidation of Morpholine Derivatives with
Ruthenium (VIII) Oxide”, Synthesis, (1976), pp. 598–600.
[3] G. Bettoni, C. Franchini, F. Morlacchi, N. Tangari and V. Tortorella: “Reactions of
Nitrogen Compounds with Ruthenium Tetroxide. 2. Oxidation of Tertiary Amines
as a Convenient Alternative to von Braun Degradation”, J. Org. Chem., Vol. 41,
(1976), pp. 2780–2782.
[4] G. Bettoni, G. Carbonara, C. Franchini and V. Tortorella: “Oxidation of Tertiary
Polycyclic Amines by RuO4”, Tetrahedron, Vol. 31, (1981), pp. 4159–4164.
[5] N. Tangari, M. Giovine, F. Morlacchi and C. Vetuschi: “Oxidation of Organic Ni-
trogen Compounds by Means of Ruthenium Tetroxide: Selective Preparation of N -
substituted Lactams”, Gazz. Chim. Ital., Vol. 115, (1985), pp. 325–328.
[6] C. Vetuschi, N. Tangari, M. Giovine, C. Franchini and V. Tortorella: “Selective
Oxidation of Piperazine Derivatives with Ruthenium Tetroxide”, Farmaco, Vol. 47,
(1992), pp. 599–605.
[7] H. Petride, C. Draghici, C. Florea and A. Petride: “RuO4-Mediated Oxidation of N -
Benzylated Tertiary Amines. Are Amine N -Oxides and Iminium Cations Reaction
Intermediates?”, Cent. Eur. J. Chem., Vol. 2, (2004), pp. 302–322.
[8] H. Petride, O. Costan, C. Draghici, C. Florea and A. Petride: “RuO4-Mediated
Oxidation of N -Benzylated Tertiary Amines. 2. Regioselectivity for N,N -Dimethyl-
and N,N -Diethylbenzylamine as Substrates”, Arkivoc, Part X, (2005), pp. 18–32,
http://www.arkat-usa.org/arkat/journal/2005/I10 Balaban/1275/AB-1275B.pdf.
[9] H. Petride, O. Costan, C. Draghici, C. Florea and A. Petride: “RuO4-Mediated
Oxidation of N -Benzylated Tertiary Amines. 4. The Case of 1-Benzylpiperazine”,
work in progress.
[10] H. Petride, O. Costan, C. Florea and S. Udrea: “Competitive Hydrolysis of Mono-
and Diformylated Ethylenediamine Derivatives”, Rev. Roum. Chim., Vol. 51, (2006),
pp. 245–253.
[11] H. Petride, C. Draghici, C. Florea, M. Maganu and S. Udrea: “E/Z Isomerism of
Some Diformamides”, Rev. Roum. Chim., Vol. 51, (2006), in press.
[12] H. Petride, A. Corbu, O. Costan, C. Florea, V. Marin, A. Petride and E. Serban:
“N -Oxides of Some N -Benzyl Azacycloalkanes. Preferred Conformation by NMR
Spectroscopy”, Rev. Roum. Chim., Vol. 50, (2005), pp. 633–640.
[13] C. Chapuis, A. Gauvreau, A. Klaebe, A. Lattes and J. J. Perie: “Condensation de

H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694 693
Diamines-1,2 sur les Composes Carbonyles. Syntheses d’imidazolidines: Mecanisme
de la Reaction”, Bull. Soc. Chim. France, (1973), pp. 977–985.
[14] Compound 20 might be present as at least two E/Z stereoisomers. The correspond-
ing 1H-NMR spectrum could contain (the values belonging to the major isomer are
underlined): 4.17+4.23+4.47+4.54 (s+s+s+s, 4H, N2-CH2+N2-Bn), 4.80+5.01 (s+s,
2H, N1-Bn), 8.26+8.35 (s+s, 1H, N2-CHO), 9.07+9.16 (s+s, 1H, N1-CHO). Some of
these signals were observed also in the 1-oxidation mixtures (work-up A), but a re-
liable quantification was not possible because of extensive signal superposition (4-5
and 8.1-8.4 ppm) and gross errors caused by its relatively small amount (9.0-9.2
ppm).
[15] H. Tanaka: “Oxidation Reaction of Steroid Alcohols by Ruthenium Tetroxide”, Tetra-
hedron, Vol. 19, (1969), pp. 1959–1963.
[16] P.E. Morris, Jr. and D.I. Kielly: “Ruthenium Tetraoxide Phase-Transfer-Promoted
Oxidation of Secondary Alcohols to Ketones”, J. Org. Chem., Vol. 52, (1987), pp.
1149–1152.
[17] S. Giddins and A. Mills: “Optimization of a Simple System for the Oxidation of
Octan-2-ol With Sodium Bromate, Mediated by Ruthenium Tetraoxide Generated
in situ“, J. Org. Chem., Vol. 53, (1988), pp. 1103–1107.
[18] A. Mills and C. Holland: “Effect of Ultrasound on the Kinetics of Oxidation of Octan-
2-ol and Other Secondary Alcohols with Sodium Bromate, Mediated by Ruthenium
Tetraoxide in a Biphasic System”, Ultrason. Sonochem., Vol. 2, (1995), pp. 533–538.
[19] S. Rajendran and D. C. Trivedi: “Ruthenium Tetroxide as a Phase Transfer Cata-
lyst in Biphasic System and Its in situ Electrochemical Regeneration: Oxidation of
Aromatic Primary Alcohols and Aldehydes”, Synthesis, (1995), pp. 153–154.
[20] S. Torii, T. Inokuchi and K. Kondo: “A Facile Procedure for Oxidative Cleavage of
Enolic Olefins to the Carbonyl Compounds with Ruthenium Tetraoxide (RuO4)”, J.
Org. Chem., Vol. 50, (1985), pp. 4980–4982.
[21] A. Dornow and H. Thies: “Uber die Umsetzung von Nitroessigsaureathylester mit
Mannich-Basen”, Liebigs Ann. Chem., Vol. 581, (1953), pp. 219–224.
[22] Compounds similar to 20 (i.e., 2, 5, 31, 34) were stable during their (attempted)
RuO4-oxidation reactions.
[23] Cation 25 is less stable than 26 owing to electron withdrawal of the carbonyl bond.
Thus, both AM1 and PM5 semiempirical MO methods (J. J. P. Stewart: MOPAC
2002 2.3, Fujitsu Ltd., Tokyo, 2002) indicated 25 as being less stable than 26 by 17.6
and 14.3 kJ/mol, respectively. As expected, the inverse situation held for the respec-
tive anions: that corresponding to 25 is more stable than the anion corresponding
to 26, by 75 kJ/mol (AM1).
[24] A black amorphous solid is often formed during the RuO4-catalyzed oxidations per-
formed in CCl4/water mixtures. See for instance, P.H.J. Carlsen, T. Katsuki, V.S.
Martin and K.B. Sharpless: “A Greatly Improved Procedure for Ruthenium Tetraox-
ide Catalyzed Oxidations of Organic Compounds”, J. Org. Chem., Vol. 46, (1981),
pp. 3936–3938.

694 H. Petride et al. / Central European Journal of Chemistry 4(4) 2006 674–694
[25] N.J. Leonard and F.P. Hauck, Jr.: “Unsaturated Amines. X. The Mercuric
Acetate Route to Substituted Piperidines, Δ2-Tetrahydropyridines and Δ2-
Tetrahydroanabasines”, J. Am. Chem. Soc., Vol. 79, (1957), pp. 5279–5292.
[26] D.S. Gierson, M. Harris and H.P. Husson: “Synthesis and Chemistry of 5,6-
Dihydropyridinium Salt Adducts. Synthons for General Electrophilic and Nucle-
ophilic Substitution of the Piperidine Ring System”, J. Am. Chem. Soc., Vol. 102,
(1980), pp. 1064–1082.
[27] B. Ho and N. Castagnoli, Jr.: “Trapping of metabolically generated electrophilic
species with cyanide ion: metabolism of 1-benzylpyrrolidine”, J. Med. Chem., Vol.
23, (1980), pp. 133–139.
[28] A. Koskinen and M. Lounasmaa: “Regiospecific Functionalisation of Carbon Atoms
α to Heterocyclic Nitrogen”, Tetrahedron, Vol. 39, (1983), pp. 1627–1633.
[29] L.M. Sayre, D.A. Engelhart, B. Venkataraman, M.K.M. Babu and G.D. McCoy:
“Generation and Fate of Enamines in the Microsomal Metabolism of Cyclic Tertiary
Amines”, Biochem. Biophys. Res. Commun., Vol. 179, (1991), pp. 1368–1376.
[30] M. Caproiu, C. Florea, C. Galli, A. Petride and H. Petride: “Oxidation of N -Benzyl
Aziridine by Molecular Iodine: Competition of Electron Transfer and Heterolytic
Pathways”, Eur. J. Org. Chem., (2000), pp. 1037–1043.
[31] H. Petride, A. Corbu, C. Florea, A. Petride and S. Udrea: “N -α-Cyano Derivatives
of Some N -Benzyl Azacycloalkanes”, Rev. Roum. Chim., Vol. 51, (2006), in press.
[32] D.W. Henry: “A Facile Synthesis of Piperazines from Primary Amines”, J. Hetero-
cyclic Chem., Vol. 3, (1966), pp. 503–511.
[33] Yu.S. Tsizin, N.L. Sergovskaya and S.A. Tcherniak: “Synthesis of 1-Alkyl (aralkyl)-
4-acyl-2-piperazinones”, Khim. Geterotsikl., (1986), pp. 514–517.
[34] J.C. Craig: “Preparation of 1-Benzylpiperazine”, J. Chem. Soc., (1959), pp. 3634–
3635.
[35] J. Cymerman-Craig, W.P. Rogers and M.E. Tate: “Chemical Constitution and An-
thelmintic Activity. III. Preparation of Substituted Phenothiazines and Some Mono-
and Dicyclic Analogues”, Aust. J. Chem., Vol. 9, (1956), pp. 397–405.
[36] S. Groszkowski, A. Serper, M. Ionesco, A. Soresco, A. Hacic and D. Panaitesco:
“Piperazine Derivatives and their Anthelmintic Effectiveness in Vitro”, Ann. Pharm.
Franc., Vol. 16, (1958), pp. 517–525; Chem. Abstr., Vol. 53, (1959), p. 8151e.
[37] J. Speziale and E.G. Jaworski: “N-Substituted Glycinate and Alalinate Esters”, J.
Org. Chem., Vol. 25, (1960), pp. 728–732.
[38] T. Yamazaki and M. Nagata: “Electrolytic Reduction of 2,5-dioxopiperazine Deriva-
tives by Tafel’s Method”, Yakugaku Zasshi, Vol. 79, (1959), pp. 1222–1224.; Chem.
Abstr., Vol. 54, (1960), p. 4596f.
[39] C. J. Pouchert and J. Behnke: The Aldrich Library of 13C and 1H FT NMR Spectra,
Vol. 2: [39a] n. 932B; [39b] n. 1063B, Aldrich Chemical Company, Inc., 1993.
[40] H.E. Gottlieb, V. Kotlyar and A. Nudelman: “NMR Chemical Shifts of Common
Laboratory Solvents as Trace Impurities”, J. Org. Chem., Vol. 62, (1997), pp. 7512–
7515.








![Synthesis and evaluation of conformationally restricted N-[2-(3,4-dichlorophenyl)ethyl]-N-methyl-2-(1-pyrrolidinyl)ethylamines at .sigma. receptors. 2. Piperazines, bicyclic amines,](https://static.fdokumen.com/doc/165x107/631289503ed465f0570a465c/synthesis-and-evaluation-of-conformationally-restricted-n-2-34-dichlorophenylethyl-n-methyl-2-1-pyrrolidinylethylamines.jpg)











