
Role of RyR2 Phosphorylation at S2814 During Heart Failure Progression
37
Wehrens Wisløff, Thomas Wieland, Xun Ai, Steven M. Pogwizd, Dobromir Dobrev and Xander H.T. deAlmeida, Niels Voigt, William S. Lawrence, Darlene G. Skapura, Kristine Skårdal, Ulrik Jonathan L. Respress, Ralph J. van Oort, Na Li, Natale Rolim, Sayali S. Dixit, Angela Role of RyR2 Phosphorylation at S2814 During Heart Failure Progression Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2012 American Heart Association, Inc. All rights reserved. is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231 Circulation Research doi: 10.1161/CIRCRESAHA.112.268094 2012;110:1474-1483; originally published online April 17, 2012; Circ Res. http://circres.ahajournals.org/content/110/11/1474 World Wide Web at: The online version of this article, along with updated information and services, is located on the http://circres.ahajournals.org/content/suppl/2012/04/17/CIRCRESAHA.112.268094.DC1.html Data Supplement (unedited) at: http://circres.ahajournals.org//subscriptions/ is online at: Circulation Research Information about subscribing to Subscriptions: http://www.lww.com/reprints Information about reprints can be found online at: Reprints: document. Permissions and Rights Question and Answer about this process is available in the located, click Request Permissions in the middle column of the Web page under Services. Further information Editorial Office. Once the online version of the published article for which permission is being requested is can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Circulation Research in Requests for permissions to reproduce figures, tables, or portions of articles originally published Permissions: at Universitaet Heidelberg on May 30, 2012 http://circres.ahajournals.org/ Downloaded from
Transcript of Role of RyR2 Phosphorylation at S2814 During Heart Failure Progression

WehrensWisløff, Thomas Wieland, Xun Ai, Steven M. Pogwizd, Dobromir Dobrev and Xander H.T.deAlmeida, Niels Voigt, William S. Lawrence, Darlene G. Skapura, Kristine Skårdal, Ulrik
Jonathan L. Respress, Ralph J. van Oort, Na Li, Natale Rolim, Sayali S. Dixit, AngelaRole of RyR2 Phosphorylation at S2814 During Heart Failure Progression
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2012 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/CIRCRESAHA.112.2680942012;110:1474-1483; originally published online April 17, 2012;Circ Res.
http://circres.ahajournals.org/content/110/11/1474World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2012/04/17/CIRCRESAHA.112.268094.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

Integrative Physiology
Role of RyR2 Phosphorylation at S2814 During HeartFailure Progression
Jonathan L. Respress,* Ralph J. van Oort,* Na Li, Natale Rolim, Sayali S. Dixit, Angela deAlmeida,Niels Voigt, William S. Lawrence, Darlene G. Skapura, Kristine Skårdal, Ulrik Wisløff,Thomas Wieland, Xun Ai, Steven M. Pogwizd, Dobromir Dobrev, Xander H.T. Wehrens
Rationale: Increased activity of Ca2�/calmodulin-dependent protein kinase II (CaMKII) is thought to promoteheart failure (HF) progression. However, the importance of CaMKII phosphorylation of ryanodine receptors(RyR2) in HF development and associated diastolic sarcoplasmic reticulum Ca2� leak is unclear.
Objective: Determine the role of CaMKII phosphorylation of RyR2 in patients and mice with nonischemic andischemic forms of HF.
Methods and Results: Phosphorylation of the primary CaMKII site S2814 on RyR2 was increased in patientswith nonischemic, but not with ischemic, HF. Knock-in mice with an inactivated S2814 phosphorylation site wererelatively protected from HF development after transverse aortic constriction compared with wild-typelittermates. After transverse aortic constriction, S2814A mice did not exhibit pulmonary congestion and hadreduced levels of atrial natriuretic factor. Cardiomyocytes from S2814A mice exhibited significantly lowersarcoplasmic reticulum Ca2� leak and improved sarcoplasmic reticulum Ca2� loading compared with wild-typemice after transverse aortic constriction. Interestingly, these protective effects on cardiac contractility were notobserved in S2814A mice after experimental myocardial infarction.
Conclusions: Our results suggest that increased CaMKII phosphorylation of RyR2 plays a role in thedevelopment of pathological sarcoplasmic reticulum Ca2� leak and HF development in nonischemic forms of HFsuch as transverse aortic constriction in mice. (Circ Res. 2012;110:1474-1483.)
Key Words: calcium � heart failure � ryanodine receptor � sarcoplasmic reticulum
Heart failure (HF) is a leading cause of morbidity andmortality and is responsible for one of every nine deaths
in the United States alone.1 Recent studies have revealed thatincreased activity of the enzyme Ca2�/calmodulin-dependentprotein kinase II (CaMKII) plays a major role in the devel-opment of HF.2–4 CaMKII can promote pathological cardiacremodeling by increasing cell death,5 stimulating cardiacdilatation,3 promoting cardiac arrhythmias,6 and interferingwith excitation–contraction coupling.2
Editorial, see p 1398In This Issue, see p 1391
Excitation–contraction coupling is initiated by the influxof extracellular Ca2� via voltage-gated Ca2� channels, which
triggers a much greater release of Ca2� from the sarcoplasmicreticulum (SR).7 In the normal heart, the amplitude of SRCa2� release can be dynamically increased by activation ofCaMKII at faster heart rates, leading to a frequency-dependent enhancement of cardiac contractility.8 In contrast,it also has been suggested that chronic elevation of CaMKIIactivity in diseased hearts can cause diastolic Ca2� leak fromthe SR associated with a loss of contractility.6,9 However, itremains unclear which Ca2� handling proteins downstream ofCaMKII are responsible for SR Ca2� release abnormalities,although the ryanodine receptor (RyR2) and phospholambancan be functionally altered by CaMKII.10,11
In previous work, we have demonstrated that CaMKIIpredominantly regulates RyR2 by phosphorylation of residue
Original received February 28, 2012; revision received April 3, 2012; accepted April 5, 2012. In March 2012, the average time from submission tofirst decision for all original research papers submitted to Circulation Research was 13.2 days.
From the Department of Molecular Physiology and Biophysics (J.L.R., R.J.v.O., N.L., S.S.D., A.d.A., D.G.S., X.H.T.W.), and Department of Medicine(X.H.T.W.), Baylor College of Medicine, Houston, TX; K.G. Jebsen Center of Exercise in Medicine (N.R., U.W.), FUGE Molecular Imaging Center(K.S.), Department of Circulation and Medical Imaging, Faculty of Medicine, Norwegian University of Science and Technology, Trondheim, Norway;Division of Experimental Cardiology (N.V., D.D.) and Institute of Experimental and Clinical Pharmacology and Toxicology (T.W.), MedicalFaculty Mannheim, Heidelberg University, Mannheim, Germany; Department of Medicine (X.A., S.M.P.), University of Alabama at Birmingham,Birmingham, AL.
*These authors contributed equally to this work.The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.112.
268094/-/DC1.Correspondence to Xander H.T. Wehrens, MD, PhD, Baylor College of Medicine, One Baylor Plaza, BCM335, Houston, TX 77030. E-mail
[email protected]© 2012 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.112.268094
1474 at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

S2814.6,8 This site is near, but distinct from, the primaryprotein kinase A (PKA) phosphorylation site S2808, whichalso modulates gating properties of the channel.12 We re-cently demonstrated that CaMKII phosphorylation of RyR2 issufficient to increase SR Ca2� leak in mice with constitu-tively phosphorylated RyR2 attributable to mutationS2814D.6 Moreover, this RyR2-mediated SR Ca2� leak leadsto the development of late-onset cardiomyopathy in S2814Dmice, suggesting that chronic phosphorylation of S2814might promote the development of HF.6 These data areconsistent with studies showing that pharmacological orgenetic inhibition of CaMKII can prevent or delay the onsetof HF in animal models.4,13 However, it also has been shownthat PKA hyperphosphorylation of RyR2 at S2808 occurs infailing hearts and that genetic inhibition of S2808 phosphor-ylation prevents development of ischemic HF in some mousemodels.14,15 Therefore, the contribution of these two phos-phorylation sites on RyR2 in the development of HF remainscontroversial.
Our studies revealed increased phosphorylation of S2814on RyR2 in patients with nonischemic dilated cardiomyopa-thy (DCM), but not in patients with ischemic cardiomyopathy(ICM). Knock-in mice with a genetically inactivated S2814phosphorylation site (S2814A mutation) were relatively pro-tected from HF development after transverse aortic constric-tion (TAC) compared to wild-type (WT) littermates. Theseeffects were associated with a decline in the amount ofspontaneous SR Ca2� release events after TAC in S2814Amice, attributable to prevention of enhanced S2814 phosphor-ylation. Interestingly, S2814A mice were not protected fromthe development of ischemic HF after myocardial infarction(MI), consistent with our data obtained in ICM humansamples. Thus, our findings suggest that increased CaMKIIphopshorylation of RyR2 plays a critical role in the develop-ment of pathological SR Ca2� leak and HF progression innonischemic forms of HF in both humans and mice withtransverse aortic constriction.
MethodsDetailed Methods are provided in the Online Supplement andprovide expanded details of surgical procedures, echocardiography,MRI, hemodynamic measurements, histology, Western blot analysis,quantitative reverse-transcriptase polymerase reaction, andcalcium imaging.
Surgical ProceduresGeneration of RyR2-S2814A knock-in mice has been described.16
All animal studies were performed according to protocols approvedby the Institutional Animal Care and Use Committee of BaylorCollege of Medicine, conforming to the Guide for the Care and Useof Laboratory Animals published by the United States NationalInstitutes of Health (National Institutes Health Publication No.85–23, revised 1996). TAC and MI were performed as described.15,17
Statistical AnalysisAll data are represented as average�SEM. Statistical significanceof differences between experimental groups was determined usingStudent paired t test or ANOVA, followed by Tukey posttestwhen appropriate. A value of P�0.05 was considered statisticallysignificant.
ResultsIncreased S2814 Phosphorylation of RyR2 inPatients With NonischemicDilated CardiomyopathyIncreased activity of CaMKII has been suggested to contrib-ute to contractile dysfunction and hypertrophy developmentassociated with HF in patients and in animal models.2,3,18 Todetermine whether RyR2 phosphorylation by CaMKII isaltered in patients with HF, we measured RyR2 phosphory-lation levels of S2814 (the principal CaMKII site) as well asS2808 (the principal PKA site and potential secondaryCaMKII site) using phosphoepitope-specific antibodies. Pon-ceau staining confirmed that similar amounts of heart lysatewere loaded on the protein gels. Western blots revealed asignificant increase in S2814 phosphorylation in nonischemicDCM but not in ICM patients, compared with healthycontrols (Figure 1). In contrast, there were no significantchanges in S2808 phosphorylation of RyR2 in both patientgroups. The finding that S2814 phosphorylation of RyR2 iselevated in patients with DCM was confirmed in a secondcohort of nonischemic DCM patients (Online Figure IA, B,and Online Table I). In these samples, increased RyR2phosphorylation of S2814 may be attributed to enhancedCaMKII activity, because CaMKII T286 autophosphorylationwas increased in nonischemic DCM patients compared withcontrols (Online Figure IC, D).
S2814A Knock-In Mice Exhibit Reduced HFDevelopment in Response to Pressure OverloadBecause previous studies have suggested that CaMKII activ-ity plays an important role in development of HF after
Non-standard Abbreviations and Acronyms
CaMKII Ca2�/calmodulin-dependent protein kinase II
DCM dilated cardiomyopathy
EDD end-diastolic diameter
EF ejection fraction
HF heart failure
HW heart weight
ICM ischemic cardiomyopathy
LuW lung weight
LVPWd left ventricular posterior wall diameter during diastole
MI myocardial infarction
MRI magnetic resonance imaging.
PCR polymerase chain reaction
PKA protein kinase A
Pmin end-diastolic pressure
RyR2 ryanodine receptor type 2
SCR spontaneous calcium release
SERCA2a sarco/endoplasmic reticulum Ca2�-ATPase 2a
SR sarcoplasmic reticulum
TAC transverse aortic constriction
TL tibia length
WT wild-type
Respress et al RyR2 Phosphorylation in HF Progression 1475
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

pressure overload in mice,13,18 we tested whether CaMKII-mediated phosphorylation of RyR2 is important for HFdevelopment. We studied RyR2-S2814A knock-in mice(S2814A) in which the CaMKII phosphorylation site wasgenetically inactivated. At baseline, cardiac dimensions and
function in S2814A mice were similar to WT mice up to atleast 12 months of age, as determined by echocardiography(Online Table II). Next, 10- to 12-week-old S2814A mice(n�9) and WT littermates (n�13) were subjected to TAC.TAC was performed by partial constriction of the transverseaorta between the right and left carotid arteries, which led topressure overload. Additional S2814A (n�9) and WT (n�8)mice were subjected to a sham procedure. One-week afterTAC, Doppler ultrasound was performed to measure flowvelocity in the right and left carotid arteries to estimate theseverity of aortic stenosis.19 The ratio between right and leftcarotid flow velocities was similar in S2814A (6.9�0.3) andWT mice (6.2�0.2), which indicates that both groups weresubjected to similar levels of pressure overload. To determinethe effects of the S2814A mutation on development ofcardiac hypertrophy and failure, cardiac geometry and func-tion were evaluated using serial echocardiography 0, 4, 8, 12,and 16 weeks after TAC (Figure 2A–C and Table 1).
Echocardiographic analysis revealed a similar hypertrophicresponse in S2814A and WT mice after TAC, evidenced by asimilar initial increase in left ventricular posterior wallthickness during diastole compared with sham mice (Figure2A). There was, however, a trend toward a lower leftventricular posterior wall diameter during diastole at 16weeks after TAC in WT mice compared with S2814A mice(0.78�0.02 mm versus 0.83�0.04 mm, respectively;P�0.20), suggesting ventricular wall thinning as a possibleresult of cardiac dilation. WT mice had development of amore pronounced cardiac dilatation after pressure overload incomparison with S2814A mice starting at 12 weeks aftersurgery (Figure 2B). The ejection fraction declined similarlyin S2814A and WT mice up to 8 weeks after TAC (Figure2C). However, subsequently only WT mice exhibited afurther decline in ejection fraction consistent with develop-ment of severe HF, whereas ejection fraction leveled off inS2814A mice. At 16 weeks after TAC, ejection fraction was
Figure 1. Increased S2814 phosphorylation on RyR2 inpatients with nonischemic dilated cardiomyopathey (DCM).A, Representative Western blots for phosphorylated RyR2-S2814 (pS2814), RyR2-S2808 (pS2808), and total RyR2 in heartlysates from healthy humans and patients with nonischemicDCM. Total amount of protein was determined by Ponceaustaining. B, Quantification revealed an increased S2814 phos-phorylation in DCM but no change in ischemic cardiomyopathy(ICM) patients. Numbers in bar indicate number of patients ana-lyzed in (B) and (D). C, Representative Western blots for phos-phorylated RyR2-S2814 (pS2814), RyR2-S2808 (pS2808), andtotal RyR2 in heart lysates from healthy humans and patientswith ICM. D, Quantification revealed no change in S2808 phos-phorylation in both patients groups. Data represented asaverage�SEM *P�0.05 vs corresponding control.
Figure 2. S2814A miceexhibit delayed progressionof heart failure after trans-verse aortic constriction(TAC). Echocardiographicanalysis of left ventricular pos-terior wall diameter (LVPWd)(A), end-diastolic diameter(EDD) (B), and ejection fraction(EF) (C) in S2814A mice orwild-type (WT) littermatesbefore, 4, 8, 12, and 16 weeksafter sham or transverse aorticconstriction (TAC), respec-tively. D, Representativepressure-volume loops at 16weeks after TAC showingreduced left ventricular diastol-ic volume and improved con-tractility in a S2814A mousecompared to WT. Representa-tive loops indicate one animalper group. Numbers in (A) indi-cate number of animals usedin (A–C). E, Bar graph showing
significant decrease in cardiac contractility (dP/dt) in WT mice after TAC, which was blunted in S2814A mice. Numbers in bar indicatenumber of animals used in (E, F). F, Unlike WT mice, S2814A mice did not have development of elevated end-diastolic left ventricularpressure at 16 weeks after TAC. *P�0.05, **P�0.01, ***P�0.001 vs corresponding sham group. #P�0.05 vs WT TAC.
1476 Circulation Research May 25, 2012
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

significantly higher in S2814A mice (43.0%�2.9%) com-pared with WT mice (32.4%�3.4%; P�0.05; Table 1).
To confirm these results, at 16 weeks after TAC, cardiaccontractility also was evaluated using left ventricular cathe-terization and pressure–volume measurements (Figure 2D,Online Table III).20,21 These hemodynamic measurementsrevealed a significant decrease in the first derivative of leftventricular pressure over time (dP/dtmax) in WT mice afterTAC (6142 mm Hg/s�655 mm Hg/s) compared with WT shammice (10 498 mm Hg/s�537 mm Hg/s; P�0.001), indicatingloss of systolic function attributable to pressure overload (Figure2E). In S2814A mice, however, dP/dtmax was not significantlydecreased after TAC (8068 mm Hg/s�506 mm Hg/s) comparedwith sham (8883 mm Hg/s�446 mm Hg/s). Quantification ofdiastolic blood pressure (Pmin or end-diastolic pressure) revealedimpaired diastolic function in WT mice after TAC(8.46 mm Hg�3.40 mm Hg), which was not seen in S2814ATAC mice (2.08 mm Hg�1.1 mm Hg; P�0.01; Figure 2F).Thus, these data suggest that inhibition of RyR2 phosphorylationby CaMKII is sufficient to reduce the development of HF and topreserve cardiac function in a mouse model of nonischemic HF.
Reduction in Cardiac Dilatation in S2814 MiceAfter TACTo further study structural changes in the heart induced by TAC,we performed postmortem analyses of the hearts at 16 weekspostsurgery. Heart weight-to-tibia length (TL) ratios were in-creased in WT mice (13.8 mg/mm�1.4 mg/mm) at 16 weeksafter TAC compared with sham-operated WT mice (7.9 mg/mm�0.4 mg/mm; P�0.01; Figure 3A). In contrast, there wasa smaller but still significant increase in heart weight/TLratios of S2814A mice after TAC (9.9 mg/mm�0.6 mg/mm)compared with sham hearts (7.6 mg/mm�0.5 mg/mm;P�0.01). In addition, the heart weight/TL ratio was smallerin S2814A compared with WT mice after TAC (Figure 3A).
Hematoxylin and eosin staining of transverse cardiac sectionsconfirmed that S2814A mice exhibited a hypertrophic re-sponse (ie, thickening of left ventricular posterior wall) 16weeks after TAC (Figure 3B). In contrast, WT mice exhibitedslightly thinner posterior walls with enlarged cavities (Figure3C), consistent with cardiac dilatation. Only hearts from WTmice but not S2814A mice exhibited a significant increase incardiac anteroposterior diameter at 16 weeks after TAC(Figure 3D), consistent with our echocardiography data(Figure 2A–C). Moreover, peak systolic ventricular pressure(end-systolic pressure, mm Hg) was decreased in WT mice(121.1�11.5) compared with S2814A mice (148.7�8.1) after16 weeks of TAC, consistent with more severe HF develop-ment in WT mice after TAC. Correlation of end-systolicpressure values with heart weight/TL ratios revealed lesscardiac enlargement despite higher pressures in S2814A miceafter TAC, consistent with a protective effect of the S2814Amutation (Online Figure II). Therefore, inhibition ofCaMKII-mediated phosphorylation of RyR2 does not preventthe hypertrophic response, but it prevents the progressionfrom hypertrophy to cardiac dilatation and failure in thesetting of pressure overload.
Reduced Cardiac Remodeling Associated With HFAfter TAC in S2814A MiceTo determine whether genetic inhibition of CaMKII phosphor-ylation of RyR2 prevented adverse remodeling of the heart afterTAC, Masson trichrome staining of cardiac sections was per-formed to measure cardiac fibrosis (Figure 4A, B). Whereasthere was a significant increase in the development of cardiacfibrosis in WT mice after TAC, the amount of fibrosis wasreduced in S2814A mice after TAC (Figure 4B). Additionally,WT mice had development of a significant increase in lungweight-to-TL ratio after TAC (14.6 mg/mm�1.7 mg/mm) com-pared with sham-operated mice (9.5 mg/mm�0.4 mg/mm;
Table 1. Echocardiographic Parameters of Wild-Type and S2814A Mice at 8 and 16 Weeks After Sham or Transverse AorticConstriction Surgery
8 Weeks 16 Weeks
Sham TAC Sham TAC
WT (n�8) S2814A (n�9) WT (n�13) S2814A (n�9) WT (n�8) S2814A (n�9) WT (n�13) S2814A (n�9)
HR (bpm) 462.1�10.5 467.9�16.1 489.2�12.3 471.3�16.0 477.4�8.8 448.9�14.4 481.1�12.5 475.0�14.9
ESD (mm) 2.76�0.08 2.75�0.07 3.36�0.14* 3.12�0.21 2.82�0.08 2.75�0.09 3.92�0.22‡ 3.25�0.16§
EDD (mm) 4.00�0.08 3.94�0.08 4.22�0.11 3.95�0.07 4.02�0.08 3.93�0.10 4.61�0.17* 4.10�0.13
EF (%) 59.2�1.5 58.0�1.0 42.1�2.6‡ 43.7�2.2‡ 57.7�1.3 57.9�1.1 32.5�3.4‡ 43.0�2.9†‡
FS (%) 31.1�1.0 30.2�0.7 20.7�1.4‡ 21.3�1.2‡ 30.0�0.9 30.1�0.7 15.5�1.7‡ 21.1�1.6‡§
IVS, diastolic (mm) 0.75�0.02 0.74�0.01 0.84�0.03 0.77�0.03 0.71�0.02 0.70�0.02 0.72�0.01 0.76�0.01*
IVSs (mm) 0.88�0.02 0.85�0.02 0.94�0.02 0.85�0.04 0.83�0.02 0.80�0.02 0.77�0.02 0.83�0.02
LVPW, diastolic (mm) 0.66�0.03 0.65�0.02 0.90�0.03‡ 0.84�0.04† 0.69�0.04 0.69�0.01 0.78�0.02 0.83�0.04*
LVPW, systolic (mm) 0.95�0.03 0.95�0.03 1.11�0.03* 1.10�0.04* 0.97�0.04 0.99�0.02 0.94�0.02 1.07�0.04§
EDD indicates end-diastolic diameter; EF, ejection fraction; ESD, end-systolic diameter; FS, left ventricular fractional shortening; HR, heart rate; IVS, intraventricularseptal wall thickness; LVPW, left ventricular posterior wall thickness; TAC, transverse aortic constriction; WT, wild-type.
Data are expressed as mean�SEM.*P�0.05 vs corresponding sham.†P�0.01 vs corresponding sham.‡P�0.001 vs corresponding sham.§P�0.05 vs WT TAC.
Respress et al RyR2 Phosphorylation in HF Progression 1477
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

P�0.05), which is indicative of pulmonary edema in the contextof congestive HF (Figure 4C). In contrast, there was no signif-icant increase in lung weight/TL ratio after TAC in S2814Amice (9.4 mg/mm�0.3 mg/mm) compared with sham-operatedS2814A mice (9.9 mg/mm�0.6 mg/mm). However, at 16 weeksafter TAC, there was a significant increase in lung weight/TL ratioin WT compared with S2814A mice, suggesting a rescue frompulmonary edema development in S2814A mice (Figure 4C).
Another major determinant of stress-induced cardiac re-modeling is reactivation of fetal cardiac genes. Therefore,mRNA levels of fetal genes nppa (atrial natriuretic factor)and nppb (brain natriuretic peptide) were determined usingquantitative polymerase chain reaction. In sham-operatedanimals, there were no differences in transcript levels com-paring S2814A and WT mice. In contrast, TAC induced asignificant increase in atrial natriuretic factor and brainnatriuretic peptide levels in WT mice, compared with sham-operated WT mice (Figure 4D, E). However, there was no
increase in atrial natriuretic factor levels and a blunted brainnatriuretic peptide response in S2814A mice compared withWT mice after TAC. Taken together, these data suggest thatinhibition of CaMKII phosphorylation of RyR2 does notsuppress the hypertrophic response after pressure overload,but it does prevent cellular signs of maladaptive HF.
Increased S2814 but Not S2808 Phosphorylation ofRyR2 After TACNext, we determined the time course of potential changes inRyR2 phosphorylation at S2814 (the principal CaMKII site) andS2808 (the principal PKA site). Western blotting of ventricularlysates using phosphoepitope-specific antibodies revealed anincrease in CaMKII phosphorylation of S2814 on RyR2 in WTmice after TAC (Figure 5A). The level of S2814 phosphoryla-tion displayed a gradual increase, which became significant at 8and 16 weeks after TAC (Figure 5B). As expected, there was nophosphorylation of this site in S2814A mice because of the
Figure 3. Reduction in cardiac dilatation inS2814A mice after transverse aortic constric-tion (TAC). A, Heart weight-to-tibia length (HW/TL)ratios at 16 weeks after TAC. Numbers in bar indi-cate number of animals used in (A–D). B, Repre-sentative photographs of mid sagittal sectionsstained with hematoxylin and eosin showingreduced cardiac enlargement in S2814A mice afterTAC. Scale bar, 1 mm. Left ventricular posteriorwall thickness (C) and anteroposterior left ventricu-lar diameters (D) measured in histological sections.*P�0.05, **P�0.01, ***P�0.001 vs correspondingsham group. #P�0.05 vs wild-type TAC.
Figure 4. Reduced cardiac remodeling associ-ated with heart failure after transverse aorticconstriction (TAC) in S2814A mice. A, Represen-tative photographs of sections at the mid sagittallevel of hearts stained with Masson trichrome(scale bar�1 mm). B, Quantification of cardiacfibrosis at 16 weeks after sham or TAC surgery. C,Postmortem measurements of lung weight-to-tibialength (LuW/TL) ratios 16 weeks after TAC. ThemRNA transcript levels of cardiac stress genesatrial natriuretic factor (D) and brain natriureticpeptide (E) by real-time polymerase chain reaction.Numbers in bar indicate number of animals.*P�0.05, **P�0.01 vs corresponding sham group.#P�0.05, ##P�0.01 vs wild-type TAC.
1478 Circulation Research May 25, 2012
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

genetic serine-to-alanine mutation of this residue. Of note, theobserved increase in S2814 phosphorylation in WT mice 8weeks after TAC coincides with the time point at which WT andS2814A mice start to diverge in terms of cardiac function(Figure 2C). Global CaMKII activity (assessed by CaMKIIT286 autophosphorylation) increased in WT mice after TAC(Online Figure IIIA, B). However, there was also a trend towardan increase in CaMKII activity in S2814A mice after TAC(P�0.16). Phosphorylation of the S2808 site on RyR2 trended toincrease in both S2814A and WT mice in the later stages of HF(Figure 5C, D). In addition, phosphorylation of phospholambanat site T17 (CaMKII site) increased in WT and S2814A mice(Online Figure IIIC, D), whereas phosphorylation of phospho-lamban at site S16 (PKA site) remained unchanged in TACgroups versus sham controls (Online Figure IIIE, F).
Inhibition of CaMKII Phosphorylation of RyR2Attenuates SR Ca2� Leak After TACTo determine the mechanisms underlying sustained cardiacfunction in S2814A mice after TAC, we next determined
whether inhibition of CaMKII-mediated phosphorylation ofRyR2 attenuated spontaneous SR Ca2� release (SCR) eventsafter pressure overload. Ventricular myocytes isolated frommice at 16 weeks after TAC or sham surgery were loadedwith a Ca2�-sensitive dye and imaged under an epifluores-cence microscope. In previous studies,22 we found a goodcorrelation between SR Ca2� leak measured using the tetra-caine protocol23 and the number of SCR events. After 1 Hzpacing to obtain steady-state, the number of SCR events wasmeasured after termination of pacing over a 40-second timeperiod (Figure 6A). The number of myocytes in which SCRevents occurred was significantly higher in WT mice afterTAC (46 events in 75 cells; �61%) compared with WT shammice (8 events in 31 cells; �26%; P�0.01; Figure 6B). Inaddition, SCR amplitude (measured as �F/Fo) was alsoincreased in WT TAC compared with WT sham mice. Incontrast, SCR amplitude was not increased in S2814A TACcompared with S2814A sham mice (Online Figure IVA). Theincrease in SCR was not attributable to increased SR Ca2�
loading, because SR content was decreased in WT TAC(�F/Fo: 1.54�0.1) compared with WT sham mice(1.97�0.2; P�0.05; Online Figure IVB). Moreover, theincrease in SCR amplitude also was not attributable toincreased SR Ca2� content, because each release event wasnormalized to SR Ca2� content (Online Figure IVC).
There were no differences in the rate of reuptake of Ca2�
into the SR, a measure of SERCA activity (tau [�]; OnlineFigure IVD). The incidence of SCR in myocytes fromS2814A mice after TAC (29.7%) was not increased comparedwith sham-operated S2814A mice (29.4%) and was signifi-cantly lower than myocytes from WT mice after TAC(61.3%; P�0.01; Figure 6B). Moreover, SR Ca2� contentremained unchanged in S2814A mice after TAC (1.88�0.1)and S2814A sham mice (1.96�0.2; Online Figure IVB).These data suggest that the increase in SCR incidence in WTmice after TAC is attributable to increased S2814 phosphor-ylation on RyR2 induced by TAC.
Similar findings were obtained when the average numberof SCR was quantified per myocyte (Figure 6C). In WT mice,there was a significant increase in SCR events per myocyteover a 40-second time period after TAC (2.0�0.3 events/cell)in comparison with WT sham (0.4�0.1; P�0.01). Thisincrease in SCR event rate attributed to TAC was blunted inS2814A mice (0.4�0.1; P�0.01). Finally, addition of theglobal CaMKII inhibitor KN93 also significantly reduced theSCR incidence and number of SCR events per cell inmyocytes from WT mice after TAC (Figure 6B, C). However,KN93 had no additional effect on SCR incidence or events inS2814A TAC cells, suggesting a specific and major role forS2814 phosphorylation on RyR2 in the development of HF.
Inhibition of S2814 Phosphorylation on RyR2 Failsto Protect Against HF Induced by MIPrevious studies demonstrated that PKA phosphorylation ofRyR2 played a role in the development of ischemic HF afterMI,15 but not after TAC-induced pressure overload.24 How-ever, these data are controversial in view of a recent studythat argued against a role for PKA phosphorylation of RyR2after MI.25 To determine the functional importance of CaM-
Figure 5. Increased CaMKII phosphorylation of RyR2 follow-ing transverse aortic constriction (TAC). A, RepresentativeWestern blots for phosphorylated RyR2-S2814 (pS2814) andtotal RyR2 in heart lysates from wild-type (WT) and S2814Amice before (0) or at 4, 8, and 16 weeks after TAC surgery,respectively. B, Quantification revealed increased S2814 phos-phorylation starting at 8 weeks after TAC. Data (n�4–8 pergroup) are represented as average�SEM. C, RepresentativeWestern blots for phosphorylated RyR2-S2808 (pS2808) andtotal RyR2 in heart lysates from WT and S2814A mice before (0)or at 4, 8, and 16 weeks after TAC surgery. D, Quantificationshowing nonsignificant increases in S2808 phosphorylation afterTAC in WT and S2814A mice. Data (n�3–4 per group) are rep-resented as average�SEM *P�0.05 **P�0.01 vs correspondingsham. ###P�0.001 vs WT TAC.
Respress et al RyR2 Phosphorylation in HF Progression 1479
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

KII phosphorylation of RyR2 during the development ofischemic HF, we subjected S2814A knock-in and WT mice toleft anterior descending coronary artery ligation to induce MI.In mice subjected to the sham procedure, cardiac dimensionsand contractility (Online Table IV) were similar in S2814Amice compared with WT mice. The effects of both mild (30%infarct area) and severe (60% infarct area) MI were deter-mined in two separate experimental groups of S2814A andWT mice (Figure 7A, B), as determined by MRI andechocardiography and confirmed by histology. Infarct sizedid not differ in the 30% group comparing S2814A(32.8%�2.2%) and WT mice (32.4%�1.0%) or in the “60%group” comparing S2814A (61.8%�2.1%) and WT(64.9%�4.0%). Echocardiography revealed that there wereno differences in the relative increase in left ventricularend-diastolic diameter comparing S2814A and WT mice afterboth 30% and 60% MI 3 weeks after surgery (Figure 7A). Asexpected, cardiac dilatation was more pronounced after 60%infarction compared with 30% infarction. Similarly, therewas an equal decline in ejection fraction in S2814A and WTmice after both 30% and 60% MI (Figure 7B). There were nosignificant differences in ejection fraction comparing S2814Aand WT mice. At the end of the experiment, ventricularmyocytes were isolated from the sham and 60% MI mice. At
3 weeks after coronary artery ligation, ventricular myocytelength was increased similarly in S2814A and WT mice(Figure 7C).
To determine the levels of CaMKII-mediated RyR2 phos-phorylation in response to MI, we performed Western blotanalysis of ventricular lysates obtained from mice subjectedto 60% MI. Using phospho-specific antibodies, the level ofS2814 phosphorylation on RyR2 was significantly decreasedin WT mice after MI (P�0.001). As expected, there was nophosphorylation of S2814 in S2814A mice attributable to thegenetic mutation of this site (Online Figure VA, B). Takentogether, these data suggest that inhibition of CaMKII phos-phorylation on S2814 of RyR2 does not prevent the devel-opment of HF induced by MI.
DiscussionThere is ample evidence that CaMKII plays a role in thedevelopment of HF. Previous studies have demonstratedincreased expression and activity levels of CaMKII in ani-mals and patients with congestive HF.10,26,27 Transgenicoverexpression of CaMKII-� causes HF in mice,3 whereasoverexpression of a peptide blocker of CaMKII delays theonset of HF in AC3-I transgenic mice.4 Moreover, knockoutof CaMKII-� was shown to limit the progression to HF.13,18 It
Figure 6. Inhibition of CaMKII phos-phorylation of RyR2 attenuates sarco-plasmic reticulum (SR) Ca2� leak aftertransverse aortic constriction (TAC). A,Representative images of spontaneousCa2� release (SCR) events in ventricularmyocytes after 1-Hz pacing. B, Incidenceof SCR events in isolated myocytes fromsham-operated and mice 16 weeks afterTAC, before and after pretreatment withKN93, respectively. C, Quantification ofSCR events per cell before and after pre-treatment with KN93. N�30 to 75 cellsfrom 3 to 6 mice per group. **P�0.01 vscorresponding sham. ##P�0.01 vs wild-type TAC.
Figure 7. Inhibition of S2814 phos-phorylation on RyR2 fails to protectagainst heart failure induced by myo-cardial infarction (MI). Echocardio-graphic analysis of wild-type (WT) andS2814A mice subjected to sham opera-tion or MI, resulting in average infarctscomprising 30% or 60% of the left ven-tricle, respectively. There were no differ-ences in left ventricular end-diastolicdiameter (LVEDD) (A) or ejection fraction(EF) (B) comparing WT and S2814A.Numbers in bars indicate number of ani-mals in (A, B). C, Quantification of iso-
lated ventricular myocyte length in WT and S2814A mice after MI. Numbers indicate number of animals and cells per group.*P�0.05 vs corresponding sham.
1480 Circulation Research May 25, 2012
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

was shown that CaMKII-� ablation reduced ventricular dila-tation and fibrosis and enhanced cardiac contractility afterTAC.13 The beneficial effects of CaMKII-� ablation havebeen attributed to a reduction in SR Ca2� leak, although thedownstream targets of CaMKII responsible for these effectswere not identified.
The results of the current study revealed increased CaMKIIphosphorylation of RyR2 in patients with nonischemic DCMbut not in patients with ICM. These findings suggest that thelevel of CaMKII activation might depend on the type of HFin patients. Previous studies have clearly demonstrated acti-vation of CaMKII-� by pressure overload in mice.13,18,28 Ourdata in mice showed that S2814 phosphorylation on RyR2 isincreased in mice subjected to TAC (pressure overload) butnot after MI. An important observation was that S2814phosphorylation increases over time after TAC in WT mice,whereas ablation of the S2814 site alone provided a beneficialeffect on deterioration toward severe HF in these mice. Thus,our data now identify RyR2 as a downstream target ofCaMKII involved in HF after pressure overload.
Moreover, our data for mice subjected to TAC revealedincreased CaMKII phosphorylation of phospholamban atT17, but not at the PKA site S16. These findings suggest thatactivated CaMKII phosphorylates multiple downstream tar-gets in failing hearts. However, the protective effects of theS2814A mutation in RyR2 in mice subjected to TAC showsthat RyR2 is a preeminent downstream target of CaMKII in thismodel of HF associated with detrimental remodeling of theheart. Thus, RyR2 phosphorylation at S2814 plays a role in theprogression of cardiac hypertrophy to HF after TAC.
Transgenic overexpression of CaMKII-� induces transientcardiac hypertrophy followed by dilated cardiomyopathy andHF in mice.3 However, hypertrophic remodeling induced bypressure overload was not attenuated in CaMKII-�–deficientmice, suggesting that CaMKII-� is not required for thedevelopment of cardiac hypertrophy in response to TAC.13
Our data are in agreement with those previous studies ofCaMKII-�–deficient mice, because S2814A mice also haddevelopment of hypertrophic remodeling similar to that ofWT mice in the early stages after TAC.
Relative Importance of RyR2 PhosphorylationSites Subsequent to HFThere has been considerable controversy about the relativeimportance of RyR2 phosphorylation sites, particularly theS2808 and S2814 sites.2,10,12,15 Our studies demonstrated thatCaMKII phosphorylation of S2814 on RyR2 was increasedafter pressure overload induced by TAC, which correlated toour findings in nonischemic DCM patients (Figure 1). Thesedata are consistent with recent studies by Ling et al.13 The factthat genetic ablation of this site only prevented the develop-ment of decompensation after TAC confirms the importanceof S2814 in the pathogenesis of congestive HF induced byTAC. Our data also show that phosphorylation of the PKAsite S2808 increases, but not significantly, after chronic TACduring later stages of HF development. Those findings are inagreement with a previous study24 showing that geneticablation of the S2808 site did not ameliorate the developmentof HF in S2808A knock-in mice after TAC.
It has been proposed that chronically elevated plasmalevels of catecholamines may promote PKA hyperphosphor-ylation of RyR2 in HF and contribute to abnormal SR Ca2�
release.14,29 Genetic inhibition of phosphorylation of the mainPKA site S2808 was shown to reduce the development ofMI-induced HF in S2808A knock-in mice.15 In addition, arecent study by Shan et al12 demonstrated that S2808Dknock-in mice with constitutively activated PKA phosphor-ylation sites on RyR2 had development of spontaneous HFwith age and also exhibited increased mortality after MI.However, our data did not reveal increased S2808 phosphor-ylation in patients with nonischemic DCM or ICM. More-over, Zhang et al25 recently reported a lack of protectiveeffects in S2808A mice after MI, adding to the controversyin the field about the importance of S2808 phosphoryla-tion in HF development. Interestingly, this article did showincreased S2808 phosphorylation after MI. One possibleexplanation for the discrepant findings is that PKA hyper-phosphorylation may only occur in advanced and more severestages of ischemic HF. Unless studies are performed duringthe same time points or the same severity of cardiac decom-pensation, phosphorylation levels of S2808 might greatlyvary between studies. In addition, there may be regionaldifferences in ischemic failing hearts.
Although previous studies had implicated activation ofCaMKII in HF development after MI,4,30 the role of CaMKIIphosphorylation of RyR2 at S2814 had not been studiedbefore in sufficient detail. Our data revealed that the level ofS2814 phosphorylation on RyR2 slightly decreased after MI.Therefore, it was not surprising that S2814A knock-in micewere not protected from development of decompensating HFafter MI. These findings are in agreement with recent work byKushnir et al,31 who also demonstrated a lack of protectionfrom MI-induced HF in S2814A mice.
Effects of CaMKII Phosphorylation of RyR2 onSR Ca2� Leak in HFEnhanced activity of CaMKII in the heart profoundly affectsSR Ca2� handling.2 Ventricular myocytes isolated fromCaMKII-�c transgenic mice exhibit increased diastolic SRCa2� release events (SR Ca2� leak) despite a lower SR Ca2�
load,2 which could be partly caused by increased CaMKIIphosphorylation of RyR2. Phosphorylation of the CaMKIIphosphorylation site S2814 on RyR2 enhances channel openprobability and Ca2� spark activity.6,8 Our recent studiesrevealed that constitutive activation of this CaMKII site inS2814D knock-in mice causes SR Ca2� leak, which is associ-ated with a mild dilated cardiomyopathy at 12 months of age.6
Moreover, S2814D mice exhibit markedly reduced survival afterTAC, suggesting that maximal phosphorylation of S2814 onRyR2 promotes development of decompensated HF.6
Our data revealed an increased incidence of spontaneousSCR events in WT mice after TAC. Pharmacological inhibi-tion of CaMKII reduced the number of SCR events, suggest-ing that CaMKII activation after pressure overload underliesCa2� release defects, as previously shown.2,13 Genetic abla-tion of the S2814 phosphorylation site on RyR2 led to asimilar decrease in SCR incidence after TAC in the absenceof exogenous CaMKII blockade, suggesting that RyR2 is a
Respress et al RyR2 Phosphorylation in HF Progression 1481
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

major downstream target of CaMKII underlying defective SRCa2� release. Thus, pressure overload induces spontaneousreleases of SR Ca2� during diastole attributable to increasedCaMKII-mediated phosphorylation of RyR2. Taken together,these data suggest that the preservation of cardiac contractil-ity, attenuation of SR Ca2� leak, and delayed development ofHF in S2814A mice are mediated by the direct prevention ofCaMKII phosphorylation of site S2814 on the RyR2 Ca2�
release channel.
ConclusionsTaken together, our present work has demonstrated thatincreased CaMKII phosphorylation of S2814 on RyR2 playsa critical role in the development of pathological SR Ca2�
leak and HF in a mouse model of pressure overload. How-ever, phosphorylation of the S2814 site does not appear toplay a role in the development of HF after MI. Moreover,there seems to be a difference in phosphorylation of RyR2sites depending on whether HF is ischemic in nature.Overall, our current findings suggest an important role forCaMKII-mediated phosphorylation of S2814 on RyR2 inthe development of nonischemic HF in mice with pressureoverload, and support its role as a potential new target forthe treatment of HF.
AcknowledgmentsThe authors thank Julia Reynolds and Claudia Liebetrau for theirexpertise and technical assistance.
Sources of FundingX.H.T.W. is a W.M. Keck Foundation Distinguished Young Scholarin Medical Research, and is supported by National Institutes ofHealth/National Heart, Lung, and Blood Institute grants R01-HL089598 and R01-R01HL091947, and Muscular Dystrophy Asso-ciation. J.L.R. is a recipient of the 2010 Ruth L. Kirschstein NationalResearch Service Award (NRSA) supported by National Institutes ofHealth/National Heart, Lung, and Blood Institute grant F31-HL099290-01. R.J.v.O. was a recipient of the American Physiolog-ical Society Postdoctoral Fellowship in Physiological Genomics.This work was also supported in part by Fondation Leducq (Alliancefor CaMKII Signaling in Heart) to X.H.T.W. and S.M.P., theEuropean North-Amarican Atrial Fibrillation Research Alliance toD.D., and by the German Center for Cardiovascular Research (toD.D. and T.W.).
DisclosuresNone.
References1. Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke
statistics–2011 update: a report from the American Heart Association.Circulation. 2011;123:e18–e209.
2. Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM.Transgenic CaMKII�C overexpression uniquely alters cardiac myocyteCa2� handling: reduced SR Ca2� load and activated SR Ca2� release.Circ Res. 2003;92:904–911.
3. Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J Jr, Bers DM, BrownJH. The deltaC isoform of CaMKII is activated in cardiac hypertrophyand induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92:912–919.
4. Zhang R, Khoo MS, Wu Y, et al. Calmodulin kinase II inhibition protectsagainst structural heart disease. Nat Med. 2005;11:409–417.
5. Khoo MS, Li J, Singh MV, et al. Death, cardiac dysfunction, and arrhyth-mias are increased by calmodulin kinase II in calcineurin cardiomyopa-thy. Circulation. 2006;114:1352–1359.
6. van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL,Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM,Wehrens XH. Ryanodine receptor phosphorylation by calcium/cal-modulin-dependent protein kinase II promotes life-threatening ven-tricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669 –2679.
7. Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release andcardiac disease. Annu Rev Physiol. 2005;67:69–98.
8. Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2�/calmodulin-dependent protein kinase II phosphorylation regulates the cardiacryanodine receptor. Circ Res. 2004;94:e61–e70.
9. Guo T, Zhang T, Mestril R, Bers DM. Ca2�/Calmodulin-dependentprotein kinase II phosphorylation of ryanodine receptor does affectcalcium sparks in mouse ventricular myocytes. Circ Res. 2006;99:398–406.
10. Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2�/calmod-ulin-dependent protein kinase modulates cardiac ryanodine receptor phos-phorylation and sarcoplasmic reticulum Ca2� leak in heart failure. CircRes. 2005;97:1314–1322.
11. Zhang T, Guo T, Mishra S, Dalton ND, Kranias EG, Peterson KL, BersDM, Brown JH. Phospholamban ablation rescues sarcoplasmic reticulumCa(2�) handling but exacerbates cardiac dysfunction in CaMKII�c
transgenic mice. Circ Res. 2010;106:354–362.12. Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A,
Dura M, Chen BX, Marks AR. Role of chronic ryanodine receptorphosphorylation in heart failure and beta-adrenergic receptor blockade inmice. J Clin Invest. 2010;120:4375–4387.
13. Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND,Peterson KL, Chen J, Bers D, Heller Brown J. Requirement for Ca2�/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest.2009;119:1230–1240.
14. Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, RosemblitN, Marks AR. PKA phosphorylation dissociates FKBP12.6 from thecalcium release channel (ryanodine receptor): defective regulation infailing hearts. Cell. 2000;101:365–376.
15. Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR.Ryanodine receptor/calcium release channel PKA phosphorylation: acritical mediator of heart failure progression. Proc Natl Acad Sci U S A.2006;103:511–518.
16. Chelu MG, Sarma S, Sood S, et al. Calmodulin kinase II-mediatedsarcoplasmic reticulum Ca2� leak promotes atrial fibrillation in mice.J Clin Invest. 2009;119:1940–1951.
17. deAlmeida AC, van Oort RJ, Wehrens XH. Transverse aortic constrictionin mice. J Vis Exp. 2010;38:pii:1729.
18. Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM,Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, Bassel-Duby R,Maier LS, Olson EN. The delta isoform of CaM kinase II is required forpathological cardiac hypertrophy and remodeling after pressure overload.Proc Natl Acad Sci U S A. 2009;106:2342–2347.
19. Reddy AK, Taffet GE, Li YH, Lim SW, Pham TT, Pocius JS, Entman ML,Michael LH, Hartley CJ. Pulsed Doppler signal processing for use in mice:applications. IEEE Trans Biomed Eng. 2005;52:1771–1783.
20. Kottam AT, Porterfield J, Raghavan K, Fernandez D, Feldman MD,Valvano JW, Pearce JA. Real time pressure-volume loops in mice usingcomplex admittance: measurement and implications. Conf Proc IEEEEng Med Biol Soc. 2006;1:4336–4339.
21. Joho S, Ishizaka S, Sievers R, Foster E, Simpson PC, Grossman W. Leftventricular pressure-volume relationship in conscious mice. Am J PhysiolHeart Circ Physiol. 2007;292:H369–H377.
22. Sarma S, Li N, van Oort RJ, Reynolds C, Skapura DG, Wehrens XH.Genetic inhibition of PKA phosphorylation of RyR2 prevents dystrophiccardiomyopathy. Proc Natl Acad Sci U S A. 107:13165–13170.
23. Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulumCa2� leak in intact ventricular myocytes from rabbits in heart failure. CircRes. 2003;93:592–594.
24. Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, JohnMC, Powers PA, Valdivia HH. Intact beta-adrenergic response andunmodified progression toward heart failure in mice with genetic ablationof a major protein kinase A phosphorylation site in the cardiac ryanodinereceptor. Circ Res. 2007;101:819–829.
25. Zhang H, Makarewich C, Kubo H, Wang W, Duran J, Li Y, Berretta R,Koch WJ, Chen X, Gao E, Valdivia H, Houser SR. Hyperphosphorylationof the cardiac ryanodine receptor at serine 2808 is not involved in cardiacdysfunction after myocardial infarction. Circ Res. 2012;110:831–840.
1482 Circulation Research May 25, 2012
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

26. Hoch B, Meyer R, Hetzer R, Krause EG, Karczewski P. Identification andexpression of �-isoforms of the multifunctional Ca2�/calmodulin-dependent protein kinase in failing and nonfailing human myocardium.Circ Res. 1999;84:713–721.
27. Hagemann D, Bohlender J, Hoch B, Krause EG, Karczewski P.Expression of Ca2�/calmodulin-dependent protein kinase II �-subunitisoforms in rats with hypertensive cardiac hypertrophy. Mol CellBiochem. 2001;220:69–76.
28. Colomer JM, Mao L, Rockman HA, Means AR. Pressure overload selec-tively up-regulates Ca2�/calmodulin-dependent protein kinase II in vivo.Mol Endocrinol. 2003;17:183–192.
29. Reiken S, Wehrens XH, Vest JA, Barbone A, Klotz S, Mancini D,Burkhoff D, Marks AR. Beta-blockers restore calcium release channelfunction and improve cardiac muscle performance in human heart failure.Circulation. 2003;107:2459–2466.
30. He BJ, Joiner ML, Singh MV, et al. Oxidation of CaMKII determinesthe cardiotoxic effects of aldosterone. Nat Med. 2011;17:1610 –1618.
31. Kushnir A, Shan J, Betzenhauser MJ, Reiken S, Marks AR. Role ofCaMKII� phosphorylation of the cardiac ryanodine receptor in the forcefrequency relationship and heart failure. Proc Natl Acad Sci U S A.2010;107:10274–10279.
Novelty and Significance
What Is Known?
● Heart failure (HF) is a leading cause of morbidity and mortality and isresponsible for one of every nine deaths in the United States.
● Ca2�/calmodulin-dependent protein kinase II (CaMKII) is upregulatedin patients with HF.
● Phosphorylation of CaMKII phosphorylation site S2814 on RyR2enhances open probability, Ca2� spark activity, and sarcoplasmicreticulum (SR) Ca2� leak.
What New Information Does This Article Contribute?
● S2814 phosphorylation of RyR2 is increased in patients with nonis-chemic dilated cardiomyopathy (DCM) and in mice after pressureoverload, but not in patients and mice with ischemic cardiomy-opathy (ICM).
● Genetic inhibition of S2814 phosphorylation of RyR2 prevents SRCa2� leak and decompensated HF after transverse aortic con-striction (TAC) in mice.
Previous studies demonstrated that increased levels of CaMKIImight contribute to enhanced SR Ca2� leak in animals andpatients with HF. Here, we tested the hypothesis that CaMKIIphosphorylation of S2814 on RyR2 underlies Ca2� cyclingdefects in HF. First, we found that S2814 phosphorylation is onlyincreased in patients with nonischemic DCM, but not in thosewith ICM. Next, genetic inhibition of S2814 phosphorylation inmice was found to prevent development of decompensated HFafter TAC. The time course of the protective effect correlatedwell with the delayed increase in S2814 phosphorylation onRyR2. Finally, SR Ca2� leak was reduced in S2814A knock-inmice after TAC, compared with WT control mice. In contrast,S2814A mice were not protected from development of HF aftermyocardial infarction, consistent with the findings in human ICMsamples. Thus, our data suggest that increased S2814 phos-phorylation of RyR2 might play an important role in developmentof nonischemic types of HF.
Respress et al RyR2 Phosphorylation in HF Progression 1483
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

1
Supplemental Material Role of RyR2 phosphorylation at S2814 during heart failure progression by Respress et al. Human heart samples - LV tissue was obtained from four failing human hearts after explantation in patients undergoing clinically indicated cardiac transplantation for end-stage idiopathic non-ischemic dilated cardiomyopathy (DCM) performed at the University of Alabama, Birmingham (UAB) or Loyola University Chicago Hospital (see Online Table I). Four non-failing (NF) human hearts (which could not be used for transplantation) were obtained from Gift of Hope (formerly the Regional Organ Bank of Illinois) or the Alabama Organ Center as described previously 1. The study was approved by the Human Studies Committees of Loyola University, Gift of Hope, UAB and the Alabama Organ Center, respectively. Another set of LV tissue samples were obtained with approval of the Institutional Review Board of the University of Mannheim, Germany. Transverse aortic constriction - Mice were anesthetized using a mixture of 1.5-2% isoflurane in 100% O2, and left anterior thoracotomy was performed to visualize the aortic arch. Transverse aortic constriction (TAC) was induced by subjecting the aorta to a defined, 27-gauge constriction using a 6-0 suture between the first and second trunk of the aortic arch, as previously described in detail 2, 3. The aorta was not constricted during the surgical sham procedure. Doppler velocities were measured to quantify the alterations in the right and left carotid arteries following TAC as previously described. Right-to-left carotid peak velocity ratios were similar in WT and S2814A TAC groups. Mice were sacrificed 16 weeks after surgery. Myocardial infarction - Myocardial infarction (MI) was induced while maintaining aseptic conditions by ligating the left anterior descending coronary artery (LAD) approximately 2 mm below the tip of the left auricle with an 7-0 silk ligature, as previously described 4. Occlusion was confirmed by observing myocardial pallor and restricted ventricular motion in the area of interest. The chest was closed in layers followed by subcutaneous administration of buprenorphine (0.1 mg/kg, 0.5 ml in volume Temgesic, Reckitt and Coleman, Hull, UK) to reduce postoperative acute pain. The LAD was not ligated during the sham procedure. Infarct size was estimated as the extent of akinesis/dyskinesis of the left ventricular free wall on MRI or of the left ventricle wall. The final determination of infarct size was made on histological sections. Transthoracic echocardiography - Echocardiography was performed using the VisualSonics VeVo 770 Imaging System (VisualSonics, Toronto, Canada) equipped with high-frequency 30 MHz probe, as described 5. All measurements were made while the mouse had a heart rate of 450 ± 50 beats per minute. In addition, body temperature was maintained between a narrow range (37.0 ± 0.5 °C) to avoid confounding effects of hypothermia. Invasive hemodynamic measurements - Cardiac catheterization was used to assess left ventricular (LV) contractility using a 1.4F high-fidelity micromanometer catheter (Millar Instruments, Houston, TX) advanced into the left ventricle via the right carotid artery, as described 2. Data were digitized and analyzed using the IOX data acquisition system (Emka Technologies, Falls Church, VA). The first derivative of LV pressure over time is expressed as dP/dtmax. The pressure inside of the LV during diastole is expressed as Pmin. The pressure inside of the LV during systole is expressed as Pmax. MRI - MRI was performed on a 7T Bruker Biospec Avance 70/20 system (Bruker Biospin, Ettlingen, Germany) with a 72 mm volume coil for transmission and an actively decoupled rat head surface coil for receive only (RAPID MR International, LLC, Ohio, USA). Anesthetized animals, 1-2% isoflurane in 1:2 O2:N2, were positioned prone atop of the surface coil in a dedicated animal bed and anaesthesia level was
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

2
monitored using a pressure sensitive pillow under the animal’s abdomen. Respiration was maintained at 80-120 breaths/min and rectal temperature was kept stable at 36-38°C.
Following localization of the heart, 7-9 contiguous short axis black blood cine movies were acquired for complete coverage of the left ventricle using a self-gated fast low flip angle shot (FLASH) sequence with an oblique navigator echo. Acquisition parameters were repetition time (TR) 6.9 ms, echo time (TE) 2.5 ms, flip angle 10°, slice thickness 1 mm, field of view 4×4 cm2 and image matrix 128x128. The sequence was repeated 300 times. In all slices, end-diastole and end-systole were visually identified and the area of the lumen was traced manually using ImageJ (ImageJ 1.44p, National Institute of Health, USA). Papillary muscles were not included in the left ventricular lumen segmentation. By summing up for all slices, LVESV and LVEDV were extracted and SV and EF calculated. Histology – Whole hearts were excised, washed briefly in normal saline, dried, weighed, and sectioned into three pieces. The middle transverse section was fixed in 4% buffered formaldehyde for 48 hours. After paraffin embedding and sectioning, 5 mm sections were stained with hematoxylin-eosin (H&E) for cell morphology and Masson’s trichrome (MT) for fibrosis. Infarct size was calculated as total infarct circumference divided by total left ventricle circumference 4. Sections were examined on an Olympus BH-2 epifluorescence microscope. MT staining was quantified using Adobe Photoshop 6 and ImageJ (National Institute of Health, Bethesda, MD). Infarct size was measured at the papillary muscle level on the transverse plane, and quantified as the length of the arc corresponding to the segment of the MI scar in relation to the total perimeter of the endocardial border of the left ventricle. Western blot analysis (mouse) - Cardiac homogenates were prepared as described previously 7. Mouse heart homogenates were subjected to electrophoresis on 6% (for RyR2) acrylamide gels, and transferred onto polyvinyl difluoride (PVDF) membranes. Membranes were probed with monoclonal anti-RyR2 (1:5,000; Thermo Fisher Scientific, Waltham, MA), polyclonal anti-pS2814-RyR2 (1:1,000), 8 polyclonal anti-pS2808-RyR2 (1:1,000), 7 monoclonal anti-pT286- CaMKII (1:1,000; Cayman Chemicals, Ann Arbor, MI), polyclonal anti-CaMKII (1:1000), polyclonal anti-pT17-PLN (1:5,000; Badrilla, Leeds, UK), polyclonal anti-pS16-PLN (1:5000; Badrilla, Leeds, UK), monoclonal anti-PLN (1:5000; Badrilla, Leeds, UK), and monoclonal anti-GAPDH (1:10,000; Millipore, Temecula, CA) antibodies at room temperature (RT) for 4 hours or overnight at 4 °C. Membranes were then incubated with secondary anti-mouse and anti-rabbit antibodies conjugated to Alexa-Fluor 680 (Invitrogen Molecular Probes, Carlsbad, CA) and IR800Dye (Rockland Immunochemicals, Gilbertsville, PA), respectively, and bands were quantified using infrared visualization (Odyssey System, Lincoln, NE) and densitometry (ImageJ Data Acquisition Software, National Institute of Health, Bethesda, MD). Western blot analysis (human) - The protein levels of total RyR2 (1:500, Affinity BioReagents, Golden, CO), Ser2808 and Ser2814 phosphorylated RyR2 (1:2,000 and 1:1,000, respectively) 7, 8 were quantified by Western blotting as described 9, 10. The RyR2-Ser2808 and RyR2-Ser2814 phosphoepitope-specific antibodies were custom generated using the peptide C-RTRRI-(pS)-QTSQV corresponding to the PKA phosphorylation site region at Ser2808 on RyR2 and peptide CSQTSQV-(pS)-VD corresponding to CaMKII phosphorylated RyR2 at Ser2814. Peroxidase-conjugated goat anti-rabbit (1:5,000, Sigma-Aldrich), goat anti-mouse (Sigma-Aldrich) and donkey anti-goat (Santa-Cruz) were used as secondary antibodies and visualized by chemifluorescense (GE Healthcare, Chalifont St. Giles, UK). AIDA Software (raytest, Straubenhardt, Germany) was used for quantification. The total amount of protein in each blot was determined by Ponceau staining. Quantitative RT-PCR - One µg of RNA from indicated mice was reverse-transcribed using Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA), as described 11. Real-time PCR using the Eppendorf qPCR machine and fluorescence detection was performed in 96-well plates using SYBR Green. PCR amplification was performed as a singleplex reaction in a total reaction volume of 25 ml, consisting of
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

3
400 nM forward and reverse primers, 1 ml cDNA, 12.5 ml 2x SYBR Green PCR master mix (Invitrogen, Carlsbad, CA). The PCR was cycled between 95°C /30 s and 60°C /30 s for 40 cycles, following an initial denaturation step at 95°C for 2 min. Primers were specific for mouse sequences and designed as described 11. Transcript quantities were compared using the relative Ct method, where the amount of target normalized to the amount of endogenous control (L7) and relative to the control sample is given by 2–ΔΔCt. Calcium imaging in isolated ventricular myocytes - Single ventricular myocytes were isolated by a modified collagenase method, as described 2. Ventricular myocytes were loaded with 5 mmol/L Fluo-4 AM (Invitrogen, Carlsbad, CA) for 30 min at room temperature (RT), ventricular myocytes were perfused with 1.8 mM Ca2+ normal Tyrode (NT) solution to wash out extra fluorescent. Intracellular Ca2+ concentrations ([Ca2+]i) were measured using an illumination device (model Lambda DG-4, Sutter Instruments, Novato, CA), and an electro-multiplier intensified back-illuminated charge coupled device (CCD) camera (model Cascade 512B, Photometrics, Tucson, AZ). After precondition, myocytes were paced at 20 V, 1 Hz for 20 seconds, followed by a 40 second pause. Caffeine (10 mM) was applied in 0 Ca2+ NT to estimate steady-state SR Ca2+ content. The amplitude and number of spontaneous Ca2+ release (SCR) events during the 40-second pause were quantified. As negative control experiments, myocytes from WT TAC and S14A TAC mice were pretreated with the CaMKII inhibitor KN93 (1 µmol/L) at least 10 min before pacing. Curve fittings of single isolated transients under steady state were used to determine SERCA activity (tau, τ) during the 20-second pacing phase. Cell length measurements - Microscopic pictures of cardiomyocytes were captured and analyzed using a Nikon Eclipse E400 Microscope with a DSFil camera (Nikon NIS-Elements Basic Research Version 3.00 software, Nikon Instruments Inc., Melville, NY). Myocytes were measured along their long axis to determine cell lengths. At least 3 mice were analyzed per group. Data on cell lengths were analyzed in a blinded manner.
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

4
Online References 1. Ai X, Pogwizd SM. Connexin 43 downregulation and dephosphorylation in nonischemic heart
failure is associated with enhanced colocalized protein phosphatase type 2a. Circ Res. 2005;96:54-63
2. van Oort RJ, Respress JL, Li N, Reynolds C, De Almeida AC, Skapura DG, De Windt LJ, Wehrens XH. Accelerated development of pressure overload-induced cardiac hypertrophy and dysfunction in an ryr2-r176q knockin mouse model. Hypertension. 2010;55:932-938
3. deAlmeida AC, van Oort RJ, Wehrens XH. Transverse aortic constriction in mice. J Vis Exp. 2010;38:1729
4. Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel pka phosphorylation: A critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103:511-518
5. Respress JL, Wehrens XH. Transthoracic echocardiography in mice. J Vis Exp. 2010:1738 6. Dahab GM, Kheriza MM, El-Beltagi HM, Fouda AM, El-Din OA. Digital quantification of
fibrosis in liver biopsy sections: Description of a new method by photoshop software. J of Gastroenterol Hepatol. 2004;19:78-85
7. Sood S, Chelu MG, van Oort RJ, Skapura D, Santonastasi M, Dobrev D, Wehrens XH. Intracellular calcium leak due to fkbp12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm. 2008;5:1047-1054
8. Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D, Wehrens XH. Calmodulin kinase ii-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940-1951
9. El-Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, Ravens U, Dobrev D. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation. 2006;114:670-680
10. Greiser M, Halaszovich CR, Frechen D, Boknik P, Ravens U, Dobrev D, Luckhoff A, Schotten U. Pharmacological evidence for altered src kinase regulation of I(Ca,L) in patients with chronic atrial fibrillation. Naunyn Schmiedebergs Arch Pharmacol. 2007;375:383-392
11. van Oort RJ, van Rooij E, Bourajjaj M, Schimmel J, Jansen MA, van der Nagel R, Doevendans PA, Schneider MD, van Echteld CJ, De Windt LJ. Mef2 activates a genetic program promoting chamber dilation and contractile dysfunction in calcineurin-induced heart failure. Circulation. 2006;114:298-308
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

5
Online Table I. Characteristics of human heart failure subject (non-ischemic DCM cohort) Patient Population Demographics & Regimen control HF No. patients 3 4
Age, y 43.7±8.8 41.5±8.1
Male/female, n 2/1 2/2
Beta blockers 1 2
Diuretics 1 3
ACE inhibitors/ ARB 0 3
Ca2+ channel blockers 0 3
Statins 0 3
Risk Factors
Hypertension 0 4
Myocardial infarction (prior) 0 0
Diabetes 0 3
Cardiac Function
Ejection fraction, % 50±5.0 11.7±1.7 ***
Data are expressed as mean ± SEM or number of patients. HF indicates non-ischemic dilated cardiomyopathy (DCM). ***P<0.001 versus control. All patients were of Caucasian descent.
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from
6
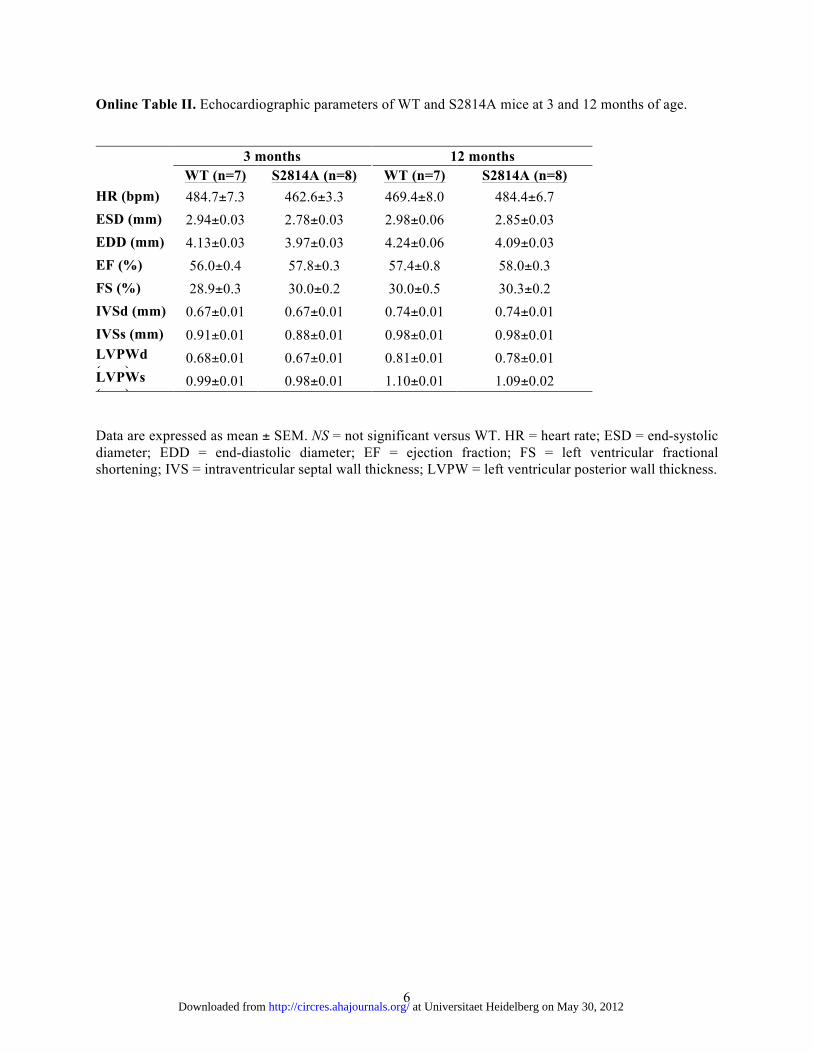
Online Table II. Echocardiographic parameters of WT and S2814A mice at 3 and 12 months of age. 3 months 12 months WT (n=7) S2814A (n=8)
WT (n=7) S2814A (n=8) HR (bpm) 484.7±7.3 462.6±3.3 469.4±8.0 484.4±6.7 ESD (mm) 2.94±0.03 2.78±0.03 2.98±0.06 2.85±0.03 EDD (mm) 4.13±0.03 3.97±0.03 4.24±0.06 4.09±0.03 EF (%) 56.0±0.4 57.8±0.3 57.4±0.8 58.0±0.3 FS (%) 28.9±0.3 30.0±0.2 30.0±0.5 30.3±0.2 IVSd (mm) 0.67±0.01 0.67±0.01 0.74±0.01 0.74±0.01 IVSs (mm) 0.91±0.01 0.88±0.01 0.98±0.01 0.98±0.01 LVPWd (mm)
0.68±0.01 0.67±0.01 0.81±0.01 0.78±0.01 LVPWs (mm)
0.99±0.01 0.98±0.01 1.10±0.01 1.09±0.02 Data are expressed as mean ± SEM. NS = not significant versus WT. HR = heart rate; ESD = end-systolic diameter; EDD = end-diastolic diameter; EF = ejection fraction; FS = left ventricular fractional shortening; IVS = intraventricular septal wall thickness; LVPW = left ventricular posterior wall thickness.
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

7
Online Table III. Pressure-volume loop parameters of WT and S2814A mice at 16 weeks after sham or TAC surgery. 16 weeks sham TAC WT (n=6) S2814A (n=8) WT (n=6) S2814A (n=8) Pmax (mmHg) 100.6±3.1 97.7±4.4 121.1±11.5 148.7±8.1 Pmin (mmHg) -0.8±0.5 0.0±0.9 8.5±3.4** 2.1±1.1 HR (bpm) 513.6±19.6 509.7±8.6 522.9±24.3 506.8±18.5 SV (µL) 7.2±0.6 7.0±0.8 3.2±0.3*** 5.8±0.3# CO (µL/min) 3726.6±292.5 3546.1±425.8 1722.0±168.4*** 2918.0±163.6### dP/dtmax (mmHg/s) 10497.8±536.9 8883.2±446.2 6141.6±655.1*** 8068.1±505.9 dP/dtmin (mmHg/s) -8606.9±424.0 -8271.7±471.5 -6354.9±723.4* -8349.8±655.9 SW (mmHg) 562.5±62.4 559.3±75.4 239.9±27.2*** 595.2±42.2### Data are expressed as mean ± SEM. *P<0.05, **P<0.01, ***P<0.001 versus corresponding sham, #P<0.05, ###P<0.001 versus WT TAC. Pmax = maximum rate of ventricular pressure change; Pmin = minimum rate of ventricular pressure change; HR = heart rate; SV = stroke volume; CO = cardiac output (HR x SV); dP/dtmax = maximum derivative of left ventricular pressure (contractility); dP/dtmin = minimum derivative left ventricular pressure (relaxation); SW = stroke work.
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

8
Online Table IV. MRI parameters of WT and S2814A mice before and after MI. sham 60% MI WT (n=5) S2814A (n=3)
WT (n=2) S2814A (n=2) LVESV (µl) 27.13±4.0 20.80±1.1 182.81±20.3* 168.0±43.0* LVEDV (µl) 63.38±6.8 50.91±5.9 230.42±17.0* 192.68±51.0* EF (%) 57.0±4.0 57.0±8.0 20.88±3.0* 12.64±1.0* SV (µl) 36.25±4.6 30.11±7.0 48.0±3.0* 24.85±8.3* Data are expressed as mean ± SEM. NS = not significant versus WT counterpart. LVESV = end-systolic volume; LVEDV = end-diastolic volume; EF = ejection fraction; SV = stroke volume. * P<0.05 versus corresponding sham.
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

9
Online Figure I
Online Figure I. Increased CaMKII auto-phosphorylation and its phosphorylation of RyR2 at site S2814 in human patients with non-ischemic heart failure. A. Representative Western blots for phosphorylated RyR2-S2814 (pS2814) and total RyR2 in heart lysates from biopsies of human patients with non-ischemic heart failure (non-ischemic DCM) in comparison to non-HF controls. B. Quantification revealed increased S2814 phosphorylation in patients with HF. C. Representative Western blots for phosphorylated CaMKII-T286 (pT286), total CaMKII, and GAPDH in heart lysates from biopsies of human patients with non-ischemic heart failure. D. Quantification shows a tend towards increased T286 phosphorylation normalized to GAPDH in patients with HF. ** P<0.01 vs. control. Numbers in bars indicate number of patients analyzed in panels B and D.
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

10
Online Figure II Online Figure II. Correlation between heart weight normalized to tibia length and peak systolic pressure. This graph shows the correlation between heart weight normalized to tibia length (HW/TL) and peak systolic ventricular pressure (Pmax) measured at 16 weeks after transverse aortic constriction (TAC). Although the average Pmax was greater in S2814A mice, the HW/TL ratio was lower consistent with a protective effect of the S2814A mutation following pressure overload-induced remodeling. N=6-8 animals per group.
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

11
Online Figure III
Online Figure III. Increased CaMKII auto-phosphorylation and its phosphorylation of PLN at site T17. A. Representative Western blots for phosphorylated CaMKII-T286 (pT286), total CaMKII, and GAPDH in heart lysates from WT and S2814A mice 16 weeks after TAC surgery, respectively. B. Quantification revealed increased T286 phosphorylation in WT mice with a trend in S2814A mice after TAC. N=4 animals per group. C. Representative Western blots for phosphorylated PLN-T17 (pT17) and total PLN in heart lysates from WT and S2814A mice 16 weeks after TAC. D. Quantification showing increase in T17 phosphorylation following TAC in WT mice with a trend in S2814A mice. N=4-7 animals per group. E. Representative Western blots for phosphorylated PLN-S16 (pS16) and total PLN in heart lysates from WT and S2814A mice 16 weeks after TAC. F. Quantification revealed no change in S16 phosphorylation following TAC in either group. * P<0.05 vs. corresponding sham. N=4 animals per group.
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

12
Online Figure IV
Online Figure IV. Inhibition of CaMKII phosphorylation of RyR2 attenuates SR Ca2+ Leak after
TAC. A. Quantification of spontaneous calcium release (SCR) amplitude in isolated myocytes from WT and S2814A mice revealed an increase in WT mice after TAC. B. Quantification of SR Ca2+ content in WT and S2814A mice revealed a decrease in WT mice after TAC. C. Quantification of SCR amplitude normalized to SR Ca2+ content revealed a significant increase in WT TAC cells. D. SERCA2a activity was estimated by calculating the time course of Ca2+ transient decay (tau). An average of >25 cells from 3 or more mice per group were analyzed. * P<0.05, *** P<0.001 versus corresponding sham, # P<0.05 versus WT TAC.
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

13
Online Figure V Online Figure V. Myocardial infarction does not increase S2814 phosphorylation on RyR2 in mice. A. Representative Western blots for phosphorylated RyR2-S2814 (pS2814) and total RyR2 in heart lysates from WT and S2814A mice subjected to sham or MI surgery resulting in 60% infarcts. B. Quantification of RyR2 phosphorylation indicates decreased S2814 after 60% MI in WT mice. Numbers in bars indicate number of animals. *** P<0.001 versus corresponding sham.
at Universitaet Heidelberg on May 30, 2012http://circres.ahajournals.org/Downloaded from

1
Supplemental Material Role of RyR2 phosphorylation at S2814 during heart failure progression by Respress et al. Human heart samples - LV tissue was obtained from four failing human hearts after explantation in patients undergoing clinically indicated cardiac transplantation for end-stage idiopathic non-ischemic dilated cardiomyopathy (DCM) performed at the University of Alabama, Birmingham (UAB) or Loyola University Chicago Hospital (see Online Table I). Four non-failing (NF) human hearts (which could not be used for transplantation) were obtained from Gift of Hope (formerly the Regional Organ Bank of Illinois) or the Alabama Organ Center as described previously 1. The study was approved by the Human Studies Committees of Loyola University, Gift of Hope, UAB and the Alabama Organ Center, respectively. Another set of LV tissue samples were obtained with approval of the Institutional Review Board of the University of Mannheim, Germany. Transverse aortic constriction - Mice were anesthetized using a mixture of 1.5-2% isoflurane in 100% O2, and left anterior thoracotomy was performed to visualize the aortic arch. Transverse aortic constriction (TAC) was induced by subjecting the aorta to a defined, 27-gauge constriction using a 6-0 suture between the first and second trunk of the aortic arch, as previously described in detail 2, 3. The aorta was not constricted during the surgical sham procedure. Doppler velocities were measured to quantify the alterations in the right and left carotid arteries following TAC as previously described. Right-to-left carotid peak velocity ratios were similar in WT and S2814A TAC groups. Mice were sacrificed 16 weeks after surgery. Myocardial infarction - Myocardial infarction (MI) was induced while maintaining aseptic conditions by ligating the left anterior descending coronary artery (LAD) approximately 2 mm below the tip of the left auricle with an 7-0 silk ligature, as previously described 4. Occlusion was confirmed by observing myocardial pallor and restricted ventricular motion in the area of interest. The chest was closed in layers followed by subcutaneous administration of buprenorphine (0.1 mg/kg, 0.5 ml in volume Temgesic, Reckitt and Coleman, Hull, UK) to reduce postoperative acute pain. The LAD was not ligated during the sham procedure. Infarct size was estimated as the extent of akinesis/dyskinesis of the left ventricular free wall on MRI or of the left ventricle wall. The final determination of infarct size was made on histological sections. Transthoracic echocardiography - Echocardiography was performed using the VisualSonics VeVo 770 Imaging System (VisualSonics, Toronto, Canada) equipped with high-frequency 30 MHz probe, as described 5. All measurements were made while the mouse had a heart rate of 450 ± 50 beats per minute. In addition, body temperature was maintained between a narrow range (37.0 ± 0.5 °C) to avoid confounding effects of hypothermia. Invasive hemodynamic measurements - Cardiac catheterization was used to assess left ventricular (LV) contractility using a 1.4F high-fidelity micromanometer catheter (Millar Instruments, Houston, TX) advanced into the left ventricle via the right carotid artery, as described 2. Data were digitized and analyzed using the IOX data acquisition system (Emka Technologies, Falls Church, VA). The first derivative of LV pressure over time is expressed as dP/dtmax. The pressure inside of the LV during diastole is expressed as Pmin. The pressure inside of the LV during systole is expressed as Pmax. MRI - MRI was performed on a 7T Bruker Biospec Avance 70/20 system (Bruker Biospin, Ettlingen, Germany) with a 72 mm volume coil for transmission and an actively decoupled rat head surface coil for receive only (RAPID MR International, LLC, Ohio, USA). Anesthetized animals, 1-2% isoflurane in 1:2 O2:N2, were positioned prone atop of the surface coil in a dedicated animal bed and anaesthesia level was

2
monitored using a pressure sensitive pillow under the animal’s abdomen. Respiration was maintained at 80-120 breaths/min and rectal temperature was kept stable at 36-38°C.
Following localization of the heart, 7-9 contiguous short axis black blood cine movies were acquired for complete coverage of the left ventricle using a self-gated fast low flip angle shot (FLASH) sequence with an oblique navigator echo. Acquisition parameters were repetition time (TR) 6.9 ms, echo time (TE) 2.5 ms, flip angle 10°, slice thickness 1 mm, field of view 4×4 cm2 and image matrix 128x128. The sequence was repeated 300 times. In all slices, end-diastole and end-systole were visually identified and the area of the lumen was traced manually using ImageJ (ImageJ 1.44p, National Institute of Health, USA). Papillary muscles were not included in the left ventricular lumen segmentation. By summing up for all slices, LVESV and LVEDV were extracted and SV and EF calculated. Histology – Whole hearts were excised, washed briefly in normal saline, dried, weighed, and sectioned into three pieces. The middle transverse section was fixed in 4% buffered formaldehyde for 48 hours. After paraffin embedding and sectioning, 5 mm sections were stained with hematoxylin-eosin (H&E) for cell morphology and Masson’s trichrome (MT) for fibrosis. Infarct size was calculated as total infarct circumference divided by total left ventricle circumference 4. Sections were examined on an Olympus BH-2 epifluorescence microscope. MT staining was quantified using Adobe Photoshop 6 and ImageJ (National Institute of Health, Bethesda, MD). Infarct size was measured at the papillary muscle level on the transverse plane, and quantified as the length of the arc corresponding to the segment of the MI scar in relation to the total perimeter of the endocardial border of the left ventricle. Western blot analysis (mouse) - Cardiac homogenates were prepared as described previously 7. Mouse heart homogenates were subjected to electrophoresis on 6% (for RyR2) acrylamide gels, and transferred onto polyvinyl difluoride (PVDF) membranes. Membranes were probed with monoclonal anti-RyR2 (1:5,000; Thermo Fisher Scientific, Waltham, MA), polyclonal anti-pS2814-RyR2 (1:1,000), 8 polyclonal anti-pS2808-RyR2 (1:1,000), 7 monoclonal anti-pT286- CaMKII (1:1,000; Cayman Chemicals, Ann Arbor, MI), polyclonal anti-CaMKII (1:1000), polyclonal anti-pT17-PLN (1:5,000; Badrilla, Leeds, UK), polyclonal anti-pS16-PLN (1:5000; Badrilla, Leeds, UK), monoclonal anti-PLN (1:5000; Badrilla, Leeds, UK), and monoclonal anti-GAPDH (1:10,000; Millipore, Temecula, CA) antibodies at room temperature (RT) for 4 hours or overnight at 4 °C. Membranes were then incubated with secondary anti-mouse and anti-rabbit antibodies conjugated to Alexa-Fluor 680 (Invitrogen Molecular Probes, Carlsbad, CA) and IR800Dye (Rockland Immunochemicals, Gilbertsville, PA), respectively, and bands were quantified using infrared visualization (Odyssey System, Lincoln, NE) and densitometry (ImageJ Data Acquisition Software, National Institute of Health, Bethesda, MD). Western blot analysis (human) - The protein levels of total RyR2 (1:500, Affinity BioReagents, Golden, CO), Ser2808 and Ser2814 phosphorylated RyR2 (1:2,000 and 1:1,000, respectively) 7, 8 were quantified by Western blotting as described 9, 10. The RyR2-Ser2808 and RyR2-Ser2814 phosphoepitope-specific antibodies were custom generated using the peptide C-RTRRI-(pS)-QTSQV corresponding to the PKA phosphorylation site region at Ser2808 on RyR2 and peptide CSQTSQV-(pS)-VD corresponding to CaMKII phosphorylated RyR2 at Ser2814. Peroxidase-conjugated goat anti-rabbit (1:5,000, Sigma-Aldrich), goat anti-mouse (Sigma-Aldrich) and donkey anti-goat (Santa-Cruz) were used as secondary antibodies and visualized by chemifluorescense (GE Healthcare, Chalifont St. Giles, UK). AIDA Software (raytest, Straubenhardt, Germany) was used for quantification. The total amount of protein in each blot was determined by Ponceau staining. Quantitative RT-PCR - One µg of RNA from indicated mice was reverse-transcribed using Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA), as described 11. Real-time PCR using the Eppendorf qPCR machine and fluorescence detection was performed in 96-well plates using SYBR Green. PCR amplification was performed as a singleplex reaction in a total reaction volume of 25 ml, consisting of

3
400 nM forward and reverse primers, 1 ml cDNA, 12.5 ml 2x SYBR Green PCR master mix (Invitrogen, Carlsbad, CA). The PCR was cycled between 95°C /30 s and 60°C /30 s for 40 cycles, following an initial denaturation step at 95°C for 2 min. Primers were specific for mouse sequences and designed as described 11. Transcript quantities were compared using the relative Ct method, where the amount of target normalized to the amount of endogenous control (L7) and relative to the control sample is given by 2–ΔΔCt. Calcium imaging in isolated ventricular myocytes - Single ventricular myocytes were isolated by a modified collagenase method, as described 2. Ventricular myocytes were loaded with 5 mmol/L Fluo-4 AM (Invitrogen, Carlsbad, CA) for 30 min at room temperature (RT), ventricular myocytes were perfused with 1.8 mM Ca2+ normal Tyrode (NT) solution to wash out extra fluorescent. Intracellular Ca2+ concentrations ([Ca2+]i) were measured using an illumination device (model Lambda DG-4, Sutter Instruments, Novato, CA), and an electro-multiplier intensified back-illuminated charge coupled device (CCD) camera (model Cascade 512B, Photometrics, Tucson, AZ). After precondition, myocytes were paced at 20 V, 1 Hz for 20 seconds, followed by a 40 second pause. Caffeine (10 mM) was applied in 0 Ca2+ NT to estimate steady-state SR Ca2+ content. The amplitude and number of spontaneous Ca2+ release (SCR) events during the 40-second pause were quantified. As negative control experiments, myocytes from WT TAC and S14A TAC mice were pretreated with the CaMKII inhibitor KN93 (1 µmol/L) at least 10 min before pacing. Curve fittings of single isolated transients under steady state were used to determine SERCA activity (tau, τ) during the 20-second pacing phase. Cell length measurements - Microscopic pictures of cardiomyocytes were captured and analyzed using a Nikon Eclipse E400 Microscope with a DSFil camera (Nikon NIS-Elements Basic Research Version 3.00 software, Nikon Instruments Inc., Melville, NY). Myocytes were measured along their long axis to determine cell lengths. At least 3 mice were analyzed per group. Data on cell lengths were analyzed in a blinded manner.

4
Online References 1. Ai X, Pogwizd SM. Connexin 43 downregulation and dephosphorylation in nonischemic heart
failure is associated with enhanced colocalized protein phosphatase type 2a. Circ Res. 2005;96:54-63
2. van Oort RJ, Respress JL, Li N, Reynolds C, De Almeida AC, Skapura DG, De Windt LJ, Wehrens XH. Accelerated development of pressure overload-induced cardiac hypertrophy and dysfunction in an ryr2-r176q knockin mouse model. Hypertension. 2010;55:932-938
3. deAlmeida AC, van Oort RJ, Wehrens XH. Transverse aortic constriction in mice. J Vis Exp. 2010;38:1729
4. Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel pka phosphorylation: A critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103:511-518
5. Respress JL, Wehrens XH. Transthoracic echocardiography in mice. J Vis Exp. 2010:1738 6. Dahab GM, Kheriza MM, El-Beltagi HM, Fouda AM, El-Din OA. Digital quantification of
fibrosis in liver biopsy sections: Description of a new method by photoshop software. J of Gastroenterol Hepatol. 2004;19:78-85
7. Sood S, Chelu MG, van Oort RJ, Skapura D, Santonastasi M, Dobrev D, Wehrens XH. Intracellular calcium leak due to fkbp12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm. 2008;5:1047-1054
8. Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D, Wehrens XH. Calmodulin kinase ii-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940-1951
9. El-Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, Ravens U, Dobrev D. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation. 2006;114:670-680
10. Greiser M, Halaszovich CR, Frechen D, Boknik P, Ravens U, Dobrev D, Luckhoff A, Schotten U. Pharmacological evidence for altered src kinase regulation of I(Ca,L) in patients with chronic atrial fibrillation. Naunyn Schmiedebergs Arch Pharmacol. 2007;375:383-392
11. van Oort RJ, van Rooij E, Bourajjaj M, Schimmel J, Jansen MA, van der Nagel R, Doevendans PA, Schneider MD, van Echteld CJ, De Windt LJ. Mef2 activates a genetic program promoting chamber dilation and contractile dysfunction in calcineurin-induced heart failure. Circulation. 2006;114:298-308

5
Online Table I. Characteristics of human heart failure subject (non-ischemic DCM cohort) Patient Population Demographics & Regimen control HF No. patients 3 4
Age, y 43.7±8.8 41.5±8.1
Male/female, n 2/1 2/2
Beta blockers 1 2
Diuretics 1 3
ACE inhibitors/ ARB 0 3
Ca2+ channel blockers 0 3
Statins 0 3
Risk Factors
Hypertension 0 4
Myocardial infarction (prior) 0 0
Diabetes 0 3
Cardiac Function
Ejection fraction, % 50±5.0 11.7±1.7 ***
Data are expressed as mean ± SEM or number of patients. HF indicates non-ischemic dilated cardiomyopathy (DCM). ***P<0.001 versus control. All patients were of Caucasian descent.

6
Online Table II. Echocardiographic parameters of WT and S2814A mice at 3 and 12 months of age. 3 months 12 months WT (n=7) S2814A (n=8)
WT (n=7) S2814A (n=8) HR (bpm) 484.7±7.3 462.6±3.3 469.4±8.0 484.4±6.7 ESD (mm) 2.94±0.03 2.78±0.03 2.98±0.06 2.85±0.03 EDD (mm) 4.13±0.03 3.97±0.03 4.24±0.06 4.09±0.03 EF (%) 56.0±0.4 57.8±0.3 57.4±0.8 58.0±0.3 FS (%) 28.9±0.3 30.0±0.2 30.0±0.5 30.3±0.2 IVSd (mm) 0.67±0.01 0.67±0.01 0.74±0.01 0.74±0.01 IVSs (mm) 0.91±0.01 0.88±0.01 0.98±0.01 0.98±0.01 LVPWd (mm)
0.68±0.01 0.67±0.01 0.81±0.01 0.78±0.01 LVPWs (mm)
0.99±0.01 0.98±0.01 1.10±0.01 1.09±0.02 Data are expressed as mean ± SEM. NS = not significant versus WT. HR = heart rate; ESD = end-systolic diameter; EDD = end-diastolic diameter; EF = ejection fraction; FS = left ventricular fractional shortening; IVS = intraventricular septal wall thickness; LVPW = left ventricular posterior wall thickness.

7
Online Table III. Pressure-volume loop parameters of WT and S2814A mice at 16 weeks after sham or TAC surgery. 16 weeks sham TAC WT (n=6) S2814A (n=8) WT (n=6) S2814A (n=8) Pmax (mmHg) 100.6±3.1 97.7±4.4 121.1±11.5 148.7±8.1 Pmin (mmHg) -0.8±0.5 0.0±0.9 8.5±3.4** 2.1±1.1 HR (bpm) 513.6±19.6 509.7±8.6 522.9±24.3 506.8±18.5 SV (µL) 7.2±0.6 7.0±0.8 3.2±0.3*** 5.8±0.3# CO (µL/min) 3726.6±292.5 3546.1±425.8 1722.0±168.4*** 2918.0±163.6### dP/dtmax (mmHg/s) 10497.8±536.9 8883.2±446.2 6141.6±655.1*** 8068.1±505.9 dP/dtmin (mmHg/s) -8606.9±424.0 -8271.7±471.5 -6354.9±723.4* -8349.8±655.9 SW (mmHg) 562.5±62.4 559.3±75.4 239.9±27.2*** 595.2±42.2### Data are expressed as mean ± SEM. *P<0.05, **P<0.01, ***P<0.001 versus corresponding sham, #P<0.05, ###P<0.001 versus WT TAC. Pmax = maximum rate of ventricular pressure change; Pmin = minimum rate of ventricular pressure change; HR = heart rate; SV = stroke volume; CO = cardiac output (HR x SV); dP/dtmax = maximum derivative of left ventricular pressure (contractility); dP/dtmin = minimum derivative left ventricular pressure (relaxation); SW = stroke work.

8
Online Table IV. MRI parameters of WT and S2814A mice before and after MI. sham 60% MI WT (n=5) S2814A (n=3)
WT (n=2) S2814A (n=2) LVESV (µl) 27.13±4.0 20.80±1.1 182.81±20.3* 168.0±43.0* LVEDV (µl) 63.38±6.8 50.91±5.9 230.42±17.0* 192.68±51.0* EF (%) 57.0±4.0 57.0±8.0 20.88±3.0* 12.64±1.0* SV (µl) 36.25±4.6 30.11±7.0 48.0±3.0* 24.85±8.3* Data are expressed as mean ± SEM. NS = not significant versus WT counterpart. LVESV = end-systolic volume; LVEDV = end-diastolic volume; EF = ejection fraction; SV = stroke volume. * P<0.05 versus corresponding sham.

9
Online Figure I
Online Figure I. Increased CaMKII auto-phosphorylation and its phosphorylation of RyR2 at site S2814 in human patients with non-ischemic heart failure. A. Representative Western blots for phosphorylated RyR2-S2814 (pS2814) and total RyR2 in heart lysates from biopsies of human patients with non-ischemic heart failure (non-ischemic DCM) in comparison to non-HF controls. B. Quantification revealed increased S2814 phosphorylation in patients with HF. C. Representative Western blots for phosphorylated CaMKII-T286 (pT286), total CaMKII, and GAPDH in heart lysates from biopsies of human patients with non-ischemic heart failure. D. Quantification shows a tend towards increased T286 phosphorylation normalized to GAPDH in patients with HF. ** P<0.01 vs. control. Numbers in bars indicate number of patients analyzed in panels B and D.

10
Online Figure II Online Figure II. Correlation between heart weight normalized to tibia length and peak systolic pressure. This graph shows the correlation between heart weight normalized to tibia length (HW/TL) and peak systolic ventricular pressure (Pmax) measured at 16 weeks after transverse aortic constriction (TAC). Although the average Pmax was greater in S2814A mice, the HW/TL ratio was lower consistent with a protective effect of the S2814A mutation following pressure overload-induced remodeling. N=6-8 animals per group.

11
Online Figure III
Online Figure III. Increased CaMKII auto-phosphorylation and its phosphorylation of PLN at site T17. A. Representative Western blots for phosphorylated CaMKII-T286 (pT286), total CaMKII, and GAPDH in heart lysates from WT and S2814A mice 16 weeks after TAC surgery, respectively. B. Quantification revealed increased T286 phosphorylation in WT mice with a trend in S2814A mice after TAC. N=4 animals per group. C. Representative Western blots for phosphorylated PLN-T17 (pT17) and total PLN in heart lysates from WT and S2814A mice 16 weeks after TAC. D. Quantification showing increase in T17 phosphorylation following TAC in WT mice with a trend in S2814A mice. N=4-7 animals per group. E. Representative Western blots for phosphorylated PLN-S16 (pS16) and total PLN in heart lysates from WT and S2814A mice 16 weeks after TAC. F. Quantification revealed no change in S16 phosphorylation following TAC in either group. * P<0.05 vs. corresponding sham. N=4 animals per group.

12
Online Figure IV
Online Figure IV. Inhibition of CaMKII phosphorylation of RyR2 attenuates SR Ca2+ Leak after
TAC. A. Quantification of spontaneous calcium release (SCR) amplitude in isolated myocytes from WT and S2814A mice revealed an increase in WT mice after TAC. B. Quantification of SR Ca2+ content in WT and S2814A mice revealed a decrease in WT mice after TAC. C. Quantification of SCR amplitude normalized to SR Ca2+ content revealed a significant increase in WT TAC cells. D. SERCA2a activity was estimated by calculating the time course of Ca2+ transient decay (tau). An average of >25 cells from 3 or more mice per group were analyzed. * P<0.05, *** P<0.001 versus corresponding sham, # P<0.05 versus WT TAC.

13
Online Figure V Online Figure V. Myocardial infarction does not increase S2814 phosphorylation on RyR2 in mice. A. Representative Western blots for phosphorylated RyR2-S2814 (pS2814) and total RyR2 in heart lysates from WT and S2814A mice subjected to sham or MI surgery resulting in 60% infarcts. B. Quantification of RyR2 phosphorylation indicates decreased S2814 after 60% MI in WT mice. Numbers in bars indicate number of animals. *** P<0.001 versus corresponding sham.




















