
Manual for the laboratory diagnosis of measles and rubella ...
Upload
independentCategory
view
2download
0
VIRAL IMMUNOLOGYVolume 15, Number 3, 2002© Mary Ann Liebert, Inc.Pp. 399–416
Role for Heat Shock Proteins in the Immune Response toMeasles Virus Infection
MICHAEL J. OGLESBEE, MARY PRATT, and THOMAS CARSILLO
ABSTRACT
Heat shock proteins (HSPs) are recognized for their support of protein metabolism. Inter-action with viral proteins also enhances the development of innate and adaptive immune re-sponses against the infecting agent. At the level of the infected cell, HSPs are uniquely ex-pressed on the cell surface, where they represent targets of lymphokine activated killer cells.Necrosis of the infected cell releases complexes of HSP and viral protein, which, in turn,binds antigen-presenting cells (APCs). One effect of binding is to stimulate APC maturationand the release of proinflammatory cytokines, an adjuvant effect that prepares the way foradaptive immune responses. A second effect of binding is to direct the antigenic cargo of theHSP into endogenous MHC presentation pathways for priming of naive cytotoxic T cells(CTL) or activation of antigen-specific CTLs. This alternate pathway of antigen presenta-tion is essential to CTL priming following primary brain infection. Using heat shock to el-evate brain levels of HSP in a mouse model of measles virus (MV) persistent infection, weprovide evidence supporting a role for HSPs in promoting cell-mediated viral clearance frombrain. The findings highlight the probable relevance of HSPs to anti-MV immunity, sug-gesting novel routes of both therapeutic intervention and preventative measures.
INTRODUCTION
GENERAL PROPERTIES of heat shock proteins. Cells respond to protein-denaturing injuries such as hy-perthermia by the selective production of heat shock proteins (HSPs). These HSPs promote cell sur-
vival by maintaining solubility of denatured protein, which they do by preventing the interaction of exposedhydrophobic domains that would lead to the formation of large insoluble complexes. The term “proteinchaperone” was initially coined to reflect this functional attribute of HSPs, to constrain the desire of twoproteins from coming together in an inappropriate manner. Interactions between HSP and the denaturedprotein substrate allow the latter to reacquire the native conformation, whereas prolonged HSP–substrateinteractions typical of irreversible denaturation result in trafficking to proteasomal or lysosomal degrada-tive pathways. These same types of HSP–target interactions also support maturation of nascent protein ob-served in cells under steady state conditions or those undergoing sudden increases in the level of proteinproduction (e.g., cells undergoing metabolic activation). Expression profiles of HSP reflect cellular demandsfor these functions, with HSP production being constitutive and/or inducible. The fundamental role HSPs
399
Department of Veterinary Biosciences, The Ohio State University, Columbus, Ohio.

play in cellular protein metabolism is reflected in both their abundance, where HSPs represent 2–15% ofintracellular protein, and the exceptional degree of sequence conservation amongst HSPs of both eukary-otes and prokaryotes. The determinants of HSP–target interaction that support protein metabolism also im-pact a great diversity of other cellular functions, functions that may be common amongst or specific to par-ticular classes of HSPs defined by mass or cellular compartmentalization. This review focuses upon HSPswhose function, abundance, and/or degree of sequence conservation influences the development of immuneresponses to cellular or viral targets (Table 1).
The best-characterized HSPs belong to the 60-, 70-, and 90-kDa families and include the major inducible70-kDa HSP (hsp72), the constitutively expressed 70-kDa HSP (hsp73, also known as hsc70), hsp90, andhsp60. Hsp72, hsc70, and hsp90 exhibit both cytoplasmic and nuclear localization, whereas hsp60 is pre-dominantly located within mitochondria. The 60- and 70-kDa HSPs act cooperatively to provide the chap-erone function essential to protein folding (31). This function reflects a propensity of the HSPs to bindpatches of surface hydrophobicity on target proteins. For 70-kDa HSPs, protein-binding is mediated by acleft exhibiting broad substrate recognition for linear sequences of seven to eight amino acids, where bind-ing constraints are mediated by the distribution of hydrophobic side groups and the nature and quantity ofcharged groups (23). Similar characteristics define the interaction between peptide and major histocompat-ibility complex (MHC) antigens. However, peptide binding by HSP differs from MHC in that affinity forthe substrate changes dramatically in an ATP-dependent manner. The 60- and 70-kDa HSPs have a nu-cleotide binding site and exhibit ATPase activity. For hsp70 bound to ATP, the peptide binding pocket isopen, a conformation that promotes rapid dissociation/association with ligand (52). Conversion of ATP toADP causes closure of the pocket and retention of peptide, with ADP–ATP nucleotide exchange favoringrepeated cycles of binding–release. It is this cycle of binding–release that allows HSPs to occupy exposedhydrophobic patches that mediate undesirable protein aggregation, while allowing these same protein do-mains to engage in more stable intramolecular hydrophobic interactions that contribute to native proteinconformation.
Protein conformation is also altered as a direct consequence of HSP binding, providing the driving forcerequired for protein maturation and renaturation. The major difference between 60- and 70-kDa HSPs isthat the latter act as monomers or oligomers on discrete protein segments, whereas 60-kDa HSPs aggregateto form barrel-like structures inside of which more global folding events are chaperoned. These features ofHSP–protein interaction influence much more than just protein folding. HSPs chaperone the conformational
OGLESBEE ET AL.
400
TABLE 1. CELLULAR CHAPERONE PROTEINS IMPLICATED IN IMMUNE RESPONSES
CellGroup as defined Chaperone Alternate Cellular MV surfaceby mass member nomenclature localization induction expressiona
90 kDa Hsp90 Cytosol 6 1
Grp94 Gp96 ER lumen 1 1
Calnexin p88 ER membrane 1 2
70 kDa Hsp70 Hsp72 Cytosol, nucleus 1 1
Hsc70 Hsp73 Cytosol, nucleus 2 NDGrp78 BiP ER lumen 1 1
50–60 kDa Hsp60 Mitochondria 2 1
Calreticulin ER lumen 1 1
(60 kDa)Protein disulfide ER ND 1
isomerase (PDI;55 kDa)
aThe potential for cell-surface expression is based primarily upon analysis of virus-infected or neoplastic cells, sug-gesting that cell-surface expression is a profile unique to abnormal cells.
ND, not determined.

changes that attend transmembrane protein transport (e.g., protein transport into mitochondria), assemblyof otherwise insoluble protein monomers into multimeric complexes, and trafficking of binding partnersinto cell compartments such as the nucleus, this trafficking mediated by localization motifs on the HSP.The binding of HSP to accessible patches of hydrophobicity on soluble native protein may also alter theactivity of that substrate, a phenomenon known as HSP-mediated activity control (26). Binding between90-kDa HSPs and substrate is also ATP-dependent, and binding mediates conformational changes in thesubstrate. However, in contrast to 60- and 70-kDa HSPs, the impact of 90-kDa HSP on substrate functionand cellular distribution eclipses its role in protein maturation (71). The cytosolic 90-kDa HSPs are thusbest known for their role in signal transduction mediated by interactions with cellular kinases and tran-scription factors.
The 70- and 90-kDa HSP family members are also localized to the endoplasmic reticulum (ER) and pro-vide the same chaperone function outlined for their cytoplasmic counterparts. Glucose deprivation induces70- and 90-kDa HSPs in the ER due to the impact of this treatment on glycoproteins whose maturation isglycosylation-dependent. Accordingly, these HSPs are known as glucose regulated proteins (grps). Promi-nent members include grp78, also known as BiP and a member of the 70-kDa family, and grp94, a 90-kDafamily member alternatively referred to as gp96. Retention in the ER is mediated by the highly conservedC-terminal amino acid sequence KDEL and a KDEL receptor. Substrate binding determinants for grp78and grp94 are similar to that outlined for hsp70 and hsp90, as is the tendency for grp94 to function as anoligomer (45). Their broad substrate recognition and ability to reversibly bind peptides has led to the sug-gestion that these chaperones, particularly grp94, play either a direct role in MHC I peptide charging orserve as antigen presenting molecules in their own right. In support of this view, the interaction betweengrp94 and an immunodominant peptide peptide ligand from vesicular stomatitis virus was used to identifyand characterize the binding domain of grp94, demonstrating structural similarities with MHC I based uponsurface probability plots and electrostatic surface potential distribution (44). Moreover, grp94-peptide bind-ing can be dependent upon the MHC I transporter associated with antigen processing (TAP) complex, al-though TAP-independent binding is also observed (3). Although ATP binding and ATPase activity are present in grp94 (43), binding reactions involving VSV peptides do not appear to be influenced by theseproperties (90).
Other ER peptide binding chaperones include protein disulfide isomerase (PDI), calreticulin, and cal-nexin. PDI catalyzes the formation and rearrangement of disulfide bones within ER proteins, thereby func-tioning as a folding catalyst. In addition, PDI diminishes aggregation of ER proteins by acting as a chap-erone and, like BiP or grp94, exhibits broad substrate recognition (80). Calreticulin is a lectin-like chaperonewith Ca21 binding activity, serving a role in both ER Ca21 homeostasis and the maturation of a broad ar-ray of glycoproteins (51). Despite a binding preference for glycosylated substrates (80), in vitro analysesshow that calreticulin suppresses the aggregation of both glycosylated and nonglycosylated proteins in anATP-dependent manner, features of a classical molecular chaperone (75). Calnexin is an integral type Imembrane protein dedicated to the retention, in the ER lumen, of unfolded or unassembled N-linked gly-coproteins. Together with calreticulin, calnexin supports assembly of MHC I-b2 microglobulin dimers priorto association with the TAP complex and subsequent peptide loading (74). It is the TAP complex—con-sisting of calreticulin, calnexin, TAP transporters TAP1 and TAP2, tapasin, class I heavy chain, and b2 mi-croglobulin—that mediates peptide transport from the cytosol to the ER lumen for MHC charging with asubset of the transported peptides. Thus, the function of ER chaperones is diverse and includes direct sup-port of folding pathways for proteins/glycoproteins, peptide trafficking, and MHC loading.
Measles virus–heat shock protein interaction. Measles virus infection induces both cytosolic and ER lu-menal molecular chaperones (Table 1). Induction of hsp70 is one of the earliest changes in cellular gene ex-pression following infection with both RNA and DNA viruses. It is not a general response of the cell to thestress of infection, but rather a highly specific response mediated by unknown signaling pathways. The speci-ficity of this response is illustrated by in vitro infections with adenovirus type 5 and herpes simplex type 1,where induction of hsp70 but not hsc70 is observed, and by infections with human immunodeficiency type 1(HIV-1), where hsp70 but not hsp90 or hsp60, is observed at early times postinfection (68,89). For HIV-1,initial induction of hsp70 does not even require viral transcription, but simply the binding of gp120 to targetcell membranes (25). Later increases in hsp70 reflect the magnitude of viral gene expression and associated
HEAT SHOCK PROTEINS AND IMMUNE RESPONSE TO MEASLES VIRUS
401

cytopathic effect (89). Similar observations have been made in vitro with measles virus (MV; M.J. Oglesbeeet al., unpublished observation). Infection of murine neuroblastoma cells with the Edmonston strain of MV(Ed-MV) results in the induction of hsp70 within 24h, a time when levels of nucleocapsid protein (N) arebarely detectable (Fig. 1). After this, levels of hsp70 and N rise in parallel through 2 days postinfection (PI),the time of maximal cell-free infectious viral progeny release, and 4 days PI, the time of maximal virus-in-duced cytopathic effect. Levels of hsc70 and hsp60 are unaffected through the course of infection, and in-creases in hsp90 are inconsistent from trial to trial. Infection also fails to enhance expression of the small heatshock proteins hsp25 and a B crystallin. Canine distemper virus (CDV), a morbillivirus closely related to MV,also induces hsp70 in brain following naturally occurring infections (61). Here, induction is observed bothwithin virus-infected cells and adjacent uninfected cells. The expression of hsp70 in the latter may be medi-ated by inflammatory cytokines or contact of uninfected cells with viral proteins expressed on neighboring in-fected cells, in a manner analogous to the induction of hsp70 by HIV-1 gp120 (25).
For both MV and CDV, virus-induced hsp70 interacts with N protein and/or nucleocapsid. Nucleocap-sid represents the self-contained means of viral gene expression and is composed of genomic (or antige-nomic) RNA encapsidated by the N protein, this serving as template for the virus-encoded RNA-dependentRNA polymerase (L) and polymerase cofactor P. Dual-label confocal immunofluorescence microscopy ofhsp70 and CDV N shows colocalization of N and hsp70 within brain astrocytes, with cytoplasmic to nu-clear transport of N protein mediated by the interaction with hsp70 (61). Similar colocalization is lackingfor hsp70 and the P protein. Interactions between hsp70 and viral capsid protein have been suggested topromote nucleocapsid morphogenesis (47). However, hsp70 interacts with preformed CDV and MV nu-cleocapsid in an ATP-dependent manner, and hsp70-nucleocapsid complex formation is associated with in-creased nucleocapsid transcriptional activity, cellular levels of vial transcripts and proteins, and virus-in-duced cytopathic effect (62,87,88). Elevating hsp70 prior to infection increases hsp70-nucleocapsid complexformation, resulting in increased viral gene expression and cytopathic effect. The ability of hsp70 to in-fluence nucleocapsid transcriptional activity is consistent with HSP-dependent activity control, a phenom-enon that extends beyond the morbilliviruses. Our work with the human T-lymphotrophic virus type 1 showsthat stress response induction enhances basal viral gene expression through the core promoter region of the long terminal repeat, resulting in enhanced viral glycoprotein expression within infected cells (1,2).
OGLESBEE ET AL.
402
FIG. 1. Induction of the major inducible 70-kDa heat shock protein (hsp70) in murine neuroblastoma cells (N2A)infected with Ed-MV (MOI 5 1.0), based upon Western blot analysis of total cell protein. The top panel represents useof a primary antibody recognizing hsp70 (hsp72). Vero cell total protein was used as a positive control since Vero cellsexhibit basal expression of hsp72 as well as hsp73, typical of primate cell lines. Hsp72 is detected only within infectedN2A cells between 1 and 4 days PI. This induction parallels expression of the MV N protein, with N being barely de-tectable by 1 day PI, readily detectable at 2 days PI, the time of maximal cell-free infectious viral progeny release, withmaximal expression at 3–4 days PI. Maximal cytopathic effect was present at 4 days PI.
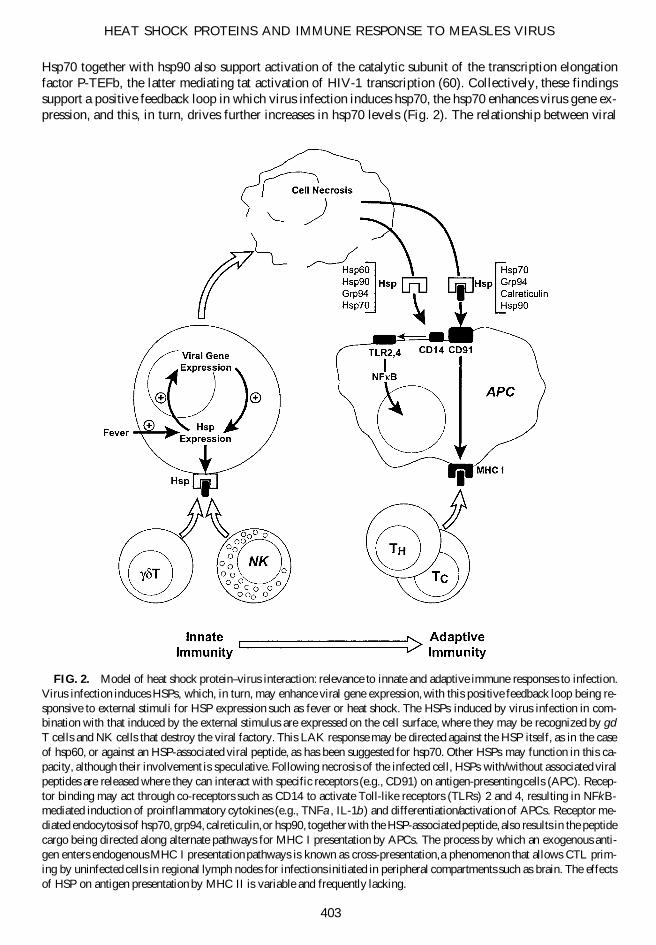
Hsp70 together with hsp90 also support activation of the catalytic subunit of the transcription elongationfactor P-TEFb, the latter mediating tat activation of HIV-1 transcription (60). Collectively, these findingssupport a positive feedback loop in which virus infection induces hsp70, the hsp70 enhances virus gene ex-pression, and this, in turn, drives further increases in hsp70 levels (Fig. 2). The relationship between viral
HEAT SHOCK PROTEINS AND IMMUNE RESPONSE TO MEASLES VIRUS
403
FIG. 2. Model of heat shock protein–virus interaction: relevance to innate and adaptive immune responses to infection.Virus infection induces HSPs, which, in turn, may enhance viral gene expression, with this positive feedback loop being re-sponsive to external stimuli for HSP expression such as fever or heat shock. The HSPs induced by virus infection in com-bination with that induced by the external stimulus are expressed on the cell surface, where they may be recognized by gd
T cells and NK cells that destroy the viral factory. This LAK response may be directed against the HSP itself, as in the caseof hsp60, or against an HSP-associated viral peptide, as has been suggested for hsp70. Other HSPs may function in this ca-pacity, although their involvement is speculative. Following necrosis of the infected cell, HSPs with/without associated viralpeptides are released where they can interact with specific receptors (e.g., CD91) on antigen-presenting cells (APC). Recep-tor binding may act through co-receptors such as CD14 to activate Toll-like receptors (TLRs) 2 and 4, resulting in NFkB-mediated induction of proinflammatory cytokines (e.g., TNFa, IL-1b) and differentiation/activation of APCs. Receptor me-diated endocytosis of hsp70, grp94, calreticulin, or hsp90, together with the HSP-associated peptide, also results in the peptidecargo being directed along alternate pathways for MHC I presentation by APCs. The process by which an exogenous anti-gen enters endogenous MHC I presentation pathways is known as cross-presentation, a phenomenon that allows CTL prim-ing by uninfected cells in regional lymph nodes for infections initiated in peripheral compartments such as brain. The effectsof HSP on antigen presentation by MHC II is variable and frequently lacking.

gene expression and hsp70 expression is responsive to hsp70 induced by external stimuli (e.g., transient ex-posure to hyperthermia). Accordingly, increasing hsp70 levels by heat shock or fever can further promoteviral gene expression. The ability of HSPs to enhance viral gene expression and cytopathic effect wouldincrease both the amount of antigenic material generated during the course of infection and the release ofimmunogenic HSP–peptide complexes from necrotic cells (7).
Measles virus infection also induces the expression of the ER chaperones grp78, grp94, calreticulin, andcalnexin (11). Induction of grp78, grp94, and calreticulin is viewed as a response to an increased burdenof unfolded proteins in the ER lumen, triggering transcription of these chaperones through a common ERstress response element (93). These chaperones are induced following infections by other viruses as well(92). The mechanism of calnexin induction is unclear, although it is also thought to be a response to un-folded proteins. The induced grp78 binds MV fusion (F) and hemagglutinin (H) glycoproteins based upontheir coimmunoprecipitation (11). Similarly, calnexin binds both F and H whereas binding to calreticulinoccurs only for the H glycoprotein. These findings are consistent with the role of ER chaperones in thestructural maturation of nascent polypeptides, where grp78 binds the polypeptide backbone and calnexinand calreticulin interact through the carbohydrate moieties of the glycoprotein. Such transient interactionshave been documented in the functional and antigenic maturation of glycoproteins for viruses from multi-ple other families and include HIV (66), rabies virus (27), and influenza virus (67).
HEAT SHOCK PROTEINS AND INNATE IMMUNITY
Heat shock proteins induced by virus infection or hyperthermic treatment can be expressed on the cellsurface, including HSPs normally localized to the ER lumen or cytosol (Table 1). This pattern of expres-sion positions the HSP to serve as either an antigen presenting molecule or primary target of lymphokineactivated killer (LAK) cells in neoplastic and virus-infected cells (14,81). Measles virus infection of humanepidermoid carcinoma cells (Hep-2) induces cell surface expression of calreticulin and grp78, overridingER retention mechanisms mediated by KDEL motifs on the HSP (11). Suggested means by which theseproteins escape the ER lumen include a virus-induced excess of calreticulin/grp78 relative to availableKDEL receptors or KDEL masking due to binding of calreticulin/grp78 to viral glycoproteins. Cell surfaceexpression of grp94 has not been examined in the context of MV infection, although cell surface expres-sion can occur, again, despite the presence of a KDEL ER localization motif (20). Calnexin is induced byMV expression but not displayed on the cell surface consistent with its being an integral ER membraneprotein. Subcellular localization of MV-induced hsp70 has not been performed. However, cell surface ex-pression of hsp70 has been demonstrated in tumor cells following thermal stress or virus infection (54),where it represents up to 15–20% of the total cellular levels of this stress protein (12,21).
Cell surface expression of hsp70 can enhance cell sensitivity to non-MHC restricted lysis by NK cells.The majority of work in this area has been performed in tumor cells, where NK-mediated tumor cell lysisis correlated to cell surface expression of hsp70 (12,70). That the phenomenon is HSP-specific is basedupon the ability of hsp70, but not hsc70, to enhance proliferation and cytolytic activity of NK cells in vitroin both the presence and absence of IL-2 (55). The relevance of NK cell responses to cell surface hsp70has been convincingly demonstrated in SCID/beige mice to which human NK cells were adoptively trans-ferred. The NK cells inhibited growth and metastasis of transplanted tumors expressing hsp70 on the plasmamembrane but not congenic tumors lacking this hsp70 expression profile (56). Much of the data from thesetumor systems suggests that killing is mediated by NK–target cell interaction through killer cell activatoryreceptors. For example, NK-mediated cell lysis can be inhibited by hsp70-specific antibody (12). However,other mechanisms of killing may also be operative. Transient heat shock or chronic HIV-1 infection en-hances hsp70-specific antibody-dependent cellular cytotoxicity in lymphoma cells, an NK cell function me-diated by the Fc receptor CD16 (21).
Cell surface expression of hsp60 mediates reactivity by gd T cells in a non–MHC restricted manner sim-ilar to that described for the interaction between NK cells and hsp70 (22,37). Although our findings sug-gest that MV infection does not induce hsp60, similar to infection by Sendai virus or vaccinia virus, in-duction is observed following infection by influenza virus (69). A gd T cell hybridoma established from
OGLESBEE ET AL.
404

influenza-infected mice responds to both recombinant hsp60 and cells productively infected with influenzaA or B, but not cells infected with vaccinia or Sendai virus, indicating that virus-induced hsp60 is the tar-get. A gd T cell hybridoma established from uninfected mice also responded to influenza virus–infectedcells, indicating that hsp60-reactive gd T cells preexist in normal mice. These populations of T cells arelikely induced and sustained by repeated recognition of epitopes on microbial hsp60 that are shared withendogenous hsp60 (37). The importance of hsp60-reactive gd T cells in protection during the early stagesof viral infection was illustrated in a murine cytomegalovirus (MCMV) infection model (59). MCMV in-fection induced an hsp60-specific gd T cell phenotype (i.e., cellular expression of Vg1), the induced cellsexpressed interferon g in response to purified hsp60, and depletion of gd T cells using an anti-T cell re-ceptor gd monoclonal antibody enhanced viral titers and decreased IFN g levels in tissues of challengedmice. The general mechanism of antiviral activity mediated by the gd T cells involves not only direct cy-tolytic activity, but also release of antiviral cytokines and chemokines based on work involving the simianimmunodeficiency virus (SIV) (42).
Whether it is the HSP or an HSP-associated peptide that is recognized by the NK and gd T cells is asubject of debate. In systems supporting NK cell lysis of tumor but not nontumor targets, HSPs do not dif-fer between tumor cells and their nontransformed counterparts (81). Thus, the enhanced susceptibility oftumor cells to lysis reflects either HSP-mediated presentation of tumor-specific peptides or the fact thatHSPs are preferentially expressed on the surface of tumor cells. Similar considerations would apply to thedifferential recognition of virus infected and uninfected cells. The potential of HSP to present peptide onthe cell surface is derived from the fact that hsp70 binds peptide and the hsp70 protein domain mediatingNK cell lysis and peptide binding both localize to the C-terminus (12). In addition, in vitro studies supportthe ability of cell surface expressed grp75, a mitochondrial 70-kDa HSP, to present antigen to LAK cells.In this work, human gd T cells that recognize an autologous B cell lymphoma are able to recognize andlyse heterologous cells transfected to express the tumor immunoglobulin lambda chain. The lambda chainis recognized as processed peptide, does not involve classical MHC molecules, but is blocked by antibod-ies against grp75 (38). Hsp70 may provide a similar antigen presentation function to generate an antigen-specific gd T cell response that contributes to mucosal immunity against SIV (42). If hsp70 can stimulateboth NK and gd T cell responses, then one must entertain the possibility that other peptide-binding HSPsmight support LAK cell stimulation when expressed on the cell surface. However, published work exam-ining the roles of grp78 (BiP), calreticulin, or grp94 in this aspect of innate immunity is lacking. Hsp60 re-quires special consideration.
Despite the fact that hsp60 reversibly binds protein, it does not exhibit the peptide binding activity typ-ical of hsp70, suggesting that it is recognition of the HSP per se that is responsible for driving the hsp60-dependent immune responses. In fact, hsp60 gd T cell responses can be stimulated in vivo through the simple administration of peptides based upon hsp60 sequence, these responses being independent of anti-gen-specific ab T cell reactivity (24). Such recognition is used to explain how molecular mimicry betweenviral structural proteins and hsp60 could give rise to autoimmune disease (30). Mechanisms by which cell-surface HSPs are recognized by LAK cells need not be mutually exclusive, so that epitopes shared betweenMV F glycoprotein and a 79-kDa HSP have the potential to drive autoimmune reactions (77). The identityof this 79-kDa HSP was not originally confirmed, although consistent with grp78 based upon its localiza-tion to the cytoplasm and induction with treatments that inhibit the maturation of glycoproteins, includingtunicamycin and 2-deoxyglucose.
Death of the infected cell represents the second phase in which innate immune responses are elicited.Basu and others showed that necrotic but not apoptotic cell death leads to the release of grp94, calreticulin,hsp70, and hsp90 (7). Murine dendritic cells exposed to grp94, hsp90, or hsp70 are stimulated to releasethe proinflammatory cytokines TNF-a and IL-1b, in addition to IL-12, a cytokine promoting the develop-ment of subsequent TH1 adaptive immune responses. Also induced are MHC II and the costimulatory mol-ecule B7-1 (CD80), the latter required for activation of naive T cells. Differences in the relative activitiesof grp94, hsp70, and hsp90 are observed, with the lowest activity exhibited by hsp70. However, estimatesof HSP concentration released from necrotic cells supports the relevance of each in acting as an endoge-nous stress signal to antigen presenting cells, with hsp70 and hsp90 being released in greatest abundance.Hsp70, but not hsc70, also induces maturation of human dendritic cells from monocyte precursors, and this
HEAT SHOCK PROTEINS AND IMMUNE RESPONSE TO MEASLES VIRUS
405

maturation is associated with enhanced ability to present peptide antigen to specific cytotoxic lymphocytes(CTLs) (40). A similar stimulation of CTL activity is observed following treatment of peritonealmacrophages with hsp60 (53), with this activity being correlated to the induction of IL-12 but not antigen-specific IL-2 production or T cell proliferation. These findings highlight the role of HSPs in stimulating innate responses in antigen presenting cells (APCs) that, in turn, promote the development of adaptive immune responses. In this capacity, HSPs function as adjuvants. The APCs involved in this process includethose found in the central nervous system, with phagocytic activity and cytokine release being observed inhuman microglia following treatment with hsp70 or hsp90 (36).
The HSP-mediated stimulation of APCs is mediated by activation of one or more of the Toll-like re-ceptors (TLRs). Toll-like receptors are the human homologue of Drosophila Toll protein and belong to theIL-1 receptor family, utilizing a signaling pathway that involves the IL-1 receptor–associated kinase andNF-kB. A constitutively active mutant of human Toll transfected into human cells causes the NFkB-de-pendent expression of IL-1, IL- 6, IL-8, and the costimulatory molecule B7-1 (CD80) (49). Stress signal-ing through TLRs was first shown for hsp60, and the specific contribution of the TLR-4 was establishedin macrophages deficient in TLR-4, where macrophages failed to mount a proinflammatory response fol-lowing hsp60 treatment (29,65). A second TLR (TLR-2) is involved in hsp60 signaling, and both TLR-2and TLR-4 also transduce the proinflammatory signal elicited by hsp70 and hsp90 (5,86).
Although these TLRs mediate signaling, they are unlikely to be primary mediators of HSP binding. Bind-ing of hsp60 to mouse macrophages or hsp70 to human monocytes was independent of TLR-4 expression,occurred at submicromolar concentrations, and was responsive to competitive inhibition, characteristic of clas-sical ligand–receptor interactions (29,46). These cell surface receptors may be HSP-specific or common toseveral HSPs. Examination of the binding reaction between hsp60 and mouse macrophages suggests that thehsp60 receptor is distinct from that of hsp90 and hsp70, since excess of hsp90 and hsp70 cannot competitivelyinhibit hsp60 binding (29). Specific receptor utilization may vary according to species, based upon analysisof human monocytes, where it appears that hsp60 and hsp70 share a common receptor (46). Regardless ofspecies, the a2 macroglobulin receptor CD91 serves as a common binding site for hsp70, hsp90, grp94, andcalreticulin, where binding results in CD91 receptor mediated endocytosis of the HSP ligand (6). Co-recep-tors such as CD14 also contribute to proinflammatory signaling by both hsp70 and hsp60 (4). A picture thusemerges wherein multiple receptors support HSP–target cell interactions, receptors that are either HSP-spe-cific or inclusive, receptors that may differ in their affinity for the HSP ligand and that may function throughco-receptors. It is the interaction with co-receptors that may provide the link between HSP binding to CD91and stimulation of TLRs with NFkB activation. The importance of this signaling pathway to innate immunityagainst virus infection was recently illustrated using a mouse model of respiratory syncytial virus (RSV) in-fection. Infected mice deficient in TLR-4 had deficient NK cell pulmonary trafficking and function, decreasedmonocyte pulmonary trafficking, impaired IL-12 production, and impaired viral clearance relative to RSV-challenged mice expressing TLR-4 (32). It is postulated that the RSV F glycoprotein serves as ligand for TLRsignaling in this system, although a role for HSPs has not been investigated.
HEAT SHOCK PROTEINS AND ADAPTIVE IMMUNITY
The classical paradigm of antigen presentation involves processing of endogenous and exogenous anti-gen along distinct presentation pathways (13). For the expression of endogenous antigen, peptide fragmentsare generated in the proteasome and transported into the lumen of the ER by the TAP complex. Here, asubset of the peptide pool combines with MHC I for subsequent cell surface expression and priming ofCTL in lymphoid organs or recognition by antigen-specific CTL in peripheral sites. Exogenous antigen, onthe other hand, is phagocytosed and processed in the lysosome, with derivative peptides presented in thecontext of MHC II for CD41 T helper cell recognition and subsequent humoral immune responses. It is be-coming increasingly clear, however, that these pathways are not mutually exclusive and that exogenousantigens can be internalized by APCs and expressed on MHC I for CTL priming and/or recognition by anti-gen-specific CTLs (82). The process is referred to as cross-priming or cross-presentation, in contrast to di-rect presentation that follows the classical endogenous pathway (16).
OGLESBEE ET AL.
406

Cross-presentation is effectively illustrated using tumor-derived hsp70 and grp94 to induce anti-tumorCTL responses. Hsp70-peptide complexes isolated from Meth A sarcoma cells can induce an immune re-sponse that protects mice against subsequent challenge with the same neoplastic cells from which the HSPswere isolated (84). The protection is tumor-specific, HSP-dosage dependent, and associated with tumor-specific CTL responses. Priming of the latter is independent of CD41 T cells but dependent uponmacrophages; depletion of macrophages by treatment of mice with carrageenan during the priming phaseresults in loss of the HSP-dependent immune response (85). Immunogenicity is also observed for hsp90isolated from tumor cells, although differences in the degree of activity relative to other HSPs are observed.Peptides bound to either grp96 or hsp70 are highly and equally immunogenic whereas the activity supportedby hsp90 is approximately 90% less (85). The basis for the latter is unclear, and Udono and others suggestthat it may reflect the low ATPase activity of hsp90, a characteristic that could influence peptide bind-ing/exchange reactions. Heat shock proteins from non-tumor cells do not elicit these protective responses,and the immunogenicity of hsp70 is lost when peptide is first dissociated by treating the samples with anexcess of ATP, proving that it is the hsp70-associated peptide and not hsp70 that mediates the effect (84,85).These findings illustrate the inherently poor immune response to free peptide.
The basis for lack of immunogenicity of free peptide appears, at least in part, to be a function of intra-cellular processing. Introduction of free antigenic peptides or peptides complexed with serum albumin intothe cytosol are poorly acquired by MHC I molecules, in contrast to peptides bound to grp94, hsp90, orhsp70 (8). Furthermore, treatments that sequester intracellular pools of hsp90 and hsp70, such as adminis-tration of the immunosuppressant deoxyspergualin, eliminates HSP-mediated MHC I expression of pep-tides, an effect that can be reversed with supplemental hsp70. The effect of HSPs on trafficking antigeninto this alternate presentation pathway primarily influences MHC I loading, with variable if any impactupon MHC II. This differential effect on MHC presentation pathways is illustrated by use of a DNA vac-cine encoding a nonstructural protein of Japanese encephalitis virus. The construct fails to elicit strong cel-lular immunity in mice whereas co-administration of a plasmid encoding hsp70 enhances both T cell pro-liferation and cytotoxic effects but not anti-viral antibody responses (18). These hsp70-mediated effectsrender the vaccine protective against lethal viral challenge. Similar results were obtained using grp94 iso-lated from cells infected with a Semliki Forest virus replicon expressing influenza nucleocapsid protein(NP). The grp96 preparation induced cellular but not humoral immune responses (33).
Immunogenic HSP–peptide complexes can also be generated in vitro, providing the most compelling ev-idence that HSPs direct exogenous peptide antigens into alternate pathways of MHC I presentation. In thecontext of antitumor immunity, both hsp70 and grp94 bind a wide variety of synthetic peptides with thebroad yet selective substrate recognition characteristic of HSPs. These HSP–peptide complexes elicit pep-tide-specific CTL responses, where it has been shown that grp94–peptide complexes formed in vitro aretaken up by APCs and the peptide expressed in a manner identical to that of grp94–peptide complexesformed within the cell (10). The approach can be exploited in viral systems, where immunodominant pep-tide epitopes of both lymphocytic choriomeningitis virus and herpes simplex virus have been used to in-duce protective cell-mediated immunity when presented as complexes with hsp70 (19,39). Bacterial ana-logues of hsp70 have also been used to enable MHC I presentation of exogenous peptide antigen. Complexesbetween an immunodominant peptide from the influenza NP, bound to mycobacterial hsp70, induce pep-tide-specific CTL responses in mouse spleen that were not observed with hsp70 alone (73).
The ability to form immunogenic HSP–peptide complexes is not restricted by the MHC haplotype of thecell from which the HSP–peptide complex is isolated (48). However, peptide presentation by MHC I is stillrestricted to the structural limitations of that haplotype of MHC. The term “cross-presentation” is thus alsodefined by differential MHC restriction, illustrated by documenting the associations between an immuno-dominant peptide of vesicular stomatitis virus (VSV) and grp94. The peptide is naturally presented by theH-2Kb haplotype of MHC I yet it associates with grp94 in VSV infected cells regardless of MHC haplo-type of that host cell (58). Given the broad substrate recognition of HSPs, the HSP-bound peptides suitablefor assembly onto a specific class I haplotype represent only a subset of the total number of HSP-interact-ing species. It is easy to see how cytosolic chaperones might have access to any or all antigenic peptidesformed within that compartment, but the ER lumenal HSPs could theoretically be limited to peptides trans-ported by the TAP complex. Isolating grp94 from cells with and without a functional TAP defect and us-
HEAT SHOCK PROTEINS AND IMMUNE RESPONSE TO MEASLES VIRUS
407

ing the resultant grp94 as immunogen in mice disproved this view (3). Results showed that peptide asso-ciation with grp94 was both TAP-dependent and TAP-independent, with this being in part a function of theprotein yielding the antigenic peptides. It thus appears that grp94 can associate with all ER lumenal pep-tides independent of transport mechanism.
Other HSPs released from necrotic cells can mediate cross-presentation. Recent work shows that cal-reticulin binds peptide from numerous tumor cells or stably transfected cells expressing ovalbumin, and thatcalreticulin–peptide complexes elicit tumor specific or ovalbumin-specific CTL responses in mice (57). Theimmunogenicity of the calreticulin–peptide complexes are comparable to grp94, whereas little to no activ-ity is observed for the ER chaperones grp78 (BiP) or PDI. Collectively, it appears that grp94, calreticulin,and hsp70 can play important roles in cross-presentation, with lesser activity supported by hsp90 and neg-ligible activity supported by PDI or grp78. Notably absent from this list are hsp60 and hsc70. Hsp60 lacksthe peptide binding activity required to support cross-presentation. However, the basis by which hsc70 isnot included is not immediately apparent. This constitutively expressed 70-kDa HSP exhibits peptide-bind-ing reactions similar to that described for hsp70, suggesting that the difference must lay in cellular uptakeand/or intracellular processing of the HSP–peptide complex.
Heat shock protein–dependent trafficking of exogenous antigen into endogenous MHC I presentationpathways involves HSP receptor mediated endocytosis. This mode of entry is contrasted to other postulatedmechanisms of exogenous antigen processing (16). These other mechanisms include phagocytosis of anti-gen that is then degraded, and the peptide fragments released (i.e., regurgitated) for association with cellsurface MHC. Alternatively, phagocytosed antigen is degraded, and peptide fragments exit the endosometo directly enter MHC I presentation pathways by unknown means. Finally, peptides may enter antigen pre-senting cells by a process called macropinocytosis, where fluid phase uptake bypasses endosomal com-partments for direct entry into antigen presenting pathways. Using grp94 as the peptide carrier, it has beenshown that only professional APCs such as dendritic cells and macrophages of humans and mice bind grp94,and that receptor mediated endocytosis of the grp94–peptide complex is required for peptide processing bythe alternate MHC presentation pathway (78). HSP–peptide complexes internalized nonspecifically do notresult in cross-presentation, and adding an excess of grp94 lacking relevant peptide can competitively in-hibit the process. The receptor mediating grp94-peptide transport is the a2 macroglobulin receptor CD91,and grp94-mediated cross-presentation can be competitively inhibited using a2 macroglobulin (9). The in-ternalized grp94 is localized to the early endosome but does not reach the stage of the lysosome (78). Theintracellular pathway for presentation beyond this point involves two possibilities. First, like MHC II mol-ecules, MHC I can be internalized and bind antigen in endosomal compartments, providing a means bywhich to load exogenous antigen (28). Co-localization of internalized grp94 and MHC I within endosomalcompartments supports this view (78). Alternatively, grp94-antigen complexes may enter the cytosol di-rectly from the early endosome, analogous to the fate of antibody-antigen complexes following endocyto-sis through Fc receptors in dendritic cells (72). In this latter pathway, endosome-to-cytosol transport is re-stricted to dendritic cells, specific to internalized antigens and selective for the size of the transportedmolecules. Support for the latter pathway is based upon the observation that presentation of the grp94-com-plexed peptide requires a functional proteasome complex, since the proteasome inhibitor lactacystin inhibitspeptide presentation (6). Also required is a functional TAP complex, based upon the observation that APCsisolated from TAP1/1 mice treated with grp96–antigen complexes could stimulate CD81 antigen-specificT cell lines, whereas APCs isolated from TAP2/2 could not. However, the observed dependence of anti-gen presentation on TAP may be a function of the antigen used in the experimental systems (3).
It is assumed that peptide cross-presentation mediated by hsp70 and calreticulin utilize similar intracel-lular pathways, since they use the same portal of entry, that is, receptor mediated endocytosis through CD91(6). For hsp70, it has been shown that receptor-mediated endocytosis of hsp70–peptide complexes utilizesboth intracellular presentation pathways described for grp94: a cytosolic proteasome- and TAP-dependentroute and an endosomal proteasome- and TAP-independent route (17). The pathway used is determined bythe peptide cargo of the HSP, and may relate to the relationship of the peptide’s MHC I binding sequenceto the surrounding protein structure. This was demonstrated by analyzing functional hsp70-mediated pre-sentation of synthetic peptide antigens, making use of proteasome inhibition with lactacystin, and TAP2/2or TAP1/1 macrophages. The antigen was an ovalbumin epitope presented by H-2Kb MHC haplotypes.
OGLESBEE ET AL.
408

The peptide antigen was extended on either the amino (N) or carboxy (C) terminus by adding sequenceknown to associate with hsp70. The epitope is efficiently processed in an endosomal (i.e., proteasome andTAP-independent) route when it occupies the extreme C-terminus, whereas processing is proteasome andTAP-dependent when the C-terminus of the epitope is internal. Mechanistic insights have also been gainedby examining the immunogenicity of fusion constructs between hsp70 and peptide. Using separate domainsof the mycobacterial hsp70 as a fusion partner, it was shown that the ability to induce CD81 CTLs inde-pendent of CD4 help in mice did not require the C-terminal domain that normally provides chaperone func-tion and peptide binding by the intact hsp70 molecule; intracellular trafficking is dictated by N-terminal se-quences (35). Although the hsp70-dependent cross-presentation targets endogenous MHC I pathways, thereare exceptions where MHC II–dependent humoral immune responses are also observed. This was shownby vaccinating mice with a fusion construct consisting of the mycobacterial hsp70 coupled to the HIV cap-sid protein p24. Vaccinated mice elicited both a CTL and humoral responses to p24 for over a year, withno corresponding immunity evoked in mice injected with soluble p24 alone (83). Priming of both endoge-nous and exogenous pathways would seem to be most likely for loading of recycled MHC in the endoso-mal compartment. Support for endosomal MHC loading comes from grp94-mediated peptide presentation,where colocalization of grp94, MHC I, and MHC II to the early endosome was shown (78).
HEAT SHOCK PROTEINS AND IMMUNITY TO MEASLES VIRUS
A working model summarizing the impact of HSPs on antiviral immunity whereby HSPs promote innateimmune responses against the virus-infected cells and also the subsequent development of adaptive immuneresponses through their effects on APCs is presented in Figure 2. HSPs expressed on the surface of virusinfected cells trigger NK and gd T cell responses, whereas HSPs released following necrosis interacts withTLR receptors on APCs to induce cytokines and costimulatory molecules needed to initiate the adaptiveimmune response. Heat shock proteins acting in this latter manner may be viewed as innate immune me-diators sending danger signals as to the presence of stressed or damaged (e.g., virus-infected) cells. Themacrophage/dendritic cell response is innate in that it represents activation through nonspecific recognitionsystems, and is not enhanced by repeated exposure to the stimulus. In this manner, HSPs with or withoutpeptide cargo function as adjuvants, similar to bacterial lipopolysarccharide. Effects of TLR signaling mayreduce dependency of CTL priming and/or activation on CD41 T cell help so that a much greater range ofcell types can act as APCs (50), including microglia of the central nervous system (CNS).
Receptor-mediated endocytosis of HSP–antigen complexes by APCs results in processing of the peptideantigen by alternate pathways for presentation by MHC I, stimulation both CTL priming and reactivation.This alternate pathway likely plays a significant role in the induction of antiviral CTLs since it allows anti-gen presentation by uninfected cells. Cross-presentation may be particularly important in peripheral com-partments such as brain since T cell priming is restricted to lymphoid organs (41). Naive T cells traffic be-tween secondary lymphoid organs but not into peripheral nonlymphoid tissues. Priming must take place inregional lymph nodes and spleen as only primed T cells are known to traffic into peripheral tissue (15). Inthe event of a primary brain infection, both cell debris and mononuclear cells enter the cerebrospinal fluidvia the perivascular space (i.e., space of Virchow-Robin), and from there enter either the systemic circula-tion through arachnoid granulations or regional lymphatics via spinal nerve roots. Material thus passes tothe spleen and cervical lymph nodes where priming events can take place. In these lymphoid organs, par-ticulate antigen can mediate cross-presentation in resident APCs and both resident and brain-derived APCswill have the necessary access to naive T cells. Relative to other means of uptake of exogenous antigen,the HSP-dependent alternate pathway operates efficiently at low concentrations of antigen; hsp70-receptoraffinity is in the submicromolar range of binding affinity (79). The integral role of HSPs in such a host de-fense mechanism is suggested by the fact that a locus encoding the major inducible 70-kDa HSP (hsp70)is located in the class III region of the human MHC complex (76).
This model could be exploited therapeutically to enhance clearance of persistent virus infection of brain.Transient hyperthermia is a potent inducer of hsp70 both in vitro and in vivo. In cell culture, our work withMV and CDV has shown that transient heat stress prior to lytic infection or during persistent infection re-
HEAT SHOCK PROTEINS AND IMMUNE RESPONSE TO MEASLES VIRUS
409
sults in increased viral gene expression and increased physical association between hsp70 and nucleocap-sid-associated viral structural proteins (62,64,87,88). Heat treatment would thus enhance the generation ofimmunogenic complexes between hsp70 and viral peptide antigens. Using whole body hyperthermia in thedog, we have also shown induction of elevated hsp70 protein levels in the brain, with immunohistochem-ical staining showing that induced hsp70 is localized to neurons as well as glia (63). Thus, heat shock in-duction of hsp70 in brain may enhance both innate immune responses against MV or CDV infected cellsand also promote priming of CTLs responsible for viral clearance (91).
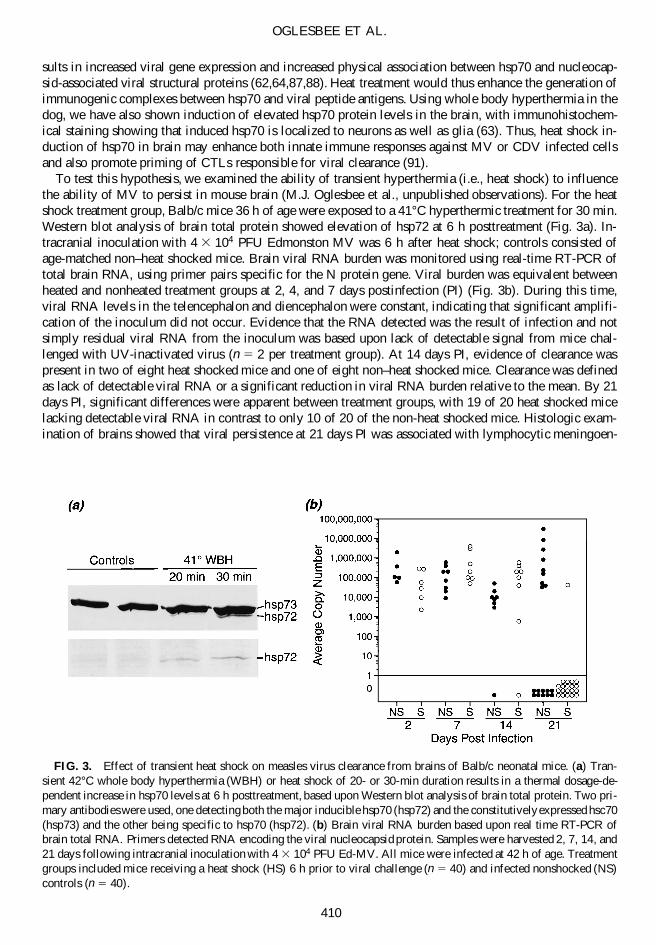
To test this hypothesis, we examined the ability of transient hyperthermia (i.e., heat shock) to influencethe ability of MV to persist in mouse brain (M.J. Oglesbee et al., unpublished observations). For the heatshock treatment group, Balb/c mice 36 h of age were exposed to a 41°C hyperthermic treatment for 30 min.Western blot analysis of brain total protein showed elevation of hsp72 at 6 h posttreatment (Fig. 3a). In-tracranial inoculation with 4 3 104 PFU Edmonston MV was 6 h after heat shock; controls consisted ofage-matched non–heat shocked mice. Brain viral RNA burden was monitored using real-time RT-PCR oftotal brain RNA, using primer pairs specific for the N protein gene. Viral burden was equivalent betweenheated and nonheated treatment groups at 2, 4, and 7 days postinfection (PI) (Fig. 3b). During this time,viral RNA levels in the telencephalon and diencephalon were constant, indicating that significant amplifi-cation of the inoculum did not occur. Evidence that the RNA detected was the result of infection and notsimply residual viral RNA from the inoculum was based upon lack of detectable signal from mice chal-lenged with UV-inactivated virus (n 5 2 per treatment group). At 14 days PI, evidence of clearance waspresent in two of eight heat shocked mice and one of eight non–heat shocked mice. Clearance was definedas lack of detectable viral RNA or a significant reduction in viral RNA burden relative to the mean. By 21days PI, significant differences were apparent between treatment groups, with 19 of 20 heat shocked micelacking detectable viral RNA in contrast to only 10 of 20 of the non-heat shocked mice. Histologic exam-ination of brains showed that viral persistence at 21 days PI was associated with lymphocytic meningoen-
OGLESBEE ET AL.
410
FIG. 3. Effect of transient heat shock on measles virus clearance from brains of Balb/c neonatal mice. (a) Tran-sient 42°C whole body hyperthermia (WBH) or heat shock of 20- or 30-min duration results in a thermal dosage-de-pendent increase in hsp70 levels at 6 h posttreatment, based upon Western blot analysis of brain total protein. Two pri-mary antibodies were used, one detecting both the major inducible hsp70 (hsp72) and the constitutively expressed hsc70(hsp73) and the other being specific to hsp70 (hsp72). (b) Brain viral RNA burden based upon real time RT-PCR ofbrain total RNA. Primers detected RNA encoding the viral nucleocapsid protein. Samples were harvested 2, 7, 14, and21 days following intracranial inoculation with 4 3 104 PFU Ed-MV. All mice were infected at 42 h of age. Treatmentgroups included mice receiving a heat shock (HS) 6 h prior to viral challenge (n 5 40) and infected nonshocked (NS)controls (n 5 40).

cephalitis, neuronal necrosis, and reactive gliosis. Histologic changes in heat shocked mice were restrictedto mild lymphocytic inflammatory infiltrates of the meninges.
At the same time points in which viral RNA burden was examined, splenic lymphocytes were isolatedand tested for blastogenic responsiveness to gradient purified MV antigen. Serum was harvested and MV-specific immunoglobulin measured in an enzyme-linked immunoadsorbant assay. The temporal onset ofclearance (i.e., 14 days PI) was correlated to the detection of MV-specific splenocyte blastogenesis. No re-sponses were observed at 7 days PI, whereas five of eight heat shocked mice and three of eight nonshockedmice supported MV-specific responses. All mice from both treatment groups exhibited responses at 21 daysPI. Production of MV-specific antibody was not correlated to clearance with all mice from both treatmentgroups producing antibody at 14 days PI.
Collectively, our results show that a heat shock capable of inducing hsp70 in brain promotes MV clear-ance and that this clearance is correlated to the onset of MV-specific cell-mediated immune responses. Lackof difference in viral burden between treatment groups at 2–14 days PI suggests that the effects of hsp70or other heat-induced HSPs on innate immune recognition of virus-infected cells was negligible. IncreasedHSP levels in the context of viral infection would promote cross-priming of CTL responses, and our re-sults are consistent with the ability of HSPs to support such responses. Since brain was the primary site ofinfection, cross-presentation must underlie the development of cell-mediated responses known to clear MVfrom this peripheral compartment. Greater mechanistic insights will require that we evaluate qualitative dif-ferences in the cell mediated immune responses that can effect such significant differences in the rate ofviral clearance, and that we define the specific role of hsp70 relative to other heat-induced HSPs in pro-moting cross-presentation. That HSPs are at least in part responsible for the differential rate of viral clear-ance is supported by the observation that the brain is intrinsically thermotolerant, such that the response totransient hyperthermia is elevated tissue levels of HSPs but not alteration in cytokine production, MHC ex-pression, blood–brain barrier permeability, or other evidence of structural and/or functional alterations (63).In the context of viral pathogenesis, these observations support the importance of fever as an antiviral de-fense mechanism, inducing HSPs that can facilitate antiviral immune responses in uninfected cells.
Despite the wealth of evidence suggesting a role for HSPs in MV pathogenesis and/or immunity, thereis only a limited amount of available information on this topic. Basic questions such as the role of feverand fever-induced HSPs on the induction of anti-MV immunity, MV-induced immune suppression, or MV-mediated autoimmune reactions have yet to be addressed, nor has the significance of an afebrile state inpromoting the establishment of viral persistence or in limiting the response to vaccination been investigated.Thus, the probable biological significance of MV-HSP interaction represents fertile ground for future re-search that should yield tangible benefits in terms of therapeutic and preventative measures.
ACKNOWLEDGMENTS
Data presented in this manuscript was based upon work supported by a grant from the National Instituteof Neurological Disorders and Stroke (R01 NS31693). Data from serologic analyses presented in this re-view were performed in the laboratory of Dr. Stefan Niewiesk.
REFERENCES
1. Andrews, J.M., G.C. Newbound, M. Oglesbee, et al. 1997. The cellular stress response enhances human T-celllymphotropic virus type 1 basal gene expression through the core promoter region of the long terminal repeat. J.Virol. 71:741–745.
2. Andrews, J.M., M.J. Oglesbee, A.V. Trevino, et al. 1995. Enhanced human T-cell lymphotropic virus type I ex-pression following induction of the cellular stress response. Virology 208:816–820.
3. Arnold, D., C. Wahl, S. Faath, et al. 1997. Influences of transporter associated with antigen processing (TAP) onthe repertoire of peptides associated with the endoplasmic reticulum-resident stress protein gp96. J. Exp. Med.186:461–466.
HEAT SHOCK PROTEINS AND IMMUNE RESPONSE TO MEASLES VIRUS
411

4. Asea, A., S.K. Kraeft, E.A. Kurt-Jones, et al. 2000. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 6:435–442.
5. Asea, A., M. Rehli, E. Kabingu, et al. 2002. Novel signal transduction pathway utilized by extracellular HSP70:Role of TLR2 and TLR4. J. Biol. Chem. 277:15028–15034.
6. Basu, S., R.J. Binder, T. Ramalingam, et al. 2001. CD91 is a common receptor for heat shock proteins gp96, hsp90,hsp70, and calreticulin. Immunity 14:303–313.
7. Basu, S., R.J. Binder, R. Suto, et al. 2000. Necrotic but not apoptotic cell death releases heat shock proteins, whichdeliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int. Immunol.12:1539–1546.
8. Binder, R.J., N.E. Blachere, and P.K. Srivastava. 2001. Heat shock protein–chaperoned peptides but not free pep-tides introduced into the cytosol are presented efficiently by major histocompatibility complex I molecules. J. Biol.Chem. 276:17163–17171.
9. Binder, R.J., D.K. Han, and P.K. Srivastava. 2000. CD91: a receptor for heat shock protein gp96. Nat. Immunol.1:151–155.
10. Blachere, N.E., Z. Li, R.Y. Chandawarkar, et al. 1997. Heat shock protein-peptide complexes, reconstituted in vitro,elicit peptide-specific cytotoxic T lymphocyte response and tumor immunity. J. Exp. Med. 186:1315–1322.
11. Bolt, G. 2001. The measles virus (MV) glycoproteins interact with cellular chaperones in the endoplasmic reticu-lum and MV infection upregulates chaperone expression. Arch. Virol. 146:2055–2068.
12. Botzler, C., G. Li, R.D. Issels, et al. 1998. Definition of extracellular localized epitopes of Hsp70 involved in anNK immune response. Cell Stress Chaperones 3:6–11.
13. Braciale, T.J., L.A. Morrison, M.T. Sweetser, et al. 1987. Antigen presentation pathways to class I and class IIMHC-restricted T lymphocytes. Immunol. Rev. 98:95–114.
14. Brenner, B.G., and M.A. Wainberg. 1999. Heat shock protein–based therapeutic strategies against human immu-nodeficiency virus type 1 infection. Infect. Dis. Obstet. Gynecol. 7:80–90.
15. Butcher, E.C., and L.J. Picker. 1996. Lymphocyte homing and homeostasis. Science 272:60–66.
16. Carbone, F.R., C. Kurts, S.R. Bennett, et al. 1998. Cross-presentation: a general mechanism for CTL immunity andtolerance. Immunol. Today 19:368–373.
17. Castellino, F., P.E. Boucher, K. Eichelberg, et al. 2000. Receptor-mediated uptake of antigen/heat shock proteincomplexes results in major histocompatibility complex class I antigen presentation via two distinct processing path-ways. J. Exp. Med. 191:1957–1964.
18. Chen, W., Y. Lin, C. Liao, et al. 2000. Modulatory effects of the human heat shock protein 70 on DNA vaccina-tion. J. Biomed. Sci. 7:412–419.
19. Ciupitu, A.M., M. Petersson, C.L. O’Donnell, et al. 1998. Immunization with a lymphocytic choriomeningitis viruspeptide mixed with heat shock protein 70 results in protective antiviral immunity and specific cytotoxic T lym-phocytes. J. Exp. Med. 187:685–691.
20. Csermely, P., T. Schnaider, C. Soti, et al. 1998. The 90-kDa molecular chaperone family: structure, function, andclinical applications. A comprehensive review. Pharmacol. Ther. 79:129–168.
21. Di Cesare, S., F. Poccia, A. Mastino, et al. 1992. Surface expressed heat-shock proteins by stressed or human im-munodeficiency virus (HIV)–infected lymphoid cells represent the target for antibody-dependent cellular cytotox-icity. Immunology 76:341–343.
22. Fisch, P., M. Malkovsky, S. Kovats, et al. 1990. Recognition by human V gamma 9/V delta 2 T cells of a GroELhomolog on Daudi Burkitt’s lymphoma cells. Science 250:1269–1273.
23. Fourie, A.M., J.F. Sambrook, and M.J. Gething. 1994. Common and divergent peptide binding specificities of hsp70molecular chaperones. J. Biol. Chem. 269:30470–30478.
24. Fu, Y.X., R. Cranfill, M. Vollmer, et al. 1993. In vivo response of murine gamma delta T cells to a heat shock protein–derived peptide. Proc. Natl. Acad. Sci. USA 90:322–326.
OGLESBEE ET AL.
412

25. Furlini, G., M. Vignoli, M.C. Re, et al. 1994. Human immunodeficiency virus type 1 interaction with the mem-brane of CD41 cells induces the synthesis and nuclear translocation of 70K heat shock protein. J. Gen. Virol.75:193–199.
26. Gamer, J., G. Multhaup, T. Tomoyasu, et al. 1996. A cycle of binding and release of the DnaK, DnaJ and GrpEchaperones regulates activity of the Escherichia coli heat shock transcription factor sigma32. EMBO J. 15:607–617.
27. Gaudin, Y. 1997. Folding of rabies virus glycoprotein: epitope acquisition and interaction with endoplasmic retic-ulum chaperones. J. Virol. 71:3742–3750.
28. Gromme, M., F.G. Uytdehaag, H. Janssen, et al. 1999. Recycling MHC class I molecules and endosomal peptideloading. Proc. Natl. Acad. Sci. USA 96:10326–10331.
29. Habich, C., K. Baumgart, H. Kolb, et al. 2002. The receptor for heat shock protein 60 on macrophages is saturable,specific, and distinct from receptors for other heat shock proteins. J. Immunol. 168:569–576.
30. Harkonen, T., M. Puolakkainen, M. Sarvas, et al. 2000. Picornavirus proteins share antigenic determinants withheat shock proteins 60/65. J. Med. Virol. 62:383–391.
31. Hartl, F.U. 1996. Molecular chaperones in cellular protein folding. Nature 381:571–579.
32. Haynes, L.M., D.D. Moore, E.A. Kurt-Jones, et al. 2001. Involvement of toll-like receptor 4 in innate immunity torespiratory syncytial virus. J. Virol. 75:10730–10737.
33. Heikema, A., E. Agsteribbe, J. Wilschut, et al. 1997. Generation of heat shock protein–based vaccines by intra-cellular loading of gp96 with antigenic peptides. Immunol. Lett. 57:69–74.
34. Heller, M., D. Vasconcelos, J. Cummins, et al. 1998. Interferon-alpha inhibits the emergence of cellular stress response-dependent morbillivirus large plaque variants. Antiviral Res. 38:195–207.
35. Huang, Q., J.F. Richmond, K. Suzue, et al. 2000. In vivo cytotoxic T lymphocyte elicitation by mycobacterial heatshock protein 70 fusion proteins maps to a discrete domain and is CD41 T cell independent. J. Exp. Med.191:403–408.
36. Kakimura, J., Y. Kitamura, K. Takata, et al. 2002. Microglial activation and amyloid-beta clearance induced byexogenous heat-shock proteins. FASEB J. 16:601–603.
37. Kaur, I., S.D. Voss, R.S. Gupta, et al. 1993. Human peripheral gamma delta T cells recognize hsp60 molecules onDaudi Burkitt’s lymphoma cells. J. Immunol. 150:2046–2055.
38. Kim, H.T., E.L. Nelson, C. Clayberger, et al. 1995. Gamma delta T cell recognition of tumor Ig peptide. J. Im-munol. 154:1614–1623.
39. Kumaraguru, U., M. Gierynska, S. Norman, et al. 2002. Immunization with chaperone-peptide complex induceslow-avidity cytotoxic T lymphocytes providing transient protection against herpes simplex virus infection. J. Vi-rol. 76:136–141.
40. Kuppner, M.C., R. Gastpar, S. Gelwer, et al. 2001. The role of heat shock protein (hsp70) in dendritic cell matu-ration: hsp70 induces the maturation of immature dendritic cells but reduces DC differentiation from monocyteprecursors. Eur. J. Immunol. 31:1602–1609.
41. Kurts, C., W.R. Heath, F.R. Carbone, et al. 1996. Constitutive class I–restricted exogenous presentation of self anti-gens in vivo. J. Exp. Med. 184:923–930.
42. Lehner, T., E. Mitchell, L. Bergmeier, et al. 2000. The role of gammadelta T cells in generating antiviral factorsand beta-chemokines in protection against mucosal simian immunodeficiency virus infection. Eur. J. Immunol.30:2245–2256.
43. Li, Z., and P.K. Srivastava. 1993. Tumor rejection antigen gp96/grp94 is an ATPase: implications for protein fold-ing and antigen presentation. EMBO J. 12:3143–3151.
44. Linderoth, N.A., A. Popowicz, and S. Sastry. 2000. Identification of the peptide-binding site in the heat shock chap-erone/tumor rejection antigen gp96 (Grp94). J. Biol. Chem. 275:5472–5477.
45. Linderoth, N.A., M.N. Simon, N.A. Rodionova, et al. 2001. Biophysical analysis of the endoplasmic reticulum–res-ident chaperone/heat shock protein gp96/GRP94 and its complex with peptide antigen. Biochemistry 40:1483–1495.
HEAT SHOCK PROTEINS AND IMMUNE RESPONSE TO MEASLES VIRUS
413

46. Lipsker, D., U. Ziylan, D. Spehner, et al. 2002. Heat shock proteins 70 and 60 share common receptors which areexpressed on human monocyte-derived but not epidermal dendritic cells. Eur. J. Immunol. 32:322–332.
47. Macejak, D.G., and P. Sarnow. 1992. Association of heat shock protein 70 with enterovirus capsid precursor P1in infected human cells. J. Virol. 66:1520–1527.
48. Matzinger, P., and M.J. Bevan. 1977. Induction of H-2-restricted cytotoxic T cells: in vivo induction has the ap-pearance of being unrestricted. Cell. Immunol. 33:92–100.
49. Medzhitov, R., P. Preston-Hurlburt, and C.A. Janeway, Jr. 1997. A human homologue of the Drosophila Toll pro-tein signals activation of adaptive immunity. Nature 388:394–397.
50. Metlay, J.P., E. Pure, and R.M. Steinman. 1989. Control of the immune response at the level of antigen-present-ing cells: a comparison of the function of dendritic cells and B lymphocytes. Adv. Immunol. 47:45–116.
51. Michalak, M., E.F. Corbett, N. Mesaeli, et al. 1999. Calreticulin: one protein, one gene, many functions. Biochem.J. 344:281–292.
52. Misselwitz, B., O. Staeck, and T.A. Rapoport. 1998. J proteins catalytically activate Hsp70 molecules to trap awide range of peptide sequences. Mol. Cell. 2:593–603.
53. More, S.H., M. Breloer, and A. von Bonin. 2001. Eukaryotic heat shock proteins as molecular links in innate andadaptive immune responses: Hsp60-mediated activation of cytotoxic T cells. Int. Immunol. 13:1121–1127.
54. Multhoff, G., C. Botzler, M. Wiesnet, et al. 1995. A stress-inducible 72-kDa heat-shock protein (HSP72) is ex-pressed on the surface of human tumor cells, but not on normal cells. Int. J. Cancer 61:272–279.
55. Multhoff, G., L. Mizzen, C.C. Winchester, et al. 1999. Heat shock protein 70 (Hsp70) stimulates proliferation andcytolytic activity of natural killer cells. Exp. Hematol. 27:1627–1636.
56. Multhoff, G., K. Pfister, C. Botzler, et al. 2000. Adoptive transfer of human natural killer cells in mice with se-vere combined immunodeficiency inhibits growth of Hsp70-expressing tumors. Int. J. Cancer 88:791–797.
57. Nair, S., P.A. Wearsch, D.A. Mitchell, et al. 1999. Calreticulin displays in vivo peptide-binding activity and canelicit CTL responses against bound peptides. J. Immunol. 162:6426–6432.
58. Nieland, T.J., M.C. Tan, M. Monne-van Muijen, et al. 1996. Isolation of an immunodominant viral peptide that isendogenously bound to the stress protein GP96/GRP94. Proc. Natl. Acad. Sci. USA 93:6135–6139.
59. Ninomiya, T., H. Takimoto, G. Matsuzaki, et al. 2000. Vgamma11 gammadelta T cells play protective roles at anearly phase of murine cytomegalovirus infection through production of interferon-gamma. Immunology 99:187–194.
60. O’Keeffe, B., Y. Fong, D. Chen, et al. 2000. Requirement for a kinase-specific chaperone pathway in the produc-tion of a Cdk9/cyclin T1 heterodimer responsible for P-TEFb–mediated tat stimulation of HIV-1 transcription. J. Biol. Chem. 275:279–287.
61. Oglesbee, M., and S. Krakowka. 1993. Cellular stress response induces selective intranuclear trafficking and ac-cumulation of morbillivirus major core protein. Lab. Invest. 68:109–117.
62. Oglesbee, M., S. Ringler, and S. Krakowka. 1990. Interaction of canine distemper virus nucleocapsid variants with70K heat-shock proteins. J. Gen. Virol. 71:1585–1590.
63. Oglesbee, M.J., S. Alldinger, D. Vasconcelos, et al. 2002. Intrinsic thermal resistance of the canine brain. Neuro-science 113:55–64.
64. Oglesbee, M.J., H. Kenney, T. Kenney, et al. 1993. Enhanced production of morbillivirus gene-specific RNAs fol-lowing induction of the cellular stress response in stable persistent infection. Virology 192:556–567.
65. Ohashi, K., V. Burkart, S. Flohe, et al. 2000. Cutting edge: heat shock protein 60 is a putative endogenous ligandof the toll-like receptor-4 complex. J. Immunol. 164:558–561.
66. Otteken, A., and B. Moss. 1996. Calreticulin interacts with newly synthesized human immunodeficiency virus type1 envelope glycoprotein, suggesting a chaperone function similar to that of calnexin. J. Biol. Chem. 271:97–103.
67. Peterson, J.R., A. Ora, P.N. Van, et al. 1995. Transient, lectin-like association of calreticulin with folding inter-mediates of cellular and viral glycoproteins. Mol. Biol. Cell 6:1173–1184.
68. Phillips, B., K. Abravaya, and R.I. Morimoto. 1991. Analysis of the specificity and mechanism of transcriptionalactivation of the human hsp70 gene during infection by DNA viruses. J. Virol. 65:5680–5692.
OGLESBEE ET AL.
414

69. Ponniah, S., P.C. Doherty, and M. Eichelberger. 1996. Selective response of gamma delta T-cell hybridomas to or-thomyxovirus-infected cells. J. Virol. 70:17–22.
70. Ponomarev, E.D., T.N. Tarasenko, and A.M. Sapozhnikov. 2000. Splenic cytotoxic cells recognize surface HSP70on culture-adapted EL-4 mouse lymphoma cells. Immunol. Lett. 74:133–139.
71. Richter, K., and J. Buchner. 2001. Hsp90: chaperoning signal transduction. J. Cell. Physiol. 188:281–290.
72. Rodriguez, A., A. Regnault, M. Kleijmeer, et al. 1999. Selective transport of internalized antigens to the cytosolfor MHC class I presentation in dendritic cells. Nat. Cell. Biol. 1:362–368.
73. Roman, E., and C. Moreno. 1996. Synthetic peptides non-covalently bound to bacterial hsp 70 elicit peptide-specific T-cell responses in vivo. Immunology 88:487–492.
74. Sadasivan, B., P.J. Lehner, B. Ortmann, et al. 1996. Roles for calreticulin and a novel glycoprotein, tapasin, in theinteraction of MHC class I molecules with TAP. Immunity 5:103–114.
75. Saito, Y., Y. Ihara, M.R. Leach, et al. 1999. Calreticulin functions in vitro as a molecular chaperone for both gly-cosylated and non-glycosylated proteins. EMBO J. 18:6718–6729.
76. Sargent, C.A., I. Dunham, J. Trowsdale, et al. 1989. Human major histocompatibility complex contains genes forthe major heat shock protein HSP70. Proc. Natl. Acad. Sci. USA 86:1968–1972.
77. Sheshberadaran, H., and E. Norrby. 1984. Three monoclonal antibodies against measles virus F protein cross-react with cellular stress proteins. J. Virol. 52:995–999.
78. Singh-Jasuja, H., R.E. Toes, P. Spee, et al. 2000. Cross-presentation of glycoprotein 96-associated antigens on major histocompatibility complex class I molecules requires receptor-mediated endocytosis. J. Exp. Med.191:1965–1974.
79. Sondermann, H., T. Becker, M. Mayhew, et al. 2000. Characterization of a receptor for heat shock protein 70 onmacrophages and monocytes. Biol. Chem. 381:1165–1174.
80. Spee, P., and J. Neefjes. 1997. TAP-translocated peptides specifically bind proteins in the endoplasmic reticulum,including gp96, protein disulfide isomerase and calreticulin. Eur. J. Immunol. 27:2441–2449.
81. Srivastava, P.K. 1993. Peptide-binding heat shock proteins in the endoplasmic reticulum: role in immune responseto cancer and in antigen presentation. Adv. Cancer Res. 62:153–177.
82. Suto, R., and P.K. Srivastava. 1995. A mechanism for the specific immunogenicity of heat shock protein-chaper-oned peptides. Science 269:1585–1588.
83. Suzue, K., and R.A. Young. 1996. Adjuvant-free hsp70 fusion protein system elicits humoral and cellular immuneresponses to HIV-1 p24. J. Immunol. 156:873–879.
84. Udono, H., and P.K. Srivastava. 1993. Heat shock protein 70–associated peptides elicit specific cancer immunity.J. Exp. Med. 178:1391–1396.
85. Udono, H., and P.K. Srivastava. 1994. Comparison of tumor-specific immunogenicities of stress-induced proteinsgp96, hsp90, and hsp70. J. Immunol. 152:5398–5403.
86. Vabulas, R.M., P. Ahmad-Nejad, S. Ghose, et al. 2002. HSP70 as endogenous stimulus of toll/interleukin-1 receptor signal pathway. J. Biol. Chem. 277:15107–15112.
87. Vasconcelos, D., E. Norrby, and M. Oglesbee. 1998. The cellular stress response increases measles virus–inducedcytopathic effect. J. Gen. Virol. 79:1769–1773.
88. Vasconcelos, D.Y., X.H. Cai, and M.J. Oglesbee. 1998. Constitutive overexpression of the major inducible 70-kDaheat shock protein mediates large plaque formation by measles virus. J. Gen. Virol. 79:2239–2247.
89. Wainberg, Z., M. Oliveira, S. Lerner, et al. 1997. Modulation of stress protein (hsp27 and hsp70) expression inCD41 lymphocytic cells following acute infection with human immunodeficiency virus type–1. Virology233:364–373.
90. Wearsch, P.A., and C.V. Nicchitta. 1997. Interaction of endoplasmic reticulum chaperone GRP94 with peptide sub-strates is adenine nucleotide–independent. J. Biol. Chem. 272:5152–5156.
91. Weidinger, G., S. Czub, C. Neumeister, et al. 2000. Role of CD41 and CD81 T cells in the prevention of measlesvirus–induced encephalitis in mice. J. Gen. Virol. 81:2707–2713.
HEAT SHOCK PROTEINS AND IMMUNE RESPONSE TO MEASLES VIRUS
415

92. Xu, A., A.R. Bellamy, and J.A. Taylor. 1998. BiP (GRP78) and endoplasmin (GRP94) are induced following ro-tavirus infection and bind transiently to an endoplasmic reticulum–localized virion component. J. Virol.72:9865–9872.
93. Yoshida, H., K. Haze, H. Yanagi, et al. 1998. Identification of the cis-acting endoplasmic reticulum stress responseelement responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basicleucine zipper transcription factors. J. Biol. Chem. 273:33741–33749.
Address reprint requests to:Michael Oglesbee, D.V.M., Ph.D.
Department of Veterinary BiosciencesThe Ohio State University
1925 Coffey RoadColumbus, OH 43210
E-mail: [email protected]
OGLESBEE ET AL.
416
Copyright © 2022 FDOKUMEN




















