
Restrictive dermopathy: a rare laminopathy
8
Arch Gynecol Obstet DOI 10.1007/s00404-008-0676-6 123 REVIEW ARTICLE Restrictive dermopathy: a rare laminopathy Marc Thill · Thuy Duong Nguyen · Manfred Wehnert · Dorothea Fischer · Ingrid Hausser · Susanne Braun · Christian Jackisch Received: 18 December 2007 / Accepted: 22 April 2008 © Springer-Verlag 2008 Abstract Background Restrictive dermopathy (RD) belongs to the laminopathies and mostly shows an autosomal recessive heredity pattern. This rare genetic disorder is lethal for the newborn in the neonatal period. Clinical and pathological Wndings are distinctive and allow for a speciWc diagnosis in most cases. Furthermore, polyhydramnios, decreased foetal movement, facial dysmorphisms and arthrogryposis are characteristic of RD. Respiratory insuYciency leads to an early neonatal death. Methods We present the case of an aVected infant and a review of the previously reported cases in the literature. Results The infant showed thin, shiny skin with exfoliat- ing desquamation, a small, round and open mouth, low-set ears, a small pinched nose, joint contractures at all four extremities and distinctive pulmonic atelectasis. It died 3 h and 20 min post-partum. Histologically, the skin showed the typical pattern of an RD with the epidermis covered by an exfoliated, hyperkeratotic horn layer, clearly hypoplastic hair follicles and a considerably reduced dermis thickness, although it had a massive subcutaneous adipose tissue. Electron microscopically, the diagnosis was conWrmed. Conclusions It is important to know about this disease and to distinguish it from others like keratinization mal- functions such as ichtyosis, congenital, developmental and akinesia disturbance, etc., to know the prognosis for the aVected newborn and to provide suYcient (genetic) coun- selling to the families. This disorder is caused by dominant mutations of the LMNA (primary laminopathy) or reces- sive mutations of the ZMPSTE24 (FACE1) (secondary laminopathy) genes. Keywords Restrictive dermopathy · Laminopathy · Foetal akinesia deformation sequence Introduction Recently, a multitude of perinatal lethal syndromes has been deWned by molecular genetic analysis in addition to the ones already known. Gynaecologists and neonatologists must be in a position to bring these syndromes into the right context as the pre- and peripartal responsibility lies in their hands. The restrictive dermopathy (RD), described as geno- dermatosis with an unclear structural gene defect (even 3 years ago), has turned out to present a laminopathy with mostly autosomal recessive inheritance and has a fatal end for the newborn. The typical characteristics are unusually tight skin, joint contractures and characteristic facial M. Thill · C. Jackisch Department of Gynecology and Obstetrics, Klinikum OVenbach, Starkenburgring 66, 63069 OVenbach, Germany M. Thill (&) · D. Fischer Department of Gynecology and Obstetrics, University Schleswig-Holstein, Campus Luebeck, Ratzeburger Allee 160, 23538 Luebeck, Germany e-mail: [email protected] T. D. Nguyen · M. Wehnert Institute of Human Genetics, Ernst-Moritz-Arndt-University Greifswald, Fleischmannstrasse 42/44, 17487 Greifswald, Germany I. Hausser Department of Dermatology, Ruprecht-Karls-University Heidelberg, Voßstrasse 2, 69115 Heidelberg, Germany S. Braun Institute of Pathology, Klinikum OVenbach, Starkenburgring 66, 63069 OVenbach, Germany
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Restrictive dermopathy: a rare laminopathy

Arch Gynecol Obstet
DOI 10.1007/s00404-008-0676-6REVIEW ARTICLE
Restrictive dermopathy: a rare laminopathy
Marc Thill · Thuy Duong Nguyen · Manfred Wehnert · Dorothea Fischer · Ingrid Hausser · Susanne Braun · Christian Jackisch
Received: 18 December 2007 / Accepted: 22 April 2008© Springer-Verlag 2008
AbstractBackground Restrictive dermopathy (RD) belongs to thelaminopathies and mostly shows an autosomal recessiveheredity pattern. This rare genetic disorder is lethal for thenewborn in the neonatal period. Clinical and pathologicalWndings are distinctive and allow for a speciWc diagnosis inmost cases. Furthermore, polyhydramnios, decreased foetalmovement, facial dysmorphisms and arthrogryposis arecharacteristic of RD. Respiratory insuYciency leads to anearly neonatal death.Methods We present the case of an aVected infant and areview of the previously reported cases in the literature.Results The infant showed thin, shiny skin with exfoliat-ing desquamation, a small, round and open mouth, low-set
ears, a small pinched nose, joint contractures at all fourextremities and distinctive pulmonic atelectasis. It died 3 hand 20 min post-partum. Histologically, the skin showedthe typical pattern of an RD with the epidermis covered byan exfoliated, hyperkeratotic horn layer, clearly hypoplastichair follicles and a considerably reduced dermis thickness,although it had a massive subcutaneous adipose tissue.Electron microscopically, the diagnosis was conWrmed.Conclusions It is important to know about this diseaseand to distinguish it from others like keratinization mal-functions such as ichtyosis, congenital, developmental andakinesia disturbance, etc., to know the prognosis for theaVected newborn and to provide suYcient (genetic) coun-selling to the families. This disorder is caused by dominantmutations of the LMNA (primary laminopathy) or reces-sive mutations of the ZMPSTE24 (FACE1) (secondarylaminopathy) genes.
Keywords Restrictive dermopathy · Laminopathy · Foetal akinesia deformation sequence
Introduction
Recently, a multitude of perinatal lethal syndromes hasbeen deWned by molecular genetic analysis in addition tothe ones already known. Gynaecologists and neonatologistsmust be in a position to bring these syndromes into the rightcontext as the pre- and peripartal responsibility lies in theirhands. The restrictive dermopathy (RD), described as geno-dermatosis with an unclear structural gene defect (even3 years ago), has turned out to present a laminopathy withmostly autosomal recessive inheritance and has a fatal endfor the newborn. The typical characteristics are unusuallytight skin, joint contractures and characteristic facial
M. Thill · C. JackischDepartment of Gynecology and Obstetrics, Klinikum OVenbach, Starkenburgring 66, 63069 OVenbach, Germany
M. Thill (&) · D. FischerDepartment of Gynecology and Obstetrics, University Schleswig-Holstein, Campus Luebeck, Ratzeburger Allee 160, 23538 Luebeck, Germanye-mail: [email protected]
T. D. Nguyen · M. WehnertInstitute of Human Genetics, Ernst-Moritz-Arndt-University Greifswald, Fleischmannstrasse 42/44, 17487 Greifswald, Germany
I. HausserDepartment of Dermatology, Ruprecht-Karls-University Heidelberg, Voßstrasse 2, 69115 Heidelberg, Germany
S. BraunInstitute of Pathology, Klinikum OVenbach, Starkenburgring 66, 63069 OVenbach, Germany
123

Arch Gynecol Obstet
aberrations such as micrognathia, low set ears, an amotile,open mouth, a small pinched nose and hypertelorism [9, 21,44, 46]. The joint contractures presented by the newbornare caused by the missing skin elasticity [46] with consecu-tive intrauterine decreased foetal movement (also known asFADS—foetal akinesia deformation sequence) [47].
The term restrictive dermopathy was used for the Wrsttime in 1986 by Witt et al. [46]. It was presumed that thiswas a new syndrome, which needed to be distinguishedfrom other congenital and lethal syndromes like the Neu-Laxova syndrome (NLS), the Pena Shokeir syndrome or theaplasia cutis congenital (ACC); however, after performingtheir own research, Witt et al. [46] Wnally named the syn-drome restrictive dermopathy. As of today, there are only62 published cases including these cases. We refer to theoverview in Table 1, which is based on 49 case reports.
Case report
Anamnesis and course
We report on a 21-year-old gravida 1, para 1 in week 28+6
of gestation from Guinea, who was hospitalised in ourfacility with preterm ruptured membranes 16 h prior to pre-term uterine contractions, isthmocervical insuYciency andpolyhydramnios. Consanguinity (Wrst cousins) existedbetween the parents. The mother did not feel reduced foetalmovement and the ultrasound examination showed polyhy-
dramnios (AFI = 22.5) and an asymmetrically growth-retarded foetus as related to its week of gestational age. Theorgan screening, anterior wall placenta and Doppler mea-surements were inconspicuous and the face showed a Wxed,O-shaped mouth. The vaginal ultrasound presented a cervixof 1.5 cm length with a small inner funnel.
Beginning amnion infection syndrome and progredient,therapy-resistant and increased uterine contractions indi-cated a caesarean section at day 6 of hospitalisation (week29+4 of gestation). A male hypotrophy, premature infant(Fig. 1a, b) was delivered with a birth weight of 1,020 g,38 cm length, 28 cm head circumference, APGAR 2/2/5and arterial umbilical cord blood pH 7.38. The umbilicalcord was remarkably short. The child showed thin, shinyand tight skin ulceration of both elbow bends and hyperker-atosis of the body. Furthermore, there was hypertelorism,hypoplasia of the eyelids, lack of eyebrows and eyelashes, aslight antimongoloid slant, a Xat, hypoplastic pinched nose,a very small, O-shaped and Wxed mouth (microstomia),micrognathia, low-set malformed ears, multiple joint con-tractures especially of the great but also of the Wnger joints(arthrogryposis multiplex), little spontaneous motor func-tion, missing facial expression and no body hair.
X-ray examination of the thorax (Fig. 2) diagnosed abilateral white lung, bilateral hypoplastic clavicles, a dys-plastic humerus on the right side and a bilateral hypoplasticmandible. Primary respiratory insuYciency required intu-bation, and mechanical respiration was required only 3 minafter delivery. A blood sample was taken for chromosome
Table 1 Frequency of abnor-mal clinicopathologic Wndings in restrictive dermopathy, based on 58 cases (including our case)
Finding Number of cases reporting Wnding (%)
Number of cases with no reference to Wnding (%)
Number of cases with absence of Wnding (%)
Thick tight skin 58 (100) 0 0
Flexion contractures of extremities 58 (100) 0 0
Micrognathia 52 (90) 5 (8) 1 (2)
Small or “O” mouth (microstomia) 52 (90) 6 (10) 0
Hypoplastic nose 46 (79) 12 (21) 0
Wide sutures/large fontanelle 37 (64) 20 (34) 1 (2)
Low set/malformed ears 41 (71) 16 (28) 1 (2)
Skeletal abnormalities 42 (72) 15 (26) 1 (2)
Premature rupture of membranes 24 (41) 28 (48) 6 (10)
Hypertelorism/blepharophimosis 29 (50) 29 (50) 0
Polyhydramnios 21 (36) 32 (55) 5 (9)
Rocker bottom feet 27 (47) 30 (52) 1 (2)
Hair abnormalities/lack of eyebrows/lashes
18 (31) 36 (62) 4 (7)
Consanguinity 14 (24) 15 (26) 29 (50)
Decreased foetal movement 16 (28) 38 (66) 4 (7)
Abnormal nails 11 (19) 27 (47) 9 (16)
Ectropion 14 (24) 43 (74) 1 (2)
Short umbilical cord 7 (12) 51 (88) 0
References 2, 4–6, 8–11, 13–16, 18–22, 24, 27–30, 32, 33, 36–41, 43, 46
123

Arch Gynecol Obstet
analysis. The child’s death followed despite IPPV respira-tion at the Wrst day and 3 h and 20 min after birth due torespiratory insuYciency.
Light microscopy of the skin
The epidermis, which was covered by an exfoliating hyper-keratotic horn layer, showed a complete loss of the rete
ridges and a reduction of the skin appendages with clearlyhypoplastic hair follicles. The thickness of the dermis wasstrongly reduced and was found lying very compact abovethe massive subcutaneous fat tissue. Within the dermal con-nective tissue, the collagen bundles were closely packed(tendon-like) and directed parallel to the epidermis. ElasticWbres could not be identiWed (Fig. 3).
Electron microscopy of the skin
We found an orthokeratotic, slightly exfoliating and other-wise massive horn layer with many melanin granules (eth-nic background). In the granular layer, there were irregularand blurrily distributed aggregations of keratohyalin gran-ules in the stratum granulosum that showed little connec-tion to the clearly rareWed tonoWlaments. Only very tinyelastic Wbres could be found sporadically between theclosely packed collagen bundles and consisting mostly ofelastotubules but not containing identiWable elastin (Fig. 4).
Pathological anatomical expertise and result of the chromosome analysis
The inner organs presented themselves without abnormality.We found a distinctive lung atelectasis with a negative swim-ming test and therefore conWrmed the clinical diagnosis ofrespiratory insuYciency. The 470 g placenta with dissociatedvillus maturation malfunction was, in relation to week 30 ofgestational age, above the norm (normal: P10 230 g, P50
335 g, P90 450 g). With three vessels and 20 cm in length, theumbilical cord was conspicuously short. The chromosomalanalysis was normal and showed a male karyotype 46, XY.
Family analysis
The parents were consanguineous (direct cousins) (Fig. 5a).Besides the presented case (II-1), in an ensuing pregnancy,
Fig. 1 (a, b) Child with typical multiple joint contractures (week 30 of gestation). These photographs are published with permission of the parents
Fig. 2 Chest X-ray with hypoplastic clavicles, hypoplastic mandiblesand dysplastic right-sided humerus
123

Arch Gynecol Obstet
the mother (I-2) delivered another child of unknown sex(II-2) by caesarean section after early rupture of mem-branes at week 29 of gestational age. The newborn wasproved histologically to be aVected by restrictive dermopathy.A third pregnancy resulted in a healthy boy (II-3). Thereare reportedly no further aVected children in the family.
Genetic analysis
Methods
Genomic DNA was extracted from paraYn-embedded skinof the index case and from peripheral blood of the mother.The healthy father and brother of the index case were notavailable for investigation. The DNA was scanned formutations in LMNA and ZMPSTE24 (FACE1) according toNavarro et al. [26] by amplifying 12 exons of the LMNAgene and 10 exons of the FACE1 gene including the intron/
exon boundaries using polymerase chain reaction (PCR).The PCR products were tested for changes by heteroduplexanalysis [45] and directly sequenced by a cycle-sequencingprocedure using a Taq Dye Deoxy Terminator Cyclesequencing kit (PE/Applied Biosystems, Foster City, CA,USA). The PCR primers were also used for sequencing.
Results
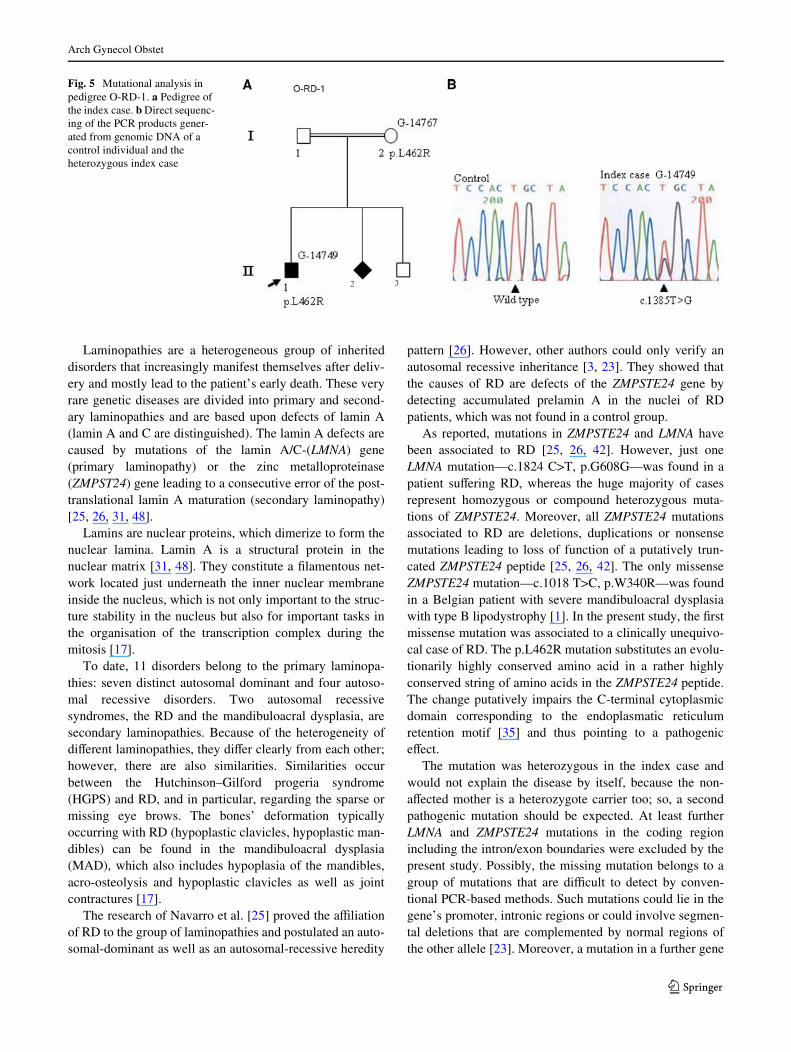
In both the index case and the mother, a heterozygousgenomic sequence variation c.1385T>G was found in exon10 of FACE1 resulting in a putative amino acid substitutionp.L462R (Fig. 5b). The mutation was conWrmed by diges-tion using the restriction enzyme TspRI. The p.L462Rmutation leads to a nonsynonymous substitution of an evo-lutionarily highly conserved leucin.
Discussion
RD is a rare and lethal, monogenic disorder, which aVectsthe development of skin, bones and lungs. The underlyingpathogenetic mechanism was unclear for a long time andseemed to be primarily a defect in the collagen metabolismthat would lead to a malfunction or would stop the foetalskin development.
DiVerent hypotheses were developed [6, 15, 46], but thecausal genetic defect could Wnally be described and there-upon the RD was identiWed as a laminopathy by Navarroet al. [25].
Fig. 3 Light microscopy of the skin: epidermis with rare skin append-ages (i.e. hair follicle, eccrine and sebaceous glands), Xat dermal-sub-cutaneous junction, absence of rete ridges (£40)
Fig. 4 Electron microscopy of the skin: dermal connective tissue,compact and dense collagen Wbres, oriented parallel with epidermis,resembling in a scar or tendon; £13,500
123
Arch Gynecol Obstet
Laminopathies are a heterogeneous group of inheriteddisorders that increasingly manifest themselves after deliv-ery and mostly lead to the patient’s early death. These veryrare genetic diseases are divided into primary and second-ary laminopathies and are based upon defects of lamin A(lamin A and C are distinguished). The lamin A defects arecaused by mutations of the lamin A/C-(LMNA) gene(primary laminopathy) or the zinc metalloproteinase(ZMPST24) gene leading to a consecutive error of the post-translational lamin A maturation (secondary laminopathy)[25, 26, 31, 48].
Lamins are nuclear proteins, which dimerize to form thenuclear lamina. Lamin A is a structural protein in thenuclear matrix [31, 48]. They constitute a Wlamentous net-work located just underneath the inner nuclear membraneinside the nucleus, which is not only important to the struc-ture stability in the nucleus but also for important tasks inthe organisation of the transcription complex during themitosis [17].
To date, 11 disorders belong to the primary laminopa-thies: seven distinct autosomal dominant and four autoso-mal recessive disorders. Two autosomal recessivesyndromes, the RD and the mandibuloacral dysplasia, aresecondary laminopathies. Because of the heterogeneity ofdiVerent laminopathies, they diVer clearly from each other;however, there are also similarities. Similarities occurbetween the Hutchinson–Gilford progeria syndrome(HGPS) and RD, and in particular, regarding the sparse ormissing eye brows. The bones’ deformation typicallyoccurring with RD (hypoplastic clavicles, hypoplastic man-dibles) can be found in the mandibuloacral dysplasia(MAD), which also includes hypoplasia of the mandibles,acro-osteolysis and hypoplastic clavicles as well as jointcontractures [17].
The research of Navarro et al. [25] proved the aYliationof RD to the group of laminopathies and postulated an auto-somal-dominant as well as an autosomal-recessive heredity
pattern [26]. However, other authors could only verify anautosomal recessive inheritance [3, 23]. They showed thatthe causes of RD are defects of the ZMPSTE24 gene bydetecting accumulated prelamin A in the nuclei of RDpatients, which was not found in a control group.
As reported, mutations in ZMPSTE24 and LMNA havebeen associated to RD [25, 26, 42]. However, just oneLMNA mutation—c.1824 C>T, p.G608G—was found in apatient suVering RD, whereas the huge majority of casesrepresent homozygous or compound heterozygous muta-tions of ZMPSTE24. Moreover, all ZMPSTE24 mutationsassociated to RD are deletions, duplications or nonsensemutations leading to loss of function of a putatively trun-cated ZMPSTE24 peptide [25, 26, 42]. The only missenseZMPSTE24 mutation—c.1018 T>C, p.W340R—was foundin a Belgian patient with severe mandibuloacral dysplasiawith type B lipodystrophy [1]. In the present study, the Wrstmissense mutation was associated to a clinically unequivo-cal case of RD. The p.L462R mutation substitutes an evolu-tionarily highly conserved amino acid in a rather highlyconserved string of amino acids in the ZMPSTE24 peptide.The change putatively impairs the C-terminal cytoplasmicdomain corresponding to the endoplasmatic reticulumretention motif [35] and thus pointing to a pathogeniceVect.
The mutation was heterozygous in the index case andwould not explain the disease by itself, because the non-aVected mother is a heterozygote carrier too; so, a secondpathogenic mutation should be expected. At least furtherLMNA and ZMPSTE24 mutations in the coding regionincluding the intron/exon boundaries were excluded by thepresent study. Possibly, the missing mutation belongs to agroup of mutations that are diYcult to detect by conven-tional PCR-based methods. Such mutations could lie in thegene’s promoter, intronic regions or could involve segmen-tal deletions that are complemented by normal regions ofthe other allele [23]. Moreover, a mutation in a further gene
Fig. 5 Mutational analysis in pedigree O-RD-1. a Pedigree of the index case. b Direct sequenc-ing of the PCR products gener-ated from genomic DNA of a control individual and the heterozygous index case
123

Arch Gynecol Obstet
not yet known to be involved in the pathogenesis of RDcould be anticipated. Unfortunately, there was not morepatient material available to test this hypothesis or to dofurther experiments to Wnd the second mutation from thepatient.
RD becomes clinically manifest in rigid and inelasticskin that leads to foetal akinesia or hypokinesia andinvolves multiple joint contractures (arthrogryposis multi-plex) and characteristic craniofacial abnormalities; hence,Verloes et al. compared the appearance with that of an“Asian porcelain doll” [41]. Typical face alterations that aredescribed in the literature are an “O”-shaped Wxed mouth,hypertelorism, blepharophimosis, small pinched nose,ectropion, lack of eyelashes and eyebrows, micrognathiaand low-set ears. Sometimes, one can observe a pretermtooth penetration [21, 44]. The feet can be rocking-chair-like deformed (rocker-bottom feet) [46]. The bones’ altera-tions present themselves as badly mineralised bones withwide fontanelles, hypo- or dysplastic mandibles, hypoplas-tic clavicles, irregular ribs and in an “overtubulation” of thehumeral shafts and the forearm bones [41] (refer toTable 1). The lung can be hypoplastic [27]. Other associ-ated visceral anomalies are rare and can be hepatospleno-megaly or ureter duplex. Even a transposition of the bigvessels and a microcolon has been described [2]. The skinis thin, rigid, tight and partially eroded and shows generallyrips at the neck, in the elbows and the groins due to themechanical exposure during delivery. The shallow vesselscan be seen easily through the translucent skin; the nipplesare prominent [20].
The rigidity of the skin causes Xexion contractures andconsecutive akinesia of the foetus in utero, which leads toan intrauterine growth retardation. This phenomenon is alsoknown as “foetal akinesia deformation sequence”(FADS)[12]. Furthermore it is responsible for polyhydramnios dueto reduced mouth movements (swallowing and sucking),and at last also for a possible lung hypoplasia due to miss-ing reduced breath excursions [21, 24, 34, 41, 44]. Polyhy-dramnios can be found in most cases. In all cases, there wasan early delivery before week 35 of gestation. Regularly,preterm rupture of membranes complicates the pregnancy.Because of the lack of elastic Wbre, the amnion fragilitytypical for RD is responsible for this feature [21, 24, 34, 41,44].
The course of pregnancy is similar in almost all cases:polyhydramnios, preterm rupture of membranes and animminent foetal asphyxia followed by a consecutive caesar-ean section [27]. The placenta is enlarged and shows ahigher weight than usual and the umbilical cord is short[27]. The children usually die immediately after delivery ofrespiratory insuYciency, caused by thorax stiVness and theinvolved lung compression. An intubation is often not pos-sible due to a possible temporomandibular ankylosis [41].
The longest survival has been declared with 120 days in lit-erature [40]. Based on the autosomal recessive inheritance,the recurrency risk amounts to 25%.
Light microscopically showing, a clear reduction of thedermis thickness is found and a lack of elastic Wbres inthe corium. For the skin rigidity, the lack of elastic Wbres inthe dermis and the tightly packed, wavelike collagen bundlesdirected parallel to the skin surface are decisive [27]. Theyshow a regularly tendon-like pattern [43]. The lack of elas-tic Wbres in the amnion will be responsible for the regularlyobserved rupture of the membranes [10, 14, 46]. Elastin isonly synthesised from week 26–28 of gestation; the generalgrowth stop can therefore be causative. The hypodermicadipose tissue is generally inconspicuous. Apart from theskin also the skin appendages are altered. Hair follicles andeccrine sweat glands are present in normal amounts; how-ever, they are positioned variably in the dermis. The secre-tory part is missing. The hair follicle suspends in thedevelopmental level of the late Wrst trimester. The hair—with exception of the capitulum—is missing [6, 10, 14, 15,41].
Electron microscopically, an orthokeratotic slightly des-quamating horn layer of the epidermis can be found. In thestratum granulosum, rough-blurrily spread and increasedkeratohyalin granules sometimes without connection to theclearly reduced tonoWlaments occur [10, 14, 46]. In the der-mis, there are compact collagen bundles composed of smallcalibre collagen Wbrils with clearly degenerated Wbroblastsin between. Most conspicuous are tiny elastic Wbres [10,27].
Autopsies showed all other organs without indication forchanges. Only aorta ascendens, truncus pulmonalis and theaorta descendens presented themselves with a remarkablyrigid vessel wall [27].
With regard to the clinical results, it is important pre-and peripartally for the gynaecologist and postpartally forthe neonatologist to be able to distinguish other similar con-ditions. DiVerentially, the RD has to be distinguishedamong others from the ichtyosis congenita gravis (harle-quin foetus), the Neu-Laxova syndrome, the Pena-Shokeirsyndrome and the cerebrooculo-facio-skeletal syndrome, asthose syndromes have characteristic features for growthdisorders as arthrogryposis, facial abnormalities and a shortlifetime. Furthermore, the aplasia cutis congenita, the scle-rema neonatorum, the stiV skin syndrome and the gaucherdisease have to be distinguished [7, 41, 43].
RD can be diagnosed genetically in amniotic Xuid cellsor chorionic villi biopsies by verifying mutations in theZMPSTE24 and the LMNA gene [26, 42]. A continuinglyopen mouth is the most apparent characteristic found Wrst inultrasonography and can be an indicator for the disease nextto polyhydramnios and increasing foetal hypokinesia foundin late pregnancy.
123

Arch Gynecol Obstet
A therapy with farnesyltransferase inhibitors as consid-ered in the HGPS therapy and already performed in animalexperiments cannot be used for RD due to the neonatallethality. The idea to treat HGPS with farnesyltransferaseinhibitors successfully is based on the fact that the HGPSphenotype is based on a farnesylated, mutated prelamin Athat could be undone through the inhibition of farnesyl-transferase. In vitro studies are partly promising [31].Another therapeutical approach aims the removal of the pri-mary defect on the mRNA level, but is practically not avail-able yet. However, for each therapeutical approach, theneonatal lethality of the children with RD is again the limit-ing factor.
Conclusions
RD is a rare primary but mainly secondary laminopathywith a fatal outcome for the newborn. Because of theinfaust prognosis for the newborn, the knowledge aboutthis disease is of considerable importance in terms of thepostpartal management and the resulting counselling for theparents, as the recurrence risk for an ill child amounts to25% for this autosomal recessively inherited disorder.
ConXict of interest statement The authors declare no conXict ofinterest relevant to this article.
References
1. Agarwal AK, Fryns JP, Auchus RJ, Garg A (2003) Zinc metallo-proteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia.Hum Mol Genet 12:1995–2001
2. Armbrust S, HoVmann R, Jochum F, Neumann LM, Fusch C(2005) Restrictive dermopathy associated with transposition of thegreat arteries and microcolon. Arch Dermatol 141:611–613
3. Broers JLV, Ramaekers FCS, Bonne G, Ben Yaou R, HutchinsonCJ (2006) Nuclear lamins: laminopathies and their role in prema-ture ageing. Physiol Rev 86:967–1008
4. Dale BA, Holbrook KA, Witt DR, Toriello HV (1987) Abnormalkeratinization in restrictive dermopathy. Curr Probl Dermatol17:45–51
5. Das-Kundu S, Klunemann HH, Mieth D, Spycher M, Stallmach T,Schinzel A (1996) Case of the month: a newborn with tight skinand joint contractures. Eur J Pediatr 155:987–989
6. Dean JCS, Gray ES, Stewart KN, Brown T, Lloyd DJ, Smith NC,Pope FM (1993) Restrictive dermopathy: a disorder of skin diVeren-tiation with abnormal integrin expression. Clin Genet 44:287–291
7. FelderhoV-Mueser U, Uhl J, Penzel R, van Landeghem F, VogelM, Obladen M, Kopitz J (2004) Intrauterine onset of acute neuro-pathic type 2 Gaucher disease: identiWcation of a novel insertionsequence. Am J Med Genet A 128:138–143
8. Gillerot Y, Koulischer L (1987) Restrictive dermopathie. Am JMed Genet 27:239–240
9. Graham J, Esterly NB (1999) What syndrome is this? Pediatr Der-matol 16(2):151–153
10. Groß C, Haußer I, von der Wense A, Langner C, Simoens W, BauO, Rompel R, Meyer W, RüschoV J (1999) Restriktive dermopat-hie. Pathologe 20:365–370
11. Hamel BCJ, Happle R, Steylen PM, Kollée LAA, SchuurmansStekhoven JH, Nijhuis JG, Rauskolb R, Anton-Lamprecht I (1992)False-negative prenatal diagnosis of restrictive dermopathy. Am JMed Genet 44:824–826
12. Hammond E, Donnenfeld AE (1995) Fetal akinesia. Obstet Gyne-col Surv 50:240–249
13. Happle R, Stekhoven JH, Hamel BC, Kollée LA, Nijhuis JG, An-ton-Lamprecht I, Steijlen PM (1992) False-negative prenatal diag-nosis of restrictive dermopathy in two brothers. Arch Dermatol128:232–235
14. HoVmann R, Lohner M, Böhm N, Leititis J, Helwig H (1993)Restrictive dermopathy: a lethal congenital skin disorder. Eur JPediatr 152:95–98
15. Holbrook KA, Dale BA, Witt DR, Hayden MR, Toriello HV(1987) Arrested epidermal morphogenesis in three newborninfants with fatal genetic disorder (restrictive dermopathy).J Invest Dermatol 88:330–339
16. Hou J-W, Chien-Fang M (2003) Restrictive dermopathy in twosisters. Chang Gung Med J 7:510–513
17. Jacob KN, Garg A., Laminopathies: multisystem dystrophy syn-dromes (2006) Mol Genet Metabol 87:289–302
18. Lenz W, Meschede D (1993) Historical note on restrictive dermopa-thy and report of two new cases. Am J Med Genet 47:1235–1237
19. Lowry RB, Machin GA, Morgan K, Marx L (1985) Congenital con-tractures, edema, hyperkeratosis and IUGR: a fatal syndrome in Hut-terite and Mennonite kindreds. Am J Med Genet 22:531–543
20. Mahadevan B, Karthikeyan K, Vishnu Bhat B, Thappa DM (2002)Restrictive dermopathy—a case report. Indian Pediatrics39:1149–1152
21. Mau U, Kendziorra H, Kaiser P, Enders H (1997) Restrictivedermopathy: report and review. Am J Med Genet 71:179–185
22. Mok Q, Curley R, Tolmie JL, Marsden RA, Patton MA, DaviesEG (1990) Restrictive dermopathy: a report of three cases. J MedGenet 27:315–319
23. Moulson CL, Gloriosa G, van der Wal AC, Sillevis Smitt JH, vanHagen JM, Miner JH (2005) Homocygous and compound hetero-zygous mutations in ZMPSTE24 cause the laminopathy restrictivedernopathy. J Invest Dermatol 125(5):913–919
24. Mulder EJH, Beemer FA, Stoutenbeek P (2001) Restrictive der-mopathy and fetal behaviour. Prenat Diagn 21:581–585
25. Navarro CL, De Sandre-Giovannoli A, Bernard R, Boccaccio I,Boyer A, Geneviève D, Hadj-Rabia S, Gaudy-Marqueste C, SmittHS, Vabres P, Faivre L, Verloes A, Van Essen T, Flori E, Henne-kam R, Beemer FA, Laurent N, Le Merrer M, Cau P, Lévy N(2004) Lamin A and ZMPSTE24 (FACE-1) defects cause nucleardisorganization and identify restrictive dermopathy as a lethal neo-natal laminopathy. Hum Mol Genet 13:2493–2503
26. Navarro CL, Cadinanos J, De Sandre-Giovannoli A, Bernard R,Courrier S, Boccaccio I, Boyer I, Kleijer WJ, Wagner A, GiulianoF, Beemer FA, Freije JM, Cau P, Hennecam RC, López-Otin C,Badens C, Lévy N (2005) Loss of ZMPSTE24 (FACE-1) causesautosomal recessive restrictive dermopathy and accumulation oflamin A precursors. Hum Mol Genet 14:1503–1513
27. Nijsten TEC, De Moor A, Colpaert CG, Robert K, Mathieu LM,Lambert J (2002) Restrictive dermopathy: a case report and a criticalreview of all hypotheses of its origin. Pediatr Dermatol 19(1):67–72
28. Paige DG, Lake BD, Ramani P, Harper JI (1992) Restrictive der-mopathy: a disorder of Wbroblasts. Br J Dermatol 127:630–634
29. Pierard-Franchimont C, Pierard GE, Hermanns-Le T, Estrada JA,Verloes A, Mulliez N (1992) Dermatopathological aspects ofrestrictive dermopathy. J Pathol 167:223–228
30. Reed MH, Chudley AE, Kroeker M, Wilmont DM (1993) Restric-tive dermopathy. Pediatr Radiol 23:617–619
31. Rusinol AE, Sinensky MS (2006) Farnesylated lamins, progeroidsyndromes and farnesyl transferase inhibitors. J Cell Sci119(Pt16):3265–3272
123

Arch Gynecol Obstet
32. Schnur RE, Ashmead J, Kelley RI (1985) A lethal ichthyosis var-iant with arthrogyposis. Am J Hum Genet A 37:76
33. Sergi C, Poeschl J, Graf M, Linderkamp O (2001) Restrictive der-mopathy: case report, subject review with Kaplan–Meier analysis,and diVerential diagnosis of the lethal congenital contractural syn-dromes. Am J Perinatol 18(1):39–47
34. Sillevis Smitt JH, van Asperen CJ, Niessen CM, Beemer FA, vanEssen AJ, Hulsman RFHJ, Oranje AP, Steiijlen PM, Wesby-vanSwaay E, Tamminga P, Breslau-Siderius EJ (1998) Restrictivedermopathy. Report of 12 cases. Dutch Task Force on Genoderma-tology. Arch Dermatol 134:577–579
35. Teasdale RD, Jackson MR (1996) Signal-mediated sorting ofmembrane proteins between the endoplasmic reticulum and theGolgi apparatus. Annu Rev Cell Dev Biol 12:27–54
36. Thakur S, Pal L, Phadke SR (2004) Lethal arthrogryposis withichtiosis: overlap with Neu-Laxova syndrome, restrictive der-mopathy and harlequin fetus. Clin Dysmorphol 13(2):117–119
37. Toriello HV, Higgins JV, Waterman DF (1983) Autosomal reces-sive aplasia cutis congenita: a report of two aVected sibs. Am JMed Genet 15:153–156
38. Toriello HV (1986) Invited editorial comment: restrictive der-mopathy and report of another case. Am J Med Genet 24:625–629
39. Van der Stege JG, van Straaten HLM, van der Wal AC, van Eyck J(1997) Restrictive dermopathy and associated prenatal ultrasoundWndings: case report. Ultrasound Obstet Gynecol 10:140–141
40. Van Hoestenberghe M, Legius E, Vandevoorde W, Eykens A,Jaeken J, Eggermont E, Devos R, De Wolf-Peeters C, Fryns JP
(1990) Restrictive dermopathy with distinct morphological abnor-malities. Am J Med 36:297–300
41. Verloes A, Mulliez N, Gonzales M, Laloux F, Hermanns-Le T, Pi-érard GE, Koulischer L (1992) Restrictive dermopathy, a letal formof arthogryposis multiplex with skin and bone dysplasias: three newcases and review of literature. Am J Med Genet 43:539–547
42. Wehnert M, Nguyen TD, Bethmann C, Hausser I, Albrecht B(2006) Mutational analysis of LMNA and ZMPSTE24 in restric-tive laminopathy. Eur J Hum Genet 14(Suppl 1):232
43. Welsh KM, Smoller BR, Kolbrook KA, Johnston K (1992)Restrictive dermopathy: report of two aVected siblings and a re-view of literature. Arch Dermatol 128:228–231
44. Wesche WA, Cutlan RT, Khare V, Chesney T, Shanklin D (2001)Restrictive dermopathy: report of a case and review of literature. JCutan Pathol 28:211–218
45. White MB, Carvalho M, Derse D, O’Brien SJ, Dean M (1992)Detecting single base substitutions as heteroduplex polymor-phisms. Genomics 12:301–306
46. Witt DR, Hayden MR, Holbrook KA, Dale BA, Baldwin VJ,Taylor GP (1986) Restrictive dermopathy: a newly recognisedautosomal recessive skin dysplasia. Am J Med Genet 24:631–648
47. Witters I, Moerman P, Fryns JP (2002) Fetal akinesia deformationsequence: a study of 30 consecutive in utero diagnoses. Am J MedGenet 113:23–28
48. Young SG, Meta M, Yang SH, Fong LG (2006) Prelamin A farn-esylation and progeroid syndromes. J Biol Chem 281(52):39741–39745
123




















