
Ammonium removal by erythrocyte‑bioreactors based ... - Nature

lable at ScienceDirect
Solid State Sciences 12 (2010) 1822e1830
Contents lists avai
Solid State Sciences
journal homepage: www.elsevier .com/locate/ssscie
Reactivity of MgeAl hydrotalcites in solid and delaminated forms in ammoniumcarbonate solutions
Georgiana Stoica a, Marta Santiago a, Sònia Abelló a, Javier Pérez-Ramírez a,b,c,*
a Institute of Chemical Research of Catalonia (ICIQ), Avinguda dels Països Catalans 16, 43007 Tarragona, SpainbCatalan Institution for Research and Advanced Studies (ICREA), Passeig de Lluís Companys 23, 08010 Barcelona, Spainc Institute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, HCI E 125, Wolfgang-Pauli-Strasse 10, CH-8093 Zurich, Switzerland
a r t i c l e i n f o
Article history:Received 24 December 2009Received in revised form20 July 2010Accepted 4 August 2010Available online 13 August 2010
Keywords:HydrotalciteLDHDelaminationColloidAmmonium carbonateCompositeDawsonite
* Corresponding author. Institute for Chemical andof Chemistry and Applied Biosciences, ETH Zurich,Strasse 10, CH-8093 Zurich, Switzerland. Fax: þ41 44
E-mail address: [email protected] (J. Pérez-Ramíre
1293-2558/$ e see front matter � 2010 Elsevier Masdoi:10.1016/j.solidstatesciences.2010.08.003
a b s t r a c t
Treatment of MgeAl hydrotalcites (LDHs, layered double hydroxides) in aqueous (NH4)2CO3 at 298 Kleads to composites of dawsonite, hydrotalcite, and magnesium ammonium carbonate. The mechanismand kinetics of this transformation, ultimately determining the relative amounts of these components inthe composite, depend on the treatment time (from 1 h to 9 days), the Mg/Al ratio in the hydrotalcite (2-4), and on the starting layered double hydroxide (solid or delaminated form). The materials at variousstages of the treatment were characterized by inductive coupled plasma-optical emission spectroscopy,X-ray diffraction, transmission electron microscopy, infrared spectroscopy, thermogravimetry, andnitrogen adsorption at 77 K. The progressive transformation of hydrotalcite towards crystallinedawsonite and magnesium ammonium carbonate phases follows a dissolutioneprecipitation mecha-nism. A gradual decrease of the Mg/Al ratio in the resulting solids was observed in time due tomagnesium leaching in the reacting medium. Dawsoniteehydrotalcite composite formation is favored athigh aluminum contents in the starting hydrotalcite, while the formation of magnesium ammoniumcarbonate is favored at high Mg/Al ratios. The synthetic strategy comprising hydrotalcite delamination informamide prior to aqueous (NH4)2CO3 treatment is more reactive towards composite formation thanstarting from the bulk solid hydrotalcite.
� 2010 Elsevier Masson SAS. All rights reserved.
1. Introduction
Hydrotalcite-like compounds (HTlcs) or layered double hydrox-ides (LDHs) have a broad spectrum of applications, as catalystprecursor or support, in separation and membrane technologies,filtration, scavenging and controlled release of anions, etc. LDHsconsist of layers of positively charged nanosheets with brucite-typestructure neutralized by anions in the interlayer space, wherewatermolecules are also present [1].
In addition to the direct synthesis by coprecipitation from saltprecursors, HTlcs have been subject of many post-synthesis trans-formations involving their well-known memory effect propertyand ion-exchange capacity [2e7]. That is, by recovering of thelayered structure through calcination of the starting hydrotalciteand subsequent contact of the derived mixed oxide with solutions
Bioengineering, DepartmentHCI E 125, Wolfgang-Pauli-633 14 05.z).
son SAS. All rights reserved.
containing many different anions, or by direct exchange of theanions located in the interlayer space by others, an impressivevariety of hydrotalcite-derived compounds have been synthesized.
Focusing on the layered nature of these compounds, LDHs actlike the host structure for polymers, bio-active compounds, pillar-ing agents, etc., leading to nanocompositematerials [7,8]. The use ofintercalating agents by ion-exchange, like sodium dodecyl sulfateor alkyl carboxylates [7], followed by exfoliation induces theseparation of the layers into unilamellar positive nanosheets(colloids) [9e11]. Polar [9,12e16] or nonpolar [17,18] solvents aidedby heat [18], ultrasounds [18], or electric current [19] were alsoapplied for delamination of HTlcs. Hibino and Jones [12] introducedthe exfoliation of intercalated LDHs in formamide without the needof heat or reflux treatments, based on a two-stage mechanism:rapid swelling due to the ability of HCONH2 for hydrogen bondingfollowed by slow exfoliation [20e22].
The subsequent restacking of the hydrotalcite nanosheetsenables to fabricate nanostructured materials, offering an elegantalternative to the traditional intercalation of anionic species in theirinterlayer space by ion-exchange [23]. The restacking of colloidalLDHs, leading to the recovery of the native hydrotalcite structure,

G. Stoica et al. / Solid State Sciences 12 (2010) 1822e1830 1823
has been practiced by physical (solvent evaporation [24,25], freeze-drying [8]) or chemical methods (treatment in aqueous solutionsof Na2CO3 [26] or NaCl [8,10], and ethanol [26]). Grounded on thedelamination-stacking concept, more sophisticated materials wererecently reported. Venugopal et al. [25] prepared a randomly cos-tacked MgeAl/CoeAl LDH compound starting from colloidaldispersions of the individual hydrotalcites. Li et al. [27] applieda layer-by-layer assembly to obtain interstratified thin films ofpositively charged MgeAl LDH nanosheets and negatively chargedoxide nanosheets (Ti0.91O2 and Ca2Nb3O10). In the cases reported sofar, the fabrication of multicomponent composites requires startingfrom all individual components in delaminated form.
Rather than the typical delamination and restacking of thehydrotalcite phase, we endeavor the transformation of the LDH intoother structured compounds. Herein, the reactivity of MgeAlhydrotalcites in solid or colloidal (delaminated) forms in aqueousammonium carbonate at ambient conditions was investigated.Delamination was carried out by treatment of the as-synthesizedhydrotalcites in formamide. Composite materials consisting ofmixtures of hydrotalcite, dawsonite, and magnesium ammoniumcarbonate were obtained. Based on detailed characterizationstudies, it is concluded that the composition of the resultingcomposite is determined by the Mg/Al ratio of the starting hydro-talcite and the treatment time. The terms ‘delamination’ and‘exfoliation’ are used indistinctly along the manuscript.
2. Experimental
2.1. Materials and treatments
Synthesis of hydrotalcites. MgeAl hydrotalcites (HTx) with molarMg/Al ratios (x) of 2, 3, and 4 were synthesized by continuouscoprecipitation using the ILDP (in-line dispersioneprecipitation)method [28,29]. Briefly, aqueous solutions of Mg(NO3)2$6H2O(0.25$xM) and Al(NO3)3$9H2O (0.25 M) and the precipitating agent(NaOHþNa2CO3,1 M of each) were pumped into a 6 cm3-reactor toa high-shear homogenizer rotating at 13,500 rpm. The pH of theslurry was measured and controlled by an in-line probe directly atthe outlet of the precipitation chamber. The syntheses were carriedout at pH10with an average residence time in the reactorof 18 s. Theprecipitate slurries were aged at 298 K for 12 h under mechanicalstirring (500 rpm), followed by filtration, washing, and drying at333 K for 12 h.
As detailed below, the synthetic strategy for the formation ofcomposites followed two different experimental approaches, thefirst one by subjecting the hydrotalcite to delamination andsubsequent treatment in (NH4)2CO3 solutions and the second one,by treating directly the as-synthesized hydrotalcites with the samebasic solution.
Delamination. A dispersion of the as-synthesized hydrotalcite inpure formamide (purity 99.5%) was prepared at a concentration of5 g l�1. In order to accelerate the exfoliation process, the suspensionwas ultrasonicated six times for a period of 30 min with an intervalof 60 min between treatments. Then, the sample was left to standfor 12 h. The system remained turbid and was centrifuged at4400 rpm for 30 min and two phases were observed: a colloidalsuspension and the sediment. The supernatant colloidal dispersion,referred to as HTDx was collected and used for further treatment.
Treatment in (NH4)2CO3. The colloidal suspension containing thedelaminated hydrotalcite was treated in aqueous 1 M (NH4)2CO3 atroom temperature in a 1:3 volume ratio. After stirring for 1 h, themixture was left to rest at ambient conditions for periods of timeranging from 1 h to 9 days. The resulting solid was centrifuged at4400 rpm for 30 min and redispersed in ethanol or water andcentrifuged again at 4400 rpm for 30 min each. The washing
procedure was repeated three times and the final solids were driedat 333 K for 12 h. The as-synthesized hydrotalcites in solid formwere also treated in 1 M aqueous solution of (NH4)2CO3 at roomtemperature using the same solid-to-liquid ratio as in the treat-ment of the colloid, stirred for 1 h, and let to stand for 1e3 days. Theresulting solids were filtered, washed with deionized water, anddried at 333 K for 12 h.
Along the manuscript, samples were designated by the codesHTx-t or Rx-t, where HT refers to the protocol initiated from thesolid hydrotalcite, R identifies the solids attained via the colloidalsolutions, x is the Mg/Al ratio of the starting hydrotalcite, and t isthe treatment time expressed in hours (h) or days (d).
Reference samples. Reference dawsonite (DW) and magnesiumammonium carbonate (MAC) were synthesized in order to supportassignments during transformation of the hydrotalcites. DW(NH4AlCO3(OH)2)was prepared at constant pHbyprecipitation of Al(NO3)3$9H2O (1.1 M) and (NH4)2CO3 (2 M) aqueous solutions usingthe ILDP method described above. The slurry was then filtered,washed with water, and dried at 333 K for 12 h. Calcination of theresulting solid at 723 K for 2 h, and further treatment in 1 M(NH4)2CO3 were applied to obtain reformed dawsonite [30]. Theprecipitate was then filtered, washedwith water, and dried at 333 Kfor 12 h. MAC ((NH4)2Mg(CO3)2$4H2O) was prepared by ILDP,adapting the recipe by Erdös et al. [31]. An aqueous solution ofMgSO4 (0.5 M) was precipitated with an excess of 5 M (NH4)2CO3
aqueous solution at pH 8.5. The precipitate was washed withmethanol and dried at 333 K for 12 h.
2.2. Techniques
Magnesium and aluminum in the as-synthesized samples andsome of the filtrates upon (NH4)2CO3 treatment were determinedby inductive coupled plasma-optical emission spectroscopy (ICP-OES) (PerkineElmer Optima 3200RL (radial)). Powder X-raydiffraction (XRD) patterns were measured in a Bruker AXS D8Advance diffractometer equippedwith a Cu tube, a Ge(111) incidentbeam monochromator, and a Vantec-1 PSD. Data were recorded inthe range of 5e70� 2q with an angular step size of 0.016� anda counting time of 6 s per step. Fourier transform infrared (FTIR)spectroscopy was carried out in a Bruker Optics Tensor 27 spec-trometer equipped with a Golden Gate Diamond ATR unit. Spectrawere collected in the range 650e4000 cm�1 by co-addition of 32scans at a nominal resolution of 4 cm�1, taking the spectrum of theempty cell as the background. Thermogravimetric analysis (TGA)was carried out in a Mettler Toledo TGA/SDTA851e microbalance.Analyses were performed in dry air flow of 50 cm3 min�1 from 298to 1173 K using a heating rate of 5 K min�1. Transmission ElectronMicroscopy was carried out in a JEOL JEM-1011 microscope oper-ated at 100 kV and equipped with a SIS Megaview III CCD camera. Afew droplets of the sample suspended in ethanol were placed ona carbon-coated copper grid followed by evaporation at ambientconditions. Nitrogen isotherms at 77 K were measured on a Quan-tachrome Autosorb-1MP analyzer. Prior to the analysis, the sampleswere degassed in vacuum at 373 K for 16 h. The BET method [32]was applied to calculate the total surface area and the t-plotmethod [33] was used to discriminate between micro- and meso-porosity.
3. Results and discussion
3.1. Parent samples and exfoliation
The chemical composition of the as-synthesized hydrotalcitesmeasured by ICP-OES indicated that the Mg/Al molar ratio in thesolids were 1.8, 2.9, and 3.3, i.e. close to the nominal ratio in the
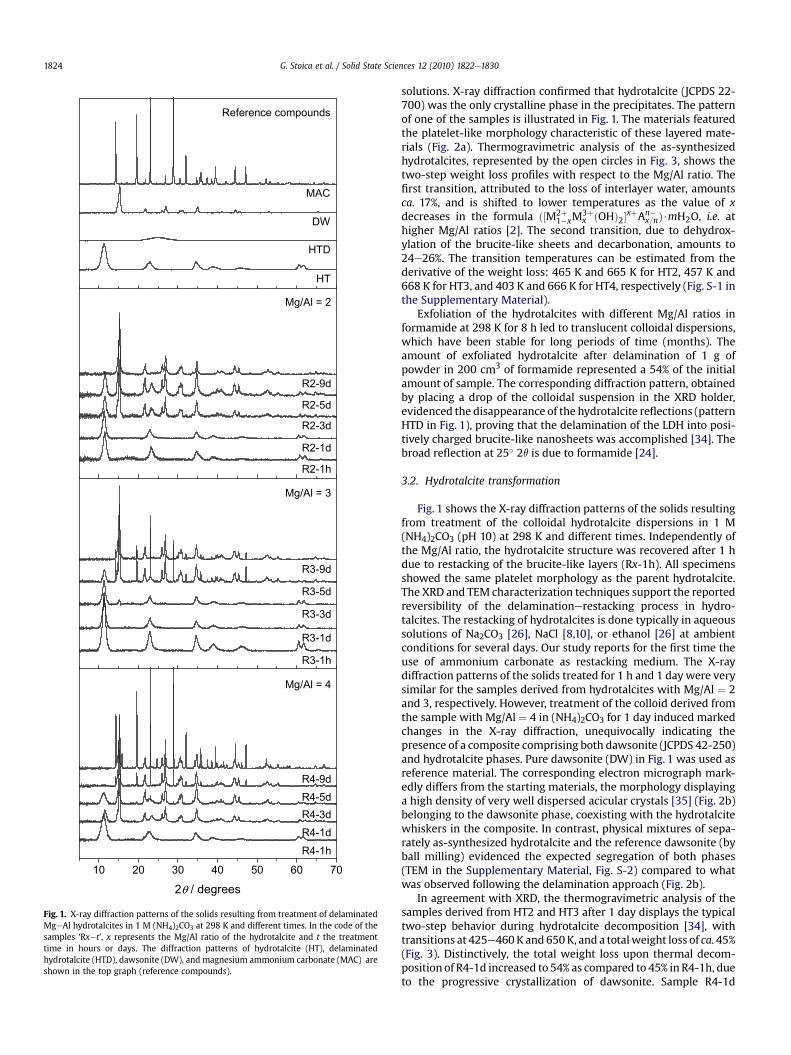
10 20 30 40 50 60 70
2θ / degrees
DW
MAC
HT
Reference compounds
R3-5d
R3-9d
R3-3d
R3-1h
R3-1d
Mg/Al = 3
R4-5dR4-9d
R4-3d
R4-1hR4-1d
Mg/Al = 4
R2-5d
R2-9d
R2-3d
R2-1h
R2-1d
Mg/Al = 2
HTD
Fig. 1. X-ray diffraction patterns of the solids resulting from treatment of delaminatedMgeAl hydrotalcites in 1 M (NH4)2CO3 at 298 K and different times. In the code of thesamples ‘Rx�t’, x represents the Mg/Al ratio of the hydrotalcite and t the treatmenttime in hours or days. The diffraction patterns of hydrotalcite (HT), delaminatedhydrotalcite (HTD), dawsonite (DW), and magnesium ammonium carbonate (MAC) areshown in the top graph (reference compounds).
G. Stoica et al. / Solid State Sciences 12 (2010) 1822e18301824
solutions. X-ray diffraction confirmed that hydrotalcite (JCPDS 22-700) was the only crystalline phase in the precipitates. The patternof one of the samples is illustrated in Fig. 1. The materials featuredthe platelet-like morphology characteristic of these layered mate-rials (Fig. 2a). Thermogravimetric analysis of the as-synthesizedhydrotalcites, represented by the open circles in Fig. 3, shows thetwo-step weight loss profiles with respect to the Mg/Al ratio. Thefirst transition, attributed to the loss of interlayer water, amountsca. 17%, and is shifted to lower temperatures as the value of xdecreases in the formula ð½M2þ
1�xM3þx ðOHÞ2�xþAn�
x=nÞ$mH2O, i.e. athigher Mg/Al ratios [2]. The second transition, due to dehydrox-ylation of the brucite-like sheets and decarbonation, amounts to24e26%. The transition temperatures can be estimated from thederivative of the weight loss: 465 K and 665 K for HT2, 457 K and668 K for HT3, and 403 K and 666 K for HT4, respectively (Fig. S-1 inthe Supplementary Material).
Exfoliation of the hydrotalcites with different Mg/Al ratios informamide at 298 K for 8 h led to translucent colloidal dispersions,which have been stable for long periods of time (months). Theamount of exfoliated hydrotalcite after delamination of 1 g ofpowder in 200 cm3 of formamide represented a 54% of the initialamount of sample. The corresponding diffraction pattern, obtainedby placing a drop of the colloidal suspension in the XRD holder,evidenced the disappearance of the hydrotalcite reflections (patternHTD in Fig. 1), proving that the delamination of the LDH into posi-tively charged brucite-like nanosheets was accomplished [34]. Thebroad reflection at 25� 2q is due to formamide [24].
3.2. Hydrotalcite transformation
Fig. 1 shows the X-ray diffraction patterns of the solids resultingfrom treatment of the colloidal hydrotalcite dispersions in 1 M(NH4)2CO3 (pH 10) at 298 K and different times. Independently ofthe Mg/Al ratio, the hydrotalcite structure was recovered after 1 hdue to restacking of the brucite-like layers (Rx-1h). All specimensshowed the same platelet morphology as the parent hydrotalcite.The XRD and TEM characterization techniques support the reportedreversibility of the delaminationerestacking process in hydro-talcites. The restacking of hydrotalcites is done typically in aqueoussolutions of Na2CO3 [26], NaCl [8,10], or ethanol [26] at ambientconditions for several days. Our study reports for the first time theuse of ammonium carbonate as restacking medium. The X-raydiffraction patterns of the solids treated for 1 h and 1 daywere verysimilar for the samples derived from hydrotalcites with Mg/Al ¼ 2and 3, respectively. However, treatment of the colloid derived fromthe sample with Mg/Al ¼ 4 in (NH4)2CO3 for 1 day induced markedchanges in the X-ray diffraction, unequivocally indicating thepresence of a composite comprising both dawsonite (JCPDS 42-250)and hydrotalcite phases. Pure dawsonite (DW) in Fig. 1 was used asreference material. The corresponding electron micrograph mark-edly differs from the starting materials, the morphology displayinga high density of very well dispersed acicular crystals [35] (Fig. 2b)belonging to the dawsonite phase, coexisting with the hydrotalcitewhiskers in the composite. In contrast, physical mixtures of sepa-rately as-synthesized hydrotalcite and the reference dawsonite (byball milling) evidenced the expected segregation of both phases(TEM in the Supplementary Material, Fig. S-2) compared to whatwas observed following the delamination approach (Fig. 2b).
In agreement with XRD, the thermogravimetric analysis of thesamples derived from HT2 and HT3 after 1 day displays the typicaltwo-step behavior during hydrotalcite decomposition [34], withtransitions at 425e460K and 650K, and a totalweight loss of ca. 45%(Fig. 3). Distinctively, the total weight loss upon thermal decom-position of R4-1d increased to 54% as compared to 45% in R4-1h, dueto the progressive crystallization of dawsonite. Sample R4-1d

Fig. 2. Transmission electron micrographs of representative samples: (a) starting hydrotalcite (sample HT2), (b) dawsonite-hydrotalcite composite (sample R2-5d), (c) puredawsonite (sample R2-9d), (d) dawsonite-magnesium ammonium carbonate composite (sample R3-9d). The same scale bar applies to all the micrographs.
G. Stoica et al. / Solid State Sciences 12 (2010) 1822e1830 1825
displays a two-step weight loss profile with a significant decom-position temperature centered at ca. 450 K [30,35], matching thedecomposition temperature of NH4-dawsonite (dashed line inFig. S-1). Concomitantly, the weight loss of the second transition(>450 K), which is exclusively attributed to the HT phase, decreasesdue to the gradual conversion of hydrotalcite into dawsonite. Wehave quantified the relative amount of both phases from thermog-ravimetry (Fig. 4) as detailed in Table S-1 and Fig. S-3 in theSupplementary Material. Dawsonite is formed at the expense ofhydrotalcite and the percentage of the former phase in the R4-1dsample is 53%.
Characterization by infrared spectroscopy (Fig. 5) substantiatesX-ray diffraction in the time scale at which phases appear in thesamples. For the sake of conciseness, we illustrate the spectra forthe R2-t system as equivalent observations were obtained for theother two Mg/Al ratios. The infrared spectrum of R2-1h featurescharacteristic bands of hydrotalcite [34,36]: 3480 cm�1 (OHstretching), 3050 cm�1 (bridging mode of carbonate and water inthe interlayer),1370 cm�1 (n3mode of the carbonate, antisymmetricstretching), 855 cm�1 (n2 mode of the carbonate, out-of-planedeformation), and 655 cm�1 (Mg-related OH translation modes).The vibration at 1640 cm�1 is due to the bendingmode of water. Theappearance of the n3 mode of the carbonate at 1370 cm�1 (being1415 cm�1 in the ‘free’ carbonate anion) is related to its reorgani-zation in the interlayer space due to electrostatic interaction withthe nearby brucite-like layers [37]. Due to the loss of symmetry ofthe carbonate ion (from D3h to C2v), the n1 mode of the carbonate(symmetric stretching) becomes activated, thus leading to theshoulder at 1045 cm�1 [37]. This vibration mode is inactive whenthe carbonate ion retains its full symmetry [38].
The infrared spectrum of the sample after 1 day reveals newbands characteristic of NH4-dawsonite, although the relativelysmall amount of this phase was not discernible by XRD. The most
relevant ones are marked by solid dots in Fig. 5 and belong to OH�
(dOH at 985 cm�1), NH4þ (nNH at 3180 cm�1), and CO3
2� (n3 at 1560 and1450 cm�1, n2 at 855 cm�1, and n1 at 1105 cm�1) [30,39]. Forcomparison, the spectrum of a pure dawsonitewas also added, thusconfirming the presence of dawsonite in the composite. As expec-ted, treatment in ammoniumcarbonate for longer times (3 days) ledto the presence of the characteristic dawsonite reflectionswith different intensities in all the samples, clearly indicating theformation of the dawsonite-hydrotalcite composite (Fig. 1). Besides,sample R4-3d contains low-intensity reflections assigned tomagnesium ammonium carbonate hydrated (MAC, (NH4)2Mg(CO3)2$4H2O, JCPDS 33-66), as it can be seen bycomparisonwith thereferencematerial in Fig.1. Extension of the treatment for 5 days ledto the progressive hydrotalcite disappearance in R2-5d and R3-5d,respectively, which is complete in R4-5d. The higher theMg/Al ratioin the parent material, the higher the amount of MAC formed.
The reflections of both dawsonite and magnesium ammoniumcarbonate hydrate phases become more intense in Rx-9d asa consequence of the enhanced crystallization at longer treatmenttimes. These changes were also observed by transmission electronmicroscopy. After 9 days, typical dawsonite particles are predomi-nant in R2-9d (Fig. 2c), while samples R3-9d and R4-9d still displaya mixed morphology of the porous dawsonite particles and MAC(Fig. 2d). The composites containing MAC could be purified bysimply washing with water, since the latter phase is highly solublein water.
In agreementwith X-ray diffraction, the infrared analysis revealsnew bands in R2-5d characteristic to dawsonite (solid dots in Fig. 5)at 3070 cm�1 (nNH). R2-9d displays more developed and additionalcharacteristic dawsonite bands at 2836 cm�1 (nNH) and 1830 and1720 cm�1 (dNH), 735 cm�1 (n4 mode of carbonate), and 635 cm�1
(AleO stretching vibration). The spectra also show that the nOH andthe n3(CO3
2�) modes in MgeAl hydrotalcite (3480 and 1370 cm�1,
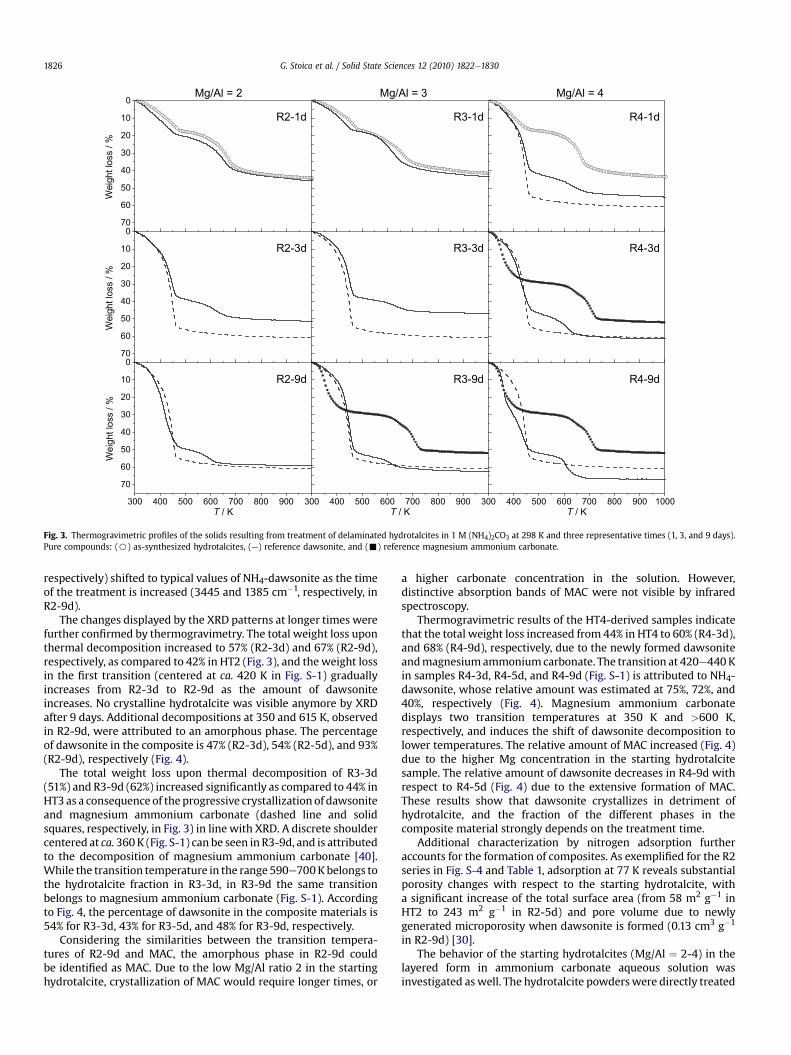
300 400 500 600 700 800 900T / K
70
60
50
40
30
20
10
0% / ssol thgie
W
70
60
50
40
30
20
10
0
% / ssol thgieW
300 400 500 600 700 800 900
70
60
50
40
30
20
10
0
% / ssol thgieW
T / K300 400 500 600 700 800 900 1000
T / K
Mg/Al = 2 Mg/Al = 3 Mg/Al = 4
R2-9d
R2-3d
R2-1d
R3-9d
R3-3d
R4-9d
R4-3d
R4-1dR3-1d
Fig. 3. Thermogravimetric profiles of the solids resulting from treatment of delaminated hydrotalcites in 1 M (NH4)2CO3 at 298 K and three representative times (1, 3, and 9 days).Pure compounds: (B) as-synthesized hydrotalcites, (—) reference dawsonite, and (-) reference magnesium ammonium carbonate.
G. Stoica et al. / Solid State Sciences 12 (2010) 1822e18301826
respectively) shifted to typical values of NH4-dawsonite as the timeof the treatment is increased (3445 and 1385 cm�1, respectively, inR2-9d).
The changes displayed by the XRD patterns at longer times werefurther confirmed by thermogravimetry. The total weight loss uponthermal decomposition increased to 57% (R2-3d) and 67% (R2-9d),respectively, as compared to 42% in HT2 (Fig. 3), and theweight lossin the first transition (centered at ca. 420 K in Fig. S-1) graduallyincreases from R2-3d to R2-9d as the amount of dawsoniteincreases. No crystalline hydrotalcite was visible anymore by XRDafter 9 days. Additional decompositions at 350 and 615 K, observedin R2-9d, were attributed to an amorphous phase. The percentageof dawsonite in the composite is 47% (R2-3d), 54% (R2-5d), and 93%(R2-9d), respectively (Fig. 4).
The total weight loss upon thermal decomposition of R3-3d(51%) and R3-9d (62%) increased significantly as compared to 44% inHT3 as a consequence of the progressive crystallization of dawsoniteand magnesium ammonium carbonate (dashed line and solidsquares, respectively, in Fig. 3) in line with XRD. A discrete shouldercentered at ca. 360K (Fig. S-1) can be seen in R3-9d, and is attributedto the decomposition of magnesium ammonium carbonate [40].While the transition temperature in the range 590e700Kbelongs tothe hydrotalcite fraction in R3-3d, in R3-9d the same transitionbelongs to magnesium ammonium carbonate (Fig. S-1). Accordingto Fig. 4, the percentage of dawsonite in the composite materials is54% for R3-3d, 43% for R3-5d, and 48% for R3-9d, respectively.
Considering the similarities between the transition tempera-tures of R2-9d and MAC, the amorphous phase in R2-9d couldbe identified as MAC. Due to the low Mg/Al ratio 2 in the startinghydrotalcite, crystallization of MAC would require longer times, or
a higher carbonate concentration in the solution. However,distinctive absorption bands of MAC were not visible by infraredspectroscopy.
Thermogravimetric results of the HT4-derived samples indicatethat the total weight loss increased from 44% in HT4 to 60% (R4-3d),and 68% (R4-9d), respectively, due to the newly formed dawsoniteandmagnesiumammoniumcarbonate. The transition at 420e440Kin samples R4-3d, R4-5d, and R4-9d (Fig. S-1) is attributed to NH4-dawsonite, whose relative amount was estimated at 75%, 72%, and40%, respectively (Fig. 4). Magnesium ammonium carbonatedisplays two transition temperatures at 350 K and >600 K,respectively, and induces the shift of dawsonite decomposition tolower temperatures. The relative amount of MAC increased (Fig. 4)due to the higher Mg concentration in the starting hydrotalcitesample. The relative amount of dawsonite decreases in R4-9d withrespect to R4-5d (Fig. 4) due to the extensive formation of MAC.These results show that dawsonite crystallizes in detriment ofhydrotalcite, and the fraction of the different phases in thecomposite material strongly depends on the treatment time.
Additional characterization by nitrogen adsorption furtheraccounts for the formation of composites. As exemplified for the R2series in Fig. S-4 and Table 1, adsorption at 77 K reveals substantialporosity changes with respect to the starting hydrotalcite, witha significant increase of the total surface area (from 58 m2 g�1 inHT2 to 243 m2 g�1 in R2-5d) and pore volume due to newlygenerated microporosity when dawsonite is formed (0.13 cm3 g�1
in R2-9d) [30].The behavior of the starting hydrotalcites (Mg/Al ¼ 2-4) in the
layered form in ammonium carbonate aqueous solution wasinvestigated aswell. The hydrotalcite powderswere directly treated
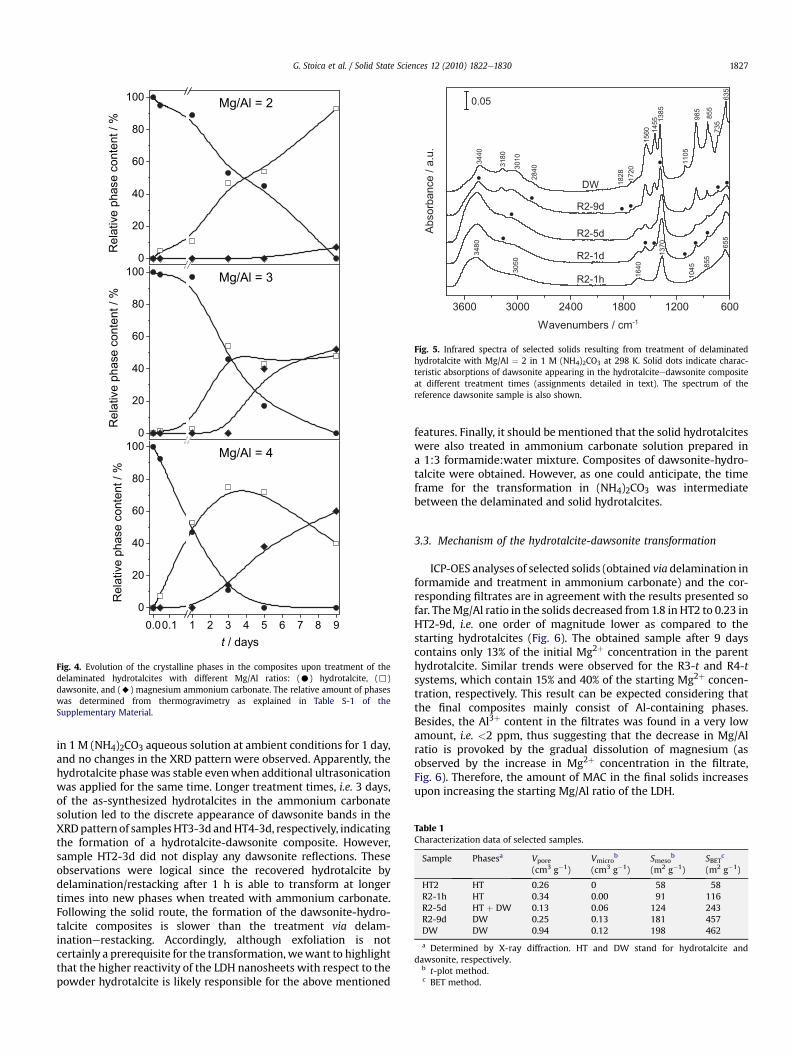
0
20
40
60
80
100% / tnetnoc esahp evitale
R
0
20
40
60
80
100
% / tnetnoc esahp evitaleR
0.00.1 1 2 3 4 5 6 7 8 90
20
40
60
80
100
% / tnetnoc esahp evitaleR
t / days
Mg/Al = 2
Mg/Al = 3
Mg/Al = 4
Fig. 4. Evolution of the crystalline phases in the composites upon treatment of thedelaminated hydrotalcites with different Mg/Al ratios: (C) hydrotalcite, (,)dawsonite, and (A) magnesium ammonium carbonate. The relative amount of phaseswas determined from thermogravimetry as explained in Table S-1 of theSupplementary Material.
3600 3000 2400 1800 1200 600
.u.a / ecnabrosbA
Wavenumbers / cm-1
R2-9d
DW
R2-5d
R2-1h
R2-1d
0443
5831
0813
0651
5011
5541
589
558
82810271
0482
537536
0103
0461
558
0503
0843
5401
5560731
0.05
Fig. 5. Infrared spectra of selected solids resulting from treatment of delaminatedhydrotalcite with Mg/Al ¼ 2 in 1 M (NH4)2CO3 at 298 K. Solid dots indicate charac-teristic absorptions of dawsonite appearing in the hydrotalciteedawsonite compositeat different treatment times (assignments detailed in text). The spectrum of thereference dawsonite sample is also shown.
Table 1Characterization data of selected samples.
Sample Phasesa Vpore
(cm3 g�1)Vmicro
b
(cm3 g�1)Smeso
b
(m2 g�1)SBET
c
(m2 g�1)
HT2 HT 0.26 0 58 58R2-1h HT 0.34 0.00 91 116R2-5d HT þ DW 0.13 0.06 124 243R2-9d DW 0.25 0.13 181 457DW DW 0.94 0.12 198 462
a Determined by X-ray diffraction. HT and DW stand for hydrotalcite anddawsonite, respectively.
b t-plot method.c BET method.
G. Stoica et al. / Solid State Sciences 12 (2010) 1822e1830 1827
in 1 M (NH4)2CO3 aqueous solution at ambient conditions for 1 day,and no changes in the XRD pattern were observed. Apparently, thehydrotalcite phase was stable evenwhen additional ultrasonicationwas applied for the same time. Longer treatment times, i.e. 3 days,of the as-synthesized hydrotalcites in the ammonium carbonatesolution led to the discrete appearance of dawsonite bands in theXRDpattern of samplesHT3-3d andHT4-3d, respectively, indicatingthe formation of a hydrotalcite-dawsonite composite. However,sample HT2-3d did not display any dawsonite reflections. Theseobservations were logical since the recovered hydrotalcite bydelamination/restacking after 1 h is able to transform at longertimes into new phases when treated with ammonium carbonate.Following the solid route, the formation of the dawsonite-hydro-talcite composites is slower than the treatment via delam-inationerestacking. Accordingly, although exfoliation is notcertainly a prerequisite for the transformation, wewant to highlightthat the higher reactivity of the LDH nanosheets with respect to thepowder hydrotalcite is likely responsible for the above mentioned
features. Finally, it should be mentioned that the solid hydrotalciteswere also treated in ammonium carbonate solution prepared ina 1:3 formamide:water mixture. Composites of dawsonite-hydro-talcite were obtained. However, as one could anticipate, the timeframe for the transformation in (NH4)2CO3 was intermediatebetween the delaminated and solid hydrotalcites.
3.3. Mechanism of the hydrotalcite-dawsonite transformation
ICP-OES analyses of selected solids (obtained via delamination informamide and treatment in ammonium carbonate) and the cor-responding filtrates are in agreement with the results presented sofar. TheMg/Al ratio in the solids decreased from1.8 in HT2 to 0.23 inHT2-9d, i.e. one order of magnitude lower as compared to thestarting hydrotalcites (Fig. 6). The obtained sample after 9 dayscontains only 13% of the initial Mg2þ concentration in the parenthydrotalcite. Similar trends were observed for the R3-t and R4-tsystems, which contain 15% and 40% of the starting Mg2þ concen-tration, respectively. This result can be expected considering thatthe final composites mainly consist of Al-containing phases.Besides, the Al3þ content in the filtrates was found in a very lowamount, i.e. <2 ppm, thus suggesting that the decrease in Mg/Alratio is provoked by the gradual dissolution of magnesium (asobserved by the increase in Mg2þ concentration in the filtrate,Fig. 6). Therefore, the amount of MAC in the final solids increasesupon increasing the starting Mg/Al ratio of the LDH.
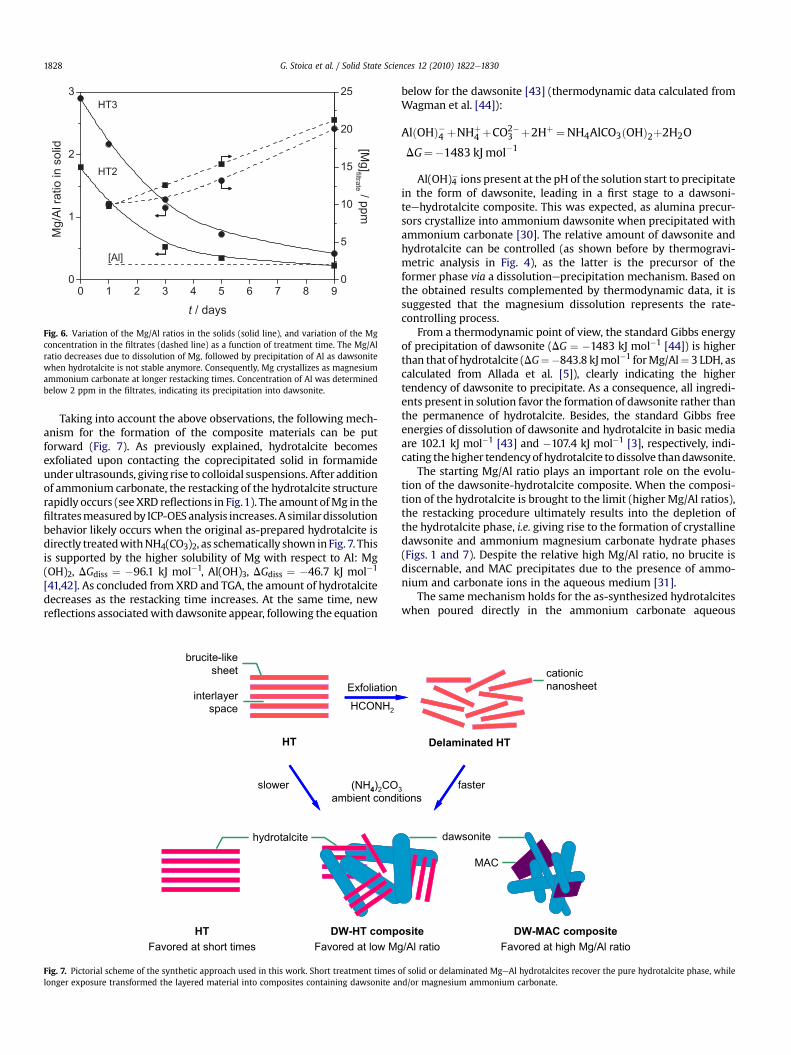
0 1 2 3 4 5 6 7 8 9 0
1
2
3
] g
M
[ e
t a
r t l i
f m
p
p /
d i l o s n i
o i t a r l A / g M
t / days
0
5
10
15
20
25
[Al]
HT3
HT2
Fig. 6. Variation of the Mg/Al ratios in the solids (solid line), and variation of the Mgconcentration in the filtrates (dashed line) as a function of treatment time. The Mg/Alratio decreases due to dissolution of Mg, followed by precipitation of Al as dawsonitewhen hydrotalcite is not stable anymore. Consequently, Mg crystallizes as magnesiumammonium carbonate at longer restacking times. Concentration of Al was determinedbelow 2 ppm in the filtrates, indicating its precipitation into dawsonite.
G. Stoica et al. / Solid State Sciences 12 (2010) 1822e18301828
Taking into account the above observations, the following mech-anism for the formation of the composite materials can be putforward (Fig. 7). As previously explained, hydrotalcite becomesexfoliated upon contacting the coprecipitated solid in formamideunderultrasounds, giving rise to colloidal suspensions. After additionof ammonium carbonate, the restacking of the hydrotalcite structurerapidly occurs (see XRD reflections in Fig.1). The amount ofMg in thefiltratesmeasuredby ICP-OESanalysis increases.A similardissolutionbehavior likely occurs when the original as-prepared hydrotalcite isdirectly treatedwithNH4(CO3)2, as schematically shown inFig. 7. Thisis supported by the higher solubility of Mg with respect to Al: Mg(OH)2, DGdiss ¼ �96.1 kJ mol�1, Al(OH)3, DGdiss ¼ �46.7 kJ mol�1
[41,42]. As concluded from XRD and TGA, the amount of hydrotalcitedecreases as the restacking time increases. At the same time, newreflections associatedwith dawsonite appear, following the equation
HT
brucite-like sheet
Exfoliation HCONH 2
(NH 4 ) 2 COambient condi
interlayer space
DW-HT comp
Favored at low Mg
hydrotalcite
slower
HT
Favored at short times
4
Fig. 7. Pictorial scheme of the synthetic approach used in this work. Short treatment timeslonger exposure transformed the layered material into composites containing dawsonite an
below for the dawsonite [43] (thermodynamic data calculated fromWagman et al. [44]):
AlðOHÞ�4 þNHþ4 þCO2�
3 þ2Hþ ¼NH4AlCO3ðOHÞ2þ2H2O
DG¼�1483 kJmol�1
Al(OH)4� ions present at the pH of the solution start to precipitatein the form of dawsonite, leading in a first stage to a dawsoni-teehydrotalcite composite. This was expected, as alumina precur-sors crystallize into ammonium dawsonite when precipitated withammonium carbonate [30]. The relative amount of dawsonite andhydrotalcite can be controlled (as shown before by thermogravi-metric analysis in Fig. 4), as the latter is the precursor of theformer phase via a dissolutioneprecipitation mechanism. Based onthe obtained results complemented by thermodynamic data, it issuggested that the magnesium dissolution represents the rate-controlling process.
From a thermodynamic point of view, the standard Gibbs energyof precipitation of dawsonite (DG ¼ �1483 kJ mol�1 [44]) is higherthan that of hydrotalcite (DG¼�843.8 kJmol�1 forMg/Al¼ 3 LDH, ascalculated from Allada et al. [5]), clearly indicating the highertendency of dawsonite to precipitate. As a consequence, all ingredi-ents present in solution favor the formation of dawsonite rather thanthe permanence of hydrotalcite. Besides, the standard Gibbs freeenergies of dissolution of dawsonite and hydrotalcite in basic mediaare 102.1 kJ mol�1 [43] and �107.4 kJ mol�1 [3], respectively, indi-cating thehigher tendencyof hydrotalcite to dissolve thandawsonite.
The starting Mg/Al ratio plays an important role on the evolu-tion of the dawsonite-hydrotalcite composite. When the composi-tion of the hydrotalcite is brought to the limit (higher Mg/Al ratios),the restacking procedure ultimately results into the depletion ofthe hydrotalcite phase, i.e. giving rise to the formation of crystallinedawsonite and ammonium magnesium carbonate hydrate phases(Figs. 1 and 7). Despite the relative high Mg/Al ratio, no brucite isdiscernable, and MAC precipitates due to the presence of ammo-nium and carbonate ions in the aqueous medium [31].
The same mechanism holds for the as-synthesized hydrotalciteswhen poured directly in the ammonium carbonate aqueous
Delaminated HT
cationic nanosheet
3 tions
osite
/Al ratio DW-MAC composite
Favored at high Mg/Al ratio
MAC
dawsonite
faster
of solid or delaminated MgeAl hydrotalcites recover the pure hydrotalcite phase, whiled/or magnesium ammonium carbonate.
1.8 2.1 2.4 2.7 3.0 3.3 0
20
40
60
80
100
%
/ s e s a h p f o t n u o m
a e v i t a l e
R
Mg/Al ratio 1.8 2.1 2.4 2.7 3.0 3.3
0
20
40
60
80
100
%
/ s e s a h p f o t n u o m
a e v i t a l e
R
Mg/Al ratio
a b
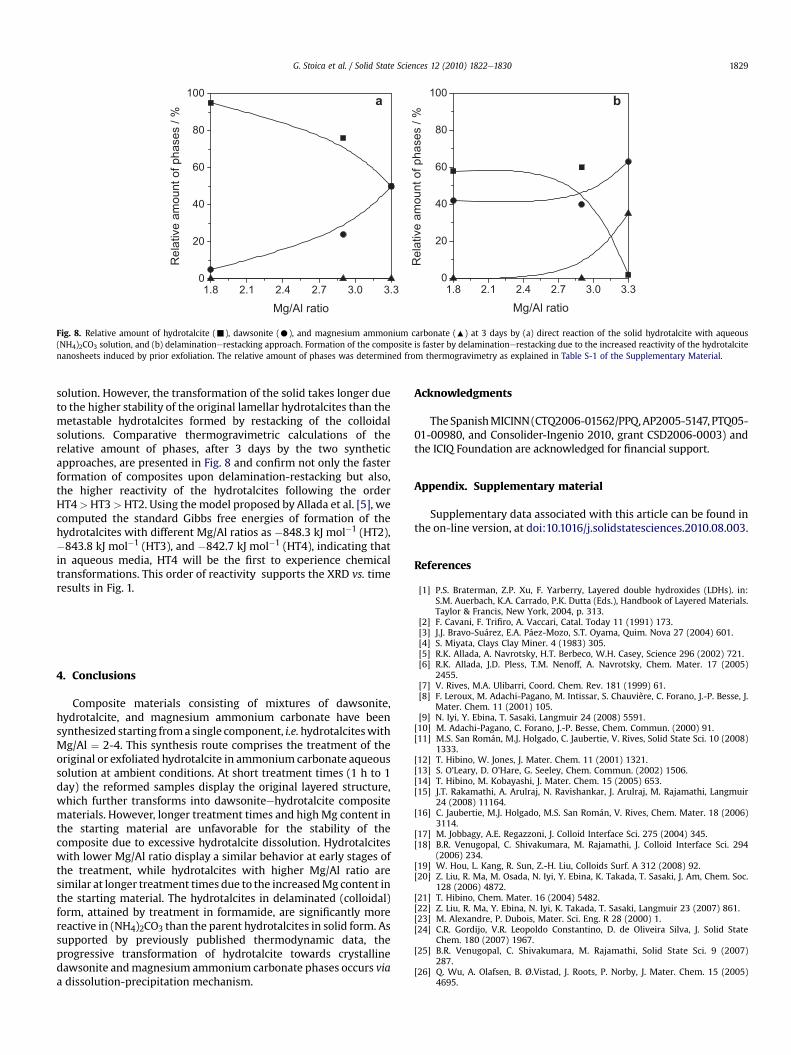
Fig. 8. Relative amount of hydrotalcite (-), dawsonite (C), and magnesium ammonium carbonate (:) at 3 days by (a) direct reaction of the solid hydrotalcite with aqueous(NH4)2CO3 solution, and (b) delaminationerestacking approach. Formation of the composite is faster by delaminationerestacking due to the increased reactivity of the hydrotalcitenanosheets induced by prior exfoliation. The relative amount of phases was determined from thermogravimetry as explained in Table S-1 of the Supplementary Material.
G. Stoica et al. / Solid State Sciences 12 (2010) 1822e1830 1829
solution. However, the transformation of the solid takes longer dueto the higher stability of the original lamellar hydrotalcites than themetastable hydrotalcites formed by restacking of the colloidalsolutions. Comparative thermogravimetric calculations of therelative amount of phases, after 3 days by the two syntheticapproaches, are presented in Fig. 8 and confirm not only the fasterformation of composites upon delamination-restacking but also,the higher reactivity of the hydrotalcites following the orderHT4>HT3>HT2. Using themodel proposed by Allada et al. [5], wecomputed the standard Gibbs free energies of formation of thehydrotalcites with different Mg/Al ratios as �848.3 kJ mol�1 (HT2),�843.8 kJ mol�1 (HT3), and �842.7 kJ mol�1 (HT4), indicating thatin aqueous media, HT4 will be the first to experience chemicaltransformations. This order of reactivity supports the XRD vs. timeresults in Fig. 1.
4. Conclusions
Composite materials consisting of mixtures of dawsonite,hydrotalcite, and magnesium ammonium carbonate have beensynthesized starting froma single component, i.e.hydrotalciteswithMg/Al ¼ 2-4. This synthesis route comprises the treatment of theoriginal or exfoliated hydrotalcite in ammonium carbonate aqueoussolution at ambient conditions. At short treatment times (1 h to 1day) the reformed samples display the original layered structure,which further transforms into dawsoniteehydrotalcite compositematerials. However, longer treatment times and highMg content inthe starting material are unfavorable for the stability of thecomposite due to excessive hydrotalcite dissolution. Hydrotalciteswith lower Mg/Al ratio display a similar behavior at early stages ofthe treatment, while hydrotalcites with higher Mg/Al ratio aresimilar at longer treatment times due to the increasedMg content inthe starting material. The hydrotalcites in delaminated (colloidal)form, attained by treatment in formamide, are significantly morereactive in (NH4)2CO3 than the parent hydrotalcites in solid form. Assupported by previously published thermodynamic data, theprogressive transformation of hydrotalcite towards crystallinedawsonite andmagnesium ammonium carbonate phases occurs viaa dissolution-precipitation mechanism.
Acknowledgments
TheSpanishMICINN(CTQ2006-01562/PPQ,AP2005-5147, PTQ05-01-00980, and Consolider-Ingenio 2010, grant CSD2006-0003) andthe ICIQ Foundation are acknowledged for financial support.
Appendix. Supplementary material
Supplementary data associated with this article can be found inthe on-line version, at doi:10.1016/j.solidstatesciences.2010.08.003.
References
[1] P.S. Braterman, Z.P. Xu, F. Yarberry, Layered double hydroxides (LDHs). in:S.M. Auerbach, K.A. Carrado, P.K. Dutta (Eds.), Handbook of Layered Materials.Taylor & Francis, New York, 2004, p. 313.
[2] F. Cavani, F. Trifiro, A. Vaccari, Catal. Today 11 (1991) 173.[3] J.J. Bravo-Suárez, E.A. Páez-Mozo, S.T. Oyama, Quim. Nova 27 (2004) 601.[4] S. Miyata, Clays Clay Miner. 4 (1983) 305.[5] R.K. Allada, A. Navrotsky, H.T. Berbeco, W.H. Casey, Science 296 (2002) 721.[6] R.K. Allada, J.D. Pless, T.M. Nenoff, A. Navrotsky, Chem. Mater. 17 (2005)
2455.[7] V. Rives, M.A. Ulibarri, Coord. Chem. Rev. 181 (1999) 61.[8] F. Leroux, M. Adachi-Pagano, M. Intissar, S. Chauvière, C. Forano, J.-P. Besse, J.
Mater. Chem. 11 (2001) 105.[9] N. Iyi, Y. Ebina, T. Sasaki, Langmuir 24 (2008) 5591.
[10] M. Adachi-Pagano, C. Forano, J.-P. Besse, Chem. Commun. (2000) 91.[11] M.S. San Román, M.J. Holgado, C. Jaubertie, V. Rives, Solid State Sci. 10 (2008)
1333.[12] T. Hibino, W. Jones, J. Mater. Chem. 11 (2001) 1321.[13] S. O’Leary, D. O’Hare, G. Seeley, Chem. Commun. (2002) 1506.[14] T. Hibino, M. Kobayashi, J. Mater. Chem. 15 (2005) 653.[15] J.T. Rakamathi, A. Arulraj, N. Ravishankar, J. Arulraj, M. Rajamathi, Langmuir
24 (2008) 11164.[16] C. Jaubertie, M.J. Holgado, M.S. San Román, V. Rives, Chem. Mater. 18 (2006)
3114.[17] M. Jobbagy, A.E. Regazzoni, J. Colloid Interface Sci. 275 (2004) 345.[18] B.R. Venugopal, C. Shivakumara, M. Rajamathi, J. Colloid Interface Sci. 294
(2006) 234.[19] W. Hou, L. Kang, R. Sun, Z.-H. Liu, Colloids Surf. A 312 (2008) 92.[20] Z. Liu, R. Ma, M. Osada, N. Iyi, Y. Ebina, K. Takada, T. Sasaki, J. Am, Chem. Soc.
128 (2006) 4872.[21] T. Hibino, Chem. Mater. 16 (2004) 5482.[22] Z. Liu, R. Ma, Y. Ebina, N. Iyi, K. Takada, T. Sasaki, Langmuir 23 (2007) 861.[23] M. Alexandre, P. Dubois, Mater. Sci. Eng. R 28 (2000) 1.[24] C.R. Gordijo, V.R. Leopoldo Constantino, D. de Oliveira Silva, J. Solid State
Chem. 180 (2007) 1967.[25] B.R. Venugopal, C. Shivakumara, M. Rajamathi, Solid State Sci. 9 (2007)
287.[26] Q. Wu, A. Olafsen, B. Ø.Vistad, J. Roots, P. Norby, J. Mater. Chem. 15 (2005)
4695.

G. Stoica et al. / Solid State Sciences 12 (2010) 1822e18301830
[27] L. Li, R. Ma, Y. Ebina, K. Fukuda, K. Takada, T. Sasaki, J. Am. Chem. Soc. 129(2007) 8000.
[28] S. Abelló, J. Pérez-Ramírez, Adv. Mater. 18 (2006) 2436.[29] M. Santiago, M.S. Yalfani, J. Pérez-Ramírez, J. Mater. Chem. 16 (2006) 2886.[30] G. Stoica, J. Pérez-Ramírez, Chem. Mater. 19 (2007) 4783.[31] E. Erdös, H. Altorfer, J. Witt, J. Appl. Cryst. 12 (1979) 611.[32] S. Brunauer, P.H. Emmett, E. Teller, J. Am. Chem. Soc. 60 (1938) 309.[33] B.C. Lippens, J.H. de Boer, J. Catal. 4 (1965) 319.[34] J.T. Kloprogge, R.L. Frost, J. Solid State Chem. 146 (1999) 506.[35] G. Stoica, J.C. Groen, S. Abelló, R. Manchanda, J. Pérez-Ramírez, Chem. Mater.
20 (2008) 3973.[36] J. Pérez-Ramírez, G. Mul, F. Kapteijn, J.A. Moulijn, J. Mater. Chem. 11 (2001)
821.
[37] V. Rives, Mater. Chem. Phys. 75 (2002) 19.[38] J.I. Di Cosimo, V.K. Diez, M. Xu, E. Iglesia, C.R. Apesteguia, J. Catal. 178 (1998)
499.[39] C.J. Serna, J.V. Garcia-Ramos, M.J. Peña, Spectrochim. Acta 41A (1985) 697.[40] R.M. Dell, S.W. Weller, Trans. Faraday Soc. 55 (1959) 2203.[41] H. Tamura, J. Chiba, M. Ito, T. Takeda, S. Kikkawa, Y. Mawatari, M. Tabata, J.
Colloid Interface Sci. 300 (2006) 648.[42] U. Costantino, F. Marmottini, M. Nocchetti, R. Vivani, Eur. J. Inorg. Chem. 10
(1998) 1439.[43] P. Bénézeth, D.A. Palmer, L.M. Anovitz, J. Horita, Geoch. Cosmoch. Acta 71
(2007) 4438.[44] D.D. Wagman, W.H. Evans, V.B. Parker, R.H. Schumm, I. Halow, S.M. Bailey,
K.L. Churney, R.L. Nuttal, J. Phys. Chem. Ref. Data 11 (1982) 1.
Copyright © 2022 FDOKUMEN




















