
Rationalization of Solvation and Stabilization of Palladium Nanoparticles in Imidazolium-Based Ionic...
10
DOI: 10.1002/cphc.201200087 Rationalization of Solvation and Stabilization of Palladium Nanoparticles in Imidazolium-Based Ionic Liquids by DFT and Vibrational Spectroscopy Sergey A. Katsyuba,* [a] Elena E. Zvereva, [a] Ning Yan, [b] Xiao Yuan , [b, c] Yuan Kou, [c] and Paul J. Dyson [b] 1. Introduction Metal nanoparticles (M-NPs) attract interest across diverse areas of science due to their unique physicochemical proper- ties, which are attributable to quantum size effects and single- electron transitions. [1] The precise control of the size of NPs and their size distribution, together with a better understand- ing of their chemical behaviour, is becoming increasingly im- portant for expanding the utility of NPs in various applications. There appear to be a number of interesting features of NPs dispersed in ionic liquids (ILs) compared to other solvents, and amalgamation of ILs with nanotechnology could result in ad- vances in materials science. NPs suspended in ILs have been demonstrated in various instances to be active catalysts for hy- drogenation and hydroformylation reactions, and they are also implicated in C C cross-coupling reactions. [2] Small M-NPs are only kinetically stable and combine to form thermodynamically favoured larger metal particles by agglom- eration. This tendency of M-NPs to aggregate is due to their high surface energy and their relatively large surface area. To avoid this agglomeration, M-NPs must be stabilised with pro- tective layers, for example, polymers and surfactants, which provide electrostatic, coordination and/or steric protection. [3] ILs can actively participate in this stabilization process, thereby being far more than innocent solvents, and ILs, in particular imidazolium-derived salts (Scheme 1), can be used to generate and stabilise M-NPs in situ. [4] The inclusion of M-NPs within a supramolecular IL network provides the required stabilization through formation of an ion layer around the M-NPs. The nature of this ion layer, and A combined DFT/vibrational spectroscopy approach is used to determine the interactions of the 1,3-dimethylimidazolium ([Mmim] + ) and 1-ethyl-3-methylimidazolium ([Emim] + ) cations and [BF 4 ] anions with each other in the liquid state and with Pd nanoparticles (NPs) immersed in ionic liquids (ILs) com- posed of these ions. Formation of aggregates of the counter- ions in the liquid does not have a strong influence on the in- teraction energy of the IL components with the palladium clus- ters used to model NPs, which is smaller than the energy of addition of a Pd atom to the cluster. Stronger Pd–Pd interac- tions in comparison to the interactions of the palladium cluster with the IL suggest kinetic stabilisation of Pd-NPs in 1,3-dial- kylimidazolium tetrafluoroborates rather than thermodynamic stabilisation. Moreover, the palladium clusters interact more strongly with the anions than with the cations, and this sug- gests an important role of the anions in formation and stabili- sation of Pd-NP in ILs. At the same time, binding between an isolated Pd atom and [Mmim] + cation is stronger than Pd–[BF 4 ] binding. IR and Raman spectral simulations reveal that surface-enhanced Raman spectroscopy is much less sensi- tive to interactions of Pd-NPs with the [BF 4 ] anion compared to interactions with [Mmim] + cations. In contrast, IR spectros- copy is better suited to study the anion–metal interactions, whereas IR spectral manifestations of the cation–metal interac- tions are rather modest. The splitting of the strong n as (BF 4 ) IR band into three components appears to be a convenient spec- troscopic marker for Pd–[BF 4 ] interactions, which is confirmed by actual spectra of Pd-NPs stabilised by [Emim][BF 4 ]. The IR spectra and their assignment with quantum chemical compu- tations suggest that both the anions and cations of [Emim]- [BF 4 ] interact with the Pd-NP surface in the IL. The cation ring orientation close to the surface normal appears to be the dom- inant interaction. The anion is bound to the surface through either two or three fluorine atoms. [a] Prof. S. A. Katsyuba, Dr. E. E. Zvereva Laboratory of Optical Spectroscopy A. E. Arbuzov Institute of Organic and Physical Chemistry Kazan Scientific Centre of the Russian Academy of Sciences Arbuzov str. 8, 420088 Kazan (Russia) Fax: (+ 7) 843-273-18-72 E-mail : [email protected] [email protected] [b] Dr. N. Yan, X. Yuan, Prof. P. J. Dyson Laboratory of Organometallic and Medicinal Chemistry Institut des Sciences et IngȖnierie Chimiques Ecole Polytechnique FȖdȖrale de Lausanne (EPFL) EPFL–BCH, 1015 Lausanne (Switzerland) [c] X. Yuan, Prof. Y. Kou Beijing National Laboratory for Molecular Sciences College of Chemistry and Molecular Engineering PKU Green Chemistry Centre Peking University Beijing 100871 (China) Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cphc.201200087. ChemPhysChem 2012, 13, 1781 – 1790 # 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 1781
Transcript of Rationalization of Solvation and Stabilization of Palladium Nanoparticles in Imidazolium-Based Ionic...

DOI: 10.1002/cphc.201200087
Rationalization of Solvation and Stabilization of PalladiumNanoparticles in Imidazolium-Based Ionic Liquids by DFTand Vibrational SpectroscopySergey A. Katsyuba,*[a] Elena E. Zvereva,[a] Ning Yan,[b] Xiao Yuan ,[b, c] Yuan Kou,[c] andPaul J. Dyson[b]
1. Introduction
Metal nanoparticles (M-NPs) attract interest across diverseareas of science due to their unique physicochemical proper-ties, which are attributable to quantum size effects and single-electron transitions.[1] The precise control of the size of NPsand their size distribution, together with a better understand-ing of their chemical behaviour, is becoming increasingly im-portant for expanding the utility of NPs in various applications.There appear to be a number of interesting features of NPsdispersed in ionic liquids (ILs) compared to other solvents, andamalgamation of ILs with nanotechnology could result in ad-vances in materials science. NPs suspended in ILs have beendemonstrated in various instances to be active catalysts for hy-drogenation and hydroformylation reactions, and they are alsoimplicated in C�C cross-coupling reactions.[2]
Small M-NPs are only kinetically stable and combine to formthermodynamically favoured larger metal particles by agglom-eration. This tendency of M-NPs to aggregate is due to theirhigh surface energy and their relatively large surface area. Toavoid this agglomeration, M-NPs must be stabilised with pro-tective layers, for example, polymers and surfactants, whichprovide electrostatic, coordination and/or steric protection.[3]
ILs can actively participate in this stabilization process, therebybeing far more than innocent solvents, and ILs, in particular
imidazolium-derived salts (Scheme 1), can be used to generateand stabilise M-NPs in situ.[4]
The inclusion of M-NPs within a supramolecular IL networkprovides the required stabilization through formation of an ionlayer around the M-NPs. The nature of this ion layer, and
A combined DFT/vibrational spectroscopy approach is used todetermine the interactions of the 1,3-dimethylimidazolium([Mmim]+) and 1-ethyl-3-methylimidazolium ([Emim]+) cationsand [BF4]� anions with each other in the liquid state and withPd nanoparticles (NPs) immersed in ionic liquids (ILs) com-posed of these ions. Formation of aggregates of the counter-ions in the liquid does not have a strong influence on the in-teraction energy of the IL components with the palladium clus-ters used to model NPs, which is smaller than the energy ofaddition of a Pd atom to the cluster. Stronger Pd–Pd interac-tions in comparison to the interactions of the palladium clusterwith the IL suggest kinetic stabilisation of Pd-NPs in 1,3-dial-kylimidazolium tetrafluoroborates rather than thermodynamicstabilisation. Moreover, the palladium clusters interact morestrongly with the anions than with the cations, and this sug-gests an important role of the anions in formation and stabili-sation of Pd-NP in ILs. At the same time, binding between an
isolated Pd atom and [Mmim]+ cation is stronger thanPd–[BF4]� binding. IR and Raman spectral simulations revealthat surface-enhanced Raman spectroscopy is much less sensi-tive to interactions of Pd-NPs with the [BF4]� anion comparedto interactions with [Mmim]+ cations. In contrast, IR spectros-copy is better suited to study the anion–metal interactions,whereas IR spectral manifestations of the cation–metal interac-tions are rather modest. The splitting of the strong nas(BF4
�) IRband into three components appears to be a convenient spec-troscopic marker for Pd–[BF4]� interactions, which is confirmedby actual spectra of Pd-NPs stabilised by [Emim][BF4] . The IRspectra and their assignment with quantum chemical compu-tations suggest that both the anions and cations of [Emim]-[BF4] interact with the Pd-NP surface in the IL. The cation ringorientation close to the surface normal appears to be the dom-inant interaction. The anion is bound to the surface througheither two or three fluorine atoms.
[a] Prof. S. A. Katsyuba, Dr. E. E. ZverevaLaboratory of Optical SpectroscopyA. E. Arbuzov Institute of Organic and Physical ChemistryKazan Scientific Centre of the Russian Academy of SciencesArbuzov str. 8, 420088 Kazan (Russia)Fax: (+ 7) 843-273-18-72E-mail : [email protected]
[b] Dr. N. Yan, X. Yuan , Prof. P. J. DysonLaboratory of Organometallic and Medicinal ChemistryInstitut des Sciences et Ing�nierie ChimiquesEcole Polytechnique F�d�rale de Lausanne (EPFL)EPFL–BCH, 1015 Lausanne (Switzerland)
[c] X. Yuan , Prof. Y. KouBeijing National Laboratory for Molecular SciencesCollege of Chemistry and Molecular EngineeringPKU Green Chemistry CentrePeking UniversityBeijing 100871 (China)
Supporting information for this article is available on the WWW underhttp://dx.doi.org/10.1002/cphc.201200087.
ChemPhysChem 2012, 13, 1781 – 1790 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 1781

hence the mode of stabilization of M-NPs in ILs, is still a matterof debate.[4–6] With the exception of certain functionalised ILsbearing groups that can interact directly with an M-NP surface,for example, thiol, ether, carboxyl, amino, and hydroxyl, it isnecessary to distinguish whether the IL cation or anion inter-acts with the NP surface. Additionally, M-NPs prepared in non-functionalised ILs can be stabilised by formation of ionic ag-gregates of the type {[(C)x(A)x�n]n + [(C)x�n(A)x]
n�}m, where C isthe 1,3-dialkylimidazolium cation and A the anion.[4] It hasbeen suggested that growth of Ru NPs in ILs is controlled bythe non-polar domains created by grouping of the lipophilicalkyl chains of 1,3-dialkylimidazolium cations.[5] It seems thatwhether the growth of NPs is controlled by the polar or non-polar domain in ILs is determined by the polarity of the precur-sor, that is, ionic precursors (e.g. RhCl3) tend to accumulate inthe polar domain and NP size is related to the volume of theanion of the imidazolium-based ILs,[7] whereas neutral organo-metallic precursor stay in the non-polar domain and thereforeNP size is related to the length of the side chain attached tothe cation.
Various experimental data indicate that non-functionalisedILs interact with the surface of M-NPs. For example, X-ray pho-toelectron spectroscopic (XPS) analysis of isolated Ir- and Pd-NPs prepared in ILs containing PF6
� and BF4� anions shows the
presence of M–F interactions.[8, 9] F 1s, P (in the case of PF6�)
and B (in the case of BF4�) contributions are also observed in
the XP spectra.[8] These data support the idea that the stabiliza-tion of M-NPs is essentially due to a positive charge on themetal surface, which is counter-balanced by adsorption of theanionic IL species onto the coordinatively unsaturated, elec-tron-deficient metal surface. The first anionic solvation shell ispostulated to be followed by a less well-ordered layer of cat-ions, and together they form an electric double layer, which isin line with the Derjaugin–Landau–Verwey–Overbeek (DLVO)model of colloid stability.[10]
Other studies suggest that there are close interactions be-tween the NPs and the cations, through deuterium exchangeon imidazolium rings,[11] and through surface-enhanced Ramanspectroscopy (SERS) on gold nanoparticles dispersed in imid-azolium-based ILs, which point to electrostatic stabilization ofa negatively charged Au surface counter-balanced by interac-tions with imidazolium cations.[12] No evidence of interactionsof the MeSO3
� anion with the NP surface was found in theSERS study.[12] These results are consistent with a SERS studyon Ag-NPs dispersed in [Bmim][BF4] , which suggests that theBmim+ cation interacts with the Ag-NP surface in a flat config-uration, forming a positively charged layer, while the BF4
�
anion is somewhat further from the surface.[13] SERS experi-
ments carried out during the electrochemical deposition ofgold from AuI complexes in 1-ethyl-3-methylimidazolium bis-(trifluoromethylsulfonyl)amide ([Emim][Tf2N]) indicate co-ad-sorption of both the anions and the cations of the IL.[14] Densi-ty functional calculations on a gas-phase model favour interac-tions between Au clusters and IL anions, such as BF4
� , insteadof imidazolium cations,[15, 16] and thus suggest Au···F interac-tions and anionic Aun stabilization in fluorous ILs. Overall, sus-pensions of M-NPs in an IL are governed by three kinds of mo-lecular interaction: ion–ion, metal–metal and metal–ion.
Herein, we consider Pd-NPs in ILs composed of 1,3-dimeth-ylimidazolium ([Mmim]+) or 1-ethyl-3-methylimidazolium([Emim]+) cations and BF4
� anions. This particular system waschosen for study as it has been shown to have many applica-tions in catalysis.[6, 17] We employed DFT methods to determinethe following interactions: 1) between ionic IL components,namely, [Mmim]+ cations and [BF4]� anions; 2) between Pdatoms in Pdn clusters (n = 1–6, 13, 14); 3) between [Mmim]+
and Pdn clusters ; 4) between [BF4]� and Pdn clusters; and 5) be-tween aggregates of IL counter-ions, [BF4]� and [Mmim]+ , andPdn clusters. DFT with the Perdew, Burke and Ernzerhof (PBE)functional,[18] which appears to give consistent results for theconsidered systems[19] and is convenient for comparison withsimilar PBE computations of Aun clusters interacting with ILanions and cations,[15, 16] was chosen for this study. DFT compu-tations were used also to simulate IR and Raman spectra ofthe systems under study and validated with spectroscopic ex-periments. These studies allow us to discuss the possiblemodes of interaction between IL ions and NPs in ILs.
2. Results and Discussion
2.1. Possible Associations of the Counterions in the IL
For computations of ILs, the 6-31G* basis set in conjunctionwith the B3LYP functional was demonstrated to be sufficientto produce reasonably accurate DFT predictions of geometryand vibrational spectra of imidazolium-based ILs.[20, 21] To testwhether the basis set is applicable to the systems under studyin the case of the PBE functional, we simulated the IR spec-trum of [Mmim][BF4] using double-zeta (6-31G*, 6-31 + G*,6-31 + + G** and aug-cc-pVDZ) and triple-zeta (6-311G*,6-311 + G** and 6-311 + + G**) basis sets. As additional basisfunctions are added to the 6-31G* basis, the vibrational fre-quencies and IR intensities converge to 6-31 + G*. Neither in-clusion of polarisation p functions on hydrogen atoms norswitching from double-zeta to triple-zeta quality of basis setssignificantly influences the simulated spectra (Figure 1S, Sup-porting Information). Moreover, only simulated IR bands of theBF4
� anion are sensitive to the choice of basis set. All othercomputed spectroscopic, structural and energetic parametersof the [Mmim][BF4] ion pair remain practically the same forboth 6-31G* and 6-31 + G* basis sets. Thus, the more compact6-31G* basis set was mainly used in the computations, exceptfor the simulation of IR or Raman bands of the [BF4]� anion, forwhich the 6-31 + G* basis set was applied.
Scheme 1. Examples of 1-alkyl-3-methylimidazolium-based ILs and atomnumbering scheme for the imidazolium ring.
1782 www.chemphyschem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 2012, 13, 1781 – 1790
S. A. Katsyuba et al.
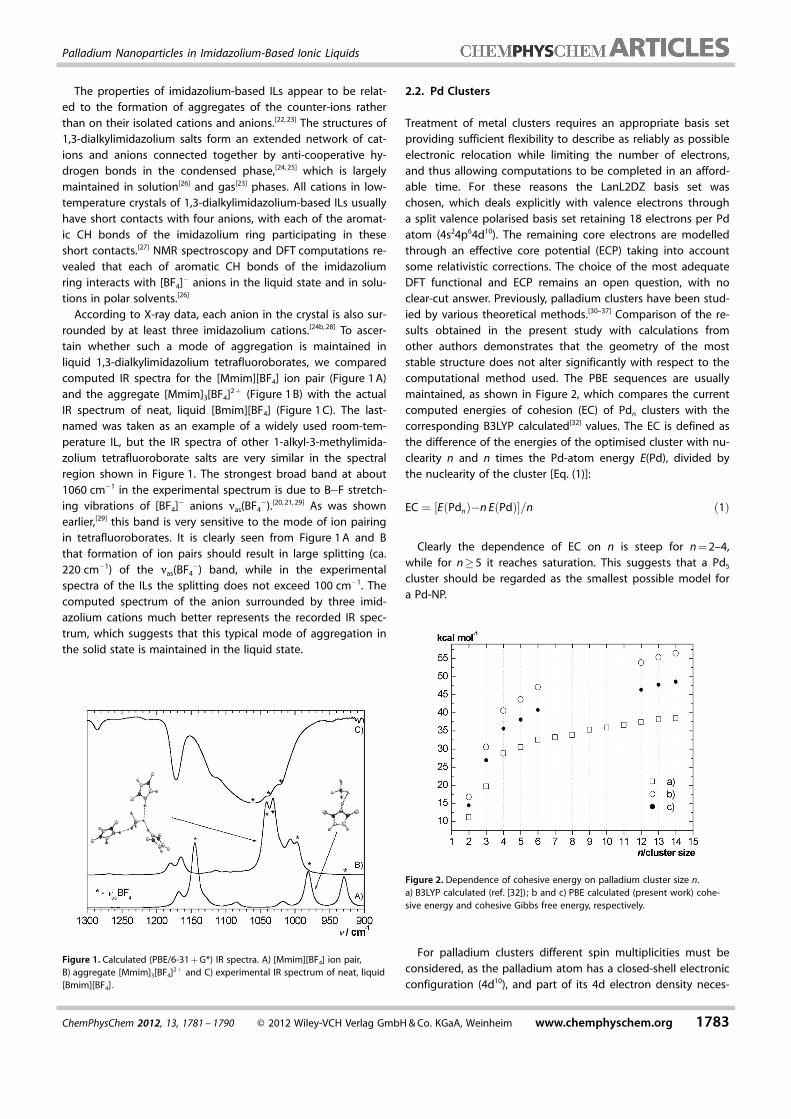
The properties of imidazolium-based ILs appear to be relat-ed to the formation of aggregates of the counter-ions ratherthan on their isolated cations and anions.[22, 23] The structures of1,3-dialkylimidazolium salts form an extended network of cat-ions and anions connected together by anti-cooperative hy-drogen bonds in the condensed phase,[24, 25] which is largelymaintained in solution[26] and gas[23] phases. All cations in low-temperature crystals of 1,3-dialkylimidazolium-based ILs usuallyhave short contacts with four anions, with each of the aromat-ic CH bonds of the imidazolium ring participating in theseshort contacts.[27] NMR spectroscopy and DFT computations re-vealed that each of aromatic CH bonds of the imidazoliumring interacts with [BF4]� anions in the liquid state and in solu-tions in polar solvents.[26]
According to X-ray data, each anion in the crystal is also sur-rounded by at least three imidazolium cations.[24b, 28] To ascer-tain whether such a mode of aggregation is maintained inliquid 1,3-dialkylimidazolium tetrafluoroborates, we comparedcomputed IR spectra for the [Mmim][BF4] ion pair (Figure 1 A)and the aggregate [Mmim]3[BF4]2+ (Figure 1 B) with the actualIR spectrum of neat, liquid [Bmim][BF4] (Figure 1 C). The last-named was taken as an example of a widely used room-tem-perature IL, but the IR spectra of other 1-alkyl-3-methylimida-zolium tetrafluoroborate salts are very similar in the spectralregion shown in Figure 1. The strongest broad band at about1060 cm�1 in the experimental spectrum is due to B�F stretch-ing vibrations of [BF4]� anions nas(BF4
�).[20, 21, 29] As was shownearlier,[29] this band is very sensitive to the mode of ion pairingin tetrafluoroborates. It is clearly seen from Figure 1 A and Bthat formation of ion pairs should result in large splitting (ca.220 cm�1) of the nas(BF4
�) band, while in the experimentalspectra of the ILs the splitting does not exceed 100 cm�1. Thecomputed spectrum of the anion surrounded by three imid-azolium cations much better represents the recorded IR spec-trum, which suggests that this typical mode of aggregation inthe solid state is maintained in the liquid state.
2.2. Pd Clusters
Treatment of metal clusters requires an appropriate basis setproviding sufficient flexibility to describe as reliably as possibleelectronic relocation while limiting the number of electrons,and thus allowing computations to be completed in an afford-able time. For these reasons the LanL2DZ basis set waschosen, which deals explicitly with valence electrons througha split valence polarised basis set retaining 18 electrons per Pdatom (4s24p64d10). The remaining core electrons are modelledthrough an effective core potential (ECP) taking into accountsome relativistic corrections. The choice of the most adequateDFT functional and ECP remains an open question, with noclear-cut answer. Previously, palladium clusters have been stud-ied by various theoretical methods.[30–37] Comparison of the re-sults obtained in the present study with calculations fromother authors demonstrates that the geometry of the moststable structure does not alter significantly with respect to thecomputational method used. The PBE sequences are usuallymaintained, as shown in Figure 2, which compares the currentcomputed energies of cohesion (EC) of Pdn clusters with thecorresponding B3LYP calculated[32] values. The EC is defined asthe difference of the energies of the optimised cluster with nu-clearity n and n times the Pd-atom energy E(Pd), divided bythe nuclearity of the cluster [Eq. (1)]:
EC ¼ ½EðPdnÞ�n EðPdÞ�=n ð1Þ
Clearly the dependence of EC on n is steep for n = 2–4,while for n�5 it reaches saturation. This suggests that a Pd5
cluster should be regarded as the smallest possible model fora Pd-NP.
For palladium clusters different spin multiplicities must beconsidered, as the palladium atom has a closed-shell electronicconfiguration (4d10), and part of its 4d electron density neces-
Figure 1. Calculated (PBE/6-31 + G*) IR spectra. A) [Mmim][BF4] ion pair,B) aggregate [Mmim]3[BF4]2 + and C) experimental IR spectrum of neat, liquid[Bmim][BF4] .
Figure 2. Dependence of cohesive energy on palladium cluster size n.a) B3LYP calculated (ref. [32]) ; b and c) PBE calculated (present work) cohe-sive energy and cohesive Gibbs free energy, respectively.
ChemPhysChem 2012, 13, 1781 – 1790 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemphyschem.org 1783
Palladium Nanoparticles in Imidazolium-Based Ionic Liquids

sarily must be promoted into the nearest 5s orbital to enableformation of stable metal–metal bonds.[32] Thus, all palladiumclusters were optimised for the various accessible spin multi-plicities. In agreement with the results of others[32, 36] the funda-mental spin multiplicities were higher than singlet. Clusterswith 2�n�6 showed a triplet ground state, while n = 12, 13and 14 gave a septet ground state. Only ground-state clustersare further discussed. The lowest-energy optimised structuresof the Pdn clusters together with their PBE/LanL2DZ computedelectronic energies and Gibbs free energies are given in Fig-ure 2S (Supporting Information).
The dependence of addition energy (AE) of a single Pd atomto a Pdn cluster on n is shown in Figure 3 A. The AE is definedas the difference of the energies of the optimised Pdn clusterand single Pd atom to the energy of the optimised Pdn + 1 clus-ter [Eq. (2)]:
AE ¼ EðPdnÞ þ EðPdÞ�EðPdnþ1Þ ð2Þ
The addition Gibbs free energies (AGFEs), defined in thesame manner, are shown in Figure 3 B.
The AE to a single Pd atom is quite low and almost doublesfor Pd2. Increasing the cluster size does not change the AE sub-stantially any further, that is, the AE is practically saturated forPd2 ; the same is true for the AGFE (Figure 3). This dependenceis qualitatively different to the behaviour of gold clusters Aun
(n = 1–3, 6 and 19) found by using similar DFT computationswith the same PBE functional :[16] the AE to a single Au atom(ca. 53 kcal mol�1) drops to about 29 kcal mol�1 on addition ofa second Au atom to form the Au2 cluster, and then grows toabout 73 kcal mol�1 as n increases.
2.3. Interaction between Palladium Clusters and IL Ions
The binding energy (BE) of different IL entities, namely,[Mmim]+ , [BF4]� , the ion pair [Mmim][BF4] and the aggregate[Mmim]3[BF4]2+ , to palladium clusters of various sizes was stud-ied. Different spin multiplicities were considered to investigatepossible effects of binding on the clusters spin state. As theground-state multiplicities found for the isolated Pdn clustersremained energetically preferable for the Pdn adducts with theIL entities (Table 1S, Supporting Information), the same spinmultiplicities were used for the Pdn clusters and for their ad-ducts with the above-mentioned stabilising substrates for com-putation of the BEs. The BE is defined as the difference of theenergies of the optimised stabilizing substrates (see above)and the Pdn clusters relative to the energy of their adducts[Eq. (3)]:
BE ¼Eðstabilising substrateÞ þ EðPdnÞ�Eðstabilising substrate=Pdn adductÞ
ð3Þ
Therefore the BE and the binding Gibbs free energy (BGFE),defined in the same manner, can be directly compared to theaddition energies of a single Pd atom to the Pdn cluster (AEand AGFE, respectively).
The optimised adducts of the clusters connected to theabove-mentioned IL components were derived from variousstarting configurations. As a result, three optimised structuresof [Mmim]+ associated with a Pd atom were found (Figure 4).The most energetically stable structures correspond to the Pdatom lying above the plane of imidazolium ring and havingshort contacts with either the C4 and C5 atoms (a) or C2 atom(b) of the latter. The energy of the third stable form of associa-tion (c) is much higher. The only b-type structure, found byB3LYP/LanL2DZ optimisation, was described in the literaturefor association of palladium with the [Mmim]+ and 1,2,3-trime-thylimidazolium cations.[38] According to our DFT computations,structure b is about 10 kcal mol�1 less stable than structure a.Similar structures were obtained after optimisation of [Mmim]+
/Pdn adducts for n = 2, 5, 6 and 13. The main difference is thatapart from a,b,c-type structures, an additional type of cluster–[Mmim]+ association occurs in which the Pd atoms havea short contact with the (C2)H proton of the imidazolium ring(d). This structure is also shown in Figure 4 for the case ofa Pd5 cluster representing a minimal-size model for a Pd-NP, asdiscussed above.
Figure 5 shows the Pd5–[BF4]� binding configurations, whichare almost isoenergetic: the energy difference between struc-tures g, e and f is less than 3 kcal mol�1. In Figure 3 A the varia-tion of the binding and addition energies with cluster size n isshown. All of the clusters, except for the case of n = 1, demon-strate similar behaviour, that is, BEs for anions and cations vary
Figure 3. Dependence on n of a) addition energy of a single Pd atom to Pdn
cluster, b) binding energy of [BF4]� to Pdn and c) binding energy of [Mmim]+
to Pdn. A) electron energy, B) Gibbs free energy.
1784 www.chemphyschem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 2012, 13, 1781 – 1790
S. A. Katsyuba et al.

by about 40–50 and 10–30 kcal mol�1, respectively, and remainessentially smaller than the corresponding AEs, which amount
to about 70 kcal mol�1. Qualitatively similar dependences arefound for binding and addition Gibbs free energies (Fig-ure 3 B).
As discussed above, the properties of 1,3-dialkylimidazoliumtetrafluoroborates are based on aggregates of the counter-ionsrather than on their isolated cations and anions. Thus, the ad-ducts of the clusters connected to the ion pair [Mmim][BF4]and to the aggregate [Mmim]3[BF4]2 + were regarded as morerealistic models for the Pd-NP/IL interface. Several optimisedstructures of the adducts are shown in Figure 6 together withthe corresponding BE and BGFE values.
The latter values demonstrate that, although aggregation ofthe ionic components of the IL slightly weakens their bindingto palladium clusters, it does not change the qualitative pic-ture presented in Figure 3. Thus, these computations suggestthat the cations mainly influence the first step of Pd-NP forma-tion (the reaction Pd + Pd!Pd2), while the anions play themajor role in stabilisation of larger palladium clusters. Never-theless, BEs and BGFEs for the IL components do not exceedthe corresponding addition energies of an isolated Pd atom toPdn cluster.[39] Thus, the Pd–Pd interaction is stronger than theinteractions of Pdn with the IL, and hence 1,3-dialkylimidazoli-um tetrafluoroborates stabilise Pd-NPs kinetically rather thanthermodynamically. Qualitatively similar results were obtainedon the basis of DFT modelling of Aun clusters interacting with
Figure 4. Optimised structures of the associations of the [Mmim]+ cationwith a Pd atom and a Pd5 cluster, including their electronic energiesE [a.e.u.] , binding energies (BE) [kcal mol�1] , Gibbs free energies G [a.e.u.] andbinding Gibbs free energies (BGFE) [kcal mol�1] . Short contacts are shown bydotted lines.
Figure 5. Optimised structures for the association of the [BF4]� anion withthe Pd5 cluster, including their electronic energies E [a.e.u.] , binding energies(BE) [kcal mol�1] , electronic energies and Gibbs free energies relative to themost stable form (DE and DG [kcal mol�1]), Gibbs free energies G [a.e.u.] andbinding Gibbs free energies (BGFE) [kcal mol�1] .
Figure 6. Optimised structures of the associations of IL aggregates with a Pdatom and a Pd5 cluster, including their electronic energies E [a.e.u.] , bindingenergies (BE) [kcal mol�1] , Gibbs free energies G [a.e.u.] and binding Gibbsfree energies (BGFE) [kcal mol�1] .
ChemPhysChem 2012, 13, 1781 – 1790 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemphyschem.org 1785
Palladium Nanoparticles in Imidazolium-Based Ionic Liquids
isolated [Mmim]+ or [BF4]� ions,[15, 16] in spite of the differencesin the nature of interactions on the surface of gold and palladi-um particles,[40] which result in larger BEs for Pdn clusters rela-tive to Aun clusters.
2.4. Spectroscopic Markers for the Interactions betweenPd-NPs and ILs
As far as we are aware, spectroscopic studies on Pd-NPs in ILshave not been reported, although SERS on gold and silver NPsin imidazolium-based ILs provided direct evidence for interac-tions between imidazolium cations and surface Au atoms ofthe NPs,[12, 13] and no evidence for interactions of the anionswith the NP surface was found.[12, 13] These results are consis-tent with SERS studies on a copper electrode in [Bmim]-[BF4][41, 42] and a silver electrode in [Bmim][PF6] ,[43] which dem-onstrate that the [Bmim]+ cation interacts with the electrodesurface, while suggesting that the anions do not interact withthe electrode surface, even at potentials less negative than thepotential of zero charge.
A comparison of the reflection-absorption infrared spectra(RAIRS) of thin films of [Emim][Tf2N] spin-coated on a Ag sur-face with the corresponding transmission IR spectra of thebulk IL[44] shows more pronounced differences for the bandsassigned to the [Tf2N]� anion than for the bands of the[Emim]+ cation, which probably means that the anions interactmore strongly with the metal surface than the cations. RAIRSof thin films of [Bmim][Tf2N] deposited on Pd-NPs grown on anordered alumina film[45] also showed strong shifts of some ofthe [Tf2N]� bands, attributed to direct interaction of the anionwith the Pd-NPs. Because of the very low intensity of the IRfeatures of the cation, a corresponding analysis of these bandsfor the submonolayer films was not feasible.
Both cation and anion vibrational bands of several ILs ofgeneral formula [Bmim][X] (X = BF4
� , PF6� , N(CN)2
� , Tf2N�) in-teracting with a Pt electrode were analysed by surface-specificvibrational spectroscopy sum frequency generation (SFG),which can probe the metal/liquid interface without interfer-ence from the bulk electrolyte.[46] Because of specific limita-tions of this technique only high-frequency CH stretching vi-brations of the [Bmim]+ cation and CN stretches of the[N(CN)2]� anion were studied. The results from this study sug-gest that both anions and cations are oriented at the Pt sur-face, and that the SFG signal from the [N(CN)2]� anion be-comes stronger at positive surface charge, while the cationsignal is related to the negative surface charge. At a positivesurface charge the cation ring is repelled from the Pt surfaceand tilts along the surface normal, albeit still remaining in con-tact with the platinum electrode.
To elucidate possible reasons for these seemingly contradic-tory spectroscopic studies we performed quantum-chemicalsimulations of IR and Raman spectra of the ionic componentsof [Mmim][BF4] interacting with Pdn clusters of various sizes. Inkeeping with the dependence of the EC on n (Figure 1S, Sup-porting Information) the dependence of the simulated spectraon n reaches saturation for n�5 (Figures 3S and 4S, Support-ing Information). Thus, the Pd5 cluster was taken as a minimum
size to model a Pd-NP for further simulations of IR and Ramanspectra of the IL/NP system.
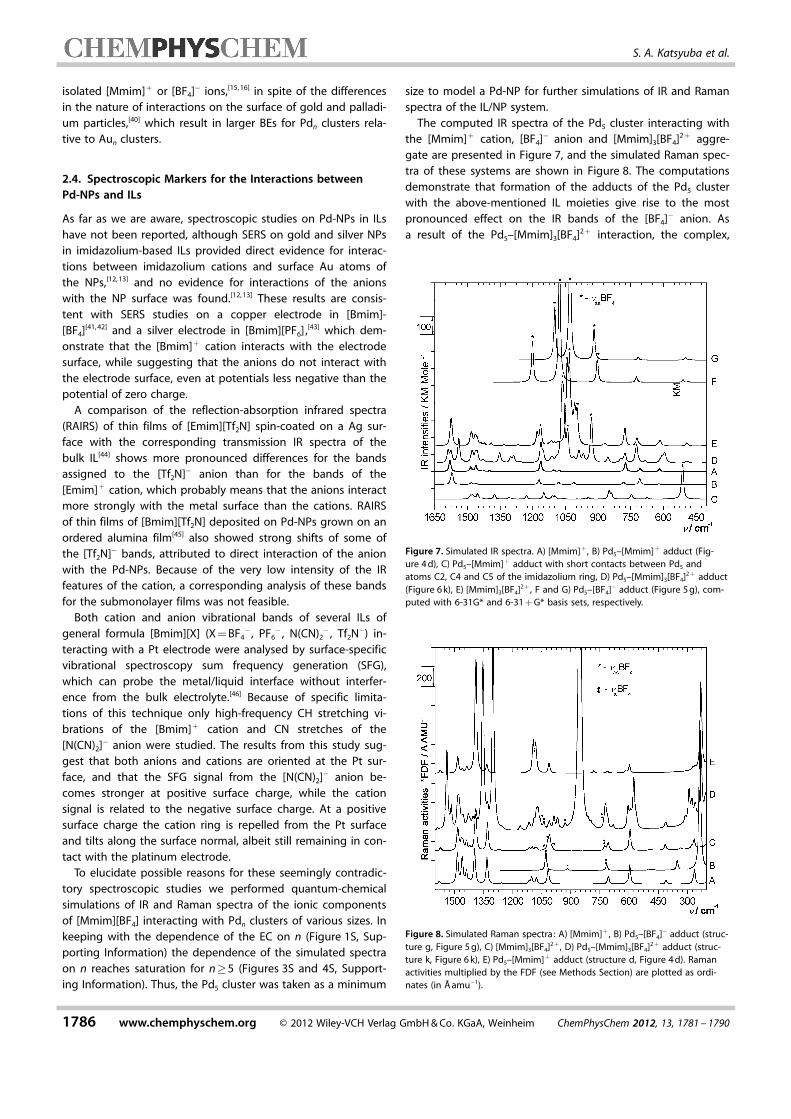
The computed IR spectra of the Pd5 cluster interacting withthe [Mmim]+ cation, [BF4]� anion and [Mmim]3[BF4]2+ aggre-gate are presented in Figure 7, and the simulated Raman spec-tra of these systems are shown in Figure 8. The computationsdemonstrate that formation of the adducts of the Pd5 clusterwith the above-mentioned IL moieties give rise to the mostpronounced effect on the IR bands of the [BF4]� anion. Asa result of the Pd5–[Mmim]3[BF4]2+ interaction, the complex,
Figure 7. Simulated IR spectra. A) [Mmim]+ , B) Pd5–[Mmim]+ adduct (Fig-ure 4 d), C) Pd5–[Mmim]+ adduct with short contacts between Pd5 andatoms C2, C4 and C5 of the imidazolium ring, D) Pd5–[Mmim]3[BF4]2 + adduct(Figure 6 k), E) [Mmim]3[BF4]2 + , F and G) Pd5–[BF4]� adduct (Figure 5 g), com-puted with 6-31G* and 6-31 + G* basis sets, respectively.
Figure 8. Simulated Raman spectra: A) [Mmim]+ , B) Pd5–[BF4]� adduct (struc-ture g, Figure 5 g), C) [Mmim]3[BF4]2 + , D) Pd5–[Mmim]3[BF4]2 + adduct (struc-ture k, Figure 6 k), E) Pd5–[Mmim]+ adduct (structure d, Figure 4 d). Ramanactivities multiplied by the FDF (see Methods Section) are plotted as ordi-nates (in � amu�1).
1786 www.chemphyschem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 2012, 13, 1781 – 1790
S. A. Katsyuba et al.
the strong nas(BF4�) band at about 1000–1040 cm�1 (Figure 7 E),
is predicted to be split into three components at about 930,about 1030–1050 and about 1060–1200 cm�1 (depending onthe mode of the Pd5–[BF4]� interaction and basis set used; seeFigure 7 D, F and G). The cation bands are expected to under-go much smaller changes, except when all carbon atoms ofthe imidazolium ring interact with the palladium cluster, whichshould result in a separate and very strong IR band for out-of-plane bending of the ring at about 510 cm�1 (Figure 7 C).
The predicted qualitative picture of the influence of Pd-NP/IL interactions on the Raman spectra of the ILs (Figure 8) is es-sentially the reverse of that described above for the IR spectra(Figure 7). Raman bands of the [Mmim]+ cation are stronglyenhanced by the interaction, whereas the ns(BF4
�) and nas(BF4�)
bands remain very weak. Probably, this result can explain theabsence of any signs of [BF4]�–metal interactions in SERS stud-ies on a copper electrode in [Bmim][BF4] .[41, 42] It is widely ac-cepted that the SERS effect is composed of contributions fromboth electromagnetic-field and chemical enhancements.[47, 48]
The present DFT computations cannot predict electromagneticenhancement, but chemical enhancement effects, usually re-sponsible for changes in relative intensities of Raman bands,[49]
appear to be well reproduced in Figure 8. In particular,a strong enhancement of the band at about 1390 cm�1 relativeto other bands found in the SERS spectra of all studied metal–IL systems[12, 13, 41–43] is properly predicted (Figure 8 E) for thestructure shown in Figure 4 d. It is noteworthy that a similarorientation of the imidazolium ring relative to surface of a plati-num electrode was deduced on the basis of SFG experiments,as discussed above.[46]
On the basis of these IR and Raman spectral simulations, it ispossible to infer that: 1) the latter method is not sensitive tothe interaction of the [BF4]� anion with the Pd-NPs, and IRspectroscopy is better suited to the study of anion–metal inter-actions; 2) the splitting of the strong nas(BF4
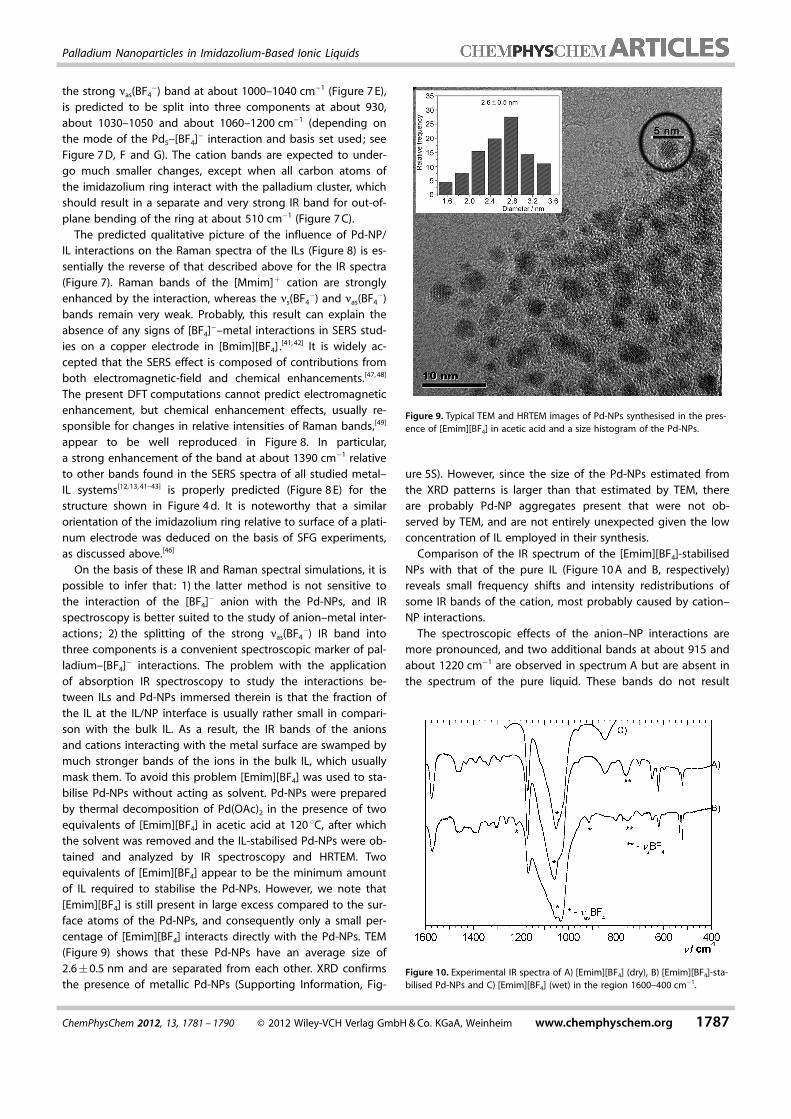
�) IR band intothree components is a convenient spectroscopic marker of pal-ladium–[BF4]� interactions. The problem with the applicationof absorption IR spectroscopy to study the interactions be-tween ILs and Pd-NPs immersed therein is that the fraction ofthe IL at the IL/NP interface is usually rather small in compari-son with the bulk IL. As a result, the IR bands of the anionsand cations interacting with the metal surface are swamped bymuch stronger bands of the ions in the bulk IL, which usuallymask them. To avoid this problem [Emim][BF4] was used to sta-bilise Pd-NPs without acting as solvent. Pd-NPs were preparedby thermal decomposition of Pd(OAc)2 in the presence of twoequivalents of [Emim][BF4] in acetic acid at 120 8C, after whichthe solvent was removed and the IL-stabilised Pd-NPs were ob-tained and analyzed by IR spectroscopy and HRTEM. Twoequivalents of [Emim][BF4] appear to be the minimum amountof IL required to stabilise the Pd-NPs. However, we note that[Emim][BF4] is still present in large excess compared to the sur-face atoms of the Pd-NPs, and consequently only a small per-centage of [Emim][BF4] interacts directly with the Pd-NPs. TEM(Figure 9) shows that these Pd-NPs have an average size of2.6�0.5 nm and are separated from each other. XRD confirmsthe presence of metallic Pd-NPs (Supporting Information, Fig-
ure 5S). However, since the size of the Pd-NPs estimated fromthe XRD patterns is larger than that estimated by TEM, thereare probably Pd-NP aggregates present that were not ob-served by TEM, and are not entirely unexpected given the lowconcentration of IL employed in their synthesis.
Comparison of the IR spectrum of the [Emim][BF4]-stabilisedNPs with that of the pure IL (Figure 10 A and B, respectively)reveals small frequency shifts and intensity redistributions ofsome IR bands of the cation, most probably caused by cation–NP interactions.
The spectroscopic effects of the anion–NP interactions aremore pronounced, and two additional bands at about 915 andabout 1220 cm�1 are observed in spectrum A but are absent inthe spectrum of the pure liquid. These bands do not result
Figure 9. Typical TEM and HRTEM images of Pd-NPs synthesised in the pres-ence of [Emim][BF4] in acetic acid and a size histogram of the Pd-NPs.
Figure 10. Experimental IR spectra of A) [Emim][BF4] (dry), B) [Emim][BF4]-sta-bilised Pd-NPs and C) [Emim][BF4] (wet) in the region 1600–400 cm�1.
ChemPhysChem 2012, 13, 1781 – 1790 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemphyschem.org 1787
Palladium Nanoparticles in Imidazolium-Based Ionic Liquids

from interactions between the IL and water introduced insmall amounts into the IL together with the Pd-NPs, which isseen by comparison of spectra A and B with the spectrum ofthe wet IL (Figure 10 C). In full agreement with the computa-tional predictions (Figure 7 D, F, G) these additional bands canbe assigned to nas(BF4
�) stretching vibrations of the anion in-teracting with the Pd-NP surface. They are rather weak relativeto the main broad band at about 1050 cm�1 because the in-tensity of the latter is a sum of the intensities of two overlap-ping nas(BF4
�) bands of the bulk IL and of the IL/palladium in-terface. Nevertheless, the IR spectra together with the compu-tational results provide direct evidence of interactions of the ILanions with Pd-NPs immersed in the IL.
Note that no additional IR bands at about 500 cm�1 are ob-served in spectrum B, where a spectroscopic marker indicativeof a parallel interaction between the imidazolium cation andthe surface of Pd-NP should be expected according to our sim-ulations (see Figure 7 C and the corresponding discussionabove). Hence, parallel binding of the imidazolium cation inwhich all three carbon atoms are in contact with the metal sur-face, proposed on the grounds of SERS studies on Au-NPs inimidazolium-based ILs,[12] does not appear to take place withthe Pd-NPs in this ILs. As mentioned above, the cation ring ori-entation close to the surface normal (structure shown in Fig-ure 4 d) seems to be more probable.
3. Conclusions
A combined DFT/vibrational spectroscopy approach was usedto determine the interactions of the 1,3-dimethylimidazoliumand 1-ethyl-3-methylimidazolium cations and [BF4]� anionswith each other in the liquid state and with Pd-NPs immersedin the IL. Formation of aggregates of the counter-ions in theliquid has no strong influence on the energy of the interactionof the IL components with model palladium clusters, which isshown to be smaller than the energy for addition of a Pd atomto the cluster. Stronger Pd–Pd interactions in comparison tothe interactions of the palladium cluster with the IL suggest ki-netic stabilisation of Pd-NPs in 1,3-dialkylimidazolium tetra-fluoroborates rather than thermodynamic stabilisation. Thisfeature could explain why Pd-NP catalyst reservoirs that reactto form catalytically active mononuclear palladium species incross-coupling reactions are particularly active in ILs.[50] More-over, the palladium clusters interact more strongly with theanions than with the cations, and this suggests an importantrole of the anions in Pd-NP formation and stabilisation in ILs.At the same time, the binding between isolated Pd atoms andthe 1,3-dimethylimidazolium cation is stronger than Pd–[BF4]�
binding, provided that the Pd atom is bound to the C atomsof the imidazolium ring. This finding is also important for un-derstanding the relative importance of the IL components incross-coupling reactions, as mononuclear palladium speciescleaved from the Pd-NPs appear to be the actual active cata-lysts, and thus the Pd-NPs can be regarded as catalyst reser-voirs.
Raman and IR spectral simulations revealed that SERS ismuch less sensitive to interactions of [BF4]� with Pd-NPs com-
pared to the interactions of the latter with the 1,3-dimethyl-imidazolium cation. In contrast, IR spectroscopy is bettersuited to study the anion–metal interactions, whereas IR spec-tral manifestations of the cation–metal interactions are rathermodest. The splitting of the strong nas(BF4
�) IR band into threecomponents appears to be a convenient spectroscopic markerfor palladium–[BF4]� interactions, which is confirmed by actualspectra of Pd-NPs stabilised with [Emim][BF4] .
The IR spectra and their assignment with quantum chemicalcomputations suggest that both the anions and cations of[Emim][BF4] interact with the Pd-NP surface in the IL. The orien-tation of the cation ring close to the surface normal appears tobe the dominant interaction. Similar conclusions were ob-tained on the basis of recent molecular dynamics studies onRu-NPs in [Bmim][Tf2N].[51] The anion is bound to the surfacethrough two or three fluorine atoms (Figures 5 and 6), al-though the spectra do not allow the subtle differences be-tween these modes of binding to be discerned.
Methods Section
Materials and Spectroscopic Methods: [Emim][BF4] was synthesisedaccording to a literature procedure.[52] All other chemicals were ob-tained from commercial sources and were used as received. IRspectra were recorded on a FTIR spectrophotometer (Tensor 27,PerkinElmer) with a resolution of 2 cm�1 in transmission mode.HRTEM measurements were carried out on a Philips Tecnai F30electron microscope operated at 300 kV. XRD analysis was per-formed on a PANalytical X’Pert PRO MPD system using CuKa radia-tion. The 2 q/w scans were carried out in the range from 30 to1008 in 2 q.For the synthesis of IL-stabilised Pd NPs, Pd(OAc)2 (2.2 mg,0.01 mmol) and [Emim][BF4] (5.1 mg, 0.02 mmol) were dissolved inacetic acid (3 mL) and the mixture was heated at 120 8C under N2
for 3 h. After cooling, the solvent was removed under vacuum for12 h. The remaining black viscous semi-solid was analysed withoutfurther treatment by IR spectroscopy and XRD. For HRTEM analysis,the Pd NPs were dispersed in ethanol following their separationfrom the IL by centrifugation. One drop of the solution was placedonto a copper grid coated with a carbon film, dried under vacuumat 343 K for 24 h, and subjected to HRTEM measurements. Theaverage particle sizes of the Pd NPs were determined from about300 NPs.Methods of Calculation: All calculations were carried out with theGaussian 03 suite of programs.[53] The gradient-corrected correla-tion functional of Perdew, Burke and Ernzerhof (PBE)[18] was usedtogether with LanL2DZ[54] (for Pd atom) and 6-31G*[55, 56] (for allother atoms) basis sets. All stationary points were characterised asminima by analysis of the Hessian matrices. Infrared spectra of theion pair [Mmim][BF4] were calculated with use of the double-zeta(6-31G*,[55, 56] 6-31 + G*,[55–57] 6-31 + + G**[55–60] and Dunning’s “cor-relation-consistent” aug-cc-pVDZ[61]) and triple-zeta (6-311G*,[55, 62, 63]
6-311 + G*[55, 57, 62, 63] and 6-311 + + G**[57, 62–66]) basis sets. The simu-lated spectra were obtained by convolution of computed band in-tensities with a Lorentzian lineshape function (f.w.h.m. = 10 cm�1)without preliminary frequency scaling procedure. In the case ofRaman spectra, in which the Raman intensities were not comput-ed, the calculated Raman activities Sk ¼ a0ð Þ2kþ7 g0ð Þ2k
� ��45 were
multiplied by the frequency dependent factor [Eq. (4)]:
1788 www.chemphyschem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 2012, 13, 1781 – 1790
S. A. Katsyuba et al.

FDF ¼ ð~n0 � ~nkÞ4
~nk 1� exp �hc~nk
kT
� �h i ð4Þ
where ~n0 is the exciting laser wavenumber, ~nk the wavenumber ofthe vibrational mode, c the speed of light, h and k are the Planckand Boltzmann constants, respectively, and T is the temperature;~n0 = 15 798 cm�1 (l= 633 nm) was tentatively taken as a value typi-cal for laser sources used for SERS excitation on palladium NPs.
Acknowledgements
Financial support from the Switzerland-Russia S&T CooperationProgram and the Swiss National Science Foundation are grateful-ly acknowledged. N.Y. thanks the EU for a Marie Curie Interna-tional Incoming Fellowship (Number: 252125-TCPBRCBDP).
Keywords: density functional calculations · ionic liquids ·nanoparticles · palladium · vibrational spectroscopy
[1] For recent reviews, see: a) A. Roucoux, J. Schulz, H. Patin, Chem. Rev.2002, 102, 3757 – 3778; b) M. L. Brongersma, Nat. Mater. 2003, 2, 296 –297; c) B. L. Cushing, V. L. Kolesnichenko, C. J. O’Connor, Chem. Rev.2004, 104, 3893 – 3946; d) M.-C. Daniel, D. Astruc, Chem. Rev. 2004, 104,293 – 346; e) M. Antonietti, D. Kuang, B. Smarsly, Y. Zhou, Angew. Chem.2004, 116, 5096 – 5100; Angew. Chem. Int. Ed. 2004, 43, 4988 – 4992.
[2] a) R. R. Deshmukh, R. Rajagopal, K. V. Srinivasan, Chem. Commun. 2001,1544 – 1545; b) J. Dupont, G. S. Fonseca, A. P. Umpierre, P. F. P. Fichtner,S. R. Teixeira, J. Am. Chem. Soc. 2002, 124, 4228 – 4229.
[3] a) D. Astruc, F. Lu, J. R. Aranzaes, Angew. Chem. 2005, 117, 8062 – 8083;Angew. Chem. Int. Ed. 2005, 44, 7852 – 7872; b) C. Pan, K. Pelzer, K. Phil-ippot, B. Chaudret, F. Dassenoy, P. Lecante, M.-J. Casanove, J. Am. Chem.Soc. 2001, 123, 7584 – 7593; c) J. D. Aiken III, R. G. Finke, J. Am. Chem.Soc. 1999, 121, 8803 – 8810.
[4] For recent reviews, see: a) J. Dupont, J. D. Scholten, Chem. Soc. Rev.2010, 39, 1780 – 1804; b) C. Vollmer, C. Janiak, Coord. Chem. Rev. 2011,255, 2039 – 2057; c) M.-A. Neouze, J. Mater. Chem. 2010, 20, 9593 – 9607.
[5] P. S. Campbell, C. C. Santini, D. Bouchu, B. Fenet, K. Philippot, B. Chau-dret, A. A. H. Padua, Y. Chauvin, Phys. Chem. Chem. Phys. 2010, 12,4217 – 4223.
[6] L. S. Ott, R. G. Finke, Coord. Chem. Rev. 2007, 251, 1075 – 1100.[7] E. Redel, R. Thomann, C. Janiak, Inorg. Chem. 2008, 47, 14 – 16; P. Mi-
gowski, D. Zanchet, G. Machado, M. A. Gelesky, S. R. Teixeira, J. Dupont,Phys. Chem. Chem. Phys. 2010, 12, 6826 – 6833.
[8] G. S. Fonseca, G. Machado, S. R. Teixeira, G. H. Fecher, J. Morais, M. C. M.Alves, J. Dupont, J. Colloid Interface Sci. 2006, 301, 193 – 204.
[9] A. P. Umpierre, G. Machado, G. H. Fecher, J. Morais, J. Dupont, Adv.Synth. Catal. 2005, 347, 1404 – 1412.
[10] B. W. Ninham, Adv. Colloid Interface Sci. 1999, 83, 1 – 17.[11] L. S. Ott, M. L. Cline, M. Deetlefs, K. R. Seddon, R. G. Finke, J. Am. Chem.
Soc. 2005, 127, 5758 – 5759.[12] H. S. Schrekker, M. A. Gelesky, M. P. Stracke, C. M. L. Schrekker, G. Macha-
do, S. R. Teixeira, J. C. Rubim, J. Dupont, J. Colloid Interface Sci. 2007,316, 189 – 195.
[13] J. C. Rubim, F. A. Trindade, M. A. Gelesky, R. F. Aroka, J. Dupont, J. Phys.Chem. C 2008, 112, 19670 – 19675.
[14] B. Bozzini, E. Tondo, A. Bund, A. Ispas, C. Mele, J. Electroanal. Chem.2011, 651, 1 – 11.
[15] E. Redel, M. Walter, R. Tomann, C. Vollmer, L. Hussein, H. Scherer, M.Krueger, C. Janiak, Chem. Eur. J. 2009, 15, 10047 – 10059.
[16] E. Redel, M. Walter, R. Tomann, L. Hussein, M. Krueger, C. Janiak, Chem.Commun. 2010, 46, 1159 – 1161.
[17] a) G. Ou, L. Xu, B. He, Y. Yuan, Chem. Commun. 2008, 4210 – 4212; b) L.Dur�n Pach�n, C. J. Elsevier, G. Rothenberg, Adv. Synth. Catal. 2006, 348,1705 – 1710; c) P. Migowski, J. Dupont, Chem. Eur. J. 2007, 13, 32 – 39;d) N. Yan, C. Xiao, Y. Kou, Coord. Chem. Rev. 2010, 254, 1179 – 1218.
[18] J. P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 1996, 77, 3865 –3868.
[19] R. Craciun, A. J. Vincent, K. H. Shaughnessy, D. A. Dixon, Inorg. Chem.2010, 49, 5546 – 5553.
[20] S. A. Katsyuba, P. J. Dyson, E. E. Vandyukova, A. V. Chernova, A. Vidis,Helv. Chim. Acta 2004, 87, 2556 – 2565.
[21] S. A. Katsyuba, E. E. Zvereva, A. Vidis, P. J. Dyson, J. Phys. Chem. A 2007,111, 352 – 370.
[22] F. C. Gozzo, L. S. Santos, R. Augusti, C. S. Consorti, J. Dupont, M. N. Eber-lin, Chem. Eur. J. 2004, 10, 6187 – 6193.
[23] B. A. DaSilveira Neto, L. S. Santos, F. M. Nachtigall, M. N. Eberlin, J.Dupont, Angew. Chem. 2006, 118, 7409 – 7412; Angew. Chem. Int. Ed.2006, 45, 7251 – 7254.
[24] a) V. Ermolaev, V. Miluykov, D. Krivolapov, I. Rizvanov, E. Zvereva, S. Kat-syuba, O. Sinyashin, R. Schmutzler, Dalton Trans. 2010, 39, 5564 – 5571;b) E. E. Zvereva, S. A. Katsyuba, Russ. Chem. Bull. 2009, 58, 1812 – 1816.
[25] S. Kossmann, J. Thar, B. Kirchner, P. A. Hunt, T. Welton, J. Chem. Phys.2006, 124, 174506.
[26] S. A. Katsyuba, T. P. Griaznova, A. Vidis, P. J. Dyson, J. Phys. Chem. B2009, 113, 5046 – 5051.
[27] E. E. Zvereva, S. A. Katsyuba, P. J. Dyson, Phys. Chem. Chem. Phys. 2010,12, 13780 – 13787, and references cited therein.
[28] J. Dupont, J. Braz. Chem. Soc. 2004, 15, 341 – 350.[29] R. Holomb, A. Martinelli, L. Albinsson, J. C. Lassegues, P. Johansson, P.
Jacobsson, J. Raman Spectrosc. 2008, 39, 793 – 805.[30] P. Nava, M. Sierka, R. Ahlrichs, Phys. Chem. Chem. Phys. 2003, 5, 3372 –
3381.[31] C. Luo, J. Wu, T. J. D. Kumar, N. Balakrishnan, R. C. Forrey, H. Cheng, Int.
J. Quantum Chem. 2007, 107, 1633 – 1641.[32] G. Zanti, D. Peeters, J. Phys. Chem. A 2010, 114, 10345 – 10356.[33] M. Moseler, H. Hakkinen, R. N. Barnett, U. Landman, Phys. Rev. Lett.
2001, 86, 2545 – 2548.[34] G. Zacarias, M. Castro, M. Tour, M. Seminario, J. Phys. Chem. A 1999, 103,
7692 – 7700.[35] F. Aguilera-Granja, A. Vega, J. Rogan, G. Garcia, Nanotechnology 2007,
18, 365706.[36] T. Futschek, M. Marsman, J. Hafner, J. Phys. Condens. Matter 2005, 17,
5927 – 5963.[37] H. Zhang, D. Tian, J. Zhao, J. Chem. Phys. 2008, 129, 087101.[38] D. S. McGuinness, K. J. Cavell, B. F. Yates, B. W. Skelton, A. H. White, J.
Am. Chem. Soc. 2001, 123, 8317 – 8328.[39] Small overlap of BE and BGFE values with AE and AGFE, respectively,
for the case of the isolated Pd atom (n = 1 in Figure 3) is, probably, anartifact. Exact computation of bond energies for the smallest palladiumclusters Pd and Pd2 is a very demanding task (e.g. see Ref. [19] and N. E.Schultz, B. F. Gherman, C. J. Cramer, D. G. Truhlar, J. Phys. Chem. B 2006,110, 24030 – 24046, and references cited herein) and is beyond thescope of the present paper.
[40] I. A. Pasti, S. V. Mentus, Electrochim. Acta 2010, 55, 1995 – 2003.[41] C. R. R. Brand¼o, L. A. F. Costa, H. S. Breyer, J. C. Rubim, Electrochem.
Commun. 2009, 11, 1846 – 1848.[42] L. A. F. Costa, H. S. Breyer, J. C. Rubim, Vib. Spectrosc. 2010, 54, 103 – 106.[43] V. O. Santos, Jr. , M. B. Alves, M. S. Carvalho, P. A. Z. Suarez, J. C. Rubim, J.
Phys. Chem. B 2006, 110, 20379 – 20385.[44] O. Hofft, S. Bahr, V. Kempter, Langmuir 2008, 24, 11562 – 11566.[45] M. Sobota, M. Schmid, M. Happel, M. Amende, F. Maier, H.-P. Steinruck,
N. Paape, P. Wasserscheid, M. Laurin, J. M. Gottfried, J. Libuda, Phys.Chem. Chem. Phys. 2010, 12, 10610 – 10621.
[46] S. Baldelli, Acc. Chem. Res. 2008, 41, 421 – 431, and references therein.[47] M. Moskovits, J. Raman Spectrosc. 2005, 36, 485 – 496.[48] R. K. Chang, T. E. Furtak, Surface Enhanced Raman Scattering, Plenum
Press, New York, 1982.[49] D.-Y. Wu, J.-F. Li, B. Ren, Z.-Q. Tian, Chem. Soc. Rev. 2008, 37, 1025 – 1041.[50] Ionic Liquids: Applications and Perspectives (Ed. : A. Kokorin), InTech,
Rijeka, Croatia, 2011, chap. 16.[51] A. S. Pensado, A. A. H. Padua, Angew. Chem. 2011, 123, 8842 – 8846;
Angew. Chem. Int. Ed. 2011, 50, 8683 – 8687.[52] J. L. Reynolds, K. R. Erdner, P. B. Jones, Org. Lett. 2002, 4, 917 – 919.[53] Gaussian 03 (Revision D.02), M. J. Frisch, G. W. Trucks, H. B. Schlegel,
G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr. , T.Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V.
ChemPhysChem 2012, 13, 1781 – 1790 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemphyschem.org 1789
Palladium Nanoparticles in Imidazolium-Based Ionic Liquids

Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H.Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M.Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E.Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts,R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Och-terski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg,V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K.Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui,A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko,P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham,C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W.Chen, M. W. Wong, C. Gonzalez, J. A. Pople, Wallingford CT, Gaussian,Inc. , 2004.
[54] a) P. J. Hay, W. R. Wadt, J. Chem. Phys. 1985, 82, 270; b) W. R. Wadt, P. J.Hay, J. Chem. Phys. 1985, 82, 284; c) P. J. Hay, W. R. Wadt, J. Chem. Phys.1985, 82, 299.
[55] W. J. Hehre, R. Ditchfield, J. A. Pople, J. Chem. Phys. 1972, 56, 2257.[56] P. C. Hariharan, J. A. Pople, Theor. Chim. Acta. 1973, 28, 213 – 222.[57] a) T. Clark, J. Chandrasekhar, G. W. Spitznagel, P. v. R. Schleyer, J. Comput.
Chem. 1983, 4, 294 – 301; b) M. J. Frisch, J. A. Pople, J. S. Binkley, J.Chem. Phys. 1984, 80, 3265.
[58] J. D. Dill, J. A. Pople, J. Chem. Phys. 1975, 62, 2921.[59] M. M. Francl, W. J. Petro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. De-
Frees, J. A. Pople, J. Chem. Phys. 1982, 77, 3654.[60] V. A. Rassolov, J. A. Pople, M. Ratner, T. L. Windus, J. Chem. Phys. 1998,
109, 1223.[61] D. E. Woon, T. H. Dunning, J. Chem. Phys. 1993, 99, 3730.[62] A. D. McLean, G. S. Chandler, J. Chem. Phys. 1980, 72, 5639.[63] R. Krishnan, J. S. Binkley, R. Seeger, J. A. Pople, J. Chem. Phys. 1980, 72,
650.[64] J.-P. Blaudeau, M. P. McGrath, L. A. Curtiss, L. Radom, J. Chem. Phys.
1997, 107, 5016.[65] L. A. Curtiss, M. P. McGrath, J.-P. Blandeau, N. E. Davis, R. C. Binning, Jr. ,
L. Radom, J. Chem. Phys. 1995, 103, 6104.[66] M. N. Glukhovtsev, A. Pross, M. P. McGrath, L. Radom, J. Chem. Phys.
1995, 103, 1878.
Received: February 1, 2012
Published online on March 23, 2012
1790 www.chemphyschem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 2012, 13, 1781 – 1790
S. A. Katsyuba et al.




















