
Rapid conversion of spirostans into furostan skeletons at room temperature
8
Rapid conversion of spirostans into furostan skeletons at room temperature Omar Viñas-Bravo a , Roxana Martinez-Pascual a , José Luis Vega-Baez b , Víctor Gómez-Calvario b , Jesús Sandoval-Ramírez b , Sara Montiel-Smith b,⇑ , Socorro Meza-Reyes b,⇑ , Alejandra López-De Rosas b , Mónica Martínez-Montiel b , Mayra Reyes c , José A. Ruiz c a Universidad del Papaloapan, Circuito Central # 200, Colonia Parque Industrial, Tuxtepec, Oax., México C.P. 68301, Mexico b Benemérita Universidad Autónoma de Puebla, Facultad de Ciencias Químicas, Ciudad Universitaria, Puebla, Pue., México C.P. 72570, Mexico c Centro de Química Biomolecular, Ciudad de La Habana, Cuba C.P. 16042, Cuba article info Article history: Received 21 June 2011 Received in revised form 20 September 2011 Accepted 13 October 2011 Available online 20 October 2011 Keywords: Sapogenins 22-Substituted furostans Pseudosapogenins Acetic–trifluoroacetic mixed anhydride abstract We report a facile protocol to obtain 22-substituted furostans and pseudosapogenins in high yields from (25R)- and (25S)-sapogenins. This method involves the treatment of the sapogenin with acetic–trifluoro- acetic mixed anhydride and BF 3 OEt 2 at room temperature, followed by the addition of a nucleophile (H 2 O, MeOH or KSeCN). In the case of 22-hydroxyfurostans, they can be transformed to pseudosapoge- nins by treatment with p-toluensulfonic acid. Ó 2011 Elsevier Inc. All rights reserved. 1. Introduction In recent years, steroidal saponins isolated from diverse plants have attracted scientific attention because of their structural diver- sity and significant biological activities [1]. Several plant extracts with a high level of steroidal saponins have been used in traditional medicine as anticancer (Dioscorea colletii var. hypoglauca), cardio-, cerebrovascular-, and gastropa- thy-protective (Dioscorea panthaica), and anti-rheumatism agents (Dioscorea nipponica, Dioscorea futschauensis) [2]. In particular, C- 22 oxygenated furostan saponins from the above mentioned genus and other natural sources have been subjected to pharmacological assays and shown to exhibit diverse activities, including cytotoxic [3], antifungal, antispasmodic, and antithrombotic [4]. Protodio- scin (1, Fig. 1) is a representative example of a 22-hydroxyfurostan saponin that has been identified in a large number of medicinal plants and vegetables. Compound 1 has shown cytotoxic effects on leukemia cell lines [5], a strong growth inhibitory effect on HL-60 cells [6], and recently, antihyperlipidemic effects [7]. The methyl protodioscin (2, Fig. 1), which is structurally close to 1, was reported to have cytotoxic activity and might act through a novel anticancer mechanism [8]. Icogenin (3, Fig. 1) was found to be cytotoxic towards HL-60 cells via the induction of apoptosis [9]. Both 22-alkoxyfurostan saponins 1 and 2 have a b-D-glucopyr- anose substituent at the 26-OH and a chacotriose sugar moiety at the 3b-OH; the difference in the biological activity reveals the importance of the substituent attached at the C-22 position of the aglycon. All 22-substituted furostans have important medicinal activity, even if they are not glycosylated at 26-OH as in 3. Due to the scarcity of these important steroidal saponins from natural sources, synthetic chemistry has been used as a practical alternative to derive them. Nevertheless, the synthesis of the 22- substituted furostan aglycon is a difficult task. The first protocol to obtain the 22-methoxyfurostan moiety from the diosgenin was developed by Yu et al. [10] by means of the oxidative opening of E and F rings and involves nine stages. Since then, Cheng et al. [11] synthesized the methyl protodioscin (2) from the same starting material by a shorter and more efficient approach: the construction of 22-methoxyfurostan aglycon of methyl protodioscin was obtained in six stages. The total syntheses of 3 and its analogs have been performed by Lei et al. [12] from diosgenin, applying a similar methodology as that used by Cheng et al. [11] for the construction of the 22-methoxyfurostan moiety. The pseudospirostans are important starting materials for the preparation of a number of biologically active steroidal compounds [12–16]. Many procedures have been reported to open the spiro- stan F ring to obtain furostan skeletons, but they use either high temperatures or very low ones; none of them can be accomplished at room temperature. The results published by Koreeda [17] and Fuchs [18] using mixed anhydrides are attractive for the selective 0039-128X/$ - see front matter Ó 2011 Elsevier Inc. All rights reserved. doi:10.1016/j.steroids.2011.10.004 ⇑ Corresponding authors. Tel.: +52 222 2295500x7382; fax: +52 222 2295584. E-mail addresses: [email protected] (S. Montiel-Smith), maria.me- [email protected] (S. Meza-Reyes). Steroids 77 (2012) 59–66 Contents lists available at SciVerse ScienceDirect Steroids journal homepage: www.elsevier.com/locate/steroids
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Rapid conversion of spirostans into furostan skeletons at room temperature

Steroids 77 (2012) 59–66
Contents lists available at SciVerse ScienceDirect
Steroids
journal homepage: www.elsevier .com/locate /s teroids
Rapid conversion of spirostans into furostan skeletons at room temperature
Omar Viñas-Bravo a, Roxana Martinez-Pascual a, José Luis Vega-Baez b, Víctor Gómez-Calvario b,Jesús Sandoval-Ramírez b, Sara Montiel-Smith b,⇑, Socorro Meza-Reyes b,⇑, Alejandra López-De Rosas b,Mónica Martínez-Montiel b, Mayra Reyes c, José A. Ruiz c
a Universidad del Papaloapan, Circuito Central # 200, Colonia Parque Industrial, Tuxtepec, Oax., México C.P. 68301, Mexicob Benemérita Universidad Autónoma de Puebla, Facultad de Ciencias Químicas, Ciudad Universitaria, Puebla, Pue., México C.P. 72570, Mexicoc Centro de Química Biomolecular, Ciudad de La Habana, Cuba C.P. 16042, Cuba
a r t i c l e i n f o a b s t r a c t
Article history:Received 21 June 2011Received in revised form 20 September2011Accepted 13 October 2011Available online 20 October 2011
Keywords:Sapogenins22-Substituted furostansPseudosapogeninsAcetic–trifluoroacetic mixed anhydride
0039-128X/$ - see front matter � 2011 Elsevier Inc. Adoi:10.1016/j.steroids.2011.10.004
⇑ Corresponding authors. Tel.: +52 222 2295500x73E-mail addresses: [email protected] (
[email protected] (S. Meza-Reyes).
We report a facile protocol to obtain 22-substituted furostans and pseudosapogenins in high yields from(25R)- and (25S)-sapogenins. This method involves the treatment of the sapogenin with acetic–trifluoro-acetic mixed anhydride and BF3�OEt2 at room temperature, followed by the addition of a nucleophile(H2O, MeOH or KSeCN). In the case of 22-hydroxyfurostans, they can be transformed to pseudosapoge-nins by treatment with p-toluensulfonic acid.
� 2011 Elsevier Inc. All rights reserved.
1. Introduction
In recent years, steroidal saponins isolated from diverse plantshave attracted scientific attention because of their structural diver-sity and significant biological activities [1].
Several plant extracts with a high level of steroidal saponinshave been used in traditional medicine as anticancer (Dioscoreacolletii var. hypoglauca), cardio-, cerebrovascular-, and gastropa-thy-protective (Dioscorea panthaica), and anti-rheumatism agents(Dioscorea nipponica, Dioscorea futschauensis) [2]. In particular, C-22 oxygenated furostan saponins from the above mentioned genusand other natural sources have been subjected to pharmacologicalassays and shown to exhibit diverse activities, including cytotoxic[3], antifungal, antispasmodic, and antithrombotic [4]. Protodio-scin (1, Fig. 1) is a representative example of a 22-hydroxyfurostansaponin that has been identified in a large number of medicinalplants and vegetables. Compound 1 has shown cytotoxic effectson leukemia cell lines [5], a strong growth inhibitory effect onHL-60 cells [6], and recently, antihyperlipidemic effects [7].
The methyl protodioscin (2, Fig. 1), which is structurally close to1, was reported to have cytotoxic activity and might act through anovel anticancer mechanism [8]. Icogenin (3, Fig. 1) was found tobe cytotoxic towards HL-60 cells via the induction of apoptosis
ll rights reserved.
82; fax: +52 222 2295584.S. Montiel-Smith), maria.me-
[9]. Both 22-alkoxyfurostan saponins 1 and 2 have a b-D-glucopyr-anose substituent at the 26-OH and a chacotriose sugar moiety atthe 3b-OH; the difference in the biological activity reveals theimportance of the substituent attached at the C-22 position ofthe aglycon. All 22-substituted furostans have important medicinalactivity, even if they are not glycosylated at 26-OH as in 3.
Due to the scarcity of these important steroidal saponins fromnatural sources, synthetic chemistry has been used as a practicalalternative to derive them. Nevertheless, the synthesis of the 22-substituted furostan aglycon is a difficult task.
The first protocol to obtain the 22-methoxyfurostan moietyfrom the diosgenin was developed by Yu et al. [10] by means ofthe oxidative opening of E and F rings and involves nine stages.Since then, Cheng et al. [11] synthesized the methyl protodioscin(2) from the same starting material by a shorter and more efficientapproach: the construction of 22-methoxyfurostan aglycon ofmethyl protodioscin was obtained in six stages. The total synthesesof 3 and its analogs have been performed by Lei et al. [12] fromdiosgenin, applying a similar methodology as that used by Chenget al. [11] for the construction of the 22-methoxyfurostan moiety.
The pseudospirostans are important starting materials for thepreparation of a number of biologically active steroidal compounds[12–16]. Many procedures have been reported to open the spiro-stan F ring to obtain furostan skeletons, but they use either hightemperatures or very low ones; none of them can be accomplishedat room temperature. The results published by Koreeda [17] andFuchs [18] using mixed anhydrides are attractive for the selective

Fig. 1. Structures of some important natural 22-oxygenated furostans.
60 O. Viñas-Bravo et al. / Steroids 77 (2012) 59–66
opening of the F ring of sapogenin. Diosgenin is treated by benzoyltrifluoromethanesulfonate (BzOTf) at �70 �C for 15 min or trifluo-roacetyl trifluoromethanesulfonate (TFAT) at �40 �C, producingpseudodiosgenin at 64% and 96% yields, respectively. Those condi-tions demonstrate that the use of a highly active mixed anhydride,such as those supporting the trifluoromethanesulfonate moiety,promotes the F ring opening at significantly lower temperatures.
As we were interested in developing fast-acylation methodolo-gies at room temperature [19], we prepared various mixed anhy-drides containing the trifluoroacetate group, a remarkably goodnucleofuge moiety [20].The mixed anhydrides were prepared veryeasily, and the reactions can be carried out at room temperaturerapidly and in excellent yields. Further, our group reported selec-tive E or F ring opening through a controlled acetolysis using aceticanhydride and BF3�OEt2 as a catalyst to obtain 23-acetyl-22,26-epoxycholestanes in good yields at room temperature [21].
Based on these results, we hypothesized that the acetic–trifluo-roacetic anhydride (ATFAA) combined with BF3�OEt2 could producea new pair of reagents to induce a regioselective F-ring opening.
In this paper, we report the efficient regioselective F ringopening of sapogenins of the 25R and 25S series to obtain 22-substituted furostans and the transformation of the 22-hydroxy-furostans in pseudosapogenins at room temperature.
2. Experimental methods
2.1. General methods
NMR spectra (1D and 2D, 1H and 13C, DEPT, COSY, NOESY, HSQC,HMBC) were recorded on a VARIAN Mercury spectrometer(400 MHz for 1H, 100 MHz for 13C). Chemical shifts are stated in
ppm (d) and are referred to the residual 1H signal (d = 7.27) or tothe central 13C triplet signal (d = 77.0) for CDCl3 or to the 8.73 sig-nal when pyridine-d5 was the solvent. Coupling constants (J) arequoted in Hz. IR spectra were acquired on a Nicolet Magna FT-IR750 spectrophotometer using KBr pellets (mmax, cm�1). FAB-MSand high resolution mass (HRMS) spectra were recorded on aJeol-JMS-SX102A mass spectrometer. Optical rotations were deter-mined on a Perkin Elmer 241 polarimeter at room temperatureusing chloroform solutions. Melting points were measured froma Mel-Temp apparatus and were not corrected. Analytical TLCwas performed on silica gel ALUGRAM� SIL G/UV254 plates, andchromatographic columns were carried out on Davisil™ grade633 silica gel (200–425 mesh).
2.2. General procedure for the formation of the acetic–trifluoroaceticanhydride (ATFAA)
In a dry 10 mL round flask equipped with a magnetic stir barand inert atmosphere, 1.6 mL of glacial acetic acid (27.9 mmol),3.1 mL (21.8 mmol) of trifluoracetic anhydride were placed andthe solution was stirred for 2 min [20].
2.3. General procedure for the formation of 22-hydroxyfurostans 6a,6b and 6c
We placed 2.0 mmol of the sapogenin 4a, 4b, or 4c in a 25 mLround bottom flask and added 10 mL of CH2Cl2, 1.0 mL of BF3�OEt2
(7.96 mmol) and 4.7 mL of recently prepared ATFAA (21.8 mmol)[20]. The mixture was magnetically stirred at room temperatureand monitored by TLC until complete conversion of the startingmaterial (approximately 5 min). The reaction mixture was poured

O. Viñas-Bravo et al. / Steroids 77 (2012) 59–66 61
over ground ice (20 g) and vigorously shaken. The crude productwas washed with brine (1 � 50 mL) and then a saturated solutionof NaHCO3 (2 � 30 mL) until neutralization, and finally with brine(30 mL) and distilled water (30 mL). The organic phase was driedusing anh. Na2SO4 and concentrated to dryness under vacuum.The crude product was purified by column chromatography andeluted with a mixture of hexane/ethyl acetate (4:1), affording 6a(80%), 6b (80%), or 6c (75%).
2.3.1. (22R, 25S)-22-Hydroxy-5b-furostane-3b,26-diyl diacetate (6a)Colorless oil; [a]D +2.5� (c 1.0, CHCl3); IR �mmax: 3482 (mO–H),
1728 (mC@O ester), 1375 (dCH3), 1236 (mC–O). 1H NMR (pyridine-d5) d: 5.22 (1H, br-s, H-3a), 4.97 (1H, ddd, J16�17 = 8.0 Hz, J16�15
a = J16�15b = 7.6 Hz, H-16a), 4.06 (1H, dd, J26b�26a = 10.8 Hz,J26b�25 = 6.0 Hz, H-26b), 3.95 (1H, dd, J26a�26b = 10.8 Hz,J26a�25 = 6.4 Hz, H-26a), 2.25 (1H, dc, J20�21 = J20�17 = 6.0 Hz, H-20), 2.07 (3H, s, CH3COO-3), 1.96 (3H, s, CH3COO-26), 1.34 (3H, d,J = 6.4 Hz, CH3-21), 0.94 (3H, s, CH3-19), 0.93 (3H, d, J27�25 =7.2 Hz, CH3-27), 0.91 (3H, s, CH3-18). 13C NMR (pyridine-d5) d:31.0 (C-1), 25.4 (C-2), 70.9 (C-3), 31.4 (C-4), 37.8 (C-5), 26.9(C-6), 26.8 (C-7), 35.7 (C-8), 40.2 (C-9), 35.4 (C-10), 21.4 (C-11),40.5 (C-12), 41.4 (C-13), 56.4 (C-14), 32.6 (C-15), 81.3 (C-16),64.1 (C-17), 17.0 (C-18), 24.2 (C-19), 40.9 (C-20), 16.7 (C-21),110.5 (C-22), 37.3 (C-23), 28.4 (C-24), 33.5 (C-25), 69.6 (C-26),17.2 (C-27), 21.6 (CH3COO-3), 21.0 (CH3COO-26), 170.2 (CH3COO-3), 170.6 (CH3COO-26). HRMS-FAB (m/z): [M]+ calcd for C31H50O6,518.3607. Found, 518.3599.
2.3.2. (22R, 25S)-22-Hydroxyfurost-5-ene-3b,26-diyl diacetate (6b)Colorless oil; [a]D �3.5� (c 1.0, CHCl3); IR �mmax: 3525 (mO–H),
1730 (mC@O ester), 1375 (dCH3), 1236 (mC–O). 1H NMR (pyridine-d5) d: 5.32 (1H, br-d, H-6), 4.92 (1H, ddd, J16�17 = 8.0 Hz,J16�15a = 7.4 Hz, J16�15b = 7.0 Hz, H-16a), 4.76 (1H, m, H-3a), 4.09(1H, dd, J26b�26a = 10.8 Hz, J26b�25 = 5.6 Hz, H-26b), 3.97 (1H, dd,J26a�26b = 10.8 Hz, J26a�25 = 6.4 Hz, H-26a), 2.24 (1H, dc,J20�21 = J20�17 = 6.8 Hz, H-20), 2.05 (3H, s, CH3COO-3), 1.96 (3H, s,CH3COO-26), 1.33 (3H, d, J21�20 = 6.8 Hz, CH3-21), 0.98 (3H, s,CH3-19), 0.92 (3H, s, CH3-18), 0.91 (3H, d, J27�25 = 6.8 Hz, CH3-27). 13C NMR (pyridine-d5) d: 37.3 (C-1), 28.3 (C-2), 74.1 (C-3),38.7 (C-4), 139.9 (C-5), 122.6 (C-6), 32.5 (C-7), 31.8 (C-8), 50.4(C-9), 37.2 (C-10), 21.4 (C-11), 40.1 (C-12), 41.0 (C-13), 56.7 (C-14), 32.7 (C-15), 81.2 (C-16), 63.9 (C-17), 16.8 (C-18), 19.6 (C-19),40.9 (C-20), 16.7 (C-21), 110.5 (C-22), 37.4 (C-23), 28.5 (C-24),33.5 (C-25), 69.5 (C-26), 17.3 (C-27), 21.5 (CH3COO-3), 21.0(CH3COO-26), 170.1 (CH3COO-3), 170.7 (CH3COO-26). HRMS-FAB(m/z): [M]+ calcd for C31H48O6, 516.3451. Found, 516.3450.
2.3.3. (22R, 25S)-22-Hydroxy-12-oxo-5a-furostane-3b,26-diyldiacetate (6c)
Colorless oil; [a]D �6.4� (c 1.0, CHCl3); IR �mmax: 1732 (mC@O es-ter), 1705 (mC@O ketone), 1375 (dCH3), 1236 (mC–O). 1H NMR (pyr-idine-d5) d: 4.84 (1H, ddd, J16–17 = 7.2 Hz, J16–15a = J16–15b =6.8 Hz, H-16a), 4.75 (1H, m, H-3a), 4.07 (1H, dd, J26b-26a = 10.8 Hz, J26b-25 = 5.6 Hz, H-26b), 3.96 (1H, dd, J26a-26b =10.8 Hz, J26a-25 = 6.8 Hz, H-26a), 2.87 (1H, dd, J17–20 = 8.0 Hz,J17–16 = 6.8 Hz, H-17), 2.42 (1H, dd, J11ax-11 eq = J11ax-9ax = 13.6 Hz, H-11ax), 2.25 (1H, dd, J11 eq-11ax = 13.6 Hz,J11 eq-9ax = 8.4 Hz, H-11 eq), 2.20 (1H, m, H-20), 2.03 (3H, s,CH3COO-3), 1.96 (3H, s, CH3COO-26), 1.52 (3H, d, J21–20 =7.2 Hz, CH3–21), 1.15 (3H, s, CH3–18), 0.91 (3H, d, J27–25 =6.4 Hz, CH3–27), 0.78 (3H, s, CH3–19).13C NMR (pyridine-d5) d:36.5 (C-1), 27.7 (C-2), 73.4 (C-3), 34.3 (C-4), 44.6 (C-5), 28.4 (C-6), 31.81 (C-7), 34.5 (C-8), 55.5 (C-9), 36.3 (C-10), 38.1 (C-11),212.5 (C-12), 55.8 (C-13), 55.9 (C-14), 31.83 (C-15), 79.8 (C-16),55.2 (C-17), 16.4 (C-18), 11.9 (C-19), 41.4 (C-20), 15.4 (C-21),110.5 (C-22), 37.2 (C-23), 28.6 (C-24), 33.5 (C-25), 69.5 (C-26),
17.3 (C-27), 21.5 (CH3COO-3), 21.0 (CH3COO-26), 170.1 (CH3
COO-3), 170.6 (CH3COO-26). HRMS-FAB (m/z): [M]+ calcd forC31H48O7, 532.3400. Found, 532.3388.
2.4. General procedure for the formation of pseudosapogenins 7a, 7band 7c
Method A: In a 25 mL round bottom flask, 1.0 mmol of the sapo-genin 4a, 4b, or 4c were dissolved in 5.0 mL of CH2Cl2, and then0.7 mL of BF3�OEt2 (5.4 mmol) and 2.3 mL of recently prepared AT-FAA (10.9 mmol) [20] were slowly added. The mixture was mag-netically stirred at room temperature and monitored by TLC untilcomplete disappearance of the starting material (5 min). The reac-tion mixture was poured over cold H2O (10 mL) and vigorouslyshaken; immediately, 100 mg of p-TsOH was added and stirredfor 10 min. After this, the organic phase was extracted with25 mL of CH2Cl2, and then the organic phase was washed withbrine (1 � 10 mL), followed by a saturated solution of NaHCO3
(2 � 15 mL) to obtain neutralization, and finally with water(1 � 15 ml). The resulting organic phase was dried under anh.Na2SO4 and concentrated to dryness under vacuum. The crudeproduct was separated by column chromatography and elutedwith hexane/ethyl acetate (8:2) to afford 7a(92%), 7b(92%), or7c(90%).
Method B: In a 25 mL round bottom flask, 1.0 mmol of the 22-hydroxyfurostan 6a, 6b, or 6c were dissolved in 5.0 mL of CH2Cl2,and then 100 mg of p-TsOH was added and stirred for 10 min. Afterthis, the reaction mixture was diluted with 25 mL of CH2Cl2, andthen the organic phase was washed with brine (1 � 10 mL) fol-lowed by a saturated solution of NaHCO3 (2 � 15 mL) to obtainneutralization, and finally with water (1 � 15 ml). The resultingorganic phase was dried to Na2SO4 and concentrated to drynessunder vacuum. The crude product was separated by column chro-matography and eluted with hexane/ethyl acetate (8:2) to afford7a (90%), 7b (88%), or 7c (88%).
2.4.1. (25S)-5b-Furost-20(22)-ene-3b,26-diyl diacetate (7a)Colorless oil; [a]D +8.4� (c 0.28, CHCl3) (Lit. �5.8� [22]); IR �mmax:
1747, 1732 (mC@O ester), 1694 (mC@C–O), 1448 (dCH2), 1374(dCH3), 1236 (mC–O). 1H NMR (CDCl3) d: 5.05 (1H, br-s, H-3a),4.71 (1H, ddd, J16�17 = 9.8 Hz, J16�15a = 6.0 Hz, J16�15b = 7.4 Hz, H-16a), 3.93 (1H, dd, J26a�26b = 10.8 Hz, J26b�25 = 6.0 Hz, H-26b),3.86 (1H, dd, J26b�26a = 10.8 Hz, J26a�25 = 6.0 Hz, H-26a), 2.46 (1H,d, J17�16 = 9.8 Hz, H-17), 2.05 (3H, s, CH3COO-3), 2.04 (3H, s,CH3COO-26), 1.57 (3H, s, CH3-21), 0.98 (3H, s, CH3-19), 0.93 (3H,d, J27�25 = 6.8 Hz,CH3-27), 0.66 (3H, s, CH3-18).13C NMRd: 30.6 (C-1), 25.1 (C-2), 70.7 (C-3), 30.8 (C-4), 37.3 (C-5), 26.6 (C-6), 26.5(C-7), 35.1 (C-8), 40.0 (C-9), 35.0 (C-10), 21.1 (C-11), 39.9 (C-12),43.7 (C-13), 54.9 (C-14), 34.1 (C-15), 84.3 (C-16), 64.3 (C-17),14.3 (C-18), 23.91 (C-19), 103.8 (C-20), 11.7 (C-21), 151.1 (C-22),23.3 (C-23), 30.8 (C-24), 32.2 (C-25), 69.2 (C-26), 16.7 (C-27),21.0 (CH3COO-3), 21.6 (CH3COO-26), 170.5 (CH3COO-3), 171.0(CH3COO-26).HRMS-FAB (m/z): [M]+ calcd for C31H48O5,500.3502. Found, 500.3507.
2.4.2. (25R)-Furosta-5, 20(22)-diene-3b,26-diyl diacetate (7b)Colorless solid, mp 99–100 �C; [a]D �35.6� (c 0.1, CHCl3) (Lit.
�31� [23a], �39� [23b]); IR �mmax: 1732 (mC–O ester), 1694 (mC@C–O), 1375 (dCH3), 1236 (mC–O). 1H NMR (CDCl3) d: 5.36 (1H, d,J = 4.8 Hz, H-6), 4.72 (1H, ddd, J16�17 = 10.4 Hz, J16�15a = 8.0 Hz,J16�15b = 5.6 Hz, H-16a), 4.58 (1H, m, H-3a), 3.93 (1H, dd,J26b�26a = 10.6 Hz, J26b�25 = 5.6 Hz, H-26b), 3.87 (1H, dd, J26a�26b =10.6 Hz, J26a�25 = 5.6 Hz, H-26a), 2.47 (1H, d, J17�16 = 10.4 Hz, H-17), 2.04 (3H, s, CH3COO-26), 2.02 (3H, s, CH3COO-3), 1.58 (3H, s,CH3-21), 1.03 (3H, s, CH3-19), 0.94 (3H, d, J27�25 = 7.2 Hz, CH3-27),

62 O. Viñas-Bravo et al. / Steroids 77 (2012) 59–66
0.69 (3H, s, CH3-18). 13C NMR d: 37.0 (C-1), 27.7 (C-2), 73.7 (C-3),38.1 (C-4), 139.4 (C-5), 122.1 (C-6), 32.1 (C-7), 31.2 (C-8), 49.9 (C-9), 36.7 (C-10), 21.0 (C-11), 39.4 (C-12), 43.2 (C-13), 54.9 (C-14),34.1 (C-15), 84.2 (C-16), 64.1 (C-17), 14.0 (C-18), 19.4 (C-19),103.6 (C-20), 11.7 (C-21), 151.1 (C-22), 23.2 (C-23), 30.8 (C-24),32.1 (C-25), 69.7 (C-26), 16.8 (C-27), 21.5 (CH3COO-3), 21.0(CH3COO-26), 170.2 (CH3COO-3), 171.0 (CH3COO-26).HRMS-FAB(m/z): [M]+ calcd for C31H46O5, 498.3345. Found, 498.3344.
2.4.3. (25R)-12-Oxo-5a-furost-20(22)-ene-3b,26-diyl diacetate (7c)Colorless solid, mp 94–95 �C; [a]D +66.8� (c 0.1, CHCl3) (Lit. +69�
[24]); IR �mmax: 1732 (mC@O ester), 1708 (mC@O ketone), 1692(mC = C–O), 1375 (dCH3), 1236 (mC–O). 1H NMR (CDCl3) d: 4.69(1H, m, H-16a), 4.67 (1H, m, H-3a), 3.94 (1H, dd, J26b�26a = 10.4 Hz,J26b�25 = 6.0 Hz, H-26b), 3.87 (1H, dd, J26a�26b = 10.4 Hz, J26a�25 =6.6 Hz, H-26a), 3.25 (1H, d, J17�16 = 10.4 Hz, H-17), 2.44 (1H, dd,J11ax�11eq = J11ax�9ax = 13.8, Hz, H-11ax) 2.05 (3H, s, CH3COO-26),2.02 (3H, s, CH3COO-3), 1.58 (3H, s, CH3-21), 0.94 (3H, d,J27�25 = 6.6 Hz, CH3-27), 0.93 (3H, s, CH3-18), 0.93 (3H, s, CH3-19).13C NMR d: 36.3 (C-1), 27.2 (C-2), 73.0 (C-3), 33.8 (C-4), 44.4(C-5), 28.1 (C-6), 31.6 (C-7), 34.1 (C-8), 55.3 (C-9), 36.1 (C-10),37.9 (C-11), 213.1 (C-12), 57.2 (C-13), 54.1 (C-14), 33.5 (C-15),82.5 (C-16), 55.6 (C-17), 11.9 (C-18), 14.0 (C-19), 103.2 (C-20),11.3 (C-21), 151.8 (C-22), 23.2 (C-23), 30.7 (C-24), 32.1 (C-25),69.0 (C-26), 16.7 (C-27), 21.4 (CH3COO-3), 21.0 (CH3COO-26),170.2 (CH3COO-3), 170.9 (CH3COO-26).HRMS-FAB (m/z): [M]+
calcd for C31H46O6, 514.3294. Found, 514.3289.
2.5. General procedure for the generation of 22-methoxyfurostans 8a,8b and 8c
The same procedure for the generation of 22-hydroxyfurostanswas used, but instead of pouring the reaction mixture over ice, itwas poured over cold methanol. The crude product was separatedby column chromatography and eluted with hexane/ethyl acetate(4:1) to afford 8a (85%), 8b (85%), or 8c (80%).
2.5.1. (22R, 25S)-22-methoxy-5b-furostane-3b,26-diyl diacetate (8a)Colorless oil; [a]D �11� (c 1.15, CHCl3); IR �mmax: 1732 (mC@O es-
ter), 1373 (dCH3), 1234 (mC–O). 1H NMR (pyridine-d5) d:5.20 (1H,br-s, H-3a), 4.49 (1H, ddd, J16�17 = 8.0 Hz, J16�15b = 7.4 Hz,J16�15a = 7.0 Hz, H-16a), 3.99 (1H, dd, J26b�26a = 10.8 Hz, J26b�25 =5.9 Hz, H-26b), 4.08 (1H, dd, J26a�26b = 10.8 Hz, J26a�25 = 6.2 Hz, H-26a), 3.24 (3H, s, CH3O-22), 2.23 (1H, dq, J20�21 = J20�17 = 6.6 Hz,H-20), 2.07 (3H, s, CH3COO-3), 2.02 (3H, s, CH3COO-26), 1.18 (3H,d, J = 6.9 Hz, CH3-21), 0.95 (3H, s, CH3-19), 0.94 (3H, d,J27�25 = 7 Hz, CH3-27), 0.84 (3H, s, CH3-18). 13C NMR (pyridine-d5) d: 31.4 (C-1), 25.4 (C-2), 70.9 (C-3), 31.1 (C-4), 37.9 (C-5),27.0 (C-6), 26.8 (C-7), 35.7 (C-8), 40.2 (C-9), 35.4 (C-10), 21.3 (C-11), 40.3 (C-12), 41.5 (C-13), 56.5 (C-14), 32.4 (C-15), 81.6 (C-16),64.5 (C-17), 16.8 (C-18), 24.2 (C-19), 40.7 (C-20), 16.6 (C-21),112.5 (C-22), 30.9 (C-23), 28.3 (C-24), 33.5 (C-25), 69.2 (C-26),17.4 (C-27), 21.6 (CH3COO-3), 21.0 (CH3COO-26), 170.1 (CH3COO-3), 170.7 (CH3COO-26), 47.5 (CH3O-22). HRMS-FAB (m/z): [M]+
calcd for C32H52O6, 532.3764. Found, 532.3770.
2.5.2. (22R, 25S)-22-methoxyfurost-5-ene-3b,26-diyl diacetate (8b)Colorless oil; [a]D �8.3� (c 1.0, CHCl3); IR �mmax: 1736 (mC@O es-
ter), 1369 (dCH3), 1242 (mC–O). 1H NMR (pyridine-d5) d: 5.34 (1H,m, H-6), 4.76 (1H, br-s, H-3a), 4.44 (1H, ddd, J16�17 = 8.0 Hz,J16�15a = 7.6 Hz, J16�15b = 7.2 Hz, H-16a), 4.10 (1H, dd, J26b�26a =10.4 Hz, J26b�25 = 5.6 Hz, H-26b), 4.01 (1H, dd, J26a�26b = 10.8 Hz,J26a�25 = 6.8 Hz, H-26a), 3.25 (CH3O-22), 2.23 (1H, dq,J20�21 = J20�17 = 6.8 Hz, H-20), 2.05 (3H, s, CH3COO-3), 2.03 (3H, s,CH3COO-26), 1.19 (3H, d, J21�20 = 6.4 Hz, CH3-21), 0.98 (3H, s,
CH3-19), 0.95 (3H, d, J27�25 = 6.8 Hz, CH3-27), 0.85 (3H, s, CH3-18). 13C NMR (pyridine-d5) d: 37.4 (C-1), 28.3 (C-2), 74.1 (C-3),38.7 (C-4), 140.0 (C-5), 122.6 (C-6), 32.4 (C-7), 31.8 (C-8), 50.31(C-9), 37.1 (C-10), 21.3 (C-11), 39.9 (C-12), 41.0 (C-13), 56.7 (C-14), 32.4 (C-15), 81.5 (C-16), 64.2 (C-17), 16.5 (C-18), 19.6 (C-19),40.6 (C-20), 16.5 (C-21), 112.5 (C-22), 30.8 (C-23), 28.3 (C-24),33.4 (C-25), 69.4 (C-26), 17.1 (C-27), 21.5 (CH3COO-3), 21.0(CH3COO-26), 170.1 (CH3COO-3), 170.69 (CH3COO-26), 47.4(CH3O-22).HRMS-FAB (m/z): [M]+ calcd for C32H50O6, 530.3607.Found, 530.3595.
2.5.3. (22R, 25S)-22-methoxy-12-oxo-5a-furostane-3b,26-diyldiacetate (8c)
Colorless oil; [a]D�20� (c 1.1, CHCl3); IR �mmax: 1732 (mC@O ester),1709 (mC@O ketone), 1366 (dCH3), 1242 (mC–O). 1H NMR(pyridine-d5) d: 4.74 (1H, br-s, H-3a), 4.36 (1H, J16�17 = 7.4 Hz,J16�15a = J16�15b = 7.0 Hz, H-16a), 4.05 (1H, dd, J26b�26a = 10.8 Hz,J26b�25 = 6.4 Hz, H-26b), 3.98 (1H, dd, J26a�26b = 10.8 Hz, J26a�25 =6.4 Hz, H-26a), 3.22 (3H, s, CH3O-22), 2.65 (1H, dd, J17�20 = 6.4 Hz,J17�16 = 7.2 Hz, H-17), 2.38 (1H, dd, J11ax�11eq = J11ax�9ax = 14.0 Hz,H-11ax), 2.21 (1H, dd, J11eq�11ax = 14.0 Hz, J11eq�9ax = 5.2 Hz, H-11 eq), 2.17 (1H, m, H-20), 2.03 (3H, s, CH3COO-3), 2.00 (3H, s,CH3COO-26), 1.37 (3H, d, J21�20 = 6.8 Hz, CH3-21), 1.08 (3H, s, CH3-18), 0.93 (3H, d, J27�25 = 6.8 Hz, CH3-27), 0.78 (3H, s, CH3-19). 13CNMR (pyridine-d5) d: 36.5 (C-1), 27.8 (C-2), 73.4 (C-3), 34.4 (C-4),44.7 (C-5), 28.3 (C-6), 31.6 (C-7), 34.6 (C-8), 55.5 (C-9), 36.4 (C-10), 38.1 (C-11), 212.3 (C-12), 55.9 (C-13), 56.0 (C-14), 31.9 (C-15),80.1 (C-16), 55.7 (C-17), 16.3 (C-18), 12.0 (C-19), 41.3 (C-20), 15.2(C-21), 112.6 (C-22), 30.8 (C-23), 28.7 (C-24), 33.5 (C-25), 69.3 (C-26), 17.1 (C-27), 21.5 (CH3COO-3), 21.0 (CH3COO-26), 170.0(CH3COO-3), 170.6 (CH3COO-26), 47.5 (CH3O-22). HRMS-FAB (m/z): [M]+ calcd for C32H50O7, 546.3557. Found, 546.3565.
2.6. (22S, 25R)-22-selenocyanatofurost-5-ene-3b,26-diyl diacetate (9b)
In a 50 mL round bottom flask, we dissolved the diosgenin (4b,500 mg, 1.20 mmol) in 20 mL of CH2Cl2.Then, 0.5 mL of BF3�OEt2
(3.94 mmol), the recently prepared ATFAA (2.4 mL) [20], andKSeCN (173 mg, 1.2 mmol) were slowly added. The mixture wasmagnetically stirred at room temperature and monitored by TLCuntil complete disappearance of the starting material (10 min).The reaction mixture was poured over ground ice (20 g) and vig-orously shaken. The organic phase was extracted with 50 mL ofCH2Cl2 and washed with brine (1 � 20 mL), followed by neutral-ization with a saturated solution of NaHCO3 (2 � 30 mL) andwater (1 � 30 ml). The resulting organic phase was dried withanh. Na2SO4 and concentrated to dryness under vacuum. Thecrude product was separated by column chromatography andeluted with hexane/ethyl acetate (9:1) to afford 9b (90%). Color-less oil; IR �mmax: 2185 (mSeCN), 1733 (mC@O ester), 1234 (mC–O–C). 1H NMR d: 5.37 (1H, d, J = 5.2 Hz, H-6), 4.59 (2H, m, H-16aand H-3a), 3.96 (1H, dd, J26b�26a = 10.8 Hz, J26b�25 = 6.8 Hz, H-26b), 3.91 (1H, dd, J26a�26b = 10.8 Hz, J26a�25 = 6.8 Hz, H-26a),2.34 (1H, d, J17�16 = 10.4 Hz, H-17), 2.06 (3H, s, CH3COO-26),2.03 (3H, s, CH3COO-3), 1.21 (3H, d, J = 7.2 Hz, CH3-21), 1.03(3H, s, CH3-19), 0.96 (3H, d, J27�25 = 6.4 Hz, CH3-27), 0.80 (3H, s,CH3-18). 13C NMR d: 36.8 (C-1), 28.3 (C-2), 73.7 (C-3), 38.0 (C-4), 139.6 (C-5), 122.1 (C-6), 31.8 (C-7), 31.5 (C-8), 49.6 (C-9),36.6 (C-10), 20.6 (C-11), 39.2 (C-12), 40.6 (C-13), 56.7 (C-14),34.6 (C-15), 84.1 (C-16), 62.8 (C-17), 16.4 (C-18), 17.0 (C-19),42.0 (C-20), 19.3 (C-21), 88.9 (C-22), 27.6 (C-23), 31.4 (C-24),32.4 (C-25), 68.6 (C-26), 16.8 (C-27), 21.4 (CH3COO-3), 20.9(CH3COO-26), 170.5 (CH3COO-3), 171.2 (CH3COO-26), 118.5(SeCN). HRMS-FAB (m/z): [M+H]+ calcd for C32H48O5NSe,606.2698. Found, 606.2702.

O. Viñas-Bravo et al. / Steroids 77 (2012) 59–66 63
3. Results and discussion
The stereoselective F ring opening of (25R)- and (25S)-sapoge-nins 4a–c was catalyzed by the action of BF3�OEt2 [25], thehydroxyl group at C-26 was immediately acetylated by the ace-tic–trifluoroacetic anhydride, resulting in the generation of theintermediate oxonium ion (5, Scheme 1). We propose that thetrifluoroacetate generated in situ is a weak base but not basic en-ough to eliminate the acidic hydrogen at C-23. The intermediate5 is stable at room temperature, and only a minor proportion ofhydrogen at C-20 was eliminated. These conditions permitted that5, which is solvated by the trifluoroacetate ions, stands in the med-ium until reaction quenching.
When the reaction was quenched with water, the 22-hydroxy-furostans 6a–c were produced at approximately 80% yields. As theintermediary 5 is flat at the oxonium moiety, the H2O moleculecould be added at the C-22 by either the a or b face, in theory pro-ducing a mixture of the alcohol diastereomers 22R and 22S. In ourexperiments only one of these was observed, that which corre-sponds to the attack of H2O by the a face. This attack is favoredbecause the 18 methyl group and the flipped cis-fused D-E ringsgenerate high steric hindrance over the b-face of the steroid inter-mediate, so the H2O molecule is added by the a side, generating a22R stereochemistry. This preferred stereochemistry is also ob-served in nature [1–7]. The stereochemistry of the products 6a–cwas also demonstrated by 1D and 2D NMR experiments. The trans-formation of 22-hydroxylfurostans 6a–c by the action of a Brön-sted–Lowry acid led to their corresponding pseudosapogenins7a–c in excellent yields. To find the best conditions for a ‘‘onepot’’ transformation of 6a–c into 7a–c, we studied different reac-tion conditions such as the proportion of the sapogenin/ATFAA,the nature of an extra protic acid, reaction time, and temperature.The results are shown in Table 1.
In this study, all trials were performed starting from 1 mmol of4a–c and catalyzed by the action of BF3�OEt2. The best result wasobtained when 4a–c was treated with 5.4 mmol of BF3�OEt2
and10 mmol of ATFAA for 5 minutes at room temperature. Thereaction crude was quenched with cold water, treated with p-TsOHand stirred for 10 min. A fast purification was run using a silica gel
Scheme 1. Synthetic route to obtain 22-substituted furostans (6a–c, 8a–c
column and provided pure compound in 92%, 92%, and 90% yields,respectively. The derivatives 7a–c showed correct spectroscopydata, and their physical properties are in agreement with those re-ported in the literature [22–26]. When we used methanol insteadof water to quench the reaction crude, we obtained the ketals8a–c at approximately 85% yield but mixed with minute quantitiesof pseudosapogenins 7a–c. Later, this methodology was applied todiosgenin (4b) but with KSeCN as the nucleophile. In this case,after the addition of the solid salt, the reaction was monitored byTLC and finally quenched with water, thereby obtaining the seleno-cyanate 9b in a good yield.
Though the ATFAA mixed anhydride can be produced using acidacetic and trifluoroacetic anhydride or trifluoroacetic acid and ace-tic anhydride [20], we observed better results using the first strat-egy. The different behavior is due to the acetate anion produced inthe second strategy. As mentioned above, this anion acts as a base,eliminating a hydrogen atom from C-23 and producing acylatedfurostans [21].
3.1. Spectroscopic analysis
The structures of compounds 6–9 were unambiguously estab-lished using two-dimensional NMR experiments DEPT, COSY, HSQCand NOESY, long-range connectivity from HMBC experiments at400 MHz (see Supplementary Information) and comparison withpreviously reported data for 7a, 7b [26] and7c [27], as well as22-hydroxy- and 22-methoxysaponins [28]. Additional informa-tion was obtained from IR and MS. Some characteristic 1H and13C NMR signals are shown in Tables 2 and 3.
The NMR spectra for compounds 6a–c were recorded using pyr-idine-d5; when CDCl3 is used the 22-hydroxyfurostans were slowlytransformed into their corresponding pseudosapogenins. The 1HNMR spectra of the 22-hydroxyfurostans at the 3.95–4.09 ppm re-gion show signals for the diasterotopic H-26 protons as an ABMsystem. In addition, the presence of an extra-singlet approximately2.0 ppm confirms the presence of the new acetate function at C-26,which corroborates the regioselective F ring opening. The Me-21signal in 6a–c appears as a doublet in the 1.33–1.52 ppm region;in 7a–c, the Me-21 signal is a singlet at 1.57–1.58 ppm.
, 9b) and pseudosapogenins (7a–c) using the couple BF3�OEt2/ATFAA.
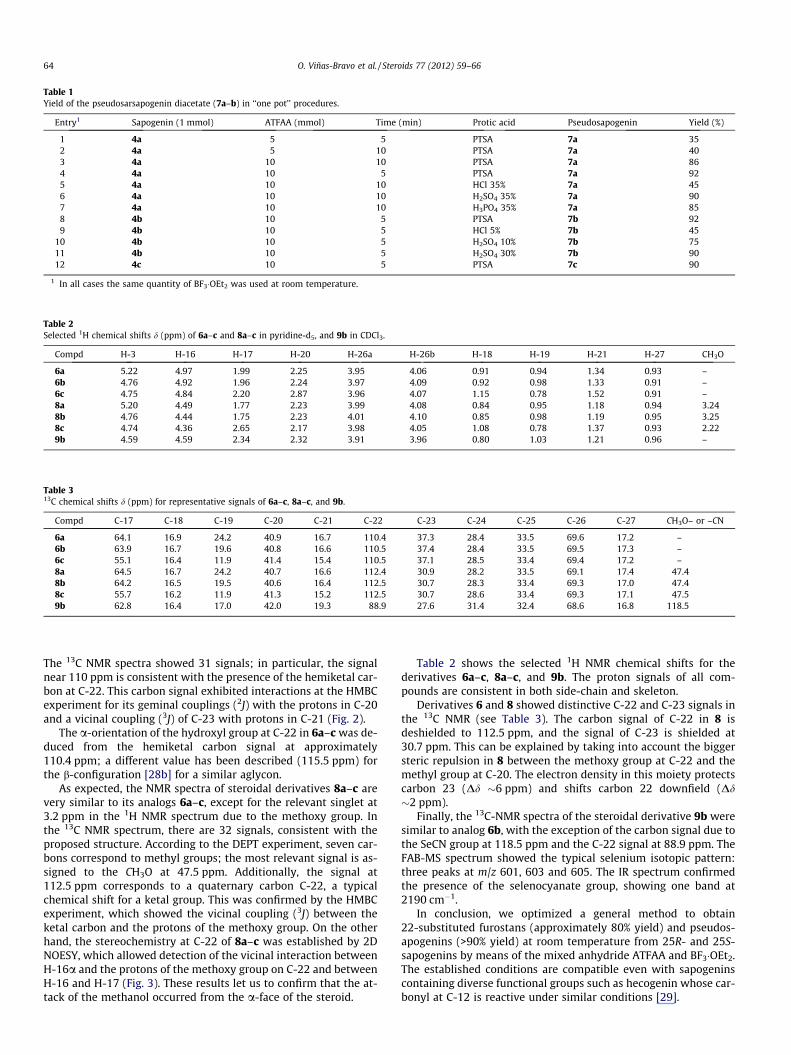
Table 1Yield of the pseudosarsapogenin diacetate (7a–b) in ‘‘one pot’’ procedures.
Entry1 Sapogenin (1 mmol) ATFAA (mmol) Time (min) Protic acid Pseudosapogenin Yield (%)
1 4a 5 5 PTSA 7a 352 4a 5 10 PTSA 7a 403 4a 10 10 PTSA 7a 864 4a 10 5 PTSA 7a 925 4a 10 10 HCl 35% 7a 456 4a 10 10 H2SO4 35% 7a 907 4a 10 10 H3PO4 35% 7a 858 4b 10 5 PTSA 7b 929 4b 10 5 HCl 5% 7b 45
10 4b 10 5 H2SO4 10% 7b 7511 4b 10 5 H2SO4 30% 7b 9012 4c 10 5 PTSA 7c 90
1 In all cases the same quantity of BF3�OEt2 was used at room temperature.
Table 2Selected 1H chemical shifts d (ppm) of 6a–c and 8a–c in pyridine-d5, and 9b in CDCl3.
Compd H-3 H-16 H-17 H-20 H-26a H-26b H-18 H-19 H-21 H-27 CH3O
6a 5.22 4.97 1.99 2.25 3.95 4.06 0.91 0.94 1.34 0.93 –6b 4.76 4.92 1.96 2.24 3.97 4.09 0.92 0.98 1.33 0.91 –6c 4.75 4.84 2.20 2.87 3.96 4.07 1.15 0.78 1.52 0.91 –8a 5.20 4.49 1.77 2.23 3.99 4.08 0.84 0.95 1.18 0.94 3.248b 4.76 4.44 1.75 2.23 4.01 4.10 0.85 0.98 1.19 0.95 3.258c 4.74 4.36 2.65 2.17 3.98 4.05 1.08 0.78 1.37 0.93 2.229b 4.59 4.59 2.34 2.32 3.91 3.96 0.80 1.03 1.21 0.96 –
Table 313C chemical shifts d (ppm) for representative signals of 6a–c, 8a–c, and 9b.
Compd C-17 C-18 C-19 C-20 C-21 C-22 C-23 C-24 C-25 C-26 C-27 CH3O– or –CN
6a 64.1 16.9 24.2 40.9 16.7 110.4 37.3 28.4 33.5 69.6 17.2 –6b 63.9 16.7 19.6 40.8 16.6 110.5 37.4 28.4 33.5 69.5 17.3 –6c 55.1 16.4 11.9 41.4 15.4 110.5 37.1 28.5 33.4 69.4 17.2 –8a 64.5 16.7 24.2 40.7 16.6 112.4 30.9 28.2 33.5 69.1 17.4 47.48b 64.2 16.5 19.5 40.6 16.4 112.5 30.7 28.3 33.4 69.3 17.0 47.48c 55.7 16.2 11.9 41.3 15.2 112.5 30.7 28.6 33.4 69.3 17.1 47.59b 62.8 16.4 17.0 42.0 19.3 88.9 27.6 31.4 32.4 68.6 16.8 118.5
64 O. Viñas-Bravo et al. / Steroids 77 (2012) 59–66
The 13C NMR spectra showed 31 signals; in particular, the signalnear 110 ppm is consistent with the presence of the hemiketal car-bon at C-22. This carbon signal exhibited interactions at the HMBCexperiment for its geminal couplings (2J) with the protons in C-20and a vicinal coupling (3J) of C-23 with protons in C-21 (Fig. 2).
The a-orientation of the hydroxyl group at C-22 in 6a–c was de-duced from the hemiketal carbon signal at approximately110.4 ppm; a different value has been described (115.5 ppm) forthe b-configuration [28b] for a similar aglycon.
As expected, the NMR spectra of steroidal derivatives 8a–c arevery similar to its analogs 6a–c, except for the relevant singlet at3.2 ppm in the 1H NMR spectrum due to the methoxy group. Inthe 13C NMR spectrum, there are 32 signals, consistent with theproposed structure. According to the DEPT experiment, seven car-bons correspond to methyl groups; the most relevant signal is as-signed to the CH3O at 47.5 ppm. Additionally, the signal at112.5 ppm corresponds to a quaternary carbon C-22, a typicalchemical shift for a ketal group. This was confirmed by the HMBCexperiment, which showed the vicinal coupling (3J) between theketal carbon and the protons of the methoxy group. On the otherhand, the stereochemistry at C-22 of 8a–c was established by 2DNOESY, which allowed detection of the vicinal interaction betweenH-16a and the protons of the methoxy group on C-22 and betweenH-16 and H-17 (Fig. 3). These results let us to confirm that the at-tack of the methanol occurred from the a-face of the steroid.
Table 2 shows the selected 1H NMR chemical shifts for thederivatives 6a–c, 8a–c, and 9b. The proton signals of all com-pounds are consistent in both side-chain and skeleton.
Derivatives 6 and 8 showed distinctive C-22 and C-23 signals inthe 13C NMR (see Table 3). The carbon signal of C-22 in 8 isdeshielded to 112.5 ppm, and the signal of C-23 is shielded at30.7 ppm. This can be explained by taking into account the biggersteric repulsion in 8 between the methoxy group at C-22 and themethyl group at C-20. The electron density in this moiety protectscarbon 23 (Dd �6 ppm) and shifts carbon 22 downfield (Dd�2 ppm).
Finally, the 13C-NMR spectra of the steroidal derivative 9b weresimilar to analog 6b, with the exception of the carbon signal due tothe SeCN group at 118.5 ppm and the C-22 signal at 88.9 ppm. TheFAB-MS spectrum showed the typical selenium isotopic pattern:three peaks at m/z 601, 603 and 605. The IR spectrum confirmedthe presence of the selenocyanate group, showing one band at2190 cm�1.
In conclusion, we optimized a general method to obtain22-substituted furostans (approximately 80% yield) and pseudos-apogenins (>90% yield) at room temperature from 25R- and 25S-sapogenins by means of the mixed anhydride ATFAA and BF3�OEt2.The established conditions are compatible even with sapogeninscontaining diverse functional groups such as hecogenin whose car-bonyl at C-12 is reactive under similar conditions [29].

Fig. 2. Some correlations observed in HMBC experiment of 6a.
Fig. 3. Some correlations observed in NOESY experiment of 8a–c.
O. Viñas-Bravo et al. / Steroids 77 (2012) 59–66 65
Acknowledgments
The authors thank CONACYT for financial support (Grant No.84380) and a scholarship to VGC. We also thank VIEP-BUAP forfinancial support (Grant No. MOSM-NAT10-G) and scholarshipsto ALR and MMM.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.steroids.2011.10.004.
References
[1] (a) Sparg SG, Light ME, van Staden J. Biological activities and distribution ofplant saponins. J Ethnopharmacol 2004;94:219–43(b) Waller GR, Yamasaki, K editors. Saponins used in traditional and modernmedicine. New York: Plenum Press; 1996.(c) Hostettmann K, Marston A. Saponins. New York: Cambridge UniversityPress; 1995.
[2] Sautour M, Mitaine-Offer AC, Lacaille-Dubois MA. The Dioscorea genus: areview of bioactive steroid saponins. J Nat Med 2007;61:91–101 [andreferences cited therein].
[3] (a) Mimaki Y, Yokosuka A, Kuroda M, Sashida Y. Cytotoxic activities andstructure–cytotoxic relationships of steroidal saponins. Biol Pharm Bull2001;24:1286–9;(b) Hua K, Yao X. The cytotoxicity of protoneodioscin (NSC-698789), afurostanol saponin from the rhizomes of Dioscorea collettii var. hypoglauca,against human cancer cells in vitro. Phytomedicine 2002;9:560–5;(c) Podolak I, Galanty A, Sobolewska D. Saponins as cytotoxic agents: a review.Phytochem Rev 2010;9:425–74;(d) Mana S, Gao W, Zhang Y, Huang L, Liu C. Chemical study and medicalapplication of saponins as anti-cancer agents. Fitoterapia 2010;81:703–14;(e) Ivanova A, Mikhova B, Batsalova T, Dzhambazov B, Kostova I. Newfurostanol saponins from Smilax aspera L. and their in vitro cytotoxicity.Fitoterapia 2011;82:282–7.
[4] (a) Lanzotti V. Bioactive saponins from Allium and Aster plants. PhytochemRev 2005;4:95–110;(b) Barile E, Bonanomi G, Antignani V, Zolfaghari B, Sajjadi SE, Scala F, et al.Saponins from Allium minutiflorum with antifungal activity. Phytochemistry2007;68:596–603;(c) Li H, Huang W, Wen Y, Gong G, Zhao Q, Yu G. Anti-thrombotic activity andchemical characterization of steroidal saponins from Dioscorea zingiberensisC.H. Wright. Fitoterapia 2010;81:1147–56.
[5] Hibasami H, Moteki H, Ishikawa K, Katsuzaki H, Imai K, Yoshioka K, et al.Protodioscin isolated from fenugreek (Trigonella foenumgraecum L) induces cell
death and morphological change indicative of apoptosis in leukemic cell lineH-60, but not in gastric cancer cell line KATO III. Int J Mol Med 2003;11:23–6.
[6] Hu K, Yao X. Protodioscin (NSC-698 796): its spectrum of cytotoxicity againstsixty human cancer cell lines in an anticancer drug screen panel. Planta Med2002;68:297–301.
[7] Wang T, Choi RCY, Li J, Li J, Bi CWC, Zang L, et al. Anti hyperlipidemic effect ofprotodioscin, an active ingredient isolated from the rhizomes of Dioscoreanipponica. Planta Med 2010;76:1642–6.
[8] (a) Hu K, Yao XS. The cytotoxicity of methyl protodiosin against human cancercell lines. Cancer Invest 2003;21:389–93;(b) Liu MJ, Yue PY, Wang Z, Wong RN. Methyl protodioscin induces G2/M arrestand apoptosis in K562 cells with the hyperpolarization of mitochondria.Cancer Lett 2005;224:229–41;(c) Wang G, Chen H, Huang M, Wang N, Zhang J, Zhang Y, et al. Methylprotodioscin induces G2/M cell cycle arrest and apoptosis in HepG2 livercancer cells. Cancer Lett 2006;241:102–9 [and references there cited].
[9] Hernández JC, León F, Quintana J, Estévez F, Bermejo J. Icogenin, a newcytotoxic steroidal saponin isolated from Dracaena draco. Bioorg Med Chem2004;12:4423–9.
[10] (a) Yu B, Liao J, Zhang J, Hui Y. The first synthetic route to furostan saponins.Tetrahedron Lett 2001;42:77–9;(b) Li M, Yu B. Facile conversion of spirostan saponin into furostan saponin:synthesis of methyl protodioscin and its 26-thio-analogue. Org Lett2006;8:2679–82.
[11] Cheng MS, Wang QL, Tian Q, Song HY, Liu YX, Li Q, et al. Total synthesis ofmethyl protodioscin: a potent agent with antitumor activity. J Org Chem2003;68:3658–62.
[12] (a) Hou S, Xu P, Zhou L, Yu D, Lei P. Synthesis and antitumor activity oficogenin and its analogue. Bioorg Med Chem Lett 2006;16:2454–8;(b) Hou SJ, Xu P, Zhou L, Yu DQ, Lei PS, Zou CC. Total synthesis of the analogueof icogenin. Chin Chem Lett 2006;17:183–6.
(a) Marker RE, Rohrmann E. Sterols LXXXI. Conversion of sarsasapogenin topregnanediol-3(a),20(a). J Am Chem Soc 1939;61:3592–3.
(b) Marker RE, Rohrmann E. Sterols LXXXVIII. Pregnanediols from sarsasapogenin. JAm Chem Soc 1940;62:518–20.
(c) Marker RE, Tsukamoto T, Turner DL. Sterols. C. Diosgenin. J Am Chem Soc1940;62:2525–32.
[14] (a) Gould DH, Staudle H, Hershberg EB. Catalytic isomerization of spirostans tofurostenols. J Am Chem Soc 1952;74:3685–8. and references therein;(b) Goswami A, Kotoky R, Rastogi RC, Ghosh AC. A one-pot efficient process for16-dehydropregnenolone acetate. Org Proc Res Dev 2003;7:306–8 [andreferences cited therein].
[15] (a) Jastrzebska I, Katrynski KS, Morzycki JW. Further study on oxidation ofpseudosapogenins. Arkivoc 2002;ix:46–54;(b) Zhang Y, Liu YY, Cheng XX, Wang XS, Zhang BW. An environmentallyfriendly process for the preparation of 16-dehydropregnenolone acetate. Chin JChem 2005;23:753–6;(c) Quan HJ, Konayagi J, Hagiwara K, Cui XR, Isshiki Y, Kondo S, Komada F, SaitoS. Reactions of 26-iodopseudodiosgenin and 26-iodopseudodiosgenone withvarious nucleophiles and pharmacological activities of the products. ChemPharm Bull 2006;54:72–9;(d) Ravindar K, Reddy MS, Lindqvist L, Pelletier J, Deslongchamps P. Synthesisof the antiproliferative agent hippuristanol and its analogues via Suarezcyclizations and Hg(II)-catalyzed spiroketalizations. J Org Chem 2011;76:1269–84.
[16] Wua JJ, Cheng KW, Zuo XF, Wang MF, Li P, Zhang LY, et al. Steroidal saponinsand ecdysterone from Asparagus filicinus and their cytotoxic activities. Steroids2010;75:734–9.
[17] Brown L, Koreeda M. Benzoyl trifluoromethanesulfonate. A mild reagent forthe benzoylation of sterically hindered hydroxyls. J Org Chem1984;49:3875–80.
[18] Lee JS, Fuchs PL. Efficient protocol for ring opening of spiroketals usingtrifluoroacetyl trifluoromethanesulfonate (TFAT). Org Lett 2003;5:3619–22[and references cited therein].
[19] Martínez-Pascual R, Viñas-Bravo O, Meza-Reyes S, Iglesias-Arteaga MA,Sandoval-Ramírez J. A fast and convenient procedure for acetylation ofalcohols. Synth Commun 2004;34:4591–6.

66 O. Viñas-Bravo et al. / Steroids 77 (2012) 59–66
[20] Montiel-Smith S, Meza-Reyes S, Viñas-Bravo O, Fernández-Herrera M A,Martínez-Pascual R, Sandoval-Ramírez J, et al. In situ preparation of mixedanhydrides containing the trifluoroacetyl moiety. Application to theesterification of cholesterol and phenol. Arkivoc 2005;vi:127–35.
[21] (a) Sandoval-Ramírez J, Castro-Méndez A, Meza-Reyes S, Reyes-Vázquez F,Santillán R, Farfán N. Preparation of 22,26-epoxycholest-22-ene steroids.Novel transformation of the side chain in sapogenins. Tetrahedron Lett1999;40:5143–6;(b) Sandoval-Ramírez J, Meza-Reyes S, del Río RE, Hernández-Linares G,Suárez-Rojas A, Rincón S, et al. Regioselective cleavage of rings E and F insarsasapogenin. Steroids 2003;68:199–204.
[22] Scheer I, Kostic RB, Mosettig E. The C-25 isomerism of smilagenin andsarsasapogenin. J Am Chem Soc 1955;77:641–6.
[23] (a) Kaufmann S, Rosenkranz G. Steroidal sapogenins. I. Transformation ofkryptogenin into diosgenin and pseudodiosgenin. J Am Chem Soc1948;70:3502–5;(b) Ziegler JB, Rosen WE, Shabica AC. The stereochemistry of steroidalsapogenins. J Am Chem Soc 1955;77:1223–9.
[24] Mueller GP, Stobaugh RE, Winniford RS. The conversion of hecogenin andmanogenin into derivatives of allopregnane-12,20-dione. J Am ChemSoc1953;75:4888–92.
[25] Jastrzebska I, Dobrogowska A, Lutostanska E, Morzycki JW. On reactions ofspirostane sapogenins with benzeneseleninic anhydride. Tetrahedron2010;66:5024–9.
[26] Tobari A, Teshima M, Koyanagi J, Kawase M, Miyamae H, Yoza K, et al.Spirostanols obtained by cyclization of pseudosaponin derivatives andcomparison of anti-platelet agglutination activities of spirostanol glycosides.Eur J Med Chem 2000;35:511–27 [and references cited therein].
[27] Morzycki JW, Paradowska K, Dabrowska-Balcerzak K, Jastrzebska I,Siergiejczyk L, Wawer I. 13C NMR study of spirostanes and furostanes insolution and solid state. J Mol Struct 2005;744-47:447–55.
[28] (a) Agrawal PK, Jain DC, Gupta RK, Thakur RS. Carbon-13 NMR spectroscopy ofsteroidal sapogenins and steroidal saponins. Phytochemistry 1985;24:2479–96;(b) Fattorusso E, Iorizzi M, Lanzotti V, Taglialatela-Scafati O. Chemicalcomposition of shallot (Allium ascalonicum Hort). J Agric Food Chem2002:50;5686–90.
[29] Perez-Diaz JO, Vega-Baez JL, Sandoval-Ramirez J, Meza-Reyes S, Montiel-SmithS, Farfan N, et al. Novel steroidal penta- and hexacyclic compounds derivedfrom 12-oxospirostan sapogenins. Steroids 2010;75:1127–36.




















