
Quantitative study of the short range order in B 2O 3 and B 2S 3 by MAS and two-dimensional...
13
SOLID STATE Nuclear Magnetic Resonance ELSEVIER Solid State Nuclear Magnetic Resonance 8 (1997) 109- 12 1 Quantitative study of the short range order in B,O, and B,S, by MAS and two-dimensional triple-quantum MAS I1 B NMR S.-J. Hwang a, C. Femandez b, J.P. Amoureux b, J. Cho ‘, S.W. Martin ‘, M. Pm&i a, * a Ames Luborutory, Ames, IA 50011, USA ’ Luborutoire de Dynumique et Structure des Mat&&.x Moltkduires, CNRS URA 801, Universith des Sciences et Technologies de Lille, F-59655 Villeneuve d’Ascq Cedex, Frunce ’ Depurtment of Muterids Science and Engineering, Iowa Stute University, Ames, IA 50011, USA Accepted 21 September 1996 Abstract Two-dimensional multiple-quantum magic angle spinning (MQMAS) NMR and MAS NMR of “B at various magnetic fields, were applied to elucidate the structure of vitreous (glassy) boron trioxide (v-B203), vitreous boron trisulfide (v-B,S,) and crystalline boron trisulfide (c-B2S3). These techniques, when combined with computer simulations of the resulting spectra, provide the isotropic chemical shifts and the quadrupole parameters, as well as a quantitative measure of the intensities of various boron resonances. The MAS NMR of v-B,O, produced overlapping anisotropic lineshapes correspond- ing to the - l/2 c, l/2 transition in two distinct types of BO, units with 3(_+0.08): 1 intensity ratio. A combination of MAS and the multiple-quantum method resulted in a better resolved, isotropic “B spectrum of v-B,O,. A remarkable enhancement of resolution of the MQMAS NMR proved instrumental in finding and identifying various impurities present in V-B,S, and c-B,S,. In addition to the resonances from boron in two types of BS, groups, four other structural units, BOS,, BO,S, BO, and BS,, were elucidated from the spectra of vitreous and crystalline samples. The effects of various experimental parameters, such as the magnitude of the B, and B, fields, on the resolution of the MAS and MQMAS techniques are also shown. 0 1997 Elsevier Science B.V. Keyworrls: Multiple-quantum NMR of quadrupolar nuclei; Boron trioxide; Boron trisulfide 1. Introduction Measurement of high-resolution NMR spectra of half-integer quadrupolar nuclei is intrinsically diffi- cult due to the highly anisotropic interaction of the nuclear quadrupole moment with the gradient of the surrounding electric field. However, remarkable ad- ’ Corresponding author. vancements have occurred in this area of solid state NMR in the past few years. Traditionally, only the central - l/2 * l/2 transition was observed be- cause it is unaffected by first order quadrupolar broadening. Even for the central transition, however, the second order quadrupolar broadening is often too large to allow observation of chemically inequivalent sites. This broadening includes both second and fourth rank spherical harmonics which can be scaled, but not completely removed, by conventional line narrowing techniques for solids, such as spinning the 0926-2040/97/$17.00 0 1997 Elsevier Science B.V. All rights reserved. P/I SO926-2040(96)0 1280-5
-
Upload
guadalajara -
Category
Documents
-
view
1 -
download
0
Transcript of Quantitative study of the short range order in B 2O 3 and B 2S 3 by MAS and two-dimensional...

SOLID STATE Nuclear Magnetic Resonance
ELSEVIER Solid State Nuclear Magnetic Resonance 8 (1997) 109- 12 1
Quantitative study of the short range order in B,O, and B,S, by MAS and two-dimensional triple-quantum MAS I1 B NMR
S.-J. Hwang a, C. Femandez b, J.P. Amoureux b, J. Cho ‘, S.W. Martin ‘, M. Pm&i a, *
a Ames Luborutory, Ames, IA 50011, USA
’ Luborutoire de Dynumique et Structure des Mat&&.x Moltkduires, CNRS URA 801, Universith des Sciences et Technologies de Lille,
F-59655 Villeneuve d’Ascq Cedex, Frunce
’ Depurtment of Muterids Science and Engineering, Iowa Stute University, Ames, IA 50011, USA
Accepted 21 September 1996
Abstract
Two-dimensional multiple-quantum magic angle spinning (MQMAS) NMR and MAS NMR of “B at various magnetic fields, were applied to elucidate the structure of vitreous (glassy) boron trioxide (v-B203), vitreous boron trisulfide (v-B,S,) and crystalline boron trisulfide (c-B2S3). These techniques, when combined with computer simulations of the resulting spectra, provide the isotropic chemical shifts and the quadrupole parameters, as well as a quantitative measure of the intensities of various boron resonances. The MAS NMR of v-B,O, produced overlapping anisotropic lineshapes correspond- ing to the - l/2 c, l/2 transition in two distinct types of BO, units with 3(_+0.08): 1 intensity ratio. A combination of MAS and the multiple-quantum method resulted in a better resolved, isotropic “B spectrum of v-B,O,. A remarkable enhancement of resolution of the MQMAS NMR proved instrumental in finding and identifying various impurities present in
V-B,S, and c-B,S,. In addition to the resonances from boron in two types of BS, groups, four other structural units, BOS,, BO,S, BO, and BS,, were elucidated from the spectra of vitreous and crystalline samples. The effects of various experimental parameters, such as the magnitude of the B, and B, fields, on the resolution of the MAS and MQMAS techniques are also shown. 0 1997 Elsevier Science B.V.
Keyworrls: Multiple-quantum NMR of quadrupolar nuclei; Boron trioxide; Boron trisulfide
1. Introduction
Measurement of high-resolution NMR spectra of half-integer quadrupolar nuclei is intrinsically diffi- cult due to the highly anisotropic interaction of the
nuclear quadrupole moment with the gradient of the surrounding electric field. However, remarkable ad-
’ Corresponding author.
vancements have occurred in this area of solid state NMR in the past few years. Traditionally, only the central - l/2 * l/2 transition was observed be- cause it is unaffected by first order quadrupolar broadening. Even for the central transition, however, the second order quadrupolar broadening is often too
large to allow observation of chemically inequivalent sites. This broadening includes both second and fourth rank spherical harmonics which can be scaled,
but not completely removed, by conventional line narrowing techniques for solids, such as spinning the
0926-2040/97/$17.00 0 1997 Elsevier Science B.V. All rights reserved.
P/I SO926-2040(96)0 1280-5

110 S.-J. Hwang et d/Solid State Nuclear Magnetic Resonance 8 (19971 109-121
sample around one rotation axis. The theoretical foundations [ 1,2] and first experimental realizations [2,3] of complete spatial averaging of second order
broadening were published in the late 1980s. These experiments involved mechanical rotation of a sam-
ple around two axes, either simultaneously in a
one-dimensional technique called double rotation
(DOR) [2], or in succession using a two-dimensional
experiment referred to as dynamic angle spinning
(DAS) [3]. Both techniques have been successfully
applied to a number of crystalline and amorphous materials, mainly inorganic solids. In spite of these
achievements, some drawbacks of DAS and DOR became apparent: (i) both methods require NMR probes of great mechanical sophistication; (ii) DAS
requires magnetization storage during the flipping of
the spinner between the two rotation axes and is therefore limited to systems with long (> - 100 ms)
spin-lattice relaxation times and relatively weak homonuclear dipolar broadening; and (iii) DOR of- ten suffers from overlapping patterns of closely
spaced sidebands resulting from the limited spinning speed.
Recently, Frydman and Harwood [4] devised a two-dimensional experiment that correlates the evo- lution of the symmetrical (- m t) m) multiple-quan- tum (MQ) transition with the evolution of the central ( - l/2 ++ l/2) transition under magic angle spin- ning (MAS) to produce highly resolved isotropic NMR spectra analogous to those obtained with DAS. The technique is referred to as two-dimensional mul-
tiple-quantum MAS (MQMAS), and its resolving power was demonstrated by using 23Na, 27Al, and ‘“Mn NMR of several model compounds [4,5].
In this work, MQMAS NMR of “B was applied to study the structure of vitreous boron trioxide (v-B,O,) as well as the crystalline and vitreous forms of boron trisulfide (c-B,S, and V-B,S,). In order to obtain accurate lineshape parameters, includ-
ing spectral intensities, we combined this technique with multi-field high-speed MAS NMR. The re- search interest in B,O, and B,S, stems from their utilization as network formers to produce binary alkali borate and thioborate glasses. When M,S (M = alkali metal) are added to modify the B,S, net- work, the resulting glasses often exhibit excellent ion conducting properties that show promising applica- tions as solid state electrolytes 16-81. The MQMAS
NMR technique proved instrumental in finding and identifying various structural impurities present in
B,S,. Because the quality of binary glasses is deter-
mined by the purity of the starting materials [9], it is essential that the nature and structure of such impuri-
ties be well understood.
2. The MQMAS method
The time averaged resonance frequencies corre-
sponding to the symmetrical ( - m e m> transition of half-integer quadrupolar nuclei in a fast rotating
sample are given by [ 101
V,, = V,,“” + v,p (‘1
where
vcs =Po[% +~,CS(%~~,s)~*(cos 011 I’ (2)
+A,( LP)%+I J$PQ) P,(cos 0)
+A,( W@(rl epPQ> ucos VI (3)
are the chemical shift and quadrupolar frequencies, p = 2m, v0 is the Larmor frequency, cisO is the isotropic chemical shift, I is the value of the nuclear
spin, Q,, is the quadrupole coupling constant, q is the asymmetry parameter, and P,(cos 0) and P,(cos 0) are the second- and fourth-order Legendre polynomials with 8 being the angle between the static field B,, and the rotation axis. The terms
A,(Z,p) and A,(Z,p) are known functions of the quantum numbers I and p, whereas B~S(acs,&s>,
@(~,ao,Po> and @(v ,cro,po) depend on the ori- entation of the chemical shift and quadrupole tensors with respect to the rotor axis [ 101. Since P 2(cos 0) and P,(cos 0) cannot simultaneously vanish, the anisotropic broadening can only be eliminated by (i) making 0 time dependent, as is the case in DOR and DAS experiments, or (ii) by spinning the sample at a fixed angle and using p as an independent variable. which is the principle of the multiple-quantum corre- lation technique used in the present work. The most convenient choice of the rotation axis is 8 = 54.7”, the magic angle, which nullifies P,(cos 0) and. therefore, completely eliminates chemical shift ani-

S.-J. Hwung et ul./Solid State Nuclrar Mqqnetic Rrsonunce 8 (19971 109-121 III
sotropy and has the additional effect of eliminating
(or reducing) the dipolar interactions [4,10]. By em- ploying the appropriate excitation sequences, the fpQ coherences can be excited and, after evolving
under the remaining quadrupole and chemical shift interactions during time I,, can be transferred to the
single-quantum coherence - 1Q and then observed
in the r,-domain. In this case, the refocusing of the isotropic interactions occurs when
(4)
Frydman and Harwood [4] demonstrated that this
multiple-quantum/single-quantum correlation
method yields purely isotropic spectra for half-in-
teger quadrupolar nuclei. Also, the experimental
strategies for efficient excitation of multiple-quan- tum coherences [ 1 1 - 141, for the production of pure-
phase 2D spectra [11,13], and for the separation of
chemical shift and quadrupole contributions [.5,13,15], were recently proposed. In spite of the recent efforts [ 13,141, achieving a uniform excitation of multiple- quantum coherences and their uniform conversion into the central transition remains a formidable task. The signal observed in MQMAS strongly depends on the quadrupole coupling constant, the asymmetry parameter, and the strength of the rf fields used to
generate the 0 * L-PQ + - 1Q coherence path- ways. In this paper we will demonstrate that the combination of MQMAS NMR with the numerical
analysis of standard, one-dimensional MAS spectra can provide full spectral information, even when these standard spectra present multiple and overlap- ping resonances. In such cases, the strategy used relies upon the MQMAS experiments to obtain high resolution and isotropic chemical shifts. This infor- mation is then used for simulation of one-dimen-
sional, quantitative MAS spectra to obtain quadrupo- lar parameters and relative intensities.
3. Experimental
3. I. Sample preparation
The ’ ’ B NMR spectra are presented for a vitreous B,O, sample which was obtained in our laboratory
by fusing boric acid. Due to the extreme sensitivity of B,O, toward hydrolysis, the sample was crushed into powder and transferred to an MAS rotor in a glove box under dried helium gas (H,O and 0, contamination level in the glove box was less than 2
ppm). The NMR spectra showed no presence of
tetrahedral BO, units in this sample. Such units had been detected earlier in wet V-B,O, [ 161. Addition-
ally, the MAS and MQMAS experiments were per- formed using B,O, obtained from Aldrich (99.99%)
and yielded analogous results.
Vitreous B,S, was synthesized following the pro- cedure developed by Martin and Bloyer [9]. In this
process, stoichiometric amounts of amorphous boron
powder (Cerac, 99.9%, 3 km particle size) and sulfur (Cerac, 99.999%, 5 mm chunks) were reacted
for 8 h under vacuum at 850°C in sealed, carbon-
coated silica tubes in a rotating furnace (5 rpm). While oxygen- and sulfur-containing compounds
of boron are known to be excellent glass formers, synthesis of the crystalline phase of B,S, is quite difficult due to its strong glass forming character. The sample of “crystalline” B,S, (c-B,S,) was produced by heat treating the glassy B,S, in a carbon-coated silica tube at 450°C for approximately 2 weeks. breaking the tube, grinding the product, and repeating the annealing process for another 2 weeks.
Again, both the synthesis and transfer of B,S, to MAS rotors were performed in a glove box to pre- vent the hydrolysis reaction.
The NMR experiments showed that in spite of
these efforts, some inclusion of oxygen into the B,S, samples occurred during the synthesis and/or han- dling and, in addition, that the c-B,S, sample may contain the glassy phase. We note that the discovery of the presence of these structural flaws is not un-
welcome. The B,S, studied in this work was later used as a starting material for the synthesis of binary glasses. In view of the fact that some oxygen con-
tamination appears inevitable, the recognition of the NMR fingerprints of all structural units present in the starting material will enhance our ability to inter-
pret the spectra of the binary thioborate glasses [17].
3.2. NMR
“B MAS NMR spectra were obtained using: (i> three home-built solid state spectrometers operating

112 S.-J. Hwung et ai./ Solid State Nuclear Magnetic Rrsonunce 8 (1997) 109-121
at 4.7 T, 5.9 T and 8.5 T equipped with a home-built 5 mm MAS NMR probe, (ii> Bruker ASX 400 and Avance 600 spectrometers operating at 9.4 T and
14.1 T, and (iii> a Chemagnetics Infinity spectrome- ter operating at 9.4 T. Samples were spun at 8-9
kHz and 12- 15 kHz in the 5 mm and 4 mm probes, respectively, which sufficed to separate the spinning
sidebands from the central portion of the spectrum. Since obtaining representative intensities was essen-
tial for this study, we measured a relative fraction of
the 4-coordinated boron atoms (the so called N,
fraction) in the binary ZO%Li,S + 80%B,S, glass as
a function of the tip angle. Under the conditions
used, an rf pulse corresponding to - 20” tip angle in a liquid was found adequate for obtaining accurate
” B intensities. The lineshape parameters and relative intensities were obtained from the MAS NMR spec- tra using a home-written simulation program,
QUASAR [ 183. T w o-dimensional triple-quantum
MAS(3QMAS)NMR of “B was performed at 9.4 T
on Bruker ASX 400 and Chemagnetics Infinity spec- trometers using an rf power sufficient to produce a
selective n/2 pulse in 0.7-0.9 ps. Several spectra were also recorded at 14.1 T on a Bruker Avance 600 spectrometer using a lower rf power (2 t_~ 1-r/2
pulse). Excitation of both the echo (- 34) and antiecho ( + 34) coherences was achieved by using the simple two-pulse (preparation and mixing) se-
quence depicted in Fig. 1. The 6-phase cycling scheme used to simultaneously select the +3Q co- herence pathways was combined with classical CY- CLOPS to eliminate the receiver artifacts and with a
12-phase TPPI cycling scheme to produce pure-phase
pq t, H t2
preparation mixing
Fig. I. Pulse sequence used for simultaneous selection of (0) +
(3) --f ( - I) and 0 + (- 3) + ( - I) multiple-quantum coherence pathways via a 6-phase cycling.
2D spectra [11,14]. The 2D spectra are shown after the shearing transformation.
All “B NMR shifts are reported using the 6 scale, with positive values being downfield, and are
referenced to BF, . O(C,H,),.
4. Results and discussion
4.1. Vitreous B, 0,
The structure of v-B,O, has been extensively studied in the past with the use of neutron and X-ray
diffraction, Raman spectroscopy, molecular dynam- ics simulations as well as NMR and NQR spec-
troscopy. A detailed review of this earlier work and a discussion of the possible structural models for this
glass has been given by Johnson et al. [19]. The previous magnetic resonance work involved I70 [20],
“B and ’ 'B [21-241 nuclei and has been reviewed by Eckert [25]. Most studies suggest that in contrast to crystalline B,O,, which is composed of infinite chains of BO, triangles (non-ring BO,) [26], the
vitreous phase contains non-ring BO, units and a significant fraction of six-membered (boroxol) rings.
However, the occurrence of boroxol rings in B,O, and the nature of the links between them have not been unambiguously established. Examples of the earlier NMR studies include lineshape analysis of I70 NMR spectra which revealed the presence of two oxygen environments with different quadrupole parameters and a 1.2: 1 intensity ratio [20]. It was concluded that the two sites represented oxygen in rigid boroxol rings and oxygen atoms that link the rings, respectively. Although the continuous wave “B and ” B NMR experiments clearly showed that
all boron is coordinated to three oxygen atoms, and that a distribution of resonance frequencies due to different asymmetry parameters and quadrupole cou-
pling constants exists in B,O, [24], multiple boron sites were not manifested in these spectra. Later efforts included the use of the MAS method to improve the resolution of “B NMR. For example, a MAS experiment in a field of 7.1 T resulted in an expected narrowing of the second-order quadrupole powder pattern, but also failed to show the presence
of multiple boron-oxygen units [23]. The presence of at least two boron evironments in B,O, glass was
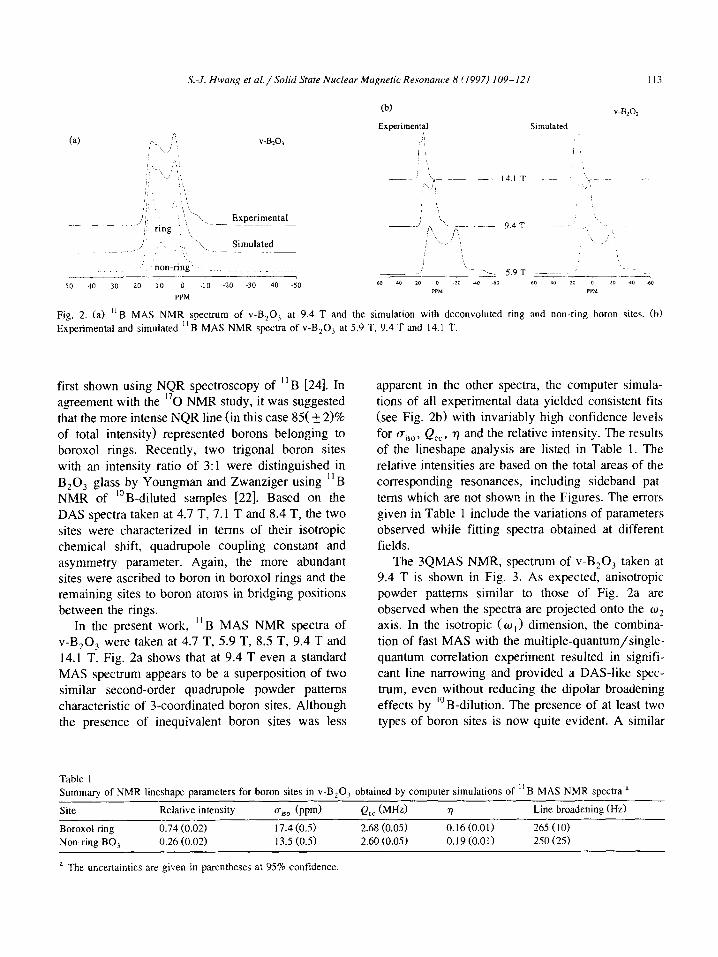
S.-J. Hwung et al./Solid Stute Nucleur Magnetic Resonance 8 (1997) 109-121 113
v-Bz03
c ,“.. “,
: \ ,I ,, v.BlOI
Simulated
’ non-ring . . ..~ _ r
50 40 30 20 LO 0 -10 -20 -30 -40 -50
PPM
(b)
Exoerimentat Simulated
Fig. 2. (a) “6 MAS NMR spectrum of V-B,O, at 9.4 T and the simulation with deconvoluted ring and non-ring boron sites. (b)
Experimental and simulated “B MAS NMR spectra of v-B,O, at 5.9 T, 9.4 T and 14.1 T.
first shown using NQR spectroscopy of “B [24]. In agreement with the I70 NMR study, it was suggested
that the more intense NQR line (in this case 85( + 2>% of total intensity) represented borons belonging to boroxol rings. Recently, two trigonal boron sites with an intensity ratio of 3:1 were distinguished in
B,O, glass by Youngman and Zwanziger using “B NMR of “B-diluted samples [22]. Based on the
DAS spectra taken at 4.7 T, 7.1 T and 8.4 T, the two sites were characterized in terms of their isotropic chemical shift, quadrupole coupling constant and asymmetry parameter. Again, the more abundant sites were ascribed to boron in boroxol rings and the remaining sites to boron atoms in bridging positions between the rings.
In the present work, ” B MAS NMR spectra of
v-B,O, were taken at 4.7 T, 5.9 T, 8.5 T, 9.4 T and 14.1 T. Fig. 2a shows that at 9.4 T even a standard MAS spectrum appears to be a superposition of two similar second-order quadrupole powder patterns characteristic of 3-coordinated boron sites. Although the presence of inequivalent boron sites was less
apparent in the other spectra, the computer simula- tions of all experimental data yielded consistent fits
(see Fig. 2b) with invariably high confidence levels
for U&T Q,,, r] and the relative intensity. The results of the lineshape analysis are listed in Table 1. The
relative intensities are based on the total areas of the corresponding resonances, including sideband pat- terns which are not shown in the Figures. The errors given in Table 1 include the variations of parameters observed while fitting spectra obtained at different
fields. The 3QMAS NMR, spectrum of v-B,O, taken at
9.4 T is shown in Fig. 3. As expected, anisotropic powder patterns similar to those of Fig. 2a are observed when the spectra are projected onto the w2 axis. In the isotropic ( w ,> dimension, the combina-
tion of fast MAS with the multiple-quantum/single- quantum correlation experiment resulted in signifi- cant line narrowing and provided a DAS-like spec- trum. even without reducing the dipolar broadening
effects by lo B-dilution The presence of at least two types of boron sites is now quite evident. A similar
Table I Summary of NMR lineshape parameters for boron sites in V-B203 obtained by computer simulations of “B MAS NMR spectra a
Site Relative intensity oisO (ppm) Q,, (MHZ) 77 Line broadening (Hz)
Boroxol ring 0.74 (0.02) 17.4 (0.5) 2.68 (0.05) 0.16(0.01) 265 (IO)
Non-ring BO, 0.26 (0.02) 13.5 (0.5) 2.60 (0.05) 0.19(0.0I) 250 (25)
a The uncertainties are given in parentheses at 95% confidence.
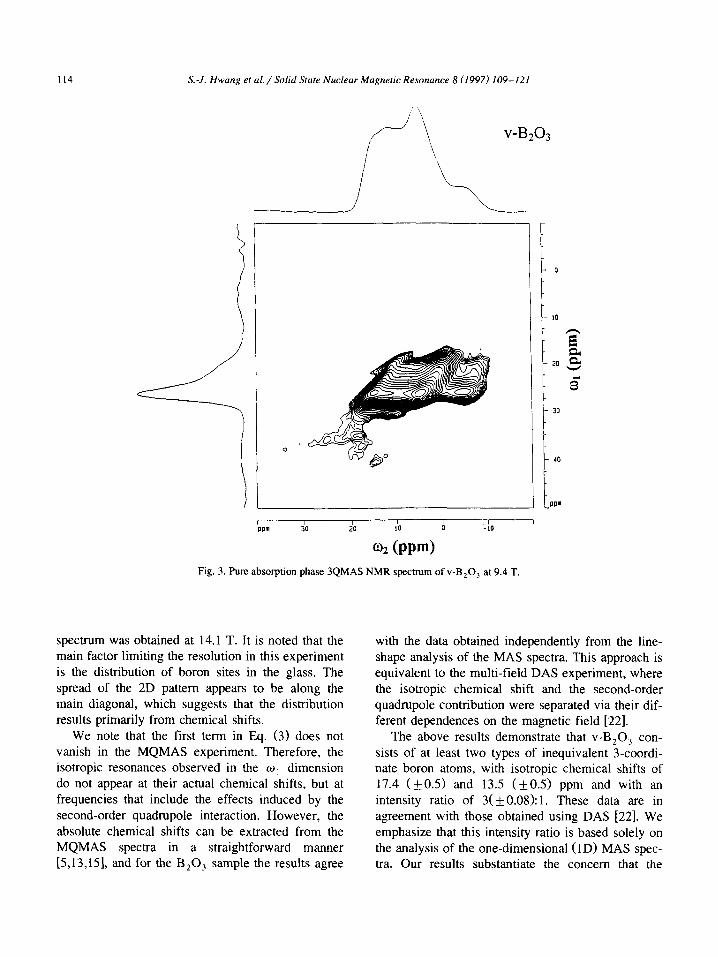
114 S.-J. Hwung et ul./Solid State Nuclear Magnetic Resonance 8 (1997) 109-121
V-B203
1 I 1
- 0
- IO
z a
- 20 ,a
c - 30
1 40
PI-
I 1 I , , / I am 30 20 IO 0 -10
02 (PPN Fig. 3. Pure absorption phase 3QMAS NMR spectrum of v-B,O, at 9.4 T.
spectrum was obtained at 14.1 T. It is noted that the main factor limiting the resolution in this experiment is the distribution of boron sites in the glass. The
spread of the 2D pattern appears to be along the main diagonal, which suggests that the distribution results primarily from chemical shifts.
We note that the first term in Eq. (3) does not vanish in the MQMAS experiment. Therefore, the isotropic resonances observed in the w, dimension do not appear at their actual chemical shifts, but at frequencies that include the effects induced by the second-order quadrupole interaction. However, the
absolute chemical shifts can be extracted from the MQMAS spectra in a straightforward manner [5,13,151, and for the B,O, sample the results agree
with the data obtained independently from the line- shape analysis of the MAS spectra. This approach is equivalent to the multi-field DAS experiment, where the isotropic chemical shift and the second-order quadrupole contribution were separated via their dif- ferent dependences on the magnetic field [22].
The above results demonstrate that v-B,O, con- sists of at least two types of inequivalent 3-coordi- nate boron atoms, with isotropic chemical shifts of 17.4 (kO.5) and 13.5 (kO.5) ppm and with an intensity ratio of 3( f 0.08): 1. These data are in agreement with those obtained using DAS [22]. We emphasize that this intensity ratio is based solely on the analysis of the one-dimensional (1D) MAS spec- tra. Our results substantiate the concern that the

X-J. Hwang et al./Solid State Nuclenr Magnetic Resonance 8 (1997) 109-121 115
MQMAS spectra may exhibit distorted intensities. Indeed, there is a clear difference between the pro- jection of the MQMAS spectrum on the w2 axis (Fig. 3) and the corresponding MAS spectrum of
Fig. 2a, in spite of the very close quadrupole interac- tions of both species.
Although the measured chemical shifts clearly
indicate that both resonances represent boron ions surrounded by three oxygen neighbors, a more spe-
cific peak assignment was done using the existing
liquid state NMR data [27]. For example, the borox- ines (RBO), exhibit resonances in the 17-34 ppm
range and thus the downfield peak at 17.4 ppm can
be reasonably assigned to boron atoms in boroxol rings (see Table XVII in ref. [27]). Moreover, the three chemical shift values available for boroxines
that include BO, units in boroxol ring environments,
(CH,OBO),, (C,H90BO), and [(CH,),SiOBO],, are in the range 17.3-19.6 ppm [27]. However, the
(a)
(b)
LO’
o boron 0 sulfur
Fig. 4. (a) A model of vitreous B,O, structure consistent with
75% of boron atoms residing in the boroxol rings. (b) The crystal
structure of B,S, according to Ref. [291.
above assignment is not unambiguous as several compounds containing isolated (non-ring) BO, groups exhibit resonances in the same range (see Table XI in ref. [27]). On the other hand, the upfield resonance at 13.5 ppm is outside of the range typical
for boroxol rings and must be assigned to non-ring
BO, groups. In summary, the above MAS and MQMAS results
confirm that v-B,O, consists of two types of boron atoms that are surrounded by three oxygen atoms in
a planar trigonal arrangement. Also, these techniques contribute an experimental argument to the boroxol ring model. Due to the quantitative nature of the
NMR method, the relative fraction of boron atoms in
boroxol rings measured here, 74 (k 2)%, and by Youngman and Zwanziger [22], 7.5 (+ 2)%, strongly suggests that vitreous B,O, consists of equal amounts
of boroxol and non-ring BO, units. This result is in agreement with earlier, although less accurate, esti-
mates obtained with “0 NMR [20], neutron diffrac- tion [ 193 and X-ray diffraction [28], but is lower than
the value of 85 ( f 2)% obtained using NQR [21]. Although more work is needed to corroborate the above assignments and to resolve the debate as to whether the boroxol and BO, units are connected in
a random or topologically ordered network, we note that the 1:l ratio of the two units is consistent with the hexagonal model depicted in Fig. 4a.
4.2. Crystalline and vitreous B2Sj
A similar set of experiments was performed on c-B,S, and V-B,S,. Examples of the MAS spectra taken at 5.9 T, 8.5 T and 14.1 T and the correspond- ing lineshape simulations are shown in Fig. 5. The spectra clearly show the presence of two superim- posed powder patterns for the 3-coordinated boron sites (labeled B’ and B* in Fig. 5a) with lineshape parameters as listed in Table 2. For both c-B,S, and V-B,S,, the simulations also included the MAS spec-
tra measured at 4.7 T and 9.4 T (not shown). The less intense spectral features located in the O-40
ppm range are not included in Table 2 and will be
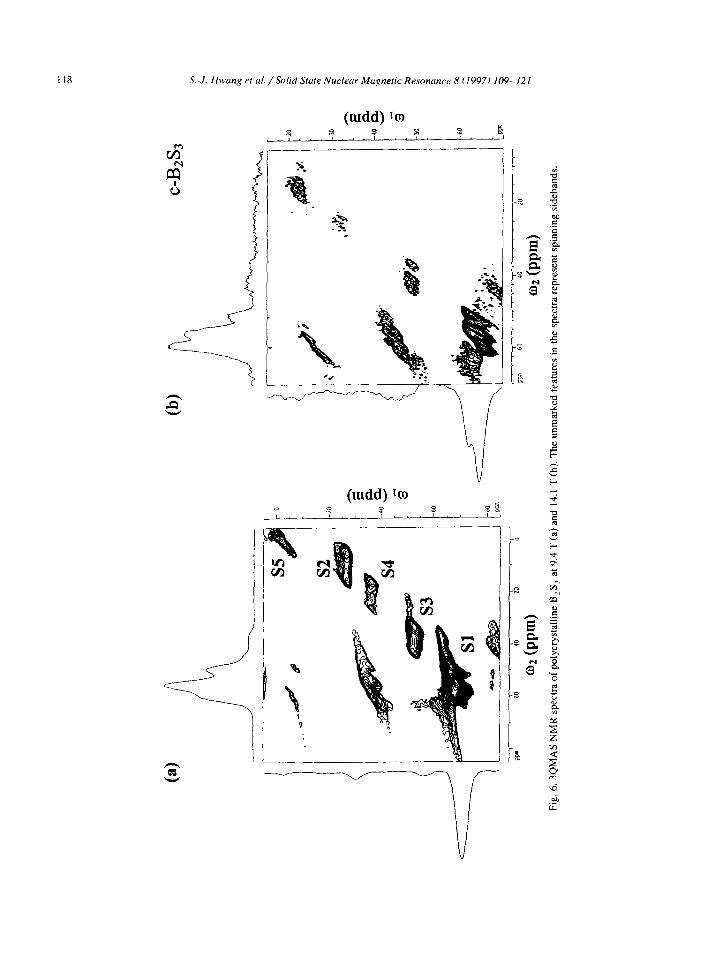
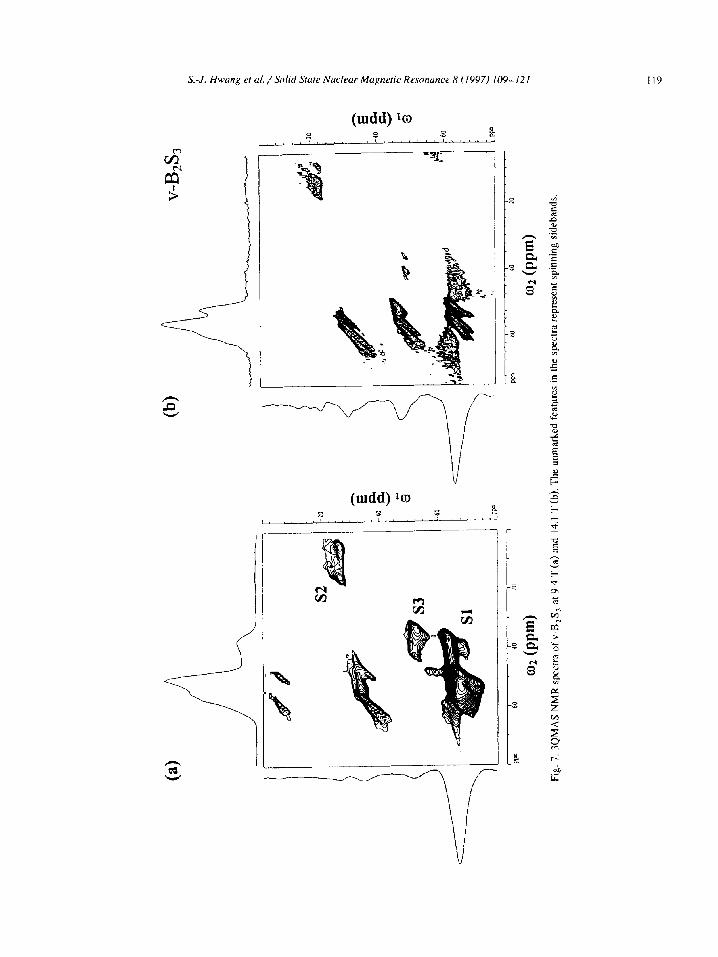
discussed further below. The 3QMAS NMR spectra of c-B2S, taken at 9.4
T and 14.1 T are shown in Fig. 6. We note that the projection of the whole isotropic spectrum is shown along the wi dimension whereas only an anisotropic
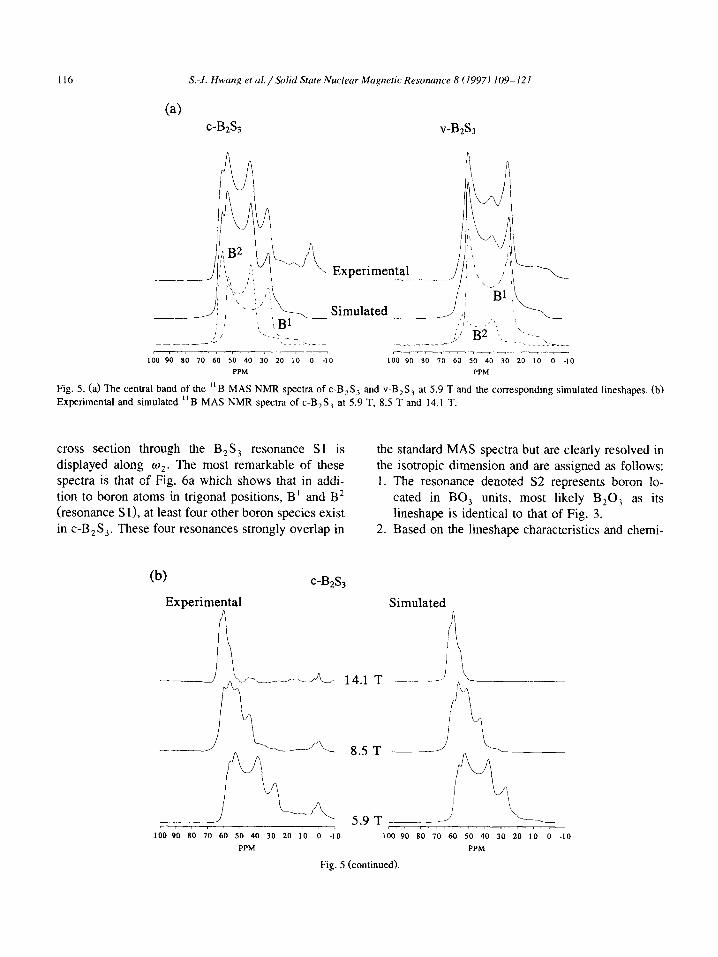
116 S.-J. Hwang et al./.%lid Stute Nuclear Magnetic Resonance 8 (1997) 109-121
c-B& v-B&
100 90 *II 70 60 50 40 30 20 10 0 -10 LOO 90 80 70 60 50 40 30 20 IO 0 -10
PPM PPM
Fig. 5. (a) The central band of the “B MAS NMR spectra of c-B,S, and V-B,S, at 5.9 T and the corresponding simulated lineshapes. (b)
Experimental and simulated “B MAS NMR spectra of c-B,S, at 5.9 T, 8.5 T and 14.1 T.
cross section through the B,S, resonance Sl is displayed along q. The most remarkable of these spectra is that of Fig. 6a which shows that in addi- tion to boron atoms in trigonal positions, B’ and B2 (resonance S 11, at least four other boron species exist in c-B,S,. These four resonances strongly overlap in
the standard MAS spectra but are clearly resolved in the isotropic dimension and are assigned as follows: 1. The resonance denoted S2 represents boron lo-
cated in BO, units, most likely B,O, as its lineshape is identical to that of Fig. 3.
2. Based on the lineshape characteristics and chemi-
(b) c-B&
Experimental Simulated 9
i\
14.1 T -.-. ._
8.5 T --_
5.9 T ___-- LOO 90 80 70 60 50 40 30 20 10 0 -10 100 90 SO 70 60 50 40 30 20 10 0 -10
PPM PPM
Fig. 5 (continued).

X-f. Hwmg et al./Solid Stute Nuclrur Mapetic Resonance 8 (19971 109%I.21 I17
3.
cal shifts we assign the two resonances labeled S3
and S4 to BS,O and BSO, species, respectively. Again, this is in agreement with the data available from liquid state NMR [27]. For example, boron atoms coordinated to two oxygen and one sulfur atoms in C,,H,B,O,S have a chemical
shift of 22.1 ppm, whereas a boron atom coordi- nated to one oxygen and two sulfur atoms in
(CH,S),B-OCH, has a chemical shift of 45.8
PPm. The upfield resonance denoted S5 must represent
boron sites of high symmetry, and is assigned to
four-fold coordinated (BS,) boron atoms. The
extended shape of this resonance along the diago- nal of the 2D spectrum suggests that a distribution
of BS, sites exists in the sample. The possible structural moieties include di-, tri-, tetra- and penta-thioborate. The presence of such units in
the c-B,S, sample was not anticipated and it is unclear how they were formed. Fig. 6 also demonstrates the effect of the B, and
B, fields on the resolution of the MQMAS tech- nique. The trigonal sites B’ and B2 in c-B,S, com-
pletely overlap in the isotropic dimension of the MQMAS spectrum at 9.4 T (Fig. 6a), but are well resolved in the 3QMAS spectrum taken at 14.1 T
(Fig. 6b). On the other hand, the overall quality of the spectra taken at 9.4 T was superior due to the rf power used, which was higher by approximately a
factor of two. As we mentioned before, the relative intensities
obtained in the MQMAS spectra are frequently dis- torted. However, a combination of MAS and MQ- MAS NMR can provide both reliable lineshape pa-
rameters and quantitative information. For example, by utilizing the spectral information from MQMAS spectra of c-B,S, we were able to integrate the one
dimensional spectra of this sample and quantify the observed boron environments: BS, units (four- and
six- membered rings) 86.70/o, BS,O units 4.5%, BSO, units 1.4%, BO, units 3.7%, and BS, units 3.7%.
The 3QMAS spectra of the V-B,S, sample are shown in Fig. 7. Again, in addition to boron reso- nances B ’ and B2, the 2D spectra reveal the presence
of BO, and BS,O units in this sample. Due to the wider distribution of isotropic shifts and the much
smaller concentration of B2 sites in this sample, the
3-coordinated sites B’ and B2 appear unresolved in
the isotropic dimension. The integration of the one
dimensional MAS spectra yielded the following con-
centrations: BS, units 95.3%, BS,O units 1.7%, and
BO, units 3.0%. We now disregard the structures involving oxygen
and consider the coordination of boron in B,S,. The structure of crystalline B,S, was established using X-ray diffraction by Diercks and Krebs [29] who
showed that it consists of borsulphol B,S, groups (the sulfur analogue of the boroxol group), and edge-sharing BS, groups which form four-membered
rings (see Fig. 4). The six- and four-membered rings form almost planar, 2D layers which bear no resem- blance to the three-dimensionally linked structure of B,O,. Earlier studies of V-B,S, provided inconsis-
tent results. For example, the variable temperature continuous wave ” B NMR study by Hendrickson and Bishop [30] and by Rubinstein [31] suggested that V-B,S, has a structure similar to that of v-B,O, and undergoes similar structural changes at elevated temperatures. A later solid state “B NMR study by Hurter et al. [32] and Sills et al. [33,34] confirmed the trigonal coordination of boron in V-B,S,. How- ever, these experiments did not reveal the exact nature of the trigonal sites. The IR studies indicated
the presence of six- and four-membered rings in
Table 2 Summary of NMR lineshape parameters for trigonal boron sites in B,S, obtained by computer simulations of “B MAS NMR spectra a
Sample Site Relative intensity b uiro (wd Q,, (MHz) 1) Line broadening (Hz)
c-B,S, B1 0.41 (0.02) 60.4 (0.5) 2.40 (0.05) 0.05 (0.0 If 210 (20) L .
B2 0.59 (0.02) 63.6 (0.5) 2.15 (0.05)
v-B@, B’ 0.79 (0.02) 60.6 (0.5) 2.41 (0.05)
B2 0.21 (0.02) 64.1 (0.5) 2.20 (0.05)
a The uncertainties are given in parentheses at 95% confidence.
b The relative intensities do not include other structural units found in these samples.
0.13 (0.01) 112(12)
0.02 (0.0 1) 200 (30) 0.16 (0.01) I65 (30)
118 S.-J. Hwang et al. / Solid Stute Nuclear Magnetic Resonance 8 (1997) 109- 12 I
---
.
II9

120 S.-J. Hwun~ et (II./ Solid State Nuclrur Magnetic Rrsonunce 8 (1997) 109-121
V-B,S,, but did not provide conclusive structural interpretation of the data [9.35,36]. It was not possi- ble from the IR spectra to determine the fraction of
six-membered rings in V-B,S,. In contrast, a recent
molecular dynamics study suggested that V-B,SI is made up exclusively of four-membered rings, bridged by sulfur atoms and arranged in a chain-like fashion
[371. To the best of our knowledge, this is the first
NMR study to reveal the presence of different types
of trigonal sites in boron trisulfide. The results of Table 2 show that, with the exception of relative
intensities, all lineshape parameters of boron sites B’
and B2 are indistinguishable between c-B,S, and
V-B2S3. Thus, these two samples contain boron in
very similar environments. As expected, the isotropic
chemical shifts observed for B’ and B2 correlate well with boron atom that are surrounded by three
sulfur atoms in a trigonal arrangement. The Q,, value is smaller in B,S, than in B,O,. Still, due to minor difference between the uisO values, the as- signment of the B’ and B* resonances is not straight-
forward. For example, borthiines, which are the pla- nar (XBS), compounds formed by adding a sub- stituent X to a heterocyclic (-BS), system, exhibit
resonances at 57.0 ppm when X = SH, and at 58.3 ppm for X = C,H, [27]. The gisO values for boron atoms in triangles are normally found between 60 and 65 ppm (see Table XX in ref. [27]). It is therefore reasonable to assign the upfield feature to boron atoms located in the six-membered (borsulphol) rings, and the resonance at - 64 ppm to those in the four-membered ring positions. However, this assignment is not compatible with the relative intensities expected for the c-B,S, sample. Accord- ing to the crystal structure proposed by Diercks and Krebs, the boron atoms in six- and four-membered
ring positions should exhibit a 3: 1 intensity ratio. The B’:B2 ratio measured here for c-B,S, is closer to 2:3, regardless of the magnitude of the magnetic
field. On the other hand, the above assignment is consistent with the MAS spectra of V-B,S, which has a much higher B’:B2 intensity ratio of 3.8: 1 (see Table 2). The above discrepancy raises several possi- bilities. (i) The structure of our c-B,S, sample dif- fers very significantly from the one described in [29]. We have already demonstrated the presence of BS,O, BSO,, BO, and BS, units in this sample which
comprise over 13% of boron sites. It is likely that the BS, sites in c-B,S, represent a mixed phase struc- ture which is more complex than the V-B,S, sample
and than the crystalline phase depicted in Fig. 4b. (ii) It is also possible that our assignment of resonances
B’ and B2 is oversimplified. According to the X-ray
diffraction study [29], the three boron atoms in the borsulphol ring are not located in equivalent posi-
tions. It is conceivable that their chemical shifts may
differ by a few ppm and that the resonances B’ and B* are a more complicated combination of contribu-
tions from both six- and four-membered rings. (iii)
Finally, the formation and presence of other B-S
compounds during the synthesis of c-B,S, is not unlikely [32].
In summary, the MAS and MQMAS spectra show that two well defined boron environments exist in
both the c-B,S, and V-B,S, samples studied here, and they support the view that the structure of
crystalline and glassy boron trisulfide is based on a network of borsulphol rings which are bridged by four-membered rings. However, the presence of other intermediate-range-order structures that involve 3- coordinated boron cannot be unambiguously ex-
cluded.
5. Conclusion
The results of this study show that a combination of MAS and 3QMAS NMR has great potential as a new tool for structural characterization of crystalline and noncrystalline solids. Whereas in chemically simple compounds having long-range structural or- der, reliable chemical shifts and quadrupole parame- ters can be extracted from the lineshape simulations of the traditional MAS NMR spectra, MQMAS proved instrumental in resolving and identifying the basic structural units in more complex samples. The NMR techniques and the conclusions of this work
are now being used to gain new insights into the structure of binary boron glasses [ 171.
Acknowledgements
This research was supported at Ames Laboratory by the U.S. Department of Energy, Office of Basic

S.-J. Hwung et al./Solid Stcrte Nuclear Mugnetic Resoncrnce 8 (1997) 109-121 121
Energy Sciences, Division of Chemical Sciences,
under Contract W-7405Eng-82. The samples were prepared using the financial support of the National
Science Foundation, grant DMR 91-04460. Thanks are due to Dr. B.C. Gerstein, Dr. J. Otaigbe, D.P.
Lang at Iowa State University and Dr. H. Eckert for reading the manuscript and helpful discussions.
References
[I] A. Llor and J. Vi&t, Chem. Phys. Lett., 152 (1988) 248.
[2] A. Samoson, E. Lipmaa and A. Pines, Mol. Phys., 65 (1988)
1013.
[3] K.T. Mueller, B.Q. Sun, G.C. Chingas, J.W. Zwanziger, T.
Terao and A. Pines, J. Magn. Reson., 86 (1990) 470.
[4] L. Frydman and J.S. Harwood, J. Am. Chem. Sot., 117 ( 1995) 5367.
[5] C. Femandez and J.P. Amoureux, Chem. Phys. Len., 242
(I 995) 449.
[6] D. Lathrop, M. Tullius, T. Tepe and H. Eckert, J. Non-Cryst.
Solids, 128 (1991) 208.
[7] A. Pradel and M. Ribes, Solid State Ionic& I8 (1986) 351.
[8] J.H. Kennedy and Y. Yang, J. Electrochem. kc., 133 (1986)
2437.
[9] S.W. Martin and D.R. Bloyer, J. Am. Ceram. Sot., 73 (1990)
3481.
[IO] J. P. Amoureux, Solid State Nucl. Magn. Reson., 2 (1993)
83.
[I I] C. Femandez and J.P. Amoureux, Solid State Nucl. Magn.
Reson., 5 (1996) 315.
[12] G. Wu, D. Rovnyank, B. Sun and R.G. Griffin, Chem. Phys.
Lett., 249 (1996) 210.
[13] D. Massiot, B. Touzo, D. Trumeau, J.P. Coutures, J. Virlet,
P. Florian and P.J. Grandinetti, Solid State Nucl. Magn.
Reson., 6 (1996) 73.
[I41 J.P. Amoureux. C. Femandez and L. Frydman, Chem. Phys.
Lett., 259 (I 996) 347.
[15] C. Femandez, J.P. Amoureux, J.M. Chezeau, L. Delmotte
and H. Kessler, Microporous Mater., 5/6 (1996) 33 I.
1161 A.H. Silver, J. Chem. Phys., 32 (1960) 959.
[17] S.-J. Hwang, J.P. Amoureux, C. Femandez, J.-W. Han, J.
Cho, SW Martin and M. Pruski, J. Chem. Phys., submitted
for publication.
[18] J.P. Amoureux, C. Femandez and Y. Dumazy, 37th Rocky
Mountain Conference, Denver, 1995, Abstract No. 264.
[19] P.A.V. Johnson, A.C. Wright and R.N. Sinclair, J. Non-Cryst.
Solids, 50 (I 982) 28 1.
[20] G.E. Jellison, Jr., L.W. Panel, P.J. Bray and G.B. Rouse, Jr.,
J. Chem. Phys., 66 (1977) 802.
[21] S.J. Gravina and P.J. Bray, J. Magn. Reson., 89 (1990) 515.
[22] R.E. Youngman and J.W. Zwanziger, J. Non-Cryst. Solids,
168 (1994) 293.
[23] S. Prabakar, K.J. Rao and C.N.R. Rao, Proc. R. Sot. London,
Ser. A, 429 (1990) I.
[24] S.J. Gravina, P.J. Bray and G.L. Petersen, J. NonCryst.
Solids, I23 (1990) l65.
[2S] H. Eckert, Prog. NMR Spectrosc., 24 (1992) 159-293.
[26] G.E. Gurr, P.W. Montgomerry, C.D. Knutson and B.T. Gor-
res, Acta Crystallogr., Sect. B, 26 (1970) 906.
[27] H. Noth and B. Wrackmeyer, Nuclear magnetic resonance
spectroscopy of boron compounds, in P. Diehl, E. Flu& and
R. Kosfeld (Eds.), NMR Basic Principles and Progress, Vol.
14, Springer-Verlag, Berlin, 1978.
[28] R.L. Mozzi and B.E. Warren, J. Appl. Cryst., 3 (1970) 25 I.
[29] H. Diercks and B. Krebs, Angew. Chem., Int. Ed. Engl., I6
(1977) 313.
[30] J.R. Hendrickson and S.G. Bishop, Solid State Commun., 17
(1975) 301.
1311 [321
[331
[341
i351
[361
[371
M. Rubinstein, Phys. Rev. B, 14 (I 976) 2778.
H.U. Hurter, B. Krebs, H. Eckert and W. Muller-Warmuth,
Inorg. Chem., 24 (1985) 1288.
I.A. Sills, S.W. Martin and D.R. Torgeson, J. Non-Cryst.
Solids, I68 ( 1994) 86.
I.A. Sills, Ph.D. Thesis, Iowa State University, Ames, IA,
1993.
I. Cho and S.W. Martin, J. Non-Cryst. Solids, 182 (1995)
248.
E.G. Zhukov and Y.K. Grinberg, Izv. Akad. Nauk SSSR,
Neorg. Mater., 5 (1969) 1646.
S. Balasubramanian and K.J. Rao, J. Phys. Chem., 98 (I 994)
9216.




















