
Purification and in vitro-phospholabeling of secretory envelope proteins E1 and E2 of hepatitis C...
13
Virus Research EL S EVIER Virus Research 45 (1996) 45-57 Purification and in vitro-phospholabeling of secretory envelope proteins E1 and E2 of hepatitis C virus expressed in insect cells Peter Hfissy a,*, Georg Schmid b, Jan Mous a, Helmut Jacobsen a ~Department of a Pharmaceutical Research-Gene Technology, Bldg 66/709, Grenzacherstrasse, 4070 Basel, Switzerland bpharmaceutical Research-Biotechnology, Bldg 66/709, Grenzacherstrasse, 4070 Basel, Switzerland Received 4 March 1996; accepted 26 June 1996 Abstract The putative envelope glycoproteins of hepatitis C virus (HCV), E1 and E2, were expressed as recombinant, secretory proteins in Sf9 insect cells through infection with recombinant baculoviruses. The influenza virus hemagglutinin signal sequence (HASS) was inserted upstream of the HCV-cDNAs in order to effect secretion. Furthermore, a hexa-histidine tag for purification on a Ni 2 + -nitrilotriacetic acid (Ni 2+ -NTA) column and a protein kinase A (PKA) recognition sequence for in vitro-phospholabeling were fused upstream of the HCV-cDNA. El-and E2 proteins lacking their carboxy-terminal, hydrophobic sequence were produced by baculovirus-infected insect cells in bioreactors of 23 1. The medium was concentrated and proteins were purified under native conditions on Ni 2 ~ -NTA columns. Purified proteins could be phospholabeled in vitro using the catalytic subunit of protein kinase A isolated from bovine heart and 7-[32p]ATP. Labeled E1 and E2 proteins expressed in insect cells could be immunoprecipitated with sera from HCV-infected patients. Co-expression of these E1 and E2 proteins led to the formation of El-E2 complexes within the insect cell and to secretion of these complexes into the medium. Keywords: Hepatitis C virus envelope proteins; Baculovirus expression system; Sf9 insect cell; Influenza virus hemagglutinin signal sequence; Ni 2 +-NTA-chromatography; Protein kinase A 1. Introduction * Corresponding author. Tel.: + 41 61 6882588; fax: + 41 61 6881448; e-mail: [email protected] Hepatitis C virus (HCV) is the major etiologic agent of transfusion-associated non-A, non-B hepatitis and has been implicated in community- acquired hepatitis (Houghton et al., 1991; Kuo et 0168-1702/96/$15.00 © 1996 Elsevier Science B.V. All rights reserved PII SO168-1702(96)01365-2
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Purification and in vitro-phospholabeling of secretory envelope proteins E1 and E2 of hepatitis C...

Virus Research
E L S E V I E R Virus Research 45 (1996) 45-57
Purification and in vitro-phospholabeling of secretory envelope proteins E1 and E2 of hepatitis C virus expressed in insect cells
Peter Hfissy a,*, Georg Schmid b, Jan Mous a, Helmut Jacobsen a
~Department of a Pharmaceutical Research-Gene Technology, Bldg 66/709, Grenzacherstrasse, 4070 Basel, Switzerland bpharmaceutical Research-Biotechnology, Bldg 66/709, Grenzacherstrasse, 4070 Basel, Switzerland
Received 4 March 1996; accepted 26 June 1996
Abstract
The putative envelope glycoproteins of hepatitis C virus (HCV), E1 and E2, were expressed as recombinant, secretory proteins in Sf9 insect cells through infection with recombinant baculoviruses. The influenza virus hemagglutinin signal sequence (HASS) was inserted upstream of the HCV-cDNAs in order to effect secretion. Furthermore, a hexa-histidine tag for purification on a Ni 2 + -nitrilotriacetic acid (Ni 2+ -NTA) column and a protein kinase A (PKA) recognition sequence for in vitro-phospholabeling were fused upstream of the HCV-cDNA. El-and E2 proteins lacking their carboxy-terminal, hydrophobic sequence were produced by baculovirus-infected insect cells in bioreactors of 23 1. The medium was concentrated and proteins were purified under native conditions on Ni 2 ~ -NTA columns. Purified proteins could be phospholabeled in vitro using the catalytic subunit of protein kinase A isolated from bovine heart and 7-[32p]ATP. Labeled E1 and E2 proteins expressed in insect cells could be immunoprecipitated with sera from HCV-infected patients. Co-expression of these E1 and E2 proteins led to the formation of E l - E 2 complexes within the insect cell and to secretion of these complexes into the medium.
Keywords: Hepatitis C virus envelope proteins; Baculovirus expression system; Sf9 insect cell; Influenza virus hemagglutinin signal sequence; Ni 2 +-NTA-chromatography; Protein kinase A
1. Introduction
* Corresponding author. Tel.: + 41 61 6882588; fax: + 41 61 6881448; e-mail: [email protected]
Hepa t i t i s C virus (HCV) is the m a j o r e t iologic agent o f t r ans fus ion-assoc ia ted non -A , non-B hepat i t i s and has been impl ica ted in c ommun i ty - acqui red hepat i t i s ( H o u g h t o n et al., 1991; K u o et
0168-1702/96/$15.00 © 1996 Elsevier Science B.V. All rights reserved PII SO 168-1702(96)01365-2

46 P. Hiissy et al. / Virus Research 45 (1996) 45 57
al., 1989). Furthermore, epidemiological data show a correlation between chronic HCV-infec- tion and the occurrence of hepatocellular car- cinoma (HCC) (Chien et al., 1992; Colombo et al., 1989; Gerber, 1993; Saito et al., 1990; Tanaka et al., 1991).
The hepatitis C virus has been classified in a separate genus in the family Flaviviridae. Its genome is a single stranded RNA of approxi- mately 9400 nt with positive polarity (Choo et al., 1989). The RNA contains an open reading frame (ORF) coding for one polyprotein which is cleaved either by cellular signalases (structural proteins core, E1 and E2) or by virus-encoded proteases (non-structural proteins NS2, NS3, NS4, NS5A, NS5B (van Doorn, 1994). The pre- dicted envelope glycoproteins E1 and E2 contain several potential N-(Asn)-linked glycosylation sites (Choo et al., 1991). The expression of glyco- sylated HCV E1 and E2 glycoproteins in Chinese hamster ovary (CHO)-ceUs (Lesniewski et al., 1995; Spaete et al., 1992), with recombinant vac- cinia viruses in BSC-40 cells (Ralston et al., 1993), in baculovirus-infected insect cells (Hsu et al., 1993; Matsuura et al., 1992; Nishihara et al., 1993), and monkey-COS-cells (Matsuura et al., 1992) has been described. The secretion and purification of hexa-histidine tagged E2 proteins containing the rabies virus G protein signal pep- tide in insect cells using recombinant bac- uloviruses has been reported (Nishihara et al., 1993). E1 and E2 form stable heterocomplexes in mammalian and insect cells (Dubuisson et al., 1994; Matsuura et al., 1994; Selby et al., 1994) which are resistant to reducing agents, high salt conditions and low concentrations of ionic deter- gents (Grakoui et al., 1993; Matsuura et al., 1994; Ralston et al., 1993).
To study the activities of the HCV envelope proteins in biological assays, labeling with ra- dioactive isotopes is still one of the most efficient methods which permits a high sensitivity of detec- tion. Protein kinase A (PKA) is an enzyme that phosphorylates proteins at serine residues and requires specific amino acid motifs (Barten- schlager et al., 1992; Kung and Bekesi, 1986; Li et al., 1989). A minimal motif for efficient labeling is arginine-arginine-(any aminoacid)-serine (Barten-
schlager et al., 1994). PKA offers the major ad- vantages that its catalytic subunit is inexpensive and that the enzyme has a relatively low target specificity, which facilitates the introduction of artificial recognition sites into proteins of interest.
2. Material and methods
2. I. Cloning o f expression plasmids
Influenza virus hemagglutinin signal sequence (HASS) was amplified from vaccinia virus expres- sion plasmid p7.5 K 131A (kindly provided by H.J. Schlicht, Ulm, Germany) by polymerase chain reaction (PCR) with sense oligo S-HASS (5'-GGC CGG ATC CTC TAG AAT GAA GAC TAT CAT CGC CCT T-3') and antisense oligo A-HASS-2 (5'-CCG GCT GCA GTT ACT TAC TTA GAA TTC ACT AGT GAT ATC CAT TGA ATT GTT GTC ATT ACC AGG TAG-3'). The resulting PCR-product contained a BamHI- and a XbaI site followed by a start codon up- stream of the HASS. Downstream of the HASS, a SpeI site, an EcoRI site, stop codons in all three reading frames and a PstI site was generated with the primers. The amplicon was cut with restriction enzymes BamHI and PstI and ligated into stan- dard baculovirus vector pVL-1393 (Invitrogen, Leek, Netherlands) cut with the same enzymes, which resulted in the new vector pVL-HASS. All transfections and cloning steps were done in the Escherichia coli strain TG-1.
A hexa-histidine-PKA-recognition-sequence- cassette was created by hybridisation of sense oligo S-hexa-his-PKA (5'-CTA GAC ATC ACC ATC ACC ATC ACG CGA GAA GGG CCA GTG TTA-3') with the antisense oligo A-hexa- his-PKA (5'-CTA GTA ACA CTG GCC CTT CTC GCG TGA TGG TGA TGG TGA TGT- 3'). Vector pVL-HASS was cut with SpeI and ligated with the untreated cassette. The 5'- and 3'-ends of the cassette have compatible ends to be ligated directly to the SpeI cut vector, but only the 3'-end reconstitutes a functional SpeI site, thus creating plasmid pVL-HASS-hexa-his- PKA.The sequence of El.7 from nucleotide posi- tion 918-1346 (amino acid (aa) positions

P. Hiissy et al. / Virus Research 45 (1996) 45 57 47
193-334) of the HCV-cDNA was amplified by PCR using the oligonucleotides S-918-BamHI (5'- GGC C GG ATC CGA AGT GCG CAA CGT ATC C GG A-3') and antisense oligo A-1346- EcoRI (5 '-GCG GAA TTC GGC TGC T G T A G G TGA CCA GTT-3'). For subcloning of E2.9, the nucleotide sequence from position 1557-2324 (amino acid position 406-660) was amplified by PCR with oligonucleotides S-1557- SpeI (5'-GGC CAC TAG T G G TCA TCC CCA GAA AAT CCA-3') and the antisense oligo A- 2324-II-EcoRI (5'-GGC CGA ATT CCT CTG ATC T G T CCC T G T CCT C-3') as primers.
All inserts were cut with SpeI and EcoRI and ligated into the vector pVL-HASS-hexa-his-PKA which had been digested with the same enzymes.
2.2. Generation, selection and titration of recombinant baculoviruses.
For co-transfection, 5 / tg linearized baculovirus BacPak 6 DNA (digested with restriction enzyme Bsu 36, CIontech, Palo Alto, CA) and 8 /tg of plasmid DNA were mixed with lipofectin (Gibco BRL, Basel, Switzerland). S f9 cells were culti- vated in TC-100 medium (Gibco BRL, Basel, Switzerland) containing 5% fetal calf serum (FCS). After incubation for 30 min, the transfec- tion mixture was added to 1.5 x 10 6 S f9 cells in TC-100 medium without FCS plated on a 35 mm dish. Twenty-four hours later, the medium was completed by 5% FCS. For selection of recombi- nant baculoviruses, 10/~1 of supernatant contain- ing baculoviruses were diluted on a 96-well plate. S f9 cells and 4 llg/ltl x-gal (Gibco-BRL, Basel, Switzerland) were added and the plate was incu- bated at 27°C for 5 days. After incubation for 5 -6 days at 27°C, the supernatant was collected and stored at 4°C. Wells which contained unre- combinated baculoviruses were colored blue due to fl-gal activity (Clontech manual for BacPak 6 DNA digest). One half of the supernatant was collected, transferred to a new 96-well plate and stored at 4°C. The remaining cells with superna- tant were lysed with an equivalent volume of 1 M NaOH and incubated 10 min at 37°C. The lysates were neutralized by addition of 0.2 volumes of 5 M ammonium acetate and blotted to a nitrocellu-
lose filter which was soaked in solution I (l M ammonium acetate, 0.02 M NaOH) with the help of a vacuum manifold (Schleicher and Schuell, Dassel, Germany). The blots were washed for 5 min in solution II (4 x SSC, 0.6 M NaC1, 0.06 M sodium citrate), air dried and incubated at 80°C for 2 h under vacuum. 32p-labeled E1/E2-DNAs were created by random priming of isolated frag- ments using the T7-Quick Prime kit (Pharmacia, Diibendorf, Switzerland). The blots were hy- bridized with the radioactive probes using the protocol of Church and Gilbert. (Church and Gilbert, 1984) Blots were analyzed by autoradiog- raphy. Dots with a positive radioactivity signal but no blue color were selected and 1-2 further purification rounds with dilution plates were car- ried out resulting in a pure recombinant bac- ulovirus population which was amplified by several rounds of infection to obtain a working stock.
For titration, the recombinant baculoviruses were diluted from 10- 2 to 10 7 in 1 ml of culture medium. 6-well plates were seeded with 1.5 x 10 ~' S f9 cells/well. For infection, the medium was removed completely and cells were treated with 100 /L1 of diluted virus/well followed by incuba- tion for 1 h at 27°C with orbital shaking every 10 min. Supernatant was replaced by 2 ml of culture medium containing 1% sea plaque agarose (FMC Bioproducts, Rockland, ME). When the agarose was solid, it was overlaid with 1 ml of culture medium. The plates were incubated in a moist box at 27°C for 5 days. For staining of the plaques, the medium was replaced by phosphate buffered saline (PBS) containing 0.5% neutral red solution (Sigma, Buchs, Switzerland). After 2~-4 h, the neutral red was removed and the plates were incubated overnight in the dark at room tempera- ture. Plaques were counted in the appropriate dilution and plaque-forming units/ml (PFU/ml) were calculated. The titers determined were be- tween l 0 7 and l0 s PFU/ml.
2.3. Insect cell culture and Jermentation.
T-flask cultures of S f9 insect cells were main- tained in TC-100 medium (Gibco BRL, Basel, Switzerland) containing 5% FCS. Inoculum and

48 P. Hfissy et al. / Virus Research 45 (1996) 4 5 - 5 7
bioreactor cultures for large-scale production of proteins were performed in SF1 medium with 1% FCS (Schlaeger et al., 1993). Sf9 cells were grown in 25 1 airlift bioreactors (Chemap, Volketswil, Switzerland) to a density of 2-3 x 10 6 cells/ml prior to infection with recombinant baculoviruses at a multiplicity of infection (m.o.i.) of 3-10. During cell growth and the subsequent infection phase (3-5 days) cells were maintained at 27°C, pH 6.2 and an oxygen partial pressure of 30% air saturation. (Schmid, 1996); (Schmid et al., 1994).
Prior to harvesting, protease inhibitors (1 mM phenyl-methyl-sulfonyl-fluoride (PMSF), 10 mM benzamidine, 20 KIU/ml aprotinin) were added to the cell suspension to minimize proteolytic degra- dation during the initial recovery steps. The biomass was separated from the culture superna- tant by continuous flow-through centrifugation (Contifuge 20RS, Heraeus, Osterode, Germany). The supernatant was then concentrated approxi- mately ten-fold by ultrafiltration (SP20 unit, Ami- con, Danvers, MA, USA) using 10 kDa cut-off membranes.
2.4. Protein purification under native conditions
All steps were carried out at 4°C. Concentrated supernatant (250 ml) from fermentation was di- alysed against 10 1 of column loading buffer (100 mM Tris pH 8.0, 100 mM NaC1, 5 mM MgC12). Three milliliters of Ni 2 +-NTA agarose (Qiagen, Chatsworth, CA) was added and the solution was shaken end-over-end for 16 h. The column was loaded, washed with 10 ml of washing buffer 1 (100 mM Tris pH 8.0, 100 mM NaC1, 5 mM MgCI2, 0.8 mM imidazole) followed by washing with 10 ml of washing buffer 2 (100 mM Tris pH 8.0, 100 mM NaC1, 5 mM MgCI> 30 mM imida- zole). Protein was eluted with elution buffer (100 mM Tris pH 8.0, 100 mM NaC1, 5 mM MgC12, 250 mM imidazole). In case of El, 1% NP-40 had to be added to column loading buffer and the two washing buffers to reduce co-elution of back- ground proteins. Aliquots of 1-ml were collected in the elution fractions. The protein was diluted with 1 volume of glycerol and stored at -70°C until use.
2.5. Prote& analys&
The silver stain kit of BioRad Laboratories (Glattbrugg, Switzerland) was used. Amounts of secreted, purified envelope proteins were esti- mated either by gel electrophoresis following coomassie blue staining and comparison with car- boanhydrase standard and in parallel with the method of Bradford (1976).
For western blot analysis, the gels were trans- ferred to nitrocellulose membranes (Schleicher and Schuell, Dassel, Germany) using a semi dry blotter. The blots were saturated for 1 h with PBS containing 5% non-fat milk. Sera were added in a dilution of 1:1000 to 1:2000 and the blots were incubated over night. The blots were washed twice with PBS, 0.05% Tween-20 for 10 min before the secondary antibody (goat anti rabbit or human, alkaline phosphatase (AP) coupled, Bio Rad Lab- oratories, Glattbrugg, Switzerland) was added in PBS containing 3% non-fat milk. The blots were incubated for 2 h and washed twice with PBS, 0.05% Tween-20 for 10 min. For the alkaline phosphatase reaction, 10 ml AP buffer (100 mM Tris, 100 mM NaC1, 5 mM MgC12) containing 0.33 tzg/Itl nitro-blue-tetrazolium (NBT) and 0.165 pg/121 5-bromo-4-chloro-3-indolyl-phos- phate (BCIP, both Promega, Ziirich, Switzerland) was added and the reaction was stopped with water after 10 min to 1 h.
2.6. Phospholabeling of El and E2 with protein kinase A
Sixteen units of the catalytic subunit of protein kinase A isolated from bovine heart (Sigma, Buchs, Switzerland) were dissolved in 5 /~1 of 39 mM dithiothreitole (DTT) and incubated at room temperature for 10 min. In a separate tube, 10-50 ng of purified E1 or E2 protein were added to 48 /11 reaction mix containing 40 /~1 of y-[32p]ATP (Redivue, specific activity greater than 5000 Ci/ mmol/185 TBq/mmol, Amersham, Z/irich, Switzerland) in TMN buffer (20 mM Tris pH 7.0, 100 mM NaC1, 10 mM MgC12). To start the reaction, 1 /tl of preincubated PKA was added and the mix was incubated for 15 min at 3°C. The second addition of 1 /tl PKA was again followed by 15 min incubation time.

P. Hiissy et al. / Virus Research 45 (1996) 45 57 49
Water and 1% bovine serum albumin (BSA) were added to the mix to a final volume of 100/tl prior to gel filtration through a Bio Spin 6 column (Bio Rad Laboratories, Glattbrugg, Switzerland). The gel filtration step resulted in a final volume of 200-500 /~1 containing the phospholabeled proteins.
2. 7. Antibodies and human sera
Hexa-his tagged E1 and E2 proteins covering the same region as the Sf9-expressed proteins were expressed in E. coli strains SG or M 15 (data not shown, Manual of Qia-expressionist, Qiagen, Chatsworth, CA.). Briefly, bacteria were lysed in lysis buffer (6 M guanidine-HC1, 0.3 M NaC1, 0.1 M NaAc, pH 6.2) and envelope proteins were bound on Ni 2 +-NTA columns. The columns were washed with 6 M guanidine-HCl, 0.3 M NaCI, 0.1 M NaAc, pH 6.0 and proteins eluted with 6 M guanidine-HC1, 0.3 M NaC1, 0.1 M NaAc, pH 5.0. After dialysis to 6 M urea, proteins were loaded on preparative sodium dodecyl sulfate- polyacrylamide gel electrophoresis (SDS-PAGE) gels and electro-eluted from the excised gel-frag- ments. Proteins were used for immunization of rabbits to raise antisera.
Human sera were collected from patients with HCV infection shown to contain antibodies which reacted with the HCV-core protein expressed in E. coli (data not shown).
2.8. Immunoprecipitation with human sera
Five microliters of human serum were preincu- bated with 20 /zl of protein G Sepharose (Phar- macia, Diibendorf, Switzerland) in 150 /21 of standard radio-immuno precipitation assay buffer (RIPA-buffer, PBS with 0.1% SDS, 1% Triton X-100 and 0.5% deoxycholate (DOC)) for 2 h at 4°C. The immobilized antibodies were washed twice with 500 /tl RIPA-buffer and 1-5 ng of phospholabeled E1 and E2 were added per assay. The tubes were shaken for 16 h at 4°C. The precipitates were washed three times with RIPA- buffer and the final complexes were heated for 10 min in SDS gel sample buffer (200 mM Tris pH 8.8, 10% glucose, 5 mM EDTA, 0.1% bromphe-
nolblue, 3% SDS and 0.3 M fl-mercapto-ethanol) prior to separation on SDS-PAGE gels followed by autoradiography.
2.9. Co-immunoprecipitation of E l and E2 proteins
S f9 cells were cultivated in TC-100 medium containing 5% FCS in 6-well plates and infected with recombinant baculoviruses at an m.o.i, of 10. Cells were labeled metabolically 36-40 h post infection with 100 /zCi [35S]methionine (Amer- sham, Ziirich, Switzerland) for 6 h after starva- tion for 2 h in methionine-free TC-100 medium (Gibco BRL, Basel, Switzerland) containing 5% dialysed FCS. Cells were lysed in TNE buffer (10 mM Tris pH 7.8, 150 mM NaCI, 1 mM EDTA, 1% NP-40, 2/zg/ml aprotinin) and centrifuged at 13 000g for 5 min.
For co-immunoprecipitation, the method of (Selby et al., 1994) was modified. Rabbit antisera (5/21) were preincubated for 2 h at 4°C with 150 /~1 RIPA-buffer and 20 #1 of protein A sepharose (Sigma, Buchs, Switzerland). The complexes were washed once with 500 /zl of RIPA-buffer and twice with 500/~1 of cold native-IP-buffer (50 mM Tris pH 7.4, 150 mM NaC1, 1 mM EDTA, 1% NP-40, 0.1% DOC, 2 /zg/ml aprotinine, 1 mM PMSF). Cell lysate was added to the washed complexes and shaken for 16 h at 4°C. Complexes were washed four times with 500 /~1 native-IP- buffer and heated for 10 min in 80/zl of SDS gel sample buffer prior to separation on SDS-PAGE gels followed by autoradiography.
3. Results
3. I. Purification of H C V envelope proteins
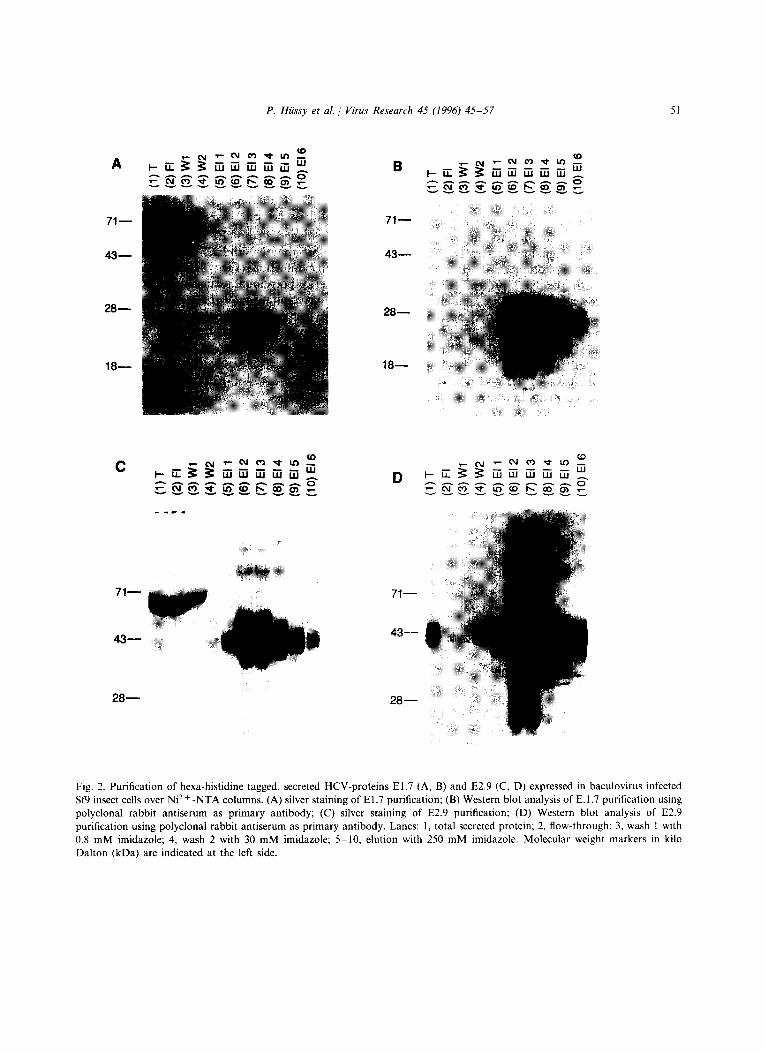
The secretory proteins El.7 and E2.9 (Fig. 1) were produced in bioreactors and purified with a Ni2+-NTA agarose column. The El.7 protein could not be detected in the total lysate by anti- bodies raised in rabbits against E1 expressed in E. coli (Fig. 2(B), lane 1). In the elution fractions 2, 3 and 4 (Fig. 2(A), lanes 6-8), the protein eluted efficiently, resulting in a broad pattern of differ-

50 P. Hiissy et al. / Virus Research 45 (1996) 45 57
ently glycosylated variants ranging in molecular weight from 24 to 35 kDa. In contrast, the E2.9 protein was detected in the total lysate by silver staining (Fig. 2(C), lane 1) and western blot anal- ysis (Fig. 2(D), lane 1) due to its higher expression level. The protein elutes in fractions 2-5 (Fig. 2(C,D), lanes 6-9) and also showed a pattern of different glycosylation variants from 40 to 50 kDa. From the silver stain analysis (Fig. 2(A,C)) compared with western blot analysis (Fig. 2(B,D)), we estimated that the purity of the El.7 and the E2.9 protein was more than 95% and therefore no further purification steps were car- ried out.
3.2. Deglycosylation analysis and yields
To prove that these pattern of bands are differ- ent glycosylation variants of the same polypeptide chain, the proteins El.7 and E2.9 were digested with N-glycosidase F (EC 3.2.2.18 and 3.5.1.52) and endoglycosidase H (EC 3.2.1.96).
N-glycosidase F, which cleaves all N-linked sugar side chains directly at the asparagine (Chu, 1986; Tarentino et al., 1985) cleaved El.7 and E2.9 efficiently. Deglycosylation of El.7 yielded a major band of 20-22 kDa and a minor band around 18 kDa (Fig. 3, lane 2).
The digestion with endoglycosidase H resulted in some completely deglycosylated EI.7 (around
a.e.: 1 192 384 746810
I con I E1 I E2 IpTJ.S2 A A A A A A A A ~ A ~ . A A A A A A
193 334 406 660 [ ~ E1.7 Iff/~A E2.: I
A A A A A A A A A A A
A potential N-linked glycosylation site [ ] signal sequence of Influenza virus hemagglutinin
IEI Hexa-histidine tag SJ protein kinase A (PKA) recognition sequence
Fig. 1. Diagram of HCV-subfragments El.7 and E2.9 (bot- tom) of the structural region (top) expressed in the baculovirus expression system. The signal sequence of influenza virus hemagglutinin was added to obtain secretory proteins, fol- lowed by six histidine residues for purification on Ni 2 + -NTA column and a protein kinase A recognition sequence (amino acids Arginine-Arginine-Alanin-Serin) for in vitro-phosphola- beling at the Serin residue. Amino acid positions of the HCV-polyprotein are indicated.
18 kDa, Fig. 3, lane 3), some incompletely di- gested forms (18-28 kDa, Fig. 3, lane 3) and to a 30-32 kDa band of digested E2.9 (Fig. 3, lane 6). The proteins were only partially digested with this enzyme. Envelope proteins incubated under the same conditions but without enzyme are shown in Fig. 3, lanes 1 and 4. The estimated yield of secreted El.7 was in the range of 3-4 mg/l con- centrated medium (9-16 mg in one bioreactor run of 23 1). The estimated yield of secreted E2.9 was 12-18 mg/1 concentrated medium (36-54 mg/23 1 bioreactor). We estimate from cell fractionating experiments (data not shown) that approximately 20% of the total recombinant glycoproteins syn- thesized were secreted into the culture medium.
3.3. In vitro-phospholabeling
The reaction with PKA was analyzed by gel electrophoresis and autoradiography (Fig. 4). As a control, reactions were done in the absence of either PKA or DTT (Fig. 4(A,B), lanes 1 and 3). The efficiency of phosphorylation in the presence of DTT was calculated using a phosphoimager for quantitation. Briefly, the area of the protein bands on the SDS-PAGE gel (Fig. 4(A,B), lanes 2) were integrated to determine the counts per minute (cpm) of the incorporated radioactivity. The specific activity (Ci/mmol) was calculated. The values for El.7 and E2.9 in several indepen- dent experiments were between 80 and 250 Ci/ mmol. Thus, the estimated proportion of labeled molecules was 10-40%.
3.4. Seroreactivity tests
To investigate if these recombinant HCV envel- ope proteins can be recognized by antibodies from HCV-seropositive patients, we performed Western blot analysis (Fig. 5) and immunoprecipitation (Fig. 6). All patient sera used in the assays were immunoreactive with recombinant HCV-core protein expressed in E. coli (data not shown). In the Western blot analysis with the purified glyco- proteins, six of eight sera, namely B14, G07, G08, G13, I01 and I02 reacted with El.7 (Fig. 5(A), lanes 3-10). In the case of E2.9 (Fig. 5(B), lanes 3-10) the seven sera B14, G07, G09, G13, I01,
P. Hiissy et al. / Virus Research 45 (1996) 45-57 51
A
7 1 ~
4 3 ~
2 8 D
1 8 ~
CO O4 O3 I.O
B
7 1 - -
43
~ - ~ ~ w w w w w w
28.
18-
C ~ N ~ w ~ N N
CO .,._ ~ ~ 0,1 03 ',~" tr'} __
D ~- ~ ~ ~ " ' " ' " ' ' " " ' ' " 0 CJ 03 ~ " LO CO I ~ CO 03
....... i!!ii!iii!~iiii 7 1 - - !i! 7 1 - -
4 3 ~ ~i~,iiii~ 4 3 - -
2 8 ~ 2 8 - -
Fig. 2. Purification of hexa-histidine tagged, secreted HCV-proteins El.7 (A, B) and E2.9 (C, D) expressed in baculovirus infected Sf9 insect cells over Ni 2 + -NTA columns. (A) silver staining of El.7 purification; (B) Western blot analysis of E. 1.7 purification using polyclonal rabbit antiserum as primary antibody; (C) silver staining of E2.9 purification; (D) Western blot analysis of E2.9 purification using polyclonal rabbit antiserum as primary antibody. Lanes: 1, total secreted protein; 2, flow-through; 3, wash l with 0.8 mM imidazole; 4, wash 2 with 30 mM imidazole; 5 10, elution with 250 mM imidazole. Molecular weight markers in kilo Dalton (kDa) are indicated at the left side.

52 P. Hiissy et al. / Virus Research 45 (1996) 45 57
I02 and SG04 showed a positive signal. In the immunoprecipitation assay with the phosphola- beled proteins (Fig. 6), only five sera (B14, G07, G13, I01 and I02) were able to precipitate El.7 (Fig. 6(A)) whereas six sera (G07, G09, G13, I01, I02 and SG04) immunoprecipitated E2.9 (Fig. 6(B), lanes 5-12). Sera of HCV-negative humans used as controls showed no signal in western blot analysis and in immunoprecipitation assay. The Western blot analysis with the El.7 protein (Fig. 5(A)) showed a higher sensitivity than the immunoprecip- itation of labeled El.7 (Fig. 6(A)), the serum G08 precipitated no detectable amounts of E 1.7 protein (Fig. 6(A), lane 7). The sera B14, G07, G13, I01 and 102 showed reactivity to El.7 in both assays. Western blot analysis of E2.9 (Fig. 5(B)) also showed a higher sensitivity in comparison with the immunoprecipitation analysis (Fig. 6(B)). The serum B 14 was reactive in the Western blot analysis (Fig. 5(B), lane 3) but no E2.9 protein could be detected in immunoprecipitation (Fig. 6(B), lane 5).
3.5. Co-immunoprecipitation
In order to investigate whether our recombinant
LL I i
r5 "r- ,,j "-r
z ,,, z ,,l
• . o ° .
71
43
28
18
!1
Fig. 3. Deglycosylation of purified proteins El.7 (lanes 1-3) and E2.9 (lanes 4 6) in western blot analysis. Lanes: 1 and 4, mock-digestions in endoglycosidase H buffer; 2 and 5, diges- tion with N-glycosidase F; 3 and 6, digestion with endoglycosi- dase H. Markers are indicated at the left side.
El.7 and E2.9 proteins were able to heteromerize (Dubuisson et al., 1994; Matsuura et al., 1994; Selby et al., 1994) we co-infected Sf9cells with E1.7 and E2.9 producing recombinant baculoviruses. Proteins were labeled metabolically with [35S]methionine (Fig. 7). Polyclonal rabbit anti- serum directed against E1 (Fig. 7, lanes 1-3) was able to precipitate both El.7 and E2.7 protein after a double infection (Fig. 7, lane 3, at 50 kDa). Vice-versa, polyclonal serum directed to E2.9 was able to precipitate E2.9 (lanes 5 and 6) and El.7 (Fig. 7, lane 6, approximately 30 kDa). Non-spe- cific precipitation of El.7 by anti-E2 antibodies or non-specific precipitation of E2.9 by anti-E1 anti- bodies in single infections could not be detected (Fig. 7, lanes 2 and 4).
4. Discussion
We have chosen the baculovirus system to ex- press secretory envelope proteins of HCV since the potential of this system to express immunoreactive HCV envelope proteins has been repeatedly demonstrated (Hsu et al., 1993; Matsuura et al., 1994; Nishihara et al., 1993).
The use of Ni 2 + -NTA columns for purification of hexa-histidine tagged proteins allows highly efficient one-step purification under native condi- tions (Janknecht et al., 1991)• The system has already been used for the purification of HCV-E2 glycoprotein (Nishihara et al., 1993). This group used the rabies virus G protein signal peptide as a secretory signal and added the hexa-histidine tag at the carboxy-terminus of the protein. Their E2 protein terminated at amino acid 683 of the HCV- polyprotein. The presence of the hydrophobic car- boxy-terminus of naturally processed E1 and E2 seems to inhibit secretion of the proteins expressed in insect and mammalian cells (Nishihara et al., 1993), P. H/issy, unpublished observation), sug- gesting that this hydrophobic stretch is membrane associated and is responsible for retaining the glycoproteins in association with the cell. The hemagglutinin signal sequence of influenza virus was used in our study to obtain secretory E1 and E2 proteins, which led to an efficient secretion of the envelope proteins lacking their carboxy-termi- nal, hydrophobic part. Our E2 protein E2.9 ends

P. Hiissy et al. / Virus Research 45 (1996) 4 5 - 5 7 53
A I: B I:: a a
,- a ~ ,-- a , + , "7" + ,
100 - - 7 1 - -
4 3 ~
2 8 - -
1 8 - -
1 0 0 - -
7 1 ~
4 3 - -
2 8 - -
Fig. 4. In vitro-phospholabeling of purified proteins El.7 (A) and E2.9 (B) with protein kinase A (PKA) in autoradiography. Lanes: 1, control reaction without PKA; 2, reaction with PKA preincubated in dithiothreitole (DTT); 3, reaction with PKA not preincubated with DTT. Markers are indicated at the left side.
at amino acid position 660. The hypervariable region HVR-1 (aa 391-400) (Hijikata et al., 1991) has been excluded in our expressed glycoprotein E2.9, thus starting at aa 406. The E1 protein El.7 ends at amino acid position 334. Based on our estimate of 0.3-0.4 mg secreted El.7 per 1 medium and including the approximate amount of intracellular El.7 protein, the total yield was 1.5 2 mg/1 total cell culture. This expression level is on the low side of the baculovirus system. The amount of secreted E2.9 was approximately 1.2- 1.8 mg/1 medium, and the total yield approxi- mately 6-9 mg/1 cell culture. The theoretically calculated molecular weight of the El.7 polyprotein chain without sugars is ca. 18 kDa. The 20-22 kDa product visible after the N-gly- cosidase F treatment (Fig. 3, lane 2) could be an intermediate which contains some protected sug- ars which are not accessible to the enzyme. This would also be consistent with the observed prod- ucts of the endoglycosidase H digest (Fig. 3, lane 3). E2.9 is converted to a major band of 30 kDa (Fig. 3, lane 5) by N-glycosidase F which is in
accordance with the calculated molecular weight of the polypeptide chain (30 kDa). There were additional bands visible at higher molecular weights on the Western blot (Fig. 3, lanes 1, 2, 4 and 5) which could represent homodimers. In the case of El.7 (Fig. 3, lane 2), we observed a putative dimer at a molecular weight of 40 kDa visible in the N-glycosidase F-treated sample. A putative E2.9-homodimer is visible in the undi- gested sample (Fig. 3, lane 4) at approximately 80 and 60 kDa in the N-glycosidase F-treated sample (Fig. 3, lane 5).
The possibility of protein-phospholabeling with protein kinase A by introduction of a PKA-recog- nition site offers an efficient, fast and convenient method for in vitro-phospholabeling (Barten- schlager et al., 1992; Kung and Bekesi, 1986; Li et al., 1989). When we calculated the specific activi- ties of our phospholabeled proteins, values dif- fered significantly in independent experiments. The differences can be explained with different activation efficiencies of the PKA with DTT in the experiments. We set the level of DTT for

54 P. Hiissy et al. / Virus Research 45 (1996) 4 5 - 5 7
activation to a minimum to avoid changing the tertiary structures of our envelope proteins by this reducing agent. Compared with the specific activi- ties reported for different mutants of phosphola- beled hepatitis B virus P-gene products ((Bartenschlager et al., 1992, 1994), 6-600 Ci/ mmol), our values for El.7 and E2.9 were in a similar range (80-250 Ci/mmol). The phosphory- lation efficiency is also influenced by the position of the PKA-recognition sequence in the protein (Bartenschlager et al., 1992, 1994). Due to the fact that we were able to bind the his-tagged envelope proteins to the Ni 2 +-NTA column under native conditions and that the PKA-recognition se- quence is directly downstream of the hexa-his box, we anticipated that the PKA-recognition se- quence should also be at the surface of the proteins and therefore well accessible for the PKA.
Labeled secretory proteins could be immuno- precipitated with sera of humans with non-A, non-B hepatitis. We pre-selected 8 sera which
.A. .,¢
.,-- 04 o
d~ ¢~ .¢ r-- co o~ eo 0
0
28--
18--
B
.,- 0.I 0
~ o o ,-- ~ oJ oO c
0
43--
28--
Fig. 5. Seroreactivity test of purified El.7 (A) and E2.9 (B) with human sera in Western blot analysis. Lanes: 1 and 2, HCV-negative control sera; 3-10, sera of HCV-infected pa- tients. Markers are indicated at the left side.
were anti HCV-core antibody positive; 6 of 8 sera (75%) reacted with El.7 and 7 of 8 sera (87%) to E2.9 in Western blot analysis. Immunoprecipita- tion of phospholabeled proteins appeared to be less sensitive. Hsu et al. (1993) tested 50 patient sera in Western blot analysis with total cellular extracts of HCV structural proteins expressed in Sf9 insect cells, 47 reacted with HCV-core protein (94%), 14 (28%) with E1 and 5 (10%) with E2. In another study (Nishihara et al., 1993) 55 of 60 tested patient sera (92%) reacted with their trun- cated, Sf9-expressed, Ni2+-NTA purified E2 protein in enzyme linked immunosorbent assay (ELISA). Our rate of anti-E2 positive sera (87%) is in accordance with the latter results. The preva- lence of anti-E1 positive sera in our study is approximately 75%. Of course, further studies are needed to generate results that are statistically representative. When combining the results of tests with El.7 and E2.9 we found 100% positive results (8/8) which suggests that our purified proteins could be useful as diagnostic tools to screen for envelope antibodies in HCV infected patients. However, antibodies which recognize the described recombinant envelope proteins are less prevalent than antibodies against recombinant core protein in the patient sera tested.
Double-infection of Sf9 cells with El- and E2- recombinant baculoviruses allowed co-immuno- precipitation of intracellular [35S]methionine labeled E1 and E2 proteins lacking their putative membrane spanning domain with polyclonal rab- bit antisera. (Fig. 7). Double-infection and fer- mentation of Sf9 cells allowed the purification of secreted El-E2 complexes which could be phos- pholabeled and then co-immunoprecipitated with polyclonal rabbit antisera (data not shown). These findings are in agreement with those of Matsuura (Matsuura et al., 1994) who also found that the interaction of co-expressed E1 and E2 is independent of the presence of the carboxy-termi- nal, hydrophobic part. Further, we can conclude that the presence of the hypervariable region 1 (HVR-1) is not required for this interaction, which is consistent with the fact that this region is extremely heterogeneous and not conserved be- tween different isolates (Hijikata et al., 1991; Kato et al., 1992, 1994) and is therefore not likely to be involved in the assembly of viral proteins.

P. Hiissy et al. / Virus Research 45 (1996) 45 57 55
A • ~ 0
• o ~ g o o
v
B
71 - - 71 - -
4 3 - - 4 3 - -
28--
18--
",'- k'~ ~ 0
• ( ~ 0 ,- ~ - . o tO O tO tD .... ~ ~
( 0 I ' ~ GO 0 ' ) ' ,-" " , - " , - v v v ~ v v v ~ v
2 8 - -
1 8 - -
Fig. 6. Autoradiography of in vitro-phospholabeled proteins El.7 (A) and E2.9 (B) immunoprecipitated with human sera. Lanes: 1, total labeled protein used in each immunoprecipitation; 2 and 3, HCV-negative control sera; 4, negative control without any serum; 5-12, sera of HCV-infected patients. Markers are indicated at the left side.
6 9 - -
4 6 ~
3 0 ~
~tE1 ~ E2 I I I I
W ILl + +
v - 0 4 T - , ~ - 0 4 W i l l W I l I LLI i l l
E2.9 intrac.
E1.7 intrac.
Fig. 7. Co-immunoprecipitation of intracellular, 35S-methion- ine-labeled proteins E1.7 and E2.9 with polyclonal rabbit antisera directed to E1 (lanes l 3) or E2 (lanes 4-6). Lanes: 1 and 4, intracellular extract of cells infected with baculovirus expressing El .7; 2 and 5, cells infected with baculovirus express- ing E2.9; 3 and 6, cells double-infected with baculoviruses expressing El.7 and E2.9. Markers are indicated at the left side.
Acknowledgement s
The authors thank N a t h a l i e Schaub, F. H o f f - m a n n - L a R o c h e Ltd, D e p a r t m e n t o f B io tech-
nology, for her con t r i bu t ion to insect cell fermen- ta t ion.
References
Bartenschlager, R., Kuhn, C. and Schaller, H. (1992) Expres- sion of the P-protein of the human hepatitis B virus in a vaccinia virus system and detection of the nucleocapsid-as- sociated P-protein by radiolabelling at newly introduced phosphorylation sites. Nucleic Acids Res. 2, 195-202.
Bartenschlager, R., Weber, M. and Schaller, H. (1994) In vitro phosphorylation of hepatitis B virus P gene product: A general method for radiolabelling of proteins. In: K.W. Adolph (Ed.), Methods in Molecular Genetics. Academic Press, San Diego, pp. 391-401.
Bradford, M.M. (1976) A rapid and sensitive method for the quantities of protein utilizing the principle of protein dye binding. Anal. Biochem. 72, 248 254.
Chien, D.Y., Choo, Q.L., Tabrizi, A., Kuo, C., McFarland, J., Berger, K., Lee, C., Shuster, J.R., Nguyen, T. and Moyer, D.L (1992) Diagnosis of hepatitis C virus (HCV) infection using an immunodominant chimeric polyprotein to capture circulating antibodies: reevaluation of the role of HCV in liver disease. Proc. Natl. Acad. Sci. USA 89(21), 10011 10015.
Choo, Q.L., Kuo, G., Weiner, A.J., Overby, L.R., Bradley, D.W. and Houghton, M. (1989) Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244(4902), 359-362.
Choo, Q.L., Richman, K.H., Han, J.H., Berger, K., Lee, C., Dong, C., Gallegos, C., Coit, D., Selby, R. and Barr, P.J. (1991) Genetic organization and diversity of the hepatitis C virus. Proc. Natl. Acad. Sci. USA 88(6), 2451-2455.

56 P. Hiissy et al. / Virus Research 45 (1996) 45 57
Chu, F.K. (1986) Requirements of cleavage of high mannose oligosaccharides in glycoproteins by peptide N-glycosi- dase F. J. Biol. Chem. 261(1), 172-177.
Church, G.M. and Gilbert, W. (1984) Genomic sequencing. Proc. Natl. Acad. Sci. USA 81(7), 1991-1995.
Colombo, M., Kuo, G., Choo, Q.L., Donato, M.F., Del, N.E., Tommasini, M.A., Dioguardi, N. and Houghton, M. (1989) Prevalence of antibodies to hepatitis C virus in Italian patients with hepatocellular carcinoma [see com- ments]. Lancet 2(8670), 1006-1008.
Dubuisson, J., Hsu, H.H., Cheung, R.C., Greenberg, H.B., Russell, D.G. and Rice, C.M. (1994) Formation and in- tracellular localization of hepatitis C virus envelope gly- coprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J. Virol. 68(10), 6147-6160.
Gerber, M.A. (1993) Relation of hepatitis C virus to hepato- cellular carcinoma. J Hepatol.
Grakoui, A., McCourt, D.W., Wychowski, C., Feinstone, S.M. and Rice, C.M. (1993) Characterization of the hep- atitis C virus-encoded serine proteinase: determination of proteinase-dependent polyprotein cleavage sites. J. Virol. 67(5), 2832 2843.
Hijikata, M., Kato, N., Ootsuyama, Y., Nakagawa, M., Ohkoshi, S. and Shimotohno, K. (1991) Hypervariable regions in the putative glycoprotein of hepatitis C virus. Biochem. Biophys. Res. Commun. 175(1), 220-228.
Houghton, M., Weiner, J., Han, J., Kuo, G. and Choo, Q.L. (1991) Molecular biology of the hepatitis C viruses: implications for diagnosis, development and control of viral diseases. Hepatology 17, 381-388.
Hsu, H.H., Donets, M., Greenberg, H.B. and Feinstone, S.M. (1993) Characterization of hepatitis C virus struc- tural proteins with a recombinant baculovirus expression system. Hepatology 17(5), 763-771.
Janknecht, R., de Martynoff, G., Lou, J., Hipskind, R.A., Nordheim, A. and Stunnenberg, H.G. (1991) Rapid and efficient purification of native histidine-tagged protein ex- pressed by recombinant vaccinia virus. Proc. Natl. Acad. Sci. USA 88(20), 8972-8976.
Kato, N., Ootsuyama, Y., Ohkoshi, S., Nakazawa, T., Sekiya, H., Hijikata, M. and Shimotohno, K. (1992) Characterization of hypervariable regions in the putative envelope protein of hepatitis C virus. Biochem. Biophys. Res. Commun. 189(1), 119-127.
Kato, N., Ootsuyama, Y., Sekiya, H., Ohkoshi, S., Nakazawa, T., Hijikata, M. and Shimotohno, K. (1994) Genetic drift in hypervariable region 1 of the viral genome in persistent hepatitis C virus infection. J. Virol. 68(8), 4776-4784.
Kung, H.F. and Bekesi, E. (1986) Phosphorylation of hu- man immune interferon (IFN-gamma). Methods Enzy- mol. 119, 296-301.
Kuo, G., Choo, Q.L., Alter, H.J., Gitnick, G.L., Redeker, A.G., Purcell, R.H., Miyamura, T., Dienstag, J.L., Alter, M.J. and Stevens, C.E. (1989) An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science 244(4902), 362-364.
Lesniewski, R., Okasinski, G., Carrick, R., Vansant, C., De- sai, S., Johnson, R., Scheffel, J. and Moore, B. (1995) Antibody to hepatitis C virus second envelope (HCV-E2) glycoprotein: A new marker of HCV infection closely associated with viremia. J. Med. Virol. 45(4), 415 422.
Li, B.L., Langer, J.A., Schwartz, B. and Pestka, S. (1989) Creation of phosphorylation sites in proteins: construc- tion of a phosphorylatable human interferon alpha. Proc. Natl. Acad. Sci. USA 86(2), 558 562.
Matsuura, Y., Harada, S., Suzuki, R., Watanabe~ Y., lnoue, Y., Saito, I. and Miyamura, T. (1992) Expression of pro- cessed envelope protein of hepatitis C virus in mam- malian and insect cells. J. Virol. 66(3), 1425-1431.
Matsuura, Y., Suzuki, T., Suzuki, R., Sato, M., Aizaki, H., Saito, I. and Miyamura, T. (1994) Processing of El and E2 glycoproteins of hepatitis C virus expressed in mam- malian and insect cells. Virology 205(1), 141-150.
Nishihara, T., Nozaki, C., Nakatake, H., Hoshiko, K., Es- umi, M., Hayashi, N., Hino, K., Hamada, F., Mizuno, K. and Shikata, T. (1993) Secretion and purification of hepatitis C virus NSI glycoprotein produced by recombi- nant baculovirus-infected insect cells. Gene 129(2), 207- 214.
Ralston, R., Thudium, K., Berger, K., Kuo, C., Gervase, B., Hall, J., Selby, M., Kuo, G., Houghton, M. and Choo, Q.L. (1993) Characterization of hepatitis C virus envel- ope glycoprotein complexes expressed by recombinant vaccinia viruses. J. Virol. 67(11), 6753-6761.
Saito, 1., Miyamura, T., Ohbayashi, A., Harada, H., Katayama, T., Kikuchi, S., Watanabe, Y., Koi, S., Onji, M. and Ohta, Y. (1990) Hepatitis C virus infection is associated with the development of hepatocellular car- cinoma. Proc. Natl. Acad. Sci. USA 87(17), 6547-6549.
Schlaeger, E.J., Foggetta, M., Vonach, J.M. and Chris- tensen, K. (1993) SF-I, a low cost culture medium for the production of recombinant proteins in baculovirus infected insect cells. Biotechnol. Lett. 7, 183-188.
Schmid, G. (1996) Insect cell cultivation: growth and kinet- ics. In: J.M. Vlak, H.G. Gooijer, H.G. Mittenburger and J. Tramper (Eds.), Insect Cell Culture: Fundamental and Applied Aspects. Kluwer Academic Publishers, Dor- drecht.
Schmid, G., Wild, N., Fountoulakis, M., Gallati, H., Gentz, R. and Ozmen, L.G. (1994) Production and characteriza- tion of soluble mouse and human interferon-gamma re- ceptors from insect cells. In: R.E. Spier, J.B. Griffiths and W. Berthold (Eds.), Animal Cell Technology: Prod- ucts of Today, Prospects for Tomorrow. Butterworth- Heinemann, Oxford, pp. 625-631.
Selby, M.J., Glazer, E., Masiarz, F. and Houghton, M. (1994) Complex processing and protein:protein interac- tions in the E2:NS2 region of HCV. Virology 204, 114- 122.
Spaete, R.R., Alexander, D., Rugroden, M.E., Choo, Q.L., Berger, K., Crawford, K., Kuo, C., Leng, S., Lee, C. and Ralston, R. (1992) Characterization of the hepatitis C virus

P. Hiissy et al. / Virus Research 45 (1996) 4 5 - 5 7 57
E2/NS1 gene product expressed in mammalian cells. Virol- ogy 188(2), 819-830.
Tanaka, E., Kiyosawa, K., Sodeyama, T., Nakano, Y., Yoshizawa, K., Hayata, T., Shimizu, S., Nakatsuji, Y., Koike, Y. and Furuta, S. (1991) Significance of antibody to hepatitis C virus in Japanese patients with viral hepati- tis: relationship between anti-HCV antibody and the prog-
nosis of non-A, non-B post-transfusion hepatitis. J. Med. Virol. 33(2), 117-122.
Tarentino, A.L., Gomez, C.M. and Plummer, T.H. (1985) Deglycosylation of asparagine-linked glycans by peptide: N-glycosidase F. Biochemistry 24(17), 4665-4671.
van Doorn, L.J. (1994) Review: molecular biology of the hepatitis C virus. J. Med. Virol. 43(4), 345 356.




















