Purification and cDNA Cloning of Porcine Brain GDP-L-Fuc:N-Acetyl-beta -D-Glucosaminide alpha...
8
Purification and cDNA Cloning of Porcine Brain GDP-L-Fuc:N-Acetyl-b-D-Glucosaminide a136Fucosyltransferase* (Received for publication, June 12, 1996, and in revised form, August 6, 1996) Naofumi Uozumi‡, Shusaku Yanagidani‡, Eiji Miyoshi‡, Yoshito Ihara‡, Takahiko Sakuma‡, Cong-Xiao Gao‡, Tadashi Teshima§, Shigeru Fujii¶, Tetsuo Shiba§, and Naoyuki Taniguchi‡i From the ‡Department of Biochemistry, Osaka University Medical School, 2-2 Yamadaoka, Suita, Osaka 565, Japan, ¶Laboratory of Chemistry, Kansai Medical University, Hirakata, Osaka 573, Japan, and the §Peptide Institute, Protein Research Foundation, 4-1-2 Ina, Minoh, Osaka 562, Japan GDP-L-Fuc:N-acetyl-b-D-glucosaminide a136fucosyl- transferase (a1– 6FucT; EC 2.4.1.68), which catalyzes the transfer of fucose from GDP-Fuc to N-linked type com- plex glycopeptides, was purified from a Triton X-100 extract of porcine brain microsomes. The purification procedures included sequential affinity chromatogra- phies on GlcNAcb1–2Mana1– 6(GlcNAcb1–2Mana1–2)- Manb1– 4GlcNAcb1– 4GlcNAc-Asn-Sepharose 4B and synthetic GDP-hexanolamine-Sepharose 4B columns. The enzyme was recovered in a 12% final yield with a 440,000-fold increase in specific activity. SDS-polyacryl- amide gel electrophoresis of the purified enzyme gave a major band corresponding to an apparent molecular mass of 58 kDa. The a1– 6FucT has 575 amino acids and no putative N-glycosylation sites. The cDNA was cloned in to pSVK3 and was then transiently transfected into COS-1 cells. a1– 6FucT activity was found to be high in the transfected cells, as compared with non- or mock- transfected cells. Northern blotting analyses of rat adult tissues showed that a1– 6FucT was highly expressed in brain. No sequence homology was found with other pre- viously cloned fucosyltransferases, but the enzyme ap- pears to be a type II transmembrane protein like the other glycosyltransferases. It has been reported that the structures of glycopeptides change during the development and differentiation of embryos (1– 4). Detailed analysis of specific antigens on the surface of various carcinoma cells revealed that carcinoma-specific sugar chains are expressed on the cell surface. A well documented phenotypic alteration of these specific sugar chains is the in- crease in the molecular weight of cell surface complex type N-linked glycan in transformed cells. This change has been observed regardless of the nature of the transforming agent: oncogenic viruses (5–9), chemical mutagens (10 –11), or DNA from unrelated tumor cells (12–14). This phenomenon was thought to reflect the deviation of carcinoma cells from the ordinary differentiation processes. a-Fucose residue attached to asparagine-linked GlcNAc also have some relationship with carcinogenesis. A difference in the binding pattern of serum a-fetoprotein with lentil lectin between hepatocellular carcino- mas and benign liver diseases has been reported (15–17). Anal- yses of the carbohydrate structure of a-fetoprotein from hepa- tocellular carcinoma cell lines have indicated that almost all of the carbohydrates of a-fetoprotein are a1– 6-fucosylated (18). a-Fetoprotein produced by germ cell tumors, such as yolk sac tumors, is also highly fucosylated (19). The activity of a1– 6FucT 1 was higher in hepatocellular carcinoma tissue than in non-tumor tissue (20) and was induced by the transfection of the ras protooncogene into 3T3 fibroblast cells (21). Schachter et al. (22, 23) first characterized a1– 6FucT in porcine liver using a partially purified enzyme extract. The special release of a1– 6FucT from platelets during blood clot- ting has been reported (24, 25), alteration of fucosylation has been reported in cystic fibrosis glycoproteins from different sources, and a1– 6FucT from human fibroblasts of cystic fibro- sis patients has been purified and characterized (26, 27). But little is known about this enzyme, and the a1– 6FucT gene has not yet been cloned. In this study we purified a novel a1– 6FucT, which is a different enzyme from the a1– 6FucTs pre- viously reported, especially in terms of the pH optimum and molecular weight. The cDNA sequence of this enzyme was also determined for the first time. A highly specific assay method involving a fluorescent reagent, 4-(2-pyridylamino)butylamine (PABA), made it possible to purify the enzyme (28). EXPERIMENTAL PROCEDURES 2-Amino pyridine was obtained from Wako Pure Chemicals. b-Ga- lactosidase (Aspergillus sp.) was obtained from Toyobo Co. TSK-gel ODS-80TM and Amide-80 columns were purchased from Tosoh. Bovine g-globulin and Pronase (Streptomyces griseus) were purchased from Sigma and Seikagaku Kogyo Co., respectively. Methods Preparation of Substrate for a1– 6FucT Fluorescence-labeled oligosaccharides were obtained from the corre- sponding oligosaccharides as described previously (28). Briefly, a gly- copeptide derived from bovine g-globulin on digestion with Pronase was coupled with PABA using water-soluble carbodimide. Further purifica- tion was performed by HPLC on a TSK-gel ODS-80TM column (4.6 3 150 mm) that had been equilibrated with 20 mM acetate buffer, pH 4.0, containing 0.1% butanol, at a flow rate of 1 ml/min isocratically. The eluted pyridylamino sugars were detected with a fluorescence spectrophotometer, the excitation and emission wavelengths being 320 and 400 nm, respectively. * This study was supported in part by a grant-in-aid for scientific research on priority areas from the Ministry of Education, Science and Culture of Japan, the Uehara Foundation. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. The nucleotide sequence(s) reported in this paper has been submitted to the GenBank TM /EBI Data Bank with accession number(s) D86723. i To whom correspondence should be addressed: Dept. of Biochemis- try, Osaka University Medical School, 2-2 Yamadaoka Suita, Osaka 565, Japan. Tel.: 81-6-879-3421; Fax: 81-6-879-3429. 1 The abbreviations used are: a1– 6FucT, GDP-L-Fuc:N-acetyl-b-D- glucosaminide a136fucosyltransferase; PABA, 4-(2-pyridylamino)bu- tyl amine; GDP-Fuc, guanosine diphosphofucopyranoside; HPLC, high performance liquid chromatography; GnGn-bi-, GlcNAcb1–2Mana1– 6- (GlcNAcb1–2Mana1–2)Manb1– 4GlcNAcb1– 4GlcNAc-; GnGnF-bi-, GlcNAcb1–2Mana1– 6(GlcNAcb1–2Mana1–2)Manb1– 4GlcNAcb1– 4- (Fuca1– 6)GlcNAc-; MES, 2-(N-morpholino)ethanesulfonic acid; PCR, polymerase chain reaction; PAGE, polyacrylamide gel electrophoresis. THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 271, No. 44, Issue of November 1, pp. 27810 –27817, 1996 © 1996 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A. 27810
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Purification and cDNA Cloning of Porcine Brain GDP-L-Fuc:N-Acetyl-beta -D-Glucosaminide alpha...

Purification and cDNA Cloning of Porcine BrainGDP-L-Fuc:N-Acetyl-b-D-Glucosaminide a136Fucosyltransferase*
(Received for publication, June 12, 1996, and in revised form, August 6, 1996)
Naofumi Uozumi‡, Shusaku Yanagidani‡, Eiji Miyoshi‡, Yoshito Ihara‡, Takahiko Sakuma‡,Cong-Xiao Gao‡, Tadashi Teshima§, Shigeru Fujii¶, Tetsuo Shiba§, and Naoyuki Taniguchi‡i
From the ‡Department of Biochemistry, Osaka University Medical School, 2-2 Yamadaoka, Suita, Osaka 565, Japan,¶Laboratory of Chemistry, Kansai Medical University, Hirakata, Osaka 573, Japan, and the §Peptide Institute, ProteinResearch Foundation, 4-1-2 Ina, Minoh, Osaka 562, Japan
GDP-L-Fuc:N-acetyl-b-D-glucosaminide a136fucosyl-transferase (a1–6FucT; EC 2.4.1.68), which catalyzes thetransfer of fucose from GDP-Fuc to N-linked type com-plex glycopeptides, was purified from a Triton X-100extract of porcine brain microsomes. The purificationprocedures included sequential affinity chromatogra-phies on GlcNAcb1–2Mana1–6(GlcNAcb1–2Mana1–2)-Manb1–4GlcNAcb1–4GlcNAc-Asn-Sepharose 4B andsynthetic GDP-hexanolamine-Sepharose 4B columns.The enzyme was recovered in a 12% final yield with a440,000-fold increase in specific activity. SDS-polyacryl-amide gel electrophoresis of the purified enzyme gave amajor band corresponding to an apparent molecularmass of 58 kDa. The a1–6FucT has 575 amino acids andno putative N-glycosylation sites. The cDNA was clonedin to pSVK3 and was then transiently transfected intoCOS-1 cells. a1–6FucT activity was found to be high inthe transfected cells, as compared with non- or mock-transfected cells. Northern blotting analyses of rat adulttissues showed that a1–6FucT was highly expressed inbrain. No sequence homology was found with other pre-viously cloned fucosyltransferases, but the enzyme ap-pears to be a type II transmembrane protein like theother glycosyltransferases.
It has been reported that the structures of glycopeptideschange during the development and differentiation of embryos(1–4). Detailed analysis of specific antigens on the surface ofvarious carcinoma cells revealed that carcinoma-specific sugarchains are expressed on the cell surface. A well documentedphenotypic alteration of these specific sugar chains is the in-crease in the molecular weight of cell surface complex typeN-linked glycan in transformed cells. This change has beenobserved regardless of the nature of the transforming agent:oncogenic viruses (5–9), chemical mutagens (10–11), or DNAfrom unrelated tumor cells (12–14). This phenomenon wasthought to reflect the deviation of carcinoma cells from theordinary differentiation processes. a-Fucose residue attachedto asparagine-linked GlcNAc also have some relationship withcarcinogenesis. A difference in the binding pattern of serum
a-fetoprotein with lentil lectin between hepatocellular carcino-mas and benign liver diseases has been reported (15–17). Anal-yses of the carbohydrate structure of a-fetoprotein from hepa-tocellular carcinoma cell lines have indicated that almost all ofthe carbohydrates of a-fetoprotein are a1–6-fucosylated (18).a-Fetoprotein produced by germ cell tumors, such as yolk sactumors, is also highly fucosylated (19). The activity of a1–6FucT1 was higher in hepatocellular carcinoma tissue than innon-tumor tissue (20) and was induced by the transfection ofthe ras protooncogene into 3T3 fibroblast cells (21).Schachter et al. (22, 23) first characterized a1–6FucT in
porcine liver using a partially purified enzyme extract. Thespecial release of a1–6FucT from platelets during blood clot-ting has been reported (24, 25), alteration of fucosylation hasbeen reported in cystic fibrosis glycoproteins from differentsources, and a1–6FucT from human fibroblasts of cystic fibro-sis patients has been purified and characterized (26, 27). Butlittle is known about this enzyme, and the a1–6FucT gene hasnot yet been cloned. In this study we purified a novel a1–6FucT, which is a different enzyme from the a1–6FucTs pre-viously reported, especially in terms of the pH optimum andmolecular weight. The cDNA sequence of this enzyme was alsodetermined for the first time. A highly specific assay methodinvolving a fluorescent reagent, 4-(2-pyridylamino)butylamine(PABA), made it possible to purify the enzyme (28).
EXPERIMENTAL PROCEDURES
2-Amino pyridine was obtained from Wako Pure Chemicals. b-Ga-lactosidase (Aspergillus sp.) was obtained from Toyobo Co. TSK-gelODS-80TM and Amide-80 columns were purchased from Tosoh. Bovineg-globulin and Pronase (Streptomyces griseus) were purchased fromSigma and Seikagaku Kogyo Co., respectively.
MethodsPreparation of Substrate for a1–6FucT
Fluorescence-labeled oligosaccharides were obtained from the corre-sponding oligosaccharides as described previously (28). Briefly, a gly-copeptide derived from bovine g-globulin on digestion with Pronase wascoupled with PABA using water-soluble carbodimide. Further purifica-tion was performed by HPLC on a TSK-gel ODS-80TM column (4.6 3150 mm) that had been equilibrated with 20 mM acetate buffer, pH4.0, containing 0.1% butanol, at a flow rate of 1 ml/min isocratically.The eluted pyridylamino sugars were detected with a fluorescencespectrophotometer, the excitation and emission wavelengths being 320and 400 nm, respectively.* This study was supported in part by a grant-in-aid for scientific
research on priority areas from the Ministry of Education, Science andCulture of Japan, the Uehara Foundation. The costs of publication ofthis article were defrayed in part by the payment of page charges. Thisarticle must therefore be hereby marked “advertisement” in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.The nucleotide sequence(s) reported in this paper has been submitted
to the GenBankTM/EBI Data Bank with accession number(s) D86723.i To whom correspondence should be addressed: Dept. of Biochemis-
try, Osaka University Medical School, 2-2 Yamadaoka Suita, Osaka565, Japan. Tel.: 81-6-879-3421; Fax: 81-6-879-3429.
1 The abbreviations used are: a1–6FucT, GDP-L-Fuc:N-acetyl-b-D-glucosaminide a136fucosyltransferase; PABA, 4-(2-pyridylamino)bu-tyl amine; GDP-Fuc, guanosine diphosphofucopyranoside; HPLC, highperformance liquid chromatography; GnGn-bi-, GlcNAcb1–2Mana1–6-(GlcNAcb1–2Mana1–2)Manb1–4GlcNAcb1–4GlcNAc-; GnGnF-bi-,GlcNAcb1–2Mana1–6(GlcNAcb1–2Mana1–2)Manb1–4GlcNAcb1–4-(Fuca1–6)GlcNAc-; MES, 2-(N-morpholino)ethanesulfonic acid; PCR,polymerase chain reaction; PAGE, polyacrylamide gel electrophoresis.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 271, No. 44, Issue of November 1, pp. 27810–27817, 1996© 1996 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
27810

a1–6FucT Assay
The enzyme activity was determined as described (28). In brief, thestandard incubation mixture for the a1–6FucT assay contained thefollowing components, in a final volume of 50 ml: 200 mM MES-NaOHbuffer, pH 7.0, 1% Triton X-100, 50 mM acceptor (GnGn-Asn-PABA), and500 mM donor (GDP-Fuc) substrates. After the mixture had been incu-bated at 37 °C for 2 h, the reaction was stopped by heating at 100 °C for1 min. The sample was centrifuged at 15,000 3 g for 10 min, and then10 ml of the supernatant was used for analyses. The product wasseparated by HPLC on a TSK-gel ODS-80TM column (4.6 3 150 mm).Elution was performed at 55 °C with 20 mM acetate buffer, pH 4.0,containing 0.1% butanol in an isocratic way. Fluorescence of the columneluate was detected with a fluorescence spectrophotometer (model RF535; Shimadzu, Japan), the excitation and emission wavelengths being320 and 400 nm, respectively. The amount of product was estimatedfrom the fluorescence intensity. The specific activity of the enzyme wasexpressed as pmol of fucose transferred/h/mg of protein. Protein wasdetermined with a Bio-Rad protein assay kit using bovine serum albu-min as a standard.
Preparation of GDP-Hexanolamine
GDP-hexanolamine was synthesized by the method of Nunez, et al.(29) with a slight modification. Briefly, GMP was activated with mor-phoridate and then coupled to 6-aminohexylphosphate. The end productwas completely purified on a TSK gel ODS TM80 column (4.6 3 150mm). The structure of the GDP-hexanolamine column was confirmed by1H NMR analyses (data not shown).
Purification and Column Chromatography of a1–6FucT
Step 1—A porcine brain (100 g) was homogenized in 4 volumes of 10mM Tris-HCl, pH 7.4, containing 0.25 M sucrose, 1 mM benzamidinehydrochloride, and 20 mM 4-(amidinophenyl)methanesulfonyl fluoride,with a Polytron homogenizer (Brinkmann Instruments).Step 2—After centrifugation at 900 3 g for 10 min, the resultant
supernatant was centrifuged at 105,000 3 g for 1 h. The pellet wasresuspended in a 1000-ml solution of 20 mM phosphate-KOH, pH 7.0, 50mM KCl, 5 mM EDTA, and 0.5% Triton X-100 buffer and then extractedby gentle stirring for 7 days using a magnetic stirrer, followed bycentrifugation at 105,000 3 g for 1 h. The supernatant was collectedand concentrated to 75 ml using a YM 30 membrane (Amicon).Step 3—The above extract was applied to a column (16 3 5 cm;
Pharmacia HR 16/10) of GnGn-bi-Asn-Sepharose 4B, which had beenequilibrated with the 20 mM phosphate-KOH, pH 7.0, 50 mM KCl, 5 mM
EDTA, and 0.5% Triton X-100 buffer. The column was washed with thesame buffer and then eluted with the washing buffer containing 1 M KCl.Step 4—Enzymatically active fractions obtained from the GnGn-bi-
Asn-Sepharose 4B column were collected, desalted against 20 mM phos-phate-KOH buffer, pH 7.0, containing 50 mM KCl, 5 mM EDTA, and0.5% Triton X-100, using a YM30 membrane (Amicon), and then ap-plied to a GDP-hexanolamine-Sepharose 4B column (16 3 5 cm; Phar-macia HR16/10), which had been equilibrated with 20 mM potassiumphosphate buffer, pH 7.0, containing 50 mM KCl, 5 mM EDTA, and0.05% Triton X-100. After washing with the same buffer sufficiently,
the column-bound enzyme was eluted with 20 mM potassium phosphatebuffer containing 50 mM KCl, 5 mM EDTA, 0.01% Triton X-100, and 100mM GDP. SDS-PAGE was performed to verify the purity of each elutedfraction according to the Laemmli method (30).
Isoelectric Focusing and SDS-PAGE
Isoelectric focusing gel electrophoresis was carried out on a pI 3–9gradient gel (Pharmacia) according to the manufacturer’s instructionsusing 1 mg of the purified enzyme. At the same time, another 1 mg of thepurified enzyme was applied to another lane of the same gel. Afterelectrophoresis, one lane was cut into pieces, and each slice was soni-cated and extracted with the assay mixture. Then a1–6FucT activitywas measured. At the same time, the remaining lane was stained withsilver utilizing Sil-Best Stain (Nacalai Tesque) according to the manu-facturer’s instructions. SDS-PAGE was performed by the method de-scribed by Laemmli (30) in the presence or absence of 2-mercaptoetha-nol. The gels were stained with Coomassie Brilliant Blue G-250 andsilver.
Determination of Partial Amino Acid Sequences of a1–6FucT
To determine the N-terminal amino acid sequences, the materialcorresponding to the 54-kDa band was excised from a polyvinylidenedifluoride membrane (Millipore) and then sequenced with an AppliedBiosystem 473A Protein Sequencer. To determine internal amino acidsequences, 13 mg of the purified protein transferred onto a polyvinyli-dene difluoride membrane was digested with 1 mg of lysyl endopepti-dase in 100 mM Tris and 5% acetonitrile, pH 8.2, at 37 °C for 12 h. Theresultant peptides were separated by reverse phase HPLC on a VydacC-18 column (250 3 2.1 mm), using a 0–60% acetonitrile gradient in0.05% trifluoroacetic acid. Five peptides were obtained and applied to aPolybrene-coated trifluoroacetic acid-activated precycled glass fiber fil-ter for amino acid sequencing and were analyzed with an Appled Bio-systems 473A protein sequencer.
Construction of the cDNA Encoding a1–6FucT cDNA
Total RNA was isolated from a porcine brain according to generalmethods. Poly(A)1 was further purified on an oligo(dT)-cellulose (Phar-macia) column. The first strand cDNA was synthesized with a cDNAsynthesis kit (Life Technologies, Inc.), using primer A11, according tothe manufacturer’s manual (see Fig. 8).
Polymerase Chain Reaction (PCR)
Four oligonucleotides were synthesized for use as primers in PCR(see Fig. 7). PCR was carried out in 50-ml reaction mixtures containingfirst strand cDNA corresponding to 1 mg of poly(A)1 RNA, 50 pmol of apair of degenerate oligonucleotides, 50 mM KCl, 10 mM Tris-HCl, pH8.3, 1.5 mM MgCl2, 0.001% gelatin, and 200 mM dNTP. After a hot startat 98 °C, 25 cycles (94 °C for 1 min, 55 °C for 2 min, 72 °C for 3 min) ofPCR were performed using 2.5 units of Thermus aquaticus (Taq) po-lymerase. The PCR products were subcloned into a pT7BlueT-Vector(Novagen).
Isolation and DNA Sequencing of Porcine a1–6FucT cDNA
A porcine brain lgt11 cDNA library (Clonetech) was screened byplaque hybridization utilizing the [a-32P]dCTP (3,000 Ci/mmol; Amer-sham) of a labeled reverse transcription-PCR product, S-A12 (see Fig.8). Five positive plaques were obtained from 4 3 105 plaques in the firstscreening. The screening was repeated 3 times, and then the inserts ofthe five isolated clones were amplified using forward (GGTGGCGAC-GACTCCTGGAGCCCG) and reversal (CGTGTATTCCTCAATGGGAT-GGAA) primers by means of PCR reaction, and the DNA sequence wasdirectly determined with a DNA Sequencing Kit of Dye TerminatorCycle Sequencing Ready Reaction (Perkin-Elmer) using forward andreversal primers as sequencing primers. Based on the determined in-sert cDNA sequence, sequencing primers were synthesized, and thespace between each sequencing primer was 200 nucleotides. Finally thefull length of the insert cDNA sequence was determined.
TABLE IDistribution of a1–6FucT in normal rat tissues
The values in parentheses indicate the number of specimens exam-ined. Each value represents the mean 6 S.D.
Tissues Specific activity
pmol/h/mg protein
Brain (3) 733.0 6 42.0Spleen (3) 192.0 6 21.0Testis (3) 149.0 6 4.7
Stomach (2) 59.4 6 9.4Lung (3) 45.3 6 9.1Kidney (3) 21.4 6 3.1
TABLE IIPurification of a1–6FucT of porcine brain
Steps Volume Protein Total activity Specific activity Purification Yield
ml mg nmol/h nmol/h/mg protein -fold %
Triton X-100 extract 75 3,680 2,220 0.6 1 100GnGn-bi-Asn-Sepharose 4B 10 0.80 1,350 1,688 2,813 61GDP- (CH2)6-NH2-Sepharose 4B 0.2 0.01 262 262,200 437,000 12
Purification and cDNA Cloning of a1–6Fucosyltransferase 27811
Transient Expression of a1–6FucT in COS-1 Cells
The coding region of cDNA of a1–6FucT was amplified using forward(ggaattccGAGTTGAAAGTCTGAAAATGCGG) and reversal (tccccc-gggggaCTTTCTCATCTGTCCGTC) primers by means of PCR reaction.The obtained PCR product was double digested by SmaI and EcoRI andsubcloned into calf intestinal alkaline phosphatase-treated pSVK3,which was previously double digested by SmaI and EcoRI. pSVK3 wasthen transiently transfected into COS-1 cells by electroporation(Bio-Rad).
Northern Blotting Analyses of a1–6FucT mRNA inRat Brain
Total RNA was prepared from rat cerebrum and cerebellum accord-ing to general methods. 20 mg of total RNA was chromatographed on a0.8% agarose gel containing 2.2 M formaldehyde. The size-fractionatedRNA was transferred to a z-Probe membrane (Bio-Rad) by capillaryaction. After hybridization with 32P-labeled S-A12 fragment (see Fig. 9)at 42 °C, the membrane was washed at 55 °C in 2 3 SSC (1 3 SSC: 15mM sodium citrate and 150 mM NaCl, pH 7.0) containing 0.1% SDS. TheKodak x-ray film was exposed for 3 days at 280 °C.
RESULTS
Purification and Column Chromatography of a1–6FucT
Steps 1 and 2—A porcine brain was chosen as a source of theenzyme for purification based on the fact that porcine and rat
brains are the most abundant sources of the enzyme (Table I).Step 3—The first chromatographic step of the purification
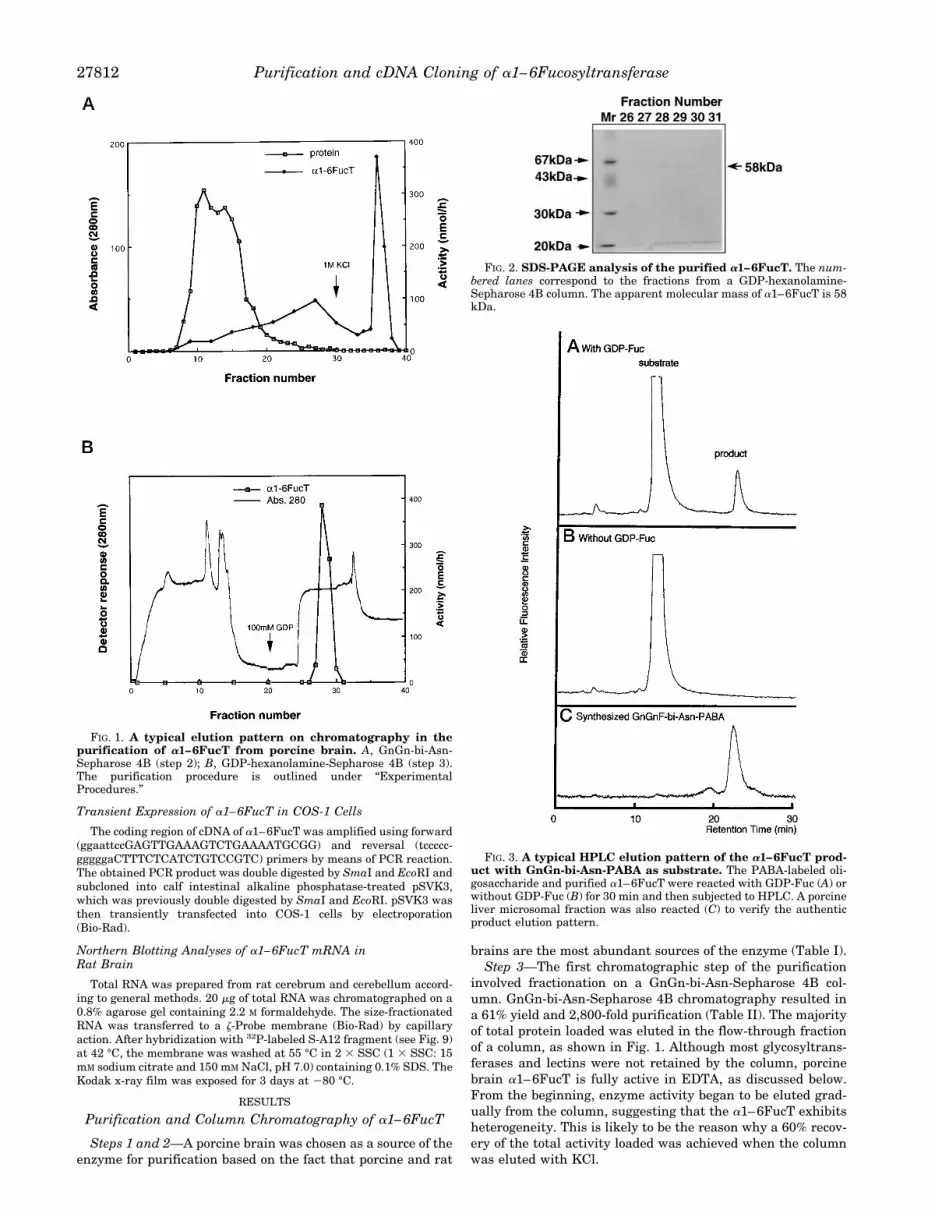
involved fractionation on a GnGn-bi-Asn-Sepharose 4B col-umn. GnGn-bi-Asn-Sepharose 4B chromatography resulted ina 61% yield and 2,800-fold purification (Table II). The majorityof total protein loaded was eluted in the flow-through fractionof a column, as shown in Fig. 1. Although most glycosyltrans-ferases and lectins were not retained by the column, porcinebrain a1–6FucT is fully active in EDTA, as discussed below.From the beginning, enzyme activity began to be eluted grad-ually from the column, suggesting that the a1–6FucT exhibitsheterogeneity. This is likely to be the reason why a 60% recov-ery of the total activity loaded was achieved when the columnwas eluted with KCl.
FIG. 1. A typical elution pattern on chromatography in thepurification of a1–6FucT from porcine brain. A, GnGn-bi-Asn-Sepharose 4B (step 2); B, GDP-hexanolamine-Sepharose 4B (step 3).The purification procedure is outlined under “ExperimentalProcedures.”
FIG. 2. SDS-PAGE analysis of the purified a1–6FucT. The num-bered lanes correspond to the fractions from a GDP-hexanolamine-Sepharose 4B column. The apparent molecular mass of a1–6FucT is 58kDa.
FIG. 3. A typical HPLC elution pattern of the a1–6FucT prod-uct with GnGn-bi-Asn-PABA as substrate. The PABA-labeled oli-gosaccharide and purified a1–6FucT were reacted with GDP-Fuc (A) orwithout GDP-Fuc (B) for 30 min and then subjected to HPLC. A porcineliver microsomal fraction was also reacted (C) to verify the authenticproduct elution pattern.
Purification and cDNA Cloning of a1–6Fucosyltransferase27812
Step 4—After the buffer had been changed using an AmiconYM30, the final purification step was accomplished on a syn-thetic GDP-hexanolamine ligand column. Chromatography onan immobilized GDP-hexanolamine column resulted in an ad-ditional 155-fold purification with a 19.7% yield compared withthe previous step. The elution profile from this column clearlyshowed that the majority of the contaminating protein was notbound to the column and that the enzyme was eluted from thiscolumn with GDP.Starting with frozen porcine brain (100 g), a1–6FucT was
purified 440,000-fold with a total yield of 12%, as summarizedin Table II.
Purity and Enzymatic Properties of a1–6FucT
In order to assess the level of purification of a1–6FucT,fractions eluted from a GDP-hexanolamine column were sub-jected to SDS-PAGE. A photograph of this gel, which wasstained with Coomassie Brilliant Blue G-250 is shown in Fig. 2.The most purified sample in lanes 28 and 29 gave one majorband corresponding to an apparent mass of 58 kDa (indicatedby an arrow on the right).The enzymatic profiles of a1–6FucT of porcine brain were
obtained utilizing the purified protein. The purified enzymewas incubated by the enzyme assay method, and an aliquot wassubjected to HPLC. A typical elution pattern is shown in Fig. 3.The substrate and product were eluted at 12.2 and 22.6 min,respectively. The product exhibits the same elution pattern asthe a1–6FucT of porcine liver (28). The reaction mixture with-out GDP-Fuc was also subjected to HPLC. Compared with theelution pattern of the reaction mixture with GDP-Fuc, no en-zymatic product was detected in the absence of GDP-Fuc in thereaction mixture. To confirm the structure of the product, thereaction was performed on a large scale, and the enzymaticproduct was analyzed with an ion spray mass spectrophotom-eter. The substrate, GnGn-bi-Asn-PABA, has a molecularweight of 1576. In contrast, the enzymatic product has one of1725. This indicated that this a1–6FucT catalyzes the transferof 1 mol of fucose to 1 mol of GnGn-bi-Asn-PABA (data notshown). The chemical shift value of the enzymatic product on1H NMR analysis was the same as synthesized GnGnF-bi-Asn-PABA, which had previously been published (28). The enzy-matic product gave the two typical signals of fucose at d 5 4.864ppm (H-1) and d 5 1.193 ppm (CH3) (Table III, boldface type).The purified enzyme was incubated under various condi-
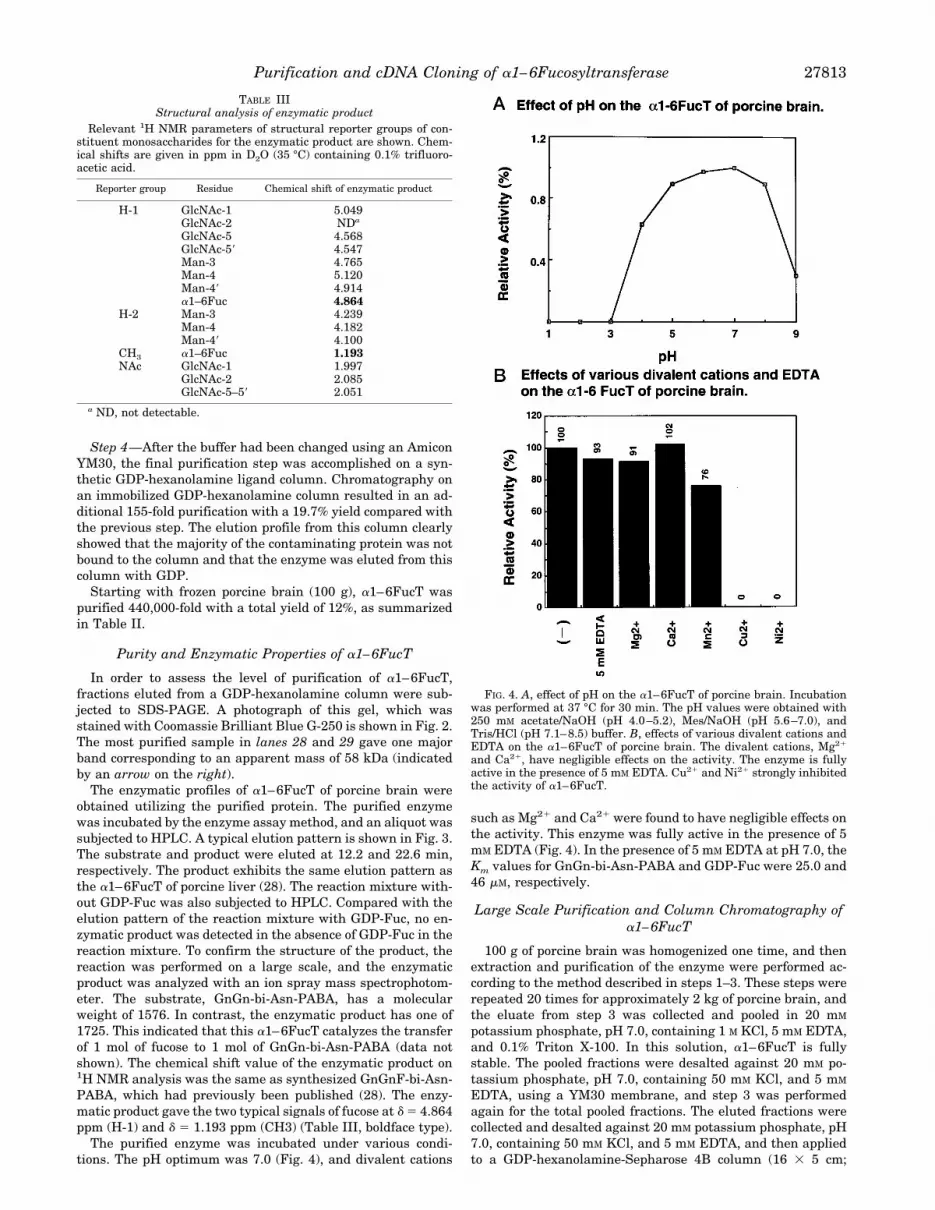
tions. The pH optimum was 7.0 (Fig. 4), and divalent cations
such as Mg21 and Ca21 were found to have negligible effects onthe activity. This enzyme was fully active in the presence of 5mM EDTA (Fig. 4). In the presence of 5 mM EDTA at pH 7.0, theKm values for GnGn-bi-Asn-PABA and GDP-Fuc were 25.0 and46 mM, respectively.
Large Scale Purification and Column Chromatography ofa1–6FucT
100 g of porcine brain was homogenized one time, and thenextraction and purification of the enzyme were performed ac-cording to the method described in steps 1–3. These steps wererepeated 20 times for approximately 2 kg of porcine brain, andthe eluate from step 3 was collected and pooled in 20 mM
potassium phosphate, pH 7.0, containing 1 M KCl, 5 mM EDTA,and 0.1% Triton X-100. In this solution, a1–6FucT is fullystable. The pooled fractions were desalted against 20 mM po-tassium phosphate, pH 7.0, containing 50 mM KCl, and 5 mM
EDTA, using a YM30 membrane, and step 3 was performedagain for the total pooled fractions. The eluted fractions werecollected and desalted against 20 mM potassium phosphate, pH7.0, containing 50 mM KCl, and 5 mM EDTA, and then appliedto a GDP-hexanolamine-Sepharose 4B column (16 3 5 cm;
FIG. 4. A, effect of pH on the a1–6FucT of porcine brain. Incubationwas performed at 37 °C for 30 min. The pH values were obtained with250 mM acetate/NaOH (pH 4.0–5.2), Mes/NaOH (pH 5.6–7.0), andTris/HCl (pH 7.1–8.5) buffer. B, effects of various divalent cations andEDTA on the a1–6FucT of porcine brain. The divalent cations, Mg21
and Ca21, have negligible effects on the activity. The enzyme is fullyactive in the presence of 5 mM EDTA. Cu21 and Ni21 strongly inhibitedthe activity of a1–6FucT.
TABLE IIIStructural analysis of enzymatic product
Relevant 1H NMR parameters of structural reporter groups of con-stituent monosaccharides for the enzymatic product are shown. Chem-ical shifts are given in ppm in D2O (35 °C) containing 0.1% trifluoro-acetic acid.
Reporter group Residue Chemical shift of enzymatic product
H-1 GlcNAc-1 5.049GlcNAc-2 NDa
GlcNAc-5 4.568GlcNAc-59 4.547Man-3 4.765Man-4 5.120Man-49 4.914a1–6Fuc 4.864
H-2 Man-3 4.239Man-4 4.182Man-49 4.100
CH3 a1–6Fuc 1.193NAc GlcNAc-1 1.997
GlcNAc-2 2.085GlcNAc-5–59 2.051
a ND, not detectable.
Purification and cDNA Cloning of a1–6Fucosyltransferase 27813

Pharmacia HR16/10), which had been equilibrated with 20 mM
potassium phosphate buffer containing 50 mM KCl, 5 mM
EDTA, and 0.05% Triton X-100. After the column had beenwashed with the same buffer sufficiently, enzymes were elutedwith 20 mM potassium phosphate buffer containing 50 mM KCl,5 mM EDTA, 0.01% Triton X-100, and 5 mM GDP. The elutedfractions were collected and used for further experiments.
SDS-PAGE and Isoelectric Focusing of Purified a1–6FucT
The most purified fraction obtained on a GDP-hexanolamineaffinity column migrates as one major component, 58 kDa, onSDS-PAGE under nonreducing conditions, and under reducingconditions the molecular mass of this component changes from58 to 54 kDa (Fig. 5). The same component gave a diffuse bandon isoelectric focusing at pI 7.0, and a1–6FucT activity wasdetected in the gel slices corresponding to this band (Fig. 6).
cDNA Cloning of a1–6FucT
The N-terminal sequence determined from a component of 54kDa on SDS-PAGE was used to design sense strand PCR prim-ers. The internal peptide sequence was determined from fourlysyl endopeptidase-derived peptides (Fig. 7) and was used todesign several antisense strand PCR primers. The five tran-script-specific oligonucleotides obtained were used to amplify acDNA that encodes the a1–6FucT (Fig. 7). Agarose gel electro-phoresis of the PCR products demonstrated that antisenseprimer A12 produced a 1.45-kilobase pair DNA fragment (Fig.8). The PCR product, S-A12, was then subcloned into pT7Blue-Vector (Novagen), and the full-length sequence was de-termined (data not shown). From a lgt11 cDNA library ofporcine brain (Clonetech), cDNA encoding a1–6FucT was also
isolated, utilizing the PCR product as a probe. Five positiveclones, C1, C2, C3, C4, and C5, were obtained as described inFig. 8. Clones C1 and C2 appeared to contain the full-lengtha1–6FucT open reading frame, and the nucleotide sequencewas determined from these two clones. The DNA sequencedetermined from the PCR product, S-A12, was completely iden-tical to that of cDNA clone isolated from the lgt11 cDNAlibrary.The translated sequence of a1–6FucT is shown in Fig. 9A.
The 1,728-base pair open reading frame encodes a 575-aminoacid polypeptide. The deduced amino acid composition wasidentical to that of the purified enzyme determined by aminoacid analysis. Both the N-terminal and internal peptide aminoacid sequences are included within the predicted sequence, asunderlined in Fig. 9A.
Transient Expression of a1–6FucT in COS-1 Cells
To verify that the cloned cDNA encodes a1–6FucT transfer-ase, the coding region of cDNA was subcloned into the mam-malian expression vector, pSVK3, and then the vector wastransfected into COS-1 cells. After 48 h, the cells were har-vested, and a1–6FucT activity was measured in the homoge-nate. Preliminary experiments indicated that untransfectedCOS-1 cells show very little enzyme activity. COS-1 cells trans-fected with an expression vector containing a1–6FucT showedhigh activity, i.e. 2,360 pmol/h/mg of protein, compared with
FIG. 6. Isoelectric focusing of the purified a1–6FucT. a1–6FucTgave a diffuse band corresponding to pI 7.0. The major a1–6FucTactivity was localized at this band.
FIG. 7. Partial amino acid sequences of lysyl endopeptidase-derived peptides and oligonucleotide sequences based on thepeptide sequences. In A, the underlined sections of the peptide se-quences were used for the design of oligonucleotide primers. In B,oligonucleotides S, A1 (A1–1 and A1–2), A2, A3, and A4 were based onN, L1, L2, L3, and L4, respectively.
FIG. 8. Schematic structure and restriction map of the a1–6FucT clone. The PCR product, S-A12, was obtained by amplificationof porcine brain first strand cDNA. Five clones, C1, C2, C3, C4, and C5,were obtained on screening of a porcine cDNA library using reversetranscription-PCR product as probes.
FIG. 5. Polyacrylamide gel electrophoresis of the purified a1–6FucT. SDS-PAGE on a 10% gel is shown under the reducing condi-tions (A) and nonreducing conditions (B). a1–6FucT was stained withCoomassie Brilliant Blue G-250 (A) and silver (B). The a1–6FucT isindicated by an arrow.
Purification and cDNA Cloning of a1–6Fucosyltransferase27814

the cells transfected with the control plasmid, mock pSVK3(Table IV).
Northern Blotting Analysis of Total RNA from Rat Brain
Northern blotting analysis of total RNA from rat brainshowed a band at 3.5 kilobases (Fig. 10). Brain showed one ofthe highest activities among various tissues, and the content ofa1–6FucT mRNA of brain was also high.
DISCUSSION
We have reported the purification of N-acetylglucosaminyl-transferases III and V (31, 32) and employed donor and accep-tor substrates for the sequential affinity chromatographies. Inthis paper we found that these chromatographies using donorand acceptor substrates as ligand were fairly effective for pu-rification of a1–6FucT from porcine brain. Electrophoresis ofthe purified a1–6FucT gave a diffuse single band on the iso-electric focusing at pI 7.0 (Fig. 6). Based on the deduced aminoacid composition from the cDNA sequence, the pI is 7.39. Noputative N-glycosylation sites were found like several glycosyl-transferases (33). On SDS-PAGE, a major band of 58 kDaappeared under nonreducing conditions. On reduction, how-ever, the molecular mass changed to 54 kDa. Because of thelimited material available for our study, the mechanism bywhich this change occurs is still unknown.Weinstein et al. (34) reported that the amino terminus of
glycosyltransferase was subjected to proteolytic cleavage due toan endogenous protease (32). According to the results of aminoacid analysis, the cleavage site is very strict in the case of theN terminus of a1–6FucT as in the case of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase (35). It seemsthat the enzyme was cleaved by an endogenous protease duringthe extraction procedure.No significant homology was found with previously cloned
aFucTs (36). But this enzyme has a domain structure similar tothose of other glycosyltransferases (33, 37); a1–6FucT also hasa short amino-terminal cytoplasmic tail, a 16-amino acid trans-membrane sequence, a “stem” region, and a long C-terminalcatalytic part, which is in the Golgi lumen (38). This enzymehas a proline-rich domain, 10 proline residues being foundamong residues 261–340 (31, 39). Based on the hydropathyanalysis, a hydrophobic region of 16 amino acids precedes theamino acid terminus and is likely to be the transmembraneregion of this enzyme (Fig. 9B).
a1–6FucT from a human fibroblast cell line required theMg21 cation and did not bind to a GDP-hexanolamine column(27). However, a1–6FucT of human serum required no cationsfor its activity and bound tightly to a GDP-hexanolamine col-umn (28). a1–6FucT in porcine liver has an optimum pH of 5.6(23, 28). In contrast, a1–6FucT in porcine brain has an opti-FIG. 9. A, nucleotide amino acid sequence of porcine a1–6FucT. Sin-
gle-letter codes are used for amino acids. The sequences of lysyl en-dopeptidase-derived peptides are underlinedwith single solid lines. Theproposed transmembrane region is underlined with a double line. B,hydropathic plot of a1–6FucT according to the method of Kyte andDoolittle (58). Various regions are identified at the bottom. T, trans-
TABLE IVExpression of porcine a1–6FucT in COS-1 cells
The a1–6FucT activities of transfected cell homogenates were deter-mined. The values in parentheses indicate the amount of plasmidused for transfection. The results are averages of the values for threeexperiments.
Conditions Activities
pmol/h/mg protein
No plasmid 13pSVK3 (10 mg) 18a1–6FucT cDNA (20 mg) 1,789a1–6FucT cDNA (40 mg) 2,360
membrane region; S, stem region; P, proline-rich region; C, catalyticdomain.
Purification and cDNA Cloning of a1–6Fucosyltransferase 27815

mum pH 7.0 (Fig. 4). Lin, A. I. et al. (40) have reported a novela1–6FucT that can catalyze the transfer of fucose to highmannose type oligosaccharide. And these results suggest thatat least two kinds of a1–6FucTs may exist and that a1–6FucTgene families may exist as observed in the b1–6GnT, a1–3FucT, and sialyltransferase gene families (41–43).Although fucose attached at the nonreducing end of GlcNAc
has been well analyzed and the interaction between the sialylLewis X and selectin families plays a significant role in themigration and homing of hematopoetic cells, little is knownabout the function of a1–6fucose attached to the reducing endof GlcNAc. In plants, the sugar chains of glycoproteins play animportant role in cell-cell recognition signals for multicellularorgans, and lectins are their partners in this event. Legumelectins serve as mediators for the synthetic interactions be-tween plants and nitrogen-fixing microorganisms, an impor-tant process in the nitrogen cycle (44), and a1–6fucose linked toN-acetylglucosamine (GlcNAc-1) greatly enhances the reactiv-ity of legume lectins of the Viciae tribe toward N-linked typeoligosaccharides (45). It is supposed that a1–6-linked fucose,which is catalyzed by a1–6FucT, may play an important role inthe regulation of intracellular interactions. In animal tissues, ithas been reported that fucose-containing glycoproteins, such aslactoferrin, are cleared rapidly from the blood into the liver(46), and recently mannose/L-fucose-mediated clearance wasalso proved in alligator liver (47). And it is generally knownthat a fucose/mannose receptor exists on the surface of macro-phages, which is different from asialoglycoprotein receptors,and mediates phagocytosis (48). Recently, there is speculationthat MRF plays a role in protection from parasites or fungi (49,50), in addition to its scavenging activity toward aging neutro-phils in inflammation (51).
a1–6FucT catalyzes the transfer of fucose to asparagine-linked GlcNAc in an early step of the processing of N-linkedcomplex type glycopeptides. Since N-linked glycosylation pro-ceeds in a stepwise manner, it is supposed that the addition offucose to asparagine-linked GlcNAc determines the posttrans-lational processing and the function of a glycopeptide (52–54).Recently, mannose-binding mammalian lectins, such as VIP36and ERGIC-53 (MR 60), were found to exhibit homology tolegume lectins and were supposed to regulate the secretion ofN-glycans (55–57). a1–6FucT was presumed to be localized inthe medial Golgi apparatus, and there is the possibility thatattachment of a-fucose to asparagine-linked GlcNAc regu-lates the interaction between intrinsic lectins and N-linkedglycoproteins. On the other hand, the commercial endo-b-N-acetylglucosaminidase F peptidase N-glycosidase/glycanylamidase (PNGase) mixture readily cleaves high mannose andoligosaccharides with a common core a1–6-linked fucose foundin thyroglobulin; however, the same endo-b-N-acetylglu-cosaminidase F mixture does not cleave the nonfucosylated
complex oligosaccharide found in human transferrin (44). a-Fu-cose is presumed to play a role in N-glycan cycle and to deter-mine the function of glycoproteins.
Acknowledgments—We thank Dr. Rujun Kang for valuable discus-sion of cell culture experiments, and Dr. Katsuhisa Noda for prepara-tion of sugar substrate. We also thank Drs. Higashiyama S., NishikawaA., Sakamoto Y., and Ozeki Y. for valuable discussion and criticism ofthis work. We also thank Dr. Kurosawa T. for technical advice.
REFERENCES
1. Solter, D., and Knowles, B. B. (1979) Proc. Natl. Acad. Sci. U. S. A. 75,5565–5569
2. Kapadia, A., Feizi, T., and Evans, M. J. (1981) Exp. Cell. Res. 131, 185–1953. Fenderson, B. A., Holmes, E. H., Fukushi, Y., and Hakomori, S. (1986) Dev.
Biol. 114, 12–214. Cummings, R. D., and Mattox, S. A. (1988) J. Biol. Chem. 263, 511–5195. Yamashita, K., Ohkura, T., Tachibana, Y., Takasaki, S., and Kobata, A. (1984)
J. Biol. Chem. 259, 10834–108406. Yamashita, K., Tachibana, Y., Ohkura, T., and Kobata, A. (1985) J. Biol.
Chem. 260, 3963–39697. Pierce, M., and Arango, J. (1986) J. Biol. Chem. 261, 10772–107778. Hubbard, S. C. (1987) J. Biol. Chem. 262, 16403–164119. Santer, U. V., and Glick, M. C. (1987) Biochemistry 18, 2533–254010. Van Beek, W. P., Emmelot, P., and Homburg, C. (1977) Br. J. Cancer 36,
157–16511. Bolscher, J. G. M., Schalier, D. C. C., Smets, L. A., Van Rooji, H., Collard, J. G.,
Bruyneel, E. A., and Mareel, M. M. K. (1986) Cancer Res. 46, 4080–408612. Santer, U. V., and Gilbert, F., and Glick, M. C. (1984) Cancer Res. 44,
3730–373513. Bolscher, J. G. M., Van der Bijl, M. M. W., Neefjes, J. J., Hall, A., Smets, L. A.,
and Ploegh, H. L. (1988) EMBO J. 7, 3361–336914. Miyoshi, E., Nishikawa, A., Ihara, Y., Saito, H., Uozumi, N., Hayashi, N.,
Fusamoto, H., Kamada, T., and Taniguchi, N. (1995) J. Biol. Chem. 270,6216–6220
15. Aoyagi, Y., Isemura, M., Yoshizawa, Z., Suzuki, Y., Sekine, C., Ono, T., andIchida, F. (1985) Biochim. Biophys. Acta. 830, 217–223
16. Taketa, K. (1990) Hepatology 12, 1420–143217. Taketa, K., Sekiya, C., Namiki, M., Akamatu, K., Ohta, Y., Endo, Y., and
Kosaka, K. (1990) Gastroenterology 99, 508–51818. Ohno, N., Nishikawa, A., Kouketsu, M., Taga, H., Endo, Y., Hada, T.,
Higashino, K., and Taniguchi, N. (1992) Int. J. Cancer 51, 315–31719. Aoyagi, Y., Suzuki, Y., Igarashi, K., Yokota, T., Mori, S., Suda, T., Naitou, A.,
Isemura, M., and Asakura, H. (1993) Cancer 72, 615–61820. Huchinson, W. L., Du, M.-Q., Johnson, P. J., and Williams, R. (1991) Hepatol-
ogy 13, 683–68821. Easton, E. W., Bolscher, J. G. M., and van den Eijnden, D. H. (1991) J. Biol.
Chem. 266, 21674–2168022. Wilson, J. R., Williams, D., and Schachter, H. (1976) Biochem. Biophys. Res.
Commun. 72, 909–91623. Longmore, G. D., and Schachter, H. (1982) Carbohydr. Res. 100, 365–39224. Koscielak, J., Pacuszka, T., Kubin, J., and Zdziechowska, H. (1987) Glycocon-
jugate J. 4, 43–4925. Koscielak, J., Antoniewicz-Papis, J., Zdebska, E., Maj, S., and Leszko, B.
(1995) Acta. Biochimica Polonica 42, 35–4026. Voynow, J. A., Scanlin, T. F., and Glick, M. C. (1988) Anal. Biochem. 169,
367–37327. Voynow, J. A., Kaiser, R. S., Scanlin, T. F., and Glick, M. C. (1991) J. Biol.
Chem. 266, 21572–2157728. Uozumi, N., Teshima, T., Yamamoto, T., Nishikawa, A., Miyoshi, E., Islam, K.
N., Fujii, S., Shiba, T., and Taniguchi, N. (1996) J. Biochem. (Tokyo) 120,385–392
29. Nunez, H. A., O’Connor, J. V., Rosevear, P. R., and Barker, R. (1981) Can. J.Chem. 59, 2086–2095
30. Laemmli, U. K. (1970) Nature 227, 680–68531. Nishikawa, A., Ihara, Y., Hatakeyama, M., Kangawa, K., and Taniguchi, N.
(1992) J. Biol. Chem. 267, 18199–1820432. Gu, J., Nishikawa, A., Tsuruoka, N., Ohno, M., Yamaguchi, N., Kangawa, K.,
and Taniguchi, N. (1993) J. Biochem. (Tokyo) 113, 614–61933. Taniguchi, N., and Ihara., Y. (1995) Glycoconjugate J. 12, 733–73834. Weinstein, J., Lee, E. U., McEntee, K., Lai, P.-H., and Paulson, J. C. (1987)
J. Biol. Chem. 262, 17735–1774335. Hagen, F. K., VanWuyckhuyse, B., and Tabak, L. (1993) J. Biol. Chem. 268,
18960–1896536. Natsuka, S., Gersten, K. M., Zenita, K., Kannagi, R., and Lowe, J. B. (1994)
J. Biol. Chem. 269, 16789–1679437. Field, M. C., and Wainwrite, L. J. (1995) Glycobiology 5, 463–47238. Paulson J. C., and Colley, K. J. (1989) J. Biol. Chem. 264, 17615–1761839. Saito, H., Nishikawa, A., Gu, J., Ihara, Y., Soejima, H., Wada, Y., Sekiya, C.,
Niikawa, N., and Taniguchi, N. (1994) Biochem. Biophys. Res. Commun.198, 318–327
40. Lin, A. I., Philipsberg, G. A., and Haltiwanger, R. S. (1994) Glycobiology 4,895–901
41. Bierhuizen, M. F. A., Mattei, M.-G., and Fukuda, M. (1993) Genes & Dev. 7,468–478
42. Weston, B. W., Smith, P. L., Kelly, R. J., and Lowe, J. B. (1992) J. Biol. Chem.267, 24575–24584
43. Wen, D. X., Livingston, B. D., Medzihradsky, K. F., Kelm, S., Burlingame, A.L., and Paulson, J. C. (1992) J. Biol. Chem. 267, 21011–21019
44. Bourne, Y., Mazurier, J., Legrand, D., Pierre, R. Montreuil, J., Spik, G., andCambillau, C. (1994) Structure 2, 209–219
FIG. 10. Northern blotting analyses of total RNA form ratbrain. Total RNA from rat brain showed a faint band at 3.5 kilobases(upper panel). Ethidium bromide staining shows a comparable amountof RNA in each lane (lower panel)
Purification and cDNA Cloning of a1–6Fucosyltransferase27816

45. Kornfeld, K., Reitman, M. L., and Kornfeld, R. (1981) J. Biol. Chem. 256,6633–6640
46. Lehrman, M. A., Pizzo, S. V., Imber, M. J., and Hill, R. L. (1986) J. Biol. Chem.261, 7412–7418
47. Lee., R. T., Yang, G. C., Kiang, J. Bingham, J. B., Golgher, D., and Lee, Y. C.(1994) J. Biol. Chem. 269, 19617–19625
48. Stahl, P. D., and Gordon, S. (1982) J. Cell. Biol. 93, 49–5649. Palatnic de Sousa, C. B., Dutra, H. S., and Borojevic, R. (1993) Acta. Trop. 53,
59–7250. Guy, R. A., and Belosevic, M. (1993) Infect. Immun. 61, 1553–155851. Hall., S. E., Savill, J. S., Henson, P. M., and Haslett, C. (1994) J. Immunol.
153, 3218–322752. Hubbard, S. C., and Ivatt, R. J. (1981) Annu. Rev. Biochem. 50, 555–58353. Schachter, H., Narasimhan, S., Gleeson, P., and Vella, G. (1983) Can. J. Bio-
chem. Cell. Biol. 61, 1049–106654. Kornfeld, R., and Kornfeld, S. (1985) Annu. Rev. Biochem. 54, 631–66455. Shinler, R., Itin, C., Zerial, M., Lottspeich., F., and Hauri, H. P. (1993) Eur. J.
Cell Biol. 61, 1–956. Arar, C., Carpentier, V., Le Caer, J.-P., Monsigny, M., Legrand, A., and Roche,
A.-C. (1995) J. Biol. Chem. 270, 3551–355357. Scheiffele, P., Peranen, J., and Simons, K. (1995) Nature 378, 96–9858. Kyte, J., and Doolittle, R. F. (1982) J. Mol. Biol. 157, 105–132
Purification and cDNA Cloning of a1–6Fucosyltransferase 27817










