
Protective effect of the heparin-derived oligosaccharide C3, on AF64A-induced cholinergic lesion in...
10
Neurobiology of Aging 24 (2003) 481–490 Protective effect of the heparin-derived oligosaccharide C3, on AF64A-induced cholinergic lesion in rats M. Rose, B. Dudas, U. Cornelli, I. Hanin ∗ Department of Pharmacology, Loyola University Chicago Stritch School of Medicine, 2160 South First Avenue, Maywood, IL 60153, USA Received 14 March 2002; received in revised form 10 June 2002; accepted 5 July 2002 Abstract Alzheimer’s disease (AD) literature indicates that glycosaminoglycans (GAGs) may prevent proteoglycan-induced amyloid- (A) aggregation, decrease A-induced tau-2 immunoreactivity, and increase the axonal growth and arborization of hippocampal neurons. However, there is no information about the impact of GAGs on cholinergic lesions. Here, AF64A was administered stereotaxically into the lateral ventricles of rats, at doses that are selective for cholinotoxicity (1 and 2 nmol). The heparin-derived oligosaccharide (HDO), C3 (25 mg/kg), was administered orally, once daily for 7 days before, and 7 days after AF64A administration. Choline acetyltransferase (ChAT) immunohistochemistry revealed that C3 administration reduced AF64A-induced cholinergic damage in the septum and cingulum bundle. Quantitative neuronal cell counts showed that C3 attenuated, by 60%, the decrease in cell number in the medial septum. Enzyme analysis showed that C3 also significantly restored ChAT (30%) and acetylcholinesterase (AChE) enzyme activity (45%), which had been diminished by AF64A. Our data suggest that, in addition to its effects of anti-A aggregation, anti-A-induced tau-2 immunoreactivity, and neurotrophic effects, C3 also effectively reduces AF64A-induced cholinergic damage; hence it may have potential therapeutic value in AD patients. © 2002 Elsevier Science Inc. All rights reserved. Keywords: Heparin-derived oligosaccharide; Glycosaminoglycan; C3; AF64A; Septum; Cingulum bundle; Cholinergic lesion 1. Introduction Classical histopathological hallmarks of Alzheimer’s dis- ease (AD) include amyloid plaques containing fragments of amyloid- (A) peptide [21,40,43,51–53] and neurofibril- lary tangles accumulating hyperphosphorylated tau protein [29,63]. In addition to these histological changes, AD is characterized by a profound deficiency in the central cholin- ergic system [1,11]. Although the pathogenesis of AD is not entirely known, several studies suggest that these lesions are functionally related to each other [17,23,45,46]. Proteoglycans have a high affinity for A and protect it against proteolysis [4,24,55]. There is a general consen- sus that this effect is antagonized by glycosaminoglycans (GAGs) [32,56,57]. Stereotaxic injection of A fragments into the amygdala of rats has been shown to increase tau-2 immunoreactivity in the hippocampus [54] and this phe- nomenon can be attenuated by administration of GAGs [8,14,62]. Furthermore, GAGs and other sulfated com- pounds inhibit the aggregation and toxicity of A itself [27,35,50]. Moreover, GAG administration increases ax- onal growth and arborization in the rat hippocampus [41], ∗ Corresponding author. Tel.: +1-708-216-3261; fax: +1-708-216-8451. E-mail address: [email protected] (I. Hanin). and stimulates the effect of growth factors in cell culture [10,30,61]. Since GAGs are natural components of the cell membrane, these findings suggest that GAGs may have a general neuroprotective effect against AD lesions and may be used as valuable adjuncts in the therapy of AD [7,9,47]. Although a cholinergic deficit demonstrated in several brain regions is believed to be responsible for some of the deficits of learning and memory processes of AD pa- tients [1,2,12,22], there is no information about the impact of GAGs on the cholinergic deficiency of AD patients. Neurotoxins influencing the cholinergic system such as ethylcholine aziridinium (AF64A) have been used to study cholinergic hypofunction in rats. The choice of locus and dose of AF64A administration is critical for cholinergic specificity [13,18,26,34,44]. Because of this, lower doses of AF64A (1–2nmol i.c.v.) are used, so as to achieve the po- tency of AF64A without compromising selectivity. AF64A is structurally similar to choline and, therefore, it is taken up by cholinergic neurons via the high-affinity choline transport system and destroys them from within [15,25,26]. This compound has also been shown to impair learning and memory in rats following i.c.v. administration [5,6,33,59]. In our studies, we used the cholinergic deficit produced by AF64A to test the putative neuroprotective effects of C3 in the rat brain. C3 is a highly sulfated ultra low molecular 0197-4580/02/$ – see front matter © 2002 Elsevier Science Inc. All rights reserved. PII:S0197-4580(02)00093-3
Transcript of Protective effect of the heparin-derived oligosaccharide C3, on AF64A-induced cholinergic lesion in...

Neurobiology of Aging 24 (2003) 481–490
Protective effect of the heparin-derived oligosaccharide C3, onAF64A-induced cholinergic lesion in rats
M. Rose, B. Dudas, U. Cornelli, I. Hanin∗Department of Pharmacology, Loyola University Chicago Stritch School of Medicine, 2160 South First Avenue, Maywood, IL 60153, USA
Received 14 March 2002; received in revised form 10 June 2002; accepted 5 July 2002
Abstract
Alzheimer’s disease (AD) literature indicates that glycosaminoglycans (GAGs) may prevent proteoglycan-induced amyloid-� (A�)aggregation, decrease A�-induced tau-2 immunoreactivity, and increase the axonal growth and arborization of hippocampal neurons.However, there is no information about the impact of GAGs on cholinergic lesions. Here, AF64A was administered stereotaxically intothe lateral ventricles of rats, at doses that are selective for cholinotoxicity (1 and 2 nmol). The heparin-derived oligosaccharide (HDO), C3(25 mg/kg), was administered orally, once daily for 7 days before, and 7 days after AF64A administration. Choline acetyltransferase (ChAT)immunohistochemistry revealed that C3 administration reduced AF64A-induced cholinergic damage in the septum and cingulum bundle.Quantitative neuronal cell counts showed that C3 attenuated, by 60%, the decrease in cell number in the medial septum. Enzyme analysisshowed that C3 also significantly restored ChAT (30%) and acetylcholinesterase (AChE) enzyme activity (45%), which had been diminishedby AF64A. Our data suggest that, in addition to its effects of anti-A� aggregation, anti-A�-induced tau-2 immunoreactivity, and neurotrophiceffects, C3 also effectively reduces AF64A-induced cholinergic damage; hence it may have potential therapeutic value in AD patients.© 2002 Elsevier Science Inc. All rights reserved.
Keywords:Heparin-derived oligosaccharide; Glycosaminoglycan; C3; AF64A; Septum; Cingulum bundle; Cholinergic lesion
1. Introduction
Classical histopathological hallmarks of Alzheimer’s dis-ease (AD) include amyloid plaques containing fragments ofamyloid-� (A�) peptide[21,40,43,51–53]and neurofibril-lary tangles accumulating hyperphosphorylated tau protein[29,63]. In addition to these histological changes, AD ischaracterized by a profound deficiency in the central cholin-ergic system[1,11]. Although the pathogenesis of AD is notentirely known, several studies suggest that these lesions arefunctionally related to each other[17,23,45,46].
Proteoglycans have a high affinity for A� and protectit against proteolysis[4,24,55]. There is a general consen-sus that this effect is antagonized by glycosaminoglycans(GAGs) [32,56,57]. Stereotaxic injection of A� fragmentsinto the amygdala of rats has been shown to increase tau-2immunoreactivity in the hippocampus[54] and this phe-nomenon can be attenuated by administration of GAGs[8,14,62]. Furthermore, GAGs and other sulfated com-pounds inhibit the aggregation and toxicity of A� itself[27,35,50]. Moreover, GAG administration increases ax-onal growth and arborization in the rat hippocampus[41],
∗ Corresponding author. Tel.:+1-708-216-3261; fax:+1-708-216-8451.E-mail address:[email protected] (I. Hanin).
and stimulates the effect of growth factors in cell culture[10,30,61]. Since GAGs are natural components of the cellmembrane, these findings suggest that GAGs may have ageneral neuroprotective effect against AD lesions and maybe used as valuable adjuncts in the therapy of AD[7,9,47].
Although a cholinergic deficit demonstrated in severalbrain regions is believed to be responsible for some ofthe deficits of learning and memory processes of AD pa-tients[1,2,12,22], there is no information about the impactof GAGs on the cholinergic deficiency of AD patients.Neurotoxins influencing the cholinergic system such asethylcholine aziridinium (AF64A) have been used to studycholinergic hypofunction in rats. The choice of locus anddose of AF64A administration is critical for cholinergicspecificity[13,18,26,34,44]. Because of this, lower doses ofAF64A (1–2 nmol i.c.v.) are used, so as to achieve the po-tency of AF64A without compromising selectivity. AF64Ais structurally similar to choline and, therefore, it is takenup by cholinergic neurons via the high-affinity cholinetransport system and destroys them from within[15,25,26].This compound has also been shown to impair learning andmemory in rats following i.c.v. administration[5,6,33,59].
In our studies, we used the cholinergic deficit producedby AF64A to test the putative neuroprotective effects of C3in the rat brain. C3 is a highly sulfated ultra low molecular
0197-4580/02/$ – see front matter © 2002 Elsevier Science Inc. All rights reserved.PII: S0197-4580(02)00093-3

482 M. Rose et al. / Neurobiology of Aging 24 (2003) 481–490
weight (MW) GAG synthesized from unfractionated heparinusing�-irradiation. C3 has been well characterized, includ-ing its ability to cross the blood–brain barrier (BBB)[38,39].C3 also has been shown to exhibit neurotrophic effects inthe rat brain[41], and to attenuate A� (25–35)-inducedtau-2 immunoreactivity in the hippocampus[14]. We uti-lized qualitative immunohistochemistry to visualize the mor-phological changes and quantitative neuronal cell counts tomeasure changes in cell number. We also employed cholineacetyltransferase (ChAT) and acetylcholinesterase (AChE)enzyme neurochemistry to quantitate cholinergic functionfollowing AF64A and C3 administration. Our results demon-strate protection by C3 from AF64A-induced cholinotoxic-ity in rats, in vivo.
2. Materials and methods
2.1. Animals
Male Fischer 344 (Harlan) rats weighing approximately250–300 g were used. The animals were group housed andmaintained in a colony room with water and rat chow readilyavailable at all times. There was also a 12 h light/dark cyclemaintained for the duration of the experiment.
2.2. Drugs and chemicals
AF64A was prepared as described previously[48].Briefly, an aqueous solution of acetylethylcholine mus-tard HCl (Research Biochemicals Inc., Wayland, MA) wasbrought to pH 11.3 with NaOH. After stirring for 30 minat room temperature, the pH was lowered to 7.4 with thegradual addition of HCl and stirred for 60 min. The amountof AF64A was then adjusted either to 2 or 1 nmol/2�l.
C3 was obtained from unfractioned porcine mucosal hep-arin by means of controlled depolymerization induced by�-irradiation (US patent #4,987,222, LDO, Trino Vercellese,Italy). Unfractioned heparin in the solid state or in solu-tion is treated with a rectilinear gamma ray beam at dosesof 2.5–20 Mrad. This irradiation is supplied in successivestages with cooling intervals between the various stages.The degraded material is then fractionated to remove highMW fractions and to obtain C3 with a narrow MW rangeof ∼2 kDa. The MW profile of C3 has been determined us-ing GPC–HPLC and it was found that C3 is a mixture ofoligosaccharides with an average MW of 2.100 kDa, and acomposition of oligosaccharides containing, primarily, 4–8dextrose units that is capable of crossing the BBB[38].
2.3. Administration of drugs and compounds
All animals were given C3 (25 mg/kg) or saline by oralgavage, once daily for 7 days before AF64A administrationand once daily for 7 days thereafter. AF64A (2 nM/2�l,1 nM/2�l, or vehicle) was infused bilaterally (i.c.v.) with
a 30-gauge needle inserted through a burr hole drilled intothe skull into both the right and left ventricles. The in-cisor bar was sunk at 3.3 mm below the interauricular line.Stereotaxic coordinates were (from the bregma): posterior0.8 mm, lateral±1.5 mm, and ventral (from dura) 3.6 mm[49]. The rate of infusion was 1.0�l/min and the needle wasleft in place for 5 min after infusion and then slowly with-drawn. The vehicle of AF64A was distilled water preparedin the same manner as the AF64A. To anesthetize the an-imals, 60 mg/kg of sodium pentobarbital was administered(i.p.).
2.4. Animal sacrifice and tissue preparation forhistochemical assays
The animals were decapitated at 7 days after AF64A ad-ministration. Following sacrifice, the brain was placed inan ice-cold bath of saline, and then on an ice-cold surfacefor dissection. The brains were either dissected for the hip-pocampus, septum, and frontal cortex, and stored at−80◦Cor stored in 4% paraformaldehyde at 4◦C for subsequentimmunohistochemistry. Coronal sections (40�m) were cuton a freezing microtome (American Optical, Buffalo, NY),placed in 0.2% sodium azide in phosphate-buffered saline(PBS) (pH 7.4) and stored at 4◦C until used for immuno-histochemistry.
2.5. Immunohistochemistry
The sections were removed from the buffer and subse-quently placed in 0.3% H2O2 in PBS for 10 min and then30 min in 0.3% Triton X-100 in PBS (all solutions at roomtemperature). The sections then were washed 3× 10 minin PBS and incubated in ChAT polyclonal rabbit antibody(Chemicon, CA, USA) at a 1:2000 dilution for 24 h at roomtemperature. The antibody diluent contained 0.2% sodiumazide and 10% normal horse serum in PBS. Subsequently,the tissue was washed 3× 10 min in PBS and then incu-bated for 1.0 h in biotinylated anti-rabbit immunoglobulin(IgG) secondary antibody (Vectastain ABC Elite kit, Vec-tor Laboratories, Burlingame, CA) diluted 1:1000 in PBScontaining 0.2% sodium azide. Following two washes inPBS for a total of 15 min, the tissue was incubated for1.0 h in a solution of avidin horseradish peroxidase (Vec-tor) diluted 1:1000 in PBS. The sections were washed2 × 10 min in PBS and 10 min in Tris-buffered saline(TBS; pH 7.6) and then reacted with 3′,3′-diaminobenzidinetetrahydrochloride (DAB), using nickel ammonium sul-fate intensification (20 mg DAB, 100 mg nickel ammoniumsulfate, and 7.0�l 30% H2O2 in 40 ml TBS). Tissue sec-tions then were washed 2× 10 min in TBS. Subsequently,the tissue sections were slide mounted, dried, defatted,and cover slipped. Immunolabeled cells in the omit sec-tions were subtracted to establish a baseline. All micro-scopic analyses were performed using a Nikon Alphaphot-2YS2.

M. Rose et al. / Neurobiology of Aging 24 (2003) 481–490 483
2.6. Neurochemistry
Before actual analysis, tissues were thawed and homog-enized in ice-cold PBS (75 mM, pH 7.4, 1/20 w/v). Theassay was performed according to Fonnum[19]. For theAChE assay, homogenate (10�l) was added to 20�l of abuffer–substrate mixture containing ice-cold sodium phos-phate 75 mM (pH 7.0) and tritium-labeled acetylcholineiodide 10 mM (8.7 mCi/mmol). After 20 min of incuba-tion at 37◦C, the tubes were placed on ice. Each tubethen received 30�l of ice-cold distilled H2O, followed by150�l of sodium tetraphenylboron solution (75 mg/ml in3-heptanone). After vortexing and centrifuging each tube,the buffer layer was frozen at−80◦C. The organic layer wasthen aspirated off, and an aliquot of 30�l from the aqueouslayer was removed and counted by liquid scintillation spec-trometry for the amount of [3H]acetic acid formed. Resultswere expressed in nanomoles per milligram protein per hour,where protein content was measured using the method ofLowry et al. [36]. The ChAT assay was performed in muchthe same way as the AChE assay, except that the substrateused was tritiated acetylcoenzyme A, only tetraphenyl-boron solution was added, 100�l of the organic layer wasextracted, and the product measured was radiolabeled acetyl-choline. The enzyme levels of different brain areas were cal-culated as percent of controls, and analyzed using one-wayANOVA followed by a Newman–Keuls post hoc analysis.
2.7. Neuronal cell counts
In the immunohistochemical sections, neurons werecounted in the area through the medial septum. The medialseptum was defined from+0.95 to −0.26 mm from theBregma, levels 14–19[58]. ChAT-immunoreactive (IR) ele-ments cells were counted from six coronal sections (40�m)in the medial septum. The number and locations of levelsexamined for each rat brain region/nucleus correspondedclosely in all groups. Neurons containing ChAT-IR werecounted at 100× magnification using a Nikon Alphaphot-2YS2. Cell counts on each section were added to obtain thetotal number of neurons showing ChAT-IR in each animal.These numbers were averaged to express the total meannumber of ChAT-IR neurons in each group.
The exact size of the neuron was not in question, only thenumber of viable cells was measured by the general integrityof the perikarya and its surrounding neurites. A neuron wasconsidered to have either high or low integrity, and onlycells with high integrity were counted. A neuron with highintegrity was defined as being intact, showing strong stain-ing, without varicosities in the neurites, without Walleriandegeneration, and with its cytoplasmic contents remaininginside the plasma membrane. If these criteria were notfulfilled, the neuron was not taken into account. A blind anal-ysis of the sections was utilized and the total number of vi-able neurons in the medial septum was expressed as percentof controls. The calculated means for neuronal counts for
each group were compared by one-way analysis of variance(ANOVA) followed by Newman–Keuls post hoc analysis.
2.8. Experimental design
For the 2-nmol study, 6–9 rats per group were utilizedin a four-way controlled experiment. The four groups wereVeh/Sal, Veh/C3, AF64A/Sal, and AF64A/C3. Saline or C3was administered once daily for 7 days before and oncedaily for 7 days after 2 nmol AF64A administration (i.c.v.).Rats were then sacrificed the day after their last drug/salineadministration for assays.
For the 1-nmol study, 5–9 rats per group were utilizedin a three-way controlled experiment. The Veh/C3 groupwas excluded from this study because there were no sig-nificant changes in enzyme level, immunohistochemistry orcell number, as compared to Veh/Sal in the 2-nmol study.Administration of drug was the same as in the 2-nmol ex-periment.
3. Results
3.1. ChAT immunohistochemistry
3.1.1. Effect of vehicle injectionStereotaxic injection of an equal amount (2�l/side) of the
vehicle for AF64A (distilled water, pH adjusted the sameway as AF64A) showed slight enlargement of the lateralventricles, when compared to virgin control group (no treat-ment, no surgery; data not shown). The ChAT-IR elementsdid not show morphological changes (Fig. 1A(a) and E).
3.1.2. Effect of C3 alone on ChAT-IR elementsC3 administration alone did not have any detectable ef-
fect on the morphology of ChAT-IR elements (Fig. 1B(b)and F). Because there were no changes in cellular integritywith either a vehicle injection or C3 administration for the2-nmol experiment, a Veh/C3 group was not included in the1-nmol study.
3.1.3. Effect of 2 nmol/2µl/side AF64ASignificant reduction of cell numbers was detected in the
medial and lateral parts of the septal area after 2 nmol/sideAF64A treatment (P < 0.05; Fig. 6), parallel with theappearance of ChAT-IR fiber varicosities in the same area(Fig. 1G). The number and morphology of the median groupof ChAT positive perikarya appeared not to be affected(Fig. 1G). The ChAT-IR fibers of the cingulum bundleshowed prominent damage usually on the dorsal part ofthe pathway; the neurites had thickened regions with inten-sive ChAT immunoreactivity (Fig. 1C(c)). The ChAT-IRelements of the striatum, nucleus of the diagonal band ofBroca, and nucleus basalis ofMeynert, did not show patho-logical changes.Fig. 1 is shown as representative sectionsfrom each group.

484M
.R
ose
et
al./N
eu
rob
iolog
yo
fA
gin
g2
4(2
00
3)
48
1–
49
0
Fig. 1. ChAT-IR elements in the cingulum bundle (A–D) and in the septum (E–H) for 6–9 rats per treatment group. The framed areas of A, B, C, and D are illustrated with higher magnification as a, b, c,and d, respectively. The i.c.v. injection of the vehicle for AF64A combined with oral saline administered once daily for 7 days before and once daily for 7 days after the stereotaxic surgery did not affectthe ChAT-IR elements (A(a) and E). Similarly, C3 given once daily for 7 days before and once daily for 7 days after i.c.v. vehicle administration did notresult in any changes in the ChAT-IR either inthe cingulum bundle or the medial septum (B(b) and F). The i.c.v. AF64A administration (2 nmol/2�l/side) resulted in damage of the ChAT-IR fibers of the cingulum bundle (C(c)) and the septum (G),and reduction of the numbers of ChAT-IR perikarya in the medial and lateral septal areas (G). Cell bodies located in the midline of the septum were not affected (G). C3 (25 mg/kg) administered 7 daysbefore and 7 days after the AF64A injection (2 nmol/2�l/side) partially reversed the AF64A-induced cholinergic damage (D(d) and H). This figure is representative of the effects observed and described.

M. Rose et al. / Neurobiology of Aging 24 (2003) 481–490 485
3.1.4. Effect of 1 nmol/2µl/side AF64AA decreased dose of AF64A injection led to lesser ven-
tricular enlargement and reduced nonspecific damage of theependymal cells. The reduction of the ChAT-IR cell numberin the lateral and medial septum was less prominent (Fig. 7).Varicosities on the ChAT-IR fibers were still detected on thelateral septal area, however, close to the ventricular surface(data not shown). The cingulum bundle did not show sig-nificant damage. Similarly to the higher dosage of AF64Aused, the ChAT-IR elements of the striatum, nucleus of di-agonal band ofBroca, and nucleus basalis ofMeynert, didnot show pathological changes.
3.1.5. Effect of C3 on AF64A-induced damageC3 significantly reduced the 2 nmol AF64A-induced le-
sion of the ChAT-IR elements. In the cingulum bundle onlysome axonal swelling was detected (Fig. 1D(d)). The reduc-tion of the numbers of perikarya in the lateral and medialseptal areas was less prominent than in the AF64A-treatedgroup, and a decreased number of varicose fibers were seenin the lateral part of the septum (Fig. 1H). Similar resultswere obtained for the 1 nmol AF64A group treated with C3(data not shown). As we have already demonstrated earlier[26], the morphology of the striatum, nucleus of diagonalband ofBroca, and nucleus basalis ofMeynertdid not showany changes.
3.2. Neurochemistry
3.2.1. Two nanomoles AF64A administration
3.2.1.1. Hippocampus.Significant reductions of bothChAT and AChE levels were observed after 2 nmol AF64Aadministration. The reduction was more significant with
Fig. 2. ChAT activity in the frontal cortex, hippocampus, and septum of rats treated with C3 or saline 7 days before and 7 days after either a 2 nmolAF64A or vehicle injection. The data are presented as percentage of control (mean± S.E.M.) for 7–9 rats per treatment group. The control rats (Veh/Sal)had enzyme activities of 85 nmol/mg protein/h (hippocampus) [F(3, 24) = 12.9; P < 0.0001], 131.5 nmol/mg protein/h (septum) [F(3, 24) = 1.93;P < 0.1544], and 55 nmol/mg protein/h (frontal cortex) [F(3, 24) = 17.3; P < 0.421]. Data were analyzed by ANOVA with Newman–Keuls analysis.∗∗P < 0.01 vs. Veh/Sal,++P < 0.01 vs. AF/Sal.
ChAT (P < 0.01; Fig. 2) than with AChE (P < 0.05;Fig. 3). After oral C3 administration (25 mg/kg), 7 daysbefore and 7 days after AF64A lesioning, there was a sig-nificant increase in ChAT (P < 0.01; Fig. 2) and AChElevels (P < 0.05; Fig. 3) as compared to AF64A animalsthat were given saline. C3 given alone did not exhibit anysignificant changes in enzyme activity. Because there wereno significant changes in enzyme levels with a vehicleinjection and C3 administration for the 2-nmol experi-ment, this group was not included in the 1-nmol AF64Astudy.
3.2.1.2. Septum.AF64A increased both ChAT and AChEenzyme activities and administration of C3 decreasedboth enzyme activities as compared to the AF64A-treatedgroup. However, this effect was not statistically signi-ficant.
3.2.1.3. Frontal cortex. There were no significant changesin either enzyme level within this region.
3.2.2. One nanomole AF64A administration
3.2.2.1. Hippocampus.Significant reductions of bothChAT and AChE activities were detected after 1 nmolAF64A administration (Figs. 4 and 5). ChAT levels in-creased significantly after C3 administration (25 mg/kg) ascompared to AF64A animals given only saline.
3.2.2.2. Septum.While there were fluctuations in the lev-els of both enzymes, the changes were insignificant.
3.2.2.3. Frontal cortex. There were no significant changesin either enzyme level within this region.

486 M. Rose et al. / Neurobiology of Aging 24 (2003) 481–490
Fig. 3. AChE activity in the frontal cortex, hippocampus, and septum of rats treated with C3 or saline 7 days before and 7 days after either 2 nmolAF64A or vehicle injection. The data are presented as percentage of control (mean± S.E.M.) for 7–9 rats per treatment group. The control rats (Veh/Sal)had enzyme activities of 1621 nmol/mg protein/h (hippocampus) [F(3, 24) = 5.26; P < 0.0073], 5434 nmol/mg protein/h (septum) [F(3, 24) = 0.793;P < 0.510], and 2164 nmol/mg protein/h (frontal cortex) [F(3, 24) = 1.25; P < 0.314]. Data were analyzed by ANOVA with Newman–Keuls analysis.∗P < 0.05 vs. Veh/Sal,+P < 0.05 vs. AF/Sal.
3.3. Neuronal cell counts
3.3.1. Two-nanomole AF64A studyThe control group injected with vehicle and given saline
for the treatment period was observed to contain 235± 58(S.E.) neurons that met the criteria for viable cells. Withthe administration of AF64A, there was a significant reduc-tion (P < 0.05) in the number of viable cells as comparedto saline controls (Fig. 6). With the administration of C3(25 mg/kg), once daily for 7 days before and once daily for7 days after the AF64A injection, there was a significant in-crease (P < 0.05) as compared to 2 nmol AF64A-lesionedanimals (Fig. 6). There was no significant change in neuronal
Fig. 4. ChAT activity in the frontal cortex, hippocampus, and septum of rats treated with C3 or saline 7 days before and 7 days after either a 1 nmol AF64Aor vehicle injection. The data are presented as percentage of control (mean± S.E.M.) for five rats per treatment group. The control rats (Veh/Sal) hadenzyme activities of 53 nmol/mg protein/h (hippocampus) [F(2, 15) = 13.7; P < 0.0006], 127.5 nmol/mg protein/h (septum) [F(2, 15) = 2.30; P < 0.139],and 40 nmol/mg protein/h (frontal cortex) [F(2, 15) = 3.53; P < 0.0595]. Data were analyzed by ANOVA with Newman–Keuls analysis.∗P < 0.05 vs.Veh/Sal,++P < 0.01 vs. AF/Sal.
number in the medial septum with C3 and vehicle-injectedrats. All neuronal counts were expressed as percent ofcontrols.
3.3.2. One-nanomole AF64A studyThe control group injected with vehicle and given saline
for the treatment period was observed to contain 184± 32(S.E.) neurons that met the criteria for viable cells. Withthe administration of AF64A, there was a reduction inthe number of viable cells as compared to saline controls(Fig. 7), however, the decrease was not statistically signifi-cant. With the administration of C3 (25 mg/kg), once dailyfor 7 days before and once daily for 7 days after the AF64A
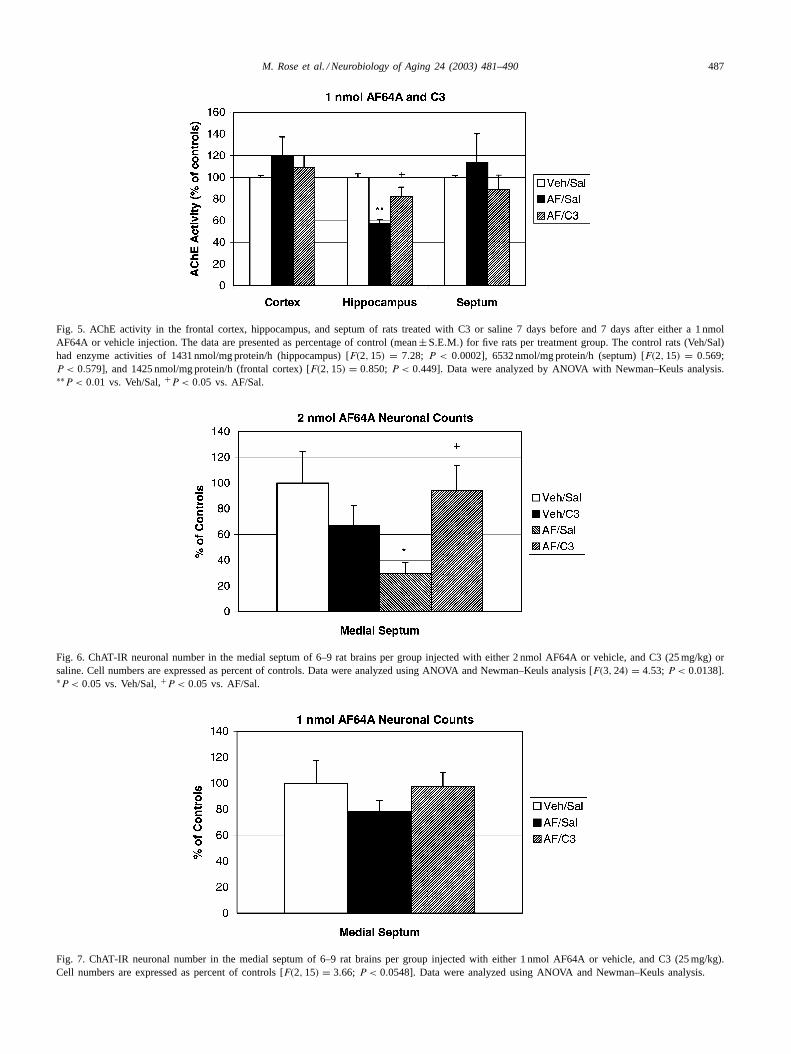
M. Rose et al. / Neurobiology of Aging 24 (2003) 481–490 487
Fig. 5. AChE activity in the frontal cortex, hippocampus, and septum of rats treated with C3 or saline 7 days before and 7 days after either a 1 nmolAF64A or vehicle injection. The data are presented as percentage of control (mean± S.E.M.) for five rats per treatment group. The control rats (Veh/Sal)had enzyme activities of 1431 nmol/mg protein/h (hippocampus) [F(2, 15) = 7.28; P < 0.0002], 6532 nmol/mg protein/h (septum) [F(2, 15) = 0.569;P < 0.579], and 1425 nmol/mg protein/h (frontal cortex) [F(2, 15) = 0.850; P < 0.449]. Data were analyzed by ANOVA with Newman–Keuls analysis.∗∗P < 0.01 vs. Veh/Sal,+P < 0.05 vs. AF/Sal.
Fig. 6. ChAT-IR neuronal number in the medial septum of 6–9 rat brains per group injected with either 2 nmol AF64A or vehicle, and C3 (25 mg/kg) orsaline. Cell numbers are expressed as percent of controls. Data were analyzed using ANOVA and Newman–Keuls analysis [F(3, 24) = 4.53; P < 0.0138].∗P < 0.05 vs. Veh/Sal,+P < 0.05 vs. AF/Sal.
Fig. 7. ChAT-IR neuronal number in the medial septum of 6–9 rat brains per group injected with either 1 nmol AF64A or vehicle, and C3 (25 mg/kg).Cell numbers are expressed as percent of controls [F(2, 15) = 3.66; P < 0.0548]. Data were analyzed using ANOVA and Newman–Keuls analysis.

488 M. Rose et al. / Neurobiology of Aging 24 (2003) 481–490
injection, there was an increase as compared to 1 nmolAF64A-lesioned animals (Fig. 7). Because there were nochanges in cell number with a vehicle injection and C3 ad-ministration for the 2-nmol experiment, a vehicle/C3 groupwas not included in the 1-nmol study. All neuronal countswere expressed as percent of controls.
4. Discussion
We have shown earlier[15] a significant decrease of bothChAT and AChE in the hippocampus, and a concurrent in-crease of these enzymes in the septum at 7 days post-AF64Asurgery (0.5–2 nmol/ventricle). These enzyme activities inthe septum showed recovery after 14 days, whereas enzymeactivities in the hippocampus persisted for >6 months. Thiseffect was interpreted as the result of a slow recovery fromthe initial cholinergic damage induced by AF64A[15,16].AF64A (i.c.v.) affects only the septum and hippocampusand now, newly shown, cingulum bundle. The lack of effectof AF64A on cortex and striatum has already been shownand reproduced extensively in this AF64A-related paradigm[26].
The present study indicates that C3 attenuates the lesionscaused by the cholinergic toxin AF64A, in the rat brain.The BBB accessibility of C3 has been shown in our previ-ous studies[38,39]. After C3 administration combined withAF64A injection, there was a definite increase in ChAT andAChE enzyme activities in the hippocampus when comparedto AF64A-lesioned animals. In the septum, there was a re-duction in the enzyme levels as compared to AF64A-treatedrats.
C3 treatment alone had a variable effect, fluctuations inChAT and AChE levels in the hippocampus; however, thesechanges were not significant. In the septum, C3 adminis-tration alone also had varying effects in ChAT and AChEactivity, but again none of these results were statisticallysignificant. C3[41] and other GAGs[10,28,30]have beenshown to have a neurotrophic effect. These data may ex-plain some of these changes in enzyme activities in thehippocampus and septum.
ChAT immunohistochemistry revealed a definite effectin septal cholinergic neurons after AF64A treatment. Thiseffect was observed as a reduction in cell number in thelateral part of the medial septum, corresponding to ourprevious studies[34]. The median sagittally located cellbodies were not affected with similar treatment. Previousstudies suggested that AF64A targets neurons with longeraxons while those with shorter neurites do not show anysigns of damage[16,42]. ChAT-IR fibers in the septumhave shown significant damage after AF64A treatment.These pathological changes included varicosities appearingalong the neurites, and axonal breakdown detected withChAT immunohistochemistry. These pathological changeswere characteristic in the cingulum bundle as well. Incontrast to these findings, cholinergic neurons in some
areas around the lateral ventricle (e.g. striatum) were notaffected by AF64A treatment. These data correspond toour previous findings[16,42] and may be interpreted bythe cholinotoxin AF64A targeting neurons only with longneurites.
The pathological changes following 2 nmol/side AF64Aadministration were more prominent in the septum thanwith 1 nmol. The cingulum bundle did not show any signsof lesion with the smaller concentration of AF64A. Thesefindings indicate that the cholinergic damage depends onthe concentration of AF64A administered.
C3 administration, when combined with AF64A injec-tion, attenuated the extent of cholinergic damage observed inboth the septum and the cingulum bundle following AF64Aadministration alone. C3 treatment alone did not show mor-phological changes in ChAT-IR elements, or statisticallysignificant changes in cell number. This could be interpretedthat cholinergic damage has to be inflicted before C3 canhave a beneficial effect. It is also interesting to note thatboth C3 and AF64A exhibit their action in the same regions.It is known that AF64A is a selective toxin for the cholin-ergic system. Because of the relationship shown here, it ispossible that certain GAGs are specific to the cholinergicsystem as well.
It is not known whether C3 acts by protecting the brainfrom damage, or whether it functions via a repair mecha-nism after injury. Further studies are necessary to identifythe mechanism by which C3 exerts its effects. It is con-ceivable that C3 acts via a NGF-mediated pathway, sinceC3 has been shown to increase dendritic arborization[41].There is also a general consensus that growth factors in-crease ChAT and AChE levels[27,37], and that GAGspromote neurite outgrowth[10,30,41,61]. Along with liter-ature showing the relationship between the cholinergic sys-tem and growth factors, other literature supports a linkagebetween cholinergic transmission and A�/plaque forma-tion [3,20,31,60]. Therefore, GAGs might have the abilityto prevent both cholinergic damage and plaque/tangleformation.
Thus, while the mechanism of C3 is still unknown, its neu-roprotective/neurotrophic effect may have therapeutic valuenot only in the treatment of AD but also, in various neu-rodegenerative disorders. In addition to mechanistic and be-havioral studies, we also plan to study similar analogs ofdermatan sulfate and chondroitin sulfate, in order to estab-lish a broad spectrum of information regarding the potentialrole of GAGs in the treatment of AD.
Acknowledgments
Thanks to Dr. Edward J. Neafsey in the Department of CellBiology, Neurobiology, and Anatomy, for allowing us theuse of his freezing microtome (Loyola University ChicagoStritch School of Medicine). Supported in part by Grant#1-R41-AG15740-02.

M. Rose et al. / Neurobiology of Aging 24 (2003) 481–490 489
References
[1] Bartus RT. Physostigmine and recent memory: effects in young andaged nonhuman primates. Science 1979;206:1087–9.
[2] Beatty WW, Carbone CP. Septal lesions, intramaze cues and spatialbehavior in rats. Physiol Behav 1980;24:675–8.
[3] Boncristiano S, Calhoun ME, Kelly PH, Pfeifer M, Bondolfi L,Stalder M, et al. Cholinergic changes in the APP23 transgenic mousemodel of cerebral amyloidosis. J Neurosci 2002;22:3234–43.
[4] Castillo GM, Ngo C, Cummings J, Wight TN, Snow AD. Perlecanbinds to the beta-amyloid proteins (A beta) of Alzheimer’s disease,accelerates A beta fibril formation, and maintains A beta fibrilstability. J Neurochem 1997;69:2452–65.
[5] Chrobak J, Hanin I, Walsh T. AF64A (ethylcholine aziridinium ion),a cholinergic neurotoxin, selectively impairs working memory in amultiple component T-maze task. Brain Res 1987;414:15–21.
[6] Chrobak J, Hanin I, Schmechel DE, Walsh TJ. AF64A-inducedworking memory impairment: behavioral, neurochemical andhistological correlates. Brain Res 1988;463:107–17.
[7] Conti L, Re F, Lazzerini F, Morey LC, Ban TA, Santini V, etal. Glycosaminoglycan polysulfate (Ateroid) in old-age dementias:effects upon depressive symptomatology in geriatric patients. ProgNeuropsychopharmacol Biol Psychiatry 1989;13:977–81.
[8] Conti L, Placidi GF, Cassano GB. Ateroid in the treatment ofdementia: results of a clinical trial. Mod Probl Pharmacopsychiatry1989;23:76–84.
[9] Cornelli U. The therapeutic approach to Alzheimer’s disease.In: Haremberg J, Casu B, editors. Non-anticoagulant action ofglycosaminoglycans. New York, NY: Plenum Press, 1996. p. 249–79.
[10] Damon D, D’Amore P, Wagner J. Sulfated glycosaminoglycansmodify growth factor-induced neurite outgrowth in PC12 cells. JCell Physiol 1988;135:293–300.
[11] Davies P, Maloney AJF. Selective loss of central cholinergic neuronsin Alzheimer’s disease. Lancet 1976;2:403.
[12] Davis KL, Mohs RC, Tinklenberg JR, Pfefferbaum A, HollisterLE, Kopell BS. Physostigmine: improvement of long-term memoryprocesses in normal humans. Science 1978;201:272–4.
[13] Dong XW, Hanin I, Lorens SA. AF64A affects septal cholineacetyltransferase but not parvalbumin immunoreactive cells. BrainRes Bull 1994;35:217–20.
[14] Dudas B, Cornelli U, Lee JM, Hejna MJ, Walzer M, Lorens SA,et al. Oral and subcutaneous administration of the glycosamino-glycan C3 attenuates A�(25–35)-induced abnormal tau proteinimmunoreactivity in rat brain. Neurobiol Aging 2002;23:97–104.
[15] El Tamer A, Corey J, Wülfert E, Hanin I. Reversible cholinergicchanges induced by AF64A in rat hippocampus and possible septalcompensatory effect. Neuropharmacology 1992;31:397–402.
[16] Fan Q, Hanin I. Effects of AF64A on gene expression of cholineacetyltransferase (ChAT) in the septo-hippocampal pathway andstriatum in vivo. Neurochem Res 1999;24:15–24.
[17] Fisher A, Pittel Z, Haring R, Brandeis R, Bar-Ner N, Sonego H, et al.In: Mizuno Y, Fisher A, Hanin I, editors. Mapping the progress ofAlzheimer’s and Parkinson’s disease. New York: Academic/PlenumPress, 2002. p. 205–10.
[18] Fisher A, Hanin I. Potential animal models for senile dementia ofAlzheimer’s type, with emphasis on AF64A-induced cholinotoxicity.Annu Rev Pharmacol Toxicol 1986;26:161–81.
[19] Fonnum F. Radiochemical micro assays for the determination ofcholine acetyltransferase and acetylcholinesterase activities. BiochemJ 1969;115:465–72.
[20] Gau JT, Steinhilb ML, Kao TC, D’Amato CJ, Gaut JR, FreyKA, et al. Stable beta-secretase activity and presynaptic cholinergicmarkers during progressive central nervous system amyloidogenesisin Tg2576 mice. Am J Pathol 2002;160:731–8.
[21] Glenner GG, Wong CW, Quaranta V, Eanes ED. The amyloid depositsin Alzheimer’s disease: their nature and pathogenesis. Appl Pathol1984;2:357–69.
[22] Glick SD, Mittag TW, Green JP. Cental cholinergic correlates ofimpaired learning. Neuropharmacology 1973;12:291–6.
[23] Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillarytangles in P301L tau transgenic mice induced by A�42 fibrils.Science 2001;293:1491–5.
[24] Gupta-Bansal R, Frederickson CA, Brunden KR. Proteoglycan-mediated inhibition of A� proteolysis. J Biol Chem 1995;270:18666–71.
[25] Hanin I. The centrally cholino-deficient animal as a model ofAlzheimer’s disease (AD). In: Giacobini E, Becker R, editors.Alzheimer’s disease: therapeutic strategies. Boston: Birkhauser, 1994.p. 367–74.
[26] Hanin I. The AF64A model of cholinergic hypofunction: an update.Life Sci 1996;58:1955–64.
[27] Hefti F, Dravid A, Hartikka J. Chronic intraventricular injections ofnerve growth factor elevate hippocampal choline acetyltransferasein adult rats with partial septo-hippocampal lesions. Brain Res1984;293:305–11.
[28] Kisilevsky R, Lemieux LJ, Fraser PE, Kong X, Hultin PG, SzarekWA. Arresting amyloidosis in vivo using small-molecule anionicsulphonates or sulphates: implications for Alzheimer’s disease. NatMed 1995;1:143–8.
[29] Ksiezak-Reding H, Liu WK, Yen SH. Phosphate analysis anddephosphorylation of modified tau associated with paired helicalfilaments. Brain Res 1992;597:209–19.
[30] Lesma E, Di Giulio AM, Ferro L, Prino G, Gorio A.Glycosaminoglycans in nerve injury: I. Low doses of glycosamino-glycans promote neurite formation. J Neurosci Res 1996;46:565–71.
[31] Leventer S, McKeag D, Clancy M, Wülfert E, Hanin I.Intracerebroventricular administration of ethylcholine mustardaziridinium ion (AF64A) reduces release of acetylcholine from rathippocampal slices. Neuropharmacology 1985;24:453–9.
[32] Leveugle B, Scanameo A, Ding W, Fillit H. Binding of heparansulfate glycosaminoglycan to beta-amyloid peptide: inhibitionby potentially therapeutic polysulfated compounds. Neuroreport1994;5:1389–92.
[33] Lim DK, Oh YH, Kim HS. Impairments of learning and memoryfollowing intracerebroventricular administration of AF64A in rats.Arch Pharm Res 2001;24:234–9.
[34] Lorens SA, Kindel G, Dong XW, Lee JM, Hanin I. Septal cholineacetyltransferase immunoreactive neurons: dose-dependent effects ofAF64A. Brain Res Bull 1991;26:965–71.
[35] Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibrilformation and is inhibited by congo red. Proc Natl Acad Sci USA1994;91:12243–7.
[36] Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein mea-surement with folin phenol reagent. J Biol Chem 1951;193:265–7.
[37] Lucas C, Czlonkowska A, Kreutzberg G. Regulation of acetyl-cholinesterase by nerve growth factor in the pheochromocytomaPC12 cell line. Neurosci Lett 1980;18:333–7.
[38] Ma Q, Dudas B, Hejna M, Cornelli U, Lee JM, Lorens S, et al. Theblood–brain barrier accessibility of a heparin-derived oligosaccharideC3. Thromb Res 2002;105:447–53.
[39] Ma Q, Dudas B, Daud A, Iqbal O, Hoppensteadt D, Jeske W,et al. Molecular and biochemical profiling of a heparin derivedoligosaccharide, C3. Thromb Res 2002;105:303–9.
[40] Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL,Beyreuther K. Amyloid plaque core protein in Alzheimer diseaseand Down syndrome. Proc Natl Acad Sci USA 1985;82:4245–9.
[41] Mervis R, McKean J, Zats S, Gum A, Reinhart R, Dudas B, etal. Neurotrophic effects of the glycosaminoglycan C3 on dendriticarborization and spines in the adult rat hippocampus: a quantitativeGolgi study. CNS Drug Rev 2000;6:44–6.
[42] Meyer EM, Judkins JH, Hardwick EO. Recovery of [3H]acetylcholinesynthesis after AF64A-treatment in primary, neuron-enriched, ratseptal-hippocampal and striatal cultures. Brain Res 1993;42:113–8.

490 M. Rose et al. / Neurobiology of Aging 24 (2003) 481–490
[43] Miller DL, Papayannopoulos IA, Styles J, Bobin SA, Lin YY,Biemann K, et al. Peptide compositions of the cerebrovascular andsenile plaque core amyloid deposits of Alzheimer’s disease. ArchBiochem Biophys 1993;301:41–52.
[44] Mouton PR, Arendash GW. Atrophy of cholinergic neurons withinthe rat nucleus basalis magnocellularis following intracortical AF64Ainfusion. Neurosci Lett 1990;111:52–7.
[45] Nitsch RM, Deng M, Tennis M, Schoenfeld D, Growdon JH. Theselective muscarinic M1 agonist AF102B decreases levels of totalAbeta in cerebrospinal fluid of patients with Alzheimer’s disease.Ann Neurol 2000;48:913–8.
[46] Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release ofAlzheimer amyloid precursor derivatives stimulated by activation ofmuscarinic acetylcholine receptors. Science 1992;258:304–7.
[47] Passeri M, Cucinotta D. Ateroid in the clinical treatment ofmulti-infarct dementia. Mod Probl Pharmacopsychiatry 1989;23:85–94.
[48] Potter PE, Tedford CE, Kindel G, Hanin I. Inhibition ofhigh affinity choline transport attenuates both cholinergic andnon-cholinergic effects of ethylcholine aziridinium (AF64A). BrainRes 1989;487:238–44.
[49] Paxinos G, Watson C. The rat brain in stereotaxic coordinates.London: Academic Press, 1986.
[50] Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE,Teplow DB, et al. Aggregation of secreted amyloid beta-protein intosodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem1995;270:9564–70.
[51] Roher AE, Lowenson JD, Clarke S, Wolkow C, Wang R, Cotter RJ, etal. Structural alterations in the peptide backbone of beta-amyloid coreprotein may account for its deposition and stability in Alzheimer’sdisease. J Biol Chem 1993;268:3072–83.
[52] Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, Gowing E,et al. Beta-amyloid-(1–42) is a major component of cerebrovascularamyloid deposits: implications for the pathology of Alzheimerdisease. Proc Natl Acad Sci USA 1993;90:10836–40.
[53] Roher AE, Palmer KC, Yurewicz EC, Ball MJ, Greenberg BD.Morphological and biochemical analyses of amyloid plaque core
proteins purified from Alzheimer disease brain tissue. J Neurochem1993;61:1916–26.
[54] Sigurdsson EM, Lorens SA, Hejna MJ, Dong XW, Lee JM. Localand distant histopathological effects of unilateral amyloid-beta 25–35injections into the amygdala of young F344 rats. Neurobiol Aging1996;17:893–901.
[55] Shaffer LM, Dority MD, Gupta-Bansal R, Frederickson RC, YounkinSG, Brunden KR. Amyloid beta protein (A beta) removal byneuroglial cells in culture. Neurobiol Aging 1995;16:737–45.
[56] Snow AD, Sekiguchi RT, Nochlin D, Kalaria RN, Kimata K. Heparansulfate proteoglycan in diffuse plaques of hippocampus but not of cer-ebellum in Alzheimer’s disease brain. Am J Pathol 1994;144:337–47.
[57] Snow AD, Kinsella MG, Parks E, Sekiguchi RT, Miller JD, KimataK, et al. Differential binding of vascular cell-derived proteoglycans(perlecan, biglycan, decorin and versican) to the beta amyloid proteinof Alzheimer’s disease. Arch Biochem Biophys 1995;320:84–95.
[58] Swanson LW. Brain maps: structure of the rat brain. 2nd ed.Amsterdam, The Netherlands: Elsevier, 1998.
[59] Tedford CE, Lorens SA, Corey JC, Lokhorst D, Kindel G, Wülfert E,et al. Behavioral and neurochemical effects of AF64A in young andold Fisher 344 male rats. In: Nagatsu T, Fisher A, Yoshida M, editors.Basic, clinical, and therapeutic aspects of Alzheimer’s disease andParkinson’s disease. New York: Plenum Press, 1990. p. 321–34.
[60] Tran MH, Yamada K, Nabeshima T. Molecular mechanisms ofcholinergic dysfunction and cognitive deficits induced by amyloidbeta-peptide. Nihon Shinkei Seishin Yakurigaku Zasshi 2001;21:125–32.
[61] Walicke P. Interactions between basic fibroblast growth factor (FGF)and glycosaminoglycans in promoting neurite outgrowth. Exp Neurol1988;102:144–8.
[62] Walzer M, Lorens S, Henja M, Fareed J, Hanin I, Cornelli U, et al.Low molecular weight glycosaminoglycan blockade of beta amyloidinduced neuropathology. Eur J Pharmacol 2002;445(3):211–20.
[63] Wischik CM, Novak M, Thogersen HC, Edwards PC, Runswick MJ,Jakes R, et al. Isolation of a fragment of tau derived from the coreof the paired helical filament of Alzheimer disease. Proc Natl AcadSci USA 1988;85:4506–10.




















