Pretreatment With TCDD Exacerbates Liver Injury From Concanavalin A: Critical Role for NK Cells
14
© The Author 2013. Published by Oxford University Press on behalf of the Society of Toxicology. All rights reserved. For permissions, please email: [email protected]. Pretreatment With TCDD Exacerbates Liver Injury From Concanavalin A: Critical Role for NK Cells Aaron M. Fullerton, Robert A. Roth, and Patricia E. Ganey 1 Department of Pharmacology & Toxicology, Center for Integrative Toxicology, Michigan State University, East Lansing, Michigan 48824 1 To whom correspondence should be addressed at Department of Pharmacology & Toxicology, Center for Integrative Toxicology, Michigan State University, 1129 Farm Lane, Room 214, East Lansing, MI 48824. Fax: (517) 432-2310. E-mail: [email protected]. Received April 24, 2013; accepted August 9, 2013 For many liver diseases, including viral and autoimmune hepatitis, immune cells play an important role in the develop- ment and progression of liver injury. Concanavalin A (Con A) administration to rodents has been used as a model of immune- mediated liver injury resembling human autoimmune hepatitis. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) has been demon- strated to alter the development of immune-mediated diseases. Mice pretreated with TCDD developed exacerbated liver injury in response to administration of a mild dose (6 mg/kg) of Con A. In the present study, we tested the hypothesis that TCDD pretreat- ment exacerbates Con A-induced liver injury by enhancing the activation and recruitment of accessory cell types including neu- trophils, macrophages, and natural killer (NK) cells. Mice were treated with 0, 0.3, 3, or 30 μg/kg TCDD and 4 days later with Con A or saline. TCDD pretreatment with doses of 3 and 30 μg/kg significantly increased liver injury from Con A administration. The plasma concentrations of neutrophil chemokines were signifi- cantly increased in TCDD-pretreated mice after Con A adminis- tration. NKT cell-deficient (CD1d KO) mice were used to examine whether NKT cells were required for TCDD/Con A-induced liver injury. CD1d KO mice were completely protected from liver injury induced by treatment with Con A alone, whereas the injury from TCDD/Con A treatment was reduced but not eliminated. However, T-cell deficient (RAG1 KO) mice were protected from liver injury induced by Con A irrespective of pretreatment with TCDD. TCDD/Con A treatment increased the percentage of NK cells expressing the activation marker CD69. Depletion of NK cells prior to treatment resulted in significant reductions in plasma interferon-γ and liver injury from TCDD/Con A treatment. In summary, exposure to TCDD exacerbated the immune-mediated liver injury induced by Con A, and our findings suggest that NK cells play a critical role in this response. Key Words: dioxin; autoimmune; inflammation; chemokines; liver. Despite increasing prevalence in the population, the etiology of autoimmune liver diseases is still not thoroughly understood (Feld and Heathcote, 2003). A number of factors appear to confer susceptibility to these diseases. In addition to known genetic risk factors, exposure to environmental xenobiotics is associated with increased incidence of autoimmune disease (Czaja and Manns, 2010; Gilbert, 2010; Longhi et al., 2010). A role for the aryl hydrocarbon receptor (AhR) in the development of these diseases has also been hypothesized. In support of this hypothesis, expo- sure of synovial tissue from patients with rheumatoid arthritis to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) led to increased production of inflammatory mediators (Kobayashi et al., 2008). TCDD and other AhR ligands also altered the onset and severity of injury in experimental animal models of autoimmune disease; however, the nature of the effect of AhR stimulation on patho- genesis is complex, with reports of both suppression and exac- erbation of responses. For example, prenatal treatment of mice with TCDD increased signs of autoimmunity and enhanced kid- ney damage in a murine model of autoimmune lupus (Holladay et al., 2011; Kobayashi et al., 2008; Mustafa et al., 2009). On the other hand, in experimental autoimmune encephalomyeli- tis, deterioration of spinal cord neurons was reduced in TCDD- treated mice (Quintana et al., 2008). TCDD also suppressed the development of autoimmune type 1 diabetes in nonobese dia- betic mice (Kerkvliet et al., 2009). Thus, exposure to TCDD can have diverse effects on immune-mediated pathogenesis. The administration of the polyclonal T-cell mitogen conca- navalin A (Con A) to mice has been used to produce a model of immune-mediated liver injury that resembles the pathophysiol- ogy of autoimmune hepatitis (Tiegs et al., 1992). In this model, errant immune cell activation and destruction of hepatic paren- chymal tissue is similar to that which occurs in patients with autoimmune hepatitis (Peters, 2002; Wang et al., 2012). We have previously reported that pretreatment with TCDD sensi- tized mice to liver injury induced by Con A (Fullerton et al., 2013). The exacerbated response was associated with increased production of interferon-γ (IFNγ) and enhanced activation of natural killer T (NKT) cells (Fullerton et al., 2013), but the effect of pretreatment with TCDD on other immune cell types in this model is unknown. toxicological sciences 136(1), 72–85 2013 doi:10.1093/toxsci/kft174 Advance Access publication August 22, 2013 by guest on July 12, 2016 http://toxsci.oxfordjournals.org/ Downloaded from
-
Upload
michiganstate -
Category
Documents
-
view
2 -
download
0
Transcript of Pretreatment With TCDD Exacerbates Liver Injury From Concanavalin A: Critical Role for NK Cells

© The Author 2013. Published by Oxford University Press on behalf of the Society of Toxicology. All rights reserved. For permissions, please email: [email protected].
Pretreatment With TCDD Exacerbates Liver Injury From Concanavalin A: Critical Role for NK Cells
Aaron M. Fullerton, Robert A. Roth, and Patricia E. Ganey1
Department of Pharmacology & Toxicology, Center for Integrative Toxicology, Michigan State University, East Lansing, Michigan 48824
1To whom correspondence should be addressed at Department of Pharmacology & Toxicology, Center for Integrative Toxicology, Michigan State University, 1129 Farm Lane, Room 214, East Lansing, MI 48824. Fax: (517) 432-2310. E-mail: [email protected].
Received April 24, 2013; accepted August 9, 2013
For many liver diseases, including viral and autoimmune hepatitis, immune cells play an important role in the develop-ment and progression of liver injury. Concanavalin A (Con A) administration to rodents has been used as a model of immune-mediated liver injury resembling human autoimmune hepatitis. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) has been demon-strated to alter the development of immune-mediated diseases. Mice pretreated with TCDD developed exacerbated liver injury in response to administration of a mild dose (6 mg/kg) of Con A. In the present study, we tested the hypothesis that TCDD pretreat-ment exacerbates Con A-induced liver injury by enhancing the activation and recruitment of accessory cell types including neu-trophils, macrophages, and natural killer (NK) cells. Mice were treated with 0, 0.3, 3, or 30 μg/kg TCDD and 4 days later with Con A or saline. TCDD pretreatment with doses of 3 and 30 μg/kg significantly increased liver injury from Con A administration. The plasma concentrations of neutrophil chemokines were signifi-cantly increased in TCDD-pretreated mice after Con A adminis-tration. NKT cell-deficient (CD1d KO) mice were used to examine whether NKT cells were required for TCDD/Con A-induced liver injury. CD1d KO mice were completely protected from liver injury induced by treatment with Con A alone, whereas the injury from TCDD/Con A treatment was reduced but not eliminated. However, T-cell deficient (RAG1 KO) mice were protected from liver injury induced by Con A irrespective of pretreatment with TCDD. TCDD/Con A treatment increased the percentage of NK cells expressing the activation marker CD69. Depletion of NK cells prior to treatment resulted in significant reductions in plasma interferon-γ and liver injury from TCDD/Con A treatment. In summary, exposure to TCDD exacerbated the immune-mediated liver injury induced by Con A, and our findings suggest that NK cells play a critical role in this response.
Key Words: dioxin; autoimmune; inflammation; chemokines; liver.
Despite increasing prevalence in the population, the etiology of autoimmune liver diseases is still not thoroughly understood (Feld and Heathcote, 2003). A number of factors appear to confer
susceptibility to these diseases. In addition to known genetic risk factors, exposure to environmental xenobiotics is associated with increased incidence of autoimmune disease (Czaja and Manns, 2010; Gilbert, 2010; Longhi et al., 2010). A role for the aryl hydrocarbon receptor (AhR) in the development of these diseases has also been hypothesized. In support of this hypothesis, expo-sure of synovial tissue from patients with rheumatoid arthritis to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) led to increased production of inflammatory mediators (Kobayashi et al., 2008). TCDD and other AhR ligands also altered the onset and severity of injury in experimental animal models of autoimmune disease; however, the nature of the effect of AhR stimulation on patho-genesis is complex, with reports of both suppression and exac-erbation of responses. For example, prenatal treatment of mice with TCDD increased signs of autoimmunity and enhanced kid-ney damage in a murine model of autoimmune lupus (Holladay et al., 2011; Kobayashi et al., 2008; Mustafa et al., 2009). On the other hand, in experimental autoimmune encephalomyeli-tis, deterioration of spinal cord neurons was reduced in TCDD-treated mice (Quintana et al., 2008). TCDD also suppressed the development of autoimmune type 1 diabetes in nonobese dia-betic mice (Kerkvliet et al., 2009). Thus, exposure to TCDD can have diverse effects on immune-mediated pathogenesis.
The administration of the polyclonal T-cell mitogen conca-navalin A (Con A) to mice has been used to produce a model of immune-mediated liver injury that resembles the pathophysiol-ogy of autoimmune hepatitis (Tiegs et al., 1992). In this model, errant immune cell activation and destruction of hepatic paren-chymal tissue is similar to that which occurs in patients with autoimmune hepatitis (Peters, 2002; Wang et al., 2012). We have previously reported that pretreatment with TCDD sensi-tized mice to liver injury induced by Con A (Fullerton et al., 2013). The exacerbated response was associated with increased production of interferon-γ (IFNγ) and enhanced activation of natural killer T (NKT) cells (Fullerton et al., 2013), but the effect of pretreatment with TCDD on other immune cell types in this model is unknown.
toxicological sciences 136(1), 72–85 2013doi:10.1093/toxsci/kft174Advance Access publication August 22, 2013
by guest on July 12, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

TCDD/CON A-INDUCED LIVER INJURY
In the model of hepatitis induced by Con A alone, activation of a number of hepatic immune cell types contributes to liver injury. The primary effector cells appear to be NKT cells, which are required for the development of injury (Kaneko et al., 2000; Takeda et al., 2000). However, conventional CD4+ T cells also contribute to Con A-mediated liver injury, and other hepatic immune cells appear to play accessory roles in the development of liver damage (Tiegs and Gantner, 1996). Con A administra-tion caused a significant increase in the number of neutrophils in the liver, and neutrophil depletion reduced Con A hepato-toxicity (Bonder et al., 2004; Hatada et al., 2005). In addition, hepatic macrophages contribute to injury through the release of inflammatory mediators including tumor necrosis alpha (TNF-α), interleukin (IL)-18, and IL-12, which act directly on paren-chymal cells or on lymphocytes to increase production of other cytokines and augment direct cytolytic activity (Faggioni et al., 2000; Nicoletti et al., 2000; Schümann et al., 2000). It is apparent that Con A-induced hepatitis results from the activity of a variety of immune cell types and that the altered response of any one type has the potential to affect the development and severity of injury.
Similarly, TCDD exposure is known to alter the activity of many types of immune cells. Suppressed immune cell function has been reported (Kerkvliet et al., 2002; Sulentic and Kaminski, 2011); however, TCDD can increase the production of inflam-matory mediators, particularly from innate immune cells. This is primarily through the activation of AhR signaling (Esser et al., 2009; Kerkvliet, 1995, 2009). In a mouse model of influenza infection, TCDD exposure increased neutrophil recruitment to the lung, resulting in exacerbated tissue injury (Teske et al., 2005). Enhanced accumulation of neutrophils in the peritoneal cavity and increased inflammatory cytokine production were observed after administration of sheep red blood cells to TCDD-treated mice (Moos et al., 1994). Furthermore, TCDD treatment augmented the response of macrophages to inflammatory stimuli such as lipopolysaccharide (Moos et al., 1997). Mice exposed to TCDD had increased mRNA expression of monocyte chem-oattractant protein-1 (MCP-1) and keratinocyte chemoattractant (KC) in spleen, kidney and liver tissues, and increased expres-sion of these chemokines was associated with enhanced accumu-lation of macrophages in those tissues (Vogel et al., 2007).
Given the effects of TCDD on neutrophils and macrophages in other models of inflammatory injury (Teske et al., 2005; Wu et al., 2011) and the contribution of these cell types to the development of liver injury after Con A administration, it was of interest to determine whether these accessory cells play a role in the increased sensitivity of TCDD-pretreated mice to Con A-induced liver injury.
MATeRIAls AND MeTHODs
Materials. All materials were purchased from Sigma-Aldrich (St Louis, MO) unless otherwise stated. TCDD was purchased from Accustandard (New Haven, CT) dissolved in dimethyl sulfoxide and diluted in olive oil to a work-ing concentration of 0.2 μg/ml.
Mice. Male C57Bl/6J, B6.129S6-Cd1d1/Cd1d2tm1Spb/J and B6.129S7-Rag1tm1Mom/J mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and acclimated for at least 1 week in a 12-h light/dark cycle with access to Global Rodent diet 2018 (Harlan Teklad, Madison, WI) and bottled spring water ad libitum. Mice were used at 10–12 weeks of age, and all procedures were carried out with the approval of the Michigan State University Institutional Animal Care and Use Committee. All experiments were performed in accord-ance with the Guide for the Care and Use of Laboratory Animals as adopted by the United States National Institutes of Health.
Experimental protocols. A single administration of 0.3, 3, or 30 μg/kg TCDD or olive oil (vehicle) was given by oral gavage on day 0, followed on day 4, 7, or 10 by IV administration of saline or 6 mg/kg Con A (Lot 096 K7011). During the course of each experiment, TCDD-treated mice were housed in separate cages from vehicle-treated mice. The activity of alanine aminotrans-ferase (ALT) in plasma was measured spectrophotometrically using Infinity ALT reagent (Thermo Fischer Scientific, Waltham, MA). Depletion of NK cells was performed using rabbit anti-mouse/rat asialoGM1 polyclonal anti-body (Cedar Lane, Burlington, Ontario, Canada) according to the manufac-turer’s instructions. Briefly, 50 uL of reconstituted asialoGM1 antibody (Lot DBJ5790) was administered IV to mice in a volume of 150 μL normal rabbit serum. AsialoGM1 antibody or control rabbit serum was given 18 h before Con A administration. We have previously demonstrated that this treatment is suf-ficient to deplete hepatic NK cells (Dugan et al., 2011).
Histopathology. For neutrophil identification, paraffin-embedded liver sections were stained with rabbit anti-mouse neutrophil antibodies by the Michigan State University Investigative Histopathology Laboratory as described previously (Yee et al., 2003). For each mouse liver section, stained neutrophils were counted in 10 randomly chosen, 40× fields, and the mean count for all fields was used as the value for 1 independent replicate.
Cytokine analysis. Plasma concentration of IL-12 was measured using an OptEIA ELISA kit purchased from BD (Franklin Lakes, NJ). A bead-based Milliplex MAP immunodetection array (Millipore, Billerica, MA) was used to measure plasma concentrations of KC, MCP-1, and macrophage inflammatory protein 2 (MIP-2) on a Bio-plex instrument (Bio-Rad Laboratories, Hercules, CA).
RNA isolation and Real Time-PCR analysis. Liver samples were homoge-nized in TRI reagent (Molecular Research Center, Cincinnati, OH), and isolation of total RNA was performed according to the manufacturer’s instructions. The quantity and quality of isolated RNA was determined using a nanodrop spectro-photometer (Thermo Scientific, Waltham, MA). Complementary DNA (cDNA) was prepared from 1 μg of RNA using iscript reverse transcription supermix for Real Time-qPCR (Bio-Rad Laboratories). Expression levels of target genes were determined on a Step-one real-time PCR system (Applied Biosystems, Foster City CA) utilizing specific DNA oligos and SYBR green PCR mas-ter mix (Applied Biosystems). Copy number was determined by comparison with standard curves for each respective gene generated from pooled cDNA. The target gene expression levels were standardized to the geometric mean of expression levels of glyceraldehyde-3-phosphate dehydrogenase (Gapdh), beta-actin (Actb), and hypoxanthine guanine phosphoribosyl transferase (Hprt). To evaluate the expression of target genes, the following PCR primers were used: Gapdh (115 bp), 5′-TCAACAGCAACTCCCACTCTTCCA-3′ (for-ward), 5′-ACCCTGTTGCTGTAGCCGTATTCA-3′ (reverse); Actb (140 bp), 5′-TGTGATGGTGGGAATGGGTCAGAA-3′ (forward), 5′-TGTGGTGCCAG ATCTTCTCCATGT-3′ (reverse); Hprt (133 bp), 5′-GGAGTCCTG-TTGA TGTTGCCAGTA-3′ (forward), 5′-GGGACGCAGCAACTGACATTTCTA-3′ (reve rse); IL-12p40, Il12b (101 bp), 5′-AAAGCTGTCTTCTGCTTGGTTGG C-3′ (forward), 5′-CTGGCTCTGCGGGCATTTAACATT-3′ (reverse); IL-27, Il27 (107 bp), 5′-GTGACAGGAGACCTTGGCTG-3′ (forward), 5′-AGCT CTTGAAGGCTCAGGG-3′ (reverse). mRNA expression data are reported as fold change of standardized treatment over standardized vehicle/saline treat-ment at time zero.
Flow cytometry. Hepatic leukocytes were isolated from mice and prepared for flow cytometry analysis as follows. Mouse livers were washed with PBS
73
by guest on July 12, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

FULLERTON, ROTH, AND GANEY
without calcium and magnesium. Livers were placed in RPMI medium sup-plemented with 5% fetal bovine serum (FBS) and 1% penicillin/streptomycin and then passed through a nylon mesh, and the resulting cell suspension was centrifuged at a speed of 50 × g for 5 min at 4°C. The supernatant was removed, and the pelleted hepatocytes were discarded. The supernatant was centrifuged at 450 × g for 5 min to pellet leukocytes. The pellet was then incubated for 4 min with red blood cell lysis buffer (BioLegend, San Diego, CA) followed by 2 washes with PBS containing 5% FBS. Lympholyte-M (Cedar Lane) was used according to manufacturer’s instructions to further purify leukocytes. For ex vivo stimulation experiments, hepatic leukocytes were cultured in RPMI medium supplemented with 5% FBS and 1% penicillin/streptomycin with or without 7.5 μg/ml Con A for 5 h. In all other instances, hepatic leukocytes were stained and prepared immediately for flow cytometric analysis.
Hepatic leukocytes were first incubated with TruStain FcX (anti-mouse CD16/CD32) to minimize nonspecific binding of staining antibodies to Fcγ receptors. Antibodies used for staining of NK, NKT, and T cells included fluo-rescein isothiocyanate or phycoerythrin-conjugated anti-NK1.1 (PK136) and allophycocyanin-cyanine dye 7-conjugated anti-CD3epsilon (145- 2c11), as well as pacific blue-conjugated anti-CD69 (H1.2F3) and allophycocyanin-con-jugated anti-NKG2d (CX5). Staining of hepatic macrophages and neutrophils was performed using fluorescein isothiocyanate or allophycocyanin-conjugated anti-F4/80 (BM8), phycoerythrin-conjugated anti-CD11b (M1/70), and phyco-erythrin-cyanine dye 7-conjugated anti-Gr-1 (RB6-8C5). Appropriate fluores-cent-conjugated isotype controls were utilized to establish positive and negative gating parameters for each antibody. Unless otherwise stated, all reagents and antibodies for flow cytometric staining were purchased from BioLegend (San Diego, CA). Hepatic leukocyte staining was performed according to manu-facturer’s directions, and stained samples were analyzed on a BD FACSCanto II with subsequent data analysis performed using Kaluza software (Beckman Coulter, Brea, CA).
Statistical analysis. Experimental results are expressed as mean ± SEM. Arcsine transformation was performed on percentile data. In some instances, a Box-Cox transformation was utilized to normalize data for further analysis. Statistical analysis of data was performed using either student’s t-test or 2-way ANOVA followed by pairwise multiple comparisons using Student–Newman–Keuls or Tukey’s method where appropriate. Analysis of nonparametric data was performed using Kruskal–Wallis 1-way ANOVA on ranks followed by pairwise multiple comparisons using Tukey’s or Dunn’s method where appro-priate. The criterion for statistical significance was p < 0.05. For all experi-ments, the term “independent replicates” refers to biological samples collected from separate mice in each treatment group.
ResulTs
TCDD Sensitization to Con A: Dose response
We have previously demonstrated that pretreatment with a nonhepatotoxic dose of 30 μg/kg TCDD exacerbates the inflammatory liver injury induced by a dose of 6 mg/kg Con A administered 4 days later (Fullerton et al., 2013). This study was undertaken to determine if smaller doses of TCDD sensitize mice to Con A-induced liver injury. When saline was adminis-tered on day 4, no liver injury developed in vehicle-pretreated mice or in TCDD-pretreated mice regardless of dose, as deter-mined by measurements of ALT activity in plasma (Fig. 1A). When Con A was administered on day 4, an increase in ALT activity in the plasma was detected in vehicle-pretreated mice, indicating moderate hepatotoxicity. Compared to pretreatment with vehicle, mice pretreated with 3 or 30 μg/kg TCDD had significantly increased ALT activity in plasma 8 h after Con A administration.
To evaluate the influence of the interval between administra-tion of TCDD and Con A, mice were given Con A 4, 7, or 10 days after pretreatment with vehicle or 30 μg/kg TCDD. They were euthanized 24 h later. At all intervals evaluated, TCDD/Con A-treated mice had increased ALT activity in plasma compared with vehicle/Con A-treated mice (Supplementary Figure 1), and the increase in ALT was greatest with the 7-day interval.
Inflammatory Chemokines in TCDD/Con A-Induced Liver Injury
Inflammatory liver injury often involves the recruitment of numerous cell types to the liver; such recruitment is mediated by various chemokines. Mice treated with TCDD/Con A devel-oped liver injury that was first evident 4 h after the adminis-tration of Con A and peaked at 8 h (Fullerton et al., 2013). In this study, the production of chemokines was measured in the plasma at times prior to the peak of TCDD/Con A-induced liver injury. TCDD pretreatment alone decreased plasma concentra-tion of KC compared with vehicle pretreatment in the absence of Con A (Fig. 2A). Concentrations of KC, MCP-1, and MIP-2 were significantly increased 2 h after Con A administration and returned to baseline by 6 h (Figs. 2A–C). TCDD pretreatment did not alter the induction of KC, or MCP-1 by Con A, at any time but led to a significant increase in the plasma concentration of MIP-2 two hours after Con A administration. In TCDD/Con A-treated mice, the concentration of KC in plasma remained elevated at 6 h compared with 0 h.
Hepatic Neutrophil Accumulation in TCDD/Con A-Induced Liver Injury
The increase in plasma KC and MIP-2 concentrations in TCDD/Con A-treated mice raised the possibility of a
FIG. 1. Dose-dependent exacerbation of Con A-induced liver injury by TCDD. Mice were treated on day 0 with 0, 0.3, 3, or 30 μg/kg of TCDD. After 4 days, they were given 6 mg/kg Con A or saline. ALT activity in plasma was measured 8 h after Con A or saline administration. a, p < 0.05 vehicle/Con A versus vehicle/saline. b, p < 0.05 TCDD/Con A versus the same TCDD dose with saline treatment. c, p < 0.05 TCDD/Con A versus vehicle/Con A. Data represent the mean ± SE of 3–6 independent replicates per treatment group.
74
by guest on July 12, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

TCDD/CON A-INDUCED LIVER INJURY
greater influx of neutrophils into livers of those animals. Accumulation of neutrophils in the liver was determined by immunohistochemical staining of liver samples collected 4 h after the administration of saline or Con A. TCDD pretreat-ment alone did not result in increased numbers of neutrophils in the liver (Figs. 3A and 3B). Con A administration signifi-cantly increased the number of neutrophils compared with vehicle/saline treatment, and TCDD pretreatment did not alter the accumulation or distribution of neutrophils in the liver.
Flow cytometry was also used to evaluate the percentage of neutrophils (Gr-1+, CD11b+ cells) in the isolated hepatic leuko-cyte population at 0, 2, 4, and 24 h after Con A administration. TCDD pretreatment did not alter the percentage of neutrophils in the liver at any time after Con A administration (data not shown).
Role of Hepatic Macrophages in TCDD/Con A-Induced Liver Injury
Macrophages play an accessory role in the development of Con A hepatitis (Hatano et al., 2008; Nakamura et al., 2001; Schümann et al., 2000). MIP-2 is produced by acti-vated macrophages, and its concentration was increased in plasma of TCDD-pretreated mice (Fig. 2C). Accordingly, the role of hepatic macrophages in the development of TCDD/Con A-induced liver injury was evaluated. The per-centage of monocytes/macrophages (F4-80+ cells) in the population of intrahepatic leukocytes isolated after Con A administration was evaluated by flow cytometry at times before the development of liver injury. Compared with vehicle, TCDD pretreatment 4 days earlier did not alter the percentage of macrophages measured in the liver at 0 h (Fig. 4A). The percentage of macrophages observed was not affected by Con A administration in either vehicle- or TCDD-pretreated mice.
In addition to the quantification of macrophages by flow cytometry, the effect of TCDD pretreatment on the production of macrophage-derived cytokines in response to Con A was evaluated. The plasma concentration of IL-12 was measured at times before the development and at the peak of liver injury. Con A administration resulted in an increased concentration of IL-12 in plasma at 3, 6, and 8 h after treatment (Fig. 4B). TCDD pretreatment decreased the concentration of IL-12 in plasma measured 8 h after Con A administration. The hepatic expres-sion of IL-12 and IL-27 mRNA was assessed by real-time PCR. Expression of both was increased in Con A-treated mice at 3 and 6 h after administration, respectively, but expression was not affected by pretreatment with TCDD (Figs. 4C and 4D).
NKT and T Cells in TCDD-Induced Sensitization of Mice to Con A Hepatotoxicity
Both NKT and conventional CD4+ T cells have been implicated as major effector cells in the development of Con A-induced liver injury, so the requirement for these cell types in the sensitization
FIG. 2. Concentrations of KC (A), MCP-1 (B), and MIP-2 (C) in plasma after Con A administration. Mice were treated on day 0 with vehicle (gray bars) or 30 μg/kg TCDD (black bars) and on day 4 with 6 mg/kg Con A. Plasma samples were collected at various times after the administration of Con A. a, p < 0.05 versus vehicle pretreatment at 0 h. b, p < 0.05 versus TCDD pretreatment at 0 h. c, p < 0.05 TCDD pretreatment versus vehicle pretreatment at the same time point. Data represent the mean ± SE of 5–10 independent replicates per treatment group. Data were combined from 2 sep-arate experiments.
75
by guest on July 12, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

FULLERTON, ROTH, AND GANEY
to Con A hepatotoxicity induced by TCDD pretreatment was evaluated. NKT cell-deficient B6.129S6-Cd1d1/Cd1d2tm1Spb/J (CD1d KO) mice were used for this purpose. In addition, B6.129S7-Rag1tm1Mom/J (RAG1 KO) mice lacking mature T cells but with intact NK cells were used to determine if CD4+ T cells are required for injury and if the presence of NK cells alone is sufficient to cause injury following TCDD/Con A treatment.
TCDD treatment alone did not cause liver injury in CD1d KO, Rag1 KO, or C57Bl/6J (wild type) mice (data not shown). TCDD-pretreated wild-type mice given Con A had increased plasma ALT activity compared with vehicle-pre-treated, wild-type mice (Fig. 5A). Plasma ALT activity was reduced in CD1d KO mice regardless of TCDD treatment; however, ALT activity in CD1d KO mice treated with TCDD/Con A was reduced only to the level in wild-type mice treated
with Con A. RAG1 KO mice were completely protected from liver injury induced by either vehicle/Con A or TCDD/Con A treatments.
As previously stated, enhanced IFNγ production is required for liver injury after TCDD/Con A treatment. In vehicle/Con A-treated mice, the plasma concentration of IFNγ was similar in wild-type and CD1d KO mice but undetectable in RAG1 KO mice (Fig. 5B). In mice treated with Con A, TCDD-pretreated, wild-type mice had increased concentrations of IFNγ compared with wild-type mice given Con A alone. The plasma concentration of IFNγ in TCDD/Con A-treated CD1d KO mice was not different from wild-type mice given the same treatment or from vehicle/Con A-treated CD1d KO mice. In RAG1 KO mice treated with TCDD/Con A, IFNγ was not detectable in plasma.
FIG. 3. Hepatic neutrophil accumulation after Con A administration. Mice were treated as described in the legend to Figure 2 with vehicle/saline, TCDD/saline, vehicle/Con A, or TCDD/Con A. A, Mice were euthanized 4 h after saline or Con A administration. Paraffin-embedded liver sections were stained for neutrophils. Representative liver sections were photographed at ×40 magnification. Examples of positive neutrophil staining are indicated by arrows. B, Immunohistochemical staining of neutrophils in the livers at 4 h was quantified as described in Materials and Methods. a, p < 0.05 vehicle/Con A versus vehicle/saline. b, p < 0.05 TCDD/Con A versus TCDD/saline. Data represent the mean ± SEM of 3–4 independent replicates per treatment group.
76
by guest on July 12, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

TCDD/CON A-INDUCED LIVER INJURY
Effect of TCDD Pretreatment on In Vivo and Ex Vivo Activation of Hepatic Lymphocytes by Con A
Lymphocytes were isolated from livers of mice pretreated with vehicle or TCDD 2 h after administration of either saline
or Con A. Expression of the early lymphocyte activation marker CD69 was evaluated by flow cytometry. TCDD pretreatment alone increased the percentage of activated CD69-positive NK (NK1.1+, CD3ε−) cells compared with vehicle pretreatment
FIG. 5. Liver injury (A) and plasma IFNγ concentration (B) after TCDD/Con A treatment in CD1d KO and RAG1 KO mice. Wild-type (open bars), B6.129S6-Cd1d1/Cd1d2tm1Spb/J (CD1d KO) (striped bars), and B6.129S7-Rag1tm1Mom/J (RAG1 KO) (cross-hatched bars) mice were treated as described in the legend to Figure 2 with vehicle/Con A or TCDD/Con A. Samples were collected 8 h after Con A administration. c, p < 0.05 TCDD/Con A versus vehicle/Con A in the same mouse genotype. f, p < 0.05 versus the same treatment in wild-type mice. Abbreviation: ND, not detected (value below the limit of detection). Data represent the mean ± SEM of 4–10 independent replicates per treatment group. Data were combined from at least 2 separate experiments.
FIG. 4. Hepatic macrophages and macrophage-associated cytokines after Con A administration. Mice were treated as described in the legend to Figure 2 with vehicle/Con A (gray bars) or TCDD/Con A (black bars). A, percentage of F4/80-positive hepatic leukocytes. B, concentration of IL-12 in plasma. C, hepatic expres-sion of IL-12 mRNA after Con A administration. D, hepatic expression of IL-27 mRNA after Con A administration. a, p < 0.05 versus vehicle at 0 h. b, p < 0.05 versus TCDD at 0 h. c, p < 0.05 TCDD pretreatment versus vehicle pretreatment at the same time. Abbreviation: ND, not detected (value below the limit of detection; 3.2 pg/ml). Data represent the mean ± SEM of 4–6 independent replicates per treatment group. Data were combined from at least 2 separate experiments.
77
by guest on July 12, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

FULLERTON, ROTH, AND GANEY
(Fig. 6A). Con A administration also increased the percent-age of CD69-positive NK cells, and this response was further increased by TCDD pretreatment. An increase in the percent-age of CD69-positive NKT (NK1.1+, CD3ε+) cells was only observed in the TCDD/Con A group. Con A administration
increased the percentage of CD69-positive T (NK1.1−, CD3ε+) cells, but this response was not altered by TCDD pretreatment.
Increased activation of NK and NKT cells after treatment with TCDD/Con A could arise from exposure of these cells in vivo to danger signals released from damaged parenchy-mal cells. To investigate the effect of TCDD on the response of hepatic leukocytes to Con A in the absence of hepatocel-lular damage (Fig. 1), hepatic leukocytes were isolated from mice treated 4 days earlier with TCDD or saline, and ex vivo activation of lymphocytes by Con A was evaluated. Cells were exposed in culture to PBS (vehicle) or to Con A (7.5 μg/ml) for 5 h and analyzed by flow cytometry. A greater percentage of NK cells isolated from TCDD-pretreated mice were CD69-positive compared with those isolated from vehicle-pretreated mice (Fig. 6B). Con A stimulation of NK cells isolated from TCDD-pretreated mice, but not vehicle-pretreated mice, resulted in significantly increased percentage of CD69-positive cells. Con A activation increased the percentage of CD69-positive NKT cells, and pretreatment with TCDD increased the percentage of CD69-positive NKT cells in the absence and presence of Con A stimulation. Exposure of T cells isolated from vehicle-pre-treated mice to Con A led to an increased percentage of CD69-positive T cells, and this response was further increased in T cells isolated from TCDD-pretreated mice.
Activation of NK Cells in TCDD/Con A-induced Liver Injury
Flow cytometry was used to develop a more extensive time course of the activation of NK cells after TCDD/Con A treat-ment. Hepatic lymphocytes were isolated from mice at 0, 2, 3, 4, or 8 h after Con A administration. In vehicle/Con A-treated mice, increased percentages of CD69-positive NK cells were detected at 3, 4, and 8 h (Figs. 7A and 7B). TCDD pretreatment increased the percentage of CD69-positive NK cells compared with vehicle pretreatment at all times evaluated. The percent-age of NK cells expressing the activating receptor NKG2d was increased 3 h after Con A administration (Fig. 7C). This response to Con A was not altered by pretreatment with TCDD.
The percentage of CD69-positive NK cells was decreased in RAG1 KO mice compared with wild-type or CD1d KO mice in both vehicle/Con A and TCDD/Con A treatments at 8 h (Fig. 8). In each genotype, TCDD/Con A treatment sig-nificantly increased the percentage of CD69-positive NK cells compared with vehicle/Con A treatment.
Role of NK Cells in Exacerbation of Con A Hepatotoxicity by TCDD Pretreatment
NK cells are not considered to be important effectors of liver injury caused by large doses (15–25 mg/kg) of Con A (Takeda et al., 2000; Toyabe et al., 1997); however, the TCDD-mediated activation of NK cells prompted the investigation of the impor-tance of these cells in the development of liver injury after TCDD/Con A treatment. Pretreatment with TCDD led to increased plasma ALT activity in Con A (6 mg/kg)-treated mice given
FIG. 6. Effect of pretreatment with TCDD on in vivo and ex vivo activation of hepatic lymphocytes by Con A. A, Mice were pretreated with vehicle (white bars) or TCDD (gray bars) as described in the legend to Figure 2, and intra-hepatic leukocytes were isolated 2 h after saline (open bars) or Con A (striped bars) administration. Cells were stained for expression of activation marker CD69 and analyzed by flow cytometry. NK cells were identified as (NK1.1+, CD3ε−), NKT cells as (NK1.1+, CD3ε+), and T cells as (NK1.1−, CD3ε+). B, Intrahepatic leukocytes were isolated from mice treated 4 days earlier with vehicle (white bars) or 30 μg/kg TCDD (gray bars) and were treated ex vivo with either PBS (open bars) or 7.5 μg/ml Con A (striped bars) in culture for 5 h. NK cells, NKT cells, and T cells were stained for the lymphocyte activa-tion marker CD69 and analyzed by flow cytometry. a, p < 0.05 vehicle/Con A versus vehicle/saline in the same cell type. b, p < 0.05 TCDD/Con A versus TCDD/saline in the same cell type. c, p < 0.05 TCDD/Con A versus vehicle/Con A in the same cell type. d, p < 0.05 TCDD/saline versus vehicle/saline in the same cell type. Data represent the mean ± SEM of 6–7 independent repli-cates per treatment group. Data were combined from 3 separate experiments.
78
by guest on July 12, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

TCDD/CON A-INDUCED LIVER INJURY
control serum (Fig. 9A). Anti-asialoGM1 depletion of NK cells significantly reduced injury in vehicle- and TCDD-pretreated mice administered 6 mg/kg Con A. In these experiments, a sepa-rate group of mice was pretreated with vehicle and then given a larger dose (20 mg/kg) of Con A to compare the effects of anti-asialoGM1 treatment in our studies with results previously reported in the literature (Toyabe et al., 1997). The plasma ALT activity in control serum-treated mice given this larger dose of Con A was comparable with ALT activity in TCDD/Con A-treated mice given control serum. Anti-asialoGM1 treatment did not protect against injury in mice given 20 mg/kg Con A.
In addition, compared with mice treated with control serum, anti-asialoGM1 treatment significantly reduced the concentra-tion of IFNγ in plasma 8 h after Con A administration in all treatment groups (Fig. 9B).
DIsCussION
TCDD is disproportionately distributed to hepatic tissue after exposure, resulting in relatively large hepatic concentra-tions, and the liver is a major target organ for TCDD toxicity in
FIG. 7. Activation of NK cells after TCDD/Con A treatment. A, Representative histograms showing fluorescence intensity of CD69 staining on NK cells 4 h after Con A or saline administration. Treatments are indicated in the panels. The horizontal bar represents the area of positive staining based on the isotype control. In each histogram, total NK cells refer to the number of NK cells measured in the treatment group sample. B, The percentage of NK cells staining positive for CD69 at 0, 2, 3, 4, and 8 h after Con A administration. a, p < 0.05 versus vehicle pretreatment at 0 h. b, p < 0.05 versus TCDD pretreatment at 0 h. c, p < 0.05 TCDD/Con A versus vehicle/Con A at the same time point. Data represent the mean ± SEM of 4–7 independent replicates per treatment group. Data were collected from at least 2 separate experiments. C, The percentage of NK cells staining positive for NKG2d at 0, 3, and 8 h after Con A or saline administration. a, p < 0.05 versus vehicle pretreatment at 0 h. b, p < 0.05 versus TCDD pretreatment at 0 h. Data represent the mean ± SEM of 3–5 independent replicates per treatment group. Data were combined from 2 separate experiments.
79
by guest on July 12, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

FULLERTON, ROTH, AND GANEY
many species (Abraham et al., 1988; Birnbaum and Tuomisto, 2000; Diliberto et al., 1995; Poland and Knutson, 1982; Thoma et al., 1990). In the liver, TCDD induces prolonged activation of the AhR, resulting in extensive changes in gene expression, and altered expression of many AhR-regulated genes associ-ated with immune cell activity has been identified (Dere et al., 2011; Kerkvliet, 2009; Stevens et al., 2009). However, despite the importance of the liver in AhR-mediated gene expression after TCDD treatment and the increasing experimental evi-dence that TCDD treatment alters the development of various immune-mediated diseases, the effect of TCDD exposure on the development of autoimmune liver disease has not been addressed thoroughly.
We previously determined that TCDD increased the sensitiv-ity of mice to liver injury in a model of autoimmune hepatitis induced by the administration of Con A (Fullerton et al., 2013). Here, we present data demonstrating that a 10-fold smaller dose of TCDD produced an equivalent degree of liver injury in Con A-treated mice (Fig. 1A). Decreasing the TCDD dose to 0.3 μg/kg resulted in loss of the TCDD-induced sensitization. These results suggest that the threshold dose for the exacerbation of Con A-induced liver injury is between 0.3 and 3 μg/kg TCDD. Toxicokinetic studies revealed that accumulation of TCDD in the liver peaks 4 days following treatment, and the elimination half-life of TCDD is approximately 8 days in mice (Birnbaum, 1986). TCDD exacerbated the hepatotoxic response to Con A when given 4, 7, or 10 days earlier (Supplementary Figure 1), and the greatest response was seen with Con A administration 7 days after TCDD pretreatment. Because liver concentration decreases from 4 to 7 days after a single TCDD administration
(Birnbaum, 1986; Kopec et al., 2008), our results suggest that the degree of injury in TCDD/Con A-treated mice is not sim-ply related to the hepatic concentration of TCDD at the time of Con A administration but rather is likely the result of other changes that follow TCDD treatment. It has been reported that peak induction of chemokines such as KC and MCP-1 occurs at 1 and 7 days, respectively, after a single administration of TCDD, and various batteries of genes are altered in expres-sion differently during this same time period (Boverhof et al., 2005; Nault et al., 2013). Additionally, the effect of TCDD on
FIG. 8. Activation of NK cells in CD1d KO and RAG1 KO mice after TCDD/Con A treatment. Wild-type (open bars), B6.129S6-Cd1d1/Cd1d2tm1Spb/J (CD1d KO; striped bars), and B6.129S7-Rag1tm1Mom/J (RAG1 KO; cross-hatched bars) mice were treated as described in the legend to Figure 2 with vehicle/Con A or TCDD/Con A. Intrahepatic leukocytes were isolated from mice 8 h after Con A administration, and NK cells were stained for CD69 expression. c, p < 0.05 TCDD/Con A versus vehicle/Con A in the same mouse genotype. f, p < 0.05 versus the same treatment in wild-type. Data represent the mean ± SEM of 3–5 independent replicates per treatment group.
FIG. 9. The effect of NK cell depletion on TCDD/Con A-mediated liver injury (A) and IFNγ production (B). Mice were treated as described in the legend to Figure 2 with vehicle (white bars) or TCDD (gray bars) on day 0, then treated with either normal rabbit serum (open bars) or rabbit anti-mouse/rat asialoGM1 polyclonal antibody (striped bars), as described in Materials and Methods section, 18 h prior to the administration of 6 mg/kg (white and gray bars) or 20 mg/kg (black bars) Con A. c, p < 0.05 versus vehicle/6 mg/kg Con A in the same control serum or anti-asialo treatment. f, p < 0.05 versus the same treatment with control serum. Data represent the mean ± SEM of 7–11 inde-pendent replicates for all groups treated with 6 mg/kg Con A and 3–4 independ-ent replicates for all groups treated with 20 mg/kg Con A. Data were combined from 2 separate experiments.
80
by guest on July 12, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

TCDD/CON A-INDUCED LIVER INJURY
the development of inflammatory liver injury varies greatly with the time between TCDD exposure and administration of the inflammagen lipopolysaccharide (Patterson et al., 2003). Although the mechanism is unknown, the TCDD-induced sen-sitization to Con A is a persistent effect.
Several chemokines are responsible for the recruitment of inflammatory cells into the liver during the development of injury. KC and MIP-2 both belong to the CXC-type chemokine family and act on CXC receptor 2. These chemokines are syn-thesized by activated tissue macrophages. KC and MIP-2 per-form similar functions to increase neutrophil egress from the bone marrow and mediate transmigration of these cells into the peripheral tissues (De Filippo et al., 2008; Lee et al., 1995; Sadik et al., 2011). MCP-1 is a CCL-type chemokine respon-sible for the recruitment of monocytes to sites of inflamma-tion (Zimmermann et al., 2012). TCDD treatment enhanced the expression of MCP-1 and KC and increased the recruitment of neutrophils and monocytes to sites of tissue injury (Vogel et al., 2007). Con A treatment induced the production of MIP-2 in a TNF-α-dependent manner, and neutralization of MIP-2 decreased the accumulation of neutrophils in the liver and reduced hepatocellular injury resulting from Con A adminis-tration (Nakamura et al., 2001). In the studies presented here, administration of Con A induced KC, MIP-2, and MCP-1 pro-duction. Although TCDD treatment alone did not affect the plasma concentration of these chemokines, in Con A-treated mice, TCDD increased the concentration of MIP-2 in plasma (Figs. 2A–C).
Neutrophils contribute to the development of injury and to the production of IFNγ by lymphocytes in Con A-treated mice (Hatada et al., 2005), and neutrophil depletion reduced the severity of liver injury (Bonder et al., 2004). Despite the increased concentration of MIP-2 in plasma in TCDD/Con A-treated mice, TCDD pretreatment did not alter the accumu-lation of neutrophils in the liver in response to Con A (Fig. 3). Based on these results, it is unlikely that neutrophils play an important role in the exacerbation of Con A-induced liver injury by TCDD.
Macrophages are another hepatic immune cell known to play an accessory role in hepatitis and liver damage induced by large doses of Con A. Hepatic macrophages contribute to injury via the production of inflammatory mediators such as TNF-α, which can induce hepatic parenchymal cell death (Gantner et al., 1996; Schümann et al., 2000). In addition, stimulated macrophages produce IL-12 and IL-27, which acti-vate hepatic lymphocytes and enhance cytolytic activity of NK and NKT cells while promoting Th1 polarization of T cells. IL-12 and IL-27 also increase the production of IFNγ from NK and CD4+ T cells (Pflanz et al., 2002; Vignali and Kuchroo, 2012). IFNγ and TNF-α can act synergistically to kill hepatic parenchymal cells (Adamson and Billings, 1993). In studies presented here, TCDD pretreatment of mice given Con A did not change the percentage of macrophages (F4/80-positive cells) recovered from the liver (Fig. 4A). In addition, there
was no difference in plasma concentration of IL-12 in TCDD-pretreated and vehicle-pretreated mice at 3 h (Fig. 4B), a time before the development of liver injury (Fullerton et al., 2013). Furthermore, TCDD pretreatment decreased IL-12 produc-tion induced by Con A. TCDD pretreatment also did not alter hepatic mRNA expression of IL-12 or IL-27 (Figs. 4C and 4D). Collectively, these results suggest that while macrophages are likely involved in the development of injury, they do not play an important role in the increased sensitivity to Con A observed in TCDD-pretreated mice.
The importance of NKT cells and conventional CD4+ T cells has been well documented in the development of Con A-induced liver injury. In particular, NKT cells are required for the development of injury and directly contribute to the killing of hepatic parenchymal cells via expression of cytolytic effec-tors such as Fas ligand (FasL; Seino et al., 1997; Tagawa et al., 1998). A number of studies have demonstrated protection from Con A-induced liver injury in mice deficient in NKT cells and in Rag1 KO mice lacking mature T cells (Kaneko et al., 2000; Takeda et al., 2000). Using a smaller dose of Con A (6 mg/kg), we saw results similar to those previously reported with larger Con A doses (Fig. 5A). TCDD/Con A-induced liver injury was abolished in RAG1 KO mice, confirming the essential role of T cells in the pathogenesis. Pretreatment with TCDD increases the activation of NKT cells and promotes expression of FasL in these cells after Con A administration (Fullerton et al., 2013). However, CD1d KO mice were only partially protected from TCDD/Con A-induced liver injury. In fact, ALT activity in the plasma of TCDD/Con A-treated CD1d KO mice was compa-rable with the ALT activity measured in the plasma of vehicle/Con A-treated wild-type mice. These results suggested that a cell type in addition to NKT cells contributes to TCDD-induced sensitization to Con A hepatotoxicity.
To identify this cell type, the activation of hepatic lympho-cytes was assessed. TCDD/Con A treatment in vivo resulted in a greater percentage of activated NK and NKT cells than either treatment alone (Fig. 6A). Interestingly, this response did not require exposure to Con A in vivo; a greater percentage of NK and NKT cells isolated from TCDD-treated mice than from vehicle-treated mice became activated upon exposure to Con A ex vivo (Fig. 6B).
Lymphocyte activation occurs 2 h after Con A administration (Fig. 6A) prior to any increase in plasma ALT activity (Fullerton et al., 2013). This suggests that lymphocyte activation occurs before initial hepatocyte injury. However, endogenous alarm-ins such as high-mobility group box 1 released from damaged hepatocytes and sinusoidal endothelial cells can activate lym-phocytes and exacerbate hepatic injury (Gong et al., 2010). Furthermore, Con A given at larger doses induces hepatic sinu-soidal endothelial cell damage within 15 min. Such damage can induce lymphocyte activation (Knolle et al., 1996). The obser-vation that a greater percentage of lymphocytes isolated from TCDD-treated mice became activated upon ex vivo stimulation with Con A compared with cells from vehicle-treated mice
81
by guest on July 12, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from
FULLERTON, ROTH, AND GANEY
(Fig. 6) suggests that TCDD alters lymphocyte activation by Con A independent of products released by dying cells.
One interesting result was that TCDD alone increased acti-vation of NK cells (Fig. 6). Upon further investigation, it was observed that TCDD pretreatment increased the activation of NK cells at all times examined (0–8 h) after Con A administra-tion (Figs. 7A and 7B). These results were unexpected because the role of NK cells as effectors in Con A-induced liver injury has been discounted (Dong et al., 2007; Kaneko et al., 2000; Takeda et al., 2000). Despite not being associated with Con A-induced liver injury, NK cells are known to play important roles in human autoimmune disease and inflammatory liver injury (Schleinitz et al., 2010). For example, NK-cell activation is increased in the livers of patients with primary biliary cir-rhosis (Chuang et al., 2006; Shimoda et al., 2011). In addition, a clear role for NK cells has been demonstrated in other animal models of immune-mediated liver injury. The administration of alpha-galactoceramide causes liver injury that is mediated by both NK and NKT cells (Trobonjaca et al., 2002). In alpha-galactoceramide-induced liver injury, NKT cells are responsi-ble for activating NK cells by producing IFNγ (Carnaud et al., 1999; Eberl and MacDonald, 2000). In studies presented here, there was no difference in the percentage of CD69-positive NK cells detected after TCDD/Con A treatment of wild-type and CD1d KO mice. This result indicates that increased NK-cell
activation is independent of the presence of NKT cells in this model. Interestingly, after either vehicle/Con A or TCDD/Con A treatment, the percentage of CD69-positive NK cells in RAG1 KO mice was decreased compared with wild-type and CD1d KO mice indicating a role for conventional T cells in the activation of NK cells following TCDD/Con A administration.
To determine if increased NK-cell activity could be a con-tributor to injury in TCDD/Con A-treated mice, NK cells were depleted with anti-asialoGM1 prior to the administration of Con A. As previously mentioned, NK-cell activity is reported to be inconsequential in the development of hepatitis from large doses of Con A (Toyabe et al., 1997). Our results are consist-ent with this finding (Fig. 9A): anti-asialoGM1 did not dimin-ish injury in mice treated with 20 mg/kg Con A. This treatment resulted in injury comparable with TCDD-pretreated mice given only 6 mg/kg Con A. However, in vehicle- or TCDD-pretreated mice administered 6 mg/kg Con A, NK-cell depletion by anti-asialoGM1 significantly protected against the development of injury. The reduction in hepatotoxicity was accompanied by a decrease in the concentration of IFNγ (Fig. 9B), which is criti-cal to the development of liver injury.
These results clearly demonstrate a role for increased NK-cell activation by TCDD pretreatment in the development of hepa-totoxicity from TCDD/Con A administration. Although the mechanisms underlying this response are not yet determined, a
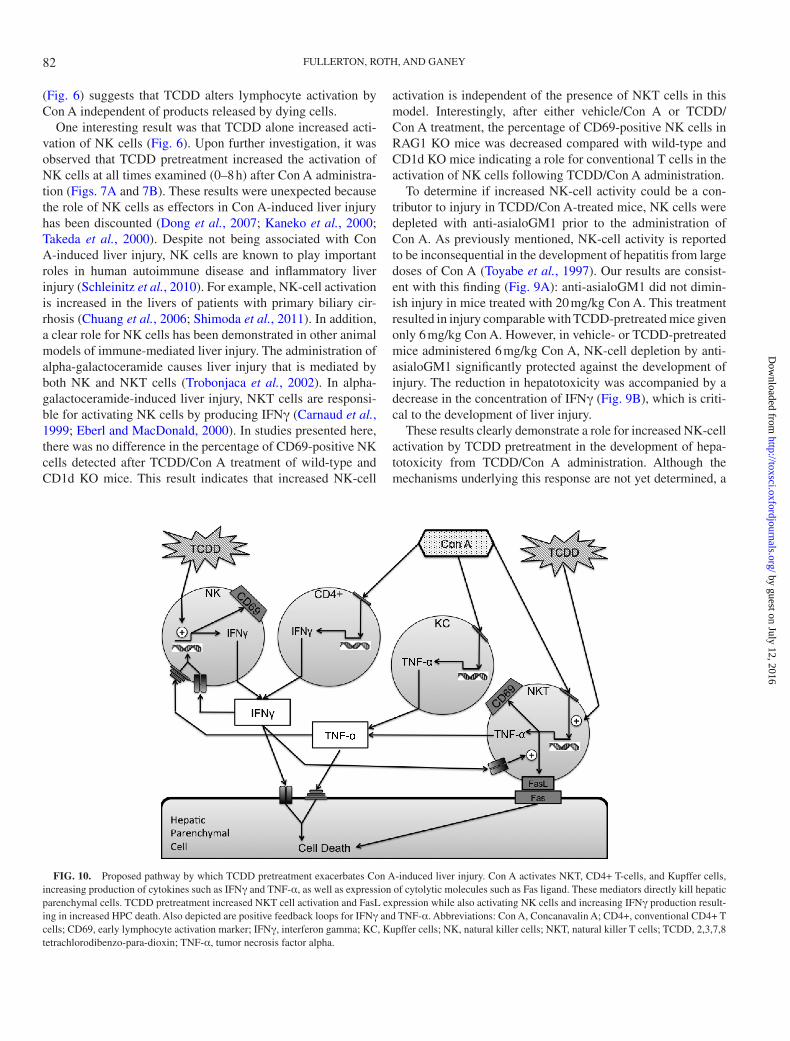
FIG. 10. Proposed pathway by which TCDD pretreatment exacerbates Con A-induced liver injury. Con A activates NKT, CD4+ T-cells, and Kupffer cells, increasing production of cytokines such as IFNγ and TNF-α, as well as expression of cytolytic molecules such as Fas ligand. These mediators directly kill hepatic parenchymal cells. TCDD pretreatment increased NKT cell activation and FasL expression while also activating NK cells and increasing IFNγ production result-ing in increased HPC death. Also depicted are positive feedback loops for IFNγ and TNF-α. Abbreviations: Con A, Concanavalin A; CD4+, conventional CD4+ T cells; CD69, early lymphocyte activation marker; IFNγ, interferon gamma; KC, Kupffer cells; NK, natural killer cells; NKT, natural killer T cells; TCDD, 2,3,7,8 tetrachlorodibenzo-para-dioxin; TNF-α, tumor necrosis factor alpha.
82
by guest on July 12, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

TCDD/CON A-INDUCED LIVER INJURY
number of possibilities exist. TCDD treatment of mice increases the activity of NK cells in the spleen and blood (Funseth and Ilback, 1992). In studies of human cohorts exposed occu-pationally to TCDD, an increase in the number of NK cells was observed in peripheral blood. In addition, TCDD treat-ment alters the expression of numerous immune-related genes through the activation of AhR signaling pathways that include coregulatory NK-cell receptors (Kerkvliet, 2009; Sun et al., 2004). TCDD pretreatment did not alter the expression of the stimulatory receptor NKG2d in NK cells after Con A treatment (Fig. 7C), but there are many other coregulatory receptors that were not investigated in this model. In addition, TCDD pretreat-ment did not alter the production of IL-12 after Con A admin-istration; however, TCDD can increase the expression of IL-12 receptor β2 and might increase the sensitivity of NK cells to the stimulatory effects of IL-12 via that mechanism (Kerkvliet, 2009). These studies did not directly investigate the role of AhR signaling in the effect of TCDD on NK cells in this model; this represents an important topic for future research given the vast array of functions that NK cells play in many diseases.
In summary, the results presented here demonstrate that pretreatment with TCDD exacerbates liver injury in a model of autoimmune hepatitis induced by Con A administration. Furthermore, TCDD pretreatment increased the activation of NK cells by Con A, and NK cells play an important role in the development of TCDD/Con A-induced liver injury (Fig. 10). This mechanism is distinctly different from the development of comparable injury obtained by administration of a larger dose of Con A. As such, the enhanced immune response induced by TCDD treatment warrants further investigation into mechanisms of NK cell activation and the larger role that exposure to TCDD and other environmental xenobiotics that influence AhR signal-ing might play in the development of autoimmune liver disease.
suppleMeNTARy DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FuNDING
National Institutes of Health grant (ES004911); National Institute of Environmental Health Sciences training grant (T32 ES007255 to A.M.F.).
ACKNOwleDGMeNTs
The authors thank Nicole Crisp for experimental support regarding flow cytometry and Dr Christine Dugan for assis-tance in developing protocols for intrahepatic immune cell isolation. The authors also thank Ryan Albee for technical assistance.
ReFeReNCes
Abraham, K., Krowke, R., and Neubert, D. (1988). Pharmacokinetics and bio-logical activity of 2,3,7,8-tetrachlorodibenzo-p-dioxin. 1. Dose-dependent tissue distribution and induction of hepatic ethoxyresorufin O-deethylase in rats following a single injection. Arch. Toxicol. 62, 359–368.
Adamson, G. M., and Billings, R. E. (1993). Cytokine toxicity and induc-tion of NO synthase activity in cultured mouse hepatocytes. Toxicol. Appl. Pharmacol. 119, 100–107.
Birnbaum, L. S. (1986). Distribution and excretion of 2,3,7,8-tetrachlorod-ibenzo-p-dioxin in congenic strains of mice which differ at the Ah locus. Drug Metab. Dispos. 14, 34–40.
Birnbaum, L. S., and Tuomisto, J. (2000). Non-carcinogenic effects of TCDD in animals. Food Addit. Contam. 17, 275–288.
Bonder, C. S., Ajuebor, M. N., Zbytnuik, L. D., Kubes, P., and Swain, M. G. (2004). Essential role for neutrophil recruitment to the liver in concanavalin A-induced hepatitis. J. Immunol. 172, 45–53.
Boverhof, D. R., Burgoon, L. D., Tashiro, C., Chittim, B., Harkema, J. R., Jump, D. B., and Zacharewski, T. R. (2005). Temporal and dose-dependent hepatic gene expression patterns in mice provide new insights into TCDD-Mediated hepatotoxicity. Toxicol. Sci. 85, 1048–1063.
Carnaud, C., Lee, D., Donnars, O., Park, S. H., Beavis, A., Koezuka, Y., and Bendelac, A. (1999). Cutting edge: Cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J. Immunol. 163, 4647–4650.
Chuang, Y. H., Lian, Z. X., Tsuneyama, K., Chiang, B. L., Ansari, A. A., Coppel, R. L., and Gershwin, M. E. (2006). Increased killing activity and decreased cytokine production in NK cells in patients with primary biliary cirrhosis. J. Autoimmun. 26, 232–240.
Czaja, A. J., and Manns, M. P. (2010). Advances in the diagnosis, pathogen-esis, and management of autoimmune hepatitis. Gastroenterology 139, 58–72.e4.
De Filippo, K., Henderson, R. B., Laschinger, M., and Hogg, N. (2008). Neutrophil chemokines KC and macrophage-inflammatory protein-2 are newly synthesized by tissue macrophages using distinct TLR signaling path-ways. J. Immunol. 180, 4308–4315.
Dere, E., Lo, R., Celius, T., Matthews, J., and Zacharewski, T. R. (2011). Integration of genome-wide computation DRE search, AhR ChIP-chip and gene expression analyses of TCDD-elicited responses in the mouse liver. BMC Genomics 12, 365.
Diliberto, J. J., Akubue, P. I., Luebke, R. W., and Birnbaum, L. S. (1995). Dose-response relationships of tissue distribution and induction of CYP1A1 and CYP1A2 enzymatic activities following acute exposure to 2,3,7,8-tet-rachlorodibenzo-p-dioxin (TCDD) in mice. Toxicol. Appl. Pharmacol. 130, 197–208.
Dong, Z., Wei, H., Sun, R., and Tian, Z. (2007). The roles of innate immune cells in liver injury and regeneration. Cell. Mol. Immunol. 4, 241–252.
Dugan, C. M., Fullerton, A. M., Roth, R. A., and Ganey, P. E. (2011). Natural killer cells mediate severe liver injury in a murine model of halothane hepa-titis. Toxicol. Sci. 120, 507–518.
Eberl, G., and MacDonald, H. R. (2000). Selective induction of NK cell pro-liferation and cytotoxicity by activated NKT cells. Eur. J. Immunol. 30, 985–992.
Esser, C., Rannug, A., and Stockinger, B. (2009). The aryl hydrocarbon recep-tor in immunity. Trends Immunol. 30, 447–454.
Faggioni, R., Jones-Carson, J., Reed, D. A., Dinarello, C. A., Feingold, K. R., Grunfeld, C., and Fantuzzi, G. (2000). Leptin-deficient (ob/ob) mice are pro-tected from T cell-mediated hepatotoxicity: Role of tumor necrosis factor alpha and IL-18. Proc. Natl. Acad. Sci. U.S.A. 97, 2367–2372.
Feld, J. J., and Heathcote, E. J. (2003). Epidemiology of autoimmune liver disease. J. Gastroenterol. Hepatol. 18, 1118–1128.
83
by guest on July 12, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

FULLERTON, ROTH, AND GANEY
Fullerton, A. M., Roth, R. A., and Ganey, P. E. (2013). 2,3,7,8-TCDD enhances the sensitivity of mice to concanavalin A immune-mediated liver injury. Toxicol. Appl. Pharmacol. 266, 317–327.
Funseth, E., and Ilback, N. G. (1992). Dioxin (2,3,7,8-tetrachlorodibenzo-p-dioxin) increases blood and spleen natural killer cell activity in the mouse. Chemosphere. 25, 7–10.
Gantner, F., Leist, M., Küsters, S., Vogt, K., Volk, H. D., and Tiegs, G. (1996). T cell stimulus-induced crosstalk between lymphocytes and liver macrophages results in augmented cytokine release. Exp. Cell Res. 229, 137–146.
Gilbert, K. M. (2010). Xenobiotic exposure and autoimmune hepatitis. Hepat. Res. Treat. 2010, 248157.
Gong, Q., Zhang, H., Li, J. H., Duan, L. H., Zhong, S., Kong, X. L., Zheng, F., Tan, Z., Xiong, P., Chen, G., et al. (2010). High-mobility group box 1 exacerbates concanavalin A-induced hepatic injury in mice. J. Mol. Med. (Berl). 88, 1289–1298.
Hatada, S., Ohta, T., Shiratsuchi, Y., Hatano, M., and Kobayashi, Y. (2005). A novel accessory role of neutrophils in concanavalin A-induced hepatitis. Cell. Immunol. 233, 23–29.
Hatano, M., Sasaki, S., Ohata, S., Shiratsuchi, Y., Yamazaki, T., Nagata, K., and Kobayashi, Y. (2008). Effects of Kupffer cell-depletion on Concanavalin A-induced hepatitis. Cell. Immunol. 251, 25–30.
Holladay, S. D., Mustafa, A., and Gogal, R. M., Jr. (2011). Prenatal TCDD in mice increases adult autoimmunity. Reprod. Toxicol. 31, 312–318.
Kaneko, Y., Harada, M., Kawano, T., Yamashita, M., Shibata, Y., Gejyo, F., Nakayama, T., and Taniguchi, M. (2000). Augmentation of Valpha14 NKT cell-mediated cytotoxicity by interleukin 4 in an autocrine mechanism resulting in the development of concanavalin A-induced hepatitis. J. Exp. Med. 191, 105–114.
Kerkvliet, N. I. (1995). Immunological effects of chlorinated dibenzo-p-diox-ins. Environ. Health Perspect. 103(Suppl. 9), 47–53.
Kerkvliet, N. I. (2009). AHR-mediated immunomodulation: The role of altered gene transcription. Biochem. Pharmacol. 77, 746–760.
Kerkvliet, N. I., Shepherd, D. M., and Baecher-Steppan, L. (2002). T lym-phocytes are direct, aryl hydrocarbon receptor (AhR)-dependent targets of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): AhR expression in both CD4+ and CD8+ T cells is necessary for full suppression of a cytotoxic T lympho-cyte response by TCDD. Toxicol. Appl. Pharmacol. 185, 146–152.
Kerkvliet, N. I., Steppan, L. B., Vorachek, W., Oda, S., Farrer, D., Wong, C. P., Pham, D., and Mourich, D. V. (2009). Activation of aryl hydrocarbon recep-tor by TCDD prevents diabetes in NOD mice and increases Foxp3+ T cells in pancreatic lymph nodes. Immunotherapy 1, 539–547.
Knolle, P. A., Gerken, G., Loser, E., Dienes, H. P., Gantner, F., Tiegs, G., Meyer zum Buschenfelde, K. H., and Lohse, A. W. (1996). Role of sinusoi-dal endothelial cells of the liver in concanavalin A-induced hepatic injury in mice. Hepatology 24, 824–829.
Kobayashi, S., Okamoto, H., Iwamoto, T., Toyama, Y., Tomatsu, T., Yamanaka, H., and Momohara, S. (2008). A role for the aryl hydrocarbon receptor and the dioxin TCDD in rheumatoid arthritis. Rheumatology (Oxford). 47, 1317–1322.
Kopec, A. K., Boverhof, D. R., Burgoon, L. D., Ibrahim-Aibo, D., Harkema, J. R., Tashiro, C., Chittim, B., and Zacharewski, T. R. (2008). Comparative toxicogenomic examination of the hepatic effects of PCB126 and TCDD in immature, ovariectomized C57BL/6 mice. Toxicol. Sci. 102, 61–75.
Lee, J., Cacalano, G., Camerato, T., Toy, K., Moore, M. W., and Wood, W. I. (1995). Chemokine binding and activities mediated by the mouse IL-8 receptor. J. Immunol. 155, 2158–2164.
Longhi, M. S., Ma, Y., Mieli-Vergani, G., and Vergani, D. (2010). Aetiopathogenesis of autoimmune hepatitis. J. Autoimmun. 34, 7–14.
Moos, A. B., Baecher-Steppan, L., and Kerkvliet, N. I. (1994). Acute inflamma-tory response to sheep red blood cells in mice treated with 2,3,7,8-tetrachlo-rodibenzo-p-dioxin: The role of proinflammatory cytokines, IL-1 and TNF. Toxicol. Appl. Pharmacol. 127, 331–335.
Moos, A. B., Oughton, J. A., and Kerkvliet, N. I. (1997). The effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on tumor necrosis factor (TNF) production by peritoneal cells. Toxicol. Lett. 90, 145–153.
Mustafa, A., Holladay, S. D., Goff, M., Witonsky, S., Kerr, R., Weinstein, D. A., Karpuzoglu-Belgin, E., and Gogal, R. M. Jr. (2009). Developmental expo-sure to 2,3,7,8-tetrachlorodibenzo-p-dioxin alters postnatal T cell pheno-types and T cell function and exacerbates autoimmune lupus in 24-week-old SNF1 mice. Birth Defects Res. A. Clin. Mol. Teratol. 85, 828–836.
Nakamura, K., Okada, M., Yoneda, M., Takamoto, S., Nakade, Y., Tamori, K., Aso, K., and Makino, I. (2001). Macrophage inflammatory protein-2 induced by TNF-alpha plays a pivotal role in concanavalin A-induced liver injury in mice. J. Hepatol. 35, 217–224.
Nault, R., Kim, S., and Zacharewski, T. R. (2013). Comparison of TCDD-elicited genome-wide hepatic gene expression in Sprague-Dawley rats and C57BL/6 mice. Toxicol. Appl. Pharmacol. 267, 184–191.
Nicoletti, F., Di Marco, R., Zaccone, P., Salvaggio, A., Magro, G., Bendtzen, K., and Meroni, P. (2000). Murine concanavalin A-induced hepatitis is prevented by interleukin 12 (IL-12) antibody and exacerbated by exogenous IL-12 through an interferon-gamma-dependent mechanism. Hepatology 32(Pt 1), 728–733.
Patterson, R. M., Stachlewitz, R., and Germolec, D. (2003). Induction of apop-tosis by 2,3,7,8-tetrachlorodibenzo-p-dioxin following endotoxin exposure. Toxicol. Appl. Pharmacol. 190, 120–134.
Peters, M. G. (2002). Animal models of autoimmune liver disease. Immunol. Cell Biol. 80, 113–116.
Pflanz, S., Timans, J. C., Cheung, J., Rosales, R., Kanzler, H., Gilbert, J., Hibbert, L., Churakova, T., Travis, M., Vaisberg, E., et al. (2002). IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces prolif-eration of naive CD4(+) T cells. Immunity 16, 779–790.
Poland, A., and Knutson, J. C. (1982). 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: Examination of the mechanism of toxicity. Annu. Rev. Pharmacol. Toxicol. 22, 517–554.
Quintana, F. J., Basso, A. S., Iglesias, A. H., Korn, T., Farez, M. F., Bettelli, E., Caccamo, M., Oukka, M., and Weiner, H. L. (2008). Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71.
Sadik, C. D., Kim, N. D., and Luster, A. D. (2011). Neutrophils cascading their way to inflammation. Trends Immunol. 32, 452–460.
Schleinitz, N., Vély, F., Harlé, J. R., and Vivier, E. (2010). Natural killer cells in human autoimmune diseases. Immunology 131, 451–458.
Schümann, J., Wolf, D., Pahl, A., Brune, K., Papadopoulos, T., van Rooijen, N., and Tiegs, G. (2000). Importance of Kupffer cells for T-cell-dependent liver injury in mice. Am. J. Pathol. 157, 1671–1683.
Seino, K., Kayagaki, N., Takeda, K., Fukao, K., Okumura, K., and Yagita, H. (1997). Contribution of Fas ligand to T cell-mediated hepatic injury in mice. Gastroenterology 113, 1315–1322.
Shimoda, S., Harada, K., Niiro, H., Shirabe, K., Taketomi, A., Maehara, Y., Tsuneyama, K., Nakanuma, Y., Leung, P., Ansari, A. A., et al. (2011). Interaction between Toll-like receptors and natural killer cells in the destruc-tion of bile ducts in primary biliary cirrhosis. Hepatology 53, 1270–1281.
Stevens, E. A., Mezrich, J. D., and Bradfield, C. A. (2009). The aryl hydro-carbon receptor: A perspective on potential roles in the immune system. Immunology 127, 299–311.
Sulentic, C. E., and Kaminski, N. E. (2011). The long winding road toward understanding the molecular mechanisms for B-cell suppression by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Sci. 120(Suppl. 1), S171–S191.
Sun, Y. V., Boverhof, D. R., Burgoon, L. D., Fielden, M. R., and Zacharewski, T. R. (2004). Comparative analysis of dioxin response elements in human, mouse and rat genomic sequences. Nucleic Acids Res. 32, 4512–4523.
Tagawa, Y., Kakuta, S., and Iwakura, Y. (1998). Involvement of Fas/Fas ligand system-mediated apoptosis in the development of concanavalin A-induced hepatitis. Eur. J. Immunol. 28, 4105–4113.
84
by guest on July 12, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

TCDD/CON A-INDUCED LIVER INJURY
Takeda, K., Hayakawa, Y., Van Kaer, L., Matsuda, H., Yagita, H., and Okumura, K. (2000). Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc. Natl. Acad. Sci. U.S.A. 97, 5498–5503.
Teske, S., Bohn, A. A., Regal, J. F., Neumiller, J. J., and Lawrence, B. P. (2005). Activation of the aryl hydrocarbon receptor increases pulmonary neutro-philia and diminishes host resistance to influenza A virus. Am. J. Physiol. Lung Cell. Mol. Physiol. 289, L111–L124.
Thoma, H., Mücke, W., and Kauert, G. (1990). Comparison of the polychlorin-ated dibenzo-p-dioxin and dibenzofuran in human tissue and human liver. Chemosphere 20, 433–442.
Tiegs, G., and Gantner, F. (1996). Immunotoxicology of T cell-dependent experimental liver injury. Exp. Toxicol. Pathol. 48, 471–476.
Tiegs, G., Hentschel, J., and Wendel, A. (1992). A T cell-dependent experi-mental liver injury in mice inducible by concanavalin A. J. Clin. Invest. 90, 196–203.
Toyabe, S., Seki, S., Iiai, T., Takeda, K., Shirai, K., Watanabe, H., Hiraide, H., Uchiyama, M., and Abo, T. (1997). Requirement of IL-4 and liver NK1+ T cells for concanavalin A-induced hepatic injury in mice. J. Immunol. 159, 1537–1542.
Trobonjaca, Z., Kröger, A., Stober, D., Leithäuser, F., Möller, P., Hauser, H., Schirmbeck, R., and Reimann, J. (2002). Activating immunity in the liver. II.
IFN-beta attenuates NK cell-dependent liver injury triggered by liver NKT cell activation. J. Immunol. 168, 3763–3770.
Vignali, D. A., and Kuchroo, V. K. (2012). IL-12 family cytokines: Immunological playmakers. Nat. Immunol. 13, 722–728.
Vogel, C. F., Nishimura, N., Sciullo, E., Wong, P., Li, W., and Matsumura, F. (2007). Modulation of the chemokines KC and MCP-1 by 2,3,7,8-tet-rachlorodibenzo-p-dioxin (TCDD) in mice. Arch. Biochem. Biophys. 461, 169–175.
Wang, H. X., Liu, M., Weng, S. Y., Li, J. J., Xie, C., He, H. L., Guan, W., Yuan, Y. S., and Gao, J. (2012). Immune mechanisms of Concanavalin A model of autoimmune hepatitis. World J. Gastroenterol. 18, 119–125.
Wu, D., Nishimura, N., Kuo, V., Fiehn, O., Shahbaz, S., Van Winkle, L., Matsumura, F., and Vogel, C. F. (2011). Activation of aryl hydrocarbon receptor induces vascular inflammation and promotes atherosclerosis in apolipoprotein E-/- mice. Arterioscler. Thromb. Vasc. Biol. 31, 1260–1267.
Yee, S. B., Hanumegowda, U. M., Hotchkiss, J. A., Ganey, P. E., and Roth, R. A. (2003). Role of neutrophils in the synergistic liver injury from mono-crotaline and bacterial lipopolysaccharide exposure. Toxicol. Sci. 72, 43–56.
Zimmermann, H. W., Trautwein, C., and Tacke, F. (2012). Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Front. Physiol. 3, 56.
85
by guest on July 12, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from