
PREPARATION, CHROMATOGRAPHIC-CHEMICAL ANALYSIS AND PHARMACOLOGICAL ACTIVITY OF EXTRACTS FROM HUMAN...
12
Journal of Neurochemistry, 1964, Vol. 11, pp. 461 to 472. Pergamon Press Ltd. Printed in Northern Ireland PREPARATION, CHROMATOGRAPHIC-CHEMICAL ANALYSIS AND PHARMACOLOGICAL ACTIVITY OF EXTRACTS FROM HUMAN BRAIN* JOSEPH V. AUDITORE and HERMAN SMITH Department of Pharmacology Meharry Medical College, Nashville, Tennessee (Received 23 August 1963) THE CHEMICAL isolation of acetycholine from vertebrate tissue has been achieved by very few investigators. DALE and DUDLEY (1929) isolated and identified acetylcholine as a constituent of horse spleen by an elaborate experimental procedure. They treated the tissue with large quantities of ethanol and relied upon a mercuric chloride frac- tional precipitation to isolate the base, which was characterized as its chloroplatinate. Other investigators, notably STEDMAN and STEDMAN (1937), and later BANISTER, WHITTAKER and WIJESUNDERA (1953), used the method of KAPFHAMMER and BISCHOFF (1 930), which permits the isolation of quaternary nitrogeneous bases as their insoluble reineckates. The method obviates the mercuric chloride precipitation of DALE and DUDLEY (1929) or EWINS (1914) and is reasonably selective. The reineckate precipita- tion procedure was also applied to rat brain by HOSEIN, PROULX and ARA (1962), who reported the presence of choline, acetylcholine, propionylcholine, butyrylcholine, the CoA esters of crotonobetaine, carnitine and y-butyrobetaine. These workers con- cluded that most of the acetylcholine-like activity of the reineckate extracts resided in the CoA ester compounds. PEPEU, SCHMIDT and GIARMAN (1963), however, recently deduced from their experiments with radioactive acetylcholine in the reineckate isolation procedure that much of the activity of rat brain is attributable to acetyl- choline. The earlier investigations on the isolation of acetylcholine in vertebrate tissues involved the use of large volumes of extracting medium. DALE and DUDLEY (1929), for example, treated approximately 30 kg of horse spleen with more than 150 1. ethanol to obtain quantities of the desired compound. While such quantities of tissue and fluid volume are not prerequisite for the isolation of tissue bases, as indi- cated by the report of HOSEIN et al. (1962), we felt that our initial investigations should involve as much tissue as practicable. We attempted first to devise an extract& procedure which would be free of the cumbersome volumes described by earlier investigators. To the authors’ knowledge, the isolation of nitrogeneous bases of the acetyl- choline type from the human central nervous system has not been attempted. This report describes the application of a modified extraction procedure to human brain and to present chemical, chromatographic and preliminary pharmacological analyses of the extracts obtained. MATERIALS AND METHODS Reagents and tissue Except where indicated, all chemicals were of reagent grade. Normal tissue, obtained at autopsy not more than 2-5 hours after death, was examined. The * This investigation was supported in whole by Public Health Service Research Grant, B-3520, from the National Institute of Neurological Diseases and Blindness. 46 1
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of PREPARATION, CHROMATOGRAPHIC-CHEMICAL ANALYSIS AND PHARMACOLOGICAL ACTIVITY OF EXTRACTS FROM HUMAN...

Journal of Neurochemistry, 1964, Vol. 11, pp. 461 to 472. Pergamon Press Ltd. Printed in Northern Ireland
PREPARATION, CHROMATOGRAPHIC-CHEMICAL ANALYSIS AND PHARMACOLOGICAL ACTIVITY
OF EXTRACTS FROM HUMAN BRAIN* JOSEPH V. AUDITORE and HERMAN SMITH
Department of Pharmacology Meharry Medical College, Nashville, Tennessee
(Received 23 August 1963)
THE CHEMICAL isolation of acetycholine from vertebrate tissue has been achieved by very few investigators. DALE and DUDLEY (1929) isolated and identified acetylcholine as a constituent of horse spleen by an elaborate experimental procedure. They treated the tissue with large quantities of ethanol and relied upon a mercuric chloride frac- tional precipitation to isolate the base, which was characterized as its chloroplatinate.
Other investigators, notably STEDMAN and STEDMAN (1937), and later BANISTER, WHITTAKER and WIJESUNDERA (1953), used the method of KAPFHAMMER and BISCHOFF (1 930), which permits the isolation of quaternary nitrogeneous bases as their insoluble reineckates. The method obviates the mercuric chloride precipitation of DALE and DUDLEY (1929) or EWINS (1914) and is reasonably selective. The reineckate precipita- tion procedure was also applied to rat brain by HOSEIN, PROULX and ARA (1962), who reported the presence of choline, acetylcholine, propionylcholine, butyrylcholine, the CoA esters of crotonobetaine, carnitine and y-butyrobetaine. These workers con- cluded that most of the acetylcholine-like activity of the reineckate extracts resided in the CoA ester compounds. PEPEU, SCHMIDT and GIARMAN (1963), however, recently deduced from their experiments with radioactive acetylcholine in the reineckate isolation procedure that much of the activity of rat brain is attributable to acetyl- choline.
The earlier investigations on the isolation of acetylcholine in vertebrate tissues involved the use of large volumes of extracting medium. DALE and DUDLEY (1929), for example, treated approximately 30 kg of horse spleen with more than 150 1. ethanol to obtain quantities of the desired compound. While such quantities of tissue and fluid volume are not prerequisite for the isolation of tissue bases, as indi- cated by the report of HOSEIN et al. (1962), we felt that our initial investigations should involve as much tissue as practicable. We attempted first to devise an extract& procedure which would be free of the cumbersome volumes described by earlier investigators.
To the authors’ knowledge, the isolation of nitrogeneous bases of the acetyl- choline type from the human central nervous system has not been attempted. This report describes the application of a modified extraction procedure to human brain and to present chemical, chromatographic and preliminary pharmacological analyses of the extracts obtained.
MATERIALS A N D METHODS Reagents and tissue
Except where indicated, all chemicals were of reagent grade. Normal tissue, obtained at autopsy not more than 2-5 hours after death, was examined. The
* This investigation was supported in whole by Public Health Service Research Grant, B-3520, from the National Institute of Neurological Diseases and Blindness.
46 1
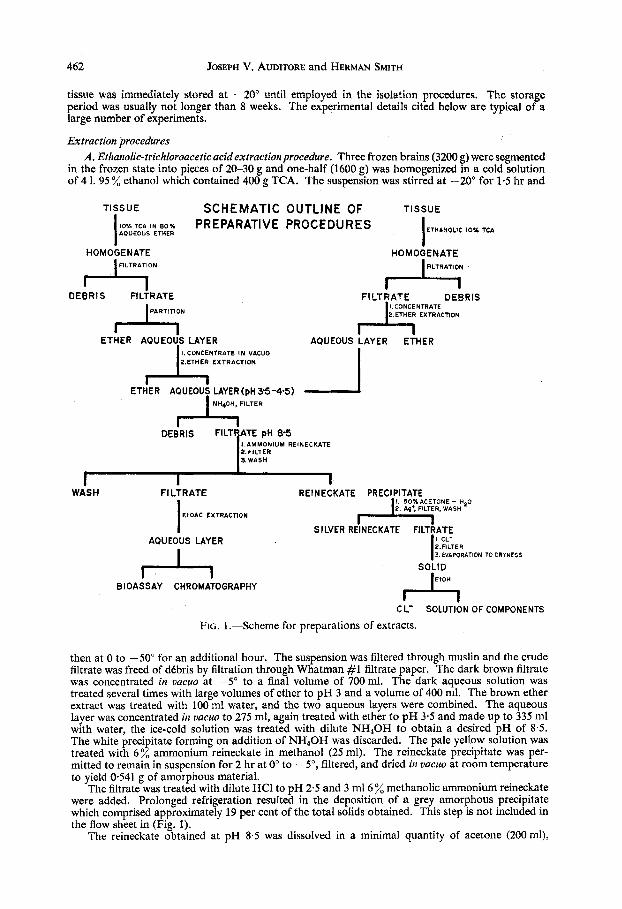
462 JOSEPH V. AUDITORE and HERMAN SMITH
I AMMONIUM REINECKATE 2 FILTER 3 WASH
tissue was immediately stored at -20" until employed in the isolation procedures. The storage period was usually not longer than 8 weeks. The experimental details cited below are typical of a large number of experiments.
Extraction procedures A . Ethanolic-trichloroacetic acid extraction procedure. Three frozen brains (3200 g) were segmented
in the frozen state into pieces of 20-30 g and one-half (1600 g) was homogenized in a cold solution of 4 1. 95 % ethanol which contained 400 g TCA. The suspension was stirred at -20" for 1.5 hr and
TISSUE SCHEMATIC OUTLINE OF TISSUE
ETHANOLIC 10% TCA I 10-AKA IN 80.h PREPARATIVE PROCEDURES AOUEOUS ETHER I
then at 0 to -50" for an additional hour. The suspension was filtered through muslin and the crude filtrate was freed of dCbris by filtration through Whatman #1 filtrate paper. The dark brown filtrate was concentrated in uucuo at -5" to a final volume of 700ml. The dark aqueous solution was treated several times with large volumes of ether to pH 3 and a volume of 400 ml. The brown ether extract was treated with 100 ml water, and the two aqueous layers were combined. The aqueous layer was concentrated in uacuo to 275 ml, again treated with ether to pH 3.5 and made up to 335 nil with water, the ice-cold solution was treated with dilute NH,OH to obtain a desired pH of 8.5. The white precipitate forming on addition of NH,OH was discarded. The pale yellow solution was treated with 6 % ammonium reineckate in methanol (25 ml). The reineckate precipitate was per- mitted to remain in suspension for 2 hr at 0" to -5", filtered, and dried in uacuo at room temperature to yield 0541 g of amorphous material.
The filtrate was treated with dilute HCl to pH 2.5 and 3 m16 % methanolic ammonium reineckate were added. Prolonged refrigeration resulted in the deposition of a grey amorphous precipitate which comprised approximately 19 per cent of the total solids obtained. This step is not included in the flow sheet in (Fig. 1).
The reineckate obtained at pH 8.5 was dissolved in a minimal quantity of acetone (200 ml),

Analysis and pharmacological activity of extracts from human brain 463
and 50 ml H 2 0 were added to the solution. A small amount of debris was filtered to yield a deep red-purple solution. The solution was treated drop-wise with AgC104 (800 mg in 15 ml water) and the precipitated silver reineckate was filtered. The excess silver ion was removed from the clear colourless solution by the addition of saturated NaCl solution to provide an excess of chloride ion. The silver chloride was filtered, and the solution of regenerated bases was concentrated in vacuo to dryness. The solid residue was dried in uacuo over phosphorous pentoxide and triturated with three successive 5 mi portions of absolute alcohol. The ethanolic solution of tissue bases was filtered free of NaCl and stored at -40". The residue was later assayed, and no measurable activity was observed. The reineckate obtained at pH 2-5 was treated similarly.
B. Aqueousether-TCA extractionprocedure. The remaining 1600 g of frozen brain were added to a suspension prepared by the addition of TCA(400 g) to a mixture of ether (3200 ml) and water (800 ml). The suspension was homogenized for 1.5 hr at -20" and at 0" to -5" for an additional 1 hr. The suspension was filtered through muslin and the filtrate was permitted to partition. The dark brown ether layer and the yellow aqueous layer (1250 ml) were separated. The latter was treated with four 250 ml portions of ether and the solution stored at 0" for 36 hr, after which it was concentrated to 335 ml in vacuo at 0". The solution was treated with ether to pH 3.5, cooled to O", and made alkaline to pH 8.5 with dilute NH40H. A white precipitate formed and was removed by filtration. The filtrate was treated with 6 % ammonium reineckate in methanol (25 ml), and precipitation was permitted to proceed for 4 hr. The precipitate was collected by vacuum filtration and dried over phosphorous pentoxide to yield 0.732 g tissue reineckate. The reineckate was decomposed as described in pro- cedure A, and the ethanol solution of tissue bases was stored at -40" until employed for bio assay and chromatography (cf. Fig. 1).
The reineckate precipitation precedure was modified to limit coprecipitation of ninhydrin- reactive material at pH 8.5. The principal advantage gained by modifying the procedure was the excellent chromatographic separation of the materials in the extract. The chromatogram was practically free of ninhydrin-positive substances below R, 0.10. This area usually contained abundant ninhydrin-reactive substances. The modified experimental procedures are as follows :
1 . Precipitation at pH 4.5. Precipitation at this pH was performed essentially as described for pH 8.5 precipitation. The flocculent white precipitate otserved on addition of NH40H did not form.
2. Precipitation with ethereal reineckate acid. Ammonium reineckate was dissolved in ~N-H,SO& and the reineckate acid was extracted with ether. The ether layer was washed with water and used for the precipitation. The brain extract (300 ml) corresponding to 200 g tissue was adjusted to pH 1.0 with H,SOd, and 4% ethereal reineckate acid (125 ml) was added. The mixture was vigorously stirred for 20 min at 0". The precipitate was filtered, washed with water and ether, and dried over P,O, to yield a pink amorphous reineckate (0-059 g). The filtrate was partitioned, the aqueous layer adjusted to pH 5.0, and the ether reineckate layer added with vigorous stirring. The precipitate which formed was filtered, washed with water until the water was colourless, and dried over P,O, to yield a purple reineckate (0.135 g). The filtrate from the pH 5.0 precipitation was partitioned, adjusted to pH 8.5 with NaOH and refrigerated overnight. A blue-grey, acid-soluble, precipitate was obtained. A portion (10 ml) of wash fluid from the pH 5.0 precipitate was treated with 3.5 g IRA 400 resin (Cl-form) and stirred until the supernatant was colourless. The resin was removed by filtration and treated with HCI (30 ml). The resin filtrates were evaporated to dryness in uacuo at 40". The red mother liquor from the precipitation was treated with ethyl acetate until colourless. The pH 1 and pH 5 precipitated reineckates were decomposed in the usual manner.
Recovery of acetylcholine from brain protein The solid residue obtained from 1 kg brain tissue was washed with water, ethanol, acetone and
ether to remove any residual soluble material. To a suspension of 11.3 g of the residue was added a solution of acetylcholine iodide (50 mg) in water (50 ml). The suspension was agitated for 15 min, and a solution of TCA (50 g) in 400 ml ether was added. The ether-water suspension was homo- genized for 15 min at 0 to 5" and agitated for an additional 15 min. The suspended material was filtered and the residue washed with water (70 ml). The aqueous layer was separated by partition and treated with successive portions of ether to pH 3.5 and a total volume of 132 ml. An additional I5 ml of the aqueous layer emulsified and resisted separation. The aqueous solution was cooled to 0" and treated with NaOH to pH 8.0; a small amount of residue was removed by filtration. Ammonium reineckate (150 mg) in methanol (5 ml) was added dropwise, and the precipitate was collected by vacuum filtration and dried in vacuo to give 57 mg of reineckate (66 per cent yield based on acetyl- choline). The reineckate was decomposed by silver perchlorate, and the regenerated material was dissolved in water (50 ml). Portions of this solution were assayed on the non-eserinized frog rectus abdominus muscle; the results indicated that 35-51 per cent of added acetylcholine was recovered by this method.

464 JOSEPH V. AUDITORE and HERMAN SMITH
Paper chromatographic methods Except where indicated, all paper chromatographic analyses were performed on acid-washed
Whatman #1 chromatography paper by descending method with the following solvents in a volume to volume ratio:
A. Butano1:acetic acid:water, 4: 1 : 5 (BUCHANAN, DEKKER and LONG 1950). B. 5 % Ammonium sulphate:isopropanol, 1 : 2 (ANAND, CLARK, HALL and TODD, 1952). C. Ethano1:ammonium hydroxide, 95:5 (KENNEDY and BARKER, 1951). D. Butano1:ethanol:acetic acid: water, 8:2: 1 : 3 (AUGUSTINSSON and GRAHN, 1953). E. Butanol:water, 100:9 (MELLON, KORN and HOOVER, 1953). F. 1sopropanol:ammonium hydroxide: water, 10: 1 : 1 (BENNET-CLARK, TAMBIAH and KEFFORD,
G. Isopropanol: hydrochloric acid :water, 68 : 165 : 15 : 5 (WYATT, 1951). H. 2 % Acetic acid. The mixtures to be chromatographed were applied by a spotting or streaking technique and the
paper (15 X 57 cm) was irrigated at 22-26' for 19-24 hr during which time the solvent front ran an average distance of 3 5 4 5 cm. Previously described visualization techniques were employed (BRANTE, 1949; CHARGAFF, LEVINE and GREEN, 1948; BREGOFF, ROBERTS and DELWICHE, 1953; PAULY, 1904; HESTRIN (1949).
Preparation of tetrachloroaureates Ethanol eluates of the desired Chromatographic components were evaporated to dryness, each
residue was dissolved in a minimum volume of 0.1~-HCl, and a few crystals of auric chloride were added. Precipitation was permitted to proceed for several hours, and the precipitate was filtered, washed and dried in vacao over phosphorous pentoxide. All samples were recrystallized from 0.1~- HCI prior to melting point determination or infra-red spectroscopy.
Infra-red spectroscopy The infra-red spectra were obtained with KBr sampling technique. The tetrachloroaureates were
incorporated into the KBr pellet in the conventional manner and the spectra were recorded with a Perkin-Elmer (Model 137B) infra-red spectrometer over a range of 700-2000 cm. When greater resolution was desired a Model 237 Perkin-Elmer double grating spectrometer was employed. Microsampling, when employed, involved the use of a microsampling KBr die, a beam condensing accessory in conjunction with a Perkin-Elmer Model 137B infra-red spectrometer.
Estimation of acetylcholine-like activity The crude aqueous extract, reineckate processed extract, and the dried eluates from chromato-
grams were assayed for acetylcholine-like activity on the non-eserinized preparations of frog rectus abdominus muscle in a manner similar to that described by CHANG and GADDUM (1933). The volume of the muscle bath was 2 0 ml and the volume of added sample to be tested never exceeded 0.4 ml. The muscle was placed under a constant tension of 500 mg. The bathing medium was Frog- Ringer's solution (25"). Each contraction was recorded for 2 min on smoked paper.
Under these experimental conditions, 05-1 p g acetylcholine produced a submaximal contraction in a great number of muscle preparations. Whenever possible, the unknown samples were diluted or resuspended in Frog-Ringer's solution, and the pH was adjusted to approximately 7.0 before testing. Care was taken to carry out appropriate controls for each experiment. Though the majority of our assays were done without eserine in the bathing solution, several experiments were performed in Frog-Ringer's solution containing eserine sulphate and D-tubocurarine.
1952).
RESULTS
Chromatographic projles, derivatives and colour development Solvent A produced the best resolution of iodine-staining components and was
considered the solvent of choice for subsequent experiments (Table 1). Although the components reacted with several different detection reagents, we chose to locate them by using the iodine staining technique for two principal reasons: the simplicity of the method and the fact that the components could be recovered essentially unaltered for pharmacological assay. Three major iodine-staining bands readily appeared on the chromatograms developed in solvent A with the following Rf values: 0.08, 0.1 1 and
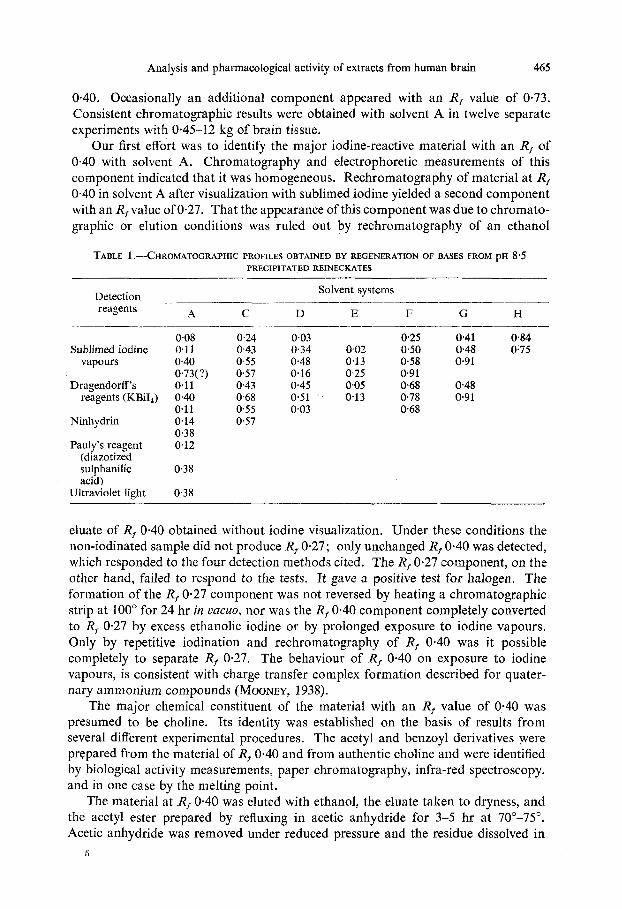
Analysis and pharmacological activity of extracts from human brain 465
0.40. Occasionally an additional component appeared with an R, value of 0.73. Consistent chromatographic results were obtained with solvent A in twelve separate experiments with 0.45-12 kg of brain tissue.
Our first effort was to identify the major iodine-reactive material with an R, of 0.40 with solvent A. Chromatography and electrophoretic measurements of this component indicated that it was homogeneous. Rechromatography of material at R, 0.40 in solvent A after visualization with sublimed iodine yielded a second component with an R, value of 0.27. That the appearance of this component was due to chromato- graphic or elution conditions was ruled out by rechromatography of an ethanol
TABLE 1 .-CHROMATOGRAPHIC PROFILES OBTAINED BY REGENERATION OF BASES FROM PH 8 5 PRECIPITATED REINECKATES
Solvent systems
A C D E F G H
008 0.24 0.03 0.25 041 0.84 Sublimed iodine 0.1 1 0.43 034 0.02 0.50 0.48 0.75
Detection __ reagents
vapours 0.40 0.55 0.48 0.13 058 0.91
Dragendorff's 0.11 0.43 0.45 0.05 0.68 0.48 reagents (KBiI,) 0.40 0.68 0-51 0-13 0.78 0.91
0.73(?) 0.57 0-16 0-25 0.91
0.1 1 0.55 0.03 0.68 Ninhydrin 0.14 0.57
0 3 8 Pauly's reagent 0.12
(diazotized sulphanilic 0.38 acid)
Ultraviolet light 0.38
eluate of R, 0.40 obtained without iodine visualization. Under these conditions the non-iodinated sample did not produce Rf 0.27; only unchanged R, 0.40 was detected, which responded to the four detection methods cited. The Rf 0.27 Component, on the other hand, failed to respond to the tests. It gave a positive test for halogen. The formation of the R, 0.27 component was not reversed by heating a chromatographic strip at 100" for 24 hr in uucuo, nor was the R, 0.40 component completely converted to R, 0.27 by excess ethanolic iodine or by prolonged exposure to iodine vapours. Only by repetitive iodination and rechromatography of R, 0.40 was it possible completely to separate R, 0.27. The behaviour of R, 0.40 on exposure to iodine vapours, is consistent with charge transfer complex formation described for quater- nary ammonium compounds (MOONEY, 1938).
The major chemical constituent of the material with an R, value of 0.40 was presumed to be choline. Its identity was established on the basis of results from several different experimental procedures. The acetyl and benzoyl derivatives were prepared from the material of R, 0.40 and from authentic choline and were identified by biological activity measurements, paper chromatography, infra-red spectroscopy. and in one case by the melting point.
The material at R, 0.40 was eluted with ethanol, the eluate taken to dryness, and the acetyl ester prepared by refluxing in acetic anhydride for 3-5 hr at 70"-75". Acetic anhydride was removed under reduced pressure and the residue dissolved in
5
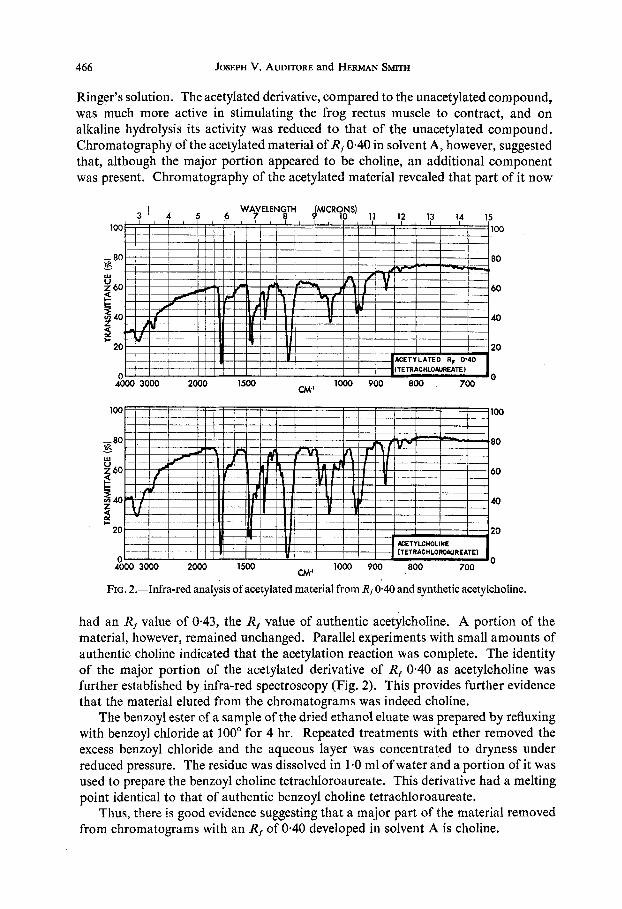
466 JOSEPH V. AUDITORE and HERMAN SMITH
Ringer's solution. The acetylated derivative, compared to the unacetylated compound, was much more active in stimulating the frog rectus muscle to contract, and on alkaline hydrolysis its activity was reduced to that of the unacetylated compound. Chromatography of the acetylated material of R,O.40 in solvent A, however, suggested that, although the major portion appeared to be choline, an additional component was present. Chromatography of the acetylated material revealed that part of it now
WAVELENGTH
- . I tw-I 1--1-- I I
100
- 80 80 1
Y 60 60
5 40 40
- Ly
3
20 20 * 3
0 0 CM' 4000 3000 2000 1500
100 100
- 80 80 kx
! 60 60
1000 900 800 700
-
40 40 Z
20 20
0 0 CM' 4000 3000 2000 1500
s
1000 900 800 700
FIG. 2.-Infra-red analysis of acetylated material from Rt 0.40 and synthetic acetylcholine.
had an R, value of 0.43, the R, value of authentic acetylchoIine. A portion of the material, however, remained unchanged. Parallel experiments with small amounts of authentic choline indicated that the acetylation reaction was complete. The identity of the major portion of the acetylated derivative of Rf 0.40 as acetylcholine was further established by infra-red spectroscopy (Fig. 2). This provides further evidence that the material eluted from the chromatograms was indeed choline.
The benzoyl ester of a sample of the dried ethanol eluate was prepared by refluxing with benzoyl chloride at 100" for 4 hr. Repeated treatments with ether removed the excess benzoyl chloride and the aqueous layer was concentrated to dryness under reduced pressure. The residue was dissolved in 1.0 ml of water and a portion of it was used to prepare the benzoyl choline tetrachloroaureate. This derivative had a melting point identical to that of authentic benzoyl choline tetrachloroaureate.
Thus, there is good evidence suggesting that a major part of the material removed from chromatograms with an R, of 0.40 developed in solvent A is choline.

Analysis and pharmacological activity of extracts from human brain 467
Zon exchange chromatography It is difficult to prepare quantities of the various components found by paper
chromatography sufficiently large for chemical and spectroscopic analysis. For this reason ion exchange chromatography was selected as an alternative purification procedure. The results from these experiments revealed the presence of an easily detected quaternary nitrogen compound in the reineckate extract after passage over a Dowex 50 ion exchange resin. The compound was identified as choline by paper chromatography and chemical and spectroscopic measurements, There follows a brief description of the experiments that enable us to arrive at this conclusion. A modification of the method of FRIEDMAN, MCFARLANE, BATTACHARYYA and FRAENKEL (1955) was employed to decompose the tissue reineckate. A portion (10 ml) of the IRA-400 regenerated bases, which contained 202 pmoles of choline equivalents according to the method of HAYASHI, UNEMOTO and MIYAKI (1962), was placed on a Dowex 50 (Hi form) column (1.8 x 60 cm). The column was treated with O-IN-HCI (818 ml) followed by 2~-(450 ml), 3~-(450 ml), and 6~-(140 ml) HCI.
Fractions 121-1 51 (484-620 ml) formed periodides (WALL, CHRISTIANSON and SENTI, 1960) as did the 6~ eluate. No other fractions exhibited this test for quater- nary nitrogen compounds. The former fractions were evaporated in YQCUO to dryness and found to contain 163 pmoles of choline equivalents (79.2 per cent). The latter fractions yielded 22 pmoles of choline equivalents (10.4 per cent) by the method of HAYASHI et al. (1962); to date, this material has not been identified.
Chromatographically, fractions 121-151 corresponded to choline chloride. Chromatography of the acetylation and benzoylation products of each confirmed that choline was the only component present. Tetrachloroaureates prepared from fractions 121-1 51 and its acetylation and benzoylation products yielded crystalline aureates with melting points of 258-258-5", 165", and 182-184". They showed no depression of melting point on admixture with authentic aureates of choline, acetylcholine, and benzoyl choline, respectively.
The aureates of fractions 121-151 and their derivatives were incorporated into KBr pellets in the usual manner, and infra-red spectra of these compounds were indistinguishable from those of choline, acetylcholine, and benzoyl choline.
Thus, it may be concluded that the easily detected quaternary nitrogen compound of the reineckate extract isolated by ion exchange chromatography is choline. This study furnishes additional support to results obtained from paper chromatography studies.
Since HOSEIN (1963) reported the presence of certain betaines in similar extracts made from rat brains, our next logical step was to search for similar compounds in our extracts. Several attempts were made, but without success. A description of some of these experimental procedures follows.
A sample of IRA 400 regenerated bases was placed on a Dowex 50 ion exchange resin column and treated with ~ N - H C ~ . Paper chromatography in solvent A revealed that the collected fractions contained at least two components with R, values of 0.08 and 0.1 1 detected by the iodine technique, Chargaff-Levine reagent, ninhydrin, and Dragendorff's reagent. That these components were not betaines was shown as follows: (a) precipitation of these components was performed at pH 8.5; (b) the elution position from Dowex 50 ion exchange resin was inconsistent with carnitine, y-butyrobetaine, or betaine; (c) the R, values (solvent A) were lower than those
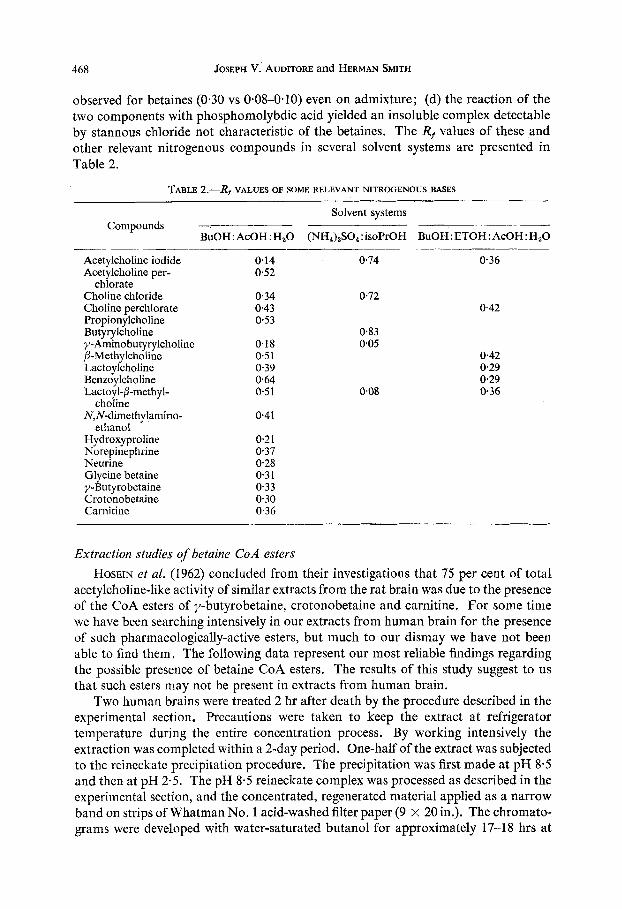
468 JOSEPH V. AUDITORE and HERMAN SMITH
observed for betaines (0.30 vs 0.08-0.10) even on admixture; (d) the reaction of the two components with phosphomolybdic acid yielded an insoluble complex detectable by stannous chloride not characteristic of the betaines. The Rf values of these and other relevant nitrogenous compounds in several solvent systems are presented in Table 2.
TABLE 2.-Rf VALUES OF SOME RELEVANT NITROGENOUS BASES
Solvent systems Compounds _______- -________ ___-__--____
BuOH: AcOH: H20 (NH,),SO,:isoPrOH BuOH:ETOH:AcOH:H,O
Acetylcholine iodide 0.14 0.74 0.36 Acetylcholine per- 0.52
Choline chloride 0.34 0.72 Choline perchlorate 0.43 0.42 Propionylcholine 0.53 Butyrylcholine 0.83 y- Amino butyrylcholine 0.18 0.05 P-Methylcholine 0.51 0.42 Lactoylcholine 0.39 029 Benzoylcholine 0.64 0 29 Lactoy I-P-methyl- 0.51 008 0.36
N,N-dimethy lamino- 0.41
chlorate
choline
ethanol Hy droxy proline 0.21 Norepinephrine 037 Neurine 0.28 Glycine betaine 0.3 1 y-Butyrobetaine 0.33 Crotonobetaine 0.30 Carnitine 0.36
Extraction studies of betaine CoA esters HOSEIN et al. (1962) concluded from their investigations that 75 per cent of total
acetylcholine-like activity of similar extracts from the rat brain was due to the presence of the CoA esters of y-butyrobetaine, crotonobetaine and carnitine. For some time we have been searching intensively in our extracts from human brain for the presence of such pharmacologically-active esters, but much to our dismay we have not been able to find them, The following data represent our most reliable findings regarding the possible presence of betaine CoA esters. The results of this study suggest to us that such esters may not be present in extracts from human brain.
Two human brains were treated 2 hr after death by the procedure described in the experimental section. Precautions were taken to keep the extract at refrigerator temperature during the entire concentration process. By working intensively the extraction was completed within a 2-day period. One-half of the extract was subjected to the reineckate precipitation procedure. The precipitation was first made at pfl 8.5 and then at pH 2.5. The pH 8.5 reineckate complex was processed as described in the experimental section, and the concentrated, regenerated material applied as a narrow band on strips of Whatman No. 1 acid-washed filter paper (9 x 20 in.). The chromato- grams were developed with water-saturated butanol for approximately 17-1 8 hrs at

Analysis and pharmacological activity of extracts from human brain 469
25". Eight chromatograms were processed. With iodine vapour two major bands with R, values of 0.02-0.15 and 0.25-0.28 and a third questionable band with an R, 0.50-0-70 were detected. We were unable to obtain a positive hydroxylamine ferric chloride test on any band. The two major iodine bands of seven chromatograms and the questionable 0.50-0-70 regions were carefully marked, eluted with water (pH 4-5), taken to dryness, hydrolysed with ~ N - H C ~ for 12 hr, and analysed for beta-alanine, a constituent of the CoA moiety, with a Beckman amino acid analyser. Beta-alanine was absent in the R, 0.50-0.70 region and insignificant amounts were found in the lower R, regions (Table 3).
TABLE 3 .-@-ALANINE ANALYSIS OF HYDROLYSED ELUATES
P-Alanine Rf p mole
0'02-0.1 5 0.006 0'25-0-28 <0.004 0.50-0.70 absent
Because of difficulties encountered in obtaining a constant source of fresh human brain and the possible post-mortem effects on pharmacologically-active substances, we chose to examine calf fresh brain extracts. According to a personal communication from HOSEIN, calf brain also contains betaine CoA esters. Several reineckate extracts from calf brain were prepared by following precisely the methods employed by HOSEIN et al. (1962). The resultsfrom this study also suggest that such compounds may not be present in the reineckate extract from calf brain. This conclusion was reached as the result of paper chromatography and bioassay studies.
The final reineckate extract as processed by procedures of HOSEIN et al. (1962) con- tained much inorganic salt. Although methanol fractionation was employed to remove the high concentrations of salts necessary to decompose the reineckate complex, all of the salt cannot be removed from the final extract even with the best grade of methanol. This is not surprising, for pure methanol dissolves many inorganic salts.
Chromatography of these extracts revealed the absence of an iodine-staining or ultraviolet-absorbing band in the region of the chromatogram between R, 0-50-0.70 where the CoA esters supposedly are located. The region between R, 0.00-0.40 stained intensely with ninhydrin, and numerous amino acids were found in the extract analysed with a Beckman amino acid analyser. A characteristic feature of the chromatograms developed in butanol-water was the obvious distortion of com- ponents, probably due to the presence of inorganic salts in the extract.
The most significant chromatographic findings to date with the extract from calf brain is the consistent appearance of four definite Dragendorff-reactive bands (N- methylated compounds) on chromatograms developed in butanol :ethanol :acetic acid:water. The R, values of these bands are 0.00,0-10,0.38, and 0-68. The orange- coloured band with an R, value of 0.10 did not appear immediately as did the orange- purple bands with the R, values of 0.00, 0.38, and 0.68. R, 0.10 appeared severaI hours after spraying. The material at 0.00 was not investigated further, but the substance with an R, 0.10 which contains phosphorus is thought to be phosphoryl- choline. Synthetic phosphorylcholine has an R, value of 0.09 & 0.02 and shows late colour development. The material at R, 0.38 is no doubt chiefly choline. The faster

470 JOSEPH V. AUDITORE and HERMAN SMITH
moving component may also be choline complexed with some unknown material. Small amounts of an unidentified acetylcholine-complex also may be present. It has been suggested by some investigators that it may be acetylcholine-trichloroacetic acid complex.
Pharmacological studies
The crude aqueous extract from human brain contained substances which induced a contraction of the non-eserinized, isolated, frog rectus abdominus muscle. The contraction produced by the ‘extract was not significantly potentiated by eserine and only slightly blocked by D-tubocurarine. Reineckate treatment of this aqueous extract removed approximately 40-50 per cent of the acetylcholine-like activity. The acetylcholine-like activity of the reineckate extract, however, was largely abolished by D-tubocurarine.
In our laboratory, Whatman # I paper washed several days with acid-water contained material which induced a contraction of the non-eserinized and eserinized frog rectus preparations. Consequently, we found it difficult to obtain precise data concerning the acetylcholine-like activity of material eluted from the chromatograms. When appropriate controls were carried out, material isolated from R, 0.00-0.1 1 and 0-40 with solvent A, however, appeared to possess acetylcholine-like activity. The activity of material at R, 0.40 was presumed to be due largely to choline, but the activity in the lower R, range still remains unidentified. Material isolated by HOSEIN’S (1963) elution technique from an area of the chromatogram with an Rf range of 0.50-0.70 stimulated the frog rectus muscle to contract, but so did material isolated from control chromatograms. These studies are still in progress.
DISCUSSION
This investigation reveals that an extract made from human brain by employing the reineckate precipitation procedure is extremely complex and difficult to resolve. The extract contains many amino acids as a result of coprecipitation. This can be circumvented to some extent by modifying the reineckate procedure. Choline has been unequivocally identified as the major quaternary nitrogen compound in the extract. Other nitrogenous bases are also present but in small concentrations and to date they have not been identified. Our more recent experiments suggest that one of the compounds in extracts of calf brain may be phosphorylcholine. These data suggest to us an absence of those betaines reported by HOSEIN (1963), in our extract from human brain. It is conceivable, however, that the procedures employed in the present studies may not be precise enough to detect the trace amounts of betaines reported by HOSEIN (1963). High voltage electrophoresis is being applied to detect betaines in the reineckate fractions and crude extracts. This technique in our labora- tory is presenting clear resolution of small quantities of authentic betaines and choline esters.
We have also been unable to find the betaine CoA esters in extracts from human brain, According to the report of HOSEIN’S et al. (1962) these esters were discovered by employing the reineckate-regenerated material, chromatography of this material in butanol-water, and eluting the iodine-positive bands on paper chromatograms with an R, range of 0.50-0.70. It was stated that the material isolated gave a positive reaction with the hydroxylamine-ferric chloride test indicative of an ester, and that

Analysis and pharmacological activity of extracts from human brain 471
hydrolysed material gave positive tests for the complete constituents of the CoA moiety. Furthermore, the presence of a thiol ester linkage was inferred from UV data. Experimental data confirming such findings were not reported.
The main criticism of our findings is the possible implication of post-mortem effects on pharmacologically-active substances. No doubt some destruction of substances such as choline esters occurs as a result of post-mortem tissue changes, but aqueous extracts from brain possessed strong acetylcholine-like activity after storage for many months at -30". DALE and DUDLEY (1929) reported that spleens kept intact overnight contained as much acetylcholine activity as those investigated immediately after death. If the spleen was minced, however, acetylcholine activity rapidly disappeared within a few minutes. Admittedly, we are more concerned with the possibility of acid hydrolytic decomposition of labile esters during the extraction period. The pH of the extraction fluid is 1.0, and the pH of maximal stability of esters such as acetylcholine lies between 3.8 and 4-5. Other extraction procedures and fluids are now being tried. The fact that the betaine CoA esters were not detected in extracts from calf fresh brain suggests that our failure to locate these esters in the extracts from human brain may not be attributed to post-mortem effects.
Much of the activity observed by HOSEIN et al. (1962) in the material isolated from the high R, region may be due to substances present in Whatman #1 paper, impurities arising from reagents employed in the extraction procedure, or to small amounts of a fast-moving unknown acetylcholine complex as described by several workers (PEPEU, et al. 1963; MCLENNAN, CURRY and WALKER, 1963; WHITTAKER, 1956). We readily admit, however, that our pharmacological studies of this region of the chro- matogram have never been consistent. In some experiments the amount of activity extracted from a control chromatogram equalled that of the experimental one, while in other experiments the findings were reversed.
It appears that a large part of the acetylcholine-like activity of the original aqueous extract from human brain may not be due to any known or unknown choline ester or to the reported CoA esters, The substance(s) largely responsible for this activity are yet to be identified. The activity associated with the reineckate extract from human brain we believe to be due largely to choline and trace amounts of acetylcholine.
SUMMARY
A procedure is described for preparing an aqueous extract from large quantities of human brain. The extract contains substances which stimulate the frog rectus muscle 'to contract. The contraction is only slightly reduced when the extract is tested on the curarized rectus muscle. A reineckate precipitation procedure extracted approximate- ly one-half of the total acetylcholine-like activity of the original aqueous extract. This activity, however, was largely abolished by D-tubocurarine.
Several modifications have been introduced into the reineckate precipitation procedure used to prepare a concentrate of nitrogenous bases. Chromatographic and chemical analyses of several reineckate extracts are reported. The best chromato- graphic resolution of the iodine-staining components in these extracts was achieved on paper chromatograms developed in BuOH: AcOH: H,O. Two major iodine- positive areas with R, values of 0.08-0.11 and 0.40 were consistently found. A questionable third component had an R, value of 0.73. Choline was presumed to be the major constituent in the R, 0.40 component. It was identified by paper

472 JOSEPH V. AUDITORE and HERMAN Smni
chro matography, melting point determination, infra-red spectroscopy and biological assay.
Column chromatography with Dowex 50 (Hf form) was also employed to resolve the various components of the reineckate extract. On the basis of chromatographic behaviour, melting point determinations, and infra-red analysis, choline again was identified as the major quaternary nitrogenous component. Failure to detect various betaines may be due to the limitations of the techniques employed, for these bases have been reported to occur only in minute quantities.
An attempt to isolate the betaine CoA esters from the human brain reineckate extract proved unsuccessful. This failure again may be due to the techniques employed or to the presence of trichloroacetic acid which may form unidentifiable complexes with the CoA esters. The CoA esters are also suspected to be extremely unstable and may not survive the reineckate procedure.
REFERENCES ANAND N., CLARK V. M., HALL R. H. and TODD A. R. (1952) J. chem. SOC. 3665. AUGUSTINSSON K. B. and GRAHN M. (1953) Acta chem. scand. 7, 906. BANISTER J., WHITTAKER V. P. and WIJESUNDERA S. (1953) J. Physiol. (Lond.) 121, 5 5 . BENNET-CLARK T. A., TAMBIAH M. S. and KEFFORD N. P. (1952) Nature (Lond.) 169, 452. BRANTE G. (1949) Nature (Lond.) 163, 651. BREGOFF H. M., ROBERTS E. and DELWICHE C. C. (1953) J. biol. Chem. 205, 565. BUCHANAN J. G., DEKKER C. A. and LONG A. G. (1950) J. chem. SOC. 3162. CHANG C . H. and GADDUM H. J. (1933) J. Physiol. (Lond.) 79, 255. CHARGAFF E., LEVINE C. and GREEN C. (1948) J. biol. Chem. 175, 67. DALE H. H. and DUDLEY H. W. (1929) J. Physiol. (Lond.) 68,97. EWINS A. J. (1914) Biochem. J . 44,8. FRJEDMAN S., MCFARLANE J. E., BHATTACHARYYA P. K. and FRAENKEL G. (1955) Arch. Biochem.
HAYASHI M., UNEMOTO T. and MIYAKI K. (1962) Chem.pharm. Bull. 10, 533. HESTRIN S. (1949) J . biol. Chem. 180, 249. HOSEIN E. A. (1963) Arch. Biochem. 100, 32. HOSEIN E. A., PROULX P. and ROUSHAN ARA (1962) Biochem. J. 83, 341. KAPFHAMMER J. and BISCHOFF C. (1930) Hoppe-Seylers Z. physiol. Chem. 191, 179. KENNEDY E. P. and BARKER H. A. (1951) Analyt. Chem. 23,1033. MCLENNAN H., CURRY L. and WALKER R. (1963) Biochem. J. 89,163. MELLON E. F., KORN A. H. and HOOVER S. R. (1953) J. Amer. chem. SOC. 75, 1675. MOONEY R. C. L. (1938) Physiol. Rev. 53, 851. PAULY H. (1904) Hoppe-Seylers 2. physiol. Chem. 42, 508. PEPEU G., SCHMIDT K. F. and GIARMAN N. J. (1963) Biochem. Pharmacol. 12, 385. STEDMAN E. and STEDMAN E. (1937) Biochem. J. 31, 817. WALL J. S., CHRISTIANSON D. D. and SENTI R. J. (1960) Analyt. Chem. 32, 870. WHITTAKER V. P. (1956) Biochem. biophys. Acfa 22, 590. WYATT G. R. (1951) Biochem. J. 48, 584.
59,484.




















