
available-power prediction of lithium-ion batteries in electric ...
Upload
independentCategory
view
0download
0
Preparation, Characterization, and Electrochemical Performance of Lithium TrivanadateRods by a Surfactant-Assisted Polymer Precursor Method for Lithium Batteries
A. Sakunthala,†,‡ M. V. Reddy,*,† S. Selvasekarapandian,*,‡,§ B. V. R. Chowdari,*,† andP. Christopher Selvin|
Department of Physics, National UniVersity of Singapore, Singapore 117542, DRDO-BU, Centre for LifeSciences, Bharathiar UniVersity, Coimbatore 641046, India, Kalasalingam UniVersity,Krishnankoil, Virudhunagar 626190, Tamil Nadu, India, and NGM College, Pollachi, Tamil Nadu, India
ReceiVed: January 20, 2010; ReVised Manuscript ReceiVed: March 30, 2010
Lithium trivanadate (LiV3O8) compound was prepared under different conditions and characterized by theRietveld-refined X-ray diffraction, scanning electron microscopy (SEM), and Brunauer-Emmett-Teller (BET)surface area and density techniques. The electrochemical performances of the LiV3O8 compounds preparedunder different conditions were compared. LiV3O8 rods prepared by the surfactant-assisted polymer precursormethod were found to perform well, delivering a discharge capacity of 230 ((5) mA ·h/g at the end of thesecond cycle, with an excellent capacity retention of 99.52% at the end of the 20th cycle, for a current densityof 30 mA/g. LiV3O8 rods delivered a discharge capacity of 135 ((5) mA ·h/g at the end of 350 cycles, fora current density of 240 mA/g, and reasonably high capacity values were achieved at the different currentrates. Impedance spectroscopic studies during the first and eighth cycles at various voltages are analyzed anddiscussed.
1. Introduction
Nanostructured materials play an important role in advanc-ing electrochemical energy storage and conversion technolo-gies such as lithium ion batteries and fuel cells, offering greatpromise to address rapidly growing environmental concernsand the increasing global demand for energy.1 For example,literature studies show that V2O5 nanostrips obtained by thepolyol method have an improved initial discharge capacityowing to the nanostructured morphology of the compound.2
Nanosized and rodlike LiMn2O4 has been found to achievegood electrochemical performance.3 LiFePO4 microstructureswith self-assembled nanoplates synthesized by a solvothermalmethod deliver better cell performance than the correspondingcommercial material.4
Li1+xV3O8 has been investigated as a cathode material forthe past 20 years. Its crystal structure5 consists of a layeredmonoclinic structure belonging to the space group P21/m. Thestructure is made up of (V3O8)(1+x)- layers in the b-c planes,stacked one above the other along the a axis, which, in turn,consists of two basic structural units, VO6 octahedra and VO5
distorted trigonal bipyramids interconnected to each other bycorner-sharing oxygen atoms to form V-O layers. Between theV-O layers, there are different octahedral and tetrahedral sitesfor the lithium ions. Unlike in other layered structures, wherethe layers are held together only by weak van der Waals forces,in this compound, each layer is held together by Li+ ions at theoctahedral sites present in the interlayer. The immobile lithiumions play a pinning role between the layers and keep the layersstrongly connected. The excess lithium corresponding to the
amount x is accommodated at the tetrahedral sites between thelayers and takes part in charge/discharge processes.6-9 Despiteits structural advantages, the electrochemical performance ofthe above compound was found to be mainly influenced by thesynthesis method.10 Synthesis conditions such as temperatureand raw materials used and morphological characteristics of thefinal product such as size, shape, texture (agglomerations), andcrystallinity were found to influence its electrochemicalperformance.11,12 The compound LiV3O8 has many advantagessuch as low cost, high energy density, and good safetycharacteristics.10,11 The moderate working voltage (2.5-3.5 vsLi)7 and high-temperature stability of this compound whencompared to those of other cathode materials such as LiCoO2
(4.0 V vs Li),13 LiMn2O4 (4.0 V vs Li),1 and LiFePO4 (3.4 Vvs Li)1 make it more suitable for use in lithium polymerbatteries. Intensive research has been carried out on this cathodematerial as reported by many authors.10,14,15 The preparation ofLiV3O8 by a sol-gel method, other oxides, and electrochemicalproperties were discussed in detail by Fu et al.16
The LiV3O8 compound has been prepared and studied bymany different methods such as a sol-gel process,12,17-19
electrospinning combined with a sol-gel process,20,21 hydro-thermal process,22-24 freeze-drying,25 a citric acid and peroxidesol-gel method,26 spray drying,11 a rheological phase reactionmethod,27 an ultrasonic method,28 a flame pyrolysis method,29
a low-heating solid-state method,30 a microwave sol-gel route,31
a microwave solid-state synthesis,14 a low-temperature reactionroute,32 and an ethylenediaminetetraacetic acid (EDTA) sol-gelmethod.33 In the present work, LiV3O8 nanostructures wereprepared by a simple preparation method within a shortcalcination time of 2 h. The structural and energy storageproperties of the material were analyzed, and detailed electro-chemical impedance spectroscopy studies were performed.
* Corresponding authors. E-mail: [email protected] (M.V.R.),[email protected] (S.S.), [email protected] (B.V.R.C.). Tel.:65-651662605 (M.V.R.). Fax: 65-67776126 (M.V.R.).
† National University of Singapore.‡ Bharathiar University.§ Kalasalingam University.| NGM College.
J. Phys. Chem. C 2010, 114, 8099–8107 8099
10.1021/jp1005692 2010 American Chemical SocietyPublished on Web 04/14/2010
2. Experimental Section
2.1. Preparation of Materials. The compound LiV3O8 wasprepared under different preparation conditions (with samplecodes LiV3O8-I, LiV3O8-II, and LiV3O8-III) as follows:
(i) Surfactant-Assisted Polymer Precursor Method(LiV3O8-I). Stoichiometric amounts of lithium acetate (0.54g, 98% purity, Aldrich) and ammonium metavanadate (1.76 g,98% purity, Fluka) were mixed with the polymer poly(vinylpyrrolidone) (PVP K40, 99% purity, Aldrich, molecular weight40000) dissolved in 100 mL of distilled water, and the mixturewas refluxed at 100 °C for 2 h. The total molar ratio metal ionsto PVP was maintained at 1:1.5. The resultant solution wasevaporated on a hot plate until the volume was reduced by one-half, and then it was added to 15 mL of the surfactant Igepal(Aldrich) and stirred well. The surfactant-containing solutionwas transferred in a crucible without any prior drying on a hotplate and heated at a temperature of 550 °C in a Carbolite boxfurnace for 2 h at a heating rate of 3 °C/min. For comparison,the preparation was repeated, and the resultant solution stirredwith Igepal was heat treated at 500 °C for 2 h.
To understand the role of the polymer and surfactant, LiV3O8
was prepared using either polymer or surfactant as follows:(ii) LiV3O8-II. Stoichiometric amounts of lithium acetate
(0.54 g) and ammonium metavanadate (1.76 g) were mixed withthe polymer PVP K40 (Aldrich) dissolved in 100 mL of distilledwater and refluxed at 100 °C for 2 h. The resultant solutionafter reflux was evaporated on a hot plate, and the dry residuewas ground and further heat-treated at 550 °C at a heating rateof 3 °C/min for about 2 h in air to obtain the LiV3O8 product.No surfactant was used during this preparation.
(iii) LiV3O8-III. The raw materials lithium acetate (0.54 g)and ammonium metavanadate (1.76 g) were dissolved in distilledwater (100 mL) in the absence of the polymer and refluxed for2 h at 100 °C. After reflux, the resultant solution was evaporatedto one-half its original volume and then mixed with 15 mL ofsurfactant, stirred well, and heat-treated in a Carbolite boxfurnace at a temperature of 550 °C at a heating rate of 3 °C/min for 2 h in air.
2.2. Characterization Techniques. Thermogravimetric analy-sis (TGA) and differential thermal analysis (DTA) studies wereperformed using SDT Q600 TGA/DTA instruments at a heatingrate of 20 °C/min in air. The as-prepared samples were character-ized by X-ray diffraction (XRD) using a Philips X’PERT MPDunit with Cu KR radiation. The unit-cell lattice parameters wereobtained by Rietveld refinement of the powder XRD data usingthe commercially available software TOPAS, version 2.1. Themorphology of the powders was examined by scanning electronmicroscopy (SEM) measurements using a JEOL JSM-6700Finstrument. A Tristar 3000 apparatus (Micromeritics, Norcross, GA)and an AccuPyc 1330 pycnometer (Micromeritics, Norcross, GA)were used to study the Brunauer-Emmett-Teller (BET) surfacearea and density, respectively, of the compounds.
2.3. Electrode Fabrication and Characterization Tech-niques. Electrochemical studies were carried out with 2016-type coin cells. The electrodes were made of 70:15:15 wt % ofactive material LiV3O8, conductive carbon black (Super P), andpoly(vinylidene fluoride) (PVDF) Kynar 2801 as a binder,respectively. N-Methylpyrrolidone (NMP) was used as thesolvent. Lithium foil was used as the counter and referenceelectrodes. Cyclic voltammetry studies at a scan rate of 0.058mV/s and galvanostatic cycling studies at different current rateswere made in the voltage range of 2.0-4.0 V vs Li, cycled atroom temperature. Impedance spectroscopy were carried outwith a Solartron Impedance/gain-phase analyzer (model SI 1255)
coupled to a potentiostat (SI 1268) at room temperature (25°C). The frequency was varied from 0.18 MHz to 3 mHz withan alternating-current signal amplitude of 10 mV. Nyquist plots(Z′ vs -Z′′) were collected and analyzed using ZPlot and ZViewsoftware (version 2.2, Scribner Associates Inc., Southern Pines,NC). Detailed descriptions of coin cell fabrication and instru-mentation are given in our previous reports.34,35
3. Results and Discussion
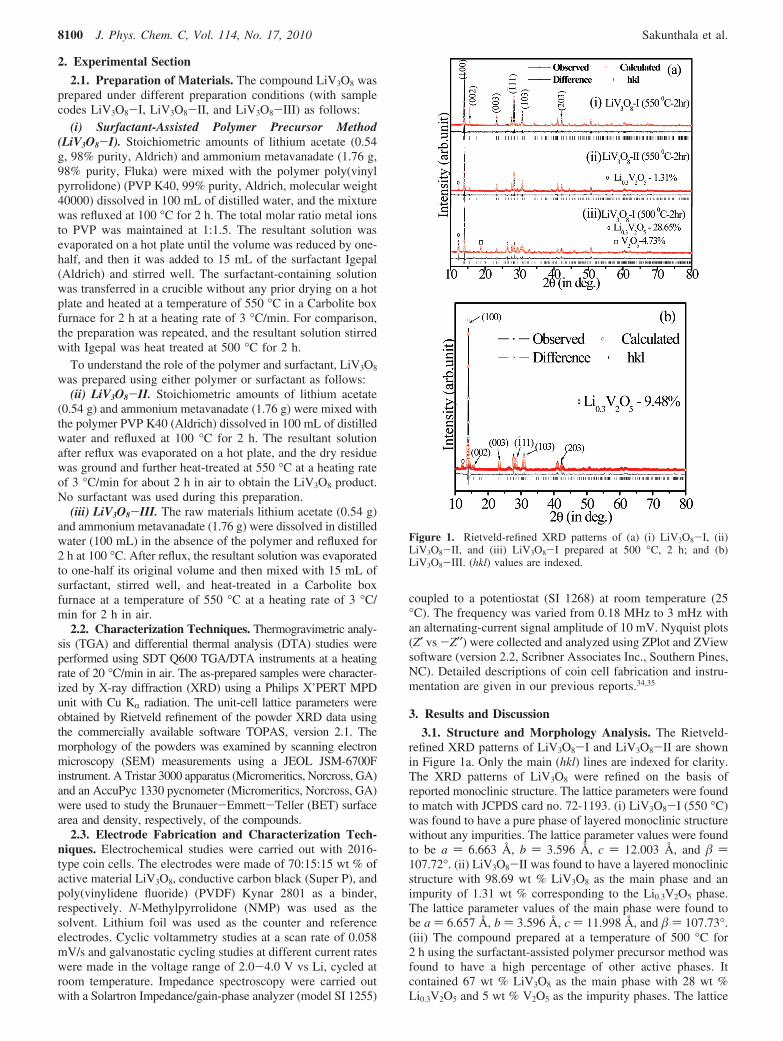
3.1. Structure and Morphology Analysis. The Rietveld-refined XRD patterns of LiV3O8-I and LiV3O8-II are shownin Figure 1a. Only the main (hkl) lines are indexed for clarity.The XRD patterns of LiV3O8 were refined on the basis ofreported monoclinic structure. The lattice parameters were foundto match with JCPDS card no. 72-1193. (i) LiV3O8-I (550 °C)was found to have a pure phase of layered monoclinic structurewithout any impurities. The lattice parameter values were foundto be a ) 6.663 Å, b ) 3.596 Å, c ) 12.003 Å, and )107.72°. (ii) LiV3O8-II was found to have a layered monoclinicstructure with 98.69 wt % LiV3O8 as the main phase and animpurity of 1.31 wt % corresponding to the Li0.3V2O5 phase.The lattice parameter values of the main phase were found tobe a ) 6.657 Å, b ) 3.596 Å, c ) 11.998 Å, and ) 107.73°.(iii) The compound prepared at a temperature of 500 °C for2 h using the surfactant-assisted polymer precursor method wasfound to have a high percentage of other active phases. Itcontained 67 wt % LiV3O8 as the main phase with 28 wt %Li0.3V2O5 and 5 wt % V2O5 as the impurity phases. The lattice
Figure 1. Rietveld-refined XRD patterns of (a) (i) LiV3O8-I, (ii)LiV3O8-II, and (iii) LiV3O8-I prepared at 500 °C, 2 h; and (b)LiV3O8-III. (hkl) values are indexed.
8100 J. Phys. Chem. C, Vol. 114, No. 17, 2010 Sakunthala et al.

parameters of the main phase were found to be a ) 6.633 Å, b) 3.599 Å, c ) 11.978 Å, and ) 107.76°.
The Rietveld-refined XRD pattern of LiV3O8-III is shownin the Figure 1b. The compound was found to have a very high-intensity (100) plane over the other planes. This is due to thepreferred orientation of the particles along the (100) plane,leading to a longer diffusion path for the lithium ions.36 Thecompound was found to have 9.5 wt % Li0.3V2O5 phase asthe impurity phase and 91.5 wt % LiV3O8 as the main phase.The lattice parameter values of the main phase were found tobe a ) 6.648 Å, b ) 3.596 Å, c ) 11.997 Å, and ) 107.77°.
The XRD results showed 550 °C as the optimum temperaturefor the formation of the single-phase LiV3O8 compound,irrespective of the preparation conditions. Electrochemicalstudies were performed only for the compound prepared at 550°C. A similar type of impurity peak corresponding to Li0.3V2O5
was observed in many reports17,21,28,31,37,38 and considered as anactive phase.17 A few reports have shown the presence of otheractive phases to degrade electrochemical performance. Zhou etal.33 reported the cyclic performance to be degraded by thepresence of Li0.3V2O5 and LiV2O5 impurities. A similar pointwas made by Wu et al.31 A few other reports have shown animproved performance of LiV3O8 in the presence of other activephases. Dubarry et al.15 studied LiV3O8/Li0.3V2O5/C nanocom-posites by a carbothermal reduction method with rapid heatingand found the capacity retention of LiV3O8 to be improved. Tranet al.11 prepared LiV3O8 through a spray-drying method, wherethey observed the Li0.3V2O5 phase by using carbon in thepreparation process, and found better electrochemical perfor-mance at a high current rate when compared to the phase-purecompound.
Figure 2a-d shows SEM images of the LiV3O8 productsprepared under different conditions. Bundles of a needlelikemorphology were observed for the LiV3O8 compound prepared
by the surfactant-assisted polymer precursor method at atemperature of 500 °C for 2 h (Figure 2a). With an increase inthe calcination temperature to 550 °C (LiV3O8-I), the needlelikeparticles were found to grow in length into LiV3O8 rods withless agglomeration (Figure 2b). The observed nanostructureswere due to the combined effect of the polymer PVP and thesurfactant Igepal, as described in the schematic diagram shownin Figure 3. The raw materials lithium acetate and ammoniummetavanadate become finely dispersed and are readily adsorbedon the polymer chains during reflux. The polymer PVP isdecomposed at a higher temperature of around 500 °C and actsas a good capping agent in controlling the growth of theparticles. It is well-known that the polymer PVP is capable ofreadily adsorbing metal ions and metal oxides.39 The additionof the surfactant to the polymer mixture aids in faster growthof the particles through the additional thermal energy createdby its decomposition. These combined effects of Igepal and thepolymer lead to bundles of nanosized needles. With increasingcalcination temperature, the needles were found to grow intoLiV3O8 rods.
Figure 2. SEM images of (a) LiV3O8 prepared by the surfactant-assisted polymer precursor method at a temperature of 500 °C for 2 h; (b)LiV3O8-I, (c) LiV3O8-II, (d) LiV3O8-III. Scale bar: 1 µm.
Figure 3. Schematic diagram of the formation of LiV3O8 rods by thesurfactant-assisted polymer precursor method.
Lithium Trivanadate Rods for Lithium Batteries J. Phys. Chem. C, Vol. 114, No. 17, 2010 8101
The combustion nature of the polymer around 500 °C issupported by thermal studies of the precursor or dry residuecorresponding to LiV3O8-II, which was collected beforecalcination. The results are discussed in the Supporting Informa-tion (SI 1). No thermal studies on the precursor related toLiV3O8-I were performed. This is due to the experimentallimitations, where the refluxed solution after the addition ofsurfactant was as such calcinated without any prior drying onthe hot plate. It is well understood that the decomposition ofthe surfactant occurs at a relatively lower temperature comparedto polymer PVP. The specific roles of the polymer PVP andthe surfactant Igepal on morphology are clearly seen in SEMstudies of compounds LiV3O8-II and LiV3O8-III (Figure 2c,d).The particles were found to be highly agglomerated with noproper crystal shape in the case of LiV3O8-II (prepared usingpolymer in the absence of surfactant) (Figure 2c). The compoundLiV3O8-III (prepared using the surfactant Igepal in the absenceof polymer) was found to form larger, agglomerated, micrometer-sized rods (Figure 2d). The rodlike particles obtained within ashort reaction time showed faster growth of the particles in thesurfactant medium. The larger particle size is due to uncontrolledgrowth in the absence of the polymer.
Better electrochemical performance could be expected forLiV3O8-I (Figure 2b), because the size and aggregation of thegrains were found to play a major role in the electrochemicalproperties. Literature studies showed the shape of the LiV3O8
compound to be a main factor influencing the cyclability.40
Experimental densities of LiV3O8-I, LiV3O8-II, andLiV3O8-III were found to be 3.5566, 3.5862, and 3.6828 g/cm3,respectively. The errors between experimental and calculateddensities were within 5-8% for all of the compounds. The BETsurface areas of LiV3O8-I, LiV3O8-II, and LiV3O8-III werefound to be 0.85, 0.53, and 0.84 m2/g, respectively.
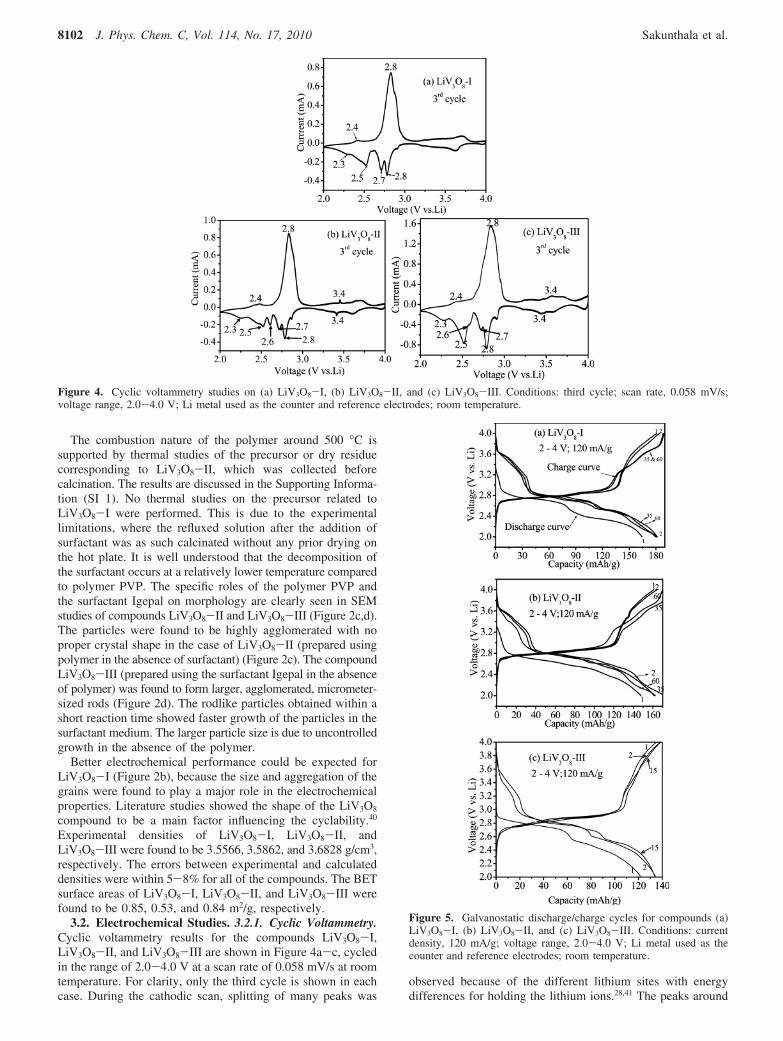
3.2. Electrochemical Studies. 3.2.1. Cyclic Voltammetry.Cyclic voltammetry results for the compounds LiV3O8-I,LiV3O8-II, and LiV3O8-III are shown in Figure 4a-c, cycledin the range of 2.0-4.0 V at a scan rate of 0.058 mV/s at roomtemperature. For clarity, only the third cycle is shown in eachcase. During the cathodic scan, splitting of many peaks was
observed because of the different lithium sites with energydifferences for holding the lithium ions.28,41 The peaks around
Figure 4. Cyclic voltammetry studies on (a) LiV3O8-I, (b) LiV3O8-II, and (c) LiV3O8-III. Conditions: third cycle; scan rate, 0.058 mV/s;voltage range, 2.0-4.0 V; Li metal used as the counter and reference electrodes; room temperature.
Figure 5. Galvanostatic discharge/charge cycles for compounds (a)LiV3O8-I, (b) LiV3O8-II, and (c) LiV3O8-III. Conditions: currentdensity, 120 mA/g; voltage range, 2.0-4.0 V; Li metal used as thecounter and reference electrodes; room temperature.
8102 J. Phys. Chem. C, Vol. 114, No. 17, 2010 Sakunthala et al.
3.5, 2.8, 2.7, and 2.3-2.4 V are due to the single-phase reactionwith lithium ions occupying empty tetrahedral sites,40,41 and thepeak at ∼2.5 V is related to the filling of octahedral sites formedupon intercalation during which a two-phase transition fromLi3V3O8 to Li4V3O8 takes place.18,41 The last step at ∼2.35 V isattributed to the slower kinetic insertion process where thesingle-phase transition corresponding to the Li4V3O8 phase takesplace and all of the lithium ions are octahedrally coordinated.42,43
Both of the peaks at 2.5 and 2.3 V were considered as primaryreasons for the origin of capacity fading during the cycling ofLiV3O8.40 The ∼2.35 V peak is also correlated with the reactivitybetween the electrolyte and the Li4+xV3O8 phase, leading topassive film formation or material dissolution, which, in turn,leads to capacity fading.42 A cathodic peak around 2.6 V andan anodic/cathodic peak around 3.4 V were noticed only in thecase of compounds LiV3O8-II and LiV3O8-III. These twopeaks belong to the active phase Li0.3V2O5 as observed in theXRD analysis of these compounds.
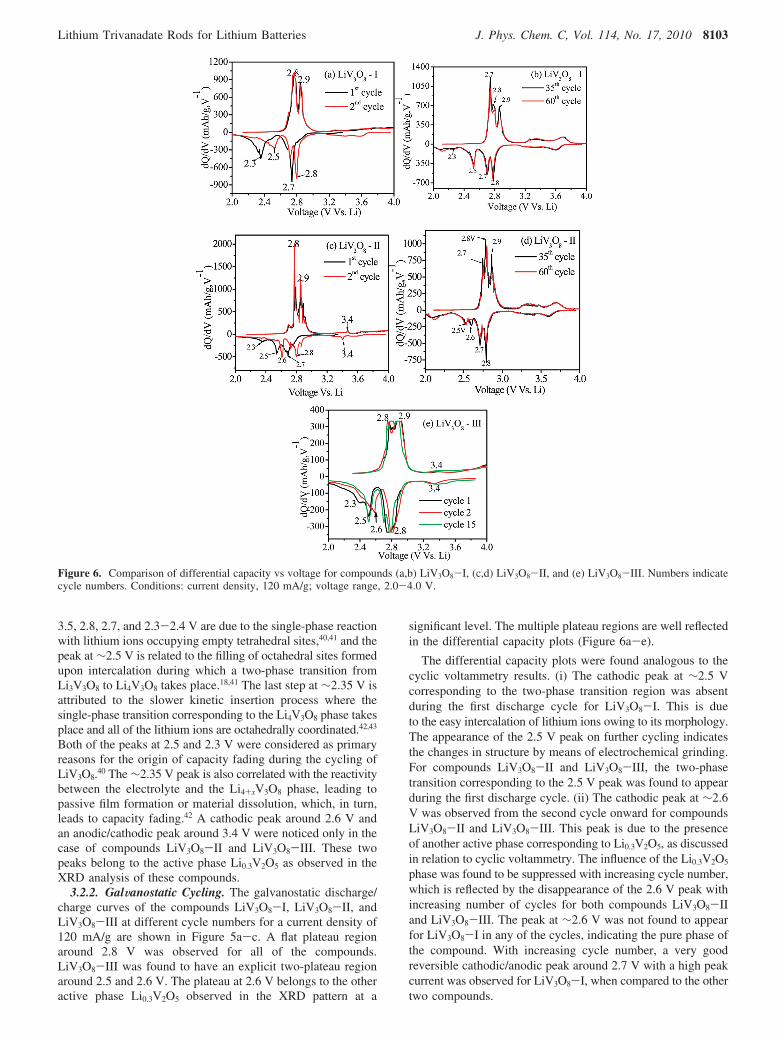
3.2.2. GalWanostatic Cycling. The galvanostatic discharge/charge curves of the compounds LiV3O8-I, LiV3O8-II, andLiV3O8-III at different cycle numbers for a current density of120 mA/g are shown in Figure 5a-c. A flat plateau regionaround 2.8 V was observed for all of the compounds.LiV3O8-III was found to have an explicit two-plateau regionaround 2.5 and 2.6 V. The plateau at 2.6 V belongs to the otheractive phase Li0.3V2O5 observed in the XRD pattern at a
significant level. The multiple plateau regions are well reflectedin the differential capacity plots (Figure 6a-e).
The differential capacity plots were found analogous to thecyclic voltammetry results. (i) The cathodic peak at ∼2.5 Vcorresponding to the two-phase transition region was absentduring the first discharge cycle for LiV3O8-I. This is dueto the easy intercalation of lithium ions owing to its morphology.The appearance of the 2.5 V peak on further cycling indicatesthe changes in structure by means of electrochemical grinding.For compounds LiV3O8-II and LiV3O8-III, the two-phasetransition corresponding to the 2.5 V peak was found to appearduring the first discharge cycle. (ii) The cathodic peak at ∼2.6V was observed from the second cycle onward for compoundsLiV3O8-II and LiV3O8-III. This peak is due to the presenceof another active phase corresponding to Li0.3V2O5, as discussedin relation to cyclic voltammetry. The influence of the Li0.3V2O5
phase was found to be suppressed with increasing cycle number,which is reflected by the disappearance of the 2.6 V peak withincreasing number of cycles for both compounds LiV3O8-IIand LiV3O8-III. The peak at ∼2.6 V was not found to appearfor LiV3O8-I in any of the cycles, indicating the pure phase ofthe compound. With increasing cycle number, a very goodreversible cathodic/anodic peak around 2.7 V with a high peakcurrent was observed for LiV3O8-I, when compared to the othertwo compounds.
Figure 6. Comparison of differential capacity vs voltage for compounds (a,b) LiV3O8-I, (c,d) LiV3O8-II, and (e) LiV3O8-III. Numbers indicatecycle numbers. Conditions: current density, 120 mA/g; voltage range, 2.0-4.0 V.
Lithium Trivanadate Rods for Lithium Batteries J. Phys. Chem. C, Vol. 114, No. 17, 2010 8103

Figure 7a shows plots of capacity vs cycle number forLiV3O8-I at current densities of 30 and 120 mA/g and forLiV3O8-II and LiV3O8-III at a current density of 120 mA/gin the voltage range 2.0-4.0 V vs Li. Figure 7b shows a similarplot for LiV3O8-I at a higher current density of 240 mA/g. Forthe current density of 120 mA/g, LiV3O8-I was found to havean initial discharge capacity of 164 mA ·h/g, and during thesecond cycle, its capacity increased to 182 mA ·h/g. Thedischarge capacity then steadily increased and reached amaximum value of 191 mA ·h/g at the 25th cycle. LiV3O8-Imaintained a discharge capacity value of 180 mA ·h/g at theend of the 60th cycle, which corresponds to 1.94 mol of lithiumper mole of LiV3O8. The capacity retention at the end of the60th cycle from the maximum was found to be 94%. The overallcapacity retention after 60 cycles calculated from the secondcycle was 99%. For LiV3O8-II, the initial and second dischargecapacity values were the same, 161 mA ·h/g (for 120 mA/g).The discharge capacity slowly decreased until the 19th cycle(158 mA ·h/g), corresponding to a 1.86% capacity loss, afterwhich it started to increase and reached a maximum of 172mA ·h/g at the 33rd cycle. Then, the discharge capacitystabilized, reaching a value of 163 mA ·h/g at the end of the60th cycle. The capacity retention from the maximum was foundto be 95%. The overall capacity retention after 60 cycles fromthe second cycle was 100%.
The preliminary electrochemical cycling studies ofLiV3O8-III (Figures 5c and 7a), with the larger particle size,revealed a lower discharge capacity of 114 mA ·h/g during theinitial cycle and a stable discharge capacity of 130 mA ·h/g atthe end of the 15th cycle. The lower capacity value is due tothe larger particles, which lead to longer diffusion paths orkinetic limitations of the material.
At current densities of 30 and 240 mA/g, the initial dischargecapacities of LiV3O8-I were found to be 206 and 120 mA ·h/g, respectively. At the second cycle, the capacity values werefound to increase to 230 and 151 mA ·h/g, respectively. At thelower current rate (30 mA/g), the discharge capacity was foundto be stabilized from the second cycle onward, with a capacityretention of 99.5% from the second to the 16th cycle and 96%from the second to the 40th cycle. For the higher current densityof 240 mA/g, the maximum discharge capacity of 160 mA ·h/gwas attained at the 33rd cycle, and the capacity retention at theend of the 60th cycle was 94%. The overall capacity retentionafter 60 cycles from the second cycle was 100%, and after 350cycles, it still remained at 88%, calculated from the second cycle(Figure 7b).
The electrochemical performance of LiV3O8-I in the presentwork was compared with literature reports, as summarized inTable 1. Although the capacity retention of the LiV3O8-polypyrrole composite prepared by a low-temperature solutionroute was found to be good, the discharge capacity value wasfound to be very low for the applied current density (40 mA/g).44 LiV3O8 prepared by the spray-drying technique was foundto have a high discharge capacity value with capacity retentionof 84% in the 2.0-3.3 V range, but it was reported that, whenthe same sample was cycled between 2.0-3.7 V, the capacityfading was >2% per cycle.11 For the LiV3O8 nanorods preparedby a hydrothermal method followed by solid-state reaction,which could not be treated as either an anode or a cathodematerial, the higher discharge capacity values were due to alower cutoff discharge voltage of 1.5 V. Moreover, no well-defined plateau region was observed during charge/dischargeexcept for the first cycle, as reflected in the cyclic voltammeteryresults.42
In the present work, good discharge capacity values wereobtained at a lower current rate, and moderate values wereobtained at a high current rate because of kinetic limitations.The cyclic stability of LiV3O8-I was found to be better in thevoltage range of 2.0-4.0 V vs Li, with a good flat plateau regionaround 2.8 V, corresponding to the working voltage. ForLiV3O8-I, at current densities of 30, 120, and 240 mA/g, theamounts of lithium inserted during the second discharge were2.46, 1.96, and 1.63 mol of Li, respectively, with Coulombicefficiencies of around 94-96% (plots are shown in the Sup-porting Information, SI 2). In comparison, the electrochemicalperformance of LiV3O8-I in our present study showed the best
Figure 7. Plots of capacity vs cycle number for (a) LiV3O8-I at currentdensities of (*) 30 and (]) 120 mA/g, (O) LiV3O8-II at 120 mA/g,and (3) LiV3O8-III at 120 mA/g and (b) (x) LiV3O8-I at a currentdensity of 240 mA/g. Voltage range: 2.0-4.0 V.
TABLE 1: Literature Reports on LiV3O8 Prepared by Different Methods and Its Electrochemical Properties
synthesismethodref composition
temperature(°C)/time (h) particle morphology
current density(mA/g)
capacity (mA ·h/g)(cycle number)
capacityretention (%)
hydrothermal24 LiV3O8 400/12 rods; diameter 200-500 nm, length 2-5 µm 120 212.8 (1)-152.1 (18) 72sol-gel45 Li1.2V3O8 350/10 - 60 281 (2)-200 (40) 71sol-gel46 Li0.96Ag0.04V3O8 300/16 - 150 328 (1)-252.7 (50) 77flame pyrolysis29 LiV3O8 350/30 min agglomerated nanoparticles, 50 nm 100 271 (1)-180 (50) 66solution route44 LiV3O8-polypyrrole
(PPy) composite480/12 amorphous PPy and LiV3O8 particles 40 184 (1)-183 (100) 99
spray drying11 Li1.1V3O8 320/1 spherical aggregates consisting of nanorods 116 260 (2)-220 (60) 84hydrothermal42 LiV3O8 450/10 single-crystalline nanorods 100 stabilized at 236 (100) 96present work:
LiV3O8-ILiV3O8 550/2 rods; diameter < 1 µm 30 230 (2)-219 (40) 96
120 182 (2)-180 (60) 99240 152 (2)-152 (60) 100
152 (2)-135 (350) 89
8104 J. Phys. Chem. C, Vol. 114, No. 17, 2010 Sakunthala et al.
results. Therefore, the LiV3O8 nanostructure prepared within avery short-duration (i.e., 2-h) calcination using surfactant andpolymer in the preparation step was found to have goodelectrochemical performance.
Figure 8 shows the observed discharge capacities at differentcurrent rates. The second discharge capacity for the currentdensity of 30 mA/g was found to be 220 mA ·h/g. Dischargecapacity values of 201, 189, 176, 172, 164, 142, 113, 96, and76 mA ·h/g were observed for current densities of 120, 180,240, 300, 360, 420, 540, 620, and 740 mA/g respectively. Slightdifferences in the capacity values when compared to long-term-cycled electrodes were due to the slow current charging stepand to the 4-h relaxation.
3.3. Electrochemical Impedance Spectroscopy (EIS) Stud-ies. EIS is a well-established technique for studying the electrodekinetics of cathode and anode materials because of its inherentlynondestructive nature and its ability to distinguish variousphenomena at different time scales by frequency modulation
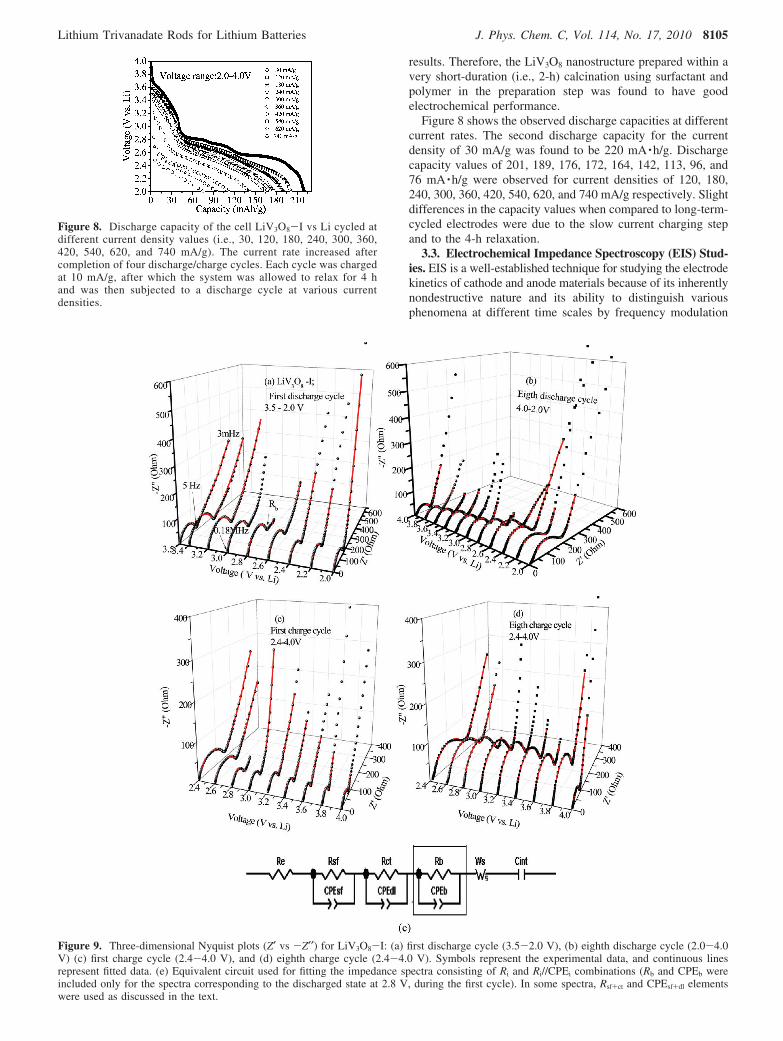
Figure 8. Discharge capacity of the cell LiV3O8-I vs Li cycled atdifferent current density values (i.e., 30, 120, 180, 240, 300, 360,420, 540, 620, and 740 mA/g). The current rate increased aftercompletion of four discharge/charge cycles. Each cycle was chargedat 10 mA/g, after which the system was allowed to relax for 4 hand was then subjected to a discharge cycle at various currentdensities.
Figure 9. Three-dimensional Nyquist plots (Z′ vs -Z′′) for LiV3O8-I: (a) first discharge cycle (3.5-2.0 V), (b) eighth discharge cycle (2.0-4.0V) (c) first charge cycle (2.4-4.0 V), and (d) eighth charge cycle (2.4-4.0 V). Symbols represent the experimental data, and continuous linesrepresent fitted data. (e) Equivalent circuit used for fitting the impedance spectra consisting of Ri and Ri//CPEi combinations (Rb and CPEb wereincluded only for the spectra corresponding to the discharged state at 2.8 V, during the first cycle). In some spectra, Rsf+ct and CPEsf+dl elementswere used as discussed in the text.
Lithium Trivanadate Rods for Lithium Batteries J. Phys. Chem. C, Vol. 114, No. 17, 2010 8105

in conjunction with electrode kinetics.35,47-51 In the present work,electrochemical impedance spectra were recorded at roomtemperature on the cells with LiV3O8-I vs Li during the firstand eighth discharge-charge cycles. The data were collectedat selected voltages in the range of 2.0-4.0 V at a currentdensity of 30 mA/g. The cells were kept under equilibrium for3 h before measurements. Figure 9a-d shows three-dimensionalNyquist plots for the first and eighth discharge-charge cycles.The impedance spectra were fitted using the reported equivalentelectrical circuit35 shown in Figure 9e.
The impedance spectra corresponding to the first cycle in thecharged state (4.0-2.8 V) and the spectra corresponding to theeighth cycle in both the charged and discharged states (4.0-2.4V) were fitted using two equivalent circuit models correspondingto surface-film and charge-transfer resistance, that is, using Rsf,CPEsf and Rct, CPEdl, respectively. For all other impedancespectra, the surface-film and the charge-transfer resistances areindistinguishable, and therefore, they were fitted using oneequivalent circuit model with Rsf+ct and CPEsf+dl elements. Rsf+ct
is the combined impedance of surface film and charge transfer,and CPEsf+dl is the corresponding constant phase element.
For the impedance spectra of the fresh cell, the high- tomedium-frequency region corresponds to the surface film (Rsf),followed by a line inclined at an angle of 45° attributed toWarburg resistance. This indicates a surface-film resistance (Rsf)value of 128 Ω with a corresponding constant phase element[i.e., surface-film capacitance (CPEsf)] value of 23 µF.
During the initial discharge cycle, the width of the semicirclevaries, indicating an increase in impedance at 3.4 V comparedto the open-circuit voltage (OCV), and remains more or lessthe same until 3.0 V. From 2.6 to 2.0 V, a decrease in theimpedance value was observed which corresponds to thecombination of surface-film and charge transfer resistance.The corresponding impedance values are given in Table 2.
The impedance spectra at the discharge voltage of 2.8 Vduring the first cycle showed a small semicircle in the low-frequency region corresponding to the bulk resistance (Rb) ofthe electrode, arising from the electronic conductivity of theactive material and ionic conductivity in the pores of thecomposite electrode.35,52 In all cases, the straight line inclinedat an angle of 45° corresponds to the finite-length Warburgresistance, related to solid-state diffusion. The line followingthe Warburg resistance at very low frequency corresponds tothe intercalation capacitance (Cint). The resistance due to the
electrolyte and the cell components was less than 5 ((2) Ω,and the Cint value was ∼0.1 F. The CPE values correspondingto the surface film and the double layer were in the range ofmicrofarads, and that of the bulk was in the millifarad range.The fitted impedance values obtained are shown in Table 2. Itwas observed that the impedance during discharge was muchreduced for the eighth cycle when compared to that of the firstcycle. However, the charge spectra show an increase inimpedance, and the reason for this behavior is not wellunderstood at present. In the discharged state, for voltages lessthan 2.8 V, a decreasing trend of impedance values was observedfor the first cycle. In contrast, an increasing trend was observedfor the eighth cycle, which is due to the two-phase reactionformed after the first cycle as discussed in relation to the cyclicvoltammetery and galvanostatic studies.
4. Conclusions
The surfactant-assisted polymer precursor method was foundto be simple and suitable for the preparation of phase-pureLiV3O8 rods. The resulting materials were characterized by avariety of physical and electroanalytical techniques. Galvano-static and cyclic voltammetry studies showed average dis-charge-charge voltages of 2.8 V vs Li. Galvanostatic cyclingstudies of LiV3O8-I showed a stable capacity of 135 mA ·h/gat the end of the 350th cycle at a current rate of 240 mA/g withnegligible capacity fading.
Acknowledgment. The authors thank Prof. G. V. Subba Rao,Department of Physics, NUS, for helpful discussions. A.S.thanks the Defence Research and Development Organisation(DRDO), India, for the grant of a Senior Research Fellowship.M.V.R. and B.V.R.C. thank the National Research Foundation(NRF), Singapore.
Supporting Information Available: Thermal analysis of theprecursor corresponding to LiV3O8-II prepared by the polymerprecursor method and plots of Coulombic efficiency with cyclenumber for LiV3O8. This material is available free of chargevia the Internet at http://pubs.acs.org.
References and Notes
(1) Manthiram, A.; Vadivel Murugan, A.; Sarkar, A.; Muraliganth, T.Energy EnViron. Sci. 2008, 1, 621.
TABLE 2: Impedance Values during First and Eighth Discharge-Charge Cyclesa,b
first cycle eighth cycle
discharge charge discharge charge
voltage Rsf+ct CPEsf+dl Rsf Rct CPEsf CPEdl Rsf Rct CPEsf CPEdl Rsf Rct CPEsf CPEdl
4.0 - - 11 54 232 67 - - - - 12 60 459 653.8 - - 14 40 121 36 12 69 699 69 5 180 49 443.6 - - 13 44 225 50 11 95 708 91 2 169 11 483.4 193 20 13 48 257 54 10 97 555 88 1 187 4 523.2 197 21 14 51 253 59 10 83 567 80 2 197 5 503.0 199 26 10 55 462 70 5 109 119 41 1 203 2 562.8 207 29 5 60 105 88 4 139 12 50 4 167 131 39
67c 17d
2.6 170 26 116e 48f 5 159 34 47 5 197 24 752.4 143 38 144e 41f 5 152 16 56 3 177 1 1032.2 143 35 - - - - 131e 100f - - - -2.0 141 42 - - - - 166e 120f - - - -
a Resistance values in ohms, and capacitance values in farads. b Error values: Rsf, (3 Ω; Rct, (5 Ω; Rsf+ct, (5 Ω; CPEsf, (3 µF; CPEdl, (3µF; CPEsf+dl, (5 µF. c Bulk resistance, Rb, in ohms (error: Rsf, (3 Ω). d Bulk capacitance, CPEb, in millifarads (error: CPEb, (3 mF). e Rsf+ct.f CPEsf+ct.
8106 J. Phys. Chem. C, Vol. 114, No. 17, 2010 Sakunthala et al.

(2) Ragupathy, P.; Shivakumara, S.; Vasan, H. N.; Munichandraiah,N. J. Phys. Chem. C 2008, 112, 16700.
(3) Shaju, K. M.; Bruce, P. G. Chem. Mater. 2008, 20, 5557.(4) Yang, H.; Wu, X. L.; Cao, M. H.; Guo, Y. G. J. Phys. Chem. C
2009, 113, 3345.(5) Wadsley, A. D. Acta Crystallogr. 1957, 10, 261.(6) kawakita, J.; Mori, H.; Miura, T.; Kishi, T. Solid State Ionics 2000,
131, 229.(7) Zhang, X.; Frech, R. Electrochim. Acta 1998, 43, 861.(8) kawakita, J.; Katayama, Y.; Miura, T.; Kishi, T. Solid State Ionics
1998, 107, 145.(9) Kawakita, J.; Miura, T.; Kishi, T. Solid State Ionics 1999, 120,
109.(10) Kannan, A. M.; Manthiram, A. J. Power Sources 2006, 159, 1405.(11) Tran, N.; Bramnik, K. G.; Hibst, H.; Prolss, J.; Mronga, N.;
Holzapfel, M.; Scheifele, W.; Novak, P. J. Electrochem. Soc. 2008, 155,A384.
(12) Jouanneau, S.; Verbaere, A.; Lascaud, S.; Guyomard, D. Solid StateIonics 2006, 177, 311.
(13) Tan, K. S.; Reddy, M. V.; Subba Rao, G. V.; Chowdari, B. V. R.J. Power Sources 2005, 147, 241.
(14) Yang, G.; Wang, G.; Hou, W. H. J. Phys. Chem. B 2005, 109,11186.
(15) Dubarry, M.; Gaubicher, J.; Moreau, P.; Guyomard, D. J. Elec-trochem. Soc. 2006, 153, A295.
(16) Fu, L. J.; Liu, H.; Li, C.; Wu, Y. P.; Rahm, E.; Holze, R.; Wu,H. Q. Prog. Mater. Sci. 2005, 50, 881.
(17) Deptula, A.; Dubarry, M.; Noret, A.; Gaubicher, J.; Olczak, T.;Lada, W.; Guyomard, D. Electrochem. Solid State Lett. 2006, 9, A16.
(18) Jouanneau, S.; La Salle, A. L. G.; Verbaere, A.; Deschamps, M.;Lascaud, S.; Guyomard, D. J. Mater. Chem. 2003, 13, 921.
(19) Cao, X. Y.; Xie, L. L.; Zhan, H.; Zhou, Y. H. Mater. Res. Bull.2009, 44, 472.
(20) Gu, Y. X.; Chen, D. R.; Jiao, X. L.; Liu, F. F. J. Mater. Chem.2006, 16, 4361.
(21) Lee, K. P.; Manesh, K. M.; Kim, K. S.; Gopalan, A. I. J. Nanosci.Nanotechnol. 2008, 8, 1.
(22) Liu, H. W.; Yang, H. M.; Huang, T. Mater. Sci. Eng. B 2007, 143,60.
(23) Xu, H. Y.; Wang, H.; Song, Z. Q.; Wang, Y. W.; Yan, H.;Yoshimura, M. Electrochim. Acta 2004, 49, 349.
(24) Xu, J.; Zhang, H. L.; Zhang, T.; Pan, Q. Y.; Gui, Y. H. J. AlloysCompd. 2009, 467, 327.
(25) Brylev, O. A.; Shlyakhtin, O. A.; Egorov, A. V.; Tretyakov, Y. D.J. Power Sources 2007, 164, 868.
(26) Feng, J.; Liu, X.; Zhang, X. M.; Jiang, J. Z.; Zhao, J.; Wang, M.J. Electrochem. Soc. 2009, 156, A768.
(27) Liu, Q. Y.; Liu, H. W.; Zhou, X. W.; Cong, C. J.; Zhang, K. L.Solid State Ionics 2005, 176, 1549.
(28) Liu, Y. M.; Zhou, X. C.; Guo, Y. L. J. Power Sources 2008, 184,303.
(29) Patey, T. J.; Ng, S. H.; Buchel, R.; Tran, N.; Krumeich, F.; Wang,J.; Liu, H. K.; Novak, P. Electrochem. Solid State Lett. 2008, 11, A46.
(30) Yang, H.; Li, J.; Zhang, X. G.; Jin, Y. L. J. Mater. Process. Technol.2008, 207, 265.
(31) Wu, F.; Wang, L.; Wu, C.; Bai, Y.; Wang, F. Mater. Chem. Phys.2009, 115, 707.
(32) Liu, L.; Jiao, L. F.; Sun, J. L.; Zhang, Y. H.; Zhao, M.; Yuan,H. T.; Wang, Y. M. J. Alloys Compd. 2009, 471, 352.
(33) Zhou, Y.; Yue, H. F.; Zhang, X. Y.; Deng, X. Y. Solid State Ionics2008, 179, 1763.
(34) Reddy, M. V.; Sundara Manoharan, S.; John, J.; Singh, B.; SubbaRao, G. V.; Chowdari, B. V. R. J. Electrochem. Soc. 2009, 156, A652.
(35) Reddy, M. V.; Subba Rao, G. V.; Chowdari, B. V. R. J. Phys.Chem. C 2007, 111, 11712.
(36) West, K.; Zachau-Christiansen, B.; Skaarup, S.; Saidi, Y.; Barker,J.; Olsen, I. I.; Pynenburg, R.; Koksbang, R. J. Electrochem. Soc. 1996,143, 820.
(37) Liu, X. H.; Wang, J. Q.; Zhang, J. Y.; Yang, S. R. J. Mater. Sci.2007, 42, 867.
(38) Liu, Y. M.; Zhou, X. C.; Guo, Y. L. Electrochim. Acta 2009, 54,3184.
(39) Christina, G.; Dembski, S.; Hofmann, A.; Ruhl, E. Langmuir 2006,22, 5604.
(40) Jouanneau, S.; La Salle, A. L. G.; Verbaere, A.; Guyomard, D. J.Electrochem. Soc. 2005, 152, A1660.
(41) Bonino, F.; Panero, S.; Pasquali, M.; Pistoia, G. J. Power Sources1995, 56, 193.
(42) Liu, H. M.; Wang, Y. G.; Wang, K. X.; Wang, Y. R.; Zhou, H. S.J. Power Sources 2009, 192, 668.
(43) Benedek, R.; Thackeray, M. M.; Yang, L. H. Phys. ReV. B 1999,60, 6335.
(44) Chew, S. Y.; Feng, C. Q.; Ng, S. H.; Wang, J. Z.; Guo, Z. P.; Liu,H. K. J. Electrochem. Soc. 2007, 154, A633.
(45) Xie, J. G.; Li, J. X.; Zhan, H.; Zhou, Y. H. Mater. Lett. 2003, 57,2682.
(46) Sun, J. L.; Jiao, L. F.; Yuan, H. T.; Liu, L.; Wei, X.; Miao, Y. L.;Yang, L.; Wang, Y. M. J. Alloys Compd. 2009, 472, 363.
(47) Shaju, K. M.; Subba Rao, G. V.; Chowdari, B. V. R. J. Electrochem.Soc. 2004, 151, A1324.
(48) Reddy, M. V.; Yu, T.; Sow, C. H.; Shen, Z. X.; Lim, C. T.; SubbaRao, G. V.; Chowdari, B. V. R. AdV. Funct. Mater. 2007, 17, 2792.
(49) Das, B.; Reddy, M. V.; Krishnamoorthi, C.; Tripathy, S.; Mahendi-ran, R.; Subba Rao, G. V.; Chowdari, B. V. R. Electrochim. Acta 2009,54, 3360.
(50) Levi, M. D.; Aurbach, D. J. Phys. Chem. B 2004, 108, 11693.(51) Nobili, F.; Tossici, R.; Marassi, R.; Croce, F.; Scrosati, B. J. Phys.
Chem. B 2002, 106, 3909.(52) Reddy, M. V.; Madhavi, S.; Subba Rao, G. V.; Chowdari, B. V. R.
J. Power Sources 2006, 162, 1312.
JP1005692
Lithium Trivanadate Rods for Lithium Batteries J. Phys. Chem. C, Vol. 114, No. 17, 2010 8107
Copyright © 2022 FDOKUMEN















![High capacity Li[Ni0.8Co0.1Mn0.1]O2 synthesized by sol–gel and co-precipitation methods as cathode materials for lithium-ion batteries](https://static.fdokumen.com/doc/165x107/6336e10720d9c9602f0b0e64/high-capacity-lini08co01mn01o2-synthesized-by-solgel-and-co-precipitation.jpg)




