
evaluación de las prácticas de aseguramiento financiero de las
Upload
independentCategory
view
6download
0
VALORACIÓN DE UNA SOLUCIÓN DE NaOH CON FAP
OBJETIVO: Conocer la concentración exacta de unadisolución NaOH 0.1 N que se ha preparado apartir de una sosa comercial que tiene unariqueza del 96 %.
FUNDAMENTO TEÓRICO:
El ftalato ácido de potasio es el patrónprimario ácido más común; la sal utilizadatiene generalmente una pureza muy elevada (99,95%) y debe secarse a 110 oC durante 2 horas.
El indicador que se va a utilizar en lareacción es fenolftaleína y la valoración delNaOH sigue la siguiente reacción deneutralización:
KOOC – C6H4 – COOH + NaOH KOOC –C6H4- COONa + H2O
En esta volumetría la solución a valorar secoloca en la bureta. Datos de ftalato ácido depotasio:
- Peso molecular = 204,22 g/mol
- Valencia = 1
MATERIAL NECESARIO:
2 Vidrios de reloj.
Matraz aforado de 250ml.
Probeta e 100ml.
Pipeta de 20ml.

Prepipeta.
Bureta de 50ml.
Soporte universal.
Frasco lavador.
Varilla.
2 Embudos.
Vasos de precipitado de 100ml y 250ml.
Pipeta pasteur.
Matraz erlenmeyer de 250ml.
REACTIVOS:
Fenolftaleína.
0,4g de FAP.
1,02 g de NaOH.
PROCEDIMIENTO:
1. Realizar los cálculos para preparar 250 mlde la solución de NaOH 0,1 N con una riqueza del98% (Pesar 1,02g de NaOH en un vidrio de reloj)
2. Preparar la solución de NaOH aproximada enmatraz aforado de 250 ml, cerrar con tapón yhomogeneizar por inversión varias veces. Etiquetarcorrectamente.
3. Después, llenar la bureta, utilizando unembudo, y enrasar a cero perfectamente. Retirarel embudo.

4. Pesar en balanza electrónica 0,4 g exactosde ftalato ácido de potasio.
5. Colocar en un matraz Erlenmeyer (de 50 mL),limpio y seco, 20 ml perfectamente medidos, conpipeta graduada de agua destilada y disolverlos 0,4 g del patrón primario. Añadir dos gotasde fenolftaleína al 2 % en etanol comoindicador y homogeneizar por rotación.
6. Abrir la llave de la bureta con la manoizquierda y con la derecha mover por rotaciónligera el matraz hasta que los líquidos cambiena un color rosa muy pálido que permanezca.Este es el punto final de la valoración.
7. Anotar el volumen gastado y repetir lavaloración de nuevo. Después, sacar la mediaaritmética.
8. Hacer los cálculos oportunos para conocerlos eq-g de NaOH exactos que tiene la soluciónvalorada.
9. Sabiendo que el peso molecular del NaOH =40 g/mol y su valencia = 1, expresar laconcentración de la solución en g/l, N, M y mg/l.
CÁLCULOS, RESULTADOS Y CONCLUSIONES:
Primero hacemos los cálculos para la solución de250ml 0,1N de NaOH con una riqueza del 98%:
0,1N = gNaOH/40g/mol/1 , gNaOH = 1g
0,25l
19,4ml
19,7ml

1g ---------- 98% Xg = 1,02 g
Xg ---------- 100%
Calculamos la media aritmética de ml gastados deNaOH en la valoración:
19,55ml
Calculamos los eq-g de NaOH que contiene lasolución:
N°equiv.FAP = 0,4g =1,96 x 10-3
204,22g/mol
Convertimos a mlequivalentes multiplicando por 1000
Luego hallamos la normalidad de la SOSA:
19,55 x NSOSA = 1,95867202
NSOSA = 0,100187827N
OBSERVACIONES:
La normalidad nos salió exacta, esto ayudó a que ladeterminación de la acidez del vinagre también fueraexacta.

DETERMINACIÓN DE LA ACIDEZ DE UN VINAGRE
OBJETIVO:
Determinar los gramos de ácido acético (CH3 –COOH) que tienen 100 mL de vinagre problema(acidez de vinagre), a partir de los mLgastados de una disolución valorada de hidróxido

sódico 0,1 M necesarios para neutralizar unacantidad pequeña del mismo, el cual se hacolocado en un matraz Erlenmeyer con unas gotasde fenolftalelína como indicador del punto final de lareacción.
FUNDAMENTO TEÓRICO:
Determinación del contenido total de ácido envinagres: Valoración ácido base con hidróxido sódico:
Como hemos visto al explicar qué es el vinagre,cabe esperar que su pH varíe, aproximadamente,entre 2,35 y 2,55. Sin embargo, para ladeterminación exacta de la acidez del vinagre, hayque proceder a su análisis químico en laboratorio.
Para los métodos de análisis, el Real Decreto661/2012 indica: “La comprobación analítica de lascaracterísticas del vinagre conforme a losparámetros fijados en la presente normativa, sepracticará mediante la aplicación de métodos depreparación de muestra y de análisis establecidosen la legislación comunitaria, y en su defecto, porlos métodos validados por la OrganizaciónInternacional de la Viña y del Vino, los fijadospor el Codex Alimentarius y aquellos métodos deorganismos nacionales e internacionales dereconocida solvencia.”
Una sencilla valoración ácido-base con una basefuerte es el método generalmente recomendado y dereconocida solvencia. Así, el contenido total deácido de un vinagre se puede determinar fácilmente

en el laboratorio mediante una volumetría con unabase patrón, generalmente el hidróxido sódico,NaOH. El contenido de ácido de un vinagre sueleexpresarse como contenido de ácido acético, ya que,como hemos dicho, es el principal componente ácido,aunque también están presentes otros ácidosorgánicos habituales. También hemos indicado que lanormativa española establece un valor en g/l, porlo que este sería el modo más adecuado de realizarnuestros cálculos.
Contenido de ácido acético de un vinagre: cómodeterminarlo en el laboratorio:
Para determinar la acidez de un vinagre en ellaboratorio emplearemos una valoración base fuerte– ácido débil. Es adecuado hacer primero unadilución del vinagre. Así, se toman, por ejemplo,25 mL y se enrasan en un matraz aforado hasta 250mL. Una vez que se ha mezclado correctamente estanueva disolución diluida, se toman 50 mL de lamisma, se introducen en un Erlenmeyer y se procedea su valoración, añadiendo dos gotitas delindicador ácido-base fenolftaleína. Se titulará conuna disolución no muy concentrada de NaOH, porejemplo 0,1M, que estará en la bureta. Lavaloración habrá terminado cuando se alcance elprimer color rosado permanente.
MATERIAL NECESARIO:
Soporte completo para bureta.
Papel de filtro.

Bureta de 50 ml.
Embudo.
Matraz Erlenmeyer de 250 ml.
Vinagre problema.
Pipeta graduada de 5 ml.
Prepipeta.
REACTIVOS:
Solución valorada de NaOH 0,1M.
Fenolftaleína.
REALIZACIÓN:
1. Colocar una bureta de 50 ml sujeta a la pinza de unsoporte universal adecuado.
2. Abrir la llave de la bureta y añadir conembudo una cantidad pequeña de la disoluciónvalorada de NaOH 0,1 M, con el fin de lavarel interior de la bureta, Recoger los líquidossobre un recipiente y cerrar la llave de la bureta.
3. Llenar la bureta con la solución valoradade NaOH 0,1 M, abrir la llave y cebar la zonainferior, cerrar la llave y enrasas a cero. Retirar elembudo.
4. Introducir en un matraz Erlenmeyer de 250ml de capacidad, limpio y seco, 5 mL devinagre problema medido exactamente con pipeta

graduada (filtrar previamente el vinagre si estáturbio).
5. Añadir aproximadamente 100 ml de aguadestilada y homogeneizar por rotación loslíquidos.
6. Añadir tres gotas de fenolftaleína. Loslíquidos quedarán transparentes. Luego, colocar elmatraz sobre una hoja de color blanco sobre labase del soporte de la bureta, con el fin de ver
bien el viraje delindicador.
7. A continuación, con la mano izquierda, abrir muypoco la llave de la bureta para ir dejando caer gota agota la solución valorada de sosa 0,1 M, e ir moviendopor rotación, con la mano derecha, el matrazErlenmeyer, hasta que los líquidos se tornen de uncolor rosa muy pálido que permanezca.
8. Este será el punto final. Anotar los ml gastados deNaOH 0,1 M.
9. Repetir la valoración y obtener otro resultado.
10. Con el volumen de la disolución valorada dehidróxido sódico 0,1 M gastado, se podrá conocer laacidez del vinagre en gramos de ácido acético por 100mL de vinagre.
CÁLCULOS, RESULTADOS E INTERPRETACIÓN:
VÁCIDO x N ÁCIDO = VBASE x NBASE
N = Número de equiv. sol = masa sol.(g)/(masa mol/val.)
vol. disolución

Primero sacamos la media aritmética de los mlgastados en la valoración:
50, 375ml
Luego hallamos la normalidad del Ácido Acético:
5ml x NÁCIDO = 10, 375ml x 0,1 N
NÁCIDO = 1,0075N
Con la N del Ácido calculamos los g en 1000ml:
1,0075N = gácido / (60 / 1) gácido =60,45 g
1l
Pasamos los g/l a g en 100ml:
60, 45 g -------------- 1000mlX = 6,045 g
Xg --------------- 100ml
Conclusión:
50, 35ml
50, 40ml

Por cada 100ml de vinagre hay 6,045g de Ácido Acético.
OBSERVACIONES:
Los resultados en general fueron exactos.
BIBLIOGRAFÍA:
http://www.quimitube.com/como-se-determina-el- contenido-acido-de-un-vinagre

DETERMINACIÓN DE CLORUROS (MÉTODO DE MOHR)
OBJETIVO: Realizar la determinación de clorurosen una muestra de agua por medio de unavaloración de precipitación.
FUNDAMENTO TEÓRICO:
Las aguas naturales tienen contenidos muy variables encloruros dependiendo de las características de losterrenos que atraviesen pero, en cualquier caso, estacantidad siempre es menor que las que se encuentran enlas aguas residuales, ya que el ClNa es común en ladieta y pasa inalterado a través del aparatodigestivo.
El aumento en cloruros de un agua puede tenerorígenes diversos. Si se trata de una zona costerapuede deberse a infiltraciones de agua del mar. En elcaso de una zona árida el aumento de cloruros en unagua se debe al lavado de los suelos producido porfuertes lluvias. En último caso, el aumento decloruros puede deberse a la contaminación del agua poraguas residuales.
Los contenidos en cloruros de las aguas naturales nosuelen sobrepasar los 50-60 mg/l. El contenido encloruros no suele plantear problemas de potabilidad a

las aguas de consumo. Un contenido elevado de clorurospuede dañar las conducciones y estructuras metálicas yperjudicar el crecimiento vegetal.
La reglamentación técnico-sanitaria españolaestablece como valor orientador de calidad 250 mg/l deCl y, como límite máximo tolerable, 350 mg/l de Cl, yaque no representan en un agua de consumo humano másinconvenientes que el gusto desagradable del agua.
La determinación de cloruros puede hacerse mediantetres métodos.
El método argentométrico o volumétrico esrecomendable para agua con concentraciones entre 1,5 y100 mg/l de cloruros.
Este método es aplicable para la determinación decloruros en aguas potables o superficiales, siempreque no tengan excesivo color o turbidez. Se basa en elmétodo de Mohr. Sobre una muestra ligeramentealcalina, con pH entre 7 y 10, se añade disolución deAgNO3 valorante, y disolución indicadora K2CrO4. LosCl- precipitan con el ión Ag+ formando un compuestomuy insoluble de color blanco. Cuando todo el productoha precipitado, se forma el cromato de plata, de colorrojo ladrillo, que es menos insoluble que el anteriory nos señala el fin de la valoración.
CLORUROS (Cl‾): El ion cloruro (Cl‾), es uno de losaniones inorgánicos principales en el agua naturaly residual. Los contenidos de cloruros de las aguasson variables y se deben principalmente a lanaturaleza de los terrenos atravesados.

Habitualmente, el contenido de ion de cloruro delas aguas naturales es inferior a 50 mg/L. En elagua potable, el sabor salado producido por el Cl‾es variable y depende de la composición química delagua.
VOLUMETRIAS DE PRECIPITACION: En las volumetrías deprecipitación se mide el volumen de solución tipo,necesario para precipitar completamente un catión oanión del compuesto que se analiza. Los métodos delMohr y Volhard son ejemplos de volumetrias deprecipitación:
_Método de Mohr: El método se utiliza para determinariones cloruro y
bromuro de metales alcalinos, magnesio y amonio.
La valoración se hace con solución patrón de AgNO3. Elindicador es el ion cromato CrO4 =, que comunica a lasolución en el punto inicial una coloración amarilla yforma en el punto final un precipitado rojo ladrillode cromato de plata,Ag2CrO4. Las reacciones queocurren en la determinación de iones cloruro son:
Cl ‾ + Ag+ → AgCl ↓ (Precipitado blanco)
CrO4= + 2Ag+ → Ag 2CrO4 ↓ (Precipitado rojo ladrillo)
La solución debe tener un pH neutro o cercano a laneutralidad. Un pH de 8.3 es adecuado para ladeterminación.
La solución patrón de AgNO3 se puede preparar por elmétodo directo dado que el nitrato de plata es unreactivo tipo primario; con el objeto de compensar los

errores en la precipitación del punto final seprefiere el método indirecto y la solución se valoracon NaCl químicamente puro. Cuando la solución tipo seprepara por el método indirecto no es necesario elensayo en blanco, porque el exceso empleado en lavaloración de la sustancia problema se compensa con elempleado en la valoración del AgNO3.
_Método de Volhard: Este método de titulación se usapara la determinación de plata y compuestos de plata,aniones que se precipitan con plata como Cl‾,Br ‾, I‾, SCN‾ y AsO4 -4.
Para el caso de determinación de un anión, se acidulacon HNO3, se agrega un exceso de solución tipo deAgNO3 para precipitar el anión y se valora porretroceso el exceso de Ag+,con solución patrón detiocianato de potasio; el indicador es el Fe+3 , queproporciona color rojo a la solución.
Las reacciones que ocurren en la determinación deiones cloruro son:
Ag++ Cl‾ → AgCl ↓
Ag+ + SCN‾ → AgSCN
Fe+3 + SCN‾ → FeSCN+2
MATERIAL NECESARIO:
Matraces erlenmeyer de 250 ml.
Matraces aforados de 250 y 100 ml.
Vasos de precipitados de 100 ml.
Bureta de 50 ml.

Pipetas de 1 y 10 mL.
Vidrios de reloj.
Probeta de 100ml.
Varilla agitadora.
Cintas de PH.
Soporte universal.
Frasco Lavador.
Pipetas pasteur.
REACTIVOS:
Disolución valorante (patrón secundario):Solución de nitrato de plata 0,01 N.
Indicador: Solución al 5% de cromato depotasio (K2CrO4).
Patrón primario: Solución de cloruro sódico0,01 N. a partir del p.a., NaCL desecado enestufa 105-110 grados, 1h previamente.
Disoluciones para llevar la muestra a pHentre 7 y 8,3:
- Solución de acido sulfúrico 0,05 M.
- Carbonato de sódio (Na2CO3) 0,1 M.
PROCEDIMIENTO:
Estandarización de la disolución patrón denitrato de plata 0,01M:

1. Preparar 250 mL de una disolución denitrato de plata aproximadamente 0,01 M apartir de una disolución del laboratorio.
2. Preparar 50 ml de una disolución de clorurode sodio 0,01 M (patrón primario).
3. Pipetear una alícuota de 10 mL de clorurosódico 0,01 M en un matraz erlenmeyer.
4. Añadir 50 mL de agua destilada aproximada(medido con probeta) y 0,5 mL (unas cuatrogotas) de la disolución indicadora de cromatode potasio al 5% la disolución tomara coloramarillo.
5. Valorar con nitrato de plata 0,01M, que seañade desde una bureta gota a gota hasta queaparezca un precipitado rojo ladrillo. Anotar elvolumen gastado. Se observa primero elprecipitado blanco de cloruro de plata, y porúltimo un precipitado rojo ladrillo de cromatode plata. (intervalo de viraje de amarillo arojo ladrillo). 6. Repetir la valoración ydeterminar el volumen gastado de nitrato deplata como el promedio de las dos valoraciones.
Valoración del blanco:
1. Pipetear una alícuota de 10 mL de aguadestilada exenta de cloruros y llevarlos a unmatraz de 100 ml.
2. Diluir con aproximadamente, 50 ml de aguadestilada y añadir como indicador unas tres

gotas de cromato de potasio, la disolucióntomara color amarillo valorar con nitrato deplata hasta que la disolución empiece a cambiarde amarillo a rojo.
3. Repetir la valoración del blanco ydeterminar el volumen gastado de nitrato deplata para el blanco como el promedio de las dosvaloraciones.
Valoración del cloruro:
1. Introducir 10 mL de la muestra de aguaproblema en un matraz erlenmeyer de 100mL ydiluir con aproximadamente, 50 ml de aguadestilada.
2. Estimar el pH de la muestra con un trozo de papelde tornasol. Si el pH de la muestra se encuentra entre6,5 y 10 ir directamente al paso siguiente de adicióndel indicador cromato de potasio. Si el pH noestuviera entre los valores indicados añadir a lamuestra 1 mL de disolución de carbonato sódico 0,1 My dos gotas de disolución de fenolftaleína, ladisolución tomará color rosa. Añadir a la disolucióngota a gota de ácido sulfúrico 0,05M hasta quedesaparezca el color rosa, para asegurar que el pHestá del orden de 7,0 y 8,3. A continuación añadir0,5 mL del indicador, cromato potásico la disolucióntomara color amarillo, y valorar con la disoluciónpatrón de nitrato de plata hasta observar la apariciónde un precipitado rojo ladrillo de cromato de plataque permanecerá por lo menos 30 segundos. El matrazdebe de mantenerse sobre un fondo blanco.

3. Determinar el volumen gastado de nitrato de platacomo promedio de dos o tres valoraciones que nodifieran en más de 0,2 mL. Realizar la misma operacióncon agua destilada (blanco).
CÁLCULOS:
Para preparar el AgNO3 0,01M a partir de ladisolución AgNO3 0,1M:
Xml x 1M = 250ml x 0,01M
Xml = 2,5ml
Calculamos las medias de los ml de AgNO3 gastados enla estandarización y el blanco:
- Vol. Estandarización:- Vol. Blanco:
10,65ml0,5ml
Determinamos la normalidad del AgNO3:
NAgNO3 = 10ml x 0,01 =0,009852216749N
Vi x Ci =
Cloruros en mg/l = Vagua prob. - Vblanco x 0,0098 x 35,45 x 1000
10,6ml
10,7ml
0,4ml
0.6ml
NAgNO3 = VClNa x 0,01 / VAgNO3 -VBLANCO

10,65ml - 0,5ml
Luego determinamos los cloruros del agua problema ydel agua de grifo:
Primero sacamos las medias de los ml de AgNO3 gastadosen las aguas problema:
- Agua Problema1: - Agua Problema 2:- Agua de Grifo:
0,55ml1,825ml 1,3ml
Agua Problema 1:
Cloruros en mg/l = 0,55ml - 0.5ml x 0,0098x 35,45 x 1000
10ml
= 1,73705 mg/l
Agua Problema 2:
Cloruros en mg/l = 1,825ml - 0,5ml x 0,0098x 35,45 x 1000
10ml
= 46,0318 mg/l
0,5ml
0.6ml
1.95ml
1,7ml
1,3ml
1,3ml

Agua de grifo:
Cloruros en mg/l = 1,3ml - 0,5ml x 0,0098x 35,45 x 1000
10ml
= 27,7928 mg/l
OBSERVACIONES:
Las disoluciones de plata se guardaron enrecipientes de color topacio, debido a lafacilidad de reducción de las sales de plataa plata metálica.
No usamos la disolución de Na2CO3 debido a que el PHde las aguas que evaluamos era 7 y se encontrabadentro de los parámetros normales.
Todas las muestras de agua estuvieron dentro de losparámetros normales.
BIBLIOGRAFÍA:
http://www.fisicanet.com.ar/quimica/analitica/ lb01_mohr_volhard.php
http://www.ambientum.com/ enciclopedia_medioambiental/aguas/Determinacion_de_cloruro.asp

DETERMINACIÓN DE LA ALCALINIDAD: CARBONATOS YBICARBONATOS
OBJETIVO:
Realizar la determinación de carbonatos ybicarbonatos en una muestra de agua problema.

FUNDAMENTO TEÓRICO:
La alcalinidad en el agua tanto natural como tratada,usualmente es causada por la presencia de ionescarbonatos ( CO3= ) y bicarbonatos ( HCO3- ),asociados con los cationes Na+, K+ Ca+2 y Mg+2 .
La alcalinidad se determina por titulación de lamuestra con una solución valorada de un ácido fuertecomo el HCl, mediante dos puntos sucesivos deequivalencia, indicados ya sea por mediospotenciométricos o por medio del cambio de colorutilizando dos indicadores ácido-base adecuados.
MATERIAL NECESARIO:
Matraces erlenmeyer.
Matraces aforados de 250 mL.
Vasos de precipitados.
Bureta de 25 mL.
Pipetas graduadas de 10 mL.
Probeta.
Vidrios de reloj.
Varilla.
Soporte Universal.
Frasco Lavador.
Pipeta de 5ml.
Embudos.

Pipetas Pasteur.
REACTIVOS:
Agua destilada libre de dióxido de carbono:debe usarse para la preparación de todas lassoluciones estándar y stock. Si el aguadestilada tiene un pH menor a 6.0, debe serhervida por 15 minutos, enfriada atemperatura ambiente y usada inmediatamente.
Fenolftaleina (indicador) .
Naranja de metilo (indicador).
Disolución valorante (patrón secundario):solución de HCl 0,1 N.
Patrón primario: Solución de Na2CO3 0,1 N.
PROCEDIMIENTO:
Estandarización del HCl:
1. Preparar 250 mL de una disolución de HCl0.1 N a partir de HCl comercial.
2. Secar durante 1 hora a 110°C el Na2CO3 decalidad patrón primario, y dejar enfriar en undesecador.
3. Preparar 50 mL de una disolución deNa2CO3 0,1 N.
4. Tomar alícuotas de 10 mL de la disoluciónde Na2CO3, añadir 3 o 4 gotas de indicador(naranja de metilo) y 50 mL de agua destilada.

Añadir HCl desde la bureta hasta que elindicador vira de amarillo a anaranjado.
5. Repetir el proceso hasta obtener 2resultados coincidentes o que se diferencienúnicamente en ± 0.2 mL. Calcular la normalidadmedia del HCl.
Determinación del contenido de carbonato ybicarbonato sódico en una muestra de agua:
1. Pipetear 10 mL de la disolución problema ypasar a un erlenmeyer. Diluir con agua destiladahasta aproximadamente 50 mL.
2. Añadir 2 gotas de disolución defenolftaleína, si aparece un color rosa, valorarhasta que vire a incoloro con HCl y anotamosel volumen gastado.
3. Si no aparece color rosa, reportar carbonatosigual a cero. A continuación añadir a ladisolución 3 gotas de naranja de metilo ycontinuar la valoración hasta el primer cambioperceptible de color del nuevo indicadoranotando de nuevo el volumen de HCl gastado.Enrasando cada vez la bureta, repetir lavaloración (adicionando gota a gota el reactivoal aproximarse el punto de equivalencia), elnúmero de veces necesarias para obtener 2resultados coincidentes o que se diferencienúnicamente en ± 0.2 ml.

CÁLCULOS Y RESULTADOS:
Para preparar 250ml de HCl 0,1N: (37% de riqueza,ρ:1,19 g/ml)
0,1N = gHCl / (36,47/1) gHCl = 0,91
0,25 l
0,91 g ----- 37% Xg = 2,46g
Xg ------- 100%
- Convertimos a ml:
1,19 g -------- 1ml Y = 2,1 ml
2,46 g -------- Yml
Calculamos la normalidad del HCl:
- Promedio de ml de HCl utilizados en laestandarización:
11, 5 ml
- NHCl = 10ml x 0,1N = 0,0869N
11,5ml
- Calculamos el factor:
F = 0,1= 1,15
11,5ml
11,5ml
meq/l de CO32- = 2V x N x
1000
meq/l de HCO3- = (VTOTAL - 2VINICIAL)x N x
1000
F = NREAL
NTEÓRICA
0,3ml
0,45ml

0,0869N
Hallamos la concentración de CO32- y HCO3
- :
- Agua Problema 1 (Móstoles): (Exenta de carbonatos)
0,375ml
meq/l HCO3- = 0,375 x 0,0869 x 1000 =
3,25875 meq/l
10
Para pasarlo a ppm solo hay que multiplicar por supeso en meq que en este caso es : 61,016
3,25875 meq/l x 61,016 = 198,83589ppm
- Agua Problema 2 :
ml de cloruros:ml total:
0,1ml0,375ml
0,1ml
0,1ml
0,4ml
0,3ml
0,1ml
0,05ml

meq/l de CO32- = 0,1ml x 0,0869 x 1000 = 0,869
meq/l
10ml
0,869 meq/l x 60 = 52,14 ppm
meq/l de HCO3- = (0,35ml - 0,2ml) x 0,0869 x 1000
= 1,3035 meq/l
10ml
1,3035 meq/l x 61,016 = 79,53 ppm
- Agua de grifo : Exenta de carbonatos
0,075ml
meq/l HCO3- = 0,075ml x 0,0869 x 1000 =
0.65175 meq/l
10ml
0,65175 meq/l x 61,016 = 39,77 ppm
BIBLIOGRAFÍA:
http://arturobola.tripod.com/carbo.htm

DETERMINACIÓN DE LA DUREZA
OBJETIVO: Determinar la dureza de un agua problemacon EDTA.
FUNDAMENTO TEÓRICO:
Se entiende por dureza total la suma de las durezasindividuales debidas a los iones de calcio, magnesio,estroncio y bario en forma de carbonato o bicarbonato.
La composición química del agua y su contenido en lassales de los iones antes mencionados depende del suelodel que provienen. En los suelos de basalto, areniscay granito las aguas son muy blandas, con 1-2º d dedureza. Las aguas procedentes de suelos de cal, yeso ydolomita pueden presentar dureza de más de 100º dgrados alemanes de dureza.
El valor hidrotimétrico expresa el contenido del aguaen sales de calcio y magnesio, por tanto secorresponde con la dureza total.
La dureza puede expresarse en:
Grado alemán ºd. Todos los componentes de la durezadel agua son determinados como CaO. 10 mg de CaO/l son1ºd.
Grado francés ºf. Todos los componentes de la durezadel agua se expresan como CaCO3. 10 mg CaCO3 son 1º f.
ppm de CaCO3 mg/l de carbonato calcio; o en óxido decalcio.
La dureza total de las aguas es un componente conbastante significación en la calidad fisico-química.

No se conocen con claridad los efectos de las aguasblandas y duras sobre el organismo de losconsumidores, aunque ciertos estudios epidemiológicosparecen apuntar a que la incidencia de enfermedadescardiovasculares es mayor en las zonas de consumo deaguas blandas. Por otra parte, las aguas blandas sonagresivas y facilitan la disolución de metales de lascañerías, provocando, entre otras enfermedades,saturnismo o intoxicación por plomo en aquellosabastecimientos en que aún se conservan tuberíasantiguas de plomo.
El uso de las aguas duras tanto a nivel domésticocomo industrial tiene graves inconvenientes. En ellavado se produce precipitación del jabón por elcalcio y el magnesio, en la cocción de legumbres y enla industria pueden presentarse problemas deincrustaciones.
La reglamentación técnico-sanitaria españolaestablece como valor orientador de calidad para ladureza total mínimo en aguas ablandadas de 150 mg/lCaCO3.
En términos generales, la calidad de las aguas enfunción de su dureza es:
Calidad del agua
Las normas europeas han adoptado como concentraciónlímite 500 mg/l CaCO3, 50ºf.
La Organización Mundial de Salud, OMS, ha adopatadocomo concentración máxima deseable 100 mg/l de CaCO3 ycomo concentración máxima admisible 500 mg/l.

La presencia de calcio en las aguas naturales tienesu origen en la lixiviación de los terrenos calizosque atraviesa. El calcio, junto con el magnesio, sonelementos de la dureza del agua. El calcio seencuentra en las aguas en cantidades mucho mayores queel magnesio siendo, salvo muy raras excepciones, elcatión más abundante. A las aguas pasa, o bien porsimple disolución cuando tiene su origen en los yesoso los silicatos, o bien por ataque de las calizas odolomías, por la acción del anhídrido carbónico.
Es, después de los cuatro elementos organógenos,oxígeno, carbono, hidrógeno y nitrógeno, el másabundante en el organismo humano, necesitando ésterecibir un aporte diario de un gramo de calcio.
El contenido de calcio en las aguas puede variardesde muy pocos miligramos por litros a varios cientosde mg/l; puede presentarse en formas de bicarbonatos,sulfatos y cloruros. Aunque se ha discutido lainfluencia del calcio sobre la salud, no hay pruebasque acrediten efectos nocivos.
La reglamentación técnico-sanitaria españolaestablece como valor orientador de calidad hasta uncontenido en calcio de 100 mg/l y como límite máximotolerable 200 mg/l.
El magnesio contribuye notablemente, junto con elcalcio, a caracterizar la dureza de un agua. Elcontenido en magnesio de un agua depende casiexclusivamente de los terrenos que atraviesa, pudiendovariar desde muy pocos mg/l a varios cientos de mg/l.

La salmuera subterránea contiene un 3% de MgCl2-.
El magnesio es un elemento indispensable para elcrecimiento. El organismo humano ingiere gran cantidadde magnesio diariamente a través de los alimentos, asícomo elemento indispensable en el desarrollo deciertos sistemas enzimáticos, actuando igualmente enla constitución de los huesos.
Se sabe que concentraciones de magnesio en aguassuperiores a 125 mg/l pueden tener efectos laxantes eincluso adquirir un sabor amargo, sobre todo cuando elcontenido de ión sulfato es notable.
La reglamentación técnico-sanitaria españolaestablece como valor orientador de calidad hasta mg/ly como límite tolerable hasta 50 mg/l.
La valoración del contenido en magnesio debe hacerseconjuntamente con el contenido en sulfatos.
La OMS establece como concentración máxima deseable30 mg/l, si hay mas de 250 mg/l de sulfato. Si laconcentración de sulfatos es inferior, puedenpermitirse hasta 50 mg/l.
DETERMINACION DE LA DUREZA EN AGUAS MEDIANTEVOLUMETRIA COMPLEJOMETRICA:
Volumetrías complejométricas. En las volumetríascomplejométricas se mide el volumen de solución tipo,necesario para formar un complejo con un catiónmetálico del compuesto que se analiza.
Muchos cationes metálicos reaccionan con especiesdadoras de electrones llamadas ligandos, para formar

compuestos de coordinación o complejos. El ligandodebe tener por lo menos un par de electrones sincompartir.
Los complejos llamados quelatos, se producen por lacoordinación de un catión y un ligando, en los que elcatión (metálico) es parte de uno o varios anillos decinco o seis miembros.
Los compuestos orgánicos más conocidos que formanquelatos utilizables en análisis cuantitativo son elácido nitrilotriacético, el ácidoetilendiaminotetraacético (EDTA) y la sal disódica delEDTA; estos compuestos se conocen comercialmente conlos nombres de Titriplex I, II, y III respectivamente;también se utilizan los nombres de Complexonas,Vercenos o Secuestrenos.
El más empleado de los anteriores compuestos es lasal disódica del EDTA, por la facilidad de disoluciónen agua; la solución se prepara por el método directodado el carácter de reactivo tipo primario de la saldisódica.
El ácido etilendiaminotetraacético EDTA contienecuatro hidrógenos ácidos; por esa razón se representatambién como H4Y.
El EDTA forma complejos estables con la mayoría delos cationes y entra siempre en relación molar 1:1 enla fórmula del complejo, independiente de la carga delcatión, como se muestra en las siguientes reacciones:
Mg+2 + Y-4 MgY-2

Al+3 + Y-4 AlY-1
Ca+2 + Y-4 --------- CaY-2
Ag+ + Y-4 AgY-3
Los iones formados en las reacciones anteriores sonincoloros, de tal manera que para determinar el puntofinal se emplean indicadores llamados metalcrómicos.Estos tienen la propiedad de formar complejos concationes como el Ca+2 y el Mg+2, de distinto color alque presenta el indicador libre. Estos indicadores sonácidos débiles que se representan como Hin.
Determinación de la dureza total:
El colorante utilizado para determinar la dureza totaldel agua (debida al calcio y al magnesio), es el negrode eriocromo T. Este colorante es triprótico y existeinicialmente como anión divalente de color azul HIn-2a pH 10. A la muestra se le adiciona solución bufferde pH 10 +0.1, para mantener la estabilidad de loscomplejos formados; no puede incrementarse el pH deeste valor, por cuanto precipitan el CaCO3 o elMg(OH)2, además porque el indicador cambia de color apH elevado, obteniéndose In-3 de color naranja.
La reacción del indicador con los iones M+2 (Ca+2 +Mg+2 ) presentes en la solución que se valora es delsiguiente tipo:
M+2 + HIn-2 Metal * indicador -1 + H+
Color azul Color rojo

Al adicionar EDTA a la solución que contiene lamuestra con el indicador, el EDTA se combina primerocon el Ca+2 y luego con el Mg+2, ya que el complejoEDTA-Ca+2, es más estable que el complejo EDTA- Mg+2,mediante las siguientes reacciones:
EDTA + Ca+2 EDTA. Ca+2 K = 1010.7
EDTA + Mg+2 EDTA. Mg+2 K = 108.7
Determinación de la dureza debida al calcio:
El calcio y el magnesio son ambos acomplejados por elEDTA a pH 10; la determinación de la dureza debidaúnicamente al calcio se hace a pH elevado (12-13), eneste rango de pH, el magnesio precipita como Mg(OH)2 yno interviene en la reacción; además el indicadorutilizado para esta determinación solo se combina conel calcio.
El indicador murexida se emplea para determinar ladureza debida al Ca+2, vira de rojo claro (cuandoforma el complejo con el Ca+2) a violeta (cuando estálibre).
Determinación de la dureza debida al magnesio:
La diferencia entre la dureza total y la durezacálcica (expresada ambas como mg/L de CaCO3), dadirectamente la dureza magnésica.
MATERIALES:
Matraces erlenmeyer.
Matraces aforados de 50, 100 y 250.

Vasos de precipitados.
Bureta.
Pipetas graduadas y aforadas.
Vidrios de reloj.
Varillas.
Soporte Universal.
Pipetas Pasteur.
REACTIVOS:
Disolución reguladora de pH 10. Preparar 500 mL(para todos) Disolver 35 g de NH4Cl en 100 ml deagua destilada aproximadamente. Adicionar 285 ml deNH3 al 28-30 % (p/p) y diluir la mezcla a un litro.Proceder según indica en la estandarización.
Disolución de NaOH 2 M. Preparar 50 mL. Para todos.
Disolución valorante (patrón secundario):disolución de EDTA 0,01 M. Preparar 250 ml.
Patrón primario: Solución de CaCl2 0,01N. Disolver0,5 g de CaCO3 secado a 100 oC durante 2 horas, en10 ml de HCl 3 N. Aforarlo a 1000 ml con aguadestilada. Para todos.
Indicador negro de eriocromo T. Mezclar ypulverizar en mortero 100 mg de indicador con 10 gde NaCl y guardar en frasco bien cerrado. Yapreparado.
Murexida: Mezclar 0,20 g de murexida con 100 g deNaCl puro bien pulverizado. Ya preparado.

PROCEDIMIENTO:
Estandarización de la disolución de EDTA:
1. Pipetear una porción de 10 mL de la disolución decloruro cálcico y transferir a un matraz erlenmeyer de250 ml, añadir 5 mL de la disolución reguladora de pH10.
2. Poner indicador negro de eriocromo T (punta deespátula). Valorar cuidadosamente con disolución deEDTA hasta el punto donde el color cambia de rojo-vinoa azul. No debe quedar ninguna coloración roja en ladisolución (se puede realizar una valoración rápidapara conocer el cambio de color).
3. Repetir la valoración con otras dos alícuotas dedisolución de cloruro cálcico.
Determinación de la dureza de un agua:
Determinación de la suma de calcio y magnesio:
1. Tomar 20 ml de agua problema, llevar a unmatraz erlenmeyer de 100 ml, añadir 0,6 mL dela disolución reguladora de pH 10 y unapequeña cantidad de indicador de negro deeriocromo T (punta de espátula).
2. Valorar con EDTA hasta viraje del rojo-vinosoa azul. Una vez alcanzado el punto finalconservar un poco de disolución como referenciade color para la valoración de las otrasmuestras.

3. Repetir la valoración tres veces paracalcular el valor medio de la concentracióntotal de Ca2+ y Mg2+.
Determinación de calcio:
1. Tomar 20 mL de agua problema, llevar a unmatraz erlenmeyer de 100 mL.
2. Añadir 0.4 mL de NaOH 2 M y agitardurante 2 minutos. El pH = 12 -13 y todo elMg precipita como Mg (OH)2 (el cual puede serimperceptible).
3. Añadir una pequeña cantidad (punta deespátula) de murexida quedando la disolución decolor rosa. Valorar de forma rápida paralocalizar el punto final, color violeta.
CÁLCULOS:
Calcular la N del EDTA:
N 1 : Nde CaCl2 , N2 : N de EDTA
V1 : ml de lasol. CaCl2, V2 : ml gast. de EDTA
Dureza cálcica:
Dureza Magnésica:
N2 = V1 x N1
mgCa/l = V1 x NEDTA x PmCa x 1000
VMUESTRA
mgMg/l = (V2 - V1) x NEDTA x PmMg x1000

OBSERVACIONES:
No realizamos está práctica porque los reactivosestaban estropeados debido a su antigüedad.
BIBLIOGRAFÍA:
http://www.ambientum.com/ enciclopedia_medioambiental/aguas/Dureza_de_aguas.asp
http://html.rincondelvago.com/dureza-en- aguas_volumetria-complejometrica.html
DETERMINACIÓN DE NITRITOS
OBJETIVOS: Determinar el contenido de nitritos deuna muestra de agua mediante su transformaciónen un complejo coloreado. Aplicar laespectrofotometría para la determinación de nitritosen aguas.
FUNDAMENTO TEÓRICO:
La presencia de nitritos en aguas es un indicador decalidad de las mismas y por este motivo es necesariopoder cuantificarlo. Para ello existen una serie de

técnicas de análisis. A continuación se exponen el máscomún: el método colorimétrico.
MÉTODO COLORIMÉTRICO
El método colorimétrico es adecuado paraconcentraciones de 5 a 1000 µg de NO2- - N/L.
Principio: El nitrito (NO2-) se determina por laformación de un colorante azo púrpura rojizo producidoa pH 2,0-2,5 por acoplamiento de sulfanilamidadiazotizada con diclorhidrato de N-(1-naftil)-etilendiamina (diclorhidrato de NED). El rango deaplicación del método para medidasespectrofotométricas es de 10 a 1000 µg de NO2--N/L yse puede aplicar al de 5 a 50 µg de N/L si se usa unrecorrido de luz de 5 cm y un filtro de color verde.El sistema de color obedece a la ley de Beer hasta 180µg N/L con 1 cm de recorrido de luz a 543 nm.Diluyendo las muestras se pueden determinarconcentraciones más altas de NO2-.
Interferencias: La incompatibilidad química haceimprobable la coexistencia de NO2-, cloro libre, ytricloruro de nitrógeno (NCl3). El tricloruro denitrógeno proporciona un color rojo falso cuando seañade el reactivo cromogénico. Los iones siguientesinterfieren debido a precipitación en las condicionesde la prueba y deben estar ausentes: Sb3+, Au3+, Bi3+,Fe3+, Pb2+, Hg2+, Ag+, cloroplatinato (PtCl62-) ymetavanadato (VO32-). El ion cobre (II) puede darlugar a resultados bajos por catalizar ladescomposición de la sal de diazonio. Los ionescoloreados que alteran el sistema de color también

deben estar ausentes. Los sólidos en suspensión debeneliminarse por filtración.
Almacenamiento de la muestra: No utilizar nunca laconservación ácida en las muestras destinadas alanálisis de NO2-. Se hace la determinacióninmediatamente sobre muestras recientes para evitar laconversión bacteriana del NO2- en NO3- o NH3. Para laconservación a corto plazo, durante uno o dos días, secongela a – 20ºC o se conserva a 4ºC.
MATERIAL:
Matraces aforados de 1000, 100 y 50 ml.
Pipetas.
Espectrofotómetro de absorción uv-visible paramedir a 543 nm.
Vaso de precipitados.
Pipetas.
REACTIVOS:
Solución patrón de nitritos de 100ppm.
Solución de nitritos 5ppm.
Disolución de Sulfamilamina.
Disolución de NED.
PROCEDIMIENTO:
1. Solución patrón de nitritos de 100 ppm:Disolver 0,1500 g de nitrito sódico secadopreviamente durante una hora a 110ºC (NaNO2) en

agua destilada, y enrásese hasta 1 L. Conservarcon 1 ml de CHCl3. Preparar para uno todos.
2. Solución de trabajo de nitritos de 5 ppm(disolución patrón diluida): Preparar 50 ml apartir de la disolución anterior de 100 ppm. Apartir de esta de 5ppm se preparan lasdisoluciones para la curva de calibradocorrespondiente.
3. Disolución de sulfanilamida: Se disuelve 1 gde sulfanilamida en una mezcla de 10 mL deHCl concentrado y 60 mL de agua destilada;después se diluye hasta 100 mL con aguadestilada.
4. Disolución diclorhidrato de N-(1-naftil)-etilendiamida (NED): 0,1 g se NED se disuelvenen agua destilada, se aforan a 100 mL con aguadestilada y se guarda la solución en botellaoscura. Se debe conservar en nevera y desechar cuandocambie de color.
5. Construcción del la curva de calibrado:
A partir de la disolución de trabajo de 5 ppmprepara disoluciones de 0 (blanco); 0,1; 0,3;0,5; y 0,7 ppm en matraces aforados de 50 ml.No completar todavía en volumen con aguadestilada, echar sólo la disolución de trabajo.
6. Preparación de la muestra La muestra:
Según su procedencia necesita una dilucióndistinta. Así, por ejemplo si es agua de rio o

mar, no será necesaria su dilución, mientrasque en el caso de vertidos habrá que probardistintas diluciones. Suponiendo este último casose comienza probando a tomar 10 mL de lamuestra y ponerlo en un aforado de 50 mL. Nocompletar todavía el volumen con agua destilada.Se realiza como mínimo tres replica de lamuestra. Nosotros solo haremos dos.
7. Desarrollo del color:
Añadir en todos los aforados (muestra y curvade calibrado) 1 mL de solución de sulfanilamidaagitar y dejar reaccionar entre 2 y 8 minutos.Añadir después 1 mL de NED en todos losaforados (muestra y curva de calibrado) yaparecerá un color rojizo en aquellos aforadosen los que existan nitratos. Se homogeniza yse deja reaccionar 10 minutos. Completar ahora elvolumen de todos los aforados con aguadestilada. Finalmente se mide la absorbancia a543 nm en el espectrofotómetro ajustandopreviamente el aparato con el blanco. El color semantiene estable unas dos horas.
CÁLCULOS:
Solución de 5ppm:
ml x 100ppm = 50ml x 5ppm
2,5ml
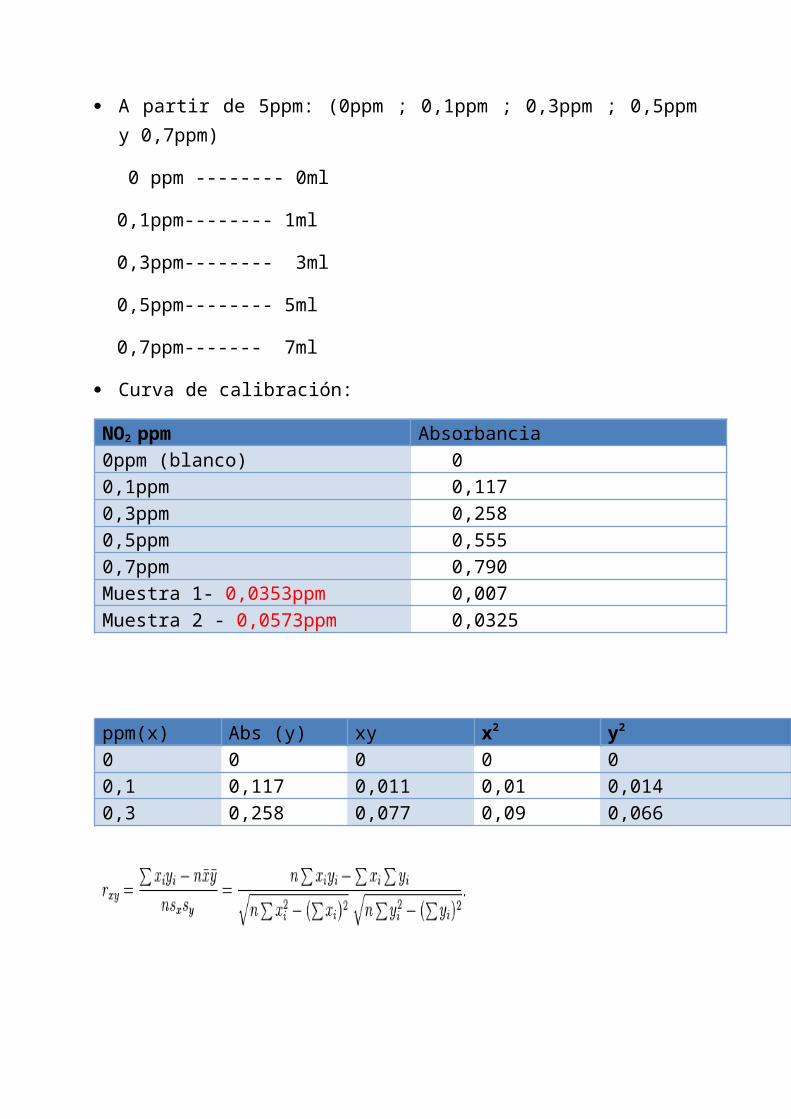
A partir de 5ppm: (0ppm ; 0,1ppm ; 0,3ppm ; 0,5ppmy 0,7ppm)
0 ppm -------- 0ml
0,1ppm-------- 1ml
0,3ppm-------- 3ml
0,5ppm-------- 5ml
0,7ppm------- 7ml
Curva de calibración:
NO2 ppm Absorbancia0ppm (blanco) 00,1ppm 0,1170,3ppm 0,2580,5ppm 0,5550,7ppm 0,790Muestra 1- 0,0353ppm 0,007Muestra 2 - 0,0573ppm 0,0325
ppm(x) Abs (y) xy x2 y2
0 0 0 0 00,1 0,117 0,011 0,01 0,0140,3 0,258 0,077 0,09 0,066

0,5 0,555 0,277 0,25 0,3180,7 0,790 0,553 0,49 0,6241,6 1,72 0,92 0,84 1,01 TOTAL
Calculamos el coeficiente de Pearson:
rxy = 0,998
Para hacer la recta de regresión, calculamos a y b:
a = ( Y)( X∑ ∑ 2) - ( X)( XY) = (1,72)(0,84) -∑ ∑
(1,6)(0,92) = - 0,034 N ( X∑ 2) - ( X)∑ 2
4(0,84) - (1,6)2
b = N( XY) - ( X)( Y) = 4(0,92) - (1,6)∑ ∑ ∑
(1,72) = 1,16 N( X∑ 2) - ( X)∑ 2 4(0,84) -(1,6)2
Luego hallamos la concentración ppm de nuestrasmuestras problemas:
Muestra 1: Abs(y)= 0,007
ppm(x) = 0,007 + 0,034 = 0,0353ppm
x = y -a

1,16
Muestra 2: Abs(y)= 0,0325
ppm(x) = 0,0325 + 0,034 = 0,0573
1,16
BIBLIOGRAFÍA:
http://www.xtec.cat/~gjimene2/llicencia/students/ 03tecnicas.html
INTRODUCCIÓN AL MANEJO DEL MICROSCOPIO
OBJETIVO:
Reconocer la importancia del microscopio
Manejar correctamente el microscopio.
Manejar correctamente el microscopio.
Señalar los componentes mecánicos y ópticos queconstituyen el microscopio.
FUNDAMENTO TEÓRICO:
Partes de un microscopio óptico

OCULAR: Lente situada cerca del ojo del observador.Amplía la imagen del objetivo.
OBJETIVO: Lente situada cerca de la preparación.Amplía la imagen de ésta.
CONDENSADOR: Lente que concentra los rayos luminosossobre la preparación.
DIAFRAGMA: Regula la cantidad de luz que entra en elcondensador.
FOCO: Dirige los rayos luminosos hacia el condensador.
Sistema mecánico
SOPORTE: Mantiene la parte óptica. Tiene dos partes:el pie o base y el brazo.
PLATINA: Lugar donde se deposita la preparación.
CABEZAL: Contiene los sistemas de lentes oculares.Puede ser monocular, binocular.
REVÓLVER: Contiene los sistemas de lentes objetivos.Permite, al girar, cambiar los objetivos.
TORNILLOS DE ENFOQUE: Macrométrico que aproxima elenfoque y micrométrico que consigue el enfoquecorrecto.
Manejo del microscopio óptico
Colocar el objetivo de menor aumento en posición deempleo y bajar la platina completamente. Si elmicroscopio se recogió correctamente en el usoanterior, ya debería estar en esas condiciones.

Colocar la preparación sobre la platina sujetándolacon las pinzas metálicas.
Comenzar la observación con el objetivo de 4x (ya estáen posición) o colocar el de 10 aumentos (10x) si lapreparación es de bacterias.
Para realizar el enfoque:
Acercar al máximo la lente del objetivo a lapreparación, empleando el tornillo macrométrico. Estodebe hacerse mirando directamente y no a través delocular, ya que se corre el riesgo de incrustar elobjetivo en la preparación pudiéndose dañar alguno deellos o ambos.
Mirando, ahora sí, a través de los oculares, irseparando lentamente el objetivo de la preparación conel macrométrico y, cuando se observe algo nítida lamuestra, girar el micrométrico hasta obtener unenfoque fino.
Pasar al siguiente objetivo. La imagen debería estarya casi enfocada y suele ser suficiente con mover unpoco el micrométrico para lograr el enfoque fino. Sial cambiar de objetivo se perdió por completo laimagen, es preferible volver a enfocar con el objetivoanterior y repetir la operación desde el paso 3. Elobjetivo de 40x enfoca a muy poca distancia de lapreparación y por ello es fácil que ocurran dos tiposde percances: incrustarlo en la preparación si sedescuidan las precauciones anteriores y mancharlo conaceite de inmersión si se observa una preparación queya se enfocó con el objetivo de inmersión.

Empleo del objetivo de inmersión:
Bajar totalmente la platina.
Subir totalmente el condensador para ver claramente elcírculo de luz que nos indica la zona que se va avisualizar y donde habrá que echar el aceite.
Girar el revólver hacia el objetivo de inmersióndejándolo a medio camino entre éste y el de x40.
Colocar una gota mínima de aceite de inmersión sobreel círculo de luz.
Terminar de girar suavemente el revólver hasta laposición del objetivo de inmersión.
Mirando directamente al objetivo, subir la platinalentamente hasta que la lente toca la gota de aceite.En ese momento se nota como si la gota ascendiera y seadosara a la lente.
Enfocar cuidadosamente con el micrométrico. Ladistancia de trabajo entre el objetivo de inmersión yla preparación es mínima, aun menor que con el de 40xpor lo que el riesgo de accidente es muy grande.
Una vez se haya puesto aceite de inmersión sobre lapreparación, ya no se puede volver a usar el objetivo40x sobre esa zona, pues se mancharía de aceite. Portanto, si desea enfocar otro campo, hay que bajar laplatina y repetir la operación desde el paso 3.
Una vez finalizada la observación de la preparación sebaja la platina y se coloca el objetivo de menoraumento girando el revólver. En este momento ya se

puede retirar la preparación de la platina. Nunca sedebe retirar con el objetivo de inmersión en posiciónde observación.
Limpiar el objetivo de inmersión con cuidado empleandoun papel especial para óptica. Comprobar también queel objetivo 40x está perfectamente limpio.
Mantenimiento y precauciones:
1. Al finalizar el trabajo, hay que dejar puesto elobjetivo de menor aumento en posición deobservación, asegurarse de que la parte mecánica de laplatina no sobresale del borde de la misma y dejarlocubierto con su funda.
2. Cuando no se está utilizando el microscopio, hayque mantenerlo cubierto con su funda para evitar quese ensucien y dañen las lentes. Si no se va a usar deforma prolongada, se debe guardar en su caja dentro deun armario para protegerlo del polvo.
3. Nunca hay que tocar las lentes con las manos. Si seensucian, limpiarlas muy suavemente con un papel defiltro o, mejor, con un papel de óptica.
4. No dejar el portaobjetos puesto sobre la platina sino se está utilizando el microscopio.
5. Después de utilizar el objetivo de inmersión, hayque limpiar el aceite que queda en el objetivo conpañuelos especiales para óptica o con papel de filtro(menos recomendable). En cualquier caso se pasará elpapel por la lente en un solo sentido y con suavidad.Si el aceite ha llegado a secarse y pegarse en el

objetivo, hay que limpiarlo con una mezcla de alcohol-acetona (7:3) o xilol. No hay que abusar de este tipode limpieza, porque si se aplican estos disolventes enexceso se pueden dañar las lentes y su sujeción.
6. No forzar nunca los tornillos giratorios delmicroscopio (macrométrico, micrométrico, platina,revólver y condensador).
7. El cambio de objetivo se hace girando el revólver ydirigiendo siempre la mirada a la preparación paraprevenir el roce de la lente con la muestra. Nocambiar nunca de objetivo agarrándolo por el tubo delmismo ni hacerlo mientras se está observando a travésdel ocular.
8. Mantener seca y limpia la platina del microscopio.Si se derrama sobre ella algún líquido, secarlo con unpaño. Si se mancha de aceite, limpiarla con un pañohumedecido en xilol.
9. Es conveniente limpiar y revisar siempre losmicroscopios al finalizar la sesión práctica y, alacabar el curso, encargar a un técnico un ajuste yrevisión general de los mismos.
MATERIAL:
Un microscopio.
Un portaobjetos.
Una hoja de papel cuadriculado.
Un bolígrafo.

Cinta adhesiva transparente.
PROCEDIMIENTO:
1.Descubrir el aumento de los oculares y decada uno de los objetivos del microscopio.Calcular el aumento obtenido con el usocombinado de los oculares y cada uno de losobjetivos. Reseñar los resultados obtenidos, en elcuadro que se adjunta a continuación:
AUMENTO DELOS OCULARES
AUMENTO DECADA OBJETIVO
AUMENTO COMBINADO
x10 X4 X10 X40 X100
x40 x100 x400 x1000
2. Recortar un trozo de papel cuadriculado, demanera que tenga la forma y el tamaño de unportaobjetos. Dibujar, en la porción media deltrozo de papel, un signo más (+), con 2trazos de 4 cuadrados cada uno. Escribir, lo máspequeño posible una letra D, en el extremo derechodel signo más, una letra IZ en el extremo izquierdo.Una letra SP, en el extremo superior del signomás. Unas letras IN, en el extremo inferiordel signo más. Fijar el trozo del papel alportaobjetos mediante una tira de cintaadhesiva, y de forma que el signo más quedehacia afuera. Situar el portaobjetos, con latira de papel hacia arriba, en la platina del

microscopio. Enfocar la zona central del signomás, con cada uno de los objetivos, exceptocon el de inmersión. En primer lugar se debeenfocar con el objetivo de menor aumento, y enúltimo lugar, se ha de enfocar con el objetivode mayor aumento.
3. Enfocar de nuevo, con el objetivo de menoraumento, la zona central del signo más. Sindejar de mirar a través de los oculares yutilizando los tornillos reguladores de laplatina:
Desplazar el campo microscópico hacia laderecha, hasta visualizar unas letras. ¿Quéletras son las visualizadas?
Letras IZ
Seguidamente, desplazar el campo microscópicohacia la izquierda, hasta visualizar otraletra. ¿Qué letra es la visualizada?
Letra D
Volver a la zona central.
Desplazar el campo microscópico hacia arriba,hasta visualizar unas letras. ¿Qué letras sonlas visualizadas?
Letras IN

Seguidamente, desplazar el campo microscópicohacia abajo, hasta visualizar otra letra. ¿Quéletra es la visualizada?
Letras SP
¿Qué consecuencia se extrae de los resultadosobtenidos?
En el microscopio la imagen es invertida, esto sedebe al efecto de refracción de la luz, quese produce cuando la luz pasa a través de mediosdiferentes al vacío.
4. Enfocar sucesivamente, con el objetivo demenor aumento, las letras rotuladas alrededor delsigno más.
Fijándose en las escalas longitudinal ytransversal de la platina, establecer laposición exacta de cada una de esas letras.Anotar los resultados obtenidos en el siguientecuadro:
LETRAS COORDENADALONGITUDINAL
COORDENADATRANSVERSAL
DIZSPIN
Quitar el portaobjetos y mover la platina.
Volver a poner el portaobjetos en laplatina.

Situando las escalas en las posicionespreviamente establecidas, localizar directamentecada una de las letras, sin necesidad de barrervisualmente la tira de papel.
OBSERVACIONES:
El ejercicio 4 no se hizo.
BIBLIOGRAFÍA:
http://www.eumed.net/libros-gratis/ciencia/ 2013/22/microscopio.html

PREPARACIÓN DE MUESTRAS PARA EL MICROSCOPIO
OBJETIVO:
Reconocer los diferentes tipos de muestra y aprenderen que caso usarlas.
FUNDAMENTO TEÓRICO:
Preparaciones en fresco
La forma más simple de preparar un espécimen para suexamen microscópico es hacer una preparación enfresco. Existen dos técnicas, una preparación enfresco simple ("entre porta y cubre") consiste encolocar una gota de líquido con los microorganismossobre un portaobjetos y a continuación cubrirla con uncubreobjetos. Una preparación en gota pendiente serealiza colocando una gota del material en uncubreobjetos y cubriéndolo con un portaobjetos(invertido) con una excavación central. Hay que sellarla preparación con vaselina alrededor de laexcavación. La ventaja de esta última técnica, es quela preparación no se seca y puede ser observadadurante un tiempo más largo.
Las preparaciones en fresco se utilizan para observarmicroorganismos vivos.
Preparación en gota pendiente:
Esta preparación se realiza colocando una gota de lasuspensión bacteriana en un cubreobjetos en el cual seha hecho previamente un círculo con vaselina oparafina (a). La vaselina actúa como sellador. (b)Sobre el cubreobjetos se coloca un portaobjetos con

una excavación central que queda adherido gracias a lavaselina (c). Se invierte la preparación y se observacolocándola en la platina del microscopio (d). Laspreparaciones en gota pendiente se utilizan paraobservar microorganismos vivos.
Preparación en seco:
Tinciones:
El microscopio de campo claro es más útil para laobservación de especímenes teñidos. Los colorantes soncompuestos químicos utilizados para aumentar elcontraste. Existen algunos, llamados colorantesvitales, que pueden añadirse directamente a unapreparación en fresco; por tanto, colorean célulasvivas. No obstante, la mayoría de los colorantes sonsolamente efectivos después de que los microorganismoshayan sido fijados, es decir, se encuentren muertos yadheridos al portaobjetos. Para la fijación por calor,se realiza una fina extensión de una gota de muestralíquida sobre un portaobjetos y se deja secar al aire;a continuación, se pasa la preparación de Forma rápidasobre la llama de un mechero. El calor de la llamamata las células microbianas por desnaturalización desus proteínas. Las proteínas coaguladas unen lascélulas al porta. Cuando se desea fijar especimenesdelicados se utiliza la fijación química, va que esmenos lesiva que el calor. Para ello se añade una gotadel fijador, por ejemplo, ácido ósmico, formaldehído,o glutaraldehído, sobre la muestra líquida con losmicroorganismos.

La fijación posee algunos inconvenientes. Por ejemplo,a menudo distorsiona la apariencia real de lascélulas, lo cual dificulta la identificación; además,no permite la observación del movímiento de losmicroorganismos. Después de la fijación, se añade elcolorante, que debe permanecer el tiempo suficiente encontacto con el espécimen, para que pueda serabsorbido. A continuación, se retira el exceso decolorante, normalmente lavando con agua.
Tipos de colorantes Casi todos los colorantes sonsales, compuestos formados por iones cargados. Loscolorantes básicos son aquellos en los cuales elagente que tiñe es el ion cargado positivamente,mientras que en los ácidos, el colorante es el ioncargado negativamente. Los colorantes más utilizadosson los de tipo básico, va que la mayor parte de lascélulas microbianas Poseen cargas débilmente negativasen su superficie, lo cual facilita su unión. Entre loscolorantes básicos más comunes se encuentran lasafranina, la fucsina básica, el cristal violeta y elazul de metileno. Los colorantes ácidos se unen a laspartes de las células cargadas positivamente. Seutilizan para teñir tejidos animales infectados conmicroorganismos. Entre los más frecuentes están laeosina, la fucsina ácida y el rojo Congo.
Tinción Simple:
Un colorante; proporciona contraste para observarmejor un organismo completo. Se tiñe con un colorantebásico (azul de metileno, cristal violeta, o fucsinabásica) durante unos 5 minutos. Aclarar brevemente con

agua. Se tiñen casi todas las bacterias; la mayoría delos tejidos no se tiñen.
Tínciones diferenciales:
Tinción de Gram:
Dos o más colorantes; distingue entre bacterias Grampositivas y Gram negativas. Se cubre la preparación debacterias fijadas con cristal violeta y después conuna solución de iodo (mordiente). Todas las célulasquedan tenidas de color violeta oscuro. Se decoloracon acetona al 95%.
Las células Gram positivas permanecen tenidas, perolas negativas pierden el colorante. Se tiñe consafranina (contraste). El color violeta de las Grampositivas se vuelve más oscuro y las Gram negativas setiñen de rosa.
Tinción de ácido-alcohol resistencia (Ziehl-Neelsen):
Dos colorantes; distingue entre las micobacterias(ácido-alcohol resistentes) y el resto de lasbacterias Se tiñen las células con fucsina básica yse calienta a emisión de vapores durante 5 minutos.Todas las bacterias se tinen de rojo. Se decolorabrevemente con una mezcla de alcohol-HCl. Lasbacterias resistentes permanecen teñidas de rojo;todas las demás se decoloran. Se trata con elcolorante de contraste azul de metileno. Las bacteriasácido-alcohol resistentes continúan tenidas de rojo,las otras se tiñen de azul.
Tinciones especificas:

Tinción de esporas de Wirtz-Cortitlin:
Tiñe selectivamente las endosporas. Se cubre lapreparación con verde de malaquita y se calienta aemisión de vapores durante 60 segundos. Se lava conagua durante 30 segundos y se tiñe con safranina. Lasendosporas retienen el color verde; el resto de lacélula toma el color rosa.
Tinción de flagelos de Leifson:
Permite observar los flagelos. A las célulaspreviamente fijadas, se le añade una mezcla de ácidotánico (mordiente) y del colorante rosanilina. Elmordiente engruesa los flagelos y el colorante losfine.
Tinción negativa Revela la presencia de cápsulas:
Se utiliza tinta china o nigrosina para tenir unapreparación en fresco del espécimen. Las particulas decolorante no pueden penetrar en la cápsula, que seobserva como una región clara alrededor de la célula.
MATERIAL:
Cristalizador.
2 varillas.
Cinta.
Pipetas Pasteur.
Mechero.
Frasco lavador.
Pinzas de madera.

Portaobjetos.
Cubreobjetos.
Asa bacteriológica.
REACTIVOS:
Azul de Metileno(colorante).
Violeta de Cristal(colorante).
LUGOL(reactivo).
Alcohol - Acetona 7:3(reactivo).
Safranina(colorante).
PROCEDIMIENTO:
1. Muestra Fresca:
Con una pipeta Pateur tomar una muestra líquida quecontenga microorganismos, luego con mucho cuidadoverter una gota en un portaobjetos etiquetadopreviamente, luego con otra pipeta Pasteur agregar unagota de agua destilada al portaobjetos, con un asabact. esterilizada mezclamos muy bien, por último secubre el portaobjetos con un cubreobjetos.
Una vez terminada la preparación ya se puede ver bajoel microscopio con todos los objetivos, excepto el deinmersión.
2. Tinción Simple:
Calentamos un asa bacteriológica en el mechero y unavez esterilizada y fría tomamos un poco de muestra y

la colocamos con mucho cuidado en un portaobjetospreviamente etiquetado, luego volvemos a esterilizardel asa bact. y la apartamos. Con la ayuda de unaspinzas de madera sujetamos el porta y lo pasamosvarias veces por encima de la llama el mechero hastaque la muestra este completamente seca. Con las 2varillas y un poco de cinta hacemos un soporte quecolocamos encima de un cristalizador, en ese soportecolocamos el portaobjetos con la muestra ya fijada,luego con una pipeta pasteur le agregamos gotas deAzul de Metileno hasta cubrirlo totalmente, cuandoeste cubierto esperamos de 2 a 3 minutos y procedemosa lavarlo muy bien con agua destilada de un frascolavador, luego lo dejamos secar y ya se encuentralisto para ser observado en el microscopio, en estecaso si podemos usar hasta el objetivo de inmersión.
3. Tinción de Gram:
Calentamos un asa bacteriológica en el mechero y unavez esterilizada y fría tomamos un poco de muestra yla colocamos con mucho cuidado en un portaobjetospreviamente etiquetado, luego volvemos a esterilizarel asa bact. y la apartamos. Con la ayuda de unaspinzas de madera sujetamos el porta y lo pasamosvarias veces por encima de la llama del mechero hastaque la muestra este completamente fijada. Con las 2varillas y un poco de cinta hacemos un soporte quecolocamos encima de un cristalizador, en ese soportecolocamos el portaobjetos con la muestra ya fijada,luego con una pipeta pasteur le agregamos gotas deVioleta de Cristal hasta cubrirlo totalmente, cuandoeste cubierto esperamos 1:30 minutos y lo enjuagamos

muy bien con agua destilada, con otra pipeta pasteuragregamos gotas de LUGOL hasta cubrirlo totalmente,esperamos 30 seg y enjuagamos , repetimos el mismoproceso pero con Alcohol-Acetona 7:3, dejamos otros 30segundos y lo enjuagamos, luego lo volvemos a lavarmuy bien con agua destilada, por último lo cubrimoscon Safranina y esperamos 3 minutos para volver aenjuagarlo, dejamos secar y ya estará listo para elmicroscopio, en este caso también se puede usar elobjetivo de inmersión.
BIBLIOGRAFÍA:
http://aulavirtual.usal.es/aulavirtual/demos/ microbiologia/unidades/documen/uni_02/56/cap304.htm
Copyright © 2022 FDOKUMEN




















