
Cardioprotection: A radical view:: Free radicals in pre and postconditioning

Basic Res Cardiol 101: 180 – 189 (2006)DOI 10.1007/s00395-006-0584-5 ORIGINAL CONTRIBUTION
Claudia PennaRaffaella RastaldoDaniele MancardiStefania RaimondoSandra CappelloDonatella GattulloGianni LosanoPasquale Pagliaro
Post-conditioning induced cardioprotection
requires signaling through a redox-sensitive
mechanism, mitochondrial ATP-sensitive
K+ channel and protein kinase C activation
� Abstract Post-conditioning (Post-C) induced cardioprotection involvesactivation of guanylyl-cyclase. In the ischemic preconditioning scenario, thedownstream targets of cGMP include mitochondrial ATP-sensitive K+
(mKATP) channels and protein kinase C (PKC), which involve reactive oxygenspecies (ROS) production. This study tests the hypothesis that mKATP, PKCand ROS are also involved in the Post-C protection. Isolated rat hearts under-went 30 min global ischemia (I) and 120 min reperfusion (R) with or withoutPost-C (i.e., 5 cycles of 10 s R/I immediately after the 30 min ischemia). In 6groups (3 with and 3 without Post-C) either mKATP channel blocker, 5-hydroxydecanoate (5-HD), or PKC inhibitor, chelerythrine (CHE) or ROSscavenger, N-acetyl-cysteine (NAC), were given during the entire reperfusion(120 min). In other 6 groups (3 with and 3 without Post-C), 5-HD, CHE orNAC were infused for 117 min only starting after 3 min of reperfusion not tointerfere with the early effects of Post-C and/or reperfusion. In an additionalgroup NAC was given during Post-C maneuvers (i.e., 3 min only). Myocar-dial damage was evaluated using nitro-blue tetrazolium staining and lactatedehydrogenase (LDH) release. Post-C attenuated myocardial infarct size (21± 3% vs. 64 ± 5% in control; p < 0.01). Such an effect was abolished by 5-HDor CHE given during either the 120 or 117 min of reperfusion as well as byNAC given during the 120 min or the initial 3 min of reperfusion. However,delayed NAC (i.e., 117 min infusion) did not alter the protective effect of Post-C (infarct size 32 ± 5%; p < 0.01 vs. control, NS vs. Post-C). CHE, 5-HD or NACgiven in the absence of Post-C did not alter the effects of I/R. Similar resultswere obtained in terms of LDH release. Our data show that Post-C inducedprotection involves an early redox-sensitive mechanism as well as a persist-ent activation of mKATP and PKC, suggesting that the mKATP/ROS/PKC path-way is involved in post-conditioning.
� Key words ischemia/reperfusion – mitochondrial KATP channels – post-conditioning – protein kinase C – redox signaling
BRC
584
C. Penna · D. Mancardi · S. Raimondo D. Gattullo · Dr. P. Pagliaro MD, PhD (�) Dipartimento di Scienze Cliniche eBiologiche Università di TorinoOspedale S. LuigiRegione Gonzole10043 Orbassano (TO) ItalyTel.: +39-11/6705430/7710Fax: +39-11/9038639E-Mail: [email protected]
R. Rastaldo · S. Cappello · G. LosanoSezione di Fisiologia del Dipartimento di NeuroscienzeUniversità di Torino, Italy
Received: 1 September 2005Returned for 1. revision: 19 September 20051. Revision received: 29 November 2005Returned for 2. revision: 15 December 20052. Revision received: 19 December 2005Accepted: 20 December 2005Published online: 6 February 2006
Introduction
It is well known that both ischemia and reperfusion mayinduce myocardial injuries [3, 4, 10, 20, 26, 32, 46]. Recent
in vivo studies in dogs [46], rabbits [44] and rats [19] aswell as in isolated hearts [8, 19, 31, 39] indicate thatmarked attenuation of reperfusion injuries may beobtained by application of a series of brief coronaryartery reocclusions and reperfusions performed during

C. Penna et al. 181Post-conditioning and redox-sensitive signaling
the early minutes of coronary artery reflow following theischemic event. This phenomenon was termed post-con-ditioning (Post-C) [46]. Post-C has been also described inisolated rat cardiomyocytes [38]. All of these studies havere-attracted scientific and clinical interest in the reperfu-sion phase, deserving further investigations due to thepotential for even more attractive clinical applicationthan ischemic preconditioning (IP) [15, 17, 19, 41]. Infact, Post-C has also been very recently tested in humans[37]. Since IP triggers protective pathways beforeischemia, whereas Post-C alters events after ischemia, ithas been hypothesized that the mechanisms by whichpre-conditioning and post-conditioning confer myocar-dial protection should be different [46]. However, datafrom various laboratories suggest that this may not be thecase. In fact some reports indicate that components ofthe so called RISK (reperfusion injury salvage kinase)pathway [e.g., phosphoinositide 3-kinase (PI3K)-PKB(Akt) and mitogen-activated ERK kinase (MEK1/2)/extracellular regulated kinase (ERK)] play a role both inpre- and in post-conditioning [5, 14, 30, 39, 45], suggest-ing that these two phenomena may share common path-ways.
We recently demonstrated a central role for guanylylcyclase (GC) activation in post-conditioning protection[31]. In the IP triggering mechanisms the activation ofGC is followed by the activation of mitochondrial ATP-sensitive K+ (mKATP) channels and protein kinase C(PKC), which involves reactive oxygen species (ROS)production [14, 23].
In a recent editorial Tsang et al. [40] state: “Othermechanisms by which postconditioning activates theRISK pathway need to be further investigated, for exam-ple, the possible role of PKC or ROS as upstream media-tors.” However, it is interesting that ROS are consideredresponsible for part of reperfusion injuries [3, 4, 10, 20,26, 32, 46]. Therefore, in the present study we investi-gated whether mKATP channels, PKC and redox-sensitivemechanism are also involved in the signaling cascadeleading to Post-C protection. We found that these threecomponents are indeed involved in Post-C.
Materials and methods
� Animals
Male Wistar rats (n = 112; body weight 450–550 g)received care in compliance with Italian law (DL-116,Jan. 27, 1992) and in accordance with the Guide for theCare and Use of Laboratory Animals published by the USNational Institutes of Health (NIH Publication No. 85-23,revised 1996).
� Isolated heart perfusion
The methods were similar to those previously described[27, 31]. In brief, each animal was anesthetized withurethane (1 g/kg i.p.). The chest was opened 10 min afterheparin treatment and the heart was rapidly excised.Isolated rat hearts were weighed, attached to the perfu-sion apparatus and retrogradely perfused with oxy-genated Krebs-Henseleit buffer (127 mM NaCl, 17.7 mMNaHCO3, 5.1 mM KCl, 1.5 mM CaCl2, 1.26 mM MgCl2, 11 mM D-glucose and gassed with 95% O2 and 5% CO2).A constant flow was adjusted with a proper pump (Wat-son-Marlow 313, Falmouth, Cornwall, UK) to obtain atypical coronary perfusion pressure of 80–85 mmHg dur-ing the initial part of stabilization. Thereafter the sameflow level (9 ± 1 ml/min/g) was maintained throughoutthe experiment.
A small hole in the left ventricular wall alloweddrainage of the thebesian flow. A polyvinyl-chloride bal-loon was placed into the left ventricle via the left atriumand connected to an electromanometer (Monitoring Kitmk 5-02 DTBNVF, Abbott, Milan, Italy) to record leftventricular pressure (LVP). The balloon was filled withsaline to achieve an end-diastolic LVP of 5 mmHg. There-after the same balloon volume was maintained through-out the experiment.
The hearts were electrically paced at 280 bpm and keptin a temperature-controlled chamber (37 °C). Coronaryperfusion pressure, coronary flow and LVP were moni-tored to assess the preparation conditions. LVP was ana-lyzed offline with LabView software (National Instru-ments). Care was taken to measure LVP in a period(15–20 s) without arrhythmias. All chemicals were pur-chased from Sigma (USA) with the exception of thereagents required to assess myocardial infarction thatwere purchased from Merck (USA).
� Experimental protocols (Fig. 1)
Each heart was allowed to stabilize for 20–30 min, afterwhich time baseline parameters were recorded. At theend of the stabilization period, hearts were randomlyassigned to one of the 14 treatment groups describedbelow and then subjected to a specific protocol, whichincluded 30 min of global no-flow ischemia followed by120 min of reperfusion (Rep) in all groups (see below andFig. 1). Pacing was discontinued on initiation of ischemiaand restarted after the third minute of reperfusion in allgroups [19, 27, 30, 31].
After stabilization the hearts of Group 1 (Control group,n = 12) were exposed to ischemia and reperfusion only.
In Group 2 (Post-C group, n = 13), after stabilizationand the 30 min ischemia, the hearts immediately under-went a Post-C protocol. This consisted of five cycles of 10 s reperfusion and 10 s occlusion [31].

In the following six groups (from group 3 to group 8)the drugs 5-hydroxydecanoate (5-HD), chelerythrine(CHE) or N-acetyl-L-cysteine (NAC) were given for theentire period of reperfusion.
In particular, in Group 3 (Post-C + 5-HD, n = 9), after30 min ischemia the hearts underwent the same protocolfor Post-C as in group 2, and were perfused with a solu-tion containing the mKATP channel inhibitor (5-HD, 100 μM) [25] for the entire period of reperfusion, includ-ing the Post-C maneuvers.
In Group 4 (Post-C + CHE, n = 8) the hearts, whichunderwent the same protocol for Post-C as in groups 2and 3, were perfused with a solution containing the PKC
inhibitor (CHE, 5 μM) [30] for the entire period of reper-fusion, including the Post-C maneuvers.
In Group 5 (Post-C + NAC, n = 9) the hearts, whichunderwent the same protocol for Post-C as in the previ-ous groups, were perfused with a solution containing theROS scavenger (NAC, 4 mM) [7, 9, 27], for the entireperiod of reperfusion, including Post-C maneuvers.
In Group 6 (Rep + 5-HD, n = 6), Group 7 (Rep + CHE,n = 7) and Group 8 (Rep + NAC, n = 6), infusions of 5-HD, CHE and NAC were performed, respectively, dur-ing the entire period of reperfusion in the absence ofPost-C protocol to determine the effects of the drugsthemselves.
182 Basic Research in Cardiology, Vol. 101, No. 2 (2006)© Steinkopff Verlag 2006
Fig. 1 Time-lines and protocols for allexperimental groups. After stabiliza-tion, all hearts underwent 30 minglobal ischemia and 120 min reperfu-sion, here represented with a sequenceof white/black/white horizontal bars.Group 1, control hearts (n = 12) wereexposed to ischemia (30 min) and thenreperfusion (120 min) only. Group 2,after ischemia the hearts (n =13) wereexposed to a protocol of Post-condi-tioning (Post-C). This consisted in fivecycles of 10 s reperfusion (R) andischemia (I), here represented withwhite space and black vertical lines,respectively. Group 3 hearts (n = 9),Group 4 hearts (n = 8) and Group 5hearts (n = 9) were treated with 5-hydroxydecanoate (5-HD, 100 �M),chelerythrine (CHE, 5 �M) and N-acetyl-L-cysteine (NAC, 4 mM), respec-tively, during the entire period of reper-fusion, including the Post-C protocol.Groups 6 hearts (n = 6), Group 7 hearts(n = 7) and Groups 8 hearts (n = 6)were treated with 5-HD, CHE, and NAC,respectively, during the entire period ofreperfusion infusion, without Post-C.Group 9 hearts (n = 6), Group 10 hearts(n = 6) and Group 11 hearts (n = 8)were treated with Post-C and withdelayed infusion (i.e., after the thirdmin of reperfusion) of 5-HD (100 �M),CHE (5 �M), and NAC (4 mM), respec-tively. Group 12 hearts (n = 6), Group13 hearts (n = 6) and Group 14 hearts (n= 6) were treated with delayed infusionof 5-HD, CHE, and NAC, respectively,after the third min of reperfusion, with-out any Post-C protocol

C. Penna et al. 183Post-conditioning and redox-sensitive signaling
In the following six groups (groups 9 to 14) the infu-sions of 5-HD, CHE or NAC were started only after thethird minute of reperfusion (delayed drug infusion).
In Group 9 (Post-C + delayed 5-HD, n = 6), Group 10(Post-C + delayed CHE, n = 6) and Group 11 (Post-C +delayed NAC, n = 8) infusions of 5-HD, chelerythrine andNAC were started, respectively, only after the thirdminute of reperfusion, i.e., when the Post-C protocol hadalready been performed. In this situation 80 s had elapsedbetween the last 10 s occlusion and the beginning of druginfusion.
In the absence of Post-C protocol, in Group 12 (Rep + delayed 5-HD, n = 6), Group 13 (Rep + delayed CHE, n = 6) and Group 14 (Rep + delayed NAC, n = 6) theinhibitors 5-HD, CHE and NAC were infused, respec-tively, starting after the third minute of reperfusion, todetermine the effects of the delayed drug infusion.
In four additional hearts (Post-C with early NAC),immediately after the 30 min of ischemia NAC (4 mM)was given only during the initial three minutes of reper-fusion, meanwhile performing the Post-C protocol.
The doses of the drugs used were selected becausethey have commonly been used successfully by otherinvestigators and by us, and because these drugs haveshown to be potent antagonists of mKATP channel andPKC activation [25, 30, 45] or ROS scavenger [7, 9, 27].
At the end of experiments all the hearts were removedfrom the apparatus for the analysis of the infarct size (seebelow).
� Assessment of myocardial injury
Since in isolated rat hearts, both IP and Post-C [27, 31]are known to reduce the production of lactate dehydro-genase (LDH) during reperfusion, the release of thisenzyme was tested. Samples of coronary effluent (2 ml)were withdrawn with a catheter inserted into the rightventricle via the pulmonary artery. Samples wereextracted immediately before ischemia and at 3, 6, 10 and20 min of reperfusion. Thereafter samples were collectedevery 20 min until the end of reperfusion. LDH releasewas measured as previously described [27, 31]. We didnot collect samples during Post-C due to the technicaldifficulty of drawing effluent while performing this pro-cedure. Data are expressed as cumulative values for theentire reperfusion period.
Infarct size was assessed at the end of the experiment[24, 27, 30, 31]. Immediately after reperfusion each heartwas rapidly removed from the perfusion apparatus andthe left ventricle (LV) was dissected into 2–3 mm thickcircumferential slices. Following 20 min of incubation at37 °C in 0.1% solution of nitro-blue tetrazolium in phos-phate buffer, unstained necrotic tissue was carefully sep-arated from stained viable tissue by a blinded independ-ent observer. The necrotic and non-necrotic tissues were
then weighed, and the necrotic mass was expressed as apercentage of total left ventricular mass. Although in thisis model the whole heart underwent normothermicischemia, only the LV had a fixed volume and pre-load;therefore only the LV mass was considered as the riskarea.
� Statistical analysis
All data are expressed as means ± S.E.M. One-WayANOVA and Tukey’s HSD (honestly significant differ-ence) post-ANOVA tests were used to evaluate the sta-tistical significance of the differences among groups. Allgroups were analyzed together. Cardiac function param-eters were analyzed by two way ANOVA for repeatedmeasures and Bonferroni correction test as post hoc test.A p value < 0.05 was considered statistically significant.
Results
Cardiac weight (1351 ± 12; range 1155–1662 mg, n = 112)and the cardiac to body weight ratio (2.67 ± 0.011; range2.03–2.99 mg/g) were similar in all groups. The risk area,i.e., LV mass, was also similar in all groups (LV weightwas 791 ± 4.49; range 691–910 mg). Out of 112 hearts, 22did not restart cardiac rhythm during reperfusion (2 ineach of groups 1, 3, 4, 5, 6, 7, 9, 13; and 1 in each of groups2, 8, 10, 11, 12 and 14).
� LDH release
In the control Group 1 the cumulative LDH from all thesamples taken during reperfusion was 1425 ± 154 U/g wetwt. In Post-C Group 2 the release of LDH was 634 ± 61 U/gwet wt; a value significantly (p < 0.01) lower than that ofcontrol Group 1. The data of these two groups arereported in Fig. 2A and B.
Figure 2A also represents LDH data of the six groups(three with and three without Post-C) to which the drugs(5-HD, CHE or NAC) were given for the entire period ofreperfusion. In these six groups (Post-C+drug andRep+drug), the release of LDH was similar (p = NS vs.each other). It was also similar to that of the controlGroup 1 (p = NS for all), but significantly (p range < 0.05–0.01) greater than the Post-C Group 2, regardlessof the presence or the absence of Post-C.
Figure 2B shows LDH data of the six groups (threewith and three without Post-C) in which the inhibitors (5-HD, CHE or NAC) were given 3 min after the beginningof reperfusion (delayed drug). In particular, in Post-C +delayed 5-HD and in Post-C + delayed CHE groups, therelease of LDH was similar to that of the control Group 1,

but significantly higher than Post-C Group 2 (p < 0.05 forboth). However, in Post-C + delayed NAC, the release ofLDH was 650 ± 22 U/g wet wt; a value which was signifi-cantly lower than that of control Group 1, of Post C +delayed 5-HD and of Post C + delayed CHE (p range
< 0.05–0.01), but not different from that of Post-C Group2.
In Rep + delayed 5-HD, Rep + delayed CHE and Rep+ delayed NAC groups, in the absence of Post-C maneu-vers, the release of LDH was similar (p = NS vs. each
184 Basic Research in Cardiology, Vol. 101, No. 2 (2006)© Steinkopff Verlag 2006
Fig. 2 Lactate dehydrogenase (LDH) release. Postischemic LDH leakage during 120min reperfusion following 30 min of global ischemia in the isolated rat hearts. Dataof Control and Post-C groups are reported both in A and B. A also represents LDHdata of the six groups (three with and three without Post-C) in which the testeddrugs were given for the entire period of reperfusion. B shows LDH data of the six
Groups (3 with and 3 without Post-C) in which the tested inhibitors were given 3 minafter beginning reperfusion (delayed drug infusion). Data are means � SEM.**p < 0.01 vs. Control; #p < 0.05 vs. Post-C; ##p < 0.01 vs. Post-C; §p < 0.05 vs. Post-C + delayed NAC; NS not significant. Rep reperfusion; other abbreviations as in Fig. 1
Fig. 3 Infarct size at the end of the experiments, expressed as percentage of leftventricle (LV) mass. Experimental groups representation in A and B as in Fig. 2.Data are means � SEM.**p < 0.01 vs. Control; #p < 0.05 vs. Post-C; ##p < 0.01 vs.
Post-C; §p < 0.05 vs. Post-C + delayed NAC; NS not significant. Rep reperfusion;other abbreviations as in Fig. 1
C. Penna et al. 185Post-conditioning and redox-sensitive signaling
other). It was also similar to that of control Group 1 (p =NS for all), but was significantly higher than Post-CGroup 2 and Post-C + delayed NAC (p < 0.05 for all).
In the four additional hearts in which NAC was givenduring Post-C maneuvers only, the release of LDH was1109 ± 56 U/g wet wt (p = NS vs. control Group 1; p < 0.05vs. Post-C Group 2 and Post-C + delayed NAC).
� Infarct size
Total infarct size, expressed as a percentage of left ven-tricular mass, was 64 ± 5% in control Group 1. In Post-CGroup 2 the infarct size was 21 ± 3%; a value significantly(p < 0.01) lower than that of control Group 1. Data ofthese two groups are reported in Fig. 3A and B.
Figure 3A also represents the infarct size data of thesix groups (three with and three without Post-C) to whichthe drugs (5-HD, CHE or NAC) were given for the entireperiod of reperfusion. In all these groups infarct size wassimilar (p = NS vs. each other). Infarct size was also sim-ilar to that of the control Group 1 (p = NS for all), but sig-nificantly (p range < 0.05–0.01) greater than that of thePost-C Group 2, regardless of the presence or the absenceof Post-C.
Figure 3B shows the infarct size data of the six groups(three with and three without Post-C) to which theinhibitors (5-HD, CHE or NAC) were given 3 min afterthe beginning of reperfusion (delayed drug). In particu-lar, in Post C + delayed 5-HD and in Post C + delayedCHE groups, infarct size was similar to that of controlGroup 1, but significantly greater than Post-C Group 2 (p < 0.05 for both). However, in Post-C + delayed NACthe infarct size was 32 ± 5% of LV; a value that was sig-nificantly lower than that of control Group 1, of Post C +delayed 5-HD and of Post C + delayed CHE (p range < 0.05–0.01), but not different from that of Post-C Group2.
In the Rep + delayed 5-HD, Rep + delayed CHE andRep + delayed NAC groups, in the absence of Post-Cmaneuvers, the infarct size was similar (p= NS vs. eachother). It was also similar to that of control Group 1 (p =NS for all), but was significantly greater than that of Post-C Group 2 and Post-C + delayed NAC (p < 0.05 for all).
In the four additional hearts in which NAC was givenduring Post-C maneuvers only, the infarct size was 62 ±2% of LV (p = NS vs. control Group 1; p < 0.05 vs. Post-C Group 2 and Post-C + delayed NAC).
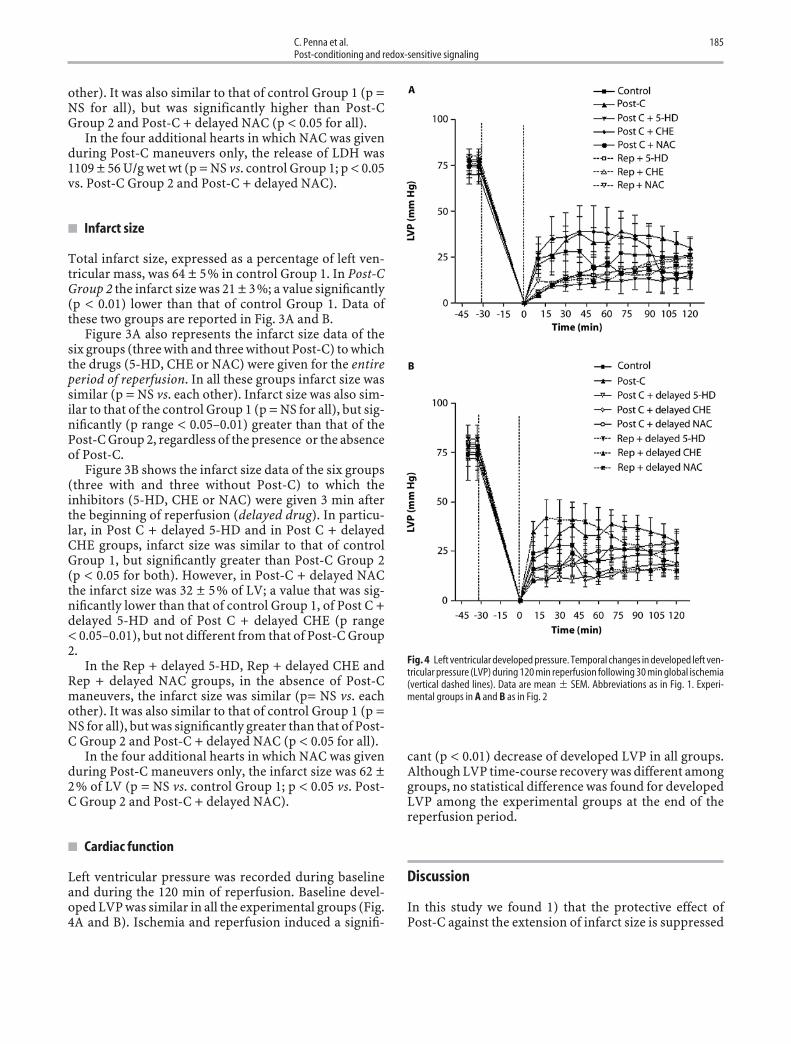
� Cardiac function
Left ventricular pressure was recorded during baselineand during the 120 min of reperfusion. Baseline devel-oped LVP was similar in all the experimental groups (Fig.4A and B). Ischemia and reperfusion induced a signifi-
cant (p < 0.01) decrease of developed LVP in all groups.Although LVP time-course recovery was different amonggroups, no statistical difference was found for developedLVP among the experimental groups at the end of thereperfusion period.
Discussion
In this study we found 1) that the protective effect ofPost-C against the extension of infarct size is suppressed
Fig. 4 Left ventricular developed pressure. Temporal changes in developed left ven-tricular pressure (LVP) during 120 min reperfusion following 30 min global ischemia(vertical dashed lines). Data are mean � SEM. Abbreviations as in Fig. 1. Experi-mental groups in A and B as in Fig. 2

if, during reperfusion, mKATP channels are blocked by 5-HD or if PKC is inhibited by CHE, and 2) that this pro-tection is abolished if during Post-C maneuvers redoxenvironment is altered by NAC.
The results indicate that mKATP channels and PKCare involved in cardioprotection by Post-C. They alsosuggest that this protection can be triggered through aredox-sensitive mechanism. It is intriguing that,although ROS are considered responsible for part of thereperfusion injuries, their scavenger NAC given duringPost-C maneuvers prevents the infarct sparing effect ofPost-C.
Although Post-C protects against the extension ofinfarct size, it neither significantly increases nor reducesthe post-ischemic developed ventricular pressure in ourmodel. This finding is similar to that reported for pre-conditioned rabbit hearts [11].
As for preconditioning [11, 16], developed pressuremay not be an appropriate end-point for studies of Post-C protection. The improvement of function duringreperfusion in preconditioned rat hearts has been attrib-uted to the reduction of adenosine release during thisphase [11]. On the contrary, in post-conditioned heartsan accumulation of adenosine has been reported [18].These differences may explain our finding. Nevertheless,it is hard to distinguish the impairment of function dueto necrosis and/or to stunning of viable tissue. To under-stand the role of Post-C on myocardial stunning, appro-priate studies are required. Therefore we focused ourattention on myocardial damages evaluated by infarctsize and LDH release.
� The signaling pathway in post-conditioning
Recently, in isolated rabbit [44] and rat [31] hearts, sol-uble GC and cGMP have been observed to play a role inPost-C induced protection. Moreover, in another study ithas been suggested that mKATP channel activation may beinvolved in Post-C protection [45]. In the present inves-tigation we confirm the involvement of mKATP channelsand show for the first time that Post-C signaling alsorequires PKC activation and a redox sensitive mecha-nism operating in the very early phase of reperfusion. Allthese signaling elements have been previously describedin the triggering phase of pre-conditioning [14, 35, 22, 21,27, 30].
The sharing of signaling-pathways is in line with theobservation that protection from IP and Post-C are notadditive [12, 19, 31, 39]. It is interesting that many of the RISK elements (e.g., PI3K-Akt and MEK1/2-ERK)involved in the signaling pathway in pre-conditioningprotection against reperfusion injuries have recentlybeen documented also in Post-C [14, 39, 40, 42, 45].
Some differences, however, may exist between pre-and post-conditioning. Darling et al. [8] showed an
increase of phospho-ERK, but not of PI3-kinase/Akt inPost-C, while Yang et al. [45] showed that ERK isinvolved in post-, but not in pre-conditioning. Thesefindings may explain a certain degree of additive protec-tion between IP and Post-C, as observed by Yang et al.[45]. Yet in contrast with Yang et al. [45], Cao et al. [5]reported that ERK is present in preconditioning triggerpathway. The reasons for the differences are not clear.Different species and/or protocols may play a role [42].A role may be also played by different way of tissue sam-pling [8].
Our findings also suggest that the Post-C protectiontakes place only if mKATP and PKC activation persistsduring later periods of reperfusion. In fact, when themKATP channel blocker 5-HD, or the PKC antagonistCHE were given after Post-C maneuvers, we found thatthe Post-C induced protection was suppressed. Thesefindings are similar to those observed in IP when RISKkinases are inhibited during reperfusion (i.e., kinasephosphorylations may persist at least 15–30 min duringreperfusion) [14]. Apparently, not only in IP [14, 35] butalso in Post-C, the opening of mKATP channels and theactivation/translocation of PKC may persist during thereperfusion.
� Role of redox environment in cardioprotection
ROS play an essential, though double-edged, role in car-dioprotection. The beneficial effect of moderate concen-trations of ROS is supported by the observation that theprotective mKATP channel opener diazoxide induces anincrease in ROS production, which is abolished by addi-tion of either NAC or N-mercaptopropionylglycine inboth adult cardiomyocytes [9] and a human atrium-derived cell line [6]. The signaling role of ROS is also con-sistent with the fact that in isolated rat hearts the protec-tive effects of IP and diazoxide are significantly bluntedby NAC (4 mM) [7, 9]. On the other hand, ROS generatedat the time of reperfusion may lethally injure viable, pre-viously ischemic myocardium [3, 20, 34]. Yet, positive[e.g. 3, 4, 20] and negative [e.g. 3, 10, 20, 26, 32] resultsagainst reperfusion injuries are obtained with ROS scav-engers given during reperfusion.
During the whole reperfusion phase an attenuation ofROS generation and ROS-mediated lipid peroxidationby Post-C has been observed in both in vivo and in vitrosettings [38, 46]. The reduced oxygen delivery duringPost-C flow interruption may contribute to this attenua-tion [38]. However, a ROS production occurs in the ini-tial phase of reperfusion even when oxygen delivery isexperimentally reduced (hypoxic reperfusion). In par-ticular, in the isolated heart, Serviddio et al. [36] haveshown that 3 min of initial hypoxic reperfusion blunt, butdo not suppress, ROS production during this period. Onthe contrary, Angelos et al. [1], in the same model, report
186 Basic Research in Cardiology, Vol. 101, No. 2 (2006)© Steinkopff Verlag 2006

C. Penna et al. 187Post-conditioning and redox-sensitive signaling
an increased ROS production during the initial 5 min ofhypoxic reperfusion. It is thus likely that the reduceddelivery of oxygen during Post-C maneuvers (shortischemias and reperfusions) may alter, but not suppress,ROS production during early reperfusion [1, 36, 45].Therefore, the protection is achieved only if such a pro-duction is not reduced by a ROS scavenger. In otherwords, our data suggest that one of the triggers of Post-Cprotection is the ROS availability occurring during Post-C protocol, which may contribute to the activation ofintrinsic mechanisms against the deleterious effects ofthe subsequent reperfusion.
Our data suggest that Post-C maneuvers allow a favor-able redox state environment. However, whether a vari-ation of the amount of ROS, and/or a different intracel-lular ROS sources (compartmentalization) and/or a dif-ferent radical species unique to Post-C contributes toPost-C triggering remains to be clarified.
In conclusion, not only elements of the RISK pathway[14, 40, 42], but also triggering mechanisms are similar inpre- and post-conditioning, two phenomena elicited bysimilar maneuvers: i.e., multiple, brief (few minutes orseconds) coronary occlusions.
All these findings stress the importance of the reper-fusion phase for the myocardial salvage by properlytimed protective interventions.
� Methodological considerations
In isolated hearts of different species (mouse, rabbit andrat), constant pressure perfusion has been successfullyused in Post-C maneuvers [8, 19, 31, 39]. In our previousstudy [31] we showed that if the isolated rat heart is per-fused at constant flow the protection by Post-C is greaterthan if perfused at constant pressure. For this reason weused the constant flow model in the present investiga-tion.
Regarding the role of ROS as triggers of Post-C in theisolated heart, it must be considered that in this prepa-ration ROS are generated by endothelial cells and car-diomyocytes only, while in vivo they derive also fromneutrophils and other inflammatory oxidant sources [3,4, 20, 26, 32]. Moreover, in buffer perfused preparationsthere are no soluble lipid pro-inflammatory mediatorsthat may be involved in the amplification of the inflam-matory and oxidant responses. Therefore, ROS genera-tion in this model may have occurred to a much lesserextent than in vivo [3]. It is, then, possible that the ROS
level may have been sufficient to contribute to the pro-tection, but not to the I/R injuries. In fact, in this studythe highest concentration of NAC suggested in the liter-ature for the isolated rat heart [7, 9, 27] (i) did not pre-vent reperfusion injuries (Rep+NAC), (ii) suppressedany protective effect if given during Post-C (Post-C+NACand additional hearts), (iii) but did not blunt the pro-tection if given only after Post-C maneuvers (Post-C +delayed NAC).
Yet, NAC may affect intra and extra-cellular ROS. Infact, NAC is readily hydrolyzed to cysteyne and is able toexpand natural antioxidant defenses by increasing intra-cellular reduced glutathione concentration. Importantly,NAC has sulfhydryl groups that can directly scavengefree radicals under acidotic conditions which exist in theearly post-ischemic phase of the experiments [2]. As amatter of fact just 3 min of NAC infusion was able to pre-vent Post-C protection. Nevertheless, in vivo studies arenecessary to corroborate the importance of ROS as trig-ger mechanism of Post-C.
Regarding the role of mKATP channel our results con-firm another study, in which 5-HD was also used as achannel blocker [45]. However, this drug has significantmetabolic effects [13], which may limit our conclusion onthe role of these channels. Unfortunately, at the momentthere is no other blocker available that may be moreadvantageous.
Conclusion
Our study conducted on isolated rat hearts shows thatpost-conditioning is a mechanism of intrinsic protectionto the heart that can be abolished by mKATP channelblockade or by PKC inhibition during reperfusion, aswell as by ROS scavenger during the Post-C maneuvers.Therefore the mKATP/ROS/PKC pathway, which plays arole in pre-conditioning, is also involved in Post-C.Whether PKC is downstream or upstream mKATPremains to be established. Nevertheless, like the pre-con-ditioning, a redox-sensitive mechanism seems necessaryin the protection triggered by post-conditioning.
Acknowledgments The authors are grateful to Jennifer M. Lee for Eng-lish revision and Dr. Francesca Tullio for technical assistance. We thank“Compagnia di San Paolo”, Torino, the Regione Piemonte, the IstitutoNazionale per la Ricerca Cardiovascolare (INRC) and the Italian Min-istry of Education, University and Research (MIUR) for the financialcontributions.

1. Angelos MG, Kutala VK, Torres CA, He G,Stoner JD, Mohammad M, Kuppusamy P(2005) Hypoxic Reperfusion of theIschemic Heart and Oxygen Radical Gen-eration. Am J Physiol Heart Circ PhysiolIn Press DOI:10.1152/ajpheart.00223.2005
2. Atmaca G (2004) Antioxidant effects ofsulfur-containing amino acids. YonseiMed J 45:776–488
3. Becker LB (2004) New concepts in reac-tive oxygen species and cardiovascularreperfusion physiology. Cardiovasc Res61:461–470
4. Bolli R, Patel BS, Jeroudi MO, Lai EK,McCay PB (1988) Demonstration of freeradical generation in “stunned” myocar-dium of intact dogs with the use of thespin trap a-phenyl-N-tertbutyl nitrone. JClin Invest 82:476–485
5. Cao Z, Liu L, Van Winkle DM (2005)Met5-enkephalin-induced cardioprotec-tion occurs via transactivation of EGFRand activation of PI3K. (2005) Am J Phys-iol Heart Circ Physiol 288:H1955–H1964
6. Carroll R, Gant VA, Yellon DM (2001)Mitochondrial KATP channels protects ahuman atrial-derived cell line by a mech-anism involving free radical generation.Cardiovasc Res 51:691–700
7. Chen W, Gabel S, Steenbergen C, MurphyE (1995) A redox-based mechanism forcardioprotection induced by ischemicpreconditioning in perfused rat heart.Circ Res 77:424–429
8. Darling CE, Jiang R, Maynard M,Whittaker P, Vinten-Johansen J,Przyklenk K (2005) ‘Postconditioning’ viaStuttering Reperfusion Limits Myocar-dial Infarct Size in Rabbit Hearts: Role ofERK 1/2. Am J Physiol Heart Circ Physiol289:H1618–H1626
9. Forbes RA, Steenbergen C, Murphy E(2001) Diazoxide-induced cardiopro-tection requires signaling through aredox-sensitive mechanism. Circ Res88:802–809
10. Forman MB, Puett DW, Cates CU,McCroskey DE, Beckman JK, Greene HL,et al. (1988) Glutathione redox pathwayand reperfusion injury. Effect of N-acetyl-cysteine on infarct size and ventricularfunction. Circulation 78:202–213
11. Gelpi RJ, Morales C, Cohen MV, DowneyJM (2002) Xanthine oxidase contributesto preconditioning’s preservation of leftventricular developed pressure in iso-lated rat heart: developed pressure maynot be an appropriate end-point for stud-ies of preconditioning. Basic Res Cardiol97:40–46
12. Halkos ME, Kerendi F, Corvera JS, WangNP, Kin H, Payne CS, Sun HY, GuytonRA, Vinten-Johansen J, Zhao ZQ (2004)Myocardial protection with postcon-ditioning is not enhanced by ischemicpreconditioning. Ann Thorac Surg78:961–969
13. Hanley PJ, Drose S, Brandt U, Lareau RA,Banerjee AL, Srivastava DK, Banaszak LJ,Barycki JJ, Van Veldhoven PP, Daut J(2005) 5-Hydroxydecanoate is metabo-lised in mitochondria and creates a rate-limiting bottleneck for beta-oxidation offatty acids. J Physiol 562:307–318
14. Hausenloy DJ, Tsang A, Yellon DM (2005)The reperfusion injury salvage kinasepathway: a common target for bothischemic preconditioning and postcondi-tioning. Trends Cardiovasc Med 15:69–75
15. Heusch G. Postconditioning: old wine ina new bottle? (2004) J Am Coll Cardiol44:1111–1112
16. Lochner A, Genade S, Moolman JA (2003)Ischemic preconditioning: infarct size is amore reliable endpoint than functionalrecovery. Basic Res Cardiol 98:337–346
17. Kerendi F, Kin H, Halkos ME, Jiang R,Zatta AJ, Zhao ZQ, Guyton RA,Vinten-Johansen J (2005) Remote postcondition-ing Brief renal ischemia and reperfusionapplied before coronary artery reperfu-sion reduces myocardial infarct size viaendogenous activation of adenosinereceptors. Basic Res Cardiol 100:404–412
18. Kin H, Zatta AJ, Lofye MT, Amerson BS,Halkos ME, Kerendi F et al. (2005) Post-conditioning reduces infarct size viaadenosine receptor activation by endo-genous adenosine. Cardiovasc Res67:124–133
19. Kin H, Zhao ZQ, Sun HY, Wang NP,Corvera JS, Halkos ME et al. (2004) Post-conditioning attenuates myocardialischemia-reperfusion injury by inhibitingevents in the early minutes of reperfu-sion. Cardiovasc Res 62:74–85
20. Kloner RA, Jennings RB (2001) Conse-quences of brief ischemia: stunning, pre-conditioning and their clinical implica-tions. Circulation 104:2981–2989
21. Krenz M, Oldenburg O, Wimpee H,Cohen MV, Garlid KD, Critz SD, DowneyJM, Benoit JN (2002) Opening of ATP-sensitive potassium channels causesgeneration of free radicals in vascularsmooth muscle cells. Basic Res Cardiol97:365–373
22. Krieg T, Cohen MV, Downey JM (2003)Mitochondria and their role in precondi-tioning’s trigger phase. Basic Res Cardiol98:228–234
23. Krieg T, Qin Q, Philipp S, Alexeyev MF,Cohen MV, Downey JM (2004) Acetyl-choline and bradykinin trigger precondi-tioning in the heart through a pathwaythat includes Akt and NOS. Am J PhysiolHeart Circ Physiol 287:H2606–H2611
24. Ma XL, Gao F, Liu GL, Lopez BL, Christo-pher TA, Fukuto JM et al. (1999) Oppositeeffects of nitric oxide and nitroxyl onpostischemic myocardial injury. ProcNatl Acad Sci USA 96:14617–14622
25. Okawa H, Horimoto H, Mieno S, NomuraY, Yoshida M, Shinjiro S (2003) Preis-chemic infusion of alpha-human atrialnatriuretic peptide elicits myoprotectiveeffects against ischemia reperfusion inisolated rat hearts. Mol Cell Biochem248:171–177
26. Pabla R, Buda AJ, Flynn DM, Blesse SA,Shin AM, Curtis MJ et al. (1996) Nitricoxide attenuates neutrophil-mediatedmyocardial contractile dysfunction afterischemia and reperfusion. Circ Res78:65–72
27. Pagliaro P, Mancardi D, Rastaldo R,Penna C, Gattullo D, Miranda KM et al.(2003) Nitroxyl affords thiol-sensitivemyocardial protective effects akin to earlypreconditioning Free Radic Biol Med34:33–43
28. Pain T, Yang XM, Critz SD, Yue Y,Nakano A, Liu GS et al. (2000) Opening ofmitochondrial KATP channels triggersthe preconditioned state by generatingfree radicals. Circ Res 87:460–466
29. Patel HH, Gross GJ (2001) Diazoxideinduced cardiprotecion: what comes first,KATP channels or reactive oxygenspecies? Cardiovasc Res 51:633–636
30. Penna C, Alloatti G, Cappello S, GattulloD, Berta G, Mognetti B et. al. (2005a)Platelet-activating factor induces cardio-protection in isolated rat heart akin toischemic preconditioning: role of phos-phoinositide 3-kinase and protein kinaseC activation. Am J Physiol Heart CircPhysiol 288:H2512–H2520
31. Penna C, Cappello S, Mancardi D,Raimondo S, Rastaldo R, Gattullo D et al.(2005b) Post-Conditioning ReducesInfarct Size in the Isolated Rat Heart: Roleof Coronary Flow and Pressure and theNitric Oxide/cGMP Pathway. Basic ResCardiol In Press [DOI: 10.1007/s00395-005-0543-6]
32. Przyklenk K, Kloner RA (1989) “Reperfu-sion injury” by oxygen-derived free radi-cals? Effect of superoxide dismutase pluscatalase, given at the time of reperfusion,on myocardial infarct size, contractilefunction, coronary microvasculature,and regional myocardial blood flow. CircRes 64:86–96
33. Sasaki N, Sato T, Ohler A, O’Rourke B,Marban E (2000) Activation of mitochon-drial ATP-dependent potassium channelsby nitric oxide. Circulation 101:439–445
References
188 Basic Research in Cardiology, Vol. 101, No. 2 (2006)© Steinkopff Verlag 2006

C. Penna et al. 189Post-conditioning and redox-sensitive signaling
34. Sato H, Jordan JE, Zhao ZQ, SarvothamSS, Vinten-Johansen J (1997) Gradualreperfusion reduces infarct size andendothelial injury but augments neu-trophil accumulation. Ann Thorac Surg64:1099–1107
35. Sato T, Marban E (2000) The role of mito-chondrial K(ATP) channels in cardiopro-tection. Basic Res Cardiol 95:285–289
36. Serviddio G, Di Venosa N, Federici A,D’Agostino D, Rollo T, Prigigallo F,Altomare E, Fiore T, Vendemiale G (2005)Brief hypoxia before normoxic reperfu-sion (postconditioning) protects theheart against ischemia-reperfusion injuryby preventing mitochondria peroxideproduction and glutathione depletion.FASEB J 19:354–361
37. Staat P, Rioufol G, Piot C, Cottin Y, CungTT, L’Huillier I, Aupetit JF, Bonnefoy E,Finet G, Andre-Fouet X, Ovize M (2005)Postconditioning the human heart. Cir-culation 112:2143–2148
38. Sun HY, Wang NP, Kerendi F, Halkos M,Kin H, Guyton RA et al. (2005) Hypoxicpostconditioning reduces cardiomyocyteloss by inhibiting ROS generation andintracellular Ca2+ overload. Am J PhysiolHeart Circ Physiol 288:H1900–H1908
39. Tsang A, Hausenloy DJ, Mocanu MM,Yellon DM (2004) Postconditioning: aform of “modified reperfusion” protectsthe myocardium by activating the phos-phatidylinositol 3-kinase-Akt pathway.Circ Res 95:230–232
40. Tsang A, Hausenloy DJ, Yellon DM (2005)Myocardial postconditioning: reperfu-sion injury revisited. Am J Physiol HeartCirc Physiol 289:H2–H7
41. Valen G, Vaage J (2005) Pre- and post-conditioning during cardiac surgery.Basic Res Cardiol 100:179–186
42. Vinten-Johansen J, Zhao ZQ, Zatta AJ,Kin H, Halkos ME, Kerendi F (2005) Post-conditioning A new link in nature’sarmor against myocardial ischemia-reperfusion injury. Basic Res Cardiol100:295–310
43. Wang Y, Takashi E, Xu M, Ayub A, AshrafM (2001) Downregulation of proteinkinase C inhibits activation of mitochon-drial K(ATP) channels by diazoxide. Cir-culation 104:85–90
44. Yang XM, Philipp S, Downey JM, CohenMV (2005) Postconditioning’s protectionis not dependent on circulating bloodfactors or cells but involves adenosinereceptors and requires PI3-kinase andguanylyl cyclase activation. Basic ResCardiol 100:57–63
45. Yang XM, Proctor JB, Cui L, Krieg T,Downey JM, Cohen MV (2004) Multiple,brief coronary occlusions during earlyreperfusion protect rabbit hearts by tar-geting cell signaling pathways. J Am CollCardiol 44:1103–1110
46. Zhao ZQ, Corvera JS, Halkos ME, KerendiF, Wang NP, Guyton RA et al. (2003) Inhi-bition of myocardial injury by ischemicpostconditioning during reperfusion:comparison with ischemic precondition-ing. Am J Physiol Heart Circ Physiol285:H579–588
Copyright © 2022 FDOKUMEN




















