
Possible mechanisms responsible for absence of a retrotransposon family on a plant Y chromosome
17
Possible mechanisms responsible for absence of a retrotransposon family on a plant Y chromosome Zdenek Kubat 1,2 , Jitka Zluvova 1 , Ivan Vogel 2 , Viera Kovacova 1 , Tomas Cermak 1 , Radim Cegan 1 , Roman Hobza 1,3 , Boris Vyskot 1 and Eduard Kejnovsky 1,2 1 Department of Plant Developmental Genetics, Institute of Biophysics ASCR, Kralovopolska 135, Brno, 61200, Czech Republic; 2 Laboratory of Genome Dynamics, CEITEC – Central European Institute of Technology, Masaryk University, Kamenice 5, Brno, 62500, Czech Republic; 3 Institute of Experimental Botany, Centre of the Region Han a for Biotechnological and Agricultural Research, Sokolovska 6, Olomouc, 77200, Czech Republic Author for correspondence: Zdenek Kubat Tel: +420 541 517 203 Email: [email protected] Received: 13 September 2013 Accepted: 25 November 2013 New Phytologist (2014) doi: 10.1111/nph.12669 Key words: epigenetics, genome size, long terminal repeat (LTR) retrotransposon, plant sex chromosomes, silencing, Silene latifolia (white campion), small RNA. Summary Some transposable elements (TEs) show extraordinary variance in abundance along sex chromosomes but the mechanisms responsible for this variance are unknown. Here, we stud- ied Ogre long terminal repeat (LTR) retrotransposons in Silene latifolia, a dioecious plant with evolutionarily young heteromorphic sex chromosomes. Ogre elements are ubiquitous in the S. latifolia genome but surprisingly absent on the Y chromosome. Bacterial artificial chromosome (BAC) library analysis and fluorescence in situ hybridization (FISH) were used to determine Ogre structure and chromosomal localization. Next generation sequencing (NGS) data were analysed to assess the transcription level and abundance of small RNAs. Methylation of Ogres was determined by bisulphite sequencing. Phylogenetic analysis was used to determine mobilization time and selection forces acting on Ogre elements. We characterized three Ogre families ubiquitous in the S. latifolia genome. One family is nearly absent on the Y chromosome despite all the families having similar structures and spreading mechanisms. We showed that Ogre retrotransposons evolved before sex chromo- somes appeared but were mobilized after formation of the Y chromosome. Our data suggest that the absence of one Ogre family on the Y chromosome may be caused by 24-nucleotide (24-nt) small RNA-mediated silencing leading to female-specific spreading. Our findings highlight epigenetic silencing mechanisms as potentially crucial factors in sex- specific spreading of some TEs, but other possible mechanisms are also discussed. Introduction Transposable elements (TEs) are ubiquitous in all eukaryotes. In plants, long terminal repeat (LTR) retrotransposons are the most widespread TEs (Finnegan, 1989). These TEs use a ‘copy and paste’ mode of spread and are among the main drivers that increase the size of plant genomes in the evolutionary short term (Kumar & Bennetzen, 1999) leading to extraordinary variations in genome size within even closely related species (Vitte & Bennetzen, 2006). The evolutionary dynamics of TEs alternate between transposition bursts and periods when the mobility of TEs is very low (Naito et al., 2006; Bergman & Bensasson, 2007). Bursts of retrotransposition are well documented in multiple mutant lines of Arabidopsis thaliana (Tsukahara et al., 2009), and demonstrate the stochastic and independent ability of each TE type to be activated. Factors that can account for varia- tion in the stability and activity of TEs include: dependence on the host’s genetic background (Vu & Nuzhdin, 2011), changes in host silencing mechanisms, the ability of TEs to escape host regulation, and the degree of TE self-regulation ( Agren & Wright, 2011). As has been shown in chromoviruses (Gao et al., 2008) and yeast (Boeke & Devine, 1998), TEs usually colonize various chromosomal niches, which is the likely consequence of TEs targeting specific chromosomal regions. Specific distribution of some TE families has been observed on heteromorphic sex chromosomes in plants (Steinemann & Steinemann, 2005). For example, large accumulations of LINE (long interspersed nuclear element) elements on the Y chromosome were observed in hemp (Cannabis sativa) (Sakamoto et al., 2000). In the dioecious plant Silene latifolia, copia retrotransposons showed accumulation on the large Y chromosome compared with other chromosomes, while Ogre and Retand (Tat) retro- transposons which are very abundant in the genome were absent or underrepresented on the Y chromosome (Kejnovsky et al., 2006a; Cermak et al., 2008; Filatov et al., 2009). A simi- lar depletion of Tat and Athila elements on the Y chromosome was observed in Rumex acetosa with the XY1Y2 system (Stefl- ova et al., 2013). Overall, this pattern of absence of some TEs on Y chromosomes contradicts the hypothesis that repetitive elements tend to accumulate on Y chromosomes because of a lack of recombination (Charlesworth, 1991). These patterns of Ó 2014 The Authors New Phytologist Ó 2014 New Phytologist Trust New Phytologist (2014) 1 www.newphytologist.com Research
Transcript of Possible mechanisms responsible for absence of a retrotransposon family on a plant Y chromosome

Possible mechanisms responsible for absence of aretrotransposon family on a plant Y chromosome
Zdenek Kubat1,2, Jitka Zluvova1, Ivan Vogel2, Viera Kovacova1, Tomas Cermak1, Radim Cegan1, Roman Hobza1,3,
Boris Vyskot1 and Eduard Kejnovsky1,2
1Department of Plant Developmental Genetics, Institute of Biophysics ASCR, Kralovopolska 135, Brno, 61200, Czech Republic; 2Laboratory of Genome Dynamics, CEITEC – Central
European Institute of Technology, Masaryk University, Kamenice 5, Brno, 62500, Czech Republic; 3Institute of Experimental Botany, Centre of the Region Han�a for Biotechnological and
Agricultural Research, Sokolovska 6, Olomouc, 77200, Czech Republic
Author for correspondence:Zdenek Kubat
Tel: +420 541 517 203Email: [email protected]
Received: 13 September 2013
Accepted: 25 November 2013
New Phytologist (2014)doi: 10.1111/nph.12669
Key words: epigenetics, genome size, longterminal repeat (LTR) retrotransposon, plantsex chromosomes, silencing, Silene latifolia(white campion), small RNA.
Summary
� Some transposable elements (TEs) show extraordinary variance in abundance along sex
chromosomes but the mechanisms responsible for this variance are unknown. Here, we stud-
iedOgre long terminal repeat (LTR) retrotransposons in Silene latifolia, a dioecious plant with
evolutionarily young heteromorphic sex chromosomes. Ogre elements are ubiquitous in the
S. latifolia genome but surprisingly absent on the Y chromosome.� Bacterial artificial chromosome (BAC) library analysis and fluorescence in situ hybridization
(FISH) were used to determine Ogre structure and chromosomal localization. Next generation
sequencing (NGS) data were analysed to assess the transcription level and abundance of small
RNAs. Methylation of Ogres was determined by bisulphite sequencing. Phylogenetic analysis
was used to determine mobilization time and selection forces acting onOgre elements.� We characterized three Ogre families ubiquitous in the S. latifolia genome. One family is
nearly absent on the Y chromosome despite all the families having similar structures and
spreading mechanisms. We showed that Ogre retrotransposons evolved before sex chromo-
somes appeared but were mobilized after formation of the Y chromosome. Our data suggest
that the absence of one Ogre family on the Y chromosome may be caused by 24-nucleotide
(24-nt) small RNA-mediated silencing leading to female-specific spreading.� Our findings highlight epigenetic silencing mechanisms as potentially crucial factors in sex-
specific spreading of some TEs, but other possible mechanisms are also discussed.
Introduction
Transposable elements (TEs) are ubiquitous in all eukaryotes. Inplants, long terminal repeat (LTR) retrotransposons are the mostwidespread TEs (Finnegan, 1989). These TEs use a ‘copy andpaste’ mode of spread and are among the main drivers thatincrease the size of plant genomes in the evolutionary short term(Kumar & Bennetzen, 1999) leading to extraordinary variationsin genome size within even closely related species (Vitte &Bennetzen, 2006). The evolutionary dynamics of TEs alternatebetween transposition bursts and periods when the mobility ofTEs is very low (Naito et al., 2006; Bergman & Bensasson,2007). Bursts of retrotransposition are well documented inmultiple mutant lines of Arabidopsis thaliana (Tsukahara et al.,2009), and demonstrate the stochastic and independent ability ofeach TE type to be activated. Factors that can account for varia-tion in the stability and activity of TEs include: dependence onthe host’s genetic background (Vu & Nuzhdin, 2011), changesin host silencing mechanisms, the ability of TEs to escape hostregulation, and the degree of TE self-regulation (�Agren &Wright, 2011).
As has been shown in chromoviruses (Gao et al., 2008) andyeast (Boeke & Devine, 1998), TEs usually colonize variouschromosomal niches, which is the likely consequence of TEstargeting specific chromosomal regions. Specific distribution ofsome TE families has been observed on heteromorphic sexchromosomes in plants (Steinemann & Steinemann, 2005).For example, large accumulations of LINE (long interspersednuclear element) elements on the Y chromosome wereobserved in hemp (Cannabis sativa) (Sakamoto et al., 2000). Inthe dioecious plant Silene latifolia, copia retrotransposonsshowed accumulation on the large Y chromosome comparedwith other chromosomes, while Ogre and Retand (Tat) retro-transposons which are very abundant in the genome wereabsent or underrepresented on the Y chromosome (Kejnovskyet al., 2006a; Cermak et al., 2008; Filatov et al., 2009). A simi-lar depletion of Tat and Athila elements on the Y chromosomewas observed in Rumex acetosa with the XY1Y2 system (Stefl-ova et al., 2013). Overall, this pattern of absence of some TEson Y chromosomes contradicts the hypothesis that repetitiveelements tend to accumulate on Y chromosomes because of alack of recombination (Charlesworth, 1991). These patterns of
� 2014 The Authors
New Phytologist� 2014 New Phytologist TrustNew Phytologist (2014) 1
www.newphytologist.com
Research

TE distribution on the Y chromosome indicate that other fac-tors and mechanisms shaping the genomic landscape of thesechromosomes are in action.
Ogre elements represent a widespread family of giant (up to23 kb long) gypsy-type LTR retrotransposons present in manyplant species (Macas & Neumann, 2007). Their gag/pol geneproduct ratio is probably regulated by splicing of the primarytranscript, as has been shown in pea (Pisum sativum) andMedicago truncatula (Neumann et al., 2003; Steinbauerov�a et al.,2008). Another interesting feature of Ogre retrotransposons is thepresence of relatively well-conserved extra open reading frames(eORFs) of unknown function downstream or upstream of theirgag–pol genes (Neumann et al., 2003; Steinbauerov�a et al., 2012).In the dioecious plant species S. latifolia, Ogre elements accountfor c. 13% of the genome and significantly enlarged its size(Macas et al., 2011; Cegan et al., 2012). Similar levels of Ogre ele-ment amplification have also been observed in Vicia panonica(Neumann et al., 2006). There are numerous other cases whereamplification of mobile elements has significantly enlarged thegenomes of one species compared with the genomes of its rela-tives (Hawkins et al., 2006; Piegu et al., 2006; Ungerer et al.,2006).
Several mechanisms could explain the absence of Ogre ele-ments on the Y chromosome and are examined here. First, Ogreelements could have expanded in the genome before the origin ofsex chromosomes and remained inactive during the evolution ofsex chromosomes. Secondly, there may be a mechanism for Ogreremoval from the Y chromosome. Thirdly, because they colonizeonly recombining parts of the genome, the spread of Ogre ele-ments could be connected in some way with the recombinationprocess. Fourthly, interaction with some female-specific cellularproteins could be crucial for Ogre retrotransposition. Lastly, Ogrecould be active only in female individuals because of eitherfemale-specific activation or male-specific silencing.
Materials and Methods
All plant species of the genus Silene used in this study came fromthe collection at the Institute of Biophysics in Brno, CzechRepublic. Full-length Ogre elements have been isolated from thebacterial artificial chromosome (BAC) library characterized byCegan et al. (2010). Ogre element copy numbers in the S. latifoliaPoir. genome were estimated by hybridization of respective LTRprobes with the BAC library and by counting reads in the geno-mic DNA 454 and Illumina libraries. To examine the transcrip-tion, splicing and diversity of Ogre elements, total RNA andDNA were isolated from male and female leaves and flower budsand used as a template for amplification reactions. To prevent theformation of chimeric PCR products, we used an emulsion PCRprotocol (Williams et al., 2006). The mapping of transcriptionstarts and ends was performed using the SMARTer RACE cDNAamplification kit (Clontech, Mountain View, CA, USA). Fluo-rescence in situ hybridization experiments were performedaccording to Lengerova et al. (2004) with slight modifications. Insitu detection of Ogre mRNAs in cryosections of S. latifoliaanthers and pistils was perfomed according to Brewer et al.
(2006). RNA-seq and small RNA-seq Illumina sequencedlibraries were prepared from purified pollen grains, pistils andyoung male and female leaves of the same S. latifolia populationfrom Brno Bystrc, Czech Republic. Transcript level and smallRNA abundance were estimated by counting reads and normal-izations as stated in the Supporting Information Methods S1.The methylation level of Ogre elements was examined by bisulph-ite sequencing of Ogre genomic copies isolated from pollengrains, flower buds and leaves; primers were designed usingBisprimer (Kovacova & Janousek, 2012). To distinguish Ogrecopies from vegetative cells and sperm cells of the pollen grain,hierarchical cluster analysis was performed. Ancestral state recon-structions, mobilization times and selection analyses of Ogreelements were performed by phylogenetic analyses as described indetail in Methods S1. Primers used in this study are summarizedin Table S1. All methods are described in detail in Methods S1.
Results
Structure and chromosomal localization ofOgre families inS. latifolia
To examine the structure of Ogre elements, we utilized sequencesobtained from similarity-based clustering of 454 reads ofS. latifolia genomic DNA (Macas et al., 2011). We identifiedthree abundant Ogre families represented by three 454 clusters:CL5, CL6 and CL11. Reconstructed elements contained gag andpol genes as well as eORFs typical for Ogre LTR retrotransposons(Steinbauerov�a et al., 2012). Ogre CL5 was identical to the Ogreidentified by Cermak et al. (2008) and SlOgre1 partially charac-terized by Filatov et al. (2009), while Ogre CL6 and Ogre CL11were newly identified in this study. To obtain full-length Ogresequences, we screened the BAC library of S. latifolia. In 32 BACclones, we found four full-length or nearly complete Ogre ele-ments (accession numbers KC206272–KC206275; Table 1), and83 partial Ogre sequences.
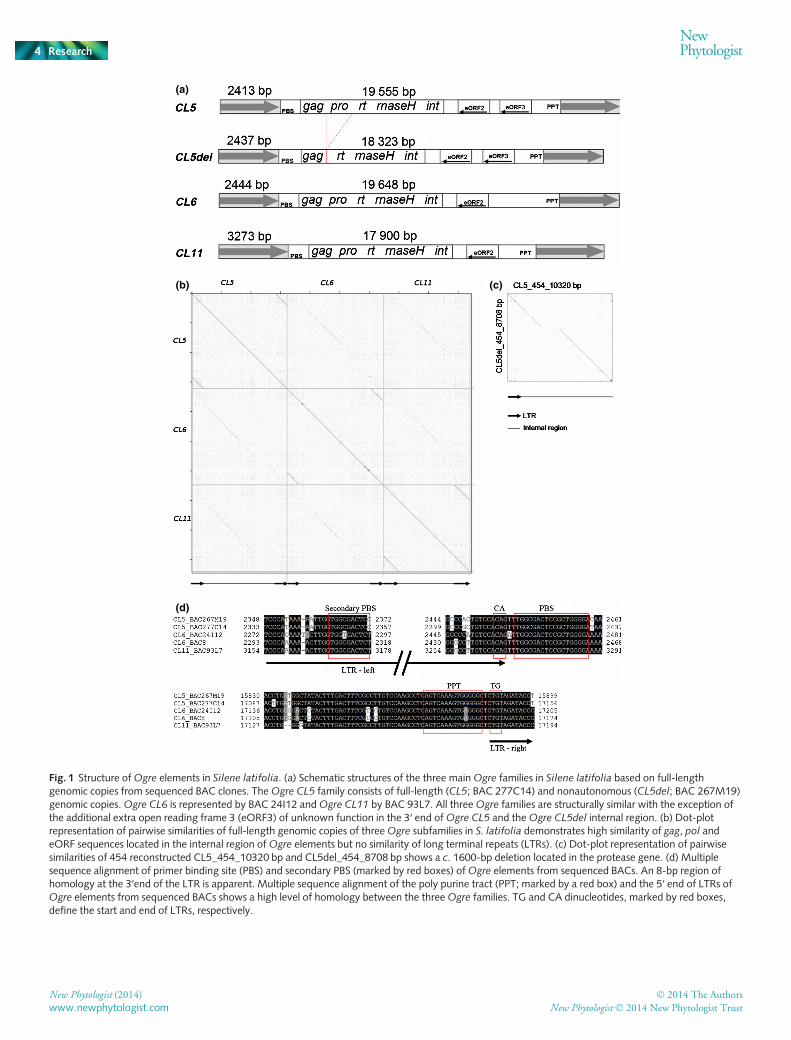
The structure of typical elements of the three Ogre families(Ogre CL5, Ogre CL6 and Ogre CL11), including nonautono-mous Ogre CL5del, is depicted in Fig. 1(a). Despite similarlengths and high structural similarity, the three Ogre families dif-fered significantly in their sequences. Pairwise homology was70% in gag–pol genes, but much lower in other parts of the ele-ments (Fig. 1b). The nonautonomous Ogre CL5del lacked theprotease domain (Fig. 1c) and Ogre CL5 had two eORFs withopposite orientations, while Ogre CL6 and Ogre CL11 had onlyone eORF. These eORFs showed protein similarity to ORF1described in pea Ogre elements, but their function is stillenigmatic (Neumann et al., 2003; Macas & Neumann, 2007;Steinbauerov�a et al., 2012). Short regions (c. 8–10 bp) at the 3′and 5′ ends of the LTRs of all studied Ogre elements exhibitedhigh conservation (Fig. 1d). These sites are known to bind integr-ase before element integration (Hindmarsh & Leis, 1999;Cherepanov et al., 2011), which suggests that all three Ogre fami-lies utilize the same integration mechanism. Poly purine tracts(PPTs) within a 70-bp conserved region were also the same forall Ogre families.
New Phytologist (2014) � 2014 The Authors
New Phytologist� 2014 New Phytologist Trustwww.newphytologist.com
Research
NewPhytologist2

A striking feature of all three Ogre families was the presence ofa fully conserved secondary primer binding site (PBS) located80–90 bp upstream of the primary PBS inside the left LTR(Fig. 1d). Both PBS sites were complementary to the 3′ end oftRNA-Arg, which is typical for Ogre elements in other plant spe-cies (Neumann et al., 2003; Macas & Neumann, 2007). Thehigh conservation of the secondary PBS indicates its functionalimportance, possibly as an alternative site for reverse transcriptioninitiation.
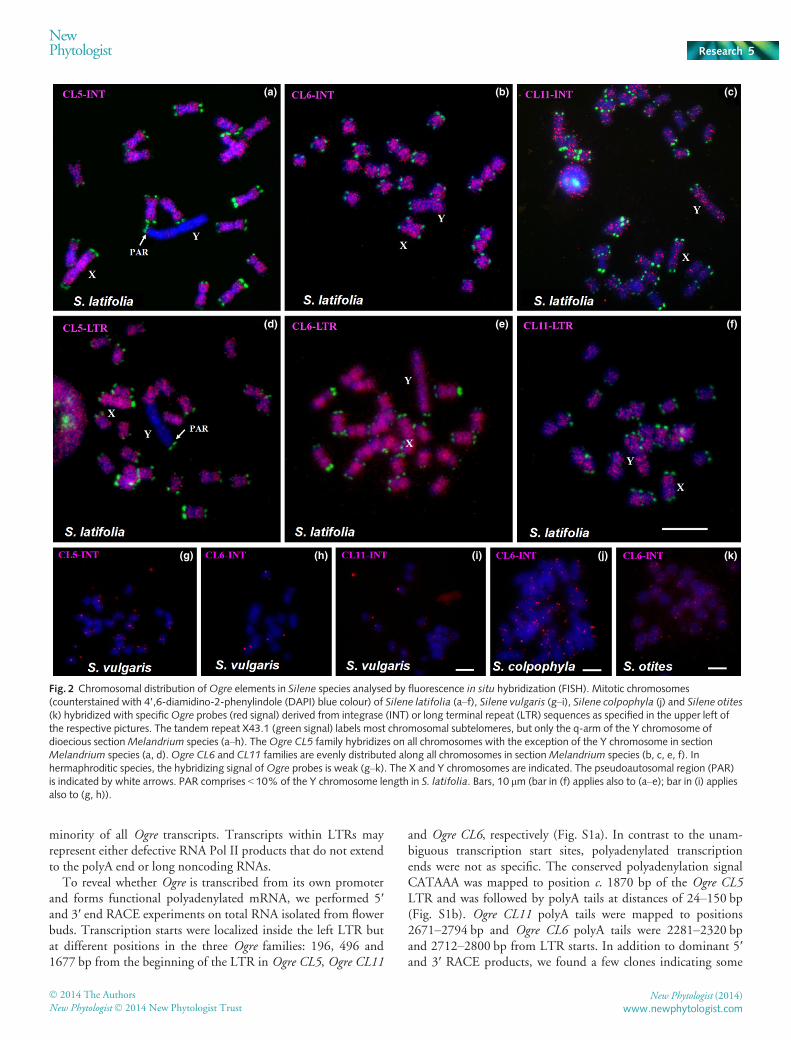
The chromosomal distribution of Ogre CL5 has been studiedin S. latifolia, Silene diclinis and Silene dioica by Cermak et al.(2008), Filatov et al. (2009) and Howell et al. (2009) using theinternal region of the elements. In all cases, Ogre CL5 has beenfound to be very abundant in the whole genome, with the excep-tion of the nonrecombinig part of the Y chromosome. Here, wealso used integrase and LTR regions as probes for fluorescence insitu hybridization (FISH) and mapped not only Ogre CL5, butalso Ogre CL6 and CL11 (Fig. 2). As expected, Ogre CL5 wasubiquitously localized with the exception of the Y chromosomebut, surprisingly, Ogre CL6 and CL11 were localized on all chro-mosomes including the Y chromosome. We used degenerateprimers to amplify Ogre elements on genomic DNA ofS. vulgaris, Silene colpohylla and Silene otites and used them asFISH probes on chromosomes of these Silene species (Table S1c).We found that the copy number of Ogre elements in these specieswas much lower, indicating specific and extensive amplificationof Ogre elements (CL5, CL6 and CL11) in the ancestor ofMelandrium section. The phylogenetic relationships within thegenus Silene are explained in Fig. 3. Copy numbers of Ogre ele-ments in the S. latifolia genome as estimated by hybridization ofLTR probes with the BAC library are 4990, 4220 and 2880 forOgre CL5, CL6 and CL11, respectively. Similar results wereobtained by counting reads in the Illumina and 454 genomiclibraries (Table S2).
In order to distinguish possible differences in the Ogre neigh-bourhood (heterochromatin or euchromatin), we analysed flank-ing sequences in Illumina genomic reads mapped onto Ogreelements. We found no differences in GC content or nucleotidecomposition (data not shown), suggesting that Ogre elements donot prefer any specific chromosomal loci (sequence context) forinsertion. This result is consistent with the regular spatial distri-bution of Ogre elements along chromosomes.
Ogre elements are transcribed and spliced
We studied the transcription of Ogre elements by RT-PCR usingtotal RNA from roots, leaves and various floral tissues, includingwhole male and female flower buds and separated pistils and pol-len grains, as templates. Primers were designed on the basis of454 reconstructed elements (Table S1a,b). We found that allthree Ogre families were constitutively transcribed in all tested tis-sues (data not shown). Further, we determined the transcript levelby counting RNA-seq reads mapped to Ogre LTRs and GAGdomains (Fig. 4a,b) in flower buds, leaves, pistils and pollen. Thetranscript level was much higher in LTRs than in GAG domains.This suggests that potential full-length Ogre transcripts form aT
able1Gen
omiccopiesofOgre
elem
entsin
Silenelatifolia
BACnam
eCluster
Complete?
TElength
(bp)
LTR–left(bp)
LTR–right(bp)
TSD
(left/right)
LTRsimilarity,%
PBS
SecondaryPBS
Note
277C14
CL5
Yes
19555
2413
2412
TAATC/TAATC
99
tRNA-A
rgYes
267M19
CL5del
Yes
18323
2076
2437
GAACT/G
AACT
98
tRNA-A
rgYes
LeftLT
Rdeletion(1235–1
589)
78D8
CL5
No
n/a
n/a
n/a
n/a
n/a
tRNA-A
rgYes
241I12
CL6
Yes
19648
2444
2457
ATTAC/A
TTAC
98
tRNA-A
rgYes
93L7
CL11
No
c.17900
3267(cons)
3267(cons)
n/a
n/a
tRNA-A
rgYes
TE,
tran
sposableelem
ent;LT
R,longterm
inalrepeat;TSD
,target
site
duplication;PBS,
primer
bindingsite;cons,consensusbased
ontw
oincomplete
leftan
drightLT
Rs.So
meinform
ationisnot
available(n/a).
� 2014 The Authors
New Phytologist� 2014 New Phytologist TrustNew Phytologist (2014)
www.newphytologist.com
NewPhytologist Research 3
(a)
(b)
(d)
(c)
Fig. 1 Structure ofOgre elements in Silene latifolia. (a) Schematic structures of the three mainOgre families in Silene latifolia based on full-lengthgenomic copies from sequenced BAC clones. TheOgre CL5 family consists of full-length (CL5; BAC 277C14) and nonautonomous (CL5del; BAC 267M19)genomic copies.Ogre CL6 is represented by BAC 24I12 andOgre CL11 by BAC 93L7. All threeOgre families are structurally similar with the exception ofthe additional extra open reading frame 3 (eORF3) of unknown function in the 3′ end ofOgre CL5 and theOgre CL5del internal region. (b) Dot-plotrepresentation of pairwise similarities of full-length genomic copies of threeOgre subfamilies in S. latifolia demonstrates high similarity of gag, pol andeORF sequences located in the internal region ofOgre elements but no similarity of long terminal repeats (LTRs). (c) Dot-plot representation of pairwisesimilarities of 454 reconstructed CL5_454_10320 bp and CL5del_454_8708 bp shows a c. 1600-bp deletion located in the protease gene. (d) Multiplesequence alignment of primer binding site (PBS) and secondary PBS (marked by red boxes) ofOgre elements from sequenced BACs. An 8-bp region ofhomology at the 3′end of the LTR is apparent. Multiple sequence alignment of the poly purine tract (PPT; marked by a red box) and the 5′ end of LTRs ofOgre elements from sequenced BACs shows a high level of homology between the threeOgre families. TG and CA dinucleotides, marked by red boxes,define the start and end of LTRs, respectively.
New Phytologist (2014) � 2014 The Authors
New Phytologist� 2014 New Phytologist Trustwww.newphytologist.com
Research
NewPhytologist4
minority of all Ogre transcripts. Transcripts within LTRs mayrepresent either defective RNA Pol II products that do not extendto the polyA end or long noncoding RNAs.
To reveal whether Ogre is transcribed from its own promoterand forms functional polyadenylated mRNA, we performed 5′and 3′ end RACE experiments on total RNA isolated from flowerbuds. Transcription starts were localized inside the left LTR butat different positions in the three Ogre families: 196, 496 and1677 bp from the beginning of the LTR in Ogre CL5, Ogre CL11
and Ogre CL6, respectively (Fig. S1a). In contrast to the unam-biguous transcription start sites, polyadenylated transcriptionends were not as specific. The conserved polyadenylation signalCATAAA was mapped to position c. 1870 bp of the Ogre CL5LTR and was followed by polyA tails at distances of 24–150 bp(Fig. S1b). Ogre CL11 polyA tails were mapped to positions2671–2794 bp and Ogre CL6 polyA tails were 2281–2320 bpand 2712–2800 bp from LTR starts. In addition to dominant 5′and 3′ RACE products, we found a few clones indicating some
(a) (b) (c)
(d)
(g) (h) (i) (j) (k)
(e) (f)
Fig. 2 Chromosomal distribution ofOgre elements in Silene species analysed by fluorescence in situ hybridization (FISH). Mitotic chromosomes(counterstained with 4’,6-diamidino-2-phenylindole (DAPI) blue colour) of Silene latifolia (a–f), Silene vulgaris (g–i), Silene colpophyla (j) and Silene otites(k) hybridized with specificOgre probes (red signal) derived from integrase (INT) or long terminal repeat (LTR) sequences as specified in the upper left ofthe respective pictures. The tandem repeat X43.1 (green signal) labels most chromosomal subtelomeres, but only the q-arm of the Y chromosome ofdioecious sectionMelandrium species (a–h). TheOgre CL5 family hybridizes on all chromosomes with the exception of the Y chromosome in sectionMelandrium species (a, d).Ogre CL6 and CL11 families are evenly distributed along all chromosomes in sectionMelandrium species (b, c, e, f). Inhermaphroditic species, the hybridizing signal ofOgre probes is weak (g–k). The X and Y chromosomes are indicated. The pseudoautosomal region (PAR)is indicated by white arrows. PAR comprises < 10% of the Y chromosome length in S. latifolia. Bars, 10 lm (bar in (f) applies also to (a–e); bar in (i) appliesalso to (g, h)).
� 2014 The Authors
New Phytologist� 2014 New Phytologist TrustNew Phytologist (2014)
www.newphytologist.com
NewPhytologist Research 5

minor alternative transcription starts and ends. Taken together,the RACE experiments showed that all Ogre families are drivenby their own promoters localized within their LTRs and thatOgre elements produce full-length polyadenylated mRNA tran-scripts.
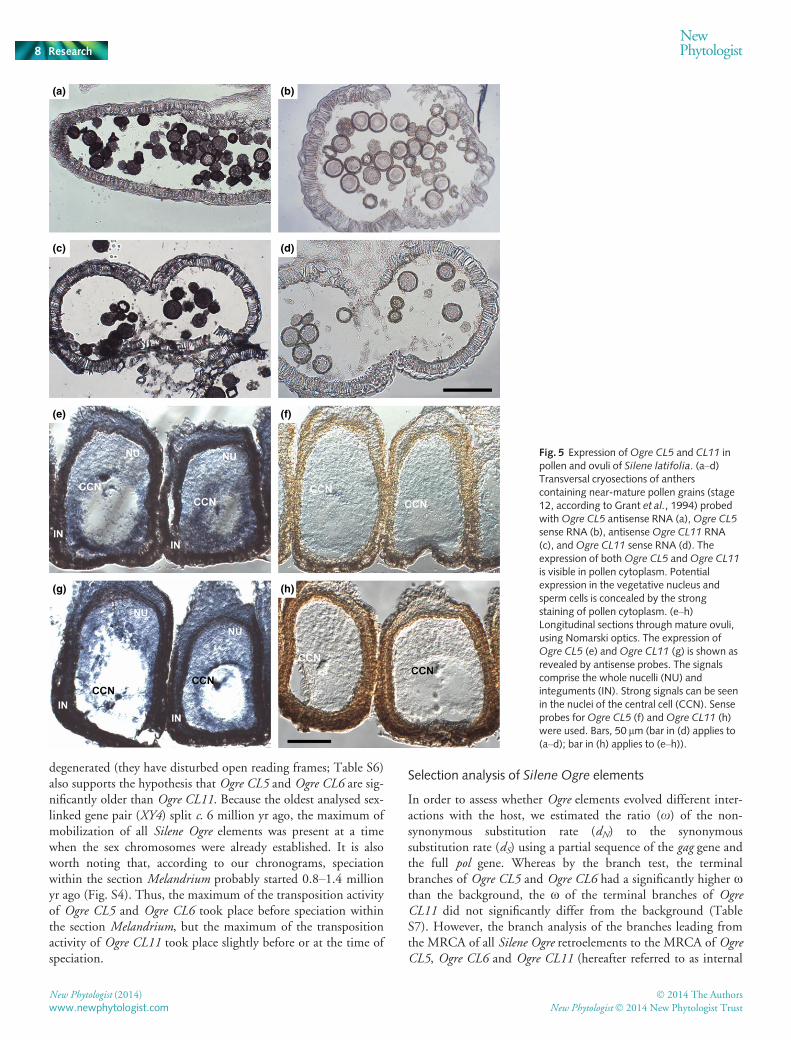
The presence of Ogre transcripts prompted us to perform insitu hybridization of Ogre-derived probes on cryosections ofanthers and ovaries of S. latifolia. As shown in Fig. 5, expressionof Ogre CL5 and Ogre CL11 was visible in pollen (Fig. 5a,c) aswell as in cells inside female ovules: integuments, the nucellusand the diploid secondary nucleus of the central cell (Fig. 5e,g).The lack of hybridization signal with sense probes indicates no ora very low level of cotranscription of Ogre elements (Fig. 5b,d,f,h). This supports the previous conclusion that Ogre elements areactively transcribed from their own promoters.
In 454 data, we found a variant of Ogre CL5 with the proteasedomain deleted: Ogre CL5del (Fig. 1a). PCR with primers bridg-ing the potential intron showed that the short (spliced, 627-bp-long) fragment dominated in RNA, while longer (unspliced,2227-bp-long) fragments prevailed in genomic DNA (Fig. S1a,b). These fragments were cloned and sequenced. Multiplesequence alignment revealed specific splicing sites bordering theprotease gene, indicating that autonomous Ogre CL5 transcripts
can be spliced (Fig. S1c). Clones containing Ogre CL5del werehighly divergent in the 100-bp region around splicing sites. Forthis reason, Ogre CL5del is probably an ancient spliced copy,which is nonautonomous but active. We counted the autono-mous Ogre CL5, its spliced variant and nonautonomous OgreCL5del in Illumina genomic data sets and discovered that theymake up 83.5, 3.5, and 13% of genomic copies, respectively.Open reading frames in the majority of nonautonomous andspliced Ogre elements remained intact, which implies that trun-cated copies can produce polyproteins containing all proteinsexcept protease.
Branching order of Silene Ogre retrotransposon groups
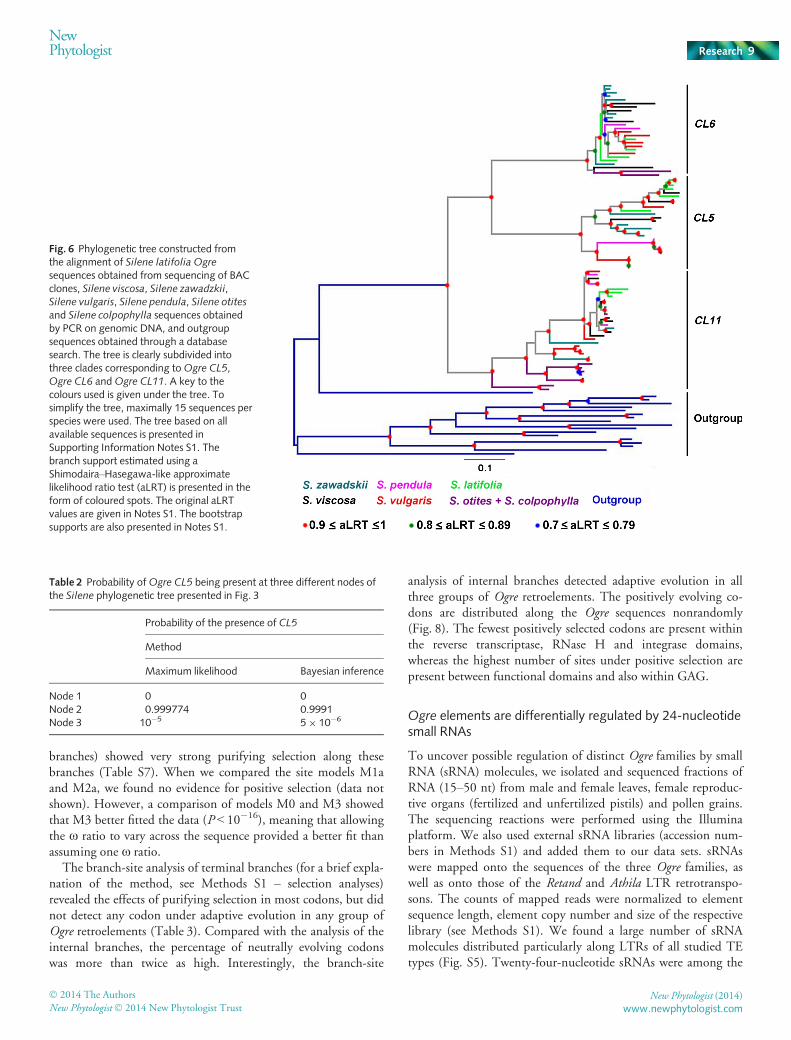
To assess the pattern of Ogre evolution in the genus Silene, weconstructed phylogenetic trees from the alignment of Silene Ogreelements as the ingroup and several Ogre elements from distantlyrelated species as the outgroup (alignment A and translated align-ment A). Fig. 6 and Notes S1 show the results of the maximumlikelihood analysis of the nucleotide data sets. The phylogenetictree clearly shows that the Silene Ogre retrotransposons are subdi-vided into three families that correspond to Ogre CL5, Ogre CL6and Ogre CL11. The Shimodaira–Hasegawa-like approximatelikelihood ratio test (aLRT; Anisimova & Gascuel, 2006) valuesthat support the grouping of each family of Silene Ogre retro-transposons were within the range 0.96–1.00 (Notes S1), andthus the subdivision into the three families is strongly supported.Apparently, the branch leading to Ogre CL11 first evolved from ahypothetical ancestor, and the sequences Ogre CL5 and Ogre CL6evolved later. Interestingly, S. otites and S. colpophylla, representa-tives of the subgenus Silene, contain only Ogre CL6 and OgreCL11 elements, whereas species of the subgenus Behenantha con-tain all three Ogre families (Fig. 6, Notes S1). The results of phy-logenetic analyses performed by Bayesian inference and on thetranslated alignment confirm the results obtained by the maxi-mum likelihood approach (Notes S1, S2).
Approximate timing of the Silene Ogre emergence
To assess the timing of the evolution of all three groups of SileneOgre retrotransposons, we constructed a phylogram based onsequences of four nuclear genes (fructose-2,4-bisphosphatase(Atanassov et al., 2001), spermidine synthase (Atanassov et al.,2001), CCLS1 (Barbacar et al., 1997; Zluvova et al., 2010), andeIF4A (Zluvova et al., 2005, 2006); Fig. 3, Table S3). The phylo-gram was used as an input in the ancestral state reconstruction tocalculate the probability of the presence of Ogre CL5 at threeinternal nodes: (1) the node corresponding to the ancestor of theSilene subgenus Silene, (2) the node corresponding to the ancestorof the sections Melandrium, Conoimorpha, Behenantha andPhysolychnis within the Silene subgenus Behenantha, and (3) thenode corresponding to the ancestor of the genus Silene. Theresults are summarized in Table 2. The results of both maximumlikelihood and Bayesian inference show that the ancestor of thesubgenus Silene probably did not contain Ogre CL5, whereasOgre CL5 was present in the ancestor of the sectionsMelandrium,
Fig. 3 Phylogram used for ancestral state reconstruction. The genus Sileneis comprised of c. 700 species, and is further divided into two subgenera,Silene and Behenantha, of approximately equal size. Silene subgenusSilene includes the species Silene colpophylla, Silene otites, andSilene saxifraga. The subgenus Behenantha includes the sectionMelandrium which comprises Silene latifolia, and other dioecious species(e. g. Silene dioica and Silene diclinis). The close relatives of the sectionMelandrium are the section Conoimorpha, with the species Silene conica,the section Physolychnis, with the species Silene zawadzkii andSilene viscosa, and the section Behenantha, with the speciesSilene vulgaris and Silene pendula. The species of the current sectionMelandrium were previously part of the section Elisanthewhich alsoincluded the species Silene noctiflora. Petrocoptis pyrenaicawas used asan outgroup. Values show bootstrap support/Bayesian posteriorprobability values for the corresponding clade; bootstrap values above 60and posterior probabilities above 0.9 are shown. The arrows point to thenodes used in the ancestral state reconstruction.
New Phytologist (2014) � 2014 The Authors
New Phytologist� 2014 New Phytologist Trustwww.newphytologist.com
Research
NewPhytologist6

Conoimorpha, Behenantha and Physolychnis. The probability thatOgre CL5 was present in the ancestor of the genus Silene is 10�5
(as calculated by maximum likelihood) or even less (as calculatedby Bayesian inference). Thus, the presence of Ogre CL5 in theancestor of the genus Silene is unlikely. We used the chronogram(prepared from the same nucleotide data set as the phylogram) toassess the approximate timing of the emergence of the Silene Ogrefamilies (Fig. S2). According to our results, both Ogre CL6 andOgre CL11 probably appeared before the split of the subgeneraSilene and Behenantha and thus were probably present at the ori-gin of the genus Silene, that is, they are older than 17 million yr.Ogre CL5 appeared between the emergence of the most recentcommon ancestor (MRCA) of the genus Silene and the MRCAof the sectionsMelandrium, Conoimorpha and Physolychnis withinthe subgenus Behenantha. Thus, Ogre CL5 probably emergedbetween 13.4 and 17 million yr ago.
Estimation ofOgremobilization time
To determine the expansion period of Ogre elements in the Silenegenome, we used two independent approaches to calculate thelength of the terminal branches within the Ogre tree: the termi-nal branch lengths in the chronogram calculated in BEAST(Drummond & Rambaut, 2007) and the length of terminal
branches for synonymous substitutions calculated in PAML(Yang, 2007). BEAST analysis suggested that Ogre CL5 activelyexpanded through the genome c. 1.75 million yr ago, Ogre CL6c. 3 million yr ago, and Ogre CL11 c. 1.5 million yr ago (Fig. 7a).The Mann–Whitney test showed that the difference betweenCL5 and CL6 was not significant (Table S4).
The distribution of branch lengths for synonymoussubstitutions (calculated in PAML) in Ogre CL5 was found topeak at c. 7.5% of synonymous nucleotide changes per codon, inOgre CL6 at c. 6%, and in Ogre CL11 at c. 3% (Fig. 7b). The dif-ference between Ogre CL5 and Ogre CL6 was not statistically sig-nificant (Table S4). To convert the percentage of synonymoussubstitutions to time, we estimated the relative time of mobiliza-tion maxima using four chronograms of sex-linked genes (XY4,Atanassov et al., 2001; DD44, Moore et al., 2003; Cyp, Bergeroet al., 2007; XY1, Delich�ere et al., 1999; Rautenberg et al., 2008).The chronograms are presented in Fig. S3, and the results aresummarized in Table S5 and Fig. S4. The predicted mobilizationmaxima are c. 2.7 million yr ago in the case of Ogre CL5, c. 2.2million yr ago in the case of Ogre CL6, and c. 1.1 million yr agoin the case of Ogre CL11.
Thus, Ogre CL5 and Ogre CL6 were massively mobilized first,followed by Ogre CL11. The fact that most of the analysed OgreCL5 and Ogre CL6, but not Ogre CL11 BAC clones are already
(a)
(b)Fig. 4 Transcriptional activity ofOgre
elements in Silene latifolia. (a) Transcriptlevel inOgre long terminal repeats (LTRs). (b)Transcript level in GAG domains. Transcriptlevel is significantly higher in LTRs than in theinternal region (GAG). Transcript levels areestimated from Illumina RNA-seq reads thatwere mapped to LTR sequences of genomiccopies from BACs (Table 1) as described inSupporting Information Methods S1.Transcript levels are normalized to reflect thelibrary size, transposable element (TE) copynumber and the reference sequence length.The column height represents the mean.Error bars, � SD of three to four biologicalreplicates (see Methods S1). Green, leaves;pink, pistils; orange, pollen; blue, male flowerbuds; red, female flower buds.
� 2014 The Authors
New Phytologist� 2014 New Phytologist TrustNew Phytologist (2014)
www.newphytologist.com
NewPhytologist Research 7
degenerated (they have disturbed open reading frames; Table S6)also supports the hypothesis that Ogre CL5 and Ogre CL6 are sig-nificantly older than Ogre CL11. Because the oldest analysed sex-linked gene pair (XY4) split c. 6 million yr ago, the maximum ofmobilization of all Silene Ogre elements was present at a timewhen the sex chromosomes were already established. It is alsoworth noting that, according to our chronograms, speciationwithin the section Melandrium probably started 0.8–1.4 millionyr ago (Fig. S4). Thus, the maximum of the transposition activityof Ogre CL5 and Ogre CL6 took place before speciation withinthe section Melandrium, but the maximum of the transpositionactivity of Ogre CL11 took place slightly before or at the time ofspeciation.
Selection analysis of Silene Ogre elements
In order to assess whether Ogre elements evolved different inter-actions with the host, we estimated the ratio (x) of the non-synonymous substitution rate (dN) to the synonymoussubstitution rate (dS) using a partial sequence of the gag gene andthe full pol gene. Whereas by the branch test, the terminalbranches of Ogre CL5 and Ogre CL6 had a significantly higher xthan the background, the x of the terminal branches of OgreCL11 did not significantly differ from the background (TableS7). However, the branch analysis of the branches leading fromthe MRCA of all Silene Ogre retroelements to the MRCA of OgreCL5, Ogre CL6 and Ogre CL11 (hereafter referred to as internal
(a) (b)
(c) (d)
(e) (f)
(g) (h)
Fig. 5 Expression ofOgre CL5 and CL11 inpollen and ovuli of Silene latifolia. (a–d)Transversal cryosections of antherscontaining near-mature pollen grains (stage12, according to Grant et al., 1994) probedwithOgre CL5 antisense RNA (a),Ogre CL5sense RNA (b), antisenseOgre CL11 RNA(c), andOgre CL11 sense RNA (d). Theexpression of bothOgre CL5 andOgre CL11
is visible in pollen cytoplasm. Potentialexpression in the vegetative nucleus andsperm cells is concealed by the strongstaining of pollen cytoplasm. (e–h)Longitudinal sections through mature ovuli,using Nomarski optics. The expression ofOgre CL5 (e) andOgre CL11 (g) is shown asrevealed by antisense probes. The signalscomprise the whole nucelli (NU) andinteguments (IN). Strong signals can be seenin the nuclei of the central cell (CCN). Senseprobes forOgre CL5 (f) andOgre CL11 (h)were used. Bars, 50 lm (bar in (d) applies to(a–d); bar in (h) applies to (e–h)).
New Phytologist (2014) � 2014 The Authors
New Phytologist� 2014 New Phytologist Trustwww.newphytologist.com
Research
NewPhytologist8
branches) showed very strong purifying selection along thesebranches (Table S7). When we compared the site models M1aand M2a, we found no evidence for positive selection (data notshown). However, a comparison of models M0 and M3 showedthat M3 better fitted the data (P < 10�16), meaning that allowingthe x ratio to vary across the sequence provided a better fit thanassuming one x ratio.
The branch-site analysis of terminal branches (for a brief expla-nation of the method, see Methods S1 – selection analyses)revealed the effects of purifying selection in most codons, but didnot detect any codon under adaptive evolution in any group ofOgre retroelements (Table 3). Compared with the analysis of theinternal branches, the percentage of neutrally evolving codonswas more than twice as high. Interestingly, the branch-site
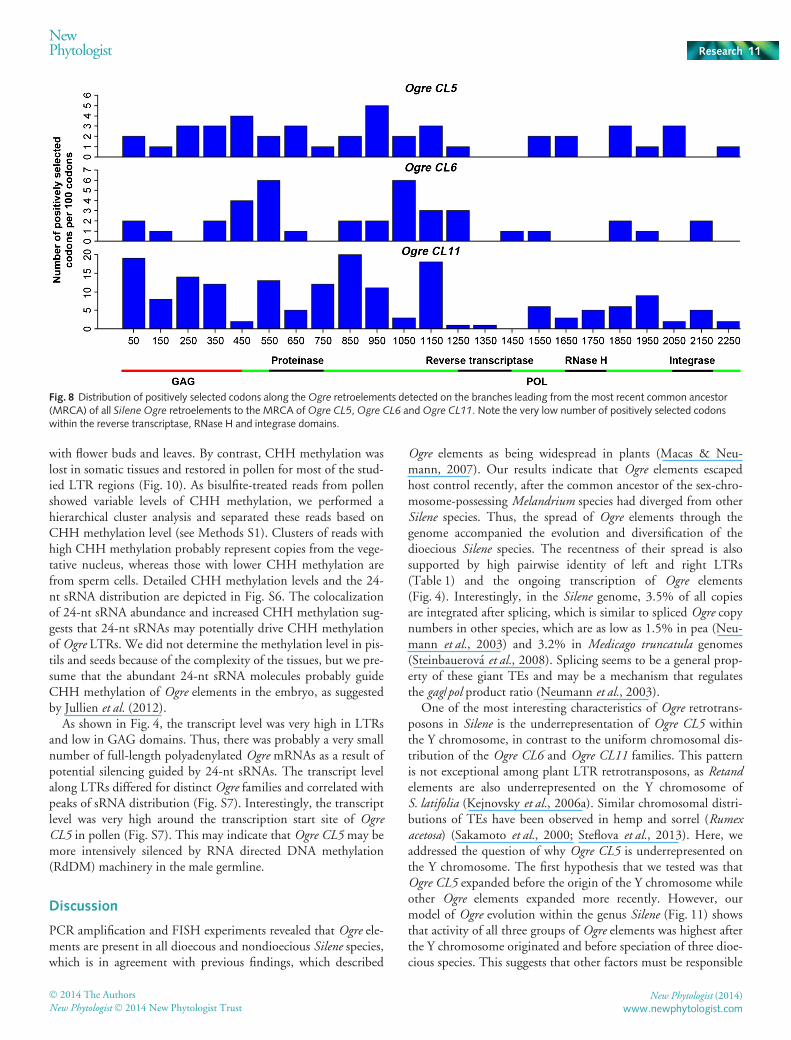
analysis of internal branches detected adaptive evolution in allthree groups of Ogre retroelements. The positively evolving co-dons are distributed along the Ogre sequences nonrandomly(Fig. 8). The fewest positively selected codons are present withinthe reverse transcriptase, RNase H and integrase domains,whereas the highest number of sites under positive selection arepresent between functional domains and also within GAG.
Ogre elements are differentially regulated by 24-nucleotidesmall RNAs
To uncover possible regulation of distinct Ogre families by smallRNA (sRNA) molecules, we isolated and sequenced fractions ofRNA (15–50 nt) from male and female leaves, female reproduc-tive organs (fertilized and unfertilized pistils) and pollen grains.The sequencing reactions were performed using the Illuminaplatform. We also used external sRNA libraries (accession num-bers in Methods S1) and added them to our data sets. sRNAswere mapped onto the sequences of the three Ogre families, aswell as onto those of the Retand and Athila LTR retrotranspo-sons. The counts of mapped reads were normalized to elementsequence length, element copy number and size of the respectivelibrary (see Methods S1). We found a large number of sRNAmolecules distributed particularly along LTRs of all studied TEtypes (Fig. S5). Twenty-four-nucleotide sRNAs were among the
Fig. 6 Phylogenetic tree constructed fromthe alignment of Silene latifolia Ogre
sequences obtained from sequencing of BACclones, Silene viscosa, Silene zawadzkii,Silene vulgaris, Silene pendula, Silene otitesand Silene colpophylla sequences obtainedby PCR on genomic DNA, and outgroupsequences obtained through a databasesearch. The tree is clearly subdivided intothree clades corresponding toOgre CL5,Ogre CL6 andOgre CL11. A key to thecolours used is given under the tree. Tosimplify the tree, maximally 15 sequences perspecies were used. The tree based on allavailable sequences is presented inSupporting Information Notes S1. Thebranch support estimated using aShimodaira–Hasegawa-like approximatelikelihood ratio test (aLRT) is presented in theform of coloured spots. The original aLRTvalues are given in Notes S1. The bootstrapsupports are also presented in Notes S1.
Table 2 Probability ofOgre CL5 being present at three different nodes ofthe Silene phylogenetic tree presented in Fig. 3
Probability of the presence of CL5
Method
Maximum likelihood Bayesian inference
Node 1 0 0Node 2 0.999774 0.9991Node 3 10�5 59 10�6
� 2014 The Authors
New Phytologist� 2014 New Phytologist TrustNew Phytologist (2014)
www.newphytologist.com
NewPhytologist Research 9

most abundant sRNA species. They were ten times more abun-dant than 23-nt, 22-nt and 21-nt sRNAs (Table S8, Fig. 9).Interestingly, Ogre CL5 and Retand expressed c. 10-fold moresRNAs than Ogre CL6, Ogre CL11 and two Athila families(Fig. 9) in all tissues. Thus, the Ogre CL5 and Retand elements,both of which are underrepresented on the Y chromosome, are
probably more intensively regulated by sRNAs than the otherelements.
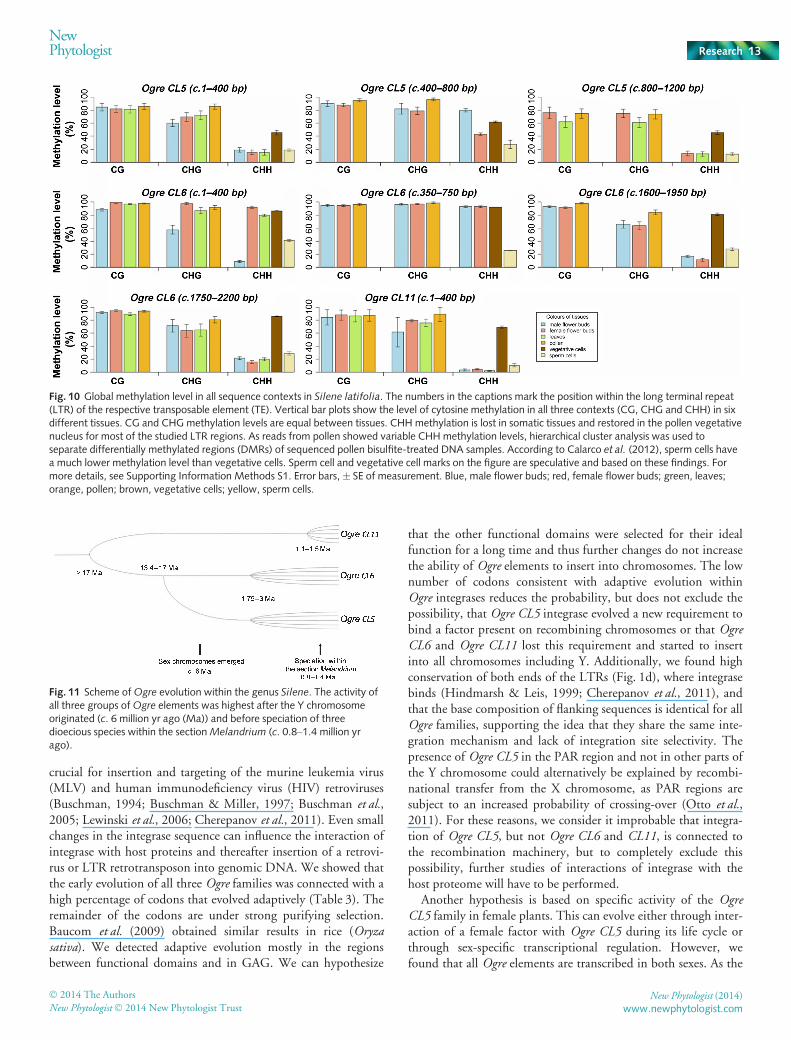
To reveal whether highly abundant 24-nt sRNAs drive themethylation of Ogre elements, we performed bisulphite sequenc-ing of Ogre LTRs. We found that CG and CHG methylation(H = A, T, or C) was largely retained in mature pollen compared
(a)
(b) Fig. 7 Expansion period ofOgre elements inSilene latifolia. (a) Frequency histogram ofterminal branch length for age in millionyears shows thatOgre CL5 activelyexpanded through the genome c. 1.75million yr ago,Ogre CL6 c. 3 million yr ago,andOgre CL11 c. 1.5 million yr ago. (b)Frequency histogram of terminal branchlength for synonymous sites per codon (ds)shows that the distribution of branch lengthsfor synonymous substitution inOgre CL5
was found to peak at c. 7.5% ofsynonymous nucleotide changes per codon,inOgre CL6 at c. 6%, and inOgre CL11 at c.3%. Blue,Ogre CL5; pink,Ogre CL6; green,Ogre CL11.
Table 3 Branch-site analysis of terminal and internal branches ofOgre elements in Silene latifolia
Branch-site analysis of terminal branches
Percentage of codons consistent with
Purifying selection (%) Neutral evolution (%) Adaptive evolution (%)
CL5 81.97 18.03 0CL6 81.41 18.59 0CL11 88.62 11.38 0
Branch-site analysis of internal branches
Percentage of codons consistent with
Purifying selection (%) Neutral evolution (%) Adaptive evolution (%)
CL5 85.80 5.65 8.55 (x = 142.5; P < 10�16)CL6 86.13 5.43 8.44 (x = 119.1; P < 10�16)CL11 81.67 5.24 13.09 (x = 31.9; P < 10�16)
Note that the percentage of codons consistent with neutral evolution is higher in terminal branches, whereas a high percentage of codons consistent withadaptive evolution was detected in the internal branches.
New Phytologist (2014) � 2014 The Authors
New Phytologist� 2014 New Phytologist Trustwww.newphytologist.com
Research
NewPhytologist10
with flower buds and leaves. By contrast, CHH methylation waslost in somatic tissues and restored in pollen for most of the stud-ied LTR regions (Fig. 10). As bisulfite-treated reads from pollenshowed variable levels of CHH methylation, we performed ahierarchical cluster analysis and separated these reads based onCHH methylation level (see Methods S1). Clusters of reads withhigh CHH methylation probably represent copies from the vege-tative nucleus, whereas those with lower CHH methylation arefrom sperm cells. Detailed CHH methylation levels and the 24-nt sRNA distribution are depicted in Fig. S6. The colocalizationof 24-nt sRNA abundance and increased CHH methylation sug-gests that 24-nt sRNAs may potentially drive CHH methylationof Ogre LTRs. We did not determine the methylation level in pis-tils and seeds because of the complexity of the tissues, but we pre-sume that the abundant 24-nt sRNA molecules probably guideCHH methylation of Ogre elements in the embryo, as suggestedby Jullien et al. (2012).
As shown in Fig. 4, the transcript level was very high in LTRsand low in GAG domains. Thus, there was probably a very smallnumber of full-length polyadenylated Ogre mRNAs as a result ofpotential silencing guided by 24-nt sRNAs. The transcript levelalong LTRs differed for distinct Ogre families and correlated withpeaks of sRNA distribution (Fig. S7). Interestingly, the transcriptlevel was very high around the transcription start site of OgreCL5 in pollen (Fig. S7). This may indicate that Ogre CL5 may bemore intensively silenced by RNA directed DNA methylation(RdDM) machinery in the male germline.
Discussion
PCR amplification and FISH experiments revealed that Ogre ele-ments are present in all dioecous and nondioecious Silene species,which is in agreement with previous findings, which described
Ogre elements as being widespread in plants (Macas & Neu-mann, 2007). Our results indicate that Ogre elements escapedhost control recently, after the common ancestor of the sex-chro-mosome-possessing Melandrium species had diverged from otherSilene species. Thus, the spread of Ogre elements through thegenome accompanied the evolution and diversification of thedioecious Silene species. The recentness of their spread is alsosupported by high pairwise identity of left and right LTRs(Table 1) and the ongoing transcription of Ogre elements(Fig. 4). Interestingly, in the Silene genome, 3.5% of all copiesare integrated after splicing, which is similar to spliced Ogre copynumbers in other species, which are as low as 1.5% in pea (Neu-mann et al., 2003) and 3.2% in Medicago truncatula genomes(Steinbauerov�a et al., 2008). Splicing seems to be a general prop-erty of these giant TEs and may be a mechanism that regulatesthe gag/pol product ratio (Neumann et al., 2003).
One of the most interesting characteristics of Ogre retrotrans-posons in Silene is the underrepresentation of Ogre CL5 withinthe Y chromosome, in contrast to the uniform chromosomal dis-tribution of the Ogre CL6 and Ogre CL11 families. This patternis not exceptional among plant LTR retrotransposons, as Retandelements are also underrepresented on the Y chromosome ofS. latifolia (Kejnovsky et al., 2006a). Similar chromosomal distri-butions of TEs have been observed in hemp and sorrel (Rumexacetosa) (Sakamoto et al., 2000; Steflova et al., 2013). Here, weaddressed the question of why Ogre CL5 is underrepresented onthe Y chromosome. The first hypothesis that we tested was thatOgre CL5 expanded before the origin of the Y chromosome whileother Ogre elements expanded more recently. However, ourmodel of Ogre evolution within the genus Silene (Fig. 11) showsthat activity of all three groups of Ogre elements was highest afterthe Y chromosome originated and before speciation of three dioe-cious species. This suggests that other factors must be responsible
Fig. 8 Distribution of positively selected codons along theOgre retroelements detected on the branches leading from the most recent common ancestor(MRCA) of all Silene Ogre retroelements to the MRCA ofOgre CL5,Ogre CL6 andOgre CL11. Note the very low number of positively selected codonswithin the reverse transcriptase, RNase H and integrase domains.
� 2014 The Authors
New Phytologist� 2014 New Phytologist TrustNew Phytologist (2014)
www.newphytologist.com
NewPhytologist Research 11

for the differential chromosomal patterns of distinct Ogre familieson the Y chromosome.
Another hypothesis was that Ogres were removed from thenonrecombining part of the Y chromosome. Rapid DNA losscounterbalances genome expansion in plants (Hawkins et al.,2009). If current transposition of Ogre CL5 is slowed as a resultof silencing and the DNA removal is faster in the Y chromosomethan in others, Ogre CL5 elements would appear underrepre-sented in the Y chromosome. This explanation is in compliancewith DNA loss from Y chromosomes at later evolutionary stages(for a review, see Bachtrog, 2013). However, it is contrary to pre-vious findings that the Y chromosome of S. latifolia accumulatesrepetitive sequences, microsatellites and chloroplast DNA(Hobza et al., 2006; Kejnovsky et al., 2006b; Kubat et al., 2008),and also to the hypothesis that TEs tend to accumulate in Y chro-mosomes as a result of the lack of recombination with X chromo-somes (Charlesworth, 1991; Steinemann & Steinemann, 2005).Importantly, the histogram of terminal branch lengths in thechronogram obtained in BEAST (Fig. 7) shows that the numberof recent insertions is about the same for all Ogre families. Filatovet al. (2009) also reported the age distribution of Ogre insertions
in several Silene species and found that, although the maximumof transposition activity was present several millions of years ago,all species contain recent insertions. Thus, the mechanism remov-ing Ogre CL5 from the Y chromosome would have to be very effi-cient and specific for this TE family. For example, recombinationbetween the left and right LTRs of a single element shouldremove the internal region of the element, whereas solo LTRsshould remain in the genome (Shirasu et al., 2000; Devos et al.,2002). Nevertheless, FISH experiments revealed that the hybrid-izing signal seems to be identical for both LTR and RT domainprobes (Fig. 2). Taken together, these results suggest that loss ofOgre elements on the Y chromosome is not likely to be a suffi-cient explanation.
As Ogre CL5 is present only in the recombining pseudoautoso-mal region (PAR) of the Y chromosome, another hypothesisexplaining this pattern is that Ogre CL5 spread is in some wayconnected with recombination. It is known that integrases ofsome Ty3/gypsy LTR retrotransposons can interact with histonesand direct integration of elements into specific chromatin loca-tions (Gao et al., 2008; Neumann et al., 2011). Similar tetheringmechanisms, where integrase interacts with various factors, are
(a)
(b)
Fig. 9 Abundance of 24-nucleotide (24-nt)(a) and 21-nt (b) small RNAs (sRNAs) inSilene latifolia tissues. Counts of sRNA readscomplementary to LTRs were normalized tothe size of the library, LTR length and copynumber of the respective element. Counts of24-nt sRNAs are c. 10 times as high as countsof 21-nt sRNAs in all tissues. The columnheight represents the mean. Error bars, � SDof two to three biological replicates. (Formore details, see Supporting InformationMethods S1.) Green, leaves; pink, pistils;orange, pollen.
New Phytologist (2014) � 2014 The Authors
New Phytologist� 2014 New Phytologist Trustwww.newphytologist.com
Research
NewPhytologist12
crucial for insertion and targeting of the murine leukemia virus(MLV) and human immunodeficiency virus (HIV) retroviruses(Buschman, 1994; Buschman & Miller, 1997; Buschman et al.,2005; Lewinski et al., 2006; Cherepanov et al., 2011). Even smallchanges in the integrase sequence can influence the interaction ofintegrase with host proteins and thereafter insertion of a retrovi-rus or LTR retrotransposon into genomic DNA. We showed thatthe early evolution of all three Ogre families was connected with ahigh percentage of codons that evolved adaptively (Table 3). Theremainder of the codons are under strong purifying selection.Baucom et al. (2009) obtained similar results in rice (Oryzasativa). We detected adaptive evolution mostly in the regionsbetween functional domains and in GAG. We can hypothesize
that the other functional domains were selected for their idealfunction for a long time and thus further changes do not increasethe ability of Ogre elements to insert into chromosomes. The lownumber of codons consistent with adaptive evolution withinOgre integrases reduces the probability, but does not exclude thepossibility, that Ogre CL5 integrase evolved a new requirement tobind a factor present on recombining chromosomes or that OgreCL6 and Ogre CL11 lost this requirement and started to insertinto all chromosomes including Y. Additionally, we found highconservation of both ends of the LTRs (Fig. 1d), where integrasebinds (Hindmarsh & Leis, 1999; Cherepanov et al., 2011), andthat the base composition of flanking sequences is identical for allOgre families, supporting the idea that they share the same inte-gration mechanism and lack of integration site selectivity. Thepresence of Ogre CL5 in the PAR region and not in other parts ofthe Y chromosome could alternatively be explained by recombi-national transfer from the X chromosome, as PAR regions aresubject to an increased probability of crossing-over (Otto et al.,2011). For these reasons, we consider it improbable that integra-tion of Ogre CL5, but not Ogre CL6 and CL11, is connected tothe recombination machinery, but to completely exclude thispossibility, further studies of interactions of integrase with thehost proteome will have to be performed.
Another hypothesis is based on specific activity of the OgreCL5 family in female plants. This can evolve either through inter-action of a female factor with Ogre CL5 during its life cycle orthrough sex-specific transcriptional regulation. However, wefound that all Ogre elements are transcribed in both sexes. As the
Fig. 10 Global methylation level in all sequence contexts in Silene latifolia. The numbers in the captions mark the position within the long terminal repeat(LTR) of the respective transposable element (TE). Vertical bar plots show the level of cytosine methylation in all three contexts (CG, CHG and CHH) in sixdifferent tissues. CG and CHG methylation levels are equal between tissues. CHH methylation is lost in somatic tissues and restored in the pollen vegetativenucleus for most of the studied LTR regions. As reads from pollen showed variable CHH methylation levels, hierarchical cluster analysis was used toseparate differentially methylated regions (DMRs) of sequenced pollen bisulfite-treated DNA samples. According to Calarco et al. (2012), sperm cells havea much lower methylation level than vegetative cells. Sperm cell and vegetative cell marks on the figure are speculative and based on these findings. Formore details, see Supporting Information Methods S1. Error bars, � SE of measurement. Blue, male flower buds; red, female flower buds; green, leaves;orange, pollen; brown, vegetative cells; yellow, sperm cells.
Fig. 11 Scheme ofOgre evolution within the genus Silene. The activity ofall three groups ofOgre elements was highest after the Y chromosomeoriginated (c. 6 million yr ago (Ma)) and before speciation of threedioecious species within the sectionMelandrium (c. 0.8–1.4 million yrago).
� 2014 The Authors
New Phytologist� 2014 New Phytologist TrustNew Phytologist (2014)
www.newphytologist.com
NewPhytologist Research 13

Ogre CL5 transcript level along its LTR was found to be spatiallydifferent and higher than the transcript levels of other Ogre fami-lies (Fig. S7), the promoter strength of Ogre families probablydiffers or there may be different mechanisms regulating Ogrefamilies at the transcriptional or posttranscriptional level. To dis-tinguish between coding and noncoding transcripts is difficult;thus, the excess of RNA transcripts from LTRs over transcriptswithin the internal region (Fig. 4) can mirror either aberrantRNA Pol II transcripts or long noncoding RNAs that are pro-duced by Pol IV and Pol V involved in RdDM. In this respect,an interesting question emerged: why do some TEs producemuch more sRNAs than do others? In Arabidopis thaliana, theproduction of 24-nt sRNA molecules is dependent on RNA PolIV synthesizing long noncoding RNAs (lncRNAs) that are madedouble-stranded by RNA-dependent RNA polymerase 2 (RDR2)and processed by Dicer-like nucleases (Haag et al., 2012; Wierzb-icki, 2012; Wierzbicki et al., 2012). The mechanism controllingRNA Pol IV targeting is still not fully understood. In A. thaliana,recent findings demonstrate that methylated H3K9 is recognizedby SAWADEE homeodomain homolog 1 (SHH1), an RNA PolIV-interacting protein, and that 44% of RNA Pol IV-dependentsRNAs are also SHH1 dependent (Law et al., 2013). Thus, wecan speculate that Ogre CL5 and Retand LTRs are targeted byRNA Pol IV more than the other elements, leading to a higherproduction of sRNAs. This hypothesis is supported by the highertranscript level in Ogre CL5 and Retand LTRs matching the dis-tribution of sRNAs (Fig. S7), and by the excess of transcripts inLTRs over internal regions (Fig. 4). Nevertheless, the reducedtranscript level within internal region can alternatively beexplained by aberrant RNA Pol II products or their rapid degra-dation.
The high level of sRNAs complementary to Ogre CL5 andRetand elements indicates a possible role of epigenetic mecha-nisms in regulating these TEs. Their conspicuous chromosomalpattern suggests that the mechanism regulating their spread hasto be conserved through many generations. Thus, epigeneticchanges in the germline and during development may provide anexplanation. Recent findings suggest that, in A. thaliana, themechanism for transposon control has evolved in reproductivecells where TEs are reactivated by genome-wide loss of DNAmethylation during both male and female gametogenesis andembryonal development. In the vegetative nucleus of pollengrains, TEs are demethylated as a result of the active effect ofDemeter (DME) and down-regulation of the heterochromatinremodeller decrease in DNA methylation 1 (DDM1), whichleads to TE transcription and subsequent production of 21-ntsRNA molecules. These sRNAs then travel to the transcription-ally silent sperm cells to reinforce TE silencing (Slotkin et al.,2009; Ibarra et al., 2012). As Ogre CL5 accumulates much higherlevels of 21-nt sRNAs than the other elements, we can speculatethat it is silenced more intensively in pollen.
Whereas DDM1 is required for asymmetric DNA methylationof internal regions of long TEs and is mediated bychromomethylase 2 (CMT2) separately from RdDM (Zemachet al., 2013), TE edges (LTRs) are also methylated by the RdDMpathway mediated by domains rearranged methyltransferase 2
(DRM2). During pollen development, CHH methylation isgradually lost from microspores and sperm cells, while it isrestored in the vegetative nucleus where DRM2 is substantiallyaccumulated (Calarco et al., 2012). We showed that there are twoclusters of Ogre elements with low and high CHH methylationlevels in pollen. We presume that the highly methylated elementsrepresent copies from the vegetative nucleus, while the demethy-lated elements are from sperm cells. This suggests that the mecha-nism of epigenetic reprogramming is probably similar in A.thaliana and Silene. The increased CHH methylation of Ogre ele-ments is probably guided by 24-nt sRNAs, as indicated by thecolocalization of sRNAs and CHH methylation level in pollen(Fig. S6). As the transcript level inside Ogre elements is very lowin pollen (Fig. 4), all Ogre elements may be silent and unable toretrotranspose in the male germline. Alternatively, the Ogre CL6and CL11 families are not fully silenced because of lower 24-ntsRNA levels (Fig. 9), and only Ogre CL5 is restricted to paternalgenome and Y chromosome transposition. However, Retand pro-duces more complete transcripts, suggesting that this elementremains active in pollen. This would explain its significantlylower presence on the Y chromosome.
A similar phenomenon is thought to occur in the female game-tophyte embryo sac of A. thaliana (Gehring et al., 2009; Hsiehet al., 2009) and can be used to explain the absence of Ogre CL5on the Y chromosome. During female gametogenesis, the activityof the maintenance DNA methyltransferases methyltransferase 1(MET1) and CMT3 required for CG and CHG methylation isundetectable and this leads to progressive demethylation, activa-tion and transposition of TEs in the maternal genome. By con-trast, both maintenance and de novo DNA methyltransferasesDRM1 and DRM2 are expressed strongly in the embryo, whilethe same genes remain inactive in the central cell and endosperm.Reactivated TEs in the endosperm then serve as a source ofmigrating sRNAs that guide gradual remethylation of TEs afterfertilization and throughout embryogenesis (Calarco & Martiens-sen, 2012; Jullien et al., 2012). Relatively small changes in thelevel of 24-nt sRNAs should be enough to reactivate TEs, asshown in cotton (Gossypium hirsutum) (Romanel et al., 2012).Thus, we hypothesize that high levels of 24-nt sRNAs derivedfrom Ogre CL5 and Retand elements ensure that these two TEfamilies are preferentially methylated in CHH sequence contextby the de novo methyltransferases DRM1 and DRM2, while TEswith lower 24-nt sRNA levels remain active for some time duringembryogenesis. Silencing in pollen together with silencing in theembryo lead to complete protection of the paternal genome frominsertions of Ogre CL5. Nevertheless, further studies of sRNAabundance, transcription and methylation level will have to beperformed in pollen and seed (embryonal) tissues.
In summary, we found that three Ogre families escaped hostregulation recently and spread intensively in the dioeciousMelandrium species, resulting in significant genome sizeincreases. We suggest that LTRs coding for regulatory sequencesare remethylated by the RdDM pathway in pollen and assumethat they may also be methylated in the embryo. The 10-foldgreater number of sRNAs targeting one Ogre family (Ogre CL5)probably results in rapid silencing of this particular element in
New Phytologist (2014) � 2014 The Authors
New Phytologist� 2014 New Phytologist Trustwww.newphytologist.com
Research
NewPhytologist14

the male germline and in the embryo after fertilization. This pre-sumed silencing mechanism may have resulted in the absence ofOgre CL5 on the Y chromosome. We also hypothesize that simi-lar epigenetic mechanisms may account for the absence of TEson the sex chromosomes of other plant species. Nevertheless,other mechanisms, such as TE insertion connected with recombi-nation or the need for female factors to allow TE spreading, can-not be excluded. Last but not least, a similar mechanism mayinfluence maternal genome-specific spreading of TEs in otherplant species without sex chromosomes.
Acknowledgements
We thank Dr Jiri Siroky for performing in situ hybridizationexperiments on cryosections of anthers and ovaries and Eleni Mi-chu for providing genic sequences. The access to computing andstorage facilities owned by parties and projects contributing tothe National Grid Infrastructure MetaCentrum, provided underthe programme ‘Projects of Large Infrastructure for Research,Development, and Innovations’ (LM2010005), is greatly appre-ciated. This work was supported by grants from the Czech Sci-ence Foundation (grant nos. P501/10/P483, P501/10/0102,P305/10/0930 and P501/12/2220), by the project ‘CEITEC –Central European Institute of Technology’ (CZ.1.05/1.1.00/02.0068) from the European Regional Development Fund andby the project OPVK (CZ.1.07/2.3.00/20.0045). The fundershad no role in study design, data collection and analysis, decisionto publish, or preparation of the manuscript.
References�Agren JA, Wright SI. 2011. Co-evolution between transposable elements and
their hosts: a major factor in genome size evolution? Chromosome Research 19:777–786.
Anisimova M, Gascuel O. 2006. Approximate likelihood-ratio test for branches:
a fast, accurate, and powerful alternative. Systematic Biology 55: 539–552.Atanassov I, Delich�ere C, Filatov DA, Charlesworth D, Negrutiu I,
Moneger F. 2001. Analysis and evolution of two functional Y-linked loci
in a plant sex chromosome system. Molecular Biology and Evolution 18:
2162–2168.Bachtrog D. 2013. Y-chromosome evolution: emerging insights into processes of
Y-chromosome degeneration. Nature Reviews Genetics 14: 113–124.Barbacar N, Hinnisdaels S, Farbos I, Mon�eger F, Lardon A, Delich�ere C,
Mouras A, Negrutiu I. 1997. Isolation of early genes expressed in reproductive
organs of the dioecious white campion (Silene latifolia) by subtraction cloning
using an asexual mutant. Plant Journal 12: 805–817.Baucom RS, Estill JC, Leebens-Mack J, Bennetzen JL. 2009.Natural selection
on gene function drives the evolution of LTR retrotransposon families in the
rice genome. Genome Research 19: 243–254.Bergero R, Forrest A, Kamau E, Charlesworth D. 2007. Evolutionary strata on
the X chromosomes of the dioecious plant Silene latifolia: evidence from new
sex-linked genes. Genetics 175: 1945–1954.Bergman CM, Bensasson D. 2007. Recent retrotransposon insertion contrasts
with waves of non-LTR insertion since speciation in Drosophila melanogaster.Proceedings of the National Academy of Sciences, USA 104: 11340–11345.
Boeke JD, Devine SE. 1998. Yeast retrotransposons: finding a nice quiet
neighborhood. Cell 93: 1087–1089.Brewer PB, Heisler MG, Hejatko J, Friml J, Benova E. 2006. In situhybridization for mRNA detection in Arabidopsis tissue sections. NatureProtocols 1: 1462–1467.
Buschman F, Lewinski M, Ciuffi A, Barr S, Leipzig J, Hannenhalli S, Hoffmann
C. 2005. Genome-wide analysis of retroviral DNA integration. Nature ReviewsMicrobiology 3: 848–858.
Buschman F, Miller MD. 1997. Tethering human immunodeficiency virus type
1 preintegration complexes to target DNA promotes integration at nearby sites.
Journal of Virology 71: 458–464.Buschman FD. 1994. Tethering human immunodeficiency virus 1 integrase to a
DNA site directs integration to nearby sequences. Proceedings of the NationalAcademy of Sciences, USA 91: 9233–9237.
Calarco JP, Borges F, Donoghue MTA, Van Ex F, Jullien PE, Lopes T, Gardner
R, Berger F, Feij�o JA, Becker JD et al. 2012. Reprogramming of DNA
methylation in pollen guides epigenetic inheritance via small RNA. Cell 151:194–205.
Calarco JP, Martienssen RA. 2012. Imprinting: DNA methyltransferases
illuminate reprogramming. Current Biology 22: R929–R931.Cegan R, Marais GAB, Kubekova H, Blavet N, Widmer A, Vyskot B, Dolezel J,
Saf�ar J, Hobza R. 2010. Structure and evolution of Apetala3, a sex-linked genein Silene latifolia. BMC Plant Biology 10: 180.
Cegan R, Vyskot B, Kejnovsky E, Kubat Z, Blavet H, �Saf�a�r J, Dole�zel J, Blavet N,
Hobza R. 2012.Genomic diversity in two related plant species with and without
sex chromosomes – Silene latifolia and S. vulgaris. PLoS ONE 7: e31898.
Cermak T, Kubat Z, Hobza R, Koblizkova A, Widmer A, Macas J, Vyskot B,
Kejnovsky E. 2008. Survey of repetitive sequences in Silene latifolia withrespect to their distribution on sex chromosomes. Chromosome Research 16:961–976.
Charlesworth B. 1991. The evolution of sex chromosomes. Science 251: 1030–1033.
Cherepanov P, Maertens GN, Hare S. 2011. Structural insights into the
retroviral DNA integration apparatus. Current Opinion in Structural Biology 21:249–256.
Delich�ere C, Veuskens J, Hernould M, Barbacar N, Mouras A, Negrutiu I,
Mon�eger F. 1999. SlY1, the first active gene cloned from a plant Y
chromosome, encodes a WD-repeat protein. EMBO Journal 18: 4169–4179.
Devos KM, Brown JKM, Bennetzen JL. 2002. Genome size reduction through
illegitimate recombination counteracts genome expansion in Arabidopsis.Genome Research 12: 1075–1079.
Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by
sampling trees. BMC Evolutionary Biology 7: 214.Filatov DA, Howell EC, Groutides C, Armstrong SJ. 2009. Recent spread of a
retrotransposon in the Silene latifolia genome, apart from the Y chromosome.
Genetics 181: 811–817.Finnegan DJ. 1989. Eukaryotic transposable elements and genome evolution.
Trends in Genetics 5: 103–107.Gao X, Hou Y, Ebina H, Levin HL, Voytas DF. 2008. Chromodomains direct
integration of retrotransposons to heterochromatin. Genome Research 18: 359–363.
Gehring M, Bubb KL, Henikoff S. 2009. Extensive demethylation of repetitive
elements during seed development underlies gene imprinting. Science 324:1447–1451.
Grant S, Hunkirchen B, Saedler H. 1994. Developmental differences between
male and female flowers in the dioecious plant Silene latifolia. Plant Journal 6:471–480.
Haag JR, Ream TS, Marasco M, Nicora CD, Norbeck AD, Pasa-Tolic L,
Pikaard CS. 2012. In vitro transcription activities of Pol IV, Pol V, and RDR2
reveal coupling of Pol IV and RDR2 for dsRNA synthesis in plant RNA
silencing.Molecular Cell 48: 811–818.Hawkins JS, Kim H, Nason JD, Wing RA, Wendel JF. 2006. Differential
lineage-specific amplification of transposable elements is responsible for
genome size variation in Gossypium. Genome Research 16: 1252–1261.Hawkins JS, Proulx SR, Rapp RA, Wendel JF. 2009. Rapid DNA loss as a
counterbalance to genome expansion through retrotransposon proliferation in
plants. Proceedings of the National Academy of Sciences, USA 106: 17811–17816.
Hindmarsh P, Leis J. 1999. Retroviral DNA integration.Microbiology andMolecular Biology Reviews 63: 836–843.
� 2014 The Authors
New Phytologist� 2014 New Phytologist TrustNew Phytologist (2014)
www.newphytologist.com
NewPhytologist Research 15

Hobza R, Lengerova M, Svoboda J, Kubekova H, Kejnovsky E, Vyskot B. 2006.
An accumulation of tandem DNA repeats on the Y chromosome in Silenelatifolia during early stages of sex chromosome evolution. Chromosoma 115:376–382.
Howell EC, Armstrong SJ, Filatov DA. 2009. Evolution of neo-sex
chromosomes in Silene diclinis. Genetics 182: 1109–1115.Hsieh TF, Ibarra CA, Silva P, Zemach A, Eshed-Williams L, Fischer RL,
Zilberman D. 2009. Genome-wide demethylation of Arabidopsis endosperm.
Science 324: 1451–1454.Ibarra CA, Feng X, Schoft VK, Hsieh T-F, Uzawa R, Rodrigues JA, Zemach A,
Chumak N, Machlicova A, Nishimura T et al. 2012. Active DNA
demethylation in plant companion cells reinforces transposon methylation in
gametes. Science 337: 1360–1364.Jullien PE, Susaki D, Yelagandula R, Higashiyama T, Berger F. 2012. DNA
methylation dynamics during sexual reproduction in Arabidopsis thaliana.Current Biology 22: 1825–1830.
Kejnovsky E, Kubat Z, Hobza R, Lengerova M, Sato I, Tabata S, Fukui K,
Matsunaga S, Vyskot B. 2006b. Accumulation of chloroplast DNA sequences
on the Y chromosome of Silene latifolia. Genetica 128: 167–175.Kejnovsky E, Kubat Z, Macas J, Hobza R, Mracek J, Vyskot B. 2006a. Retand: anovel family of gypsy-like retrotransposons harboring an amplified tandem
repeat.Molecular Genetics and Genomics 276: 254–263.Kovacova V, Janousek B. 2012. Bisprimer – a program for the design of primers
for bisulfite-based genomic sequencing of both plant and mammalian DNA
samples. Journal of Heredity 103: 308–312.Kubat Z, Hobza R, Vyskot B, Kejnovsky E. 2008.Microsatellite accumulation
on the Y chromosome in Silene latifolia. Genome 51: 350–356.Kumar A, Bennetzen JL. 1999. Plant retrotransposons. Annual Review of Genetics33: 479–532.
Law JA, Du J, Hale CJ, Feng S, Krajewski K, Palanca AM, Strahl BD, Patel DJ,
Jacobsen SE. 2013. Polymerase IV occupancy at RNA-directed DNA
methylation sites requires SHH1. Nature 498: 385–389.Lengerova M, Kejnovsky E, Hobza R, Macas J, Grant SR, Vyskot B. 2004.
Multicolor FISH mapping of the dioecious model plant, Silene latifolia.Theoretical and Applied Genetics 108: 1193–1199.
Lewinski MK, Yamashita M, Emerman M, Ciuffi A, Marshall H, Crawford G,
Collins F, Shinn P, Leipzig J, Hannenhalli S et al. 2006. Retroviral DNA
integration: viral and cellular determinants of target-site selection. PLoSPathogens 2: e60.
Macas J, Kejnovsk�y E, Neumann P, Nov�ak P, Kobl�ı�zkov�a A, Vyskot B. 2011.Next generation sequencing-based analysis of repetitive DNA in the model
dioecious plant Silene latifolia. PLoS ONE 6: e27335.
Macas J, Neumann P. 2007. Ogre elements – a distinct group of plant Ty3/gypsy-like retrotransposons. Gene 390: 108–116.
Moore RC, Kozyreva O, Lebel-Hardenack S, Siroky J, Hobza R, Vyskot B,
Grant SR. 2003. Genetic and functional analysis of DD44, a sex-linked genefrom the dioecious plant Silene latifolia, provides clues to early events in sex
chromosome evolution. Genetics 163: 321–334.Naito K, Cho E, Yang G, Campbell MA, Yano K. 2006. Dramatic amplification
of a rice transposable element during recent domestication. Proceedings of theNational Academy of Sciences, USA 103: 17620–17625.
Neumann P, Koblkov A, Navr�atilov�a A, Macas J. 2006. Significant expansion of
Vicia pannonica genome size mediated by amplification of a single type of giant
retroelement. Genetics 173: 1047–1056.Neumann P, Navr�atilov�a A, Kobl�ı�zkov�a A, Kejnovsk�y E, H�ribov�a E, Hobza R,
Widmer A, Dole�zel J, Macas J. 2011. Plant centromeric retrotransposons: a
structural and cytogenetic perspective.Mobile DNA 2: 4.
Neumann P, Po�z�arkov�a D, Macas J. 2003.Highly abundant pea LTR
retrotransposon Ogre is constitutively transcribed and partially spliced. PlantMolecular Biology 53: 399–410.
Otto SP, Pannell JR, Peichel CL, Ashman TL, Charlesworth D, Chippindale
AK, Delph LF, Guerrero RF, Scarpino SV, McAllister BF. 2011. About PAR:
The distinct evolutionary dynamics of the pseudoautosomal region. Trends inGenetics 27: 358–367.
Piegu B, Guyot R, Picault N, Roulin A, Sanyal A, Kim H, Collura K, Brar DS,
Jackson S, Wing RA et al. 2006. Doubling genome size without
polyploidization: dynamics of retrotransposition-driven genomic expansion in
Oryza australiensis, a wild relative of rice. Genome Research 16: 1262–1269.Rautenberg A, Filatov D, Svennblad B, Heidari N, Oxelman B. 2008.
Conflicting phylogenetic signals in the SlX1/Y1 gene in Silene. BMCEvolutionary Biology 8: 299.
Romanel E, Silva TF, Correa RL, Farinelli L, Hawkins JS, Schrago CE, Vaslin
MF. 2012. Global alteration of microRNAs and transposon-derived small
RNAs in cotton (Gossypium hirsutum) during cotton leafroll dwarf polerovirus
(CLRDV) infection. Plant Molecular Biology 80: 443–460.Sakamoto K, Ohmido N, Fukui K, Kamada H, Satoh S. 2000. Site-specific
accumulation of a LINE-like retrotransposon in a sex chromosome of
the dioecious plant Cannabis sativa. Plant Molecular Biology 44:
723–732.Shirasu K, Schulman AH, Lahaye T, Schulze-Lefert P. 2000. A contiguous
66-kb barley DNA sequence provides evidence for reversible genome
expansion. Genome Research 10: 908–915.Slotkin RK, Vaughn M, Borges F, Tanurd�zi�c M, Becker JC, Feij�o JA,
Martienssen RA. 2009. Epigenetic reprogramming and small RNA silencing of
transposable elements in pollen. Cell 136: 461–472.Steflova P, Tokan V, Vogel I, Lexa M, Macas J, Novak P, Hobza R, Vyskot B,
Kejnovsky E. 2013. Contrasting patterns of transposable element and satellite
distribution on sex chromosomes (XY1Y2) in the dioecious plant Rumexacetosa. Genome Biology and Evolution 5: 769–782.
Steinbauerov�a V, Neumann P, Macas J. 2008. Experimental evidence for
splicing of intron-containing transcripts of plant LTR retrotransposon Ogre.Molecular Genetics and Genomics 280: 427–436.
Steinbauerov�a V, Neumann P, Nov�ak P, Macas J. 2012. A widespread occurence
of extra open reading frames in plant Ty3/gypsy retrotransposons. Genetica 139:1543–1555.
Steinemann S, Steinemann M. 2005. Retroelements: tools for sex chromosome
evolution. Cytogenetics and Genome Research 110: 134–143.Tsukahara S, Kobayashi A, Kawabe A, Mathieu O, Miura A, Kakutani T.
2009. Bursts of retrotransposition reproduced in Arabidopsis. Nature 461:423–426.
Ungerer MC, Strakosh SC, Zhen Y. 2006. Genome expansion in three hybrid
sunflower species is associated with retrotransposon proliferation. CurrentBiology 16: 872–873.
Vitte C, Bennetzen JL. 2006. Analysis of retrotransposon diversity uncovers
properties and propensities in angiosperm genome evolution. Proceedings of theNational Academy of Sciences, USA 103: 17638–17643.
Vu W, Nuzhdin SV. 2011. Genetic variation of copia suppression in Drosophilamelanogaster. Heredity 106: 207–217.
Wierzbicki AT. 2012. The role of long non-coding RNA in transcriptional gene
silencing. Current Opinion in Plant Biology 15: 517–522.Wierzbicki AT, Cocklin R, Mayampurath A, Lister R, Rowley MJ, Gregory BD,
Ecker JR, Tang H, Pikaard CS. 2012. Spatial and functional relationships
among Pol V-associated loci, Pol IV-dependent siRNAs, and cytosine
methylation in the Arabidopsis epigenome. Genes and Development 26: 1825–1836.
Williams R, Peisajovich SG, Miller OJ, Magdassi S, Tawfik DS, Griffiths AD.
2006. Amplification of complex gene libraries by emulsion PCR. NatureMethods 3: 545–550.
Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood.MolecularBiology and Evolution 24: 1586–1591.
Zemach A, Kim MY, Hsieh PH, Coleman-Derr D, Eshed-Williams L, Thao K,
Harmer SL, Zilberman D. 2013. The Arabidopsis nucleosome remodeler
DDM1 allows DNA methyltransferases to access H1-containing
heterochromatin. Cell 153: 193–205.Zluvova J, Lengerova M, Markova M, Hobza R, Nicolas M, Vyskot B,
Charlesworth D, Negrutiu I, Janousek B. 2005. The inter-specific hybrid
Silene latifolia 9 S. viscosa reveals early events of sex chromosome evolution.
Evolution and Development 7: 327–336.Zluvova J, Nicolas M, Berger A, Negrutiu I, Mon�eger F. 2006. Premature arrest
of the male flower meristem precedes sexual dimorphism in the dioecious plant
Silene latifolia. Proceedings of the National Academy of Sciences, USA 103:
18854–18859.
New Phytologist (2014) � 2014 The Authors
New Phytologist� 2014 New Phytologist Trustwww.newphytologist.com
Research
NewPhytologist16

Zluvova J, Zak J, Janousek B, Vyskot B. 2010. Dioecious Silene latifoliaplants show sexual dimorphism in the vegetative stage. BMC Plant Biology10: 208.
Supporting Information
Additional supporting information may be found in the onlineversion of this article.
Fig. S1 RACE mapped transcription starts and polyA signals ofOgre CL5 and Ogre CL5del and putative splice site of Ogre CL5and Ogre CL5del in the Silene latifolia genome.
Fig. S2 Chronogram showing divergence times estimated inBEAST based on sequences of four genes in species of the genusSilene.
Fig. S3 Chronograms showing divergence times estimated inBEAST for species of the genus Silene.
Fig. S4 Linear regression of X–Y split time in millions of yearsand percentage of synonymous substitutions in Silene latifolia.
Fig. S5 The spatial distribution and abundance of 24-nucleotide(24-nt) small RNAs (sRNAs) along four genomic copies of Ogreelements in Silene latifolia.
Fig. S6 Microview of CHH methylation of the right LTR in theTE Ogre CL5 in Silene latifolia.
Fig. S7 Detailed view of the spatial distribution of 24-nucleotide(24-nt) small RNAs (sRNAs) along LTRs of Retand and Ogre ele-ments (first three rows) in Silene latifolia.
Table S1 Primers used in this study
Table S2 Copy numbers of TEs in the Silene latifolia genome(1C) estimated by two methods – BAC library hybridization andin silico read mapping
Table S3 List of sequences used in the phylogenetic analyses ofOgre elements in Silene including their accession numbers
Table S4 P-values for the Mann–Whitney test in Silene latifolia
Table S5 Percentages of synonymous substitutions per codonand time of X–Y chromosome split into four sex-linked Silenelatifolia genes
Table S6 Percentages of disturbed open reading frames (ORFs)in each group of retrotransposons in Silene latifolia
Table S7 Branch analysis of terminal and internal branches inthe Silene latifolia genome
Table S8 Abundance of 19–24-nucleotide small RNAs (sRNAs)complementary to LTRs in sense (+) and antisense (�) orienta-tion in Silene latifolia
Methods S1Materials and Methods explained in detail.
Notes S1 Original tree files.
Notes S2 Branching order of Silene Ogre retrotransposon groups.
Please note: Wiley Blackwell are not responsible for the contentor functionality of any supporting information supplied by theauthors. Any queries (other than missing material) should bedirected to the New Phytologist Central Office.
� 2014 The Authors
New Phytologist� 2014 New Phytologist TrustNew Phytologist (2014)
www.newphytologist.com
NewPhytologist Research 17




















