
PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell...
41
1 PDK1 Signaling Towards PLK1-Myc Activation Confers Oncogenic Transformation and Tumor Initiating Cell Activation and Resistance to mTOR-targeted Therapy Jing Tan 1 , Zhimei Li 1 , Puay Leng Lee 1 , Peiyong Guan 2 , Mei Yee Aau 1 , Shuet Theng Lee 1 , Min Feng 1 , Cheryl Zihui Lim 1 , Eric Yong Jing Lee 1 , Zhen Ning Wee 1,3 Yaw Chyn Lim 4 , R. K. Murthy Karuturi 2 and Qiang Yu 1, 4, 5,* 1 Cancer Therapeutics and Stratified Oncology, Genome Institute of Singapore, A*STAR (Agency for Science, Technology and Research), Biopolis, Singapore. 2 Information and Mathematical Science, Genome Institute of Singapore, A*STAR (Agency for Science, Technology and Research), Biopolis, Singapore. 3 Graduate School for Integrative Sciences and Engineering, National University of Singapore 4 Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore 5 Cancer and Stem Cell Biology, DUKE-NUS Graduate Medical School of Singapore Running Title: PDK1- PLK1-Myc signaling in transformation, cancer stem cells and drug resistance. *Correspondence: [email protected] Qiang Yu, M.D. Ph.D. Cancer Therapeutics and Stratified Oncology Genome Institute of Singapore Email: [email protected] Fax: 65-6808-9003 Disclosure of Potential Conflicts of Interest No potential conflicts of interests were disclosed. Research. on June 8, 2016. © 2013 American Association for Cancer cancerdiscovery.aacrjournals.org Downloaded from Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell...

1
PDK1 Signaling Towards PLK1-Myc Activation Confers Oncogenic Transformation and Tumor Initiating Cell Activation and Resistance to mTOR-targeted Therapy Jing Tan1, Zhimei Li1, Puay Leng Lee1, Peiyong Guan2, Mei Yee Aau1, Shuet Theng Lee1, Min Feng1, Cheryl Zihui Lim1, Eric Yong Jing Lee1, Zhen Ning Wee1,3 Yaw Chyn Lim4, R. K. Murthy Karuturi2 and Qiang Yu1, 4, 5,*
1 Cancer Therapeutics and Stratified Oncology, Genome Institute of Singapore, A*STAR (Agency for Science, Technology and Research), Biopolis, Singapore. 2 Information and Mathematical Science, Genome Institute of Singapore, A*STAR (Agency for Science, Technology and Research), Biopolis, Singapore.
3Graduate School for Integrative Sciences and Engineering, National University of Singapore
4Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore 5Cancer and Stem Cell Biology, DUKE-NUS Graduate Medical School of Singapore Running Title: PDK1- PLK1-Myc signaling in transformation, cancer stem cells and drug resistance. *Correspondence: [email protected] Qiang Yu, M.D. Ph.D. Cancer Therapeutics and Stratified Oncology Genome Institute of Singapore Email: [email protected] Fax: 65-6808-9003 Disclosure of Potential Conflicts of Interest No potential conflicts of interests were disclosed.
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

2
ABSTRACT
Although 3-Phosphoinositide–dependent protein kinase-1 (PDK1) has been
predominately linked to PI3K-AKT pathway, it may also evoke additional signaling
outputs to promote tumorigenesis. Here we report that PDK1 directly induces
phosphorylation of Polo-like kinase 1 (PLK1), which in turn induces Myc
phosphorylation and protein accumulation. We show that PDK1-PLK1-Myc signaling is
critical for cancer cell growth and survival and small molecule inhibition of PDK1/PLK1
provides an effective approach for therapeutic targeting Myc-dependency. Intriguingly,
PDK1-PLK1-Myc signaling induces an embryonic stem cell-like gene signature
associated with aggressive tumor behaviors and is a robust signaling axis driving cancer
stem cell (CSC) self renewal. Finally, we show that PLK1 inhibitor synergizes with
mTOR inhibitor to induce synergistic anti-tumor effect in colorectal cancer by
antagonizing a compensatory Myc induction. These findings identify a novel pathway in
human cancer and CSC activation and provide a therapeutic strategy for targeting Myc-
associated tumorigenesis and therapeutic resistance.
Significance: This work identifies PDK1-PLK1-Myc signaling as a new oncogenic
pathway driving oncogenic transformation and cancer stem cell self-renewal. Targeted
inhibition of PDK1/PLK1 is robust in targeting Myc dependency in cancer cells. Thus,
our findings provide important insights into cancer and cancer stem cell biology and have
significant therapeutic implications.
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

3
INTRODUCTION
Phosphatidylinositol 3’-kinase (PI3K)-AKT pathway is one of the most commonly
deregulated signaling pathways in human cancers (1). Genetic aberrations affecting this
pathway, such as activating mutations of PIK3CA or inactivation of PTEN, have been
identified in virtually all epithelial tumors (2). The 3-phosphoinositide-dependent protein
kinase-1 (PDK1) is known to be activated as a result of the accumulation of the PI3K
product phosphatidylinositol-3,4,5-trisphosphate (PIP3) and thus considered as an
important component of the PI3K pathway. PDK1 is a master regulator of AGC kinase
members, including AKT, p70 ribosomal S6 kinase (S6K), serum- and glucocorticoid-
induced protein kinase (SGK) and protein kinase C (PKC) family members, thus having
multiple roles in various physiological processes such as metabolism, growth,
proliferation and survival (3). In human cancers, PDK1 is thought to be constitutively
activated upon elevation of PIP3 owing to the loss of PTEN or gain of PIK3CA activity.
In addition, PDK1 deregulation in human malignancy can also be caused by gene
amplification or abnormal phosphorylation in cytosol and nucleus, such as colon cancer
and invasive breast cancer (4, 5).
One of the most defined PDK1 targets relevant in human cancer is AKT.
Specifically, PDK1 directly phosphorylates AKT on T308, but requires mTORC2-
induced AKT phosphorylation on S473 to confer a full activation (6). Given its
connection to AKT, PDK1 has been pursued as a critical anti-cancer target (7). However,
in view of the diversity of PDK1 substrates, additional downstream targets of PDK1 may
confer aberrant signaling heterogeneity and complexity in human malignance. Indeed, it
has been recently shown that inhibition of PDK1 has no significant effect on AKT
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

4
signaling in a PTEN-deficient transgenic tumor mouse model (8) or breast tumor growth
(9), and oncogenic functions of PDK1 through substrates other than AKT, such as SGK3
(10), MAPK (11), or PKC� (12), have also been reported. In addition, our recent work
has shown that PDK1 is required for Myc protein accumulation in colon cancer cells
treated with mTOR inhibitor rapamycin (5), indicating a potential functional link of
PDK1 with Myc in oncogeneiss.
Myc is implicated in both cancer and stem cell self-renewal. The relationship
between stem cell and human cancers has become an important issue in cancer research
given that self-renewal is a hallmark of both cell types (13). Genes associated with
embryonic stem cell (ESC) identity, including pluripotency transcription factors,
Polycomb targets and Myc targets, have been observed in aggressive human cancers and
are associated with poor disease outcome (14). Moreover, the Myc associated molecular
network is strikingly similar between ESC and human cancer transcription programs (15),
and ectopic overexpression of Myc in differentiated somatic cells can induce both ESC
gene signature and properties of cancer stem cells (CSC) (16). These findings suggest
that activation of an ESC-like gene expression program in adult cells may confer self-
renewal to cancer cells or cancer stem cells. Notably, although the cancer associated
ESC–like gene regulation by transcription factors has been well documented, its
regulation by a druggable kinase-driven signaling pathway has yet to be identified.
In the present studies, we investigated PDK1-evoked key signaling events required
for oncogenic transformation. We identified that PDK1-PLK1-Myc pathway is a major
driver pathway conferring PDK-induced transformation and its existence is readily
evident in human cancers. We further show that PDK1-PLK1-Mys signaling drives an
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

5
ESC-like gene expression signature relevant in human cancers and is robust in inducing
CSC phenotype. It also involves in resistance to mTOR inhibitor in colorectal cancer
cells. These findings provide important insights into the cancer and cancer stem cell
biology and potential new treatment for targeting Myc-dependency in human cancers
RESULTS
PDK1-Induced Myc Protein Induction Confers Oncogenic Transformation
As the first step to investigate the differential signaling pathways activated by PDK1 or
PI3K in tumorigenesis, we compared the transforming capacity of PDK1 and PI3K by
using the in vitro transformation assay that measures the anchorage-independent growth
in soft agar. We began with semi-transformed human embryonic kidney epithelial cells
(HEK) that express a low level of activated HRasV12 (HEK-TERV)(17) and infected
them with retroviral vectors expressing PDK1, Myc, a constitutively activating mutant of
PIK3CA (E545K) or PTEN small hairpin RNA (shRNA), resulting in stable cell lines
designated as HEK-PDK1, HEK-Myc, HEK-E545K or HEK-shPTEN cells, respectively.
The transformation assay results showed that they were all able to induce cellular
transformation, although PDK1- or Myc-induced colonies appeared to be larger in size as
compared with that of E545K- or shPTEN-expressing cells (Fig. 1A and Supplementary
Fig. S1A). Consistent with our previous report showing a post-translational Myc
induction by PDK1 (5), we detected a marked protein accumulation of Myc in HEK-
PDK1 cells but not in HEK-E545K or HEK-shPTEN cells (Fig. 1B) which was not due to
the induction of Myc mRNA level (Supplementary Fig. S1B). We also show that the
kinase activity of PDK1 is required for transformation as well as Myc protein induction
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

6
as a kinase-dead mutant of PDK1 (PDK1 K100N) (9) induced neither the transformation
nor the Myc accumulation (Supplementary Fig. S1C).
A survey of known AGC substrates of PDK1 revealed that PDK1 also induced a
strong phosphorylation of PKC� and a modest increase of phosphorylated AKT (T308).
The other known PDK1 substrates, including SGK1/3 and S6K, were not activated, nor
AKT phosphorylated at S473, which is required for a full activation of AKT. In contrast,
E545K overexpression induced strong phosphorylations of AKT (at both T308 and S473)
as well as the downstream substrates FOXO1 and FOXO3A (Fig. 1B). Thus, the
remarkable observation that PDK1 induces transformation in the presence of a weak
AKT activation suggests a potential more functional role of Myc in this process. Indeed,
RNA interference-mediated knockdown of Myc resulted in much reduced transformation
of HEK-PDK1 cells, but not in HEK-E545K cells (Fig. 1C), demonstrating a Myc-
dependency for PDK1-induced transformation. Moreover, in a series of dose-response
analysis (see Supplementary Fig. S1D&E), HEK-PDK1 cells, compared with HEK-
E545K cells, were much more sensitive to small molecule PDK1 inhibitors BX795 and
BX912 (Fig. 1D, Left and Supplementary Fig. S1D). In contrast, E545K-transformed
cells were much more sensitive to the PI3K inhibitor GDC-0941 and the AKT inhibitor
MK2206 and GSK690693 (Fig. 1D, Left and Supplementary Fig. S1E). Consistent with
these effects, BX795 reduced Myc accumulation but had only a modest effect on AKT.
By contrast, GDC-0941 or MK2206 easily abolished phosphorylations of AKT in HEK-
E545K cells, but had no such effects on Myc inhibition (Fig. 1D, Right). These results
demonstrated the differential pathway dependency for the two transformed cell systems.
Interestingly, Myc-transformed cells were also sensitive to BX795 (Fig. 1D, Left), which
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

7
is consistent with the observation that BX795 was able to eliminate the exogenous Myc
in these cells (Fig. 1D, Right). Altogether, these data show that PDK1-induced
transformation depends more on Myc, but less on AKT signaling, when compared with
E545K-driven transformation. The data also suggest that Myc-dependent cells become
sensitive to the PDK1 inhibitor, regardless of PDK1 status, which reveals a PDK1-
dependency in Myc-driven cells. PDK1-induced Myc activation upon transformation was
also observed in immortalized human mammary epithelial cells (HMEC) and prostate
epithelial cells (RWPE-1) (Fig. 1E and F), suggesting that the Myc activation by PDK1 is
not restricted to HEK but occurs in multiple epithelial lineages.
To demonstrate the physiological relevance of PDK1-Myc connection in human
cancers, we show that PDK1 knockdown was able to eliminate Myc expression in a
variety of human cancer cell lines (Fig. 1G). Moreover, in a panel of breast cancer cell
lines in which the Myc-dependent viability has been previously characterized (18),
BX795 treatment resulted in similar Myc depletion in all these cells (Fig. 1H) but
preferentially reduced the cell viability of Myc-dependent breast cancer cell lines (MDA-
MB-231, Hs578T, and SUM159PT) as compared to Myc-independent breast cancer cell
lines (T47D and BT474)(Fig. 1I). Of notice, in these cell lines BX795 seemed to inhibit
AKT and FOXO3A phosphorylations in a cell-dependent manner (Fig. 1H). Taken
together, these results show a potential role of PDK1 activity towards Myc regulation
which is therapeutically implicated for Myc-driven tumors.
Synthetic Lethal Screening Identifies PLK1 as a Crucial Downstream Effector of
PDK1 for Myc Induction and Cancer Cell Survival
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

8
To investigate whether or not there are downstream kinase(s) of PDK1 that is crucial for
Myc induction and cell transformation, we performed a screen for kinases that, when
pharmacologically inhibited, selectively kill PDK1-transformed cells. Among 60 small
molecule protein kinase inhibitors we have screened, we found that two PLK1 inhibitors
(BI2536 and GW843682X), one MEK inhibitor (PD0325901), and one ALK inhibitor
(NVP-TAE684), PD180970, one Brc-Abl inhibitor (PD180970) and one tyrosine kinase
inhibitor (Sunitinib) showed a preferential inhibitory effect on the viability of HEK-
PDK1 cell as compared to the control cells (Supplementary Fig. S2A). The two PLK1
inhibitors were further validated in a secondary screen and were thus chosen for further
study (data not shown). Further analyses in all the three epithelial systems showed that
PDK1-transformed cells were much more sensitive to the PLK1 inhibitors compared to
vector control or E545K-transformed cells (Fig. 2A and B). This finding reveals a
possible role of PLK1 in PDK1-induced transformation. Indeed, western blot analysis
showed an induction of PLK1 phosphorylation in all the three PDK1-transformed cell
lines, but not in E545K- or Myc-transformed cells (Fig. 2C). Similar to PDK1 inhibitor
BX795, BI2536 treatment resulted in strong colony growth inhibition in both PDK1-and
Myc-transformed cells, but not in E545K-transformed cells (Fig. 2D and Supplementary
Fig. S2B). Furthermore, like BX795, PLK1 inhibitor BI2536 or GW843682X was able to
eliminate endogenus Myc in HEK-PDK1 cells but also the exogenous Myc in HEK-Myc
cells (Fig. 2E). This finding suggests that the exogenous Myc is also sensitive to the
perturbation of the basal level of PDK1-PLK1 signaling. Accordingly, in both HEK-
PDK1 and HEK-Myc cells, but not in HEK-E545K cells, BI2536 treatment resulted in
strong apoptosis, as demonstrated by both FACS analysis (Fig. 2F) and increased caspase
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

9
3 activity (Supplementary Fig. S2C), while E545K-transformed cells mainly displayed
G2/M arrest, a typical feature related to a mitotic effect following PLK1 inhibition
(Supplementary Fig. S2D). Furthermore, to confirm the PLK1-specific effect of BI2536,
PLK1 depletion by three independent siRNAs gave rise to similar effects on endogenous
and exogenous Myc and apoptosis in PDK1 or Myc-driven cells (Fig. 2G). These
findings suggest a crucial role for PDK1-PLK1 signaling in regulating Myc and cancer
cell survival. Consistent with the in vitro data, xenograft tumors derived from HEK-
PDK1 cells were highly sensitive to BI2536 treatment and displayed a strong tumor
regression following just two dosages, while the same treatment only induced tumor
growth inhibition in E545K-derived xenograft tumors (Fig. 2H).
We further show that the PLK1 regulation of Myc is not limited to transformed
cells but physiologically relevant in human cancers as either PLK1 knockdown or BI2536
treatment resulted in endogenous Myc protein depletion in various cancer cell lines
without changing Myc mRNA level (Supplementary Fig. S3A-C). In addition, a time
course analysis indicates that BI2536 treatment can result in Myc depletion as early as 8
hrs, concomitant with an early G2/M arrest (Supplementary Fig S3D), indicating that
Myc downregulation is unlikely to be a result of the secondary effect of cell cycle
change. BI2536 treatment also resulted in more effective growth inhibition in Myc-
dependent breast cancer cell lines compared to Myc-independent cells (except MDA-
MB-231) (Fig. 2I). These results further support a role of PDK1-PLK1 signaling in
supporting Myc-driven tumorigenesis.
PDK1 Induces PLK1 Phosphorylation in Human Cancer Cells
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

10
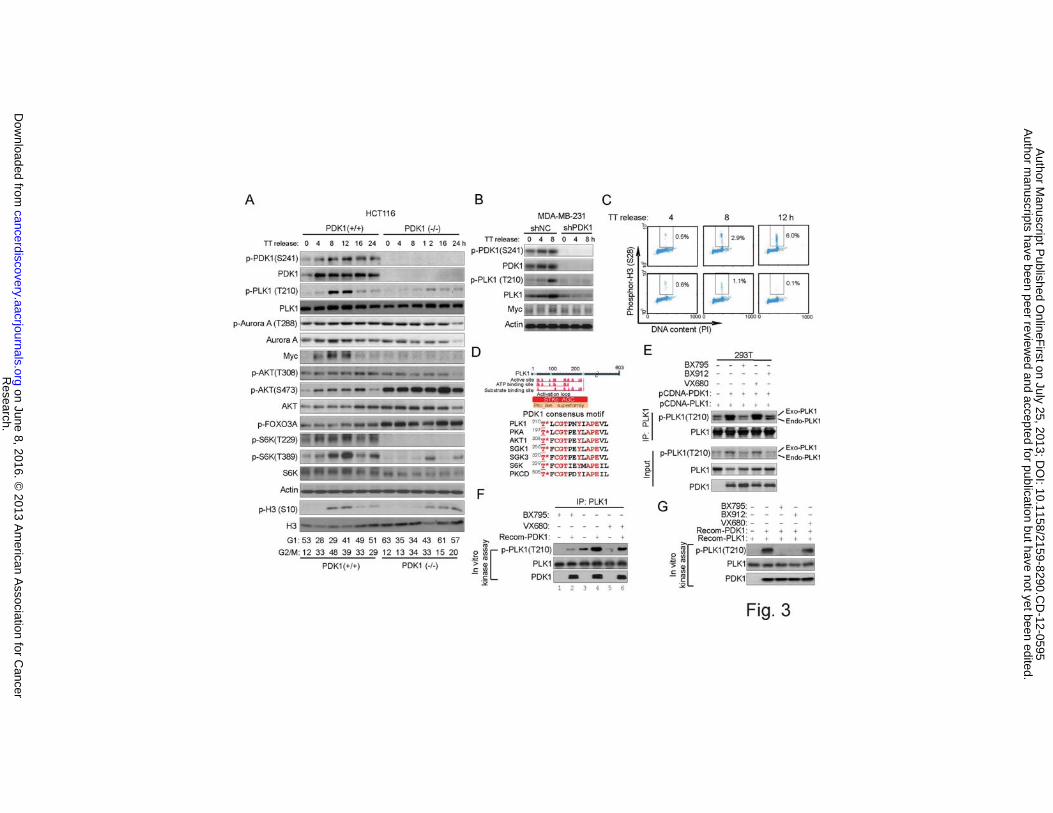
We next sought to determine whether or not the PLK1 activation by PDK1 seen in
transformed cells represents a finding that is physiologically relevant in human cancer
cells. To achieve this, we first used the colon cancer HCT116 and DLD1 cells in which
PDK1 is genetically knocked out (19). To facilitate the detection of phosphorylation of
PLK1, which is a mitotic kinase, cells were first synchronized by double-thymidine block
and then released into the cell cycle progressively (Fig. 3A and Supplementary Fig. S4A).
In PDK1 wild-type cells, we noticed progressive induction of PDK1 phosphorylation
upon cell cycle progression into mitosis as indicated by elevated levels of phosphor-
histone H3, which was accompanied by a similar pattern of PLK1 phosphorylation,
whereas in PDK1-/- counterparts we detected a much more deficient PLK1
phosphorylation and Myc accumulation but not the phosphorylation of the PLK1-related
kinase Aurora A (20, 21)(Fig. 3A and Supplementary Fig. S4A).
We also probed the changes of AKT-mTOR pathway in these cellular contexts.
Of notice, compared with p-PLK1, p-AKT (T308) was only modestly changed in this
condition in PDK1-/- cells, while p-AKT( S473) and p-FOXO3A were even enhanced in
both PDK1-/- cell lines (Fig. 3A). This could be due to the inhibition of S6K in PDK1-/-
cells, leading to a feedback activation of p-AKT (S473). In contrast, in a different
condition where cells were serum starved and then stimulated with growth factors for
early time points, we saw a clear p-AKT-(T308) inhibition in HCT116 PDK-/- cells
(Supplementary Fig. S4B). Thus, PDK1 regulates p-PLK1 and p-AKT (T308) in different
growth conditions. To further consolidate the data, we also performed PDK1 knockdown
by shRNA in MDA-MB-231 cells. The result shows again that the PDK1 knockdown
resulted in ablation of PLK1 phosphorylation and Myc accumulation (Fig. 3B), as well as
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

11
deficient entry into mitosis (Fig. 3C). These data consolidated a role of PDK1 in driving
PLK1 and Myc activation not just in a confined system but also in cancer cells.
Furthermore, in multiple cancer cell lines treated with BX795, GDC0941 or
MK2206 upon double-thymidine block and release (Supplementary Fig. S4C), we saw
that BX795 always blocked PLK1 phosphorylation and Myc accumulation, but inhibited
AKT phosphorylation in a cell line dependent manner (for example, AKT
phosphorylation is not affected by BX795 in MDA-MB-231 cells). By contrast,
GDC0941 and MK2206 consistently inhibited AKT phosphorylation in each of these cell
lines, but had little effect on PLK1 and Myc. Together, these data demonstrate the
physiological relevance of AKT-independent PDK1-PLK1-Myc signaling in cancer cells.
We next investigated whether or not PLK1 is a potential substrate of PDK1.
PDK1 is known to regulate AGC kinases. Protein domain analysis indicates that the
kinase domain of PLK1 is part of the AGC kinase family (Fig. 3D). Interestingly, the
amino acid sequence surrounding the Thr210 contains a consensus motif for PDK1 which
is found in many known PDK1 substrates (Fig. 3D), thus enhancing the possibility that
PLK1 could be a potential substrate of PDK1. Indeed, co-transfection of PDK1 and
PLK1 into 293T cells, followed by PLK1 immunoprecipitation, showed that PDK1
enhanced the phosphorylation of both the endogenous and exogenous PLK1, which was
abolished when cells were treated with BX795, BX912, but not Aurora A inhibitor
VX680 (Fig. 3E). This suggests that PDK1-induced PLK1 phosphorylation was unlikely
to be an indirect effect of Aurora A, which might be co-immunoprecipiated with PLK1.
Furthermore, in an in vitro kinase assay using endogenous PLK1 immunoprecipitated
from DLD1 as a substrate, recombinant PDK1 added in the kinase assay induced the
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

12
PLK1 phosphorylation at T210, which was markedly reduced in cells treated with BX795
(Fig. 3F), indicating that the recombinant PDK1 can directly induce endogenous PLK1
phosphorylation in vitro. Importantly, in cells treated with VX680, where the PLK1
phosphorylation was greatly reduced as expected, recombinant PDK1 still boosted the
PLK1 phosphorylation in the in vitro kinase assay (Fig. 3F). This further excludes the
possibility that PDK1 may induce PLK1 phosphorylation indirectly through Aurora A
kinase. Finally, in an in vitro kinase assays using both PDK1 and PLK1 as recombinant
proteins, we show that PDK1 induced a strong PLK1 phosphorylation, which was
blocked by BX795, BX912, but not by VX680 (Fig. 3G). Collectively, these experiments
provided evidence that PDK1 directly regulates PLK1 in human cancer cells.
PLK1 Directly Interacts with Myc and Induces Myc Phosphorylation in a PDK1-
Dependent Manner
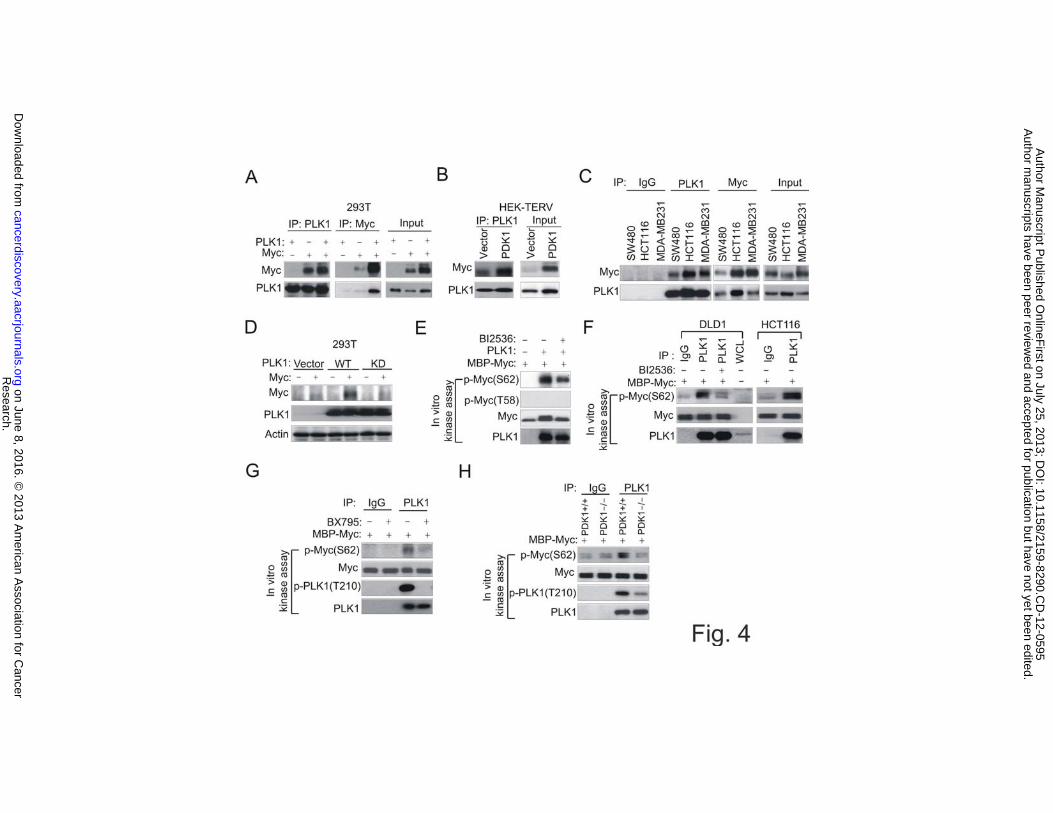
We next investigated whether PLK1 directly regulates Myc. Through co-
immunoprecipitation (co-IP) assay, we demonstrated an interaction between both
exogenous PLK1 and Myc in 293T cells (Fig. 4A). We also showed that the endogenous
interaction between the two proteins occurs in HEK-PDK1 cells as well as in various
cancer cell lines (Fig. 4B and C). We further showed that PLK1 kinase activity is
required for the Myc protein accumulation as the wild type PLK1, but not the kinase dead
mutant, induced strong Myc accumulation (Fig. 4D). Crucially, in vitro kinase assays
using either recombinant PLK1 (Fig. 4E) or endogenous PLK1 pulled down from the
cancer cells (Fig. 4F) demonstrated a robust induction of S62 phosphorylation of
recombinant Myc but not T58 phosphorylation, which was reduced in the presence of
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

13
BI2536. Importantly, the endogenous PLK1 kinase activity towards Myc phosphorylation
was strongly abolished in cells treated with BX795 (Fig. 4G) or in PDK1-/- cells (Fig.
4H). Thus, these results not only showed a direct phosphorylation of Myc by PLK1, but
also showed that PLK1 activity on Myc is crucially dependent on PDK1. Together, these
data reiterate the operation of PDK1-PLK1-Myc signaling in cancer cells.
PDK1-PLK1-Myc Signaling Drives Cancer Initiating Cell Maintenance and Self-
Renewal
During culturing of these transformed cells, we noticed that HEK-PDK1 cells, and to a
lesser extent, HEK-Myc cells, displayed distinct morphologies from HEK-E545K cells
and once they became confluent in culture, started to form semi-attached 3D clusters on
the plate (Fig. 5A, Upper), suggesting that they displayed tumorigenic and stem cell-like
properties. This feature, however, was not observed in E545K-transformed cells (Fig.
5A). Given a known role of Myc in inducing ESC- or CSC-like phenotypes in
differentiated somatic cells (16), it raises a possibility that PDK1, which activates Myc,
may have a similar capacity in inducing CSC-like behavior. This hypothesis was first
tested by using an in vitro assay for spheroids formation in serum-free suspension culture,
a property associated with cancer stem/progenitor cells (22). We observed that PDK1-or
Myc transformed HEK or HMEC cells formed large and abundant non-adherent
tumorspheres after 7 days of growth in suspension culture, whereas E545K-transformed
cells were only able to generate low number of small spheres (Fig. 5A, Bottom and
Supplementary Fig. S5A). These spheres were able to reform a monolayer when placed
back to a tissue culture plate containing serum-rich medium (Fig. 5B). Furthermore, after
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

14
dispersion into single cells, PDK1 or Myc-transformed HEK or HMEC cells reformed
spheres with increasing enrichments for at least 4 passages (Fig. 5C and Supplementary
Fig. S5B), indicating a gain of self-renewal capacity that resembles a stem cell-like
property.
To demonstrate the tumor-initiating capacity of these transformed cells in vivo,
we next injected these cells at different numbers into the flank of BALB/c nude mice.
Strikingly, 1000 HEK-PDK1 cells were sufficient to generate tumors in all 6 mice as
early as 2 weeks (Fig. 5D, left). HEK-Myc cells seemed to be less tumorigenic and
required 10,000 cells to generate a similar size of tumors. By contrast, 10,000 HEK-
E545K cells were unable to induce tumors in the mice (Fig. 5D). A further experiment
shows that as low as 100 PDK1 cells were sufficient to give rise to xenograft tumors,
whereas 3 x 106 E545K cells were required to generate observable tumors by 28 days
(Fig. 5D, right). Importantly, PDK1-associated primary xenograft tumors were self-
renewable, as determined by the ability to form secondary and tertiary tumors using as
low as 100 xenograft tumor cells (Fig. 5E). These in vitro and in vivo data demonstrated a
strong tumorigenicity of PDK1-transformed cells with self-renewal capacity.
We also tested the ability of PDK1 in inducing mouse embryonic fibroblasts
(MEFs) reprogramming. To achieve this, we used p53–deficient MEFs, as
immortalization by p53 inactivation has been shown to enhance MEF reprogramming
efficiency (23, 24). Again, PDK1 but not E545K was also able to induce Myc activation,
as well as tumorsphere formation in immortalized p53-/- MEFs (Figure S5C and S5D). In
the PDK1-sphere populations we detected strongly increased expressions of ES
pluripotency factors Sox2 and Oct4 as assessed by both qPCR and confocal imaging
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

15
compared to the monolayer growth (Figure S5E and S5F). When these MEF-PDK1cells
were cultured in ESC medium containing the differentiation inhibitor LIF (leukemia
inhibitory factor), MEF-PDK1 cells formed colonies resembling the ESC-like
morphology and were alkaline-phosphatase (AP) positive (Figure S5G), though we found
that these colonies were unable to maintain the ES-like morphology in the subsequent
passages, probably due to an incomplete reprogramming. Thus, in both human epithelial
cells and MEFs, PDK1 is able to induce PLK1 and Myc activation and ESC-like
property.
Aberrant high PDK1 activity has been shown in invasive and metastatic breast
tumor samples (25). To demonstrate the capacity of the PDK1-PLK1-Myc pathway in
regulating cancer stem cells, we utilized the highly invasive breast cancer MDA-MB-231
and SUM159PT cells that contain a high percentage of CD44+/CD24-low CSC-like
population (26). Intriguingly, PDK1-PLK1-Myc signaling was found to be enriched in
CD44+/CD24-low cells compared to the non-CD44+/CD24-low cells (Fig. 5F).
Knockdown of PDK1/PLK1 or treatment with their corresponding inhibitors
BX795/BI2536, resulted in marked reduction of CD44+/CD24-low populations (Fig. 5G,
H and I). By contrast, PI3K/AKT inhibitors GDC-0941 and MK-2206 were unable to do
so (Fig. 5I). Corresponding to the reduced CD44+/CD24-low cells, PDK1 or PLK1
inhibition either by gene knockdown or inhibitor treatment resulted in marked inhibition
of tumorsphere formation in MDA-MB-231 cells (Fig. 5J).
PDK1 Activates ES or CSC-like Transcriptional Programs
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

16
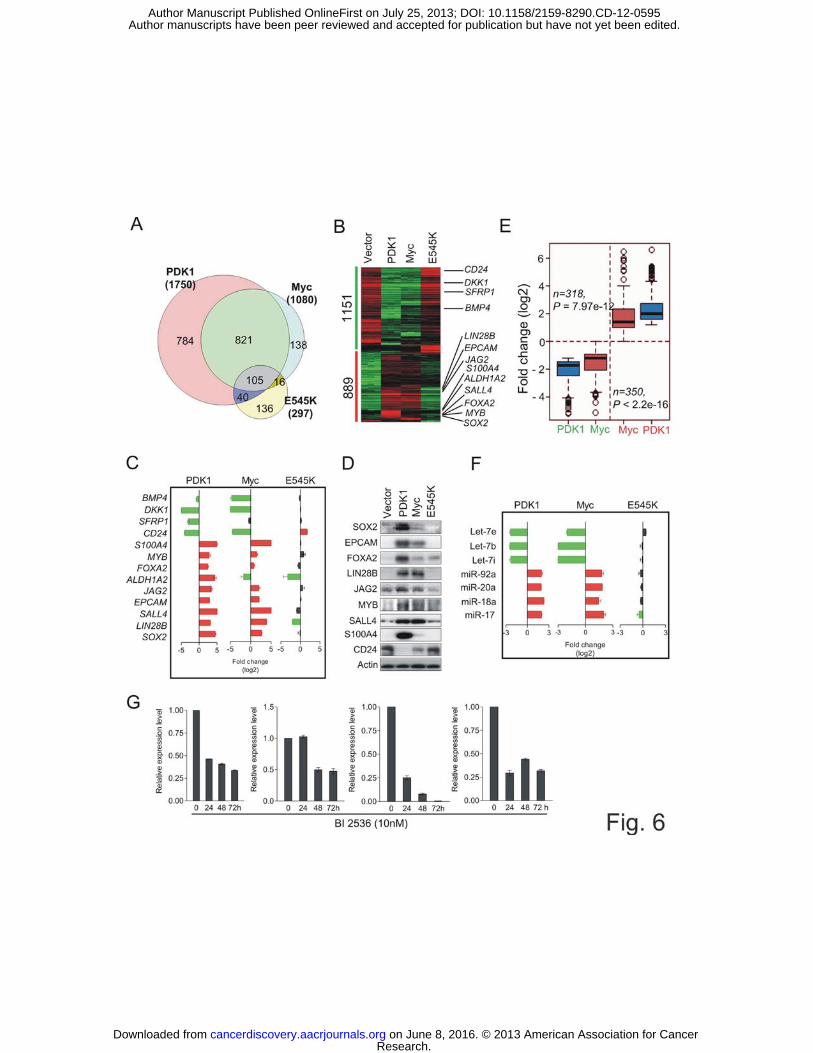
It is known that Myc is able to activate ESC-like transcriptional programs in adult
epithelial cells, resulting in a CSC-like phenotype in the appropriate genetic context (16).
To characterize the transcriptional program underlying the PDK1-induced CSC-like
behavior, we compared the gene expression profiles in HEK-PDK1, -Myc or -E545K
cells. Significant Analysis of Microarray (SAM) identified 1750, 1080 and 297
differentially expressed genes in these transformed cells when compared to non-
transformed control cells, respectively (FDR < 0.05, p < 0.01; Supplementary Table S1-
3). Gene Venn Diagram analysis shows that HEK-PDK1 shared a robust transcriptional
program with HEK-Myc, but had little overlap with HEK-E545K (Fig. 6A). In addition
to the PDK1 and Myc common gene set, PDK1 also regulates a unique set of 784 genes.
We further stratified the PDK1- or Myc-regulated genes into 889 upregulated and 1151
downregulated genes via gene cluster analysis (Fig. 6B). Notably, a number of well-
known genes implicated in ESC pluripotency or maintenance, including SOX2, LIN28B,
SALL4 and EZH2 (27), or CSC, including EPCAM, ALDH1A and S100A4 (28), were
upregulated in both HEK-PDK1 and HEK-Myc cells, but not in HEK-E454K cells (Fig.
6B). JAG2, which was recently shown to be a Myc target (29) with a role in modulating
CSCs, was also markedly induced by PDK1 and Myc but not by E545K. In addition, a set
of secreted inhibitors of autocrine signaling, including DKK1, SFRP1, and BMP4, whose
reduction has been recently shown by Weinberg’s group to enable self-renewal of
epithelial cells (30), were strongly repressed in PDK1 and Myc cells but not in E545K
cells. CD24, a negative selection marker for CSCs (31), was also selectively repressed in
PDK1 and Myc cells. The array results of selected genes were further validated by qRT-
PCR (Fig. 6C) and Western blotting (Fig. 6D). Notably, many genes co-regulated by
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

17
PDK1 and Myc, including SOX2, EPCAM, JAG2 and S100A4, were more affected by
PDK1 than Myc. In total, we identified 668 genes showing such a pattern (Fig. 6E and
Supplementary Table S4), which is consistent with a more robust role of PDK1 than Myc
in tumorigenesis.
We also investigated the changes of microRNAs that are Myc-associated and
implicated in ESC self-renewal. LIN28B is a known Myc target and is able to inhibit the
biogenesis of the let-7 family microRNAs (32). Inhibition of let-7 miRNAs has been
shown to enhance reprogramming of somatic cells to induced pluripotent (iPS) cells (33).
Myc also transactivates the mir-17-92 cluster, which is also implicated in ESC
maintenance (34). Consistent with Myc and LIN28B elevation, we detected a marked
upregulation of miR-17-92 and down-regulation of let-7s in PDK1 and Myc cells, but not
in E545K cells (Fig. 6F). Since let-7 suppresses its own negative regulator LIN28B (33),
it is likely that PDK1 enforces a feedback loop via Myc-LIN28B-mediated Let-7
downregulation to support the self-renewal program. Lastly, we show that BI2536
treatment of HEK-PDK1 cells resulted in reduced expression of some ESC or CSC-
related genes, including EPCAM, SOX2, SALL4, and JAG2 (Fig. 6G), validating a role of
PLK1 in the PDK1-mediated CSC gene signature. These findings indicate that PDK1 is
able to evoke multiple transcriptional programs that coordinately induced a remarkable
reprogramming towards a state resembling CSCs. In this process, Myc is one important
factor but not the only one that modulates the reprogramming.
PDK1-Induced CSC-like Gene Signature is Relevant to Human Cancers and is
Associated with Aggressive Tumor Behavior
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

18
Aberrant gene expression associated with ES cell identity, including ESC genes, Myc
targets and Polycomb targets, have been found in poorly differentiated tumors (14, 16,
35). By further interrogating several previously published datasets collectively, we show
that the above ESC-related genes were significantly enriched in the PDK1-dependent
transcriptome, including upregulation of 97 ESC-expressed genes and downregulaton of
182 Polyccomb targets (PRC genes) (Supplementary Fig. S6A and Table S5). In contrast,
the E545K-associated transcriptional program displayed a distinct gene set that is not
significantly associated with ESCs (Supplementary Fig. S6A).
We next determined whether PDK1-driven ESC-like gene expression is of clinical
relevance to human malignance. Gene set enrichment analysis (GSEA)(36) of several
previously published data sets showed that PDK1-induced ESC-like genes were found to
be significantly enriched in colon and lung tumor samples as compared to the normal
controls, while the Polycomb targets were reversely correlated in these samples
(Supplementary Fig. S6B). Moreover, in breast cancer, deregulation of these genes was
significantly correlated with the high-grade tumors as compared to the low-grade tumors
(Supplementary Fig. S6C). This indicates that aberrant expression of these PDK1-
regulated ESC genes is associated with malignant progression from normal to aggressive
tumors. In addition, we also demonstrated that the PDK1-regulated ESC-like gene
signature was associated with poor disease outcome as shown in the survival analysis of
breast and lung cancer cohorts (Supplementary Fig. S6D), providing a prognostic value
of these genes. Together, these findings demonstrate that the PDK1-activated ESC-like
gene signature we identified from the in vitro culture system is clinically relevant to
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

19
human cancers arising in distinct tissues and support a link of PDK1-Myc signaling to
aggressive cancer behavior.
BI2536 Synergizes with PI3K-mTOR Inhibitor BEZ235 to Induce Robust Apoptosis
and Tumor Growth Inhibition in CRC
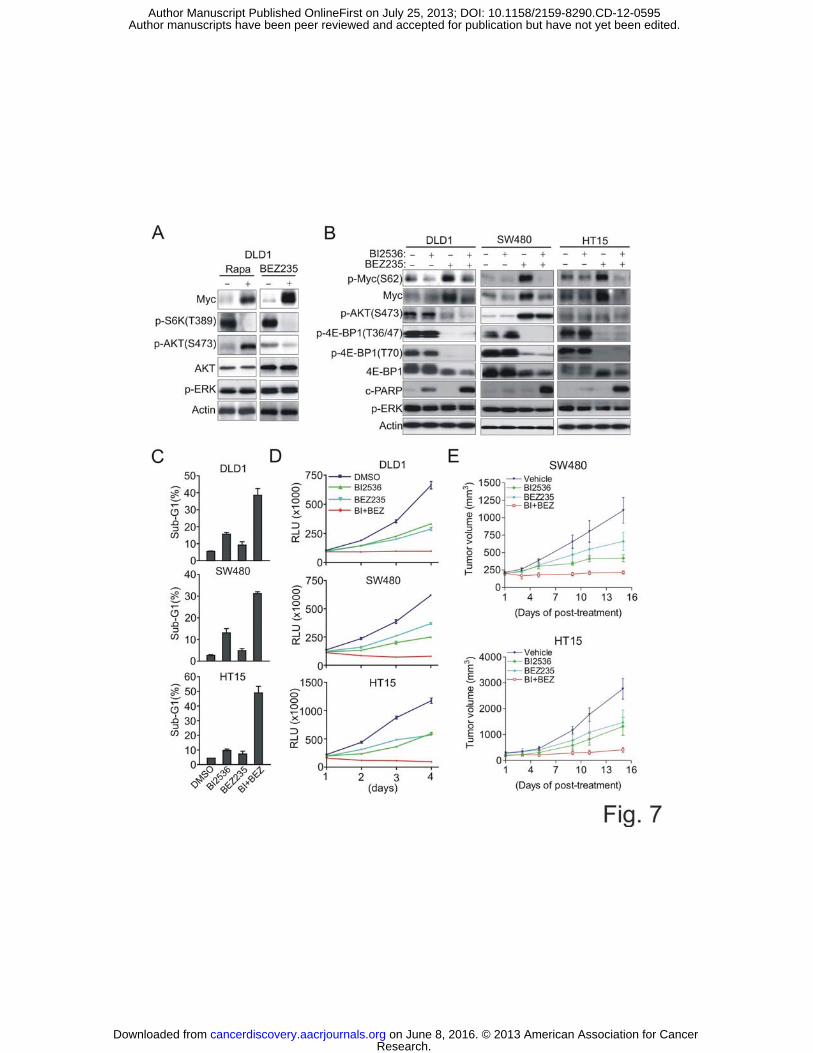
We have previously shown that mTOR inhibition by rapamycin or mTOR/Raptor
knockdown induces Myc accumulation in CRC, which can be inhibited by PDK1
inhibition, resulting in rapamycin sensitization (5). As our data now indicate that PLK1 is
required for PDK1-Myc signaling, together with our further observation that PLK1 is
highly expressed in CRC tumors compared to adjunct normal regions (Supplementary
Fig. S7A, B), we hypothesized that the PLK1 inhibitor could also sensitize CRC cells to
mTOR inhibitors through abolishing mTOR inhibitor-induced Myc activation. Classical
mTOR inhibitors like rapalogs are known to induce compensatory feedback activation of
PI3K-AKT due to S6K inhibition. BEZ235, a dual PI3K-mTOR kinase inhibitor is able
to overcome the feedback AKT activation and is currently being tested in several clinical
trials either as a single agent or in combination with other therapeutics. Unlike
Rapamycin treatment which induced both AKT and Myc activation in CRC cells,
BEZ235 did not induce AKT activation but retained the ability to induce Myc (Fig. 7A).
Of notice, neither drugs induced ERK activation in CRC, which is however often seen in
breast cancer cells (37). As expected, BI2536 co-treatment effectively removed BEZ235-
induced Myc induction (Fig. 7B). In these cells, BI2536 or BEZ235 alone failed to
induce significant apoptosis, but their combination, which resulted in inhibition of both
Myc and p-4E-BP1, induced a massive apoptosis, as evidenced by strong detection of
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

20
PARP cleavage (Fig. 7B), cells in Sub-G1 (Fig. 7C), and caspase 3 activation
(Supplementary Fig. S7C). The combinatorial effect was synergistic as shown by
combination index analysis (Supplementary Fig. S7D) and further confirmed by time-
course analysis of cell viability (Fig. 7D) and long term colony formation assay
(Supplementary Fig. S7E). Finally, to assess the potential of the combination strategy in
vivo, SW480 and HT15 cells were injected subcutaneously into nude mice to establish
tumor xenografts. We demonstrated that BEZ235 also induced Myc accumulation in the
xenograft tumors which can be inhibited via combination with BI2536 (Supplementary
Fig. S7F). Accordingly, the combination treatment induced synergistic tumor growth
inhibition compared to the single agent treatment, validating the in vitro findings (Fig.
7E).
Like BEZ235, a specific mTOR inhibitor PP242 (38) also generated similar
results on Myc, p-4EBP1 and apoptosis when combined with BI2536 (Supplementary
Fig. S8A, B). On the contrary, BI2536, though it also blocked Rapamycin-induced Myc
accumulation (Supplementary Fig. S8C), did not enhance apoptosis (Supplementary Fig.
S8D), only potentiated the anti-proliferation effect and xenograft tumor growth inhibition
(Supplementary Fig. S8E, F). This is probably due to the inability of rapamycin to block
4EBP1 phosphorylation (Supplementary Fig. S8C) as previously shown. 4EBP1, but not
S6K, has been recently shown to be the key effector of the mTOR pathway responsible
for cell proliferation and survival (39) and additional inhibition of 4EBP1
phosphorylation seems to be required for apoptosis induction in response to the AKT
inhibitor (40). Thus, the simultaneous inhibition of both Myc and 4EBP1 phosphorylation
upon combination of BI2536 and BEZ235 seemed to be crucial for apoptosis induction of
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

21
CRC cells. Taken all the data together, we conclude that the combination of BI2536 and
PI3K-mTOR dual inhibitors like BEZ235 may represent a promising treatment strategy
for CRC.
DISCUSSION
PDK1 Regulation of PLK1-Myc Signaling in Human Cancer Cells
Although PI3K-AKT signaling has been considered to be the main signaling pathway
associated with PDK1 in oncogenesis, our study uncovers another arm of signaling that
routs to PLK1-Myc to confer malignant phenotypes. Importantly, the pathway we
identified using a chemical genetic approach with a PDK1-transformed cell line has been
validated to be relevant in human cancers as demonstrated in multiple cancer cell lines
derived from various tissue types. Although in our system we detected AKT
phosphorylation at T308 by PDK1, we did not see AKT phosphorylation at S473 which
is required for a full AKT activation (6). This is in contrast to PI3K-transformed cells
where both AKT phosphorylations are strongly induced. This observation is consistent
with a recent report showing that PDK1-deficiency in colon cancer cells has only a
modest effect on p-AKT (T308) and had no effect on p-AKT (S473) (19). Our data thus
indicate that PDK1 signaling might be wired differentially in certain oncogenic contexts
to confer growth advantage that becomes less dependent on AKT. Indeed, previous
reports have shown that PDK1 is unlinked to PI3K signaling in a PTEN-deficient tumor
model (8) and can route through the AKT-independent pathway for cell survival in some
cancer cell lines (10). As PDK1 has attracted much attention as a potential therapeutic
target in cancer, we propose that Myc can be an alternative pharmacodynamic marker for
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

22
the evaluation of small molecule PDK1 inhibitors under preclinical and clinical
development.
Therapeutic Targeting of PDK1-PLK1 Signaling in Myc-dependent Tumors
An intriguing finding of this study is the identification of the crucial role of PDK1-PLK1-
Myc signaling for cancer cell survival. We provide evidence that PDK1 induces PLK1
phosphorylation and PLK1 binds to and induces Myc phosphorylation and protein
accumulation widely in cancer cells. A previous report has shown that PLK1-induced
Myc phosphorylation is required for SCF�TrCP-mediated Myc protein stabilization during
late stage cell cycle progression (41). We further show here that PLK1 can directly bind
to Myc. Regardless of whether or not PDK1-PLK1 signaling regulates Myc stability
through a similar or distinct mechanism, the direct regulation of Myc by PDK1-PLK1
signaling immediately suggested a therapeutic approach targeting Myc-driven tumors.
Indeed, our data show a preferentially killing of small molecule inhibitor of PDK1 or
PLK1 in Myc-dependent breast cancer cells compared with Myc-independent breast
cancer cells. Given that a clinical inhibitor of Myc is not available, small molecule
inhibitors like BI2536, which is currently in late stage of clinical trial, may provide an
alternative anti-Myc strategy. Given PLK1 is often found to be overexpressed in human
cancers (42), therapeutic targeting of PLK1 may yield a more favorable therapeutic index
in Myc-associated tumors.
Role of PDK1-PLK1-Myc Signaling in Driving Tumor Initiating Cells
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

23
The main characteristic of the PDK1-induced transformation is that it is able to induce
both genotype and phenotype of CSCs that has been proposed to account for tumor
initiation, progression and chemo-resistance (13, 31). We show that as low as 500 PDK1-
transformed cells can induce robust tumorigenicity and the PDK1 activates clinically-
relevant transcriptional programs associated with poor disease outcome. In addition,
PDK1 or PLK1 inhibition also resulted in disruption of both embryonic and adult stem
cell self-renewal while inducing differentiation. Activation of an ESC-like signature and
an ESC-like phenotype in differentiated somatic cells also indicates that the ES program
can be reactivated during the course of tumor progression and is not necessarily inherited
from a stem cell-of-origin. This notion is consistent with recent report from Weinberg’s
group showing spontaneous CSC-generation from mammary epithelial cells (43).
Furthermore, consistent with a previous report that PDK1 is hyperactivated in
invasive and metastatic breast cancer (25), we show that PDK1 or PLK1 inhibition in
highly invasive breast cancer MDA-MB-231 cells resulted in depletion of CSC-like
CD44+/CD24-low populations and accordingly strongly reduced tumorsphere formation,
while PI3K-AKT inhibition did not have such effects. Thus, small molecule inhibition of
PDK1-PLK1-Myc signaling for elimination of CSCs may provide a targeted therapy to
overcome recurrence of aggressive breast tumors following chemotherapy.
Combination of PLK Inhibitor and PI3K-mTOR Inhibitor for CRC
An additional therapeutic application of our studies is the identification of strategies to
overcome resistance to mTOR-targeted therapy in CRC. Drug resistance and tumor
recurrence is the main cause of patient relapse, possibly owing to recurrence of cancer
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

24
stem cells. We have previously discovered that mTOR inhibition induces Myc activation,
a compensatory effect mitigating the anti-proliferative effect of mTOR inhibitors in CRC
(5). We now show that PLK1 inhibitor blocks mTOR inhibitor-induced Myc activation,
providing a rational approach to develop a new combination therapy for CRC.
Specifically, low dose of PLK1 inhibitor BI2536 plus PI3K-mTOR dual inhibitor
BEZ235 induced massive apoptosis in CRC cells and a synergistic loss of colony
formation, suggesting that this strategy might be useful in CRC. Given both drugs are in
late stage clinical trials, we hope that our findings could spur clinical trials in CRC for
combination of BEZ235 and BI2536 to improve the therapeutic outcome.
MATERIALS AND METHODS
Constructs and Reagents
Human full-length PDK1, Myc, PIK3CA-E545K and PLK1 were cloned into PMN-
IRES-GFP retroviral vector and introduced into human epithelial cells and MEFs. All
kinase inhibitors used in this study were obtained from Axon Medchem. Information for
plasmid DNA vectors and stable cell line construction are provided in Supplemental
Materials and Methods
Cell Cultures
Cell cultures and various cellular assays are described in Supplementary Materials and
Methods. All cancer cell lines were purchased from American Type Culture Collection
(ATCC) (Manassas, VA) and No authentication of cell lines was done by the authors.
Mouse Experiments
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

25
All of the experiments in xenografts are described in Supplemental Materials and
Methods.
Immunobloting, immunoprecipitation and in vitro kinase assays Details are described in Supplemental Materials and Methods
Gene Expression, Data Analysis and Real-Time PCR Analysis
The microarray hybridization was performed using the Illumina Gene Expression Sentrix
BeadChip HumanHT-12_V4 (Illumina) and the data was analyzed using the GeneSpring
GX 11.0.2 (Agilent Technologies). Detailed information can be found in Supplemental
Materials and Methods. Primers used in real-time PCR analysis are described in Table
S6.
Statistical Analysis
PDK1 regulated ESC-like and PRC gene signature definition was described in
Supplementary Experimental Procedures. Gene Set Enrichment Analysis (GSEA) (36)
was conducted to assess the degree of correlation between PDK1-regulated gene
signatures and cancer phenotypes on different human patient. Survival curves were
calculated using the Kaplan-Meier survival analyses and the quantiles-rank test. Detailed
statistical analysis is included in Supplemental Information. Data are presented as mean ±
SEM, unless otherwise stated. A student’s t test was used to compare two groups for
statistical significance analysis.
Accession number
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

26
The microarray data are deposited into the Gene Expression Omnibus (GEO) with the
accession number GSE30669.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Materials and Methods, 8 figures, and
six tables.
ACKNOWLEDGEMENTS
We thank Dr. William C. Hahn for the HEK-TERV cells. We thank Dr. Fu Zheng for the human
PLK1 plasmids and Dr. Luca Primo for the PDK1 kinase-dead mutant construct. This work was
supported by the Agency for Science, Technology and Research of Singapore.
REFERENCES 1. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627-44. 2. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550-62. 3. Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15:161-70. 4. Maurer M, Su T, Saal LH, Koujak S, Hopkins BD, Barkley CR, et al. 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res. 2009;69:6299-306. 5. Tan J, Lee PL, Li Z, Jiang X, Lim YC, Hooi SC, et al. B55beta-associated PP2A complex controls PDK1-directed myc signaling and modulates rapamycin sensitivity in colorectal cancer. Cancer Cell. 2010;18:459-71. 6. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098-101. 7. Peifer C, Alessi DR. Small-molecule inhibitors of PDK1. ChemMedChem. 2008;3:1810-38. 8. Ellwood-Yen K, Keilhack H, Kunii K, Dolinski B, Connor Y, Hu K, et al. PDK1 Attenuation Fails to Prevent Tumor Formation in PTEN-Deficient Transgenic Mouse Models. Cancer Res. 2011;71:3052-65.
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

27
9. Gagliardi PA, di Blasio L, Orso F, Seano G, Sessa R, Taverna D, et al. 3-phosphoinositide-dependent kinase 1 controls breast tumor growth in a kinase-dependent but akt-independent manner. Neoplasia. 2012;14:719-31. 10. Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16:21-32. 11. Sato S, Fujita N, Tsuruo T. Involvement of 3-phosphoinositide-dependent protein kinase-1 in the MEK/MAPK signal transduction pathway. J Biol Chem. 2004;279:33759-67. 12. Zeng X, Xu H, Glazer RI. Transformation of mammary epithelial cells by 3-phosphoinositide-dependent protein kinase-1 (PDK1) is associated with the induction of protein kinase Calpha. Cancer Res. 2002;62:3538-43. 13. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-11. 14. Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499-507. 15. Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, et al. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell.143:313-24. 16. Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2:333-44. 17. Chen W, Possemato R, Campbell KT, Plattner CA, Pallas DC, Hahn WC. Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell. 2004;5:127-36. 18. Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 2011;335:348-53. 19. Ericson K, Gan C, Cheong I, Rago C, Samuels Y, Velculescu VE, et al. Genetic inactivation of AKT1, AKT2, and PDPK1 in human colorectal cancer cells clarifies their roles in tumor growth regulation. Proc Natl Acad Sci U S A. 2010;107:2598-603. 20. Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, et al. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008;455:119-23. 21. Seki A, Coppinger JA, Jang CY, Yates JR, Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science. 2008;320:1655-8. 22. Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506-11. 23. Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460:1140-4.
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

28
24. Marion RM, Strati K, Li H, Murga M, Blanco R, Ortega S, et al. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460:1149-53. 25. Xie Z, Yuan H, Yin Y, Zeng X, Bai R, Glazer RI. 3-phosphoinositide-dependent protein kinase-1 (PDK1) promotes invasion and activation of matrix metalloproteinases. BMC Cancer. 2006;6:77. 26. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983-8. 27. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663-76. 28. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555-67. 29. Yustein JT, Liu YC, Gao P, Jie C, Le A, Vuica-Ross M, et al. Induction of ectopic Myc target gene JAG2 augments hypoxic growth and tumorigenesis in a human B-cell model. Proc Natl Acad Sci U S A. 2010;107:3534-9. 30. Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell States in the breast. Cell. 2011;145:926-40. 31. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755-68. 32. Rybak A, Fuchs H, Smirnova L, Brandt C, Pohl EE, Nitsch R, et al. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nat Cell Biol. 2008;10:987-93. 33. Melton C, Judson RL, Blelloch R. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature. 2010;463:621-6. 34. Smith KN, Singh AM, Dalton S. Myc represses primitive endoderm differentiation in pluripotent stem cells. Cell Stem Cell. 2010;7:343-54. 35. Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, et al. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010;143:313-24. 36. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545-50. 37. Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065-74. 38. Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR, et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010;17:249-61. 39. Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science. 2010;328:1172-6.
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

29
40. She QB, Halilovic E, Ye Q, Zhen W, Shirasawa S, Sasazuki T, et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010;18:39-51. 41. Popov N, Schulein C, Jaenicke LA, Eilers M. Ubiquitylation of the amino terminus of Myc by SCF(beta-TrCP) antagonizes SCF(Fbw7)-mediated turnover. Nat Cell Biol. 2010;12:973-81. 42. Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer. 2010;10:825-41. 43. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011;108:7950-5. Figure Legends
Figure 1 PDK1 induces cell transformation through Myc induction
(A) Soft-agar growth of HEK-TERV cells infected with retroviral constructs expressing
empty vector, PDK1, Myc, shPTEN, or PIK3CA-E545K.
(B) Immunoblot analysis of indicated proteins in HEK-TERV-derived cell lines.
(C) Soft-agar growth of HEK-PDK1 and HEK-E545K cells transfected with non-
targeting siRNA (siNC) or Myc siRNA, respectively. * P<0.01.
(D) Soft-agar growth of HEK-PDK1, HEK-E545K and HEK-Myc cells treated with
BX795 (2.5 μM), GDC-0941 (0.5 μM), or MK2206 (0.5 μM) for 14 days. Right panel
shows the changes of Myc and AKT after indicated drug treatments.
(E) Soft-agar growth of HMEC and RWPE-1 cells expressing retroviral empty vector,
PDK1 or E545K.
(F) Immunoblot analysis of indicated proteins in HMEC and RWPE-1-derived cell lines.
(G) Immunoblot analysis of indicated cancer cell lines treated with PDK1 siRNA.
(H) Immunoblot analysis of indicated breast cancer cell lines treated with BX795 (2.5
�M) for 24h.
(I) Cell viability assay showing the dose response of a panel of breast cancer cell lines
that are Myc-dependent (MDA-MB-231, SUM159PT and Hs578T) and Myc-independent
(T47D and BT474) to BX795 treatment.
All the data in the graph bars represent mean ± SEM, n=3.
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

30
Figure 2 PLK1 is a crucial downstream effector of PDK1 for Myc activation and cell
survival
(A) Cell viability of HEK-vector, HEK-PDK1 and HEK-E545K cell treated with the
indicated concentrations of BI2536 and GW843682X for 4 days.
(B) Cell viability of RWPE-1 and HMEC-derived cell lines treated with 10 nM BI2536
for 4 days.
(C) Immunoblot analysis of PLK1 in indicated cell lines.
(D) Soft-agar growth of indicated cell lines treated with 10 nM BI2536 for 14 days.
(E) Immunoblot analysis in HEK-PDK1 and HEK-Myc cells treated with BI2536 and
GW843682X as indicated concentration for 24 hr.
(F) Apoptosis by sub-G1 analysis of indicated cell lines treated with 10 nM BI2536 for
48 hr.
(G) Apoptosis of indicated cell lines treated with NC or PLK1 siRNAs for 48 hr (Upper)
and immunoblot analysis of indicated proteins (Bottom).
(H) Xenograft tumor growth of HEK-PDK1 and HEK-E545K cells in nude mice treated
with 50 mg/kg BI2536 twice per week as described in Experimental Procedures. Data are
means ± SEM (n=5 for each group).
(I) Cell viability assay showing the dose response of a panel of breast cancer cell lines
that are Myc-dependent (MDA-MB-231, SUM159PT and Hs578T) and Myc-independent
(T47D, BT474, MCF-10A and HMEC) to BI2536 treatment.
Figure 3 PDK1 regulates PLK1 in vivo and in vitro
(A) Immunoblot analysis of indicated proteins in HCT116 PDK1 wild-type (PDK1+/+)
and knockout (PDK1-/-) cells. Cells were synchronized by double-thymidine block and
released into cell cycle at indicated times.
(B) Immunoblot analysis of indicated proteins in MDA-MB-231 shNC and PDK1
knockdown (shPDK1) cells. Cells were synchronized by double-thymidine block and
released into cell cycle at indicated times.
(C) Cells were synchronously released from double-thymidine arrest (TT) and harvested
at the indicated times for FACS analysis. Percentages of cells positive for phosphor-H3
(S28) are indicated.
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

31
(D) PLK1 protein domain analysis (upper) and PDK1 consensus motif alignment with
other known PDK1 substrates (bottom).
(E) Immunoblot analysis of immunoprecipitated PLK1 in 293T cells transfected with
PLK1, or-cotransfected with PDK1, with or without 2.5 �M BX795, 5.0 �M BX912 and
1.0 μM VX680 treatment for 24 hr.
(F) In vitro immunoprecipitation-kinase assay using recombinant PDK1 and
immunoprecipitated endogenous PLK1 from DLD1 cells as substrate. Cells were
synchronized by a double-thymidine arrest and released in the presence or absence of 2.5
μM BX795 or 1.0 μM VX680 for 8 hr. PLK1 IP-kinase assay was performed and the
phosphorylation of PLK1 was assessed by using p-T210 PLK1 antibody.
(G) In vitro kinase assay using recombinant PDK1 and recombinant PLK1 with or
without 1.0 μM BX795, 1.0 μM BX912 and 1.0 μM VX680.
Figure 4 PLK1 interacts with Myc and induces Myc phosphorylation
(A) Co-immunoprecipitation analysis in 293T cells transfected with ectopic PLK1, Myc,
or both.
(B) Co-immunoprecipitation analysis of endogenous PLK1 and Myc in HEK-Vector and
HEK-PDK1 cells.
(C) Co-immunoprecipitation analysis of endogenous PLK1 and Myc in cancer cell lines
(D) Immunoblot analysis of Myc protein expression in 293T cells transfected with empty
vector, PLK1 WT or kinase dead mutant of PLK1 (KD) in the absence or presence
ectopic Myc.
(E) Immnoblot analysis of in vitro kinase assay using recombinant PLK1 and
recombinant Myc proteins in the presence or absence of BI2536. Phosphorylation of Myc
was assessed by indicated Myc antibodies.
(F) Immnoblot analysis of in vitro kinase assay using immunoprecipitated PLK1 and
recombinant Myc proteins in the presence or absence of BI2536.
(G) Immnoblot analysis of in vitro kinase assay using immunoprecipitated PLK1 from
DLD1 cells treated with or without 2.5 �M BX795.
(H) Immnoblot analysis of in vitro kinase assay using immunoprecipitated PLK1 from
DLD1 and DLD1 PDK1-/- cells.
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

32
Figure 5 PDK1-PLK1-Myc signaling drives CSC-like phenotypes
(A) Representative phase-contrast images of HEK-vector, PDK1, Myc or E545K cells
grown in monolayer culture in upper panel. Lower panel shows tumorsphere formation in
suspension culture without serum. Scale bar represents 100 μm.
(B) Spheres formed in suspension culture reattached when transferred back to gelatin-
coated culture plates in DMEM, 10% FBS and the sphere reformed a monolayer for 48
hr. Scale bar represents 100 μm.
(C) Self-renewal capacity of PDK1 and Myc-transformed cells. Primary tumorspheres
were trypsinized into single cells and reformed spheres 7 days later for 4 passages.
(D) Xenograft tumor growth in nude mice. HEK-PDK1, Myc or E545K injected with
indicated cell numbers were shown. Data are means ± SEM.
(E) Xenograft tumor formation frequencies of tumor-initiating cells derived from the
first, second, and third passage tumors arising from HEK-PDK1 cells.
(F) Immunoblot analysis showing the PDK1-PLK1-Myc signaling in CD44+/CD24-low
or non-CD44+/CD24-low populations.
(G) Representative FACS profiles for CD44+/CD24-low or non-CD44+/CD24-low
populations in MDA-MB-231 and MDA-MB-231-PDK1 KD cells. Inset: Isotype control.
(H) Bar graphs showing the percentages of CD44+/CD24-low cells in MDA-MB-231
cells depleted of PDK1 or PLK1. * P<0.005.
(I) Bar graphs showing the percentages of CD44+/CD24-low cells in MDA-MB-231 cells
treated with indicated inhibitors. * P<0.01, ** P<0.005.
(J) Bar graphs showing the number of tumorspheres of MDA-MB-231 cells depleted of
PDK1/PLK1 (Left) or treated with BX795/BI2536. * P<0.01.
Data are means ± SEM (n=3).
Figure 6 PDK1 evokes ESC-like gene expression profile
(A) Venn diagram showing the overlapping of differentially expressed genes in HEK-
PDK1, Myc or E545K as compared with HEK-vector control cells. .
(B) Heat map of differentially expressed genes in HEK-PDK1, Myc or E545K cells.
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

33
(C) qRT-PCR analysis of representative genes in HEK-transformed cells. Data are shown
as gene expression fold change (log 2) relative to HEK-vector cells. Red and green bars
indicate upregulation and downregulation, respectively. Black bars indicate <0.6-fold
change in log 2 (1.5 fold in linear scale). Data are means ± SEM, n=3.
(D) Immunoblot analysis of indicated proteins.
(E) 318 upregulated and 350 downregulated genes show significant differences between
PDK1 and Myc regulation. Average gene expression levels indicating a higher impact of
PDK1 on these genes.
(F) qRT-PCR analysis of indicated miRNAs in HEK-PDK1,-Myc and -E545K cells. Data
are presented as (c).
(G) qRT-PCR analysis of indicated genes in HEK-PDK1 cells treated with 10 nM
BI2536 at indicated times.
Data are means ± SEM (n=3).
Figure 7 BI2356 synergizes with BEZ235 to induce synthetic lethality in CRC both in vitro and in vivo (A) Immunoblot analysis of DLD1 cells treated with 100 nM Rapamycin or 100 nM
BEZ235 for 48 hr.
(B) Immunoblot analysis of DLD1, SW480 and HT15 cells treated with 10 nM BI2536,
100 nM BEZ235 alone or combination for 48 hr.
(C) Sub-G1 detection of apoptosis in DLD1, SW480 and HT15 cells treated as (B).
(D) The growth curves of DLD1, SW480 and HT15 cells treated with 10 nM BI2536,
100 nM BEZ235 single or combination for 4 days. RLU means relative luminescence
units.
(E) Xenograft tumor growth of SW480 and HT15 cells in nude mice treated with BI2536
at 50 mg/kg or BEZ235 at 35 mg/kg or both, every other day as described in
Experimental Procedures. Error bars represent ± SEM (n=6 per group).
Data are means ± SEM (n=3).
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

Research. on June 8, 2016. ©
2013 Am
erican Association for C
ancercancerdiscovery.aacrjournals.org
Dow
nloaded from
Author m
anuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Author M
anuscript Published O
nlineFirst on July 25, 2013; D
OI: 10.1158/2159-8290.C
D-12-0595

Research. on June 8, 2016. ©
2013 Am
erican Association for C
ancercancerdiscovery.aacrjournals.org
Dow
nloaded from
Author m
anuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Author M
anuscript Published O
nlineFirst on July 25, 2013; D
OI: 10.1158/2159-8290.C
D-12-0595
Research. on June 8, 2016. ©
2013 Am
erican Association for C
ancercancerdiscovery.aacrjournals.org
Dow
nloaded from
Author m
anuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Author M
anuscript Published O
nlineFirst on July 25, 2013; D
OI: 10.1158/2159-8290.C
D-12-0595
Research. on June 8, 2016. ©
2013 Am
erican Association for C
ancercancerdiscovery.aacrjournals.org
Dow
nloaded from
Author m
anuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Author M
anuscript Published O
nlineFirst on July 25, 2013; D
OI: 10.1158/2159-8290.C
D-12-0595

Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595

Published OnlineFirst July 25, 2013.Cancer Discovery Jing Tan, Zhimei Li, Puay Leng Lee, et al. and Resistance to mTOR-targeted TherapyOncogenic Transformation and Tumor Initiating Cell Activation PDK1 Signaling Towards PLK1-Myc Activation Confers
Updated version
10.1158/2159-8290.CD-12-0595doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerdiscovery.aacrjournals.org/content/suppl/2013/07/26/2159-8290.CD-12-0595.DC1.htmlAccess the most recent supplemental material at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
Research. on June 8, 2016. © 2013 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on July 25, 2013; DOI: 10.1158/2159-8290.CD-12-0595




















