
Origin of the Differential Nanoscale Reactivity of Biologically and Chemically Formed Green Rust...
10
Origin of the Differential Nanoscale Reactivity of Biologically and Chemically Formed Green Rust Crystals Investigated by Chemical Force Spectroscopy Asfaw Zegeye, †,‡,⊥,# Marjorie Etique, †,‡,# Ce ́ dric Carteret, †,‡ Christian Ruby, †,‡ Pierre Schaaf, §,∥ and Gre ́ gory Francius* ,†,‡ † Universite ́ de Lorraine, Laboratoire de Chimie Physique et Microbiologie pour l’Environnement, UMR 7564, Villers-le ̀ s-Nancy F-54601, France ‡ CNRS, Laboratoire de Chimie Physique et Microbiologie pour l’Environnement, UMR 7564, Villers-le ̀ s-Nancy F-54601, France § Inserm/Universite ́ de Strasbourg, UMR S-1121, 11 rue Humann, 67085 Strasbourg, France ∥ Institut Charles Sadron, CNRS UPR 22, BP 84749, F-67034 Strasbourg Cedex 2, France * S Supporting Information ABSTRACT: Iron-containing nanoparticles, such as green rusts, can be formed by either chemical (c-GR) or biological (b-GR) pathways. It is known that c-GRs display very high reactivity toward organic and inorganic contaminants and thus have great potential for the remediation of contaminated environments, whereas b-GRs are very weakly reactive. This reactivity difference is usually attributed to much higher surface/volume ratio of c-GR compared to b-GR. Using atomic and chemical force microscopy to probe the reactivity at the nanoscale of both types of nanoparticles, we are able to show that the primary reason for the low reactivity of b-GR is not the low surface/volume ratio but the passivation of the surface due to the presence of biological exopolymers (EPS). This conclusion should hold true for many biological nanoparticles and allows us to explain their often observed low, yet unexplained, reactivity. ■ INTRODUCTION Iron oxides/hydroxides are some of the most abundant and reactive natural minerals. 1 The reactivity of the iron oxide− aqueous solution interface is due to its acid−base, ligand exchange, and redox chemistry involving protons, hydroxyl groups, and iron cations that constitute the mineral surface. 2 As a result, iron oxides/hydroxides play a fundamental role in elemental cycling in oceanic and terrestrial environments through nutrient and contaminant uptake and release, 3 act as electron acceptors or donors in anaerobic respiration by microorganisms, 4 and buffer soil pH and redox potential among other roles. Given the importance of the iron oxide/hydroxide surfaces in these processes, there is an urgent need to probe their reactivity at the nanoscale to gain a better understanding of the mechanisms governing their environmental significance. Among the iron hydroxide groups, green rusts (GRs) have been recognized as promising minerals in environmental remediation processes. 5 GRs, [Fe II (1−x) Fe III x (OH) 2 ] x+• [(x/n)- A n− (m)H 2 O] x− , are Fe(II−III) double hydroxyl salt minerals with a layered structure consisting of positively charged hydroxide layers, [Fe II (1−x) Fe III x (OH) 2 } x+ ], separated by an interlayer containing intercalated anions, A n− (e.g., CO 3 2− , SO 4 2− , Cl − ), and water molecules. 6 The redox flexibility of GRs 7 enables them to reduce relevant organic and inorganic pollutants. This redox flexibility depends on the Fe 2+ content of the mineral and the subsequent structural modification. 8 The ability of GRs to reduce relevant pollutants such as nitrate, their environmental significance, 9 and their potential role in remediation processes 10 has naturally attracted considerable interest from the scientific community. 11 While the mechanisms of the reactivity of GRs are still under investigation, published data highlight the role that hydrated interlayer spacing plays during their oxidation by pollutants such as nitrate and chromate. 12 Two modes of GR oxidation have been identified: 13 (i) a dissolution−recrystallization mechanism involving the mineralogical transformation of GRs into Fe III oxyhydroxides (i.e., ferrihydrite, magnetite, goethite, etc.) and (ii) a solid state reaction where GRs are transformed in situ into a ferric homologue called “ferric GR” (GR*). 14 This latter mechanism involves electron transfer and a progressive deprotonation of the hydroxyl groups of the Fe−(O−H) octahedral sheets. 15 However, only the reactivity of chemically formed GRs (c-GR) against several pollutants has been reported, even though their biological counterpart displays the same mineralogical characteristics but exhibits a more important stability in air and in aqueous media. Biological GRs (b-GR) induced by iron respiring bacteria, such as Shewanella putrefaciens, 16 are intimately intermixed with Received: January 15, 2014 Revised: February 26, 2014 Article pubs.acs.org/JPCC © XXXX American Chemical Society A dx.doi.org/10.1021/jp500462r | J. Phys. Chem. C XXXX, XXX, XXX−XXX
-
Upload
univ-lorraine -
Category
Documents
-
view
5 -
download
0
Transcript of Origin of the Differential Nanoscale Reactivity of Biologically and Chemically Formed Green Rust...

Origin of the Differential Nanoscale Reactivity of Biologically andChemically Formed Green Rust Crystals Investigated by ChemicalForce SpectroscopyAsfaw Zegeye,†,‡,⊥,# Marjorie Etique,†,‡,# Cedric Carteret,†,‡ Christian Ruby,†,‡ Pierre Schaaf,§,∥
and Gregory Francius*,†,‡
†Universite de Lorraine, Laboratoire de Chimie Physique et Microbiologie pour l’Environnement, UMR 7564, Villers-les-NancyF-54601, France‡CNRS, Laboratoire de Chimie Physique et Microbiologie pour l’Environnement, UMR 7564, Villers-les-Nancy F-54601, France§Inserm/Universite de Strasbourg, UMR S-1121, 11 rue Humann, 67085 Strasbourg, France∥Institut Charles Sadron, CNRS UPR 22, BP 84749, F-67034 Strasbourg Cedex 2, France
*S Supporting Information
ABSTRACT: Iron-containing nanoparticles, such as green rusts, can be formed by eitherchemical (c-GR) or biological (b-GR) pathways. It is known that c-GRs display very highreactivity toward organic and inorganic contaminants and thus have great potential for theremediation of contaminated environments, whereas b-GRs are very weakly reactive. Thisreactivity difference is usually attributed to much higher surface/volume ratio of c-GRcompared to b-GR. Using atomic and chemical force microscopy to probe the reactivity atthe nanoscale of both types of nanoparticles, we are able to show that the primary reasonfor the low reactivity of b-GR is not the low surface/volume ratio but the passivation of thesurface due to the presence of biological exopolymers (EPS). This conclusion should holdtrue for many biological nanoparticles and allows us to explain their often observed low, yetunexplained, reactivity.
■ INTRODUCTION
Iron oxides/hydroxides are some of the most abundant andreactive natural minerals.1 The reactivity of the iron oxide−aqueous solution interface is due to its acid−base, ligandexchange, and redox chemistry involving protons, hydroxylgroups, and iron cations that constitute the mineral surface.2 Asa result, iron oxides/hydroxides play a fundamental role inelemental cycling in oceanic and terrestrial environmentsthrough nutrient and contaminant uptake and release,3 act aselectron acceptors or donors in anaerobic respiration bymicroorganisms,4 and buffer soil pH and redox potential amongother roles. Given the importance of the iron oxide/hydroxidesurfaces in these processes, there is an urgent need to probetheir reactivity at the nanoscale to gain a better understandingof the mechanisms governing their environmental significance.Among the iron hydroxide groups, green rusts (GRs) have
been recognized as promising minerals in environmentalremediation processes.5 GRs, [FeII(1−x)Fe
IIIx(OH)2]
x+•[(x/n)-An−(m)H2O]
x−, are Fe(II−III) double hydroxyl salt mineralswith a layered structure consisting of positively chargedhydroxide layers, [FeII(1−x)Fe
IIIx(OH)2}
x+], separated by aninterlayer containing intercalated anions, An− (e.g., CO3
2−,SO4
2−, Cl−), and water molecules.6 The redox flexibility ofGRs7 enables them to reduce relevant organic and inorganicpollutants. This redox flexibility depends on the Fe2+ content ofthe mineral and the subsequent structural modification.8 The
ability of GRs to reduce relevant pollutants such as nitrate, theirenvironmental significance,9 and their potential role inremediation processes10 has naturally attracted considerableinterest from the scientific community.11
While the mechanisms of the reactivity of GRs are still underinvestigation, published data highlight the role that hydratedinterlayer spacing plays during their oxidation by pollutantssuch as nitrate and chromate.12 Two modes of GR oxidationhave been identified:13 (i) a dissolution−recrystallizationmechanism involving the mineralogical transformation of GRsinto FeIII oxyhydroxides (i.e., ferrihydrite, magnetite, goethite,etc.) and (ii) a solid state reaction where GRs are transformedin situ into a ferric homologue called “ferric GR” (GR*).14 Thislatter mechanism involves electron transfer and a progressivedeprotonation of the hydroxyl groups of the Fe−(O−H)octahedral sheets.15 However, only the reactivity of chemicallyformed GRs (c-GR) against several pollutants has beenreported, even though their biological counterpart displaysthe same mineralogical characteristics but exhibits a moreimportant stability in air and in aqueous media.Biological GRs (b-GR) induced by iron respiring bacteria,
such as Shewanella putrefaciens,16 are intimately intermixed with
Received: January 15, 2014Revised: February 26, 2014
Article
pubs.acs.org/JPCC
© XXXX American Chemical Society A dx.doi.org/10.1021/jp500462r | J. Phys. Chem. C XXXX, XXX, XXX−XXX

biopolymers produced by bacteria and known as exopolymericsubstances (EPS). These macromolecules are often associatedwith mineral surfaces and are involved in many mineralinterfacial processes such as dissolution,17,18 biomineraliza-tion,19 and pollutant distribution.20 However, few studiesrelated to the interactions of EPS and mineral surfaces arereported in the literature,21 despite the important issue tounderstand the mechanisms and the role of EPS in formation,reactivity, and protection of mineral particles.In the present study, we demonstrate the protective role of
the EPS that prevents the transformation of b-GR toward athermodynamically more stable mineral such as magnetite. Dueto the interaction between the surface functional groups of GRsand organic polymers, such as cellular debris, one canhypothesize that the surface properties of b-GR are dramaticallymodified in comparison with the purely synthetic c-GR. Withincharacterization techniques, chemical force microscopy (CFM)has the advantage of allowing the quantification ofphysicochemical properties22,23 such as thermodynamic fea-tures,24 molecular dynamic processes,25 and surface reactivity26
of mineral and biological samples27 in combination with high-resolved images. We will monitor green rust oxidation and theresulting deprotonation kinetics of GR particles in the presenceand absence of EPS with this technique to correlate the stabilityof c-GR and b-GR particles with the presence of biopolymers.Infrared spectroscopy, Raman experiments, and atomic forcemicroscopy (AFM) will also be used to support CFMexperiments to quantify and understand at the nanoscale thechemical modifications of GR particles during nitrate oxidation.We will also prove the presence of EPS covering the b-GRsurfaces and their protective effect in respect with oxidation.Besides, CFM experiments performed with c-GR coated withEPS will emphasize the role played by these biopolymers on thestability and reactivity of the mineral particles. This studyrepresents the first attempt to characterize the physicochemicalproperties of c-GR and b-GR at a molecular level forapplications in heterogeneous oxidation. It is critical tounderstand the molecular interactions of these particles withenvironmentally relevant pollutants such as nitrate and hence topredict their behavior in natural and engineered systems forremediation purposes. In addition, this is also an importantissue for the understanding of bacterial involvement and impactwith respect to iron geochemical cycle, biocorrosion, andbiofouling processes, which are relevant in the waterdistribution industry.
■ EXPERIMENTAL SECTIONChemical and Biochemical Synthesis of Hydroxycar-
bonated Green Rust and Sample Preparation. Thechemical green rust c-GR was synthesized using a coprecipi-tation method described by Bocher et al.28 by mixing a solutionof NaOH and Na2CO3 with a solution of FeII and FeIII salts. Inthe anoxic chamber (N2/H2, 95%/5%), ferrous sulfateheptahydrate FeSO4·7H2O and ferric sulfate pentahydrateFe2(SO4)3·5H2O were dissolved in 30 mL of demineralizedand deaerated water in a 100 mL flask. The ferric molar fractionx = [FeIII]/([FeII] + [FeIII]) was set at 0.33. Then 30 mL of abasic solution of NaOH and Na2CO3 was added to the FeII−FeIII solution, corresponding to a [OH−]/([FeII] + [FeIII]) ratioof 2 and [CO3
2−]/([FeII] + [FeIII]) ratio of 7/6. To avoid theformation of sulfated green rust GR2(SO4
2−), carbonate anionswere in slight excess compared with sulfate anions. A bluish-green precipitate appeared immediately. The flask was sealed
with a thick butyl rubber stopper and purged for 30 min bybubbling Ar to outgas atmospheric N2.The biological green rust b-GR was generated using the
bioreduction of γ-FeOOH as reported by Zegeye et al.29 Briefly,Shewanella putrefaciens CIP 8040T grown aerobically to astationary growth phase (24 h) in tryptic soy broth washarvested by centrifugation, washed twice with sterile 0.7%NaCl, concentrated in the same medium, and purged for 30min by bubbling with N2. An inoculum of 9.0 × 109 cells mL−1
was used to inoculate the basal medium containing 300 mM γ-FeOOH as an electron acceptor, sodium methanoate (160 mMHCOONa, final concentration) as the electron donor, andsodium AQDS (100 μM, final concentration) as an electronshuttle. The pH measured after all components had been mixedwas 6.8. The culture was incubated at 30 °C in darkness, andthe precipitated b-GR was recovered by centrifugation after onemonth of the nitrate exposure. The same procedure describedby Zegeye et al.30 was used to wash and remove bacterial cellsfrom the biogenic green rust. Transmission electron micros-copy analyses confirmed that the washing procedure did notinduce the GR transformation.
Infrared Spectroscopy. Infrared measurements werecarried out on a Nicolet 8700 spectrometer continuouslypurged with ultrapure N2, equipped with a KBr beam splitterand an MCT detector. The spectral resolution and the totalacquisition time were, respectively, 4 cm−1 and 2 min. TheFTIR spectra in diffuse reflectance mode were collected usingHarrick Praying Mantis equipment. To perform the analysis,the samples were first diluted in a KBr matrix (5 wt %). Thesamples were mixed very gently with KBr in an agate mortar, sothat these mixtures were not subjected to any elevatedpressures. It should be noted that we took care to avoidoxygen: the samples were prepared in an anaerobic chamber(Coy Laboratory products) and enclosed in an airtight HarrickHVC-DRP-4 cell with KBr windows. The spectrum wasrecorded at room temperature in the following five minutes.Reflectances Rs of the sample and Rr of pure KBr, used as anonabsorbing reference powder, were measured under thesame conditions. The oxide-hydroxide reflectance is defined asR = Rs/Rr. The spectra are shown in pseudoabsorbance (−logR) mode.
AFM Imaging and Chemical Force MicroscopyMeasurements. AFM images and force−distance curveswere obtained at room temperature in liquid, using acommercial microscope (MFP3D-BIO, Asylum ResearchTechnology, Atomic Force F&E GmbH, Mannheim, Ger-many). Silicon nitride AFM tips (MLCT-Au, Bruker AXS,Palaiseau, France) with low spring constants of about 8−12pN/nm were used for imaging. Prior to each experiment, thegeometry of the tip was systematically controlled using acommercial grid for 3-D visualization (TGT1, NT-MTDCompany, Moscow, Russia). Experiments were performed inpotassium nitrate solution (KNO3) at 1 mM with pH = 3, 7,and 10 at room temperature. Chemical force microscopy(CFM) measurements were performed in Milli-Q water withgold-coated AFM tips (NPG-10, Bruker AXS, Palaiseau,France) functionalized with 11-mercapto-undecanoic acid(HS-C11H22-COOH) as the reactive probe. The quality ofAFM tip modification was systematically verified by forcemeasurements performed on reference surfaces (see moredetails in the Supporting Information). Force mappings wererealized with a loading rate of about 10 000 pN/scorresponding to a pulling speed of 850−1250 nm/s.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp500462r | J. Phys. Chem. C XXXX, XXX, XXX−XXXB

Experiments testing the reactivity of the chemical and biologicalGRs in the presence of nitrates were carried out with a closedfluid cell (CCELL) for the MFP3D AFM, completely enclosedand sealed. In an anoxic chamber (95%/5%, N2/H2), theparticles of the GR were deposited on a freshly split mica stripcovering the glass disk of a CCELL, then dried. The closed fluidcell dish was filled up with demineralized and anoxic water (pH= 6.5), and the totality of the CCELL was put together inanaerobic conditions. During CFM measurements, 6 mL ofanoxic NaNO3 0.16 M at pH = 6.5 was injected to replacewater.
■ RESULTS AND DISCUSSION
Morphological Analysis and Surface Properties. AFMimaging of GRs deposited onto mica surfaces in deionized anddeaerated water shows single and stacked regular hexagonalparticles (Figure 1). These observations are in agreement withTEM analyses in the Supporting Information (Figure S1). Thecell parameters calculated from the electron diffraction patternof c-GR and b-GR crystals were around a = b = 3.154 Å, whichare characteristic of the carbonated green rust structure.31 Adistinct c-GR crystal displayed a height h of ∼25 nm and adiameter d of ∼150 nm (Figures 1c and 1d). These values areconsistent with the measurements performed by TEM (i.e.,∼100 nm ≤ d ≤ ∼500 nm, ∼15 nm ≤ h ≤ ∼100 nm, and ∼5 ≤d/h ≤ ∼6). On the other hand, a single b-GR crystal displayeda larger height and diameter of ∼150 nm and ∼4 μm,respectively (Figures 1e and 1h), which is in agreement withprevious studies.29 The difference in GR crystal size originatesmainly from their differential growth and kinetic formation.Indeed, c-GRs are smaller because of high Fe(II) kinetics in thesolution that leads to an instantaneous crystal formation.Conversely, b-GR particles are larger because the kinetic ofFe(II) in the solution is low and leads to a slow germinationand the nucleation of larger crystals as previously described byZegeye et al.29
Furthermore, if chemical reactions are taking place at theparticle surface, the mass-based reactivity of the mineralincreases for smaller particles. Therefore, the morphologicalcharacterization suggests that c-GR may be likely more reactivethan b-GR. To validate this hypothesis, we conducted batchexperiments with c-GR and b-GR with the same Fe3+/Fetot ratio(e.g., ≈0.33) in the presence of an excess of nitrate (e.g.,[NO3
−]/[Fe2+] > 1/8]). After 4 h of nitrate exposure, theresults clearly indicate a significant oxidation of the c-GRevidenced by the increase of Fe3+/Fetot ratio (e.g., from ≈0.33to ≈0.63), while no oxidation of the b-GR was observed(Figure 2). This difference of reactivity was awaited because ofthe significant difference in the surface/volume ratio for the twotypes of particles. As a complementary approach tomorphological characterization and batch experiments and to
Figure 1. AFM imaging and height profiles of c-GR and b-GR. (a) Height images of c-GR particles performed in contact mode and in deaerated anddeionized water (z-scale = 50 nm). (b) Deflection images of c-GR particles performed in contact mode and in deaerated and deionized water. (c)Tridimensional images of c-GR particles observed in deaerated and deionized water. (d) Height profile taken on a cross section (white dashed line inb). (e) Height images of b-GR particles performed in contact mode and in deaerated and deionized water (z-scale = 2 μm). (f) Deflection images ofb-GR particles performed in contact mode and in deaerated and deionized water. (g) Tridimensional images of b-GR particles observed in deaeratedand deionized water. (h) Height profile taken on a cross section (white dashed line in f).
Figure 2. Oxidation rate of chemical green rust versus biological overtime. The x ratio stands for [Fe(III)]/[Fetot] and at the beginning isequal at ∼0.33 where GRs present 2 Fe(II) for 1 Fe(III). At the end, x∼ 0.67 only for the c-GR, which was oxidized into magnetite.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp500462r | J. Phys. Chem. C XXXX, XXX, XXX−XXXC

investigate deeper into nanoscale observations, we used CFMto assess the reactivity of GR particles in the presence of nitrate.In particular, we recorded spatially resolved force curves acrossthe GR surface using AFM tips, which we functionalized with acarboxyl motif that interacts with hydroxyl groups at theloading rate of 10 000 pN s−1. The validation of the CFMtechnique with model surfaces is reported in the SupportingInformation and described below. Figure S2a (SupportingInformation) displays the analysis of the force−distance curvesrecorded between the mica substrate and the modified probebefore and after nitrate injection. We did not detect anyinteraction between carboxylic groups grafted on the tip andthe mica surface, which indicates that the mica substrate is aninert surface in our experimental setup (see Figure S2a,Supporting Information) mainly because the substrate is notcharged at its isoelectric point (pH 6.5). Typical force−distancecurves recorded on the mica substrates, respectively, covered byc-GR and b-GR particles were reported in Figures S2b and S2c(Supporting Information). The force curve recorded on c-GRshowed a very different behavior (linear dependence of therupture force with the surface/tip distance) than on the micasubstrate: indeed a major part of the force curves displayed asingle well pronounced maximum at 200 pN on a shortseparation distance (≤5 nm) before nitrate injection whichcorresponds to the interaction of the mineral surface with thecarboxylic groups present on the AFM tip (Figure S2b,Supporting Information). The nature and specificity of thisinteraction were confirmed by the appearance of a similaradhesion pattern that occurred when performing the experi-
ments at pH = 4.2 on a gold model surface functionalized withHS-C11H22-COOH (pKa = 10.3) with the modified AFM tip(Figures S3a,b, Supporting Information). Given these results,we concluded that the interaction between the AFM tip and thesurface is due both to the hydroxyl groups (−OH) present onthe mineral surface and to the carboxyl moieties (−COOH)grafted on the AFM tip. Moreover, the dramatic loss ofinteraction at pH = 11.5 (Figures S3c,d, SupportingInformation) indicates that this linear dependence of thepulling force with the separation distance over short distances(≤5 nm) can be related to a H-bond interaction. The energyfor these nonspecific interactions can be directly estimated fromthe retraction force curves by integrating the area under theforce curves.32 The relation between the force and thecorresponding energy is given by eq 1, which also theoreticallyestimates the force needed to rupture one H-bond
=·Δ
FE
N zH
A (1)
where F corresponds to the deprotonating or adhesion force;EH stands for the H-bond energy; NA is Avogadro’s number;and Δz is the distance between the two molecules. In water, EHwas previously evaluated to be ∼23 kJ mol−1 for an averagebinding length of 0.2 nm33 (i.e., the energy was about 3.5 ×10−20 J per H-bond). Considering eq 1, the H-bond energy canbe transposed into a rupture force of about 175 pN.28 Thistheoretical value is consistent with the values observed in ourstudy (e.g., ∼160−200 pN) for a loading rate of 10 000 pN/s
Figure 3. Kinetic investigation of nitrate reduction by c-GR using CFM. Histograms represent the statistic distribution of forces during c-GRdeprotonation by NO3
− at different times: (a) 0 min, (b) 63 min, (c) 93 min, and (d) 116 min. Force mappings (F-scale = 100 pN) were included tofollow c-GR deprotonation coupling with topography maps. c-GR particles are located by the red hexagon.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp500462r | J. Phys. Chem. C XXXX, XXX, XXX−XXXD
(Figure S4, Supporting Information) and further supports ourhypothesis that the measured force corresponds to a ruptureforce of one or several H-bond interactions. Indeed, theamplitude of H-bond rupture force remained at the same valuewhatever the pulling rate or rupture speed (Figure S4,Supporting Information); i.e., the necessary energy to breakthe bond between a proton and the surface is independent withthe breaking speed. According to CFM experiments performedon the COOH model surface (see Figure S3, SupportingInformation), we can also suggest that the measured force, atthe molecular scale, corresponds to the rupture of a single H-bond. This result is close to the force observed in earlierstudies27 and validates our methodology to detect and localizeH-bonds or protons on the GR surface. We used a weakloading rate to avoid dynamic effects such as an apparentenhancement of the adhesion force observed in DFS (dynamicforce spectroscopy) experiments performed on ligand−receptorbond rupture34 that lead to an overestimation of the measuredforces.In contrast to c-GR, multiple interactions were detected with
maximal amplitude of about 200 pN with b-GR (Figure S2c,Supporting Information). On a short separation distance (≤5nm), we observed a linear behavior of the pulling force with theseparation distance similar to that detected for c-GR. Thislinear behavior could be attributed to a single H-bondinteraction. Additionally, the nonlinear dependence of thepulling force on a longer separation distance (≤100 nm) canlikely be assigned to molecular uncoiling of sorbed macro-molecules.27
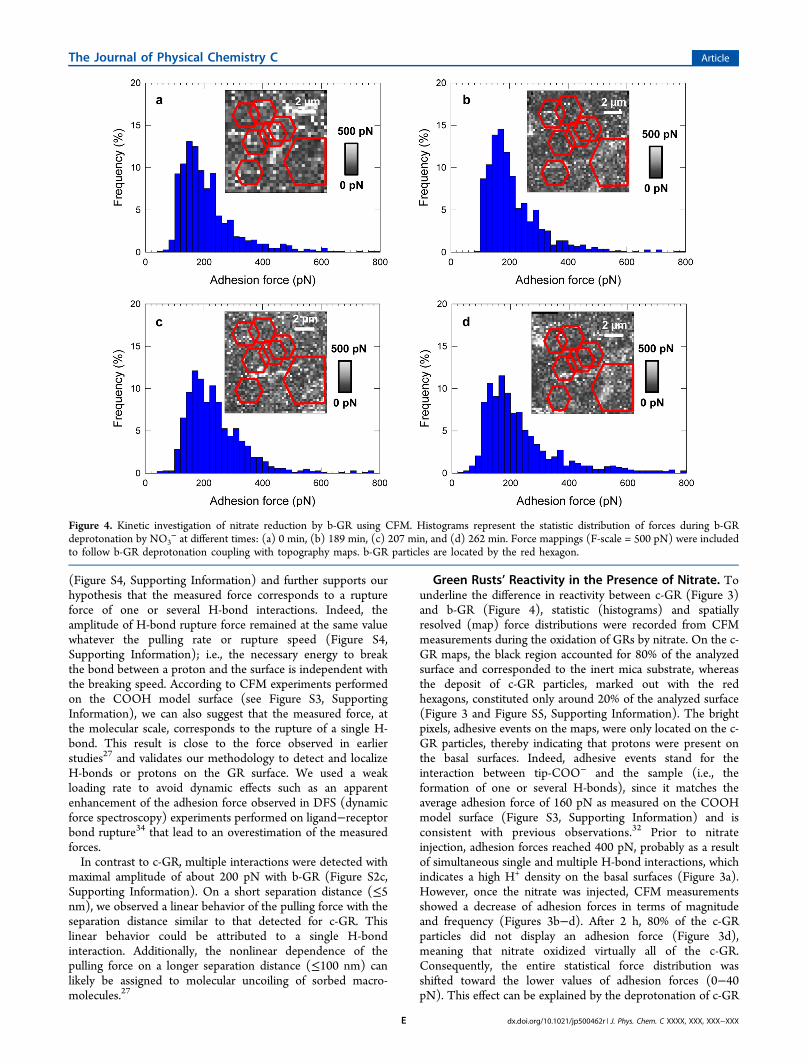
Green Rusts’ Reactivity in the Presence of Nitrate. Tounderline the difference in reactivity between c-GR (Figure 3)and b-GR (Figure 4), statistic (histograms) and spatiallyresolved (map) force distributions were recorded from CFMmeasurements during the oxidation of GRs by nitrate. On the c-GR maps, the black region accounted for 80% of the analyzedsurface and corresponded to the inert mica substrate, whereasthe deposit of c-GR particles, marked out with the redhexagons, constituted only around 20% of the analyzed surface(Figure 3 and Figure S5, Supporting Information). The brightpixels, adhesive events on the maps, were only located on the c-GR particles, thereby indicating that protons were present onthe basal surfaces. Indeed, adhesive events stand for theinteraction between tip-COO− and the sample (i.e., theformation of one or several H-bonds), since it matches theaverage adhesion force of 160 pN as measured on the COOHmodel surface (Figure S3, Supporting Information) and isconsistent with previous observations.32 Prior to nitrateinjection, adhesion forces reached 400 pN, probably as a resultof simultaneous single and multiple H-bond interactions, whichindicates a high H+ density on the basal surfaces (Figure 3a).However, once the nitrate was injected, CFM measurementsshowed a decrease of adhesion forces in terms of magnitudeand frequency (Figures 3b−d). After 2 h, 80% of the c-GRparticles did not display an adhesion force (Figure 3d),meaning that nitrate oxidized virtually all of the c-GR.Consequently, the entire statistical force distribution wasshifted toward the lower values of adhesion forces (0−40pN). This effect can be explained by the deprotonation of c-GR
Figure 4. Kinetic investigation of nitrate reduction by b-GR using CFM. Histograms represent the statistic distribution of forces during b-GRdeprotonation by NO3
− at different times: (a) 0 min, (b) 189 min, (c) 207 min, and (d) 262 min. Force mappings (F-scale = 500 pN) were includedto follow b-GR deprotonation coupling with topography maps. b-GR particles are located by the red hexagon.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp500462r | J. Phys. Chem. C XXXX, XXX, XXX−XXXE

particles resulting from their oxidation by nitrate. Similarly, thedeprotonation of hydroxyl groups that occurs with theoxidation of ferrous iron within the GR structure has beenpreviously evidenced by direct spectroscopic analysis.15
Previous observations have shown that the oxidation of c-GRleads to the formation of various oxidation products includingmagnetite (Fe3O4), ferric oxyhydroxides such as ferrihydrite(Fe8.2O8.5(OH)7.4),
35 goethite (α-FeOOH),16 or “ferric greenrust” (GR*) (Fe6(OH)8O4CO3).
15 The nature of the endproduct is strongly dependent on the kinetics of the reaction.On one hand, the use of strong oxidants, such as hydrogenperoxide (H2O2), chromate (Cr2O7
2−), and persulfate(S2O8
2−), leads to the rapid formation of FeIII compounds(i.e., ferrihydrite or GR*). On the other hand, the formation ofa partially oxidized FeII−FeIII magnetite compound is reportedduring the slow oxidation of GR conducted either in partiallydeaerated water13 or in the presence of nitrate.5 All theseoxidation reactions induce a strong decrease of the H/Fe ratio,ranging from 2 for the initial c-GR (FeII4Fe
III2(OH)12CO3) to
1.3 for GR*, 1 for goethite, and 0 for magnetite.13 The quasidisappearance of the adhesion forces in the range 100−400 pNcould be an indicator that the oxidation end product is fullydeprotonated as it is in the case of magnetite. In fact, thecharacterization of the samples from our batch experiments(e.g., Figure 1) by Raman spectroscopy indicates the formationof magnetite as the end product of c-GR oxidation by nitrate(Figure S6, Supporting Information). This observation isconsistent with previous studies5 where the oxidation of anabiotic GR by nitrate led to the formation of magnetite (eq 2)
+ +
⇄ + + +
− −
+ −
4Fe Fe (OH) CO NO 6OH
8Fe O NH CO 25H O
II4
III2 12 3(s) 3
3 4 4 32
2 (2)
Contrary to what we observed with c-GR, we only recorded aslight decrease in the frequency of adhesive events ranging from100% to 87% after 4 h during the exposure of nitrates with b-GR (Figure 4). We did not notice any significant evolution ofadhesion force distribution during the oxidation process (i.e., itremained centered near 200 pN). Similarly, when analyzing thespatial distribution of the forces measured on the surface, wedid not observe any significant modification of pixel intensityinside the red hexagons (Figure 4). Comparing CFMmeasurements performed on the COOH model surface, itwas clear that the rupture force over a short distance
(characterized by a linear rupture) was due to b-GR surface’sprotons and not to a polymeric or soft component (nonlinearrupture). To discriminate the H-bond contribution, we inferredthat force curves that had only one adhesive event andcharacterized by a linear rupture force for a short separationdistance (<5−10 nm) correspond to the detection of H-bonds(Figure S2b, Supporting Information). Thus, on the b-GR weidentified a number of force curves (around 25%) with only asingle interaction for an average adhesion force of about 100−200 pN. Moreover, these force curves were very similar to thosemeasured on the model surface (see Figure S3, SupportingInformation), which indicates that they are related to H-bondinteractions. Because they do not evolve over time during thenitrate exposure, this further suggests a weaker reactivity of b-GR in comparison to c-GR, within the time’s range of ourexperimental setup. Indeed, H-bond interactions were reducedby a factor of 3 for the c-GR particles in 2 h exposure bycontrast to b-GR particles where these interactions were notimpacted or remained the same after 2 and 4 h nitrate exposure.We can therefore confidently conclude that b-GR is more stableover time than c-GR in the presence of nitrate. Thisobservation is consistent with our Raman analysis of thesamples from our batch experiments (e.g., Figure 1) where notransformation of the b-GR was observed at the end of theexposure (Figure S6, Supporting Information). Biosynthesis ofGR particles might probably induce surface properties differentfrom those obtained by a chemical way and thus modify theirsurface reactivity. Moreover, it is generally accepted that thelarge difference in reactivity between c-GR and b-Gr should bedue to the difference in surface/volume ratio. So, most of thesestudies only argued that particles obtained by the biologicalpathway are less reactive than their synthetic homologuewithout solid explanation or nanoscale investigation of thecovering biomolecules.
Enhancement of GR Stability by Bacterial Macro-molecules. The surface of b-GR particles is probably coveredby the biomacromolecules excreted by bacteria. The influenceof such macromolecules on mineral surfaces and interfacialprocesses such biomineralization19 and pollutant distribution20
has been already addressed. In the case of GR particles, doesthe presence of EPS significantly impact the particle surfacereactivity? To further identify chemical modifications of c-GRand b-GR particles during nitrate oxidation and to demonstratethe major role played by the bacterial macromolecules or EPS
Figure 5. (a) IR spectra of the c-GR particles before nitrate exposure (1) and 4 h after nitrate injection (2). c-GR particles were synthesized in thepresence of bacterial EPS (3) and incubated 4 h with nitrate solution (4). (b) IR spectra of b-GR particles before nitrate exposure (1) and 4 h afternitrate injection (2).
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp500462r | J. Phys. Chem. C XXXX, XXX, XXX−XXXF

on the stability and reactivity of the particles, infrared spectrawere recorded before and after the oxidation process (Figure5). The infrared spectra (Figures 5a1 and 5b1) of c-GR and b-GR minerals exhibit the typical features of carbonate greenrust:36−38 bands arising to the brucite-like sheets at 770, 850,1550, and 3000−3600 cm−1 and bands due to the intercalatedcarbonate at 1350−1410 cm−1. In addition to these GRabsorptions, the b-GR spectrum reveals the presence oflepidocrocite especially with the sharp absorption at 1022cm−1 and the absorptions in the range 1200−1250 cm−1 in thec-GR spectrum revealing the presence of adsorbed orprecipitated sulfate on the GR surface.36
After 4 h of nitrate exposure, the bands associated to OH-brucite-like sheets have almost disappeared for the c-GRmineral. On the contrary, we observe no spectral change for b-GR (Figure 5b) proving the high stability of this mineral withrespect to nitrate oxidation. The c-GR and the oxidationproduct were analyzed by Raman spectroscopy (Figure S6,Supporting Information). The spectrum of c-GR (Figure S6a,Supporting Information) exhibits the characteristic features ofcarbonate green rust mineral with bands at 155, 220, 260, 432,512, and 680 cm−1.37 The Raman spectrum of the oxidationproduct (Figure S6b, Supporting Information) is characteristicof magnetite, as revealed by bands around 310, 540, and 670cm−1.39 When the c-GR particles were synthesized in thepresence of bacterial EPS minor changes were observed after 4h in nitrate solution (Figures 5a3 and 5a4) confirming the keyrole of macromolecules or EPS upon the oxidation process ofgreen rust minerals.
Furthermore, vibrational spectroscopy measurements per-formed during nitrate oxidation of b-GR and c-GR particles inthe presence or the absence of EPS correlate with forcespectroscopy experiments. Contrary to the c-GR where theadhesion force curves exhibited only one linear dependencewith the separation distance (<5 nm) (Figure S2b, SupportingInformation), the b-GR displays multiple adhesive interactionswith nonlinear behavior (Figure S2c, Supporting Information),which is a typical signature of macromolecular stretchings27 andwhich suggests the presence of biological exopolymers on theb-GR surface. These force curves were modeled using thetheoretical approach free-joined-chain (FJC) that describes thebehavior of polysaccharidic chains under a pulling force (FigureS7a, Supporting Information).40 For each peak, with theexception of the first one, which is attributed to a linearbehavior, the red lines correspond to the theoretical fitting ofdifferent kinds of exopolymers. Three major fit parameters (thecontour length LC, the number of monomers N, and the Kuhnlength lK) reflecting the conformational analysis of the polymersassociated with b-GR were extracted from the theoretical model(Figure S7b−d, Supporting Information). According to thestatistical data, 90% of the polysaccharides detected on the b-GR show a contour length LC of about 20−200 nm (FigureS5c, Supporting Information). These detected polysaccharidescould be constituted by N = 120 to 650 monomers (sugars)(Figure S7b, Supporting Information) with a wide Kuhn lengthlK distribution in the range of 0.20−1.40 nm (Figure S7d,Supporting Information). Previous studies have established thata Kuhn length of ∼0.23 nm corresponds to the elongation of a
Figure 6. Kinetic investigation of nitrate reduction by c-GR incubated with bacterial polymer extract using CFM. Histograms represent the statisticdistribution of forces during c-GR deprotonation by NO3
− at different times: (a) 0 min, (b) 43 min, (c) 89 min, and (d) 147 min. Force mappings(F-scale = 500 pN) were included to follow c-GR deprotonation coupling with topography maps. c-GR particles are located by the red hexagon.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp500462r | J. Phys. Chem. C XXXX, XXX, XXX−XXXG

single sugar molecule (glucose, mannose, rhamnose, etc.).41 Wenote that the lK interval in our experiments (0.20−1.40 nm)could represent a wide distribution of polysaccharide monomerlengths. Indeed, prior investigations performed on severalbacterial exopolysaccharides (EPS) revealed that the monomersconsist of the repetition of 6 or 7 sugars.27,42 In our case, thepolysaccharidic molecules present on the b-GR are shorter (lessthan 1 μm length) with various monomers that could becomposed of 4 to 7 sugars (Table S1 and Table S2, SupportingInformation). On the basis of these observations, the importantdifference between the two GRs (c-GR and b-GR) in terms ofreactivity toward the presence of nitrate can be explained by thepresence of biopolymers on the b-GR surface, which hinderedits reactivity. Indeed, it has been shown that bacterial materials(e.g., polymers, membrane debris) or phosphate28 stabilize theb-GR and inhibit its mineralogical transformation toward athermodynamically more stable mineral, yet the origin of thisinhibition remains unknown.To confirm if the low degree of reactivity of b-GR is indeed
related to the presence of the bacterial biopolymers and to theparticle size difference between the two GRs, we incubated c-GR particles for a few hours with an extract of bacterialbiopolymers, and we investigated their reactivity after nitrateinjection (Figure 6). The time evolution of the statisticdistribution of the number of rupture events per force curve forb-GR particles and c-GR particles incubated with bacterialpolymers is illustrated in Figure 7. We observed a similar trendbetween the kinetics of adhesion of b-GR (Figure 3) and c-GRembedded with biopolymers (Figure 6). The statisticdistribution of adhesive forces is characterized by a widedistribution with a maximum in the range of 150−200 pN. Thisdistribution does not seem to be impacted by the nitrate asexpected due to the presence of biopolymers added after thesynthesis of the c-GR. To further confirm this hypothesis, we
isolated the H-bound contributions from the other interactionsuch as polymer uncoiling (characterized by one or morenonlinear rupture forces) and analyzed the force curves thathad one adhesive event (characterized by a linear ruptureforce), as was done in the case of b-GR (Figure 6a−c andFigure S7, Supporting Information). For the c-GR incubatedwith an extract of bacterial polymer, 40% of the curvesdisplayed only one adhesive event. These force curves werevery similar to what was measured on the model surface(Figures 6d−f and Figure S7, , Supporting Information).Therefore, they could be related to the detection of protons orH-bonds present on the mineral surface. These curves did notevolve during the nitrate exposure indicating a protective roleof bacterial polymers. These results indicate clearly that thepresence of biopolymers decreases the reactivity of c-GRparticles even though they have the same particle size,confirming our hypothesis on the protective role of thebacterial polymers in relation to the GR reactivity towardnitrate.
■ CONCLUSIONS
The chemical force microscopy experimental setup allows thedirect mapping of the deprotonation process of the particles inreal time, while the collected data demonstrate the crucial rolethat bacterial exopolymers (EPS) play in defining the nanoscalesurface properties and the deprotonation capacity of chemically(c-GR) versus biologically (b-GR) formed green rusts. Wehighlighted their stability difference under nitrate exposure,despite their mineralogical similarities and the same Fe2+/Fetotratio. The c-GR was deprotonated almost completely after 2 hof nitrate exposure, whereas the b-GR and c-GR coated withbiopolymers were not affected. Nevertheless, AFM imagingindicated that the surface/volume ratio for c-GR and b-GR wasapproximately 108 m−1 and 5 × 106 m−1, respectively. This
Figure 7. Evolution of the number of adhesive events per force measurements during the nitrate reduction of b-GR (a,b,c) and c-GR incubated withbiopolymers (d,e,f). Histograms represent the statistic distribution of the number of adhesive events per force curves at different times: (a) 0 min,(b) 189 min, and (d) 262 min for the b-GR particles and at (d) 0 min, (e) 43 min, and (f) 147 min for c-GR particles embedded into bacterialpolymers. Insets show typical force curves located at the b-GR and c-GR particle surface.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp500462r | J. Phys. Chem. C XXXX, XXX, XXX−XXXH

discrepancy should have been promoting higher stability for thesmaller particles. This is not the case, and we prove that thedifference of reactivity between b-GR and c-GR particles is notonly due to the difference in their surface/volume ratio but alsoto the presence of bacterial biopolymers (EPS) that increasessignificantly the GR particles stability in the presence of nitrate.The line of evidence highlighted in this study argues for animportant protective role of microbial exopolymers against theinorganic pollutant. The above demonstration explains why theb-GR particles are more stable than c-GR ones under nitrateexposure. This indicates that the reactivity of b-GR particles islikely to be variable and should depend on the nature andbiopolymer properties (ad)sorbed on their surface. In turn, thisdepends on the conditions under which b-GR forms (species/strain of iron reducing or oxidizing bacteria involved and theirabundance). The bacterial densities in natural systems where b-GR may precipitate are likely much lower than the bacterialdensities used for b-GR preparation for this study. Given thecomplexity of biogeochemical processes in natural subsurfaces,additional insight is clearly needed to assess the contribution ofb-GR to nitrate oxidation for practical applications.
■ ASSOCIATED CONTENT*S Supporting InformationMaterials and methods, Figures S1−S7, and Tables S1 and S2.This material is available free of charge via the Internet athttp://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected]. Phone: (33) 03 8368 52 36.Present Address⊥Universite de Lorraine, Laboratoire Interdisciplinaire desEnvironnements Continentaux, UMR 7360, BP 70239,Vandœuvre-les-Nancy, F-54506, France (A.Z.).Author Contributions#A.Z. and M.E. have equally contributed to the manuscript.Author ContributionsA.Z. and M.E. performed the synthesis of GR particles; AFMand CFM experiments were carried out by G.F. and Raman andInfrared spectroscopies measurements by C.C. A.Z., G.F., C.R.,C.C., and P.S. contributed to the writing and datainterpretation. A.Z and G.F. supervised the project, and M.E.contributed to the analysis and the writing. All authors tookactive part in scientific discussions and the finalization of themanuscript.NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSThe authors acknowledge Prof. F.P.A. Jorand (Universite deLorraine, LCPME) for kindly providing an extract oflyophilized EPS and Dr. J. Ghanbaja for TEM analysis(Universite de Lorraine, SCMEM).
■ REFERENCES(1) Cornell, R. M.; Schwertmann, U. The Iron Oxides: Structure,Properties, Reactions, Occurences and Uses; WILEY-VCH Verlag GmbH& Co: Weinheim, Germany, 2003.(2) Brown, D. A.; Kamineni, D. C.; Sawicki, J. A.; Beveridge, T. J.Minerals Associated with Biofilms Occuring on Exposed Rock in a
Granitic Underground Research Laboratory. Appl. Environ. Microbiol.1994, 60, 3182−3191.(3) Taylor, K. G.; Konhauser, K. O. Iron in Earth Surface Systems: AMajor Player in Chemical and Biological Processes. Elements 2011, 7,83−87.(4) Konhauser, K. O.; Kappler, A.; Roden, E. E. Iron in MicrobialMetabolisms. Elements 2011, 7, 89−93.(5) Hansen, H. C. B.; Koch, C. B.; NanckeKrogh, H.; Borggaard, O.K.; Sorensen, J. Abiotic Nitrate Reduction to Ammonium: Key Role ofGreen Rust. Environ. Sci. Technol. 1996, 30, 2053−2056.(6) Simon, L.; Francois, M.; Refait, P.; Renaudin, G.; Lelaurain, M.;Genin, J.-M. R. Structure of the Fe(II-III) Layered DoubleHydroxysulphate Green Rust Two from Rietveld Analysis. SolidState Sci. 2003, 5, 327−334.(7) Ruby, C.; Upadhyay, C.; Gehin, A.; Ona-Nguema, G.; Genin, J.M. R. In Situ Redox Flexibility of FeII-III Oxyhydroxycarbonate GreenRust and Fougerite. Environ. Sci. Technol. 2006, 40, 4696−4702.(8) Wander, M. C. F.; Rosso, K. M.; Schoonen, M. A. A. Structureand Charge Hopping Dynamics in Green Rust. J. Phys. Chem. C 2007,111, 11414−11423.(9) Trolard, F.; Genin, J. M. R.; Abdelmoula, M.; Bourrie, G.;Humbert, B.; Herbillon, A. Identification of a Green Rust Mineral in aReductomorphic Soil by Mossbauer and Raman Spectroscopies.Geochim. Cosmochim. Acta 1997, 61, 1107−1111.(10) Della Rocca, C.; Belgiorno, V.; Meric, S. Heterotrophic/Autotrophic Denitrification (HAD) of Drinking Water: ProspectiveUse for Permeable Reactive Barrier. Desalination 2007, 210, 194−204.(11) Hansen, H. C. B., Environmental Chemistry of Iron(II)−Iron(III) LDHs (Green rusts). In Layered Double Hydroxides: Presentand Future; Rives, V., Ed.; Nova Science Publishers, Inc.: Huntington,NY, 2001; pp 469−493.(12) Legrand, L.; Mazerolles, L.; Chausse, A. The Oxidation ofCarbonate Green Rust into Ferric Phases: Solid-state Reaction orTransformation via Solution. Geochim. Cosmochim. Acta 2004, 68,3497−3507.(13) Ruby, C.; Abdelmoula, M.; Naille, S.; Renard, A.; Khare, V.;Ona-Nguema, G.; Morin, G.; Genin, J. M. R. Oxidation Modes andThermodynamics of FeII-III Oxyhydroxycarbonate Green Rust:Dissolution-precipitation versus In Situ Deprotonation. Geochim.Cosmochim. Acta 2010, 74, 953−966.(14) Refait, P.; Benali, O.; Abdelmoula, M.; Genin, J. M. R.Formation of ’Ferric Green Rust’ and/or Ferrihydrite by FastOxidation of Iron(II-III) Hydroxychloride Green Rust. Corros. Sci.2003, 45, 2435−2449.(15) Mullet, M.; Guillemin, Y.; Ruby, C. Oxidation andDeprotonation of Synthetic Fe-II-Fe-III (oxy)HydroxycarbonateGreen Rust: An X-ray Photoelectron Study. J. Solid State Chem.2008, 181, 81−89.(16) Pantke, C.; Obst, M.; Benzerara, K.; Morin, G.; Ona-Nguema,G.; Dippon, U.; Kappler, A. Green Rust Formation during Fe(II)Oxidation by the Nitrate-Reducing Acidovorax sp Strain BoFeN1.Environ. Sci. Technol. 2012, 46, 1439−1446.(17) Welch, S. A.; Ullman, W. J. The Effect of Microbial GlucoseMetabolism on Bytownite Feldspar Dissolution Rates between 5degrees and 35 degrees. Geochim. Cosmochim. Acta 1999, 63, 3247−3259.(18) Welch, S. A.; Barker, W. W.; Banfield, J. F. MicrobialExtracellular Polysaccharides and Plagioclase Dissolution. Geochim.Cosmochim. Acta 1999, 63, 1405−1419.(19) Chang, V. T. C.; Williams, R. J. P.; Makishima, A.; Belshawl, N.S.; O’Nions, R. K. Mg and Ca Isotope Fractionation during CaCO3Biomineralisation. Biochem. Biophys. Res. Commun. 2004, 323, 79−85.(20) Wolfaardt, G. M.; Lawrence, J. R.; Headley, J. V.; Robarts, R. D.;Caldwell, D. E. Microbial Exopolymers Provide a Mechanism forBioaccumulation of Contaminants. Microb. Ecol. 1994, 27, 279−291.(21) Kwon, K. D.; Vadillo-Rodriguez, V.; Logan, B. E.; Kubicki, J. D.Interactions of Biopolymers with Silica Surfaces: Force Measurementsand Electronic Structure Calculation Studies. Geochim. Cosmochim.Acta 2006, 70, 3803−3819.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp500462r | J. Phys. Chem. C XXXX, XXX, XXX−XXXI

(22) Dague, E.; Alsteens, D.; Latge, J. P.; Verbelen, C.; Raze, D.;Baulard, A. R.; Dufrene, Y. F. Chemical Force Microscopy of SingleLive Cells. Nano Lett. 2007, 7, 3026−3030.(23) Jones, C. A.; Grasley, Z. C.; Ohlhausen, J. A. Measurement ofElastic Properties of Calcium Silicate Hydrate with Atomic ForceMicroscopy. Cem. Concr. Compos. 2012, 34, 468−477.(24) Akhlaghi, S.; Sharif, A.; Kalaee, M.; Nouri, A.; Manafi, M.Morphology, Nanomechanical and Thermodynamic Surface Charac-teristics of Nylon 6/Feather Keratin Blend Films: an Atomic ForceMicroscopy Investigation. Polym. Int. 2012, 61, 646−656.(25) Getfert, S.; Reimann, P. Hidden Multiple Bond Effects inDynamic Force Spectroscopy. Biophys. J. 2012, 102, 1184−1193.(26) Ito, T.; Grabowska, I.; Ibrahim, S. Chemical Force Microscopyfor Materials Characterization. TrAC, Trends Anal. Chem. 2010, 29,225−233.(27) Francius, G.; Lebeer, S.; Alsteens, D.; Wildling, L.; Gruber, H. J.;Hols, P.; De Keersmaecker, S.; Vanderleyden, J.; Dufrene, Y. F.Detection, Localization, and Conformational Analysis of SinglePolysaccharide molecules on Live Bacteria. ACS Nano 2008, 2,1921−1929.(28) Bocher, F.; Gehin, A.; Ruby, C.; Ghanbaja, J.; Abdelmoula, M.;Genin, J. M. R. Coprecipitation of Fe(II-III) Hydroxycarbonate GreenRust Stabilised by Phosphate Adsorption. Solid State Sci. 2004, 6, 117−124.(29) Zegeye, A.; Mustin, C.; Jorand, F. Bacterial and Iron OxideAggregates Mediate Secondary Iron Mineral Formation: Green Rustversus Magnetite. Geobiology 2010, 8, 209−222.(30) Zegeye, A.; Bonneville, S.; Benning, L. G.; Sturm, A.; Fowle, D.A.; Jones, C.; Canfield, D. E.; Ruby, C.; MacLean, L. C.; Nomosatryo,S.; Crowe, S. A.; Poulton, S. W. Green Rust Formation ControlsNutrient Availability in a Ferruginous Water Column. Geology 2012,40, 599−602.(31) Aissa, R.; Francois, M.; Ruby, C.; Fauth, F.; Medjahdi, G.;Abdelmoula, M.; Genin, J.-M. R. Formation and CrystallographicalStructure of Hydroxysulphate and Hydroxycarbonate Green RustsSynthetised by Coprecipitation. J. Phys. Chem. Solids 2006, 67, 1016−1019.(32) Boland, T.; Ratner, B. D. Direct Measurement of Hydrogen-Bonding in DNA Nucleotide Bases by Atomic-Force Microscopy. Proc.Natl. Acad. Sci. U.S.A. 1995, 92, 5297−5301.(33) Suresh, S. J.; Naik, V. M. Hydrogen Bond ThermodynamicProperties of Water from Dielectric Constant Data. J. Chem. Phys.2000, 113, 9727−9732.(34) Lo, Y. S.; Zhu, Y. J.; Beebe, T. P. Loading-Rate Dependence ofIndividual Ligand-Receptor Bond-Rupture Forces Studied by AtomicForce Microscopy. Langmuir 2001, 17, 3741−3748.(35) Michel, F. M.; Ehm, L.; Antao, S. M.; Lee, P. L.; Chupas, P. J.;Liu, G.; Strongin, D. R.; Schoonen, M. A. A.; Phillips, B. L.; Parise, J. B.The structure of Ferrihydrite, a Nanocrystalline Material. Science 2007,316, 1726−1729.(36) Ona-Nguema, G.; Carteret, C.; Benali, O.; Abdelmoula, M.;Genin, J. M.; Jorand, F. Competitive Formation of HydroxycarbonateGreen Rust 1 versus Hydroxysulphate Green Rust 2 in ShewanellaPutrefaciens Cultures. Geomicrobiol. J. 2004, 21, 79−90.(37) Legrand, L.; Sagon, G.; Lecomte, S.; Chausse, A.; Messina, R. ARaman and Infrared Study of a New Carbonate Green Rust Obtainedby Electrochemical Way. Corros. Sci. 2001, 43, 1739−1749.(38) Peulon, S.; Legrand, L.; Antony, H.; Chausse, A. Electro-chemical Deposition of Thin Films of Green Rusts 1 and 2 on InertGold Substrate. Electrochem. Commun. 2003, 5, 208−213.(39) Bellot-Gurlet, L.; Neff, D.; Reguer, S.; Monnier, J.; Saheb, M.;Dillmann, P. Raman Studies of Corrosion Layers Formed onArchaeological Irons in Various Media. J. Nano Res. 2009, 8, 147−156.(40) Janshoff, A.; Neitzert, M.; Oberdoerfer, Y.; Fuchs, H. ForceSpectroscopy of Molecular Systems - Single Molecule Spectroscopy ofPolymers and Biomolecules. Angew. Chem., Int. Ed. 2000, 39, 3212−3237.
(41) Camesano, T. A.; Abu-Lail, N. I. Heterogeneity in BacterialSurface Polysaccharides, Probed on a Single-Molecule Basis.Biamacromolecules 2002, 3, 661−667.(42) Landersjo, C.; Yang, Z. N.; Huttunen, E.; Widmalm, G.Structural Studies of The Exopolysaccharide Produced by LactobacillusRhamnosus Strain GG (ATCC 53103). Biamacromolecules 2002, 3,880−884.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp500462r | J. Phys. Chem. C XXXX, XXX, XXX−XXXJ




















