
novel strategies for improving the pharmacological - Wake ...
568
NOVEL STRATEGIES FOR IMPROVING THE PHARMACOLOGICAL PROPERTIES OF PLATINUM-ACRIDINE ANTICANCER AGENTS BY SONG DING A Dissertation Submitted to the Graduate Faculty of WAKE FOREST UNIVERSITY GRADUATE SCHOOL OF ARTS AND SCIENCES in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY Chemistry December 2015 Winston-Salem, North Carolina Approved By: Ulrich Bierbach, Ph.D, Advisor Martin Guthold, Ph.D, Chair Patricia C. Dos Santos, Ph.D S. Bruce King, Ph.D Mark E. Welker, Ph.D
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of novel strategies for improving the pharmacological - Wake ...

NOVEL STRATEGIES FOR IMPROVING THE PHARMACOLOGICAL
PROPERTIES OF PLATINUM-ACRIDINE ANTICANCER AGENTS
BY
SONG DING
A Dissertation Submitted to the Graduate Faculty of
WAKE FOREST UNIVERSITY GRADUATE SCHOOL OF ARTS AND SCIENCES
in Partial Fulfillment of the Requirements
for the Degree of
DOCTOR OF PHILOSOPHY
Chemistry
December 2015
Winston-Salem, North Carolina
Approved By:
Ulrich Bierbach, Ph.D, Advisor
Martin Guthold, Ph.D, Chair
Patricia C. Dos Santos, Ph.D
S. Bruce King, Ph.D
Mark E. Welker, Ph.D

II
ACKNOWLEDGEMENTS
The work shown in this dissertation would become immeasurably difficult without the
help from many people. First of all, I would like to thank my research advisor, Professor
Ulrich Bierbach, who constantly inspired me and firmly supported my every research
project. His real dedication to science is contagious and encouraged me to be a scientist
in the future.
I also want to express my sincere appreciation to my committee members Dr. Patricia
Dos Santos, Dr. Stephen Bruce King, Dr. Martin Guthold and Dr. Mark Welker for
taking the time to read my dissertation and offering me valuable suggestions during my
Ph.D. study. I would like to thank Dr. Marcus Wright and Dr. Julie Reisz Haines for
support with NMR and mass spectrometry.
I have been fortunate to be able to work with great lab members: Dr. Leigh Ann
Graham, Dr. Jimmy Suryadi, Dr. Amanda Pickard, Dr. Xin Qiao, Ye Zheng, Mu Yang,
and Xiyuan Yao. I also had the opportunity to work with talented undergraduate students:
Ben Fontaine, Zach Hood, Thomas Bartenstein, Layne Raborn and Chris Hackett.
Working with you has been a great experience. To all of my friends in Winston-Salem,
thank you for your constant support over the last five years.
Many thanks to my wife and my parents.
Lastly, I would like to thank Wake Forest University and the National Cancer Institute
(Grant R01 CA101880) for supporting my Ph.D. research, and Wake Forest Innovations
for protecting my intellectual property.

III
TABLE OF CONTENTS
LIST OF FIGURES AND TABLES XIII
LIST OF ABBREVIATIONS XIX
ABSTRACT XXIV
CHAPTER 1. INTRODUCTION 1
1.1. Platinum Based Cancer Chemotherapy 1
1.2. Platinum(II) Based Anticancer Agents 6
1.2.1. Traditional platinum(II) complexes 6
1.2.2. Non-traditional platinum(II) based anticancer agents 9
1.2.2.1. Transplatin analogues 9
1.2.2.2. Monofunctional platinum complexes 12
1.2.2.3. Polynuclear platinum(II) complexes 14
1.2.2.4. A new “lead”: platinum-acridines 17
1.3. Drug Delivery Systems for Platinum Based Anticancer Drugs 20
1.3.1. Polymeric micelles 21
1.3.2. Polymer-based prodrugs 23
1.3.3. Liposomes 24
1.3.4. Nanotubes 30
1.4. Target-Selective Activation of Platinum-Based Anticancer Agents 31

IV
1.4.1. Controlling solution reactivity 32
1.4.2. Activation by ligand exchange 34
1.4.3. Activation by pH-sensitive release 38
1.4.4. Photochemical activation 41
1.4.5. Activation by bioreduction 46
1.5. Goals of the doctoral research 50
CHAPTER 2. USING A BUILDING-AND-CLICK APPROACH
FOR PRODUCING STRUCTURAL AND FUNCTIONAL DIVERSITY
IN DNA-TARGETED HYBRID ANTICANCER AGENTS 53
2.1. Introduction and Design Rationale 54
2.2. Results and Discussion 56
2.2.1. Design of a combinatorial screen assay: assembly and
characterization 56
2.2.2. Validation of the combinatorial screen assay: biological
evaluation and SAR analysis 59
2.3. Conclusion 66
2.4. Experimental Section 66
2.4.1. Reagents and Instrumentation 66
2.4.2. Synthesis and characterization of acridine building blocks 68
2.4.3. Synthesis and characterization of platinum building blocks 74

V
2.4.4. Experimental procedure for “click” reactions 75
2.4.5. Typical procedure for the preparation of platinum–acridine
derivatives 76
2.4.6. Cell proliferation assay 79
CHAPTER 3. USING FLUORESCENT POST-LABELING TO PROBE
THE SUBCELLULAR LOCALIZATION OF DNA-TARGETED
PLATINUM 81
3.1. Introduction and Design Rationale 82
3.2. Results and Discussion 83
3.2.1. Post-labeling method 84
3.2.2. Post-labeling using copper-free click chemistry 92
3.2.3. Pre-labeling method using copper-free click chemistry 95
3.3. Conclusion 100
3.4. Experimental Section 101
3.4.1. Materials and methods 101
3.4.2. Synthetic procedures and product characterization 102
3.4.3. In-gel Alexa Fluor 488 fluorescence detection 104
3.4.4. Stability of compound P1-A9 in buffers 107
3.4.5. Plasmid unwinding experiments 107
3.4.6. CD experiments 107

VI
3.4.7. Cell-based assays based on post-labeling method 108
3.4.7.1. Cell culture maintenance 108
3.4.7.2. Cell treatment and Cu(I)-catalyzed azide-alkyne
cycloaddition (CuAAC) ligation 109
3.4.7.3. Confocal microscopy and fluorescence intensity analysis 110
3.4.8. Cell based assays based on pre-labeling method 111
3.4.8.1. Labeling of compound P1-A9 with Alexa Fluor
488-DIBO to generate P1-A9* 111
3.4.8.2. Cell culture and incubations 111
3.4.8.3. Confocal microscopy 113
CHAPTER 4. SYNTHESIS AND EVALUATION OF A
PLATINUM-ACRIDINE-ENDOXIFEN CONJUGATE 114
4.1. Introduction and Design Rationale 115
4.2. Results and Discussion 117
4.2.1. Efforts to synthesize platinum-acridine-endoxifen conjugates 118
4.2.2. Biological evaluation of target conjugates 119
4.3. Conclusion 122
4.4. Experimental Section 124
4.4.1. Synthesis of target conjugates 124

VII
4.4.2. Stability study of compound P4-A12 133
4.4.3. Cytotoxicity studies and cell viability assays 133
4.4.3.1. Sample preparation and cell culture 133
4.4.3.2. Cytotoxicity studies 134
CHAPTER 5. DESIGN OF ENZYMATICALLY CLEAVABLE
PRODRUGS OF A POTENT PLATINUM-CONTAINING
ANTICANCER AGENT 136
5.1. Introduction and Design Rationale 137
5.2. Results and Discussion 139
5.2.1. Design and chemistry 139
5.2.2. Metal-assisted ester hydrolysis 143
5.2.3. Ester cleavage by human carboxylesterase-2 (hCES-2) 149
5.2.4. Cytotoxicity studies 152
5.3. Conclusion 156
5.4. Experimental Section 157
5.4.1. Materials, general procedures, and instrumentation 157
5.4.2. Synthesis and characterization of target prodrugs 159
5.4.3. Time-dependent NMR spectroscopy 168
5.4.4. Chemical hydrolysis assay 169

VIII
5.4.5. Enzymatic cleavage assay 169
5.4.6. Determination of partition coefficients (logD) 170
5.4.7. Cell based assays 171
5.4.7.1. Cell culture maintenance 171
5.4.7.2. Cytotoxicity assay 172
CHAPTER 6. DEVELOPMENT OF A CHEMICAL TOOLBOX FOR
THE DISCOVERY OF MULTIFUNCTIONAL PLATINUM-ACRIDINE
AGENTS 173
6.1 Introduction 174
6.2 Results and Discussion 177
6.2.1 Optimization of coupling reaction 177
6.2.2. Assembly of fragment library for multifunctional
platinum-acridine conjugates 182
6.2.3 Aqueous conditions for the synthesis platinum-acridine based
conjugates 194
6.2.4. “One-tube” assembly of multifunctional platinum-acridine
conjugates 197
6.2.5 Platinum mediated ester hydrolysis 201
6.3. Conclusions 206

IX
6.4. Experimental Section 208
6.4.1.Materials, general procedures, and instrumentation 208
6.4.2. Synthetic procedures and product characterization 209
6.4.3. Assembly of multifunctional platinum-acridine agents through
a library approach 211
6.4.4. Chemical hydrolysis assay 212
CHAPTER 7. LIPOSOMAL DELIVERY OF
PLATINUM-ACRIDINE ANTICANCER FOR NON-SMALL
CELL LUNG CANCER 214
7.1. Design Rationale 215
7.2. Results and Disscussion 216
7.2.1. Aqueous stability of platinum-acridine analogues 216
7.2.2. Optimization of conditions for drug loading 218
7.2.2.1. Selection of lipids 218
7.2.2.2. The importance of freeze-thaw cycles for drug loading 222
7.2.2.3. The influence of loading buffers on drug loading 223
7.2.2.4. The influence of drug feeds on drug loading 224
7.2.2.5. The influence of loading methods on drug loading 225
7.2.3. Characterization of liposomes 226

X
7.2.4. Stability of liposomes in PBS and during long-term storage 231
7.2.5. In vitro cellular uptake and localization study 235
7.2.6. Acute toxicity studies of liposomal P8-A1 238
7.2.7. in vivo antitumor efficacy 239
7.3. Conclusion 241
7.4. Experimental Section 245
7.4.1. Materials and instruments 245
7.4.2. Optimization of the encapsulation conditions for P8-A1 245
7.4.3. Preparation of liposomes using ethanol injection method 246
7.4.4. Preparation of liposomes using reverse-phase evaporation method 247
7.4.5. Preparation of liposomes for biological evaluation 247
7.4.6. Characterizations of liposomes 248
7.4.6.1. Size and zeta potential of liposomes 248
7.4.6.2. Transmission electron microscopy (TEM) 248
7.4.6.3. Encapsulation efficiency of liposomes 249
7.4.6.4. Determination of lipid composition 249
7.4.7. In vitro stability assay 250
7.4.7.1. Drug leakage under physiological relevant conditions 250
7.4.7.2. In vitro drug release of P8-A1 at elevated temperature 250

XI
7.4.7.3. Storage stability 250
7.4.8. Cellular uptake study of Lipo-1 in NCI-H460 lung cancer cells 251
7.4.9. Animal studies 215
7.4.9.1. Evaluation of maximum tolerated dose (MTD) 252
7.4.9.2. A549 xenograft study 252
CHAPTER 8. SUMMARY AND OUTLOOK 254
REFERENCES 259
APPENDIX A. NMR SPECTROSCOPY 316
APPENDIX B. LC-MS SPECTROSCOPY 380
APPENDIX B1. LC-MS ANALYSIS OF THE REACTIONS
BETWEEN FRAGMENTS “A”AND “P” 380
APPENDIX B2. LC-ESMS ANALYSIS OF PURIFIED
COMPOUNDS 441
APPENDIX B3. LC-ESMS ANALYSIS OF
STABILITY OF CONJUGATES IN BUFFERS 477
APPENDIX B4. METAL-ASSISTED ESTER HYDROLYSIS 485
APPENDIX B5. hCES-2 MEDIATED ESTER HYDROLYSIS 509
APPENDIX B6. LC-MS ANALYSIS OF THE
COUPLING REACTIONS FOR MULTIFUNCTIONAL

XII
PLATINUM-ACRIDINES 516
APPENDIX C. CD SPECTRA 529
APPENDIX D. CONFOCAL MICROSCOPY IMAGES 530
APPENDIX E. CELL PROLIFERATION ASSAYS 533
APPENDIX F. COMPUTATIONAL STUDIES 535
APPENDIX G. MTD STUDIES 536
APPENDIX H. SIZE DISTRIBUTION OF LIPOSOMES 537
APPENDIX I. COUPLING REAGENTS USED IN CHAPTER 6 539
APPENDIX J. LIPIDS USED IN CHAPTER 7 540
SCHOLASTIC VITA 541

XIII
LIST OF FIGURES, SCHEMES AND TABLES
CHAPTER ONE
Figure 1.1. Clinically relevant platinum-containing anticancer agents. 2
Table 1.1. Platinum Complexes and Formulations in Clinical Use and
Clinical Trials 3
Figure 1.2. Cellular uptake, detoxification and active efflux of cisplatin. 6
Figure 1.3. Examples of traditional cisplatin-type complexes. 10
Figure 1.4. Structures of transplatin and active analogues. 11
Figure 1.5. Examples of inactive and active monofunctional platinum agents. 14
Figure 1.6. Examples of polynuclear platinum agents. 17
Figure 1.7. New lead: “Platinum-Acridine”. 20
Figure 1.8. Examples of polymeric prodrugs for platinum drugs. 24
Table 1.2. FDA Approved Liposomal Formulations 27
Table 1.3. Liposomal Formulations for Platinum Drugs 28
Figure 1.9. Examples of nanotube as delivery vectors for platinum drugs. 31
Figure 1.10. Activation of polymer–platinum conjugates by ligand exchange. 36
Table 1.4. Platinum-Based Formulations Activated by Ligand Exchange 37
Figure 1.11. Representative examples of pH-sensitive platinum complexes. 38
Table 1.5. Platinum-Based Formulations Activated by Lysosomal Degradation 40
Figure 1.12. Examples of photochemically active Pt(IV) complexes. 43

XIV
Figure 1.13. Self-assembly of polymeric photoactivated Pt (IV). 44
Table 1.6. Examples and Characteristics of Photoactivatable Pt(IV) Prodrugs 45
Figure 1.14. Delivery of Pt(IV) prodrug with nanoparticles. 47
Table 1.7. Targeted and Multifunctional Cisplatin-Derived Pt(IV) Prodrugs
Activated by Bioreduction 48
Figure 1.15. The schematic illustration of platinum-acridine based conjugates. 50
Figure 1.16. Strategies to constructe platinum-acridine conjugates. 52
CHAPTER TWO
Figure 2.1. Structures of cisplatin and platinum-acridine derivatives. 55
Figure 2.2. Design of the combinatorial screening assay. 55
Figure 2.3. Fragments library. 57
Figure 2.4. High-throughput LC-ESMS analysis of “click” reaction mixtures. 60
Figure 2.5. Biological activity profiles for the 60 compounds. 63
Figure 2.6. Relative potencies of fragments in hybrid agents. . 64
Table 2.1. Biological Activity for Selected Library Members 64
Scheme 2.1. Synthesis of acridine (A) building blocks. 69
Scheme 2.2. Synthesis of platinum (P) building blocks. 74
Scheme 2.3. Synthesis of platinum-acridine derivatives. 76

XV
CHAPTER THREE
Figure 3.1. Bioorthogonal fluorescent labeling of platinum–acridine agents. 84
Figure 3.2. Unwinding studies. 85
Figure 3.3. In vitro evaluation of post-labeling method. 88
Figure 3.4. Post-labeling of platinum-acridine in NCI-H460 lung cancer
cells using Alexa Fluor 488-alkyne. 89
Figure 3.5. Intracellular distribution of P1-A9. 91
Figure 3.6. Post-labeling of platinum-acridine in NCI-H460 lung cancer
cells using copper-free click chemistry. 93
Figure 3.7. Schematic illustration of simultaneous detection of
platinum–acridine using preassembly and post-labeling methods. 94
Figure 3.8. LC-MS analysis of P1-A9*. 98
Figure 3.9. Simultaneous detection of platinum–acridine using preassembly
and post-labeling methods in NCI-H460 cells. 99
Scheme 3.1. Synthesis of precursors 3-3 and 3-5. 102
Table 3.1. Composition of CuAAC Reactions Analyzed by Gel Electrophoresis 106
CHAPTER FOUR
Figure 4.1. Structures of tamoxifen and platinum–acridine hybrids. 116
Figure 4.2. Synthesis of target molecules. 119

XVI
Table 4.1. Cytotoxicity Data in Human Breast Cancer Cell Lines 121
Figure 4.3. Differential response of compounds in MCF-7 cancer cells. 122
Figure 4.4. Synthesis of precursor 4-3. 124
Figure 4.5. Synthesis of precursor A11. 127
CHAPTER FIVE
Figure 5.1. General structure platinum–acridine hybrid agents. 137
Scheme 5.1. Synthesis of target compounds. 139
Figure 5.2. Cleavage of ester moieties in compounds P9-A1─P15-A1. 145
Figure 5.3. LC-MS analysis of reaction products resulting from ester
cleavage in compound P9-A1 in PB, PBS and by hCES-2. 147
Figure 5.4. Kinetics of ester hydrolysis in compound P9-A1. 148
Figure 5.5. LC-MS analysis of reaction products resulting from ester cleavage
in P13-A1 by hCES-2. 151
Figure 5.6. Results of the chemosensitivity screen of compounds
P3-A1 ─ P15-A1 in A549 and NCI-H1435 cancer cells. 155
Scheme 5.2. The synthesis of precursors 5-7 ─ 5-10. 159
CHAPTER SIX
Figure 6.1. General structure of platinum-acridine hybrid agents. 175
Figure 6.2. Structures of ester based platinum-acridine analogues. 178

XVII
Scheme 6.1. Synthesis of target compounds. 180
Table 6.1. Conditions of Coupling Reactions with Model Amines 183
Figure 6.3. Schematic representation of coupling reaction and library members. 185
Figure 6.4. LC-ESMS analysis for the reaction of P16-A1 and 6-7 187
Figure 6.5. Schematic illustration of high throughput drug discovery screen. 190
Table 6.2. Conversion Yield and HR-ESMS Results of Platinum-Acridine Based
Conjugates 191
Scheme 6.2. “Two-step” synthesis of platinum-acridine conjugates under
aqueous conditions. 194
Figure 6.6. LC-ESMS analysis of the reaction mixture for preparation of 6-27. 195
Figure 6.7. Schematic illustration of “one-tube” assembly of multifunctional
platinum-acridine conjugates. 199
Figure 6.8. LC-ESMS analysis of “one-tube” reaction. 200
Figure 6.9. LC-ESMS analysis of the hydrolytic products of 6-11. 202
Figure 6.10. Cleavage of ester moieties in selected conjugates. 204
Figure 6.11. Proposed mechanism of chemical hydrolysis. 205
CHAPTER SEVEN
Figure 7.1. Aqueous stability analysis of P2-A1 at 60 ºC for 4 hours 218
Table 7.1. Conditions for the Liposomal Encapsulation of P8-A1 221

XVIII
Figure 7.2. Factors that influence the encapsulation efficiency of P8-A1 223
Table 7.2. Characterization of Four Liposomal Formulations Prepared for P8-A1 229
Figure 7.3. TEM images of liposomes. 230
Figure 7.4. TEM image of a single liposome and the bilayer discs. 231
Figure 7.5. Sephadex G-25 elution profiles for the mixture of Lipo-1 and P8-A1. 232
Figure 7.6. In vitro leakage of P8-A1 from liposomes. 234
Figure 7.7. In vitro release of P8-A1 from Lipo-1 at elevated temperatures. 234
Figure 7.8. Confocal microscopy studies of subcellular distribution. 237
Figure 7.9. Colocalization images of cells treated with Lipo-1 and P8–A1
co-stained with LysoTracker Red. 238
Table 7.3. MTD Study of P8-A1. 242
Table 7.4. MTD study of liposomal formulations of P8-A1 243
Figure 7.10. Antitumor efficacy and mouse weights in A549 xenograft mice. 244
Table 7.5. Average ng Pt in Organs. 245

XIX
LIST OF ABBREVIATION
A Aden(os)ine
ADME Adsorption, distribution, metabolism, and excretion
BER Base excision repair
Carboplatin cis-Diammine-1,1-cyclobutanedicarboxylatoplatinum(II)
CBDCA cyclobutane-1,1-dicarboxylate
CD Circular dichroism
CDI 1,1'-Carbonyldiimidazole
CHOL Cholesterol
Cisplatin cis-Diamminedichloroplatinum(II)
CMC Critical micelle concentration
CNTs Carbon nanotubes
COMU (1-Cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-
morpholino-carbenium hexafluorophosphate
CT Calf thymus
CTR1 Copper transporter 1
DACH trans-1,2-diaminnocyclohexane
DCM Dichloromethane
DCC N,N'-Dicyclohexylcarbodiimide

XX
DIPEA N,N-Diisopropylethylamine
DLS Dynamic light scattering
dsDNA Double-stranded DNA
DMAP 4-Dimethylaminopyridine
DMF Dimethylformamide
DMSO Dimethylsulfoxide
DMPC 1-α-Dimyristoylphosphatidylcholine
DMPG l-α-Dimyristoylphosphatidylglycerol
DNA Deoxyribonucleic acid
DPPG 1,2-Dipalmitoyl-sn-glycero-3-phospho-(1'-rac-glycerol)
DOPE Dioleoylphosphatidylethanolamine
DOPC Dioleoylphosphatidylcholine
DSPG Distearoylphosphatidylglycerol
DSPE-mPEG2k 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy
(polyethylene glycol)-2000]
EDC 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide
EPC Egg phosphatidylcholine
EPR effect Enhanced permeability and retention effect
ER Endoplasmic reticulum

XXI
FBS Fetal bovine serum
G Guan(os)ine
GMP 5´-Guanosine monophosphate
GSH Glutathione
GST Glutathione S-transferase
HBTU (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium
hexafluorophosphate)
hCES-2 Human carboxylesterase 2
HPLC High-performance liquid chromatography
HPMA N-(2-Hydroxypropyl) methacrylamide
HR-ESMS High-resolution electrospray-ionization mass-spectrometry
HSPC L-α-phosphatidylcholine, hydrogenated (Soy)
i.p. Intraperitoneal
i.v. Intravenous
IC50 Inhibitory concentration necessary to reduce cell proliferation by
50% of control
ICD Induced circular dichroism or immunogenic cell death
ICP-MS Inductively-coupled plasma mass spectrometry
LC-ESMS Liquid chromatography-electrospray mass spectrometry
Lobaplatin cis-[Trans-1,2-cyclobutanebis(methylamine)] [(S) lactatoO¹,O¹]
platinum(II)

XXII
m/z Mass-to-charge ratio
MDR Multifactorial drug resistance
MMR Mismatch repair pathway
MPS Mononuclear phagocyte system
MRI Magnetic resonance imaging
MRP Multidrug resistance protein
MTD Maximum tolerated dose
MTS 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-
sulfophenyl)-2H-tetrazolium
MWCO Molecular weight cutoff
Nedaplatin cis-Diammine(glycolato-O¹,O²)platinum(II)
NER Nucleotide excision repair
NMR Nuclear magnetic resonance
NSCLC Non-small-cell lung cancer
nt Nucleotides
OCTs Organic cation transporters
Oxaliplatin trans-L-Diaminocyclohexaneoxalatoplatinum(II)
PDT Photodynamic therapy
PEG Polyethylene glycol
Picoplatin cis-Amminedichlor(id)o-2-methylpyridineplatinum(II)
PyBOP Benzotriazol-1-yl-oxytripyrrolidinophosphonium
hexafluorophosphate

XXIII
PK Pharmacokinetics
PT-ACRAMTU [Pt(en)(ACRAMTU-S)Cl](NO3)2: en = ethane-1,2-diamine,
ACRAMTU = 1-[2-(acridine-9-ylamino)ethyl]-1,3-
dimethylthiourea, acridinium cation
RES Reticuloendothelial system
RNA pol II RNA polymerase II
SAR Structure–activity relationships
Satraplatin Bisacetatoamminedichlorocyclohexylamineplatinum(IV)
SCLC Small cell lung cancer
SEM Standard error of the mean
SWNTs Single-walled nanotubes
TEM Transmission electron microscopy
TMS Tetramethylsilane
TSTU O-(N-Succinimidyl)-N,N,N′,N′-tetramethyluronium
Tetrafluoroborate
XPF Xerodermapig mentosum complementation group F

XXIV
NOVEL STRATEGIES FOR IMPROVING THE PHARMACOLOGICAL
PROPERTIES OF PLATINUM-ACRIDINE ANTICANCER AGENTS
Song Ding
Dissertation under the direction of Ulrich Bierbach, Ph.D.
Professor of Chemistry
ABSTRACT
Unlike traditional cisplatin-type platinum-based anticancer drugs, platinum-acridine
hybrid agents were designed as dual platinating/intercalating DNA-targeted cytotoxics,
which are able to cause cancer cell death at low-nanomolar concentrations.
Unfortunately, the preclinical development of these agents has been hampered by their
severe systemic toxicity. The goal of this dissertation was to devise strategies that
improve the drug-like properties of platinum-acridines and allow their safe delivery. To
achieve this, several classical and newly developed synthetic methodologies have been
used to generate functionally unique hybrid agents. Several model systems, whole-cell
assays, and animal studies have been used in this dissertation to validate their design and
demonstrate their utility as anticancer agents. A modular screening platform was
developed, based on a platinum-mediated amine-to-nitrile addition reaction, for rapid
identification of functionalized platinum-acridine agents. These pre-screening assays
produced functionalized “warheads” while providing insight into structure–activity
relationships (SAR). Using several library members, we set out to explore synthetic

XXV
approaches to construct platinum-acridine-based conjugates. A chemically robust azide-
modified platinum-acridine was selected to validate the feasibility of copper-mediated
and copper-free click chemistry as platinum-compatible conjugation reactions. This
chemistry was used to attach fluorescent molecules to detect platinum-acridines in cancer
cells by confocal microscopy. Both fluorophore tagging prior to incubation with cells and
post-labeling methods were explored. In addition, a hydroxyl-modified warhead was
conjugated with endoxifen via a chemically stable carbamate bond to produce a highly
active hybrid agent in estrogen receptor positive (ER+) breast cancer. In another study,
lipophilic ester-based prodrugs of platinum–acridines were generated showing improved
drug-like properties (e. g., partition coefficients, logD). Two distinct pathways by which
the target compounds can be activated have been confirmed by LC-ESMS and/or NMR
techniques: (i) a platinum-assisted, self-immolative ester cleavage in a low-chloride
environment, and (ii) enzymatic cleavage by human carboxylesterase-2 (hCES-2). Highly
efficient amide coupling reactions in platinum complexes were also developed. This
modular approach can be used to assemble a diverse library of platinum-acridines
containing other bioactive components, such as molecularly targeted therapies, targeted
ligands, and chemosensitizers. Finally, liposomal encapsulation of platinum-acridine was
achieved, and the formulations were evaluated in A549 lung cancer xenograft models in
mice. Improved anticancer efficacy of one of the liposomal formulations compared with
the free drug was observed in this assay. In conclusion, the research in this dissertation
has laid the foundation for the future preclinical development of platinum-acridines as
oncology drugs by devising new synthetic methodology and providing proof-of-concept
data in clinically relevant models of cancer.

1
CHAPTER 1
INTRODUCTION
1.1 Platinum based cancer chemotherapy
Since the serendipitous discovery of the anticancer activity of cisplatin, or cis-
diamminedichloroplatinum(II), platinum based drugs became the mainstay of
chemotherapy for years. Table 1.1 and Figure 1.1 summarize relevant drugs and their
liposomal formulations in routine clinical use, as well as agents that are currently being,
or have been, evaluated in clinical trials. Cisplatin was the first platinum complex
approved for the treatment of a broad range of cancers including non-small cell lung
cancer, small cell lung cancer, testicular, ovarian and bladder cancer [1]. It is widely
accepted that the intracellular activation of platinum drugs through aquation, which is
sensitive to the concentration of physiological chloride ions, is important for subsequent
DNA platination, with the preference to coordinate at the N7 position of guanine [2]
(Figure 1.2). The induced conformational distortions in duplex DNA are recognized by
many proteins that are involved in replication, transcription, and DNA damage repair,
leading to aberrant signal transduction and consequential cell apoptosis [2, 3]. Aside from
DNA associated cytotoxicity, recent evidence indicates that oxaliplatin, but not cisplatin,
may be involved in modulating immune response by triggering immunogenic cell death
(ICD) [4, 5]. It has been proposed that such a mechanism, in which the cancer cells killed
by oxaliplatin act as cancer vaccines, may eliminate the residual, highly resistant cancer

2
cells and exert a long-lasting anticancer effect. These compelling findings prompted a
new therapeutic paradigm for platinum based anticancer drugs [6, 7].
Figure 1.1. Clinically relevant platinum-containing anticancer agents. For current status,
refer to Table 1.1.

3
Table 1.1. Platinum Complexes and Formulations in Clinical Use and Clinical Trials [1,
8]
Complex/Formulation Status Major Indications
Cisplatin Worldwide clinical use Metastatic testicular and ovarian
tumors, advanced bladder cancer
Carboplatin Worldwide clinical use Advanced ovarian carcinoma
Oxaliplatin Worldwide clinical use Metastatic colorectal cancer
Nedaplatin Clinical use in Japan Small and non-small cell lung
cancer, head and neck tumors,
esophageal and bladder tumors,
cervix carcinomas
Lobaplatin Clinical use in China Breast, testicular, ovarian, small
cell lung and gastric carcinomas,
chronic myeloid leukemia
Heptaplatin Clinical use in South
Korea
Gastric cancer
Picoplatin Phase II clinical trials
Phase III clinical trials
Metastatic colorectal cancer,
metastatic castration-resistant
prostate cancer refractory or
resistant ovarian cancer,
refractory or progressed SCLC
BBR3464 Phase II clinical trials Gastric and esophageal
adenocarcinoma
Satraplatin Phase II clinical trials Metastatic, androgen-
independent prostate cancer
Lipoplatin (cisplatin
liposome)
Phase III clinical trials NSCLC, breast cancer, gastric
cancer
SPI-77 (cisplatin
liposome)
Phase II clinical trials Advanced NSCLC, refractory
ovarian cancer
MBP-426 (oxaliplatin
liposome)
Phase II clinical trials Gastric and esophageal
adenocarcinomas

4
Unfortunately, the efficacy of cisplatin is limited by its notorious systemic toxicities
such as nephrotoxicity, neurotoxicity, ototoxicity and myelosuppression [9]. The reasons
for these symptoms are multifold, but generally can be attributed to a lack of tumor
selectivity of current platinum drugs. Additional limitations of platinum-based drugs are
intrinsic and acquired tumor resistance [3]. Lack of responsiveness of platinum drugs can
be caused by insufficient cellular accumulation [10]. Specific pathways for platinum
drugs to enter into cancer cells include passive diffusion and uptake by cell membrane
transporters [10] (Figure 1.2). Multiple transporters were found to participate in the active
influx of platinum drugs, such as copper transporter 1 (CTR1) [11] and organic cation
transporters (OCTs) [12]. Factors that impede cellular uptake result in reduced levels of
platinum in cancer cells, which is correlated with the ineffectiveness of these drugs [10].
Active efflux, likewise, plays an important role in platinum drug resistance. Cancer cells
with overexpressed efflux proteins, including multidrug resistance 1 (MDR1), multidrug
resistance protein-1 (MRP1, also known as ABCC1), multidrug resistance protein-2
(MRP2, also known as ABCC2) and copper efflux transporters ATP7A and ATP7B, may
lose their sensitivity to platinum drugs [10]. Once platinum complexes have entered into
cancer cells, detoxification by various cytoplasmic thiol-containing nucleophiles is
another barrier for them to overcome before reaching their cellular target, nuclear DNA
[13]. The major scavengers in this category is glutathione, the tripeptide that avidly binds
to platinum center via cysteine sulfur, which is likely facilitated by glutathione S-
transferase (GST) [14]. Likewise, elevated levels of thiol-containing proteins, such as
metallothioneins, may lead to platinum drug resistance as well [3].

5
The resistance to platinum drugs may also be triggered after the formation of platinum-
DNA adducts. In the case of cisplatin, the major adducts are intrastrand 1,2-crosslinks,
which bend the DNA duplex significantly towards the major groove. This distortion
produces a wide minor groove, which is preferentially recognized by DNA damage
recognition proteins [3]. An extensive body of evidence suggests the involvement of
DNA repair machineries in causing increased tolerance to platinum-induced DNA
damage by removing platinum-DNA adducts, including nucleotide-excision repair (NER),
base excision repair (BER), mismatch repair (MMR) and double-strand-break repair.
NER is the major mechanism responsible for the removal of cisplatin induced DNA
lesions. As a result, the levels of the key proteins in this pathway, such as ERCC1
(excision repair cross-complementing-1) and XPF (xerodermapig mentosum
complementation group F), are being used as predicative biomarkers of cisplatin
resistance in the clinic [15, 16]. Cancer cells with deficient DNA repair systems were
found to be hypersensitive to platinum drugs. Abnormal apoptotic signaling pathways,
moreover, which are associated with various proteins such as p53, anti-apoptotic and pro-
apoptotic proteins, may result in platinum drug resistance in cancer cells [17].
To overcome the abovementioned limitations, several strategies have been proposed for
safer and more effective platinum based anticancer agents. The first approach is based on
the assumption that certain types of platinum-DNA adducts, which cause unique
conformational distortions, may be biologically more significant than others. To tune the
structural distortions produced by platinum(II) in dsDNA, variation was made to (i) the
ligand sets (non-leaving groups), (ii) the stereochemistry, (iii) the nuclearity, (iv) and the
ability of the complexes to form cross-links vs. monofunctional adducts (see 1.2.1).

6
Thousands of derivatives have been synthesized and tested since the FDA approval of
cisplatin. In another approach, cisplatin-type agents have been linked to tumor selective
carriers, such as folic acid, polymers and nanoparticles. In addition, multifunctional
platinum agents, which contain other bioactive molecules may help to overcome
resistance mechanisms and sensitize tumors to these drugs. Some examples of these
strategies will be discussed in the later parts of this dissertation.
Figure 1.2. Cellular uptake, detoxification and active efflux of cisplatin.
1.2. Platinum(II) Based Anticancer agents
1.2.1 Traditional platinum (II) complexes
Inspired by cisplatin, a large number of cisplatin analogues have been produced and
evaluated as effective anticancer candidates with reduced systemic toxicity [1]. Like

7
cisplatin, these analogues contain two leaving ligands, which leads to the formation of
similar intrastrand or interstrand crosslinks in DNA, thereby inhibiting DNA replication
and transcription. Modifications in the coordination sphere of cisplatin include variations
of non-leaving ammine groups and leaving ligands. The replacement of leaving ligands
affects the aquation kinetics of platinum complexes (will be discussed in detail in 1.4.2),
which, in turn, has a significant impact on their tissue distributions and side effects. This
idea led to the development of carboplatin (Figure 1.1), a second-generation cisplatin
analogue bearing dicarboxylate as the leaving groups. Carboplatin shows less toxic side
effect than cisplatin, even though it produces identical platinum-DNA adducts. The
success of carboplatin spurred the design of many platinum complexes containing
(di)carboxylate leaving groups [18]. Alternative leaving ligands such as acetylacetonate
(acac) and β-diketonates (Figure 1.3, 1-1, 1-2) have also been reported in platinum
anticancer agents [19].
The anticancer efficacy of platinum drugs is closely associated with the platinum-DNA
adducts they produce. Variations of the non-leaving ammine groups of cisplatin can lead
to platinum-DNA adducts with disparate steric hindrance and conformational changes in
target DNA, which might not be recognized by the proteins that contribute to cisplatin
induced cytotoxicity. Therefore, replacement of one or two of the non-leaving ligand may
help to create platinum complexes that maintain sensitivity in tumors resistant to cisplatin.
One successful example is oxaliplatin (Figure 1.1), in which DACH (trans-1,2-
diaminnocyclohexane) was selected as the non-leaving group. The bulky DACH ring in
oxaliplatin occupies most of the DNA major groove, leaving a narrow groove surface and
thus resulting in platinum-DNA adducts processed differently from cisplatin [3].

8
Oxaliplatin has been approved worldwide for the treatment of colorectal cancer and
showed no cross-resistance with cisplatin. It should be noted that the non-leaving ligands
also influence the lipophilicity [18] and the aquation rate [18, 20] of platinum complexes,
which overall can aid to circumvent platinum drug resistance. In particular, picoplatin
(Figure 1.1) was found to overcome glutathione-mediated detoxification due to its bulky
2-methyl pyridine (picoline) ring. In picoplatin, the methyl group is situated right above
the metal center, which protects platinum from attack from intracellular sulfur
nucleophile. Several examples of traditional cisplatin type complexes are summarized in
Figure 1.3.
Lack of tumor selectivity is a major reason for the severe systemic toxicity of cisplatin.
To produce tumor targeted platinum-based anticancer agents, molecules with a high
affinity for cancer cells have been incorporated into platinum complexes as non-leaving
ligands. These ligands include peptides [21] (Figure 1.3, 1-8), estradiol (Figure 1.3, 1-9
and 1-10) [22-24], and folic acid (Figure 1.3, 1-11) [25]. The presence of these ligands
allows the platinum complexes to be recognized by receptors overexpressed on the cancer
cell surfaces and enhance their uptake into tumors. Targeting moieties can also be
introduced as leaving groups in platinum complexes. Since most of them are bulky, the
dissociation of the platinum moiety from the tumor targeting ligands in cancer cells
assures that the carrier does not interfere with the DNA binding of the platinum
“warheads”. Examples of these platinum complexes are shown in Figure 1.3. Apart from
these tumor selective carriers, bioactive ligands, such as tarmoxifen analogues [26]
(Figure 1.3, 1-12), were installed in platinum complexes, which were expected to act

9
synergistically with platinum complexes to sensitize resistant breast cancers to platinum-
based agents.
1.2.2. Non-traditional platinum(II)-based anticancer agents
1.2.2.1. Transplatin analogues
The reasons for the poor anticancer properties of the trans isomer of cisplatin,
transplatin (Figure 1.4), have been investigated for decades [27]. Unlike its cis congener,
transplatin is unable to form stable intrastrand 1,2-crosslinks in the DNA helix due to
geometric constrains [28], while the formation of intrastrand 1,3-crosslinks has been
observed [29]. The major platinum-DNA adducts produced by transplatin are
monofunctional, and only a small amount of them slowly convert into interstrand
crosslinks [27]. This can be explained by the solution chemistry of transplatin. The
replacement of the first chloride with water in transplatin is kinetically favored due to a
mutual trans effect of the chloride ligands, however, the removal of the second chloride
ligand trans to a ligand, low in the trans-effect series such as water or DNA nitrogen
appears to be difficult [30]. The ineffectiveness of transplatin, despite its ability to
modify DNA, emphasizes the importance of the type of platinum-DNA adduct for the
biological outcome of platinum-based anticancer drugs. The higher chemical reactivity of
transplatin also facilitates the interaction with many intracellular nucleophiles, such as
GSH, which accelerates the deactivation process and results in poor biological activity
[28].

10
Fig
ure
1.3
. E
xam
ple
s of
trad
itio
nal
cis
pla
tin
-type
com
ple
xes
.

11
Active trans-platinum complexes can be generated by substituting one or two NH3
ligands with steric hindered amines [27]. These bulky ligands are able to reduce the
reactivity of transplatin analogues in blood and stabilize the platinum-DNA adducts they
produce. Representative examples of active transplatin analogues are shown in Figure 1.4.
The first cytotoxic transplatin analog reported (trans-[PtCl2(NH3)(pyridine)]) contains a
pyridine ring and exhibits comparable cytotoxicity with cisplatin [31], which inspired the
synthesis of numerous platinum complexes with trans geometry bearing planar
heterocyclic ligands, such as quinoline, isoquinoline, and thiazole [27]. The platinum-
DNA adducts produced by these analogues include monofunctional adducts, interstrand
and intrastrand crosslinks [27, 28], which elicit biological responses distinct from
cisplatin and in cell lines overcome resistance to platinum drugs used in the clinic [28].
Other ligands containing N-donor groups, for instance, iminoethers [32], aliphatic amines
[33] and heterocyclic aliphatic amines [34], have been used in the design of transplatin
analogues as well, most of which display cytotoxicity in the micromolar range even in
cell lines resistant to cisplatin [35]. Another intriguing feature of transplatin analogues
lies in their ability to induce ternary DNA-protein adducts, which also potentially
contribute to their cytotoxicity [35].

12
Figure 1.4. Structures of transplatin and active analogues (ipa = isopropyl amine).
1.2.2.2. Monofunctional platinum complexes
Early empirical structure-activity relationships (SAR) ruled out the possibility of using
monofunctional platinum complexes as clinically useful anticancer agents [36]. Most of
the monofunctional platinum complexes share the general formula cis-[Pt(Am)2(L)Cl]+,
in which one chloride ligand is the only labile ligand, which leads to the formation of
monofunctional adducts with DNA. Early efforts to develop monofunctional platinum
complexes resulted in many complexes unfortunately inactive both in vivo and in vitro
[37-40]. Some of them are listed in Figure 1.5 (complexes 1-20, 1-21, 1-22, 1-23). The
concept of monofunctional platinum complexes, however, was recently revisited. The

13
cationic complex cis-[Pt(NH3)2(Pyridine)Cl]+, also known as pyriplatin, was used in a
study to demonstrate that the cytotoxicity of cationic platinum complexes correlates with
the level of organic cation transporters (OCTs) expressed by colon cancer cells [41].
Although much less potent than cisplatin in a number of cell lines, pyriplatin exhibited a
distinct spectrum of activity [41, 42]. The DNA adduct formed by pyriplatin (1-24),
revealed in a crystal structure of a site-specifically platinated DNA, cause less local
distortions in dsDNA compared with crosslinking platinum anticancer agents. It was
proposed that this feature may protect the adducts from removal by the DNA repair
machinery [41, 43] and overcome tumor resistance. Additional mechanistic studies
revealed a relationship between the cytotoxicity of pyriplatin and its capacity to stall
RNA polymerase II (RNA pol II), subsequently inducing transcription inhibition and cell
death [41]. The crystallographic analysis suggested the increase in the steric bulk of the
amine ligand may increase the transcription inhibition. This finding prompted a drug
screen based on a small library of monofunctional platinum complexes bearing
heterocyclic ammine ligands with various steric hindrance [44]. In this study, the
phenanthridine derivative, cis-[Pt(NH3)2(phenanthridine)Cl]+ (NO3-), (phenanthriplatin
(1-25)), was the most potent analogue identified, and its unique anticancer spectrum was
confirmed in the NCI-60 cancer cell line panel [44-46]. The cell killing ability of
phenanthriplatin, on the basis of IC50 values, is 4-40 times higher than that of cisplatin.
The discovery of phenanthriplatin led to the development of other monofunctional
platinum complexes, such as SA-PT(1-26) [47] shown in Figure 1.5.

14
Figure 1.5. Examples of inactive and active monofunctional platinum agents.
1.2.2.3. Polynuclear platinum(II) complexes
Polynuclear platinum(II) complexes represent a novel class of platinum based drugs
with structural features derived from cisplatin [48, 49]. BBR3464 (Figure 1.1) is one of
the most successful analogues in this class, which is a trinuclear drug composed by two
separated monofunctional trans-[PtCl(NH3)2]+ moieties bridged by a tetra-amine trans-

15
[Pt(NH3)2(NH2(CH2)6NH2)]2+ linker[49]. Long range (Pt,Pt) inter and intrastrand
crosslinks were found in the DNA adducts produced by BBR3464, in which the two
platinated DNA bases are separated by up to 4 base pairs. In particular, the intrastrand
crosslinks are formed in both 3’→3’ and 5’→5’ directions, which are distinctly different
from other known platinum-DNA adducts [50]. This novel DNA binding mode finally
leads to Z-DNA like structure around the platinated sites. This type of distortion is not
recognized by high mobility group HMG1 proteins, which are known to contribute
substantially to the cytotoxicity of cisplatin [50]. BBR3464 showed higher potency than
cisplatin when tested in a broad range of tumor cell lines and overcomes resistance to the
clinical drugs [51]. The effective doses in xenografts in mice ranged from 0.3-0.6 mg/kg,
which caused significant tumor regression in several tumor models [51]. BBR3464 is the
only non-cisplatin platinum complex that entered into phase II clinic trials with
manageable toxicity,however, its narrow therapeutic index prevented it from advancing
to the next stage [48].
Variations in the tetramine platinum linkers in BBR3464 with polyamines gave rise to
exceptionally cytotoxic dinuclear analogues [49, 50]. Notably, the positive charges and
the ability to form hydrogen bonds of these connectors seemed to play important roles in
determining the anticancer efficacy. One lead complex is BBR 3610 (1-27,Figure 1.6)
with a spermine analogue as the linker, which was designed to perform in a similar way
to BBR3464 [52]. Other alternative bridging ligands include steric hindered polyamine
(1-29) [53],aromatic (1-30) [54] and heterocyclic ring systems (1-31 and 1-32,Figure
1.6) [55]. Replacement of the leaving ligand chloride with NH3 or other dangling
amines resulted in a new class of substitution-inert platinum complexes [48]. The lead

16
complex in this class is TriplatinNC, of which the square planar tetraamine platinum
moiety is able to form “bidentate” N-O-N hydrogen bonds with the oxygen in phosphate.
This distinct non-covalent binding mode, known as “phosphate clamps” [56], displays
high affinity with DNA and leads to nucleic acid compaction [57]. The prototype
TriplatinNC (1-33,Figure 1.6) shows comparable cytotoxicity to cisplatin in a panel of
tumor cell lines independent of p53 status and the level of GSH in cells [58, 59]. High
cellular accumulation acts as another parameter contributing to the cytotoxicity of
TriplatinNC [60]. The uptake of the substitution-inert, highly charged platinum
complexes is mediated by heparin sulfate proteoglycan (HSPG) [60, 61], since HSPG is
overexpressed in tumor cells, this unique recognition mechanism may confer tumor
selectivity to TriplatinNC analogues. Moreover, the nucleolar localization of TriplatinNC
was also determined [62, 63], linking its anticancer effect to the inhibition of RNA
polymerase I mediated synthesis of ribosomal RNA (rRNA) [62].

17
Figure 1.6. Examples of polynuclear platinum agents.
1.2.2.4. A new lead: platinum-acridines
The idea to generate DNA adducts distinct from the traditional bifunctional 1,2-
intrastrand cross-links has led to the discovery of many new types of non-classical
platinum anticancer complexes with different DNA binding modes. Inspired by these
designs, attempts to combine DNA intercalation and platination in one molecule led to
the creation of platinum-acridine agents [64-66]. One prototypical complex in this class is
PT-ACRAMTU ([PtCl(en)(ACRAMTU)](NO3)2, en = ethane-1,2-diamine, ACRAMTU
= 1-[2-(acridin-9-ylamino)ethyl]-1,3-dimethylthiourea, Figure 1.7, 1-34) [65], which

18
features a single chloro leaving group and a sulfur donor containing linker that connects
the intercalator to the metal, allowing the formation of monofunctional adducts. The best
biological activity was achieved when N-methylethylenediamine was selected as the
main component of the linker (~ 4 fold more potent than cisplatin in NCI-H460 cells),
which is flexible enough in this case to platinate DNA base pairs adjacent to interaction
sites, and thus favor the dual-binding mode involving both DNA intercalation and
platination [67]. Extension of the linker chain reduces the rate of platination step, and
results in reduced cytotoxicity [68]. Other properties of PT-ACRAMTU, such as the
cationic nature and DNA affinity of the acridinium moiety, seem to allow efficient uptake
into cancer cells and in nuclear accumulation and rapid DNA binding. PT-ACRAMTU
proved to be more active than cisplatin confirmed by clonogenic growth and cell
proliferation assays, with IC50 in the low-micromolar and submicromolar range across a
broad range of cancer cells, including ovarian, lung, colon, breast, pancreatic, and brain
cancers [65, 69-71]. Submicromolar activity of PT–ACRAMTU was also found in cancer
cells with aberrant p53 and k-ras gene status, which are typically resistant to cisplatin
[72].
Unfortunately, PT-ACRAMTU was unable to slow tumor growth in a mouse xenograft
model. This prompted extensive SAR studies to identify second generation platinum-
acridines with enhanced activity in vitro and in vivo. Structural modifications were made
to the platinum–intercalator linker, the acridine moiety itself, the non-leaving groups, and
the donor group through which the intercalator is attached to the metal (Figure 1.7B) [70,
73-75]. The most striking cytotoxic enhancement was observed when the thiourea moiety
in PT-ACRAMTU was replaced by amidine as the donor group, resulting in an up to 500-

19
fold enhanced potency compared with clinical drug cisplatin in NSCLC cell line NCI-
H460 (Figure 1.7C, 1-35) [76]. Low nanomolar activity was also found in other cell
lines[68]. The significantly improved activity of these hybrids in vitro has been attributed
to their ability to produce adducts in double-stranded DNA more rapidly than either PT–
ACRAMTU or cisplatin [77]. In addition, unlike bifunctional cisplatin type complexes,
only minor DNA conformational perturbations were induced by the DNA adducts of
platinum-acridines, as confirmed in an NMR solution structural study [78], suggesting
that these DNA adducts are recognized and processed differently by intracellular proteins.
Although the superior cytotoxicity of the second-generation platinum-acridines finally
translated into inhibition of tumor growth in vivo, like other platinum-based anticancer
agents, they remained quite toxic to test animals [76]. This drawback obviously restricts
the development of platinum-acridines as anticancer drugs. To address this limitation,
tumor-targeted approaches and prodrug concepts will be explored in this dissertation in
an attempt to turn the platinum–acridine hybrid agents into systemically less toxic tumor
selective warheads.

20
Figure 1.7. (A) Structure of PT-ACRAMTU. (B) Variations in platinum-acridine
analogues for structure-activity relationship (SAR) studies. (C) Cytotoxcity enhancement
in the first and second generation of platinum-acridines.
1.3. Drug Delivery Systems for Platinum Based Anticancer Drugs
Like other cytotoxics, the “platinums” often show unfavorable pharmacokinetic
properties, which result in inefficient delivery to the diseased tissue and undesired off-
target effects. To overcome these drawbacks, various drug delivery carriers have been
developed in which platinum drugs are chemically attached to tumor-targeting carriers or
physically encapsulated inside nano-sized particles. Targeted delivery vehicles developed
for platinum-based anticancer drugs have recently been reviewed in the literature [79, 80].
The effectiveness of a drug delivery system depends on the nature of the carrier materials,
which alter the pharmacokinetic properties of the cytotoxic payload and may promote

21
selective accumulation in tumor tissue. A wide range of carriers have been introduced to
deliver platinum-based agents, including peptides, receptor ligands, and monoclonal
antibodies, which are actively recognized by cancer cells. Polymers and nanoparticles
enhance the passive accumulation of platinum drugs at the tumor site by exploiting the
enhanced permeability and retention (EPR) effect. This effect, termed “passively targeted”
in the literature, has been confirmed in animal models [81] and observed in patients who
are treated with intravenous liposomal doxorubicin (Doxil) [82]. It should be noted
though that the exact role of the EPR effect in the treatment in humans is still being
debated [81]. To further improve tumor uptake of cytotoxic platinum, tumor-targeting
ligands have been engineered on the surface of nano-sized drug delivery vectors. The
selective interactions between the ligands and the receptors, which are commonly
overexpressed in tumor sites, are proposed to mediate endocytosis, and improve the
therapeutic index [83]. Nevertheless, the advantages of targeted over non-targeted
nanoparticles in vivo are not conclusive and several studies have generated conflicting
results [84].
1.3.1. Polymeric Micelles
Polymeric micelles are self-assembled core-shell structures composed of amphiphilic
block copolymers in aqueous solution at the concentration above their critical micelle
concentrations (CMC) [85]. Unlike those based on small molecule surfactants, the CMCs
of polymeric micelles, generally, are much lower, which indicates higher stability against
dissociation caused by dilution in a large volume of body fluid in vivo [85, 86].
Polymeric micelles as drug carriers have been employed to deliver many anticancer drugs,

22
such as doxorubicin, paclitaxel, camptothecin, which resulted in improved anticancer
efficacy and reduced adverse effects [87]. In particular, due to hydrophobic interactions
with the core of micelles, drugs with poor water solubility can be encapsulated in
micelles with high loading capacity; as a result, micellar encapsulation became one of the
most useful strategies to solubilize hydrophobic drugs and improve their bioavailability
[87]. By contrast, the micellation of hydrophilic compounds in the hydrophobic core has
been problematic due to the weak interactions between the drugs and lipophic segments
[86, 87]. Stability is another concern when designing a micellar formulation. Accelerated
dissociation of micelles may take place under high ionic strength conditions, such as in
blood, or in the presence of serum proteins, inducing pre-mature release of payloads
before they reach tumor sites. This limitation can be partly overcome through cross-
linking amongst unimers [87]. Notably, the extent of crosslinking not only influences the
stability, but can be also used to tune the rate and profile of drug release.
Several micellar formulations for platinum drugs are in various phases of clinic trials
and show promising anticancer efficacy [8]. Successful micellation of platinum
complexes with conventional amphiphilic block copolymers, such as PEG-b-PCL [88],
requires hydrophobic modification, which can be achieved by incorporating lipophilic
ligands as leaving groups in Pt(II) analogues or as the axial ligands in Pt(IV) prodrugs
[89]. Polymers with polyionic segments [90, 91], as alternative delivery vectors, can
spontaneously form polyionic complex micelles (PICs) with cationic platinum complexes
in aqueous solution as a result of electrostatic interactions and coordination bonds [92,
93]. Due to electrostatic neutralization, platinum complexes were found to localize to the
core of micelles [93, 94]. Moreover, the bifunctional cisplatin type platinum complexes

23
can act as cross-linkers in PICs, which may further stabilize the micelles in circulation
and lead to drug release in a controllable manner [93]. The examples and relevant
chemistry of platinum loaded micelles will be discussed in detail in sections 1.4.2 and
1.4.3.
1.3.2. Polymeric Based Prodrugs
The covalent attachment of anticancer agents to the backbones of polymers has been
demonstrated to be an effective strategy to improve their bioavailability [95, 96]. Several
promising polymer-drug conjugates are currently undergoing clinical studies [8]. The
tissue selectivity of the loaded drugs is dominated by the hydrophilic polymers, which
prolong circulation and are able to accumulate at tumor sites by taking advantage of the
EPR effect [97]. To further improve their anticancer efficacy, tumor specific moieties can
be incorporated into prodrugs, allowing enhanced tumor uptake [97]. The concept of
polymeric prodrugs has already been applied in the delivery of platinum drugs [98].
Platinum complexes can be conjugated to polymers through an extended linker, the
cleavage of which not only determines the stability of the conjugate in circulation, but
also allows selective drug release. In addition, platinum complexes can be attached to
polymers containing carboxylate and amino groups. The most successful polymeric
platinum prodrug so far is Prolindac [99-101], in which DACHPt, the [Pt (DACH)]2+
fragment of oxaliplatin, is attached to N-(2-Hydroxypropyl) methacrylamide (HPMA)
[96] through an amidomalonate chelating group (Figure 1.8) [100]. This conjugate
demonstrated ability to induce tumor regression in several xenograft models [101], and
was well tolerated in phase I clinic studies without significant toxicity [102]. Other

24
polymers exploited for this purpose include polyphosphazenes [103], cyclodextrins [104],
polyamino acids [105] and polysaccharides [106]. Additionally, dendrimers [107],
representing a unique class of dendritic polymers, can be used in a similar way [108-110].
Attractive features of dendrimers that make them promising drug delivery systems
include highly uniform structures, narrow size distribution and sufficient surface
functional groups available for drug tethering.
Figure 1.8. Examples of polymeric prodrugs for platinum drugs.
1.3.3. Liposomes
Liposomes are nano-sized, spherical and enclosed bilayer structures with superior
biocompatibility and low toxicity [84, 111]. They are mainly composed of phospholipids
and cholesterol and can encapsulate hydrophilic small molecules in their aqueous cores
and hydrophobic drugs within their phospholipid bilayers [84, 111]. Biomacromolecules,

25
such as therapeutic proteins [84] and nucleic acids [112], which are susceptible to
enzymatic degradation and poorly penetrate membranes, can also be effectively loaded
and delivered by liposomal formulations, leading to enhanced in vivo efficacy. In addition
to the therapeutics, magnetic resonance imaging (MRI) contrast agents can be
incorporated into liposomes as well for disease diagnosis and MRI-guided drug delivery
[113, 114]. Currently, several liposomal formulations have already been approved by the
US Food and Drug Administration (FDA) (Table 1.2), and many more are in various
phases of clinic trials. The first successful product is Doxil, the liposomal formulation of
doxorubicin used in the treatment of Kaposi's sarcoma, breast cancer, and ovarian cancer
with significantly reduced cardiotoxicity compared to the free drug.
Liposomal formulations of platinum-based agents, such as cisplatin and carboplatin,
have been extensively studied, with several promising candidates currently tested in
clinical trials (Table 1.3). One challenge in generating liposomal formulations of
platinum drugs is to achieve a high drug-to-lipid ratio [115]. Since most platinum drugs
are barely soluble in organic solvents, they cannot be loaded by the common procedure
used for paclitaxel [116], according to which the lipophilic drug is first dissolved along
with the lipids in an organic solvent such as chloroform. Platinum complexes are
normally encapsulated by passive loading [117, 118]. In this process the aqueous solution
containing platinum complex is used to hydrate a pre-formed lipid film to produce
platinum loaded liposomes. However, this method usually suffers from low loading
capacity as the result of limited water solubility (for example, the water solubility of
cisplatin is ~2 mg/ml) and poor membrane permeability of platinum complexes [119]. To
increase their water solubility, platinum complexes can be converted to aqua-species by

26
reacting them with silver nitrate, in which the chloride ligand is replaced by water [120].
This conversion also renders the cisplatin-type complexes cationic, which improves their
ability to interact with negatively charged lipids, such as DPPG, through electrostatic
interactions [115]. Liposomes developed according to this strategy showed an
unprecedented drug-to-lipid ratio of 2.5 and 1000-fold higher cytotoxicity in cell based
assays [115]. Lipophilic modification of platinum complexes is an alternative strategy to
improve drug loading. In this case, lipophilic ligands, including fatty acids [121],
phospholipids [122] and carboxylic acid-modified cholesterol [123], are incorporated
through ligand-exchange reactions, producing amphibious platinum complexes that can
self-assemble into phospholipid bilayers.
Leakage of the entrapped molecules from the liposomes during circulation would
produce undesired systemic toxicity. To avoid premature release in vivo, structural
modifications are made to the lipid bilayers. For example, incorporation of cholesterol
can reduce the fluidity of the membranes, leading to decreased leakage of the payloads
from the liposomes [124]. Alternatively, phospholipids with high phase transition
temperatures, which undergo gel-to-liquid-crystalline phase changes above physiological
temperature, also stabilize the bilayers [117]. Furthermore, drug retention is also affected
by the type of payload. Excellent drug retention properties are observed in Doxil, for
example, in which doxorubicin is encapsulated with a high loading capacity due to
loading technique that takes advantage of a trans-membrane pH/salt gradient [82]. The
exploitation of trapping agents, such as EDTA [125] and dextran sulfate [126], which
form inclusion complexes with the payload inside the liposomes, can improve the drug
retention as well.

27
Tab
le 1
.2. F
DA
Appro
ved
Lip
oso
mal
Fo
rmula
tions.
Bra
nd N
ame
Dru
g
Lip
id c
om
posi
tion (
mola
r
rati
o)
Indic
atio
ns
Ref
eren
ce
Am
bis
om
e A
mphote
rici
n B
H
SP
C,
DS
PG
, ch
ole
ster
ol
(2:0
.8:1
)
Fun
gal
infe
ctio
ns
[12
7]
Dau
noX
om
e D
aunoru
bic
in
DS
PC
and c
hole
ster
ol
(2:1
)
Blo
od t
um
ors
[1
28]
Dox
il
Dox
oru
bic
in
HS
PC
, ch
ole
ster
ol,
and
PE
G2
000-D
SP
E (
56:3
9:5
)
Kap
osi
’s s
arco
ma,
Ovar
ian/b
reas
t ca
nce
r
[82]
Myoce
t D
ox
oru
bic
in
EP
C a
nd c
hole
ster
ol
(55:4
5)
Com
bin
atio
n t
her
apy w
ith
cycl
ophosp
ham
ide
in m
etas
tati
c
bre
ast
cance
r
[129]
Dep
ocyt
Cyta
rabin
e
Chole
ster
ol,
Tri
ole
in, D
OP
C,
and D
PP
G (
11:1
:7:1
)
Neo
pla
stic
men
ingit
is a
nd
lym
phom
atous
men
ingit
is
[130]
Epax
al
Inac
tivat
ed h
epat
itis
A
vir
us
(str
ain R
G-S
B)
DO
PC
and D
OP
E
Hep
atit
is A
[1
31]
Infl
exal
V
Inac
tivat
ed h
emag
luti
nin
e
of
Infl
uen
za v
irus
stra
ins
A
and B
DO
PC
and D
OP
E
Infl
uen
za
[132]

28
Table 1.3. Liposomal Formulations for Platinum Drugs.
Formulation Drug Status Indications Reference
Lipoplatin Cisplatin Phase III NSCLC, breast cancer,
gastric cancer
[133]
SPI-077 Cisplatin Phase II Advanced NSCLC,
refractory ovarian cancer
[134]
Aroplatin
(L-NDDP)
aroplatin Phase II Refractory colorectal
cancer, malignant pleural
mesothelioma
[135]
Lipoxal Oxaliplatin Phase I Advanced gastrointestinal
cancer
[136]
MBP-426 Oxaliplatin Phase II Gastric, gastroesophageal,
esophageal
adenocarcinomas
[137]
Another factor that influences the therapeutic responses of liposomes is their release
properties at the disease sites. Formulations with poor release rates are unable to deliver
adequate amount of drug to the diseased tissues, which limits their bioavailability. This
has led to the discontinuation of clinical trials of SPI-077 (Table 1.3), a liposomal
formulation of cisplatin lacking in vivo antitumor efficacy due to insufficient drug release,
in spite of improved pharmacokinetics and reduced side effects [138]. This issue has been
resolved in lipoplatin, another liposomal cisplatin, which uses DPPG as one of the lipid
components. Because of the fusogenic DPPG, lipoplatin delivers platinum drug into
cancer cells through a membrane-fusion mechanism [133]. In addition, controlled release
from liposomes can be achieved when they are incorporated with various triggers in
response to environmental stimuli, such as heat, light, ultrasound, enzymes, and local pH
[111].

29
Short circulation times are frequently observed in conventional liposomes. This
drawback can be attributed to the rapid recognition of liposomes by the mononuclear
phagocyte system (MPS), such as hepatic Kupffer cells and splenic macrophages, which
leads to rapid clearance and reduced accumulation of drugs in targeted organs [83, 139].
The non-specific uptake by MPS is likely to be associated with opsonin, the serum
protein that can bind with liposomes, causing uptake into liver and spleen [139]. The
half-life of liposomes in circulation is to a large extent determined by the
physicochemical properties of lipid bilayers. For example, small liposomes with sizes
less than 100 nm have been demonstrated to evade scavange by MPS and show
prolonged circulation time [140]. Liposomes bearing negative charges interact more
readily with serum proteins than the cationic or neutral ones, showing rapid recognition
by MPS, and are normally more toxic [141]. Long circulation time can be achieved by
hydrophilic modification with polyethylene glycol (PEG). This leads to so-called “stealth”
liposomes, in which PEG provides a protective, hydrophilic shield that prevents the
interaction of liposomes with proteins in blood [83, 142]. The ability of long-circulating
liposomes to accumulate in tumors has been documented in thousands of publications,
and has been attributed to enhance permeability and retention (EPR) effect. Nevertheless,
it should be noted that rapid blood clearance may occur if the dose of PEGylated
liposomes is very low, which is caused by an undesired immunogenic response, known as
the “accelerated blood clearance (ABC) phenonomon” [142].

30
1.3.4. Nanotubes
Carbon nanotubes (CNTs) have been used as drug delivery systems in an attempt to
overcome the dose-limiting toxicity of platinum-based drugs [143, 144]. They are tubular
structures on the nanometer scale with high surface area, and relatively large interior
cavities. CNTs can be further categorized into single-walled nanotubes (SWNTs) and
multi-walled nanotubes (MWNTs) [144]. Although most nanotubes show little
cytotoxicity, their biocompatibility remains as major concern for in vivo applications
[145]. Ultra-short CNTs, 20–80 nm in length and 1.4 nm in diameter, seemed to be more
bio-compatible than the full-length CNTs [146, 147]. Both the exterior wall surface and
the interior space in CNTs can be used to load various drugs for improved efficacy [143].
However, to avoid cytosolic detoxification by intracellular nucleophiles, the interior
cavity is most likely a better location for platinum complexes, where they can be
encapsulated via capillary action [148]. This approach actually turns out to be
challenging for loading hydrophilic platinum complexes, such as cisplatin, which are
inherently incompatible with the hydrophobic cavities of CNTs. In this context, high
loading capacity would be achieved by selecting the corresponding more lipophilic Pt(IV)
prodrugs as alternatives [149, 150] (Figure 1.9, 1-38). To prevent rapid drug leakage of
nanotubes from occurring in circulation, the openings of CNTs can be blocked by
molecular caps; this also allows the release of platinum complexes in a controlled way at
disease sites [151]. In addition, platinum complexes can be tethered to CNTs through
covalent bonds, which requires surface modification of CNTs with carboxylic acid [151]
(Figure 1.9, 1-39), in which case the release of platinum complexes relies on ligand
exchange. This will be discussed in more detail in section 1.4.2. Apart from their

31
extraordinary capacity to retain payloads, the transmembrane penetration of CNTs was
found to occur via a “nanoneedle” mechanism as a complementary pathway to
endocytosis [152], which may help minimize undesired entrapment of platinum drugs in
the lysosomes. Incorporation of PEG [153] (Figure 1.9, 1-40) as a surface modification
not only improves the biocompatibility of CNTs and prolong their circulation times, by
evading the recognition of MPS, but also may improve their dispersity in biological
media. CNTs containing targeting moieties [154, 155], such as folic acid (Figure 1.9, 1-
41), have been demonstrated to preferentially accumulate at tumor sites, and result in
significant tumor regression in mice xenograft compared with controls.
Figure 1.9. Examples of nanotube as delivery vectors for platinum drugs.

32
1.4. Target-Selective Activation of Platinum-Based Anticancer Agents
A successful drug delivery system requires structural integrity of the formulation in
circulation and selective release of cytotoxic payload in tumor tissue. Several validated
strategies for the targeted release of non-metal-based anticancer agents can also be used
for delivering platinum complexes. These involve installation of pH-sensitive [156, 157],
hypoxia-labile [158, 159], and enzymatically cleavable linkers [160, 161]. In platinum-
containing targeted therapies, additional metal-centered reactivities exist such as ligand
exchange, redox reactions, and electrophilic/Lewis-acidic bond activation, which can be
exploited for drug delivery applications.
1.4.1. Controlling solution reactivity
The reactivity of platinum-based agents in biological matrices depends on the nature
and stereoisomerism of the ligands in the metal coordination sphere and on the relative
nucleophilicity of competing bioligands. As mentioned above, the coordinative Pt–N
bonds between cisplatin and DNA require intracellular aquation. By contrast, undesired
reaction of cisplatin with thiol-containing molecules, such as GSH, which causes off-
target effects and tumor resistance, proceeds without aquation [162, 163]. Relative
ligand affinities can generally be predicted from the hard and soft acids and bases
principle (HSAB) [164]. The bonds formed between Pt(II) and cysteine thiol can be
considered resistant to cleavage by any intracellular nucleophile on a biologically
relevant time scale [162, 165]. On the other hand, coordination to methionine thioether
was found to be slowly reversed by DNA nitrogen [166-169]. Formation of reversible

33
Pt(II)–thioether bonds with methionine-rich proteins, such as copper transporter protein
(CTR1), has been implicated in the cellular trafficking of platinum complexes [170, 171].
Inspired by these findings, thioethers have been proposed as alternative donor groups in
carrier ligands that allow controlled release and transfer of Pt(II) to DNA [170]. Finally,
the stereochemistry of substitution reactions in square-planar platinum complexes and
susceptibility of specific ligands to substitution are modulated by the trans-effect [172].
Sulfur ligands, for example, show a relatively strong trans-effect that may promote
(undesired) substitution of the ammine nonleaving groups in cisplatin [172]. Conversely,
even weakly nucleophilic chloride ion has been shown to substitute sulfur donors, if the
latter has been installed trans to phosphine ligands [173], which are highest in the trans-
effect series.
Antitumor active platinum compounds contain leaving groups of intermediate lability,
such as chloride or dicarboxylates, to assure the (pro)drug remains intact during
circulation but sufficiently reactive in target tissue to produce its biological effect.
Aquation of cisplatin occurs in a chloride-ion dependent manner, which is largely
suppressed in the blood [3], but favored in the cytosol where the concentration of
chloride is 30–50-fold lower. The aquation products of cisplatin rapidly react with DNA
nitrogen and can be considered the therapeutically active form of the drug. Aquation also
enhances the water solubility of cisplatin [174] and confers positive charge to the
complex [115, 164], which allows encapsulation of the activated form of the drug in the
hydrophilic core of nanoliposomes [115, 174] and polymeric nanoparticles [8, 175].
Replacement of the chlorido ligands in cisplatin with CBDCA (cyclobutane-1,1-
dicarboxylate), which produces a relatively inert six-membered O,O-chelate, affords

34
carboplatin, a second-generation cisplatin derivative with improved toxicity profile. Loss
of the leaving group in aqueous media in carboplatin is significantly slower than in
cisplatin (half-lives at 37 ºC and pH 7 of 450 h and 2 h, respectively [176, 177]). Unlike
chlorido and carboxylato ligands, N-donor ligands become leaving groups only in the
presence of trans-activating sulfur donors. While this reactivity is generally undesired
because it causes deactivation of platinum drug, it has been exploited in platinum-
containing linkers used to conjugate kinase inhibitors to a tumor-specific carrier for
improved selectivity. In this case, intracellular thiols allow controlled release of active
inhibitor by cleavage of a Pt–N bond [178].
1.4.2. Activation by ligand exchange
Various platforms have been developed for the targeted delivery of a cytotoxic
platinum payload in which platinum has been attached to carboxylic acid-modified
polymeric materials. Polyaspartic acid and polyglutamic acid contain cis-
diam(m)ineplatinum(II) moieties coordinated to monodentate carboxylate or chelated in a
bidentate fashion (Figure 1.10). Platinum in these formulations decreases the negative
charge and increases the hydrophobicity of the polymers. Because of their amphiphilic
character, the platinum-modified polymers self-assemble into micelles, in which the most
hydrophobic segments of platinum-modified polymer form the core and unmodified
polycarboxylic acid the outer shell of the particle. Bifunctional platinum produces cross-
links between polymer chains, which increases the stability of the micellar structure in
circulation [80, 179-181]. The release of active platinum species from these formulations
is triggered by simple ligand exchange with ubiquitous nucleophiles such as chloride and

35
water [8, 80, 181]. Acidic conditions may accelerate the release of the platinum payload
due to competitive protonation of carboxylate at low pH (discussed in detail in the
following section). Carboxylic acid-modified peptide residues were also introduced as
side chains in polymers to allow modification with platinum complexes. Terminal Ɛ-
carboxylate groups installed adjacent to an amide nitrogen form a stable five-membered
N,O-chelate with cis-diammineplatinum(II) [182].
The release kinetics of platinum complexes from polymer can be tuned by
incorporation of multiple donors with varying affinity for platinum. For instance,
biopolymers “doped” with tellurium release platinum complex triggered by competitive
ligand displacement on a time scale of several days up to a month [183]. Carriers
containing N-donors have been used as macromolecular non-leaving groups to generate
substitution-inert polymer–platinum conjugates [184]. In this case, the inactive polymer
would require degradation to produce freely diffusible, monomeric fragments able to
form cytotoxic DNA adducts similar to cisplatin. However, a mechanism by which intact
polymer enters the nucleus to damage chromatin has also been proposed [185]. As
alternative carriers to polymers, other systems such as dendrimers [186], mesoporous
silica nanoparticles (MSN) [187, 188], carbon nantotubes [146], and gold nanoparticles
[189, 190], as well as hybrid materials, have also been used to deliver platinum
complexes (see also section 1.3). Selective representative systems are summarized in
Table 1.4.

36
Fig
ure
1.1
0.
Act
ivat
ion o
f poly
mer
–pla
tinum
conju
gat
es b
y l
igan
d e
xch
ange.
Note
the
dis
tinct
ion b
etw
een
pla
tinum
-coord
inat
ing c
arbox
yla
te i
n t
he
inte
rior
and s
olu
bil
izin
g c
arbox
yla
te o
n t
he
surf
ace
of
the
par
ticl
e.

37
Tab
le 1
.4.
Pla
tinum
-Bas
ed F
orm
ula
tions
Act
ivat
ed b
y L
igan
d E
xch
ange
Dru
g
Pla
tfo
rm
Co
mponen
ts
Pre
clin
ical
and c
linic
al s
tatu
s R
efer
ence
s
Cis
pla
tin
Mic
ella
r
form
ula
tio
n
(NC
-60
04
)
Po
ly(e
thyle
neg
lyco
l)-b
-
po
ly(g
luta
mic
aci
d)
NC
-6004 s
how
s sa
me
pote
ncy
as
cisp
lati
n i
n m
ou
se x
eno
gra
fts
and r
educe
s oto
toxic
ity a
nd n
ephro
toxic
ity i
n m
ice
and g
uin
ea
pig
; dow
nre
gula
tes
gen
es r
elat
ed t
o t
um
or
invas
ion a
nd
met
asta
sis;
is
curr
entl
y b
eing e
val
uat
ed i
n p
has
e II
cli
nic
al t
rial
s
in t
he
US
agai
nst
pan
crea
tic
cance
r an
d N
SC
LC
.
[19
1-1
93
]
Oxal
ipla
tin
Mic
ella
r
form
ula
tio
n
(NC
-40
16
)
Po
ly(e
thyle
neg
lyco
l)-b
-
po
ly(g
luta
mic
aci
d)
NC
-4016 s
how
s im
pro
ved
eff
icac
y i
n x
eno
gra
ft m
ou
se m
od
els
com
par
ed t
o o
xal
ipla
tin, unli
ke
oxal
ipla
tin
, d
oes
not
ind
uce
acu
te
cold
hyper
sensi
tivit
y i
n r
ats;
sel
ecti
vel
y r
elea
ses
pay
load
wit
hin
late
en
do
som
es n
ear
the
nucl
eus;
fo
rmula
tion i
s cu
rren
tly b
eing
eval
uat
ed i
n p
has
e I
clin
ical
tri
als.
[19
4-1
96
]
Oxal
ipla
tin
Po
lym
eric
pro
dru
g
Po
lyp
hosp
haz
ene
The
pro
dru
g s
how
s hig
h c
yto
toxic
ity i
n a
wid
e ra
nge
of
hu
man
cance
r ce
ll l
ines
and im
pro
ved
eff
icac
y a
nd
lo
wer
tox
icit
y t
han
oxal
ipla
tin i
n x
enogra
ft m
odel
s; i
s bio
deg
radab
le a
nd
accu
mula
tes
in t
um
or
tiss
ue.
[19
7,
198
]
Cis
pla
tin
Cro
ss-l
inked
po
lym
er
ves
icle
s
P(O
NB
An) m
-b-P
(ON
B-
PE
G2
k) n
; te
trak
is(3
-
mer
capto
pro
pio
nat
e)a
The
self
-ass
emble
d v
esic
le a
llow
s co
ntr
oll
ed r
elea
se o
f ci
spla
tin
over
day
s in
vit
ro.
[19
9]
Oxal
ipla
tin
Po
lym
eric
met
allo
som
es
Y-s
hap
ed c
opoly
mer
, -
cho
lest
eroyl-
poly
(L-
glu
tam
ic a
cid)
and
po
ly(e
thyle
neg
lyco
l)
(PE
Gas
us-
PL
GA
-
Ch
ole
)
The
form
ula
tion s
how
s pro
longed
blo
od
cir
cula
tio
n, se
lect
ive
tum
or
accu
mula
tion, an
d i
mpro
ved
eff
icac
y i
n v
itro
an
d i
n v
ivo
.
[20
0]
Cis
pla
tin
Den
dri
mer
P
oly
(am
idoam
ine)
(PA
MA
M)
den
dri
mer
s
Den
dri
mer
ic f
orm
ula
tion s
how
s re
duce
d t
oxic
ity a
nd
co
mp
arab
le
effi
cacy
to c
ispla
tin i
n A
2780 x
enogra
ft m
od
el.
[18
6]
Cis
pla
tin
MS
N
Sil
ica
Nan
opar
ticl
es p
rom
ote
upta
ke
of
cisp
lati
n i
nto
HeL
a ce
lls;
sho
w
hig
her
anti
tum
or
acti
vit
y t
han
fre
e ci
spla
tin
agai
nst
MC
F-7
an
d
HeL
a ce
lls.
[18
8]
aO
NB
An =
ox
anorb
orn
enyl
anh
yd
ride;
ON
B-P
EG
2K =
ox
anorb
orn
enyl
po
ly(e
thyle
neg
lyco
l).

38
1.4.3. Activation by pH-sensitive release
The extracellular matrix of tumor tissue is slightly more acidic than that found in
normal tissue. This has been exploited for the design of several pH-sensitive drug
delivery systems, in which the low pH (6.0–6.5) [201] triggers rapid drug release and
uptake into target cells [202]. Drugs with pH-dependent cytotoxicity profiles reduce
severe toxicities in normal tissues, which may lead to an improved therapeutic index.
Thioplatins [203], a group of platinum xanthate derivatives, are significantly more
cytotoxic if treated cancer cell cultures are maintained at pH 6.8 instead of pH 7.4. They
show potent antitumor activity similar to cisplatin in a xenograft model, but are better
tolerated than the clinical drug by the test animals. In structure–activity relationship
(SAR) studies a correlation was observed between biological activity and the basicity
(based on pKa values of the conjugate acids) of the pH-responsive leaving group.
Likewise, aminoalcohol [201] and 1,3-dihydroxyacetone oxime [204] have been
introduced with similar results (Figure 1.11). Both leaving groups form five-membered
N,O-chelates, which are inert at neutral physiological pH but become reactive under
slightly acidic conditions.
Figure 1.11. Representative examples of pH-sensitive platinum complexes.

39
Targeting acidic intracellular organelles, such as endosome (pH ≈ 5.5) and lysosome
(pH ≈ 5.0) [156], is another strategy intensively investigated for targeted delivery of
anticancer agents. Unlike the acidic environment in the tumor extracellular matrix, the
low pH in these organelles accelerates hydrolysis of acid-labile functional groups,
including hydrazones, acetals, and amides, which are frequently used to conjugate
organic drugs to carrier molecules and materials [205]. Similar to platinum payloads
targeted at the low-pH extracellular matrix, acid-labile platinum–carboxylate
coordination has also been exploited to trigger release of platinum drugs by an
intracellular pH differential. Micellar vehicles [8] composed of polymers bearing
carboxylic acids, such as polypeptides, poly(methacrylic acid), and acid-modified
polysaccharides have been developed. These micelles are stabilized in circulation by
platinum-mediated cross-links between polymer strands, but disintegrate as they
accumulate in acidic intracellular endosomal/lysosomal vesicles [159]. Five-membered
N,O-chelates containing carboxylate show enhanced stability at neutral physiological pH
compared to carboxylate coordination alone. This observation has led to the
development of ProlindacTM, a poly(N-(2-hydroxypropyl)methacrylamide) (pHPMA)
based polymer–platinum conjugate, which is currently undergoing phase II clinical
trials[206].
A major problem with the delivery of anticancer agents whose major biological targets
are located in the cytosol or nucleus is their sequestration into, and degradation by, acidic
organelles [207]. Incorporation of agents capable of disrupting the endosomal membrane,
like pore-forming proteins and peptides, pH-buffering molecules, or fusogenic molecules,
into drug delivery systems may overcome this obstacle. An example of this concept is

40
the use of pH-sensitive liposomes containing fusogenic lipid
dioleoylphosphatidylethanolamine (DOPE), which promotes endosomal escape [208].
The cisplatin-based liposomal formulation showed increased activity in a human lung
cancer cell line compared with free cisplatin and was able to overcome resistance to the
clinical drug. On the other hand, a recent study [209] suggests that the release of
platinum in acidic subcellular compartments may lead to improved potency. When
oxaliplatin-based (1R,2R-diaminocyclohexane) platinum (II) was delivered with
poly(ethyleneglycol)-b-poly(glutamic acid)-based polymer, improved antitumor activity
was observed for the micellar formulation. This effect was ascribed to selective drug
release in the lysosomes, which allows the cytotoxic payload to bypass cytoplasmic
detoxification and accumulate in the perinuclear region. Three representative
formulations activated in a pH-dependent manner intracellularly are summarized in Table
1.5.
Table 1.5. Platinum-Based Formulations Activated by Lysosomal Degradation
Platform Components Preclinical and clinical
observations
References
Nanoparticle Succinic acid-
decorated dextran,
cisplatin,
doxorubicin
Prolonged circulation, decreased
accumulation in normal tissues,
enhanced therapeutic efficacy in
tumor-bearing mice
[210]
Dendrimer mPEGylated
peptide dendrimer
w/ peripheral
carboxyl groups,
oxaliplatin
Formulation suppresses tumor
growth more efficiently than
oxaliplatin without inducing
toxicity in a SKOV-3 human
ovarian tumor xenograft
[174]
Polymeric
prodrug
(ProLindac™)
Poly(hydroxyprop
ylmethacrylamide)
w/triglycine side
chains, oxaliplatin
Safety and efficacy have been
demonstrated in phase I/II clinical
trials, superior efficacy of
ProLindac in several tumor
models, acts synergistically with
other cytotoxic agents in vitro
[100, 206]

41
1.4.4. Photochemical activation
The concept of spatially selective activation of anticancer agents at the tumor site with
light, known as photodynamic therapy, has also been applied to photochemically active
metal complexes. Irradiation of transition metal complexes with ultraviolet or visible
light generates excited electronic states, which lead to altered chemical reactivity [211].
As a consequence, specific metal–ligand bonds are weakened, giving rise to accelerated
bond dissociation and ligand exchange. Octahedral Pt(IV) complexes undergo various
photochemical transformations, including ligand substitution, isomerization, and
reductive elimination to the corresponding square-planar Pt(II) complexes [211]. This
provides a means of caging the reactive Pt(II)-based drugs in their inert and nontoxic
Pt(IV) form and activating them as prodrugs selectively in tumors, which reduces damage
to normal tissue and systemic toxicity [212, 213]. A major advantage of light-activated
Pt(IV) complexes over traditional, singlet oxygen-producing photodynamic therapy (PDT)
is that the former mechanism is oxygen independent [214], which has advantages in
treating hypoxic tumors. Photochemical activation of Pt(IV) complexes typically
requires short-wavelength (UVA) radiation, which does not penetrate tissue deeply
enough for treatment of non-topical lesions [215]. (Two-photon excitation has been
proposed as a technique to overcome this drawback [216], see Table 1.6.) Representative
Pt(IV) complexes explored for applications in PDT are presented in Figure 1.12.
One of the first light-sensitive Pt(IV) complexes studied was trans,cis-[PtCl2I2(en)] (1-
20), which readily loses its photolabile iodido ligands. The photoreductive loss of the
iodido ligands generates Pt(II) complexes, which are able to form DNA cross-links [214,
217, 218]. Unfortunately, complex 1-42 suffers from unexpected instability under cell

42
culture conditions even in the absence of light, presumably due to intracellular reduction
by glutathione [214, 217, 218]. As a consequence, light had no effect on the inhibition of
cancer cell proliferation by this complex [217]. Structural modifications were made to
reduce the dark reactivity of the prototypical Pt(IV) complex in the presence of
intracellular reducing agents. This was achieved by introducing axial ligands (Y, see
Figure 1.12) that allow tuning the lipophilicity and redox potential of the metal. For
example, cis,trans-[PtI2(OAc)2(en)] (1-43) [217], which contains axial acetato instead of
chlorido ligands, behaves inert when incubated with calf-thymus DNA for 6 h in the dark,
but readily forms DNA adducts in the presence of light. By contrast, use of hydroxide
ligands in cis,trans-[PtI2(OH)2(en)] (1-44) gave a complex that was relatively unreactive
with DNA in the presence of light [219].
Regardless of the nature of axial ligands none of the iodide complexes showed
sufficient dark stability in the presence of biological reducing agents. To address this
problem, the iodido ligands were replaced with photolabile azido ligands. The Pt(IV)–
azido complexes undergo two-electron reductions to generate Pt(II) species via release of
azide radicals, which further decompose into N2 [220]. Pt(IV)–diazidodiam(m)ine
complexes 1-45 and 1-46 are stable in serum for several weeks in the dark, and only their
photoreduction products react with DNA. The Pt(IV)–azido complexes overcome
cisplatin resistance in 5637 human bladder cancer cells [221].
The complex trans,trans,trans-[Pt(N3)2(OH)2(NH3)2] (1-47), a geometric isomer of
compound 1-45, was as cytotoxic as cisplatin in a number of cell lines [222, 223].
Replacement of one NH3 ligand with pyridine (py) to produce trans,trans,trans-
[Pt(N3)2(OH)2(NH3)(py)] (1-48) resulted in markedly enhanced phototoxicity [224]. The

43
compound showed an advantage over cisplatin in cisplatin-sensitive and cisplatin-
resistant A2780 ovarian cancer cells of 80-fold and 15-fold, respectively [225]. NH3 in
1-48 undergoes undesired photodissociation leading to the species with reduced
cytotoxicity [226, 227]. This reactivity is not observed in trans,trans,trans-
[Pt(N3)2(OH)2(py)2] (1-49), which rapidly induces DNA cross-links and shows greatly
enhanced cytotoxicity [228-230].
Figure 1.12. Examples of photochemically active Pt(IV) complexes.\
Finally, to further improve the pharmacokinetic properties of the Pt(IV)–azido
complexes, the prodrugs were attached to an amphiphilic block copolymer through amide
linkages formed between the amine groups of the polymer and the dangling carboxylate
groups of succinato ligands installed in the axial positions of Pt(IV) [231]. The
polymeric prodrug self-assembles into micellar nanoparticles in aqueous solution, which
are stable in the dark but spontaneously release active Pt(II) species photoreductively
(Figure 1.13).

44
Fig
ure
1.1
3.
Sel
f-as
sem
bly
of
poly
mer
ic p
rodru
g t
hat
rel
ease
s ac
tive
Pt(
II)
spec
ies
trig
ger
ed b
y U
VA
irr
adia
tion.

45
Tab
le 1
.6. E
xam
ple
s an
d C
har
acte
rist
ics
of
Photo
acti
vat
able
Pt(
IV)
Pro
dru
gs
Pla
tinum
der
ivat
ive
Pla
tform
P
recl
inic
al a
nd c
linic
al o
bse
rvat
ions
Ref
eren
ces
1-4
5
Pro
dru
g
Form
s ci
spla
tin-t
ype
guan
ine–
gu
anin
e cr
oss
-lin
ks
wit
h D
NA
up
on
irra
dia
tion;
is a
s cyto
tox
ic a
s ci
spla
tin i
n a
cel
l m
odel
when
photo
acti
vat
ed.
[23
2, 233]
1-4
6,1
-47
P
rod
rug
T
he
com
ple
xes
wer
e m
ore
photo
cyto
tox
ic t
han
the
corr
espondin
g
cis-
isom
ers.
[230, 234, 235]
1-4
8
Pro
dru
g
Form
s in
tras
tran
d D
NA
cro
ss-l
inks;
is
up
to 8
0-f
old
more
cyto
tox
ic
than
cis
pla
tin;
is a
ctiv
e in
cis
pla
tin
-res
ista
nt
hum
an o
var
ian c
ance
r
cell
s; a
nti
tum
or
acti
vit
y i
n x
enogra
ft i
s en
han
ced b
y v
isib
le l
ight.
[222, 225]
1-4
9
Pro
dru
g
Com
ple
x c
an b
e ac
tivat
ed o
ver
a w
ide
wav
elen
gth
ran
ge,
incl
udin
g
vis
ible
lig
ht;
pro
duce
s ce
ll k
ill
pote
nti
ally
thro
ugh
pla
tinat
ion o
f
DN
A a
nd/o
r pro
tein
s.
[236]
trans,
tran
s,tr
ans-
[Pt(
N3) 2
(OH
) 2(N
H2M
e)(p
y)]
Pro
dru
g
Ox
idiz
es g
uan
ine
wh
en i
rrad
iate
d d
ue
to f
orm
atio
n o
f si
ngle
t
ox
ygen
.
[237]
cis-
[PtC
l 2(M
OP
EP
a) 2
]
Pro
dru
g
The
firs
t P
t(IV
) co
mple
x t
hat
can
be
acti
vat
ed b
y t
wo
-photo
n
abso
rpti
on;
ligan
d e
xch
ange
can b
e tr
igger
ed b
y f
emto
seco
nd
las
er
irra
dia
tion a
t 600–740 n
m.
[216]
Pt(
IV)–
azid
e co
mple
xes
b;
Mic
elle
Ir
radia
tion o
f m
icel
lar
form
ula
tion r
apid
ly l
iber
ates
act
ive
Pt(
II)
spec
ies
that
pro
duce
DN
A c
ross
-lin
ks
in v
itro
; in
viv
o s
tud
y s
how
ed
impro
ved
eff
icac
y a
nd r
educe
d s
yst
emic
tox
icit
y.
[231]
aM
OP
EP
= 4
-[2-(
4-m
ethox
yphen
yl)
- et
hyn
yl]
pyri
din
e. b
Co
-form
ula
ted w
ith m
ethox
yl-
poly
(eth
yle
ne
gly
col)
-blo
ck-p
oly
(ε-
capro
lact
one)
-poly
(L-l
ysi
ne)

46
1.4.5. Activation by bioreduction
Reactions of square-planar Pt(II) anticancer agents with biological nucleophiles other
than DNA are likely responsible for their severe systemic toxicity. To overcome this
drawback, Pt(IV) complexes have been proposed early on as non-toxic prodrugs that can
be activated by reduction to the corresponding Pt(II) species [238]. Because octahedral
Pt(IV) complexes are coordinatively saturated and kinetically inert, ligand exchange
reactions with bionucleophiles in the blood can be suppressed to minimize off-target
toxicity. As another potential benefit, Pt(IV) complexes show pharmacokinetic and
toxicity profiles favorable for oral delivery [239, 240].
Satraplatin [218] (cis,trans,cis-[PtCl2(OAc)2(NH3)(cyclohexylamine)], also known as
JM216), is the paradigm of a clinically evaluated oral platinum complex. Satraplatin
loses its axial acetato ligands in cells by reductive elimination. Modification of the axial
ligands with lipophilic moieties is an important means of tuning the
lipophilicity/hydrophilicity balance of the metal complexes [241], which affords
molecules with both favorable solubility and membrane permeability[242]. Lipophilic
groups are also beneficial for nanoencapsulation of the complexes (Figure 1.14A) [89,
243].
The rate of reduction of Pt(IV) prodrugs mainly depends on the nature of the axial
ligands and correlates well with Pt(IV)/Pt(II) reduction potentials [242, 244, 245].
Carboxylato and hydroxido ligands typically give complexes with lower reduction
potentials and slower reduction rates than chlorido ligands [246, 247]. Strongly electron-
withdrawing ligands may render the Pt(IV) complexes too reactive. Mitaplatin [248], for
example, a Pt(IV) compound containing dichloroacetate (DCA) as axial ligands, which

47
was designed to target both nuclear DNA and the mitochondria, undergoes rapid ester
hydrolysis (t1/2 = 2 h) when incubated in biological relevant media in the absence of
reducing agents [249]. This observation suggests that the complex may not enter cancer
cells intact, which would affect the synergistic action of its components.
Importantly, the axial ligands can be turned into chemical handles for conjugating the
inert Pt(IV) prodrugs to macromolecules [250, 251] and nano-sized particles [150, 154,
252-254] (Figure 1.14B). Other bioactive molecules [255, 256], such as valproic acid [89,
257, 258], estrogen [258] and peptides [259-261], have also been introduced as axial
ligands, yielding multifunctional Pt(IV) complexes that damage cancer cells by
synergistic mechanisms or selectively target tumor tissue. Table 1.7 summarizes
technologies developed for the delivery of Pt(IV) prodrugs and their incorporation into
multifunctional therapies.
Figure 1.14. (A) Encapsulation of lipophilic Pt(IV) prodrug within nanoparticles; (B)
covalent attachment of Pt(IV) prodrug to nanoparticles.

48
Tab
le 1
.7.
Tar
get
ed a
nd M
ult
ifunct
ion
al C
ispla
tin-D
eriv
ed P
t(IV
) P
rodru
gs
Act
ivat
ed b
y B
iore
duct
ion.
Pla
tform
C
om
ponen
ts
Pre
clin
ical
and c
linic
al o
bse
rvat
ions
Ref
eren
ces
Tar
get
ed c
onju
gat
e
Mit
och
ondri
a-pen
etra
tin
g
pep
tide,
Pt(
IV)
com
ple
x
The
conju
gat
e is
equ
ally
eff
ecti
ve
in w
ild
-typ
e an
d
cisp
lati
n-r
esis
tant
ovar
ian c
ance
r ce
lls;
cyto
tox
icit
y
corr
elat
es w
ith d
amag
e to
mit
och
ondri
al D
NA
(m
tDN
A).
[262]
Dual
-funct
ion
conju
gat
e
α-T
oco
ph
erol
succ
inat
e,
Pt(
IV)
com
ple
x
The
conju
gat
e ca
use
s both
dam
age
to n
ucl
ear
DN
A a
nd
mit
och
ondri
al d
ysf
un
ctio
n;
the
conju
gat
e is
more
cyto
tox
ic
than
cis
pla
tin.
[263]
Nan
opar
ticl
e P
LG
A-b
-PE
G,
Pt(
IV)
pro
dru
g,
pro
stat
e-sp
ecif
ic m
embra
ne
anti
gen
Pro
longed
syst
emic
blo
od c
ircu
lati
on a
nd d
ecre
ased
accu
mula
tion o
f P
t in
the
kid
ney
s le
ad t
o a
rem
ark
able
impro
vem
ent
in t
her
apeu
tic
index
.
[264]
Nan
otu
be
Sin
gle
-wal
led
car
bon
nan
otu
be,
Pt(
IV)
pro
dru
g,
foli
c ac
id
The
nan
otu
bes
incr
ease
cel
lula
r upta
ke
of
pla
tinum
and
show
enhan
ced c
yto
tox
icit
y.
[150, 154]
Gold
nan
opar
ticl
e A
min
e-fu
nct
ional
ized
,
poly
val
ent
oli
gonucl
eoti
de
gold
nan
op
arti
cles
,
Pt(
IV)
pro
dru
g
The
cyto
tox
icit
y o
f th
e P
t(IV
) co
mp
lex
incr
ease
s
signif
ican
tly i
n s
ever
al c
ance
r ce
ll l
ines
.
[252]
Dual
-funct
ion
conju
gat
e
Dic
hlo
roac
etat
e,
Pt(
IV)
pro
dru
g
The
conju
gat
e kil
ls c
ance
r ce
lls
by d
amag
ing n
ucl
ear
DN
A
and d
isru
pti
ng m
itoch
on
dri
al f
unct
ion.
[265]
Du
al-f
un
ctio
n,
targ
eted
conju
gat
e
Pt(
IV)
pro
dru
g,
FP
R1/2
-
targ
eted
pep
tide
Sel
ecti
vel
y k
ills
the
cance
r ce
lls
and
in
du
ces
imm
un
e
resp
onse
in c
ell
model
s.
[26
6]

49
Tab
le 1
.7 (
conti
nued
)
Mic
elle
P
OE
GM
EM
A3
5-b
-PM
AA
200
and P
OE
GM
EM
A2
6-b
-
PM
AA
90, P
t(IV
) p
rodru
g,
foli
c ac
id
Mic
elle
s en
ter
cance
r ce
lls
by f
ola
te-r
ecep
tor-
med
iate
d e
ndocyto
sis.
The
cyto
tox
icit
y i
n c
ance
r
cell
lin
es i
s in
feri
or
to t
he
dru
g a
lone.
[267]
Mic
elle
P
t(IV
) co
mple
xes
as
bu
ild
ing
blo
cks
for
coord
inat
ion
poly
mer
s
Red
uct
ion o
f P
t(IV
) le
ads
to d
egra
dat
ion o
f
poly
mer
; fo
rmula
tion s
how
s pro
longed
blo
od
circ
ula
tion a
nd i
mpro
ved
tum
or
accu
mula
tion i
n
mic
e.
[250]
Mic
elle
M
PE
G5
000-b
-P(L
A1
400-c
o-
MC
C100
0),
pac
lita
xel
,
pla
tinu
m(I
V)
pro
dru
g
Form
ula
tion s
how
s sy
ner
gis
tic
acti
on i
n v
itro
and
reduce
d s
yst
emic
tox
icit
y a
nd e
nhan
ced a
nti
tum
or
effi
cacy i
n v
ivo.
[268]

50
1.5. Goals of Doctoral Research
While platinum–acridine agents outperform cisplatin in cultured cells, they suffer from
poor pharmacokinetic properties, which result in limited efficacy and dose-dependent
systemic toxicity. The overall goal of this research is to develop chemistries that will
enable new technologies to accelerate the preclinical development of platinum-acridines
with improved drug-like properties. Toward this end, we set out to establish the
methodologies that allow the chemical conjugation of platinum-acridine derivatives to
other bioactive molecules, such as tumor-targeted ligands, so that the consequent
conjugates are rendered with tumor selectivity, enhanced efficacy and reduced side
effects. To achieve this goal, conjugatable platinum-acridines with excellent cytotoxicity
should be firstly identified (Figure 1.15).
Figure 1.15. The schematic illustration of platinum-acridine based conjugates.
In chapter 2, we have designed a high-throughput library approach to search for potent
functionalized platinum-acridines by taking advantage of a highly efficient platinum-
mediated amine-nitrile addition reaction. This approach enables us to rapidly assemble

51
platinum-acridines in a fragment-based manner and test them in cytotoxic assays. In
addition, the structure-activity relationship established in this study suggests that the
optimal sites for further chemical conjugations are the side chains in nitrile moiety (R1,
Figure 1.15) and acridine fragment (R2, Figure 1.15), the modications of which cause
minimal interference as platinum-acridines bind to DNA.
Next, we explored the conjugation chemistries for the attachment of functionalized
platinum-acridines to other molecules. Two synthetic strategies have been developed in
this dissertation (Figure 1.16). In chapter 4, the estrogen receptor (ER) inhibitor
endoxifen was tethered to hydroxyl-modified platinum-acridine to sensitize ER positive
breast cancer to platinum drugs. The synthesis of the conjugates were achieved via the
“pre-assembly” method, in which the platinum moiety is introduced after all other
functional fragments were assembled into organic ligands. However, synthesis of
platinum complexes according to this method appears to be highly customized, which
prompted us to develop “post-modification” method for platinum-acridines. In this
approach, the conjugatable platinum-acridine analogues were ligated to other components
through “platinum-compatible” conjugation reactions. The copper-mediated click
chemistry was used in chapter 3 to map the subcellular distribution of an azide-modified
platinum-acridine derivative with fluorescent molecular probe containing an alkyne
handle, which allows us to gain more mechnastic information about platinum-acridines.
In addition, we have developed an efficient coupling reaction in chapter 6 to generate an
amide bond between carboxylic acid modified platinum-acridines and other bioactive
ligands containing amino group. The modularity of this approach enables the parallel
synthesis of a library of multifunctional platinum-acridine conjugates for drug screening.

52
For platinum-acridine conjugates, we reasoned that the controlled release of cytotoxic
warheads in tumor cancer is also important to their efficacy. A class of ester-based
prodrugs of platinum-acridine were produced in chapter 5. The installation of ester group
is supposed to improve the lipophilicity of the hydrophilic platinum complexes, and thus
presenting improved drug-like properties. These prodrugs were found to be activated
through two pathways: (1) metal-assisted, chloride-triggered ester cleavage and (2)
hCES-2 mediated enzymatic cleavage.
Different from the approaches relying on platinum-acridine conjugates, another strategy
in this dissertation is to deliver platinum-acridines using nanoliposomes, which have been
exploited to improve the therapeutical indices of many other conventional
chemotherapeutic agents. We optimized the procedures to encapsulate the most cytotoxic
derivative and expected this delivery vehicle for platinum-acridine analogues to able to
overcome the drawbacks they currently experienced.
Figure 1.16. (A) Construction of platinum-acridine conjugates with “pre-assembly”
method. (B) Construction of platinum-acridine conjugates with “post-modification”
method.

53
CHAPTER 2
USING A BUILDING-AND-CLICK APPROACH FOR PRODUCING
STRUCTURAL AND FUNCTIONAL DIVERSITY IN DNA-TARGETED
HYBRID ANTICANCER AGENTS
The work contained in this chapter was initially published in Journal of Medicinal
Chemistry in 2012. The manuscript, including figures and schemes, was drafted by Song
Ding and Dr. U. Bierbach and edited by Dr. U. Bierbach before submission to the journal.
Since the publication, changes in both format and content have been made to maintain a
consistent format throughout this work. The experiments described were performed by
Song Ding, except for the the cytotoxicity assays which were contributed by Dr. Xin
Qiao.

54
2.1. Introduction and Design Rationale
Platinum-based anticancer drugs continue to be the cornerstone of many chemotherapy
regimens despite their clinical drawbacks and the recent advent of more tolerable
molecularly targeted therapies[3] [269]. DNA-targeted agents acting by novel
mechanisms at the molecular level show considerable promise as potential treatments for
intractable tumors [270]. In particular, we and others have demonstrated that DNA
adducts that do not mimic the cytotoxic lesions produced by the anticancer agent cisplatin
(cis-diamminedichloridoplatinum(II), Figure 2.1) may overcome tumor resistance to the
clinical platinum drugs [271-273]. Platinum–acridine agents, represented by the
prototypical structures in Figure 2.1, unlike cisplatin, do not induce DNA cross-links but
form monofunctional–intercalative adducts [274]. The unique structural and
thermodynamic features of this dual binding mode and the rapid formation of the hybrid
adducts in cellular DNA result in an up to 500-fold higher cytotoxic potency compared to
cisplatin in aggressive, rapidly proliferating cancers [275-279]. While the cytotoxic
properties of the platinum–acridines translate into promising tumor growth inhibition in
vivo, the dose-limiting systemic toxicity observed in mice suggests that modifications of
these agents are necessary to improve their pharmacological properties [279].
Specifically, we are interested in attaching the platinum–acridines as cytotoxic
“warheads” to receptor- or tumor tissue-targeted vehicles. To achieve this goal, it is
necessary to install chemically or enzymatically reversible linkers in the original
structures. This is not a trivial task as many modifications made to a cytotoxic agent for
the purpose of delivering it may compromise its target interactions and decrease its
potency [280].

55
Figure 2.1. Structures of cisplatin and platinum-acridine derivatives.
Figure 2.2. Design of the combinatorial screening assay. (A) Synthesis of fragment
libraries containing variable and functionalizable groups R1–R3. For the color-coding of
residues, Figure 2.3. (B) Microscale coupling (“click”) reactions. (C) Qualitative and
quantitative LC-ESMS analysis. (D) High-throughput screening in cancer cells on 96-
well plates. (E) SAR data analysis and hit identification.

56
To accelerate the preclinical development of our technology, we have developed new
methodology based on efficient platinum-mediated click chemistry to generate and screen
a modular library of functionalized, conjugatable platinum–acridines. Fragment-based
approaches using simple condensation and click chemistries have demonstrated great
utility for the design of enzyme-targeted inhibitors [281, 282]. Here, we demonstrate, for
the first time, that modular library screening can be applied to DNA-interacting
anticancer agents. Proof-of-concept data is presented demonstrating that the new assay
provides a powerful tool for establishing structure–activity relationships (SAR) and
identifying candidates for the desired biological application.
2.2. Results and Discussion
2.2.1. Design of the combinatorial screen assay: assembly and characterization
To generate the desired compound library, sets of suitably modified platinum
complexes and 9-aminoacridine derivatives, the two building blocks of the hybrid agents,
were synthesized. We then used highly efficient amine addition to platinum-coordinated
nitrile ligand, a classical metal-catalyzed reaction for forming CN bonds [283], to
generate the amidine-linked hybrids (based on the prototype with X = NH in Figure 2.1).
The clean conversion to hybrid agent without the formation of stoichiometric by-
products, which is reminiscent of organic click chemistry [284], provides an ideal platform
for generating diversity within the library of platinum–acridines (for details see the
Experimental Section).

57
Figure 2.3. Library of platinum (P) and acridine (A) building blocks with design
elements highlighted.
A total of 6 nitrile-modified platinum complexes and 10 acridines were synthesized as
building blocks for the modular click assembly of 60 hybrids (Figure 2.3). Structural
diversity in this small library was achieved by varying the nonleaving group(s) (R1) and
nitrile ligands (R2) in the platinum moieties, as well as the geometry of the acridine side
chain (R3). Several of the unmodified parent compounds studied previously were also

58
generated. Conjugatable functional groups were placed strategically within the platinum
and acridine moieties as attachment points for targeted carriers. These include hydroxyl
(in P5, P6, A3, and A4) and carboxylic acid (in A5–A7) groups, which lend themselves
as coupling partners in (reversible) ester, carbamate, and amide linkages [285]. In
addition, terminal azide groups were installed in extended side chains using amide (A8,
A9) and carbamate (A10) coupling chemistry. These derivatives are also of interest for
their use as imaging tools in bioorthogonal reactions[286] with alkyne-modified
fluorophores to study the subcellular distribution of the hybrid agents.
To demonstrate the utility of this approach, 60 micro-scale reactions were assembled
containing stoichiometric amounts of platinum (P) and acridine (A) and allowed to
incubate in DMF under conditions that produce hybrid agent without any detectable side
products (see experimental details and Appendix B1). Automated in-line high-
performance liquid chromatography–electrospray mass spectrometry (LC-ESMS) was
used to monitor the progress of the reactions and to calculate the yield of each hybrid.
This was possible by integrating the HPLC traces recorded at an acridine-specific
wavelength assuming the chromophores in the acridine precursors (A) and in the newly
formed hybrids show the same absorptivity. An example of an LC profile along with
ESMS characterization of the reaction mixture is given in Figure 2.4 for hybrid P6-A1
(for a complete set of 60 LC-MS profiles see APPENDIX B1). Note the absence of side
products and high yield (80%) of the conversion. Baseline separation of the two fractions
corresponding to unreacted acridine and product platinum–acridines was observed for all
60 samples analysed, which greatly facilitated quantification and mass spectrometric
characterization of the hybrids.

59
Species in solution were identified by their molecular-ion peaks and fragments
generated by in-source collision-induced dissociation (CID). The reaction mixtures were
diluted with appropriate amounts of media so that each sample contained the same
concentration of hybrid agent prior to incubations with cancer cells. NCI-H460 cells
were then exposed to a fixed concentration of 50 nM of each platinum–acridine and the
viability of the cancer cells relative to untreated control was assessed after 72 h of
incubation using a colorimetric cell proliferation assay. The assay was also performed
with the platinum and acridine precursors, which proved to be at least two orders of
magnitude less cytotoxic than the hybrids. These control experiments were necessary to
demonstrate that unreacted precursors in reactions that did not go to completion did not
significantly contribute to the inhibition of cell proliferation (see APPENDIX E. Figure
E.2. for an example). Finally, to further validate the pre-screening approach, selected
compounds of interest were resynthesized and their IC50 values determined in the same
cell line (Figure 2.2).
2.2.2. Validation of the combinatorial screen assay: biological evaluation and SAR
analysis.
To validate our combinatorial approach as a tool for establishing SAR in platinum–
acridines, the data set generated was examined by plotting cell viabilities for the 60
samples (P1-A1 to P6-A10). To gain insight into the relative importance of the
fragments P and A in each compound, samples were numerically sorted into groups of
hybrids containing the same platinum moiety and groups sharing the same acridine ligand
(Figure 2.5). The former alignment demonstrates that for hybrids containing the same

60
Figure 2.4. High-throughput LC-ESMS analysis of “click” reaction mixtures. (A)
Reverse-phase HPLC trace for the reaction of platinum precursor P6 with acridine A1.
(B) ESMS spectrum of A1 (HPLC fraction with retention time 8.8 min) recorded in
positive-ion mode. Characteristic molecular and fragment ions: [M+H]+ (m/z 252.2) (C)
ESMS spectrum of P6-A1 (HPLC fraction with retention time 9.6 min) recorded in
positive-ion mode. Characteristic molecular and fragment ions: [M]+ (m/z 613.2),
[M+H]2+ (m/z 307.1, z = 2), [M-OH+H]+ (m/z 596.2), [M-[PtCl(pn2-OH)]]+ (free acridine–
amidine ligand, m/z 293.3) [M-2H-Cl]+ (m/z 575.3), [M-H-Cl]2+ (m/z 288.3, z = 2), [M-
pn2-OH-Cl]+ (m/z 485.2). The structures of A1 and P6-A1 are shown as insets in (B) and
(C).

61
platinum moiety (P1–P6) the biological activity usually decreases significantly with
increasing degree of modification and length of the (functionalized) 9-aminoacridine side
chain (Figure 2.5A). Analysis of the global activity profiles for P1–P6 also shows that
variation of the nitrile ligand (R2 = Et in P1, P3, and P5 vs. R2 = Me in P2, P4, and P6)
leads to little variation in activity and produces pairs of most similar compounds. By
contrast, replacement of the ethane-1,2-diaminoethane (en) non-leaving group with
ammine (NH3) ligands (P1/P2 vs. P3/P4) enhances the cytotoxicity of hybrids containing
acridines A5–A7. For several of the less active derivatives introduction of rac-1,3-
diaminopropan-2-ol (pn2-OH) as a nonleaving group (in P5 and P6) resulted in improved
cancer cell kill (average cell viabilities in Figure 2.5A were 55% for P1/P2, 45% for
P3/P4, and 42% for P5/P6, relative to control cells). Likewise, a plot of cell viabilities
for the 10 types of hybrids defined by common acridine moieties (Figure 2.5B) gives
important clues about SAR in this library of compounds. In general, the derivatives
containing ethylene groups in the acridine side chain performed better than those
containing extended propylene chains, based on average cell viabilities of 15% and 30%
calculated for the pairs A1/A3 and A2/A4, respectively. Derivatives A6 and A7, which
contain extended, carboxylic acid-modified side chains that only differ in the positioning
of the secondary amino function (Figure 2.3) produce an average reduction in cell
viability of merely 50% while showing similar activity profiles. Extension of the side
chains to contain azide functionalities (A8–A10) follows the same trend and leads to a
further decrease in cytotoxicity of the corresponding hybrids, which form a cluster of
highly similar profiles (Figure 2.5B). On the basis of mean cell viabilities calculated

62
across the entire set of 60 library members, hybrids containing A1, A3, and P6 resulted in
the strongest cell kill effect (Figure 2.6).
To assess the utility of the library screen as a tool for target compound identification, a
structurally diverse subset of four analogues of interest was resynthesized and tested in
NCI-H460 cells. IC50 values were calculated from the corresponding dose–response
curves (see Appendix E, Figure E.1) and are summarized along with data acquired
previously for the parent compounds in Table 2.1. Of the hybrids chosen, the azide-
functionalized compound P1-A8 required the highest concentration (110 ± 8 nM) to
inhibit cell proliferation by 50%, consistent with its relatively poor performance in the
prescreen. (It should be noted, however, that this derivative is still an order of magnitude
more potent than cisplatin in this cell line [276].) Compounds that reduced cell viability
to levels of 20% of control in the high-throughput assay resulted in significantly lower
IC50 values. The hydroxy-modified hybrids P4-A3 and P6-A1 showed the highest cell-
kill potential with the latter reaching cytotoxicity levels in the low-nanomolar
concentration range similar to the unmodified platinum–acridines P1-A1 and P2-A1
[277, 279].

63
Fig
ure
2.5
. B
iolo
gic
al a
ctiv
ity p
rofi
les
for
the
60 c
om
pounds
bas
ed o
n t
he
via
bil
itie
s of
trea
ted
cel
ls r
elat
ive
to u
ntr
eate
d
contr
ol
det
erm
ined
in a
colo
rim
etri
c ce
ll p
roli
fera
tion a
ssay.
The
test
com
pounds
are
sort
ed a
nd c
olo
r-co
ded
by c
om
mon
pla
tinum
moie
ties
in (
A)
and b
y c
om
mon a
crid
ine
moie
ties
in (
B).
E
rror
bar
s in
dic
ate
± s
tandar
d d
evia
tions
for
sets
of
thre
e
dat
a poin
ts f
or
each
com
pound.

64
Figure 2.6. Relative potencies of acridine (A) and platinum (B) fragments as building
blocks in hybrid agents expressed as ± % deviations from the mean cell viability (M.V.)
determined across the entire set of 60 library members.
Table 2.1. Summary of structural elements and biological activity for selected library
members.
Compound R1 R2 Functional group attached to R1 or R3 IC50 (nM)c
P1-A1a en Et b 12 ± 2
P1-A8 en Et N3 110 ± 8
P2-A1a en Me b 2.8 ± 0.3
P3-A7 (NH3)2 Et COOH 90 ± 5
P4-A3 (NH3)2 Me OH 45 ± 9
P6-A1 pn2-OH Me OH 8.6 ± 0.5
a References [277] and [278]. b Unmodified derivative. c IC50 values are reported as the
mean ± standard deviation for at least 2 individual experiments performed in triplicate.

65
The pre-screening method provides insight into the relative importance of the two
functionally interdependent components, based on the similarity of activity profiles and
clustering of individual library members. Because the method is based on testing
reaction mixtures without purification steps at an arbitrarily fixed concentration of test
compound, the cell viabilities extracted from it do not correlate with IC50 values, which
have to be calculated from the corresponding drug–response curves. Potential
complications in evaluating the biological activity of library members may arise in cases
in which incomplete conversion of the building blocks to hybrid is observed. Under such
circumstances, unreacted platinum or acridine precursors may contribute to the cell kill
observed in the pre-screen. Furthermore, synergistic effects between multiple
components, which might lead to enhanced cell kill, cannot be completely ruled out.
Generally these mixtures resulted in poor inhibition of cancer cell proliferation,
indicating that both the precursors and the hybrid agent were only marginally cytotoxic.
However, in some cases, such as P3-A7, which showed only 50% conversion but reduced
cell viability by more than 70%, it had to be confirmed that the hybrid and not unreacted
precursor caused the cell kill, which proved to be the case (see the Appendix E, Figure
E.2). Finally, in addition to demonstrating the feasibility of our library design and its
validity as a prescreening tool, we have identified promising hydroxyl-modified hybrids
P4-A3 and P6-A1 as potential “warheads” for the desired application of targeted prodrug
delivery.

66
2.3. Conclusion
In summary, we have demonstrated the potential of library screening for delineating
SAR in a class of DNA-targeted hybrid agents and for lead discovery. The modular click
approach in conjunction with the facile preparation of building blocks provides a
powerful tool for efficient screening of second-generation derivatives of these promising
cytotoxics. The use of metal-based coupling chemistry as an efficient screening tool for
the discovery of metal-containing (hybrid) pharmacophores is a previously unexplored
approach. Here, we provided proof-of-concept data by generating 60 hybrid agents and
testing them in one cancer cell line. We envision that this assay can easily be extended to
multi-step library synthesis, for instance by combining the platinum-mediated additions
with other forms of click or coupling chemistries. Likewise, parallel testing in multiple
cell lines would add another dimension to this assay. Such an approach would ultimately
lend itself to cluster analysis of extended databases with the ultimate goal of designing
personalized oncology drugs that can be targeted to specific cancer types.
2.4 Experimental Section
2.4.1. Reagents and Instrumentation.
All reagents were used as obtained from commercial sources without further
purification unless indicated otherwise. 1H NMR spectra of the target compounds and
intermediates were recorded on a Bruker Advance 300 MHz instrument. Proton-
decoupled 13C NMR spectra were recorded on a Bruker Advance 300 instrument

67
operating at 75 MHz. Chemical shifts (δ) are given in parts per million (ppm) relative to
tetramethylsilane (TMS). 1H NMR data are reported in the conventional form including
chemical shifts (δ, ppm), multiplicities (s = singlet, d = doublet, t = triplet, q = quartet, m
= multiplet, br = broad), coupling constants (Hz), and integral intensities). 13C{1H} NMR
data are reported as chemical shifts (δ, ppm). HPLC-grade solvents were used for all
HPLC and mass spectrometry experiments. LC-ESMS analysis was performed on an
Agilent 1100LC/MSD ion trap mass spectrometer equipped with an atmospheric pressure
electrospray ionization source. Eluent nebulization was achieved with a N2 pressure of 50
psi, and solvent evaporation was assisted by a flow of N2 drying gas (350 °C). Positive-
ion mass spectra were recorded with a capillary voltage of 2800 V and a mass-to-charge
scan range of 150 to 2200 m/z. To establish the purity of target compounds, samples
were diluted in methanol containing 0.1 % formic acid and analyzed using a 4.6 mm
150 mm reverse-phase Agilent ZORBAX SB-C18 (5 µm) analytical column at 25 °C.
The following solvent system was used for the characterization of “click” reactions:
solvent A, optima water, and solvent B, methanol/0.1% formic acid, with a flow rate of
0.5 mL/min and a gradient of 95% A to 5% A over 12 minutes. The purity of the
resynthesized compounds (> 95%) was confirmed using the following solvent system:
solvent A, optima water, and solvent B, methanol/0.1% formic acid, with a flow rate of
0.5 mL/min and a gradient of 95% A to 5% A over 25 minutes. A wavelength range of
363–463 nm was used to monitor the reactions and to determine reaction yields. Peak
integration was done using the Agilent LC/MSD Trap Control 4.0 data analysis software.
The synthetic precursors [PtCl2(en)], cisplatin , and rac-1,3-diaminopropan-2-ol [287], 9-
phenoxyacridine (2-1) [65], and building blocks N1-(acridin-9-yl)-N2-methylethane-1,2-

68
diamine (A1) [65], and N1-(acridin-9-yl)-N3-methylpropane-1,3-diamine (A2) [288] were
synthesized according to the cited methods.
2.4.2. Synthesis and characterization of acridine building blocks.
The synthesis of 2-((2-(acridin-9-ylamino)ethyl)amino)ethan-1-ol (A3). A mixture
of phenoxyacridine (2-1) (2.71 g, 0.01 mol) and 2-(2-aminoethylamino)ethanol (2-4)
(1.14 g, 0.011 mol) in 15 mL of anhydrous THF was refluxed for 16 h. The solvent was
evaporated off and the residue was dissolved in 30 mL of acetone. To this solution were
added 5 mL of concentrated HCl and the mixture was stirred at 4 C for 3 hours. A
yellow precipitate formed which was recovered by filtration, resuspended in 50 mL of 2
M ammonium hydroxide, and stirred at room temperature for 30 min. The aqueous phase
was extracted with CH2Cl2 and the organic phase was collected, dried over Na2SO4, and
concentrated using rotary evaporation, affording 2.57 g of the free base as an yellow solid
(Yield: 92%). 1H NMR (300 MHz, CDCl3) δ 8.27 (d, J = 8.5 Hz, 2H), 7.69 (d, J = 7.6 Hz,
2H), 7.59 (t, J = 7.3 Hz, 1H), 7.26 (t, J = 7.5 Hz, 2H), 4.50 (s, 1H), 3.88 (t, J = 6.2 Hz,
2H), 3.47 (t, J = 5.7 Hz, 1H), 2.90 (t, J = 6.2 Hz, 2H), 2.63 (t, J = 5.7 Hz, 2H). 13C NMR
(75 MHz, CDCl3) δ 151.70, 129.74, 124.84, 121.30, 60.39, 51.27, 50.18, 40.90. MS (ESI,
positive-ion mode): calculated for C17H20N3O ([M+H]+), 282.15; found: 282.3.

69
Sch
eme
2.1
. S
ynth
esis
of
acri
din
e (A
) buil
din
g b
lock
s.

70
N1-(acridin-9-yl)-N3-methylpropane-1,3-diamine (A2) was prepared using the
procedure described for A3. Yield: 94%. 1H NMR (300 MHz, CDCl3) δ 8.12 (d, J = 8.7
,2H), 8.02 (d, J = 8.7, 2H), 7.61 (t, J = 6.7 Hz, 2H), 7.25 (t, J = 7.5 Hz,2H), 4.01 (t, J
= 5.9 Hz, 2H), 2.96 - 2.75 (m, 2H), 2.53 (s, 3H), 1.94-1.68 (p, J = 5.9 Hz , 2H). MS (ESI,
positive-ion mode): calculated for C17H20N3 ([M+H]+), 266.36; found: 266.2.
3-((2-(acridin-9-ylamino)ethyl)amino)propan-1-ol (A4) was prepared using the
procedure described for A3. Yield: 86%. 1H NMR (300 MHz, CDCl3) δ 8.11 (d, J = 8.5
Hz, 2H), 8.02 (d, J = 8.6 Hz, 2H), 7.72 - 7.54 (m, 2H), 7.35-7.29 (m, 2H), 3.87-3.82 (m,
4H), 3.16 - 2.64 (m, 4H), 1.79 (p, J = 6.5 Hz, 2H). 13C-NMR (75 MHz, CDCl3) δ 151.97,
147.93, 130.25, 127.65, 123.25, 122.70, 116.33, 62.15, 49.48, 48.91, 47.68, 31.98. MS
(ESI, positive-ion mode): calculated for C18H22N3O ([M+H]+), 296.38; found: 296.3.
The synthesis of (2-(acridin-9-ylamino)ethyl)glycine (A5). A mixture of
phenoxyacridine (2-1) (2.71 g, 0.01 mol) and 2-((2-aminoethyl)glycine (2-6) (1.3 g,
0.011 mol) in 20 mL of dry MeOH was refluxed for 3 h. The yellow solid that
precipitated during the reaction was collected by filtration, washed with hot THF and
ether, and dried in a vacuum, affording 2.55 g of the product as a yellow solid (Yield: 86
%). 1H NMR (300 MHz, MeOH-d4) δ 8.22 (m, 2H), 7.55 (t, J = 7.6 Hz, 2H), 7.43 (m,
2H), 7.15 (t, J = 7.6 Hz, 2H), 4.08 (d, J = 6.0 Hz, 2H), 3.22 (s, 2H), 3.16 (t, J = 5.9 Hz,
2H). 13C NMR spectrum of this compound was not obtained due to limited solubility of
the compound. MS (ESI, positive-ion mode): calculated for C17H18N3O2 ([M+H]+),
296.34; found: 296.3.
(3-(acridin-9-ylamino)propyl)glycine (A6) was prepared using the procedure
described for A5. Yield: 91%. 1H NMR (300 MHz, CDCl3) δ 8.21 (d, J = 8.4 Hz, 2H),

71
7.54 (m, 3H), 7.12-7.18 (m, 4H), 6.93-6.62 (m, 2H), 3.91 (t, J = 6.4 Hz, 2H), 3.21 (s,
2H), 3.01 (t, J = 7.2 Hz, 2H), 2.03 (p, J = 6.5 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ
166.95, 157.31, 152.15, 130.13, 129.21, 125.84, 120.79, 118.56, 115.13, 49.87, 48.74,
45.42, 27.92. MS (ESI, positive-ion mode): calculated for C18H20N3O2 ([M+H]+), 310.37;
found: 310.2.
3-((2-(acridin-9-ylamino)ethyl)amino)propanoic acid (A7) was prepared using the
procedure described for A5. Yield: 84%. 1H NMR (300 MHz, D2O) δ 8.04 (d, J = 8.7
Hz, 2H), 7.83 (dd, J = 8.4, 7.0 Hz, 2H), 7.58 - 7.34 (m, 4H), 4.33 (t, J = 6.1 Hz, 2H), 3.60
(t, J = 5.8 Hz, 1H), 3.33 (t, J = 6.3 Hz, 2H), 3.33 (t, J = 6.3 Hz, 2H). 13C NMR (300
MHz, D2O) δ 166.95, 157.31, 152.15, 130.13, 129.21, 125.84, 118.56, 115.13, 49.87,
48.80, 45.42, 27.92. MS (ESI, positive-ion mode): calculated for C18H20N3O2 ([M+H]+),
310.37; found: 310.3.
The Boc-protected acridine derivative (2-9) (1.36 g, 4.6 mmol) was synthesized as
follows. A5 was suspended in 30 mL of anhydrous methanol, to which was added Boc2O
(1.3 g, 6 mmol) in 5 mL of anhydrous MeOH at 0-5 ºC maintained with an ice bath. The
mixture was then stirred at room temperature for 4 h. The solvent was removed by rotary
evaporation and residue was dissolved in 10 mL of dichloromethane and precipitated
with 200 mL of anhydrous diethyl ether. The solid was recovered by filtration and dried
in a vacuum affording 1.79 g (99%) of the product as a yellow solid, which was used in
the next step without further purification.
Compound 2-9 (1 g, 2.52 mmol) and 1,1'-carbonyldiimidazole (CDI, 533 mg, 3.28
mmol) were combined in 20 mL of anhydrous DMF. The mixture was heated to 40-50 ºC
and stirred for 6 h. Then the solution was cooled to 0-5 º C in an ice bath and 264 mg of

72
2-azidoethanamine dissolved in 3 mL of anhydrous DMF were added. The mixture was
stirred at 0-5 º C for 4 h. DMF was removed by vacuum distillation at 35-40 ºC, and the
residue was redissolved in 40 mL of dichloromethane and washed with 1 M HCl (3 × 20
mL). The organic phase was collected, dried with anhydrous Na2SO4, and concentrated to
afford an orange oil. To remove the Boc group, the residue was dissolved in 6 mL of a
1:1 mixture of anhydrous dichloromethane and trifluoroacetic acid and stirred at room
temperature for 3 h. The reaction was quenched by adding 10 mL of 1 M NaOH solution.
The crude product was extracted from NaOH solution with DCM, dried over anhydrous
Na2SO4, and concentrated. The product was purified by flash chromatography (Al2O3,
DCM:MeOH, 30:1). Yield: 0.59 g (64 %). 1H NMR (300 MHz, CDCl3) δ 8.10 (d, J = 8.7
Hz, 2H), 7.97 (d, J = 8.6 Hz, 2H), 7.60 (t, J = 8.3, 6.8 Hz, 2H), 7.40 - 7.14 (m, 3H), 3.89
(t, J = 5.6 Hz, 2H), 3.50 - 3.23 (m, 6H), 2.99 (t, J = 5.6 Hz, 2H). 13C NMR (75 MHz,
CDCl3) δ 172.56, 152.98, 146.04, 131.21, 125.75, 123.56, 122.97, 115.48, 50.72, 48.69,
48.47, 44.85, 38.82, 36.22. MS (ESI, positive-ion mode): calculated for C19H22N7O
([M+H]+), 364.42; found: 364.3.
3-((2-(acridin-9-ylamino)ethyl)amino)-N-(2-azidoethyl)propanamide (A9) was
prepared using the procedure described for A8. Yield: 64%. 1H NMR (300 MHz, CDCl3)
δ 8.09 (d, J = 8.7 Hz, 2H), 8.02 (d, J = 8.7 Hz, 2H), 7.59 (t, J = 7.6 Hz, 2H), 7.41 (s, 1H),
7.27 (t, J = 7.5 Hz, 2H), 4.95 (brs, 2H), 3.92 (t, J = 5.6 Hz, 2H), 3.44 (s, 4H), 2.91-3.14
(m, 4H), 2.53 (t, J = 6.0 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 152.09, 149.60, 129.65,
129.23, 123.60, 121.91, 115.75, 51.37, 51.32, 36.51, 29.15. MS (ESI, positive-ion mode):
calculated for C20H24N7O ([M+H]+), 378.45; found: 378.3.

73
The synthesis of 2-((2-(acridin-9-ylamino)ethyl)amino)ethyl (2-
azidoethyl)carbamate (A10). The Boc-protected acridine derivative (2-11) was prepared
as described for 2-9 starting with compound A3. Compound 2-11 (1 g, 2.62 mmol), TEA
(793 mg, 7.85 mmol) and 4-nitrobenzyl chloroformate (732 mg, 3.4 mmol) were
dissolved in 20 mL of anhydrous DCM. The mixture was stirred at room temperature for
16 h. Then 271 mg of 2-azidoethanamine dissolved in 5 mL of anhydrous DCM was
added and the reaction was stirred for another 8 h. The solvent was removed using
vacuum distillation and the residue was redissolved in 40 mL of DCM and washed with 1
M HCl (3 × 20 mL). The organic phase was collected, dried with anhydrous Na2SO4, and
concentrated to afford an orange oil. To remove the Boc group, the orange oil was
dissolved in 6 mL of a 1:1 mixture of anhydrous dichloromethane and trifluoroacetic acid
and stirred at room temperature for 3 h. The reaction was quenched by adding 10 mL of 1
M NaOH solution. The crude product was extracted from NaOH solution with DCM,
dried over anhydrous Na2SO4, and concentrated. The product was further purified by
flash chromatography (Al2O3, DCM:MeOH, 30:1). Yield: 0.73 g (71 %). 1H NMR (300
MHz, CDCl3) δ 8.12 - 7.96 (m, 4H), 7.59 (t, J = 7.8 Hz, 2H), 7.23 (t, J = 7.6 Hz, 2H),
5.65 (brs, 1H), 4.27 (t, J = 4.8 Hz, 2H), 3.92 (t, J = 5.7 Hz, 1H), 3.61 - 3.20 (m, 4H), 3.01
(t, J = 5.7 Hz, 2H), 2.96 (t, J = 4.9 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 156.53,
152.69, 145.93, 131.22, 125.98, 123.3, 123.00, 115.07, 64.48, 50.97, 48.35, 48.17, 47.83,
40.45. MS (ESI, positive-ion mode): calculated for C20H24N7O2 ([M+H]+), 394.45; found:
394.3.

74
Scheme 2.2. Synthesis of platinum (P) building blocks. en = ethylenediamine; pn2-OH = 2-
hydroxy-1,3-propanediamine.
2.4.3. Synthesis and characterization of platinum building blocks.
Synthesis of precursor [PtCl(EtCN)(en)]Cl (P1) [279, 289]. The complex [PtCl2(en)]
(2-12) (0.50 g, 1.54 mmol) was heated under reflux in 25 mL of dilute HCl (pH 4) with
propionitrile (6.85 mL, 98.5 mmol) until the yellow suspension turned into a colorless
solution (~2 h). Solvent was removed by rotary evaporation, and the pale-yellow residue
was redissolved in 10 mL of dry methanol. A small amount of an insoluble yellow solid
was removed by membrane filtration and the colorless filtrate was added directly into 250
mL of vigorously stirred dry diethyl ether, affording P1 as an off-white, extremely
hygroscopic microcrystalline precipitate. Yield: 0.48 g (83%). 1H NMR (300 MHz, D2O)
δ 5.72, 5.64 (2 br s,0.7 H, HD exchange), 2.87(q, J=7.5 Hz, 2H), 2.48 - 2.75 (m, Pt
satellites, 4 H), 1.3 (t, J=7.5 Hz, 3H,).
Synthesis of precursor [PtCl(MeCN)(en)]Cl (P2). P2 was prepared using the similar
procedure as described for P1 as the white solid with the yield 89%. 1H NMR (300 MHz,

75
D2O) δ 5.80, 5.65 (2 br s,1H, HD exchange), 2.87(q, J=7.5 Hz, 2H), 2.55 - 2.65 (m, Pt
satellites, 4 H), 2.53 (s, 3H).
Synthesis of precursor [PtCl(EtCN)(NH3)2]Cl (P3). P3 was prepared using the
procedure described for P1. Yield: 74%. 1H NMR (300 MHz, D2O) δ 2.89 (t, J = 7.6,
2H), 1.30 (t, J = 7.5, 3H).
Synthesis of precursor [PtCl(MeCN)(NH3)2]Cl (P4). P4 was prepared using the
procedure described for P1. Yield: 63%. 1H NMR (300 MHz, D2O) δ 2.53 (3 H, m, Pt
satellites), 4.35, 4.48 (4 H, HD exchange, 2 br s).
Synthesis of precursor [PtCl(EtCN)(pn2-OH)]Cl (P5). P5 was prepared using the
procedure described for P1. Yield: 91%. 1H NMR (300 MHz, D2O) δ 4.26 (m, 1H), 2.99 -
2.54 (m, 6H), 1.39 - 0.96 (t, J = 7.5 Hz, 3H).
Synthesis of precursor [PtCl(MeCN)(pn2-OH)]Cl (P6). P6 was prepared using the
procedure described for P1. Yield: 77%. 1H NMR (300 MHz, D2O) δ 4.28 (m, 1H), 2.85 -
2.53 (m, 4H), 2.53 (s, 3H).
2.4.4. Experimental procedure for “click” reactions.
Stock solutions of the platinum–nitrile complexes P1–P6 (20 mM) and acridine
derivatives A1–A10 (20 mM) were prepared in anhydrous DMF. Concentrations of A1–
A10 were determined spectrophotometrically (λmax = 413 nm, ε = 10,000 M-1 cm-1). The
reactions between platinum complexes and acridine derivatives were carried out in 1.5-
mL Eppendorf tubes by mixing equal volumes (100 μL) of platinum–nitrile complex and
acridine derivative. The reaction mixtures were placed in a shaker and incubated at 4 C

76
for 5 days. To detect and characterize the hybrid agents and to determine conversion
yields, 1-μL samples were removed from each reaction and diluted with 1000 μL of
methanol containing 0.1% formic acid prior to in-line LC-ESMS analysis. Sample
injections into the LC unit were accomplished via a thermostatted (4 C) autosampler.
Chromatographic separations were performed with a 4.6 mm 150 mm reverse-phase
Agilent ZORBAX SB-C18 (5 µm) analytical column with the column temperature
maintained at 25 °C. The binary mobile phase consisted of: solvent A, optima water, and
solvent B, methanol/0.1% formic acid delivered at a gradient of 95% A to 5% A over 12
minutes and a flow rate of 0.5 mL/min. The formation and the extent of conversion of the
platinum–acridines was monitored in the corresponding chromatograms using the
LC/MSD Trap Control 4.0 data analysis software.
Scheme 2.3. Synthesis of platinum-acridine derivatives. en = ethylenediamine; pn2-OH =
2-hydroxy-1,3-propanediamine.

77
2.3.5. Typical Procedure for the Preparation of Platinum–Acridine Derivatives.
Synthesis of [PtCl(en)C20H25N4O](NO3)2 (P1-A3). Complex P1 (381 mg, 1.00 mmol)
was converted to its nitrate salt by reaction with AgNO3 (162 mg, 0.950 mmol) in 7 mL
of anhydrous DMF. AgCl was removed by syringe filtration, and the filtrate was cooled
to -10 °C. Acridine precursor A3 (282 mg, 0.100 mmol) was added to the solution, and
the suspension was stirred at -10 °C for 24 h. After treatment with activated carbon, the
reaction mixture was added into 300 mL of vigorously stirred diethyl ether. The yellow
precipitate was stirred for approximately 30 min and then recovered by membrane
filtration and dried in a vacuum overnight. The solid was dissolved in anhydrous
methanol containing one equivalent of HNO3 (1 M) and stirred at room temperature for
30 min, and the crude dinitrate salt was precipitated with 300 mL of anhydrous diethyl
ether. The product was further purified by recrystallization from hot ethanol to give 606
mg of the dinitrate salt as a yellow microcrystalline solid (Yield: 82%). 1H NMR (300
MHz, MeOH-d4) δ 8.42 (d, J = 8.4 Hz, 2 H), 7.88 (t d, J = 6.9, 1.2 Hz, 2 H), 7.74 (d, J =
8.4 Hz, 2 H), 7.51 (t d, J = 7.2, 1.2 Hz, 2H 1 H), 6.12 (br s, 1 H), 5.32 (s, 2 H), 5.11 (s, 2
H), 4.32 (t, J = 6.7 Hz, 2 H), 3.89 (t, J = 6.7 Hz, 2H), 3.63 (t, J = 5.0 Hz, 2 H), 3.47 (t, J =
5.1 Hz, 2 H), 2.94 (q, J = 7.2 Hz, 1 H), 2.47 (m, 4 H), 1.18 (t, J = 7.2 Hz, 3 H). 13C NMR
(75 MHz, MeOH-d4) δ 171.73, 160.21, 141.38, 136.63, 126.48, 125.35, 119.73, 114.13,
60.70, 29.26, 11.85. MS (ESI, positive-ion mode): m/z for C22H32ClN6OPt ([M]+),
627.06; found: 627.3.
Synthesis of [PtCl(en)C22H27N8O](NO3)2 (P1-A8). P1-A8 was prepared using the
same procedure as described for P1-A3 and was recovered with a yield of 81%.1H NMR
(300MHz, DMF-d7) δ 13.90 (s, 1H), 9.99 (s, 1H), 8.83 - 8.52 (m, 3H), 8.28 - 7.92 (m,

78
4H), 7.62 (td, J = 6.6, 1.5 Hz 2H), 6.73 (s, 1H), 5.84 (s, 2H), 5.51 (s, 2H), 4.54 (q, J = 5.7
Hz, 2H), 4.44 (s, 2H), 4.15 (t, J = 5.9 Hz, 2H), 3.44 - 3.52 (m , 4H), 3.06 (q, J = 7.9 Hz,
2H), 2.67 (s, 4H), 1.33 (t, J = 7.5 Hz, 3H). 13C NMR (75 MHz, DMF-d7) δ 170.37,
169.56, 158.62, 140.17, 135.27, 125.92, 123.80, 118.90, 112.94, 50.32, 48.97, 48.79,
46.34, 38.78, 28.67, 11.00. MS (ESI, positive-ion mode): for C24H34ClN10OPt ([M]+),
709.13; found: 708.5.
Synthesis of [PtCl(NH3)2C19H23N4O](NO3)2 (P4-A3). P4-A3 was prepared using the
same procedure as described for P1-A3 and was recovered with a yield of 87%. 1H NMR
(300 MHz, MeOH-d4) δ 8.40 (d, J = 8.8 Hz, 2H), 7.87 (t, J = 6.8, 2H), 7.72 (dd, J = 8.7,
1.2 Hz, 1H), 7.49 (d, J = 8.4 Hz, 2H), 4.31 (t, J = 7.0 Hz, 2H), 4.10 (brs, 2H), 3.91 (t, J =
6.8 Hz, 1H), 3.75 (s, 2H), 3.65 (t, J = 4.9 Hz, 2H), 3.49 (t, J = 4.9 Hz, 2H), 2.58 (s, 3H).
13C NMR (75 MHz, MeOH-d4) δ 167.49, 160.02, 141.40, 136.55, 126.49, 125.30, 119.77,
114.14, 60.73, 49.86, 48.16, 47.39, 23.02. MS (ESI, positive-ion mode): for
C19H28ClN6OPt ([M]+), 587.00; found: 585.2.
Synthesis of [PtCl(pn2-OH)C18H21N4](NO3)2 (P6-A1). P6-A1 was prepared using the
same procedure as described for P1-A3 and was recovered with a yield of 92%. 1H NMR
(300 MHz, DMF-d7) δ 13.92 (s, 1H), 9.89 (s, 1H), 8.68 (d, J = 8.7 Hz, 2H), 7.9 -8.11 (m,
4H), 7.62 (td, J = 6.4, 3.1 Hz, 2H), 6.21 (s, 1H), 5.65 (s, 1H), 5.60 (s, 1H), 5.13 (s, 2H),
4.87 (s, 1H), 4.50 (s, 2H),4.12 (t, J = 6.3 Hz 2H), 4.06 (s, 1H), 3.50 (s, 4H), 3.18 (s, 3H),
2.59-2.97 (m, 4H). 13C NMR (75 MHz, DMF-d7) δ 165.94, 158.64, 139.96, 135.33,
125.29, 123.91, 118.98, 112.80, 65.84, 48.38, 47.80, 33.79, 28.66. MS (ESI, positive-ion
mode): for C21H30ClN6OPt ([M]+), 613.04; found: 612.3.

79
Synthesis of [PtCl(NH3)2C21H24N4O2](NO3) (P3-A7). Platinum complex P3 (354 mg,
1 mmol) was converted to its nitrate salt by reaction with AgNO3 (162 mg, 0.95 mmol) in
7 mL of anhydrous DMF. AgCl was removed by syringe filtration, and the filtrate was
cooled to -10 °C. Acridine precursor A7 (310 mg, 0.1 mmol) was added to the solution,
and the suspension was stirred at 4 °C for 5 days. The mixture was poured into 300 mL
of vigorously stirred diethyl ether, and the precipitate was recovered by membrane
filtration and dried in a vacuum overnight. The product was further purified by
recrystallization from hot methanol to give 461.7 mg of the product as a yellow solid
(Yield: 67%). 1H NMR (300 MHz, MeOH-d4) δ 8.29 (d, J = 8.6 Hz, 2H), 7.92 - 7.58 (m,
4H), 7.41 (t, J = 7.7 Hz, 2H), 4.06 (t, J = 6.5 Hz, 2H), 3.74 (t, J = 6.4 Hz, 2H), 3.63 (t, J =
6.5 Hz, 2H), 3.02 (t, J = 7.9 Hz, 2H), 2.35 (t, J = 6.4 Hz, 2H), 1.23 (t, J = 8.0 Hz, 2H).
MS (ESI, positive-ion mode): for C21H30ClN6O2Pt ([M]+), 629.04; found: 629.2.
2.4.6. Cell proliferation assay
The human non-small cell lung cancer cell line, NCI-H460, was obtained from the
American Type Culture Collection (Rockville, MD, USA) and was cultured in RPMI-
1640 media (HyClone) containing 4.5 g/L glucose, 1.5 g/L sodium bicarbonate, 10 mM
HEPES, and 110 mg/L sodium pyruvate supplemented with 10% fetal bovine serum
(FBS), 10% penstrep (P&S), and 10% L-glutamine. Cells were incubated at a constant
temperature at 37 °C in a humidified atmosphere containing 5% CO2 and were
subcultured every 2 to 3 days in order to maintain cells in logarithmic growth.

80
Cell viability was assessed using the CellTiter 96 Aqueous Non-Radioactive Cell
Proliferation Assay (Promega, Madison, WI, USA) as described previously [277-278].
Briefly, NCI-H460 cell suspensions were harvested and seeded into 96-well microplates
at a density of 750 cells/well. The cells were preincubated at 37 °C overnight and then
treated with 50 nM compound from the library reactions or serial dilutions of selected
purified compounds (the DMF content in the mixtures was less than 1% at the highest
incubation concentration), as well as control samples containing platinum and acridine
precursors and samples containing media/DMF. After an incubation period of 72 h, 20
μL of MTS/PMS solution was added to each well and incubated at 37 °C for 4 h. The
absorbance of tetrazolium dye was measured at 490 nm using a plate reader. The fraction
of viable cells was calculated as a percentage of untreated control and is reported as the
mean ± standard deviation for 3 incubations of each compound. IC50 values were
calculated from non-linear curve fits using a sigmoidal dose–response equation in
GraphPad Prism (GraphPad Software, La Jolla, CA) and are averages of two individual
experiments performed in triplicate.

81
CHAPTER 3
USING FLUORESCENT POST-LABELING TO PROBE THE SUBCELLULAR
LOCALIZATION OF DNA-TARGETED PLATINUM
The work contained in this chapter was initially published in Angew.Chem.Int.Ed. in
2013 and J.Biol.Inorg.Chem. in 2014. The manuscript, including figures and schemes,
was drafted by Song Ding and Dr. U. Bierbach and edited by Dr. U. Bierbach before
submission to the journal. Since the publication, changes in both format and content have
been made to maintain a consistent format throughout this work. The experiments
described were performed by Song Ding, except for the the confocal microscopy and
cytotoxicity assays which were contributed by Dr. Xin Qiao and Dr. Fang Liu, and the
DNA unwinding experments was performed by Dr. Jimmy Suryadi.

82
3.1. Introduction and Design Rationale
Adducts in the nuclear DNA are the major cause of cancer-cell death triggered by
platinum-based anticancer drugs [290]. Thus, cellular uptake and accumulation,
distribution, and trafficking between subcellular compartments and, ultimately,
localization to the nucleus are crucial parameters in the mechanism of these agents.
Several techniques have been used to monitor the intracellular distribution of platinum
drugs. These include element-specific analytical methods and nondestructive absorption-
or emission-based imaging techniques, as well as electron microscopy [291, 292].
Fluorophore-tagged derivatives have provided insight into the uptake, distribution, and
intracellular transformation of platinum drugs [291]. Such an approach has to take into
consideration the organelle selectivity of the fluorophore, which may vary widely
depending on parameters such as molecular weight, partition coefficient (log P),
amphiphilic character, and pKa value [293]. Thus, one drawback of modifying platinum
drugs with organic fluorophores is that such conjugates may, at least in part, mimic the
properties of the reporter molecule [294]. This would be an undesired feature unless the
fluorescent group itself is a functionally important part of the bioactive molecule.
Likewise, bulky fluorophores may interfere with the DNA-binding mechanism of
platinum drugs. To circumvent these problems, we have developed a method based on
bioorthogonal ligation chemistry, which allowed us to fluorescently label platinum-
acridine hybrid agents in lung cancer cells. Herein, we report the development of this
technique and demonstrate, for the first time, that post-labeling is a powerful tool for
detecting DNA-targeted platinum drugs in subcellular and subnuclear structures.

83
Platinum-acridines, represented by the prototypical P1-A1 (Figure 3.1), are up to 500-
times more cytotoxic to non-small-cell lung cancer cells than the drug cisplatin [295].
Studies in NCI-H460 cells suggest that the cytotoxicity produced by P1-A1 is triggered
by high levels of monofunctional-intercalative hybrid adducts in cellular DNA [275],
which is supported by recent results from chemogenomic screening in S. cerevisiae [296].
Unfortunately, direct fluorescent imaging of platinum-acridines in cancer cells is difficult
because the fluorescence of the 9-aminoacridine is severely quenched when intercalated
into DNA (Appendix D, Figure D.1) [297]. We therefore developed a method that allows
for the labeling of platinum-acridines, in particular their DNA adducts, intracellularly
using a high-performance fluorescent dye in conjunction with a copper-catalyzed azide–
alkyne cycloaddition (CuAAC, click chemistry) [298, 299].
3.2. Results and Discussion
3.2.1. Post-labeling method
This strategy required modification of P1-A1 with an alkyne or azide group in a way
that would not interfere with the DNA- binding mode of the agent. Molecular models
derived from the NMR solution structure of the hybrid adduct [274] (Appendix F, Figure
F.1) suggest that the addition of a clickable group at the amidine N1-methyl group in P1-
A1 (Figure 3.1), which is protruding out of the DNA major groove, should satisfy this
requirement. While direct attachment of either functional group to this position using
polymethylene linkers was incompatible with the metal and resulted in complex
decomposition, our search identified a stable amide-linked azide derivative (P1-A9,

84
Figure 3.1), which also showed excellent stability in aqueous media (see Appendix B3).
Using gel-shift experiments to monitor DNA unwinding (Figure 3.2) and CD
spectropolarimetry, we confirmed that the azide-containing linker does not affect the
geometry of the hybrid adduct (APPENDIX C). Thus, P1-A9 was chosen for our labeling
technique in combination with the alkyne-modified form of the bright green-fluorescent
dye Alexa Fluor 488 (Figure 3.1).
Figure 3.1. Structures of compounds and reaction scheme for bioorthogonal fluorescent
labeling of DNA-targeted platinum–acridine hybrid agents.

85
Fig
ure
3.2
. G
el el
ectr
ophore
sis
of
pla
tinum
–ac
ridin
e-m
odif
ied,
neg
ativ
ely su
per
coil
ed pla
smid
D
NA
. pU
C19 m
odif
ied w
ith
com
pound P
1-A
9 (
A)
wit
h c
om
pound P
1-A
1 (
B).
Eac
h g
el i
s sh
ow
n w
ith t
he
corr
espond
ing p
lot
of
rela
xed
DN
A b
and
inte
nsi
ties
vs.
rb.
The
unw
indin
g a
ngle
s (ϕ
) ca
lcula
ted f
rom
ϕ =
18σ
/ r b
(c),
wher
e σ
is
the
super
hel
ical
den
sity
of
the
pla
smid
(
0.0
50)
and
r b(c
) is
the
max
imum
of
the
quad
rati
c poly
nom
ial
curv
e fi
ts, fo
r co
mpound
s 1 a
nd
2 a
re 1
7 ±
1
and 1
6 ±
1,
res
pec
tivel
y.

86
We established conditions for labeling the DNA adducts of P1-A9 using a CuAAC
reaction in a randomly platinated linearized plasmid. The procedure involved incubation
of the DNA with P1-A9 at varying platinum-to-nucleotide ratios (rb) and reaction of the
platinated DNA with Alexa Fluor 488-alkyne in the presence of a copper-based click-
reaction buffer, followed by purification of the samples and analysis of the labeled DNA
on agarose gels. A fluorescent image of the gel (at 520 nm) along with the ethidium
bromide-stained gel is shown in Figure 3.3 A. The bands on the gel show a pronounced
increase in Alexa Fluor 488 fluorescence intensity with an increasing platinum-to-
nucleotide ratio (lanes 3–5, top gel). To test whether the Alexa Fluor 488-alkyne was
ligated to covalently bound platinum-acridine on the plasmid, a sample of fluorescently
labeled DNA was incubated with NaCN, which reverses Pt-DNA adducts [300]. The
complete disappearance of Alexa Fluor fluorescence in lane 6 (Figure 3.3A, top gel) and
conversion of the platinated DNA into unmodified plasmid (Figure 3.3 A, bottom gel,
band of highest mobility) confirm that the platinum adducts were modified by the green-
fluorescent dye.
CD spectra were recorded of native DNA globally modified with P1-A9 before and
after undergoing ligation with the Alexa Fluor dye (Figure 3.3 B). The adducts formed by
P1-A9 give rise to positive Cotton effects in the 300–500 nm range, characteristic of the
induced circular dichroism (ICD) of the intercalated acridine chromophore.[301] After
labeling with Alexa Fluor 488-alkyne, the CD spectrum shows an additional negative
ICD band at 498 nm, which mimics the absorbance spectrum of the dye, a consequence
of the DNA chirality experienced by the ligated fluorophore. The hypsochromic shift of
four nanometers observed for the acridine-based long-wavelength ICD bands in the

87
labeled adduct relative to the unlabeled adduct of P1-A1 suggests that ligation of the
Alexa Fluor dye perturbs the hybrid binding mode, leading to partial unstacking of the
acridine moiety [274]. By contrast, a comparison of the ICD signatures of DNA-bound
compounds P1-A1 and P1-A9 shows that addition of the azide-containing side chain does
not compromise acridine intercalation. This observation corroborates the notion that P1-
A9, but not its fluorophore-tagged form, is a faithful mimic of P1-A1, which validates the
post-labeling approach.

88
Figure 3.3. (A) Electrophoretic analysis of copper-catalyzed ligation reactions between
linearized (BamH I), platinated pUC19 plasmid DNA and Alexa Fluor 488-alkyne with
reaction conditions indicated for each incubation (for details see Experimental Section).
The top gel shows Alexa Fluor fluorescence monitored at 520 nm. Note the increase in
band intensities and mobility shift with increasing platinum content. The latter band shift
is caused by accumulation of positive charge on the plasmid. The bottom panel depicts
the same gel stained with ethidium bromide. Saturation of DNA binding sites by Alexa
Fluor-tagged platinum–acridine causes inefficient staining by ethidium at rb = 0.2. (B)
CD spectra recorded in the ICD region for unmodified DNA (green trace), DNA
modified with P9-A1 (blue trace), and DNA modified with P9-A1 after ligation with
Alexa Fluor 488-alkyne (red trace).

89
Figure 3. 4. Imaging of P1-A9 (5 µM) in fixed NCI-H460 lung cancer cells using Alexa
Fluor 488-alkyne. Cells were co-stained with Hoechst 33342. (A) Single confocal image
planes of Alexa Fluor 488 (green), Hoechst 33342 (blue), the green and blue channels
merged, and corresponding bright field image are shown for P1-A9 treatment and control
conditions. Control groups were treated with Alexa Fluor 488-alkyne and copper-based
ligation buffer. No-copper controls were treated with Alexa Fluor 488-alkyne in the
absence of copper. (B) Single confocal image plane of the cellular distribution of
platinum–azide-related fluorescence in NCI-H460 cells during mitosis (labeled a) and
interphase (example labeled b).

90
Next, we tested if the labeling strategy developed in cell-free DNA could be applied to
whole cancer cells. For confocal fluorescence microscopy studies, NCI-H460 cells were
incubated with P1-A9 under conditions that were expected to produce high intracellular
levels of platinum drug without killing the cells, as confirmed by the absence of the
morphological hallmarks of apoptotic cell death [302]. Briefly, cultured cells were treated
with three different doses (1, 5, or 25 μm) of P1-A9 for three hours, washed, fixed in
formalin, permeabilized, and incubated with CuAAC reaction buffer in the presence of
Alexa Fluor 488-alkyne. Control experiments without platinum drugs were treated with
fluorescent dye in the presence or absence of copper ions. Following exhaustive washing
to remove excess labeling mixture and co-staining with a nuclear dye (Hoechst 33342),
cells were imaged by confocal fluorescence microscopy.
Images of platinum-treated NCI-H460 cells incubated with Alexa Fluor 488-alkyne and
copper ions clearly demonstrate that this labeling method can be applied to whole cells
(Figure 3.4 A). Cells treated with P1-A9 show a pronounced intensity increase in
fluorescence emission relative to condition-matched controls, as a result of selective
intracellular azide–alkyne ligation chemistry. Alexa Fluor 488 emission intensities in the
nuclei of the treated cells (n > 50) show a clear correlation with increasing incubation
concentrations of P1-A9, confirming that intracellular reaction with the azide-modified
platinum/DNA adduct is the source of the fluorescence (see Appendix D).

91
Fig
ure
3.5
. D
eter
min
atio
n o
f in
trac
ellu
lar
dis
trib
uti
on o
f P
1-A
9 i
n a
NC
I-H
460 c
ell
in i
nte
rphas
e b
y c
o-s
tain
ing w
ith n
ucl
ear
(Hoec
hst
33342)
and m
itoch
ondri
al (
Mit
oT
rack
er D
eep R
ed)
dyes
. T
he
nucl
eolu
s, w
hic
h i
s re
adil
y i
den
tifi
ed i
n t
he
bri
ght-
fiel
d
imag
e an
d a
s a
nu
clea
r re
gio
n u
nst
ained
by H
oec
hst
dye,
appea
rs a
s an
are
a of
hig
h f
luo
resc
ent
inte
nsi
ty i
n t
he
Ale
xa
Flu
or
488
and m
erged
chan
nel
im
ages
.

92
Platinum-azide-associated fluorescence was observed at high intensity within the nuclei
and at a much lower intensity within the cytoplasm (for a detailed analysis of the distinct
cellular regions, see the Appendix D). Figure 3.4 B shows co-localization images of
Alexa Fluor 488 and the nuclear stain Hoechst 33342 within NCI-H460 cells during
different stages of the cell cycle. During mitosis, the highest levels of green fluorescence
were observed in the condensed, replicated chromatin, whereas cells in interphase show
the strongest fluorescence signal across the entire nucleus. The highest fluorescence
intensity in the nucleus during interphase was observed in the nucleoli, which are the
sites of ribosomal RNA (rRNA) transcription during interphase [303]. An NCI-H460 cell
imaged at high magnification shows the relative distribution of Alexa Fluor within the
cell and confirms that P1-A9 accumulates in the nucleus and nucleolus (Figure 3.4).
Analysis of a large number of cells (n = 40–50) shows that the emission in the 488 nm
excitation channel has 50 % higher relative intensity within the nucleolar regions
compared to the surrounding chromatin (see Appendix D, Figure D.3).
3.2.2. Post-labeling using copper-free click chemistry
To investigate if the detection of intracellular platinum–acridine can also be achieved
by copper-free click chemistry, similar labeling experiments were performed with Alexa
Fluor 488-DIBO, which contains an azide-reactive dibenzocyclooctyne group (Figure.
3.6B) [304]. The confocal images obtained from these experiments (Figure. 3.7) show
unfavorable high background fluorescence in all cytosolic organelles of cells not treated
with compound P1-A9. In addition, the DIBO-based fluorophore, unlike simple
Cu/alkyne, was unable to label platinum adducts in the DNA of dividing cells. This

93
observation suggests that the steric bulk of DIBO may prohibit reaction with platinum-
based azide in the compacted chromatin. Thus, copper-free click chemistry using DIBO-
modified dye was dismissed as a post-labeling technique in experiments requiring
orthogonal reactions with two fluorophores.
Figure 3.6. (A) Structures of probes used in this study. (B) Detection of azide-modified
platinum–acridine (DNA adducts) with complementary alkyne-modified fluorescent dye.
The star indicates Alexa Fluor 488 (green) and Alexa Fluor 647 (red) fluorophore. (C)
Simultaneous detection of platinum–acridine in cells incubated with a mixture of
compound P1-A9 and its Alexa Fluor 488-DIBO conjugate, P1-A9*. Post-labeling of P1-
A9 was performed with the alkyne form of Alexa Fluor 647 (red star).

94
Fig
ure
3.7
. C
onfo
cal
imag
es o
f untr
eate
d N
CI-
H460 l
ung c
ance
r ce
lls
(co
ntr
ol)
and c
ells
tre
ated
wit
h c
om
pound P
1-A
9 (
5 µ
M).
Both
pla
tinum
-tre
ated
and c
ontr
ol
cell
s w
ere
trea
ted w
ith A
lex
a F
luor
488
-DIB
O (
gre
en).
Hoec
hst
33342 (
blu
e),
mer
ged
, an
d
bri
ght
fiel
d ch
ann
el im
ages
ar
e al
so sh
ow
n.
T
he
arro
ws
indic
ate
com
pac
ted ch
rom
atin
in
div
idin
g ce
lls,
w
hic
h is
st
ained
effi
cien
tly b
y n
ucl
ear
dye
but
not
by t
he
DIB
O-c
onta
inin
g f
luoro
phore
. S
cale
bar
= 1
0 µ
M.

95
3.2.3. Pre-labeling method
In section 3.2.1, we have developed and validated the post-labeling technique using
quantitative sampling of fluorescence intensities in treated cells and appropriate
condition-matched controls. We have also used this technique to compare the subcellular
localization of post-labeled platinum with that of fluorescently (pre-)labeled compound
P1-A9* ( Figure 3.6A) in NCI-H460 lung cancer cells during interphase and mitosis. We
have also combined the detection of platinum with a second orthogonal click reaction.
These experiments represent the first successful case of orthogonal post-labeling
chemistry in whole cells involving a platinum-based pharmacophore.
To determine the effects of tagging the azide-modified complex P1-A9 with a
fluorophore prior to treatment of cancer cells, we attempted to label this compound with
Alexa Fluor-alkyne dye using CuAAC chemistry. Unfortunately, these reactions, which
had to be performed in (partly) aqueous media, led to substantial decomposition of
compound P1-A9 and products indicating aquation of complex and undesired substitution
of platinum-coordinated chloride by the alkyne moiety (data not shown). By contrast,
Alexa Fluor 488-DIBO reacted cleanly with compound P1-A9 in DMF solution to
produce the desired conjugate P1-A9* (Figure. 3.6A). When the cyclooctyne-modified
dye was reacted with excess platinum (1:10) at room temperature, complete conversion to
cycloadduct was observed within 3 h, as confirmed by in-line LC–MS analysis (Figure
3.8). Reaction mixtures generated this way, in which no unreacted Alexa Fluor 488-
DIBO was detectable, were directly used for colocalization studies in NCI-H460 cells.
Because conjugate P1-A9* was generated in situ on a microgram scale, it was not

96
possible to isolate sufficient amounts of the pure conjugate for imaging and cell
proliferation assays.
Cells exposed to a mixture of P1-A9 and P1-A9* (~9:1) were imaged after
permeabilization and treatment with Alexa Fluor 647-alkyne in the absence or presence
of copper. The former conditions were chosen to selectively detect green-fluorescent P1-
A9* and as a negative control of non-platinum associated Alexa Fluor-647 (red)
fluorescence. Copper-based click chemistry with the red-fluorescent dye was used to
detect Pt-azide (P1-A9) and P1-A9* simultaneously. Confocal images taken under both
conditions show the highest level of green fluorescence in the nucleolar regions of the
cells, confirming that the fluorophore-labeled platinum–acridine (P1-A9*) accumulates
in this subnuclear structure (Figure 3.9). This is an important observation, since Alexa
Fluor 488-DIBO itself shows no significant affinity for the nucleoli in cells in interphase
(see controls for DIBO post-labeling experiments in APPENDIX D). By comparison,
nuclear and cytosolic levels of P1-A9* are considerably lower. As expected, under
copper-free conditions only insignificant background levels of red fluorescence are
observed in the Alexa Fluor 647 channel (Figure 3.9, top panel). When copper is
introduced for the purpose of post-labeling intracellular compound P1-A9, high levels of
red fluorescence are observed across the entire nuclear region of the cells (Figure 3.9,
bottom panel). This observation is consistent with the previously observed (Figure 3.4)
accumulation of the hybrid agent in the nuclear DNA of both cells in interphase and
mitosis. A high degree of co-localization is observed for compounds P1-A9 and P1-A9*
in the merged-channel image, suggesting that labeled and unlabeled platinum–acridine
share qualitatively similar subcellular distribution patterns. However, closer inspection

97
of individual cells reveals that the fluorescence level in the chromatin (relative to that
observed in the nucleolus and cytosol) is significantly higher in the red channel than in
the green channel (see Figure D.4 in APPENDIX D for an enlarged image of a single
cell). A possible explanation for this effect would be the increased steric bulk in P1-A9*
introduced by the Alexa Fluor-DIBO moiety, which can be expected to affect the ability
of the conjugate to associate and form adducts with double-stranded DNA of the
chromatin.

98
Figure 3.8. Labeling of compound P1-A9 with Alexa Fluor 488-DIBO in DMF solution
monitored by in-line LC-MS. HPLC traces are shown for an Alexa Fluor-specific (λmax =
488 nm) and an acridine-specific (λmax = 413 nm) wavelength range. HPLC fractions and
the corresponding negative-ion electrospray mass spectra are labeled ‘1’ and ‘2’ for the
two constitutional isomers of cycloadduct P1-A9* and ‘3’ for compound P1-A9,
respectively. Molecular ions ([M–nH]-) are observed for each species at m/z values of
1556, 1557, and 721, respectively. Note the absence of unreacted Alexa Fluor 488-DIBO
under these reaction conditions (10-fold excess compound P1-A9).

99
Fig
ure
3.9
. C
olo
cali
zati
on s
tud
y o
f pla
tinum
–ac
ridin
e P
1-A
9,
post
-lab
eled
wit
h A
lex
a F
luor
64
7-a
lkyn
e (r
ed c
han
nel
), a
nd i
ts
Ale
xa
Flu
or
488
-DIB
O-l
abel
ed d
eriv
ativ
e, P
1-A
9*,
in N
CI-
H46
0 c
ells
. C
ells
wer
e co
-sta
ined
wit
h H
oec
hst
33342 n
ucl
ear
dye
(blu
e ch
ann
el).
N
ote
the
hig
hes
t deg
ree
of
colo
cali
zati
on o
f th
e tw
o s
pec
ies
in t
he
nucl
eola
r re
gio
ns
of
post
-lab
eled
cel
ls,
whic
h
appea
r as
bri
ght-
yel
low
spots
in t
he
mer
ged
-ch
ann
el i
mag
e. S
cale
bar
s re
pre
sent
a le
ngth
of
10 µ
m.

100
3.3. Conclusions
In conclusion, the current study demonstrates that copper-mediated click chemistry
based post-labeling strategy is a powerful method for mapping the subcellular
localization of a platinum-containing pharmacophore in fixed cancer cells. This was
achieved by modifying a potent platinum-acridine anticancer agent so that a fluorophore
could be ligated to it within cells. The post-labeling approach is likely to truly reflect the
subcellular distribution of platinum-acridines due to less steric hindrance may involve in
the technology, which outperformed other strategies in this study, including post-labeling
with copper-free click chemistry and pre-assembly method. P1-A9 represents the first
example of a platinum-based anticancer agent compatible with alkyne–azide ligation
chemistry. The rapid accumulation of P1-A9 in chromatin in the nucleus after only three
hours of incubation is consistent with the high levels of platinum-DNA adducts seen
previously in NCI-H460 cells by ICP-MS [275]. The imaging results support the notion
that damage to nuclear DNA by platinum drugs is a major trigger of S-phase arrest and
apoptosis in NCI-H460 cells [296, 305]. The biological consequences of platinum-
acridine localization to the nucleoli, on the other hand, remain elusive. Potential targets
for platinum adduct formation in this dynamic, non-membrane-bound structure include
transcriptionally active rRNA genes (rDNA) and nascent rRNA itself [303]. Nucleolar
rRNA synthesis is an emerging target in anticancer drug development, and certain DNA
intercalators have shown selective inhibition of rRNA transcription [306, 307]. Notably,
in deletion strains of S. cerevisiae, platinum-acridines elicit responses from genes
involved in RNA metabolism, which suggests that inhibition of rRNA synthesis may
contribute to the cell death produced by these agents [296]. Finally, we anticipate that this

101
new imaging method could be extended to more biocompatible and ultrafast copper-free
click chemistry, which could monitor platinum drug levels and localization in real-time
within live cells [308].
3.4 Experimental Section
3.4.1. Materials and Methods.
The synthetic precursors 3-2 and 3-4 were synthesized according to the methods
described previously. All reagents were used as obtained from commercial sources
without further purification unless indicated otherwise. 1H NMR spectra of the target
compounds and intermediates were recorded on a Bruker Advance 300 MHz instrument.
Chemical shifts (δ) are given in parts per million (ppm) relative to internal standard
tetramethylsilane (TMS). 1H NMR data is reported in the conventional form including
chemical shift (δ, ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m =
multiplet, br = broad), coupling constants (Hz), and signal integrations. 13C{1H} NMR
data are reported as chemical shift listings (δ, ppm). The NMR spectra were processed
and analyzed using the MestReNova software package. HPLC-grade solvents were used
for all HPLC and mass spectrometry experiments. LC-ESMS analysis was performed on
an Agilent 1100LC/MSD ion trap mass spectrometer equipped with an atmospheric
pressure electrospray ionization source. Eluent nebulization was achieved with a N2
pressure of 50 psi and solvent evaporation was assisted by a flow of N2 drying gas
(350 °C). Positive-ion mass spectra were recorded with a capillary voltage of 2800 V
and a mass-to-charge scan range of 150 to 2200 m/z. To establish the purity of target

102
compounds, samples were diluted in methanol containing 0.1 % formic acid and
separated using a 4.6 mm x 150 mm reverse-phase Agilent ZORBAX SB-C18 (5 µm)
analytical column at 25 °C. The purities of compound S3 and compound S5 were
assessed using the following solvent system: solvent A, optima water, and solvent B,
methanol/0.1% formic acid, with a flow rate of 0.5 mL/min and a gradient of 95% A to
5%. A over 15 minutes. The purity of compound P1-A9 was assessed using the
following solvent system: solvent A, optima water, and solvent B, methanol/0.1% formic
acid, with a flow rate of 0.5 mL/min and a gradient of 95% A to 5% A over 30 minutes.
HPLC traces were recorded with a monitoring wavelength range of 363-463 nm. Peak
integration was done using the LC/MSD Trap Control 4.0 data analysis software.
Scheme 3.1. Synthesis of precursor 3-3 and 3-5.
3.4.2. Synthetic procedures and product characterization
The synthesis of N1-(acridin-9-yl)-N2-(2-azidoethyl)ethane-1,2-diamine (3-3). A
mixture of phenoxyacridine (3-1) (2.71 g, 0.01 mol) and N1-(2-azidoethyl) ethane-1,2-
diamine (3-2) (1.42 g, 0.011 mol) in 25 mL of anhydrous THF was refluxed for 16 h. The
solvent was evaporated off and the residue was dissolved in 30 mL of acetone. To this

103
solution were added 5 mL of conc. HCl, and the mixture was stirred at 4 ˚C for 3 hours.
A yellow precipitate formed which was recovered by filtration, resuspended in 50 mL of
2 M ammonium hydroxide, and stirred at room temperature for 30 min. The aqueous
phase was extracted with CH2Cl2 and the organic phase was collected, dried over Na2SO4,
and concentrated using rotary evaporation, affording 2.57 g of the free base (3-3) as an
yellow solid (Yield: 87%). 1H NMR (300 MHz, CDCl3) δ = 8.16 (d, J = 7.2 Hz, 2H),
8.06 (d, J = 8.4 Hz, 2H), 7.65 (t, J = 7.8 Hz, 2H), 7.35 (t, J = 8.1 Hz, 2H), 6.24 (br s, 1H),
3.82 (t, J = 5.6 Hz, 2H), 3.46 (t, J = 5.6 Hz, 2H), 2.93 (t, J = 6.4 Hz, 2H), 2.87 (t, J = 6.3
Hz, 2H), 1.39 (br s, 1H). 13C NMR (75 MHz, CDCl3) δ 151.65, 149.51, 129.80, 122.97,
117.11, 51.57, 49.19, 49.09, 47.91. MS (ESI, positive-ion mode): calculated for
C17H18N6 ([M+H]+), 306.16; found: 307.2.
The synthesis of N1-(acridin-9-yl)-N2-(3-azidopropyl)ethane-1,2-diamine (3-5). 3-5
was prepared using the procedure described for 3-3 starting from phenoxyacridine (3-1)
(2.71 g, 0.01 mol) and N1-(3-azidopropyl)ethane-1,2-diamine (3-4). 1H NMR (300 MHz,
CDCl3) δ 8.15 (d, J = 8.7 Hz, 2H), 8. 06 (d, J = 8.9 Hz, 2H), 7.65 (t, J = 8.5 Hz, 2H),
7.34 (d, J = 8.4 Hz, 2H), 6.26 (brs, 1H), 3.81 (t, J = 5.4 Hz, 2H), 3.40 (t, J = 6.6, 2H),
2.90 (t, J = 5.7 Hz, 2H), 2.75 (t, J = 6.8, 2H), 1.80 (p, J = 6.7, 2H), 1.12 (brs, 1H). 13C
NMR (75 MHz, CDCl3) δ 151.70, 149.47, 129.82, 129.78, 123.00, 122.84, 116.87,
49.50, 49.43, 49.09, 46.28, 29.42. MS (ESI, positive-ion mode): calculated for C18H20N6
([M+H]+), 320.17; found: 321.2.
The synthesis of [PtCl(NH3)2C23H28N8O](NO3)2 (P1-A9). Platinum complex A1 (381
mg, 1 mmol) was converted to its nitrate salt by reaction with AgNO3 (162 mg, 0.95
mmol) in 7 mL of anhydrous DMF. AgCl was removed by syringe filtration, and the

104
filtrate was cooled to -10 °C. Acridine precursor A9 (378 mg, 1 mmol) was added to the
solution, and the suspension was stirred at -10 °C for 24 h. After treatment with activated
carbon, the reaction mixture was added into 300 mL of diethyl ether. The yellow slurry
was stirred for 30 min, then the precipitate was recovered by membrane filtration and
dried in a vacuum overnight. The solid was dissolved in anhydrous methanol containing
one equivalent of 1 M HNO3, stirred at room temperature for 30 minutes and precipitated
with 300 mL of anhydrous diethyl ether. The product was further purified by
recrystallization from hot ethanol to give 432 mg of a yellow microcrystalline solid
(Yield: 51%). 1H NMR (300 MHz, MeOH-d4) δ 8.45 - 8.39 (m, 2H), 7.89 (ddd, J=8.3,
6.9, 1.2 Hz, 2H), 7.78 - 7.70 (m, 2H), 7.52 (ddd, J=8.8, 6.9, 1.3 Hz, 1H), 5.37 - 5.15 (m,
4H), 4.27 (t, J=6.8 Hz, 2H), 3.84 (t, J=6.8 Hz, 2H), 3.59 (t, J=6.6 Hz, 2H), 3.30 - 3.22 (m,
4H), 2.94 (q, J=7.6, 2H), 2.58 - 2.26 (m, 7H), 1.18 (t, J=7.6, 3H). 13C NMR (75 MHz,
MeOH-d4) δ 173.27, 171.13, 160.17, 141.38, 136.67, 125.45, 119.80, 114.17, 51.38,
49.86, 39.96, 29.19, 11.99.
3.4.3. In-gel Alexa Fluor 488 fluorescence detection
Plasmid DNA (pUC19, 2686 bp) was generated by transforming a chemically
competent E. coli (NovaBlue, Novagen) with pUC19 and extracted from cells using
Qiagen’s MegaPrep Plasmid kit (Qiagen, Valencia, CA). The plasmid was linearized
with restriction endonuclease BamH I (New England BioLabs, Ipswich, MA) at 37 °C
overnight. A small aliquot was removed and analyzed on an agarose gel to confirm
complete conversion to the linear form. After heat-inactivation of enzyme at 65 °C, the
DNA was purified with the QIAquick PCR Purification kit (Qiagen, Valencia, CA).

105
DNA concentrations and purity were determined spectrophotometrically at 260 nm with
= 6500 M1 cm1 (nucleotides, n.t.). The linearized pUC19 was incubated with
compound P1-A9 at drug-to-nucleotide ratios (rb) of 0.2, 0.1, and 0.05 in 5 µL of 10 mM
PBS at 37 °C for 24 hours. To the samples were then added 45 µL of click reaction buffer
(CuSO4, TBTA (tris-[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine), Alexa Fluor 488-
alkyne (Invitrogen, Carlsbad, CA) and sodium ascorbate in 10 mM PBS, pH 7.4) to
produce the final concentrations listed in Table 3.1. These mixtures were incubated for 3
hours at 37 °C in the dark. To remove the low-molecular-weight compounds in the click
reaction mixture, the clicked plasmid DNA were purified by using the MinElute PCR
purification columns (Qiagen, Valencia, CA). To further support that the fluorescent dye
was ligated with to platinum complexes covalently bound to the DNA, 3 mM NaCN was
incubated with the purified Alexa Fluor 488-modified plasmid at 37 °C for 12 hours to
remove the platinum complex from DNA, followed by purification on MinElute PCR
purification column. The samples and control plasmid were analyzed by agarose gel (1 %)
electrophoresis and visualized through in-gel fluorescence using a Typhoon Trio Variable
Mode imager (GE Healthcare, Piscataway, NJ). Images were collected with a 488 nm
excitation laser and a 520 bp40 emission filter at 600 V PMT. The same gels were further
stained with ethidium bromide and documented using the Kodak Electrophoresis
Documentation and Analysis System 290 (Rochester, NY).

106
Tab
le 3
.1. C
om
posi
tion o
f C
uA
AC
Rea
ctio
ns
An
alyze
d b
y G
el E
lect
ropho
resi
s
Lan
e r b
P
uc1
9
(nucl
eoti
des
, µ
M)
P1-A
9
(µM
)
CuS
O4
(µM
)
TB
TA
(µM
)
Sodiu
m A
scorb
ate
(µM
)
Ale
xa
Flu
or
488
(µM
)
NaC
N
(mM
)
1
0
1.8
2
0
0
0
0
0
0
2
0
1.8
2
0
0.1
82
0.1
82
9.1
0.9
1
0
3
0.0
5
1.8
2
0.0
91
0.0
91
0.0
91
4.5
5
0.4
55
0
4
0.1
1.8
2
0.1
82
0.1
82
0.1
82
9.1
0.9
1
0
5
0.2
1.8
2
0.3
64
0.3
64
0.3
64
18.2
1.8
2
0
6
0.1
1.8
2
0.1
82
0.1
82
0.1
82
9.1
0.9
1
3

107
3.4.4. Stability of Compound P1-A9 in buffers
10 mM solutions of compound P1-A9 were prepared in the following buffers: 10 mM
phosphate-buffered saline (PBS), pH 7.4; 10 mM phosphate (PB), pH 7.4; acetate, pH 5.0.
Each sample was heated at 37 °C in the dark for 24 h and then analyzed by LC-ESMS.
The chromatographic separations were performed with a 4.6 mm x 150 mm reverse-
phase Agilent ZORBAX SB-C18 (5 µm) analytical column with the column temperature
maintained at 25 °C and a binary mobile phase system consisting of: solvent A, optima
water, and solvent B, methanol/0.1% formic acid, a gradient of 95% A to 5% A over 30
minutes and a flow rate of 0.5 mL/min.
3.4.5. Plasmid Unwinding Experiments.
Unwinding experiments were performed according to a previously published procedure
[309].
3.4.6. CD experiments
Solutions of calf thymus DNA (lyophilized powder, Sigma, St. Louis, MO) were
prepared in 10 mM phosphate buffer (pH 7.4). The DNA samples were annealed by slow
cooling from 90 ˚C to room temperature, and the final concentration was determined
spectrophotometrically at 260 nm with ε = 6500 M−1·cm−1 (nucleotides, n.t.).
To modify the DNA with platinum agent, calf thymus DNA (1 mM, n.t.) was incubated
in phosphate buffer (10 mM, pH 7.4) overnight at 37 ºC with P1-A1 and P1-A9 at a
platinum-to-nucleotide ratio (rb) of 0.05. To perform the click reaction, calf thymus DNA

108
(1 mM n.t.) was incubated with platinum complex P1-A9 with rb 0.05 at 37 ºC overnight.
The sample was then incubated with click reaction buffer at 37 °C for 2 hour with the
following final concentrations: CuSO4 (50 µM), TBTA (50 µM), Alexa Fluor 488 Alkyne
(50 µM) and sodium ascorbate (500 µM)).
CD experiments were performed on an AVIV Model 215 spectrophotometer equipped
with a thermoelectrically-controlled cell-holder. All the experiments were performed in
quartz cells with 0.5 cm path length. Spectra were recorded in the range 200-600 nm at
25 °C in 1-nm increments with an averaging time of 1 s. CD profiles were base-line
adjusted by subtracting buffer background.
3.4.7. Cell based assays based on post-labeling method
3.4.7.1. Cell culture maintenance
The human non-small cell lung cancer cell line, NCI-H460, was obtained from the
American Type Culture Collection (Rockville, MD, USA) and was cultured in RPMI-
1640 media (HyClone) containing 4.5 gL1glucose, 1.5 gL1 sodium bicarbonate, 10
mM HEPES, and 110 mgL1sodium pyruvate supplemented with 10% fetal bovine
serum (FBS), 10% penstrep (P&S), and 10% L-glutamine. Cells were incubated at a
constant temperature at 37 °C in a humidified atmosphere containing 5% CO2 and were
subcultured every 2 to 3 days in order to maintain cells in logarithmic growth.

109
3.4.7.2. Cell treatment and Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC)
ligation
NCI-H460 cells were seeded into poly-D-lysine coated glass bottom Petri dishes
(MatTeck Corporation, Ashland, MD, USA) with 105 cellsmL1 suspended in 2 mL of
medium per dish. Cells were incubated overnight and then treated with compound P1-A9
(5 μM) (or media for controls) for 3 h. After rinsing with chilled PBS (3), cells were
fixed by treatment with 3.7% formaldehyde solution (in PBS, pH 7.4) for 15 min at room
temperature and subsequently washed with a 3% solution of bovine serum albumin (BSA;
Sigma) in PBS (pH 7.4) twice for 10 min. For colocalization experiments using
mitochondrial stain, cells were treated with 100 nM MitoTracker Deep Red TM
(Invitrogen) in 2 mL pre-warmed phenol red-free media at 37 °C for 30 min prior to
fixing them using the abovementioned procedure. Cells were then permeabilized by
treatment with 0.5% Triton X-100 (in PBS, pH 7.4) at room temperature for 20 min.
Permeabilization buffer was quenched with 3% BSA in PBS (2 10 min) and the cells
were incubated with 250 μL of click reaction mixture (1 mM CuSO4; 0.5 μM Alexa Fluor
488-alkyne (Invitrogen); 10 mM sodium ascorbate; 50 mM Tris-HCl, pH 7.3) at room
temperature for 30 min. For the no-copper control, the copper solution in the click buffer
was replaced with water. Before co-staining with organelle-specific dyes, cells were
subjected to extensive washes with gentle agitation: 1) 3% BSA in PBS (5 min); 2) 0.5%
Triton X-100 in PBS (2 10 min); 3) PBS for (3 10 min). Nuclei were stained by 5
μgmL1 Hoechst 33342 (Sigma) in PBS for 5 min. Three final PBS washes were
performed immediately prior to image capture.

110
3.4.7.3. Confocal microscopy and fluorescence intensity analysis
Images were collected using a Zeiss LSM 710 confocal microscope (Carl Zeiss
MicroImaging, Thornwood, NY) using either a 40x (PLAN APO, 0.95 NA) or a 63x
(PLAN APO, 1.2 NA) objective lens. All images were acquired in multi-track
configuration mode to minimize excitation cross talk and emission bleed-through. We
utilized a 405 nm laser line (for Hoechst 33342) with an emission range of 424466 nm,
a 488 nm laser line (for Alexa Fluor 488) with an emission range of 489553 nm, and a
633 nm laser line (for MitoTracker Deep Red TM) with an emission range of 645740
nm. For comparative fluorescence intensity analysis, great care was taken to equalize
excitation power, pinhole settings, PMT gain, and offset values across and within
imaging sessions for each respective channel. For all images, the pinhole value was kept
at or below 1.2 airy units, and images were acquired with 8x line averaging at 1024 x
1024 pixels. Images were collected at 12-bit sampling to provide a wide dynamic
intensity range for analysis (0–4096). Zen software was used for image acquisition.
The intensity of platinum–azide-related Alexa Fluor 488 fluorescence in nuclei and
nucleoli was compared quantitatively using Zen software. The fluorescence signals from
the nuclei of labeled cells were measured by drawing a region of interest (ROI) around
each nucleus, with the Hoechst 33342 image used to verify nucleus location. The
fluorescence intensity of each object, Fobj, was calculated using the following formula:
Fobj = IROI Ibkgd,

111
where IROI and Ibkgd are the averaged signal intensities from all the pixels in the ROI and
background, respectively. Background was determined by drawing an ROI of the same
size in an empty region of the image. The fluorescence intensity in the nucleoli was
determined in the same manner. A total of 40–50 cells in 8 different fields were analyzed
in this manner for each treatment (cell counts were analyzed by one-way ANOVA,
indicating there was no statistical difference between fields; P = 0.54). The relative
enhancement of Pt-Alexa Fluor 488 fluorescence in the nucleoli was determined from the
ratio of intensities in nucleoli and in the nuclei for each group. Statistical analysis was
done using one-way ANOVA (GraphPad Prism). P < 0.05 was considered significant.
3.4.8. Cell based assays based on pre-labeling method
3.4.8.1. Labeling of compound P1-A9 with Alexa Fluor 488-DIBO to generate P1-
A9*
The copper-free click reaction was performed by combining solutions of compound
P1-A9 (2.5 µL, 25 mM) and Alexa Fluor 488-DIBO (2.5 µL, 2.5 mM, purchased from
Invitrogen) in DMF, followed by adjustment of the volume to 200 µL, vortexing, and
incubation of the mixture at room temperature for 3 h. The progress of the reaction was
monitored by reverse-phase high-performance liquid chromatography (HPLC) using the
LC module of an Agilent Technologies 1100 LC/MSD trap system equipped with a
multi-wavelength diode-array detector. Analytical separations were performed on a 4.6
mm × 150 mm reverse-phase Agilent ZORBAX SB-C18 (5 μm) column at 25° C using

112
the following conditions: solvent A: optima water/0.1% ammonium formate, solvent B:
methanol/acetonitrile (1:1, v/v), flow rate – 0.5 mL/min, gradient 0-5 min, 95% A; 5-20
min 95% A to 5% A; 20-30 min, 5% A. HPLC traces were recorded over a wavelength
range of 363–463 nm (for compound P1-A9) and 480–496 nm (for labeled P1-A9*).
Electrospray mass spectra (ESMS) of the LC fractions were recorded in negative-ion
mode. All data were processed with the LC/MSD Trap Control 4.0 data analysis
software.
3.4.8.2. Cell culture and incubations
The human non-small cell lung cancer cell line NCI-H460 (American Type Culture
Collection, Rockville, MD) was maintained as described previously. Cells were seeded
into poly-D-lysine coated glass bottom cell culture dishes (MatTeck Corporation,
Ashland, MD) with 105 cells·mL1 suspended in 2 mL of medium per dish. Cells were
incubated overnight and then treated with compound P1-A9 (5 μM, synthesized
according to a published procedure [[310]]), compound P1-A9 in the presence of 10% of
its labeled form (P1-A9*), or medium for controls, at 37 °C for 3 h. After incubation,
cells were washed with PBS (3 2 mL). Finally, cells were fixed by treatment with 3.7%
formaldehyde solution (in PBS, pH 7.4) for 15 min at room temperature, washed with a
solution of bovine serum albumin (3 % BSA in PBS; Sigma) twice for 10 min, and
permeabilized by treatment with 0.5% Triton X-100 (in PBS, pH 7.4) at room
temperature for 20 min. The reaction was quenched with 3% BSA in PBS (2 10 min)
and the cells were incubated with 250 μL of click reaction mixture (1 mM CuSO4; 0.5
μM Alexa Fluor 488-alkyne or Alexa Fluor 647-alkyne (Invitrogen); 10 mM sodium

113
ascorbate; 50 mM Tris-HCl, pH 7.3) at room temperature for 30 min. Before adding
nuclear stain in colocalization experiments, cells were subjected to extensive washes: 1)
3% BSA in PBS (5 min); 2) 0.5% Triton X-100 in PBS (2 10 min); 3) PBS for (3 10
min). Nuclei were stained with 5 μg·mL1 Hoechst 33342 (Sigma) in PBS (5 min),
followed by three additional PBS washes prior to image acquisition.
3.4.8.3. Confocal microscopy
Images were recorded on a Zeiss LSM 710 confocal microscope (Carl Zeiss,
Thornwood, NY) using either a 40 (PLAN APO, 0.95 NA) or a 63 (PLAN APO, 1.2
NA) objective lens. All images were acquired in multi-track configuration mode to
minimize excitation cross talk and emission bleed-through. A 405 nm laser line with an
emission range of 410–478 nm was used for Hoechst 33342, a 488 nm laser line
(emission: 489–553 nm) for Alexa Fluor 488. For all images, the pinhole value was kept
at or below 1.2 airy units. Images were acquired with 8 line averaging at 1024 1024
pixel resolution and 12-bit sampling to provide a wide dynamic intensity range for
analysis (0–4096). Zen software was used for image acquisition and processing.
Confocal image planes for each channel were not contrast-adjusted or otherwise changed
and assembled in Adobe Photoshop CS without further manipulation.

114
CHAPTER 4
SYNTHESIS AND EVALUATION OF A PLATINUM-ACRIDINE-ENDOXIFEN
CONJUGATE
The work contained in this chapter was initially published in Chemical Communications
in 2013. The manuscript, including figures and schemes, was drafted by Song Ding and
Dr. U. Bierbach and edited by Dr. U. Bierbach before submission to the journal. Since the
publication, changes in both format and content have been made to maintain a consistent
format throughout this work. The experiments described were performed by Song Ding,
except for the the cytotoxicity assays which were contributed by Dr. Xin Qiao.

115
4.1. Introduction and design rationale
Breast cancer continues to be a major cause of cancer-related mortality in women
[311]. Anti-estrogens such as tamoxifen (Figure 4.1) have become the mainstay in the
management of the early-stage hormone-dependent form of the disease [311].
Unfortunately, a significant population of patients does not respond to this drug or
develops resistance to endocrine therapy during the course of treatment, and systemic
chemotherapies often become the only armamentarium to combat recurrent disease [312].
Current platinum-based drugs have shown limited utility as first-line or salvage therapies
and no survival benefits have been reported for regimens based on these agents in
combination with targeted therapies [313]. The general lack of responsiveness of breast
cancers to platinum has been attributed to unusually high levels of acquired resistance to
this treatment including detoxification by elevated levels of cytosolic glutathione and
repair of DNA adducts [313]. One common strategy to overcome several of these
problems and provide more effective drugs for hormone-dependent breast cancer is to
combine an estrogen receptor (ER)-binding molecule with a classical cytotoxic agent.
The rationale behind this approach is to improve the pharmacological properties of
common cytotoxics by taking advantage of the targeting, antiestrogenic, and
chemosensitizing effects of ER-interacting ligands [314]. Various conjugates containing
ER-affinic carriers have been designed with the goal of targeting ER-positive breast
cancers more efficiently with the drug cisplatin [258, 315-317] and other cytotoxic agents
[318-321].
While several of the cis-platinum-based compounds have shown promising activity in
vitro and in mouse models [317], no attempts of extending this concept to the newer

116
nonclassical platinum anticancer agents have been reported. One such class of
compounds are platinum–acridine agents (Figure 4.1), which, unlike cisplatin, do not
induce cross-links but target DNA through a dual binding mode involving intercalation
and monofunctional platination of nucleobase nitrogen [289]. In non-small cell lung
cancer (NSCLC), the hybrid agents produce more severe DNA damage than cisplatin and
are significantly more cytotoxic than the clinical agent [275, 277, 301, 305]. Given the
superior cell kill of these agents and their ability to circumvent resistance to cisplatin, we
embarked on generating a breast cancer targeted agent containing our platinum–acridine
hybrid as a cytotoxic component. Here, we report on the design of the first conjugates
featuring a suitably functionalized platinum–acridine warhead coupled to an active
metabolite of the ER modulator and anticancer drug tamoxifen. The synthetic
methodology developed to generate these molecules is presented as well as screening
results in breast cancer cells.
Figure 4.1. Structures of tamoxifen and platinum–acridine hybrids.

117
4.2. Results and Discussion
4.2.1. Efforts to synthesize platinum-acridine-endoxifen conjugates
The strategy for generating the conjugates was to modify our platinum–acridine at the
N1 position (Figure 4.1) with a hydroxyl-functionalized side chain and attach it to the
secondary amino group of (E/Z)-4-hydroxy-N-desmethyltamoxifen [(E/Z)-endoxifen, 4-3,
Figure 4.2], the monohydroxylated/N-demethylated metabolite of tamoxifen [322].
Molecular models (not shown) were used to confirm that linking the two pharmacophores
at this position does not interfere with the binding of the components to their
pharmacological targets [274, 323]. The 4-hydroxo form of the pharmacophore was
chosen because of its superior binding affinity for ER and exquisite potency as an
inhibitor of hormone-dependent breast cancer cell proliferation [322] (confirmed in this
study). In addition, since the E- and Z-isomers of 4-hydroxytamoxifen (but not those of
tamoxifen) interconvert under physiologically relevant conditions [324], it was not
necessary to generate the stereochemically pure forms of the target compounds.
First, we designed the conjugatable platinum–acridine (P4-A3, Figure 4.2) based on the
potent derivatives identified previously (Figure 4.1). The target cytotoxic warhead
features simple ammine (NH3) nonleaving groups to maximize the aqueous solubility
[289] of the conjugate and an acetamidine donor group (R = Me), which has proven to
produce a 3-4 times more cytotoxic hybrid than propionamidine (R = Et) [277]. The
synthesis of P4-A3 involved addition of the linker secondary amino group in 2-((2-
(acridin-9- ylamino)ethyl)amino)ethanol (A3) and its ethylene glycol extended derivative
(A11) to the nitrile ligand in [PtCl(NH3)2(MeCN)]NO3 (Figure 4.2). Attempts to directly

118
attach the endoxifen moiety to the hydroxyl group in P4-A3 using carbamate coupling
chemistry failed because the reaction conditions were incompatible with the metal-
containing fragment. Instead, it was necessary to introduce platinum as the last step of
the reaction sequence after preassembling the entire organic scaffold. This was achieved
by reacting N-Boc-protected and 4-nitrophenyl carbonate-activated acridine–amines 4-1
and 4-2 with endoxifen (4-3) to generate the carbamate-coupled conjugates A12 and A13,
followed by addition of platinum nitrile complex to install the amidine-linked platinum
moiety (Figure 4.2). Conjugates P4-A12 and P4-A13 were isolated as 1:1 mixtures of the
E- and Z-isomers (for details of the product characterization, see the Experimental section
and Appendix A, Figure A.35-Figure A.37).
The carbamate linkage in the conjugates was chosen for its chemical robustness to
assure stability in circulation and delivery of the intact conjugate to the target site [325].
To test the reactivity of the carbamate group in aqueous media, conjugate P4-A12 was
incubated at 37 ˚C for 48 h in buffers mimicking the environments in serum, cytosol, and
the lysosomes. In-line high-performance liquid chromatography–mass spectrometry
(LC-MS) analysis of the samples confirmed that the carbamate linkage in compound P4-
A12 resists hydrolytic cleavage in all cases (see Appendix B3).

119
Figure 4.2. Synthesis of target molecules. Reagents and conditions: (a) 1. For P4-A3 :
[PtCl(NH3)2(MeCN)]NO3, DMF, -20 ˚C; 2. 1 M HNO3, MeOH. (b) Boc2O, DCM, rt. (c)
4-nitrophenyl chloroformate, TEA, DCM, rt. (d) 1. TEA, DCM, rt; 2. TFA, DCM, rt; 3. 1
M NaOH, DCM, rt. (e) 1. [PtCl(NH3)2(MeCN)]NO3, DMF, 4 ˚C; 2. 1 M HNO3, MeOH.

120
4.2.2. Biological evaluation of target conjugates
To assess the cytotoxic potency of the target conjugates, a colorimetric cell
proliferation assay was performed in the human breast adenocarcinoma cell lines MCF-7,
which overexpresses estrogen receptor (ER+), and the estrogen receptor-negative (ER-)
cell line MDA-MB-231.[326] Compounds P4-A12 and P4-A13 were tested along with
the two precursors, hydroxyl-modified platinum–acridine P4-A3, and (E/Z)-4-hydroxy-
N-desmethyltamoxifen (4-3). For comparison, the clinical drugs tamoxifen and cisplatin
were also included in this assay (Table 4.1). The tamoxifen metabolite, 4-3, showed the
strongest antiproliferative effect in MCF-7 with an IC50 value of 1.6±0.4 μM determined
after exposure of cells to the drug for 72 h. Under the same conditions, a 2.4-fold higher
concentration of compound P4-A3 is required to produce the same cell kill effect in this
cell line. Conjugate P4-A12 shows virtually the same potency as its cytotoxic component,
P4-A3, whereas the extended conjugate P4-A13 was the least active compound in this
assay. All compounds were significantly more cytotoxic in MCF-7 cells than in MDA-
MB-231 cells. For compounds P4-A3, 4-3 and P4-A12, for example, the cell line-
specific cytotoxic enhancements were 3-fold, 8-fold, and 6-fold, respectively. While
compounds P4-A3 and P4-A12 are equipotent in MCF-7, the conjugate proves to be
relatively less cytotoxic in MDA-MB-231. Tamoxifen, the mainstay of breast cancer
treatment, and cisplatin were the least active compounds tested in the ER-positive cell
line.

121
Table 4.1. Summary of cytotoxicity data (IC50, μM)a in human breast cancer cell lines
Compound MCF-7 MDA-MB-231
Tamoxifen 16.2 ± 1.2 20.1 ± 1.0
cisplatin 12.1 ± 1.1 40.6 ± 4.2
P4-A3 3.8 ± 0.4 11.3 ± 0.2
4-3 1.6 ± 0.4 13.2 ± 0.1
P4-A12 3.5 ± 0.1 21.4 ± 1.0
P4-A13 27.5 ± 1.4 46.5 ± 0.7 a The IC50 values are reported as the mean ± standard deviation for two individual 72h
incubations performed in triplicate.
Modification of compound P4-A3 with endoxifen (4-3) produces a conjugate (P4-A12)
that shows essentially the same cytotoxic response (based on IC50 values) as the
unmodified platinum–acridine hybrid in MCF-7 cells. While equipotent, both
compounds can be expected to induce cancer cell kill by different mechanisms. A
comparison of the drug–response profiles for compounds 5 and 10 after 48 h of
incubation with MCF-7 cells confirms this supposition. While the platinum–acridine
agent P4-A3 clearly shows an antiproliferative advantage over compound P4-A12 at low
concentrations, it is unable to kill a major fraction of the cells at the highest
concentrations tested (Figure 4.3). These findings suggest that a subpopulation of cells
may exist that are relatively insensitive to this agent, as has been observed previously for
MCF-7 cells exposed to cytotoxics.[327] Importantly, compound P4-A12 overcomes this
form of resistance, based on the considerably lower percentage of cells that remain viable
after incubation with 20 and 40 µM conjugate for 48 h.

122
Figure 4.3. Differential response of MCF-7 cancer cells to compounds P4-A3 and P4-
A12.
4.3. Conclusion
Here, we have presented the first targeted conjugate based on a nonclassical platinum-
containing hybrid structure. The synthetic approach provides a versatile platform for
incorporating a wide range of small-molecule receptor- and cancer cell-affinic ligands, as
well as enzymatically cleavable and self-immolative linkers.[328] Such modifications
may help target cancers more selectively and reduce the undesired high systemic
toxicity[289] observed for the simple platinum–acridines. Using a hormone-dependent
cancer model, we demonstrated that the ER-targeted form of platinum–acridine maintains

123
the same cytotoxicity as compound P4-A3 in MCF-7 cells. A comparison of the relative
cytotoxic enhancements observed for P4-A3 and P4-A12 in ER+ MCF-7 vs. ER- MDA-
MB-231 (Table 4.1) and the individual drug responses for each derivative suggest that
conjugate P4-A12 acts by a distinct mechanism potentially involving interactions with
ER. Further investigation into the molecular mechanism of the conjugate is necessary to
substantiate this notion. Likewise, it will be important to determine why introduction of
an extended linker in compound P4-A13 results in a greatly diminished cytotoxic
response.
In conclusion, the novel conjugate P4-A12 was generated in an attempt to marry the
cytotoxic, DNA-damaging properties of a nonclassical platinum-acridine agent with the
targeting potential of an antiestrogen. Initial tests show promising cell kill effects, which
warrant further studies into structure–activity relationships in derivatives of the
prototypical agent and their in vivo efficacy against hormone-dependent breast cancer.
Multifunctional agents like compound P4-A12 may have applications in patients who do
not respond to tamoxifen due to altered expression levels of ER or an intrinsic inability to
metabolize tamoxifen [329].

124
Figure 4.4. Synthesis of precursor 4-3. Reagents and conditions: (a) Zn dust, TiCl4,
THF, reflux, 82%. (b) Me2NCH2CH2Cl, Cs2CO3, DMF, 120 °C, 67%. (c) (1)ACE-Cl,
DCM, reflux; (2)MeOH, 6 M HCl, reflux, 81%.
4.4. Experimental Section
4.4.1 Synthesis of target conjugates
The synthesis of 1, 1-Bis(4-hydroxyphenyl)-2-phenylbut-1-ene (4-6). This
compound was synthesized according to the method reported previously[330] with
modifications. To a stirred suspension of zinc powder (8 g, 0.124 mol) in 80 mL of
anhydrous THF, TiCl4 (6 mL, 0.056 mol) was added dropwise under argon at -20 °C.
When the addition was complete, the mixture was warmed to room temperature and
refluxed for 3 h. The reaction was cooled in an ice bath and a solution of 4,4’-
hydroxybenzophenone (4-4, 2 g, 0.0092 mol) and propiophenone (4.0 g, 0.0296 mol) in
10 mL of anhydrous THF was added. The reaction mixture was refluxed in the dark for

125
12 h. The reaction was quenched with 100 mL of 10% aqueous K2CO3 and extracted
with ethyl acetate. The organic layers were collected, washed with saturated NaCl, dried
over MgSO4, and concentrated by rotary evaporation, affording a pink oil. To purify the
crude product, the oil was dissolved in 10 mL of ethanol and precipitated by adding 40
mL of water. The white precipitate was recovered by filtration and recrystallized from 35
mL of hot DCM to afford 4-6 (2.4 g, 82%) as a white solid. 1H NMR (300 MHz,
DMSO-d6) δ 9.35 (s, 1H), 9.10 (s, 1H), 7.21 – 7.04 (m, 5H), 6.97 (d, J = 8.4 Hz, 2H),
6.74 (d, J = 8.5 Hz, 2H), 6.60 (d, J = 8.5 Hz, 2H), 6.39 (d, J = 8.5 Hz, 2H), 2.38 (q, J =
7.4 Hz, 2H), 0.84 (t, J = 7.3 Hz, 3H).
The synthesis of (E,Z)-1-[4-(2-Dimethylaminoethoxy)phenyl]-1-(4-hydrox-
yphenyl)-2-phenylbut-1-ene (4-7). This compound was synthesized according to the
method[330] reported previously with slight modifications. A solution of 1,1-bis(4-
hydroxyphenyl)-2-phenylbut-1-ene (4-6, 1 g, 3.2 mmol) in DMF (15 mL) was mixed
with Cs2CO3 (3.1 g, 9.5 mmol) and refluxed for 30 min. To this mixture was added 2-
(dimethylamino)ethylchloride hydrochloride (0.56 g, 3.8 mmol) in three portions over 1 h.
The mixture was heated at 120 °C for another 6 h. After cooling to room temperature,
the mixture was poured into 20 mL of saturated NH4Cl and extracted with ethyl acetate
(3 × 20 mL). The organic phases were combined, washed with saturated NaCl (2 × 20
mL), dried over anhydrous Na2SO4, and concentrated to give brown oil. The crude
material was purified by recrystallization from 10 mL of hot ethanol to afford 0.82 g
white crystals (4-7) with a 1:1 Z/E ratio (yield: 67%). 1H NMR (300 MHz, DMSO-d6) δ
9.38 (s, 0.5H), 9.13 (s, 0.5H), 7.23 - 7.04 (m, 5H), 6.98 (d, J = 8.4 Hz, 1H), 6.92 (d, J =
8.7 Hz, 1H), 6.75 (d, J = 8.5 Hz, 1H), 6.71 (d, J = 8.7 Hz, 1H), 6.63 - 6.53 (m, 2H), 6.40

126
(d, J = 8.6 Hz, 1H), 4.05 (t, J = 5.8 Hz, 1H), 3.88 (t, J = 5.8 Hz, 1H), 2.62 (t, J = 5.8 Hz,
1H), 2.56 - 2.49 (m, 1H), 2.44 - 2.33 (m, 2H), 2.22 (s, 3H), 2.15 (s, 3H), 0.84 (t, J = 7.3
Hz, 3H).
The synthesis of (E,Z)-Endoxifen Hydrochloride (4-3). This compound was
synthesized according to the method[331] reported previously with modifications. 1-
Chloroethyl chloroformate (ACE-Cl, 668 μL, 61.9 mmol) was added dropwise to a
solution of 4-7 and 1,8-bis(dimethylamino)naphthalene (proton sponge, 249 mg, 11.6
mmol) in 9 mL of DCM at 0 ºC. The resulting solution was stirred for 30 min. at 0 ºC and
then refluxed for 12 h. The reaction mixture was cooled to room temperature and
removal of the solvent under reduced pressure gave a brown crude oil. This residue was
dissolved in a mixture of MeOH (20 mL) and 6 M HCl (5 mL) and refluxed for 3 h and
then cooled to room temperature. The MeOH was evaporated off and the residue was
dissolved in 50 mL of ethyl acetate, washed with saturated NaCl and water, dried over
anhydrous Na2SO4, and concentrated to give 0.260 g of 4-3 as white solid, (yield: 81%),
as 1:1 mixture of Z/E Endoxifen isomers, which was used in the next step without further
purification. 1H NMR (300 MHz, DMSO-d6) δ 9.30 (br s, 2H), 7.25 - 7.06 (m, 5H), 7.00
(d, J = 2.7 Hz, 1H), 6.96 (d, J = 2.7 Hz, 1H), 6.79 - 6.72 (m, 2H), 6.65 (d, J = 8.5 Hz, 1H),
6.60 (d, J = 8.5 Hz, 1H), 6.42 (d, J = 8.5 Hz, 1H), 4.26 (t, J = 4.6 Hz, 1H), 4.08 (d, J =
5.7 Hz, 1H), 3.19 (d, J = 5.7 Hz, 1H), 2.60 (s, 1.5H), 2.54 (s, 1.5H), 2.40 (m, 2H), 0.85 (t,
J = 7.3 Hz, 3H).

127
F
igu
re 4
.5.
Synth
esis
of
pre
curs
or
A11.
Rea
gen
ts a
nd c
ondit
ions:
(a)
K2C
O3,
TH
F,
refl
ux
, 87%
. (
b)
9-p
hen
ox
yac
ridin
e, T
HF
,
refl
ux
, 86%
.

128
Synthesis of 2-(2-(2-((2-aminoethyl)amino)ethoxy)ethoxy)ethanol (4-9). The
mixture of ethylene diamine (3.05g, 0.05 mol) and anhydrous potassium carbonate (1.15
g, 0.0083 mol) in 30 ml anhydrous THF was heated under reflux, to which was added
monotosylated triethylene glycol (4-8, 1.71g, 0.0056 mol) in 15 ml anhydrous THF
dropwisely over 1 h, then the reaction was refluxed for another 16 hours. When the
reaction was completed, the white solids were filtered off after the mixture was cooled to
room temperature. To remove THF and unreacted ethylene diamine, the filtrate was then
concentrated under reduced pressure followed by dried under vacume at 45 ºC overnight
to give a pale yellow crude oil (1.34 g, yield 87%), which can be used in the next step
without any purification. 1H NMR (300 MHz, CDCl3) δ 3.85 - 3.51 (m, 10H), 2.92 -
2.63 (m, 10H), 2.31 (s, 4H). 13C NMR (75 MHz, CDCl3) δ 72.86, 70.46, 70.29, 70.12,
61.30, 52.17, 48.94, 41.46.
Synthesis of 2-(2-(2-((2-(acridin-9-ylamino)ethyl)amino)ethoxy)ethoxy)ethan-1-ol
(A11). A mixture of 9-phenoxyacridine (2.71 g, 0.01 mol) and 2-(2-(2-((2-aminoethyl)
amino) ethoxy) ethoxy) ethanol (4-9, 2.31 g, 0.011 mol) in 30 mL of anhydrous THF was
refluxed for 16 h. The solvent was evaporated off and the residue was dissolved in 30 mL
of acetone. To this solution were added 5 mL of concentrated HCl and the mixture was
stirred at 4 ºC for 3 hours. A yellow precipitate formed which was recovered by filtration,
resuspended in 50 mL of 1 M sodinum hydroxide, and stirred at room temperature for 30
min. The aqueous phase was extracted with CH2Cl2 and the organic phase was collected,
dried over Na2SO4, and concentrated using rotary evaporation, affording 3.18 g of the
free base as an orange oil (Yield: 86%). 1H NMR (300 MHz, CDCl3) δ 8.17 (d, J = 8.8
Hz, 1H), 8.05 (d, J = 8.7 Hz, 1H), 7.69 – 7.59 (m, 1H), 3.84 (t, J = 5.5 Hz, 1H), 3.74 (d, J

129
= 4.7 Hz, 1H), 2.93 (t, J = 5.5 Hz, 1H), 2.84 (t, J = 5.0 Hz, 1H). 13C NMR (75 MHz,
CDCl3) δ 151.98, 149.25, 141.24, 133.06, 129.86, 129.11, 126.91, 123.21, 122.70, 121.31,
120.94, 116.86, 116.77, 72.58, 70.50, 70.34, 70.23, 61.51, 49.32, 49.02, 48.45. MS (ESI,
positive mode): for C26H35N3O5 ([M+H]+), 370.26; found: 370.5.
Synthesis of tert-butyl (2-(acridin-9-ylamino)ethyl)(2-hydroxyethyl)carbamate
(Boc protected A3). The Boc protected A3 was synthesized as follows. 2-((2-(Acridin-
9-ylamino) ethyl)amino)ethanol (A3) (2.82 g, 0.01 mol) was suspended in 30 mL of
anhydrous dichloromethane, to which was added Boc2O (2.18 g, 0.01 mol) in 5 mL of
DCM at 0-5 ºC, maintained with an ice bath. The mixture was then stirred at room
temperature for 4 h. The solvent was removed by rotary evaporation and the residue was
dissolved in 10 mL of DCM and precipitated with 200 mL of anhydrous diethyl ether.
The solid was recovered by filtration and dried in a vacuum, affording Boc protected A3
as yellow solid 3.57 g (yield: 93%), which was used in the next step without purification.
1H NMR (300 MHz, CDCl3) δ 8.08 (d, J = 8.7 Hz, 2H), 7.78 (d, J = 7.8 Hz, 2H), 7.45 (t,
J = 7.8 Hz, 2H), 7.12 (t, J = 7.8 Hz, 2H), 4.15 - 3.95 (m, 2H), 3.78 - 3.54 (m, 4H), 3.42 -
3.24 (m, 2H), 1.43 - 1.10 (m, 9H).
Synthesis of 2-((2-(acridin-9-ylamino)ethyl)amino)ethyl (2-(4-(1-(4-
hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)ethyl)(methyl)carbamate (A12).
Boc protected A3 (1 g, 3.55 mmol), TEA (1.79 g, 17.7 mmol) and 4-nitrophenyl
chloroformate (0.99 g, 4.6 mmol) were dissolved in 20 mL of anhydrous
dichloromethane. The mixture was stirred at room temperature for 16 h. To the resulting
solution of the 4-nitrophenyl-activated form of the acridine (4-1) was added 4-3 (1.74 g,
4.23 mmol) in 5 mL of anhydrous DCM and the reaction was stirred for another 8 h. The

130
solvent was removed in a high vacuum and the residue was redissolved in 40 mL of
DCM and washed with 1 M HCl (3 × 20 mL). The organic phase was collected, dried
over anhydrous Na2SO4, and concentrated to afford orange oil. To remove the Boc group,
the orange oil was dissolved in 6 mL of a 1:1 mixture of anhydrous DCM and TFA and
stirred at room temperature for 3 h. The reaction was quenched by adding 10 mL of 1 M
NaOH solution. The free base was extracted from NaOH solution with DCM, dried over
anhydrous Na2SO4, and concentrated. The product was purified by flash chromatography
(Al2O3, DCM:MeOH, 30:1) to give 1.35 g of A12 as an orange oil with a 1:1 Z/E ratio
(yield: 56%). 1H NMR (500 MHz, CDCl3, 60 ºC) δ 8.13 - 8.05 (m, 4H), 7.51 (s, 2H), 7.27
- 6.41 (m, 16H), 4.34 - 4.29 (t, J = 5.4 Hz, 1H), 4.28 (t, J = 5.3 Hz, 1H), 4.17 - 3.94 (m,
4H), 3.69 (t, J = 5.4 Hz, 1H), 3.58 (t, J = 5.3 Hz, 1H), 3.11 - 2.94 (m, 7H), 2.55 - 2.40 (m,
2H), 1.00 – 0.84 (m, 3H). MS (ESI, positive mode): for C43H44N4O4 ([M+H]+), 681.3;
found: 681.5.
Compound 2-(2-(2-((2-(acridin-9-ylamino)ethyl)amino)ethoxy)ethoxy)ethyl (2-(4-(1-
(4-hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)ethyl)(methyl)carbamate (A13) was
prepared according to the procedure described for A12, by using A11 as the precursor
with the yield of 37%. 1H NMR (500 MHz, CDCl3, 60 ºC ) δ 8.38 - 8.01 (m, 3H), 7.77 -
7.24 (m, 3H), 7.22 - 6.98 (m, 6H), 6.86 - 6.25 (m, 6H), 4.33 - 3.39 (m, 16H), 3.07 - 2.79
(m, 6H), 2.62 - 2.37 (m, 2H), 1.79 - 1.66 (m, 2H), 1.31 (s, 1H), 0.95 (m, 3H). 13C NMR
(126 MHz, CDCl3, 25 ºC) δ 156.33, 152.38, 148.61, 141.17, 137.80, 132.09, 132.06,
130.67, 130.67, 130.30, 129.73, 127.86, 125.90, 122.83, 122.83, 116.48, 115.36, 115.28,
114.62, 114.56, 113.96, 113.79, 113.23, 113.04, 70.62, 70.36, 69.58, 48.91, 48.70, 48.58,

131
48.21, 47.91, 35.98, 28.99, 26.40, 13.65, 1.03. MS (ESI, positive mode): for C47H52N4O6
([M+H]+), 769.39; found: 769.8.
Synthesis of conjugate [PtCl(NH3)2C19H22N4O](NO3)2 (P4-A3). Platinum complex
P4 (341 mg, 1 mmol) was converted to its nitrate salt by reaction with AgNO3 (162 mg,
0.95 mmol) in 7 mL of anhydrous DMF. AgCl was removed by syringe filtration, and the
filtrate was cooled to -10 °C. Acridine precursor A3 (282 mg, 0.1 mmol) was added to
the solution, and the suspension was stirred at -10 °C for 24 h. After treatment with
activated carbon, the reaction mixture was added into 300 mL of diethyl ether. The
yellow slurry was stirred for 30 min, and the precipitate was recovered by membrane
filtration and dried in a vacuum overnight. The solid was dissolved in anhydrous MeOH
and treated with 1 equivalent of 1 M HNO3, stirred at room temperature for 30 minutes
and precipitated by 300 mL anhydrous diethyl ether. The product was further purified by
recrystallization from hot ethanol to give 618 mg of hybrid P4-A3 a microcrystalline
orange material (yield: 87%). 1H NMR (300 MHz, MeOH-d4) δ 8.40 (d, J = 8.8 Hz, 2H),
7.87 (t, J = 6.8, 2H), 7.72 (dd, J = 8.7, 1.2 Hz, 1H), 7.49 (d, J = 8.4 Hz, 2H), 4.31 (t, J =
7.0 Hz, 2H), 4.10 (brs, 2H), 3.91 (t, J = 6.8 Hz, 1H), 3.75 (s, 2H), 3.65 (t, J = 4.9 Hz, 2H),
3.49 (t, J = 4.9 Hz, 2H), 2.58 (s, 3H). 13C NMR (75 MHz, MeOH-d4) δ 167.49, 160.02,
141.40, 136.55, 126.49, 125.30, 119.77, 114.14, 60.73, 49.86, 48.16, 47.39, 23.02. MS
(ESI, positive mode): for C19H28ClN6OPt ([M]+), 587.00; found: 585.2.
Synthesis of conjugate [PtCl(NH3)2C45H47N5O4](NO3)2 (P4-A12). Platinum complex
A4 (75 mg, 0.22 mmol) was converted to its nitrate salt by reaction with AgNO3 (35.6 mg,
0.21 mmol) in 7 mL of anhydrous DMF. AgCl was removed by syringe filtration, and the
filtrate was cooled to -10 °C. Acridine precursor A12 (150 mg, 0.22 mmol) was added to

132
the solution, and the mixture was stirred at 4 °C for 5 days. After treatment with
activated carbon, the reaction mixture was added into 300 mL of diethyl ether. The
yellow slurry was stirred for ~30 min, and the precipitate was recovered by membrane
filtration, washed by 10 mL of anhydrous DCM and dried in a vacuum overnight. The
solid was dissolved in anhydrous MeOH containing 1 equivalent of 1 M HNO3, stirred at
room temperature for 30 minutes and precipitated with 300 mL of anhydrous diethyl
ether. The product was further purified by recrystallization from hot ethanol to give 116
mg of P4-A12 (yield 47 %). 1H NMR (500 MHz, DMF-d7) δ 14.28 (s, 0.5H), 10.61 (s,
0.5H), 9.64 (s, 0.5H), 9.40 (s, 0.5H), 8.81 (s, 2H), 7.10 (s, 2H), 7.28 - 6.00 (m, 15H), 4.74
(s, 3H), 4.50 (s, 3H), 4.59 - 3.87 (m, 10H), 3.65 (t, J = 5.6 Hz, 1H), 3.55 (t, J = 6.3 Hz,
1H), 3.02 (s, 2H), 2.96 (s, 3H), 2.78 (s, 3H), 2.54 - 2.37 (m, 2H), 0.97 - 0.82 (m, 3H). 13C
NMR (126 MHz, DMF-d7) δ 165.54, 161.88, 158.26, 157.48, 156.61, 156.01, 155.67,
142.56, 140.32, 138.34, 136.31, 135.06, 131.60, 130.41, 129.75, 127.93, 125.71, 123.69,
118.93, 115.13, 113.87,113.36, 65.84, 62.39, 48.38, 21.99, 12.96. MS (ESI, positive
mode): for C45H54ClN7O4 Pt([M - 2H]+), 986.37; found: 985.6.
Synthesis of conjugate [PtCl(NH3)2C49H55N5O6](NO3)2 (P4-A13). P4-A13 was
prepared according to the procedure described for P4-A12, by using A13 as the precursor
with the yield of 84%. 1H NMR (500 MHz, DMF-d7) δ 9.65 (s, 0.5H), 9.39 (s, 0.5H),
8.64 (s, 2H), 8.06 - 7.92 (m, 3H), 7.78 - 6.35 (m, 16H), 4.67 - 3.87 (m, 14H), 3.87 - 3.35
(m, 22H), 3.01- 2.98 (m, 1H), 2.59 - 2.36 (m, 2H), 1.79 (m, 6H), 0.90 (s, 3H). 13C NMR
(126 MHz, DMF-d7) δ 165.75, 161.87, 156.85, 156.00, 142.79, 140.58, 140.30, 138.43,
136.29, 134.49, 131.81, 130.48, 129.74, 127.92, 126.02, 115.11, 114.37, 114.15, 113.33,

133
97.85, 70.12, 69.22, 67.32, 65.53, 64.56, 34.29, 34.12, 29.17, 29.00, 25.36, 21.97, 13.15.
MS (ESI, positive mode): for C49H61ClN7O6Pt ([M+H]+), 1074.40; found: 1074.8.
4.4.2. Stability Study of Compound P4-A12
10 µM solutions of compound P4-A12 were prepared in the following buffers: 10 mM
phosphate-buffered saline (PBS), pH 7.4; 10 mM phosphate (PB), pH 7.4; acetate, pH 5.0.
Each sample was heated at 37 °C in the dark for 48 h and then analyzed by LC-ESMS.
The chromatographic separations were performed with a 4.6 mm × 150 mm reverse-
phase Agilent ZORBAX SB-C18 (5 µm) analytical column with the column temperature
maintained at 25 °C and a binary mobile phase system consisting of: solvent A, optima
water, and solvent B, methanol/0.1% formic acid, a gradient of 95% A to 5% A over 20
minutes and a flow rate of 0.5 mL/min.
4.4.3 Cytotoxicity studies and cell viability assays
4.4.3.1. Sample preparation and cell culture
Stock solutions of compound P4-A3, compound P4-A12 and compound P4-A13 were
prepared in DMF and concentrations were determined spectrophotometrically by ε413 =
10000 M-1 cm-1. Cisplatin, endoxifen and estradiol were purchased from Sigma. Stock
solution of cisplatin was prepared in phosphate-buffered saline (PBS) and the

134
concentration was determined by ε300 = 132 M-1 cm-1. Stock solutions of endoxifen and
tamoxifen were prepared in DMSO.
The human breast adenocarcinoma cells MCF-7 (ER-positive), and MDA-MB-231
(ER-negative) were obtained from American Type Culture Collection (Rockville, MD,
USA). MCF-7 cells were maintained in phenol red DMEM/F-12 media (Gibco)
containing 2.4 g/L sodium bicarbonate and 2.5 mM L-glutamine supplemented with 10%
fetal bovine serum (FBS), 10% penstrep (P&S), and 10 μg/mL insulin. MDA-MB-231
cells were cultured in high glucose DMEM media (Gibco) containing 4.5 g/L D-glucose,
110 mg/L sodium pyruvate supplemented with 10% fetal bovine serum (FBS), 10%
penstrep (P&S), and 10% L-glutamine. All cultures were grown at a constant temperature
at 37 °C in a humidified atmosphere containing 5% CO2, and cells were subcultured once
a week in order to maintain cells in logarithmic growth.
4.4.3.2. Cytotoxicity studies
The cytotoxicity studies of selected compounds were carried out by the CellTiter 96
Aqueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI, USA) as
described previously.[277] Briefly, cells suspensions were harvested and seeded into 96-
well microplates at a certain density (MCF-7, 10000 cells/well; MDA-MB-231, 12000
cells/well). After overnight incubation, cells cells were treated with varying
concentrations (starting concentrations were 80 or 100 μM, and DMF or DMSO amount
was less than 0.5%) of test compound for 72 h at 37 ºC in an atmosphere of 5% CO2.
After incubation periods of 72 h, 20 μL of MTS/PMS solution was added to each well
and incubated at 37 °C for 4 h. The absorbance of tetrazolium dye was measured at 490

135
nm using an enzyme-linked immunesorbent assay (ELISA) reader. The reported IC50 data
were calculated from non-linear curve fits using a sigmoidal dose-response equation in
GraphPad Prism (GraphPad Software, La Jolla, CA) and are averages of two individual
experiments performed in triplicate.

136
CHAPTER 5
DESIGN OF ENZYMATICALLY CLEAVABLE PRODRUGS OF A POTENT
PLATINUM-CONTAINING ANTICANCER AGENT
The work contained in this chapter was initially published in Chem. Eur. J in 2014. The
manuscript, including figures and schemes, was drafted by Song Ding and Dr. U.
Bierbach and edited by Dr. U. Bierbach before submission to the journal. Since the
publication, changes in both format and content have been made to maintain a consistent
format throughout this work. The experiments described were performed by Song Ding,
except for the the confocal microscopy and cytotoxicity assays which were contributed
by Dr. Amanda J. Pickard

137
5.1. Introduction and Design Rationale
Traditional chemotherapies often suffer from high systemic toxicity and a narrow
therapeutic window. To improve the pharmacokinetics (PK) and toxicity profiles of
anticancer drugs, various avenues are being pursued, such as nano-sized delivery
platforms, receptor-targeted conjugates, and prodrug designs [332, 333]. The rationale
behind the latter approach is to generate a precursor molecule, which is converted post
administration to the bioactive form of the drug either enzymatically or in response to a
chemical stimulus. Bioactivation may occur during absorption, circulation, or at the
tumor site [334]. Potential benefits of lipophilic prodrugs include efficient retention in,
and absorption from, circulation, as well as improved penetration of membranes and
accumulation in target tissues [332].
Figure 5.1. General structure of first- and second-generation platinum–acridine hybrid
agents.
PT-ACRAMTU, (Figure 5.1; ACRAMTU = 1-[2-(acridin-9-ylamino)ethyl]-1,3-
dimethylthiourea) represents the prototype of a class of DNA-targeted platinum–acridine

138
hybrid agents, which have shown exquisite potency in several solid tumor models [295].
Non-small cell lung cancer (NSCLC) cells prove to be particularly sensitive to this
pharmacophore, with the newer derivatives showing IC50 values in NSCLC cell lines in
the low-nanomolar range and activity in tumor xenografts [279, 295]. Unlike cisplatin
[cis-diamminedichloroplatinum(II)] and its analogues, platinum–acridines derived from
PT-ACRAMTU do not cross-link DNA bases but produce structurally unique hybrid
adducts that are an intrinsically more severe form of DNA damage than the former
bifunctional adducts [275, 296, 305]. The classical structure–activity relationship (SAR)
approach based on modular library screening has previously been used to tune the
chemical stability and reduce the off-target reactivity of the pharmacophore [76]. The
desired improvements were achieved essentially by modifying the ligand and donor sets
around the electrophilic metal [295]. These efforts have led to the development of a
derivative (P8-A1, Figure 5.1) that shows three orders of magnitude higher potency than
cisplatin (the IC50 values for P8-A1 in NCI-H460 and A549 lung cancer cells were 1.3
and 3.9 nM, respectively; see the Figure E.3. in APPENDIX E for dose–response curves).
Despite their promising cell kill in chemoresistant, intractable cancers, platinum–
acridines show unfavorable ADME [332] (absorption, distribution, metabolism, and
excretion) properties, which slow their preclinical development. P3-A1, for instance,
while inhibiting the growth of xenografted NCI-H460 tumors in mice, showed signs of
severe toxicity in the test animals, resulting in a low maximum tolerated dose (MTD)
[279]. Mice necropsied after treatment with P3-A1 showed high levels of platinum in
normal tissues, but insufficient accumulation in tumors, as well as discoloration of the
kidneys, a possible sign of hepatotoxicity or nephrotoxicity [279]. To improve the

139
pharmacological properties of platinum–acridines and to potentially open the therapeutic
window for systemic treatment with these agents, we have now designed lipophilic ester-
based derivatives that can be specifically tuned to undergo enzymatic hydrolysis. This
concept has resulted in the first case of a platinum-containing agent that is recognized as
a substrate by human carboxylesterase-2 (hCES-2), a key enzyme involved in the
activation of several anticancer prodrugs [332].
Scheme 1. Synthesis of target compounds. Reagents and conditions: (i) AgNO3, DMF, rt;
(ii) appropriate nitrile–ester (5-7 ─ 5-10), DMF, 60 °C, 4 h, (iii) (1) A1, DMF, 4 °C, (2) 1
M HNO3. Abbreviations: en = ethane-1,2-diamine, pn = propane-1,3-diamine.
5.2. Results and Discussion
5.2.1. Design and chemistry
Platinum–acridines show excellent cytotoxicity but unfavorable drug-like properties.
The most cytotoxic derivatives exist in their fully protonated [pKa (9-aminoacridine) ≈
9.5–10], dicationic form [295]. Because these hybrids are too hydrophilic, they show

140
poor tissue distribution and are most likely removed too rapidly from circulation via renal
clearance [335]. Thus, the goal of the structural modifications introduced here was to
increase the lipophilicity of the agents while maintaining good water solubility. The
alkyl residue of the amidine donor group (residue R, for Y = NH, see Figure 5.1) was
chosen as the site of attachment for the carboxylic acid ester groups. The design involved
installation of a hydroxymethyl (MeOH) or an extended 3-hydroxypropyl (Pr3-OH) group as
R in place of simple Me and Et in prototypes and masking of the terminal OH function as
lipophilic esters (see Scheme 5.1). A primary carboxylic acid, butanoic (butyric) acid,
and a bulkier, branched derivative, 2-propylpentanoic (valproic) acid, were introduced as
acyl components (see Scheme 5.1). Use of the latter residue was inspired by an
analogous valproic amide-based prodrug of the anticancer drug gemcitabine, which is
activated by hCES-2 [336].
Two distinct mechanisms of ester cleavage have to be considered: chemical and
enzymatic hydrolysis. The former mechanism has previously been observed in
chemically related carboxylic acid-modified platinum–acridines containing a reversed
ester linkage (Pt-linker-C(O)OR´, instead of Pt-linker-OC(O)R´ used in this study) [68].
It is dominated by platinum-assisted ester cleavage in a chloride-ion concentration
dependent manner. Low intracellular chloride favors aquation of platinum, which serves
as a Lewis-acidic metallohydrolase and accelerates ester cleavage. The second mode of
activation involves carboxylesterase isoenzymes, in particular hCES-2, which is not only
expressed at high levels in the gastrointestinal tract and the liver, but also in tumor tissue
[332]. The choice of hCES-2 rather than hCES-1 as the target enzyme was based on the
well-documented substrate selectivity of the two forms, according to which hCES-2

141
preferentially recognizes esters containing bulky alcohols (here, the hydroxyl-modified
hybrid agent itself) in combination with relatively smaller acyl moieties [337].
A total of seven ester-protected hybrids were synthesized. Structural diversity in this
set of compounds was achieved by varying the chain length, n, and the nature of the acyl
residue, R´ (Scheme 1). In addition, different non-leaving amines (L) were introduced to
tune the reactivity of the platinum moiety (see discussion below). All Compounds P9-A1
─P15-A1 were generated from the platinum precursors (P9─P15) containing the
appropriate ester-modified nitrile ligands (5-7 ─ 5-10, see the Experimental Section).
The ligand substitution reactions leading to the precursor complexes containing short-
chain nitrile–esters (n = 1, A9 ─ A12) produced significantly lower yields than reactions
performed with the long-chain derivatives (n = 3, A13─A15) (20–30 % vs. 80–90 %).
This previously observed effect can be attributed to the high CH acidity and reactivity of
the nitrile ligands in the former set of derivatives [68]. (Attempts to generate analogous
derivatives with n = 2 failed due to unexpected β-elimination of the hydroxyl group; data
not shown). The final step affording hybrid agents P9-A1─P15-A1 (yield > 75 %,
analytical purity > 95 %) involved addition of the NHMe group in N-(acridin-9-yl)-N´-
methylethane-1,2-diamine (A1) across the metal-activated nitrile triple bond [338],
producing the desired amidine linkage, and subsequent protonation to generate the
dinitrate salts.
An anomaly in the stereochemistry of the addition reactions leading to hybrids P9-
A1─P12-A1 was observed. The extended-chain ester derivatives (P13-A1, P14-A1, and
P15-A1) and the prototype (P3-A1) almost exclusively (> 90 %) exist as E-isomers in
which the platinum moiety and amidine-NMe group adopt a trans configuration with

142
respect to the N(imino)=C double bond, as typically observed in amidine ligands formed
from secondary amines.[339] By contrast, hybrids P9-A1─P12-A1 form a relatively high
amount of the Z-isomer (> 25 %, based on 1-D and 2-D NMR analysis, see Figure
A.47.─ Figure A.49. in APPENDIX A). We attribute this outcome to the increased steric
hindrance produced by the short-chain (n = 1) acyl groups around the nitrile group.
Intramolecular hydrogen bonding between the imino proton and the ester group
(NH•••O=C-O) may also contribute to this effect (see Figure A.47 ─ Figure A.49 in
APPENDIX A) [340].
All newly synthesized hybrids maintain excellent solubility of > 10 mg/mL in relevant
aqueous media. To demonstrate the effect of the pendant ester groups on the
hydrophilicity/lipophilicity balance of the compounds, we studied the partitioning of
selected derivatives between octanol and phosphate-buffered saline (PBS) (expressed as
log[Coctanol/CPBS] = logD, the distribution coefficient for protonable pharmacophores
[334]). The experiment was performed in PBS at pH 7.4 rather than water to suppress
complex aquation and platinum-mediated ester hydrolysis as described in the following
section. This setup also takes into consideration the pH dependence of logD to faithfully
mimic conditions in plasma. For the unmodified, hydrophilic agent, P3-A1, a logD of –
0.98 (±0.19) was determined. By contrast, compound P15-A1, the presumably most
lipophilic derivative (n = 3, valproic ester, L = pn), preferentially partitions into the
octanol phase with a logD of 0.73 (±0.06), which reflects an increase in lipophilicity by
50-fold relative to compound P3-A1. An intermediate logD of –0.31 (±0.06) was
determined for compound P14-A1 (n = 3, L = en, butyric ester). The logD value
generated for this compound has to be interpreted with caution because of minor ester

143
hydrolysis, which was unavoidable under the conditions of the experiment (< 10 %, see
the following section).
5.2.2. Metal-assisted ester hydrolysis.
One of the proposed mechanisms of activation of compounds P9-A1─P15-A1 as
prodrugs involves platinum-promoted ester cleavage. To mimic the chloride ion
concentration differential that exists between serum during circulation and after uptake
into target cells, compounds were incubated at 37 ºC in PBS (~150 mM NaCl, pH 7.4)
and in phosphate buffer (PB, pH 7.4), respectively. The reaction mixtures were analyzed
at appropriate time points by in-line high-performance liquid chromatography–
electrospray mass spectrometry (LC-ESMS). Reaction products were identified as 1+ or
2+ charged molecular ions in mass spectra recorded in positive-ion mode and quantified
by integrating UV-visible HPLC traces at an acridine-specific wavelength (see the
APPENDIX B.4 for a complete set of data).
The time course of ester hydrolysis yielding hydroxyl-modified platinum–acridine and
butyric/valproic acid is summarized for both media in Figure 5.2. Generally, in sets of
analogues characterized by common spacers linking the platinum and ester moieties,
(CH2)n, the valproic esters show significantly slower conversion than the butyric esters,
or no conversion at all (P10-A1─P12-A1 vs. P9-A1, and P13-A1, P14-A1 vs. P15-A1).
In phosphate-buffered solution in the absence of chloride (Figure 5.2A), the most
extensive hydrolysis is observed for butyric ester-based compounds P9-A1 (n = 1) and
P14-A1 (n = 3), with the former producing approximately two-fold higher levels of
cleaved product after 36 h of continuous incubation. Hydrolytic activity is also observed

144
for the valproic ester derivatives P10-A1, P11-A1, and P12-A1 (all n = 1), but at a much
slower rate. Most strikingly, hybrids P13-A1 and P15-A1, which contain the same
secondary acyl moiety but on an extended linker (n = 3), are completely resistant to
cleavage under these conditions. When incubations were performed in buffer
supplemented with physiological chloride, a major reduction in ester hydrolysis of up to
75 % was observed (Figure 5.2B) compared to reactions in chloride-free media,
consistent with the notion that (reversible) aquation of the platinum moiety plays a role in
the cleavage mechanism.

145
Figure 5.2. Cleavage of ester moieties in compounds P9-A1─P15-A1 monitored by
quantitative HPLC for hydrolysis reactions in phosphate buffer, PB, pH 7.4 (panel A),
phosphate-buffered saline, PBS, pH 7.4 (panel B), and in PBS in the presence of hCES-2
(panel C). Plotted data are the mean of three incubations ± standard deviations. Yields of
conversion for compound P14-A1 in panel C represent the sum of chemical (minor) and
enzymatic (major) cleavage, which produced indistinguishable products. Reactions were
performed at 37 ºC.

146
The LC-ESMS profiles of compounds P10-A1─P12-A1 share common features and
support the proposed mechanism of platinum-mediated ester cleavage. We have chosen
compound P9-A1, which was also suitable for a kinetic study by 1H NMR spectroscopy,
for a detailed discussion (for complete sets of LC-ESMS data for all other analogues, see
the Supporting Information). The only hydrolysis product formed in incubations of
compound P9-A1 in PB was identified as a chelate in which the chloro leaving group has
been replaced with the unprotected hydroxyl oxygen of the cleaved butyric ester (see
peak labeled ‘a’ in Figure 5.3A and the corresponding mass spectrum/structure in Figure
5.3D/E, respectively). A minor amount of chemically unchanged P9-A1 and a product
resulting from substitution of chloride by phosphate containing intact ester are also
observed after 36 h of incubation (peaks b and c). The same array of products was
observed when compound P9-A1 was incubated in the presence of sodium chloride in
PBS, but ester cleavage and chelate formation were significantly suppressed based on the
relative abundances of each species (Figure 5.3B). The absence of an opened-chelate
form in this high-chloride environment attests to the inertness of the five-membered
amidine-N/hydroxyl-O chelate. This was also confirmed in incubations with biological
nitrogen and sulfur model nucleophiles, in which this product was completely unreactive
(data not shown). Unlike compounds P10-A1─P12-A1 (n = 1), compound P14-A1 (n =
3) exclusively forms hydrolysis products containing a dangling hydroxyl group,
confirming that a seven- membered, presumably less stable, chelate does not form (see
the APPENDIX B.4).

147
Fig
ure
5.3
. R
ever
se-p
has
e H
PL
C t
race
s fo
r th
e se
par
atio
n o
f re
acti
on p
roduct
s re
sult
ing f
rom
est
er c
leav
age
in c
om
pound P
9-A
1
in P
B (
A),
in P
BS
(B
), a
nd b
y h
CE
S-2
(C
).
Mas
s sp
ectr
a re
cord
ed i
n p
osi
tive-i
on m
ode
for
frac
tions
lab
eled
a–
d a
nd t
he
corr
espondin
g s
tru
cture
s an
d m
/z v
alues
of
the
mole
cula
r io
ns
are
giv
en i
n p
anel
s D
and E
, re
spec
tivel
y.

148
The dramatic effect of chloride ion on the kinetics of ester hydrolysis was confirmed
for compound P9-A1 in arrayed 1H NMR experiments (for an analysis of 1H NMR
spectra, see the Appendix A). Cleavage of the butyric ester follows a (pseudo-) first-
order rate law with rate constants of k = 3.9 ± 10-5 s-1 in PB and k = 1.2 ± 10-5 s-1 in PBS,
which corresponds to half-lives of 5 h and 16 h, respectively (Figure 5.4). Thus, chloride
slows cleavage of the pendant ester in complex P9-A1 by approximately 70 %.
Figure 5.4. Kinetics of ester hydrolysis in compound P9-A1 monitored by 1H NMR
spectroscopy [D2O, pH* 7.0, 2 mM Pt in 10 mM PB (black trace) or PBS (red trace)] at
37 ºC for 24 h. The data sets were fitted to the first-order exponential decay function y =
y0 + A e-x/t, where t-1 is the rate constant, k [h-1], and x is the reaction time, t [h]. Data
points represent means of at least three integrated signal intensities ± standard deviations.
The experiment was performed in duplicate with similar results.

149
On the basis of the above product analysis, a mechanism of platinum-mediated,
intramolecular ester cleavage is proposed (Scheme 5.2). Cleavage is triggered by
(reversible) aquation of the platinum complex. The Lewis-acidic metal assists in the
deprotonation of the aqua to a hydroxo ligand, which undergoes a nucleophilic attack on
the acyl carbon to promote cleavage of the ester linkage. Cleavage results in the loss of
the acyl protecting group as carboxylic acid and in a dangling or chelated
alcohol/alkoxide moiety. High chloride concentrations shift the aquation reaction toward
the chloro-substituted hybrid, which quenches ester cleavage. An intramolecular attack
by platinum-associated hydroxide is also supported by the following observations: (i)
hydrolysis of ester is approximately twice as efficient for complex P11-A1 bearing
ammine (NH3) non-leaving groups than for the en-substituted analogue P10-A1. This
effect is consistent with the lower pKa value of the aqua ligands in Pt–ammine than in Pt–
en complexes [20], which produces higher concentrations of the more reactive hydroxo
ligand. (ii) For derivatives containing the same acyl moiety, ester cleavage is
dramatically reduced for n = 3 vs. n = 1. This effect can be rationalized with the fact that
internal nucleophilic attack by the hydroxo ligand in the former compound requires
formation of a 9-membered, thermodynamically less favorable, macrocyclic intermediate.
5.2.3. Ester Cleavage by human carboxylesterase-2 (hCES-2)
To determine if the compounds, especially those not undergoing chemical ester
cleavage (P13-A1 and P15-A1), are susceptible to deesterification by a
pharmacologically relevant prodrug-converting enzyme, incubations we also performed

150
with recombinant human carboxylesterse-2 (hCES-2). The particular engineered form of
the protein used was sufficiently robust to provide enzymatic activity over long reaction
times (> 30 min), which allows for identification of cleavage products for non-classical,
slowly metabolized substrates [341]. Reactions were performed in a buffer supplemented
with chloride to suppress non-enzymatic hydrolysis and monitored for a relatively short
period of time to minimize the effects of loss of enzyme activity over time and chemical
hydrolysis. Unavoidable contributions from the latter pathway have been subtracted,
where possible, from the data presented in Figure 2C. LC-ESMS analysis of the reaction
mixtures shows minimal or no enzyme-mediated cleavage of ester for compounds P9-
A1─P12-A1, respectively, which contain short spacers (n = 1). By contrast, derivatives
P13-A1─P15-A1(n = 3) show deesterification under these conditions with yields of ~20
% at the 10-h time point. Importantly, the bulky valproic esters in compounds P13-A1
and P15-A1, which are completely resistant to chemical hydrolysis, are efficiently
cleaved by the enzyme. The HPLC trace recorded for compound P9-A1 after 10 h of
incubation with enzyme (Figure 5.3C) shows the same reaction products formed in PB
and PBS, as well as an additional peak (d, ~5 %) not observed for chemical hydrolysis.
Positive-ion mass spectra unequivocally identified this product as the platinum–chloro
complex containing a dangling hydroxyl group (Figure 5.3D,E), consistent with a non-
metal-mediated cleavage mechanism.

151
Figure 5.5. Reverse-phase HPLC traces for the separation of reaction products resulting
from ester cleavage in compound P13-A1 by hCES-2 (A). Mass spectra recorded in
positive-ion mode for fractions labeled a and b and the corresponding structures and m/z
values of the molecular ions are given in panels B and C, respectively.

152
The fact that no opened chelate was detected for chemical hydrolysis of derivatives
with n = 1, but only five-membered N,O chelate, can be taken as evidence that the former
must be an enzyme-specific product. The synthesis of cisplatin derivatives containing
chelated aminoalcoholato ligands requires basic conditions to help deprotonate the
alcohol, while opening of such chelates only occurs under acidic conditions [342]. This is
in agreement with the observation that the two forms (peaks a and d, Figure 5.3C) do not
interconvert under the conditions and on the time scale of the assay. Unlike the butyric
ester derivative P14-A1, which shows dual cleavage reactivity resulting in the
danglinghydroxyl form of the hybrid agent, the valproic ester derivatives P13-A1 and
P15-A1 only undergo enzyme-mediated cleavage to produce the corresponding
deesterified form (shown for compound P13-A1 in Figure 5.5). It can be concluded that
only esters installed on an extended linker are viable substrates for hCES-2 and that only
the chemically robust valproic ester confers true selectivity for enzymatic cleavage.
5.2.4. Cytotoxicity Studies
The cell kill properties of the newly generated compounds P9-A1─P15–A1 were
assessed using a colorimetric cell proliferation assay and compared to that of prototype
P3-A1 (chloride salt). (Note: attempts to synthesize the deesterified, proposed active
hydroxyl forms of P9-A1─P15–A1 in pure form for screening in this assay have been
unsuccessful due to the chemical instability of the platinum–nitrile precursor complexes.)
Two NSCLC adenocarcinoma cell lines were screened for chemosensitivity: A549,
which responds well to treatment with platinum–acridines, and NCI-H1435, a DNA
repair-proficient and highly chemoresistant form of this cancer [343]. The latter cell line

153
has also been shown to express significantly higher levels (3–4-fold) of hCES-2 enzyme
than A549 and other lung adenocarcinomas [344]. Thus, cytotoxicity levels observed in
the two cell lines may also provide hints about potential intracellular prodrug activation.
Both cell lines were dosed with hybrid agents for 72 h at three selected concentrations,
which were based on relative chemosensitivities established for compound P3-A1. A549
was dosed at 5.0, 50, and 500 nM, whereas 20-fold higher concentrations, 0.1, 1.0, and 10
µM, were chosen for NCI-H1435 to account for the relatively higher resistance observed
in this cell line. The dose–response data resulting from the chemosensitivity screen are
presented in Figure 5.6.
As a general trend, the ester-modified derivatives show reduced cytotoxicity levels
compared to the unmodified hybrid, P3-A1, except for compound P14-A1, which shows
a response similar to that of the prototype in both cell lines. Because the butyrate-
protected derivative is efficiently cleaved both chemically and enzymatically on a time
scale relevant to the cell culture assay (see previous section), it is proposed that the
hydroxyl form of this agent is the major contributor to the cell kill. It also suggests that
the n-Pr3-OH residue in deesterified P14-A1 in place of the Et residue in P3-A1 does not at
the DNA level. By contrast, the derivatives containing bulky valproic ester show
compromise the potency of the pharmacophore, possibly pointing to a similar mechanism
greatly reduced activity. Compound P13-A1, for instance, which would generate the
same active hydroxyl form as compound P14-A1, and compound P15-A1 show no cell
kill in A549 at the highest dose and significantly reduced activity in NCI-H1435. This
observation is in agreement with the less efficient intracellular cleavage of the bulkier
valproic esters P13-A1 and P15-A1, which, in their intact form, are less cytotoxic,

154
possibly due to their reduced reactivity with cellular DNA. The same trend is observed
for compounds P10-A1─P12–A1, which proved to be relatively resistant to chemical and
enzymatic hydrolysis. For compounds sharing the same linkers and ester moieties, the
nature of the non-leaving group had only minor or no effects on the activity. Finally,
compound P9-A1, although cleaved very efficiently in chloride-free media (see previous
section), showed activity inferior to the prototype. This outcome was expected because
of the inertness of the 5-membered chelate generated in the process.
Compounds P13-A1 and P15-A1, which are cleaved by hCES-2, show a more
pronounced enhancement in activity relative to compound P3-A1 in NCI-H1435 (high
hCES-2) than in A549 (low hCES-2). The opposite is the case for compound P14-A1.
As a preliminary measure of chemoselectivity, we defined the selectivity index, S: S =
[CVi,NCI-H1435/CV1´,NCI-H1435]/[CVi,A549/CV1´,A549], where CVi and CV1´ are the % viabilities
of the ester-based compounds and compound P3-A1 for their highest doses, respectively.
CV1´ was introduced to normalize for differences in chemosensitivities between the two
cell lines. Assuming that CV1´ is the highest cell kill that can be achieved with any of the
cleaved esters, S values smaller than ‘1’ would indicate a relative advantage of a given
derivative in NCI-H1435, and vice versa. For compounds P13-A1, P14-A1, and P15-A1,
S values of 0.6, 1.4, and 0.5 were calculated, respectively. It can be concluded that the
esters that are not cleaved chemically (P13-A1, P15-A1) but show hCES-2-mediated
cleavage perform relatively better in NCI-H1435. Conversely, compound P14-A1, which
efficiently converts to its active form via an enzyme-independent pathway, appears to
have a relative advantage in A549. These findings may indicate that compounds P13-A1
and P15-A1 act as prodrugs at high levels of hCES-2, which confers sensitivity to NCI-

155
H1435 cells. It should be noted, however, that a direct comparison of cytotoxic
responses between two different cell lines has to be interpreted with caution. Because
cellular uptake and efflux, among other factors, may also contribute to the observed
differences, additional experiments in hCES-2 knockdown or hCES-2 transfected cells11
of the same type are necessary.
Figure 5.6. Results of the chemosensitivity screen of compounds P3-A1─P15-A1 in
A549 (A) and NCI-H1435 (B) cancer cells. Relative cell viabilities are means of two
independent experiments performed in triplicate ± the standard deviation.

156
5.3. Conclusions
Lipophilic, ester-modified derivatives of platinum–acridines have been synthesized and
evaluated with the goal of improving the drug-like properties of these highly cytotoxic
anticancer agents. On the basis of their physicochemical properties, chemical reactivities,
their ability to serve as hCES-2 substrates, and their performance in cell lines,
compounds P13-A1 and P15-A1 can be considered candidates for slow cleavage by
hCES-2. Compound P14-A1 holds promise of providing a dual mode of cleavage, in
which chemical activation may be necessary for applications requiring extended
circulation of precursor in plasma and self-immolative activation in cells lacking hCES-2.
By contrast, the more lipophilic and chemically less reactive analogues P13-A1 and P15-
A1, which should achieve even longer half-lives in circulation, would require activation
by target enzyme expressed in the liver (similar to the anticancer prodrug irinotecan) or in
tumor tissue. Whether the butyric and valproic esters are prone to hydrolysis by other
ubiquitous serum esterases remains to be determined. To this end, incubations of P9-
A1─P15–A1 with fetal bovine serum (FBS, Thermo Scientific HyClone), which is
commonly used as a model for human serum [345], have shown no evidence for this
unwanted reactivity (data not reported).
Another potential advantage of bulky prodrugs of platinum–acridines may be their poor
DNA binding properties. If ester cleavage occurs primarily intracellularly in tumor tissue,
differential DNA recognition by the ester-protected (inactive) and hydroxo (active) forms
of the hybrid agent might confer prodrug selectivity to cancer cells while sparing normal
cells. In addition to an improved ADME profile, lipophilic prodrugs also have the
potential benefit of improving drug safety [332]. To test if this supposition holds for our

157
prodrug design, dose escalation studies were performed with the chemically robust
derivative P15-A1 in Swiss Webster mice. Mice tolerated this analogue without showing
signs of toxicity and weight loss when injected intraperitoneally (i.p.) once a day for five
consecutive days (qd × 5) at a dose of 1.6 mg/kg (see the Supporting Information). For
comparison, compound P3-A1 was significantly more toxic and showed an MTD of 0.1
mg/kg when the same dosing schedule was applied [279].
In conclusion, we have developed a versatile platform for tuning the pharmacological
parameters of potent platinum–acridines and have demonstrated for the first time that a
metal-based pharmacophore is compatible with the classical concept of carboxylesterase-
mediated prodrug activation. This feature may provide a strategy of improving the
ADME and safety of platinum–acridines and other metallopharmacophores.
5.4. Experimental Section
5.4.1. Materials, general procedures, and instrumentation.
All reagents were used as obtained from commercial sources without further
purification unless indicated otherwise. Compound P3-A1[279] (chloride salt) and N-
(acridin-9-yl)-N´-methylethane-1,2-diamine[346] (A1) were prepared according to
published procedures. The platinum-nitrile precursors (P9-P15) were synthesized from
the corresponding nitriles and silver-ion activated diam(m)inedichloroplatinum(II)
complexes. 1H NMR spectra of the target compounds and intermediates were recorded on
Bruker Advance DRX-500 and 300 MHz instruments. Proton-decoupled 13C NMR

158
spectra were recorded on a Bruker DRX-500 instrument operating at 125.8 MHz. 2-D
1H-13C gradient-selected Heteronuclear Multiple Bond Coherence (gsHMBC)
experiments and temperature-dependent spectra were acquired on a Bruker DRX-500
instrument equipped with a TBI probe and a variable-temperature unit. 2-D HMBC
spectra were collected with 2048 pts in t2 (sw = 6510 Hz), 256 pts in t1 (sw = 27670 Hz),
128 scans per t1 increment, and a recycle delay (d1) of 1.5 s. Chemical shifts (δ) are
given in parts per million (ppm) relative to internal standard tetramethylsilane (TMS). 1H
NMR data is reported in the conventional form including chemical shift (δ, ppm),
multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad),
coupling constants (Hz), and signal integrations. 13C NMR data are reported as chemical
shift listings (δ, ppm). The NMR spectra were processed and analyzed using the
MestReNova software package. HPLC-grade solvents were used for all HPLC and mass
spectrometry experiments. LC-ESMS analysis was performed on an Agilent
1100LC/MSD ion trap mass spectrometer equipped with an atmospheric pressure
electrospray ionization source. Eluent nebulization was achieved with a N2 pressure of 50
psi and solvent evaporation was assisted by a flow of N2 drying gas (350 °C). Positive-
ion mass spectra were recorded with a capillary voltage of 2800 V and a mass-to-charge
scan range of 150 to 2200 m/z. To establish the purity of target compounds, samples
were diluted in methanol containing 0.1 % formic acid and separated using a 4.6 mm ×
150 mm reverse-phase Agilent ZORBAX SB-C18 (5 mm) analytical column at 25 °C, by
using the following solvent system: solvent A, optima water, and solvent B,
methanol/0.1 % formic acid, with a flow rate of 0.5 mL/min and a gradient of 95 % A to
5 % A over 30 minutes. HPLC traces were recorded with a monitoring wavelength range

159
of 363-463 nm. Peak integration was done using the LC/MSD Trap Control 4.0 data
analysis software. Analytical purity of greater than 95 % was confirmed this way for all
target compounds prior to analytical and biological experiments (see the Supporting
Information for spectroscopic and analytical data).
Scheme 5.2. The synthesis of precursors 5-7 ─ 5-10.
5.4.2. Synthesis and characterization of target prodrugs
Synthesis of cyanomethyl butyrate (5-7). To the mixture of butyric acid (10.6 g, 0.12
mol), anhydrous potassium carbonate (20.7 g, 0.15 mol) and sodium iodide (22.5 g, 0.15
mol) in 80 ml of acetonitrile was added 2-chloroacetonitrile (7.5 g, 0.1 mol) in 5 ml
acetonitrile at room temperature. When the addition was complete, the mixture was
refluxed for 16 h. The acetonitrile was then removed by rotary evaporation and the
residue was redissolved in 100 ml CH2Cl2, washed with 10% aqueous K2CO3 and brine,
respectively. The organic layers were collected, dried over MgSO4, and concentrated.
This crude material was connected to the vacumm at 45°C overnight to remove unreacted
2-chloroacetonitrile, affording 11.2 g colorless oil (yield: 88%). 1H NMR (300 MHz,

160
CDCl3) δ 4.73 (s), 2.40(t, J = 7.3 Hz, 2H), 1.68(h, J = 7.6 Hz, 3H), 0.98 (t, J = 7.4 Hz,
2H). 13C NMR (75 MHz, Chloroform-d) δ 171.94, 114.58, 48.12, 35.22, 18.09, 13.46.
Synthesis of cyanomethyl 2-propylpentanoate (5-8). Compound 5-8 was prepared
according to the procedure described for 5-7, by using 4-chlorobutanenitrile as the
precursor with the yield of 74%. 1H NMR (300 MHz, CDCl3) δ 4.19 (t, J = 6.0 Hz, 2H),
2.47 (t, J = 7.2 Hz, 2H), 2.31 (t, J = 7.4 Hz, 2H), 2.01 (h, J = 7.0, 5.8 Hz, 2H), 1.66 (h, J
= 7.4 Hz, 2H), 0.96 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 173.27, 118.90,
61.86, 35.89, 24.80, 18.28, 14.21, 13.58.
Synthesis of 3-cyanopropyl butyrate (5-9). Compound 5-9 was prepared according to
the procedure described for 5-8, by using valproic acid as the precursor with the yield of
84%. 1H NMR (300 MHz, CDCl3) δ 4.67 (s, 2H), 2.43 (m, 1H), 1.48 (m, 4H), 1.24 (m,
4H), 0.85 (t, J = 7.3 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 174.84, 114.55, 47.96, 44.69,
34.32, 20.49, 13.84, 0.96.
Synthesis of 3-cyanopropyl 2-propylpentanoate (5-10). Compound 5-10 was
prepared according to the procedure described for 5-9, by using 4-chlorobutanenitrile and
valproic acid as the precursors with the yield of 69%. 1H NMR (300 MHz, CDCl3) δ 4.09
(t, J = 6.0 Hz, 2H), 2.37 (t, J = 6.1 Hz, 2H), 2.30 (m, 1H), 1.92 (m, 2H), 1.40 (m, 4H),
1.18 (m, 4H), 0.81 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.10, 118.77, 61.66, 45.09,
34.50, 24.84, 20.56, 14.13, 13.88.
Synthesis of precursor [PtCl(en)C6H9NO2]Cl (P9). A mixture of 0.652 g (2.00 mmol)
of [PtCl2(en)] and 0.338 g (2.00 mmol) of AgNO3 in 10 mL of anhydrous DMF was
stirred at room temperature in the dark for 16 h. Precipitated AgCl was filtered off
through a Celite pad, 1.78 g (14 mmol) of 5-7 was added to the filtrate, and the

161
suspension was stirred at 55 °C for 3 h in the dark. The solution was evaporated to
dryness in a vacuum at 30 °C yielding a yellow residue, which was dissolved in 20 ml of
dry methanol. Activated carbon was added, and the solution was stirred for 15 min.
Carbon was filtered off, and the solution was concentrated to a final volume of 5 mL,
which is further precipitated in 200 mL anhydrous ethyl ether to afford 297 mg (yield:
32%) off-white microcrystalline precipitate. 1H NMR (300 MHz, D2O) δ 5.98 – 5.58 (m,
2H), 5.25 (s, 2H), 2.86 – 2.36 (m, 5H), 1.64 (h, J = 7.4 Hz, 2H), 0.92 (t, J = 7.6 Hz, 3H).
The 13C NMR was not taken due to decomposition.
Synthesis of precursor [PtCl(en)C10H17NO2]Cl (P10). Precursor P10 was prepared
according to the procedure described for P9, by using [PtCl2(en)] and 5-8 as the
precursors with the yield of 37%. 1H NMR (300 MHz, D2O) δ 5.95 – 5.43 (m, 2H), 5.22
(s, 1H), 2.79 – 2.42 (m, 5H), 1.65 – 1.39 (m, 4H), 1.23 (h, J = 7.5 Hz, 4H), 0.83 (t, J =
7.3 Hz, 6H).
Synthesis of precursor [PtCl(NH3)2C10H17NO2]Cl (P11). Precursor P11 was prepared
according to the procedure described for P9, by using [(NH3)2PtCl2] and 5-8 as the
precursors with the yield of 29%. 1H NMR (300 MHz, MeOH-d4) δ 4.77 (s, 2H), 2.46 -
2.35 (m, 1H), 1.62 – 1.33 (m, 4H), 1.32 – 1.13 (m, 4H), 0.82 (t, J = 7.3 Hz, 6H).
Synthesis of precursor [PtCl(pn)C10H17NO2]Cl (P12). Precursor P12 was prepared
according to the procedure described for P9, by using [PtCl2(Pn)] and 5-8 as the
precursors with the yield of 39%. 1H NMR (300 MHz, MeOH-d4) δ 5.95 – 5.43 (m, 2H),
5.22 (s, 1H), 2.83 – 2.38 (m, 5H), 1.78 –1.66 (m, 2H), 1.65 – 1.39 (m, 4H), 1.23 (h, J =
7.5 Hz, 4H), 0.83 (t, J = 7.3 Hz, 6H).

162
Synthesis of precursor [PtCl(en)C12H21NO2]Cl (P13). Precursor P13 was prepared
according to the procedure described for P9, by using [PtCl2(en)] and 5-10 as the
precursors with the yield of 84%. 1H NMR (300 MHz, MeOH-d4) δ 4.21 (t, J = 6.1 Hz,
2H), 3.08 (t, J = 7.1 Hz, 1H), 2.69 – 2.41 (m, 5H), 2.19 – 2.03 (m, 2H), 1.72 – 1.39 (m,
4H), 1.42 – 1.24 (m, 4H), 0.94 (t, J = 7.2 Hz, 6H).
Synthesis of precursor [PtCl(en)C8H13NO2]Cl (P14). Precursor P14 was prepared
according to the procedure described for P9, by using [PtCl2(en)] and 5-9 as the
precursors with the yield of 86%. 1H NMR (300 MHz, MeOH-d4) δ 4.21 (t, J = 6.0 Hz,
2H), 3.08 (t, J = 7.0 Hz, 2H), 2.82 – 2.48 (m, 2H), 2.42 (t, J = 7.4 Hz, 2H), 2.19 – 1.99
(m, 2H), 1.68 (h, J = 7.4 Hz, 2H), 0.98 (t, J = 7.4 Hz, 3H).
Synthesis of precursor [PtCl(pn)C12H21NO2]Cl (P15). Precursor P15 was prepared
according to the procedure described for P9, by using [PtCl2(pn)] and 5-10 as the
precursors with the yield of 84%. 1H-NMR (300 MHz, MeOH-d4) δ 4.76 – 3.94 (m, 2H),
3.00 – 2.87 (m, 2H), 2.81 – 2.27 (m, 5H), 2.00 (p, J = 6.9 Hz, 2H), 1.73 (p, J = 5.4 Hz,
3H), 1.62 – 1.28 (m, 4H), 1.29 – 1.16 (m, 4H), 0.83 (t, J = 7.2 Hz, 8H).
Synthesis of compound [PtCl(pn)C18H21N4](NO3)2 (P8-A1). Compound P8-A1 was
prepared according to the procedure reported previously[289]. 1H NMR (300 MHz,
DMF-d7) δ 13.89 (s, 1H), 9.89 (s, 1H), 8.77-8.67 (m, 2H), 8.19-7.91 (m, 4H),7.66-7.59
(m, 2H), 6.19 (s, 1H), 5.35-4.98 (m, 4H), 4.51 (t, J = 5.7 Hz, 2H), 4.12 (t, J = 6.5 Hz, 2H),
3.48 – 3.37 (m, 4H), 3.19 (s, 3H), 2.69 (s, 3H), 1.83 (brs, 2H). 13C NMR (75 MHz, DMF-
d7) δ 166.57 , 159.26 , 140.81 , 135.92 , 126.48 , 124.52 , 119.60 , 113.67, 65.87 , 47.62,
43.95, 43.32 , 28.59, 22.33 , 15.50.

163
Synthesis of [PtCl(en)C22H27N4O2](NO3)2 (P9-A1). Precursor complex P9 (240 mg,
0.5 mmol) was dissolved in 10 mL of anhydrous DMF and the solution was cooled to -
20 °C. Acridine–amine A1 (138 mg, 0.55 mmol) was added to the solution, and the
suspension was stirred at 4 °C for 24 h. The reaction mixture was added dropwise into
200 mL of anhydrous ethyl ether, and the resulting yellow slurry was vigorously stirred
for 30 min. The precipitate was recovered by membrane filtration, dried in a vacuum
overnight, and dissolved in 20 mL of methanol containing 1 equivalent of HNO3. After
removal of the solvent by rotary evaporation, the crude product was recrystallized from
hot ethanol, affording 2 as a 3:1 mixture of E and Z isomers. Yield: 238 mg (71 %). 1H
NMR (300 MHz, MeOH-d4) δ 8.61 – 8.51 (m, 2H, Acridine-1/8), 8.13 – 7.75 (m, 4H,
Acridine-3/4/5/6), 7.75 – 7.45 (m, 2H, Acridine-2/7), 6.87 – 6.57 (m, 1H, Pt-
NH=CCH2O- ), 5.50 (s, 1.5H, Pt-NH=CCH2O- ,E-isomer), 5.49 – 5.39 (m, 4H, -
NH2CH2CH2NH2-), 4.76 (s, 0.5H, Pt-NH=CCH2O-, Z-isomer), 4.59 (m, 0.5H, -
NHCH2CH2N(CH3)-,Z-isomer), 4.45 (t, J = 6.3 Hz, 1.5H, -NHCH2CH2N(CH3)-,E-
isomer), 4.02 (t, J = 6.3 Hz, 1.5H, -NHCH2CH2N(CH3)-,E-isomer), 3.56 – 3.39 (m, 0.5H,
-NHCH2CH2N(CH3), Z-isomer), 3.14 (s, 2.25H, -NHCH2CH2N(CH3)-,E-isomer), 2.61 –
2.57(m, 4H, -NH2CH2CH2NH2-), 2.38 – 2.06 (m, 2H, -COCH2CH2CH3), 1.76 – 1.40 (m,
2H, -COCH2CH2CH3), 1.02 – 0.75 (m, 3H, -COCH2CH2CH3). 13C NMR (75 MHz,
MeOH-d4) δ 172.50 (-COCH2CH2CH3, E-isomer), 171.76 (-COCH2CH2CH3, Z-isomer),
165.14 (Pt-NH=CCH2O-, Z-isomer), 163.38 (Pt-NH=CCH2O-, E-isomer), 158.48
(Acridine-9C), 139.87 (Acridine-11C/13C), 135.21(Acridine-3C/6C), 124.99 (Acridine-
1C/8C), 124.02(Acridine-2C/7C), 118.28 (Acridine-4C/5C), 112.63 (Acridine-10C/12C),
63.55 (Pt-NH=CCH2O-, E-isomer), 61.87 (Pt-NH=CCH2O- ,Z-isomer), 35.03 (-

164
COCH2CH2CH3), 17.69 (-COCH2CH2CH3), 12.43(-COCH2CH2CH3). MS (ESI, positive-
ion mode): m/z for C24H35ClN6O2Pt ([M-H]+), 669.22; found, 669.3.
Synthesis of [PtCl(en)C26H35N4O2](NO3)2 (P10-A1). Compound P10-A1 was
prepared according to the procedure described for P9-A1 from precursor P10 with a yield
of 69 %. 1H NMR (300 MHz, MeOH-d4) δ 8.82 – 8.40 (m, 2H, Acridine-1/8), 8.11 – 7.78
(m, 4H,Acridine-3/4/5/6), 7.77 – 7.50 (m, 2H,Acridine-2/7), 5.52 (s, 1.5H, Pt-
NH=CCH2O-, E-isomer), 5.42 – 5.13 (m, 4H, -NH2CH2CH2NH2-), 4.64 – 4.54 (m, 0.5H,
NHCH2CH2N(CH3)-, Z-isomer), 4.48 (t, J = 6.5 Hz, 1.5H, NHCH2CH2N(CH3)-, E-
isomer), 4.03 (t, J = 6.4 Hz, 1.5H, -NHCH2CH2N(CH3)-, E-isomer), 3.15 (s, 2.25H, -
NHCH2CH2N(CH3)-, E-isomer), 2.75 – 2.47 (m, 4H, -NH2CH2CH2CH2NH2-), 2.44 –
2.11 (m, 1H, -COCH(CH2CH2CH3)2), 1.54 – 1.13 (m, 8H, -COCH(CH2CH2CH3)2), 0.97
– 0.71 (m, 6H, -COCH(CH2CH2CH3)2). 13C NMR (75 MHz, MeOH-d4) δ 176.65 (-
CO(CH2CH2CH3)2, E-isomer), 176.01 (-COCH(CH2CH2CH3)2, Z-isomer), 167.30 (Pt-
NH=CCH2O-, Z-isomer), 164.85 (Pt-NH=CCH2O-, E-isomer), 159.90 (Acridine-9C),
141.38 (Acridine-11C/13C), 136.77 (Acridine-3C/6C), 126.49 (Acridine-1C/8C), 125.61
(Acridine-2C/7C), 119.88 (Acridine-4C/5C), 114.12 (Acridine-10C/12C), 66.90 (Pt-
NH=CCH2O- ,E-isomer), 65.27 (Pt-NH=CCH2O- ,Z-isomer), 49.87 (-NH2CH2CH2NH2-),
48.16 (-NHCH2CH2N(CH3)-), 46.35 (-COCH(CH2CH2CH3)2), 35.48 (-
COCH(CH2CH2CH3)2), 21.58 (-COCH(CH2CH2CH3)2), 14.31 (-COCH(CH2CH2CH3)2).
MS (ESI, positive-ion mode): m/z for C28H43ClN6O2Pt ([M-H]+), 724.28; found, 724.4.
Synthesis of [PtCl(NH3)2C26H35N4O2](NO3)2 (P11-A1). Compound P11-A1 was
prepared according to the procedure described for P9-A1 from precursor P11 with a yield
of 74 %. 1H NMR (300 MHz, MeOH-d4) δ 8.56 (d, J = 8.7, 2H, Acridine-1/8), 8.03 (ddd,

165
J = 9.2, 5.8, 1.8 Hz, 2H, Acridine-3/6), 7.95 – 7.79 (m, 2H, Acridine-4/5), 7.75 – 7.51 (m,
2H, Acridine-2/7), 5.56 (s, 1.5H, Pt-NH=CCH2O-,E-isomer), 4.67 – 4.54 (s, 0.5H, -
NHCH2CH2N(CH3)-,Z-isomer), 4.48 (t, J = 6.5 Hz, 1.5H, -NHCH2CH2N(CH3)-, E-
isomer), 4.18 (bs, 3H,NH3), 4.02 (t, J = 6.5 Hz, 1.5H, NHCH2CH2N(CH3)-, E-isomer),
3.93 (bs, 3H, NH3), 3.14 (s, 2.25, H-NHCH2CH2N(CH3)-,E-isomer), 2.36 – 2.16 (m, 1H,
-COCH(CH2CH2CH3)2), 1.56 – 1.03 (m, 8H, -COCH(CH2CH2CH3)2), 0.90 – 0.68 (m,
6H, -COCH(CH2CH2CH3)2). 13C NMR (75 MHz, MeOH-d4) δ 176.67 (-
CO(CH2CH2CH3)2, E-isomer), 176.07 (-COCH(CH2CH2CH3)2, Z-isomer), 164.74 (Pt-
NH=CCH2O-, E-isomer), 159.89 (Acridine-9C), 141.39 (Acridine-11C/13C), 136.76
(Acridine-3C/6C), 126.77 (Acridine-1C/8C), 125.60 (Acridine-2C/7C), 119.88 (Acridine-
4C/5C), 114.20 (Acridine-10C/12C), 65.32 (Pt-NH=CCH2O- ,E-isomer), 46.14 (-
COCH(CH2CH2CH3)2), 35.49 (-COCH(CH2CH2CH3)2) 21.58 (COCH(CH2CH2CH3)2),
14.29 (-COCH(CH2CH2CH3)2). MS (ESI, positive-ion mode): m/z for C26H41ClN6O2Pt
([M-H]+), 699.26; found, 699.3.
Synthesis of [PtCl(pn)C26H35N4O2](NO3)2 (P12-A1). Compound P12-A1 was
prepared according to the procedure described for P9-A1 from precursor P12 with the
yield of 77 %. 1H NMR (300 MHz, MeOH-d4) δ 8.61 (dd, J = 8.8, 4.5 Hz, 2H, Acridine-
1/8), 8.14 – 7.95 (m, 2H, Acridine-3/6), 7.87 (dd, J = 9.0, 4.2 Hz, 2H, Acridine-4/5), 7.75
– 7.53 (m, 2H, Acridine-2/7), 6.83 – 6.73 (m, 1H, Pt-NH=CCH2O-), 5.53 (s, 1.5H, Pt-
NH=CCH2O-, E-isomer), 5.29 – 4.88 (m, 4H, -NH2CH2CH2CH2NH2-), 4.68 – 4.58 (m,
0.5H, -NHCH2CH2N(CH3), Z-isomer), 4.55 (t, J = 6.6 Hz, 1.5H, -NHCH2CH2N(CH3)-,E-
isomer), 4.03 (t, J = 6.4 Hz, 1.5H, -NHCH2CH2N(CH3)-,E-isomer), 3.68 – 3.44 (m, 0.5H,
m, 0.5H, - NHCH2CH2N(CH3)-,Z-isomer), 3.15 (s, 2.25H, -NHCH2CH2N(CH3)-, E-

166
isomer), 2.95 – 2.52 (m, 4H, -NH2CH2CH2CH2NH2-), 2.41 – 2.07 (m, 1H, -
COCH(CH2CH2CH3)2), 1.92 – 1.69 (m, 2H, -NH2CH2CH2CH2NH2-), 1.59 – 0.98 (m, 8H,
-COCH(CH2CH2CH3)2), 0.98 – 0.64 (m, 6H, -COCH(CH2CH2CH3)2). 13C NMR (75
MHz, MeOH-d4) δ 176.68 (-CO(CH2CH2CH3)2, E-isomer), 176.22 (-
COCH(CH2CH2CH3)2, Z-isomer), 164.89 (Pt-NH=CCH2O-, E-isomer), 159.90
(Acridine-9C),, 141.41 (Acridine-11C/13C), 136.76 (Acridine-3C/6C), 126.56 (Acridine-
1C/8C), 125.60 (Acridine-2C/7C), 119.88 (Acridine-4C/5C), 113.48 (Acridine-10C/12C),
46.29 (-COCH(CH2CH2CH3)2), 44.42 (-NH2CH2CH2CH2NH2-), 43.63 (-
NH2CH2CH2CH2NH2-), 35.54 (-COCH(CH2CH2CH3)2) , 29.39 (-NH2CH2CH2CH2NH2-),
21.60 (-COCH(CH2CH2CH3)2), 14.31 (-COCH(CH2CH2CH3)2). MS (ESI, positive-ion
mode): m/z for C29H45ClN6O2Pt ([M-H]+), 739.29; found, 739.4.
Synthesis of [PtCl(en)C28H39N4O2](NO3)2 (P13-A1). Compound P13-A1 was
prepared according to the procedure described for P9-A1 from precursor P13 with the
yield of 83 %. 1H NMR (300 MHz, MeOH-d4) δ 8.46 (dd, J = 8.5, 4.3 Hz, 2H, Acridine-
1/8), 7.91 (m, 2H, Acridine-3/6), 7.77 (dd, J = 9.0, 4.2 Hz, 2H, Acridine-4/5), 7. 54 (m,
2H, Acridine-2/7), 6.08 (s, 1H, Pt-NH=CCH2CH2CH2O-), 5.36 – 5.11 (m, 4H, -
NH2CH2CH2NH2-), 4.33 (t, J = 6.6 Hz, 2H, -NHCH2CH2N(CH3)-), 4.03 (t, J = 6.4 Hz,
2H, -NHCH2CH2N(CH3)-), 3.88 (m, 2H, Pt-NH=CCH2CH2CH2O-), 3.03 (s, 3H, -
NHCH2CH2N(CH3)-), 2.97 (m, 2H, Pt-NH=CCH2CH2CH2O-), 2.55 – 2.37 (m, 4H, -
NH2CH2CH2NH2-), 2.10 – 2.03 (m, 3H, Pt-NH=CCH2CH2CH2O-,-
COCH(CH2CH2CH3)2), 1.59 – 0.98 (m, 8H, -COCH(CH2CH2CH3)2), 0.98 – 0.64 (m, 6H,
-COCH(CH2CH2CH3)2). 13C NMR (75 MHz, MeOH-d4) δ 178.08 (-CO(CH2CH2CH3)2),
170.02 (Pt-NH=CCH2O-), 159.98 (Acridine-9C), 141.37 (Acridine-11C/13C), 136.71

167
(Acridine-3C/6C), 126.52 (Acridine-1C/8C), 125.53 (Acridine-2C/7C), 119.85 (Acridine-
4C/5C), 114.10 (Acridine-10C/12C), 64.83 (Pt-NH=CCH2CH2CH2O-), 46.53 (-
COCH(CH2CH2CH3)2), 35.81 (-COCH(CH2CH2CH3)2), 29.39 (-NH2CH2CH2CH2NH2-),
27.15 (Pt-NH=CCH2CH2CH2O-), 21.67 (-COCH(CH2CH2CH3)2), 14.34 (-
COCH(CH2CH2CH3)2). MS (ESI, positive-ion mode): m/z for C30H47ClN6O2Pt ([M-H]+),
752.31; found, 752.4.
Synthesis of [PtCl(en)C24H31N4O2](NO3)2 (P14-A1). Compound P14-A1 was
prepared according to the procedure described for P9-A1 from precursor P14 with the
yield of 89 %. 1H NMR (300 MHz, MeOH-d4) δ 8.72 – 8.46 (m, 2H, Acridine-1/8), 8.01
(ddd, J = 8.1, 6.9, 1.0 Hz, 2H, Acridine-3/6), 7.92 – 7.80 (m, 2H, Acridine-4/5), 7.64
(ddd, J = 8.3, 6.9, 1.2 Hz, 2H, Acridine-2/7), 6.16 (s, 1H, Pt-NH=CCH2CH2CH2O-), 5.44
– 5.22 (m, 4H,-NH2CH2CH2NH2-), 4.42 (t, J = 6.5 Hz, 2H, -NHCH2CH2N(CH3)-), 4.18 (t,
J = 6.5 Hz, 2H, -NHCH2CH2N(CH3)-), 3.98 (t, J = 6.5 Hz, 2H, Pt-NH=CCH2CH2CH2O-),
3.12 – 3.02 (m, 5H, Pt-NH=CCH2CH2CH2O-,-NHCH2CH2N(CH3)-), 2.74 – 2.46 (m, 4H,
-NH2CH2CH2NH2-), 2.34 – 1.99 (m, 4H, Pt-NH=CCH2CH2CH2O-, -COCH2CH2CH3),
1.58 (d, J = 7.3 Hz, 2H, -COCH2CH2CH3), 0.90 (t, J = 7.4 Hz, 3H, -COCH2CH2CH3).
13C NMR (75 MHz, MeOH-d4) δ 175.33 (-COCH2CH2CH3), 170.11 (Pt-
NH=CCH2CH2CH2O-), 160.04 (Acridine-9C), 141.38 (Acridine-11C/13C), 136.69
(Acridine-3C/6C), 126.43 (Acridine-1C/8C), 125.50 (Acridine-2C/7C), 119.87 (Acridine-
4C/5C), 114.12 (Acridine-10C/12C), 64.7 (Pt-NH=CCH2CH2CH2O-), 36.84 (-
COCH2CH2CH3), 32.07 (Pt-NH=CCH2CH2CH2O-), 27.10 (Pt-NH=CCH2CH2CH2O-),
19.37 (-COCH2CH2CH3), 13.94 (-COCH2CH2CH3). MS (ESI, positive-ion mode): m/z
for C26H39ClN6O2Pt ([M-H]+), 697.25; found, 697.3.

168
Synthesis of [PtCl(pn)C28H39N4O2](NO3)2 (P15-A1). Complex P15-A1 was prepared
according to the procedure described for P9-A1 from precursor P15 with the yield of
76 %. 1H NMR (300 MHz, MeOH-d4) δ 8.54 (d, J = 8.6 Hz, 2H, Acridine-1/8), 8.10 –
7.76 (m, 4H, Acridine-3/4/5/6), 7.59 (t, J = 7.4 Hz, 2H, Acridine-2/7), 4.40 (t, J = 6.1 Hz,
2H, -NHCH2CH2N(CH3)-), 4.17 (t, J = 6.2 Hz, 2H, -NHCH2CH2N(CH3)-), 3.97 (t, J =
6.0 Hz, 2H, Pt-NH=CCH2CH2CH2O-), 3.21 – 2.98 (m, 5H, Pt-NH=CCH2CH2CH2O-,-
NHCH2CH2N(CH3)-), 2.94 – 2.54 (m, 4H, -NH2CH2CH2CH2NH2-), 2.40 – 1.95 (m, 3H,
Pt-NH=CCH2CH2CH2O-,-COCH(CH2CH2CH3)2), 1.79 (m, 2H, -NH2CH2CH2CH2NH2-),
1.56 – 0.99 (m, 8H, -COCH(CH2CH2CH3)2), 0.92 – 0.68 (m, 6H, -
COCH(CH2CH2CH3)2). 13C NMR (75 MHz, MeOH-d4) δ 178.02 (-CO(CH2CH2CH3)2),
169.87 (Pt-NH=CCH2CH2CH2O-), 159.85 (Acridine-9C), 141.33 (Acridine-11C/13C),
136.67 (Acridine-3C/6C), 126.61 (Acridine-1C/8C), 125.50 (Acridine-2C/7C), 119.87
(Acridine-4C/5C), 114.13 (Acridine-10C/12C), 64.86 (Pt-NH=CCH2CH2CH2O-), 46.56
(-COCH(CH2CH2CH3)2), 44.31 (-NH2CH2CH2CH2NH2-), 43.63 (-NH2CH2CH2CH2NH2-
), 35.83 (-COCH(CH2CH2CH3)2), 29.29 (-NH2CH2CH2CH2NH2-), 26.76 (Pt-
NH=CCH2CH2CH2O-), 21.69 (-COCH(CH2CH2CH3)2), 14.37 (-COCH(CH2CH2CH3)2).
MS (ESI, positive-ion mode): m/z for C31H49ClN6O2Pt ([M-H]+), 767.33; found, 767.4.
5.4.3. Time-dependent NMR spectroscopy.
NMR spectra in arrayed experiments were collected at 37 °C on a Bruker 500 DRX
spectrometer equipped with a triple-resonance broadband inverse probe and a variable
temperature unit. Reactions were performed with 2 mM platinum complex dissolved in
either 600 µL of 10 mM phosphate buffer (PB, D2O, pH* = 7.0) or in 600 mL of

169
phosphate-buffered saline (1× PBS, D2O, pH* = 7.0). The 1-D NMR kinetics
experiments were carried out as a standard Bruker arrayed 2-D experiment using a
variable-delay list. Each incremented spectrum was processed using the same procedure,
and suitable signals of the ester moiety were integrated. Data were processed with
MestReNova NMR software. The concentrations of platinum complex at each time point
were deduced from relative peak intensities, averaged over multiple signals to account for
differences in proton relaxation, and fitted to a first-order exponential decay function in
Origin 8.0 (OriginLab, Northampton, MA).
5.4.4. Chemical hydrolysis assay.
The ester hydrolysis study of compounds P9-A1─P15–A1 was carried out by
incubating 1 mM of each test compound in 10 mM phosphate buffer (pH 7.4) or 1× PBS
containing ~150 mM NaCl at 37 °C. At various time points samples were withdrawn
from the reaction mixture and analyzed by in-line LC-ESMS. Chromatographic
separations were performed with a 4.6 mm × 150 mm reverse-phase Agilent ZORBAX
SB-C18 (5 μm) analytical column with the column temperature maintained at 25 °C. The
following solvent system was used: solvent A, optima water, and solvent B,
methanol/0.1 % formic acid, at a flow rate of 0.5 mL/min and a gradient of 95% A to 5%
A over 15 min.

170
5.4.5. Enzymatic cleavage assay.
To study ester cleavage in compounds P9-A1─P15-A1 by recombinant human
carboxylesterase-2 (rhCES-2), 30 μM of each compound was incubated with 400 μg/mL
hCES-2 (BD Biosciences, San Jose, CA, USA) at 37 °C in 1× PBS. Aliquots were
withdrawn at various time points, quenched in an equal volume of methanol, and
centrifuged for 5 min at 10000 g to denature and precipitate protein. The supernatant was
collected and subjected to product separation and analysis using in-line LC-ESMS.
Chromatography was performed on a 4.6 mm × 150 mm reverse-phase Agilent ZORBAX
SB-C18 (5 μm) analytical column with the column temperature maintained at 25 °C. The
following solvent system was used: solvent A, optima water, and solvent B, methanol/
0.1% formic acid, at a flow rate of 0.5 mL/min and a gradient of 95% A to 5% A over 15
min.
5.4.6. Determination of partition coefficients (logD).
To obtain octanol-saturated water and water-saturated octanol, 100 mL of PBS was
stirred with 100 mL of octanol for 24 h, followed by centrifugation for 5 min. The
platinum complexes were dissolved in 1.0 mL of octanol-saturated PBS to a typical
concentration of 0.1 mM and then mixed with 1.0 mL water-saturated octanol.
Triplicates of each experiment were mixed in a multi-tube vortexer incubator for 16 h at
room temperature and then centrifuged for 5 min. The layers were separated carefully,
and the content of platinum–acridines was determined spectrophotometrically at 413 nm
(with λ413 = 10,000 M-1 cm-1 in octanol-saturated PBS and λ413 = 8,600 M-1 cm-1 in PBS-
saturated octanol). The partition coefficients (D) of the samples were then determined as

171
the quotient of the concentration of compound in octanol and the concentration in the
aqueous layer. Reported logD values are the mean ± standard deviations of three
determinations.
5.4.7. Cell based assays
5.4.7.1. Cell culture maintenance
The human non-small cell lung cancer cell lines, NCI-H1435 and A549
(adenocarcinomas) were obtained from the American Type Culture Collection (Rockville,
MD, USA). A549 cells were cultured in HAM’s F12K media (Gibco) supplemented with
10 % fetal bovine serum (FBS), 10 % penstrep (P&S), 10 % L-glutamine, and 1.5 g/L
NaHCO3. NCI-H1435 cells were cultured in serum-free 1:1 DMEM/F12 media (Gibco)
containing 2.436 g/L NaHCO3, 0.02 mg/mL insulin, 0.01 mg/mL transferrin, 25 nM
sodium selenite, 50 nM hydrocortisone, 1 ng/mL epidermal growth factor, 0.01 mM
ethanolamine, 0.01 mM phosphorylethanolamine, 100 pM triiodothyronine, 0.5 % (w/v)
bovine serum albumin (BSA), 10 mM HEPES, 0.5 mM sodium pyruvate, and an extra 2
mM L-glutamine (final concentration 4.5 mM). Cells were incubated at a constant
temperature at 37 °C in a humidified atmosphere containing 5% CO2 and were
subcultured every 2–3 days in order to maintain cells in logarithmic growth, except for
slowly proliferating NCI-H1435, which was subcultured every 7 days.

172
5.3.7.2. Cytotoxicity assay.
The cytotoxicity studies were carried out according to a standard protocol using the
Celltiter 96 aqueous nonradioactive cell proliferation assay kit (Promega, Madison, WI).
Stock solutions (5–10 mM) of P3-A1─P15–A1 were made in DMF and serially diluted
with media prior to incubation with cancer cells. All drugs and controls were tested at
the indicated concentrations in triplicate wells on duplicate plates. Incubations were
carried out for 72 h and cell viabilities were determined by comparing drug-treated wells
with control cells.

173
CHAPTER 6
DEVELOPMENT OF A CHEMICAL TOOLBOX FOR THE DISCOVERY OF
MULTIFUNCTIONAL PLATINUM-ACRIDINE AGENTS

174
6.1. Introduction
Platinum-based cytotoxics continue to be important component in cancer chemotherapy
regimens [3, 213] despite the progress that has been made in the field of molecularly
targeted drugs [347] in recent years. However, there is still considerable interest in safer
and more effective platinum complexes with a wider anticancer spectrum and improved
safety profile. Among them, platinum-acridine (Figure 6.1) hybrids represent a novel
class of non-classical platinum based anticancer agents [348], which, unlike cisplatin, do
not generate cross-links in DNA, but intercalate into DNA base pairs and subsequently
produce monofunctional hybrid adducts [348]. This unique mode of DNA binding has
been demonstrated to cause more severe DNA damage than classic bifunctional platinum
complexes [275, 349], as corroborated by their extraordinary cytotoxicity across a broad
range of cancer cell lines. In particular, the IC50 of P8-A1, one of the most active
analogues identified in non-small cell lung cancer cell lines, was found to be 1.3 nM in
NCI-H460 cells, which is about 1000-fold more potent than cisplatin under the same
conditions [68, 76]. Unfortunately, the narrow therapeutic window and chemoresistance
in some cancers have hampered the development of platinum-acridines as therapeutics
[76, 338, 350].
To overcome these drawbacks, we are pursuing multifunctional conjugates, in which
one or more pharmacophores would be chemically hybridized with the platinum-acridine
agents through a stable or cleavable linker [338, 351]. The rationale behind this approach
is to create molecules capable of interacting with multiple molecular targets that are
clinically relevant in a potentially synergistic way [352]. This concept may lead to more
efficacious treatments for heterogeneous and resistant cancers. Several multifunctional

175
platinum conjugates containing Pt(II) [231, 315, 351] or Pt(IV) analogues [6, 7, 258, 353-
355] have been generated, which show promising activity in vitro and in xenograft
models. In principle, the same goal can also be accomplished with drug cocktails or
formulations containing multiple components [356]. However, the drawbacks of
complicated dose regimens are poor patient compliance [357] and potential severe side
effect as a result of drug-drug interactions [358]. The latter is caused by complicated
pharmacodynamics and pharmacokinetic profiles [359].
Figure 6.1. General structure of selected platinum-acridine hybrid agents.
It should be noted that chemical hybridization would not always lead to an
enhancement in biological action. Mutual interference between the pharmacophores, both
mechanistic and structural, mechanistically or structurally are the major concerns in the
design of multifunctional agents, which may, compared with the parent drugs, lead to

176
reduced efficacy and off-target effects [360]. The goal of this study was to devise an
efficient method that allows us to rapidly screen for viable combinations of
pharmacophores and develop the chemistry by which they can be linked. We have
previously described a breast cancer-targeted platinum-acridine-endoxifen conjugate in
an effort to combine a DNA-damaging platinum agent with an antiestrogen moiety [351].
To build the conjugate, an acridine-endoxifen ligand was assembled prior to attachment
of the platinum center [351]. This may not be the most effective way to assemble a
chemically diverse library of conjugates. A better synthetic strategy would be the direct
attachment of functionalized platinum-acridine analogues to another biologically active
or carrier group. This is not a simple task due to the poor solubility of platinum
complexes in most organic solvents and the chemical reactivity of platinum that may
interfere with the subsequent coupling reaction. In this study, carboxylic acid-modified
platinum-(benz)acridine precursors were synthesized (Figure 6.2), which can be attached
to molecules containing amino groups using coupling reagents routinely exploited in
peptide chemistry [361]. Since amino groups are often found in commercially available
clinically relevant drugs, this approach would not require any additional modifications.
By contrast, Cu(I)-catalyzed click chemistry [310, 362], for instance, has the
disadvantage of depending on starting materials that are either expensive or require to
multi-step synthesis.

177
6.2. Results and discussion
6.2.1. Optimization of coupling reaction
In our previous studies, P3-A7 (Figure 6.1), a carboxylic acid modified platinum-
acridine analogue that shows promising potency in NSCLC cell line NCI-H460 [338],
was selected as a precursor for the development of multifunctional agents. To initiate the
optimizations of coupling conditions, 2-azidoethan-1-amine (Figure 6.3, 6-1) was chosen
as a model amine to produce an amide bond in platinum complexes. To our surprise, no
desired product was detected by high-performance liquid chromatography-electrospray
mass spectrometry (LC-ESMS) under any of the conditions tested. We reasoned that the
poor reactivity of P3-A7 may be caused by the bulky platinum moiety, which could
present a huge steric hindrance as other reactants approached. The possibility of installing
an extended linker in place of –(CH2)2COOH in P3-A7, which might reduce steric
hindrance, was dismissed since this modification has been demonstrateded to
compromise the biological activity of the conjugate [338]. In addition, it is unclear if the
amide linkage in conjugates derived from P3-A7 can be cleaved by protease
intracellularly to release the platinum-acridine agents from the conjugate. For the
conjugate design in this study, we selected platinum precursors that allow ester-based
conjugation, and the point of attachment of the linker was moved from the acridine
nitrogen (acidine fragment, see Figure 1.15 in Chapter 1) in P3-A7 to the alkylene chain
attached to the amidine carbon (nitrile fragment, see Figure 1.15 in Chapter 1).

178
Figure 6.2. (A) Unsuccessful coupling reactions using P3-A7 as the precursor. (B)
Structures of ester based platinum-(benz)acridine analogues.
Recently, we have reported a butyric acid-masked lipophilic prodrug (P14-A1) of
platinum-acridines. P14-A1 is activated by a platinum-mediated, self-immolative ester
cleavage mechanism [350], which generates a hydroxyl-modified, highly cytotoxic
platinum-acridine. In continuation of this work, butyric acid was replaced with succinic
acid as an alternative acyl component, leading to the new ester analogues P16-A1 and
P16-B1, which resemble the structure of P14-A1 but contain an extended carboxylic acid

179
for further chemical ligation (Figure 6.2). Benz(c)acridine was also introduced as the
intercalating moiety that has been shown to be a more selective DNA binder and produce
less severe toxicity in animals than acridine itself [363].
The synthesis of carboxylic acid precursors P16-A1 and P16-B1 was accomplished as
shown in Scheme 6.1. Since the nitrile-modified platinum intermediate is highly
hydroscopic and readily decomposes during work-up, we developed a simple three-step,
one-pot synthetic method starting from [Pt(pn)Cl2] (pn = 1,3-diaminopropane). In the
first step, this platinum precursor was reacted overnight with AgNO3 in DMF at room
temperature, which activated the platinum complexes for the subsequent ligand
substitution reaction. The resulting white precipitate in the reaction mixture (AgCl) was
filtered off, and the filtrate was incubated with the ester-modified nitrile precursor at
60 °C for 4 hours. Without any purification at this stage, the reaction mixture, containing
the key intermediate carboxylic acid-modified “platinum-nitrile” complex, was then
cooled to 4 °C and immediately mixed with N1-(acridin-9-yl)-N2-methylethane-1,2-
diamine or N1-(benzo[c]acridin-7-yl)-N2-methylethane-1,2-diamine, which was kept at
4 °C for another 48 hours to allow for formation of amidine linkage as a result of an
efficient amine addition to the platinum-activated nitrile (Scheme 6.1). Finally, the target
compounds were purified by recrystallization from hot ethanol and recovered an overall
yield of 40 - 50% after three steps.
We next explored the coupling conditions for the assembly of the amide based
multifunctional platinum-acridine agents. As a preliminary experiment, P16-A1 was
reacted with 2-azidoethan-1-amine, which is a simple, primary amine. We investigated
various solution-phase conditions with a number of coupling reagents (for the structures

180
of coupling reagents used in this study, see APPENDIX I), and the results are listed in
Table 6.1. (Note: solid-phase synthesis [364] was not attempted since the chemistry
required for removal of the coupled conjugate would be incompatible with the platinum-
acridine moiety.) The carbodiimide-based reagents [361, 365], DCC and EDC, were
unable to mediate the formation of the desired amide bond. By contrast, the target
conjugate was produced with a high conversion yield (> 90%) when uronium-based
HBTU and phosphonium-based PyBOP were used. The very different outcomes can be
attributed to the rapid coupling kinetics of the latter reagents [365]. COMU, another
uronium-based coupling reagent, although a better reagent than HBTU in peptide
synthesis [366], did not perform as effectively as anticipated in the presence of platinum.
Good conversion was also achieved by using excess (10 equivalents) CDI, which
required the removal of unreacted CDI before reaction with the amine. This additional
step may become a major disadvantage when using this method for high-throughput
library assembly.

181
Sch
eme
6.1
. S
ynth
esis
of
targ
et c
om
po
un
ds.
i)
AgN
O3,
DM
F,
rt,
ii)
HO
OC
(CH
2) 2
C(O
)O(C
H2) 3
CN
, D
MF
, 60 ⁰
C,
4 h
, ii
i) N
1-
(acr
idin
-9-y
l)-N
2-m
eth
yle
than
e-1,2
-dia
min
e, D
MF
, 4 ⁰
C, iv
) N
1-(
ben
zo[c
]acr
idin
-7-y
l)-N
2-m
eth
yle
than
e-1,2
-dia
min
e, D
MF
, 4 ⁰
C.

182
To test the influence of alkyl substituents attached to the amines on the coupling
efficiency, the secondary amino group in endoxifen was studied in conjugation reaction
with P16-A1. HBTU and PyBOP were found to mediate amide coupling with high yields
similar to the primary amine (Table 6.1). Since uronium-based reagents may react with
amines, yielding undesired guanidinium byproducts [361, 367], PyBOP was selected as
the coupling reagent for the library assembly. The optimized conditions for amide
formation with this reagent use 1 equivalent of carboxylic acid, 1.1 equivalents of amine,
1 equivalent of DIPEA and 1.5 equivalents of PyBOP. High conversion yields can be
achieved when reactions were performed at room temperature for 16 hours in DMF at a
Pt concentration of 5 mM or higher.
6.2.2 Assembly of a fragment library for multifunctional platinum-acridine
conjugates
To further evaluate the potential of the PyBOP coupling reaction for producing a
structurally diverse library of platinum-acridine conjugates for biological screening, we
designed a fragment library consisting of 2 carboxylic acid-modified platinum and 10
biological relevant amines, and performed 20 conjugation reactions in a parallel manner.
Specifically, we selected P16-A1 and P16-B1 as the carboxylic acid fragments because
of their potent but different biological behaviors found in cancer cells [363]. P16-A1
contains the prototypical platinum-acridine derivative, which preferentially binds to
duplex DNA and effectively kill NSCLC cells at low-nanomolar concentrations. The

183
benz[c]acridinepharmacophore in P16-B1, although generally not as potent as the
acridine-based agents in terms of cytotoxicity, have shown extraordinary sensitivity in
several cancer cell lines harboring p53 mutation [363], a defect known to cause platinum
drug resistance [363].
To demonstrate the scope and utility of the amide/ester-based conjugation chemistry, a
set of 10 amines with diverse structural and biological features were included in the
fragments library. They include (1) the azide linker (6-1), which allows further chemical
ligation [310], (2) fatty amines and phospholipids producing lipophilic conjugates that
can potentially self-assemble into micelles [368] (6-2) or liposomes [369] (6-3) in
aqueous solution, (3) the arginine analogue (6-4) that may enhance cell uptake due to the
interactions of the guanidinium group with phospholipids in the cell membrane [370], (4)
clinically approved drugs such as Rucaparib [371] (6-5, PARP inhibitor), endoxifen [351]
(6-6, ER antagonist), and a gefitinib analogue [372] (6-7, EGFR inhibitor), (5) the
mitochondria-targeted phosphonium moieties [373] (6-8 and 6-9), and (6) the dendrimer
PAMAMAG3.5 [374].

184
Tab
le 6
.1.
Condit
ions
for
Co
upli
ng R
eact
ions
wit
h M
odel
Pri
mar
y (
Top)
and S
econdar
y (
Bott
om
) A
min
es
Entr
ya
Coupli
ng
reag
ents
Bas
e M
ola
r ra
tio o
f re
acta
nts
(Pt:
Am
ine:
coupli
ng r
eagen
t: b
ase)
Pt
conce
ntr
atio
n i
n
reac
tion (
mM
)
Conver
sion
b (
%)
1
DC
C
--
1:1
.2:1
.2:0
5
0
2
DC
C
DIP
EA
1:1
.2:1
.2:1
5
0
3
DC
C+
NH
S
DIP
EA
1:1
.2:1
.2(D
CC
):1.2
(NH
S):
1
5
0
4
ED
C
--
1:1
.2:1
.2:0
5
0
5
ED
C
DIP
EA
1:1
.2:1
.2:1
5
0
6
ED
C+
NH
S
DIP
EA
1:1
.2:1
.2(E
DC
):1.2
(NH
S):
1
5
0
7
CD
I --
1:1
.2:1
.2:0
5
76
± 2
8c
CD
I --
1:1
0:1
.2:0
5
98
± 2
9
HB
TU
D
IPE
A
1:1
.1:1
.1:1
5
94
± 3
10
PyB
OP
D
IPE
A
1:1
.1:1
.1:1
5
99 ±
0.4
11
CO
MU
D
IPE
A
1:1
.1:1
.1:1
5
83
± 1
12
HB
TU
D
IPE
A
1:1
.1:1
.1:1
0.5
22
± 3
13
PyB
OP
D
IPE
A
1:1
.1:1
.1:1
0.5
17
± 2
14
CO
MU
D
IPE
A
1:1
.1:1
.1:1
0.5
18
± 1

185
Tab
le 6
.1 (
conti
nued
)
En
try
a
Coupli
ng
reag
ents
Bas
e M
ola
r ra
tio o
f re
acta
nts
(Pt:
Am
ine:
coupli
ng r
eagen
t: b
ase)
Pt
conce
ntr
atio
n i
n
reac
tion (
mM
)
Conver
sion
b (
%)
15
c C
DI
--
1:1
0:1
.2:0
5
67 ±
3
1
6
HB
TU
D
IPE
A
1:1
.1:1
.1:1
5
92 ±
1
17
P
yB
OP
D
IPE
A
1:1
.1:1
.1:1
5
90 ±
2
18
C
OM
U
DIP
EA
1:1
.1:1
.1:1
5
73 ±
1
19
H
BT
U
DIP
EA
1:1
.1:1
.5:1
5
88 ±
1
20
PyB
OP
D
IPE
A
1:1
.1:1
.5:1
5
99 ±
0.7
aE
ach r
eact
ion w
as p
erfo
rmed
in t
ripli
cate
and c
har
acte
rize
d b
y L
C-E
SM
S w
ithout
puri
fica
tion.
bT
he
conver
sion y
ield
was
calc
ula
ted
bas
ed o
n t
he
inte
gra
tions
of
HP
LC
tra
ces
at a
n a
crid
ine-
spec
ific
wav
elen
gth
. c C
DI
was
pre
-in
cubat
ed w
ith P
t
com
ple
xes
for
5 h
, th
en t
he
reac
tion m
ixtu
re w
as p
reci
pit
ated
in d
ieth
yl
ether
to r
emove
unre
acte
d C
DI
bef
ore
mix
ing w
ith
amin
e.

186
Fig
ure
6.3
. (A
) S
chem
atic
rep
rese
nta
tion o
f th
e co
upli
ng r
eact
ion.
(B)
Str
uct
ure
s of
carb
ox
yli
c ac
id p
recu
rsors
. (C
) S
truct
ure
s of
amin
e pre
curs
ors
.

187
To perform the parallel synthesis of 20 platinum-acridine conjugates, stock solutions
(25 mM) of each reactant were prepared in DMF. A total of 20 microscale reactions were
assembled in 1 mL Eppendorf tubes with a final Pt concentration of 5 mM and incubated
for 16 hours at room temperature. Species generated in each reaction were characterized
by automated LC-ESMS based on their molecular-ion peaks and fragments. The reactions
resulted in clean conversion to the desired conjugates without the formation of major side
products (for conversion yields and MS data, see Table 6.2). A representative HPLC
chromatogram and MS spectra for the characterization of conjugate 6-16 are shown in
Figure 6.4. Two species were found in the HPLC trace, which were identified as the
desired conjugate (peak 2) and the ethyl ester derivative of P16-A1 (peak 1), respectively.
The latter product was produced due to the reaction of unreacted P16-A1 with ethanol,
the solvent chosen for the preparation of LC-ESMS samples, in the presence of excess
coupling reagent in the mixture. To confirm the connectivity produced in the conjugates,
6-16 was synthesized as a representative example on a large scale (100-200 mg) using the
same method, purified by recrystallization and characterized by 2-D NMR spectroscopy
(see Figure A.60-Figure A.62 in APPENDIX A).

188
Figure 6.4. (A) Reverse-phase HPLC trace for the reaction of P16-A1 and 6-7. (B)
ESMS spectrum of 6-25 and target conjugate 6-16 recorded in positive-ion mode.
Characteristic molecular and fragment ions of 6-16 are [M]+, m/z 1113.6; [M+H]2+, m/z
557.3, [M+2H]3+ , m/z 371.6.
The amount of platinum-acridine precursors that were converted to the desired products
in each reaction can be calculated based on their integrations in the HPLC traces at a
chromophore-specific wavelength. In most cases, baseline separations in HPLC traces
were found under identical analytical conditions, which facilitates the determination of

189
conversion yields. Of the 20 reactions, 14 went to completion (> 95%) (see APPENDIX
B6). The coupling efficiency greatly depends on the steric hindrance of the amine
components. This was evidenced by the reactions of carboxylic acid precursors with the
bulky, positively charged triphenylphosphonium (TPP) analogues 6-8 and 6-9. Notably,
while 6-9 containing an extended 11-aminoundecanoic acid linker gave a conversion
yield >98%, derivative 6-8 produced only 70-80% conjugate. Unreacted carboxylic acid
precursors was recovered in the reactions with PAMAM dendrimer G3.5 (6-10), the
macromolecular amine that is much bulkier than any other small molecules. Furthermore,
two reactions with 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE, 6-3), a
flexible and primary amine, resulted in reaction mixtures lacking any detectable product,
which was attributed to the limited solubility of the lipid in DMF at the concentration
required for the coupling reactions.
Purification of platinum-acridines is generally challenging due to their reactivity in
aqueous media and poor solubility in most organic solvents. Pure derivatives can be
isolated by repeated recrystallization from ethanol or methanol, which is not suitable for
generating libraries. A typical purification method employed in conventional chemical
libraries is preparative HPLC [375, 376], however, when applied to platinum-acridine
analogues, partial decomposition of the platinum complexes is observed. Since the
optimized conditions we have developed generally led to coupling reactions that virtually
went to completion, to accelerate the drug screening process, only reactions with
conversion yields higher than 95% were selected in the subsequent studies. Only these
products were finally confirmed by HR-ESMS (Table 6.2).

190
In summary, we have developed an efficient coupling reaction to produce amide
linkages in the presence of platinum with high conversion yields, which allowed facile
and rapid generation of a library of multifunctional platinum-acridine agents. This
approach can be readily translated to a 96-well plate-based high-throughput screening
platform (Figure 6.5). All the reactions would be carried out in 96 well plates,
subsequently serial diluted with physiological relevant buffers and then, along with all
the precursors included as controls, directly pre-screened in cell-based assays without
purification. Finally, conjugates indicating promising activity in pre-screen assays will be
synthesized and purified, and inhibition concentrations (IC50 values) determined using the
same assay.

191
Fig
ure
6.5
. S
chem
atic
ill
ust
rati
on o
f a
pla
te-b
ased
hig
h t
hro
ughput
dru
g d
isco
ver
y s
cree
n.

192
Tab
le 6
.2. T
he
Conver
sion Y
ield
and H
R-E
SM
S R
esult
s fo
r P
lati
num
-Acr
idin
e B
ased
Conju
gat
es
Com
pound
Code
Conju
gat
es
Conver
sion
yie
ld (
%)a
Cal
cula
ted
[M]+
Obse
rved
[M]+
To
lera
nce
(ppm
)
6-1
1
99 ±
1
808.2
778
808.2
786
0.9
89
6-1
2
97 ±
1
963.4
955
963.4
954
-0.1
04
6-1
3
100 ±
2
910.3
458
910.3
466
0.8
79
6-1
4
96 ±
3
1045.3
619
1045.3
645
2.4
87
6-1
5
97 ±
2
1095.4
227
1095.4
242
1.3
69

193
6-1
6
98 ±
2
1111.3
604
1111.3
615
0.9
89
6-1
7
100 ±
0.2
612.7
688
612.7
696
1.3
06
6-1
8
98 ±
2
858.2
934
858.2
940
0.6
99
6-1
9
97 ±
2
1013.5
111
1013.5
125
1.3
81
6-2
0
99 ±
1
960.3
615
960.3
610
-0.5
21

194
6-2
1
99 ±
1
1095.3
776
1095.3
784
0.7
30
6-2
2
97 ±
1
1145.4
383
1145.4
391
0.6
98
6-2
3
96 ±
3
1162.3
760
1162.3
744
-1.3
76
6-2
4
100 ±
0.5
637.7
766
(2+
)
637.7
769
(2+
) 0.4
70
aE
ach r
eact
ion w
as p
erfo
rmed
in t
ripli
cate
. T
he
conver
sion y
ield
was
cal
cula
ted b
ased
on t
he
inte
gra
tions
of
HP
LC
tra
ce a
t an
acri
din
e-sp
ecif
ic w
avel
ength
. R
eact
ions
wit
h c
onver
sion y
ield
low
er t
han
90%
are
not
report
ed.

195
6.2.3. Aqueous conditions for the synthesis platinum-acridine based conjugates
We are also pursuing to develop the coupling conditions for amide formation in
aqueous media. This would become particularly useful when the molecules, which are
only soluble in water, such as proteins, peptides, and polysaccharides, are chosen to be
conjugated as sensitizers or targeting moieties. Toward this goal, 8-aminooctanoic acid,
an amino acid that is insoluble in most organic solvents but fairly soluble in water, was
selected as a model compound for conjugation with P16-B1 through an amide bond. This
experiment will help establish coupling conditions for tethering short peptides, such as
tumor targeted RGD peptide [377]. It should be noted that the protection and deprotection
steps, which are commonly employed in peptide synthesis, may have to be avoided, as
these harsh conditions may lead to decomposition of the platinum-acridine complexes.
Another application of the extended 8-aminooctanoic acid chain would be as a linker
module for conjugation of sterically hindered molecules, such as proteins and polymers,
which, did not readily react with P16-A1 and P16-B1 (see section 6.2.2).
Scheme 6.2. “Two-step” synthesis of platinum-acridine conjugates in aqueous media.

196
Figure 6.6. (A) Reverse-phase HPLC trace of the reaction mixture for preparation of 6-
27. (B) ESMS spectrum of P16-B1 and target conjugate 6-27 recorded in positive-ion
mode. (c) Structures and characteristic molecular and fragment ions of P16-B1 and 6-27.

197
To avoid cross-reactions of unprotected amino acids in the presence of coupling
reagents, we proposed a simple two-step synthesis method (Scheme 6.2). The crucial
steps in this approach involved the activation of carboxylic acid precursor to an N-
hydroxysuccinimide (NHS) based ester, an intermediate that is known to be stable and
reactive in aqueous solution, followed by complete removal of coupling reagent before
adding the amino acid. Inspired by our previous experience that uronium-based coupling
reagents, such as HBTU, are highly efficient at producing amide bond in platinum-
acridines, TSTU [378], the NHS analogue that works in both organic and aqueous
solution, was selected in this study. To generate the active NHS ester intermediate, 1
equivalent of P16-B1, was incubated with 1.5 equivalents of TSTU in DMF for 16 h.
The product was then precipitated with diethyl ether, which subsequently underwent
centrifugation for two times. Since the platinum complexe is not soluble in diethyl ether,
but TSTU remains dissolved, unreacted TSTU can be separated from the desired product.
The orange precipitate, which is the NHS modified P16-B1 (6-26), was then reacted with
1 equivalent of 8-aminooctanoic acid in PBS (pH = 7.4). The high chloride concentration
in this buffer helps suppress aquation of the platinum complex. This step was performed
at room temperature for another 16 h, and the reaction mixture was characterized by LC-
ESMS as described previously. About 74% of P16-B1 was converted to the desired
product (Figure 6.6A, peak 2) after two steps without the detection of major side products.
Specifically, the formation of possible side products due to cross-reactivities of
unprotected amino acid was not observed. The same procedures have also been used to
conjugate 8-aminooctanoic acids with P16-A1 with similar results (data not shown). In
conclusion, we have demonstrated a two-step conjugation approach for platinum-acridine

198
derivatives that can be performed in aqueous solution with conversion yields >70%.
Future research will address the purification of the reaction mixture, which can
potentially accomplished by recrystallization or size-exclusion chromatography.
6.2.4. “One-tube” assembly of multifunctional platinum-acridine conjugates
Fragment-based drug discovery represents a novel strategy to identify promising drug
leads [378, 379]. However, this approach has not been used for the discovery of metal-
based drugs. In another set of experiments, we explored the feasibility of producing
multifunctional platinum-acridine conjugates by assembling three components without
laborious purifications. Such a method, in combination with automated synthesis, would
lead to a combinatory assay that rapidly generates a large number of platinum-acridine
analogues with diverse structural features and different sensitivities in cancer cells
derived from various types of tumors [21, 380, 381]. As a preliminary experiment, we
attempted to prepare a known platinum-acridine conjugate (6-14) by a “one-tube”
synthesis (Figure 6.7). [Pt(pn)Cl2] was activated with 0.95 equivalents of AgNO3, and the
AgCl was removed by syringe filtration. The filtrate was then combined with 8
equivalents of carboxylic acid modified nitrile precursor 4-(3-cyanopropoxy)-4-
oxobutanoic acid and the mixture reacted for another 4 h at 65 ºC. The mixture was then
cooled to 4 ºC, combined with 1 equivalent of acridine-amine (A1), and incubated at that
temperature for another 48 h. Unreacted nitrile and acridine precursors were removed
using ether precipitation as described in section 6.2.3. These procedures afforded P16-A1

199
with 85 % conversion yield, as confirmed by LC-ESMS (Figure 6.8A). The major side
product detected by LC-ESMS was assigned to complex 6-28, which formed due to the
reaction of unreacted platinum precursor with A1. To generate the target conjugate (6-14),
a solution containing 0.95 equivalents of rucaparib, 1 equivalent of Hünig's base and 1.2
equivalent of PyBOP in DMF was mixed with the solution containing P16-A1 and
allowed to react at room temperature for 16 h. The mixture was then examined
characterized by LC-ESMS, showing that the desired conjugate had formed with a yield
of 72% (Figure 6.8B). The molecular ion and fragments (Figure 6.8C, peak 3, 20.4 min)
of the conjugate were consistent with the data obtained for this compound in section 6.2.2.
In conclusion, we have demonstrated, using multifunctional platinum-acridine agent as an
example, that the synthesis of inorganic complexes can be accomplished through the
fragment-based, one-tube approach. However, the success of this method significantly
relies on the selection of reactions producing high yields at each step. Poor conversion
yield in only one step would lead to a complicated mixture of products.

200
Figure 6.7. Schematic illustration of “one-tube” assembly of multifunctional platinum-
acridine conjugates.

201
Figure 6.8. (A) Reverse-phase HPLC trace of the reaction mixture before the coupling
reaction (Reaction mixture IV). (B) Reverse-phase HPLC trace of the final reaction
mixture (Reaction mixture V). (C) ESMS spectrum of species in the reaction mixture
recorded in positive-ion mode. Characteristic molecular and fragment ions of 6-28 are
[M]+, m/z 555.4, characteristic molecular and fragment ions of P16-A1 are [M]+, m/z
741.5; [M+H]2+, m/z 371.3; characteristic molecular and fragment ions of 6-14 are [M]+,
m/z 1046.6, [M+H]2+, m/z 523.9.

202
6.2.5. Platinum mediated ester hydrolysis
In chapter 5, we demonstrated that at low chloride concentration, ester-based platinum-
acridine prodrugs undergo hydrolysis. Based on our results for P14-A1, the butyrate
analogue, we anticipated that the multifunctional platinum-acridine conjugates would
behave similarly. To evaluate their ester cleavage, seven representative conjugates were
selected and incubated in phosphate buffer (PB, pH 7.4) and phosphate buffered saline
(PBS, pH 7.4, ≈ 150 mM NaCl), which simulate the chloride ion concentration in the
cytosol of cancer cells and in serum during circulation, respectively. The selected
conjugates were generated by the microscale reactions as described before with
conversion yields of > 95%, and the byproducts were removed by precipitating in ethyl
ether followed by size-exclusion chromatography (Sephedex LH-20, 10 × 1 cm column,
eluted with methanol-acetonitrile of 1:1, v/v). All incubations were performed at 37 °C
and analyzed at various time points by LC-ESMS. A representative LC-ESMS profile is
shown for conjugate 6-11 was shown in Figure 6.9. Cleavage of the amide bond was not
detected, suggesting the ester hydrolysis predominated the drug release.The hydrolyzed
products were identified as a mixture of hydroxyl-containing platinum-acridine that had
undergone chloride-exchange reactions with water and formic acid during LC-MS
analysis. The ligand-exchange reactions were significantly suppressed in chloride-
containing PBS buffers. Other species found in the incubation mixture included the intact
conjugates and their ligand-exchange products, particularly in the absence of chloride.
Only minor decomposition was observed in PB after incubation for 48 hours for
conjugates showing slow hydrolytic rates (6-14 and 6-16).

203
Fig
ure
6.9
. (A
) R
ever
se-p
has
e H
PL
C t
race
of
the
hydro
lyti
c pro
duct
s of
6-1
1 in
PB
S (
top)
and i
n P
B (
bott
om
). (
B)
ES
MS
spec
trum
of
hyd
roly
tic
pro
duct
s re
cord
ed i
n p
osi
tive-i
on m
ode.
(C
) S
truct
ure
s an
d c
har
acte
rist
ic m
ole
cula
r io
ns
for
hydro
lysi
s
pro
duct
.

204
The time course of ester hydrolysis in PB and PBS is summarized in Figure 6.10. In
agreement with previous results (see chapter 5), with the exception of 6-11 and 6-22 that
underwent cleavage extremely rapidly, a 2-3 fold increase in ester hydrolysis occurred in
other conjugates when incubated in phosphate buffer without chloride compared with the
reactions performed in buffer supplemented with chloride. This suggests that the selected
conjugates underwent metal-assisted, chloride concentration dependent ester hydrolysis
(Figure 6.11). To our surprise, although the conjugates share common succinyl ester
linkage, -C(CH2)3OC(O)(CH2)2C(O)NH-, their hydrolysis rates vary significantly,
indicating that the amine components have an effect on the stability of the ester. In
general, rapid ester cleavage was found in the conjugates with flexible and lipophilic
primary amines (6-11, 6-12, 6-17, and 6-18) yielding more than 60% hydrolytic products
after incubation for 24 hours in PB, while only approximately 40% of P14-A1 was
cleaved in the same buffer after 36 hours [350]. The type of chromophore does not seem
to affect hydrolysis (6-11 vs 6-22, showing similar hydrolytic rates). A dramatic
reduction in ester cleavage of up to 40% was found in the conjugates with fused aromatic
heterocyclic ring systems connected through secondary amides (6-14, 6-16 and 6-22)
after incubation in PB for 48 hours. An explanation for this effect can not be given.

205
Figure 6.10. Cleavage if ester moieties in selected conjugates in phosphate buffer, PB,
pH 7.4 (A) and phosphate buffered saline, PBS, pH 7.4 (B). Each incubation was
performed in triplicate.

206
Fig
ure
6.1
1. P
ropose
d m
echan
ism
of
chem
ical
hydro
lysi
s.

207
6.3. Conclusions
To overcome the drawbacks of current platinum based chemotherapy, thousands of
platinum complexes with complicated molecular architectures have been produced for the
biological screening. Many endeavors in these platinum based anticancer agents were
made to confer them multifunctionality and tumor selectivity for better anticancer
effacacy. However, the rapid generation of multifunctional platinum complexes for drug
screening was usually impeded by many synthetic difficulties associated with
platinum(II). Due to the presence of platinum (II), even those seemly straightforward
reactions that afford quantitative yields might result in extremely complicated products.
To remove these synthetic bottlenecks for platinum complexes, our solution is to develop
highly reliable and effective platinum(II) compatible reactions as a highly generalized
approach to sequentially assemble common modular fragments. In this chapter, we
developed a highly efficient coupling reaction for the formation amide linkage in
platinum complexes. The synthetic strategy enables the construction of a library of
multifunctional platinum-acridine conjugates in a fast and economical manner, which
would be helpful for the screening of synergistic pharmacophores particularly for
platinum-acridine derivatives in various cancers. The scope and diversity of this approach
can be further expanded by the variations in modular fragments, and the same concept
can even apply to the discovery of other metal-based drugs such as ruthenium and radium,
which were known to activate nitrile for addition reactions. Furthermore, the ultimate
goal of this technology is to assemble an oncologist toolbox composed of various
multifunctional platinum-acridine conjugates that would be selectively sensitive to a
certain type of cancer cells. Since tumor in each patient is unique, this toolbox has the
potential to evolve to a personalized therapeutic option for individual patient in future.

208
6.4. Experimental Section
6.4.1. Materials, general procedures, and instrumentation.
All reagents were used as obtained from commercial sources without further
purification unless indicated otherwise. 1H NMR spectra of the target compounds and
intermediates were recorded on Bruker Advance DRX-500 and 300 MHz instruments.
Proton-decoupled 13C NMR spectra were recorded on a Bruker DRX-500 instrument
operating at 125.8 MHz. 2-D 1H-13C gradient-selected Heteronuclear Multiple Bond
Coherence (gsHMBC) experiments and temperature-dependent spectra were acquired on
a Bruker DRX-500 instrument equipped with a TBI probe and a variable-temperature unit.
2-D HMBC spectra were collected with 2048 pts in t2(sw = 6510 Hz), 256 pts in t1 (sw =
27670 Hz), 128 scans per t1 increment, and a recycle delay (d1) of 1.5 s. Chemical shifts
(δ) are given in parts per million (ppm) relative to internal standard tetramethylsilane
(TMS). 1H NMR data is reported in the conventional form including chemical shift (δ,
ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br =
broad), coupling constants (Hz), and signal integrations. 13C NMR data are reported as
chemical shift listings (δ, ppm). The NMR spectra were processed and analyzed using
the MestReNova software package. HPLC-grade solvents were used for all HPLC and
mass spectrometry experiments. LC-ESMS analysis was performed on an Agilent
1100LC/MSD ion trap mass spectrometer equipped with an atmospheric pressure
electrospray ionization source. Eluent nebulization was achieved with a N2 pressure of 50
psi and solvent evaporation was assisted by a flow of N2 drying gas (350 °C). Positive-
ion mass spectra were recorded with a capillary voltage of 2800 V and a mass-to-charge

209
scan range of 150 to 2200 m/z. HR-ESMS was performed on a Thermo Scientific LTQ
Orbitrap XL equipped with an electrospray source, and the data was processed with
Xcalibur 2.1 (Thermo Scientific). To establish the purity of target compounds, samples
were diluted in methanol containing 0.1 % formic acid and separated using a 4.6 mm ×
150 mm reverse-phase Agilent ZORBAX SB-C18 (5 mm) analytical column at 25 °C, by
using the following solvent system: solvent A, optima water, and solvent B,
methanol/0.1 % formic acid, with a flow rate of 0.5 mL/min and a gradient of 95 % A to
5 % A over 30 minutes. HPLC traces were recorded with a monitoring wavelength range
of 363-463 nm. Peak integration was done using the LC/MSD Trap Control 4.0 data
analysis software. Analytical purity of greater than 95 % was confirmed this way for all
target compounds prior to analytical and biological experiments.
6.4.2. Synthetic procedures and product characterization
Synthesis of [PtClC27H38N6O4](NO3) (P16-A1). P16-A1 was synthesized using a
two-step, one-pot approach. A mixture of 0.678 g (2.00 mmol) of [PtCl2(pn)] and 0.338 g
(2.00 mmol) of AgNO3 in 10 mL of anhydrous DMF was stirred at room temperature in
the dark for 16 h. Precipitated AgCl was filtered off through a syringe filter. 2.96 g (16
mmol) of nitrile precursor 4-(3-cyanopropoxy)-4-oxobutanoic acid was added to the
filtrate, and the suspension was stirred at 65 °C for 4 h in the dark. Then the reaction
mixture was cooled to 4 °C with ice bath. Acridine–amine A1 (0.51 g, 2.02 mmol) was
added to the solution, and the suspension was stirred at 4 °C for another 48 h. To quench
the reaction, the reaction mixture was added dropwise into 200 mL of anhydrous ethyl
ether, and the resulting yellow slurry was vigorously stirred for 30 min. The precipitate

210
was recovered by membrane filtration and recrystallized from hot ethanol, affording P16-
A1 as a yellow solid. Yield: 704 mg (43 %). 1H NMR (300 MHz, MeOH-d4) δ 8.56 (d, J
= 8.7 Hz, 2H), 7.95 (t, J = 7.5 Hz, 2H), 7.81 (d, J = 8.4 Hz, 2H), 7.57 (t, J = 7.6 Hz, 2H),
5.08 (brs, 2H), 4.80 (brs, 2H), 4.38 (t, J = 6.7 Hz, 2H), 4.32 – 4.22 (m, 3H), 3.94 (t, J =
6.6 Hz, 3H), 3.13 – 2.96 (m, 5H), 2.82 – 2.46 (m, 8H), 2.15 (s, 2H), 1.81 (s, 2H). 13C
NMR (75 MHz, MeOH-d4) δ 174.33, 172.68, 168.67, 161.75, 161.75, 154.16, 142.51,
131.29, 126.77, 121.35, 119.47, 117.88, 65.29, 63.64, 44.43, 43.58, 33.34, 32.57, 31.97,
28.99, 14.91. HR-ESMS (positive-ion mode): m/z for C27H38ClN6O4Pt ([M]+), Calculated
740.2291; found, 740.2297 (tolerance: 0.81 ppm).
Synthesis of [PtClC31H40N6O4](NO3) (P16-B1). P16-B1 was synthesized by the same
procedure as P16-A1 with 0.678 g (2.00 mmol) of [PtCl2(pn)] and benzo[c]acridine–
amine B1 (0.61 g, 2.02 mmol) as precursors. Yield: 782 mg (46 %). 1H NMR (300 MHz,
MeOH-d4) δ 9.06 (d, J = 7.9 Hz, 1H), 8.55 (d, J = 8.6 Hz, 1H), 8.27 (t, J = 9.3 Hz, 2H),
8.14 – 7.76 (m, 5H), 7.69 (t, J = 7.7 Hz, 1H), 5.08 (brs, 2H), 4.80 (brs, 2H), 4.35 (t, J =
6.5 Hz, 2H), 4.20 (d, J = 5.9 Hz, 2H), 3.93 – 3.89 (m, 2H), 3.19 – 2.90 (m, 5H), 2.88 –
2.45 (m, 8H), 2.08 (s, 2H), 1.79 (s, 2H). 13C NMR (75 MHz, MeOH-d4) δ 175.59, 173.30,
168.88, 154.31, 150.58, 143.04, 135.22, 132.17, 131.75, 130.20, 128.42, 127.56, 126.32,
125.37, 123.09, 122.94, 122.07, 116.70, 113.97, 64.04, 44.13, 43.48, 32.77, 31.75, 28. 93,
27.50, 15.44. HR-ESMS (positive-ion mode): m/z for C27H38ClN6O4Pt ([M]+),
Calculated 790.2447; found, 790.2463 (tolerance: 2.02 ppm).
Synthesis of [PtCl(pn)C43H47ClFN9O4](NO3)2 (6-14). 100 mg of P16-A1 (0.122
mmol), 71 mg of 6-7 (0.183 mmol), 127 mg of PyBOP (0.244 mmol) and 31 mg of
DIPEA were mixed in 3 ml anhydrous DMF. The reaction mixture was incubated at room

211
temperature for 16 h. To quench the reaction, the reaction mixture was added dropwise
into 200 mL of anhydrous ethyl ether, and the resulting yellow slurry was vigorously
stirred for 30 min. The precipitate was recovered by membrane filtration, which was
recrystallized from the mixture of ethanol and ethyl ethanol (3:1) to give121 mg (yield:
83.4%) yellow solid. 1H NMR (500 MHz, DMF-d7) δ 13.91 (brs, 1H), 10.05 (brs, 1H),
8.91-8.87 (m, 1H), 8.74-8.68 (m, 2H), 8.17-8.12 (m, 1H), 8.03-7.96 (m, 5H), 7.92-7.84
(m, 1H), 7.68-7.51 (m, 4H), 6.72-6.27 (m, 2H), 5.49 (brs, 2H), 5.05 ((brs, 2H), 4.58-4.50
(m, 2H), 4.37-3.99 (m, 6H), 3.86-3.74 (m, 2H), 3.50-3.47(m, 2H), 3.28-3.13 (m, 8H),
2.83-2.53 (m, 8H), 2.30-2.17 (m, 2H), 1.90-1.80 (m, 2H), 1.56-1.43 (m, 3H). 13C NMR
(126 MHz, DMF-d7) δ 172.94, 172.91, 172.74, 171.31, 168.33, 158.56, 158.31, 157.43,
157.22, 156.23, 154.28, 154.08, 146.39, 140.75, 139.82, 135.30, 135.16, 126.27, 125.99,
124.58, 123.82, 119.75, 119.60, 118.95, 116.93, 116.76, 109.17, 109.03, 99.03, 98.59,
97.20, 96.77, 65.45, 63.53, 56.69, 47.53, 45.99, 43.45, 42.82, 41.57, 41.08, 33.05, 28.15,
27.87, 14.92, 13.91. HR-ESMS (positive-ion mode): m/z for C46H57Cl2FN11O4Pt ([M]+),
Calculated 1111.3604; found, 1111.3615 (tolerance: 0.989 ppm).
6.4.3. Assembly of multifunctional platinum-acridine agents through a library
approach
Stock solutions of the carboxylic acid-modified platinum–(benz)acridines (25 mM),
coupling reagent PyBOP (25 mM), amines to be linked via amide bond formation (25
mM), and Hünig's base (25 mM) are prepared in anhydrous DMF. Microscale coupling
reactions are carried out in 1.5-mL Eppendorf tubes by mixing all the reaction
components using the following volumes: carboxylic acid platinum precursor (40 L),

212
PyBOP (60 L), Hünig's base (40 L), and selected amines (44 L). The reaction
mixtures are then replenished with 16 μL fresh DMF to adjust the final platinum
concentration to 5 mM, followed by incubation at room temperature for 16 hours. To
characterize conjugates and to determine conversion yields, 5-L samples are removed
from the PBS solutions and serial diluted with 995 L ethanol containing 0.1% formic
acid prior to LC-ESMS analysis. Chromatographic separations are performed with a 4.6
mm × 150 mm reverse-phase Agilent ZORBAX SB-C18 36 (5 m) analytical column
with the column temperature maintained at 25 °C. The binary mobile phase consisted of:
solvent A, optima water, and solvent B, methanol/0.1% formic acid delivered at a
gradient of 95% A to 5% A over 30 minutes and a flow rate of 0.5 mL/min. The
formation and the extent of conversion of the precursors was 5 monitored in the
corresponding chromatograms using the LC/MSD Trap Control 4.0 data analysis
software. To quench the reactions, 1 mL of diethyl ether is added to precipitate platinum
complexes, which are collected by centrifugation, washed with 0.5 mL of
dichloromethane, and redisolved in 50 µL of phosphate-buffered saline (PBS) (pH 7.4).
Traces of unreacted precursors and byproducts are soluble in diethyl ether, or
dichloromethane, and can be removed by washing the solid and discarding the
supernatant.
6.4.4. Chemical hydrolysis assay
All selected conjugates were produced by the microscale reactions as described before.
To purify these conjugates, the reaction mixtures were precipitated in ethyl ether,
followed by size exclusion chromatography (Sephedex LH-20, 10 × 1 cm column) eluted

213
with methanol-acetonitrile (1:1, v/v). The ester hydrolysis study of compounds was
carried out by incubating 0.1 mM of each test compound in 10 mM phosphate buffer (pH
7.4) or 1× PBS containing ~150 mM NaCl at 37 °C. At various time points samples were
withdrawn from the reaction mixture and analyzed by in-line LC-ESMS.
Chromatographic separations were performed with a 4.6 mm × 150 mm reverse-phase
Agilent ZORBAX SB-C18 (5 μm) analytical column with the column temperature
maintained at 25 °C. The following solvent system was used: solvent A, optima water,
and solvent B, methanol/0.1 % formic acid, at a flow rate of 0.5 mL/min and a gradient of
95 % A to 5 % A over 30 min.

214
CHAPTER 7
LIPOSOMAL DELIVERY OF PLATINUM-ACRIDINE ANTICANCER FOR
NON-SMALL CELL LUNG CANCER
The experiments described in this chapter were performed by Song Ding, except for the
the confocal microscopy which were contributed by Dr. Fang Liu, and animal studies,
which were contributed by Washington Biotechnology Inc. ICP-MS was performed by
the Analytic Chemistry Service facility of Research Triangle Institute, Research Triangle
Park, NC.

215
7.1. Design Rationale
Previous studies have suggested that the severe side-effects of platinum-acridine agents
are likely caused by their indiscriminate accumulation in off-target tissues, such as the
liver and kidneys [279]. To improve the toxicity profile, we hypothesized that the
liposomal formulations might help deliver platinum-acridines to tumor sites passively as
a result of the EPR effect (see chapter 1, section 1.3.3) and slow tumor growth in mice
with higher efficacy than the free drug when injected intravenously [111]. Platinum-
acridine derivatives are generally cationic [279], highly hydrophilic [279], and, like other
charged molecules, not membrane permeable. This would make it difficult to load
platinum-acridines into liposomes. Therefore, a method that allows efficient
encapsulation of platinum-acridines must be developed prior to in vivo studies.
Our search for appropriate lipid components started with the lipids that are used in
FDA-approved Doxil, the liposomal doxorubicin made from hydrogenated soybean
phosphatidylcholine (HSPC), cholesterol and polyethylene glycol-2000-
distearoylphosphatidylethanolamine (mPEG2000-DSPE) (For structures of lipid
components used in this study, see APPENDIX J) [82]. This combination has been
widely used for the encapsulation of other hydrophilic drugs [382]. Specifically, HSPC is
a lipid with a high phase transition temperature (Tm = 55 ˚C), showing an ordered
crystalline phase at physiological temperature, which reduces drug leakage during
circulation. Incorporation of cholesterol into the lipid bilayer increases the stability of the
liposomal formulation [111]. Additionally, liposomes decorated with mPEG2000-DSPE, a
polyethylene glycol-modified lipid, can extend the circulation time of liposome by
evading the mononuclear phagocyte system (MPS, see chapter 1, section 1.3.3) [84, 111].

216
The mPEG with larger molecular weight, however, may slow down the cellular uptake
and compromise in vivo efficacy [383]. Moreover, lipids bearing negative charges, such
as 1,2-dihexadecanoyl-sn-glycero-3-phospho-(1'-rac-glycerol) (DPPG), which was
chosen in liposomal cisplatin (Lipoplatin) [133], will be also added to the lipid
composition. Their electrostatic interactions with the cationic platinum complexes will
not only increase the loading capacity, but also enhance drug retention in circulation.
Another benefit conferred by DPPG to the liposomes is the ability to fuse with the cell
membrane, as demonstrated for lipoplatin [133], which would release the payloads
directly into the cytosol of cancer cells.
7.2. Result and Disscussion
7.2.1. Aqueous stability of platinum-acridine analogues
Prior to liposomal encapsulation of platinum-acridines, we selected P2-A1 (Figure
7.1A) as a representative for their aqueous stability studies in water, 0.9 % saline and
phosphate buffered saline (pH = 7.4) at 60 ºC, the temperature that was required for the
preparation of liposomes. All the samples were incubated for 4 hours, which is
significantly longer than the time required for encapsulation, and the reaction mixtures
were characterized by LC-MS. P2-A1 was found to be quite stable in saline, but undergo
aquation in Milli Q water under testing conditions. Aquation typically occurs when the
environmental chloride level is low and can be suppressed by high level chloride ions in
solution at 37 ºC (for example, 150 mM NaCl) [68]. This experiment suggested that 0.9 %

217
saline can completely inhibit this ligand exchange reaction at high temperature (60 ºC)
for at least 4h. Contrary to our expectations, the formation of a seven-membered, inactive
Pt-N chelate (7-1, Figure 7.1C) was detected in the sample incubated with PBS, even
though the chloride concentration was close to that of a saline solution. The reaction
mixture was then lyophilized and redissolved in D2O, followed by 1H-NMR analysis. The
appearance of the down-field shifted proton signal (10.5 ppm, Figure 7.1B), resulting
from the deshielding effect of platinum, further confirmed the formation of the chelate,
which had been observed previously [277]. The decomposition can be explained with the
coordination of phosphate to platinum, which is negligible at room temperature, but is
accelerated at higher temperature and competes with chloride. Thus, substitution of
chloride is accelerated by phosphate. The labile phosphate is then rapidly replaced by the
intramolecular nucleophilic 9-amino nitrogen of acridine to produce the Pt-N chelate. To
avoid the decomposition induced by the weakly coordinating buffer, we used water and
0.9% saline as the loading “buffers” for the preparation of liposomes.

218
Figure 7.1. (A) Structures of P2-A1 and P8-A1. (B) 1H-NMR characterization of the
same reaction mixture in D2O. (C) LC-ESMS analysis of P2-A1 incubated in water at 60
ºC for 4 hours. Peak 2 indicates the formation of side product (7-1) under the test
conditions.
7.2.2. Optimization of conditions for drug loading
7.2.2.1. Selection of lipids
We first screened the optimal lipid composition for the encapsulation of P8-A1 (Figure
7.1A), the most cytotoxic platinum-acridine analogue identified in NSCLC cells [350]. A

219
library of liposomes (Table 7.1) with various combination of lipid components was
prepared on a small scale (10 mg of total lipids for each) with a 1-mL mini-extruder using
the film-hydration method (see chapter 1). In the screen, all the preparations were
performed in Milli Q water with the same initial drug feeds, defined as the relative ratio
of “mg of Pt complex/mg of total lipids”. The content of mPEG was fixed at 5 mol% in
the library, which is based on the calculation for the estimated surface coverage of
mPEG-DSPE2000 for 100-nm sized liposomes [384]. Higher degrees of PEGylation will
not have a significant impact on the stabilities in circulation, but may introduce multiple
populations of nanoparticles, including mixed micelles and discs, if the content of mPEG
is more than 15 mol% [384, 385]. The liposomal encapsulations were evaluated based on
their encapsulation efficiency (EE%, Table 7.1). In initial experiments, we used
liposomes containing HSPC and cholesterol, such as liposome 1-3 (Table 7.1). No
encapsulation of P8-A1 was observed for these formulations, which can be attributed to
the poor lipid affinity of the cationic platinum complex. The addition of 30 mol% DPPG,
as expected, greatly increases EE% to 32 ± 1%, as evidenced in liposome 4, compared
with all the DPPG-free liposomes. This improvement can be explained with the
electrostatic interaction of DPPG with the cationic payload, which facilitates drug uptake
into the aqueous core. The encapsulation efficiency increased as more DPPG was
incorporated into the lipid bilayers, with the EE% reaching 53 ± 2% for a DPPG content
of 45 mol%.
No consequences to the drug loading were observed when the content of HSPC and
cholesterol was varied (Table 7.1). However, the inclusion of these components is
essential for maintaining the in vivo stability of the liposomes. For example, addition of

220
cholesterol is able to enhance the retention of payloads in the liposomes that are mainly
composed of lipids with a phase transition temperature (Tm) lower than 37 °C. They
increase the permeability of the gel phase bilayers (Tm > 37 °C) under physiological
conditions. In the latter case, incorporation of cholesterol may impede drug release [386],
leading to a reduction in anticancer efficacy. Therefore, two cholesterol free liposomes
were also included in the library. In particular, 87% of P8-A1, the highly hydrophilic
molecule, was successfully encapsulated in formulation 9 made of DPPG, HSPC and
mPEG-DSPE (80:15:5, molar ratio). In fact, high blood stability of cholesterol-free
liposomes comparable to conventional cholesterol-containing liposomes has been
demonstrated when phospholipids with a high Tm, such as DPPC, DPPG and HSPC, are
major components of the bilayers [387]. Decoration of cholesterol free liposomes with
mPEG further improves their in vivo stability and prolongs their circulation time in blood
[111].

221
Ta
ble
7.1
. C
ondit
ions
for
the
Lip
oso
mal
En
capsu
lati
on o
f P
8-A
1.
#
Lip
id C
om
po
nen
ts
Lip
ids
Rat
io
(mola
r)
Init
ial
Dru
g/L
ipid
(wt)
L
oad
ing m
edia
E
E%
1
HS
PC
/CH
OL
/mP
EG
-DS
PE
55/4
0/5
0.2
H2O
0
2
HS
PC
/CH
OL
/mP
EG
-DS
PE
73/2
4/5
0.2
H2O
0
3
HS
PC
/ m
PE
G-D
SP
E
95/5
0.2
H2O
0
4
DP
PG
/HS
PC
/CH
OL
/mP
EG
-DS
PE
30/2
5/4
0/5
0.2
H2O
32
± 1
%
5
DP
PG
/HS
PC
/CH
OL
/mP
EG
-DS
PE
45/1
5/4
0/5
0.2
H2O
53 ±
2%
6
DP
PG
/HS
PC
/mP
EG
-DS
PE
50/4
5/5
0.2
H2O
36
± 2
%
7
DP
PG
/HS
PC
/mP
EG
-DS
PE
8
0/1
5/5
0.2
H2O
87 ±
2%
8
DP
PG
/HS
PC
/mP
EG
-DS
PE
80/1
5/5
0.5
H2O
45
± 2
%
9
DP
PG
/HS
PC
/mP
EG
-DS
PE
71/2
4/5
0.2
H2O
46
± 1
%
10
D
PP
G/H
SP
C/m
PE
G-D
SP
E
47.5
/47.5
/5
0.2
H2O
39
± 1
%
11
D
PP
G/H
SP
C/m
PE
G-D
SP
E
80/1
5/5
0.2
0.9
% S
alin
e
49
± 1
%
12
D
PP
G/H
SP
C/m
PE
G-D
SP
E
80
/15/5
0.2
0.9
% S
alin
e
30 ±
0.3
%
13
D
PP
G/H
SP
C/m
PE
G-D
SP
E
80/1
5/5
0.2
5
% S
ucr
ose
, 0
.9%
Sal
ine
72
± 1
%
14
D
PP
G/H
SP
C/m
PE
G-D
SP
E
80/1
5/5
0.2
5
% S
ucr
ose
, 0
.9%
Sal
ine
40
± 2
%

222
7.2.2.2. The importance of freeze-thaw cycles for drug loading
All the liposomes in the library were prepared using 10 freeze-thaw cycles, during
which the liposomal suspensions were frozen in liquid nitrogen (−196 °C) and thawed at
a temperature above the Tm of the bulk lipids. To evaluate the impact of freeze-thaw
cycles on the encapsulation of P8-A1, a cholesterol free liposome formulation without
undergoing freeze-thaw process has also been generated with the bilayer compositions
identical to liposome 9 in the library. Results are shown in Figure 7.2A. Strikingly, about
54% more drug was encapsulated when applying the freeze-thaw technique, confirming
its importance in the drug loading process. The freeze-thaw technique has been widely
implemented to improve the encapsulation of hydrophilic drugs. It has been proposed that
the process causes a reequilibration of drug distribution inside and outside the liposomes,
most likely due to disruption of the lipid bilayers caused by the formation of ice crystals
[388, 389]. An increase in the trapping volume was also observed after freeze-thaw cycle,
which improves the encapsulation yield [389]. Moreover, since the liposomes lacking
cholesterol are very sensitive to changes in temperature and osmotic gradients, the freeze-
thaw technique is likely to be more effective for the cholesterol free liposomes than
conventional formulations [390].

223
Figure 7.2. (A) Effect of the freeze-thaw cycles on the encapsulation efficiency (EE%) of
DPPG/HSPC/mPEG2000-DSPE (80/15/5, molar ratio). The liposomes were prepared using
the film-hydration method with an initial drug/lipid ratio of 0.2 (wt/wt). (B) Effect of the
loading buffers on the EE% of DPPG/HSPC/mPEG2000-DSPE (80/15/5, molar ratio). The
liposomes were prepared using the film-hydration method with 10 freeze-thaw cycles.
The initial drug/lipid ratio was set as 0.2 (wt/wt). (C) Effect of the drug feeds on the EE%
of DPPG/HSPC/mPEG2000-DSPE (80/15/5, molar ratio). The liposomes were prepared
using the film-hydration method with 10 freeze-thaw cycles. (D) Effect of the
lyoprotectants on the EE% of DPPG/HSPC/mPEG2000-DSPE (80/15/5, molar ratio). The
liposomes were prepared using the film-hydration method with 10 freeze-thaw cycles.
The initial drug/lipid ratio was set as 0.2 (wt/wt).
7.2.2.3. The influence of loading buffers on drug loading
We next assessed the roles of the loading buffers in the liposomal encapsulation process.
Milli Q water, 0.9% saline, and 0.9% saline containing various cryoprotectants (glucose,

224
sucrose and mannitol) were used in this study to hydrate the lipid thin film composed of
DPPG, HSPC and mPEG-DSPE (80:15:5, molar ratio). The drug feeds were set as 0.2.
The EE%, obtained after 10 freeze-thaw cycles, is shown in the Figure 7.2D. The use of
water yielded the formulation with EE% of 88%, which dropped notably to about 49%
when the loading solution was replaced with 0.9% saline. This can be in part explained
by the increased ionic strength of the loading solution, which weakens the electrostatic
interactions of the lipid bilayer with the cationic platinum compounds, and therefore
reduces the amount of encapsulated drug. In addition, the high concentration of NaCl
(~150 mM) in saline inhibited the phase separation of ice and solute during freezing and
thus strongly inhibited liposomal encapsulation [391]. We found that the addition of
cryoprotectants can partly restore the EE%. Specifically, 72% of P8-A1 was encapsulated
using 0.9% saline containing 5% glucose as the loading solution. The cryoprotectants
may form multiple hydrogen bonds with the lipids. This interaction may stabilize the
bilayers, preventing them from being completely disrupted during freeze-thaw cycles,
which leads to the leakage of loaded drug. The increase in the amount of glucose from 5%
to 10% did not result in significant changes in EE. Since P8-A1 may undergo aquation in
water at high temperature, we decided to use 0.9% saline containing 5% as the loading
solution to suppress this process.
7.2.2.4. The influence of drug feeds on drug loading
To evaluate the effects of initial amounts of drug on encapsulation, drug feeds of 0.05,
0.1 and 0.2 were chosen in this study (Figure 7.2C). A formulation with the EE% of 92%
was produced with a drug feed of 0.05, which dropped to 72% as the drug feed was

225
increased to 0.2. This is as expected because with lower drug feed (higher DPPG: P8-A1
ratio), negatively charged lipids in the core can better wrap up the cationic platinum
complexes, which lead to higher EE%. In addition, about 60% of P8-A1 was not
encapsulated in the liposomes when the drug feed was increased to 0.5. This is most
likely caused by amount of dicationic P8-A1, which electrostatically neutralize the
negatively charged DPPG and lead to saturation with payload. Drug feeds higher than 0.5
were found to result in the formation of yellow precipitate regardless of the drug
concentration, possibly due to complete neutralization of lipids with platinum complexes.
7.2.2.5. The influence of loading methods on drug loading
We also compared different methods of formulating liposomes for P8-A1, including
film hydration, reverse-phase evaporation, and ethanol injection [119]. All the
formulations in this study were prepared with the lipid component DPPG, HSPC, and
mPEG-DSPE (molar ratio: 80:15:5) using 0.9% saline containing 5% sucrose as the
loading buffer, with an initial drug feed of 0.2. The film hydration method resulted in the
formulation with the highest EE% of about 80%, while a significant drop in EE% was
observed in the formulations prepared by reverse-phase evaporation (21%). This method
commonly leads to higher EE% than others for encapsulation of hydrophilic drugs
because a large internal aqueous volume is generated in the preparation. This observation
confirms our previous finding that the freeze-thaw procedure is critical for achieving the
highest EE%. Less than 5% of P8-A1 was entrapped in the liposomes using ethanol
injection under the same conditions. Because drug loading with this technique relied on
the self-assembly of the lipids with platinum complexes, poor partitioning of P8-A1

226
between lipids and aqueous solution may be responsible for the lower EE%. In addition
to EE%, the hydrodynamic size of these liposomes generated by different methods were
examined using dynamic light scattering (DLS) measurements. Liposomes prepared by
film hydration and reverse-phase evaporation method, which underwent serial extrusion
through 0.22 µM membranes, were homogeneous with a diameter of approximate 120
nm (polydispersity index, PDI < 0.2), whereas the ethanol injection method without
extrusion generated 133 nm liposomes with a broad size distribution (PDI > 0.2). As a
result, film hydration and subsequent extrusion is a better method for encapsulating
platinum-acridines and was chosen for all formulations prepared for further biological
evaluation.
7.2.3. Characterization of liposomes
Four PEGylated liposomal formulations of P8-A1, denoted as Lipo-1─Lipo-4, were
prepared for the following biological evaluations (compositions and properties are
summarized in Table 7.2). Lipo-1, which had the highest loading capacity, was a
cholesterol-free liposomal formulation with the lipid composition of DPPG, HSPC, and
mPEG-DSPE at a molar ratio of 80:15:5. Lipo-2 was inspired by lipoplatin [133],
liposomal cisplatin, which is currently in phase II/III clinical trials and is composed of
DPPG, HSPC, cholesterol, and mPEG-DSPE. Lipo-3 is composed of the identical lipid
components as Lipo-1 but with lower drug loading capacity. Lipo-4, containing the same
lipids as Lipo-1 yet with reduced level of DPPG in the bilayers, was produced to evaluate
the role of DPPG in the formulation. The average hydrodynamic diameter of all the

227
formulations, determined by dynamic light scattering (DLS), were approximately110-120
nm with small polydispersity indices (PDI) of less than 0.2, suggesting highly
homogeneous size distributions (see Appendix G). The liposomes with a size of about
100 nm are optimal for in vivo applications since they show favorable tumor retention
and long circulation time in blood [111]. We found that liposome size correlates with
drug loading capacity, with larger diameters in the formulations producing higher drug-
to-lipid ratios. In addition, due to the presence of anionic DPPG, all formulations showed
negative zeta potentials. This also confirms that the cationic platinum complexes did not
localize to the liposome surface, but remained trapped within the aqueous core.
Reduction of the zeta potential from ~ -40 mV in Lipo-1 to -29 mV in Lipo-4 was found
as the amount of DPPG decreased. Less negatively charged liposomes are thought to
have a longer circulating time, which may result in an enhanced EPR effect [111]. The
relative amounts of phospholipids and cholesterol were determined with Bartlett
assay[392] and reverse-phase HPLC, respectively. Lipo-1 containing a high level of
DPPG was found to be able to reach a drug-to-lipid ratio of 19.7% (wt/wt), while the
drug-to-lipid ratio of Lipo-2, which contained less DPPG and 9.3% (wt/wt) cholesterol,
was 6.8% (wt/wt). This value considerably dropped to 2.3% (wt/wt) in formulation Lipo-
4, which contained the lowest concentration of DPPG. This observation is consistent with
our earlier conclusion that the content of DPPG dominates the encapsulation efficiency of
P8-A1.
To further characterize the liposomes, transmission electronic microscopy (TEM) was
exploited to reveal structural details. All liposomes were negatively stained with uranyl
acetate prior to TEM image acquisition. Most of the liposomes were found to be spherical

228
with a size of less than 100 nm, which was smaller than the results obtained with DLS.
This difference in apparent size is typically observed in PEGylated liposomes, in which
the hydrophilic PEG residues form a hydration layer on the surface in solution. This
makes the particle size appear large when measured by DLS [111]. TEM also showed an
electron-dense platinum-filled core wrapped in a brighter layer in the cholesterol-free
formulations Lipo-1, Lipo-3, and Lipo-4. Careful measurement of the thickness of the
liposomal bilayer exhibited a range from 6 to 9 nm (Figure 7.4A). Because the thickness
of a double lipid bilayer should be at least 10.4 nm [391], we reasoned that our liposomes
consisted of a single bilayer of lipids.
By contrast, no fine structure was observed in Lipo-2. Ruptured liposomes and disc-
like structures (Figure 7.5B) were found in Lipo-3 and Lipo-4. This typically occurred in
the cholesterol-free liposomes containing DSPE-mPEG after undergoing freeze-thaw
cycles throughout their Tm [124]. This effect was not observed in Lipo-1, the cholesterol
free formulation with the highest loading capacity, suggesting that platinum-acridines
may help stabilize the lipid bilayer during the freeze-thaw process.

229
Figure 7.3. TEM images of Lipo-1(A), Lipo-2(B), Lipo-3(C) and Lipo-4(D). Liposomes
were negatively stained using uranyl acetate.

230
Tab
le 7
.2.
Char
acte
riza
tion o
f F
our
Lip
oso
mal
Fo
rmula
tions
Pre
par
ed f
or
P8-A
1
L
ipo
-1
Lip
o-2
L
ipo
-3
Lip
o-4
Lip
id c
om
ponen
ts
DP
PG
:HS
PC
:DS
PE
-
mP
EG
2k
(mola
r ra
tio:
80:1
5:5
)
DP
PG
:HS
PC
:Chol:
DS
PE
-
mP
EG
2k
(mola
r ra
tio:
40:3
2:2
0:8
)
DP
PG
:HS
PC
:DS
PE
-
mP
EG
2k
(mo
lar
rati
o:
80
:15:5
)
DP
PG
:HS
PC
:DS
PE
-
mP
EG
2k
(mola
r ra
tio:
20:7
5:5
)
Pay
load
a/l
ipid
b
(wt/
wt)
, (r
epo
rted
as %
paylo
ad)
19.7
± 1
.7
6.8
± 0
.4
1.7
± 0
.2
2.3
± 0
.3
Chole
ster
ol
(wt
%)c
--
9.3
± 0
.6
--
--
Siz
e (n
m)d
120.9
± 1
.1
112.3
± 1
.6
110.7
± 1
.2
102.4
± 0
.7
PD
Id
0.0
73 ±
0.0
09
0.0
37 ±
0.0
15
0.1
27 ±
0.0
27
0.1
22 ±
0.0
09
Zet
a pote
nti
al (
mV
)
d
-47.4
± 0
.8
-41.7
± 1
.0
-40.7
± 0
.7
-29.2
± 0
.1
TE
M i
mag
es
a S
pec
trophoto
met
ry. b
Bar
tlet
t as
say. c
Rev
erse
-phas
e H
PL
C.
d D
ynam
ic l
ight
scat
teri
ng (
DL
S).

231
Figure 7.4. (A) TEM image of a single liposome in Lipo-1 highlighting the width of the
lipid bilayer. (B) TEM image of Lipo-3 which was freshly prepared by cycling ten times
through Tm. White arrows indicate the formation of bilayer discs. The scale bar indicates
100 nm.
7.2.4. Stability of liposomes in PBS and during long-term storage
Pre-mature release of platinum complexes in the liposomes before reaching tumor sites
should be avoided to achieve a better therapeutic index. To test their stability under
physiologically relevant conditions, Lipo-1 ─ Lipo-4 were incubated in PBS (pH = 7.4)
at 37 ⁰C for 48 h. Samples were withdrawn at various time points and passed through a
size exclusion column to remove any released P8-A1 [393]. The purified liposomes were
then quantified by UV-Vis spectroscopy and their absorbance compared with that of
controls to determine the amount of drugs within the liposomes. To demonstrate that size
exclusion chromatography is able to effectively separate the free P8-A1 from the

232
liposomes, a mixture of the two forms was loaded onto a column (1 cm × 10 cm) filled
with Sephadex G-25. The chromatographic elution profiles of Lipo-1 and P8-A1 are
shown in Figure 7.5. As can be clearly seen in the elution profile, the liposome eluted
from the column using 5 mL of 0.9% saline, while P8-A1 at the same concentration
required about 9 mL of the same solution.
Figure 7.5. Sephadex G-25 elution profiles for the mixture of 100 µL Lipo-1(1 mg/mL,
based on P8-A1) and 100 µL P8-A1 (1 mg/mL). The sample was eluted with 0.9% saline
solution (pH 6.5 – 7.0) and collected every 0.5 mL. Fractions were quantified by UV-Vis
spectroscopy at a wavelength of 413 nm. The absorbance in each fraction was normalized
as the percentage of the maximal absorbance in the same group. Fractions were analyzed
for Lipo-1 (solid line) and P8-A1 (dash line). Note the difference in the retention times,
allowing the baseline separation of free P8-A1 from liposomes under these conditions.
We next assessed the stability of each formulation in PBS (pH 7.4) at 37 °C. All
liposomes lacking cholesterol (Lipo-1, lipo-3 and Lipo-4) showed little leakage after 48
hours (Figure 7.6), indicating excellent stability under the test conditions. This
observation is consistent with a phase transition temperature greater than 37 °C, which

233
renders the lipid bilayers impermeable to the platinum complexes at physiological
temperature. In addition, the electrostatic interactions of DPPG with P8-A1 may also
enhance the retention of platinum complex within the lipid bilayer. On the other hand, 40%
of P8-A1 was released by Lipo-2 under the same conditions, which can be explained
with the incorporation of cholesterol. Cholesterol is a modulator of membrane mobility
that permeabilizes the bilayer to release platinum complex at temperatures below the
phase transition temperature of the bulk lipid. Moreover, since the fluidity of cholesterol-
free lipid bilayers is highly sensitive to variations in temperature [124], cholesterol-free
liposomes in this study would become more permeable at higher temperature. Lipo-1,
mainly composed of DPPG with a phase transition temperature of 41 °C, was incubated
in PBS at 42 °C (Figure 7.7). 24% of P8-A1 was found to be released after 30 min, and
this amount increased after an incubation time of 1 hour (35%) or after elevating the
temperature to 44 °C (35%). A burst release of 46% P8-A1 was detected when Lipo-1
was incubated at 44 °C for 1 h. However, it should be noted that this stability study in
vitro does not faithfully mimic the in vivo behavior of liposomes. Other parameters, such
as serum proteins and components of the immune system, usually facilitate
decomposition of liposomes in blood, and thus release the payloads in off-target tissues.
The storage stability studies indicated that all liposomal formulations were highly stable
when stored in 0.9% saline at 4 ⁰C for at least 2 months without significant changes in
size and payload content.

234
Figure 7.6. In vitro leakage of P8-A1 from Lipo-1 ─ Lipo-4 during 48 h of incubation in
PBS (pH 7.4) at 37 °C.
Figure 7.7. In vitro release profile of P8-A1 from Lipo-1 in PBS (pH 7.4) incubated at
42 °C and 44 °C for 0.5 hour (red) and 1 hour (blue).

235
7.2.5. In vitro cellular uptake and localization study
Because of the intrinsic blue fluorescence of the 9-aminoacridine, cellular uptake and
intracellular accumulation of P8-A1 in liposomes can be directly observed using confocal
fluorescent microscopy [363]. In this study, P8-A1 (5 µM) and Lipo-1 (containing 5 µM
P8-A1) were incubated with non-small cell lung cancer NCI-H460 cells. Dosed cells
were then co-stained with Lyso Tracker (red fluorescence) to study the accumulation of
the P8-A1 in acidic lysosomes prior to the microscopiy sessions. As shown in Figure 7.8
(A) and (B), higher levels of intracellular blue fluorescence were detected in the Lipo-1
treated cells relative to cells dosed with P8-A1, indicating that the liposomes facilitate the
uptake of P8-A1 into cells. However, it is difficult to quantify the differential uptake,
since significant quenching of the fluorescence occurs when the 9-aminoacridine
chromophore is intercalated between DNA base pairs in the nucleus [310]. After
incubation for 4 hours, P8-A1, consistent with our previous findings [310], was found to
localize to the nucleoli in both groups. Importantly, nucleolar localization of free P8-A1
was observed after 2-hour incubation (Figure 7.8(A)), which was significantly faster than
liposomes, suggesting that they may have different mechanisms of cellular uptake.
A high degree of co-localization of P8-A1 with Lyso Tracker was found in the cells
incubated with free P8-A1 for only 15 min, confirming rapid accumulation of platinum
complexes in the lysosomes after entering into cancer cells. We reason that the
lysosomotropic properties of platinum-acridines can be attributed to their cationic nature,
which may favor interactions with the negatively charged lysosomal membranes to
facilitate sequestration [394]. The lysosomes seem to localize to the perinuclear region
and slowly release P8-A1 into the nucleus. Lack of effective lysosomal escape is the

236
primary reason for drug resistance observed for lysosomotropic agents if their major
biological targets are located in the nucleus [395]. This may be a shortcoming of P8-A1
in resistant cancer cells. To induce apoptosis in these cells, rapid accumulation in the
nucleus may be beneficial.
By contrast, Lipo-1 seemed to internalize into cells through a membrane-fusion
mechanism presumably as a result of the presence of fusogenic phospholipid DPPG [133].
Strikingly, a ring-shaped intense blue fluorescence is observed around the membranes of
cells treated with Lipo-1 after 15 minutes of incubation (Figure 7.8B). The membrane-
associated liposomes persist after 2 hours, indicating that the uptake of Lipo-1, unlike
that of free P8-A1, is a relatively slower process. After fusing with the cell membrane,
Lipo-1 releases loaded P8-A1 directly into the cytosol, as clearly evidenced by the blue
punctuations that does not co-localize with the lysosomes stained by red fluorescent Lyso
Tracker. Accumulation of P8-A1 to the lysosomes was also observed in these cells, but to
a lesser extent in cells treated with free drug at the same time points (Figure 7.8). We
reasoned that since liposomes are able to facilitate the delivery of a large amount of P8-
A1 directly into the cytosols, P8-A1 did not get sequestered by the lysosomes. Elevated
levels of blue fluorescence in vesicles distinct from the red-fluorescent lysosomes,
appeared in the perinuclear region of cells after incubation with Lipo-1 for 4 hours
(Figure 7.8B). This observation suggests that a high concentration of P8-A1 is able to
accumulate close to the nucleus, which may help the drug to reach its target and evade
cytosolic detoxification.

237
Figure 7.8. Confocal microscopy images of (A) P8-A1 (blue, 5 µM) and (B) Lipo-1(blue,
5 µM) incubated in real-time of NCI-H460. Cells were co-stained with LysoTracker Red.
Scale bars represent a distance of 5 μm. White arrow indicates that Lipo-1 internalize
into cells through a membrane-fusion mechanism.

238
Figure 7.9. Colocalization images of cells treated with Lipo-1 (A) and P8–A1 (B) for 4 h
and co-stained with LysoTracker Red. The arrows indicate blue-fluorescent intranuclear
vesicular structures that were separated from lysosome. Scale bars represent a distance of
5 μm.
7.2.6. Acute toxicity studies of liposomal P8-A1
To evaluate the acute toxicity of the formulations prepared in this study, Lipo-1─Lipo-
4 were administered intravenously to female Swiss-Webster mice. The maximum
tolerated dose (MTD) was determined based on clinical signs of toxicity, such as
piloerection, weight loss, and lethargy. Free P8-A1 was tested as a control under the
identical circumstances. P8-A1, in agreement with previous results [279], exhibited
severe systemic toxicity in mice models (Table 7.3) with MTD of 0.4 mg/kg for a single
dose or multiple doses (q4d×4). Typical strategies in the clinic to reverse the platinum-
induced toxicity include the use of hydration and chelating agents or agents which
otherwise react with platinum. Liposomal formulation of cisplatin also showed reduced
side effects. A two fold increase in MTD was observed with Lipo-2, which containing
cholesterol in the bilayer. However, the toxicity was not alleviated by encapsulation of

239
P8-A1 with cholesterol-free liposomes (Table 7.4). Lipo-1, Lipo-3 and Lipo-4 were
found to have the same MTD as the free drug. Since negatively charged liposomes are
rapidly recognized by the mononuclear phagocyte system (MPS), we reasoned that
macrophage uptake may be involved in the mechanism of toxicity of these formulations.
The data may indicate delivery of highly cytotoxic P8-A1 to liver and spleen, impairing
phagocytic activity of liver macrophages [396]. One common way to address this
problem, as suggested in other studies, is to co-inject a large amount of blank liposomes
or a similar formulation with extremely low drug/lipid ratio, so that it saturates the MPS
and protects the macrophages from the effects of cytotoxic agents [84, 392, 397].
7.2.7. In vivo antitumor efficacy
A549 (NSCLC, human adenocarcinoma, wild-type p53) was selected as the xenograft
model (athymic nude mice) to test the efficacy of P8-A1 and two formulations. Before a
decision was made in favor of this model, P8-A1 was tested in A549 cells using a
colorimetric cell proliferation assay, in which the derivative was highly active with an
IC50 value of 3.9 nM. The in vivo treatment schedule consisted of five i.v. injections at
doses of 0.4 mg/kg (days 0, 4, 8, 12, 16; q4d5; n = 8). Test animals were monitored for
weight loss and signs of toxicity three times a week. Data analysis for weight and tumor
volumes was done with the Student’s t-test. Only P values of less than 0.05 were
considered as statistically significant. One of the animals treated with Lipo-1 (group 3)
had to be euthanized prematurely because its tumor had grown approximately five times
the size (V = 2677 mm3, day 21) compared to the average of other tumors in that group

240
(passed Grubbs’ outlier test at P < 0.01). Few additional obvious outliers (P < 0.01 and P
< 0.05, confirmed by Grubbs’ outlier test) were identified, which caused major
differences in the variation of tumor sizes between the four groups and resulted in large
error bars (SEM). These artificially large error bars mask the true drug response of the
majority of tumors and were removed from the data sets.
The result of antitumor effects are presented in Figure 7.10A. While Treatment with
P8-A1 and Lipo-2 had no effect on tumor volumes compare to control, a modest, but
significant (P < 0.05), effect on tumor growth was found in the group treated with Lipo-
1, resulting in a ~40% reduction of final tumor volumes relative to untreated mice. To
further evaluate the systemic toxicity of liposomal formulations compared with free P8-
A1, the body weight change of mice were monitored and recorded in Figure 7.10A. Mice
in all treatment groups showed significant weight loss compared to control, however, the
mice dosed with liposomal formulations showed significantly less weight loss than mice
treated with unencapsulated P8-A1. In particular, weight loss was less than 20% and
appeared to be reversible after dosing was discontinued.
Tumors and organs were recovered from necropsied test animals on day 30, 14 days
after the last dose was administered. Residual platinum levels were determined by ICP-
MS to assess if differences in the tissue distribution exist between the free drug and the
two formulations. Lipo-1 (Group 3) produced significantly lower platinum levels in
liver, kidneys, and lungs than unencapsulated P8-A1 (Group 2). Nevertheless, between 1
and 6 ng Pt were detected in the tumors (similar to levels detected in the lungs; data not
corrected for tumor weight/volume; data not shown, n = 5) in all treatment groups.
Differences between the treatment groups were not significant. It also appeared that P8-

241
A1 was delivered more efficiently to large tumors, which faithfully mimic the poor
lymphatic drainage and defective blood vessels in the tumor tissue. This observation is
consistent with enhanced accumulation through EPR effect.
7.3. Conclusion
Liposomal formulations have been extensively investigated to improve the therapeutic
index of cytotoxic agents. In this study, we have developed a DPPG based liposome for
the most cytotoxic platinum-acridine analogue in non small lung cancer cells, P8-A1. A
method that ensures efficient drug loading has been optimized and established. Due to the
presence of DPPG, the liposome was found to be internalized into cells through a
membrane-fusion mechanism. To evaluate their in vivo anticancer efficacy, four
liposomal formulations were tested in A549 xenograft models in mice. Moderate
inhibition of tumor growth was found in mice dosed with Lipo-1, while the other
formulations and free P8-A1, had no effect on tumor size. Future studies will focus on
improving the anticancer efficacy of the liposomal formulations, including optimization
of lipid components, alteration of drug loading capacity and incorporation of tumor-
targeting ligands into the lipid bilayers. Alternatively, less cytotoxic platinum-acridine
analogues may show an advantage as payloads.

242
Ta
ble
7.3
. M
TD
Stu
dy f
or
P8
-A1
Co
mpound
D
osi
ng R
egim
en (
I.V
.)
Sig
ns
of
Tox
icit
y
P8
-A1
0.1
mg/k
g
No o
bvio
us
signs
of
tox
icit
y
P8
-A1
0.4
mg/k
g
Thre
e m
ice
hav
e m
ild p
iloer
ecti
on p
ost
day 6
P8
-A1
0.8
mg/k
g
All
mic
e hav
e pil
oer
ecti
on p
ost
day
11
P8
-A1
0.4
mg/k
g, qd×
5
All
mic
e hav
e pil
oer
ecti
on p
ost
day
4
P8
-A1
0.4
mg/k
g, q4d×
4
Tw
o m
ice
hav
e m
ild p
iloer
ecti
on p
ost
day 1
2
P8
-A1
0.6
mg/k
g, q4d×
4
Thre
e m
ice
hav
e m
ild p
iloer
ecti
on p
ost
day 1
2
qd×
5 :
dosi
ng o
nce
dai
ly (
i.v.)
for
5 d
ays.
q4d
×4 :
dosi
ng o
nce
eac
h (
i.v.)
on d
ays
0, 4, 8 a
nd 1
2 (
4 d
ose
s).

243
T
ab
le 7
.4.
MT
D S
tud
y o
f L
iposo
mal
Form
ula
tion
s o
f P
8-A
1.
Form
ula
tion
D
osi
ng R
egim
en (
I.V
.)
Sig
ns
of
Tox
icit
y
Lip
o-1
0.8
mg/k
g
All
mic
e hav
e pil
oer
ecti
on p
ost
day
11
Lip
o-1
0.4
mg/k
g
No o
bvio
us
signs
of
tox
icit
y
Lip
o-1
0.4
mg/k
g, qd×
5
All
mic
e hav
e pil
oer
ecti
on p
ost
day
4
Lip
o-1
0.4
mg/k
g, q4d×
4
Thre
e m
ice
hav
e m
ild p
iloer
ecti
on p
ost
day12
Lip
o-1
0.6
mg/k
g, q4d×
4
All
mic
e hav
e pil
oer
ecti
on p
ost
day
12
Lip
o-2
0.4
mg/k
g, q4d×
4
No o
bvio
us
signs
of
tox
icit
y
Lip
o-2
0.8
mg/k
g, q4d×
4
Tw
o m
ice
had
mil
d p
iloer
ecti
on p
ost
day 1
2
Lip
o-2
1.6
mg/k
g, q4d×
4
Tw
o m
ice
hav
e pil
oer
ecti
on an
d hunch
ed post
ure
, an
d th
ree
mic
e hav
e pil
oer
ecti
on p
ost
day
0.
Lip
o-3
0.4
mg/k
g, q4d×
4
One
mouse
has
mil
d p
iloer
ecti
on p
ost
day 1
2
Lip
o-3
0.8
mg/k
g,
q4d×
4
All
mic
e hav
e pil
oer
ecti
on a
nd
hu
nch
ed p
ost
ure
po
st d
ay 2
4 a
nd
two m
ice
wer
e fo
und d
ead.
Lip
o-4
0.4
mg/k
g, q4d×
4
No o
bvio
us
signs
of
tox
icit
y
Lip
o-4
0.8
mg/k
g, q4d×
4
All
mic
e hav
e pil
oer
ecti
on a
nd h
unch
ed p
ost
ure
post
day
24
qd
×5
: d
osi
ng o
nce
dai
ly (
i.v.)
for
5 d
ays.
q4d
×4
: d
osi
ng o
nce
eac
h (
i.v.)
on d
ays
0, 4, 8 a
nd 1
2 (
4 d
ose
s).

244
Fig
ure
7.1
0.
Tum
or
volu
mes
± S
EM
(A
) an
d m
ou
se w
eights
(B
) in
A5
49
tum
or
bea
rin
g n
ude
mic
e re
cord
ed f
or
con
trol
(gro
up
1)
and t
reat
men
t gro
ups
(gro
ups
2–4).
M
ice
wer
e tr
eate
d o
n d
ays
0, 4, 8
, 12
, an
d 1
6 v
ia i
.v. in
ject
ions
into
the
tail
vei
n.

245
a Recovered from euthanized mice 14 days after administration of the last dose. Data are
ng Pt per organ ± SEM determined for 5 organs per group by ICP-MS. b Limit of
detection. * P < 0.05.
7.4. Experimental Section
7.4.1. Materials and instruments.
All reagents were used as obtained from commercial sources without further
purification unless indicated otherwise. HSPC and cholesterol were purchased from
Avanti Polar Lipids, Inc. DPPG and mPEG-DSPE were purchased from Lipoid.
Polycarbonate membranes with pore size of 100, 200 and 400 nm were purchased from
Millipore. Two extrusion devices were used in this study: 1-mL mini extruder (Avanti
Polar Lipids, Inc.) and 10-mL LIPEX™ Extruder (Northern Lipids Inc).
Table 7.5. Average ng Pt in Organsa
Liver Kidneys Lungs Spleen
control 0.4810.200 0.02780.0001b 0.04630.009 0.02780.0001b
P8-A1 1316226 17534 5.030.585 10.041.53
Lipo-1 617180* 55.919.8* 2.310.31* 28.48.2
Lipo-2 976183 15141 5.380.72 97.910.6

246
7.4.2. Optimization of the encapsulation conditions for P8-A1 using film hydration
method
Stock solutions (10 mg/mL) of each lipid were prepared by dissolving in a mixture of
chloroform and methanol with the proportion of 3:1 (V/V). To produce the formulations
with varying lipid compositions, various combinations of stock solutions, the volumes of
which were determined according to the desired molar ratio of lipids, were mixed in a 5-
mL round bottom flask to result in solutions containing 10 mg total lipids. The organic
solvent was then removed by rotary evaporator and subsequently dried in a oil-pump to
allow the formation of a thin lipid film. The lipid film was hydrated at 65 °C in 1 mL of
either Milli Q water or 0.9% saline containing 10 mg/mL P8-A1 for 30 min, followed by
ten freeze-thaw cycles using liquid nitrogen and a water bath (65 °C). The liposomal
suspension was repeatedly extruded at 65 °C through polycarbonate membranes with
pore sizes of 200 and 100 nm (10 times each) using a 1-mL mini extruder (Avanti Polar
Lipids). The non-encapsulated platinum complex was removed from the liposomal
suspension by gel permeation chromatography using a PD-10 Sephadex G-25 column
(GE Healthcare, Buckinghamshire, UK), eluted with 0.9% saline solution containing 5%
glucose.
7.4.3. Preparation of liposomes using ethanol injection method
The lipids DPPG, HSPC and mPEG-DSPE (80:15:5, molar ratio) were dissolved in 0.4
mL ethanol to produce the alcoholic solution of lipids with a concentration of 25 mg/mL.
Next, the ethanol solution was added slowly into 20-times volume of a 0.9% saline
solution containing 25 mg/mL P8-A1. The solution was stirred at room temperature for 1

247
h, followed by dialysis against 0.9% saline solution containing 5% glucose at 4 °C for 24
h. The liposomes formed spontaneously as the ethanol was completely removed after
dialysis. The liposomal suspension in water was then filtered through the 220 nm filter
membrane for sterilization, and stored at 4 °C.
7.4.4. Preparation of liposomes using reverse-phase evaporation method
DPPG, HSPC and mPEG2000-DSPE at a molar ratio of 80:15:5 (the total weight of
lipids is 10 mg) were dissolved in a mixture of chloroform and diethyl ether (1:2, v/v),
which was added into an aqueous solution containing 2 mg of P8-A1 dissolved in 0.9%
saline/5% glucose solution. The ratio of aqueous to organic phase was 1:3 (V/V). The
mixture was then sonicated at room temperature for 10 min, and the organic solvents
were slowly removed by rotary evaporation. The resultant aqueous suspension was
extruded with a mini extruder following the method described in section 7.4.2.
7.4.5. Preparation of liposomes for biological evaluation
Liposomes used for biological testing were prepared using the film hydration method as
described in section 7.4.2. The formulations were homogenized using a 10-mL LIPEX™
Extruder (Northern Lipids Inc.). The unbound platinum complex was removed from
liposomal suspension by gel permeation chromatography using a PD-10 Sephadex G-25
column (GE Healthcare, Buckinghamshire, UK), eluted with 0.9% saline/5% glucose
solution, followed by dialysis against 0.9% saline/5% glucose solution at 4 °C for 24
hour. The liposomal formulation was store at 4 °C when not in use.

248
7.4.6. Characterization of liposomes
7.4.6.1. Size and zeta-potential of liposomes
Liposome suspensions (~10 mg/mL lipids, 25 μL) were diluted in 1 mL of Milli-Q
water and filtered through a membrane filter with a pore size of 220 nm. The
hydrodynamic diameter was assessed by dynamic light scattering (DLS) using a Zetasizer
ZS90 (Malvern Instruments) in automatic mode (Software version 6.34) using the
intrument’s default settings for refractive index and viscosity of the dispersant. All
measurements were run at 25 °C. The zeta-potential of was assessed in Milli-Q water at
pH 6.8–7 using the Zetasizer ZS90 instrument. Triplicate measurements were performed.
The results are presented as paticle sizes and charges ± SD.
7.4.6.2. Transmission Electron Microscopy
The liposome samples were diluted before analysis with 0.9% saline and then pipetted
on carbon-stabilized Formvar copper grids. Excess sample was removed with a filter
paper. A drop of 2% (w/v) aqueous solution of uranyl acetate was added and the sample
was allowed to stain for 1 min. The samples were dried at room temperature and then
imaged using FEI Tecnai BioTwin Transmission Electron Microscope operated at 80 kV.
The images were captured with an AMT 2vu camera capable of 12 megapixel images and
analyzed with the included AMT camera software.

249
7.4.6.3. Encapsulation efficiency of liposomes
To determine the content of P8-A1 entrapped in the liposomes, methanol was used to
lyse the liposomal lipid bilayer. Briefly, 100 µL of liposomes before and after
purification were mixed with 1900 µL of methanol. The mixtures were sonicated for 15
min and the amount of platinum-acridine was determined spectrophotometrically at 413
nm (ɛ413 = 10000 M-1cm-1). Encapsulation efficiencies (EE %) were determined using the
following equation: EE% = ([Drug]purified/[Drug]initial) × 100%.
7.4.6.4. Determination of lipid composition
Phospholipids concentration was determined using Bartlett’s assay [392]. The assay
relies on the interaction between the inorganic phosphates and ammonium molybdate,
which can be monitored by reaction with 4-amino-3-hydroxyl-1-naphtalene sulfonic acid
at 180 °C to yield a blue-colored product and quantified at 815 nm. The amount of
cholesterol was quantification by reversed phase HPLC using a UV detector with the
wavelength at 207 nm. The samples were separated using a 4.6 mm × 150 mm reverse-
phase Agilent ZORBAX SB-C18 (5 mm) analytical column at 25 °C, by using the
following solvent system: solvent A, optima water, and solvent B, methanol/0.1 % formic
acid, with a flow rate of 0.5 mL/min and a gradient of 95 % A to 5 % A over 20 minutes.

250
7.4.7. In vitro stability assay
7.4.7.1. Drug leakage under physiologically relevant conditions
Liposomal formulations (~1 mg/ml lipids) were incubated in 1 Ml PBS (pH 7.4) at
37 °C. At selected time-points, samples (in triplicate) were eluted with 0.9% NaCl
solution by size exclusion chromatography using PD-10 Sephadex G-25 columns to
remove released free P8-A1. The amount of drug retened was then measured
spectroscopically as described in 7.4.5.2.
7.3.6.2. In vitro drug release of P8-A1 at elevated temperature
Lipo-1 (~1 mg/mL lipids) was incubated in 1 mL PBS (pH 7.4) at 42 °C and 44 °C. At
selected time-points, samples (in triplicate) were eluted with 0.9% NaCl solution by size
exclusion chromatography using PD-10 Sephadex G-25 columns to remove released free
P8-A1. The amount of drug retention was then measured spectroscopically as described
in 7.4.2.
7.4.6.3. Storage stability
Purified liposomal formulations (10 mL) were sterilized by passing them through 0.22
µm membrane filters and stored at 4 °C for 1, 4, and 8 weeks. Samples were tested for
particle size and drug retention within liposomes. The particle size was evaluated by DLS
measurements. To determine the concentration of encapsulated P8-A1, samples were
passed through PD-10 Sephadex G-25 columns and eluted with 0.9% saline solution to

251
remove any free platinum complex. The amount of drug retented was then measured
spectroscopically as described above. The samples before the storage were used as
controls. The liposomes lysed by methanol were used as positive controls. Measurements
were performed in triplicate.
7.4.8. Cellular uptake of Lipo-1 in NCI-H460
NCI-H460 cells were seeded into poly-d-lysine coated glass-bottom Petri dishes
(MatTeck Corporation, Ashland, MD, USA) with 105 cells mL−1 suspended in 2 mL of
medium per dish. Cells were incubated overnight and then treated with Lipo-1 and P8–
A1 (5 μm), or medium for controls for designed time points. After treatment, medium
was removed and the cells were stained with 75 nM of LysoTracker Red DND-99
(Invitrogen) in 2 mL pre-warmed Hank’s Balanced Salt Solution (HBSS) at 37 °C for 30
min. After staining with LysoTracker, solutions were replaced with fresh medium.
Images were collected using a Zeiss LSM 710 confocal microscope (Carl Zeiss
MicroImaging, Thornwood, NY) using a 63× (PLAN APO, 1.2 NA) objective lens. All
images were acquired in multi-track configuration mode to minimize excitation cross talk
and emission bleed-through. The fluorescence of P8–A1 (blue), and Lyso–Tracker Red
DND-99, was excited/collected at 405/477 nm and 561/591 nm, respectively. ZEN
software (blue edition, Carl Zeiss, 2011) was used for image processing. Confocal image
planes for each channel were not contrast-adjusted (unless indicated) or otherwise
changed. Panels were assembled and annotated without any additional manipulation of
images in Adobe Photoshop CS2.

252
7.4.9. Animal studies
7.4.9.1. Evaluation of maximum tolerated dose (MTD)
MTD of P8-A1 and liposomeal formulations were evaluated in Swiss-Webster female
mice (5 to 6 week-old, 22-25 g). Mice (5 per group) were treated i.v. with the doses
ranging from 0.2-0.8 mg/kg every four days for four times. The acute toxicity was
assessed based on animal survival and visual signs of toxicity including body weight loss
(BWL) and clinical signs. Clinical signs observed were piloerection or hunched posture.
Animals displaying mild piloerection were considered to be indicating mild toxicity at
that dose.
7.4.9.2. A549 Xenograft study
A549 xenografts were established in nude athymic female mice via subcutaneous
injections. Treatment began when the average tumor volume was approximately 100 mm3.
The tumor-bearing mice were randomized depending on tumor volume into three groups
of eight test animals each: one control group receiving vehicle only, P8-A1 group treated
at 0.4 mg/kg q4d×4, Lipo-1 group treated at 0.4 mg/kg q4d×4, and Lipo-2 group treated
at 0.8 mg/kg q4d×4. Animal weights and tumor volumes were measured and recorded for
17 days after the first dose was administered. Tumor volumes were determined using the
formula :V (mm3) = d2 × D/2, where d and D are the shortest and longest dimension of
the tumor, respectively, and are reported as the sum of both tumors for each test animal.
At the end of the study, all animals were euthanized and disposed off according to
Standard Operating Procedures (SOPs). One tumor per test animal as well as lungs,

253
spleens, and kidneys were removed during necropsy. The concentration of Pt in these
tissues was determined by quadrupole inductively-coupled mass spectrometry (ICP-MS)
on a Thermo ICP-MS instrument at the Analytic Chemistry Service facility of Research
Triangle Institute, Research Triangle Park, NC. The xenograft study was performed by
Washington Biotechnology Inc. (Simpsonville, MD), PHS-approved Animal Welfare
Assurance, # A4912-01. Data analysis for weight and tumor volumes was done with the
Student’s t-test. Only P values of less than 0.05% were considered as statistically
significant. Few outliers were identified using Grubb’s outlier test at 90% and 95%
confidence and removed from the data set.

254
CHAPTER 8
SUMMARY AND OUTLOOK
Despite the success of cisplatin as a mainstay chemotherapeutic agent, tumor resistance
and severe side effects have inspired the development of new generations of platinum-
based agents. Platinum complexes with entirely different structural features have been
proposed to produce DNA-platinum adducts that would be recognized differently by the
proteins involved in cisplatin-induced cell death. Based on this reasoning, numerous
non-classical platinum agents have been generated. Among these are platinum-acridine
hybrid agents, which represent a unique class of monofunctional platinum complexes
capable of simultaneously intercalating and platinating DNA. While platinum-acridine
derivatives have demonstrated superior cytotoxicity in human lung cancer models, their
application as systemic cancer treatments is limited by severe systemic toxicity. The goal
of this dissertation was to develop the chemistry and devise new strategies for improving
the pharmacological properties of these agents. These include the design of lipophilic and
less reactive prodrugs, targeted and nontargeted vehicles for safer delivery of cytotoxic
payload to diseased tissue, and multifunctional agents to overcome the chemoresistance
of certain cancers. To achieve these challenging goals, new chemistry-based
methodologies had to be developed that allow platinum-acridines to be turned into
“smarter” anticancer agents. In particular, platinum-compatible chemistry was needed
that allows functionalization of platinum-acridines and their subsequent conjugation with
targeting and/or other bioactive molecules.

255
In Chapter 2, we have presented a library approach for the discovery of functionalized
platinum-acridine analogues. This method was based on a highly efficient addition
reaction between a platinum-activated nitrile precursor and a secondary amine, which
results in an amidine linkage that connects the intercalator to the metal. 60 microscale
reactions were performed in parallel, characterized and tested in cell models, which
helped us identify several conjugatable warheads that maintained potent cytotoxicity.
This assay also allowed us to study structure-activity relationships in functionalized
platinum-acridines.
Next, we explored various conjugation chemistries. To produce platinum-acridine
based conjugates, two strategies, “post-modification” and “pre-assembly”, have been
validated in this dissertation. In Chapter 3, a “post-labeling” method using a clickable
azide-modified platinum-acridine was developed. In a proof-of-concept study we
exploited this type of compound, in conjunction with copper-based and copper-free click
chemistries, to investigate the subcellular distribution of this type of platinum complex.
Cancer cells were first incubated with the azide derivative, followed by labeling of
intracellular platinum compound with an alkyne-containing fluorophore. Unlike the bulky
fluorophore-tagged derivative, the clickable azide derivative faithfully mimics the
subcellular localization of the parent platinum-acridines. The nucleolar localization of
platinum-acridine was discovered in this study, which is a previously unexplored target
for platinum-based drugs. Although very efficient, click chemistry may not be a widely
applicable method due to expensive precursors and additional steps needed to incorporate
alkyne groups into the non-platinum components.

256
Chapter 4 provides an example of the “pre-assembly” method, in which the platinum
moiety was not incorporated until the sensitizer/tumor-targeted ligand had been
assembled in its entirety. In this study, we linked a hydroxyl-modified platinum-acridine
to the estrogen receptor inhibitor endoxifen with the goal of producing a breast cancer
targeted agent. Coupling was achieved by a chemically robust carbamate bond. To
generate the conjugate, endoxifen was tethered to 9-amino acridine precursor to produce
the organic ligand that was in turn added to the platinum-nitrile precursor. Our “pre-
assembly” method holds promise of providing a universal approach for constructing
small molecule based conjugates, but may not be useful to conjugate functionalized
platinum-acridine warheads to macromolecules, such as monoclonal antibodies, proteins,
and synthetic polymers. The endoxifen-platinum-acridine conjugates were tested in breast
cancer cells where they showed the same level of cytotoxicity as that observed for the
parent “warhead”. The non-cleavable carbamate linker was identified as a potential
drawback of the design and prompted us to also pursue cleavable platinum-acridine
conjugates.
Chapter 5 described ester-based platinum-acridine prodrugs with improved drug-like
properties. Butyric acid and valproic acid were installed to confer lipohilicity to
platinum-acridines, which was expected to improve their systemic circulation and tumor
penetration. The valproate ester was found to undergo a carboxylesterase 2 (CES-2)-
mediated prodrug activation. Because CES-2 is overexpressed in many cancers, prodrug-
activation may occur tumor-selectively. However, activation by ubiquitous CES-2 in the
liver also has to be considered a possible mechanism of drug activation. In addition to
enzymatic cleavage, a platinum-mediated ester hydrolysis was also identified, in which

257
platinum(II) is able to accelerate ester hydrolysis in a low-chloride environment, such as
the cytosol.
As a continuation of the ester conjugation chemistry developed in Chapter 5, we
designed a fragment-based platform for the discovery of multifunctional platinum-
acridine conjugates (Chapter 6). Hydroxyl-modified platinum-acridines were extended
with a succinate linker, which was then conjugated with bioactive molecules containing a
primary or secondary amino group. Because of their steric hindrance, the ester groups in
the conjugates should not serve as substrates of CES-2 (and other esterases). We
demonstrated that these esters undergo metal-facilitated hydrolysis similar to the prodrug
models developed in Chapter 5. The major goal of this project was to develop
methodology that allows post-modification of the carboxylic acid modified platinum-
acridines via formation of an amide bond. We exhaustively screened various coupling
reagents and conditions that allow clean conversion of precursors to the desired products
with yields of higher than 90%. A total of 20 microscale reactions were assembled to
demonstrate the versatility and utility of this reaction. The high efficiency of this method
may allow the screening of high-throughput libraries in the future. Similar coupling
chemistries may also be an option for generating other cleavable linkers (e.g., pH-
sensitive hydrazones) for advanced applications such as conjugates containing antibodies
or targeted peptides.
Attempts to physically encapsulate platinum-acridine in nanocarriers are reported in
Chapter 7. In this chapter, we have developed a method to effectively load platinum-
acridines into negatively charged liposomes by taking advantage of the cationic nature of
the platinum complexes. One formulation shows improved anticancer efficacy relative to

258
the unencapsulated drug. Importantly, the liposomal formulations also appear to reduce
the accumulation of platinum in off-target tissues. Future efforts will focus on increasing
the MTD of the liposomal formulations by optimizing the lipid components and drug
loading and release properties, and by incorporating tumor-targeting ligands into the lipid
bilayers.
In conclusion, the research in this dissertation has established various modular
conjugation chemistries, new drug release mechanisms, and nanocarrier-based delivery,
which may be useful for the development of next-generation platinum-acridine agents
and of metallodrugs in general. In this respect, highly efficient transformations, such as
the platinum-mediated nitrile-amine addition and amide coupling chemistries developed
in Chapters 2 and 6 will prove indispensible tools.

259
REFERENCES
[1] N.J. Wheate, S. Walker, G.E. Craig, R. Oun, The status of platinum anticancer drugs
in the clinic and in clinical trials, Dalton Transactions, 39 (2010) 8113-8127.
[2] Z.H. Siddik, Cisplatin: mode of cytotoxic action and molecular basis of resistance,
Oncogene, 22 (2003) 7265-7279.
[3] L. Kelland, The resurgence of platinum-based cancer chemotherapy, Nature Reviews
Cancer, 7 (2007) 573-584.
[4] A. Tesniere, F. Schlemmer, V. Boige, O. Kepp, I. Martins, F. Ghiringhelli, L.
Aymeric, M. Michaud, L. Apetoh, L. Barault, J. Mendiboure, J.P. Pignon, V. Jooste,
P. van Endert, M. Ducreux, L. Zitvogel, F. Piard, G. Kroemer, Immunogenic death
of colon cancer cells treated with oxaliplatin, Oncogene, 29 (2010) 482-491.
[5] A.R. de Biasi, J. Villena-Vargas, P.S. Adusumilli, Cisplatin-Induced Antitumor
Immunomodulation: A Review of Preclinical and Clinical Evidence, Clinical
Cancer Research, 20 (2014) 5384-5391.
[6] D.Y.Q. Wong, W.W.F. Ong, W.H. Ang, Induction of Immunogenic Cell Death by
Chemotherapeutic Platinum Complexes, Angewandte Chemie-International Edition,
54 (2015) 6483-6487.
[7] D.Y.Q. Wong, C.H.F. Yeo, W.H. Ang, Immuno-Chemotherapeutic Platinum(IV)
Prodrugs of Cisplatin as Multimodal Anticancer Agents, Angewandte Chemie-
International Edition, 53 (2014) 6752-6756.

260
[8] H.S. Oberoi, N.V. Nukolova, A.V. Kabanov, T.K. Bronich, Nanocarriers for delivery
of platinum anticancer drugs, Advanced Drug Delivery Reviews, 65 (2013) 1667-
1685.
[9] S. Dasari, P.B. Tchounwou, Cisplatin in cancer therapy: Molecular mechanisms of
action, European Journal of Pharmacology, 740 (2014) 364-378.
[10] M.D. Hall, M. Okabe, D.W. Shen, X.J. Liang, M.M. Gottesman, The role of cellular
accumulation in determining sensitivity to platinum-based chemotherapy, Annual
Review of Pharmacology and Toxicology, 48 (2008) 495-535.
[11] C.A. Larson, B.G. Blair, R. Safaei, S.B. Howell, The role of the mammalian copper
transporter 1 in the cellular accumulation of platinum-based drugs, Molecular
Pharmacology, 75 (2009) 324-330.
[12] S. Zhang, K.S. Lovejoy, J.E. Shima, L.L. Lagpacan, Y. Shu, A. Lapuk, Y. Chen, T.
Komori, J.W. Gray, X. Chen, S.J. Lippard, K.M. Giacomini, Organic cation
transporters are determinants of oxaliplatin cytotoxicity, Cancer Research, 66 (2006)
8847-8857.
[13] P. Mistry, L.R. Kelland, G. Abel, S. Sidhar, K.R. Harrap, The relationships between
glutathione, glutathione-S-transferase and cytotoxicity of platinum drugs and
melphalan in eight human ovarian carcinoma cell lines, British Journal of Cancer,
64 (1991) 215-220.
[14] A.D. Lewis, J.D. Hayes, C.R. Wolf, Glutathione and glutathione-dependent enzymes
in ovarian adenocarcinoma cell lines derived from a patient before and after the
onset of drug resistance: intrinsic differences and cell cycle effects, Carcinogenesis,
9 (1988) 1283-1287.

261
[15] K.V. Ferry, T.C. Hamilton, S.W. Johnson, Increased nucleotide excision repair in
cisplatin-resistant ovarian cancer cells - Role of ERCC1-XPF, Biochemical
Pharmacology, 60 (2000) 1305-1313.
[16] E. Reed, ERCC1 and clinical resistance to platinum-based therapy, Clinical Cancer
Research, 11 (2005) 6100-6102.
[17] A. Gadducci, S. Cosio, S. Muraca, A.R. Genazzani, Molecular mechanisms of
apoptosis and chemosensitivity to platinum and paclitaxel in ovarian cancer:
biological data and clinical implications, European Journal of Gynaecological
Oncology, 23 (2002) 390-396.
[18] M. Galanski, B.K. Keppler, Searching for the Magic Bullet: Anticancer Platinum
Drugs Which Can Be Accumulated or Activated in the Tumor Tissue, Anti-Cancer
Agents in Medicinal Chemistry, 7 (2007) 55-73.
[19] J.J. Wilson, S.J. Lippard, In Vitro Anticancer Activity of cis-Diammineplatinum(II)
Complexes with beta-Diketonate Leaving Group Ligands, Journal of Medicinal
Chemistry, 55 (2012) 5326-5336.
[20] S. Hochreuther, R. Puchta, R. van Eldik, Thermodynamic and Kinetic Studies on
Novel Dinuclear Platinum(II) Complexes Containing Bidentate N,N-donor ligands,
Inorganic Chemistry, 50 (2011) 8984-8996.
[21] M.S. Robillard, M. Bacac, H. van den Elst, A. Flamigni, G.A. van der Marel, J.H.
van Boom, J. Reedijk, Automated parallel solid-phase synthesis and anticancer
screening of a library of peptide-tethered platinum(II) complexes, Journal of
Combinatorial Chemistry, 5 (2003) 821-825.

262
[22] K. Brasseur, V. Leblanc, F. Fabi, S. Parent, C. Descoteaux, G. Berube, E. Asselin,
ER alpha-Targeted Therapy in Ovarian Cancer Cells by a Novel Estradiol-
Platinum(II) Hybrid, Endocrinology, 154 (2013) 2281-2295.
[23] M. Kvasnica, L. Rarova, J. Oklestkova, M. Budesinsky, L. Kohout, Synthesis and
cytotoxic activities of estrone and estradiol cis-dichloroplatinum(II) complexes,
Bioorganic & Medicinal Chemistry, 20 (2012) 6969-6978.
[24] P. Saha, C. Descoteaux, K. Brasseur, S. Fortin, V. Leblanc, S. Parent, E. Asselin, G.
Berube, Synthesis, antiproliferative activity and estrogen receptor alpha affinity of
novel estradiol-linked platinum(II) complex analogs to carboplatin and oxaliplatin.
Potential vector complexes to target estrogen-dependent tissues, European Journal
of Medicinal Chemistry, 48 (2012) 385-390.
[25] O. Aronov, A.T. Horowitz, A. Gabizon, D. Gibson, Folate-targeted PEG as a
potential carrier for carboplatin analogs. Synthesis and in vitro studies,
Bioconjugate Chemistry, 14 (2003) 563-574.
[26] S. Top, E.B. Kaloun, A. Vessieres, G. Leclercq, I. Laios, M. Ourevitch, C. Deuschel,
M.J. McGlinchey, G. Jaouen, Tamoxifen derivatives for delivery of the antitumoral
(DACH)Pt group: Selective synthesis by McMurry coupling, and biochemical
behaviour, ChemBioChem, 4 (2003) 754-761.
[27] U. Kalinowska-Lis, J. Ochocki, K. Matlawska-Wasowska, Trans geometry in
platinum antitumor complexes, Coordination Chemistry Reviews, 252 (2008)
1328-1345.

263
[28] V. Brabec, J. Kasparkova, Modifications of DNA by platinum complexes - Relation
to resistance of tumors to platinum antitumor drugs, Drug Resistance Updates, 8
(2005) 131-146.
[29] E. BernalMendez, M. Boudvillain, F. GonzalezVilchez, M. Leng, Chemical
versatility of transplatin monofunctional adducts within multiple site-specifically
platinated DNA, Biochemistry, 36 (1997) 7281-7287.
[30] T.G. Appleton, A.J. Bailey, K.J. Barnham, J.R. Hall, Aspects of the solution
chemistry of trans-diammineplatinum(II) complexes, Inorganic Chemistry, 31
(1992) 3077-3082.
[31] N. Farrell, L.R. Kelland, J.D. Roberts, M. Vanbeusichem, Activation of the
transgeometry in platinum antitumor complexes-a survey of the cytotoxicity of
transcomplexes containing planar ligands in murine-L1210 and human tumr panels
and studies on their mechanism of action, Cancer Research, 52 (1992) 5065-5072.
[32] O. Novakova, J. Kasparkova, J. Malina, G. Natile, V. Brabec, DNA-protein cross-
linking by trans-[PtCl(2)(E-iminoether)(2)]. A concept for activation of the trans
geometry in platinum antitumor complexes, Nucleic Acids Research, 31 (2003)
6450-6460.
[33] M. Frybortova, O. Novakova, J. Stepankova, V. Novohradsky, D. Gibson, J.
Kasparkova, V. Brabec, Activation of trans geometry in bifunctional mononuclear
platinum complexes by a non-bulky methylamine ligand, Journal of Inorganic
Biochemistry, 126 (2013) 46-54.
[34] F.J. Ramos-Lima, O. Vrana, A.G. Quiroga, C. Navarro-Ranninger, A. Halamikova,
H. Rybnickova, L. Hejmalova, V. Brabec, Structural characterization, DNA

264
interactions, and cytotoxicity of new transplatin analogues containing one aliphatic
and one planar heterocyclic amine ligand, Journal of Medicinal Chemistry, 49
(2006) 2640-2651.
[35] S.M. Aris, N.P. Farrell, Towards Antitumor Active trans-Platinum Compounds,
European Journal of Inorganic Chemistry, (2009) 1293-1302.
[36] T.C. Johnstone, J.J. Wilson, S.J. Lippard, Monofunctional and Higher-Valent
Platinum Anticancer Agents, Inorganic Chemistry, 52 (2013) 12234-12249.
[37] T. Ren, D.P. Bancroft, W.I. Sundquist, A. Masschelein, M.V. Keck, S.J. Lippard,
Synthesis,characterization, DNA-binding properties, and solution thermochromism
of platinum(II) complexes of the ethidinium cation-regiospecificity in a DNA-
promoted reaction, Journal of the American Chemical Society, 115 (1993) 11341-
11352.
[38] S. Wu, X. Wang, C. Zhu, Y. Song, J. Wang, Y. Li, Z. Guo, Monofunctional
platinum complexes containing a 4-nitrobenzo-2-oxa-1,3-diazole fluorophore:
Distribution in tumour cells, Dalton Transactions, 40 (2011) 10376-10382.
[39] L.S. Hollis, A.R. Amundsen, E.W. Stern, Chemical and biological properties of a
new series of cis-diammineplatinum(II) antitumors agents containing 3 nitrogen
donors- Cis-Pt(NH3)2(N-dornor)Cl, Journal of Medicinal Chemistry, 32 (1989)
128-136.
[40] W.I. Sundquist, D.P. Bancroft, S.J. Lippard, Synthesis, characterization , and
biological-activity of cis-diammineplatinum (II) complexes of the DNA
intercalators 9-aminoacridine and chloroquine Journal of the American Chemical
Society, 112 (1990) 1590-1596.

265
[41] K.S. Lovejoy, R.C. Todd, S. Zhang, M.S. McCormick, J.A. D'Aquino, J.T. Reardon,
A. Sancar, K.M. Giacomini, S.J. Lippard, cis-
diammine(pyridine)chloroplatinum(II), a monofunctional platinum(II) antitumor
agent: Uptake, structure, function, and prospects, Proceedings of the National
Academy of Sciences of the U.S.A., 105 (2008) 8902-8907.
[42] K.S. Lovejoy, M. Serova, I. Bieche, S. Emami, M. D'Incalci, M. Broggini, E. Erba,
C. Gespach, E. Cvitkovic, S. Faivre, E. Raymond, S.J. Lippard, Spectrum of
Cellular Responses to Pyriplatin, a Monofunctional Cationic Antineoplastic
Platinum(II) Compound, in Human Cancer Cells, Molecular Cancer Therapeutics,
10 (2011) 1709-1719.
[43] G. Zhu, M. Myint, W.H. Ang, L. Song, S.J. Lippard, Monofunctional Platinum-
DNAAdducts Are Strong Inhibitors of Transcription and Substrates for Nucleotide
Excision Repair in Live Mammalian Cells, Cancer Research, 72 (2012) 790-800.
[44] G.Y. Park, J.J. Wilson, Y. Song, S.J. Lippard, Phenanthriplatin, a monofunctional
DNA-binding platinum anticancer drug candidate with unusual potency and
cellular activity profile, Proceedings of the National Academy of Sciences of the
U.S.A., 109 (2012) 11987-11992.
[45] N. Margiotta, S. Savino, V. Gandin, C. Marzano, G. Natile, Monofunctional
Platinum(II) Complexes with Potent Tumor Cell Growth Inhibitory Activity: The
Effect of a Hydrogen-Bond Donor/Acceptor N-Heterocyclic Ligand,
ChemMedChem, 9 (2014) 1161-1168.
[46] M.W. Kellinger, G.Y. Park, J. Chong, S.J. Lippard, D. Wang, Effect of a
Monofunctional Phenanthriplatin-DNA Adduct on RNA Polymerase II

266
Transcriptional Fidelity and Translesion Synthesis, Journal of the American
Chemical Society, 135 (2013) 13054-13061.
[47] B. Wang, Z. Wang, F. Ai, W.K. Tang, G. Zhu, A monofunctional platinum(II)-based
anticancer agent from a salicylanilide derivative: Synthesis, antiproliferative
activity, and transcription inhibition, Journal of Inorganic Biochemistry, 142 (2015)
118-125.
[48] N.P. Farrell, Progress in platinum-derived drug development, Drugs of the Future,
37 (2012) 795-806.
[49] N. Farrell, Polynuclear platinum drugs, Metal Ions in Biolgical Systems, Vol 42:
Metal Complexes in Tumor Diagnosis and as Anticancer Agents, 42 (2004) 251-
296.
[50] J.B. Mangrum, N.P. Farrell, Excursions in polynuclear platinum DNA binding,
Chemical Communications, 46 (2010) 6640-6650.
[51] C. Manzotti, G. Pratesi, E. Menta, R. Di Domenico, E. Cavalletti, H.H. Fiebig, L.R.
Kelland, N. Farrell, D. Polizzi, R. Supino, G. Pezzoni, F. Zunino, BBR 3464: A
novel triplatinum complex, exhibiting a preclinical profile of antitumor efficacy
different from cisplatin, Clinical Cancer Research, 6 (2000) 2626-2634.
[52] A. Hegmans, Y. Qu, L.R. Kelland, J.D. Roberts, N. Farrell, Novel approaches to
polynuclear platinum pro-drugs. Selective release of cytotoxic platinum spermidine
species through hydrolytic cleavage of carbamates, Inorganic Chemistry, 40 (2001)
6108-6114.

267
[53] Y. Zhao, W. He, P. Shi, J. Zhu, L. Qiu, L. Lin, Z. Guo, A positively charged
trinuclear 3N-chelated monofunctional platinum complex with high DNA affinity
and potent cytotoxicity, Dalton Transactions, (2006) 2617-2619.
[54] L. Zerzankova, H. Kostrhunova, M. Vojtiskova, O. Novakova, T. Suchankova, M.
Lin, Z. Guo, J. Kasparkova, V. Brabec, Mechanistic insights into antitumor effects
of new dinuclear cis Pt-II complexes containing aromatic linkers, Biochemical
Pharmacology, 80 (2010) 344-351.
[55] S. Komeda, Y.-L. Lin, M. Chikuma, A Tetrazolato-Bridged Dinuclear Platinum(II)
Complex Exhibits Markedly High in vivo Antitumor Activity against Pancreatic
Cancer, ChemMedChem, 6 (2011) 987-990.
[56] S. Komeda, T. Moulaei, K.K. Woods, M. Chikuma, N.P. Farrell, L.D. Williams, A
third mode of DNA binding: Phosphate clamps by a polynuclear platinum complex,
Journal of the American Chemical Society, 128 (2006) 16092-16103.
[57] J. Malina, N.P. Farrell, V. Brabec, Substitution-Inert Trinuclear Platinum Complexes
Efficiently Condense/Aggregate Nucleic Acids and Inhibit Enzymatic Activity,
Angewandte Chemie-International Edition, 53 (2014) 12812-12816.
[58] B.T. Benedetti, E.J. Peterson, P. Kabolizadeh, A. Martinez, R. Kipping, N.P. Farrell,
Effects of Noncovalent Platinum Drug-Protein Interactions on Drug Efficacy: Use
of Fluorescent Conjugates as Probes for Drug Metabolism, Molecular
Pharmaceutics, 8 (2011) 940-948.
[59] W.S. El-Deiry, The role of p53 in chemosensitivity and radiosensitivity, Oncogene,
22 (2003) 7486-7495.

268
[60] A.L. Harris, J.J. Ryan, N. Farrell, Biological consequences of trinuclear platinum
complexes: comparison of {trans-PtCl(NH3)2}2mu-(trans-Pt(NH3)2)H2N(CH2)6-
NH)2) (4+) (BBR 3464) with its noncovalent congeners, Molecular Pharmacology,
69 (2006) 666-672.
[61] J.B. Mangrum, B.J. Engelmann, E.J. Peterson, J.J. Ryan, S.J. Berners-Price, N.P.
Farrell, A new approach to glycan targeting: enzyme inhibition by oligosaccharide
metalloshielding, Chemical Communications, 50 (2014) 4056-4058.
[62] E.J. Peterson, V.R. Menon, L. Gatti, R. Kipping, D. Dewasinghe, P. Perego, L.F.
Povirk, N.P. Farrell, Nucleolar Targeting by Platinum: p53-Independent Apoptosis
Follows rRNA Inhibition, Cell-Cycle Arrest, and DNA Compaction, Molecular
Pharmaceutics, 12 (2015) 287-297.
[63] J. Malina, N.P. Farrell, V. Brabec, DNA Condensing Effects and Sequence
Selectivity of DNA Binding of Antitumor Noncovalent Polynuclear Platinum
Complexes, Inorganic Chemistry, 53 (2014) 1662-1671.
[64] H. Baruah, C.L. Rector, S.M. Monnier, U. Bierbach, Mechanism of action of non-
cisplatin type DNA-targeted platinum anticancer agents: DNA interactions of novel
acridinylthioureas and their platinum conjugates, Biochemical Pharmacology, 64
(2002) 191-200.
[65] E.T. Martins, H. Baruah, J. Kramarczyk, G. Saluta, C.S. Day, G.L. Kucera, U.
Bierbach, Design, synthesis, and biological activity of a novel non-cisplatin-type
platinum-acridine pharmacophore, Journal of Medicinal Chemistry, 44 (2001)
4492-4496.

269
[66] H. Baruah, C.G. Barry, U. Bierbach, Platinum-intercalator conjugates: From DNA-
targeted cisplatin derivatives to adenine binding complexes as potential modulators
of gene regulation, Current Topics in Medicinal Chemistry, 4 (2004) 1537-1549.
[67] M.E. Budiman, R.W. Alexander, U. Bierbach, Unique base-step recognition by a
platinum-acridinylthiourea conjugate leads to a DNA damage profile
complementary to that of the anticancer drug cisplatin, Biochemistry, 43 (2004)
8560-8567.
[68] L.A. Graham, J. Suryadi, T.K. West, G.L. Kucera, U. Bierbach, Synthesis, Aqueous
Reactivity, and Biological Evaluation of Carboxylic Acid Ester-Functionalized
Platinum-Acridine Hybrid Anticancer Agents, Journal of Medicinal Chemistry, 55
(2012) 7817-7827.
[69] S.M. Hess, J.G. Anderson, U. Bierbach, A non-crosslinking platinum-acridine
hybrid agent shows enhanced cytotoxicity compared to clinical BCNU and
cisplatin in glioblastoma cells, Bioorganic & Medicinal Chemistry Letters, 15
(2005) 443-446.
[70] R. Guddneppanavar, G. Saluta, G.L. Kucera, U. Bierbach, Synthesis, biological
activity, and DNA-damage profile of platinum-threading intercalator conjugates
designed to target adenine, Journal of Medicinal Chemistry, 49 (2006) 3204-3214.
[71] S.M. Hess, A.M. Mounce, R.C. Sequeira, T.M. Augustus, M.C. Ackley, U. Bierbach,
Platinum-acridinylthiourea conjugates show cell line-specific cytotoxic
enhancement in H460 lung carcinoma cells compared to cisplatin, Cancer
Chemotherapy and Pharmacology, 56 (2005) 337-343.

270
[72] R. Guddneppanavar, U. Bierbach, Adenine-N3 in the DNA Minor Groove - An
Emerging Target for Platinum Containing Anticancer Pharmacophores, Anti-
Cancer Agents in Medicinal Chemistry, 7 (2007) 125-138.
[73] Z. Ma, G. Saluta, G.L. Kucera, U. Bierbach, Effect of linkage geometry on
biological activity in thiourea- and guanidine-substituted acridines and platinum-
acridines, Bioorganic & Medicinal Chemistry Letters, 18 (2008) 3799-3801.
[74] R. Guddneppanavar, J.R. Choudhury, A.R. Kheradi, B.D. Steen, G. Saluta, G.L.
Kucera, C.S. Day, U. Bierbach, Effect of the diamine nonleaving group in
platinum-acridinylthiourea conjugates on DNA damage and cytotoxicity, Journal of
Medicinal Chemistry, 50 (2007) 2259-2263.
[75] M.C. Ackley, C.G. Barry, A.M. Mounce, M.C. Farmer, B.E. Springer, C.S. Day,
M.W. Wright, S.J. Berners-Price, S.M. Hess, U. Bierbach, Structure-activity
relationships in platinum-acridinylthiourea conjugates: effect of the thiourea
nonleaving group on drug stability, nucleobase affinity, and in vitro cytotoxicity,
Journal of Biological Inorganic Chemistry, 9 (2004) 453-461.
[76] Z. Ma, L. Rao, U. Bierbach, Replacement of a Thiourea-S with an Amidine-NH
Donor Group in a Platinum-Acridine Antitumor Compound Reduces the Metal's
Reactivity with Cysteine Sulfur, Journal of Medicinal Chemistry, 52 (2009) 3424-
3427.
[77] J.R. Choudhury, L. Rao, U. Bierbach, Rates of intercalator-driven platination of
DNA determined by a restriction enzyme cleavage inhibition assay, Journal of
Biological Inorganic Chemistry, 16 (2011) 373-380.

271
[78] H. Baruah, C.S. Day, M.W. Wright, U. Bierbach, Metal-intercalator-mediated self-
association and one-dimensional aggregation in the structure of the excised major
DNA adduct of a platinum-acridine agent, Journal of the American Chemical
Society, 126 (2004) 4492-4493.
[79] X. Wang, Z. Guo, Targeting and delivery of platinum-based anticancer drugs,
Chemical Society Reviews, 42 (2013) 202-224.
[80] J.S. Butler, P.J. Sadler, Targeted delivery of platinum-based anticancer complexes,
Current Opinion in Chemical Biology, 17 (2013) 175-188.
[81] C.M. Dawidczyk, C. Kim, J.H. Park, L.M. Russell, K.H. Lee, M.G. Pomper, P.C.
Searson, State-of-the-art in design rules for drug delivery platforms: Lessons
learned from FDA-approved nanomedicines, Journal of Controlled Release, 187
(2014) 133-144.
[82] Y. Barenholz, Doxil (R) - The first FDA-approved nano-drug: Lessons learned,
Journal of Controlled Release, 160 (2012) 117-134.
[83] A.A. Gabizon, H. Shmeeda, S. Zalipsky, Pros and cons of the liposome platform in
cancer drug targeting, Journal of Liposome Research, 16 (2006) 175-183.
[84] T.M. Allen, P.R. Cullis, Liposomal drug delivery systems: From concept to clinical
applications, Advanced Drug Delivery Reviews, 65 (2013) 36-48.
[85] G. Riess, Micellization of block copolymers, Progress in Polymer Science, 28 (2003)
1107-1170.
[86] V.P. Torchilin, Structure and design of polymeric surfactant-based drug delivery
systems, Journal of Controlled Release, 73 (2001) 137-172.

272
[87] C. Allen, D. Maysinger, A. Eisenberg, Nano-engineering block copolymer
aggregates for drug delivery, Colloids and Surfaces B: Biointerfaces, 16 (1999) 3-
27.
[88] X. Li, R. Lie, X. Qian, Y. Ding, Y. Tu, R. Guo, Y. Hu, X. Jiang, W. Guo, B. Liu,
Superior antitumor efficiency of cisplatin-loaded nanoparticles by intratumoral
delivery with decreased tumor metabolism rate, European Journal of Pharmaceutics
and Biopharmaceutics, 70 (2008) 726-734.
[89] T.C. Johnstone, S.J. Lippard, The Effect of Ligand Lipophilicity on the Nanoparticle
Encapsulation of Pt(IV) Prodrugs, Inorganic Chemistry, 52 (2013) 9915-9920.
[90] A. Lavasanifar, J. Samuel, G.S. Kwon, Poly(ethylene oxide)-block-poly(L-amino
acid) micelles for drug delivery, Advanced Drug Delivery Reviews, 54 (2002) 169-
190.
[91] N. Nishiyama, K. Kataoka, Preparation and characterization of size-controlled
polymeric micelle containing cis-dichlorodiammineplatinum(II) in the core, Journal
of Controlled Release, 74 (2001) 83-94.
[92] S. Bontha, A.V. Kabanov, T.K. Bronich, Polymer micelles with cross-linked ionic
cores for delivery of anticancer drugs, Journal of Controlled Release, 114 (2006)
163-174.
[93] H.S. Oberoi, F.C. Laquer, L.A. Marky, A.V. Kabanov, T.K. Bronich, Core cross-
linked block ionomer micelles as pH-responsive carriers for cis-
diamminedichloroplatinum(II), Journal of Controlled Release, 153 (2011) 64-72.

273
[94] N. Nishiyama, Y. Kato, Y. Sugiyama, K. Kataoka, Cisplatin-loaded polymer-metal
complex micelle with time-modulated decaying property as a novel drug delivery
system, Pharmaceutical Research, 18 (2001) 1035-1041.
[95] H. Ringsdorf, Structure and propertie of pharmacologically active polymers, Journal
of Polymer Science Part C-Polymer Symposium, (1975) 135-153.
[96] J. Kopecek, P. Kopeckova, T. Minko, Z.R. Lu, HPMA copolymer-anticancer drug
conjugates: design, activity, and mechanism of action, European Journal of
Pharmaceutics and Biopharmaceutics, 50 (2000) 61-81.
[97] R. Satchi-Fainaro, R. Duncan, C.M. Barnes, Polymer therapeutics for cancer:
Current status and future challenges, Polymer Therapeutics Ii: Polymers as Drugs,
Conjugates and Gene Delivery Systems2006, pp. 1-65.
[98] K.J. Haxton, H.M. Burt, Polymeric Drug Delivery of Platinum-Based Anticancer
Agents, Journal of Pharmaceutical Sciences, 98 (2009) 2299-2316.
[99] P. Sood, K.B. Thurmond, II, J.E. Jacob, L.K. Waller, G.O. Silva, D.R. Stewart, D.P.
Nowotnik, Synthesis and characterization of AP5346, a novel polymer-linked
diaminocyclohexyl platinum chemotherapeutic agent, Bioconjugate Chemistry, 17
(2006) 1270-1279.
[100] J.R. Rice, J.L. Gerberich, D.P. Nowotnik, S.B. Howell, Preclinical efficacy and
pharmacokinetics of AP5346, a novel diaminocyclohexane-platinum tumor-
targeting drug delivery system, Clinical Cancer Research, 12 (2006) 2248-2254.
[101] D. Stewart, J. Rice, P. Sood, J.S. John, S. Shevchuk, K.B. Thurmond, D. Nguyen,
G. Russell-Jones, D. Nowotnik, Preclinical development of AP5346: A
dachplatinum polymer conjugate, Journal of Controlled Release, 91 (2003) 255-256.

274
[102] D.P. Nowotnik, E. Cvitkovic, ProLindac (TM) (AP5346): A review of the
development of an HPMA DACH platinum Polymer Therapeutic, Advanced Drug
Delivery Reviews, 61 (2009) 1214-1219.
[103] Y.S. Sohn, Y.H. Cho, H. Baek, O.S. Jung, Synthesis and properties of low-
molecular-weight polyphosphazenes, Macromolecules, 28 (1995) 7566-7568.
[104] M. Ravera, E. Gabano, S. Bianco, G. Ermondi, G. Caron, M. Vallaro, G. Pelosi, I.
Zanellato, I. Bonarrigo, C. Cassino, D. Osella, Host-guest inclusion systems of
Pt(IV)-bis(benzoato) anticancer drug candidates and cyclodextrins, Inorganica
Chimica Acta, 432 (2015) 115-127.
[105] E.W. Neuse, G. Caldwell, A.G. Perlwitz, Carrier polymers for cisplatin-type
anticancer drug models, Polymers for Advanced Technologies, 7 (1996) 867-872.
[106] M. Li, Z. Tang, S. Lv, W. Song, H. Hong, X. Jing, Y. Zhang, X. Chen, Cisplatin
crosslinked pH-sensitive nanoparticles for efficient delivery. of doxorubicin,
Biomaterials, 35 (2014) 3851-3864.
[107] C.C. Lee, J.A. MacKay, J.M.J. Frechet, F.C. Szoka, Designing dendrimers for
biological applications, Nature Biotechnology, 23 (2005) 1517-1526.
[108] B.A. Howell, D. Fan, Poly(amidoamine) dendrimer-supported organoplatinum
antitumour agents, Proceedings of the Royal Society a-Mathematical Physical and
Engineering Sciences, 466 (2010) 1515-1526.
[109] V.K. Yellepeddi, A. Kumar, D.M. Maher, S.C. Chauhan, K.K. Vangara, S.
Palakurthi, Biotinylated PAMAM Dendrimers for Intracellular Delivery of
Cisplatin to Ovarian Cancer: Role of SMVT, Anticancer Research, 31 (2011) 897-
906.

275
[110] T. Kapp, A. Dullin, R. Gust, Platinum(II)-Dendrimer Conjugates: Synthesis and
Investigations on Cytotoxicity, Cellular Distribution, Platinum Release, DNA, and
Protein Binding, Bioconjugate Chemistry, 21 (2010) 328-337.
[111] G. Bozzuto, A. Molinari, Liposomes as nanomedical devices, International Journal
of Nanomedicine, 10 (2015) 975-999.
[112] B. Ozpolat, A.K. Sood, G. Lopez-Berestein, Liposomal siRNA nanocarriers for
cancer therapy, Advanced Drug Delivery Reviews, 66 (2014) 110-116.
[113] V.V. Mody, M.I. Nounou, M. Bikram, Novel nanomedicine-based MRI contrast
agents for gynecological malignancies, Advanced Drug Delivery Reviews, 61
(2009) 795-807.
[114] N. Hijnen, S. Langereis, H. Grull, Magnetic resonance guided high-intensity
focused ultrasound for image-guided temperature-induced drug delivery, Advanced
Drug Delivery Reviews, 72 (2014) 65-81.
[115] K.N.J. Burger, R. Staffhorst, H.C. de Vijlder, M.J. Velinova, P.H. Bomans, P.M.
Frederik, B. de Kruijff, Nanocapsules: lipid-coated aggregates of cisplatin with
high cytotoxicity, Nature Medicine, 8 (2002) 81-84.
[116] Z. Zhang, L. Mei, S.-S. Feng, Paclitaxel drug delivery systems, Expert Opinion on
Drug Delivery, 10 (2013) 325-340.
[117] S.H. Alavizadeh, A. Badiee, S. Golmohammadzadeh, M.R. Jaafari, The influence
of phospholipid on the physicochemical properties and anti-tumor efficacy of
liposomes encapsulating cisplatin in mice bearing C26 colon carcinoma,
International Journal of Pharmaceutics, 473 (2014) 326-333.

276
[118] A.S. Abu Lila, S. Kizuki, Y. Doi, T. Suzuki, T. Ishida, H. Kiwada, Oxaliplatin
encapsulated in PEG-coated cationic liposomes induces significant tumor growth
suppression via a dual-targeting approach in a murine solid tumor model, Journal of
Controlled Release, 137 (2009) 8-14.
[119] S. Zalba, I. Navarro, I.F. Troconiz, C. Tros de Ilarduya, M.J. Garrido, Application
of different methods to formulate PEG-liposomes of oxaliplatin: Evaluation in vitro
and in vivo, European Journal of Pharmaceutics and Biopharmaceutics, 81 (2012)
273-280.
[120] M. Hirai, H. Minematsu, Y. Hiramatsu, H. Kitagawa, T. Otani, S. Iwashita, T.
Kudoh, L. Chen, Y. Li, M. Okada, D.S. Salomon, K. Igarashi, M. Chikuma, M.
Seno, Novel and simple loading procedure of cisplatin into liposomes and targeting
tumor endothelial cells, International Journal of Pharmaceutics, 391 (2010) 274-
283.
[121] I. Vhora, N. Khatri, J. Desai, H.P. Thakkar, Caprylate-Conjugated Cisplatin for the
Development of Novel Liposomal Formulation, AAPS PharmSciTech, 15 (2014)
845-857.
[122] Q. Li, Y. Tian, D. Li, J. Sun, D. Shi, L. Fang, Y. Gao, H. Liu, The effect of
lipocisplatin on cisplatin efficacy and nephrotoxicity in malignant breast cancer
treatment, Biomaterials, 35 (2014) 6462-6472.
[123] Y. Kuang, J. Liu, Z. Liu, R. Zhuo, Cholesterol-based anionic long-circulating
cisplatin liposomes with reduced renal toxicity, Biomaterials, 33 (2012) 1596-1606.

277
[124] L.M. Ickenstein, M.C. Arfvidsson, D. Needham, L.D. Mayer, K. Edwards, Disc
formation in cholesterol-free lippsornes during phase transition, Biochimica Et
Biophysica Acta-Biomembranes, 1614 (2003) 135-138.
[125] J. Gubernator, G. Chwastek, M. Korycinska, M. Stasiuk, G. Grynkiewicz, F.
Lewrick, R. Suess, A. Kozubek, The encapsulation of idarubicin within liposomes
using the novel EDTA ion gradient method ensures improved drug retention in
vitro and in vivo, Journal of Controlled Release, 146 (2010) 68-75.
[126] R.M. Abra, R.B. Bankert, F. Chen, N.K. Egilmez, K. Huang, R. Saville, J.L. Slater,
M. Sugano, S.J. Yokota, The next generation of liposome delivery systems: Recent
experience with tumor-targeted, sterically-stabilized immunoliposomes and active-
loading gradients, Journal of Liposome Research, 12 (2002) 1-3.
[127] I.M. Hann, H.G. Prentice, Lipid-based amphotericin B: a review of the last 10 years
of use, International Journal of Antimicrobial Agents, 17 (2001) 161-169.
[128] C.E. Petre, D.P. Dittmer, Liposomal daunorubicin as treatment for Kaposi's
sarcoma, International Journal of Nanomedicine, 2 (2007) 277-288.
[129] G. Batist, G. Ramakrishnan, C.S. Rao, A. Chandrasekharan, J. Gutheil, T. Guthrie,
P. Shah, A. Khojasteh, M.K. Nair, K. Hoelzer, K. Tkaczuk, Y.C. Park, L.W. Lee, G.
Myocet Study, Reduced cardiotoxicity and preserved antitumor efficacy of
liposome-encapsulated doxorubicin and cyclophosphamide compared with
conventional doxorubicin and cyclophosphamide in a randomized, multicenter trial
of metastatic breast cancer, Journal of Clinical Oncology, 19 (2001) 1444-1454.
[130] M.J. Glantz, K.A. Jaeckle, M.C. Chamberlain, S. Phuphanich, L. Recht, L.J.
Swinnen, B. Maria, S. LaFollette, G.B. Schumann, B.F. Cole, S.B. Howell, A

278
randomized controlled trial comparing intrathecal sustained-release cytarabine
(DepoCyt) to intrathecal methotrexate in patients with neoplastic meningitis from
solid tumors, Clinical Cancer Research, 5 (1999) 3394-3402.
[131] V. D'Acremont, C. Herzog, B. Genton, Immunogenicity and safety of a virosomal
hepatitis A vaccine (Epaxal (R)) in the elderly, Journal of Travel Medicine, 13
(2006) 78-83.
[132] C. Herzog, K. Hartmann, V. Kuenzi, O. Kuersteiner, R. Mischler, H. Lazar, R.
Glueck, Eleven years of Inflexal (R) V-a virosomal adjuvanted influenza vaccine,
Vaccine, 27 (2009) 4381-4387.
[133] G.P. Stathopoulos, T. Boulikas, Lipoplatin formulation review article, Journal of
drug delivery, 2012 (2012) 581363-581363.
[134] M.L. Immordino, F. Dosio, L. Cattel, Stealth liposomes: review of the basic science,
rationale, and clinical applications, existing and potential, International Journal of
Nanomedicine, 1 (2006) 297-315.
[135] T. Dragovich, D. Mendelson, S. Kurtin, K. Richardson, D. Von Hoff, A. Hoos, A
phase 2 trial of the liposomal DACH platinum L-NDDP in patients with therapy-
refractory advanced colorectal cancer, Cancer Chemotherapy and Pharmacology,
58 (2006) 759-764.
[136] G.P. Stathopoulos, T. Boulikas, A. Kourvetaris, J. Stathopoulos, Liposomal
oxaliplatin in the treatment of advanced cancer: A phase I study, Anticancer
Research, 26 (2006) 1489-1493.
[137] K.K. Sankhala, A.C. Mita, R. Adinin, L. Wood, M. Beeram, S. Bullock, N.
Yamagata, K. Matsuno, T. Fujisawa, A. Phan, A phase I pharmacokinetic (PK)

279
study of MBP-426, a novel liposome encapsulated oxaliplatin, Journal of Clinical
Oncology, 27 (2009).
[138] D. Liu, C. He, A.Z. Wang, W. Lin, Application of liposomal technologies for
delivery of platinum analogs in oncology, International Journal of Nanomedicine, 8
(2013) 3309-3319.
[139] T. Daemen, G. Hofstede, M.T.T. Kate, I. Bakkerwoudenberg, G.L. Scherphof,
Liposomal doxorubicin-induced toxicity-depletion and impairment of phagocytic-
activity of liver macrophages, International Journal of Cancer, 61 (1995) 716-721.
[140] M.C. Woodle, K.K. Matthay, M.S. Newman, J.E. Hidayat, L.R. Collins, C.
Redemann, F.J. Martin, D. Papahadjopoulos, Versatility in lipid compositions
showing prolonged circulation with sterically stabilized liposomes, Biochimica Et
Biophysica Acta-Biomembranes, 1105 (1992) 193-200.
[141] P.I. Campbell, Toxicity of some charged lipids used in liposome preparations,
Cytobios, 37 (1983) 21-26.
[142] A.S. Abu Lila, H. Kiwada, T. Ishida, The accelerated blood clearance (ABC)
phenomenon: Clinical challenge and approaches to manage, Journal of Controlled
Release, 172 (2013) 38-47.
[143] B.S. Wong, S.L. Yoong, A. Jagusiak, T. Panczyk, H.K. Ho, W.H. Ang, G. Pastorin,
Carbon nanotubes for delivery of small molecule drugs, Advanced Drug Delivery
Reviews, 65 (2013) 1964-2015.
[144] C.R. Martin, P. Kohli, The emerging field of nanotube biotechnology, Nature
Reviews Drug Discovery, 2 (2003) 29-37.

280
[145] S.-T. Yang, X. Wang, G. Jia, Y. Gu, T. Wang, H. Nie, C. Ge, H. Wang, Y. Liu,
Long-term accumulation and low toxicity of single-walled carbon nanotubes in
intravenously exposed mice, Toxicology Letters, 181 (2008) 182-189.
[146] A. Guven, I.A. Rusakova, M.T. Lewis, L.J. Wilson, Cisplatin@US-tube carbon
nanocapsules for enhanced chemotherapeutic delivery, Biomaterials, 33 (2012)
1455-1461.
[147] J. Kolosnjaj-Tabi, K.B. Hartman, S. Boudjemaa, J.S. Ananta, G. Morgant, H.
Szwarc, L.J. Wilson, F. Moussa, In Vivo Behavior of Large Doses of Ultrashort
and Full-Length Single-Walled Carbon Nanotubes after Oral and Intraperitoneal
Administration to Swiss Mice, ACS Nano, 4 (2010) 1481-1492.
[148] S.C. Tsang, Y.K. Chen, P.J.F. Harris, M.L.H. Green, A simple chemical method of
opening and filling carbon nanotubes, Nature, 372 (1994) 159-162.
[149] J. Li, S.Q. Yap, C.F. Chin, Q. Tian, S.L. Yoong, G. Pastorin, W.H. Ang,
Platinum(IV) prodrugs entrapped within multiwalled carbon nanotubes: Selective
release by chemical reduction and hydrophobicity reversal, Chemical Science, 3
(2012) 2083-2087.
[150] R.P. Feazell, N. Nakayama-Ratchford, H. Dai, S.J. Lippard, Soluble single-walled
carbon nanotubes as longboat delivery systems for Platinum(IV) anticancer drug
design, Journal of the American Chemical Society, 129 (2007) 8438-8439.
[151] J. Li, S.Q. Yap, S.L. Yoong, T.R. Nayak, G.W. Chandra, W.H. Ang, T. Panczyk, S.
Ramaprabhu, S.K. Vashist, F.-S. Sheu, A. Tan, G. Pastorin, Carbon nanotube
bottles for incorporation, release and enhanced cytotoxic effect of cisplatin, Carbon,
50 (2012) 1625-1634.

281
[152] L. Lacerda, J. Russier, G. Pastorin, M. Antonia Herrero, E. Venturelli, H.
Dumortier, K.T. Al-Jamal, M. Prato, K. Kostarelos, A. Bianco, Translocation
mechanisms of chemically functionalised carbon nanotubes across plasma
membranes, Biomaterials, 33 (2012) 3334-3343.
[153] S. Matsumura, K. Ajima, M. Yudasaka, S. Iijima, K. Shiba, Dispersion of cisplatin-
loaded carbon nanohorns with a conjugate comprised of an artificial peptide
aptamer and polyethylene glycol, Molecular Pharmaceutics, 4 (2007) 723-729.
[154] S. Dhar, Z. Liu, J. Thomale, H. Dai, S.J. Lippard, Targeted Single-Wall Carbon
Nanotube-Mediated Pt(IV) Prodrug Delivery Using Folate as a Homing Device,
Journal of the American Chemical Society, 130 (2008) 11467-11476.
[155] A.A. Bhirde, V. Patel, J. Gavard, G. Zhang, A.A. Sousa, A. Masedunskas, R.D.
Leapman, R. Weigert, J.S. Gutkind, J.F. Rusling, Targeted Killing of Cancer Cells
in Vivo and in Vitro with EGF-Directed Carbon Nanotube-Based Drug Delivery,
ACS Nano, 3 (2009) 307-316.
[156] J. Liu, Y. Huang, A. Kumar, A. Tan, S. Jin, A. Mozhi, X.-J. Liang, pH-Sensitive
nano-systems for drug delivery in cancer therapy, Biotechnology Advances, 32
(2014) 693-710.
[157] S. Aryal, C.-M.J. Hu, L. Zhang, Polymer−Cisplatin Conjugate Nanoparticles for
Acid-Responsive Drug Delivery, ACS Nano, 4 (2009) 251-258.
[158] T. Thambi, V.G. Deepagan, H.Y. Yoon, H.S. Han, S.-H. Kim, S. Son, D.-G. Jo, C.-
H. Ahn, Y.D. Suh, K. Kim, I. Chan Kwon, D.S. Lee, J.H. Park, Hypoxia-
responsive polymeric nanoparticles for tumor-targeted drug delivery, Biomaterials,
35 (2014) 1735-1743.

282
[159] F. Perche, S. Biswas, T. Wang, L. Zhu, V.P. Torchilin, Hypoxia-Targeted siRNA
Delivery, Angewandte Chemie International Edition, 53 (2014) 3362-3366.
[160] Y. Kageyama, Y. Yamazaki, H. Okuno, Novel approaches to prodrugs of
anticancer diaminodichloroplatinum(II) complexes activated by stereoselective
enzymatic ester hydrolysis, Journal of Inorganic Biochemistry, 70 (1998) 25-32.
[161] J.R. Tauro, B.-S. Lee, S.S. Lateef, R.A. Gemeinhart, Matrix metalloprotease
selective peptide substrates cleavage within hydrogel matrices for cancer
chemotherapy activation, Peptides, 29 (2008) 1965-1973.
[162] M.A. Fuertes, C. Alonso, J.M. Pérez, Biochemical Modulation of Cisplatin
Mechanisms of Action: Enhancement of Antitumor Activity and Circumvention of
Drug Resistance, Chemical Reviews, 103 (2003) 645-662.
[163] J. Reedijk, Metal-Ligand Exchange Kinetics in Platinum and Ruthenium
Complexes significance for effectiveness as anticancer drugs, Platinum Metals
Review, 52 (2008) 2-11.
[164] E. Gabano, M. Ravera, D. Osella, The Drug Targeting and Delivery Approach
Applied to Pt-Antitumour Complexes. A Coordination Point of View, Current
Medicinal Chemistry, 16 (2009) 4544-4580.
[165] J. Reedijk, Why Does Cisplatin Reach Guanine-N7 with Competing S-Donor
Ligands Available in the Cell?, Chemical Reviews, 99 (1999) 2499-2510.
[166] G. Ma, Y. Min, F. Huang, T. Jiang, Y. Liu, Thioether binding mediates
monofunctional platinum antitumor reagents to trans configuration in DNA
interactions, Chemical Communications, 46 (2010) 6938-6940.

283
[167] C. Li, R. Huang, Y. Ding, E. Sletten, F. Arnesano, M. Losacco, G. Natile, Y. Liu,
Effect of Thioethers on DNA Platination by trans-Platinum Complexes, Inorganic
Chemistry, 50 (2011) 8168-8176.
[168] M.E. Alberto, N. Russo, Methionine ligand selectively promotes monofunctional
adducts between trans-EE platinum anticancer drug and guanine DNA base,
Chemical Communications, 47 (2011) 887-889.
[169] S.S.G.E. van Boom, B.W. Chen, J.M. Teuben, J. Reedijk, Platinum−Thioether
Bonds Can Be Reverted by Guanine−N7 Bonds in Pt(dien)2+ Model Adducts,
Inorganic Chemistry, 38 (1999) 1450-1455.
[170] C. Li, Z. Li, E. Sletten, F. Arnesano, M. Losacco, G. Natile, Y. Liu, Methionine
Can Favor DNA Platination by trans-Coordinated Platinum Antitumor Drugs,
Angewandte Chemie International Edition, 48 (2009) 8497-8500.
[171] F. Arnesano, S. Scintilla, G. Natile, Interaction between Platinum Complexes and a
Methionine Motif Found in Copper Transport Proteins, Angewandte Chemie
International Edition, 46 (2007) 9062-9064.
[172] Y. Kasherman, S. Sturup, D. Gibson, Trans labilization of am(m)ine ligands from
platinum(II) complexes by cancer cell extracts, Journal of Biological Inorganic
Chemistry, 14 (2009) 387-399.
[173] J. Mendez-Arroyo, J. Barroso-Flores, A.M. Lifschitz, A.A. Sarjeant, C.L. Stern,
C.A. Mirkin, A Multi-State, Allosterically-Regulated Molecular Receptor With
Switchable Selectivity, Journal of the American Chemical Society, 136 (2014)
10340-10348.

284
[174] D. Pan, W. she, C. Guo, K. Luo, Q. Yi, Z. Gu, PEGylated dendritic
diaminocyclohexyl-platinum (II) conjugates as pH-responsive drug delivery
vehicles with enhanced tumor accumulation and antitumor efficacy, Biomaterials,
35 (2014) 10080-10092.
[175] H. Wu, H. Cabral, K. Toh, P. Mi, Y.-C. Chen, Y. Matsumoto, N. Yamada, X. Liu,
H. Kinoh, Y. Miura, M.R. Kano, H. Nishihara, N. Nishiyama, K. Kataoka,
Polymeric micelles loaded with platinum anticancer drugs target preangiogenic
micrometastatic niches associated with inflammation, Journal of Controlled
Release, 189 (2014) 1-10.
[176] U. Frey, J.D. Ranford, P.J. Sadler, Ring-opening reactions of the anticancer drug
carboplatin: NMR characterization of cis-[Pt(NH3)2(CBDCA-O)(5'-GMP-N7)] in
solution, Inorganic Chemistry, 32 (1993) 1333-1340.
[177] D.P. Bancroft, C.A. Lepre, S.J. Lippard, Platinum-195 NMR kinetic and
mechanistic studies of cis- and trans-diamminedichloroplatinum(II) binding to
DNA, Journal of the American Chemical Society, 112 (1990) 6860-6871.
[178] M.E.M. Dolman, K.M.A. van Dorenmalen, E.H.E. Pieters, M. Lacombe, J. Pato, G.
Storm, W.E. Hennink, R.J. Kok, Imatinib-ULS-lysozyme: A proximal tubular cell-
targeted conjugate of imatinib for the treatment of renal diseases, Journal of
Controlled Release, 157 (2012) 461-468.
[179] V.T. Huynh, J.Y. Quek, P.L. de Souza, M.H. Stenzel, Block Copolymer Micelles
with Pendant Bifunctional Chelator for Platinum Drugs: Effect of Spacer Length on
the Viability of Tumor Cells, Biomacromolecules, 13 (2012) 1010-1023.

285
[180] S.-M. Lee, T.V. O’Halloran, S.T. Nguyen, Polymer-Caged Nanobins for
Synergistic Cisplatin−Doxorubicin Combination Chemotherapy, Journal of the
American Chemical Society, 132 (2010) 17130-17138.
[181] N.P.E. Barry, P.J. Sadler, Challenges for Metals in Medicine: How
Nanotechnology May Help To Shape the Future, ACS Nano, 7 (2013) 5654-5659.
[182] N. Nishiyama, S. Okazaki, H. Cabral, M. Miyamoto, Y. Kato, Y. Sugiyama, K.
Nishio, Y. Matsumura, K. Kataoka, Novel Cisplatin-Incorporated Polymeric
Micelles Can Eradicate Solid Tumors in Mice, Cancer Research, 63 (2003) 8977-
8983.
[183] W. Cao, Y. Gu, M. Meineck, T. Li, H. Xu, Tellurium-Containing Polymer Micelles:
Competitive-Ligand-Regulated Coordination Responsive Systems, Journal of the
American Chemical Society, 136 (2014) 5132-5137.
[184] M. Johnson, E. Neuse, C.J. van Rensburg, E. Kreft, Cell Growth-Inhibiting
Properties of Selected Carrier-Bound, Monoamine-Coordinated Platinum(II)
Compounds, Journal of Inorganic and Organometallic Polymers, 13 (2003) 55-67.
[185] M. Callari, J.R. Aldrich-Wright, P.L. de Souza, M.H. Stenzel, Polymers with
platinum drugs and other macromolecular metal complexes for cancer treatment,
Progress in Polymer Science, 39 (2014) 1614-1643.
[186] G.J. Kirkpatrick, J.A. Plumb, O.B. Sutcliffe, D.J. Flint, N.J. Wheate, Evaluation of
anionic half generation 3.5–6.5 poly(amidoamine) dendrimers as delivery vehicles
for the active component of the anticancer drug cisplatin, Journal of Inorganic
Biochemistry, 105 (2011) 1115-1122.

286
[187] H. He, H. Xiao, H. Kuang, Z. Xie, X. Chen, X. Jing, Y. Huang, Synthesis of
mesoporous silica nanoparticle–oxaliplatin conjugates for improved anticancer
drug delivery, Colloids and Surfaces B: Biointerfaces, 117 (2014) 75-81.
[188] J. Gu, J. Liu, Y. Li, W. Zhao, J. Shi, One-Pot Synthesis of Mesoporous Silica
Nanocarriers with Tunable Particle Sizes and Pendent Carboxylic Groups for
Cisplatin Delivery, Langmuir, 29 (2012) 403-410.
[189] S.D. Brown, P. Nativo, J.-A. Smith, D. Stirling, P.R. Edwards, B. Venugopal, D.J.
Flint, J.A. Plumb, D. Graham, N.J. Wheate, Gold Nanoparticles for the Improved
Anticancer Drug Delivery of the Active Component of Oxaliplatin, Journal of the
American Chemical Society, 132 (2010) 4678-4684.
[190] B. Duncan, C. Kim, V.M. Rotello, Gold nanoparticle platforms as drug and
biomacromolecule delivery systems, Journal of Controlled Release, 148 (2010)
122-127.
[191] R. Plummer, R.H. Wilson, H. Calvert, A.V. Boddy, M. Griffin, J. Sludden, M.J.
Tilby, M. Eatock, D.G. Pearson, C.J. Ottley, Y. Matsumura, K. Kataoka, T. Nishiya,
A Phase I clinical study of cisplatin-incorporated polymeric micelles (NC-6004) in
patients with solid tumours, British Journal of Cancer, 104 (2011) 593-598.
[192] M. Baba, Y. Matsumoto, A. Kashio, H. Cabral, N. Nishiyama, K. Kataoka, T.
Yamasoba, Micellization of cisplatin (NC-6004) reduces its ototoxicity in guinea
pigs, Journal of Controlled Release, 157 (2012) 112-117.
[193] Y. Matsumura, Poly (amino acid) micelle nanocarriers in preclinical and clinical
studies, Advanced Drug Delivery Reviews, 60 (2008) 899-914.

287
[194] T. Ueno, K. Endo, K. Hori, N. Ozaki, A. Tsuji, S. Kondo, N. Wakisaka, S. Murono,
K. Kataoka, Y. Kato, T. Yoshizaki, Assessment of antitumor activity and acute
peripheral neuropathy of 1,2-diaminocyclohexane platinum (II)-incorporating
micelles (NC-4016), International Journal of Nanomedicine, 9 (2014) 3005-3012.
[195] H. Cabral, N. Nishiyama, K. Kataoka, Optimization of (1,2-diamino-
cyclohexane)platinum(II)-loaded polymeric micelles directed to improved tumor
targeting and enhanced antitumor activity, Journal of Controlled Release, 121
(2007) 146-155.
[196] H. Cabral, N. Nishiyama, S. Okazaki, H. Koyama, K. Kataoka, Preparation and
biological properties of dichloro(1,2-diaminocyclohexane)platinum(II) (DACHPt)-
loaded polymeric micelles, Journal of Controlled Release, 101 (2005) 223-232.
[197] R. Song, Y. Joo Jun, J. Ik Kim, C. Jin, Y.S. Sohn, Synthesis, characterization, and
tumor selectivity of a polyphosphazene–platinum(II) conjugate, Journal of
Controlled Release, 105 (2005) 142-150.
[198] P.G. Avaji, H.I. Joo, J.H. Park, K.S. Park, Y.J. Jun, H.J. Lee, Y.S. Sohn, Synthesis
and properties of a new micellar polyphosphazene–platinum(II) conjugate drug,
Journal of Inorganic Biochemistry, 140 (2014) 45-52.
[199] J. Xu, Q. Fu, J.M. Ren, G. Bryant, G.G. Qiao, Novel drug carriers: from grafted
polymers to cross-linked vesicles, Chemical Communications, 49 (2013) 33-35.
[200] K. Osada, H. Cabral, Y. Mochida, S. Lee, K. Nagata, T. Matsuura, M. Yamamoto,
Y. Anraku, A. Kishimura, N. Nishiyama, K. Kataoka, Bioactive Polymeric
Metallosomes Self-Assembled through Block Copolymer–Metal Complexation,
Journal of the American Chemical Society, 134 (2012) 13172-13175.

288
[201] S. Valiahdi, A. Egger, W. Miklos, U. Jungwirth, K. Meelich, P. Nock, W. Berger,
C. Hartinger, M. Galanski, M. Jakupec, B. Keppler, Influence of extracellular pH
on the cytotoxicity, cellular accumulation, and DNA interaction of novel pH-
sensitive 2-aminoalcoholatoplatinum(II) complexes, Journal of Biological
Inorganic Chemistry, 18 (2013) 249-260.
[202] R.M. Sawant, J.P. Hurley, S. Salmaso, A. Kale, E. Tolcheva, T.S. Levchenko, V.P.
Torchilin, “SMART” Drug Delivery Systems: Double-Targeted pH-Responsive
Pharmaceutical Nanocarriers, Bioconjugate Chemistry, 17 (2006) 943-949.
[203] W. Friebolin, G. Schilling, M. Zöller, E. Amtmann, Synthesis and
Structure−Activity Relationship of Novel Antitumoral Platinum Xanthate
Complexes, Journal of Medicinal Chemistry, 47 (2004) 2256-2263.
[204] Y.Y. Scaffidi-Domianello, A.A. Legin, M.A. Jakupec, V.B. Arion, V.Y. Kukushkin,
M. Galanski, B.K. Keppler, Synthesis, Characterization, and Cytotoxic Activity of
Novel Potentially pH-Sensitive Nonclassical Platinum(II) Complexes Featuring
1,3-Dihydroxyacetone Oxime Ligands, Inorganic Chemistry, 50 (2011) 10673-
10681.
[205] C.-H. Lin, S.-H. Cheng, W.-N. Liao, P.-R. Wei, P.-J. Sung, C.-F. Weng, C.-H. Lee,
Mesoporous silica nanoparticles for the improved anticancer efficacy of cis-platin,
International Journal of Pharmaceutics, 429 (2012) 138-147.
[206] D.P. Nowotnik, E. Cvitkovic, ProLindac™ (AP5346): A review of the
development of an HPMA DACH platinum Polymer Therapeutic, Advanced Drug
Delivery Reviews, 61 (2009) 1214-1219.

289
[207] A.K. Varkouhi, M. Scholte, G. Storm, H.J. Haisma, Endosomal escape pathways
for delivery of biologicals, Journal of Controlled Release, 151 (2011) 220-228.
[208] A.D. Carvalho Júnior, F.P. Vieira, V.J. De Melo, M.T.P. Lopes, J.N. Silveira, G.A.
Ramaldes, A. Garnier-Suillerot, E.C. Pereira-Maia, M.C. De Oliveira, Preparation
and cytotoxicity of cisplatin-containing liposomes, Brazilian Journal of Medical
and Biological Research, 40 (2007) 1149-1157.
[209] M. Murakami, H. Cabral, Y. Matsumoto, S. Wu, M.R. Kano, T. Yamori, N.
Nishiyama, K. Kataoka, Improving Drug Potency and Efficacy by Nanocarrier-
Mediated Subcellular Targeting, Science Translational Medicine, 3 (2011).
[210] M. Li, Z. Tang, S. Lv, W. Song, H. Hong, X. Jing, Y. Zhang, X. Chen, Cisplatin
crosslinked pH-sensitive nanoparticles for efficient delivery of doxorubicin,
Biomaterials, 35 (2014) 3851-3864.
[211] K. Szaciłowski, W. Macyk, A. Drzewiecka-Matuszek, M. Brindell, G. Stochel,
Bioinorganic Photochemistry: Frontiers and Mechanisms, Chemical Reviews, 105
(2005) 2647-2694.
[212] P.C. Bruijnincx, P.J. Sadler, Controlling Platinum, Ruthenium and Osmium
Reactivity for Anticancer Drug Design, Advances in Inorganic Chemistry, 61
(2009) 1-62.
[213] N.P.E. Barry, P.J. Sadler, Exploration of the medical periodic table: towards new
targets, Chemical Communications, 49 (2013) 5106-5131.
[214] P.J. Bednarski, F.S. Mackay, P.J. Sadler, Photoactivatable platinum complexes,
Anti-Cancer Agents in Medicinal Chemistry, 7 (2007) 75-93.

290
[215] Y. Zhao, G.M. Roberts, S.E. Greenough, N.J. Farrer, M.J. Paterson, W.H. Powell,
V.G. Stavros, P.J. Sadler, Two-Photon-Activated Ligand Exchange in Platinum(II)
Complexes, Angewandte Chemie-International Edition, 51 (2012) 11263-11266.
[216] Y. Zhao, G.M. Roberts, S.E. Greenough, N.J. Farrer, M.J. Paterson, W.H. Powell,
V.G. Stavros, P.J. Sadler, Two-Photon-Activated Ligand Exchange in Platinum(II)
Complexes, Angewandte Chemie International Edition, 51 (2012) 11263-11266.
[217] N.A. Kratochwil, M. Zabel, K.-J. Range, P.J. Bednarski, Synthesis and X-ray
Crystal Structure of trans,cis-[Pt(OAc)2I2(en)]: A Novel Type of Cisplatin Analog
That Can Be Photolyzed by Visible Light to DNA-Binding and Cytotoxic Species
in Vitro, Journal of Medicinal Chemistry, 39 (1996) 2499-2507.
[218] H. Choy, C. Park, M. Yao, Current Status and Future Prospects for Satraplatin, an
Oral Platinum Analogue, Clinical Cancer Research, 14 (2008) 1633-1638.
[219] N.A. Kratochwil, J.A. Parkinson, P.J. Bednarski, P.J. Sadler, Nucleotide Platination
Induced by Visible Light, Angewandte Chemie International Edition, 38 (1999)
1460-1463.
[220] A. Vogler, A. Kern, J. Hüttermann, Photochemical Reductive trans-Elimination
from trans-Diazidotetracyanoplatinate(IV), Angewandte Chemie International
Edition, 17 (1978) 524-525.
[221] P.J. Bednarski, R. Grünert, M. Zielzki, A. Wellner, F.S. Mackay, P.J. Sadler, Light-
Activated Destruction of Cancer Cell Nuclei by Platinum Diazide Complexes,
Chemistry & Biology, 13 61-67.
[222] A.F. Westendorf, J.A. Woods, K. Korpis, N.J. Farrer, L. Salassa, K. Robinson, V.
Appleyard, K. Murray, R. Grünert, A.M. Thompson, P.J. Sadler, P.J. Bednarski,

291
Trans,trans,trans-[PtIV(N3)2(OH)2(py)(NH3)]: A Light-Activated Antitumor
Platinum Complex That Kills Human Cancer Cells by an Apoptosis-Independent
Mechanism, Molecular Cancer Therapeutics, 11 (2012) 1894-1904.
[223] Y. Zhao, N.J. Farrer, H. Li, J.S. Butler, R.J. McQuitty, A. Habtemariam, F. Wang,
P.J. Sadler, De Novo Generation of Singlet Oxygen and Ammine Ligands by
Photoactivation of a Platinum Anticancer Complex, Angewandte Chemie-
International Edition, 52 (2013) 13633-13637.
[224] L. Ronconi, P.J. Sadler, Photoreaction pathways for the anticancer complex
trans,trans,trans-[Pt(N3)2(OH)2(NH3)2], Dalton Transactions, 40 (2011) 262-268.
[225] F.S. Mackay, J.A. Woods, P. Heringová, J. Kašpárková, A.M. Pizarro, S.A.
Moggach, S. Parsons, V. Brabec, P.J. Sadler, A potent cytotoxic photoactivated
platinum complex, Proceedings of the National Academy of Sciences of the U.S.A.,
104 (2007) 20743-20748.
[226] A.F. Westendorf, L. Zerzankova, L. Salassa, P.J. Sadler, V. Brabec, P.J. Bednarski,
Influence of pyridine versus piperidine ligands on the chemical, DNA binding and
cytotoxic properties of light activated trans,trans,trans- Pt(N3)(2)(OH)2(NH3)(L),
Journal of Inorganic Biochemistry, 105 (2011) 652-662.
[227] H.-C. Tai, Y. Zhao, N.J. Farrer, A.E. Anastasi, G. Clarkson, P.J. Sadler, R.J. Deeth,
A Computational Approach to Tuning the Photochemistry of Platinum(IV)
Anticancer Agents, Chemistry – A European Journal, 18 (2012) 10630-10642.
[228] J. Pracharova, L. Zerzankova, J. Stepankova, O. Novakova, N.J. Farrer, P.J. Sadler,
V. Brabec, J. Kasparkova, Interactions of DNA with a New Platinum(IV) Azide

292
Dipyridine Complex Activated by UVA and Visible Light: Relationship to Toxicity
in Tumor Cells, Chemical Research in Toxicology, 25 (2012) 1099-1111.
[229] H.-C. Tai, R. Brodbeck, J. Kasparkova, N.J. Farrer, V. Brabec, P.J. Sadler, R.J.
Deeth, Combined Theoretical and Computational Study of Interstrand DNA
Guanine-Guanine Cross-Linking by trans- Pt(pyridine)2 Derived from the
Photoactivated Prodrug trans,trans,trans- Pt(N3)2(OH)2(pyridine)2, Inorganic
Chemistry, 51 (2012) 6830-6841.
[230] A.M. Pizarro, R.J. McQuitty, F.S. Mackay, Y. Zhao, J.A. Woods, P.J. Sadler,
Cellular Accumulation, Lipophilicity and Photocytotoxicity of Diazido
Platinum(IV) Anticancer Complexes, ChemMedChem, 9 (2014) 1169-1175.
[231] Z. Zhu, X. Wang, T. Li, S. Aime, P.J. Sadler, Z. Guo, Platinum(II)-Gadolinium(III)
Complexes as Potential Single-Molecular Theranostic Agents for Cancer Treatment,
Angewandte Chemie-International Edition, 53 (2014) 13225-13228.
[232] P. Müller, B. Schröder, J.A. Parkinson, N.A. Kratochwil, R.A. Coxall, A. Parkin, S.
Parsons, P.J. Sadler, Nucleotide Cross-Linking Induced by Photoreactions of
Platinum(IV)–Azide Complexes, Angewandte Chemie International Edition, 42
(2003) 335-339.
[233] F.S. Mackay, J.A. Woods, H. Moseley, J. Ferguson, A. Dawson, S. Parsons, P.J.
Sadler, A Photoactivated trans-Diammine Platinum Complex as Cytotoxic as
Cisplatin, Chemistry – A European Journal, 12 (2006) 3155-3161.
[234] N.J. Farrer, J.A. Woods, V.P. Munk, F.S. Mackay, P.J. Sadler, Photocytotoxic
trans-Diam(m)ine Platinum(IV) Diazido Complexes More Potent than Their cis
Isomers, Chemical Research in Toxicology, 23 (2009) 413-421.

293
[235] Y. Zhao, J.A. Woods, N.J. Farrer, K.S. Robinson, J. Pracharova, J. Kasparkova, O.
Novakova, H. Li, L. Salassa, A.M. Pizarro, G.J. Clarkson, L. Song, V. Brabec, P.J.
Sadler, Diazido Mixed-Amine Platinum(IV) Anticancer Complexes Activatable by
Visible-Light Form Novel DNA Adducts, Chemistry – A European Journal, 19
(2013) 9578-9591.
[236] N.J. Farrer, J.A. Woods, L. Salassa, Y. Zhao, K.S. Robinson, G. Clarkson, F.S.
Mackay, P.J. Sadler, A Potent Trans-Diimine Platinum Anticancer Complex
Photoactivated by Visible Light, Angewandte Chemie International Edition, 49
(2010) 8905-8908.
[237] Y. Zhao, N.J. Farrer, H. Li, J.S. Butler, R.J. McQuitty, A. Habtemariam, F. Wang,
P.J. Sadler, De Novo Generation of Singlet Oxygen and Ammine Ligands by
Photoactivation of a Platinum Anticancer Complex, Angewandte Chemie
International Edition, 52 (2013) 13633-13637.
[238] N. Graf, S.J. Lippard, Redox activation of metal-based prodrugs as a strategy for
drug delivery, Advanced Drug Delivery Reviews, 64 (2012) 993-1004.
[239] P. Sova, A. Mistr, A. Kroutil, F. Zak, P. Pouckova, M. Zadinova, Preclinical anti-
tumor activity of a new oral platinum(IV) drug LA-12, Anti-Cancer Drugs, 16
(2005) 653-657.
[240] K.S. Lovejoy, S.J. Lippard, Non-traditional platinum compounds for improved
accumulation, oral bioavailability, and tumor targeting, Dalton Transactions, (2009)
10651-10659.

294
[241] A.R. Khokhar, Y. Deng, Y. Kido, Z.H. Siddik, Preparation, characterization, and
antitumor activity of new ethylenediamine platinum(IV) complexes containing
mixed carboxylate ligands, Journal of Inorganic Biochemistry, 50 (1993) 79-87.
[242] M.D. Hall, S. Amjadi, M. Zhang, P.J. Beale, T.W. Hambley, The mechanism of
action of platinum(IV) complexes in ovarian cancer cell lines, Journal of Inorganic
Biochemistry, 98 (2004) 1614-1624.
[243] Y.-R. Zheng, K. Suntharalingam, T.C. Johnstone, H. Yoo, W. Lin, J.G. Brooks, S.J.
Lippard, Pt(IV) Prodrugs Designed to Bind Non-Covalently to Human Serum
Albumin for Drug Delivery, Journal of the American Chemical Society, 136 (2014)
8790-8798.
[244] S. Choi, C. Filotto, M. Bisanzo, S. Delaney, D. Lagasee, J.L. Whitworth, A. Jusko,
C. Li, N.A. Wood, J. Willingham, A. Schwenker, K. Spaulding, Reduction and
Anticancer Activity of Platinum(IV) Complexes, Inorganic Chemistry, 37 (1998)
2500-2504.
[245] L. Ellis, H. Er, T. Hambley, The Influence of the Axial Ligands of a Series of
Platinum(IV) Anti-Cancer Complexes on Their Reduction to Platinum(II) and
Reaction With DNA, Australian Journal of Chemistry, 48 (1995) 793-806.
[246] C.K.J. Chen, J.Z. Zhang, J.B. Aitken, T.W. Hambley, Influence of Equatorial and
Axial Carboxylato Ligands on the Kinetic Inertness of Platinum(IV) Complexes in
the Presence of Ascorbate and Cysteine and within DLD-1 Cancer Cells, Journal of
Medicinal Chemistry, 56 (2013) 8757-8764.
[247] J.Z. Zhang, E. Wexselblatt, T.W. Hambley, D. Gibson, Pt(IV) analogs of
oxaliplatin that do not follow the expected correlation between electrochemical

295
reduction potential and rate of reduction by ascorbate, Chemical Communications,
48 (2012) 847-849.
[248] S. Dhar, S.J. Lippard, Mitaplatin, a potent fusion of cisplatin and the orphan drug
dichloroacetate, Proceedings of the National Academy of Sciences of the U.S.A.,
106 (2009) 22199-22204.
[249] E. Wexselblatt, E. Yavin, D. Gibson, Platinum(IV) Prodrugs with Haloacetato
Ligands in the Axial Positions can Undergo Hydrolysis under Biologically
Relevant Conditions, Angewandte Chemie International Edition, 52 (2013) 6059-
6062.
[250] J. Yang, W. Liu, M. Sui, J. Tang, Y. Shen, Platinum (IV)-coordinate polymers as
intracellular reduction-responsive backbone-type conjugates for cancer drug
delivery, Biomaterials, 32 (2011) 9136-9143.
[251] X. Wang, C. Yang, Y. Zhang, X. Zhen, W. Wu, X. Jiang, Delivery of platinum(IV)
drug to subcutaneous tumor and lung metastasis using bradykinin-potentiating
peptide-decorated chitosan nanoparticles, Biomaterials, 35 (2014) 6439-6453.
[252] S. Dhar, W.L. Daniel, D.A. Giljohann, C.A. Mirkin, S.J. Lippard, Polyvalent
Oligonucleotide Gold Nanoparticle Conjugates as Delivery Vehicles for
Platinum(IV) Warheads, Journal of the American Chemical Society, 131 (2009)
14652-14653.
[253] A. Kumar, S. Huo, X. Zhang, J. Liu, A. Tan, S. Li, S. Jin, X. Xue, Y. Zhao, T. Ji, L.
Han, H. Liu, X. Zhang, J. Zhang, G. Zou, T. Wang, S. Tang, X.-J. Liang,
Neuropilin-1-Targeted Gold Nanoparticles Enhance Therapeutic Efficacy of
Platinum(IV) Drug for Prostate Cancer Treatment, ACS Nano, 8 (2014) 4205-4220.

296
[254] S.L. Yoong, B.S. Wong, Q.L. Zhou, C.F. Chin, J. Li, T. Venkatesan, H.K. Ho, V.
Yu, W.H. Ang, G. Pastorin, Enhanced cytotoxicity to cancer cells by mitochondria-
targeting MWCNTs containing platinum(IV) prodrug of cisplatin, Biomaterials, 35
(2014) 748-759.
[255] Y. Shi, S.-A. Liu, D.J. Kerwood, J. Goodisman, J.C. Dabrowiak, Pt(IV) complexes
as prodrugs for cisplatin, Journal of Inorganic Biochemistry, 107 (2012) 6-14.
[256] W. Neumann, B.C. Crews, L.J. Marnett, E. Hey-Hawkins, Conjugates of Cisplatin
and Cyclooxygenase Inhibitors as Potent Antitumor Agents Overcoming Cisplatin
Resistance, ChemMedChem, 9 (2014) 1150-1153.
[257] V. Novohradsky, L. Zerzankova, J. Stepankova, O. Vrana, R. Raveendran, D.
Gibson, J. Kasparkova, V. Brabec, Antitumor platinum(IV) derivatives of
oxaliplatin with axial valproato ligands, Journal of Inorganic Biochemistry, 140
(2014) 72-79.
[258] K.R. Barnes, A. Kutikov, S.J. Lippard, Synthesis, Characterization, and
Cytotoxicity of a Series of Estrogen-Tethered Platinum(IV) Complexes, Chemistry
& Biology, 11 (2004) 557-564.
[259] S. Abramkin, S.M. Valiahdi, M.A. Jakupec, M. Galanski, N. Metzler-Nolte, B.K.
Keppler, Solid-phase synthesis of oxaliplatin-TAT peptide bioconjugates, Dalton
Transactions, 41 (2012) 3001-3005.
[260] D.Y.Q. Wong, C.H.F. Yeo, W.H. Ang, Immuno-Chemotherapeutic Platinum(IV)
Prodrugs of Cisplatin as Multimodal Anticancer Agents, Angewandte Chemie
International Edition, 53 (2014) 6752-6756.

297
[261] N. Graf, T.E. Mokhtari, I.A. Papayannopoulos, S.J. Lippard, Platinum(IV)-
chlorotoxin (CTX) conjugates for targeting cancer cells, Journal of Inorganic
Biochemistry, 110 (2012) 58-63.
[262] Simon P. Wisnovsky, Justin J. Wilson, Robert J. Radford, Mark P. Pereira, Maria R.
Chan, Rebecca R. Laposa, Stephen J. Lippard, Shana O. Kelley, Targeting
Mitochondrial DNA with a Platinum-Based Anticancer Agent, Chemistry &
Biology, 20 (2013) 1323-1328.
[263] K. Suntharalingam, Y. Song, S.J. Lippard, Conjugation of vitamin E analog [small
alpha]-TOS to Pt(IV) complexes for dual-targeting anticancer therapy, Chemical
Communications, 50 (2014) 2465-2468.
[264] S. Dhar, N. Kolishetti, S.J. Lippard, O.C. Farokhzad, Targeted delivery of a
cisplatin prodrug for safer and more effective prostate cancer therapy in vivo,
Proceedings of the National Academy of Sciences of the U.S.A., 108 (2011) 1850-
1855.
[265] S. Dhar, F.X. Gu, R. Langer, O.C. Farokhzad, S.J. Lippard, Targeted delivery of
cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA–
PEG nanoparticles, Proceedings of the National Academy of Sciences of the U.S.A.,
105 (2008) 17356-17361.
[266] D. Montagner, S.Q. Yap, W.H. Ang, A Fluorescent Probe for Investigating the
Activation of Anticancer Platinum(IV) Prodrugs Based on the Cisplatin Scaffold,
Angewandte Chemie International Edition, 52 (2013) 11785-11789.

298
[267] W. Scarano, H.T.T. Duong, H. Lu, P.L. De Souza, M.H. Stenzel, Folate
Conjugation to Polymeric Micelles via Boronic Acid Ester to Deliver Platinum
Drugs to Ovarian Cancer Cell Lines, Biomacromolecules, 14 (2013) 962-975.
[268] H. Xiao, H. Song, Q. Yang, H. Cai, R. Qi, L. Yan, S. Liu, Y. Zheng, Y. Huang, T.
Liu, X. Jing, A prodrug strategy to deliver cisplatin(IV) and paclitaxel in
nanomicelles to improve efficacy and tolerance, Biomaterials, 33 (2012) 6507-6519.
[269] J. Graham, M. Mushin, P. Kirkpatrick, Oxaliplatin, Nature Reviews Drug
Discovery, 3 (2004) 11-12.
[270] G. Momekov, A. Bakalova, M. Karaivanova, Novel approaches towards
development of non-classical platinum-based antineoplastic agents: Design of
platinum complexes characterized by an alternative DNA-binding pattern and/or
tumor-targeted cytotoxicity, Current Medicinal Chemistry, 12 (2005) 2177-2191.
[271] R. Guddneppanavar, U. Bierbach, Adenine-N3 in the DNA minor groove - an
emerging target for platinum containing anticancer pharmacophores, Anticancer
Agents Med Chem, 7 (2007) 125-138.
[272] S. Komeda, A. Casini, Next-generation anticancer metallodrugs, Current Topics in
Medicinal Chemistry, 12 (2012) 219-235.
[273] S. Komeda, Unique platinum-DNA interactions may lead to more effective
platinum-based antitumor drugs, Metallomics, 3 (2011) 650-655.
[274] H. Baruah, M.W. Wright, U. Bierbach, Solution structural study of a DNA duplex
containing the guanine-N7 adduct formed by a cytotoxic platinum-acridine hybrid
agent, Biochemistry, 44 (2005) 6059-6070.

299
[275] X. Qiao, A.E. Zeitany, M.W. Wright, A.S. Essader, K.E. Levine, G.L. Kucera, U.
Bierbach, Analysis of the DNA damage produced by a platinum-acridine antitumor
agent and its effects in NCI-H460 lung cancer cells, Metallomics, 4 (2012) 645-652.
[276] C.L. Smyre, G. Saluta, T.E. Kute, G.L. Kucera, U. Bierbach, Inhibition of DNA
Synthesis by a Platinum-Acridine Hybrid Agent Leads to Potent Cell Kill in Non-
Small Cell Lung Cancer, ACS Med Chem Lett, 2 (2011) 870-874.
[277] L.A. Graham, G.M. Wilson, T.K. West, C.S. Day, G.L. Kucera, U. Bierbach,
Unusual Reactivity of a Potent Platinum-Acridine Hybrid Antitumor Agent, ACS
Medicinal Chemistry Letters, 2 (2011) 687-691.
[278] J.R. Choudhury, L. Rao, U. Bierbach, Rates of intercalator-driven platination of
DNA determined by a restriction enzyme cleavage inhibition assay, J Biol Inorg
Chem, 16 (2011) 373-380.
[279] Z. Ma, J.R. Choudhury, M.W. Wright, C.S. Day, G. Saluta, G.L. Kucera, U.
Bierbach, A non-cross-linking platinum-acridine agent with potent activity in non-
small-cell lung cancer, Journal of Medicinal Chemistry, 51 (2008) 7574-7580.
[280] S.C. Alley, N.M. Okeley, P.D. Senter, Antibody-drug conjugates: targeted drug
delivery for cancer, Current Opinion in Chemical Biology, 14 (2010) 529-537.
[281] R. Srinivasan, J. Li, S.L. Ng, K.A. Kalesh, S.Q. Yao, Methods of using click
chemistry in the discovery of enzyme inhibitors, Nature Protocols, 2 (2007) 2655-
2664.
[282] M. Bahta, F. Liu, S.E. Kim, A.G. Stephen, R.J. Fisher, T.R. Burke, Jr., Oxime-
based linker libraries as a general approach for the rapid generation and screening
of multidentate inhibitors, Nature Protocols, 7 (2012) 686-702.

300
[283] V.Y. Kukushkin, A.J. Pombeiro, Additions to metal-activated organonitriles,
Chemical Reviews, 102 (2002) 1771-1802.
[284] J.M. Baskin, J.A. Prescher, S.T. Laughlin, N.J. Agard, P.V. Chang, I.A. Miller, A.
Lo, J.A. Codelli, C.R. Bertozzi, Copper-free click chemistry for dynamic in vivo
imaging, Proceedings of the National Academy of Sciences of the U.S.A., 104
(2007) 16793-16797.
[285] R.E. Wang, F. Costanza, Y. Niu, H. Wu, Y. Hu, W. Hang, Y. Sun, J. Cai,
Development of self-immolative dendrimers for drug delivery and sensing, Journal
of Controlled Release, 159 (2012) 154-163.
[286] M.R. Karver, R. Weissleder, S.A. Hilderbrand, Bioorthogonal reaction pairs enable
simultaneous, selective, multi-target imaging, Angew Chem Int Ed Engl, 51 (2012)
920-922.
[287] S.C. Dhara, A rapid method for the synthesis of cis-[Pt(NH3)2Cl2], Indian Journal
of Chemistry, 8 (1970) 193-194.
[288] T.M. Augustus, J. Anderson, S.M. Hess, U. Bierbach,
Bis(acridinylthiourea)platinum(II) Complexes: Synthesis, DNA affinity, and
biological activity in ghoblastoma cells, Bioorganic & Medicinal Chemistry Letters,
13 (2003) 855-858.
[289] Z. Ma, J.R. Choudhury, M.W. Wright, C.S. Day, G. Saluta, G.L. Kucera, U.
Bierbach, A Non-Cross-Linking Platinum−Acridine Agent with Potent Activity in
Non-Small-Cell Lung Cancer, Journal of Medicinal Chemistry, 51 (2008) 7574-
7580.

301
[290] D. Wang, S.J. Lippard, Cellular processing of platinum anticancer drugs, Nature
Reviews Drug Discovery, 4 (2005) 307-320.
[291] A.V. Klein, T.W. Hambley, Platinum Drug Distribution in Cancer Cells and
Tumors, Chemical Reviews, 109 (2009) 4911-4920.
[292] L.E. Wedlock, S.J. Berners-Price, Recent Advances in Mapping the Sub-cellular
Distribution of Metal-Based Anticancer Drugs, Australian Journal of Chemistry, 64
(2011) 692-704.
[293] S. Trapp, R.W. Horobin, A predictive model for the selective accumulation of
chemicals in tumor cells, European Biophysics Journal with Biophysics Letters, 34
(2005) 959-966.
[294] X.J. Liang, D.W. Shen, K.G. Chen, S.M. Wincovitch, S.H. Garfield, M.M.
Gottesman, Trafficking and localization of platinum complexes in cisplatin-
resistant cell lines monitored by fluorescence-labeled platinum, Journal of Cellular
Physiology, 202 (2005) 635-641.
[295] J. Suryadi, U. Bierbach, DNA Metalating-Intercalating Hybrid Agents for the
Treatment of Chemoresistant Cancers, Chemistry – A European Journal, 18 (2012)
12926-12934.
[296] K. Cheung-Ong, K.T. Song, Z.D. Ma, D. Shabtai, A.Y. Lee, D. Gallo, L.E. Heisler,
G.W. Brown, U. Bierbach, G. Giaever, C. Nislow, Comparative Chemogenomics
To Examine the Mechanism of Action of DNA-Targeted Platinum-Acridine
Anticancer Agents, ACS Chemical Biology, 7 (2012) 1892-1901.

302
[297] D. Fornasiero, T. Kurucsev, The binding of 9-aminoacridine to calf thymus DNA
in aqueous solution-electronic spectral studies, Biophysical Chemistry, 23 (1985)
31-37.
[298] Z.P. Demko, K.B. Sharpless, A click chemistry approach to tetrazoles by Huisgen
1,3-dipolar cycloaddition: Synthesis of 5-sulfonyl tetrazoles from azides and
sulfonyl cyanides, Angewandte Chemie-International Edition, 41 (2002) 2110-2113.
[299] S.B. Buck, J. Bradford, K.R. Gee, B.J. Agnew, S.T. Clarke, A. Salic, Detection of
S-phase cell cycle progression using 5-ethynyl-2 '-deoxyuridine incorporation with
click chemistry an alternative to using 5-bromo-2 '-deoxyuridine antibodies,
Biotechniques, 44 (2008) 927-929.
[300] S.A. Kane, S.J. Lippard, Photoreactivity of platinum(II) in cisplatin-modified DNA
affords specific cross-links to HMG domain proteins, Biochemistry, 35 (1996)
2180-2188.
[301] H. Kostrhunova, J. Malina, A.J. Pickard, J. Stepankova, M. Vojtiskova, J.
Kasparkova, T. Muchova, M.L. Rohlfing, U. Bierbach, V. Brabec, Replacement of
a Thiourea with an Amidine Group in a Monofunctional Platinum-Acridine
Antitumor Agent. Effect on DNA Interactions, DNA Adduct Recognition and
Repair, Molecular Pharmaceutics, 8 (2011) 1941-1954.
[302] A. Saraste, K. Pulkki, Morphologic and biochemical hallmarks of apoptosis,
Cardiovascular Research, 45 (2000) 528-537.
[303] Y.W. Lam, L. Trinkle-Mulcahy, A.I. Lamond, The nucleolus, Journal of Cell
Science, 118 (2005) 1335-1337.

303
[304] X. Ning, J. Guo, M.A. Wolfert, G.J. Boons, Visualizing metabolically labeled
glycoconjugates of living cells by copper-free and fast huisgen cycloadditions,
Angew Chem Int Ed Engl, 47 (2008) 2253-2255.
[305] C.L. Smyre, G. Saluta, T.E. Kute, G.L. Kucera, U. Bierbach, Inhibition of DNA
Synthesis by a Platinum-Acridine Hybrid Agent Leads to Potent Cell Kill in
Nonsmall Cell Lung Cancer, ACS Medicinal Chemistry Letters, 2 (2011) 870-874.
[306] D. Drygin, W.G. Rice, I. Grummt, The RNA Polymerase I Transcription
Machinery: An Emerging Target for the Treatment of Cancer, Annual Review of
Pharmacology and Toxicology2010, pp. 131-156.
[307] D. Drygin, A. Lin, J. Bliesath, C.B. Ho, S.E. O'Brien, C. Proffitt, M. Omori, M.
Haddach, M.K. Schwaebe, A. Siddiqui-Jain, N. Streiner, J.E. Quin, E. Sanij, M.J.
Bywater, R.D. Hannan, D. Ryckman, K. Anderes, W.G. Rice, Targeting RNA
Polymerase I with an Oral Small Molecule CX-5461 Inhibits Ribosomal RNA
Synthesis and Solid Tumor Growth, Cancer Research, 71 (2011) 1418-1430.
[308] J.M. Baskin, J.A. Prescher, S.T. Laughlin, N.J. Agard, P.V. Chang, I.A. Miller, A.
Lo, J.A. Codelli, C.R. Bertozzi, Copper-free click chemistry for dynamic in vivo
imaging, Proceedings of the National Academy of Sciences of the United States of
America, 104 (2007) 16793-16797.
[309] L.A. Graham, J. Suryadi, T.K. West, G.L. Kucera, U. Bierbach, Synthesis, aqueous
reactivity, and biological evaluation of carboxylic Acid ester-functionalized
platinum-acridine hybrid anticancer agents, J Med Chem, 55 (2012) 7817-7827.
[310] S. Ding, X. Qiao, J. Suryadi, G.S. Marrs, G.L. Kucera, U. Bierbach, Using
Fluorescent Post-Labeling To Probe the Subcellular Localization of DNA-Targeted

304
Platinum Anticancer Agents, Angewandte Chemie-International Edition, 52 (2013)
3350-3354.
[311] A.C. Society, Cancer Facts & Figures 2012, American Cancer Society, Atlanta, GA,
2012.
[312] M.J. Higgins, V. Stearns, Understanding Resistance to Tamoxifen in Hormone
Receptor-Positive Breast Cancer, Clinical Chemistry, 55 (2009) 1453-1455.
[313] M.P. Decatris, S. Sundar, K.J. O'Byrne, Platinum-based chemotherapy in
metastatic breast cancer: current status, Cancer Treatment Reviews, 30 (2004) 53-
81.
[314] C.L. Smith, B.W. O'Malley, Coregulator function: A key to understanding tissue
specificity of selective receptor modulators, Endocrine Reviews, 25 (2004) 45-71.
[315] E. Kim, P.T. Rye, J.M. Essigmann, R.G. Croy, A bifunctional platinum(II)
antitumor agent that forms DNA adducts with affinity for the estrogen receptor,
Journal of Inorganic Biochemistry, 103 (2009) 256-261.
[316] R. Gust, W. Beck, G. Jaouen, H. Schoenenberger, Optimization of cisplatin for the
treatment of hormone dependent tumoral diseases Part 1: Use of steroidal ligands,
Coordination Chemistry Reviews, 253 (2009) 2742-2759.
[317] R. Gust, W. Beck, G. Jaouen, H. Schoenenberger, Optimization of cisplatin for the
treatment of hormone-dependent tumoral diseases Part 2: Use of non-steroidal
ligands, Coordination Chemistry Reviews, 253 (2009) 2760-2779.
[318] K. Mitra, J.C. Marquis, S.M. Hillier, P.T. Rye, B. Zayas, A.S. Lee, J.M. Essigmann,
R.G. Croy, A rationally designed genotoxin that selectively destroys estrogen

305
receptor-positive breast cancer cells, Journal of the American Chemical Society,
124 (2002) 1862-1863.
[319] P.J. Burke, T.H. Koch, Design, synthesis, and biological evaluation of doxorubicin
- Formaldehyde conjugates targeted to breast cancer cells, Journal of Medicinal
Chemistry, 47 (2004) 1193-1206.
[320] K.L. Dao, R.R. Sawant, J.A. Hendricks, V. Ronga, V.P. Torchilin, R.N. Hanson,
Design, Synthesis, and Initial Biological Evaluation of a Steroidal Anti-Estrogen-
Doxorubicin Bioconjugate for Targeting Estrogen Receptor-Positive Breast Cancer
Cells, Bioconjugate Chemistry, 23 (2012) 785-795.
[321] K.W. Peng, H. Wang, Z. Qin, G.T. Wijewickrama, M. Lu, Z. Wang, J.L. Bolton,
G.R.J. Thatcher, Selective Estrogen Receptor Modulator Delivery of Quinone
Warheads to DNA Triggering Apoptosis in Breast Cancer Celts, ACS Chemical
Biology, 4 (2009) 1039-1049.
[322] M.D. Johnson, H. Zuo, K.H. Lee, J.P. Trebley, J.M. Rae, R.V. Weatherman, Z.
Desta, D.A. Flockhart, T.C. Skaar, Pharmacological characterization of 4-hydroxy-
N-desmethyl tamoxifen, a novel active metabolite of tamoxifen, Breast Cancer
Research and Treatment, 85 (2004) 151-159.
[323] A.K. Shiau, D. Barstad, P.M. Loria, L. Cheng, P.J. Kushner, D.A. Agard, G.L.
Greene, The structural basis of estrogen receptor/coactivator recognition and the
antagonism of this interaction by tamoxifen, Cell, 95 (1998) 927-937.
[324] B.S. Katzenellenbogen, M.J. Norman, R.L. Eckert, S.W. Peltz, W.F. Mangel,
Bioactivities, estrogen-receptor interactions, and plasminogen activator-inducing

306
activities of tamoxifen an hydroxytamoxifen isomers in MCF-7 human-breast
cancer-cells, Cancer Research, 44 (1984) 112-119.
[325] S.C. Alley, N.M. Okeley, P.D. Senter, Antibody-drug conjugates: targeted drug
delivery for cancer, Current Opinion in Chemical Biology, 14 (2010) 529-537.
[326] M. Lacroix, G. Leclercq, Relevance of breast cancer cell lines as models for breast
tumours: an update, Breast Cancer Research and Treatment, 83 (2004) 249-289.
[327] M.J. Abedin, D. Wang, M.A. McDonnell, U. Lehmann, A. Kelekar, Autophagy
delays apoptotic death in breast cancer cells following DNA damage, Cell Death
and Differentiation, 14 (2007) 500-510.
[328] R.E. Wang, F. Costanza, Y. Niu, H. Wu, Y. Hu, W. Hang, Y. Sun, J. Cai,
Development of self-immolative dendrimers for drug delivery and sensing, Journal
of Controlled Release, 159 (2012) 154-163.
[329] E.A. Musgrove, R.L. Sutherland, Biological determinants of endocrine resistance in
breast cancer, Nature Reviews Cancer, 9 (2009) 631-643.
[330] D.D. Yu, B.M. Forman, Simple and Efficient Production of (Z)-4-
Hydroxytamoxifen, a Potent Estrogen Receptor Modulator, The Journal of Organic
Chemistry, 68 (2003) 9489-9491.
[331] A.H. Fauq, G.M. Maharvi, D. Sinha, A convenient synthesis of (Z)-4-hydroxy-N-
desmethyltamoxifen (endoxifen), Bioorganic & Medicinal Chemistry Letters, pp.
3036-3038.
[332] K.M. Huttunen, H. Raunio, J. Rautio, Prodrugs-from Serendipity to Rational
Design, Pharmacological Reviews, 63 (2011) 750-771.

307
[333] T. Lammers, F. Kiessling, W.E. Hennink, G. Storm, Drug targeting to tumors:
Principles, pitfalls and (pre-) clinical progress, Journal of Controlled Release, 161
(2012) 175-187.
[334] E.H. Kerns, L. Di, Pharmaceutical profiling in drug discovery, Drug Discovery
Today, 8 (2003) 316-323.
[335] K.M. H. Lullmann, A. Ziegler, D. Bieger, , Color Atlas of Pharmacology, 2ednd ed,
Thieme, Stuttgart (2000) pp.32-42.
[336] S.E. Pratt, S. Durland-Busbice, R.L. Shepard, K. Heinz-Taheny, P.W. Iversen, A.H.
Dantzig, Human Carboxylesterase-2 Hydrolyzes the Prodrug of Gemcitabine
(LY2334737) and Confers Prodrug Sensitivity to Cancer Cells, Clinical Cancer
Research, 19 (2013) 1159-1168.
[337] T. Imai, M. Taketani, M. Shii, M. Hosokawa, K. Chiba, Substrate specificity of
carboxylesterase isozymes and their contribution to hydrolase activity in human
liver and small intestine, Drug Metabolism and Disposition, 34 (2006) 1734-1741.
[338] S. Ding, X. Qiao, G.L. Kucera, U. Bierbach, Using a Build-and-Click Approach for
Producing Structural and Functional Diversity in DNA-Targeted Hybrid Anticancer
Agents, Journal of Medicinal Chemistry, 55 (2012) 10198-10203.
[339] G. Natile, F.P. Intini, R. Bertani, R.A. Michelin, M. Mozzon, S.M. Sbovata, A.
Venzo, R. Seraglia, Synthesis and characterisation of the amidine complexes trans-
PtCl(NH3) {HN=C(NH2)R}(2) Cl (R = Me, Ph, CH2Ph) derived from addition of
NH3 to the coordinated nitriles in trans- PtCl2(N CR)(2), Journal of
Organometallic Chemistry, 690 (2005) 2121-2127.

308
[340] R.A. Michelin, P. Sgarbossa, S.M. Sbovata, V. Gandin, C. Marzano, R. Bertani,
Chemistry and Biological Activity of Platinum Amidine Complexes,
ChemMedChem, 6 (2011) 1172-1183.
[341] J. Wang, E.T. Williams, J. Bourgea, Y.N. Wong, C.J. Patten, Characterization of
Recombinant Human Carboxylesterases: Fluorescein Diacetate as a Probe
Substrate for Human Carboxylesterase 2, Drug Metabolism and Disposition, 39
(2011) 1329-1333.
[342] S.M. Valiahdi, A.E. Egger, W. Miklos, U. Jungwirth, K. Meelich, P. Nock, W.
Berger, C.G. Hartinger, M. Galanski, M.A. Jakupec, B.K. Keppler, Influence of
extracellular pH on the cytotoxicity, cellular accumulation, and DNA interaction of
novel pH-sensitive 2-aminoalcoholatoplatinum(II) complexes, Journal of
Biological Inorganic Chemistry, 18 (2013) 249-260.
[343] D.A. Weaver, E.L. Crawford, K.A. Warner, F. Elkhairi, S.A. Khuder, J.C. Willey,
ABCC5, ERCC2, XPA and XRCC1 transcript abundance levels correlate with
cisplatin chemoresistance in non-small cell lung cancer cell lines, Molecular
Cancer, 4 (2005).
[344] B.L. Barthel, Z.Y. Zhang, D.L. Rudnicki, C.D. Coldren, M. Polinkovsky, H.R. Sun,
G.G. Koch, D.C.F. Chan, T.H. Koch, Preclinical Efficacy of a Carboxylesterase 2-
Activated Prodrug of Doxazolidine, Journal of Medicinal Chemistry, 52 (2009)
7678-7688.
[345] Z. Zhang, H. Hatta, K. Tanabe, S. Nishimoto, A new class of 5-fluoro-2 '-
deoxyuridine prodrugs conjugated with a tumor-homing cyclic peptide CNGRC by

309
ester linkers: Synthesis, reactivity, and tumor-cell-selective cytotoxicity,
Pharmaceutical Research, 22 (2005) 381-389.
[346] E.T. Martins, H. Baruah, J. Kramarczyk, G. Saluta, C.S. Day, G.L. Kucera, U.
Bierbach, Design, synthesis, and biological activity of a novel non-cisplatin-type
platinum-acridine pharmacophore, J Med Chem, 44 (2001) 4492-4496.
[347] J.B. Gibbs, Mechanism-based target identification and drug discovery in cancer
research, Science, 287 (2000) 1969-1973.
[348] J. Suryadi, U. Bierbach, DNA Metalating-Intercalating Hybrid Agents for the
Treatment of Chemoresistant Cancers, Chemistry-a European Journal, 18 (2012)
12926-12934.
[349] K. Cheung-Ong, K.T. Song, Z. Ma, D. Shabtai, A.Y. Lee, D. Gallo, L.E. Heisler,
G.W. Brown, U. Bierbach, G. Giaever, C. Nislow, Comparative Chemogenomics
To Examine the Mechanism of Action of DNA-Targeted Platinum-Acridine
Anticancer Agents, ACS Chemical Biology, 7 (2012) 1892-1901.
[350] S. Ding, A.J. Pickard, G.L. Kucera, U. Bierbach, Design of Enzymatically
Cleavable Prodrugs of a Potent Platinum-Containing Anticancer Agent, Chemistry
– A European Journal, 20 (2014) 16164-16173.
[351] S. Ding, X. Qiao, G.L. Kucera, U. Bierbach, Design of a platinum-acridine-
endoxifen conjugate targeted at hormone-dependent breast cancer, Chemical
Communications, 49 (2013) 2415-2417.
[352] R. Morphy, Z. Rankovic, Designed multiple ligands. An emerging drug discovery
paradigm, Journal of Medicinal Chemistry, 48 (2005) 6523-6543.

310
[353] J. Yang, X. Sun, W. Mao, M. Sui, J. Tang, Y. Shen, Conjugate of Pt(IV)-Histone
Deacetylase Inhibitor as a Prodrug for Cancer Chemotherapy, Molecular
Pharmaceutics, 9 (2012) 2793-2800.
[354] S. Dhar, S.J. Lippard, Mitaplatin, a potent fusion of cisplatin and the orphan drug
dichloroacetate, Proceedings of the National Academy of Sciences of the United
States of America, 106 (2009) 22199-22204.
[355] R.K. Pathak, S. Marrache, J.H. Choi, T.B. Berding, S. Dhar, The Prodrug Platin-A:
Simultaneous Release of Cisplatin and Aspirin, Angewandte Chemie-International
Edition, 53 (2014) 1963-1967.
[356] C.T. Keith, A.A. Borisy, B.R. Stockwell, Multicomponent therapeutics for
networked systems, Nature Reviews Drug Discovery, 4 (2005) 71-U10.
[357] S.A. Eisen, D.K. Miller, R.S. Woodward, E. Spitznagel, T.R. Przybeck, The effect
of prescribled daily dose frequency on patient medication compliance, Archives of
Internal Medicine, 150 (1990) 1881-1884.
[358] A.S. Reddy, S. Zhang, Polypharmacology: drug discovery for the future, Expert
Review of Clinical Pharmacology, 6 (2013) 41-47.
[359] R. Morphy, C. Kay, Z. Rankovic, From magic bullets to designed multiple ligands,
Drug Discovery Today, 9 (2004) 641-651.
[360] Y. Bansal, O. Silakari, Multifunctional compounds: Smart molecules for
multifactorial diseases, European Journal of Medicinal Chemistry, 76 (2014) 31-42.
[361] A. El-Faham, F. Albericio, Peptide Coupling Reagents, More than a Letter Soup,
Chemical Reviews, 111 (2011) 6557-6602.

311
[362] P. Thirumurugan, D. Matosiuk, K. Jozwiak, Click Chemistry for Drug
Development and Diverse Chemical-Biology Applications, Chemical Reviews, 113
(2013) 4905-4979.
[363] A.J. Pickard, F. Liu, T.F. Bartenstein, L.G. Haines, K.E. Levine, G.L. Kucera, U.
Bierbach, Redesigning the DNA-Targeted Chromophore in Platinum-Acridine
Anticancer Agents: A Structure-Activity Relationship Study, Chemistry – A
European Journal, 20 (2014) 16174-16187.
[364] G. Dirscherl, B. Koenig, The use of solid-phase synthesis techniques for the
preparation of peptide-metal complex conjugates, European Journal of Organic
Chemistry, (2008) 597-634.
[365] E. Valeur, M. Bradley, Amide bond formation: beyond the myth of coupling
reagents, Chemical Society Reviews, 38 (2009) 606-631.
[366] A. El-Faham, F. Albericio, COMU: A third generation of uronium-type coupling
reagents, Journal of Peptide Science, 16 (2010) 6-9.
[367] C. Najera, From alpha-amino acids to peptides: All you need for the journey,
Synlett, (2002) 1388-1403.
[368] J. Lu, C. Liu, P. Wang, M. Ghazwani, J. Xu, Y. Huang, X. Ma, P. Zhang, S. Li,
The self-assembling camptothecin-tocopherol prodrug: An effective approach for
formulating camptothecin, Biomaterials, 62 (2015) 176-187.
[369] S. Aryal, C.-M.J. Hu, L. Zhang, Synthesis of Ptsome: a platinum-based liposome-
like nanostructure, Chemical Communications, 48 (2012) 2630-2632.

312
[370] J.R. Vargas, E.G. Stanzl, N.N.H. Teng, P.A. Wender, Cell-Penetrating,
Guanidinium-Rich Molecular Transporters for Overcoming Efflux-Mediated
Multidrug Resistance, Molecular Pharmaceutics, 11 (2014) 2553-2565.
[371] T. Ekblad, E. Camaioni, H. Schuler, A. Macchiarulo, PARP inhibitors:
polypharmacology versus selective inhibition, FEBS Journal, 280 (2013) 3563-
3575.
[372] M. Yang, A.J. Pickard, X. Qiao, M.J. Gueble, C.S. Day, G.L. Kucera, U. Bierbach,
Synthesis, Reactivity, and Biological Activity of Gold(I) Complexes Modified with
Thiourea-Functionalized Tyrosine Kinase Inhibitors, Inorganic Chemistry, 54
(2015) 3316-3324.
[373] L. Milane, M. Trivedi, A. Singh, M. Talekar, M. Amiji, Mitochondrial biology,
targets, and drug delivery, Journal of Controlled Release, 207 (2015) 40-58.
[374] V.K. Yellepeddi, K.K. Vangara, S. Palakurthi, Poly(amido)amine (PAMAM)
dendrimer-cisplatin complexes for chemotherapy of cisplatin-resistant ovarian
cancer cells, Journal of Nanoparticle Research, 15 (2013).
[375] D.M. Turner, C.T.M.B. Tom, A.R. Renslo, Simple Plate-Based, Parallel Synthesis
of Disulfide Fragments using the CuAAC Click Reaction, Acs Combinatorial
Science, 16 (2014) 661-664.
[376] O. Moukha-Chafiq, R.C. Reynolds, Parallel Solution-Phase Synthesis and General
Biological Activity of a Uridine Antibiotic Analog Library, Acs Combinatorial
Science, 16 (2014) 232-237.

313
[377] S. Mukhopadhyay, C.M. Barnes, A. Haskel, S.M. Short, K.R. Barnes, S.J. Lippard,
Conjugated platinum(IV)-peptide complexes for targeting angiogenic tumor
vasculature, Bioconjugate Chemistry, 19 (2008) 39-49.
[378] W. Bannwarth, R. Knorr, Formation of carboxamides with N,N,N',N'-tetramethyl
(succinimido) uronium tetrafluoroborate in aqueous / organic solvent systems,
Tetrahedron Letters, 32 (1991) 1157-1160.
[379] M.J. Wigglesworth, D.C. Murray, C.J. Blackett, M. Kossenjans, J.W.M. Nissink,
Increasing the delivery of next generation therapeutics from high throughput
screening libraries, Current Opinion in Chemical Biology, 26 (2015) 104-110.
[380] S. van Zutphen, E.A. Stone, S. van Rijt, M.S. Robillard, G.A. van der Marel, H.S.
Overkleeft, H. den Dulk, J. Brouwer, J. Reedijk, Combinatorial discovery of new
asymmetric cis platinum anticancer complexes is made possible with solid-phase
synthetic methods, Journal of Inorganic Biochemistry, 99 (2005) 2032-2038.
[381] C.J. Ziegler, A.P. Silverman, S.J. Lippard, High-throughput synthesis and
screening of platinum drug candidates, Journal of Biological Inorganic Chemistry,
5 (2000) 774-783.
[382] A. Cern, A. Golbraikh, A. Sedykh, A. Tropsha, Y. Barenholz, A. Goldblum,
Quantitative structure - property relationship modeling of remote liposome loading
of drugs, Journal of Controlled Release, 160 (2012) 147-157.
[383] A.A. Kale, V.P. Torchilin, "Smart" drug carriers: PEGylated TATp-Modified pH-
Sensitive Liposomes, Journal of Liposome Research, 17 (2007) 197-203.
[384] C. Allen, N. Dos Santos, R. Gallagher, G.N.C. Chiu, Y. Shu, W.M. Li, S.A.
Johnstone, A.S. Janoff, L.D. Mayer, M.S. Webb, M.B. Bally, Controlling the

314
physical behavior and biological performance of liposome formulations through
use of surface grafted poly(ethylene glycol), Bioscience Reports, 22 (2002) 225-
250.
[385] V.P. Torchilin, V.G. Omelyanenko, M.I. Papisov, A.A. Bogdanov, V.S. Trubetskoy,
J.N. Herron, C.A. Gentry, Poly(ethylene glycol) on the liposome surface-on the
mechanism of polymer-coated liposome longevity, Biochimica Et Biophysica Acta-
Biomembranes, 1195 (1994) 11-20.
[386] M.-L. Briuglia, C. Rotella, A. McFarlane, D.A. Lamprou, Influence of cholesterol
on liposome stability and on in vitro drug release, Drug Delivery and Translational
Research, 5 (2015) 231-242.
[387] N. Dos Santos, L.D. Mayer, S.A. Abraham, R.C. Gallagher, R.A.K. Cox, P.G.
Tardi, M.B. Bally, Improved retention of idarubicin after intravenous injection
obtained for cholesterol-free liposomes, Biochimica Et Biophysica Acta-
Biomembranes, 1561 (2002) 188-201.
[388] J.-P. Colletier, B. Chaize, M. Winterhalter, D. Fournier, Protein encapsulation in
liposomes: efficiency depends on interactions between protein and phospholipid
bilayer, BMC biotechnology, 2 (2002) 9-9.
[389] A.P. Costa, X. Xu, D.J. Burgess, Freeze-Anneal-Thaw Cycling of Unilamellar
Liposomes: Effect on Encapsulation Efficiency, Pharmaceutical Research, 31 (2014)
97-103.
[390] N. Dos Santos, C. Allen, A.-M. Doppen, M. Anantha, K.A.K. Cox, R.C. Gallagher,
G. Karlsson, K. Edwards, G. Kenner, L. Samuels, M.S. Webb, M.B. Bally,
Influence of poly(ethylene glycol) grafting density and polymer length on

315
liposomes: Relating plasma circulation lifetimes to protein binding, Biochimica Et
Biophysica Acta-Biomembranes, 1768 (2007) 1367-1377.
[391] K.N. Burger, R.W. Staffhorst, H.C. de Vijlder, M.J. Velinova, P.H. Bomans, P.M.
Frederik, B. de Kruijff, Nanocapsules: lipid-coated aggregates of cisplatin with
high cytotoxicity, Nature Medicine, 8 (2002) 81-84.
[392] H. Shmeeda, Y. Amitay, D. Tzemach, J. Gorin, A. Gabizon, Liposome
encapsulation of zoledronic acid results in major changes in tissue distribution and
increase in toxicity, Journal of Controlled Release, 167 (2013) 265-275.
[393] K. Riviere, H.M. Kieler-Ferguson, K. Jerger, F.C. Szoka, Jr., Anti-tumor activity of
liposome encapsulated fluoroorotic acid as a single agent and in combination with
liposome irinotecan, Journal of Controlled Release, 153 (2011) 288-296.
[394] C. He, K. Lu, D. Liu, W. Lin, Nanoscale Metal-Organic Frameworks for the Co-
Delivery of Cisplatin and Pooled siRNAs to Enhance Therapeutic Efficacy in
Drug-Resistant Ovarian Cancer Cells, Journal of the American Chemical Society,
136 (2014) 5181-5184.
[395] H.K. Shete, R.H. Prabhu, V.B. Patravale, Endosomal Escape: A Bottleneck in
Intracellular Delivery, Journal of Nanoscience and Nanotechnology, 14 (2014) 460-
474.
[396] T. Daemen, G. Hofstede, M.T.T. Kate, I. Bakkerwoudenberg, G.L. Scherphof,
Liposomal doxorubicin-induced toxicity-depletion and impairment of phagocytic-
activity of liver macrophages, International Journal of Cancer, 61 (1995) 716-721.
[397] S. Kim, D.J. Kim, S.B. Howell, Modulation of the peritoneal clearance of
liposomal cytarabine by blank liposomes, Clinical Research, 35 (1987) A524-A524.

316
APPENDIX
APPENDIX A. NMR SPECTROSCOPY
Figure A.1. 1H NMR spectrum of compound A2 in CDCl3.
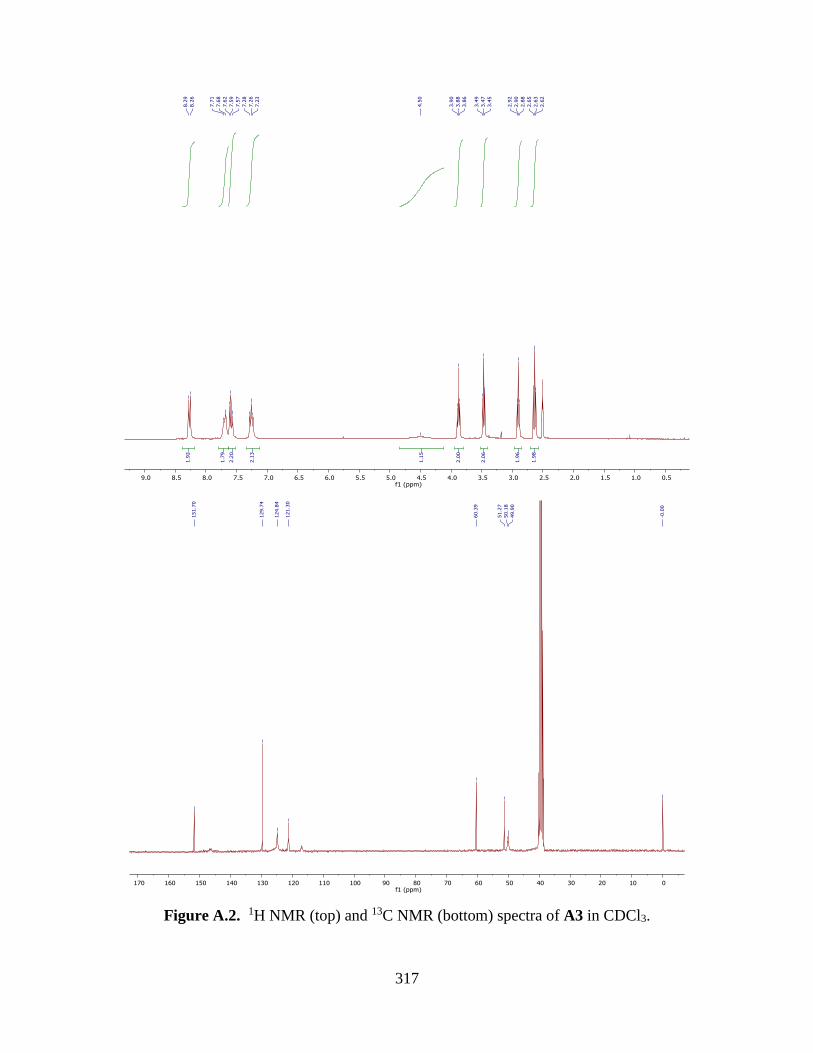
317
Figure A.2. 1H NMR (top) and 13C NMR (bottom) spectra of A3 in CDCl3.

318
Figure A.3. 1H NMR (top) and 13C NMR (bottom) spectra of A4 in CDCl3.

319
Figure A.4. 1H NMR of A5 in MeOH-d4.

320
Figure A.5. 1H NMR (top) and 13C NMR (bottom) spectra of A6 in CDCl3.

321
Figure A.6. 1H NMR (top) and 13C NMR (bottom) spectra of A7 in D2O.

322
Figure A.7. 1H NMR (top) and 13C NMR (bottom) spectra of A8 in CDCl3.

323
Figure A.8. 1H NMR (top) and 13C NMR (bottom) spectra of A9 in CDCl3.

324
Figure A.9. 1H NMR (top) and 13C NMR (bottom) spectra of A10 in CDCl3.

325
Figure A.10. 1H NMR spectra of P1 in D2O.

326
Figure A.11. 1H NMR spectra of P2 in D2O.

327
Figure A.12. 1H NMR spectra of P3 in D2O.

328
Figure A.13. 1H NMR spectra of P4 in D2O.

329
Figure A.14. 1H NMR spectra of P5 in D2O.

330
Figure A.15. 1H NMR spectra of P6 in D2O.

331
Figure A.16. 1H NMR (top) and 13C NMR (bottom) spectra of P1-A3 in DMF-d7.

332
Figure A.17. 1H NMR (top) and 13C NMR (bottom) spectra of P4-A3 in DMF-d7.

333
Figure A.18. 1H NMR (top) and 13C NMR (bottom) spectra of P6-A1 in DMF-d7.

334
Figure A.19. 1H NMR (top) and 13C NMR (bottom) spectra of P1-A8 in DMF-d7.

335
Figure A.20. 1H NMR spectra of P3-A7 in D2O.

336
Figure A.21. 1H NMR (top) and 13C NMR (bottom) spectra of 3-3 in CDCl3.

337
Figure A.22. 1H NMR (top) and 13C NMR (bottom) spectra of 3-5 in CDCl3.

338
Figure A.23. 1H NMR (top) and 13C NMR (bottom) spectra of P1-A9 in MeOH-d4.

339
Figure A.24. 1H NMR spectra of 4-6 in DMSO-d6.

340
Figure A.25. 1H NMR spectra of 4-7 in DMSO-d6.

341
Figure A.26. 1H NMR spectra of 4-3 in DMSO-d6.

342
Figure A.27. 1H NMR (top) and 13C NMR (bottom) spectra of 4-9 in CDCl3.

343
Figure A.28. 1H NMR (top) and 13C NMR (bottom) spectra of A11 in CDCl3.

344
Figure A.29. 1H NMR spectra of Boc-A3 in CDCl3.

345
Figure A.30. 1H NMR spectra of A12 in CDCl3.

346
Figure A.31. 1H NMR spectra of A13 in CDCl3.

347
Figure A.32. Variable Temperature 1H NMR spectrum of compound A12 in CDCl3 at -40 °C
(red), 0 °C (yellow), 25 °C (green), 35 °C (blue), 60 °C (purple), confirming interconversion of
the E- and Z-isomeric forms. Coalescence of endoxifen based peaks due to rapid interconversion
of E- and Z- isomers is highlighted by red, green and blue box.
a b
c

348
Figure A.33. Variable Temperature 1H NMR spectrum of compound A13 in CDCl3 at 25 °C
(red), 45 °C (green), 60 °C (blue).

349
Figure A.34. 1H NMR (top) and 13C NMR (bottom) spectra of P4-A3 in MeOH-d4.

350
Figure A.35. 1H NMR (top) and 13C NMR (bottom) spectra of P4-A12 in DMF-d7.

351
Figure A.36. 2-D 1H–13C HMBC spectra for compound P4-A12 in DMF-d7 giving
connectivities in the carbamate linker. The two sets of cross-peaks observed for several of the
proton and carbon nuclei is due to the presence of E- and Z-isomers, which were assigned in the
expanded view of the spectrum. No attempts were made to assign the two sets of signals to the
specific isomeric forms.

352
Figure A.37. 1H NMR (top) and 13C NMR (bottom) spectra of P4-A13 in DMF-d7.

353
Figure A.38. 1H NMR (top) and 13C NMR (bottom) spectra of 5-7 in CDCl3.

354
Figure A.39. 1H NMR (top) and 13C NMR (bottom) spectra of 5-8 in CDCl3.

355
Figure A.40. 1H NMR (top) and 13C NMR (bottom) spectra of 5-9 in CDCl3.

356
Figure A.41. 1H NMR (top) and 13C NMR (bottom) spectra of 5-10 in CDCl3.

357
Figure A.42. 1H NMR spectra of P9 in D2O.

358
Figure A.43. 1H NMR spectra of P10 in D2O.

359
Figure A.44. 1H NMR spectra of P11 in D2O.

360
Figure A.45. 1H NMR spectra of P12 in MeOH-d4.

361
Figure A.46. 1H NMR spectra of P13 in MeOH-d4.

362
Figure A.45. 1H NMR spectra of P14 in MeOH-d4.

363
Figure A.46. 1H NMR spectra of P15 in MeOH-d4.

364
Figure A.47. 1H NMR (top) and 13C NMR (bottom) spectra of P9-A1 in CDCl3.

365
Figure A.48. HMBC spectrum of compound P9-A1 and assignment of isomers.

366
Figure A.49. COSY spectrum of compound P9-A1.

367
Figure A.50. 1H NMR (top) and 13C NMR (bottom) spectra of P10-A1 in MeOH-d4.

368
Figure A.51. 1H NMR (top) and 13C NMR (bottom) spectra of P11-A1 in MeOH-d4.

369
Figure A.52. 1H NMR (top) and 13C NMR (bottom) spectra of P12-A1 in MeOH-d4.

370
Figure A.53. 1H NMR (top) and 13C NMR (bottom) spectra of P13-A1 in MeOH-d4.

371
Figure A.54. 1H NMR (top) and 13C NMR (bottom) spectra of P14-A1 in MeOH-d4.

372
Figure A.55. 1H NMR (top) and 13C NMR (bottom) spectra of P15-A1 in MeOH-d4.

373
Figure A.56. 1H NMR (top) and 13C NMR (bottom) spectra of P8-A1 in DMF-d7.

374
Figure A.57. Kinetic study using time-dependent 1H NMR spectra for the hydrolysis reaction of
complex P9-A1 in 10 mM PB (pH* 7.0) incubated at 37 ºC. Protons A, B and C of complex P9-
A1 were monitored and used for signal integrations.

375
Figure A.58. 1H NMR (top) and 13C NMR (bottom) spectra of P16-A1 in MeOH-d4.

376
Figure A.59. 1H NMR (top) and 13C NMR (bottom) spectra of P16-B1 in MeOH-d4.

377
Figure A.60. 1H NMR (top) and 13C NMR (bottom) spectra of 6-16 in DMF-d7.

378
Figure A.61. COSY spectrum of compound 6-16.

379
Figure A.62. HMBC spectrum of compound 6-16.

380
APPENDIX B. LC-MS SPECTROSCOPY
APPENDIX B1. LC-MS SPECTROSCOPY
Figure B1.1. LC-ESMS analysis of reaction P1 + A1.

381
Figure B1.2. LC-ESMS analysis of reaction P1 + A2.

382
Figure B1.3. LC-ESMS analysis of reaction P1 + A3.

383
Figure B1.4. LC-ESMS analysis of reaction P1 + A4.

384
Figure B1.5. LC-ESMS analysis of reaction P1 + A5.

385
Figure B1.6. LC-ESMS analysis of reaction P1 + A6.

386
Figure B1.7. LC-ESMS analysis of reaction P1 and A7.

387
Figure B1.8. LC-ESMS analysis of reaction P1 + A8.

388
Figure B1.9. LC-ESMS analysis of reaction P1 + A9.

389
Figure B1.10. LC-ESMS analysis of reaction P1 + A10.

390
Figure B1.11. LC-ESMS analysis of reaction P2 + A1.

391
Figure B1.12. LC-ESMS analysis of reaction P2 + A2.

392
Figure B1.13. LC-ESMS analysis of reaction P2 + A3.

393
Figure B1.14. LC-ESMS analysis of reaction P2 + A4.

394
Figure B1.15. LC-ESMS analysis of reaction P2 + A5.

395
Figure B1.16. LC-ESMS analysis of reaction P2 + A6.

396
Figure B1.17. LC-ESMS analysis of reaction P2 + A7.

397
Figure B1.18. LC-ESMS analysis of reaction P2 + A8.

398
Figure B1.19. LC-ESMS analysis of reaction P2 + A9.

399
Figure B1.20. LC-ESMS analysis of reaction P2 + A10.

400
Figure B1.21. LC-ESMS analysis of reaction P3 + A1.

401
Figure B1.22. LC-ESMS analysis of reaction P3 + A2.

402
Figure B1.23. LC-ESMS analysis of reaction P3 + A3.

403
Figure B1.24. LC-ESMS analysis of reaction P3 + A4.

404
Figure B1.25. LC-ESMS analysis of reaction P3 + A5.

405
Figure B1.26. LC-ESMS analysis of reaction P3 + A6.

406
Figure B1.27. LC-ESMS analysis of reaction P3 + A7.

407
Figure B1.28. LC-ESMS analysis of reaction P3 + A8.

408
Figure B1.29. LC-ESMS analysis of reaction P3 + A9.

409
Figure B1.30. LC-ESMS analysis of reaction P3 + A10.

410
Figure B1.31. LC-ESMS analysis of reaction P4 + A1.

411
Figure B1.32. LC-ESMS analysis of reaction P4 + A2.

412
Figure B1.33. LC-ESMS analysis of reaction P4 + A3.

413
Figure B1.34. LC-ESMS analysis of reaction P4 + A4.

414
Figure B1.35. LC-ESMS analysis of reaction P4 + A5.

415
Figure B1.36. LC-ESMS analysis of reaction P4 + A6.

416
Figure B1.37. LC-ESMS analysis of reaction P4 + A7.

417
Figure B1.38. LC-ESMS analysis of reaction P4 + A8.

418
Figure B1.39. LC-ESMS analysis of reaction P4 + A9.

419
Figure B1.40. LC-ESMS analysis of reaction P4 + A10.

420
Figure B1.41. LC-ESMS analysis of reaction P5 + A1.

421
Figure B1.42. LC-ESMS analysis of reaction P5 + A2.

422
Figure B1.43. LC-ESMS analysis of reaction P5 + A3.

423
Figure B1.44. LC-ESMS analysis of reaction P5 + A4.

424
Figure B1.45. LC-ESMS analysis of reaction P5 + A5.

425
Figure B1.46. LC-ESMS analysis of reaction P5 + A6.

426
Figure B1.47. LC-ESMS analysis of reaction P5 + A7.

427
Figure B1.48. LC-ESMS analysis of reaction P5 + A8.

428
Figure B1.49. LC-ESMS analysis of reaction P5 + A9.

429
Figure B1.50. LC-ESMS analysis of reaction P5 + A10.

430
Figure B1.51. LC-ESMS analysis of reaction P6 + A1.

431
Figure B1.52. LC-ESMS analysis of reaction P6 + A2.

432
Figure B1.53. LC-ESMS analysis of reaction P6 + A3.

433
Figure B1.54. LC-ESMS analysis of reaction P6 + A4.

434
Figure B1.55. LC-ESMS analysis of reaction P6 + A5.

435
Figure B1.56. LC-ESMS analysis of reaction P6 + A6.

436
Figure B1.57. LC-ESMS analysis of reaction P6 + A7.

437
Figure B1.58. LC-ESMS analysis of reaction P6 + A8.

438
Figure B1.59. LC-ESMS analysis of reaction P6 + A9.

439
Figure B1.60. LC-ESMS analysis of reaction P6 + A10.

440
Figure B1.61. Percent conversion in ‘click’ reactions for platinum-acridines. Compounds are
sorted and color-coded by common acridine moieties.

441
APPENDIX B2. LC-ESMS ANALYSIS OF PURIFIED COMPOUND
Figure B2.1. LC-MS analysis of compound A1.
1
04271219.D: UV Chromatogram, 363-463 nm
0
25
50
75
100
125
Intens.
mAU
0 5 10 15 20 25 Time [min]
195.2
252.2
+MS, 10.9min #575
0
1
2
3
4
7x10
Intens.
150 175 200 225 250 275 300 325 350 375 m/z

442
Figure B2.2. LC-MS analysis of compound A2.
1
05021201.D: UV Chromatogram, 363-463 nm
0
20
40
60
80
Intens.
mAU
0 5 10 15 20 25 Time [min]
266.2
+MS, 11.9min #661
0
1
2
3
7x10
Intens.
220 240 260 280 300 320 340 m/z

443
Figure B2.3. LC-MS analysis of compound A3.
1
04271212.D: UV Chromatogram, 363-463 nm
0
50
100
150
Intens.
mAU
0 5 10 15 20 25 Time [min]
195.3
282.3
+MS, 11.0min #616
0.0
0.2
0.4
0.6
0.8
1.0
8x10
Intens.
150 200 250 300 350 400 m/z

444
Figure B2.4. LC-MS analysis of compound A4.
1
04271213.D: UV Chromatogram, 363-463 nm
0
20
40
60
80
Intens.
mAU
0 5 10 15 20 25 Time [min]
195.2
296.3
+MS, 11.3min #625
0
2
4
6
7x10
Intens.
150 200 250 300 350 400 m/z

445
Figure B2.5. LC-MS analysis of compound A5.
1
04271214.D: UV Chromatogram, 363-463 nm
0
10
20
30
40
Intens.
mAU
0 5 10 15 20 25 Time [min]
296.3
+MS, 13.4min #734
0.0
0.5
1.0
1.5
7x10
Intens.
150 200 250 300 350 400 m/z

446
Figure B2.6. LC-MS analysis of compound A6.
1
04271221.D: UV Chromatogram, 363-463 nm
0
5
10
15
20
Intens.
mAU
0 5 10 15 20 25 Time [min]
259.3
310.2
+MS, 12.4min #659
0.00
0.25
0.50
0.75
1.00
1.25
6x10
Intens.
150 200 250 300 350 400 m/z

447
Figure B2.7. LC-MS analysis of compound A7.
1
04271216.D: UV Chromatogram, 363-463 nm
0
5
10
15
20
Intens.
mAU
0 5 10 15 20 25 Time [min]
259.3
267.3 275.3
279.3
283.3
297.3
310.3
325.4 335.3 341.3 355.3
+MS, 14.6min #794
0
1
2
3
4
5
5x10
Intens.
260 280 300 320 340 m/z

448
Figure B2.8. LC-MS analysis of compound A8.
1
04271217.D: UV Chromatogram, 363-463 nm
0
5
10
15
20
25
Intens.
mAU
0 5 10 15 20 25 Time [min]
364.3
+MS, 13.3min #715
0
2
4
6
7x10
Intens.
200 250 300 350 400 450 500 m/z

449
Figure B2.9. LC-MS analysis of compound A9.
1
05021202.D: UV Chromatogram, 363-463 nm
0
10
20
30
40
Intens.
mAU
0 5 10 15 20 25 Time [min]
378.3
+MS, 13.1min #728
0.0
0.2
0.4
0.6
0.8
1.0
8x10
Intens.
200 250 300 350 400 450 500 m/z

450
Figure B2.10. LC-MS analysis of compound A10.

451
Figure B2.11. LC-MS analysis of compound P1-A3.
1
05261207.D: UV Chromatogram, 363-463 nm
0.0
2.5
5.0
7.5
10.0
12.5
Intens.
mAU
0 5 10 15 20 25 Time [min]
169.3
195.2
207.2
221.2
250.2264.2
282.2
295.3
320.2
337.2
363.3
396.2
440.1 473.1
499.2
529.2589.3
627.3
670.2
+MS, 14.0min #756
0.0
0.2
0.4
0.6
0.8
1.0
1.2
6x10
Intens.
200 300 400 500 600 700 m/z

452
Figure B2.12. LC-MS analysis of compound P4-A3.
1
05261205.D: UV Chromatogram, 363-463 nm
0
5
10
15
20
Intens.
mAU
0 5 10 15 20 25 Time [min]
159.2
195.2
207.2
221.2
239.2
252.2
265.6279.2
305.2
327.3
344.3
363.2
391.3
413.3
453.3
463.2
474.2
497.3
513.2
524.3
539.2
561.2
585.2
616.7632.4
653.1
680.7
698.2
718.6
+MS, 10.4min #567
0.0
0.2
0.4
0.6
0.8
1.0
5x10
Intens.
200 300 400 500 600 700 m/z

453
Figure B2.13. LC-MS analysis of compound P6-A1.
1
06011211.D: UV Chromatogram, 363-463 nm
0
10
20
30
Intens.
mAU
0 5 10 15 20 25 Time [min]
195.2
221.2
252.2
288.2
307.2
361.1
391.4 415.1428.3
485.2
514.2575.3
612.3
+MS, 13.4min #737
0.0
0.5
1.0
1.5
2.0
2.5
6x10
Intens.
200 300 400 500 600 700 m/z

454
Figure B2.14. LC-MS analysis of compound P1-A8.
1
02091211.D: UV Chromatogram, 363-463 nm
0.0
2.5
5.0
7.5
10.0
12.5
Intens.
mAU
0 5 10 15 20 25 Time [min]
195.1 221.1279.2
333.4
354.8
391.3
419.4
708.5
+MS, 16.6min #949
0.0
0.2
0.4
0.6
0.8
1.0
1.2
7x10
Intens.
200 300 400 500 600 700 800 900 m/z

455
Figure B2.15. LC-MS analysis of compound P3-A7.
1
0 5 10 15 20 25 Time [min]
0
5
10
15
20
25
Intens.
mAU
06121202.D: UV Chromatogram, 363-463 nm
159.2
195.2
207.2221.2
248.2264.1
279.2
288.2
310.2
320.1 399.1
415.1
442.1458.1
485.1
501.1
557.2
575.2611.2
629.2
+MS, 14.3min #774
0.00
0.25
0.50
0.75
1.00
1.25
6x10
Intens.
200 300 400 500 600 700 m/z

456
Figure B2.16. LC-MS analysis of compound P1-A9.
102091207.D: UV Chromatogram, 363-463 nm
0
10
20
30
40
Intens.
mAU
0 5 10 15 20 25 Time [min]
195.1 221.2293.3
329.7343.8
361.8
433.4
722.4
+MS, 15.1min #870
0
1
2
3
4
5
6
7x10
Intens.
200 300 400 500 600 700 m/z

457
Figure B2.17. LC-ESMS analysis of compound P4-A3. (A) Reverse-phase HPLC trace of
compound P4-A3. (B) ESMS spectrum of compound 5 (HPLC fraction with retention time 10.0
min) recorded in positive-ion mode. Characteristic molecular and fragment ions: [M]+ (m/z
585.2).
1
05261205.D: UV Chromatogram, 363-463 nm
0
5
10
15
20
Intens.
mAU
0 5 10 15 20 25 Time [min]
159.2
195.2
207.2
221.2
239.2
252.2
265.6279.2
305.2
327.3
344.3
363.2
391.3
413.3
453.3
463.2
474.2
497.3
513.2
524.3
539.2
561.2
585.2
616.7632.4
653.1
680.7
698.2
718.6
+MS, 10.4min #567
0.0
0.2
0.4
0.6
0.8
1.0
5x10
Intens.
200 300 400 500 600 700 m/z
A
B
B

458
Figure B2.18. LC-ESMS analysis of compound A12. (A) Reverse-phase HPLC trace of
compound A12. (B) ESMS spectrum of compound 9 (HPLC fraction with retention time 34.0
min) recorded in positive-ion mode. Characteristic molecular and fragment ions: [M]+ (m/z
681.5).
681.5
459.4
552.4 283.4
A
B

459
A

460
Figure B2.19. LC-ESMS analysis of compound P4-A12. (A) Reverse-phase HPLC trace of
compound P4-A12. (B) ESMS spectrum of compound P4-A12 (HPLC fraction with retention
time 33.8 min) recorded in positive-ion mode. Characteristic molecular and fragment ions: [M-
2H]+ (m/z 985.6), [M-2NH3-Pt-Cl-MeCN]+ (m/z 681.5), [M-H]2+ (m/z 493.3, z = 2), [M-2NH3-
Cl]2+ (m/z 457.8, z = 2), [M-2NH3-Cl-Et-OH]2+ (m/z 436.9, z =2).
436.9
457.8
493.3
681.5
+MS, 33.9min #1897
0
1
2
3
4
5
7x10
Intens.
300 400 500 600 700 800 900 1000 m/z
985.6
B

461
Figure B2.20. LC-ESMS analysis of compound A13. (A) Reverse-phase HPLC trace of
compound A13. (B) ESMS spectrum of compound A13 (HPLC fraction with retention time 35.6
min) recorded in positive-ion mode. Characteristic molecular and fragment ions: [M+H]+ (m/z
769.8), [M+H]2+ (m/z 384.5, z=2).
A
B

462
1

463
Figure B2.21 continued.

464
Figure B2.21 continued.

465
Figure B2.21. LC-ESMS analysis of compound P4-A13. (1) Reverse-phase HPLC trace of
compound P4-A13. Peaks 2–5 show identical masses and can be assigned to rotational isomers
as a result of hindered rotation within the amidine and (E,Z)-endoxifen moieties. Peak 1 is an
unidentified impurity. Combined % purity for peaks 2–5 (rotamers): 97%. (2) ESMS spectrum
of one species of compound 10’ (HPLC fraction with retention time 35.2 min) recorded in
positive-ion mode. Characteristic molecular and fragment ions: [M+H]+ (m/z 1074.8), [M+H]2+
(m/z 537.5, z = 2), [M-2NH3-Cl]2+ (m/z 502.4, z = 2). (3) ESMS spectrum of one species of
compound 10’ (HPLC fraction with retention time 36.4 min) recorded in positive-ion mode.
Characteristic molecular and fragment ions: [M+H]+ (m/z 1074.8), [M]2+ (m/z 537.5, z = 2), [M-
2NH3-Cl]2+ (m/z 502.4, z = 2). (4) ESMS spectrum of one species of compound 10’ (HPLC
fraction with retention time 37.2 min) recorded in positive-ion mode. Characteristic molecular
and fragment ions: [M]+ (m/z 1073.8), [M+H]2+ (m/z 537.8, z = 2), [M-2NH3-Cl]2+ (m/z 502.4, z
= 2). (5) ESMS spectrum of one species of compound 10’ (HPLC fraction with retention time
37.2 min) recorded in positive-ion mode. Characteristic molecular and fragment ions: [M]+ (m/z
1073.8), [M]2+ (m/z 537.4, z = 2), [M-2NH3-Cl]2+ (m/z 502.4, z = 2).

466
Figure B2.22. LC-MS analysis of compound P9-A1.

467
Figure B2.23. LC-MS analysis of compound P10-A1.

468
Figure B2.24. LC-MS analysis of compound P11-A1.

469
Figure B2.25. LC-MS analysis of compound P12-A1.

470
Figure B2.26. LC-MS analysis of compound P13-A1.

471
Figure B2.27. LC-MS analysis of compound P14-A1.

472
Figure B2.28. LC-MS analysis of compound P15-A1.

473
Figure B2.29. LC-MS analysis of compound P8-A1.

474
Figure B2.30. LC-MS analysis of compound P16-A1.

475
Figure B2.31. LC-MS analysis of compound P16-B1.

476
Figure B2.32. LC-MS analysis of compound 6-16.

477
APPENDIX B3. LC-MS ANALYSIS EVALUATION OF STABILITY OF CONJUGATES IN
BUFFERS
Figure B3.1. LC-ESMS analysis of the reaction between 3-3 and P1 suggesting decomposition
of the target compound (peak 4) via loss of N2 and N-chelate formation (peak 1) along with an
unidentified platinum-containing product (peak 3).

478
Figure B3.2. LC-ESMS analysis of the reaction mixture between 3-5 and P1 suggesting
decomposition of the target compound (peak 4) via loss of N2 and N-chelate formation (peak 1)
along with unidentified platinum-containing products (peaks 3 and 5).

479
Figure B3.3. LC-ESMS analysis of the mixture of compound P1-A9 in 10 mM PBS (pH 7.4)
after incubation for 24 h at 37 °C, showing minor aquation of the chloro leaving group. The
azide group and amide linkage are stable under these conditions.

480
Figure B3.4. LC-ESMS analysis of the mixture of compound P1-A9 in 10 mM PB (pH 7.4)
after incubation for 24 h at 37 °C, showing aquation and substitution of chloride by phosphate.

481
Figure B3.5. LC-ESMS analysis of the mixture of compound P1-A9 in 10 mM acetate buffer
(pH 5.0) incubating for 24 h at 37 °C.

482
Figure B3.6. LC-ESMS analysis of the mixture of compound P4-A12 in 10 mM acetate buffer
(pH = 5.0) incubating for 48 h at 37 °C. ESMS spectrum was recorded in positive-ion mode.
Characteristic molecular and fragment ions: [M-2NH3-Pt-Cl]+ (m/z 722.4), [M-Cl+MeCOO]2+
(m/z 505.3, z =2), [M-H]2+ (m/z 493.3, z = 2), [M-2NH3-Cl+H]2+ (m/z 458.2, z = 2), [M-2NH3-
Cl-Et-OH]2+ (m/z 436.9, z =2). (C) Compound P4-A12 is chemically stable in the incubation
condition. Only the displacement of chloride ligand with buffer was observed.
195.2436.3
458.2
505.3
722.4
+MS, 12.5min #692
0.0
0.5
1.0
1.5
2.0
7x10
Intens.
200 300 400 500 600 700 800 900 1000 m/z
C
A
B

483
Figure B3.7. LC-ESMS analysis of the mixture of compound P4-A12 in 10 mM phosphate
buffer saline (pH = 7.4) incubating for 48 h at 37 °C. ESMS spectrum was recorded in positive-
ion mode. Characteristic molecular and fragment ions: [M-Cl+H2PO4]2+ (m/z 524.3, z =2), [M-
H]2+ (m/z 493.3, z = 2), [M-2NH3-Cl]2+ (m/z 458.3, z = 2), [M-2NH3-Cl-Et-OH]2+ (m/z 436.2, z
=2). (C) Compound P4-A12 is chemically stable in the incubation condition. The displacement
of chloride ligand with buffer was observed, while dramatically inhibited by the presence of
chloride ions in buffer.
436.2
458.3
475.3
493.7
524.2
+MS, 12.6min #693
0.00
0.25
0.50
0.75
1.00
1.25
7x10
Intens.
200 300 400 500 600 700 800 900 1000 m/z
A
B
C

484
Figure B3.8. LC-ESMS analysis of the mixture of compound P4-A12 in 10 mM phosphate
buffer (pH = 7.4) incubating for 48 h at 37 °C. ESMS spectrum was recorded in positive-ion
mode. Characteristic molecular and fragment ions : [M-2NH3-Pt-Cl-MeCN]+ (m/z 681.4), [M-
Cl+H2PO4]2+ (m/z 524.3, z =2), [M-H]2+ (m/z 493.3, z = 2), [M-2NH3-Cl]2+ (m/z 457.8, z = 2),
[M-2NH3-Cl-Et-OH]2+ (m/z 436.2, z =2). (C) Compound P4-A12 is chemically stable in the
incubation condition. Only the displacement of chloride ligand with buffer was observed.
391.3419.4
436.2
457.8
475.3 493.3
524.3
537.7550.2
681.4
+MS, 12.7min #697
0.0
0.5
1.0
1.5
2.0
2.5
3.0
6x10
Intens.
350 400 450 500 550 600 650 700 750 800 m/z
A
B
C

485
APPENDIX B4. METAL-ASSISTED ESTER HYDROLYSIS
Figure B4.1. LC-ESMS analysis of the mixture of compound P10-A1 in phosphate buffer (PB,
pH 7.4) incubated at 37 °C.

486
Figure
B4.2. LC-ESMS analysis of the mixture of compound P11-A1 in phosphate buffer (PB, pH 7.4)
incubated at 37 °C.

487
Figure B4.3. LC-ESMS analysis of the mixture of compound P12-A1 in phosphate buffer (PB,
pH 7.4) incubated at 37 °C.

488
Figure B4.4. LC-ESMS analysis of the mixture of compound P13-A1 in phosphate buffer (PB,
pH 7.4) incubated at 37 °C.

489
Figure B4.5. LC-ESMS analysis of the mixture of compound P14-A1 in phosphate buffer (PB,
pH 7.4) incubated at 37 °C.

490
Figure B4.6. LC-ESMS analysis of the mixture of compound P15-A1 in phosphate buffer (PB,
pH 7.4) incubated at 37 °C.

491
Figure B4.7. LC-ESMS analysis of the mixture of compound 6-12 in phosphate buffer (PB, pH
7.4) incubated at 37 °C.

492
Figure B4.8. LC-ESMS analysis of the mixture of compound 6-14 in phosphate buffer (PB, pH
7.4) incubated at 37 °C.

493
Figure B4.9. LC-ESMS analysis of the mixture of compound 6-16 in phosphate buffer (PB, pH
7.4) incubated at 37 °C.

494
Figure B4.10. LC-ESMS analysis of the mixture of compound 6-17 in phosphate buffer (PB,
pH 7.4) incubated at 37 °C.
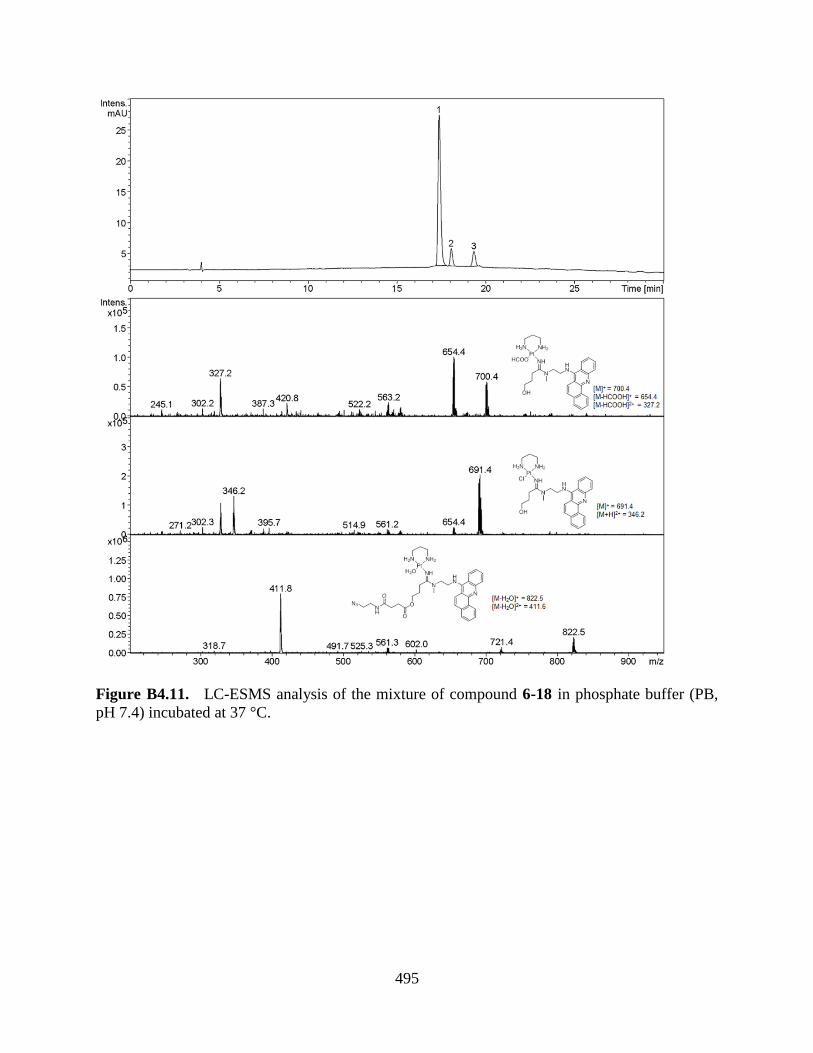
495
Figure B4.11. LC-ESMS analysis of the mixture of compound 6-18 in phosphate buffer (PB,
pH 7.4) incubated at 37 °C.

496
Figure B4.12. LC-ESMS analysis of the mixture of compound 6-22 in phosphate buffer (PB,
pH 7.4) incubated at 37 °C.

497
Figure B4.13. LC-ESMS analysis of the mixture of compound P10-A1 in phosphate buffered
saline (PBS, pH 7.4) incubated at 37 °C.

498
Figure B4.14. LC-ESMS analysis of the mixture of compound P11-A1 in phosphate buffered
saline (PBS, pH 7.4) incubated at 37 °C.

499
Figure B4.15. LC-ESMS analysis of the mixture of compound P12-A1 in phosphate buffered
saline (PBS, pH 7.4) incubated at 37 °C.

500
Figure B4.16. LC-ESMS analysis of the mixture of compound P13-A1 in phosphate buffered
saline (PBS, pH 7.4) incubated at 37 °C.

501
FigureB4.17. LC-ESMS analysis of the mixture of compound P14-A1 in phosphate buffered
saline (PBS, pH 7.4) incubated at 37 °C.

502
Figure B4.18. LC-ESMS analysis of the mixture of compound P15-A1 in phosphate buffered
saline (PBS, pH 7.4) incubated at 37 °C.

503
Figure B4.19. LC-ESMS analysis of the mixture of compound 6-12 in phosphate buffered
saline (PBS, pH 7.4) incubated at 37 °C.

504
Figure B4.20. LC-ESMS analysis of the mixture of compound 6-14 in phosphate buffered
saline (PBS, pH 7.4) incubated at 37 °C.

505
Figure B4.21. LC-ESMS analysis of the mixture of compound 6-16 in phosphate buffered
saline (PBS, pH 7.4) incubated at 37 °C.

506
Figure B4.22. LC-ESMS analysis of the mixture of compound 6-17 in phosphate buffered
saline (PBS, pH 7.4) incubated at 37 °C.

507
Figure B4.23. LC-ESMS analysis of the mixture of compound 6-18 in phosphate buffered
saline (PBS, pH 7.4) incubated at 37 °C.

508
Figure B4.24. LC-ESMS analysis of the mixture of compound 6-22 in phosphate buffered
saline (PBS, pH 7.4) incubated at 37 °C.

509
APPENDIX B5. hCES-2 MEDIATED ESTER HYDROLYSIS
Figure B5.1. LC-ESMS analysis of the enzymatic cleavage of compound P9-A1 in phosphate
buffered saline (PBS, pH 7.4) incubated at 37 °C.

510
Figure B5.2. LC-ESMS analysis of the enzymatic cleavage of compound P10-A1 in phosphate
buffered saline (PBS, pH 7.4) incubated at 37 °C.

511
Figure B5.3. LC-ESMS analysis of the enzymatic cleavage of compound P11-A1 in phosphate
buffered saline (PBS, pH 7.4) incubated at 37 °C.

512
Figure B5.4. LC-ESMS analysis of the enzymatic cleavage of compound P12-A1 in phosphate
buffered saline (PBS, pH 7.4) incubated at 37 °C.

513
Figure B5.5. LC-ESMS analysis of the enzymatic cleavage of compound P13-A1 in phosphate
buffered saline (PBS, pH 7.4) incubated at 37 °C.

514
Figure B5.6. LC-ESMS analysis of the enzymatic cleavage of compound P14-A1 in phosphate
buffered saline (PBS, pH 7.4) incubated at 37 °C.

515
Figure B5.7. LC-ESMS analysis of the enzymatic cleavage of compound P15-A1 in phosphate
buffered saline (PBS, pH 7.4) incubated at 37 °C.

516
APPENDIX B6. LC-MS ANALYSIS OF THE COUPLING REACTIONS FOR
MULTIFUNCTIONAL PLATINUM-ACRIDINES
Figure B6.1. LC-MS analysis of the reaction mixture for the preparation of 6-11.

517
Figure B6.2. LC-MS analysis of the reaction mixture for the preparation of 6-12.

518
Figure B6.3. LC-MS analysis of the reaction mixture for the preparation of 6-13.

519
Figure B6.4. LC-MS analysis of the reaction mixture for the preparation of 6-14.

520
Figure B6.5. LC-MS analysis of the reaction mixture for the preparation of 6-15.

521
Figure B6.6. LC-MS analysis of the reaction mixture for the preparation of 6-17.

522
Figure B6.7. LC-MS analysis of the reaction mixture for the preparation of 6-18.

523
Figure B6.8. LC-MS analysis of the reaction mixture for the preparation of 6-19.

524
Figure B6.9. LC-MS analysis of the reaction mixture for the preparation of 6-20.
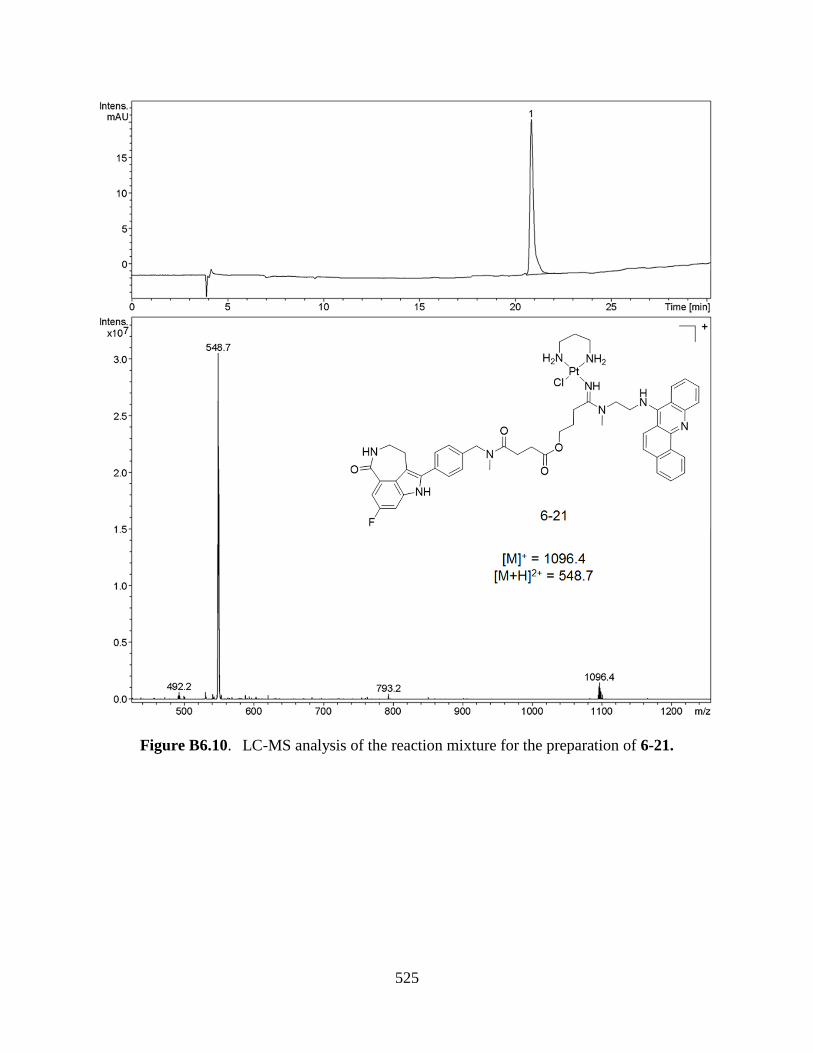
525
Figure B6.10. LC-MS analysis of the reaction mixture for the preparation of 6-21.

526
Figure B6.11. LC-MS analysis of the reaction mixture for the preparation of 6-22.

527
Figure B6.12. LC-MS analysis of the reaction mixture for the preparation of 6-23.

528
Figure B6.13. LC-MS analysis of the reaction mixture for the preparation of 6-24.

529
APPENDIX C. CD SPECTRA
Figure C.1. Circular dichroism (CD) spectra (ICD region) of calf thymus DNA modified
with platinum-acridines P1-A1 (blue trace) and P1-A9 (red trace). CD spectra were
recorded for ~1 mM (n.t) DNA in 10 mM phosphate buffer (25 °C, pH 7.4) at a drug-to-
nucleotide ratio (rb) of 0.05.

530
APPENDIX D. CONFOCAL MICROSCOPIC IMAGES
Figure D.1. Acridine fluorescence in NCI-H460 cells (excitation wavelength: 405 nm,
emission wavelength range: 414-470 nm) after treatment with 5 µM compound P1-A9
(“Pt-Azide”) for 3 h.
Figure D.2. Total background-corrected fluorescent intensities in cells treated with
compound P1-A9 (1, 5, 25 μM) and in untreated cells. Plotted data are the mean
fluorescence intensities (± S.D.) determined for n > 50 cells. The data was analyzed with
one-way ANOVA (***P < 0.001; P < 0.05 was considered significant).
******
***
0
500
1000
1500
2000
2500
3000
3500
control 1 uM 5 uM 25 uM
Flu
ore
sce
nce
at
48
8 n
m (
arb
itr.
un
its)

531
Figure D.3. (a) Background-corrected fluorescent intensities in nuclei of cells treated
with compound P1-A9 (5 μM) and in untreated cells. (b) Nucleolus-to-nucleus
fluorescence intensity ratios (Fnucleolus/Fnucleus) in cells treated with compound P1-A9 (5
μM) and in untreated cells. Plotted data are the mean fluorescence intensities (± S.D.)
determined for n = 40–50 cells. The data was analyzed with one-way ANOVA (***P <
0.001; P < 0.05 was considered significant).

532
Figure D.4. Enlarged view of a colocalization image of a single NCI-H460 cancer cell
treated with P1-A9 and P1-A9* (green), followed by post-labeling with CuAAC/Alexa
Fluor 647-alkyne (red). Hoechst 33342 (blue), merged, and bright field channel images
are also shown. Scale bar = 5 µM. Note the high degree of colocalization of the green-
and red-fluorescent dyes in the nucleolus (and in two unidentified cytosolic structures),
and the markedly reduced relative fluorescence intensity in the nucleus in the green
channel vs the red channel.

533
APPENDIX E. CELL PROLIFERATION ASSAYS
Figure E.1. Drug-response curves for cell proliferation assays in NCI-H460 cells treated
with selected compounds. Error bars indicate ± standard deviations from the mean for
two independent experiments performed in triplicate.
Figure E.2. Drug-response curves for cell proliferation assays in NCI-H460 cells treated
with P3-A7 and the corresponding acridine ligand A7. Error bars indicate ± standard
deviations from the mean for two independent experiments performed in triplicate.

534
Figure E.3. Dose-response curves for cell proliferation assays in non-small cell lung
cancer cell line (A) NCI-A549 and (B) NCI-H460 treated with compound P8-A1. Error
bars indicate ± standard deviations from the mean for two independent experiments
performed in triplicate.

535
APPENDIX F. COMPUTATION STUDIES
Figure F.1. Molecular models (AMBER) of the monofunctional–intercalative adducts at
guanine–N7 in the DNA major groove formed by compound 1 (left) and compound 2
(right) based on the NMR solution structure of the hybrid adduct (PDB 1XRW). The
platinum–acridine and the newly installed azide linker are shown in red and element-
specific colors, respectively.

536
APPENDIX G. MTD STUDIES
Figure G.1. Maximum tolerated dose (MTD) study of compound P15-A1 in 420 Swiss-
Webster (SW) mice (6-8 weeks, 18-25 g, female, n = 5). Compound P15-A1 was
administered i.p. at the dose of 1.6 mg/kg on (A) days 0, 4, 8, and 12, and (B) days 0, 1, 2,
3, and 4. No statistically significant weight loss is observed in both treatment groups.

537
APPENDIX H. SIZE DISTRIBUTION OF LIPOSOMES
Figure H.1. Size distribution of Lipo-1 determined by DLS.
Figure H.2. Size distribution of Lipo-2 determined by DLS.

538
Figure H.3. Size distribution of Lipo-3 determined by DLS.
Figure H.4. Size distribution of Lipo-4 determined by DLS.

539
APPENDIX I.CHEMICAL STRUCTURES OF COUPLING REAGENTS USED IN
CHAPTER 6

540
APPENDIX J.CHEMICAL STRUCTURES OF LIPIDS USED IN CHAPTER 7

541
SCHOLASTIC VITA
Song Ding
BORN: January 6, 1985
Nanjing, China
UNDERGRADUATE STUDY: China Pharmaceutical University
Nanjing, China
B.S. Basic Pharmacy, 2007
GRADUATE STUDY: China Pharmaceutical University
Nanjing, China
M.S. Medicinal Chemistry, 2010
GRADUATE STUDY: Wake Forest University
Winston-Salem, North Carolina
Ph.D. candidate, 2015
SCHOLASTIC AND PROFESSIONAL EXPERIENCE:
Teaching Assistant, Wake Forest University 2014-
2015
Winston-Salem, North Carolina
Research Assistant, Wake Forest University 2010-
2014
Winston-Salem, North Carolina
HORNORS AND AWARDS:
GlaxoSmithKline Graduate Research Award 2014

542
Department of Chemistry, Wake Forest University
Wake Forest Innovation and Commercialization Services, SPARK Award (for
development of a liposomal formulation for delivery of platinum–acridine anticancer
agents, Co-PI with U. Bierbach)
8/2013– 7/2014
PUBLICATIONS:
1. Ding, S.; Bierbach,U. et al, Development of a chemical toolbox for discovery of
multifunctional platinum-acridine agents. In preparation.
2. Fahrenholtz C.D., Ding S., Bernish B.W., Wright M.L., Bierbach U., Singh R.N.,
Self-assembling Platinum-acridine loaded carbon nanotubes for cancer chemotherapy.
Submitted to Scientific reports.
3. Ding, S.; Bierbach,U. et al, Liposomal delivery of platinum-acridine anticancer
agents for non-small cell lung cancer. In preparation.
4. Ding, S.; Bierbach, U. Target-selective delivery and activation of platinum-based
anticancer
agents. Future Medicinal Chemistry 2015,7(7), 911-927.
5. Ding, S.; Pickard A.J.; Kucera G.L.; Bierbach U., Design of Enzymatically Cleavable
Prodrugs of a Potent Platinum-Containing Anticancer Agent. Chemistry – A European
Journal 2014, 20 (49), 16164-16173.
6. Qiao, X.; Ding, S.; Liu, F.; Kucera, G. L.; Bierbach, U., Investigating the cellular fate
of a DNA-targeted platinum-based anticancer agent by orthogonal double-click
chemistry. Journal of Biological Inorganic Chemistry 2014, 19 (3), 415-26.
7. Ding, S.; Qiao, X.; Kucera, G. L.; Bierbach, U., Design of a platinum-acridine-
endoxifen conjugate targeted at hormone-dependent breast cancer. Chemical
Communications 2013, 49 (24), 2415-2417;
8. Ding, S.; Qiao, X.; Suryadi, J.; Marrs, G. S.; Kucera, G. L.; Bierbach, U., Using
Fluorescent Post-Labeling To Probe the Subcellular Localization of DNA-Targeted
Platinum Anticancer Agents. Angewandte Chemie-International Edition 2013, 52
(12), 3350-3354;
9. Ding, S.; Qiao, X.; Kucera, G. L.; Bierbach, U., Using a Build-and-Click Approach
for Producing Structural and Functional Diversity in DNA-Targeted Hybrid
Anticancer Agents. Journal of Medicinal Chemistry 2012, 55 (22), 10198-10203;
PATENTS:
1. Bierbach U., Ding S.: Targeted delivery and prodrug designs for platinum-acridine
anti-cancer compounds and methods thereof. PCT/US2012/053189 (filed 08/30/2012).
Nationalization in US: #US 14/241,900, July 10, 2014.
2. Bierbach, U., Ding, S.: Enzymatically activatable prodrugs of DNA-targeted
platinum-acridine agents and methods thereof. Provisional patent application #
62/015,478 (filed 06/22/2014).

543
PRESENTATION:
1. Ding, S.; Qiao, X.; Kucera, G. L.; Bierbach, U., Development and evaluation of
novel conjugation chemistries for the targeted delivery and cellular imaging of
potent platinum-acridine anticancer agents. 243th ACS National Meeting, San
Diego, CA, March 28, 2012. (Poster Presentation)
2. Ding, S.; Qiao, X.; Kucera, G. L.; Bierbach, U., Novel conjugation chemistries for
the functionalization and cellular imaging of potent platinum- acridine anticancer
agents. SERMACS 2012, Raleigh, NC, November 15, 2012. (Oral presentation)
3. Ding, S.; Kucera, G. L.; Bierbach, U., Novel strategies for improving the
pharmacological properties of platinum-acridine anticancer agents. 249th ACS
National Meeting, San Diego, CA, March 25, 2015. (Poster Presentation)




















