
Non-covalent routes to tune the optical properties of molecular materials
18
Non-covalent routes to tune the optical properties of molecular materials Sunil Varughese * Switching and tuning solid state luminescence properties of molecular materials by modulating molecular packing through non-covalent routes is an attractive prospect. This strategy further makes it feasible to expand the utility of molecules of interest by obtaining a large array of solids—polymorphs, solvates, amorphous phase, nano/micro-crystals and as multi-component systems—with distinct fluorophore arrangement and hence emission characteristics. Because non-covalent interactions that determine the fluorophore arrangements in polymorphs or supramolecular complexes are weak and flexible, their making and breaking become more realistic under ambient conditions, thus having potential to achieve reversible transformations and hence external-stimuli-responsive and switchable molecular fluorescent materials. Recent advances in this context are highlighted in this review with the aid of illustrative examples and further emphasize the scope and relevance of interdisciplinary and multitechnique approaches to unravel the structure–optical property relationships and also to augment the foundations of factual knowledge. Introduction The burgeoning world of light-emitting molecular solids and their potential utility in optical devices, sensors and biological imaging has catalysed its emergence as an area of topical research with a spectrum of prolic activities. The light-emitting behaviour of molecular solids differs from that of dilute solutions because emission in the solid state is dictated by the whole collective rather than by individual molecules. Studying molecular crystals provides essential information on how molecules are arranged with respect to their neighbours, interactions that bind them together and their conformational preferences required to correlate the structural facts with several macroscopic properties, including their emission char- acteristics. Such systematic knowledge pertaining to how complex properties emerge from collections of molecules and how sensitive these properties are to specic molecular ordering in solids, could be translated to deriving new strategies to control molecular arrangement, to tune solid state lumines- cent properties and also to obtain multi-colour emission. 1 Non- covalent routes inclusive of polymorphism, phase transition, nano/microcrystal forms and multi-component systems, have evolved as an efficient method to draw such correlations with a minimum number of variables. 2–4 Also it provides an opportu- nity to enhance the utility of a molecule of interest by generating a wide range of solid forms and with added scope for tailoring the emission characteristics. Several approaches are being sought to modify the uores- cence wavelength and intensity in the solid state, the state in which compounds are intended to be used as aggregate suspensions, thin lms or crystalline materials. Though our molecular level awareness on the structure–property relation- ship in terms of optical properties is fairly well established, the perception on factors that precisely control bulk or supramo- lecular properties is still hazy. In organic materials, modica- tion or alteration of chemical structures is a common approach to tune solid-state luminescence properties. For example, Sunil Varughese obtained his PhD in Chemistry in 2007 from National Chemical Laboratory, Pune under the guidance of Professor V. R. Pedireddi. Aer two year’s post-doctoral research (2007–2009) with Professor Syl- via M. Draper at Trinity College Dublin, he worked as a DST Young Scientist (2009–2013) in the research group of Professor Gautam R. Desiraju at Indian Institute of Science, Bangalore. Since 2013 he has been a Scientist Fellow at National Institute for Interdisciplinary Science and Technology (CSIR-NIIST), Trivan- drum. His research focuses on crystal engineering of functional solids and structure–property correlations in molecular crystals. Chemical Science and Technology Division, CSIR-National Institute for Interdisciplinary Science and Technology, Trivandrum 695 019, Kerala, India. E-mail: [email protected] Cite this: J. Mater. Chem. C, 2014, 2, 3499 Received 6th December 2013 Accepted 28th January 2014 DOI: 10.1039/c3tc32414a www.rsc.org/MaterialsC This journal is © The Royal Society of Chemistry 2014 J. Mater. Chem. C, 2014, 2, 3499–3516 | 3499 Journal of Materials Chemistry C FEATURE ARTICLE Published on 30 January 2014. Downloaded by Regional Research Laboratory (RRL_Tvm) on 11/04/2014 05:17:10. View Article Online View Journal | View Issue
Transcript of Non-covalent routes to tune the optical properties of molecular materials

Journal ofMaterials Chemistry C
FEATURE ARTICLE
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article OnlineView Journal | View Issue
Non-covalent ro
SPNPPt(vDYtGI
Since 2013 he has been a ScientistInterdisciplinary Science and Tedrum. His research focuses on crsolids and structure–property corr
Chemical Science and Technology D
Interdisciplinary Science and Technology,
E-mail: [email protected]
Cite this: J. Mater. Chem. C, 2014, 2,3499
Received 6th December 2013Accepted 28th January 2014
DOI: 10.1039/c3tc32414a
www.rsc.org/MaterialsC
This journal is © The Royal Society of C
utes to tune the optical propertiesof molecular materials
Sunil Varughese*
Switching and tuning solid state luminescence properties of molecular materials by modulating molecular
packing through non-covalent routes is an attractive prospect. This strategy further makes it feasible to
expand the utility of molecules of interest by obtaining a large array of solids—polymorphs, solvates,
amorphous phase, nano/micro-crystals and as multi-component systems—with distinct fluorophore
arrangement and hence emission characteristics. Because non-covalent interactions that determine the
fluorophore arrangements in polymorphs or supramolecular complexes are weak and flexible, their
making and breaking become more realistic under ambient conditions, thus having potential to achieve
reversible transformations and hence external-stimuli-responsive and switchable molecular fluorescent
materials. Recent advances in this context are highlighted in this review with the aid of illustrative
examples and further emphasize the scope and relevance of interdisciplinary and multitechnique
approaches to unravel the structure–optical property relationships and also to augment the foundations
of factual knowledge.
Introduction
The burgeoning world of light-emitting molecular solids andtheir potential utility in optical devices, sensors and biologicalimaging has catalysed its emergence as an area of topicalresearch with a spectrum of prolic activities. The light-emittingbehaviour of molecular solids differs from that of dilute
unil Varughese obtained hishD in Chemistry in 2007 fromational Chemical Laboratory,une under the guidance ofrofessor V. R. Pedireddi. Aerwo year’s post-doctoral research2007–2009) with Professor Syl-ia M. Draper at Trinity Collegeublin, he worked as a DSToung Scientist (2009–2013) inhe research group of Professorautam R. Desiraju at Indiannstitute of Science, Bangalore.Fellow at National Institute forchnology (CSIR-NIIST), Trivan-ystal engineering of functionalelations in molecular crystals.
ivision, CSIR-National Institute for
Trivandrum 695 019, Kerala, India.
hemistry 2014
solutions because emission in the solid state is dictated by thewhole collective rather than by individual molecules. Studyingmolecular crystals provides essential information on howmolecules are arranged with respect to their neighbours,interactions that bind them together and their conformationalpreferences required to correlate the structural facts withseveral macroscopic properties, including their emission char-acteristics. Such systematic knowledge pertaining to howcomplex properties emerge from collections of molecules andhow sensitive these properties are to specic molecularordering in solids, could be translated to deriving new strategiesto control molecular arrangement, to tune solid state lumines-cent properties and also to obtain multi-colour emission.1 Non-covalent routes inclusive of polymorphism, phase transition,nano/microcrystal forms and multi-component systems, haveevolved as an efficient method to draw such correlations with aminimum number of variables.2–4 Also it provides an opportu-nity to enhance the utility of a molecule of interest by generatinga wide range of solid forms and with added scope for tailoringthe emission characteristics.
Several approaches are being sought to modify the uores-cence wavelength and intensity in the solid state, the state inwhich compounds are intended to be used as aggregatesuspensions, thin lms or crystalline materials. Though ourmolecular level awareness on the structure–property relation-ship in terms of optical properties is fairly well established, theperception on factors that precisely control bulk or supramo-lecular properties is still hazy. In organic materials, modica-tion or alteration of chemical structures is a common approachto tune solid-state luminescence properties. For example,
J. Mater. Chem. C, 2014, 2, 3499–3516 | 3499

Journal of Materials Chemistry C Feature Article
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
number and position of alkoxy substituents and alkyl chainlength direct the molecular packing and consequently theiruorescence properties in diphenylbutadienes.5 However,several limitations inherent to covalent synthesis and solid statereactions make dynamic tuning or switching of solid-stateluminescence a formidable target. Also because solid-stateluminescence depends strongly on the intermolecular dipolecoupling, resulting from the relative arrangements of neigh-boringmolecules and orientations of their dipole moments, it isdifficult to expect similar properties in related molecules.Looking beyond the covalent routes that involve chemicalmodications (synthetic tailoring of uorophore cores), recentdevelopments in the modulation of materials properties vianon-covalent routes such as polymorphism (through confor-mational/packing changes), phase transition (structuralchanges while maintaining the molecular integrity), amorph-ization (disrupting the long-range molecular ordering) andpreparation of multi-component systems (controlling uo-rophore aggregation) look promising.
The purpose of this review is to highlight some of the recentdevelopments in non-covalent methods and their utility inattaining switchable solid state emitting molecular materials(Scheme 1). Multiple stimuli responsive materials and theirphase transition (between crystalline phases as well as crystallineand amorphous phases) will be considered in detail. Since theamorphous phase is of particular interest in the context of theconstruction of organic light-emitting diodes, special mention ismade on different approaches available in the literature to attainamorphization. Further to external shear stress or fast aggrega-tion, crystal engineering principles provide an alternate route toattain amorphization, with resultant phases having large glasstransition temperature. Also this article examines some possibledesign strategies to enable phase transformation and hencetuning of emission characteristics. Towards the latter section, theimportance of multidisciplinary and multitechnique approacheswill be highlighted, by providing a few recent examples whereinthe structural variations were examined by combining thepossibilities of diffraction, microscopy and indentation tech-niques. Such approaches are necessary to obtain complementaryinformation that enable design and synthesis of molecularmaterials with greater predictability.
Scheme 1 Schematic overview of the non-covalent routes and topicsdiscussed in this feature article.
3500 | J. Mater. Chem. C, 2014, 2, 3499–3516
A brief overview of optical behaviour ofaggregate states
Curtis and co-workers analyzed solid state packing of molecularmaterials in terms of “pitch and roll” inclinations from an“ideal” cofacial p-stack (H-aggregate). The “pitch” inclinationsdescribe translation of adjacent molecules relative to oneanother in the direction of the long molecular axis, while the“roll” inclinations that along the short molecular axis (Scheme2a).6 Moderately large pitch distortions preserve p/p interac-tions between adjacent molecules (J-aggregation), whereas rolltranslations greater than 2.5 A lead to minimum p-overlap(Herringbone-type).1 Compared to the cofacial p-stackedstructure in H-aggregates, translation of molecules along thelong molecular axis leads to a head-to-tail arrangement with anenhanced delocalization of electronic excitation over severalmolecules due to a signicant retention of p-overlap betweenneighbouring molecules. Such structural variations in molec-ular aggregates have profound inuence on their uorescenceproperties such as emission wavelength and efficiency. Kashaand co-workers explained the optical properties of interactingconjugated systems in terms of exciton coupling theory inwhich the excited state of the aggregates splits (Davydov split-ting) into two energy levels: one lower and another higher thanthat of the monomer (Scheme 2b).7 The transitions to and froma level is forbidden where the transition dipoles of the mono-mers cancel out because of their opposing directions and incontrast those transitions are allowed wherein the coupling ofthe dipoles results in a nonzero transition moment. The lower
Scheme 2 (a) Schematic representation of “pitch” and “roll” angles/distances (P/dP; R/dR) and the structural description of Herringbone aswell as H- and J-aggregates in terms of relative translation of mole-cules. Insets A and B represent pitch and roll inclinations that lead to J-aggregate and Herringbone (adapted with permission from ref. 6.Copyright © 2004, American Chemical Society). (b) Exciton splitting inJ-aggregate and H-aggregate (reproduced with permission from ref.8. Copyright © 2010, John Wiley and Sons).
This journal is © The Royal Society of Chemistry 2014

Fig. 1 Polymorphs of ROY that differ in their colours, melting pointsand conformations. (Reproduced with permission from ref. 14.Copyright © 2010, American Chemical Society).
Feature Article Journal of Materials Chemistry C
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
level of the exciton is allowed for the J-aggregate with a resultantred-shied uorescence, compared with that of the monomer,while the allowed transition in the H-aggregate is to the upperlevel with a blue shi in uorescence. Vibrational relaxationfrom the upper level to the lower one (from which radiativetransition is forbidden) explains the lower luminescence effi-ciency of H-aggregates, making it detrimental for effective solidstate uorescence.8 Thus, substantial changes in the aggrega-tion state and hence relative arrangement of the uorophoremoiety, because of polymorphism or phase transition, couldlead to distinct solid state emission characteristics.
Polymorphism
McCrone dened a polymorph as “a solid crystalline phase of agiven compound resulting from the possibility of at least twodifferent arrangements of the molecules of that compound in thesolid state”.9 This implies that there are oen a number of waysin which molecules can tessellate, while retaining near-optimalpacking. Crystallization being a remarkable process that bringsan enormous number of molecules or ions into an essentiallyordered array (entropy factor), the system seeks (kinetics allow-ing)multiple solutions to the problemofmolecular ordering withthe possible optimization of enthalpically favourable directionaland non-directional interactions—essentially an entropy–enthalpy balancing. This makes prediction of polymorphs infa-mously difficult.10 Hence polymorphism as a phenomenon moreoen than not is considered as a nemesis to crystal engineering.Nevertheless, the pervasiveness of this phenomenon is evidentfrom the fact that about one third of organicmolecules crystallizeinto more than one solid form; it is a truism that the number ofpolymorphs of a compound increases in proportion to theamount of time spent on its study.9
Yet regardless of the degree of unpredictability, poly-morphism offers an unparalleled multitude of opportunitiesin material modications and property modulations, withoutaltering the molecular integrity. Polymorphs oen differ intheir physico-chemical properties such as melting point,crystal shape, colour, chemical reactivity, dissolution rate,and solubility.11 Because crystal forms of active pharmaceu-tical ingredients (APIs) differ in their crucial properties suchas stability, bioavailability and processability, polymorphismand phase transition in pharmaceutics is a hot research topicwith signicant technological and economic impacts.12 Inmolecular materials, change in crystal lattice and morphologycould inuence several bulk properties such as charge-transfer mobility, conductivity, luminescence efficiency andON/OFF ratio.
Colour is a notable property of polymorphs and is commonlyused to identify and isolate crystal forms, especially when thecompound crystallizes as concomitant polymorphs.13 ROY is animpressive example where different polymorphs exist indifferent morphologies and colours (Fig. 1).14 Changes in thecolour of a substance15 resulting from the perturbation ofstructural environment (chromogenic effects)11,16,17 and theconcomitant crystallization of compounds with differentcolours (chromoisomerism)18,19 were known for several years.
This journal is © The Royal Society of Chemistry 2014
Yet, switching and tuning of solid state optical properties,especially uorescence, have received greater attention only inrecent years thanks to the emerging interest both in terms offundamental research and potential applications such assensors, display devices and security inks, to mention a few.Since solid-state emission in organic compounds is closelyrelated to molecular arrangement, conformational exibilityand intermolecular interactions, polymorphism makes itpossible to perturb these factors and thereby the HOMO–LUMOenergy levels of uorophores, while maintaining the molecularintegrity.20,21 Since polymorphs all have the same chemicalcompositions, and hence minimum number of variables,correlations with the differences in aggregate structures and theobserved physical properties become lucid. Some factors thatcould lead to different emission characteristics in solid formscan be broadly discerned, as concisely discussed in thefollowing section.
(i). Extent of p-overlap and conformation differences
In organic functional materials varied molecular arrangementsbased on the collective interactions, p-overlap geometries andco-planarity of adjacent p-planes could lead to different extentof electronic conjugation and hence discernible optoelectronicproperties of the bulk (Scheme 3A). For example, ve crystallineforms of 3(5)-(9-anthryl)pyrazole (ANP) exhibit distinct crystalpacking and intermolecular interactions (Fig. 2a). Further to thehydrogen bond types, the emission colours of the crystal formswere correlated with the extent of p–p interactions: that with nointeraction (blue emission); with strong interaction (greenemission), and with weak interaction (blue-green emission).22,23
An increasing p-overlapping area leads to greater electronicdelocalization (Fig. 2b) and hence a red-shi in the emissioncolour (Scheme 3B). Additionally, differences in conformationalexibility, interaction modes, structural defects, stacking modeand p-overlap also could strongly direct the emission propertiesof molecular compounds.24,25
J. Mater. Chem. C, 2014, 2, 3499–3516 | 3501

Scheme 3 Some of the possible reasons for polymorphism-inducedemission colour switching. (A): Extent of p-overlap; (B): conforma-tional differences; (C): intermolecular interactions; (D): ESIPT (part Dadapted with permission from ref. 27. Copyright © 2010, AmericanChemical Society).
Fig. 2 (a) Tetramer and trimer assemblies in ANP polymorphs andphotographs of corresponding crystals, excited under UV light (lex ¼365 nm) (adapted with permission from ref. 22. Copyright © 2006,John Wiley and Sons). (b) Variations in extent of p-overlap and cor-responding crystal emission colour in ANP derivatives (adapted withpermission from ref. 23. Copyright © 2009, American ChemicalSociety).
Fig. 3 ESIPT-induced emission switching in 2-(20-hydroxyphenyl)-imidazo[1,2-a]pyridine (adapted with permission from ref. 28. Copy-right © 2008, John Wiley and Sons).
Journal of Materials Chemistry C Feature Article
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
(ii). Interaction types
Covalent bonds, the nature of the links and the effectivedistance between donor and acceptor units play the directingrole in electron transfer processes in the molecular context.
3502 | J. Mater. Chem. C, 2014, 2, 3499–3516
However in crystal engineering of functional materials,manipulation of weak interactions is the key protocol. On top ofclassical hydrogen bonds, weak interactions such as C–H/p,halogen/p, p/p or even hydrophobic interactions could leadto changes in molecular conformations, electronic delocaliza-tion and induced dipoles and thereby different optoelectronicresponses (Scheme 3C).26 This topic is further discussed in thedesign strategy section (vide infra).
(iii). Excited state intramolecular proton transfer (ESIPT)27
Although not strictly polymorphs by denition, molecularsystems that exhibit ESIPT with resultant changes in dipolemoment are a good route to achieve switchable emission colourand intensity. Interconversion of the crystal forms of 2-(20-hydroxyphenyl)imidazo[1,2-a]pyridine by a thermal dry processrealized switching of ESIPT luminescence between blue-greenand yellow emission and also leads to differences in molecularconformations and modes of packing (Fig. 3).28 Similarly, 2-(2-hydroxyphenyl)-4(3H)-quinazolinone (HPQ) exhibits emissionby ESIPT to form dimorphs with different conformational twist,molecular packing and distinct emission colours (lmax
em ¼ 497and 511 nm) (Scheme 3D).29
Stimuli responsive phase transition andfluorescence switching
Stimuli responsive phase transition and the resultant changes inuorophore arrangements is imbued with opportunity to obtainmulti-colour emission by uorescence switching.30 Nevertheless,design and synthesis of organic materials that display efficientand reversible switching between two luminescent states in thesolid state is still a looming challenge. This can be attributed tothe fact that the interconversion of molecular structures in theconstrained environment of the solid state is oen formidable,making the controlled and reversible switching of solid stateluminescence between polymorphs rather exceptional.31 Basedon the type of stimuli, the materials are broadly classied asmechanochromic or piezochromic (mechanical shock),32 ther-mochromic (temperature),33 solvatochromic (solvent)34 andvapochromic (vapour) materials.35
(i). Temperature-responsive materials
Temperature-induced phase transition offers an excellentmethod to switch the solid state emission of molecular
This journal is © The Royal Society of Chemistry 2014

Fig. 5 Reversible thermochromic phase transition in HMPA (adaptedwith permission from ref. 42. Copyright © 2013, American ChemicalSociety).
Feature Article Journal of Materials Chemistry C
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
materials. Thoughmonotropic relations36 between crystal formscould be advantageous to achieve phase stability in APIs,enantiotropic transformations37 of polymorphic forms inmolecular compounds are valuable to achieve efficient andreversible emission switching. In systems where the crystalforms differ in their emission intensity the possible intercon-version could be an efficient and reproducible route to ON/OFFswitching of solid-state luminescence. The heat-mode inter-conversion between the plate (Form-I, emissive) and needle(Form-II, non-emissive) crystals of 2,20:60,20 0-terpyridine (TPY) isan illustrative example.20
Structural variations brought about by thermal treatmentlead to apparent solid state emission switching in octyloxy-cyano-substituted diphenylbutadiene (BC8); the interdigitationof the alkyl chains and the resultant disaggregation of uo-rophore moieties lead to a blue solid state emission. Uponmelting, the molecules move out of its interdigitated arraysmaking way to layers of unidirectional oriented molecularstacks of J-aggregates, which results in green emission.38 Simi-larly, melt cooling induced transformation of blue-emittingcrystal (Form-I) of 4-((1E,3E)-4-(4-butoxyphenyl)buta-1,3-dienyl)-pyridine (BP4) to Form-II with yellow emission involves struc-tural change from a herringbone to brickstone arrangementand a corresponding shi from monomer to J-type aggregateemission (Fig. 4a).39
Diethyl-2,5-bis(4-(triuoromethyl)phenylamino)terephthalate(AA1) and diethyl-2,5-bis(3,5-bis(triuoromethyl)phenylamino)-terephthalate (AA2) exhibit thermochromic uorescenceswitching. When heated at 144 �C (below the melting point), redemitting crystals of AAI (Form-I; lmax
em ¼ 581 nm) exhibit ther-mochromic transformation to a yellow solid (Form-II; lmax
em ¼545 nm); similarly AA2 crystals transform from red (Form-I;lmaxem ¼ 610 nm) to green emitting form (Form-II; lmax
em ¼ 503 nm)at 124 �C (Fig. 4b). In both cases the emission switchinginvolves transformation of a p-stacked H-aggregation to a cross-stacked J-typemode. The reverse transitions occur, however, onlyaer the compounds melt.31 Thermochromic transformationplus distinct packing modes and interaction types originatingfrom the conformational differences in the polymorphs of a1,8-naphthyridine derivative resulted in discernable solid state
Fig. 4 (a) Herringbone to brickstone structural transformation in BP4Chemical Society). (b) Thermochromic transformations in AA1 and AA2 (aSons).
This journal is © The Royal Society of Chemistry 2014
emission switching (Form-I, lmaxem ¼ 459 nm; Form-II, lmax
em ¼444 nm) and intensity variations (emitting intensity for II is 5times larger than for I).40 Thermal annealing of Form-I crystals ofdi(p-methoxylphenyl)dibenzofulvene at 135 �C promotes anirreversible single crystal-to-single crystal (I / II) phase transi-tion with an associated shi in the emission colour fromblue (lmax
em ¼ 466 nm;Fem¼ 0.95) to yellow-green (lmaxem ¼ 518 nm;
Fem ¼ 0.62). Amorphization (II / amorphous) by meltquenching, in contrast, leads to orange emission (lmax
em ¼583 nm; Fem ¼ 0.05).41
Conformational exibility and changes in the tightness ofpacking upon annealing or melting thermal treatments couldlead to phase transformation with apparent thermochromism.Phase transition between the dimorphs in (2-hydroxy-4-methoxyphenyl)(phenyl)methanone azine (HMPA) and subse-quent emission switching between green (Form-I; lmax
em ¼ 521nm) and yellowish-green (Form-II; lmax
em ¼ 540 nm) colour(Fig. 5), is a typical example.42
(ii). Multiple-stimuli responsive materials
Molecular compounds that undergo phase transition in responseto multiple stimuli with associated changes in emissioncharacteristics are suitable for developing stimuli-responsive
(adapted with permission from ref. 39. Copyright © 2008, Americandapted with permission from ref. 31. Copyright © 2009, JohnWiley and
J. Mater. Chem. C, 2014, 2, 3499–3516 | 3503

Journal of Materials Chemistry C Feature Article
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
light emitting materials that have potential for various applica-tions. Though several types of stimuli are possible, the presentreview will discuss a few illustrative examples for temperature,pressure and solvent vapour induced transformations and theensuing emission switching.
Non-uorescent crystals of (Z)-3-(30,50-bis(triuoromethyl)-biphenyl-4-yl)-2-((40-triuoromethyl)biphenyl-4-yl)acrylonitrile(CN(L)-TrFMBE) switch to a highly uorescent state with strongsky-blue emission under prolonged UV irradiation or with anexternal shear strain. The close pairs of stilbenic (C]C) bonds(�3.8 A) due to the antiparallel p-dimers in the pristine crystalbecome perturbed under shear force and hence uoresce at thecost of frustrated [2 + 2] cycloaddition. Similarly, the formationof sterically bulkier s-dimer due to prolonged UV irradiationalso effectively exerts an internal shear strain on the neigh-bouring p-dimer pairs resulting in uorescence (Fig. 6a).43
D–A–D-type molecule (2Z,20Z)-2,20-(1,4-phenylene)bis(3-(4-butoxyphenyl))acrylonitrile (DBDCS) with stacking and shear-sliding capabilities of two-dimensional molecular sheetspromote two-colour uorescence switching in response topressure, temperature and solvent vapour stimuli. Thermalannealing at 125 �C leads to sliding of molecular sheets alongthe shorter axis of DBDCS to initiate a phase transition withresulting shi in solid state emission from green light (lmax
em ¼533 nm) to blue (lmax
em ¼ 458 nm) (Fig. 6b). Rough and sheet-likeregular surface projections on the transformed crystals furthervalidates the layer-sliding mechanism. Form-II solid reverts toForm-I under shear stress and this two-colour uorescenceswitching allows the fabrication of a poly(methyl methacrylate)–DBDCS blend lm for rewritable uorescent optical recording.44
Thermal or mechanical induced reversible phase transitionin the polymorphic forms of dicyanodistyrylbenzene derivative,(2Z,20Z)-2,20-(1,4-phenylene)bis(3-phenylacrylonitrile) (BDCS)involve structural transformation from a slip-stack architectureto one with substantial p-overlap and associated switching ofemission colour between blue (Form-I; lmax
em ¼ 441 nm) andgreen (lmax
em ¼ 510 nm) (Fig. 7a).45 Mechanical and thermalinduced reversible crystal to amorphous phase transition in
Fig. 6 (a) Schematic representation of mechanisms for fluorescence atwith permission from ref. 43. Copyright © 2009, American Chemical Socthermo/piezochromic switching between green and blue forms. SEM imapermission from ref. 44. Copyright © 2010, American Chemical Society)
3504 | J. Mater. Chem. C, 2014, 2, 3499–3516
propeller-shaped diphenylbenzofulvene leads to emission ON/OFF switching, because of the less emissive nature of amor-phous phase (Fig. 7b). The amorphous phase reverts to greenemitting Form-I spontaneously, making it a promising self-healing optical material.46 A triuoromethyl-substitutedbenzothiadiazole-cored phenylene vinylene compound dis-played mechano-, vapo- and thermo-induced uorescencevariation in the solid state.47
Crystal to amorphous transformationand emission switching
Crystal to amorphous conversion involves the transformation ofa stable phase to one less stable. Amorphous phase being a highenergy phase, amorphization requires the free energy of thecrystal to be raised above to that of the amorphous phase andalso involves destruction of the regular lattice arrangement bymeans of mechanical energy input or quench melting. Anamorphous solid being a non-ordered phase enjoys greatermolecular mobility compared to its crystalline counterparts andhence is more susceptible to chemical degradation and phasetransformation. Mechanical grinding is essentially an ineffi-cient process, in energetic terms, since the ratio of energyconsumed in lattice bond breaking and crack formation versusmechanical energy input is of the order of a mere 1%.48 Thismethod, however, is widely employed in pharmaceuticals andalso to achieve inter/intramolecular chemical reaction, chem-ical complexation, polymorphic transition, amorphization.49
Based on the concepts of mechanical and thermodynamicdestabilization, a few mechanisms were proposed for crystal toamorphous phase transformations: (a) greater anharmonicity oflattice vibrations (phonons) under increased compression willresult in soening of the lattice vibrations. Such soening,above a critical pressure, leads to Born instability and latticecollapse. (b) Mechanical energy input could increase theconcentration of defects in a crystal lattice, and thereby theenergy of the system, to a critical limit beyond which amor-phous phase has greater thermodynamic stability than the
the cost of frustrated [2 + 2] cycloaddition in CN(L)-TrFMBE (adaptediety). (b) Different modes of slip-stacking in DBDCS molecular sheets;ges show corresponding changes on the crystal surface (adapted with.
This journal is © The Royal Society of Chemistry 2014

Fig. 7 (a) Phase transition in BDCS brought about by thermal annealing and mechanical grinding (adapted with permission from ref. 45; RoyalSociety of Chemistry). (b) Thermo/mechanochromic ON/OFF switching in diphenylbenzofulvene (adapted with permission from ref. 46.Copyright © 2011, John Wiley and Sons).
Feature Article Journal of Materials Chemistry C
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
disordered crystal. (c) Mechanical energy due to grinding couldcause a local increase in temperature beyond the melting pointof the sample. (d) Pressure-induced lattice instability couldarise when the root-mean-square thermal displacement of theconstituent atoms of a crystal lattice reaches a critical fractionof the interatomic spacing (Lindemann criteria).50,51
(i). Amorphization by shear stress
With the shear stress caused transformation of crystallinephase to a non-ordered amorphous phase, TPE-Py moleculesenjoy more rotational freedom and are further enabled torearrange and make excimeric or intermolecular charge-trans-fer species with the necessary dye–dye orientations and spac-ings, leading to redder emission (Fig. 8a).52,53 For example,yellow (lmax
em ¼ 565 nm) emitting crystals of hemicyanin dyeunder mechanical stress convert to an amorphous form withred emission (lmax
em ¼ 650 nm) (Fig. 8b).54 The stress-inducedcrystal to amorphous transition can also lead to planarization ofthe twisted molecular conformation, which in turn can lead toheightened effective conjugation lengths ECLs and henceredder emissions. Switching of emission colour from green(lmax
em ¼ 500 nm) to yellow (lmaxem ¼ 551 nm) with amorphization
in DPATPAN, is an illustrative example (Fig. 8c).55
Grinding being an inefficient process, at times it fails toeffect total conversion of phases. In such instances the residualcrystal phase acts as a nucleus for future transformation. The
Fig. 8 Stimuli-induced switching of solid-state emission by (a) TPE-Py(adapted with permission from ref. 53; Royal Society of Chemistry), (b)hemicyanin dye (adapted with permission from ref. 54; Royal Societyof Chemistry), and (c)DPATPAN (adapted with permission from ref. 55;Royal Society of Chemistry).
This journal is © The Royal Society of Chemistry 2014
amorphous phase (green emitting; lmaxem ¼ 491 nm) of tetra-
phenylethene-based luminogen (1CA), obtained by grinding thepristine Form-I crystals (deep-blue emitting; lmax
em ¼ 448 nm)reverts to its native phase upon thermal annealing at 90 �C(Fig. 9a). The residual small crystallites in the powder sampleact as the nucleus and catalyse the reversion of amorphous toForm-I phase. However, the amorphous phase obtained byquenchmelting of Form-I converts to another crystal phase withsky-blue emission (Form-II; lmax
em ¼ 462 nm), with thermalannealing (at both 90 and 115 �C).56
(ii). Amorphous phase via fast aggregation
Random agglomeration of molecules from solution by control-ling the mixing ratio of the solvent–antisolvent system offers analternate route to attain amorphous phases and also a method toeffectively tune the morphologies of aggregates from crystalline-to-amorphous phase and hence their emission colour andefficiency.57 Molecules of 2-(2,6-bis[4-(cholesteryloxymethyl)-styryl]-4H-pyran-4-ylidene) malononitrile (BCPM) form emissiveJ-aggregates in solvent systems with lower water volume. Withadditional water content, morphology of the nanoaggregateschanges from crystalline to amorphous phase, with concomitantfall in quantum yield. Also the emission maximum reverts to itsmonomer emission wavelength (Fig. 9b).58
(iii). Amorphization by thwarted crystallization
Crystal engineering principles provide one with the opportunityto identify the features that could thwart crystallization andthus to obtain stable amorphous molecular materials. Thoughless in number there are examples wherein a stable amorphousphase was achieved by design. Molecules with rigid, labyrin-thine conformations and steric congestion can prevent effectivecrystal packing, leading to a stable amorphous phase with highglass transition (Tg) temperature.59 In yet another strategy, Dasand coworkers exploited the weakened intermolecular interac-tions and enhanced molecular mobility at temperature close tothe melting point to bring about trans–cis isomerization of X12polyenes, by UV irradiation. Upon cooling down to roomtemperature, the incoherence in the mixture of trans and cisisomers hinders crystallization, thus leading to a stable glassymaterial.60 Molecules with symmetric globular structure, largemolecular weight, and small intermolecular cohesion areproposed to lead to a stable amorphous phase.61
J. Mater. Chem. C, 2014, 2, 3499–3516 | 3505

Fig. 9 (a) Phase transition and solid-state emission switching in 1CA by the grinding–heating processes (adapted with permission from ref. 56.Copyright © 2012, American Chemical Society). (b) Switching of emission characteristics by nanoaggregates of BCPM; notice the emissionquenching with formation of amorphous aggregates at 99% water content. TEM images and ED patterns of aggregates at 40 and 99% watercontent (adapted with permission from ref. 58. Copyright © 2007, American Chemical Society).
Journal of Materials Chemistry C Feature Article
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
Amorphous glassy materials are of signicant importancein optoelectronics applications for various reasons. In addi-tion to their relative ease to prepare, a stable amorphousphase is helpful to avert many factors that deteriorate theperformance of the optical device. For instance, unlike thepolycrystalline thin lms wherein the grain boundariesbetween adjacent crystallites act as traps for charge carriers,the spatial homogeneity possible in amorphous thin lmsavoids such eventualities, leading to enhanced transportproperties. Moreover, optical properties of isotropic amor-phous phase are devoid of any signicant scattering andbirefringence that are common in polycrystalline and singlecrystal materials. Because of these factors the amorphousphase of molecular materials are generally preferred for devicefabrication.61 Nevertheless, the unstable nature of this phaseand possible transformation to crystalline phase is a loomingdanger and hence manufacturing processes of amorphouslms demand a high stability of the glassy state to preventpossible degradation of devices in applications. Tg is animportant physical parameter that indicates the stability ofthe amorphous state of a given material, above which a phasetransition into the crystalline state occurs. Thus it is of utmostimportance to design low molecular weight materials with ahigh Tg value. Further, high Tg and thermal stability also assistto withstand inevitable Joule heating encountered duringOLED operation, especially at higher electric elds andcurrent densities.62
Nano/microcrystals and luminescenceswitching
Nanostructured phases of functional materials offer newopportunities in exploring unique optoelectronic properties inlower dimensions that are different from the bulk. In contrast tothe prolic literature on nanostructures in inorganic materials,that corresponding to nanoaggregates of molecular materials isrelatively rare. However, recent developments on the nano-structured functional organic small molecule materials in the
3506 | J. Mater. Chem. C, 2014, 2, 3499–3516
context of their doping properties, better processability anduorescence efficiency makes them complementary to inor-ganic materials. Therefore, the possibility of exploring opto-electronic properties of small organic compounds in thenanosize regime offers a unique strategy and hence needs amention, at least in brief. A recent review by Yao et al. provides agreat repertoire of synthetic strategies involved in nano-fabrication of low-dimensional nanoaggregates and hence isnot considered presently, to avoid tautology.63
The nanostructured phase becomes unique for organicmaterials due to several reasons:
(a) Unlike the ionic interactions in inorganic compounds,weak intermolecular interactions (such as hydrogen bonds,halogen bonds and van derWaals forces) in organic compoundsafford additional exibility in the material processability.
(b) By adjusting the nanofabrication/processing conditions,such as solvents, temperature, additives etc. it is possible toobtain different nanostructures including low-dimensionalparticles, viz., nanowires, nanorods and nanoribbons, andhence possible to derive additional size and morphologydependent properties.
(c) The anisotropy in shape, size, interactions and stackingmodes provide multiple options in the arrangement of thechromophore units in the crystal lattice, making their proper-ties more versatile and tunable; in inorganic materials the hardsphere shape of the atoms circumscribe their options for close-packing.
(d) Polymorphism and phase transition in nanoaggregatesprovides additional opportunities in understanding theirstructure–property relationships.
Nakanishi and coworkers made one of the rst reports onthe size-dependent uorescent properties of organic nano-aggregates. They observed that emission wavelength was blueshied from lmax
em ¼ 560 nm (bulk crystals) to lmaxem ¼ 482 and 470
nm (Dlem ¼ �80 nm), with reducing size of the aggregates ofperylene (to 50 and 200 nm). Variations in the emission prop-erties were attributed to changes of crystal-lattice state withincreasing surface area and effect of electric eld around thesurface of nanocrystals.64
This journal is © The Royal Society of Chemistry 2014
Feature Article Journal of Materials Chemistry C
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
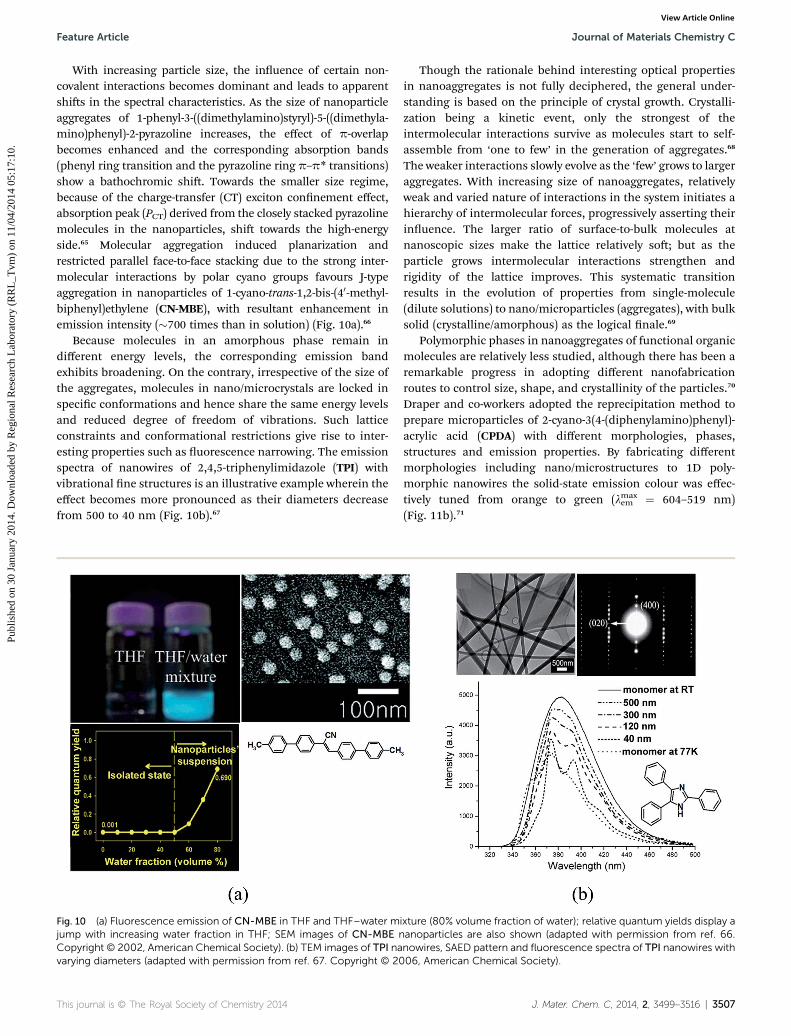
With increasing particle size, the inuence of certain non-covalent interactions becomes dominant and leads to apparentshis in the spectral characteristics. As the size of nanoparticleaggregates of 1-phenyl-3-((dimethylamino)styryl)-5-((dimethyla-mino)phenyl)-2-pyrazoline increases, the effect of p-overlapbecomes enhanced and the corresponding absorption bands(phenyl ring transition and the pyrazoline ring p–p* transitions)show a bathochromic shi. Towards the smaller size regime,because of the charge-transfer (CT) exciton connement effect,absorption peak (PCT) derived from the closely stacked pyrazolinemolecules in the nanoparticles, shi towards the high-energyside.65 Molecular aggregation induced planarization andrestricted parallel face-to-face stacking due to the strong inter-molecular interactions by polar cyano groups favours J-typeaggregation in nanoparticles of 1-cyano-trans-1,2-bis-(40-methyl-biphenyl)ethylene (CN-MBE), with resultant enhancement inemission intensity (�700 times than in solution) (Fig. 10a).66
Because molecules in an amorphous phase remain indifferent energy levels, the corresponding emission bandexhibits broadening. On the contrary, irrespective of the size ofthe aggregates, molecules in nano/microcrystals are locked inspecic conformations and hence share the same energy levelsand reduced degree of freedom of vibrations. Such latticeconstraints and conformational restrictions give rise to inter-esting properties such as uorescence narrowing. The emissionspectra of nanowires of 2,4,5-triphenylimidazole (TPI) withvibrational ne structures is an illustrative example wherein theeffect becomes more pronounced as their diameters decreasefrom 500 to 40 nm (Fig. 10b).67
Fig. 10 (a) Fluorescence emission of CN-MBE in THF and THF–water mjump with increasing water fraction in THF; SEM images of CN-MBE nCopyright © 2002, American Chemical Society). (b) TEM images of TPI navarying diameters (adapted with permission from ref. 67. Copyright © 20
This journal is © The Royal Society of Chemistry 2014
Though the rationale behind interesting optical propertiesin nanoaggregates is not fully deciphered, the general under-standing is based on the principle of crystal growth. Crystalli-zation being a kinetic event, only the strongest of theintermolecular interactions survive as molecules start to self-assemble from ‘one to few’ in the generation of aggregates.68
The weaker interactions slowly evolve as the ‘few’ grows to largeraggregates. With increasing size of nanoaggregates, relativelyweak and varied nature of interactions in the system initiates ahierarchy of intermolecular forces, progressively asserting theirinuence. The larger ratio of surface-to-bulk molecules atnanoscopic sizes make the lattice relatively so; but as theparticle grows intermolecular interactions strengthen andrigidity of the lattice improves. This systematic transitionresults in the evolution of properties from single-molecule(dilute solutions) to nano/microparticles (aggregates), with bulksolid (crystalline/amorphous) as the logical nale.69
Polymorphic phases in nanoaggregates of functional organicmolecules are relatively less studied, although there has been aremarkable progress in adopting different nanofabricationroutes to control size, shape, and crystallinity of the particles.70
Draper and co-workers adopted the reprecipitation method toprepare microparticles of 2-cyano-3(4-(diphenylamino)phenyl)-acrylic acid (CPDA) with different morphologies, phases,structures and emission properties. By fabricating differentmorphologies including nano/microstructures to 1D poly-morphic nanowires the solid-state emission colour was effec-tively tuned from orange to green (lmax
em ¼ 604–519 nm)(Fig. 11b).71
ixture (80% volume fraction of water); relative quantum yields display aanoparticles are also shown (adapted with permission from ref. 66.nowires, SAED pattern and fluorescence spectra of TPI nanowires with06, American Chemical Society).
J. Mater. Chem. C, 2014, 2, 3499–3516 | 3507

Journal of Materials Chemistry C Feature Article
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
Reprecipitation in the presence of additives is being usedto control the size and shape of nanostructures of organicsmall molecules,72 wherein the additives that adsorb onspecic crystal faces regulate the crystal growth and henceits size and shape. The nature of additives inuences thegrowth habits of nano/microcrystals, for example, of theorganic dye, 4-n-octylamino-7-nitrobenzoxadiazole and theiremission properties. While linear polymer additivesresulted in uniformly sized crystals, platelet morphology wasobtained in the presence of dendrimer additives (Fig. 12).73
Thus, the developments in the studies of low dimensionalnanomaterials herald its potential utility in understandingaggregation-induced emission, uorescence narrowing,multi-colour emission as well as tunable and switchableemission.74
Multicomponent systems to tune theemission properties
The co-crystallization approach in pharmaceutics is widelyadopted to modify several solid state properties of APIs.75 Interms of chemical reactivity, co-crystallization with appropriatetemplate molecules, that act as scaffolds, allows the bringingof olenic bridges in the reactive distance of <4.0 A to achieve[2 + 2] photodimerization reaction.76 In the context of materialsscience, this supramolecular approach offers a powerful way toexpand the utility of a molecule of interest by obtaining a largearray of related ordered solids that share the same molecule,but with distinct behaviours since the components are notarranged in the same way.77 Polymorphism is less predictable;the co-crystallization approach, in contrast, offers a moreprobable route to tune solid state luminescence. Though thereare a few interesting studies in this regard, this approachremains largely underexplored for tuning the optical propertiesof molecular solids.78
Introduction of coformers could alter the geometricarrangement of chromophores in molecular complexes,causing substantial variations in their optical properties suchas absorption, emission, colour, lifetime and quantum yield.For example, depending on the nature of interactions and
Fig. 11 (a) Rubrene microcrystals: emission spectra and fluorescence mwith scale bar of 50 mm (adapted with permission from ref. 70; Royal Somorphologies and emission colours (adapted with permission from ref.
3508 | J. Mater. Chem. C, 2014, 2, 3499–3516
chromophore arrangement, molecular complexes of 1,4-bis-p-cyanostyrylbenzene exhibited multi-colour solid state emis-sion from blue through green to yellow (Fig. 13a).79 Effect ofthe nature of interactions and the molecular reorganizationin the crystal lattice by cocrystallization is further evidentfrom the large solid state emission switching observed in themolecular complexes of 2-cyano-3(4-(diphenylamino)phenyl)-acrylic acid (CDPA) with organic amines (Fig. 13b).80,81 Thus, bythe judicious choice of coformers it is possible to alter thestacking mode and aggregation/deaggregation of a uo-rophore moiety in a crystalline array thereby enabling theswitching of emission colour, lifetimes and intensity withgreater predictability.82
Besides the emission colour switching, molecular complexesof functional molecules with appropriate coformers providenovel routes to induce properties that are supposedly impos-sible to obtain from the original compounds. Force-inducedphase separation in the co-crystals of 2,5-di(E)-distrylyfuran andN-alkyl maleimides (as uorescent donor and acceptor respec-tively) leads to switching of luminescence from the quenched tothe emitting (lmax
em ¼ 468 nm) state (Fig. 14a).83 One-dimensionalmixed stacks of two isometric distyrylbenzene- and dicyano-distyrylbenzene-based molecules exhibit strongly red-shied,bright photoluminescence as well as high and ambipolar p-/n-type eld-effect mobility up to 6.7 � 10�3 and 6.7 � 10�2 cm2
V�1 s�1, respectively, originating from an intermoleculardonor–acceptor (D–A) charge-transfer state.84
Formation of halogen bonding between organic moleculescould facilitate molecular complexes that show unique photo-emission properties in nano-dimensions85 and also be exploredas a route to direct heavy atom effect. Kim et al. reported theutility of crystal-state halogen bonding to hold chromophoreswith triplet-producing aromatic aldehydes and triplet-promoting bromine to achieve a pronounced increase inphosphorescent quantum yield (up to 55%), due to the heavyatom effect and reduction of self-quenching (Fig. 14b).86 Also itis possible to add elements of complexity to systems by adopt-ing the co-crystallization approach and such systems could evenact as prototypes of complex structures and help us to get betterinsights on structure–property relationships. For example, byco-crystallizing with achiral and racemic co-formers it is
icroscopy images of (A) ribbons, (B) rhombic and (C) hexagonal plates,ciety of Chemistry). (b) Nano/microaggregates of CDPA with differing71. Copyright © 2010, American Chemical Society).
This journal is © The Royal Society of Chemistry 2014

Fig. 12 Fluorescence and morphology tuning of 4-n-octylamino-7-nitrobenzoxadiazole microparticles by means of additive-induced nano-fabrication (adapted with permission from ref. 73. Copyright © 2009, American Chemical Society).
Feature Article Journal of Materials Chemistry C
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
possible to achieve spontaneous resolution to chiral supramo-lecular uorescent systems with circularly polarised uores-cence properties.87 The diversity and versatility of this approachopen new avenues in unravelling new ways to tune the opticalproperties of molecular solids.
Fig. 13 (a) The emission switching observed in 1,4-bis-p-cyanostyrylben79. Copyright © 2011, JohnWiley and Sons). (b) Fluorescence switching inbase interactions (adapted with permission from ref. 80; Royal Society o
This journal is © The Royal Society of Chemistry 2014
Design strategy
Although an explicit design of molecular packing and itsproperty prediction a priori based on chemical structure is nextto impossible, extra- or interpolations of structure–property
zene by means of co-crystallization (adapted with permission from ref.CDPA bymeans of formation of multicomponent systems using acid–f Chemistry).
J. Mater. Chem. C, 2014, 2, 3499–3516 | 3509

Fig. 14 (a) Piezochromic luminescence by the co-crystals of 2,5-di(E)-distrylyfuran and N-alkyl maleimides; the emitting layer mirrors theprotruding pattern of the coin (adapted with permission from ref. 83. Copyright © 2011, John Wiley and Sons). (b) Photographs of mixed crystals,each containing 1 wt% aldehyde chromophore and 99 wt% analogous host compound and the corresponding normalized photoluminescenceemission spectra (adapted by permission from Macmillan Publishers Ltd: ref. 86, copyright © 2011).
Journal of Materials Chemistry C Feature Article
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
relations of a series of related compounds could be translated tosystematizations and predictions with an added degree ofprobability or predictability. The restriction to a certain class ofcompounds or process is surmountable towards a more generalstrategy, with the accessibility to more examples and general-ization of factual foundations based on better understanding ofstructure–property relations.
(i). Flexible molecular structure
A molecular structure with a exible backbone that couldaccommodate torsional changes is an energetically feasibleroute to attain polymorphism. The energetics involved intorsional rotation being fairly low as compared to that of bondstretching, the molecules enjoy much more conformationalfreedom to seek multiple options of conformations, which arenot very far energetically from the equilibrium structure.11
This in turn results in a number of energetically closelyspaced local minima that are kinetically accessible confor-mational polymorphs. The Gibbs free energy differencebetween individual crystal forms oen being small with a lowactivation energy barrier, it is possible to achieve phasetransition between different forms in near ambient condi-tions.31,46 Different conformations adopted by the poly-morphic forms can cause differences in the extent ofconjugation in the molecules and hence their solid stateoptical behaviour.
(ii). Secondary bonding interactions
The non-bonded interactions that bind the molecules in thecrystal lattice are much weaker than the intramolecular bonds,but play a critical role in directing the nal moleculararrangement in the crystal lattice and hence polymorphism.88,89
The weaker nature of interactions makes the events of molec-ular recognition and aggregation a complex process whoseoutcome is determined by amultitude of enthalpic and entropicfactors. Weak interactions also introduce additional exibility
3510 | J. Mater. Chem. C, 2014, 2, 3499–3516
to the system that could promote energetic, steric and statisticalvulnerability to external environment/stimuli. Dicyanodistyryl-benzene polymorphs exhibit differently coloured uorescenceemission that originates from different modes of local dipolecoupling and the resultant alternation of p–p overlap andexcited state delocalization.45 In di(benzimidazol-2-yl)benzene,interconversion of an intermolecular to intramolecularhydrogen bonded isomer brings about a large change in theelectron charge density distribution of low lying singlet states.Subsequently, its solid state uorescence shows a shi fromblue to green colour (Dlem ¼ 120 nm).90 Though weak in nature,changes in patterns of C–H/p interactions are known toinuence molecular conformations, induced dipole, electronicdelocalization and intra- and intermolecular electron transfertransitions.26
Further to emission colour and intensity, weak interactionsand resulting packing rigidity are known tomodulate solid-stateluminescence quantum efficiency of materials as well. Never-theless, the available reports provide contradicting effects ofsecondary interactions on quantum yields. There are reports onthe effect of weak interactions in enhancing solid state lumi-nescence quantum efficiency of molecular materials.91–94 Forexample, in secondary amines of ammonium anthracene-2,6-disulfonate, C–H/O hydrogen bonds and packing rigidityreportedly enhanced the emission quantum efficiency from lessthan 1% to 23%.91 Similarly, in anthracene carboxamide deriv-ative, N-(3-(benzo[d]thiazol-2-yl)-4-hydroxyphenyl)anthracene-9-carboxamide, a specic N/p interaction acts as the maindriving force for the J-aggregation with exceptional quantumefficiency of 78%.93 In contrast, there are also many experi-mental studies that demonstrate the role of intermolecularinteractions in reducing the emission quantum yield.95–98 Thisdichotomy is yet to be addressed and in the absence ofsubstantial studies pertaining to this specic topic it is, ingeneral, difficult to ascertain the possible inuence of a set ofinteractions, be it strong or weak, on the solid state emissionquantum efficiency.
This journal is © The Royal Society of Chemistry 2014

Feature Article Journal of Materials Chemistry C
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
(iii). Introducing active slip planes in the crystal
Crystal-to-amorphous transition in molecular crystals could beenvisioned as a plastic deformation, generally, attained throughirreversible glide (or slip), twinning, and kinking motions ofmolecular layers, under anisotropic stress. Such amechanism isactive in specic crystallographic planes—slip planes.99 Thenature and strength of non-covalent interactions have a majorrole in directing molecular slip. While the slip in layeredcompounds takes place between networks of hydrogen bonds,that in compounds with an intricate network of interactionscould be achieved through breaking the weakest of theseinteractions. Short-range non-directional interactions (such asvan der Waals interactions), which have a large bearing on howbonds break, could inuence the plastic deformation inmolecular crystals, whereas long-range directional interactions(such as hydrogen bonds) inuence elastic deformationbecause of their restorative character.100 Shear stress promotedlong range migration in molecular crystals could alter theintermolecular interactions existing in the system and hencethe emission characteristics.101 On the contrary, the mechano-chromism can become annulled by blocking the slip planesusing bulky substituents.4
(iv). Loose molecular packing and crystal defects
Molecules with labyrinthine structure usually adopt relativelyloose molecular packing.59,102 Such ineffective packing togetherwith presence of defects and dislocations in the crystal latticecould promote plastic deformation under shear stress. Gener-ally, higher mobility of dislocations could reduce the activationenergy barrier for the phase transformation, thus making theplastic deformation more facile. Chi and co-workers proposed ageneral molecular design to obtain a piezouorochromicphenomenon in molecular compounds.103 The proposed designincorporates multiple phenyl peripheries that are linked to anolenic core by rotatable carbon–carbon single bonds to inducetwisted conformation and weak non-bonded interactions thatlead to a relatively loose molecular packing and hence suscep-tible to external shear stress.24
(v). Adjusting electron density in the central core and thelength of peripheral aliphatic tails
Quadrupolar interaction between electron-rich and electron-decient aromatic rings is an important class of aromatic–aromatic interactions. One of the well-studied stacking inter-actions is that between aryl and peruoraryl units with acalculated binding energy that ranges from 3.7 to 4.7 kcalmol�1.104 In their report on p-stacking directed self-assembly,Gdaniec et al. highlighted the need of size and shape compat-ibility between supramolecular substrates to achieve effectivearomatic–aromatic stacking.105 Prevailing quadrupolar interac-tions and extent of stacking in planarized p-conjugatedmolecular systems play a key role in determining their solidstate emission properties. Crystal structures of cyano-substituted oligo(p-phenylene vinylene) derivatives (cyano-OPVs) show pronounced p–p interactions, with electron-poor
This journal is © The Royal Society of Chemistry 2014
cyanovinylene moiety of onemolecule stacked over the electron-rich central ring of its neighbour. With a set of cyano-OPVs asexamples, Weder and coworkers proposed that piezochromicuorescent characteristics can be obtained by the variation ofelectron density in the central core and the length of peripheralaliphatic tails, and thereby balancing p–p and aliphatic inter-actions. Accordingly, compounds capable of making differentpolymorphic forms can exhibit piezochromic behaviour if oneof the polymorphs allows the formation of excimers.106 Aliphaticside chains with different length and their substituted positionsin molecular cores could further inuence the molecularpacking and emission properties.5,107
(vi). Adopting a molecular design strategy that incorporatestwo distinct and competing modes of molecular packing
Possible phase transition between crystalline forms where themolecular arrangement is governed by distinct packing princi-ples is an impressive route to achieve mechanochromism. Arakiet al. proposed a design strategy that incorporated two competingfactors that determine the mode of molecular packing; Kitai-gorodsky's close packing principle108 and Etter's rst hydrogenbond rule.109 Generally, the disk-shaped aromatics tend to close-pack while hydrogen bond-directed columnar assembliesdemand a wider interplanar distance. In 1,3,6,8-tetraphenylpyr-ene (TPPy) with four hexyl amide units, the dominant multiplehydrogen bonds direct a H-type columnar stacking mode.110 Thecolumnar packing gets disturbed with the application of a lowpressure of 2.0 MPa, with a subsequent change in emission fromblue to blue-green. Nevertheless, the enthalpically favouredhydrogen bond-directed packing gets restored by thermalannealing of the solid with concomitant reversal of emissioncolour change. With higher applied pressure (9.8 MPa) thehydrogen bond-directed structure undergoes extensive disrup-tion, resulting in an added red-shi of the luminescence to green;the resultant molecular packing is governed by factors related toclose packing (Fig. 15a) and the recovery of the initial hydrogenbond-directed structure is possible only by solution-reprecipita-tion. In the ester derivative, in the absence of any classichydrogen bonds, the packing is directed by close-packing prin-ciples and undergo structural variations only under high pres-sure conditions.111 Similarly, the polymorphic behaviour oftriphenylgermane, triphenylsilane and triphenylstannane wereinferred to arise from the conicting driving forces of long rangedipolar interactions and the stronger, but shorter-range six-foldphenyl embrace. Though the long-range dipolar interactionsbring about rapid growth of the kinetic form, the systems adopt afacile mechanism to undergo gradual transformation to themorestable and symmetrical six-fold phenyl embrace.112
(vii). D–p–A chromophore skeleton with greater dipolemoments
Molecular stacking and p–p interactions between adjacentmolecular planes generally have a centrosymmetry to attainanti-parallel orientation of dipole moments. Harima et al.proposed that under shear stress, molecules move closer tominimize the dipole–dipole interaction energy (so as to arrange
J. Mater. Chem. C, 2014, 2, 3499–3516 | 3511

Fig. 15 (a) Schematic illustration of the structural variations in TPPy at low and high pressure conditions (adapted with permission from ref. 111;Royal Society of Chemistry). (b) Proposed mechanisms of mechanofluorochromism observed with heteropolycyclic D–p–A fluorescent dyes(adapted with permission from ref. 113; Royal Society of Chemistry).
Journal of Materials Chemistry C Feature Article
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
adjacent dipole moments in head-to-tail orientation) as well asthe intermolecular p–p interaction energy, resulting inincreased densities in amorphous state. Molecules with greaterdipole moments exhibit enhanced dipole–dipole interactionenergy (Fig. 15b). Furthermore, the steric sizes of the substitu-ents can greatly inuence the degree of change in intermolec-ular p–p interactions during its phase transformation; bulkysubstituents lead to weak charge transfer and p–p interactionsand hence weaker mechanouorescence.113 In addition to theaforementioned general strategies, the literature providesvarious designs adopted to enhance the solid state emissionefficiency of molecular materials; asymmetric substitution andits effect on packing mode other than J-type aggregation,43
selecting substituents to invite cross-stacking,114 planaritybreaking of N,N-diorganoquinacridones,115 to mention a few.
Proper selection of the mode of treatment to bring aboutphase transformation could also lead to distinct emissioncharacteristics. Unlike thermal or vapour stimuli inuencedtransformation, the outcome of mechanical treatment dependsnot only on total mechanical energy input but also on the type oftreatment. For example, resultant physical and chemicalprocesses of a continuous hydrostatic compression, generally,could be different from that obtained by compressioncombined with shear. Structural transformation brought aboutby shear stress involves plastic deformation such as slipping ofmolecular layers, resulting in alternation of the packing stateand interaction types. Such shearing of molecular layers usuallyleads to a blue-shi in emission wavelength. In contrast, underhydrostatic pressure, the layers are compressed wherein theexisting non-covalent interactions become more effective andhence a red-shi in emission wavelength (Fig. 16).116,117
Future outlook
Designing functional materials for futuristic applications needscritical information on structure–property relations. Combiningthe possibilities of different characterizing techniques canprovide such vital insights on various factors that direct theoptoelectronic properties of molecular materials in the solidstate. One of the recent developments in this direction is theutilization of force microscopy or nanoindentation techniquesin tandem with X-ray diffraction and spectroscopic data to
3512 | J. Mater. Chem. C, 2014, 2, 3499–3516
establish the structure–mechano–optical relations in molecularcrystals. This multi-technique strategy is a step forward toquantify weak intermolecular interactions and crystal packingin molecular crystals for understanding their role in varioussolid state properties.118,119
Diuoroboron avobenzone (BF2AVB) exhibits interestingreversible mechanoluminescence; the three polymorphs exhibitdistinct solid state emission colours of green (Form-I; lmax
em ¼505 nm), cyan (Form-II; lmax
em ¼ 470 nm) and blue (Form-III;lmaxem ¼ 459 nm). Under slight mechanical stress the Form-IIswitches the emission to yellow (lmax
em ¼ 542 nm), which even-tually fades and transform to green.21 Reddy et al. investigatedmechanical properties of the green (Form-I) and cyan (Form-II)crystals, using nanoindentation, and correlated the trans-formation in the context of crystal packing, intermolecularinteractions and presence of slip planes. While, anisotropiccrystal packing with the orthogonal orientation of stronger(multiple C–H/F) and weaker interactions (–tert-But, –OMe)make Form-II crystals more compliant, as compared to Form-I,weak interaction (slip) planes with nonspecic van der Waalsinteractions induce reversibility of the recoverable defects andhence its mechanoluminescent behaviour (Fig. 17).120
Force microscopy in conjunction with diffraction data is aneffective tool to correlate the structural changes andmechanicalresponse of millimeter and nanometer-sized crystals thatundergo chemical transformation,121 to follow phase changes atcrystal surfaces in real time and also to understand the mech-anism by which molecules move during transformation.122
Because the interest in polymorphism is here to stay and is topermeate through a wide spectrum of academic researchelds, multi-technique and multi-disciplinary approachesshould become indispensable and ubiquitous in the nearfuture. Such a development will be benecial to derive mecha-nistic insights, to enhance conceptual understanding and alsoto appreciate complex structure–function relationships thathitherto remained elusive.
Summary
The foregoing sections have summarized some of the non-covalent synthetic approaches that are being adopted to expandthe utility of molecules of interest by obtaining different solid
This journal is © The Royal Society of Chemistry 2014
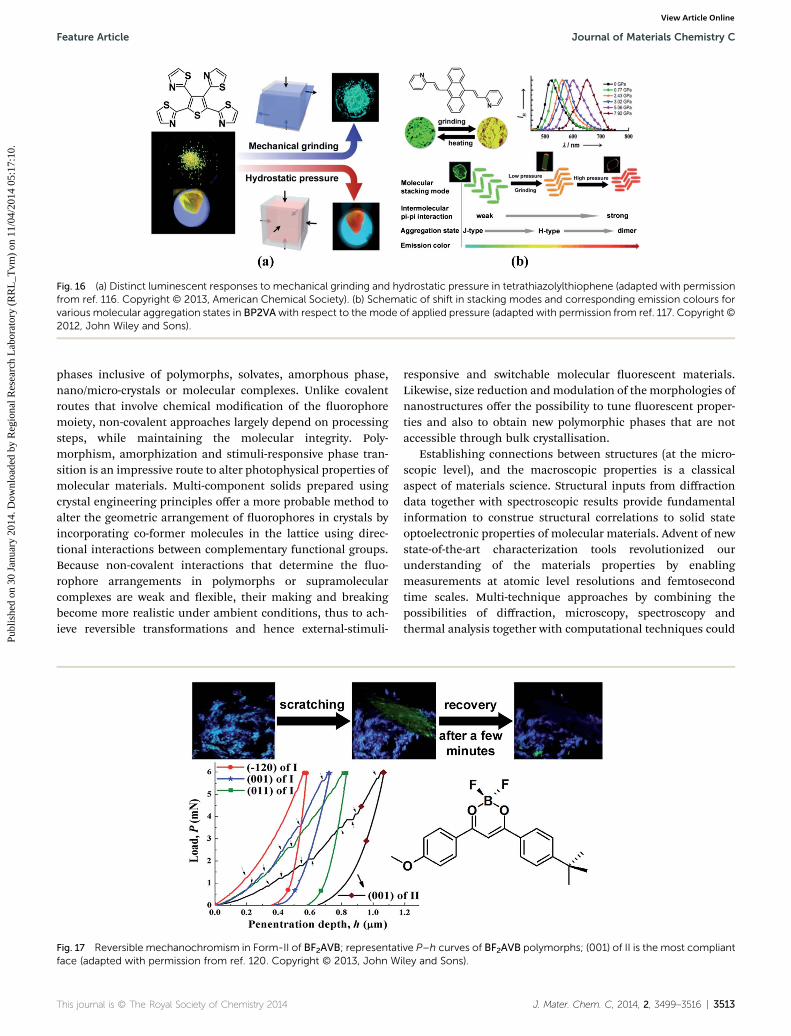
Fig. 16 (a) Distinct luminescent responses to mechanical grinding and hydrostatic pressure in tetrathiazolylthiophene (adapted with permissionfrom ref. 116. Copyright © 2013, American Chemical Society). (b) Schematic of shift in stacking modes and corresponding emission colours forvarious molecular aggregation states in BP2VAwith respect to the mode of applied pressure (adapted with permission from ref. 117. Copyright ©2012, John Wiley and Sons).
Feature Article Journal of Materials Chemistry C
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
phases inclusive of polymorphs, solvates, amorphous phase,nano/micro-crystals or molecular complexes. Unlike covalentroutes that involve chemical modication of the uorophoremoiety, non-covalent approaches largely depend on processingsteps, while maintaining the molecular integrity. Poly-morphism, amorphization and stimuli-responsive phase tran-sition is an impressive route to alter photophysical properties ofmolecular materials. Multi-component solids prepared usingcrystal engineering principles offer a more probable method toalter the geometric arrangement of uorophores in crystals byincorporating co-former molecules in the lattice using direc-tional interactions between complementary functional groups.Because non-covalent interactions that determine the uo-rophore arrangements in polymorphs or supramolecularcomplexes are weak and exible, their making and breakingbecome more realistic under ambient conditions, thus to ach-ieve reversible transformations and hence external-stimuli-
Fig. 17 Reversible mechanochromism in Form-II of BF2AVB; representatface (adapted with permission from ref. 120. Copyright © 2013, John W
This journal is © The Royal Society of Chemistry 2014
responsive and switchable molecular uorescent materials.Likewise, size reduction andmodulation of the morphologies ofnanostructures offer the possibility to tune uorescent proper-ties and also to obtain new polymorphic phases that are notaccessible through bulk crystallisation.
Establishing connections between structures (at the micro-scopic level), and the macroscopic properties is a classicalaspect of materials science. Structural inputs from diffractiondata together with spectroscopic results provide fundamentalinformation to construe structural correlations to solid stateoptoelectronic properties of molecular materials. Advent of newstate-of-the-art characterization tools revolutionized ourunderstanding of the materials properties by enablingmeasurements at atomic level resolutions and femtosecondtime scales. Multi-technique approaches by combining thepossibilities of diffraction, microscopy, spectroscopy andthermal analysis together with computational techniques could
ive P–h curves of BF2AVB polymorphs; (001) of II is the most compliantiley and Sons).
J. Mater. Chem. C, 2014, 2, 3499–3516 | 3513
Pauling
Typewriter
Pauling
Typewriter

Journal of Materials Chemistry C Feature Article
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
provide crucial complementary information that hithertoremained elusive and making design and synthesis of molec-ular materials with greater predictability, a feasible target. Suchstudies can open up new scientic vistas in materials researchand in developing stimuli-responsive luminescent materials.
Acknowledgements
The author thanks DST, New Delhi for a Start-up Research Grant(CS-210/2013) and Dr Suresh Das, The Director, CSIR-NIIST forsupport and encouragement.
References
1 S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863.2 D. Yan and D. G. Evans, Mater. Horiz., 2014, 1, 46.3 C. Wang, D. Chen, W. Chen, S. Chen, K. Ye, H. Zhang,J. Zhang and Y. Wang, J. Mater. Chem. C, 2013, 1, 5548.
4 X. Y. Shen, Y. J. Wang, E. Zhao, W. Z. Yuan, Y. Liu, P. Lu,A. Qin, Y. Ma, J. Z. Sun and B. Z. Tang, J. Phys. Chem. C,2013, 117, 7334.
5 R. Davis, N. S. S. Kumar, S. Abraham, C. H. Suresh,N. P. Rath, N. Tamaoki and S. Das, J. Phys. Chem. C, 2008,112, 2137.
6 M. D. Curtis, J. Cao and J. W. Kampf, J. Am. Chem. Soc., 2004,126, 4318.
7 M. Kasha, Radiat. Res., 1963, 20, 55.8 M. Shimizu and T. Hiyama, Chem.–Asian J., 2010, 5, 1516.9 W. C. McCrone, in Physics and Chemistry of the Organic SolidState, ed. D. Fox, M. M. Labes and A. Weissberger, WileyInterscience, New York, USA, 1965, pp. 725–767.
10 A. Gavezzotti, Acc. Chem. Res., 1994, 27, 309.11 J. Bernstein, in Polymorphism in Molecular Crystals, Oxford
Univ. Press, New York, 2002.12 Polymorphism in Pharmaceutical Solids, ed. H. G. Brittain,
Marcel Dekker, Inc., New York, 1999.13 J. Bernstein, R. J. Davey and J.-O. Henck, Angew. Chem., Int.
Ed., 1999, 38, 3440.14 L. Yu, Acc. Chem. Res., 2010, 43, 1257 and references cited
therein.15 H. Bouas-Laurent and H. Durr, Pure Appl. Chem., 2001, 73,
639.16 B. Kahr and M. McBride, Angew. Chem., Int. Ed. Engl., 1992,
31, 1.17 J.-S. Filhol, J. Descamps, S. G. Dutremez, B. Boury,
T. Barisien, L. Legrand and M. Schott, J. Am. Chem. Soc.,2009, 131, 6976.
18 P. H. Toma, M. P. Kelley, T. B. Borchardt, S. R. Byrn andB. Kahr, Chem. Mater., 1994, 6, 1317.
19 W. M. Dehn and L. McBride, J. Am. Chem. Soc., 1917, 39,1348.
20 T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685.21 G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc.,
2010, 132, 2160.22 H. Y. Zhang, Z. L. Zhang, K. Q. Ye, J. Y. Zhang and Y. Wang,
Adv. Mater., 2006, 18, 2369.
3514 | J. Mater. Chem. C, 2014, 2, 3499–3516
23 Z. Zhang, Y. Zhang, D. Yao, H. Bi, I. Javed, Y. Fan, H. Zhangand Y. Wang, Cryst. Growth Des., 2009, 9, 5069.
24 I. Hisaki, E. Kometani, H. Shigemitsu, A. Saeki, S. Seki,N. Tohnai andM.Miyata, Cryst. Growth Des., 2011, 11, 5488.
25 C. Kitamura, T. Ohara, N. Kawatsuki, A. Yoneda,T. Kobayashi, H. Naito, T. Komatsu and T. Kitamura,CrystEngComm, 2007, 9, 644.
26 Y.-H. Gao, W. Zhou, L. Li, J.-Y. Wu, H.-P. Zhou, J.-X. Yangand Y.-P. Tian, Cryst. Growth Des., 2009, 9, 253.
27 C.-C. Hsieh, C.-M. Jiang and P.-T. Chou, Acc. Chem. Res.,2010, 43, 1364.
28 T. Mutai, H. Tomoda, T. Ohkawa, Y. Yabe and K. Araki,Angew. Chem., Int. Ed., 2008, 47, 9522.
29 S. P. Anthony, Chem.–Asian J., 2012, 7, 374.30 D. R. T. Roberts and S. J. Holder, J. Mater. Chem., 2011, 21,
8256 and references therein.31 Y. Zhao, H. Gao, Y. Fan, T. Zhou, Z. Su, Y. Liu and Y. Wang,
Adv. Mater., 2009, 21, 3165.32 Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605.33 Y. Abe, S. Karasawa and N. Koga, Chem.–Eur. J., 2012, 18,
15038.34 C.-H. Zhao, A. Wakamiya, Y. Inukai and S. Yamaguchi, J.
Am. Chem. Soc., 2006, 128, 15934.35 E. Takahashi, H. Takaya and T. Naota, Chem.–Eur. J., 2010,
16, 4793.36 S. Toscani, Thermochim. Acta, 1998, 321, 73.37 L. Yu, J. Pharm. Sci., 1995, 84, 966.38 R. Davis, N. P. Rath and S. Das, Chem. Commun., 2004, 74.39 N. S. S. Kumar, S. Varghese, N. P. Rath and S. Das, J. Phys.
Chem. C, 2008, 112, 8429.40 N. Harada, Y. Abe, S. Karasawa and N. Koga,Org. Lett., 2012,
14, 6282.41 X. Gu, J. Yao, G. Zhang, Y. Yan, C. Zhang, Q. Peng, Q. Liao,
Y. Wu, Z. Xu, Y. Zhao, H. Fu and D. Zhang, Adv. Funct.Mater., 2012, 22, 4862.
42 R. Wei, P. Song and A. Tong, J. Phys. Chem. C, 2013, 117,3467.
43 J. W. Chung, Y. You, H. S. Huh, B.-K. An, S.-J. Yoon,S. H. Kim, S. W. Lee and S. Y. Park, J. Am. Chem. Soc.,2009, 131, 8163.
44 S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi,D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675.
45 S.-J. Yoon and S. Y. Park, J. Mater. Chem., 2011, 21, 8338.46 X. Luo, J. Li, C. Li, L. Heng, Y. Q. Dong, Z. Liu, Z. Bo and
B. Z. Tang, Adv. Mater., 2011, 23, 3261.47 C. Dou, D. Chen, J. Iqbal, Y. Yuan, H. Zhang and Y. Wang,
Langmuir, 2011, 27, 6323.48 E. L. Parrott, in Encyclopedia of Pharmaceutical Technology,
ed. J. Swarbrick and J. C. Boylan, Marcel Dekker, NewYork, 1990, pp. 101–121.
49 E. Boldyreva, Chem. Soc. Rev., 2013, 42, 7719 and referencescited therein.
50 K. J. Crowley and G. Zogra, J. Pharm. Sci., 2002, 91, 492.51 A. R. Sheth, J. W. Lubach, E. J. Munson, F. X. Muller and
D. J. W. Grant, J. Am. Chem. Soc., 2005, 127, 6641.52 G. Zhang, J. P. Singer, S. E. Kooi, R. E. Evans, E. L. Thomas
and C. L. Fraser, J. Mater. Chem., 2011, 21, 8295.
This journal is © The Royal Society of Chemistry 2014

Feature Article Journal of Materials Chemistry C
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
53 N. Zhao, M. Li, Y. Yan, J. W. Y. Lam, Y. L. Zhang, Y. S. Zhao,K. S. Wong and B. Z. Tang, J. Mater. Chem. C, 2013, 1, 4640.
54 N. Zhao, Z. Yang, J. W. Y. Lam, H. H. Y. Sung, N. Xie,S. Chen, H. Su, M. Gao, I. D. Williams, K. S. Wong andB. Z. Tang, Chem. Commun., 2012, 48, 8637.
55 Y. Gong, Y. Tan, J. Liu, P. Lu, C. Feng, W. Z. Yuan, Y. Lu,J. Z. Sun, G. He and Y. Zhang, Chem. Commun., 2013, 49,4009.
56 X. Luo, W. Zhao, J. Shi, C. Li, Z. Liu, Z. Bo, Y. Q. Dong andB. Z. Tang, J. Phys. Chem. C, 2012, 116, 21967.
57 H. Tong, Y. Dong, M. Haußler, J. W. Y. Lam, H. H. Y. Sung,I. D. Williams, J. Sun and B. Z. Tang, Chem. Commun., 2006,1133.
58 H. Tong, Y. Hong, Y. Dong, Y. Ren, M. Haussler,J. W. Y. Lam, K. S. Wong and B. Z. Tang, J. Phys. Chem. B,2007, 111, 2000.
59 J. N. Moorthy, P. Venkatakrishnan, P. Natarajan,D.-F. Huang and T. J. Chow, J. Am. Chem. Soc., 2008, 130,17320.
60 N. S. S. Kumar, S. Varghese, C. H. Suresh, N. P. Rath andS. Das, J. Phys. Chem. C, 2009, 113, 11927.
61 T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker andJ. Salbeck, Chem. Rev., 2007, 107, 1011.
62 A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe,Chem. Mater., 2004, 16, 4556.
63 Y. S. Zhao, H. Fu, A. Peng, Y. Ma, D. Xiao and J. Yao, Adv.Mater., 2008, 20, 2859.
64 H. Kasai, Y. Yoshikawa, T. Seko, S. Okada, H. Oikawa,H. Mastuda, A. Watanabe, O. Ito, H. Toyotama andH. Nakanishi, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A,1997, 294, 173.
65 H.-B. Fu and J.-N. Yao, J. Am. Chem. Soc., 2001, 123, 1434.66 B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem.
Soc., 2002, 124, 14410.67 Y. S. Zhao, D. Xiao, W. Yang, A. Peng and J. Yao, Chem.
Mater., 2006, 18, 2302.68 G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952.69 A. Patra, N. Hebalkar, B. Sreedhar, M. Sarkar, A. Samanta
and T. P. Radhakrishnan, Small, 2006, 2, 650.70 L. Huang, Q. Liao, Q. Shi, H. Fu, J. Ma and J. Yao, J. Mater.
Chem., 2010, 20, 159.71 S. P. Anthony and S. M. Draper, J. Phys. Chem. C, 2010, 114,
11708.72 S. J. Lee, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc.,
2008, 130, 9632.73 M. Abyan, D. de Caro and S. Fery-Forgues, Langmuir, 2009,
25, 1651.74 S. P. Anthony, ChemPlusChem, 2012, 77, 518.75 Pharmaceutical Salts and Co-crystals, ed. J. Wouters and L.
Quere, RSC Publishing, Cambridge, UK, 2012.76 L. R. MacGillivray, G. S. Papaefstathiou, T. Friscic,
T. D. Hamilton, D. K. Bucar, Q. Chu, D. B. Varshney andI. G. Georgiev, Acc. Chem. Res., 2008, 41, 280 andreferences cited therein.
77 J. D. Wuest, Nat. Chem., 2012, 4, 74.78 D. Yan, B. Patel, A. Delori, W. Jones and X. Duan, Cryst.
Growth Des., 2013, 13, 333.
This journal is © The Royal Society of Chemistry 2014
79 D. Yan, A. Delori, G. O. Lloyd, T. Friscic, G. M. Day,W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duan, Angew.Chem., Int. Ed., 2011, 50, 12483.
80 S. P. Anthony, S. Varughese and S. M. Draper, Chem.Commun., 2009, 7500.
81 S. P. Anthony, C. Delaney, S. Varughese, L. Wang andS. M. Draper, CrystEngComm, 2011, 13, 6706.
82 B. Dong, M. Wang, C. Xu, Q. Feng and Y. Wang, Cryst.Growth Des., 2012, 12, 5986.
83 J. Luo, L. Y. Li, Y. Song and J. Pei, Chem.–Eur. J., 2011, 17,10515.
84 S. K. Park, S. Varghese, J. H. Kim, S.-J. Yoon, O. K. Kwon,B.-K. An, J. Gierschner and S. Y. Park, J. Am. Chem. Soc.,2013, 135, 4757.
85 D. Yan, D.-K. Bucar, A. Delori, B. Patel, G. O. Lloyd, W. Jonesand X. Duan, Chem.–Eur. J., 2013, 19, 8213.
86 O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat.Chem., 2011, 3, 205.
87 N. Nishiguchi, T. Kinuta, Y. Nakano, T. Harada, N. Tajima,T. Sato, M. Fujiki, R. Kuroda, Y. Matsubara and Y. Imai,Chem.–Asian J., 2011, 6, 1092.
88 Z. Xie, B. Yang, G. Cheng, L. Liu, F. He, F. Shen, Y. Ma andS. Liu, Chem. Mater., 2005, 17, 1287.
89 Y. Liu, X. Tao, F. Wang, J. Shi, J. Sun, W. Yu, Y. Ren, D. Zouand M. Jiang, J. Phys. Chem. C, 2007, 111, 6544.
90 B. Jena and S. Manoharan, Chem. Commun., 2009, 4426.91 T. Hinoue, Y. Mizobe, I. Hisaki, M. Miyata and N. Tohnai,
Chem. Lett., 2008, 37, 642.92 H. Jung, K. Kim, S. Lee, H. Hwang, K. Park, J. Kim,
S. –K. Kwon and Y. Kang, Bull. Korean Chem. Soc., 2007,28, 1807.
93 M. Cai, Z. Gao, X. Zhou, X. Wang, S. Chen, Y. Zhao, Y. Qian,N. Shi, B. Mi, L. Xie andW. Huang, Phys. Chem. Chem. Phys.,2012, 14, 5289.
94 I. D. W. Samuel, G. Rumbles and C. J. Collison, Phys. Rev. B:Condens. Matter Mater. Phys., 1995, 52, R11573.
95 J. Cornil, D. Beljonne, J.-P. Calbert and J.-L. Bredas, Adv.Mater., 2001, 13, 1053.
96 S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765.97 A. Yassar, G. Horowitz, P. Valat, V. Wintgens, M. Hmyene,
F. Deloffre, P. Srivastava, P. Lang and F. Garnier, J. Phys.Chem., 1995, 268, 270.
98 G. C. Bazan, Y. J. Miao, M. L. Renak and B. J. Sun, J. Am.Chem. Soc., 1996, 118, 2618.
99 C. C. Sun and Y.-H. Kiang, J. Pharm. Sci., 2008, 97, 3456.100 S. Varughese, M. S. R. N. Kiran, U. Ramamurty and
G. R. Desiraju, Angew. Chem., Int. Ed., 2013, 52, 2701.101 H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka,
Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge andM. Sawamura, J. Am. Chem. Soc., 2008, 130, 10044.
102 H. Tong, Y. Dong, Y. Hong, M. Haussler, J. W. Y. Lam,H. H. Y. Sung, X. Yu, J. Sun, I. D. Williams,H. S. Kwok and B. Z. Tang, J. Phys. Chem. C, 2007, 111,2287.
103 Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu andJ. Xu, Chem. Soc. Rev., 2012, 41, 3878 and references citedtherein.
J. Mater. Chem. C, 2014, 2, 3499–3516 | 3515

Journal of Materials Chemistry C Feature Article
Publ
ishe
d on
30
Janu
ary
2014
. Dow
nloa
ded
by R
egio
nal R
esea
rch
Lab
orat
ory
(RR
L_T
vm)
on 1
1/04
/201
4 05
:17:
10.
View Article Online
104 F. Ponzini, R. Zagha, K. Hardcastle and J. S. Siegel, Angew.Chem., Int. Ed., 2000, 39, 2323.
105 M. Gdaniec, W. Jankowski, M. J. Milewska and T. Polonski,Angew. Chem., Int. Ed., 2003, 42, 3903.
106 J. Kunzelman, M. Kinami, B. R. Crenshaw,J. D. Protasiewicz and C. Weder, Adv. Mater., 2008, 20, 119.
107 J. Dong, K. M. Solntsev and L. M. Tolbert, J. Am. Chem. Soc.,2009, 131, 662.
108 A. I. Kitaigorodsky, in Molecular Crystals and Molecules,Academic Press, INC., New York, 1973.
109 M. C. Etter, Acc. Chem. Res., 1990, 23, 120.110 Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem.
Soc., 2007, 129, 1520.111 M. Sase, S. Yamaguchi, Y. Sagara, I. Yoshikawa, T. Mutai
and K. Araki, J. Mater. Chem., 2011, 21, 8347.112 G. S. McGrady, M. Odlyha, P. D. Prince and J. W. Steed,
CrystEngComm, 2002, 4, 271.113 Y. Ooyama and Y. Harima, J. Mater. Chem., 2011, 21, 8372.114 Z. Xie, H. Wang, F. Li, W. Xie, L. Liu, B. Yang, L. Ye and
Y. Ma, Cryst. Growth Des., 2007, 7, 2512.
3516 | J. Mater. Chem. C, 2014, 2, 3499–3516
115 M. Shimizu, Y. Asai, Y. Takeda, A. Yamatani and T. Hiyama,Tetrahedron Lett., 2011, 52, 4084.
116 K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa,H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem.Soc., 2013, 135, 10322.
117 Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv,S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem.,Int. Ed., 2012, 51, 10782.
118 M. K. Mishra, S. Varughese, U. Ramamurty andG. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 8121.
119 S. Varughese, M. S. R. N. Kiran, K. A. Solanko, A. D. Bond,U. Ramamurty and G. R. Desiraju, Chem. Sci., 2011, 2, 2236.
120 G. R. Krishna, M. S. R. N. Kiran, C. L. Fraser, U. Ramamurtyand C. M. Reddy, Adv. Funct. Mater., 2013, 23, 1422.
121 C. Karunatilaka, D.-K. Bucar, L. R. Ditzler, T. Friscic,D. C. Swenson, L. R. MacGillivray and A. V. Tivanski,Angew. Chem., Int. Ed., 2011, 50, 8642.
122 R. Thakuria, M. D. Eddleston, E. H. H. Chow, G. O. Lloyd,B. J. Aldous, J. F. Krzyzaniak, A. D. Bond and W. Jones,Angew. Chem., Int. Ed., 2013, 52, 10541.
This journal is © The Royal Society of Chemistry 2014




















