
A pharmaco-EEG study on antipsychotic drugs in healthy volunteers
Upload
independentCategory
view
1download
0
Cognitive deficits in schizophrenia are associ-
ated with prefrontal cortex (PFC) abnormali-
ties. Schizophrenic patients show a reduced
performance in tasks engaging the PFC and a
reduction of markers of cellular integrity and
function. Non-competitive N-methyl-D-
aspartate (NMDA) receptor antagonists are
widely used as pharmacological models of
schizophrenia due to their ability to exacerbate
schizophrenia symptoms in patients and to
elicit psychotomimetic actions in healthy volun-
teers. Also, these drugs evoke behavioral alter-
ations in experimental animals that resemble
schizophrenia symptoms. The PFC seems to be
a key target area for these agents. However, the
cellular and network elements involved are
poorly known. Cognitive deficits are of particu-
lar interest since an early antipsychotic-induced
improvement in cognitive performance predicts
a better long-term clinical outcome.
Here we report that the non-competitive
NMDA receptor antagonist phencyclidine (PCP)
induces a marked disruption of the activity of
PFC. PCP administration increased the activity
of a substantial proportion of pyramidal neu-
rons, as evidenced by an increase in discharge
rate and in c-fos expression. Examination of the
effects of PCP on other brain areas revealed an
increased c-fos expression in a number of corti-
cal and subcortical areas, but notably in thal-
amic nuclei projecting to the PFC. The adminis-
tration of classical (haloperidol) and/or atypical
(clozapine) antipsychotic drugs reversed PCP
effects. These results indicate that PCP induces a
marked disruption of the network activity in
PFC and that antipsychotic drugs may partly
exert their therapeutic effect by normalizing
hyperactive cortico-thalamocortical circuits.
Keywords: Antipsychotics; GABAergic interneurons; Glutamate; NMDA receptors; Prefrontal cortex; Pyramidal neurons; Oscillations; Thalamus
INTRODUCTION
Schizophrenia is associated with alterations in the anatomy and function of several cortical and sub-cortical areas (Harrison, 1999; Lewis and Lieberman, 2000). Among these, the prefrontal cortex (PFC) seems to play a key role in the pathophysiology of the illness (Lewis et al., 2005). Despite the obvious
F.P. Graham Publishing Co.
NMDA Antagonist and Antipsychotic Actions in Cortico-subcortical Circuits
LUCILA KARGIEMANa, NOEMÍ SANTANAa, GUADALUPE MENGODb, PAU CELADAa,*
and FRANCESC ARTIGASa,*
Department of Neurochemistry and Neuropharmacology, Institut d' Investigacions Biomèdiques de Barcelona
(CSIC), IDIBAPS, aCentro de Investigación Biomédica en Red de Salud Mental (CIBERSAM);
bCentro de
Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED), 08036 Barcelona, Spain.
[email protected], [email protected].
(Submitted 22 February 2008; Revised 4 August 2008; In final form 4 August 2008)
*Corresponding author: Tel.: +3493-363 8315; FAX: +3493-363 8301; E-mail: [email protected], [email protected]
ISSN 1029 8428 print/ ISSN 1476-3524 online. © 2008 FP Graham Publishing Co., www.NeurotoxicityResearch.com
Neurotoxicity Research, 2008, VOL. 14(2,3). pp. 129-140

L. KARGIEMAN et al.130
difficulty in modeling these alterations in experi-mental models of the illness, non-competitive N-methyl-D-aspartate (NMDA) receptor antago-nists such as the dissociative anaesthetics ket-amine and phencyclidine (PCP) and MK-801 (dizocilpine), have been extensively used as phar-macological models of schizophrenia due to their ability to evoke positive and negative symptoms of schizophrenia as well as the cognitive deficits of the illness in humans. These agents elicit a potent behavioral syndrome as well as cognitive and sensory deficits in experimental animals that resemble human schizophrenia symptoms (Javitt and Zukin, 1991; Krystal et al., 1994; Malhotra et
al., 1997; Newcomer et al., 1999). NMDA recep-tor antagonists also induce schizophrenia symp-toms in healthy subjects and aggravate them in schizophrenic patients (Krystal et al., 2003). Furthermore, the behavioral effects of NMDA receptor antagonists are sensitive to the treatment with antipsychotic drugs that alleviate psychotic symptoms in schizophrenic patients (Geyer et al.,2001; Krystal et al., 2003). Despite the widespread use of NMDA receptor antagonists as pharmacological models of schizo-phrenia, their neurobiological basis of action is still poorly known. Neuroimaging studies indicate that a sub-anesthetic dose of ketamine increases the activ-ity of the PFC in human volunteers (Breier et al.,1997). In experimental animals, NMDA receptor antagonists such as MK-801 or PCP increase neu-ronal activity (Suzuki et al., 2002; Jackson et al.,2004; Kargieman et al., 2007). The disrupted neuronal function in PFC is accom-panied by an increased release of various neu-rotransmitters in this area, such as glutamate (Moghaddam et al., 1997; Adams and Moghaddam, 2001; Lorrain et al., 2003; López-Gil et al., 2007), dopamine (Schmidt and Fadayel, 1996; Adams and Moghaddam, 1998; Mathé et al., 1999), serotonin (Martin et al., 1998; Millan et al., 1999; Adams and Moghaddam, 2001; Amargós-Bosch et al., 2006; López-Gil et al., 2007) and acetylcholine (Nelson et al., 2002). The disorganized increase in neuronal activity and neurotransmitter release in PFC may likely mediate the deleterious effects of NMDA receptor antagonists on cognitive processes (Krystal et al., 2003; Jackson et al., 2004). Indeed, the PFC is involved in a large number of higher brain func-
tions (Fuster, 1997; Miller and Cohen, 2001), which are altered in schizophrenic patients. In these psy-chiatric patients numerous studies have reported EEG abnormalities in different frequency oscilla-tions (Hajos et al., 2006). To provide a deeper insight of the brain areas and neuronal types affected by PCP, we examined the effect of its systemic administration on the activity of identified pyramidal neurons in the mPFC, on low frequency oscillation and on the expression of the immediate early gene c-fos in various cortical and subcortical areas. Finally, we also examined the ability of clozapine (CLZ) and haloperidol (HAL) to reverse PCP-induced changes. Some of the data reviewed have been published recently (Kargieman et al., 2007) or are to be published (Santana, Celada, Mengod, Artigas). Here we extend these observa-tions by providing a deeper insight on the cortico-subcortical circuits affected by PCP and antipsy-chotic drugs as well as changes in neuronal activity induced by PCP and antipsychotic drugs.
MATERIAL AND METHODS
Animals and Treatments
Adult male Wistar rats (250-300 g) were purchased from Iffa Credo (Lyon, France). Animals were acclimatized to standard laboratory conditions (12 hr light-dark cycle and 22 ± 2ºC room temperature) with food and water provided ad libitum. Animal procedures were performed according to the European Union regulations (O.J. of E.C. L358/1 18/12/1986) for the use of laboratory animals and were approved by the Institutional Animal Care and Use Committee. In histological experiments, rats were adminis-tered i.p. with the following treatments: saline + saline, saline + PCP (10 mg/kg), CLZ (5 mg/kg) + saline and CLZ (5 mg/kg) + PCP (10 mg/kg), respectively. Time between injections was 30 min and rats were killed by decapitation 1 h after the second injection. The rats were immediately decap-itated, the brains rapidly removed, frozen on dry ice and stored at -20ºC. Tissue sections, 14 m thick, were cut using a microtome-cryostat (Microm HM500 OM, Walldorf, Germany), thaw-mounted onto APTS (3-aminopropyltriethoxysilane, Sigma, St. Louis, MO, USA)-coated slides and kept at -20ºC until use.

NMDA ANTAGONIST & CORTICO-SUBCORTICAL CIRCUITS 131
Electrophysiology: Single Unit
and LFP Recordings
We examined the responses of mPFC pyramidal neurons to the systemic administration of PCP, alone or followed by CLZ and HAL. We also ana-lyzed the effects of these drugs on the local field potential (LFP) in mPFC. Rats were anesthetized (chloral hydrate, 400 mg/kg, i.p.) and positioned in a David Kopf stereotaxic frame. Thereafter, chloral hydrate was continuously administered i.p. at a dose of 50-70 mg/kg/h using a perfusion pump to maintain constant anesthesia. Body temperature was maintained at 37°C with a heating pad. Pyramidal neurons were recorded extracellularly with glass micropipettes pulled from 2 mm capil-lary glass (World Precision Instruments, Sarasota, FL) on a Narishige (Tokyo, Japan) PE-2 pipette puller. Microelectrodes were filled with 2 M NaCl. Typically, impedance was 4-10 M . Single-unit extracellular recordings were amplified with a Neurodata IR283 (Cygnus Technology, Delaware Water Gap, PA), postamplified and filtered with a Cibertec (Madrid, Spain) amplifier and computed on-line using a DAT 1401 plus interface system Spike2 software (Cambridge Electronic Design, Cambridge, UK). In some experiments, single units and LFPs were simultaneously recorded and fil-tered using a band pass filter (0.1-100 Hz for LFP and 30 Hz-1 kHz for action potentials). Descents in the mPFC were carried out at AP+3.2 to +3.4, L-0.5 to -1.0, DV -1.9 to -4.8 below brain surface. Stereotaxic coordinates were taken from bregma and duramater according to the atlas of Paxinos and Watson (1998). All recorded units were identified as pyramidal neurons by antidromic activation from dorsal raphe (DR) or ventral tegmental area (VTA) and collision extinction with spontaneously occur-ring spikes as described in Puig et al. (2003). Neurons without antidromic activation were dis-carded. After recording stable baseline activity for 5 min, PCP (0.25 mg/Kg in saline) was slowly (15 s) administered through the femoral vein. In rever-sal experiments, CLZ (1 mg/ Kg i.v.) or HAL (0.1 mg/kg i.v.) were injected 4-5 min after PCP admin-istration. Only one neuron per rat was recorded. At the end of experiments, rats were killed by an overdose of anesthetic. The brains were removed and frozen in dry ice before being sectioned (70
m) with a cryostat in the coronal planes. In some
cases, recording electrodes were filled with Pontamine sky blue to verify the recording site. Brain sections were then stained with neutral red, according to standard procedures.
In situ Hbridization Hstochemistry
Excitatory glutamatergic neurons were identified by hybridization with two oligonucleotides comple-mentary to bases 127-172 and 1756-1800 of the vesicular glutamate transporter 1 (vGluT1) mRNA (GenBank accession number U07609). GABAergic neurons were recognized by the presence of the enzyme synthesizing GABA, glutamate decarboxy-lase (GAD), with two oligonucleotides for each isoform of GAD mRNA (GAD65 and GAD67)complementary to bases 159-213 and 514-558 (GenBank accession number NM_012563) and to 191-235 and 1600-1653 (GenBank accession num-ber NM_017007), respectively. c-fos mRNA-con-taining cells were identified by hybridization with an oligonucleotide complementary to bases 131-178 of the rat mRNA (GenBank accession number NM_022197.1). All oligonucleotides were synthe-sized and HPLC purified by Isogen Bioscience BV (De Meern, The Netherlands).
c-fos oligonucleotide was labeled at its 3'-end with [33P]-dATP (>2500 Ci/mmol; DuPont-NEN, Boston, MA, USA) with terminal deoxynucleoti-dyltransferase (TdT, Calbiochem, La Jolla, CA, USA) and purified with ProbeQuantTM G-50 Micro Columns (GE Healthcare UK Limited, Buckinghamshire, UK). vGluT1 and GAD oligonucleotides were indi-vidually labeled with Dig-11-dUTP (Boehringer Mannheim) using TdT (Roche Diagnostics GmbH, Mannheim, Germany), and purified as above. The protocol for double-label in situ. hybridization was used as previously described (Serrats et al. 2003). Frozen tissue sections were first brought to room temperature, fixed for 20 min at 4°C in 4% para-formaldehyde in phosphate buffered saline (1× PBS: 8 mM Na2HPO4, 1.4 mM KH2PO4, 136 mM NaCl, 2.6 mM KCl), washed for 5 min in 3×PBS at room temperature, twice for 5 min each in 1× PBS and incubated for 2 min at 21°C in a solution of predigested pronase (Calbiochem, San Diego, CA) at a final concentration of 24 U/ml in 50 mM Tris-HCl pH 7.5, 5 mM EDTA. The enzymatic activity was stopped by immersion for 30 s in 2 mg/ml gly-

L. KARGIEMAN et al.132
cine in 1× PBS. Tissues were finally rinsed in 1× PBS and dehydrated through a graded series of ethanol. For hybridization, the radioactively-labeled and the non-radioactively labeled probes were diluted in a solution containing 50% formamide, 4× SSC (1× SSC: 150 mM NaCl, 15 mM sodium cit-rate), 1× Denhardt's solution (0.02% Ficoll, 0.02% polyvinylpyrrolidone, 0.02% bovine serum albu-min), 10% dextran sulfate, 1% sarkosyl, 20 mM phosphate buffer pH 7.0, 250 g/ml yeast tRNA and 500 g/ml salmon sperm DNA. The final con-centrations of radioactive and Dig-labeled probes in the hybridization buffer were in the same range ( 1.5 nM). Tissue sections were covered with hybrid-ization solution containing the labeled probe(s), overlaid with Nescofilm coverslips (Bando Chemical Ind., Kobe, Japan) and incubated over-night at 42°C in humid boxes. Sections were then washed four times (45 min each) in a buffer con-taining 0.6 M NaCl and 10 mM Tris-HCl (pH 7.5) at 60°C.
Development of Radioactive
and non-Radioactive Hybridization Signal
After washing, the hybridized slides were immersed for 30 min in a buffer containing 0.1 M Tris-HCl pH 7.5, 1 M NaCl, 2 mM MgCl2 and 0.5% bovine serum albumin (Sigma) and incubated overnight at 4°C in the same solution with alkaline-phosphate-conjugated anti-digoxigenin-F(ab) fragments (1:5000; Boehringer Mannheim). Afterwards, they were washed three times (10 min each) in the same buffer (without antibody) and twice in an alkaline buffer containing 0.1 M Tris-HCl pH 9.5, 0.1 M NaCl and 5 mM MgCl2. Alkaline phosphatase activity was developed by incubating the sections with 3.3 mg nitroblue tetrazolium and 3.3 mg bro-mochloroindolyl phosphate (Gibco BRL, Gaithersburg, MD) diluted in 10 ml of alkaline buffer. The enzymatic reaction was blocked by extensive rinsing in the alkaline buffer containing 1 mM EDTA. The sections were then briefly dipped in 70 and 100% ethanol, air-dried and exposed during 2 days to Biomax MR autoradio-graphic films (Kodak) at -80ºC with intensifying screens. Then the same sections were dipped into Ilford K5 nuclear emulsion (Ilford, Mobberly, Cheshire, UK) diluted 1:1 with distilled water, and exposed at 4°C for 5 weeks.
FIGURE 1 A) Coronal section of the rat prefrontal cortex at bregma +3.2 mm showing the localization of extracellular recordings (shaded area). B) The recorded units were identified by antidromic activation from mid-brain (stimulating electrodes were placed in the dorsal raphe nucleus or the ventral tegmental area) and the col-lision test (asterisk).
FIGURE 2 Examples of mPFC pyramidal neurons activated by the administration of PCP. A and B are two firing rate histograms showing the effect of PCP (0.25 mg/kg i.v., first arrow) on the overall firing rate. The subsequent administration of haloperidol (HAL, 0.1 mg/kg i.v., panel A) and clozapine (CLZ; 1 mg/kg i.v., panel B) reversed the PCP-induced increase in firing rate. Bar time: 1 min; ordinate in spikes/10 s.

NMDA ANTAGONIST & CORTICO-SUBCORTICAL CIRCUITS 133
Data Analysis
Changes in the firing rate or the proportion of burst firing in pyramidal neurons were assessed using ANOVA or paired Student's t test, as appropriate. Values were quantified during the last 2 min of the 5-min baseline recording and during 2 min after PCP or antipsychotic administration (omitting the two first minute after injection). Drug effect was defined as ± 30% change in firing rate from base-line. A burst episode was defined as the occurrence of two or more spikes with an interspike interval (ISI) of <45 ms, according to Laviolette et al.
(2005). Power spectra were constructed for repre-sentative 1-min periods during baseline, PCP and PCP + antipsychotics periods. Spectrograms (FIGs. 5 and 6) were constructed from normalized Power Spectral Density (PSD) with NeuroExplorer (Nex Technologies, Littleton, MA). Tissue sections were examined in a Wild 420 macroscope (Leica, Heerbrugg, Germany) and in a Nikon Eclipse E1000 microscope (Nikon, Tokyo, Japan). Micrography was performed using a digital camera (DXM1200 3.0, Nikon) and analySIS Software (Soft Imaging System GmbH, Munchen, Germany). The figures were prepared for publica-tion using Adobe Photoshop software (Adobe Software, Mountain View, CA, USA).
RESULTS
Effects of PCP on Neuronal Activity
in the mPFC
The effect of PCP was examined in a total of 80 rats (one neuron per rat). Most units were located in the prelimbic area of the mPFC. Some were located more dorsal, in the cingulate area and more ventral, in the infralimbic area (FIG. 1). Forty six neurons were antidromically activated from the DR and 34 from the VTA. Saline injections did not alter the firing rate of pyramidal neurons. PCP administra-tion (0.25 mg/kg i.v.) produced an overall increase in the firing rate of pyramidal neurons in the mPFC, from 2.2 ± 0.3 to 3.4 ± 0.4 spikes/s (n=80). (Kargieman et al., 2007). However, three distinct responses were identified. Approximately 45% of the recorded neurons were excited by PCP, with an almost 3-fold increase (from 2.1 ± 0.3 to 6.0 ± 0.7 spikes/s, n=36; p <0.01) whereas 30% were inhib-ited (from 2.8 ± 0.8 to 1.2 ± 0.4 spikes/s, n=26; p
<0.05). The rest of the neurons examined (n=18)were unaffected by PCP administration. FIG. 2 shows two examples of pyramidal neurons excited by PCP administration. The PCP-induced increase in the overall firing rate was accompanied by a simultaneous change in burst firing. This was evidenced by the total num-ber of spikes fired in bursts (from 117 ± 25 in basal conditions to 386 ± 61, p <0.01, data from 2 min) and the number of burst episodes (from 54 ± 11 to 169 ± 24, p <0.01, data from 2 min) in the neurons excited by PCP (n=36). Conversely, PCP reduced burst firing in the neurons whose firing rate was inhibited. The total number of spikes fired in bursts was reduced from 288 ± 99 to 100 ± 37 (p <0.05,data from 2 min) whereas the number of burst epi-sodes was reduced from 109 ± 31 to 39 ± 12 (p<0.05; data from 2 min). FIG. 3 shows representa-
FIGURE 3 Examples of the changes on burst firing produced by PCP in pyramidal neurons of the mPFC. Each box depicts burst trains before and after PCP administration (each vertical line corresponds to a single burst episode; bar time = 10 s). Column 1 shows typical examples of burst episodes in basal conditions. Note the inactivating nature of some of these bursts (B1 and C1;Dégenètais et al., 2002). Neuron in A shows no change on burst firing after PCP (basal: 108 burst/2 min; PCP: 95 burst/2 min). Neurons in B and D show increase on burst firing after PCP (B: basal: 44 burst/2 min; PCP: 244 burst/2 min; D: 68 burst/2 min; PCP: 98 burst/2 min) and neurons in C and E show decrease on bursting activity (C: basal: 71 burst/2 min; PCP: 33 burst/2 min; E: 328 burst/2 min; PCP: 198 burst/2min).

L. KARGIEMAN et al.134
FIGURE 4 Relationship between the degree of burst and frequency firing in mPFC neurons. Examples of two neu-rons with similar frequency firing and different bursting activity. A) Neuron showing a high degree of bursting activ-ity (69.3%). Note the peak in the ISI histogram at short (<45 ms) interspike interval (ISI). B) An example of a neuron showing mainly an irregular pattern of spike discharge with a small number of bursts (13.4 %). Note the more sym-metrical ISI distribution. The dotted line marks the cutoff for burst ISI (45 ms) as described in Laviolette et al., 2005. C) Analysis of the relationship between firing frequency and the percentage of spike events occurring in bursts indi-cates a significant but low correlation between basal neuronal firing frequency and percentage of burst events. D) The difference between baseline firing frequency and firing frequency after PCP administration is plotted as a function of the difference between baseline percentage of bursting and percentage of bursting after PCP in the same neuron.
FIGURE 5 Spectrogram showing the effects of the administration of phencyclidine (PCP, 0.25 mg/kg i.v.) and clozapine (CLZ, 1 mg/kg i.v.) on low frequency oscillations recorded in mPFC. Note the marked reduction in the power spectrum induced by PCP shortly after its administration (first arrow) and the reversal produced by CLZ. Abscissa is in s, ordinate is in Hz. The intensity of the power spectrum is color-coded (red = high intensity; blue
= low intensity).
NMDA ANTAGONIST & CORTICO-SUBCORTICAL CIRCUITS 135
tive examples of the effect of PCP in five different experiments illustrating the changes in burst firing. A full account of the effects of PCP on burst firing can be found in Kargieman et al. (2007). FIGs. 4A and 4B show individual examples of neurons with similar firing rate and different degree of burst fir-ing. The analysis of the relationship between firing frequency and the percentage of spike events occur-ring in bursts in basal conditions reveals a signifi-cant low correlation (R2=0.1451) between mPFC neuronal firing frequency and percentage of burst events (FIG. 4C). Treatment with PCP doesn't modify this low correlation (R2=0.1504, after PCP administration) (FIG. 4D). Slightly different results in the proportion of pyra-midal neurons excited or inhibited by PCP were obtained depending on the site of projection. Of 46 DR-projecting neurons, 24 were activated, 11 were inhibited and 11 were unaffected by PCP, whereas of the 34 neurons projecting to VTA, 12 were acti-vated, 15 were inhibited and 7 remained unaffected ( 2 = 3.79, p=0.15). In parallel with the effects on the discharge rate of pyramidal neurons, PCP markedly affected the amplitude of slow oscillations (0.3-4 Hz) recorded
extracellularly. The effect of PCP administration (0.25 mg/kg i.v.) on LFP recorded in prelimbic mPFC was examined in 20 rats. PCP administration reduced the power spectrum from 0.26 ± 0.04 to 0.10 ± 0.02 V2 (p <0.001, Student's t-test; n=20)(FIG. 5) and increased the percentage of spikes discharged during inactive phases of the LFP (Kargieman et al., 2007).
Effects of PCP on c-fos Expression
The administration of PCP (10 mg/kg i.p.) pro-duced a marked increase in the expression of c-fos
in most subdivisions of the PFC. Most marked effects were noted in the mPFC, particularly the prelimbic area, and in a layer of cells between laminae III and V in dorsolateral aspects of the PFC (Kargieman et al., 2007). Double in situ hybridiza-tion experiments revealed that this increase took place exclusively in pyramidal neurons (vGluT1-positive) and not in GABAergic neurons. The pro-portion of vGluT1-positive neurons expressing c-fos increased from 18 ± 2 % in rats treated with saline (two injections, 30-min apart) to 50 ± 3 % in those treated with saline + PCP (p <0.01). This was accompanied by a parallel and significant 2.2-fold
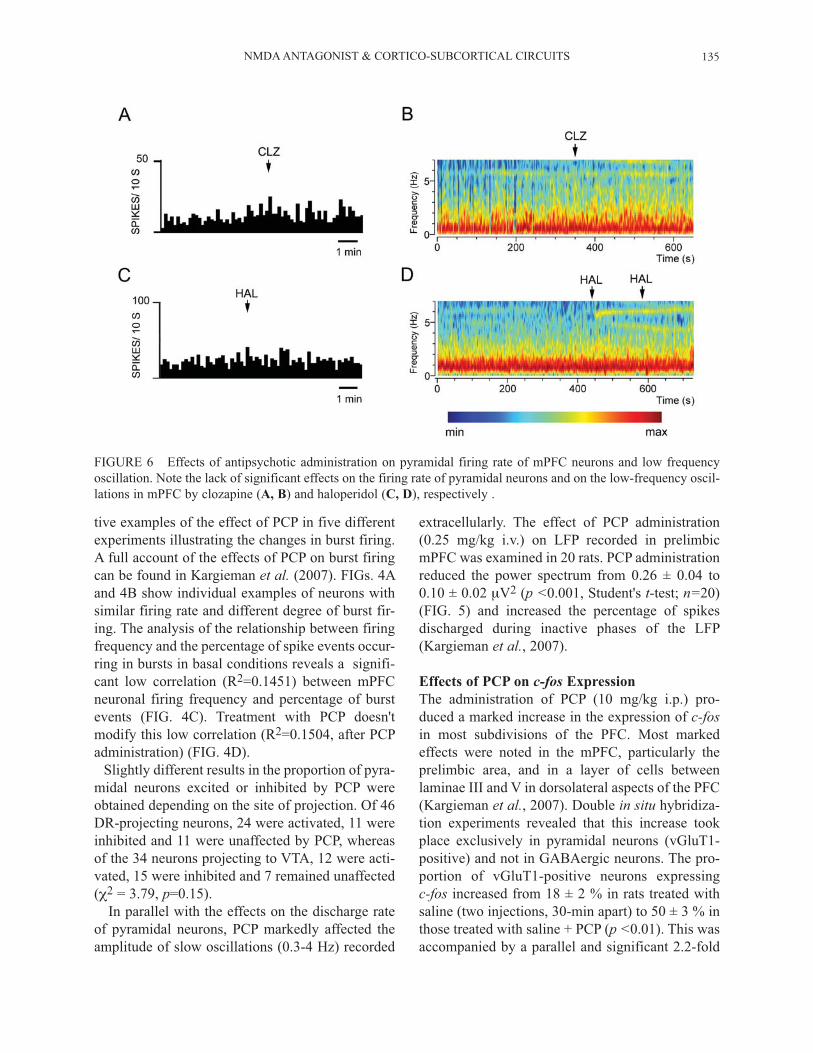
FIGURE 6 Effects of antipsychotic administration on pyramidal firing rate of mPFC neurons and low frequency oscillation. Note the lack of significant effects on the firing rate of pyramidal neurons and on the low-frequency oscil-lations in mPFC by clozapine (A, B) and haloperidol (C, D), respectively .

L. KARGIEMAN et al.136
increase in the individual expression of c-fos, as assessed by counting the number of silver grains in each individual cell (Kargieman et al., 2007). In addition to the PFC, PCP administration increased the expression of c-fos in various cortical areas, such as retrosplenial and somatosensory corti-ces, as well as in various thalamic nuclei, such as the centromedial, mediodorsal, reuniens and rhomboid nuclei (FIG. 7). Double in situ hybridization experi-ments indicated that the increase in c-fos expression was limited to excitatory projection neurons (vGluT1-positive). In contrast with these areas, PCP produced a very small increase in the expression of c-fos in the hippocampus (not shown).
Antipsychotic Reversal
The effects of PCP on neuronal activity in mPFC, both at cellular and population levels, were reversed by the subsequent administration of classical (halo-peridol, HAL, 0.1-0.2 mg/kg i.v.) and atypical antip-sycotics (clozapine, CLZ, 1-2 mg/kg i.v.) (FIGs. 2
and 5; see also Kargieman et al., 2007). Both antip-sychotic drugs induced a partial reversal of the PCP-evoked reduction in the low-frequency oscillations in mPFC (FIG. 5 and Kargieman et al., 2007). When administered alone neither antipsychotic drugs altered the mPFC low frequency oscillations nor the firing rate of mPFC pyramidal neurons. FIG. 6 shows two representative examples of the lack of effect of clozapine and haloperidol on the firing rate of two mPFC pyramidal neurons (FIG. 6A and 6C, respectively) and two spectrograms showing the lack of effect of both antipsychotic in the low-frequency oscillation. Likewise, the increase in c-fos expression in PFC and other cortical areas, as well as in subcortical structures produced by the i.p. administration of PCP was prevented by the prior administration of CLZ (5 mg/kg i.p., given 30 min before PCP). FIG. 7 shows a representative example of the effect of PCP and the CLZ-induced antagonism on c-fos
expression in cortical areas and in thalamic nuclei.
FIGURE 7 Modulation of c-fos cellular expression in PCP (A1, B1, B2) and CLZ+PCP (A2, B3, B4) treated rats. Autoradiographic localization of c-fos mRNA expression in rat coronal sections at AP -2.52 mm from Bregma after PCP (A1) and CLZ+PCP (A2) treatment. Note the high level of c-fos expression in several cortical (retrosplenial, somatosensory) and subcortical (thalamic nuclei) areas in PCP-treated rats, an effect partially prevented by CLZ. Cellular localization of c-fos mRNA in glutamatergic cells of centromedial thalamic nuclei (B1, B3) and layer IV of primary somatosensory cortex (B2, B4). B1 and B2, High-magnification bright-field microphotographs of emulsion-dipped sections of high magnification photomicrographs showing the expression of c-fos in glutamatergic cells. B1
and B2 correspond to PCP treated rats. Note the high density of silver grains, corresponding to c-fos-positive cells, in nearly all glutamatergic cells of both structures. B3 and B4 show the prevention of this effect by CLZ pretreatment. Bar: 20 m.

NMDA ANTAGONIST & CORTICO-SUBCORTICAL CIRCUITS 137
DISCUSSION
The present results indicate that the non-competi-tive NMDA receptor antagonist PCP produced a marked alteration of the activity of the PFC, as assessed by electrophysiological and histological techniques. Thus, PCP evoked a dramatic imbal-ance of the activity of excitatory transmission in PFC, increasing 3-fold the activity of nearly half of the pyramidal neurons recorded and reducing the activity of 30%. Similar effects were also reported for MK-801, which also increased, decreased or left unaffected the discharge rate of putative pyramidal neurons in the mPFC of awake rats (Jackson et al.,2004). The effect of PCP was accompanied by a remarkable reduction in the amplitude of low oscil-lations in the same area. Oscillations of different frequencies that are syn-chronized over widespread areas of the cerebral cortex are recorder in the LFP and electroencepha-logram (EEG) (Steriade et al., 1993; Steriade, 2006). Low-frequency oscillations play an impor-tant role in information processing and memory consolidation during sleep (Stickgold et al., 2001; Stickgold, 2005; Marshall, et al., 2006). Since reduced delta wave has been reported in schizo-phrenic patients during sleep (Keshavan et al.,1998; Hoffmann et al., 2000; Sekimoto et al., 2007) deciphering the dynamics of neuronal circuits involved in low oscillations, could help to under-stand brain alterations in pathological states. Histological experiments revealed that PCP evoked a similar 3-fold increase in the number of pyramidal (vGluT1-positive) neurons expressing c-fos, as well as in the individual cellular expres-sion of the gene. The increase in c-fos expression was also observed in other cortical and thalamic areas, suggesting that PCP alters information pro-cessing in cortico-thalamocortical circuits, an effect possibly involved in the psychotomimetic proper-ties of PCP. Interestingly, these effects were reversed or prevented by the administration of CLZ and HAL (the effect of HAL was examined only in elec-trophysiological experiments). We found that PCP had parallel effects on the overall discharge rate and on burst firing while a previous report using MK-801 in awake rats report-ed opposite effects on cell firing (increase) and burst firing (decrease) (Jackson et al., 2004). The
latter change was interpreted as a reduction in the efficiency of cortical information processing. The reasons for this discrepancy are unclear, but may be due to the different NMDA antagonists used and/or to the different method employed to detect burst episodes in pyramidal neurons (Jackson et al.,2004; Laviolette et al., 2005). The latter method appears to be less restrictive than others (e.g.,Nowak et al., 2003) in terms of the interspike inter-vals. However, the mean interspike interval in our study was 12.2 ± 0.8 ms (n=67) (see supplementary material 1 in Kargieman et al., 2007) which is in close agreement with the estimated firing rate within bursts in pyramidal neurons of the rat mPFC (77 ± 21 Hz for inactivating bursts, 92 ± 39 Hz for non-inactivating bursts; Dégenètais et al., 2002). Also, the different conditions on the experimental procedures, deeply anesthetized animals (this work) versus un-anesthetized animals (Jackson et al.,2005), may account for these differences, since fir-ing patterns depend on the state of the system (Steriade, 2004). Even though, our data concur with the study by Jackson et al. (2004) in that NMDA receptor blockade with PCP results in a profound loss of the efficiency of cortical information pro-cessing. This is shown by a) the dramatic change in activity patterns of PFC pyramidal neurons, b) the reduction of cortical synchrony at low frequencies (0.3-4 Hz, corresponding to slow and delta oscilla-tions) and c) by the disorganization on the pyrami-dal discharge during PCP treatment (e.g., a substan-tial percentage of pyramidal spikes occurred out of active phases of the LFP). Interestingly, the mito-toxic agent methylazoxymethanol, used as a neu-rodevelopment model of schizophrenia, also evokes a loss of cortical synchronicity in adult rats prena-tally treated with this agent (Goto and Grace, 2006). Also, the hallucinogen DOI (a preferential 5-HT2Areceptor agonist) altered the activity pattern of PFC pyramidal neurons similarly to PCP (Puig et al.,2003) and reduced cortical synchrony at 0.3-4 Hz (Celada et al., in press). Despite a large body of data on the actions of NMDA receptor antagonists, and in particular on PFC function, it is unclear whether the primary targets are PFC neuronal elements or if other areas are involved. Indeed, it may seem paradoxical that blockade of excitatory ionotropic NMDA receptors in PFC may result in an increased activity of a large

L. KARGIEMAN et al.138
population of pyramidal neurons. Hence, it has been suggested that NMDA receptor blockade of GABAergic interneurons may disinhibit pyramidal neurons resulting in an increased discharge rate (Jackson et al., 2004). However local administra-tion of PCP or MK-801 in mPFC did not increase or reduce in some instances the spontaneous dis-charge of PFC neurons (Suzuki et al., 2002; Jodo et
al., 2005), whereas NMDA blockade in ventral hip-pocampus increased the activity of mPFC neurons (Jodo et al., 2005). This suggested that NMDA receptor antagonists may drive the increase in PFC neuronal activity through the enhancement of hip-pocampal-PFC excitatory inputs. Likewise, sys-temic - but not intra-PFC-administration of ket-amine, PCP and MK-801 increased the release of 5-HT (and of glutamate, when examined) in PFC ( Moghaddam et al., 1997; Amargós-Bosch et al.,2006; López-Gil et al., 2007). In parallel with electrophysiological observations, we found that PCP markedly increased c-fos expres-sion in pyramidal neurons of the mPFC (cingulate, prelimbic and infralimbic areas) but also in orbito-frontal, motor, insular and piriform cortices (Kargieman et al., 2007). PCP also produced a dra-matic increase of c-fos expression in various thal-amic nuclei, particularly in the centromedial and mediodorsal nuclei which project heavily to mPFC (Berendse and Groenewegen, 1991; Kuroda et al.,1998). Interestingly, a dense c-fos expression was observed in a thin layer of cells between layers III and V (layer IV is absent in rat PFC) where thal-amic afferents synapse on pyramidal neurons (Berendse and Groenewegen, 1991; Kuroda et al.,1998; Vertes et al., 2006). The retrosplenial and primary somatosensory cortices, which also showed an enhanced c-fos response, are also innervated by midline thalamic nuclei (Vertes et al., 2006). In turn, these areas project to the midline thalamic nuclei, thus closing thalamocortical circuits (Groenewegen and Uylings, 2000; McKenna and Vertes, 2004; Vertes, 2004; Gabbot et al., 2005). However, contrary to Jodo et al. (2005), our observations indicate that PCP affects mainly corti-cal and thalamic areas, but not the hippocampus. Indeed, the expression of c-fos in the CA1 region and the subiculum, which project to the mPFC show a minor abundance of neurons expressing c-fos, a cellular marker of neuronal activity. Hence,
although we cannot discard an action of PCP on local circuit GABA neurons in PFC, the present results suggest that PCP mainly affects cortico-thalamocortical circuits. Interestingly, PCP increased the expression of c-fos in thalamic excitatory neurons of the centro-medial and mediodorsal nuclei, whereas the reticu-lar thalamic nucleus did not seem to be affected (Kargieman et al., 2007; Santana et al., unpub-lished). Since reticular GABAergic neurons inhibit relay thalamic neurons in the rest of thalamic nuclei, a putative blockade of NMDA receptors by PCP in reticular neurons may disinhibit projection thalamic neurons and increase of the activity of thalamic afferents to PFC. An important observation in the present study is that the effects of PCP at cellular and population levels were antagonized by antipsychotic drugs. The atypical antipsychotic drug clozapine fully or partially blocked the effect of PCP on a) pyramidal discharge, b) low frequency oscillations, and c) c-fos expression. Similarly, haloperidol and clozap-ine blocked the MK-801-induced rise in extracel-lular glutamate in the rat mPFC after systemic or local application (López-Gil et al., 2007). The phar-macological activities responsible for the effects of CLZ are presently unknown but may lie in its abil-ity to interact with various 5-HT receptors. Indeed, the 5-HT2A receptor antagonist M100907 antago-nized some neurochemical and behavioral effects of NMDA receptor blockade (Martin et al., 1997; Mirjana et al., 2004; Amargós-Bosch et al., 2007), yet discrepant results have also been reported (Adams and Moghaddam, 2001). However, halo-peridol was equally effective in reversing PCP-induced alterations in cell firing and LFP despite a comparatively lower affinity for 5-HT2A receptors, which suggests the involvement of other pharmaco-logical activities. An increase in DA-mediated transmission in PFC secondary to the blockade of DA D2 autoreceptors may be accountable since VTA stimulation inhibits pyramidal neuron activity (Tseng et al., 2006). Also, haloperidol may alter information processing in cortico-limbic circuits by blocking striatal DA D2 receptors, thus affecting the thalamic feed-back to PFC. In summary, the present results indicate that PCP causes a profound disruption of information pro-cessing in PFC, altering the normal discharge pat-

NMDA ANTAGONIST & CORTICO-SUBCORTICAL CIRCUITS 139
tern of pyramidal neurons and the oscillatory activ-ity at low frequency, two effects that likely account for the psychotomimetic actions of PCP. The rever-sal of these effects by clozapine and haloperidol indicates that, despite their differential receptor profile, both drugs share the ability to normalize the marked disruption of excitatory neurotransmission in PFC, an effect possibly involved in their thera-peutic activity.
Acknowledgements
Work supported by grant SAF2007-62378. Support from CIBERSAM and SENY Fundació is also acknowledged. The Department of Neurochemistry and Neuropharmacology is "Grup de Recerca de Qualitat" (Generalitat de Catalunya (2005-SGR00758). We also thank Judith Ballart for skilful technical assistance.
References
Adams B and B Moghaddam (1998) Corticolimbic dopamine neurotransmission is temporally dissociated from the cogni-tive and locomotor effects of phencyclidine. J. Neurosci. 18,5545-5554.
Adams BW and B Moghaddam (2001) Effect of clozapine, haloperidol or M100907 on phencyclidine-activated gluta-mate efflux in the prefrontal cortex. Biol. Psychiatry 50,750-757.
Amargós-Bosch M, X López-Gil, F Artigas and A Adell (2006) Clozapine and olanzapine, but not haloperidol, suppress serotonin efflux in the medial prefrontal cortex elicited by phencyclidine and ketamine. Int. J. Neuropsychopharmacol.
9, 565-573.Berendse HW and HJ Groenewegen (1991) Restricted cortical
termination fields of the midline and intralaminar thalamic nuclei in the rat. Neuroscience 42, 73-102.
Breier A, AK Malhotra, DA Pinals, NI Weisenfeld and D Pickar (1997) Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy vol-unteers. Am. J. Psychiatry 154, 805-811.
Celada P, MV Puig, L Díaz-Mataix and F Artigas (2008) The hallucinogen DOI reduces low-frequency oscillations in rat prefrontal cortex: reversal by antipsychotic drugs. Biol.
Psychiatry 2008 Apr 22. [Epub ahead of print].Dégenètais E, AM Thierry, J Glowinski and Y Gioanni (2002)
Electrophysiological properties of pyramidal neurons in the rat prefrontal cortex: an in vivo intracellular recording study. Cereb. Cortex 12, 1-16.
Fuster JM (1997) The Prefrontal Cortex Anatomy, Physiology
and Neuropsychology of the Frontal Lobe (Lipincott-Raven:Philadelphia/New York).
Geyer MA, K Krebs-Thomson, DL Braff and NR Swerdlow (2001) Pharmacological studies of prepulse inhibition mod-
els of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl.) 156, 17-154.
Goto Y and AA Grace (2006) Alterations in medial prefrontal cortical activity and plasticity in rats with disruption of corti-cal development. Biol. Psychiatry 60, 1259-1267.
Groenewegen HJ and HB Uylings (2000) The prefrontal cortex and the integration of sensory limbic and autonomic infor-mation. Prog. Brain Res. 126, 3-28.
Hajos M (2006) Targeting information-processing deficit in schizophrenia: a novel approach to psychotherapeutic drug discovery. Trends Pharmacol. Sci. 27, 391-398.
Harrison PJ (1999) The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain 122,593-624.
Hoffmann R, W Hendrickse, AJ Rush and R Armitage (2000) Slow-wave activity during non-REM sleep in men with schizophrenia and major depressive disorders. Psychiatry
Res. 95, 215-225.Jackson ME, H Homayoun and B Moghaddam (2004) NMDA
receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc. Natl. Acad. Sci. USA 101, 8467-8472.
Javitt DC and SR Zukin (1991) Recent advances in the phen-cyclidine model of schizophrenia. Am. J. Psychiatry 148,1301-1308. Review.
Jodo E, Y Suzuki, T Katayama, KY Hoshino, S Takeuchi, S Niwa and Y Kayama (2005) Activation of medial prefrontal cortex by phencyclidine is mediated via a hippocampo-pre-frontalpathway. Cereb. Cortex 15, 663-669.
Kargieman L, N Santana, G Mengod, P Celada and F Artigas (2007) Antipsychotic drugs reverse the disruption in pre-frontal cortex function produced by NMDA receptor block-ade with phencyclidine. Proc. Natl Acad. Sci. USA 104,14843-14848.
Keshavan MS, CF Reynolds 3rd, MJ Miewald, DM Montrose, JA Sweeney, RC Vasko Jr and DJ Kupfer (1998) Delta sleep deficits in schizophrenia: evidence from automated analyses of sleep data. Arch. Gen. Psychiatry 55, 443-448.
Krystal JH, LP Karper, JP Seibyl, GK Freeman, R Delaney, JD Bremner, GR Heninger, MB Bowers and DS Charney (1994) Subanesthetic effects of the noncompetitive NMDA antago-nist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen.
Psychiatry 51, 199-214.Krystal JH, DC D'Souza, D Mathalon, E Perry, A Belger and R
Hoffman (2003) NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a para-digm shift in medication development. Psychopharmacolo-
gy (Berl.) 169, 215-233. Review.Kuroda M, J Yokofujita and K Murakami (1998) An ultrastruc-
tural study of the neural circuit between the prefrontal cortex and the mediodorsal nucleus of the thalamus. Prog.
Neurobiol. 54, 417-458. Laviolette SR, WJ Lipski and AA Grace (2005) A subpopula-
tion of neurons in the medial prefrontal cortex encodes emotional learning with burst and frequency codes through a dopamine D4 receptor-dependent basolateral amygdala input J. Neurosci. 25, 6066-6075.
Lewis DA and JA Lieberman (2000) Catching up on schizo-

L. KARGIEMAN et al.140
phrenia: natural history and neurobiology. Neuron 28, 325-334.
Lewis DA, T Hashimoto and DW Volk (2005) Cortical inhibi-tory neurons and schizophrenia. Nat. Rev. Neurosci. 6, 312-324.
López-Gil J, Z Babot, M Amargós-Bosch, C Suñol, F Artigas and A Adell (2007) Clozapine and haloperidol differently suppress the MK-801-increased glutamatergic and seroton-ergic transmission in the medial prefrontal cortex of the rat. Neuropsychopharmacology 32, 2087-2097.
Lorrain DS, CS Baccei, LJ Bristow, JJ Anderson and MA Varney (2003) Effects of ketamine and N-methyl-D-aspartate on glutamate and dopamine release in the rat prefrontal cortex: modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience 117,697-706.
Malhotra AK, DA Pinals, H Weingartner, K Sirocco, CD Missar, D Pickar and A Breier (1994) NMDA receptor func-tion and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology 14, 301-307.
Marshall L, H Helgadottir, M Molle and J Born (2006) Boosting slow oscillations during sleep potentiates memory. Nature 444, 610-613.
Martin P, N Waters, S Waters, A Carlsson and M Carlsson (1997) MK-801-induced hyperlocomotion: differential effects of M100907, SDZ PSD 958 and raclopride. Eur.J.
Pharmacol. 335, 107-116.Martin P, ML Carlsson and S Hjorth (1998) Systemic PCP
treatment elevates brain extracellular 5-HT: a microdialysis study in awake rats. Neuroreport 9, 2985-2988.
Mathé JM, GG Nomikos, KH Blakeman and TH Svensson (1999) Differential actions of dizocilpine (MK-801) on the mesolimbic and mesocortical dopamine systems: role of neuronal activity. Neuropharmacology 38, 121-128.
McKenna JT and RP Vertes (2004) Afferent projections to nucleus reuniens of the thalamus. J. Comp. Neurol. 480,115-142.
Millan MJ, M Brocco, A Gobert, F Joly, K Bervoets, JM Rivet, A Newman-Tancredi, V Audinot and S Maurel (1999) Contrasting mechanisms of action and sensitivity to antip-sychotics of phencyclidine versus amphetamine: impor-tance of nucleus accumbens 5-HT2A sites for PCP-induced locomotion in the rat. Eur. J. Neurosci. 11, 4419-4432.
Miller EK and JD Cohen (2001) An integrative theory of pre-frontal cortex function. Annu. Rev. Neurosci. 24, 167-202.
Mirjana C, M Baviera, RW Invernizzi and C Balducci (2004) The serotonin 5-HT2A receptors antagonist M100907 pre-vents impairment in attentional performance by NMDA receptor blockade in the rat prefrontal cortex. Neuropsychopharmacology 29, 1637-1647.
Moghaddam B, B Adams, A Verma and D Daly (1997) Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci. 17, 2921-2927.
Nelson CL, JA Burk, JP Bruno and M Sarter (2002) Effects of acute and repeated systemic administration of ketamine on
prefrontal acetylcholine release and sustained attention per-formance in rats. Psychopharmacology (Berl.) 161, 168-179.
Newcomer JW, NB Farber, V Jevtovic-Todorovic, G Selke, AK Melson, T Hershey, S Craft and JW Olney (1999) Ketamine-induced NMDA receptor hypofunction as model of memory impairment and psychosis. Neuropsychopharmacology 20, 106-118.
Nowak LG, R Azouz, MV Sanchez-Vives, CM Gray and DA McCormick (2003) Electrophysiological classes of cat pri-mary visual cortical neurons in vivo as revealed by quantita-tive analyses. J. Neurophysiol. 89, 1541-1566.
Paxinos G and C Watson (1998) The Rat Brain in Stereotaxic
Coordinates, 4th Edition (Academic Press:Sydney).Puig MV, P Celada, L Díaz-Mataix and F Artigas (2003) In
vivo modulation of the activity of pyramidal neurons in the rat medial prefrontal cortex by 5-HT2A receptors. Relationship to thalamocortical afferents. Cereb. Cortex 13,1870-1882.
Sekimoto M, M Kato, T Watanabe, N Kajimura and K Takahashi (2007) Reduced frontal asymmetry of delta waves during all-night sleep in schizophrenia. Schizophr.
Bull. 33, 1307-1311.Serrats J, F Artigas, G Mengod and R Cortés (2003) GABAB
receptor mRNA in the raphe nuclei: co-expression with serotonin transporter and glutamic acid decarboxylase. J.
Neurochem. 84, 743-752. Schmidt CJ and GM Fadayel (1996) Regional effects of
MK-801 on dopamine release: effects of competitive NMDA or 5-HT2A receptor blockade. J. Pharmacol. Exp.
Ther. 277, 1541-1549.Steriade M, A Nuñez and F Amzica (1993) A novel slow (< 1
Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J. Neurosci. 13, 3252-3265.
Steriade M (2004) Neocortical cell classes are flexible enti-ties. Nat. Rev. Neurosci. 2, 121-134.
Steriade M (2006) Grouping of brain rhythms in corticotha-lamic systems. Neuroscience 137, 1087-1106.
Stickgold R (2005) Sleep-dependent memory consolidation. Nature 437, 1272-1278.
Stickgold R, JA Hobson, R Fosse and M Fosse (2001) Sleep, learning, and dreams: off-line memory reprocessing. Science
294, 1052-1057.Suzuki Y, E Jodo, S Takeuchi, S Niwa and Y Kayama (2002)
Acute administration of phencyclidine induces tonic activa-tion of medial prefrontal cortex neurons in freely moving rats. Neuroscience 114, 769-779.
Tseng KY, N Mallet, KL Toreson, C Le Moine, F Gonon and P O'Donnell (2006) Excitatory response of prefrontal corti-cal fast-spiking interneurons to ventral tegmental area stimulation in vivo. Synapse 59, 412-417.
Vertes RP (2004) Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse 51, 32-58.
Vertes RP (2006) Interactions among the medial prefrontal cortex, hippocampus and midline thalamus in emotional and cognitive processing in the rat. Neuroscience 29, 1-20.
Copyright © 2022 FDOKUMEN



















