
New challenges for R&D in coating resins
17
Progress in Organic Coatings 45 (2002) 101–117 New challenges for R&D in coating resins Emiel Staring a , Aylvin A. Dias b,∗ , Rolf A.T.M. van Benthem b,∗ a DSM Coating Resins, P.O. Box 615, 8000 AP, Zwolle, The Netherlands b DSM Research, P.O. Box 18, 6160 MD, Geleen, The Netherlands Received 1 September 2001; accepted 13 March 2002 Abstract The coatings industry has experienced a strong drive towards new products during the last couple of decades. New products must either allow lower total “system” cost and/or have better performance, and/or lower impact on the environment. A number of trends like globalisation, consolidation, specialisation and increased competitiveness can be discerned. These developments encourage a different approach towards coatings development. New elements that come into play are speed of development and decomplexation. These elements will influence the way the coating (resins) R&D is performed. The central theme for addressing these questions, at least in the area of thermosetting coatings, in DSM’s view is a better, more thorough understanding of the relationship between structure and properties: from craftsmanship to science. This relationship must be clarified on at least three levels: the molecular, the mesoscopic and the temporal level. One of the most important challenges for R&D in coating resins is to use and/or develop new characterisation techniques that relate structure (of molecules and networks) to properties. Using recent developments from DSM laboratories various approaches to meet these challenges are presented. It is our considered opinion that by having precise characterisation of raw materials with techniques that allow us to monitor temporal and spatial changes we will be able to exercise control over chemistry, network and mechanical properties to achieve performance coatings. In order to characterise coating resins with non-linear architectures we have used the combination of size-exclusion chromatography with mass spectrometry to exactly determine molecular weight distributions and molecular monomer compositions. This is illustrated with the full characterisation of a set of a priori calculated and synthesised hyperbranched polyesteramides. Compliances as well as deviations from theory have been established and explained. The improved understanding of photocured networks and their heterogeneity has been derived using solid state proton NMR T 2 relaxation, where relaxation times can be related to the modulus of photocured coatings. This technique is of high utility to the coatings industry due to the fact that it allows one to probe effects on a mesoscopic level with minimal sample preparation. Time-resolved chemical changes have been monitored using RT-FTIR (real time also for photocuring systems). These techniques allow us to relate chemical changes to a prescribed development of network topology and the eventual macroscopic properties like modulus and T g . © 2002 Elsevier Science B.V. All rights reserved. Keywords: Structure–property relations; Hyperbranched polyesteramides; Photocured networks; NMR proton relaxation; Time-resolved FTIR 1. Introduction The coatings industry has experienced a strong drive to- wards new products during the last 20 years with ever in- creasing diversity of applications. New products must either allow lower total “system” cost and/or have better perfor- mance, and/or lower impact on the environment. The golden rule turned out to be that successful new products scored better on at least one of the above drivers with two others at least equal. ∗ Corresponding authors. E-mail address: [email protected] (R.A.T.M. van Benthem). Coatings and paints are performance materials of which the relation between molecular composition and macro- scopic properties is poorly understood. In many cases, the most effective way to develop better coatings was and still is, based on systematic testing of new resins, x-linkers and/or additives in a sheer endless variety of combinations (Edisonian type of research). Most of this empirical knowl- edge is present within industrial laboratories, and thus not universally accessible. This approach has resulted in a situa- tion where coatings consist of a large number of ingredients. Furthermore, numerous grades designed for specific appli- cations have further broadened the range of raw materials used. The result is a product portfolio with ever-increasing complexity and the accompanying logistical problems. 0300-9440/02/$ – see front matter © 2002 Elsevier Science B.V. All rights reserved. PII:S0300-9440(02)00066-8
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of New challenges for R&D in coating resins

Progress in Organic Coatings 45 (2002) 101–117
New challenges for R&D in coating resins
Emiel Staringa, Aylvin A. Diasb,∗, Rolf A.T.M. van Benthemb,∗a DSM Coating Resins, P.O. Box 615, 8000 AP, Zwolle, The Netherlands
b DSM Research, P.O. Box 18, 6160 MD, Geleen, The Netherlands
Received 1 September 2001; accepted 13 March 2002
Abstract
The coatings industry has experienced a strong drive towards new products during the last couple of decades. New products musteither allow lower total “system” cost and/or have better performance, and/or lower impact on the environment. A number of trends likeglobalisation, consolidation, specialisation and increased competitiveness can be discerned. These developments encourage a differentapproach towards coatings development. New elements that come into play are speed of development and decomplexation. These elementswill influence the way the coating (resins) R&D is performed.
The central theme for addressing these questions, at least in the area of thermosetting coatings, in DSM’s view is a better, more thoroughunderstanding of the relationship between structure and properties: from craftsmanship to science. This relationship must be clarified onat least three levels: the molecular, the mesoscopic and the temporal level.
One of the most important challenges for R&D in coating resins is to use and/or develop new characterisation techniques that relatestructure (of molecules and networks) to properties. Using recent developments from DSM laboratories various approaches to meet thesechallenges are presented. It is our considered opinion that by having precise characterisation of raw materials with techniques that allow usto monitor temporal and spatial changes we will be able to exercise control over chemistry, network and mechanical properties to achieveperformance coatings.
In order to characterise coating resins with non-linear architectures we have used the combination of size-exclusion chromatographywith mass spectrometry to exactly determine molecular weight distributions and molecular monomer compositions. This is illustrated withthe full characterisation of a set of a priori calculated and synthesised hyperbranched polyesteramides. Compliances as well as deviationsfrom theory have been established and explained.
The improved understanding of photocured networks and their heterogeneity has been derived using solid state proton NMRT2 relaxation,where relaxation times can be related to the modulus of photocured coatings. This technique is of high utility to the coatings industry dueto the fact that it allows one to probe effects on a mesoscopic level with minimal sample preparation. Time-resolved chemical changeshave been monitored using RT-FTIR (real time also for photocuring systems). These techniques allow us to relate chemical changes to aprescribed development of network topology and the eventual macroscopic properties like modulus andTg.© 2002 Elsevier Science B.V. All rights reserved.
Keywords: Structure–property relations; Hyperbranched polyesteramides; Photocured networks; NMR proton relaxation; Time-resolved FTIR
1. Introduction
The coatings industry has experienced a strong drive to-wards new products during the last 20 years with ever in-creasing diversity of applications. New products must eitherallow lower total “system” cost and/or have better perfor-mance, and/or lower impact on the environment. The goldenrule turned out to be that successful new products scoredbetter on at least one of the above drivers with two others atleast equal.
∗ Corresponding authors.E-mail address: [email protected] (R.A.T.M. van Benthem).
Coatings and paints are performance materials of whichthe relation between molecular composition and macro-scopic properties is poorly understood. In many cases, themost effective way to develop better coatings was and stillis, based on systematic testing of new resins, x-linkersand/or additives in a sheer endless variety of combinations(Edisonian type of research). Most of this empirical knowl-edge is present within industrial laboratories, and thus notuniversally accessible. This approach has resulted in a situa-tion where coatings consist of a large number of ingredients.Furthermore, numerous grades designed for specific appli-cations have further broadened the range of raw materialsused. The result is a product portfolio with ever-increasingcomplexity and the accompanying logistical problems.
0300-9440/02/$ – see front matter © 2002 Elsevier Science B.V. All rights reserved.PII: S0300-9440(02)00066-8

102 E. Staring et al. / Progress in Organic Coatings 45 (2002) 101–117
Global trends like consolidation and specialisationdemand a different approach to coating development. Newelements that come into play are speed of development anddecomplexation. These elements will influence the way thecoating (resins) R&D is performed. Important challengesare: how can we develop better coating (resins) in a fasterway than done until now? And how can we develop bettercoating (resins) without increasing the complexity?
The central theme for addressing these questions, at leastin the area of thermosetting coatings, in DSM’s view isa better, more thorough understanding of the relationshipbetween structure and properties: from craftsmanship toscience. This relationship may be addressed on at least threelevels:
• The molecular level. The molecular structure of the poly-mers involved in the coatings network formation is oftennot fully known. Monomer sequences, type and amountof endgroups are known in general terms but not alwaysin sufficient detail to precisely understand their role infilm formation. The introduction of new polymer architec-tures, like stars, combs, dendrimers, hyperbranched, andsupramolecular assemblies promises much to further de-velop the general field of polymer chemistry and in par-ticular for coating resins. The determination of molecularweight distributions, however is no longer reliable withconventional calibration standards and new characterisa-tion techniques must be applied. In addition improved un-derstanding of the polymer chain topology is needed.
• The mesoscopic level. It is generally acknowledged thatthe network as formed by coating binders is a principalfeature that contributes to mechanical and some chemi-cal properties. Network topology of the resultant curingsystems with attention to mean molar mass betweencross-links, trapped and temporary entanglements, dan-gling chain ends and chain mobility as well unboundmigrateable materials are of prime importance for thecoatings performance. There are, however, not many stan-dard characterisation techniques available for studyingcross-linked materials in such physical/chemical detail.
• The temporal level. In order to understand the final per-formance of industrial products, it is important to knowtheir relation to the production processes in which theyfind their origin. Especially, the network formation duringcure of coatings is a strictly time-dependent phenomenon,and accordingly is the development of properties as afunction of processing.
Using recent developments from DSM laboratories var-ious approaches to meet these challenges are presented.Section 2deals with characterisation on the molecular levelof polymers with new architectures,Section 3deals withcharacterisation of photocured coatings in terms of networktopology and using new time-resolved techniques. It is ourconsidered opinion that by having precise characterisationof raw materials accompanied by powerful techniques thatallow us to monitor temporal and spatial changes we will be
able to exercise control over chemistry, network topologyand mechanical properties to achieve performance coatings.
2. Characterisation of hyperbranched resins
2.1. Calculated synthesis of polyesteramides
The basic concepts of the synthesis of hyperbranchedpolyesteramides as economically feasible counterparts of thewell-known dendrimers, as well as their promising applica-tions in a variety of different coating formulations have beendescribed previously[1,2] and presented at this conferencein 1999[3].
In summary, our synthetic approach bears a strong re-semblance with the classic A2/B3 approach[4] which isunsuitable for higher molecular weights. In our approach,however, one (A) group denoted (a) of the A2-compoundand one (B) group denoted (b) of the B3-component, whichis present in molar excess, are preferentially reactive towardseach other. In this way, in the pre-reaction A-a-b-B2-units(i.e. functional AB2-type units) are formed while the molarexcess of B3-units is retained in the system[5]. This excesslimits molecular weight build-up and eliminates the risk ofgel formation at higher molecular weights, contrary to theclassical A2/B3 approach. Thus, the eventual hyperbranchedpolymer is composed ofn “Aa” units and (n + 1) “B2b”units and bears(n + 3) (B) end groups.
The exothermal reaction of 1,2-cyclohexane-dicarboxylicacid anhydride and diisopropanolamine normally leadsto a molecule with one carboxylic acid group and two2-hydroxy-propylamide groups. This molecule exemplifiesthe A-a-b-B2 unit. The 2-hydroxy-alkyl groups formed (B)can react readily with the carboxylic acid groups (A). Theesterification reaction between a 2-hydroxy-alkyl group anda carboxylic acid is known to proceed much faster com-pared to normal aliphatic primary or secondary alcohols[6]. Instead of an addition–elimination reaction mechanism,the formation of an intermediate oxazolinium–carboxylateion pair is thought to prevail, which strongly facilitatesthe esterification reaction. Typically, this reaction can takeplace at temperatures from about 140◦C without a catalyst.
It also possible to introduce a monocarboxylic acid (A1)as a third component in the reaction mixture in a one-potprocedure. Depending on the molar ratio of A1 (m) to theother two components in the system, the monocarboxylicacid can be used merely as endgroup modifier(m < n + 3)
or as chain stopper(m > n + 3). In this study lauric acid(1-dodecanoic acid) was used. When the reaction betweengroups (a) and (b) is considered to occur instantaneously andcycle formation is excluded according to Flory[4], the aver-age molecular weightsMn andMw of the resulting polymerscan be directly calculated from the monomer composition asa function of the chemical conversion according to Durandand Bruneau[7], optionally treating the mono-functionalcarboxylic acid A1 as a third component.

E. Staring et al. / Progress in Organic Coatings 45 (2002) 101–117 103
Table 1Description of the three series of target molecules
Resinnumbers
D/C Lauric estermodification (DS)
Series I 1–5 1.5–1.14 –Series II 6–9 1.14 or 1.20 Partially (0.20–0.80)Series III 10–14 1.55–1.05 Fully
Experimental. Diisopropanolamine was introduced in a1 l glass reactor under a nitrogen atmosphere. The calcu-lated amount of 1,2-cyclohexanedicarboxylic acid anhydridewas then added portionwise. As a result of the immediateexothermic reaction, the temperature of the mixture raisedto ca. 120◦C. Lauric acid was optionally introduced, withthe total of the calculated amounts of raw materials amount-ing to about 600 g. The reaction mixture was then heated to180◦C. The evolving reaction water was distilled off in 6 h,ultimately under reduced pressure. At the desired acid con-centration but ultimately after 8 h, the hot resin was pouredout and cooled to room temperature.
According to a set of calculated targets, three series of hy-perbranched polyesteramides were synthesised, seeTable 1,in which the ratio diisopropanolamine/anhydride (D/C) andthe degree of substitution (DS) with lauric acid were varied.
Since carboxylic acid groups are theoretically completelyconsumed in all these polycondensation reactions, the resid-ual amount of carboxylic acid as determined by potentio-metric titration was taken as a relative quantitative measureof the total chemical conversion. The targeted and recalcu-lated moments are given inTable 2.
2.2. SEC-DV and SEC-MS for characterisingpolyesteramides
Three imminent questions have to be answered in orderto address the molecular characterisation of these hyper-
Table 2Collected data of hyperbranched polyesteramide resins
No. DS D/C TargetMna p(A) Calculated Found (SEC-DV)
Mna Mw
a Mna Mw
a
1 0 1.47 0.7 98.8 0.68 1.6 1.5 3.62 0 1.35 0.9 98.8 0.87 2.1 1.8 5.93 0 1.26 1.2 98.8 1.10 2.9 2.4 114 0 1.20 1.5 98.4 1.36 3.9 2.4 595 0 1.14 2.0 98.9 1.85 6.1 2.8 2506 0.2 1.14 2.4 98.6 2.13 6.7 2.8 547 0.4 1.14 2.8 97.1 2.11 6.7 2.1 198 0.6 1.14 3.1 94.9 1.88 6.0 2.1 129 0.8 1.14 3.4 94.4 1.88 6.1 1.8 8.5
10 1.0 1.55 1.5 91.1 0.95 2.0 1.1 2.511 1.0 1.25 2.5 91.5 1.29 3.3 1.4 3.412 1.0 1.20 3.0 96.7 2.07 5.6 2.1 6.813 1.0 1.13 4.0 89.5 1.41 4.9 1.4 4.914 1.0 1.08 6.0 91.1 1.85 8.7 1.6 5.4
a All values in 103 g mol−1.
branched polyesteramides:
• What is the exact monomer composition of each polymerchain? (Are there side-reactions or other anomalies?)
• What is the exact molecular weight distribution? (Does itfit with the calculated values?)
• What is the degree of branching or the fractal dimensionof these polymers? (Are they really hyperbranched?)
The first question has been answered by MALDI-TOFmass spectrometry[8]. The latter two questions could beanswered by size exclusion chromatography with on-linedifferential viscosimetry detection (SEC-DV) in principle.The determination of exact the molecular weight distribu-tion with conventional SEC techniques (RI or UV detec-tion) which are based on polystyrene calibration standards,is not feasible since the hydrodynamic volume of a highlybranched polymer is intrinsically different from a linear one.There are simply no suitable calibration standards for highlybranched polymers. SEC-DV relies on the universal cali-bration principle[9] which correlates the intrinsic viscos-ity of a polymer collection in one SEC-slice to its absolutemolecular weight. Although this principle was thought to bevalid for hyperbranched polymers as well, and gives indirectinformation on the hyperbranched character of these poly-mers through the Mark–Houwink relation, its validity hadto checked. We have attempted this by combined SEC-DVand (off-line) SEC-MS (ESI and MALDITOF-MS) mea-surements.
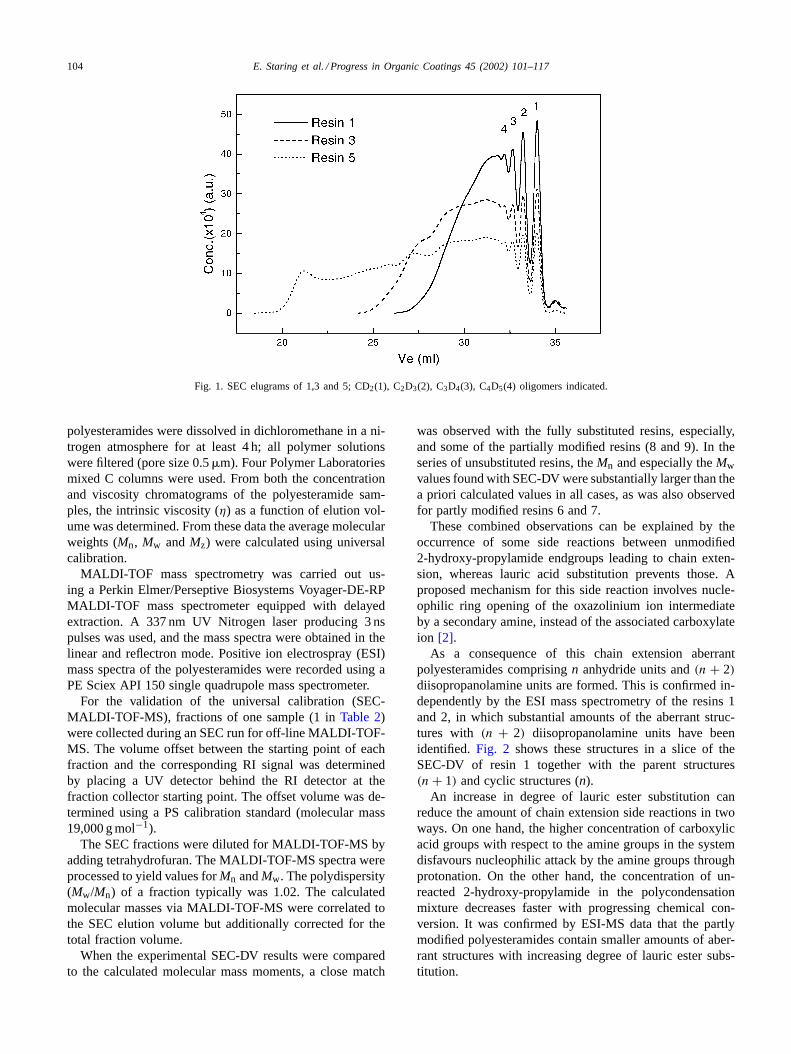
Table 2summarises the experimental SEC-DV results, incorrelation with the calculated molecular mass moments,of the three series of hyperbranched polyesteramide resinsdisplayed inTable 1. Fig. 1 illustrates the SEC elugrams ofthree unsubstituted polyesteramides.
Experimental. The SEC measurements were performedon a Hewlett-Packard chromatograph (HP 1090) equippedwith a differential refractometer (RI) and a differentialviscosimeter (DV) placed in parallel (Viscotek200). The
104 E. Staring et al. / Progress in Organic Coatings 45 (2002) 101–117
Fig. 1. SEC elugrams of 1,3 and 5; CD2(1), C2D3(2), C3D4(3), C4D5(4) oligomers indicated.
polyesteramides were dissolved in dichloromethane in a ni-trogen atmosphere for at least 4 h; all polymer solutionswere filtered (pore size 0.5�m). Four Polymer Laboratoriesmixed C columns were used. From both the concentrationand viscosity chromatograms of the polyesteramide sam-ples, the intrinsic viscosity (η) as a function of elution vol-ume was determined. From these data the average molecularweights (Mn, Mw andMz) were calculated using universalcalibration.
MALDI-TOF mass spectrometry was carried out us-ing a Perkin Elmer/Perseptive Biosystems Voyager-DE-RPMALDI-TOF mass spectrometer equipped with delayedextraction. A 337 nm UV Nitrogen laser producing 3 nspulses was used, and the mass spectra were obtained in thelinear and reflectron mode. Positive ion electrospray (ESI)mass spectra of the polyesteramides were recorded using aPE Sciex API 150 single quadrupole mass spectrometer.
For the validation of the universal calibration (SEC-MALDI-TOF-MS), fractions of one sample (1 inTable 2)were collected during an SEC run for off-line MALDI-TOF-MS. The volume offset between the starting point of eachfraction and the corresponding RI signal was determinedby placing a UV detector behind the RI detector at thefraction collector starting point. The offset volume was de-termined using a PS calibration standard (molecular mass19,000 g mol−1).
The SEC fractions were diluted for MALDI-TOF-MS byadding tetrahydrofuran. The MALDI-TOF-MS spectra wereprocessed to yield values forMn andMw. The polydispersity(Mw/Mn) of a fraction typically was 1.02. The calculatedmolecular masses via MALDI-TOF-MS were correlated tothe SEC elution volume but additionally corrected for thetotal fraction volume.
When the experimental SEC-DV results were comparedto the calculated molecular mass moments, a close match
was observed with the fully substituted resins, especially,and some of the partially modified resins (8 and 9). In theseries of unsubstituted resins, theMn and especially theMwvalues found with SEC-DV were substantially larger than thea priori calculated values in all cases, as was also observedfor partly modified resins 6 and 7.
These combined observations can be explained by theoccurrence of some side reactions between unmodified2-hydroxy-propylamide endgroups leading to chain exten-sion, whereas lauric acid substitution prevents those. Aproposed mechanism for this side reaction involves nucle-ophilic ring opening of the oxazolinium ion intermediateby a secondary amine, instead of the associated carboxylateion [2].
As a consequence of this chain extension aberrantpolyesteramides comprisingn anhydride units and(n + 2)
diisopropanolamine units are formed. This is confirmed in-dependently by the ESI mass spectrometry of the resins 1and 2, in which substantial amounts of the aberrant struc-tures with (n + 2) diisopropanolamine units have beenidentified. Fig. 2 shows these structures in a slice of theSEC-DV of resin 1 together with the parent structures(n + 1) and cyclic structures (n).
An increase in degree of lauric ester substitution canreduce the amount of chain extension side reactions in twoways. On one hand, the higher concentration of carboxylicacid groups with respect to the amine groups in the systemdisfavours nucleophilic attack by the amine groups throughprotonation. On the other hand, the concentration of un-reacted 2-hydroxy-propylamide in the polycondensationmixture decreases faster with progressing chemical con-version. It was confirmed by ESI-MS data that the partlymodified polyesteramides contain smaller amounts of aber-rant structures with increasing degree of lauric ester subs-titution.

E. Staring et al. / Progress in Organic Coatings 45 (2002) 101–117 105
Fig. 2. Slice of SEC elugram of 1 as monitored by ESI-MS.
Both MALDI-TOF and ESI MS confirmed the generallyexpected structures including the increase of lauroyl sub-stitution of the polyesteramide backbones as a function ofthe designed degree of substitution. However, next to theaberrant structures in some samples, both MS techniques re-vealed the presence of cyclic structures in all the samplesinvestigated, see alsoFig. 2. Although cyclisation reactionswere expected to occur, they could not be taken into account
Fig. 3. Mark–Houwink plots of 1–5.
in our calculative approach. Therefore, our calculated val-ues inTable 2may need some correction; further studies toestablish this are underway.
The hyperbranched character of the polyesteramides un-der investigation was confirmed in two independent ways[2]. First, Mark–Houwink plots revealed slopes (α) of be-tween 0.295 and 0.373 for unsubstituted polyesteramides(seeFig. 3) and between 0.288 and 0.335 for the substituted
106 E. Staring et al. / Progress in Organic Coatings 45 (2002) 101–117
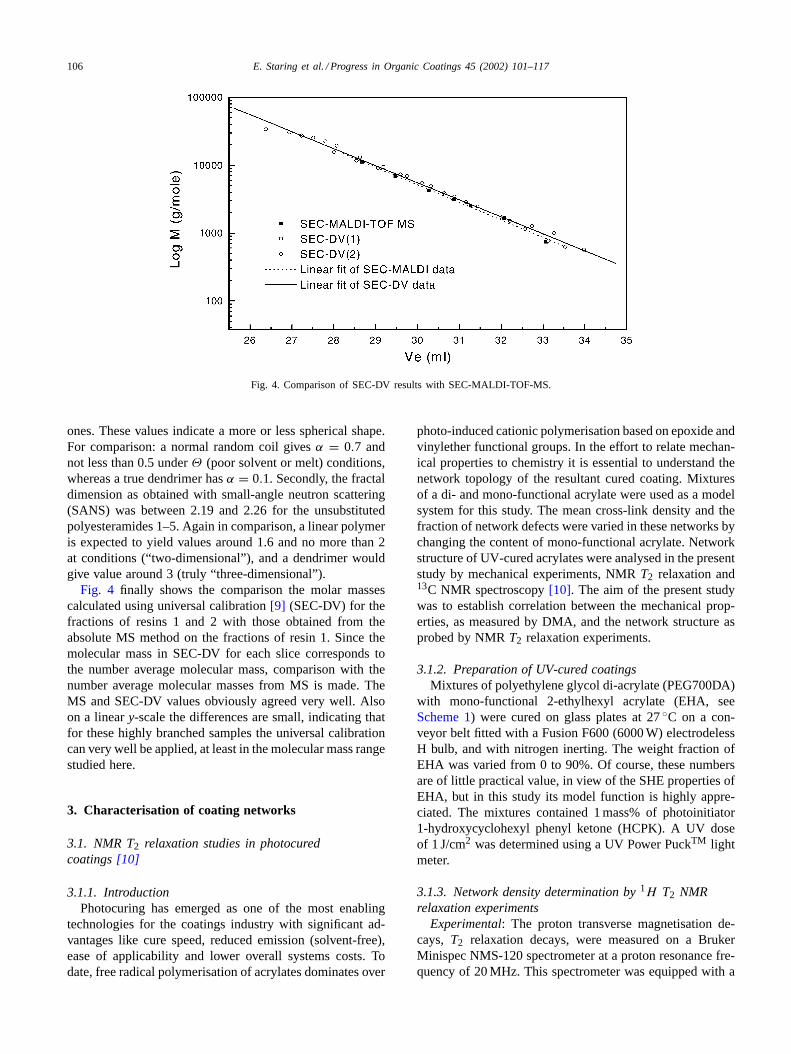
Fig. 4. Comparison of SEC-DV results with SEC-MALDI-TOF-MS.
ones. These values indicate a more or less spherical shape.For comparison: a normal random coil givesα = 0.7 andnot less than 0.5 underΘ (poor solvent or melt) conditions,whereas a true dendrimer hasα = 0.1. Secondly, the fractaldimension as obtained with small-angle neutron scattering(SANS) was between 2.19 and 2.26 for the unsubstitutedpolyesteramides 1–5. Again in comparison, a linear polymeris expected to yield values around 1.6 and no more than 2at conditions (“two-dimensional”), and a dendrimer wouldgive value around 3 (truly “three-dimensional”).
Fig. 4 finally shows the comparison the molar massescalculated using universal calibration[9] (SEC-DV) for thefractions of resins 1 and 2 with those obtained from theabsolute MS method on the fractions of resin 1. Since themolecular mass in SEC-DV for each slice corresponds tothe number average molecular mass, comparison with thenumber average molecular masses from MS is made. TheMS and SEC-DV values obviously agreed very well. Alsoon a lineary-scale the differences are small, indicating thatfor these highly branched samples the universal calibrationcan very well be applied, at least in the molecular mass rangestudied here.
3. Characterisation of coating networks
3.1. NMR T2 relaxation studies in photocuredcoatings [10]
3.1.1. IntroductionPhotocuring has emerged as one of the most enabling
technologies for the coatings industry with significant ad-vantages like cure speed, reduced emission (solvent-free),ease of applicability and lower overall systems costs. Todate, free radical polymerisation of acrylates dominates over
photo-induced cationic polymerisation based on epoxide andvinylether functional groups. In the effort to relate mechan-ical properties to chemistry it is essential to understand thenetwork topology of the resultant cured coating. Mixturesof a di- and mono-functional acrylate were used as a modelsystem for this study. The mean cross-link density and thefraction of network defects were varied in these networks bychanging the content of mono-functional acrylate. Networkstructure of UV-cured acrylates were analysed in the presentstudy by mechanical experiments, NMRT2 relaxation and13C NMR spectroscopy[10]. The aim of the present studywas to establish correlation between the mechanical prop-erties, as measured by DMA, and the network structure asprobed by NMRT2 relaxation experiments.
3.1.2. Preparation of UV-cured coatingsMixtures of polyethylene glycol di-acrylate (PEG700DA)
with mono-functional 2-ethylhexyl acrylate (EHA, seeScheme 1) were cured on glass plates at 27◦C on a con-veyor belt fitted with a Fusion F600 (6000 W) electrodelessH bulb, and with nitrogen inerting. The weight fraction ofEHA was varied from 0 to 90%. Of course, these numbersare of little practical value, in view of the SHE properties ofEHA, but in this study its model function is highly appre-ciated. The mixtures contained 1 mass% of photoinitiator1-hydroxycyclohexyl phenyl ketone (HCPK). A UV doseof 1 J/cm2 was determined using a UV Power PuckTM lightmeter.
3.1.3. Network density determination by 1H T2 NMRrelaxation experiments
Experimental: The proton transverse magnetisation de-cays, T2 relaxation decays, were measured on a BrukerMinispec NMS-120 spectrometer at a proton resonance fre-quency of 20 MHz. This spectrometer was equipped with a

E. Staring et al. / Progress in Organic Coatings 45 (2002) 101–117 107
Scheme 1. Components of the photocuring formulations.
BVT-3000 variable temperature unit. The temperature gra-dient and stability was about 1 and 0.1 K, respectively.
The decay of the transverse magnetisation was mea-sured with the Hahn-echo pulse sequence (HEPS),90x
◦–tHe–180x◦–tHe–acquisition, wheretHe = 35�s. Anecho signal is formed after the second pulse in the HEPSwith a maximum at timet = 2tHe after the first pulse. Byvarying the pulse spacing in the HEPS, the amplitude of thetransverse magnetisation,A(t), is measured as a function oftime t.
The T2 relaxation experiments were performed on sam-ples as whole and swollen samples. A certain amount of
Fig. 5. Schematic drawing ofT2 relaxation time against temperature for amorphous polymers[12].
1,1,2,2-C2D2Cl4 was added to the sample, then a Teflon plugis inserted so that the bottom is just above the sample creat-ing a slight free volume above the sample. The samples werestored for 1 day before the measurements were performed.In this case, 1,1,2,2-C2D2Cl4 was solvent of preference dueto its high boiling point, which permitted NMR experimentsat elevated temperatures.
A distinguishing feature ofT2 relaxation for thermoset-ting systems is the high-temperature plateau that is observedat temperatures well aboveTg (Fig. 5). The temperature in-dependence ofT2 is attributed to constraints which limit thenumber of possible conformations of network chain with

108 E. Staring et al. / Progress in Organic Coatings 45 (2002) 101–117
respect to those of a free chain. TheT2 at the plateau is(T2
p) quantitatively related to the mean molar mass of net-work chains with the relationships given below[10]. For aGaussian chain, in which the average, squared distance be-tween network junctions is much shorter than the contourchain length, theT2
p value is related toZ statistical segmentsbetween the network junctions:
Z = Tp2
aTrl2
(1a)
wherea is the theory coefficient, which depends on the anglebetween the segment axis and the internuclear vector forthe nearest nuclear spins at the main chain. TheT rl
2 value,which is measured for swollen samples belowTg, is relatedto the strength of intrachain proton–proton interactions inthe rigid lattice. Using the number of backbone bonds in thestatistical segment,Cω, the molar mass of network chainsbetween chemical and physical network junctions,Mc+e iscalculated:
Mc+e = ZCωMu
n(1b)
whereMu is the molar mass per elementary chain unit forthe polymer chain andn the number of backbone bonds inan elementary chain unit.
In addition to the NMR analysis, the mean molar massof network chains was calculated from the slope of the lin-ear part of the dependence of the modulus at temperaturesaboveTg. The following equation relates the slope of thedependence (E′/T) to the molar mass of network chains be-tween chemical cross-links and chain entanglements,Mc+e,in kg/kmol [4,11]:
Mc+e = 3ρRT(1 − x)
E(2)
whereρ is the specific density in kg/m3; R the gas constantthat equals 8.3 J/(mol K);E′ the modulus in Pa andx thevolume fraction of 2-ethyl hexyls fragment of EHA. Sug-gesting density additivity, a value ofx was calculated fromthe specific density of network chains (PEGDA) and alkylicfragment of EHA 1.11 and 0.86 g/cm3, respectively.Eq. (2)is applicable to affine networks of the same chain length andwithout network defects. It is recognised that the effectivenumber of elastically active network chains reduces in net-works with spatial clustering of cross-links[12–14]. Thismeans that a value ofMc+e is overestimated withEqs. (1a)and (1b)in the case of heterogeneous networks.
As expected, the glass transition temperature of the com-pounds in this study increased linearly upon an increase inthe content of di-functional monomer, as shown inFig. 6.Fox and Loshaek[15] or similar equations can describe thedependence:
Tg = T ∞g + kn
Mc(3)
whereT ∞g is theTg of a polymer of infinite molecular weight
andkn the material constant.
Fig. 6. Tg of cured acrylates against the content of EHA. The line repre-sents the result of a linear regression analysis: intercept= −39.0±0.4◦C;slope= −0.381±0.009◦C (mass%)−1. The correlation coefficient equals0.998.
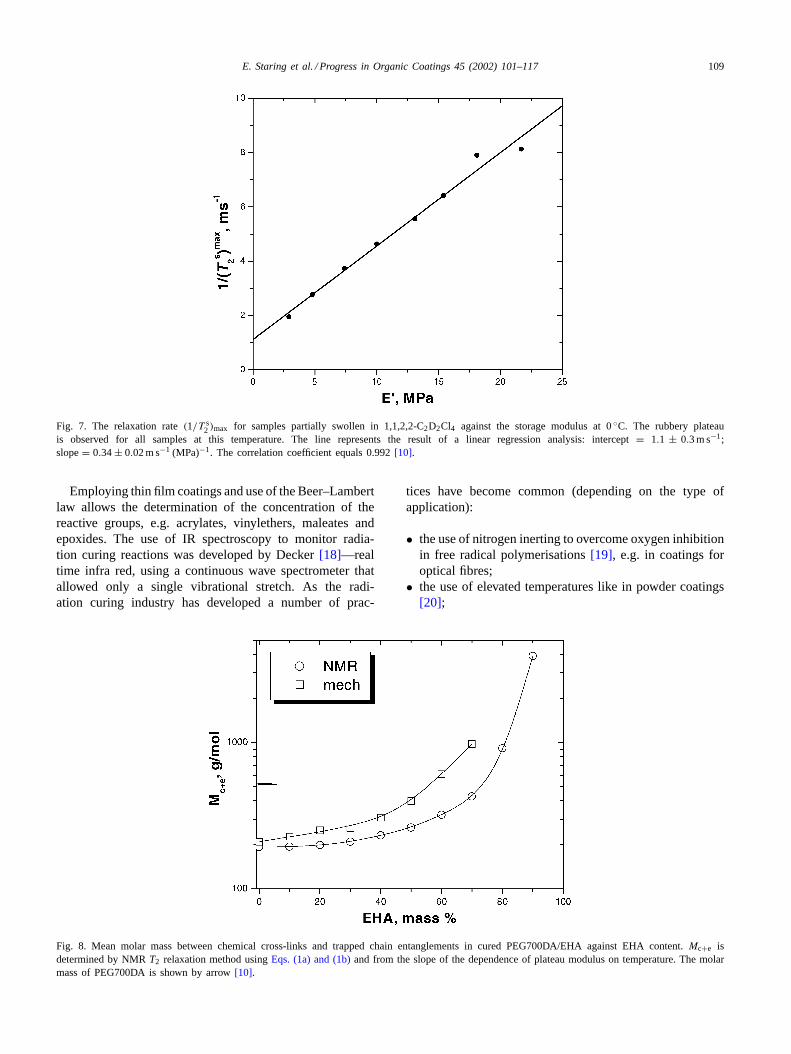
The NMR relaxation rate, 1/T2, and the storage modulusincrease proportionally with the content of difunctional acry-late (PEG700DA). The NMR relaxation rate, 1/T2, revealeda linear dependence against the storage modulus, seeFig. 7.Thus, the cross-link density in cured acrylates is inverselyproportional to the content of mono-functional monomer.
The molar mass of network chains,Mc+e, as calculatedfrom mechanical and NMRT2 relaxation data, is comparedin Fig. 8. The results of these two methods are in goodagreement, if one accounts for assumptions made for thecalculation of theMc+e from the NMR and mechanical data.The molar mass of network chains is significantly smallercompared to the molar mass of di-functional monomer forcured acrylates that is apparently caused by zip-like originof network junctions, seeFig. 9.
3.2. Real time FTIR for both liquid and powder radiationcurable systems
3.2.1. IntroductionCurrent photocuring processes vary from a multi-second
timescale to sub-second timescale, e.g. UV powder coatingsand optical fibre coatings, respectively. The techniques usedto monitor the degree of cure and the kinetics of photoiniti-ated polymerisation reactions are comprehensively reviewedby Rabek[16].
Infra Red spectroscopy has emerged as one of the mostversatile techniques in radiation curing since it yields thefollowing information about a photocuring reaction:
• the kinetics of UV-based radiation curing/polymerisation,• the degree of reactant conversion[17],• the induction time related to, for example, air or moisture
inhibition.
E. Staring et al. / Progress in Organic Coatings 45 (2002) 101–117 109
Fig. 7. The relaxation rate(1/T s2 )max for samples partially swollen in 1,1,2,2-C2D2Cl4 against the storage modulus at 0◦C. The rubbery plateau
is observed for all samples at this temperature. The line represents the result of a linear regression analysis: intercept= 1.1 ± 0.3 m s−1;slope= 0.34± 0.02 m s−1 (MPa)−1. The correlation coefficient equals 0.992[10].
Employing thin film coatings and use of the Beer–Lambertlaw allows the determination of the concentration of thereactive groups, e.g. acrylates, vinylethers, maleates andepoxides. The use of IR spectroscopy to monitor radia-tion curing reactions was developed by Decker[18]—realtime infra red, using a continuous wave spectrometer thatallowed only a single vibrational stretch. As the radi-ation curing industry has developed a number of prac-
Fig. 8. Mean molar mass between chemical cross-links and trapped chain entanglements in cured PEG700DA/EHA against EHA content.Mc+e isdetermined by NMRT2 relaxation method usingEqs. (1a) and (1b)and from the slope of the dependence of plateau modulus on temperature. The molarmass of PEG700DA is shown by arrow[10].
tices have become common (depending on the type ofapplication):
• the use of nitrogen inerting to overcome oxygen inhibitionin free radical polymerisations[19], e.g. in coatings foroptical fibres;
• the use of elevated temperatures like in powder coatings[20];

110 E. Staring et al. / Progress in Organic Coatings 45 (2002) 101–117
Fig. 9. Suggested network structure for compounds: (A) PEG700DA/EHA(80:20) and (B) PEG700DA(100)[10].
• the increase in the number of radiation curing chemistriessuch as free radical systems (acrylate, maleate–vinylether[21], thiolene[22]), cationic-photogenerated acids (withepoxide [23], vinylethers and propenylethers[24])anionic-photogenerated bases[25];
• the use of lamps with different emission spectra in the UVand visible lamps for surface and through cure and forvarious pigmented systems (often done with photoinitiatoroptimisation based on their absorption spectra)[26];
• the variety of monomers used in a given technology, e.g.N-vinyl pyrollidone has been used in acrylate formula-tions, etc.[27].
The development of UV powder curing technology[28]is an exciting development in the radiation curing industry.In UV powder curing, solid powder formulations are appliedto a substrate, melted with IR heat allowing the resin to flowand coat the substrate and then exposing the coating to UVlight for the curing/cross-linking reaction.
3.2.2. EquipmentAcknowledging these developments, a new real time infra
red apparatus that had to meet the following requirementswas built[29]:
• capable of measuring both liquid and solid radiation cur-able formulations;
• monitoring photocuring reactions under various atmo-spheres, e.g. with nitrogen inerting;
• monitoring photocuring reactions at various tempera-tures;
• allow the rates of consumption of different monomersin a formulation to be monitored simultaneously duringphotocuring, e.g. maleates and vinylethers, acrylatesandN-vinylamides, epoxides and vinylethers in cationicsystems.
The RT-FTIR instrument shown inFig. 10may be dividedinto three distinct parts: (i) the spectrophotometer, (ii) theillumination system and (iii) the sample.

E. Staring et al. / Progress in Organic Coatings 45 (2002) 101–117 111
Fig. 10. RT-FTIR experimental setup for liquid and powder samples.
i) The spectrophotometer (Bruker IFS55) has been fit-ted with a broadband MCT (mercury cadmium tel-luride) detector. The spectrophotometer can measurebetween 20 and 40 spectra/s depending on the res-olution required. Further hardware updates includedwere: acquisition processors (AQP) to store up to 600spectra in the memory of the instrument, and digitalinput/output ports controlled via the software (TTL)to control the UV illumination with high precision(1 ms).
ii) These systems are used together with liquid light guide(10 mm) to focus the UV onto the sample. The UV lightenergy delivered in the test cell was 150 mW/cm2 forthe Hg halide lamp and 75 mW/cm2 for the Hg lamp. Ashutter that is triggered by the spectrophotometer via adelay timer. The electronic delay timer was constructedat DSM Research.
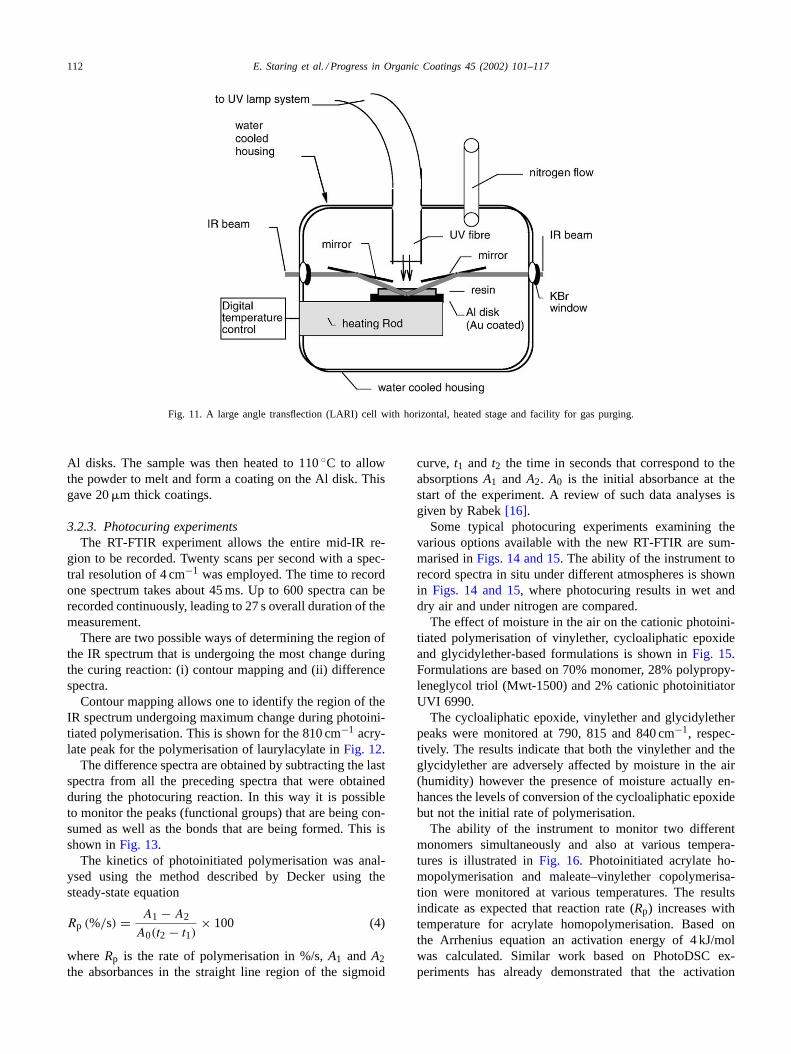
iii) A transflection (transmission–reflection) setup was cho-sen for our system. In a transflection system, the IRbeam is reflected on a mirror plate, this plate is cov-ered with the sample substance. The cell which met ourdemands as a very versatile system was a large anglereflectance infra red (LARI) cell. The construction isgiven inFig. 11.
The cell has an extra inlet that positions the optical lightguide directly above the sample, which is in a horizontal po-sition. The end of the light guide is 1 cm away from sample.The horizontal setup was used to eliminate problems withlow viscosity samples.
The features of this cell are:
• 70◦ LARI construction, to increase sensitivity.• Heatable sample holder with thermocouple (up to 500◦C)
with water cooled housing.• Digital temperature controller: this device allows to pro-
gram an initial temperature, a heating rate, and a finaltemperature, similar to a gas chromatograph.
• Gas tight housing: this allows for partial evacuation of thecell and/or purging with a suitable gas, e.g. a nitrogen.
Thin coating of 10–50�m is applied on a highly reflectiveAu-coated aluminium disk (thickness 0.5 mm) with a sizeof 7 mm× 7 mm and 8 mm× 8 mm. The surface of the Aldisk is made of polished Al oxide, which introduced broadbands in the single beam spectrum of the background inthe 3000–3700 and 1000–1800 cm−1 spectral range. Thesebands not only caused an intensity reduction of the reflectedlight, but led to baseline problems because of differences inthe reflection when the disk is coated with resin. To circum-vent this effect, the disks were gold plated with a sputteringtechnique normally used for electron microscopy.
Liquid coating sample preparation: The gold plated alu-minium disks were placed a recess (slot) of predetermineddepth (510 and 520�m) in a steel block. A few drops ofthe liquid formulation were placed beside the slot and theformulation drawn over the Al disks. In this way a coatinglayer of 10–20�m was obtained.
Powder coating sample preparation. A 45�m sieve car-rying the powder sample was shaken above the gold-coated
112 E. Staring et al. / Progress in Organic Coatings 45 (2002) 101–117
Fig. 11. A large angle transflection (LARI) cell with horizontal, heated stage and facility for gas purging.
Al disks. The sample was then heated to 110◦C to allowthe powder to melt and form a coating on the Al disk. Thisgave 20�m thick coatings.
3.2.3. Photocuring experimentsThe RT-FTIR experiment allows the entire mid-IR re-
gion to be recorded. Twenty scans per second with a spec-tral resolution of 4 cm−1 was employed. The time to recordone spectrum takes about 45 ms. Up to 600 spectra can berecorded continuously, leading to 27 s overall duration of themeasurement.
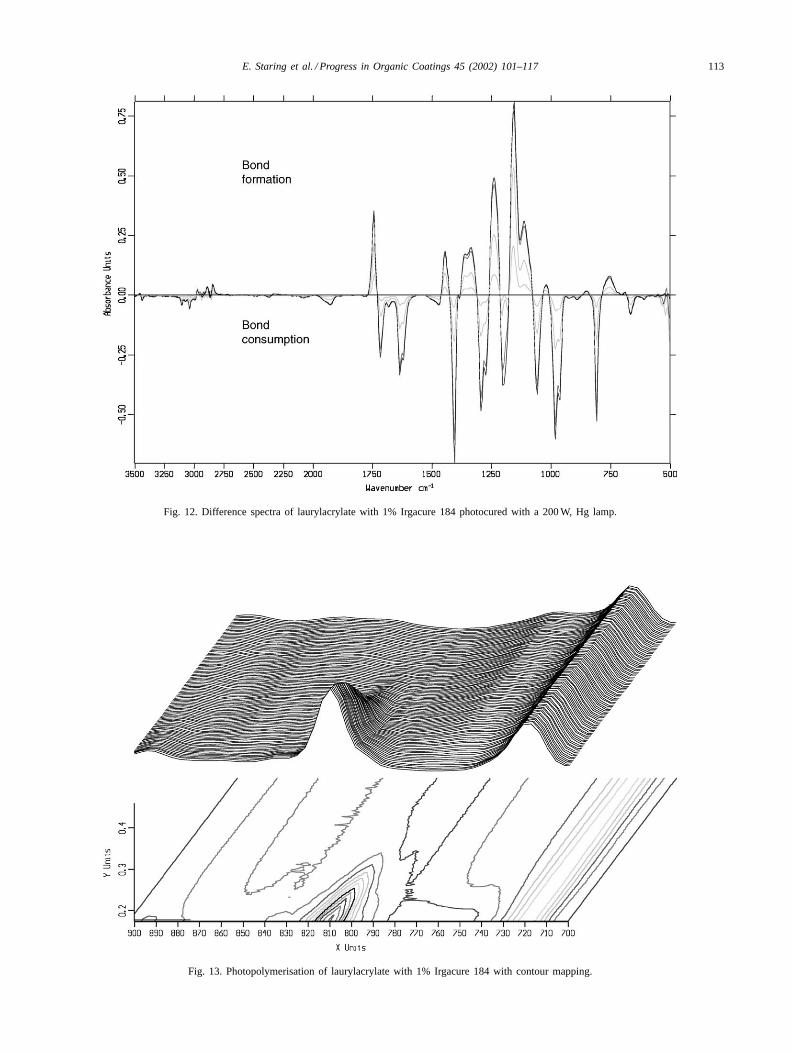
There are two possible ways of determining the region ofthe IR spectrum that is undergoing the most change duringthe curing reaction: (i) contour mapping and (ii) differencespectra.
Contour mapping allows one to identify the region of theIR spectrum undergoing maximum change during photoini-tiated polymerisation. This is shown for the 810 cm−1 acry-late peak for the polymerisation of laurylacylate inFig. 12.
The difference spectra are obtained by subtracting the lastspectra from all the preceding spectra that were obtainedduring the photocuring reaction. In this way it is possibleto monitor the peaks (functional groups) that are being con-sumed as well as the bonds that are being formed. This isshown inFig. 13.
The kinetics of photoinitiated polymerisation was anal-ysed using the method described by Decker using thesteady-state equation
Rp (%/s) = A1 − A2
A0(t2 − t1)× 100 (4)
whereRp is the rate of polymerisation in %/s,A1 and A2the absorbances in the straight line region of the sigmoid
curve, t1 and t2 the time in seconds that correspond to theabsorptionsA1 and A2. A0 is the initial absorbance at thestart of the experiment. A review of such data analyses isgiven by Rabek[16].
Some typical photocuring experiments examining thevarious options available with the new RT-FTIR are sum-marised inFigs. 14 and 15. The ability of the instrument torecord spectra in situ under different atmospheres is shownin Figs. 14 and 15, where photocuring results in wet anddry air and under nitrogen are compared.
The effect of moisture in the air on the cationic photoini-tiated polymerisation of vinylether, cycloaliphatic epoxideand glycidylether-based formulations is shown inFig. 15.Formulations are based on 70% monomer, 28% polypropy-leneglycol triol (Mwt-1500) and 2% cationic photoinitiatorUVI 6990.
The cycloaliphatic epoxide, vinylether and glycidyletherpeaks were monitored at 790, 815 and 840 cm−1, respec-tively. The results indicate that both the vinylether and theglycidylether are adversely affected by moisture in the air(humidity) however the presence of moisture actually en-hances the levels of conversion of the cycloaliphatic epoxidebut not the initial rate of polymerisation.
The ability of the instrument to monitor two differentmonomers simultaneously and also at various tempera-tures is illustrated inFig. 16. Photoinitiated acrylate ho-mopolymerisation and maleate–vinylether copolymerisa-tion were monitored at various temperatures. The resultsindicate as expected that reaction rate (Rp) increases withtemperature for acrylate homopolymerisation. Based onthe Arrhenius equation an activation energy of 4 kJ/molwas calculated. Similar work based on PhotoDSC ex-periments has already demonstrated that the activation
E. Staring et al. / Progress in Organic Coatings 45 (2002) 101–117 113
Fig. 12. Difference spectra of laurylacrylate with 1% Irgacure 184 photocured with a 200 W, Hg lamp.
Fig. 13. Photopolymerisation of laurylacrylate with 1% Irgacure 184 with contour mapping.

114 E. Staring et al. / Progress in Organic Coatings 45 (2002) 101–117
Fig. 14. Effect of nitrogen inerting with benzophenone and dimethylethanolamine as photoinitiators of ethoxylated TMPTA (Mw-607)[25].
energy for this homopolymerisation is low (2 kJ/mol)[32]. However, with the maleate–vinylether copolymerisa-tion starts levelling off at 70◦C. This may be attributedto the disruption of the ground state donor acceptorcomplex.
An experimental maleate–vinylether powder coating for-mulation was photocured at 100◦C and both maleate andvinylether peaks monitored as shown inFig. 17. This illus-
Fig. 15. Effect of moisture on the cationic photoinitiated polymerisation of cycloaliphatic, epoxide (CAE), vinylether (VE) and glycidylether (GE) basedformulations.
trates that it is possible to analyse a solid formulation in themelt phase.
Similar experiments have also been performed to look atmaleimides as photoinitiators in the polymerisation of acry-lates where the acrylate group at 810 cm−1 was monitoredsimultaneously with the maleimide group at 698 cm−1 Theimproved consumption of both maleimide and acylate whenphotocured under nitrogen was demonstrated[30].

E. Staring et al. / Progress in Organic Coatings 45 (2002) 101–117 115
Fig. 16. A comparison of the effect of temperature on the photoinitiated homopolymerisation of laurylacrylate and the copolymerisation of triethyleneg-lycoldivinylether and dioctyl fumarate (with 1% (w/w) Irgacure I84).
3.3. Real time dynamic mechanical analysis
To complement the developments in time-resolved spec-troscopy, time-resolved mechanical property measurementsfor curing systems are necessary. This is mainly due to thefact that chemical conversion does not translate readily tosimilar developments in mechanical properties. The compli-cation arises due to a number of factors, one of the mostimportant of which is the development of the network.
In recent years real time dynamic mechanical analysishas been developed[31]. Our efforts have also been devoted
Fig. 17. UV curing of powder coating based on experimental maleate–vinylether reactive groups at 110◦C with 1% (w/w) Irgacure 184.
to the modification of a dynamic mechanical analyser fittedwith UV transparent quartz parallel plates to allow simul-taneous UV illumination. Initial results suggest that this isreadily feasible for low modulus systems in the MPa range.This is attributed to torque limitations that make measure-ments prior to vitrification possible. Special modificationsare necessary to prevent the parallel plate from grinding to-gether due to shrinkage associated with cross-linking poly-merisation. Work is ongoing to examine UV powder coat-ings the results of which will be available at the time ofpresentation.

116 E. Staring et al. / Progress in Organic Coatings 45 (2002) 101–117
4. Conclusions
We have shown in this paper how new developments incharacterisation techniques on the molecular, mesoscopicand temporal level can improve our scientific understandingof such complex industrial products as coatings.
In Section 2, we have described an absolute method todetermine the molecular weight distribution of a new coat-ing resins with hyperbranched architecture in a combinationof SEC-DV and SEC-MS. It was established that fullylauric ester functionalised hyperbranched polyesteramides,synthesised in a straight-forward one-pot procedure, have amolecular weight distribution in accordance with calculatedvalues. MALDI-TOF and ESI-MS confirmed the expectedmolecular structures, but also indicated the formation ofcyclic structures that were excluded in the calculations. De-viations from the predicted molecular mass moments werefound with unfunctionalised and partly lauric acid esterfunctionalised(DS < 0.60) polyesteramides, due to sidereactions of 2-hydroxy-propylamide groups. The compar-ison between SEC-DV and SEC-MALDI-TOF-MS resultsappeared to confirm the principle of universal calibrationfor these polymers.
In Section 3, we have presented two techniques for thecharacterisation of (photocured) coatings. With the first,NMR T2 relaxation, we were able to gather relevant quanti-tative information on the network structure in a facile waywith minimal sample preparation. This method showedgood correlation with mechanical tests to determine molarmass between cross-links. An increase in the content ofmono-functional monomer in a UV-curable formulation re-sulted in the expected significant decrease in the cross-linkdensity; the mono-functional monomer acting as a chain ex-tender. Large fraction of mono-functional monomer (above80 mass%) caused significant fraction of network defects.Obtained values ofMc were significantly smaller comparedto molar mass of di-acrylate. As anticipated, it was apparentthat classical rubber elasticity theories are not applicable forcharacterisation of this type of networks containing zip-likenetwork junctions.
For the second, RT-FTIR, we have outlined a versatileexperimental setup. This instrument is capable of measuring20 spectra/s under various atmospheres and temperatures.The analyses of both liquid and solid (powder) samplescan be performed. The ability to control the atmosphere inthe transflection cell has been put to good use in demon-strating the varied behaviour of cycloaliphatic epoxides,glycidylethers and vinylethers under dry and humid con-ditions. Our future work will be directed to explain theseinitial findings.
Acknowledgements
The authors wish to thank their colleagues Eric Geladé,Chris de Koster and Theo Zwartkruis for their contributions
to the characterization of the hyperbranched polyesteramidesand Victor Litvinov, Paul Steeman and Jos Linsen for theircontributions to the NMR relaxation and real time studies,respectively. We thank the management of DSM Researchand DSM Coating Resins for the permission to publish thiswork.
References
[1] T.A. Misev, R.A.T.M. van Benthem, T.J.G. Zwartkruis, Bull. Chem.Tech. Maced. 17 (1998) 77–88;R.A.T.M. van Benthem, D. Muscat, D.A.W. Stanssens, Proc. Am.Chem. Soc. Div. PMSE 80 (1999) 72;H.J.J. Rutten, W. Grisnich, R.A.T.M. van Benthem, in: Proceedingsof the 16th Congress Federation Scandinavian Paint and VarnishTechnologists SLE, May 2000;R.A.T.M. van Benthem, Prog. Org. Coat. 40 (2000) 203;D. Muscat, R.A.T.M. van Benthem, in: Vogtle (Ed.), Topics in CurrentChemistry, Dendrimers III, Vol. 212, 2001, p. 41.
[2] R.A.T.M. van Benthem, E. Geladé, N. Meijerink, P. Froehling,P.H.M. Hendriks, D. Muscat, T.J.G. Zwartkruis, C.G. de Koster,Macromolecules 34 (2001) 3559–3566;E. Gelade, N. Meijerink, C.G. de Koster, R.A.T.M. van Benthem, K.Mortensen, R. Fokkens, N.N. Nibbering, Macromolecules 34 (2001)3552–3558.
[3] R.A.T.M. van Benthem, in: Proceedings of the 25th AthensConference on Organic Coatings, 1999, p. 345.
[4] P.J. Flory, Principles of Polymer Chemistry, Cornell University Press,Ithaca, NY, 1953.
[5] E. Malmström, A. Hult, Compare with the use of AB2 monomersin combination with B3 starter molecules in the synthesis ofhyperbranched polyesters, Macromolecules 29 (1996) 1222.
[6] D.A.W. Stanssens, R. Hermanns, H. Wories, Prog. Org. Coat. 22(1993) 379–391.
[7] D. Durand, C.-M. Bruneau, Br. Polym. J. (1981) 33–40;D. Durand, C.-M. Bruneau, Polymer 23 (1982) 69–72.
[8] D. Muscat, H. Henderickx, G. Kwakkenbos, R.A.T.M. van Benthem,C.G. de Koster, R. Fokkens, N.M.M. Nibbering, J. Am. Soc. MassSpectrom. 11 (2000) 218–227.
[9] Z. Grubisic, R. Rempp, H. Benoit, J. Chim. Phys. 63 (1966) 1507.[10] V. Litvinov, A.A. Dias, Macromolecules 34 (2001) 4051.[11] Y.Y. Gotlib, M.I. Lifshits, V.A. Shehelev, I.A. Lishanskii, I.V.
Balanina, Polym. Sci. USSR 18 (1976) 2630.[12] L.R.G. Treloar, The Physics of Rubber Elasticity, 3rd Edition,
Clarendon Press, Oxford, 1975.[13] T.A. Vilgis, G. Heinrich, Kautsch, Gummi Kunstst. 45 (1992)
1006.[14] K.-H. Schimmel, G. Heinrich, Coll. Polym. Sci. 269 (1991) 1003.[15] T.A. Vilgis, G. Heinrich, Makromol. Theory Simul. 3 (1994) 271.[16] T.G. Fox, S. Loshaek, J. Polym. Sci. 15 (1955) 371.[17] J.F. Rabek, in: J.P. Fouassier, J.F. Rabek (Eds.), Radiation Curing in
Polymer Science and Technology Fundamentals and Methods, Vol.1, Elsevier, London, 1993, p. 329.
[18] R.S. Davidson, K.S. Tranter, S. Wilkinson, in: D.R. Randell (Ed.),Radiation Curing of Polymers, Vol. II, RSC, Special Publication No.64, 1991, p. 400.
[19] C. Decker, K. Moussa, Makromol. Chem. 189 (1988) 2381;C. Decker, K. Moussa, Macromolecules 22 (1989) 4455.
[20] R. Holman, N. Arsu, E. Cockburn, R. Whiting, in: Proceedings ofthe Conference on Aspects of Photoinitiation, PRA, Egham, UK,1993, p. 245.
[21] J. Finter, I. Frischinger, T. Haug, R. Morton, in: Proceedings of theFourth Nurnberg Congress, PRA, Germany, 1997, p. 38.
[22] C. Decker, D. Decker, Polymer 38 (9) (1997) 2229.

E. Staring et al. / Progress in Organic Coatings 45 (2002) 101–117 117
[23] C.R. Morgan, F. Magnotta, A.D. Ketley, J. Polym. Sci. Polym. Chem.156 (1977) 627.
[24] S.P. Pappas, in: N.S. Allen (Ed.), Photopolymerisation andPhotoimaging Science and Technology, 1989, p. 55 (Chapter 2).
[25] J.V. Crivello, S.A. Bratlavsky, J. Polym. Sci. A 32 (1994) 2755;J.V. Crivello, S.A. Bratlavsky, J. Polym. Sci. A 32 (1994) 2919.
[26] M. Tsunuoka, Y. Shigeru, K. Ito, in: Proceedings of the Radtech,1996, p. 393.
[27] C. Armstrong, S. Herlihy, in: Proceedings of the Conference onAspects of Photoinitiation, PRA, Egham, UK, 1993, p. 1.
[28] J. Doughherty, F.J. Vara, P.K. Wolf, in: Proceedings of the Radtech,Vol. 1, Nashville, 1996, p. 80.
[29] S. Udding, F. Witte, S. de Jong, Product Finishing, April 1997,pp. 26 and 30.
[30] A.A. Dias, H. Hartwig, J.F.G.A. Jansen, Surf. Coat. Int. 83 (2000)382.
[31] A.A. Dias, J.F.G.A. Jansen, M. van Dijck, Surf. Coat. Int. JOCCA83 (2000) 502.
[32] B.S. Chiou, R.J. English, S.A. Khan, Macromolecules 29 (1996)5368.




















