
Na + Stimulated Na + /H + Exchange and an Unfavorable Ca 2+ Homeostasis Initiate the Cycloxygenase-2...
15
Oncology Research, Vol. 18, pp. 243–257 0965-0407/09 $90.00 + .00 Printed in the USA. All rights reserved. DOI: 10.3727/096504009X12596189659286 Copyright 2009 Cognizant Comm. Corp. E-ISSN 1555-3906 www.cognizantcommunication.com Na + -Stimulated Na + /H + Exchange and an Unfavorable Ca 2+ Homeostasis Initiate the Cycloxygenase-2 Inhibitors-Induced Apoptotic Signals in Colonic Epithelial Cells During the Early Stage of Colon Carcinogenesis Shailender Singh Kanwar,* 1 Karnati R. Roy,† 2 Bimla Nehru,* Pallu Reddanna,† and Sankar Nath Sanyal* *Department of Biophysics, Panjab University, Chandigarh, India †Department of Animal Sciences, School of Life Sciences, University of Hyderabad, Hyderabad, India Evidence suggests that nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit cycloxygenase (COX) and production of the proinflammatory prostaglandin, PGE 2 , and thus prevent carcinogenesis in the colon. In- deed, one of the specific COX-2 inhibitors, celecoxib, had been accepted by the US FDA for the treatment of familial adenomatous polyposis. However, the molecular mechanism of such inhibition is not clear, although apoptosis appears to be the dominant antiproliferative end effect. The present study delineates the intracellu- lar ionic milieu in the colonocytes that could generate strong apoptotic signals where DMH-induced carcino- genesis was studied in the initiation stage in rats and its regression with the COX inhibitors. While DMH treatment produced a significant elevation in the Na + /H + exchanger activity and resultant proton efflux, this was reversed by the NSAIDs, particularly so with celecoxib and etoricoxib compared to aspirin. Similarly, the intracellular pH was changed, with more alkalosis noted in DMH, which was reversed by NSAIDs. Also, an intracellular Ca 2+ build up was noted by Fura 2 AM, which was also supported by a reduced Ca 2+ ATPase and an enhanced inward movement of Ca 2+ . Further, mitochondrial dysfunction-related cyt C release, in- creased DNA ladder formation, activation of caspase-3, and cleavage product of poly (ADP-ribose) polymer- ase (PARP) were not seen in DMH but well noted in NSAIDs. Our results indicate that NSAIDs can induce apoptosis through a change in the colonic Na + /H + exchange, intracellular pH, and an unfavorable Ca 2+ homeostasis. Key words: Colon carcinogenesis; Na + /H + exchange; Intracellular pH; Ca 2+ homeostasis; Apoptosis INTRODUCTION one Na + outside. The ubiquitous NHE is the primary membrane transporter to regulate pH i and cell volume which contains a highly conserved Na + /H + binding site A sudden increase in intracellular pH (pH i ) compared to the interstitial pH (pH e ) has been shown to cause ex- and an inhibitory amiloride binding site in the extracel- lular domain (7,8). Several reports using different cell tensive biochemical and morphological changes in cells (1), which may further contribute either to the cellular lines demonstrated that if the Na + /H + exchangers were inhibited by amiloride analogs, acidification of the cells proliferation or activation of apoptotic signals. The in- terstitial environment in malignant tumors is found to be occurred with a concomitant induction of apoptosis (9– 11). In contrast, the growth factors have been shown to acidic relative to that in normal tissues (2 ). Therefore, an appropriate pH i is essential for various cellular func- increase pH i with an associated activation of cell stimu- lation and proliferation (7). Also, the phorbol esters acti- tions as well as survival and proliferation of the cells so that pH i regulatory mechanisms may be used as targets vate the exchanger nonspecifically (12). We have shown earlier that nonsteroidal anti-inflam- for cancer therapy (3). The pH i of normal growing cells is maintained at a steady state that is relatively more matory drugs (NSAIDs) such as aspirin, celecoxib, and etoricoxib can act as chemopreventive agents in experi- alkaline than the pH e that is maintained by a Na + /H + ex- changer (NHE) or called antiport in the plasma mem- mental colon cancer induced by 1,2-dimethylhydrazine (DMH) in a rat model (13–15) as they are known cyclox- brane (4–6), which exchanges one H + inside the cell for 1 Presently at John D. Dingell VA Medical Center, Department of Internal Medicine, School of Medicine, Wayne State University, Detroit, MI 48201, USA. 2 Presently at Department of Cell Biology, University of Virginia, Charlottesville, VA 22903, USA. Address correspondence to Prof Sankar N. Sanyal, Department of Biophysics, Panjab University, Chandigarh, 160 014, India. Tel: +91-172- 2534122; E-mail: [email protected] 243
Transcript of Na + Stimulated Na + /H + Exchange and an Unfavorable Ca 2+ Homeostasis Initiate the Cycloxygenase-2...

Oncology Research, Vol. 18, pp. 243–257 0965-0407/09 $90.00 + .00Printed in the USA. All rights reserved. DOI: 10.3727/096504009X12596189659286Copyright 2009 Cognizant Comm. Corp. E-ISSN 1555-3906
www.cognizantcommunication.com
Na+-Stimulated Na+/H+ Exchange and an Unfavorable Ca2+ HomeostasisInitiate the Cycloxygenase-2 Inhibitors-Induced Apoptotic Signals in Colonic
Epithelial Cells During the Early Stage of Colon Carcinogenesis
Shailender Singh Kanwar,*1 Karnati R. Roy,†2 Bimla Nehru,*Pallu Reddanna,† and Sankar Nath Sanyal*
*Department of Biophysics, Panjab University, Chandigarh, India†Department of Animal Sciences, School of Life Sciences, University of Hyderabad, Hyderabad, India
Evidence suggests that nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit cycloxygenase (COX) andproduction of the proinflammatory prostaglandin, PGE2, and thus prevent carcinogenesis in the colon. In-deed, one of the specific COX-2 inhibitors, celecoxib, had been accepted by the US FDA for the treatment offamilial adenomatous polyposis. However, the molecular mechanism of such inhibition is not clear, althoughapoptosis appears to be the dominant antiproliferative end effect. The present study delineates the intracellu-lar ionic milieu in the colonocytes that could generate strong apoptotic signals where DMH-induced carcino-genesis was studied in the initiation stage in rats and its regression with the COX inhibitors. While DMHtreatment produced a significant elevation in the Na+/H+ exchanger activity and resultant proton efflux, thiswas reversed by the NSAIDs, particularly so with celecoxib and etoricoxib compared to aspirin. Similarly,the intracellular pH was changed, with more alkalosis noted in DMH, which was reversed by NSAIDs. Also,an intracellular Ca2+ build up was noted by Fura 2 AM, which was also supported by a reduced Ca2+ ATPaseand an enhanced inward movement of Ca2+. Further, mitochondrial dysfunction-related cyt C release, in-creased DNA ladder formation, activation of caspase-3, and cleavage product of poly (ADP-ribose) polymer-ase (PARP) were not seen in DMH but well noted in NSAIDs. Our results indicate that NSAIDs caninduce apoptosis through a change in the colonic Na+/H+ exchange, intracellular pH, and an unfavorable Ca2+
homeostasis.
Key words: Colon carcinogenesis; Na+/H+ exchange; Intracellular pH; Ca2+ homeostasis; Apoptosis
INTRODUCTION one Na+ outside. The ubiquitous NHE is the primarymembrane transporter to regulate pHi and cell volumewhich contains a highly conserved Na+/H+ binding siteA sudden increase in intracellular pH (pHi) compared
to the interstitial pH (pHe) has been shown to cause ex- and an inhibitory amiloride binding site in the extracel-lular domain (7,8). Several reports using different celltensive biochemical and morphological changes in cells
(1), which may further contribute either to the cellular lines demonstrated that if the Na+/H+ exchangers wereinhibited by amiloride analogs, acidification of the cellsproliferation or activation of apoptotic signals. The in-
terstitial environment in malignant tumors is found to be occurred with a concomitant induction of apoptosis (9–11). In contrast, the growth factors have been shown toacidic relative to that in normal tissues (2 ). Therefore,
an appropriate pHi is essential for various cellular func- increase pHi with an associated activation of cell stimu-lation and proliferation (7). Also, the phorbol esters acti-tions as well as survival and proliferation of the cells so
that pHi regulatory mechanisms may be used as targets vate the exchanger nonspecifically (12).We have shown earlier that nonsteroidal anti-inflam-for cancer therapy (3). The pHi of normal growing cells
is maintained at a steady state that is relatively more matory drugs (NSAIDs) such as aspirin, celecoxib, andetoricoxib can act as chemopreventive agents in experi-alkaline than the pHe that is maintained by a Na+/H+ ex-
changer (NHE) or called antiport in the plasma mem- mental colon cancer induced by 1,2-dimethylhydrazine(DMH) in a rat model (13–15) as they are known cyclox-brane (4–6), which exchanges one H+ inside the cell for
1Presently at John D. Dingell VA Medical Center, Department of Internal Medicine, School of Medicine, Wayne State University, Detroit, MI48201, USA.2Presently at Department of Cell Biology, University of Virginia, Charlottesville, VA 22903, USA.Address correspondence to Prof Sankar N. Sanyal, Department of Biophysics, Panjab University, Chandigarh, 160 014, India. Tel: +91-172-2534122; E-mail: [email protected]
243

244 KANWAR ET AL.
ygenase (COX) enzyme inhibitors and thereby inhibit Isolation of Colonic Brush Border Membrane (BBM)and Colonocytesthe proinflammatory molecule prostaglandin E2 (16).
The role of NSAIDs in regulating the Na+/H+ antiport-The BBM of rat colon was isolated using the method
mediated pHi is an area of research that has receivedof Brasitus and Keresztes (17) and had been variously
little attention and the monitoring of this regulatorydescribed in our earlier publications (13–15). The final
mechanism is thought to be crucial to the role ofmembrane so obtained was analyzed for the membrane-
NSAIDs on the intracellular conditions during the re-specific enzyme, cysteine-sensitive alkaline phosphatase
gression of colon cancer. In the present study, therefore,(14). Colonic epithelial cells (colonocytes) were ob-
NHE activity and pHi were determined along with thetained from the freshly dissected colons by the method
Ca2+ homeostasis to identify the possible initiatingof Mouille at al. (18) (Fig. 1). The integrity of the colon-
mechanism of NSAID-induced cellular death in colonocytes was assessed by the ability of the cells to exclude
carcinogenesis.vital dye, trypan blue. The number of colonocytes resus-pended in DMEM buffered with morpholinopropane
MATERIALS AND METHODS sulfonic acid (MOPS) (pH 7.5, 25 mM) was counted onAnimal Husbandry and Drug Treatment a hemocytometer. It was determined that the isolation
procedure led to the recovery of the viable colonocytes,Male Sprague-Dawley rats weighing 140–150 g were
to the extent of at least 97%. In order to assess the effi-procured from the Central Animal House of Panjab Uni-
ciency of colonic epithelial cell retrieval, pieces of mu-versity and housed comfortably in polypropylene cages
cosa were fixed in 10% formalin saline both before andunder hygienic conditions, ambient light and tempera-
after cell separation. The tissue was processed for paraf-ture (18–22°C). They were fed with pellet diet and
fin embedding and then the sections stained with hema-drinking water ad libitum and assigned into the follow-
toxylin and eosin (19).ing groups: group 1—control (vehicle treated); group2—DMH treated; group 3—DMH + aspirin; group 4—
Sodium-Stimulated Na+-H+ Exchange in BBMDMH + celecoxib; and group 5—DMH + etoricoxib.
and Intracellular pH (pHi) Assay in ColonocytesAnimals in groups 2 to 5 were given DMH weekly at adose of 30 mg/kg body weight subcutaneously (SC) for Na+-H+ exchange (NHE) in the BBM vesicles was
followed using the pH-sensitive fluorescent dye acridine6 weeks (13–15). In addition, the animals in groups 3to 5 received a daily dose of the respective NSAIDs orange by the method of Warnock et al. (20). The acri-
dine orange technique was chosen because it can pro-orally per kg body weight as follows: aspirin, 60 mg;celecoxib, 6 mg; etoricoxib, 0.6 mg. vide the time resolution needed for the measurement of
initial transport rates. The dye is a weak base with char-DMH (Sigma, St. Louis, MO, USA) was preparedfresh immediately before injection in 1 mM EDTA sa- acteristic excitation (493 nm) and emission maxima (530
nm). Both the protonated and the nonprotonated form ofline, the pH being adjusted to 7.0 using 100 mM NaOH.Aspirin, celecoxib, and etoricoxib were generous gifts the dye can fluoresce; however, only the nonprotonated
form can pass through the membrane. This will result infrom Ranbaxy Research Lab (Gurgaon, India). Their re-ported anti-inflammatory doses were prepared fresh ev- an accumulation of protonated acridine orange in acidic
compartments. Also, the intravesicular dye is quenchedery day in 0.5% sodium carboxymethyl cellulose. Thevehicle-treated animals were administered with 0.5% so- to such an extent that it does not contribute to the mea-
sured fluorescence. Thus, if the intravesicular mediumdium carboxymethyl cellulose every day per oral and 1mM EDTA saline solution once weekly by SC injection. is more acidic than the outer one, acridine orange will
accumulate in the intravesicular space and the dye fluo-The bodyweight of the rats was recorded weekly untilthe termination of the experiments. All the animal proce- rescence will be quenched (and vice versa).
Figure 2a shows a typical fluorescence recording, us-dures were approved by Panjab University Ethical Com-mittee on Experimental Animals for Biomedical Re- ing a Perkin Elmer Luminescence Spectrometer LS-55
equipped with computer programmed applications of re-search. The guidelines were prepared according to theprinciples of animal care laid down by the National In- cording the time-derived fluorescence intensities. Re-
sults obtained are expressed as fluorescence units perstitute of Health, USA (NIH publication No. 23-85, re-vised in 1985). Animals were acclimatized for a period second percent control.
Measurement of intracellular pH (pHi) was done fol-of 1 week prior to the experimental work. They weresacrificed in overnight fasted conditions 6 weeks after lowing the method of Wakabayashi and Groschner (21)
using fluorescent indicator dye, bis carboxyethyl car-beginning of the study. They were put under an over-dose of ether anesthesia and sacrificed around 0800 h to boxyfluorescein (BCECF). In the acetoxymethyl ester
form (BCECF-AM), the dye is incorporated into the liveavoid diurnal variation in the parameters studied.
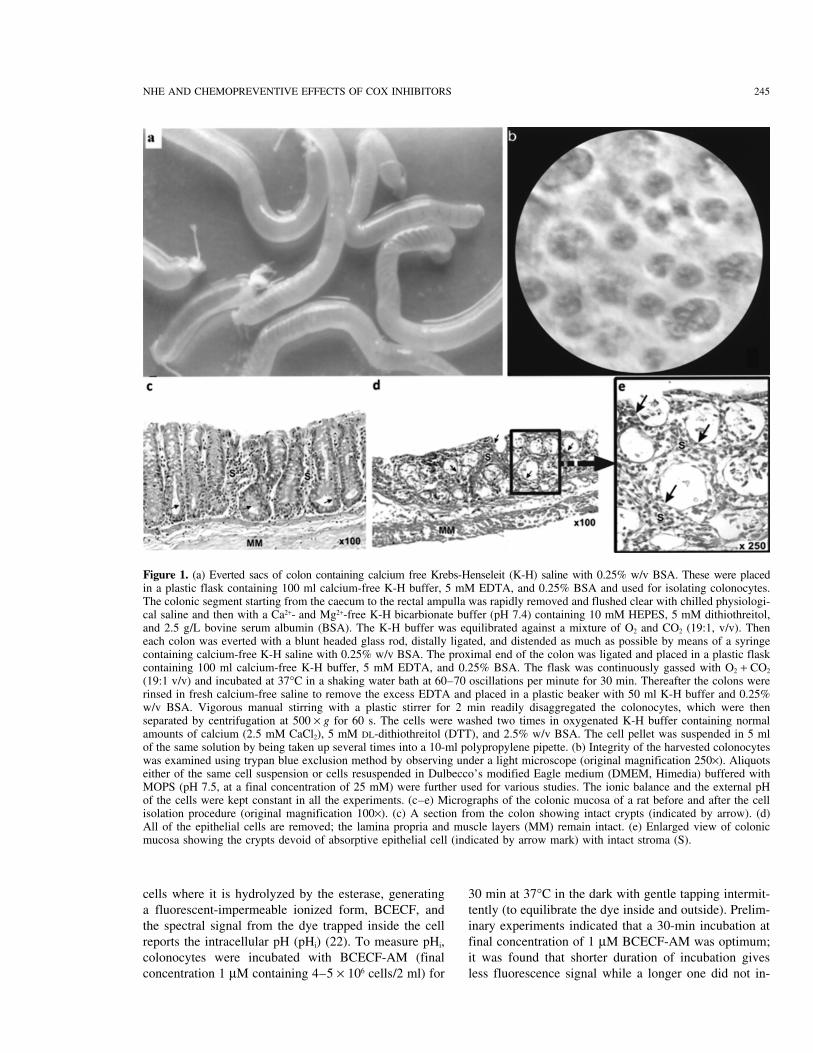
NHE AND CHEMOPREVENTIVE EFFECTS OF COX INHIBITORS 245
Figure 1. (a) Everted sacs of colon containing calcium free Krebs-Henseleit (K-H) saline with 0.25% w/v BSA. These were placedin a plastic flask containing 100 ml calcium-free K-H buffer, 5 mM EDTA, and 0.25% BSA and used for isolating colonocytes.The colonic segment starting from the caecum to the rectal ampulla was rapidly removed and flushed clear with chilled physiologi-cal saline and then with a Ca2+- and Mg2+-free K-H bicarbionate buffer (pH 7.4) containing 10 mM HEPES, 5 mM dithiothreitol,and 2.5 g/L bovine serum albumin (BSA). The K-H buffer was equilibrated against a mixture of O2 and CO2 (19:1, v/v). Theneach colon was everted with a blunt headed glass rod, distally ligated, and distended as much as possible by means of a syringecontaining calcium-free K-H saline with 0.25% w/v BSA. The proximal end of the colon was ligated and placed in a plastic flaskcontaining 100 ml calcium-free K-H buffer, 5 mM EDTA, and 0.25% BSA. The flask was continuously gassed with O2 + CO2
(19:1 v/v) and incubated at 37°C in a shaking water bath at 60–70 oscillations per minute for 30 min. Thereafter the colons wererinsed in fresh calcium-free saline to remove the excess EDTA and placed in a plastic beaker with 50 ml K-H buffer and 0.25%w/v BSA. Vigorous manual stirring with a plastic stirrer for 2 min readily disaggregated the colonocytes, which were thenseparated by centrifugation at 500 × g for 60 s. The cells were washed two times in oxygenated K-H buffer containing normalamounts of calcium (2.5 mM CaCl2), 5 mM DL-dithiothreitol (DTT), and 2.5% w/v BSA. The cell pellet was suspended in 5 mlof the same solution by being taken up several times into a 10-ml polypropylene pipette. (b) Integrity of the harvested colonocyteswas examined using trypan blue exclusion method by observing under a light microscope (original magnification 250×). Aliquotseither of the same cell suspension or cells resuspended in Dulbecco’s modified Eagle medium (DMEM, Himedia) buffered withMOPS (pH 7.5, at a final concentration of 25 mM) were further used for various studies. The ionic balance and the external pHof the cells were kept constant in all the experiments. (c–e) Micrographs of the colonic mucosa of a rat before and after the cellisolation procedure (original magnification 100×). (c) A section from the colon showing intact crypts (indicated by arrow). (d)All of the epithelial cells are removed; the lamina propria and muscle layers (MM) remain intact. (e) Enlarged view of colonicmucosa showing the crypts devoid of absorptive epithelial cell (indicated by arrow mark) with intact stroma (S).
cells where it is hydrolyzed by the esterase, generating 30 min at 37°C in the dark with gentle tapping intermit-tently (to equilibrate the dye inside and outside). Prelim-a fluorescent-impermeable ionized form, BCECF, and
the spectral signal from the dye trapped inside the cell inary experiments indicated that a 30-min incubation atfinal concentration of 1 µM BCECF-AM was optimum;reports the intracellular pH (pHi) (22). To measure pHi,
colonocytes were incubated with BCECF-AM (final it was found that shorter duration of incubation givesless fluorescence signal while a longer one did not in-concentration 1 µM containing 4–5 × 106 cells/2 ml) for
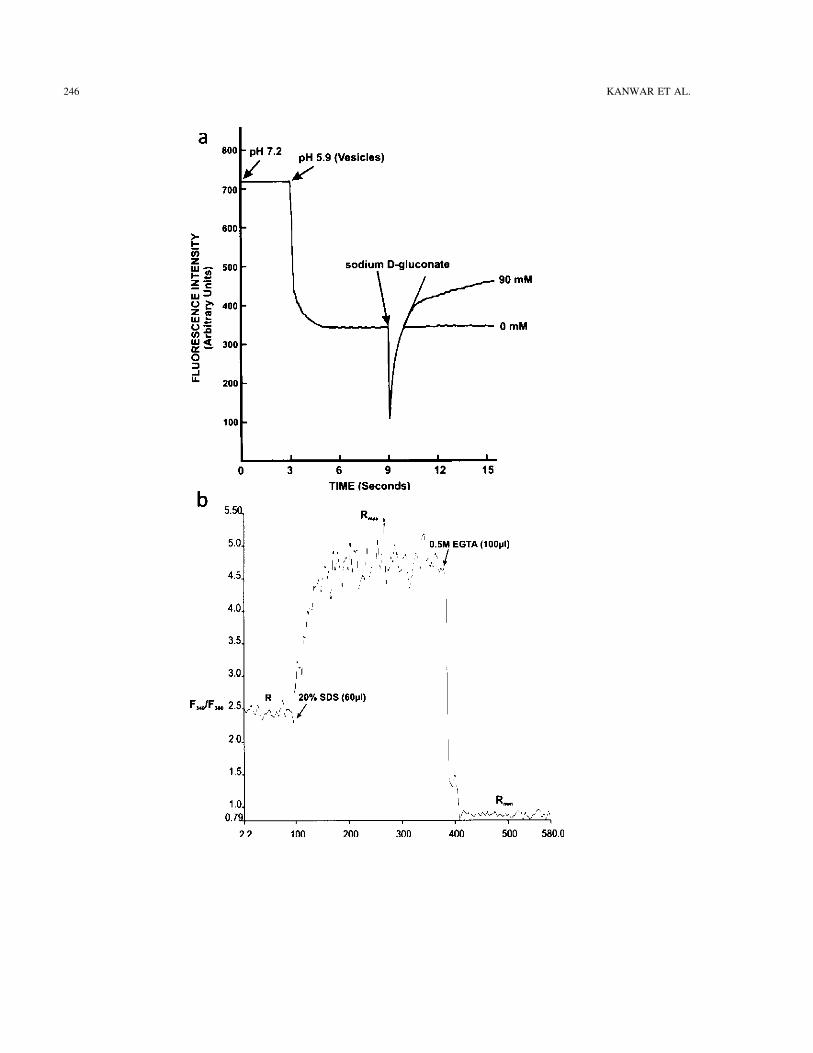
246 KANWAR ET AL.

NHE AND CHEMOPREVENTIVE EFFECTS OF COX INHIBITORS 247
crease it. The externally bound dye was washed off by of 530 nm recorded for different pH values were ana-lyzed by linear regression and the derived equation usedcentrifuging the suspension at 500 × g for 5 min and the
cells resuspended in the same volume of phosphate- to calculate the fractional pHi as described above.buffered saline (PBS) (Himedia, India; calcium andmagnesium free) buffered with MOPS, in a fluorescence Intracellular Calcium Concentration ([Ca2+]i) Assaycuvette.
Measurement of [Ca2+]i in the colonocytes was doneFluorescence measurements were carried out with a
using the modified method of Soldati et al. (24) usingdual-wavelength Luminescence Spectrometer using a 2-
the acetoxymethyl (AM) ester form of Ca2+-sensitive dyeml, 1-cm path length quartz cuvette and installed with
Fura-2. Fura-2AM is a cell-permeable fluorescent probeFL WinLab Software 4.00.02. Excitation and emission
metabolized inside the cell to the active ligand Fura-2,slits with a nominal band pass of 5 nm were used. Exci-
which then binds to free cytosolic Ca2+ and releases thetation wavelengths used were 506 nm and 455 nm, and
fluorescent AM molecule. The fluorescence intensityemission spectra collected at 530 nm. Using the Ratio
obtained as a result of dissociation of fluorophore AMData Collection method, the ratios (R) of fluorescence
from Fura-2 and the binding of later to Ca2+ in the cyto-intensity (F) of F506/F455 were determined (21). The frac-
solic environment gives the measure of [Ca2+]i as de-tional changes of pHi were obtained from R using the
scribed by Grynkiewicz et al. (25). Figure 2b shows theregression equation derived from the generated standard
typical spectra as obtained for Fura-2 studies in the co-curve as stated below.
lonocytes.Standard curve for the pH-dependent fluorescence of
BCECF in the colonocytes was generated as decribed byCa2+ Uptake in Colonocytes Using 45Ca-CaCl2Pardhasaradhi et al. (23). Thirty milliliters of 2 × 106/ml
of cells was loaded with BCECF-AM (1 µM final assay Ca2+ uptake in the isolated colonocytes was studiedin MOPS buffer, pH 7.4, as described previously (26).concentration) for 30 min at 37°C in the dark with gentle
tapping intermittently, then washed and resuspended in Incubation was started by adding colonocytes to the me-dium, which contained 5 µM CaCl2 and 45Ca-CaCl2 (530 ml of MOPS-PBS buffer. The cells were lysed with
0.1% Triton X-100 and homogenized using a probe-type µCi/100 ml) and made isotonic with the solution inwhich the colonocytes were suspended. After 10 min theultrasonicator (Sonics, USA). The homogenate was spun
at 1200 × g for 10 min to remove the cell debris and pH uptake was stopped by instant freezing and addition ofchilled MOPS buffer minus 45Ca2+ with a 10 times dilu-of the supernatant (2 ml v) adjusted to different values
between 6.2 and 8.1 using a research type pH meter. tion. The 45Ca-loaded colonocytes were collected byrapid filtration through a cellulose acetate/nitrate filterExcitation ratios (R) of F506/F455 at emission wavelength
FACING PAGE
Figure 2. (a) Typical fluorescent recording of Na+-H+ exchange activity as assayed by acridine orange. Two microliters of acridineorange (final concentration 1.5 µM) dissolved in 179 mM mannitol/18 mM Mes/47 mM Tris/79 mM HEPES, pH 7.2, waspipetted into a fluorescent cuvette. The solution was excited at 493 nm and the dye emission was registered at 530 nm. Later, atthe first break of the recording, 50 µl (150 µg protein) of colonic BBM vesicle preloaded for 1.5 h at 0°C in a medium containing191 mM mannitol, 91 mM Mes, 29 mM Tris, and 14 mM HEPES, pH 5.9, was added. At the second break of the fluorescenttrace, 20 µl of a sodium d-gluoconate stock solution was added, resulting in a final concentration of 90 mM. It was seen that thepresence of Na+ results in fluorescence recovery. This event reflects the Na+-H+ exchange activity of the membrane vesicles, theinitial rate of fluorescence recovery was calculated by using the slope of the tangent drawn through the recording obtained withinthe first 2 s after addition of Na+. (b) Representative curve of intracellular Ca2+ with Fura-2AM. Fura-2AM studies were performedin Perkin-Elmer LS-55 Luminescence Spectrometer using Ratio Data Collection application of FL WinLab software to calculateintracellular free calcium ion concentration [Ca2+]i in the isolated rat colonocytes. Isolated colonocytes (4–6 × 106 cells) wereincubated with 1 µM Fura-2AM (1 µg/µl in DMSO) for 30 min at 37°C, to allow loading of the dye. The cells were collectedby centrifugation at 500 × g for 10 min at 4°C, and resuspended in the PBS (calcium and magnesium free) buffered with MOPSbuffer (pH 7.4). The Fura 2-AM-loaded coloncytes were then transferred to a quartz cuvette and the fluorescence measured atexcitation wavelengths of 340 nm and 380 nm. An emission wavelength of 510 nm was used and emission slit width set at 5 nm.The fluorescence after sequential addition of SDS (20%) and 0.5 M EGTA (pH 8) to the cell suspension provided the maximumfluorescence ratio (Rmax) and minimum fluorescence ratio (Rmin), respectively. The average values of R, Rmax, and Rmin from thedifferent spectras were calculated with the Intracellular Biochemistry (ICBC) application of FL WinLab software package. Forevery experiment the autofluorescence of the colonocyte suspension without Fura-2 AM was measured and subtracted automati-cally from the total fluorescence of the loaded colonocytes. [Ca2+]i was calculated using the method of Grynkiewicz et al. (26):[Ca2+]i = [(R − Rmin)/(Rmax + R)] × Kd Fura-2, where R is the ratio of the fluorescence intensities measured at 340 and 380 nm, Rmax
is the same ratio when saturated with Ca2+ (after the addition of SDS), Rmin is the same ratio in the absence of Ca2+ (after thesequential addition of EGTA), and Kd is the dissociation constant of Fura-2–calcium complex, taken to be 225 nM.

248 KANWAR ET AL.
(0.45 µm pore size) (Sartorius, USA), which was pre- length of 505 nm (23). The specific activity of eachcaspase was calculated in terms of the pmol AMC pro-washed with the chilled MOPS buffer. Colonocytes
were added to the incubation medium in a ratio of 1:3 duced/mg protein/min and expressed as percentagechange with respect to the control.so that up to 2 × 106 cells were added to each filter. All
experiments were conducted at 20–22°C, which was ap-Assessment of Nucleosomal DNA Ladder Formationproximately the room temperature of the day. Immedi-by Agarose Gel Electrophoresisately after adding the sample, the filter was washed
twice with 2.5 ml of the chilled MOPS buffer as earlier. In apoptosis, genomic DNA of the cells undergoingapoptosis gets degraded into pieces of smaller base pairNonspecific binding of the radioactivity was determined
by placing 150 µl incubation medium (lacking colono- length (<180 bp ) and forms a DNA ladder that can bedetected by separating on agarose gel electrophoresis.cytes) on the filter and washing as described. The counts
on such a filter are accepted as the filter blank, which DNA from colonocytes was isolated by the method ofSambrook et al. (29). Cells were pelleted at 400 × g forwas subtracted from the sample counts. Counts of the
standards were also taken in which 45Ca-CaCl2 (5 µCi) 10 min, 500 µl of lysis buffer added, followed by 100µl of 10% SDS and mixed gently. Proteinase K (5 µl ofwas used in the scintillation cocktail.20 mg/ml stock used in 1 ml of cell suspension) wasthen added. The pellet was broken with a micropipetteCa2+ ATPase Activity Analysistip and reaction mixture agitated gently, and kept in a
Ca2+ ATPase was measured by the method of Desaiahwater bath at 56°C overnight. It was brought to room
et al. (27). The total ATPase was assayed in a reactiontemperature, equal amount of phenol-chloroform-isoa-
mixture containing 40 mM Tris-HCl buffer (pH 7.5), 5myl alcohol (PCA) mix added, and mixed gently until
mM MgCl2, 0.05 mM CaCl2, requisite amount of colon-an emulsion formed. The reaction mixture was centri-
ocyte lysate (4 × 106 colonocytes lysed in lysis bufferfuged at 10,000 × g for 15 min. The upper aqueous layer
containing 1% Triton X-100, 0.32 M sucrose, 5 mMwas carefully separated using a micropipette and to it
EDTA, 1 mM PMSF, 1 µg/ml aprotinin, 1 µg/ml leu-again equal amount of PCA was added and mixed gently
peptin, 2 mM DTT, 10 mM Tris-HCl, pH 8, for 15 min,for 15–20 min. After centrifugation, again the aqueous
4°C, followed by ultrasonication and centrifugation atlayer was transferred into a fresh vial and equal amount
20,000 × g for 5 min), and 2.5 mM ATP. The Mg2+-of chloroform-isoamyl alcohol mix added and centri-
ATPase was determined in the presence of 1 mMfuged. The aqueous phase was transferred into a fresh
EGTA, which specifically chelates Ca2+ ion and this wasvial and 1/10 volume of 3 M sodium acetate added. The
subtracted from the total activity to obtain the net Ca2+-vial was tapped gently. Then 2.5 volume of chilled
ATPase. The inorganic phosphate liberated by ATPase100% ethanol was added and kept at −20°C for 2 h for
was estimated by the method of Fiske and Subbarrowthe precipitation of DNA. The mix was centrifuged at
(28) using KH2PO4 as the standard. The results were ex-12,000 × g for 10 min to obtain the DNA pellet. This
pressed as nanomoles of Pi released/mg protein/min.pellet was washed with 70% ethanol two times. Thenthe DNA was dried, dissolved in 10 mM Tris/1 mM
Caspase-1 (ICE) and Caspase-3 (CPP32)EDTA (TE) buffer, and stored at −20°C until further
Activity Analysisuse.
DNA was dissolved in gel loading buffer and re-The activities of the caspase-1 and caspase-3-likeproteases were assayed by proteolytic cleavage of the solved by electrophoresis in 1.5 % agarose gel. Electro-
phoresis was performed by the method of Sambrook etrespective fluorogenic substrates. N-acetyl-Tyr-Val-ala-Asp-4-methylcoumaryl-7-amide (YVAD-AMC) is pref- al. (29). A 1.5% gel was cast and run in 0.5× TBE (45
mM Tris-borate buffer, pH 8.0, containing 1 mMerentially cleaved by caspase-1 while N-acteyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin (DEVD-AMC) is EDTA) at 5–10 v/cm. Six times gel loading (0.25% bro-
mophenol blue + 30% glycerol) was used for loading thecleaved by caspase-3. Colonocytes (2 × 106) were lysedin the lysis buffer as above and caspase-1 and caspase- samples (final concentration 1×). Ethidium bromide (0.5
µg/ml) was used for the staining of the sample and pho-3 activities were determined in the resulting supernatantat 50 µM final concentration of the assay buffer (100 tographed on UV transilluminator Gel-Doc (Foto Dyne,
Pharmacia, USA). A 100-bp DNA ladder (Genei, India)mM HEPES, 10% sucrose, 0.1% CHAPS, pH 7.5, 1 mMPMSF, 1 µg/ml aprotinin, 1 µg/ml leupeptin, 2 mM was used as a marker along with the samples.DTT). Standards containing AMC (0–1000 pmol) were
Morphological Evaluation of Apoptosis in Colonocytesused to determine the amount of the fluorochrome re-leased. Fluorescence recordings were made using an ex- Hoechst 33342–propidium iodide (PI) costaining is
used to stain DNA, because it is a DNA intercalatingcitation wavelength of 400 nm versus an emission wave-

NHE AND CHEMOPREVENTIVE EFFECTS OF COX INHIBITORS 249
dye and binds at A-T base pairs. Staining procedure was chrome C, rabbit polyclonal caspase-3, rabbit polyclonalPARP, rabbit polyclonal β-actin) diluted 1:2000 (1:500performed by the method of Yuan et al. (30). Briefly, 2
µl of 1 mg/ml PI was added to 100 µl of the harvested for cytochrome C) in 2% nonfat dry milk in TBST withgentle shaking for overnight at 4°C. The membrane wascells. The mixture was incubated at 37°C for 10 min
under dark conditions and 2 µl of 1 mg/ml Hoechst washed three times with TBST (1 × 15 min, 2 × 5 min)and incubated with the corresponding alkaline phospha-33342 dye added. Cell suspension was kept at room
temperature for 5 min; 50 µl was placed on a clean glass tase (ALP) conjugated secondary antibody (goat anti-mouse IgG-ALP conjugated for cytochrome C and bcl-slide and examined under a fluorescence microscope.
The percentage of apoptotic cells was calculated by 2 and goat anti-rabbit ALP conjugated for caspase-3,PARP, and β-Actin) diluted 1:2000 in 2% nonfat drycounting 100 cells in separate fields.milk TBST. The membrane was washed five times withTBST (1 × 15 min, 4 × 5 min) and the signals visualizedImmunoblottingby incubating with 10 ml ALP buffer (0.5 M Tris, pH9.5, 2 M NaCl, 5 mM MgCl2 per 100 ml of double dis-The protocol for Western blot was based on Sam-
brook et al. (29). Colonic mucosa was homogenized in tilled water) containing BCIP/NBT substrate for alkalinephosphatase (Genei, Bangalore, India). The reaction wasthe lysis buffer containing 10 mM Tris, 100 mM NaCl,
0.32 M sucrose, 5 mM EDTA, 1% Triton X-100, 1 mM stopped by washing the membrane with PBS, afterwhich the membrane was dried and stored.PMSF, 1 µg/ml aprotinin, 1 µg/ml leupeptin, 2 mM
DTT, pH 8, for 15 min at 4°C, followed by ultrasonica-Protein Estimationtion in an ice-cold environment and centrifugation
(10,000 × g, 30 min). Supernatant was used for protein The method was based on the Bradford method andestimation, SDS-PAGE, and Western blotting. The pro- the details given in the product description by the manu-tein content was determined according to the Bradford facturer (Sigma, USA). To 0.1 ml of lysate was added 3(31) method, and an equal amount of the sample (100 ml of Bradford reagent and vortexed gently for thoroughµg protein) was resolved on SDS-PAGE along with the mixing and incubated at room temperature for 45 min.molecular weight standards. Samples were read for absorbance at 595 nm. Appro-
SDS-PAGE was performed by the protocol of Laem- priate standards were also run.mli (32). The lysates were treated with 4× sample buffercontaining 1% SDS, 5% β-mercaptoethanol, 0.01% bro- Statistical Analysismophenol blue, and 20% glycerol in 0.063 M Tris-HCl,
Statistical examination of the data was performed bypH 6.8, for 5 min in a boiling water bath. Samples con-
the analysis of variances (one-way ANOVA) followingtaining 100 µg of protein were subjected to SDS-PAGE
post hoc test using the least significant difference (LSD)using 8–15% separating gel and with 5% stacking gel
method with the help of a SPSS computer softwareon 1-mm minigels. Electrophoresis was carried out at a
(SPSS Inc. Chicago, IL, USA).constant voltage (100 V) in a buffer containing 25 mMTris-HCl, 192 mM glycine, and 0.1% SDS, pH 8.3. Mo- RESULTS AND DISCUSSIONlecular weight markers were also run simultaneously.
Sodium-Stimulated Na+-H+ ExchangeWestern blotting was carried out according to theand Intracellular Alkalosisprocedure of Towbin et al. (33). The cell lysates as sepa-
rated on SDS-PAGE were transferred onto the nitrocel- Figure 3A shows the effect of DMH and DMH +NSAIDs on the sodium-stimulated NHE activity in thelulose membrane with a constant current at 70 V in a
buffer containing 25 mM Tris-HCl, 192 mM glycine, BBM during a 6-week treatment. In the DMH group theNHE activity was found to be enhanced while the inhibi-and 20% methanol for 4 h. The membrane was subse-
quently stained with 0.5% Ponceau S in 1% acetic acid tion of this antiport was observed in all the DMH +NSAID-treated animals. When compared with the con-to verify that equal amounts of protein were loaded in
each lane and transferred efficiently. trol, the increase was found to be 163.76% (p ≤ 0.001)as a result of DMH treatment. In the DMH + aspirinAfter electrophoretic transfer the nonspecific binding
sites were blocked by incubating the membrane with 5% group 81.19% (p ≤ 0.05) of inhibition was recorded andwhen DMH was coadministered with celecoxib andnonfat dry milk in Tris-buffered saline Tween (TBST)
(25 mM Tris, pH 8.0, 150 mM NaCl, 0.05% Tween 20, etoricixib, 38.14% (p ≤ 0.001) and 37.60% (p ≤ 0.001)inhibition was reported, respectively, in comparisonand 0.1% sodium azide) for overnight at room tempera-
ture. After washing three times in TBST (1 × 15 min, with the control. All the DMH + NSAIDs groups, whencompared with the DMH group alone, showed a signifi-2 × 5 min), the membrane was probed with primary anti-
body (mouse monoclonal bcl-2, mouse polyclonol Cyto- cant decline (p ≤ 0.001).
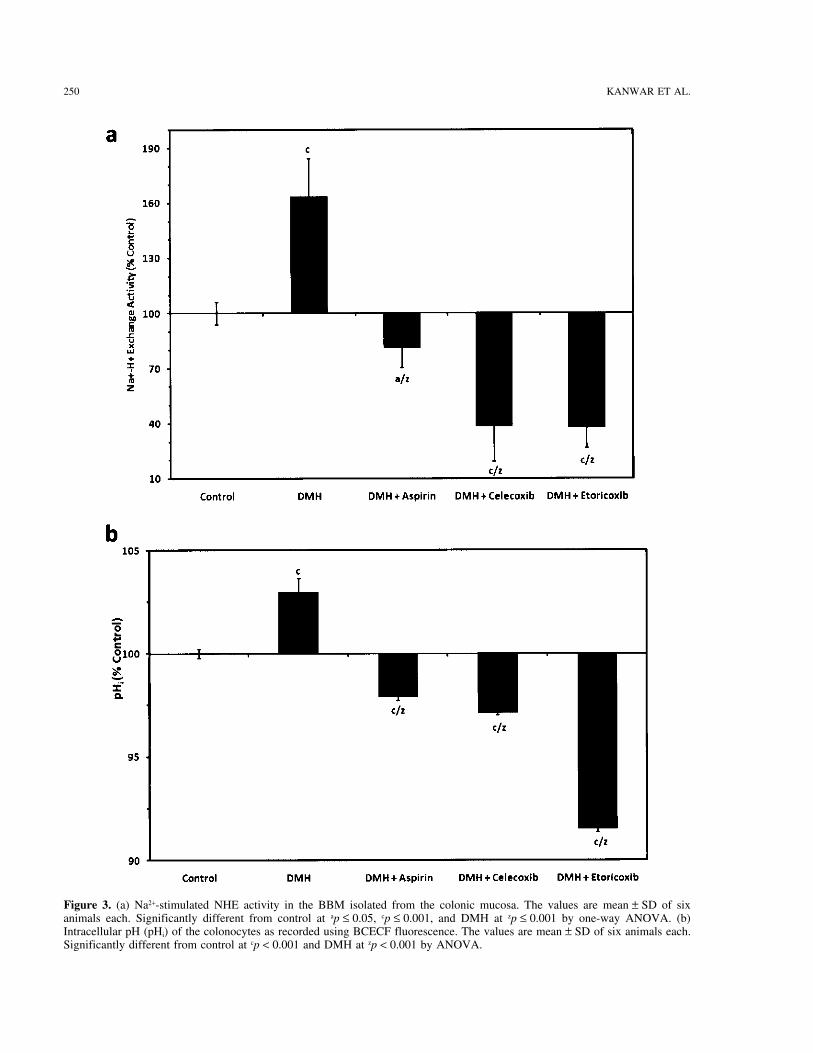
250 KANWAR ET AL.
Figure 3. (a) Na2+-stimulated NHE activity in the BBM isolated from the colonic mucosa. The values are mean ± SD of sixanimals each. Significantly different from control at ap ≤ 0.05, cp ≤ 0.001, and DMH at zp ≤ 0.001 by one-way ANOVA. (b)Intracellular pH (pHi) of the colonocytes as recorded using BCECF fluorescence. The values are mean ± SD of six animals each.Significantly different from control at cp < 0.001 and DMH at zp < 0.001 by ANOVA.
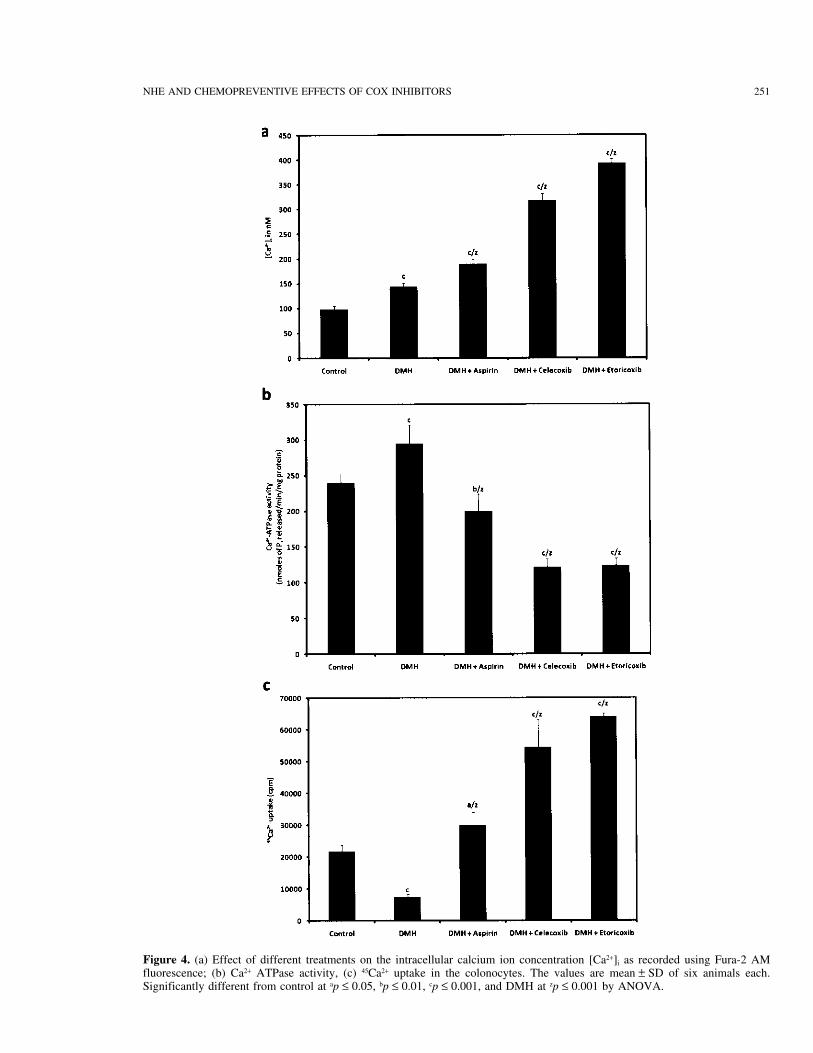
NHE AND CHEMOPREVENTIVE EFFECTS OF COX INHIBITORS 251
Figure 4. (a) Effect of different treatments on the intracellular calcium ion concentration [Ca2+]i as recorded using Fura-2 AMfluorescence; (b) Ca2+ ATPase activity, (c) 45Ca2+ uptake in the colonocytes. The values are mean ± SD of six animals each.Significantly different from control at ap ≤ 0.05, bp ≤ 0.01, cp ≤ 0.001, and DMH at zp ≤ 0.001 by ANOVA.
252 KANWAR ET AL.
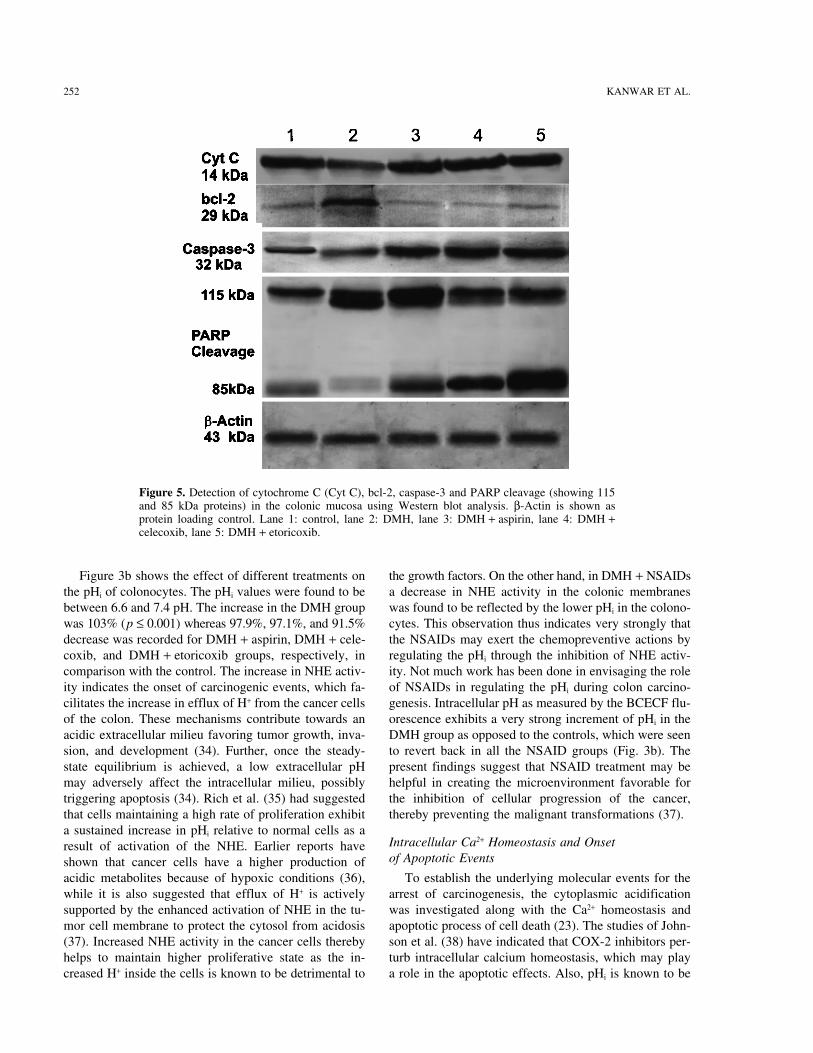
Figure 5. Detection of cytochrome C (Cyt C), bcl-2, caspase-3 and PARP cleavage (showing 115and 85 kDa proteins) in the colonic mucosa using Western blot analysis. β-Actin is shown asprotein loading control. Lane 1: control, lane 2: DMH, lane 3: DMH + aspirin, lane 4: DMH +celecoxib, lane 5: DMH + etoricoxib.
Figure 3b shows the effect of different treatments on the growth factors. On the other hand, in DMH + NSAIDsa decrease in NHE activity in the colonic membranesthe pHi of colonocytes. The pHi values were found to be
between 6.6 and 7.4 pH. The increase in the DMH group was found to be reflected by the lower pHi in the colono-cytes. This observation thus indicates very strongly thatwas 103% (p ≤ 0.001) whereas 97.9%, 97.1%, and 91.5%
decrease was recorded for DMH + aspirin, DMH + cele- the NSAIDs may exert the chemopreventive actions byregulating the pHi through the inhibition of NHE activ-coxib, and DMH + etoricoxib groups, respectively, in
comparison with the control. The increase in NHE activ- ity. Not much work has been done in envisaging the roleof NSAIDs in regulating the pHi during colon carcino-ity indicates the onset of carcinogenic events, which fa-
cilitates the increase in efflux of H+ from the cancer cells genesis. Intracellular pH as measured by the BCECF flu-orescence exhibits a very strong increment of pHi in theof the colon. These mechanisms contribute towards an
acidic extracellular milieu favoring tumor growth, inva- DMH group as opposed to the controls, which were seento revert back in all the NSAID groups (Fig. 3b). Thesion, and development (34). Further, once the steady-
state equilibrium is achieved, a low extracellular pH present findings suggest that NSAID treatment may behelpful in creating the microenvironment favorable formay adversely affect the intracellular milieu, possibly
triggering apoptosis (34). Rich et al. (35) had suggested the inhibition of cellular progression of the cancer,thereby preventing the malignant transformations (37).that cells maintaining a high rate of proliferation exhibit
a sustained increase in pHi relative to normal cells as aIntracellular Ca2+ Homeostasis and Onsetresult of activation of the NHE. Earlier reports haveof Apoptotic Eventsshown that cancer cells have a higher production of
acidic metabolites because of hypoxic conditions (36), To establish the underlying molecular events for thearrest of carcinogenesis, the cytoplasmic acidificationwhile it is also suggested that efflux of H+ is actively
supported by the enhanced activation of NHE in the tu- was investigated along with the Ca2+ homeostasis andapoptotic process of cell death (23). The studies of John-mor cell membrane to protect the cytosol from acidosis
(37). Increased NHE activity in the cancer cells thereby son et al. (38) have indicated that COX-2 inhibitors per-turb intracellular calcium homeostasis, which may playhelps to maintain higher proliferative state as the in-
creased H+ inside the cells is known to be detrimental to a role in the apoptotic effects. Also, pHi is known to be
NHE AND CHEMOPREVENTIVE EFFECTS OF COX INHIBITORS 253
involved in the upregulation of caspases while [Ca2+]i group while 191% (p ≤ 0.01) and 321% (p ≤ 0.001) in-crease was observed in the DMH + aspirin and DMH +plays an important role in activating endoplasmic reticu-
lum stress to induce cytochrome C release from the mi- celecoxib groups, respectively. The highest increase of396% (p ≤ 0.001) was observed in DMH + etoricoxib.tochondria, resulting in cell death (39). These reports
along with the results of NHE activity and pHi suggest Ca2+-ATPase was, however, highest in the DMH groupwhile lower activities were observed in the DMH +that pHi and [Ca2+]i may participate coherently in the
NSAID-induced apoptosis. The status of [Ca2+]i was in- NSAIDs. When DMH + NSAIDs were compared withDMH alone, a higher level of [Ca2+]i was found in thevestigated using the Ca2+-sensitive dye Fura-2AM and
assessing the uptake of radiolabeled Ca2+ in the colono- colonocytes accompanied by an inhibition in Ca2+-ATPase in DMH + NSAIDs. Figure 4c also shows thecytes along with the Ca2+-ATPase activity in colonic
membranes. The induction of apoptosis was assessed by uptake of 45CaCl2 in the colonocytes of the differentgroups, which shows very low 45Ca uptake in DMHmeasuring caspases in the colonocytes and also by mor-
phological evidence of apoptosis by fluorescent micros- while a much higher uptake was observed in the DMH+ NSAIDs.copy.
Figure 4a and b shows the effects of DMH and An increased level of [Ca2+]i in the DMH + NSAIDsin comparison to the DMH alone may signify that thisNSAIDs on [Ca2+]i in the colonocytes and Ca2+-ATPase
activity, respectively. [Ca2+]i was increased in the DMH- is an important step for the NSAID-activated apoptosis,which was further supported by an enhanced uptake oftreated animals; however, this increase was more promi-
nent where NSAIDs were given along with DMH. The 45Ca2+. In addition, Ca2+-ATPase was markedly inhib-ited in the NSAIDs, which is important for mitochon-increase with respect to control was 145% in the DMH
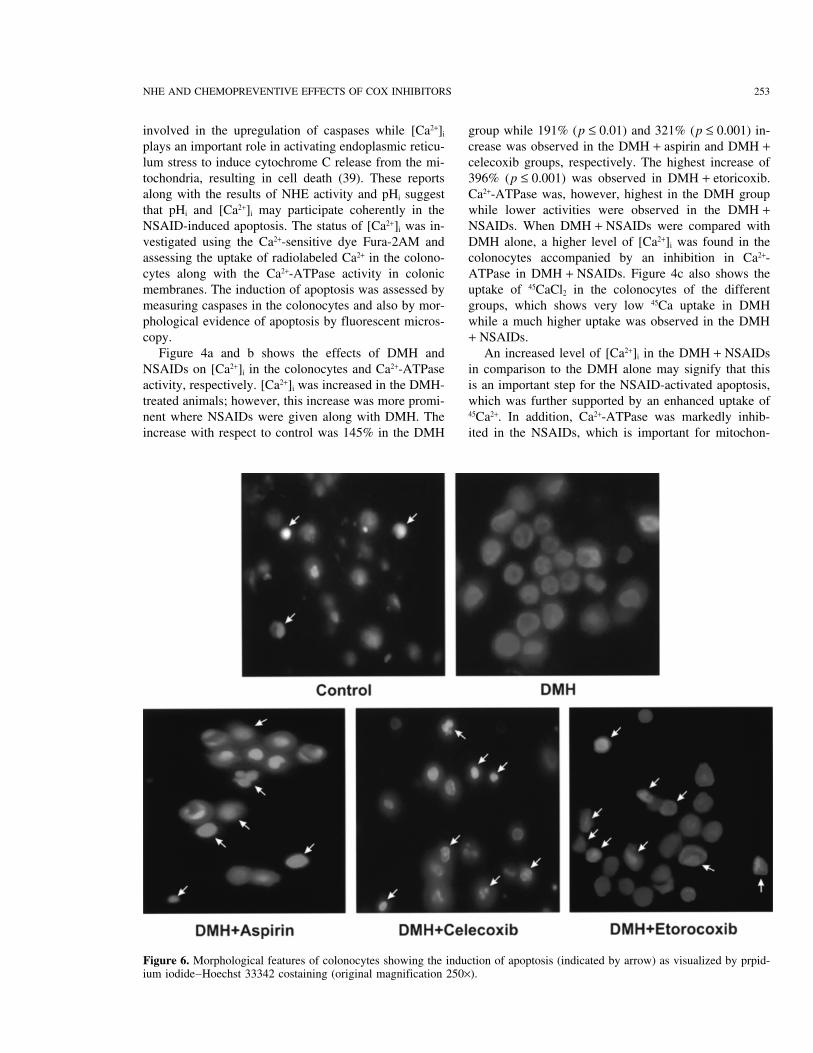
Figure 6. Morphological features of colonocytes showing the induction of apoptosis (indicated by arrow) as visualized by prpid-ium iodide–Hoechst 33342 costaining (original magnification 250×).
254 KANWAR ET AL.
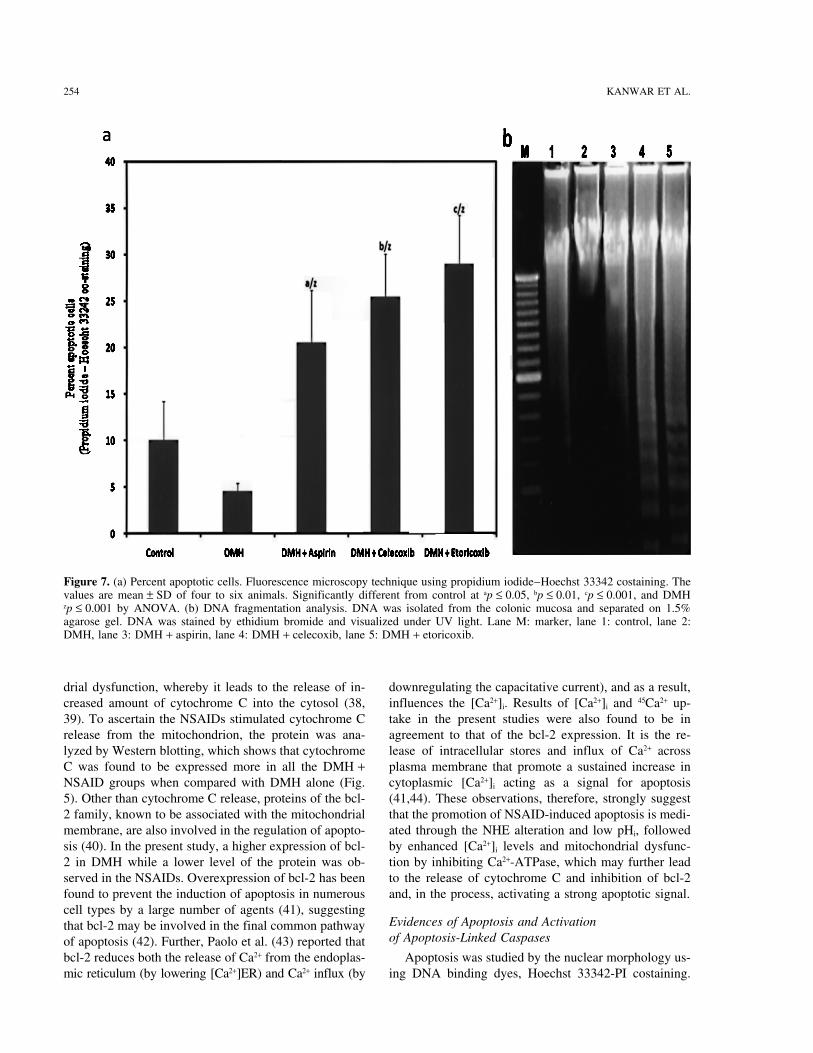
Figure 7. (a) Percent apoptotic cells. Fluorescence microscopy technique using propidium iodide–Hoechst 33342 costaining. Thevalues are mean ± SD of four to six animals. Significantly different from control at ap ≤ 0.05, bp ≤ 0.01, cp ≤ 0.001, and DMHzp ≤ 0.001 by ANOVA. (b) DNA fragmentation analysis. DNA was isolated from the colonic mucosa and separated on 1.5%agarose gel. DNA was stained by ethidium bromide and visualized under UV light. Lane M: marker, lane 1: control, lane 2:DMH, lane 3: DMH + aspirin, lane 4: DMH + celecoxib, lane 5: DMH + etoricoxib.
drial dysfunction, whereby it leads to the release of in- downregulating the capacitative current), and as a result,influences the [Ca2+]i. Results of [Ca2+]i and 45Ca2+ up-creased amount of cytochrome C into the cytosol (38,
39). To ascertain the NSAIDs stimulated cytochrome C take in the present studies were also found to be inagreement to that of the bcl-2 expression. It is the re-release from the mitochondrion, the protein was ana-
lyzed by Western blotting, which shows that cytochrome lease of intracellular stores and influx of Ca2+ acrossplasma membrane that promote a sustained increase inC was found to be expressed more in all the DMH +
NSAID groups when compared with DMH alone (Fig. cytoplasmic [Ca2+]i acting as a signal for apoptosis(41,44). These observations, therefore, strongly suggest5). Other than cytochrome C release, proteins of the bcl-
2 family, known to be associated with the mitochondrial that the promotion of NSAID-induced apoptosis is medi-ated through the NHE alteration and low pHi, followedmembrane, are also involved in the regulation of apopto-
sis (40). In the present study, a higher expression of bcl- by enhanced [Ca2+]i levels and mitochondrial dysfunc-tion by inhibiting Ca2+-ATPase, which may further lead2 in DMH while a lower level of the protein was ob-
served in the NSAIDs. Overexpression of bcl-2 has been to the release of cytochrome C and inhibition of bcl-2and, in the process, activating a strong apoptotic signal.found to prevent the induction of apoptosis in numerous
cell types by a large number of agents (41), suggestingEvidences of Apoptosis and Activationthat bcl-2 may be involved in the final common pathwayof Apoptosis-Linked Caspasesof apoptosis (42). Further, Paolo et al. (43) reported that
bcl-2 reduces both the release of Ca2+ from the endoplas- Apoptosis was studied by the nuclear morphology us-ing DNA binding dyes, Hoechst 33342-PI costaining.mic reticulum (by lowering [Ca2+]ER) and Ca2+ influx (by
NHE AND CHEMOPREVENTIVE EFFECTS OF COX INHIBITORS 255
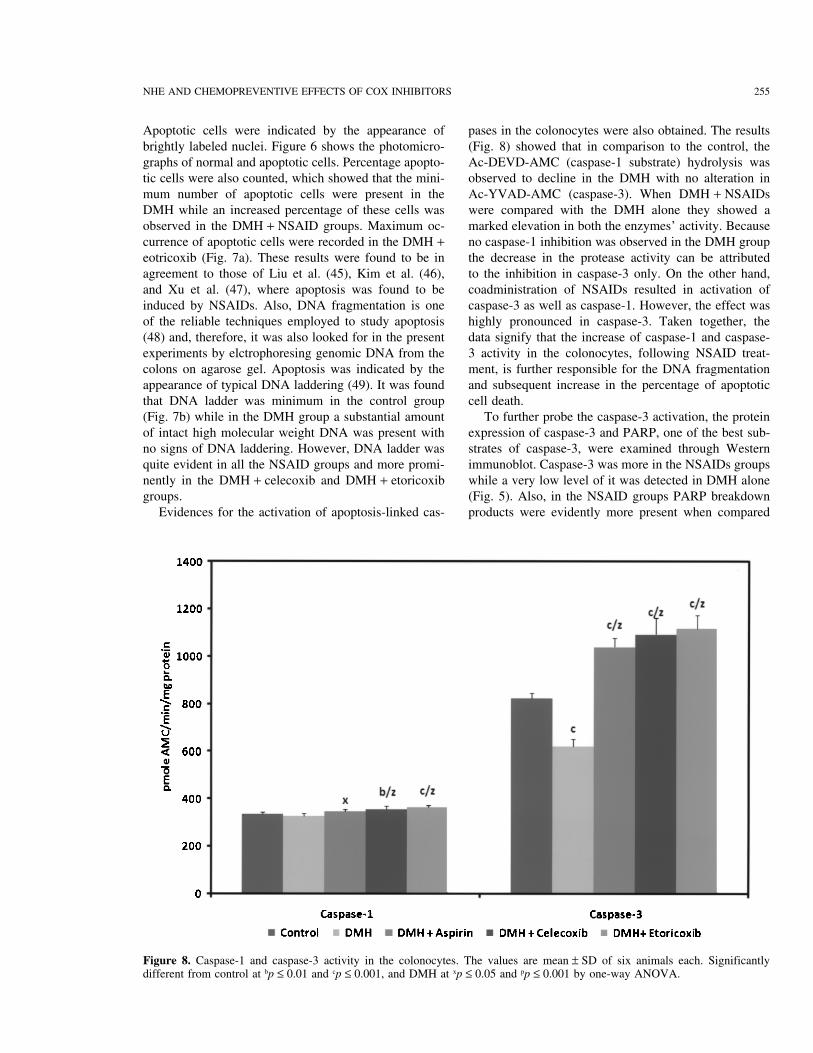
Apoptotic cells were indicated by the appearance of pases in the colonocytes were also obtained. The results(Fig. 8) showed that in comparison to the control, thebrightly labeled nuclei. Figure 6 shows the photomicro-
graphs of normal and apoptotic cells. Percentage apopto- Ac-DEVD-AMC (caspase-1 substrate) hydrolysis wasobserved to decline in the DMH with no alteration intic cells were also counted, which showed that the mini-
mum number of apoptotic cells were present in the Ac-YVAD-AMC (caspase-3). When DMH + NSAIDswere compared with the DMH alone they showed aDMH while an increased percentage of these cells was
observed in the DMH + NSAID groups. Maximum oc- marked elevation in both the enzymes’ activity. Becauseno caspase-1 inhibition was observed in the DMH groupcurrence of apoptotic cells were recorded in the DMH +
eotricoxib (Fig. 7a). These results were found to be in the decrease in the protease activity can be attributedto the inhibition in caspase-3 only. On the other hand,agreement to those of Liu et al. (45), Kim et al. (46),
and Xu et al. (47), where apoptosis was found to be coadministration of NSAIDs resulted in activation ofcaspase-3 as well as caspase-1. However, the effect wasinduced by NSAIDs. Also, DNA fragmentation is one
of the reliable techniques employed to study apoptosis highly pronounced in caspase-3. Taken together, thedata signify that the increase of caspase-1 and caspase-(48) and, therefore, it was also looked for in the present
experiments by elctrophoresing genomic DNA from the 3 activity in the colonocytes, following NSAID treat-ment, is further responsible for the DNA fragmentationcolons on agarose gel. Apoptosis was indicated by the
appearance of typical DNA laddering (49). It was found and subsequent increase in the percentage of apoptoticcell death.that DNA ladder was minimum in the control group
(Fig. 7b) while in the DMH group a substantial amount To further probe the caspase-3 activation, the proteinexpression of caspase-3 and PARP, one of the best sub-of intact high molecular weight DNA was present with
no signs of DNA laddering. However, DNA ladder was strates of caspase-3, were examined through Westernimmunoblot. Caspase-3 was more in the NSAIDs groupsquite evident in all the NSAID groups and more promi-
nently in the DMH + celecoxib and DMH + etoricoxib while a very low level of it was detected in DMH alone(Fig. 5). Also, in the NSAID groups PARP breakdowngroups.
Evidences for the activation of apoptosis-linked cas- products were evidently more present when compared
Figure 8. Caspase-1 and caspase-3 activity in the colonocytes. The values are mean ± SD of six animals each. Significantlydifferent from control at bp ≤ 0.01 and cp ≤ 0.001, and DMH at xp ≤ 0.05 and pp ≤ 0.001 by one-way ANOVA.

256 KANWAR ET AL.
7. Fliegel, L.; Frohlich, O. The Na+/H+ exchanger: An updateto DMH alone. It is the common feature of the cysteineon structure, regulation and cardiac physiology. Biochem.proteases with DEVD cleavage activity to cleave PARPJ. 296:273–285; 1993.
(Mr 115,000) enzyme into Mr 85,000 and Mr 25,000 frag- 8. Wakabayashi, I.; Groschner, K. Divergent effects of extra-ments (50). The appearance of native PARP was found cellular and intracellular alkalosis on Ca2+ entry pathways
in vascular endothelial cells. Biochem. J. 323:567–573;to be maximum in the DMH while its cleavage product1997.expressed more in NSAIDs.
9. Perez-Sala, D.; Collado-Escobar, D.; Mollinedo, F. Intra-The results in the present study provide an associa-cellular alkalinization suppresses lovastatin-induced apo-
tion between the pHi-induced apoptosis and the activa- ptosis in HL-60 cells through the inactivation of a pH-tion of caspase proteases as a result of altered NHE ac- dependent endonuclease. J. Biol. Chem. 270:6235–6242;
1995.tivity and Ca2+ homeostasis. This offers a strong molecular10. Chen, Q.; Benson, R. S.; Whetton, A. D.; Brant, S. R.;link behind the chemopreventive role of NSAIDs in co-
Donowitz, M.; Montrose, M. H.; Dive, C.; Watson, A. J.lon carcinogenesis. Further, the chemopreventive effectsRole of acid/base homeostasis in the suppression of apo-
were found to be associated more with the selective ptosis in haemopoietic cells by v-Abl protein tyrosine ki-COX-2 inhibitors like celecoxib and etoricoxib, com- nase. J. Cell. Sci. 110:379–387; 1997.
11. Tsao, N.; Lei, H. Y. Activation of the Na+/H+ antiporter,pared to the nonselective COX inhibitor, aspirin. HowNa+/HCO3−/CO32− cotransporter, or Cl−/HCO3− exchangerCOX inhibition relates to a change in NHE activity, pHi,in spontaneous thymocyte apoptosis. J. Immunol. 157:and an unfavorable Ca2+ homeostasis is a matter of con-1107–1116; 1996.
juncture. It is possible that the product of COX-2 en- 12. Owen, P. J.; Musk, P.; Evans, C. A.; Whetton, A. D. Cel-zyme, the prostaglandins (PGE2, PGJ2), which are lular signaling events elicited by v-abl associated with
growth factor independence in an interleukin-3-dependentknown ligands for the peroxisome proliferator activatorcell line. J. Biol. Chem. 268:15696–15703; 1993.receptor-γ (PPAR-γ) may upregulate it (51). PPAR-γ
13. Kanwar, S. S.; Vaiphei, K.; Nehru, B.; Sanyal, S. N.binds to the retinoic acid receptor, translocates to theChemopreventive effects of non steroidal anti-inflamma-
nucleus, and activates specific gene transcription (e.g., tory drugs on 1, 2-dimethylhydrazine-induced colon carci-Na+/H+ exchanger). PGE2 can also activate the nuclear nogenesis in rats. Toxicol. Mech. Methods 17:1–8; 2007.
14. Kanwar, S. S.; Vaiphei, K.; Nehru, B.; Sanyal, S. N.factor-κB (NF-κB), the master proinflammatory mole-Chemopreventive effects of non-steroidal anti-inflamma-cule for transcription of a number of oncogenes down-tory drugs in the membrane lipid composition and fluiditystream (52). Thus, either way PGE2 occupies a centralparameters of the 1,2-dimethylhydrazine induced colon
position and a specific COX-2 inhibitor may thus be carcinogenesis in rats. Drug Chem. Toxicol. 30:293–309;likely to bring about the apoptotic cell death as the end- 2007.
15. Kanwar, S. S.; Vaiphei, K.; Nehru, B.; Sanyal, S. N. Anti-effect in cancer cell killings.oxidative effects of non-steroidal anti-inflammatory drugs
ACKNOWLEDGMENTS: This work was partially supported (NSAIDs) during the initiation stages of experimental co-by research grants from Special Assistance Program sup- lon carcinogenesis in rats. J. Environ. Pathol. Toxicol. On-ported by University Grants Commission (UGC), New Delhi, col. 27:89–100; 2008.India to the Department of Biophysics, Panjab University, 16. Ricchi, P.; Zarrilli, R.; diPalma, A.; Acquaviva, A. M.Chandigarh, India. We also duly acknowledge Indian Council Nonsteroidal anti-inflammatory drugs in colorectal cancer:of Medical Research (ICMR), New Delhi, India for providing From prevention to therapy. Br. J. Cancer 88:803–807;financial assistance to S. S. Kanwar in the form of a Senior 2003.Research Fellowship. 17. Brasitus, T. A.; Keresztes, R. S. Protein-lipid interactions
in antipodal plasma membranes of rat coloncytes. Bio-REFERENCES chim. Biophys. Acta 773:290–300; 1984.18. Mouille, B.; Robert, V.; Blacheir, F. Adaptative increase1. Srinivas, U. K.; Revathi, C. J. Changes in cytoplasmic pH
upon heat shock in embryonic and adult rat liver cells. J. of ornithine production and decrease of ammonia metabo-lism in rat colonocytes after hyperproteic diet ingestion. J.Biosci. 18:175–186; 1993.
2. Song, C. W. Effect of local hyperthermia on blood flow Physiol. 287:G344–G351; 2004.19. Roediger, W. E.; Truelove, S. C. Method of preparing iso-and microenvironment: A review. Cancer Res. 44:4721s–
4730s; 1984. lated colonic epithelial cells (colonocytes) for metabolicstudies. Gut 20:484–488; 1979.3. Tannock, I. F.; Rotin, D. Acid pH in tumors and its poten-
tial for therapeutic exploitation. Cancer Res. 49:4373– 20. Warnock, D. G.; Reenstra, W. W.; Yee, V. J. Na+/H+ anti-porter of brush border vesicles: Studies with acridine or-4384; 1989.
4. Aronson, P. S. Kinetic properties of the plasma membrane ange uptake. Am. J. Physiol. 242:F733–F739; 1982.21. Wakabayashi, I.; Groschner, K. Divergent effects of extra-Na+/H+ exchanger. Annu. Rev. Physiol. 47:545–560; 1985.
5. Kim, G. E.; Lyons, C. J.; Song, C. W. Effects of amiloride cellular and intracellular alkalosis on Ca2+ entry pathwaysin vascular endothelial cells. Biochem. J. 323:567–573;on intracellular pH and thermosensitivity. Int. J. Radiat.
Oncol. Biol. Phys. 20:541–549; 1991. 1997.22. Rink, T. J.; Tsien, R. Y.; Pozzan, T. Cytoplasmic pH and6. Lyons, J. C.; Kim, G. E.; Song, C. W. Modification of
intracellular pH and thermosensitivity. Radiat. Res. 129: free Mg2+ in lymphocytes. J. Cell. Biol. 95:189–196;1982.79–87; 1992.

NHE AND CHEMOPREVENTIVE EFFECTS OF COX INHIBITORS 257
23. Pardhasaradhi, B. V. V.; Khar, A.; Srinivas, U. K. Effect 39. Tanaka, K.; Tomisato, W.; Hoshino, T.; Ishihara, T.;Namba, T.; Aburaya, M.; Katsu, T.; Suzuki, K.; Tsutsumi,of anti-apoptotic genes and peptide inhibitors on cyto-
plasmic acidification during apoptosis. FEBS Lett. 411: S.; Mizushima, T. Involvement of intracellular Ca2+ levelsin nonsteroidal anti-inflammatory drug-induced apoptosis.67–70; 1997.
24. Soldati, L.; Spaventa, R.; Vezzoli, G.; Zerbi, S.; Adamo, J. Biol. Chem. 280:31059–31067; 2005.40. Altunkan, A.; Aydin, O.; Ozer, Z.; Colak, T.; Bilgin, E.;D.; Caumo, A.; Rivera, R.; Bianchi, G. Characterization
of voltage-dependent calcium influx in human erythro- Oral, U. Anti-apoptotic effect of succinyl gelatine in aliver ischaemia-reperfusion injury model (Bcl-2, Bax, cas-cytes by fura-2. Biochem. Biophys. Res. Commun. 236:
549–554; 1997. pase 3)? Pharmacol. Res. 45:485–489; 2002.41. Zhang, Q. H.; Sheng, H. P.; Loh, T. T. Bcl-2 protects HL-25. Grynkiexicz, G.; Poenie, M.; Tsien, R. Y. A new genera-
tion of Ca2+ indicators with greatly improved fluorescence 60 cells from apoptosis by stabilizing their intracellularcalcium pools. Life Sci. 68:2873–2883; 2001.properties. J. Biol. Chem. 260:3440–3450; 1985.
26. Anand, R. J. K.; Kanwar, U.; Sanyal, S. N. Ca2+-transport 42. Antonsson, B.; Montessuit, S.; Lauper, S.; Eskes, R.;Martinou, J. C. Bax oligomerization is required for chan-across brush border and basal surfaces of human terms
placenta. Int. J. Exp. Biol. 34:786–793; 1996. nel-forming activity in liposomes and to trigger cyto-chrome c release from mitochondrion. Biochem. J. 345:27. Desaiah, D.; Chetty, C. S.; Prasada-Rao, K. S. Chlorde-
cone inhibition of calmodulin activated Ca2+ATPase in rat 1–8; 2000.43. Paolo, P.; Davide, F.; Elena, R.; Francesco, D. V.; Tullio,brain synaptosomes. J. Toxicol. Environ. Health 16:189–
196; 1985. P.; Rosario, R. A role for calcium in Bcl-2 action? Biochi-mie 84:195–201; 2002.28. Fiske, C. H.; Subbarow, Y. The colorimetric determina-
tion of phosphorus. J. Biol. Chem. 66:375–400; 1925. 44. Montague, J. W.; Cidlowski, J. A. Cellular catabolism inapoptosis: DNA degradation and endonuclease activation.29. Sambrook, J.; Fritsch, E. F.; Maniatis, T. Molecular clon-
ing: A laboratory manual, 2nd ed. Cold Spring Harbor, Experientia 52:957–962; 1996.45. Liu, X.; Yue, P.; Zhou, Z.; Khuri, F. R.; Sun, S. Y. DeathNY: Cold Spring Harbor Laboratory Press; 1989.
30. Yuan, Y.; Zhi-Qiang, G. E.; Jing-Chuan, L. Differentia- receptor regulation and celecoxib-induced apoptosis in hu-man lung cancer cells. J. Natl. Cancer Inst. 96:1769–1780;tion of apoptotic and necrotic cells in suspension cultures
of Taxus cuspidate by the cmbined use of fluorescent dy- 2004.46. Kim, S. H.; Song, S. H.; Kim, S. G.; Chun, K. S.; Lim,ing and histochemical staining methods. Biotechnol. Lett.
24:71–76; 2002. S. Y.; Na, H. K.; Kim, J. W.; Surh, Y. J.; Bang, Y. J.;Song, Y. S. Celecoxib induces apoptosis in cervical cancer31. Bradford, M. M. A rapid and sensitive method for the
quantitation of microgram quantities of protein utilizing cells independent of cyclooxygenase using NF-kappaB asa possible target. J. Cancer Res. Clin. Oncol. 130:551–the principle of protein-dye binding. Anal. Biochem. 72:
248–254; 1976. 560; 2004.47. Xu, Y.; Zhao, Y. M.; Huang, H. Celecoxib-induced apo-32. Laemmli, U. K. Cleavage of structural proteins during the
assembly of the head of bacteriophage T4. Nature 227: ptosis in acute promyelocytic leukemia cell line MR2 andits mechanism. Zhejiang Da Xue Xue Bao Yi Xue Ban (J.680–685; 1970.
33. Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic Zhejiang Univ. Med. Sci.) 36:319–324; 2007.48. Nath, R.; Scott, M.; Nadimpalli, R.; Gupta, R.; Wang,transfer of proteins from polyacrylamide gels to nitrocel-
lulose sheets: Procedure and some applications. Proc. K. K. W. Activation of apoptosis-linked caspases(s) inNMDA-injured brains in neonatal rats. Neurochem. Int.Natl. Acad. Sci. USA 76:4350–4354; 1979.
34. Pawel, S.; Vaughan-Jones, R.; Harris, A. Regulation of 36:119–126; 2000.49. Wyllie, A. Glucocorticoid-induced thymocyte apoptosis istumor pH and the role of carbonic anhydrase 9. Cancer
Metastasis Rev. 26:299–310; 2007. associated with endogenous endonuclease activation. Na-ture 284:555–556; 1980.35. Rich, I. N.; Worthington-White, D.; Garden, O. A.; Musk,
P. Apoptosis of leukemic cells accompanies reduction in 50. Kaufmann, S.; Desnoyers, S.; Ottaviano, Y.; Davidson,N.; Poirier, G. Specific proteolytic cleavage of poly(ADP-intracellular pH after targeted inhibition of the Na+/H+ ex-
changer. Blood 95:1427–1434; 2000. ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 53:3976–3985; 1993.36. Lyons, J. C.; Song, C. W. Killing of hypoxic cells by low-
ering the intracellular pH in combination with hyperther- 51. Brockman, J. A.; Gupta, R. A.; DuBois, R. N. Activationof PPARγ leads to anchorage-dependent growth of humanmia. Rad. Res. 141:216–218; 1995.
37. DeMilito, A.; Fais, S. Tumor acidity, chemoresistance and colorectal cancer cells. Gastroenterology 115:1049–1055;1998.proton pump inhibitors. Future Oncol. 1:779–786; 2005.
38. Johnson, A. J.; Hsu, A-L.; Lin, H-P.; Song, X.; Chen, 52. Sanchez-Alcezar, J. A.; Broadbury, D. A.; Pang, L.; Knox,A. P. Cyclooxygenase (COX) inhibitors induce apoptosisC-H. The cyclo-oxygenase-2 inhibitor celecoxib perturbs
intracellular calcium by inhibiting endoplasmic reticulum in non-small cell lung cancer cell line through cyclooxy-genase independent pathways. Lung Cancer 40:33–40;Ca2+-ATPases: A plausible link with its anti-tumor effects
and cardiovascular risks. Biochem. J. 366:831–837; 2002. 2003.




















