
Mutations in SLC34A3/NPT2c Are Associated with Kidney Stones and Nephrocalcinosis
10
CLINICAL RESEARCH www.jasn.org Mutations in SLC34A3/NPT2c Are Associated with Kidney Stones and Nephrocalcinosis Debayan Dasgupta,* Mark J. Wee,* Monica Reyes,* Yuwen Li,* Peter J. Simm, †‡ Amita Sharma, § Karl-Peter Schlingmann, | Marco Janner, ¶ Andrew Biggin, † Joanna Lazier,** Michaela Gessner, †† Dionisios Chrysis, ‡‡ Shamir Tuchman, §§ H. Jorge Baluarte, || Michael A. Levine, ¶¶ Dov Tiosano,*** Karl Insogna, ††† David A. Hanley, ‡‡‡ Thomas O. Carpenter, §§§ Shoji Ichikawa, ||| Bernd Hoppe, †† Martin Konrad, | Lars Sävendahl, ¶¶¶ Craig F. Munns, †‡ Hang Lee,**** Harald Jüppner,* § and Clemens Bergwitz* *Endocrine Unit, and § Pediatric Nephrology Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts; † Institute of Endocrinology and Diabetes, Children’ s Hospital at Westmead, Westmead, New South Wales, Australia; ‡ Discipline of Pediatrics and Child Health, University of Sydney, Sydney, Australia; | Department of General Pediatrics, University Children’ s Hospital, Münster, Germany; ¶ Division of Pediatric Endocrinology, Diabetology and Metabolism, University Children’ s Hospital, Bern, Germany; **Department of Medical Genetics, University of Calgary, Calgary, Alberta, Canada; †† Division of Pediatric Nephrology, Department of Pediatrics, University Hospital, Köln, Germany; ‡‡ Division of Endocrinology, Department of Pediatrics, University of Patras Medical School, Patras, Greece; §§ Division of Pediatric Nephrology, Children’ s National Medical Center, The George Washington University School of Medicine, Washington, District of Columbia; || University of Pennsylvania, School of Medicine, Division of Pediatric Nephrology, Children’ s Hospital of Philadelphia, Philadelphia, Pennsylvania; ¶¶ Division of Endocrinology and Diabetes, Children’ s Hospital of Philadelphia, Philadelphia, Pennsylvania; ***Division of Pediatric Endocrinology, Meyer Children’ s Hospital, Rambam Health Care Campus, Haifa, Israel; ††† Division of Endocrinology, Department of Medicine and §§§ Department of Pediatrics (Endocrinology), Yale University School of Medicine, New Haven, Connecticut; ‡‡‡ Departments of Medicine, Community Health Sciences, and Oncology, University of Calgary Faculty of Medicine, Calgary, Alberta, Canada; ||| Division of Endocrinology, Indiana University School of Medicine, Indianapolis, Indiana; ¶¶¶ Pediatric Endocrinology Unit, Department of Women’ s and Children´s Health, Karolinska Institutet, Stockholm, Sweden; and ****Biostatistics Center, Massachusetts General Hospital, Boston, Massachusetts ABSTRACT Compound heterozygous and homozygous (comp/hom) mutations in solute carrier family 34, member 3 (SLC34A3), the gene encoding the sodium (Na + )-dependent phosphate cotransporter 2c (NPT2c), cause hereditary hypophosphatemic rickets with hypercalciuria (HHRH), a disorder characterized by renal phosphate wasting resulting in hypophosphatemia, correspondingly elevated 1,25(OH) 2 vitamin D levels, hypercalciuria, and rickets/osteomalacia. Similar, albeit less severe, biochemical changes are observed in heterozygous (het) carriers and indistinguishable from those changes encountered in idiopathic hypercalciuria (IH). Here, we report a review of clinical and laboratory records of 133 individuals from 27 kindreds, including 5 previously unreported HHRH kindreds and two cases with IH, in which known and novel SLC34A3 mutations (c.1357delTTC [p.F453del]; c.G1369A [p.G457S]; c.367delC) were identified. Individuals with mutations affect- ing both SLC34A3 alleles had a significantly increased risk of kidney stone formation or medullary nephrocalcinosis, namely 46% compared with 6% observed in healthy family members carrying only the wild-type SLC34A3 allele (P=0.005) or 5.64% in the general population (P,0.001). Renal calcifications were also more frequent in het carriers (16%; P=0.003 compared with the general population) and were more likely to occur in comp/hom and het individuals with decreased serum phosphate (odds ratio [OR], 0.75, 95% confidence interval [95% CI], 0.59 to 0.96; P=0.02), decreased tubular reabsorption of phosphate (OR, 0.41; 95% CI, 0.23 to 0.72; P=0.002), and increased serum 1,25(OH) 2 vitamin D (OR, 1.22; 95% CI, 1.05 to 1.41; P=0.008). Additional studies are needed to determine whether these biochemical parameters are independent of genotype and can guide therapy to prevent nephrocalcinosis, nephrolithiasis, and potentially, CKD. J Am Soc Nephrol 25: ccc–ccc, 2014. doi: 10.1681/ASN.2013101085 Received October 16, 2013. Accepted February 16, 2014. Published online ahead of print. Publication date available at www.jasn.org. Correspondence: Dr. Clemens Bergwitz, Endocrine Unit, 50 Blossom Street, Thier Building 1055, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114. Email: [email protected] Copyright © 2014 by the American Society of Nephrology J Am Soc Nephrol 25: ccc–ccc, 2014 ISSN : 1046-6673/2510-ccc 1
Transcript of Mutations in SLC34A3/NPT2c Are Associated with Kidney Stones and Nephrocalcinosis

CLINICAL RESEARCH www.jasn.org
Mutations in SLC34A3/NPT2c Are Associated withKidney Stones and Nephrocalcinosis
Debayan Dasgupta,* Mark J. Wee,* Monica Reyes,* Yuwen Li,* Peter J. Simm,†‡
Amita Sharma,§ Karl-Peter Schlingmann,| Marco Janner,¶ Andrew Biggin,† Joanna Lazier,**Michaela Gessner,†† Dionisios Chrysis,‡‡ Shamir Tuchman,§§ H. Jorge Baluarte,||
Michael A. Levine,¶¶ Dov Tiosano,*** Karl Insogna,††† David A. Hanley,‡‡‡
Thomas O. Carpenter,§§§ Shoji Ichikawa,||| Bernd Hoppe,†† Martin Konrad,| Lars Sävendahl,¶¶¶
Craig F. Munns,†‡ Hang Lee,**** Harald Jüppner,*§ and Clemens Bergwitz*
*Endocrine Unit, and §Pediatric Nephrology Unit, Massachusetts General Hospital and Harvard Medical School, Boston,Massachusetts; †Institute of Endocrinology and Diabetes, Children’s Hospital at Westmead, Westmead, New South Wales,Australia; ‡Discipline of Pediatrics and Child Health, University of Sydney, Sydney, Australia; |Department of General Pediatrics,University Children’s Hospital, Münster, Germany; ¶Division of Pediatric Endocrinology, Diabetology and Metabolism,University Children’s Hospital, Bern, Germany; **Department of Medical Genetics, University of Calgary, Calgary, Alberta,Canada; ††Division of Pediatric Nephrology, Department of Pediatrics, University Hospital, Köln, Germany; ‡‡Division ofEndocrinology, Department of Pediatrics, University of Patras Medical School, Patras, Greece; §§Division of PediatricNephrology, Children’s National Medical Center, The George Washington University School of Medicine, Washington, Districtof Columbia; ||University of Pennsylvania, School of Medicine, Division of Pediatric Nephrology, Children’s Hospital ofPhiladelphia, Philadelphia, Pennsylvania; ¶¶Division of Endocrinology and Diabetes, Children’s Hospital of Philadelphia,Philadelphia, Pennsylvania; ***Division of Pediatric Endocrinology, Meyer Children’s Hospital, Rambam Health Care Campus,Haifa, Israel; †††Division of Endocrinology, Department of Medicine and §§§Department of Pediatrics (Endocrinology), YaleUniversity School of Medicine, New Haven, Connecticut; ‡‡‡Departments of Medicine, Community Health Sciences, andOncology, University of Calgary Faculty of Medicine, Calgary, Alberta, Canada; |||Division of Endocrinology, Indiana UniversitySchool of Medicine, Indianapolis, Indiana; ¶¶¶Pediatric Endocrinology Unit, Department of Women’s and Children´s Health,Karolinska Institutet, Stockholm, Sweden; and ****Biostatistics Center, Massachusetts General Hospital, Boston, Massachusetts
ABSTRACTCompound heterozygous and homozygous (comp/hom) mutations in solute carrier family 34, member 3 (SLC34A3), thegene encoding the sodium (Na+)-dependent phosphate cotransporter 2c (NPT2c), cause hereditary hypophosphatemicrickets with hypercalciuria (HHRH), a disorder characterized by renal phosphate wasting resulting in hypophosphatemia,correspondingly elevated 1,25(OH)2 vitamin D levels, hypercalciuria, and rickets/osteomalacia. Similar, albeit less severe,biochemical changes are observed in heterozygous (het) carriers and indistinguishable from those changes encountered inidiopathic hypercalciuria (IH). Here, we report a review of clinical and laboratory records of 133 individuals from 27kindreds, including 5 previously unreported HHRH kindreds and two cases with IH, in which known and novel SLC34A3mutations (c.1357delTTC [p.F453del]; c.G1369A [p.G457S]; c.367delC) were identified. Individuals with mutations affect-ingbothSLC34A3 alleles had a significantly increased risk of kidney stone formation ormedullary nephrocalcinosis, namely46%comparedwith 6%observed in healthy familymembers carryingonly thewild-typeSLC34A3 allele (P=0.005)or 5.64%in the general population (P,0.001). Renal calcifications were also more frequent in het carriers (16%; P=0.003 comparedwith the general population) and were more likely to occur in comp/hom and het individuals with decreased serumphosphate (odds ratio [OR], 0.75, 95% confidence interval [95%CI], 0.59 to 0.96; P=0.02), decreased tubular reabsorptionof phosphate (OR, 0.41; 95%CI, 0.23 to0.72;P=0.002), and increased serum1,25(OH)2 vitaminD (OR, 1.22; 95%CI, 1.05 to1.41; P=0.008). Additional studies are needed to determine whether these biochemical parameters are independent ofgenotype and can guide therapy to prevent nephrocalcinosis, nephrolithiasis, and potentially, CKD.
J Am Soc Nephrol 25: ccc–ccc, 2014. doi: 10.1681/ASN.2013101085
Received October 16, 2013. Accepted February 16, 2014.
Published online ahead of print. Publication date available atwww.jasn.org.
Correspondence: Dr. Clemens Bergwitz, Endocrine Unit, 50
Blossom Street, Thier Building 1055, Massachusetts GeneralHospital and Harvard Medical School, Boston, MA 02114. Email:[email protected]
Copyright © 2014 by the American Society of Nephrology
J Am Soc Nephrol 25: ccc–ccc, 2014 ISSN : 1046-6673/2510-ccc 1

Inactivating mutations on both parental alleles of the solutecarrier family 34, member 3 (SLC34A3), the gene encoding thesodium (Na+)-dependent phosphate cotransporter 2c (NPT2c),are the cause of hereditary hypophosphatemic rickets with hy-percalciuria (HHRH; OMIM: 241530)1–3—an autosomal re-cessive renal phosphate-wasting disorder that was originallydescribed by Tieder et al.4,5 Individuals affected by HHRHwho carry compound heterozygous or homozygous (comp/hom) SLC34A3/NPT2c mutations show increased urinaryphosphate excretion leading to hypophosphatemic rickets,bowing, and short stature as well as appropriately elevated1,25(OH)2D levels. Elevated 1,25(OH)2D levels, in turn, resultin hypercalciuria because of enhanced intestinal calciumabsorption and reduced parathyroid hormone (PTH) -dependent calcium reabsorption in the distal renal tubules.Even heterozygous SLC34A3/NPT2c mutations are frequent-ly associated with hypercalciuria, but none of the carriersof SLC34A3/NPT2c mutations in the originally describedHHRH patients were reported to have renal calcificationsand kidney stones.4,5 Subsequent investigations, however, re-vealed that these complications affecting the kidneys wereobserved in numerous patients with comp/hom SLC34A3/NPT2c mutations.3,6–11 However, the small size of HHRHkindreds and the relatively high prevalence of renal calcifica-tions in the general population (5.64%)12,13 have, thus far, pre-vented segregation-based statistical approaches to determinewhether SLC34A3/NPT2c mutations do increase the risk ofdeveloping kidney stones or nephrocalcinosis. Likewise, it isunknown whether loss of NPT2c can lead to additional prox-imal tubular phenotypes such as Fanconi syndrome, whichhas been described in two patients with homozygousSLC34A3/NPT2a mutations14 who developed CKD later inlife.
The presence of hypercalciuria, kidney stones, andnephrocalcinosis observed in HHRH kindreds is differentfrom the findings in fibroblast growth factor 23 (FGF23)–dependent hypophosphatemic disorders, such as X-linkedhypophosphatemia (XLH; mutant PHEX),15 autosomal dom-inant hypophosphatemic rickets (ADHR;mutant FGF23),16 orautosomal recessive hypophosphatemic rickets (ARHR; mu-tant DMP1, ENPP1, or FAM20C),17–20 in which affected indi-viduals show, before treatment with oral phosphate and 1,25(OH)2D, inappropriately normal or suppressed 1,25(OH)2Dlevels despite significant hypophosphatemia and thus, no in-crease in urinary calcium excretion. Oral phosphate supple-ments combined with active vitamin D analogs are generallyrecommended for treatment of FGF23-dependent hypophos-phatemic disorders.21 In contrast, HHRH is thought to re-quire phosphate supplements alone,4,5 in part because endog-enously elevated 1,25(OH)2D levels are predicted to preventan increase in PTH secretion triggered by intermittent eleva-tions in serum phosphate. However, long-term studies arelacking that determine whether oral phosphate supplementa-tion alone of HHRH patients is sufficient for prevention ofrenal calcifications and bone loss. It is likewise unknown
how therapy should be monitored, whether secondary hyper-parathyroidism can develop as observed in FGF23-dependenthypophosphatemic disorders,15 and whether phosphate re-quirements decrease with age, which has been reported forADHR.16
In the current study, we investigated five new HHRHkindreds and two new cases with idiopathic hypercalciuria(IH), inwhomwediscovered known andnovel homozygous orcompounded heterozygous SLC34A3/NPT2c mutations. Re-view of the clinical and laboratory findings along with thosefindings reported for 22 previously published kindreds sug-gests that renal calcifications and/or kidney stones may beimportant, often unrecognized initial findings suggestive ofcomp/hom SLC34A3/NPT2c mutations. Importantly, hetero-zygous carriers also show an increased frequency of renal cal-cifications and biochemical profiles in plasma and urine thatare intermediate to those profiles of individuals withoutSLC34A3/NPT2c mutations and comp/hom changes. Ourdata suggest that serum phosphate, tubular reabsorption ofphosphate (TRP), and serum 1,25(OH)2D levels predict thedevelopment of renal calcifications. However, additional stud-ies are needed to determine whether these biochemical param-eters are independent of genotype and can guide therapy toprevent renal calcifications and potentially, CKD.
RESULTS
Novel Mutations in the SLC34A3/NPT2c Gene inKindreds with HHRH and IHFive new kindreds were referred to us for genetic evaluation,with the index case presenting with classic HHRH duringchildhood. Metabolic bone disease was found to be associatedwith hypophosphatemia, and subsequent laboratory studieswere consistent with FGF23-independent renal phosphatewasting. In addition, two unrelated childrenwith IH presentedto their pediatrician with renal stones and/or nephrocalcinosison renal ultrasound and biochemical abnormalities that wereconsistent with those abnormalities seen in HHRH [i.e., hy-percalciuria and elevated 1,25(OH)2D levels]. Apparent bonedisease was missing, and mild hypophosphatemia was onlynoted on more careful evaluation. Nucleotide sequence anal-ysis of SLC34A3/NPT2c revealed known and novel mutationsin kindreds A and B and case G (Supplemental Figure 1, A andB, Supplemental Tables 1 and 2). A novel homozygous mis-sense mutation c.1369G.A(p.G457S) was detected in indi-viduals A/IV-1, A/IV-2, A/IV-3, and A/IV-4, whereas bothparents were heterozygous for this nucleotide change. The indexcase B/II-2 inherited the previously described intronic dele-tion c.560+27_561–38del (g.1440_1469del)22 from hismother (B/I-1) and the novel in-frame deletion c.1357delTTC(p. F453del) from his father (B/I-2); c.1369G.A (p.G457S)and c.1357delTTC (p.F453del) affect highly conserved aminoacid residues (Supplemental Table 3). Case G was found to becompound heterozygous for a novel deletion c.367delC,
2 Journal of the American Society of Nephrology J Am Soc Nephrol 25: ccc–ccc, 2014
CLINICAL RESEARCH www.jasn.org

which introduces 14 novel amino acids followed by prematuretermination of the NPT2c protein after residue p.P48(WTLPQLKDPWTLPS-Stop) and the known missense muta-tion c.575C.T(p.S192L).2 All three novel mutations are ab-sent from the 1000 Genome,23 dbSNP,24 and the NationalHeart, Lung, and Blood Institute Gene Ontology Exome Se-quencing Project (http://evs.gs.washington.edu/EVS/) data-bases, predicted by Polyphen to reduce protein function,25
and therefore, likely disease-causing. Detailed clinical andgenotyping information on kindreds A–G is in SupplementalMaterial, Supplemental Figure 1, A and B, Supplemental Ta-bles 1 and 2.
Meta-Analysis of Clinical Information, IncludingPreviously Reported Kindreds with HHRHSeven of eleven com/hom carriers of the identified SLC34A3/NPT2c mutations and one of ten heterozygous carriers pre-sented initially with renal calcifications or developed thesechanges subsequently. To better understand whether thishigh prevalence of renal calcifications in these not previouslyreported kindreds can be generalized to all carriers ofSLC34A3/NPT2c mutations and to identify possible predic-tors of renal calcifications, we performed a meta-analysis ofthe clinical information from 13 reports1–3,6–9,11,22,26–29 andtwo unpublished families (Simm P, Briody J, Reyes M, GibbonsP, Alexander S, Rauch F, Jüppner H, Bergwitz C, Munns C,manuscript in preparation). Two reports of heterozygous mu-tations in kindreds with IH and HHRH10,30 were excludedfrom this analysis, because no mutations on the second allelehad been identified; thus, segregationwith the renal phenotypein the affected individuals remains uncertain. After includingour new cases, this analysis comprised 133 individuals from 27kindreds (Supplemental Table 2). Genotype information isavailable for 117 individuals and four obligate heterozygouscarriers.
We first evaluated the prevalence of renal calcifications (i.e.,nephrolithiasis or nephrocalcinosis) in the comp/hom carriersof SLC34A3/NPT2c mutations. This was significantly in-creased (20 of 43 [46%] individuals) compared with the avail-able genotyped relatives who carried only the wild-type allele(1 of 16 [6%] individuals; P=0.005) and compared with theheterozygote carriers of SLC34A3/NPT2c mutations (10 of 61[16%] individuals; P=0.002) (Table 1); 15 of 43 (35%) comp/hom carriers of SLC34A3/NPT2c mutations presented withrenal stones (P=0.05), of whom 6 (14%) individuals alsohad evidence for nephrocalcinosis. Furthermore, renal ultra-sound studies showed medullary nephrocalcinosis in 13 of 43(30%) com/hom individuals (P=0.01) (Table 1). Seven ofthese individuals (16%) did not have stones. Importantly, 6of 61 (10%) heterozygous and 7 of 43 (16%) comp/hom car-riers of SLC34A3/NPT2c mutations presented with renal cal-cifications, whereas clinical and laboratory findings suggestiveof metabolic bone disease were absent (Figure 1).
Prevalence of renal calcifications was increased 3-fold inheterozygous carriers of SLC34A3/NPT2c, which is significant
when compared with the prevalence of 5.64% reported inlarge cohorts of healthy controls12,13 (P=0.003) (Table 1).Consistent with an intermediate frequency of renal calcifica-tions, the biochemical findings were, furthermore, intermedi-ate for heterozygous carriers of SLC34A3/NPT2cmutations. Acombined analysis of the initial biochemical findings in ournewHHRH families along with previously published kindreds(Figures 2 and 3) showed reduced mean serum P, TRP, andintact PTH in carriers of SLC34A3/NPT2cmutations, whereasserum Ca, 1,25(OH)2D, and urinary calcium excretion (uCa/uCrea) ratiowere increased. Differences were significant basedon one-way ANOVA for these analytes when comparing wild-type, heterozygous, and comp/hom carriers. Despite the smallnumber of healthy siblings, serum P was also significantly re-duced in heterozygous carriers compared with healthy siblings(P,0.03), and heterozygous means for 1,25(OH)2D and TRPwere above and below the normal range, respectively. No cleardifference in severity was observed for individual mutations(Supplemental Figure 2). Renal function was normal in all butone heterozygous and two comp/hom carriers of SLC34A3/NPT2c mutations at initial presentation (Figure 2C).
Several of the above biochemical parameters have beenimplicated as predictors of renal calcifications31 andmetabolicbone disease.32 To identify predictors in our HHRH kindreds,we first performed a univariate logistic regression analysis inindividuals with all genotypes (n=133) (Table 2). As expected,renal calcifications were more likely to occur in carriers ofSLC34A3/NPT2c mutations. Furthermore, individuals withrenal calcifications had decreased serum P and TRP but in-creased serum 1,25(OH)2D. However, only TRP remained sig-nificant after adjusting for genotype in a multivariate model,and 43 cases were not sufficient to identify predictors of renalcalcifications when limiting the univariate logistic regressionanalysis to individuals with twomutant SLC34A3/NPT2c alleles.
Genotype was likewise the strongest predictor of metabolicbone disease (Table 3). Furthermore, decreased serum P andTRP and increased serum 1,25(OH)2D and serum alkalinephosphatase (ALP) were significantly associated with boneinvolvement, even after adjusting for genotype in a multivariatemodel; 1,25(OH)2D, TRP, and ALP continued to be significantwhen limiting the univariate logistic regression analysis to indi-viduals with two mutant SLC34A3/NPT2c alleles.
DISCUSSION
Prevalence of Renal Calcifications in Carriers ofSLC34A3/NPT2c Mutations Is IncreasedRenal calcifications were not previously thought to be partof the clinical presentation of HHRH.4,5 More recently, how-ever, an increased frequency of medullary nephrocalcinosisand nephrolithiasis has been recognized3,6,7,9,11 in comp/homcarriers. These observations are further substantiated in the cur-rent report, because the index cases in three of five new kindredsshowed renal calcifications on initial presentation. Furthermore,
J Am Soc Nephrol 25: ccc–ccc, 2014 Frequency of Renal Calcifications in HHRH 3
www.jasn.org CLINICAL RESEARCH

evaluation of 27 available kindreds indicates that approxi-mately one half of all comp/hom carriers of SLC34A3/NPT2cmutations present with renal calcifications. Renal calcifications,furthermore, are the only presenting sign in 16%, whereas bonedisease is (apparently) absent. Even heterozygous carriers ofSLC34A3/NPT2c mutations show an approximately 3-foldhigher incidence of renal calcifications, which may be relatedto their intermediate biochemical profile (Figures 2 and 3). There-fore, all affected individuals and their first-degree relativesshould be examined for renal calcifications. The biochemicalprofile may help identify, in addition to genetic analysis ofSLC34A3/NPT2c, those first-degree members in HHRH fam-ilies who are at risk for developing renal complications as fur-ther discussed below.
Although prevalence of renal calcifications in heterozygouscarriers in this initial survey is only significant compared withthe prevalence reported in large cohorts of healthy controls,12,13
it may be underestimated, because mostasymptomatic individuals have not had im-aging studies. A particularly importantstrength of our study is the analysis of het-erozygous carriers of only those mutationsthat are clearly disease-causing when com-bined with another mutation on the secondallele or homozygously present. Systematicevaluation of these heterozygous individualswill be an important aspect of future inves-tigations.
Several genes that cause rare monogenicdisorders have been associated with hyper-calciuric nephrolithiasis (i.e., CLCN5,CASR, CLDN16, CLDN19 , ADCY10,SLC34A1, SLC9A3R1, GLUT2, HSPG2,and FN1),31,33 whereas variants of uromo-dulin and fetuin seem to be protective.34
Some of these genes affect tubular handlingof calcium and phosphate in a way that is similar to what isobserved in our patients with SLC34A3/NPT2c mutations.More recently, FAM20Amutations in enamel renal syndromewere found to be associated with renal calcifications, but theseindividuals do not seem to fit the biochemical profile of IH20;thus, it is more likely that loss of function of the extracellularkinase encoded by FAM20A affects local tissue factors thatcontribute to the development of renal calcifications. Fur-thermore, mutations in CYP24A1, the gene encoding the 24-hydroxylase, which is the key enzyme leading to inactivation of1,25(OH)2D, have been reported as a cause of hypercalciuricnephrolithiasis.35 It should be noted that, thus far, genome-wide association studies for uric acid nephrolithiasis,36 serumphosphate levels,37 and CKD34 have not supported an associ-ation of hypercalciuric stone disease with the SLC34A3/NPT2clocus. However, our meta-analysis clearly suggests thatSLC34A3/NPT2c should be added to the above list of
Table 1. Frequency of renal calcifications is increased in carriers of SLC34A3/NPT2c mutations
Phenotype
Genotype
Normal (n=16) Heterozygous (n=61) Comp/Hom (n=43)
N P Value N P Value N P Value
RenalSymptomatic 1 1.00 10 0.44/0.003a 20 ,0.01/0.002b/,0.001a
Nephrolithiasis 1 1.00 10 0.44/0.003a 15 0.05/0.04b/,0.001a
Nephrocalcinosis 0 3 1.00 13 0.01/,0.001b
Asymptomatic 15 51 23BoneSymptomatic 0 8 0.19 33 ,0.001Rickets/osteomalacia 0 8 0.19 32 ,0.001Adult osteopenia/osteoporosis 0 0 1 1.00Asymptomatic 16 53 10
Number (N ) of individuals with renal phenotype who presented with nephrolithiasis or nephrocalcinosis and number of individuals (N ) with bone phenotype whopresented with rickets/osteomalacia or osteopenia/osteoporosis. P values (Fisher exact t test) are shown compared with normal individuals.aAssuming average global prevalence of stones of 5.64% according to the work by Romero et al.12bP values compared with heterozygous individuals.
Figure 1. Frequency of renal calcifications is increased in carriers of SLC34A3/NPT2cmutations. Shown here is the prevalence of renal calcifications (i.e., nephrolithiasisand/or nephrocalcinosis) and bone disease (i.e., rickets/osteomalacia and/or osteo-penia/osteoporosis) among individuals with two normal alleles as well as heterozygousand comp/hom carriers of SLC34A3/NPT2c mutations. Calc., calcification; dis., dis-ease.
4 Journal of the American Society of Nephrology J Am Soc Nephrol 25: ccc–ccc, 2014
CLINICAL RESEARCH www.jasn.org

hypercalciuric stone disease genes. Our finding may have im-portant implications for the general population, becauseheterozygous nucleotide changes that alter the amino acidsequence or introduce deletions/insertions are relatively fre-quent in NPT2c, but it is currently unknownwhether they areof biologic significance. Improved clinical characterizationmay provide a rationale for genetic analysis of SLC34A3/NPT2c in a subpopulation of hypercalciuric stone patientswith clinical and biochemical profiles that resemble the pro-files of HHRH.
To increase sample size, we decided to combine nephro-calcinosis and nephrolithiasis for statistical analysis, becausethese clinical findings are both associated with the increasedurinary calcium excretion observed in HHRH. However, itshould be noted that 13 of 43 (30%) comp/hom individualsshowed nephrocalcinosis on ultrasound, which reaches sta-tistical significance, compared with normal and heterozygousfamily members (Table 1), and 7 of these individuals did nothave evidence for renal stones. Future studies may be able toevaluate the possibility that nephrocalcinosis is a unique fea-ture of the hypercalciuria and hyperphosphaturia caused byloss of NPT2c in the proximal tubule, which raises interestingpathophysiological questions.
Serum 1,25(OH)2D, Phosphate, andTRP May Be Predictors of RenalCalcifications in Carriers of SLC34A3/NPT2c MutationsIt is unclear at the moment which geneticand biochemical criteria best predict therisk for renal calcifications in our HHRHkindreds. Based on the current understand-ing of the pathophysiology of HHRH, thedefective sodium–phosphate cotransportercauses renal phosphate wasting, whichtriggers renal 1a-hydroxylase (CYP27B1)enzyme activity; this activity causes an in-crease in circulating 1,25(OH)2D levels,which in turn, increases intestinal Ca ab-sorption, leading to an increased renaltubular Ca load and hypercalciuria. Thus,hypercalciuria, hyperphosphaturia, andpossibly, tissue-specific effects of 1,25(OH)2D levels may lead to renal calcifica-tions and stones. Hypercalciuria is themostcommon risk factor of kidney stones, whichis present in 40%–50% of adults with re-current calcium stones and 75%–80% ofchildren with kidney stones. This diseaseis often referred to as IH because of in-creased intestinal absorption of calcium,and it can be associated with a mild renalphosphate leak, despite the lack of para-thyroid hyperactivity.38 Association of in-creased 1,25(OH)2D levels with renalcalcifications was observed inmousemod-
els that lack Fgf2339 or Klotho40 or individuals with CYP24A1mutations,35 even in the setting of low or normal urinaryphosphate excretion. Finally, excessive phosphaturia, evenwith normal or low urinary calcium, can cause nephrocalcinosisin humans as is seen in XLH41 or in phosphate enema–inducednephrocalcinosis.42 Consistent with the pathophysiology ofHHRH, we found that increased serum 1,25(OH)2D, lowserum P, and decreased TRP may be positive predictors ofrenal calcifications. Only TRP remained associated withrenal calcifications after controlling for genotype; however,serum P, 1,25(OH)2D, TRP, and ALP continued to be signif-icant predictors of bone involvement. This initial evalua-tion suggests that decreased serum P levels and increasedexcretion of phosphate and serum 1,25(OH)2D merit addi-tional evaluation as possible nongenetic predictors of renalcalcifications.
We wondered whether specific SLC34A3/NPT2cmutationsare associated with an increased serum 1,25(OH)2D, low se-rum P, decreased TRP, and renal calcifications (SupplementalFigure 2). However, not all individuals with a specific mutationhave developed stones, and the numbers are small. It is, there-fore, not possible to be certain about a genotype–phenotypeeffect at the present time.
Figure 2. Heterozygous carriers of SLC34A3/NPT2c mutations have intermediatebiochemical findings. Summary of biochemical values of the presented and publishedkindreds analyzed with respect to genotype. Individual values with mean695% con-fidence interval are shown for (A) normalized serum calcium, (B) normalized serumphosphorus, (C) normalized serum creatinine, and (D) serum 1,25(OH)2 vitamin D. het,heterozygous; n/a, not applicable; wt, wild-type.
J Am Soc Nephrol 25: ccc–ccc, 2014 Frequency of Renal Calcifications in HHRH 5
www.jasn.org CLINICAL RESEARCH
Our study is limitedby its relatively smallsample size, because HHRH is a rarecondition affecting, thus far, less than 50individuals worldwide. Furthermore, clin-ical and biochemical data are often onlyavailable for the initial presentation, andfamily members who are heterozygous fora SLC34A3/NPT2c mutation or carry nomutation have been studied less well. Inaddition to a systematic evaluation of theseindividuals, long-term studies are requiredthat determine whether oral phosphatesupplementation alone of HHRH patientsis sufficient for the prevention of renalcalcifications and bone loss. It is likewiseunknown how therapy should be moni-tored, whether secondary hyperparathy-roidism can develop as observed inFGF23-dependent hypophosphatemicdisorders,15 and whether phosphate re-quirements decrease with age, which hasbeen reported for ADHR.16 It will also beimportant to determine whether lack ofthe NPT2c transporter leads to additionalsymptoms, such as Fanconi syndrome orCKD, which have been shown for individ-uals with SLC34A1/NPT2a mutations,14
using either animal models of HHRH or
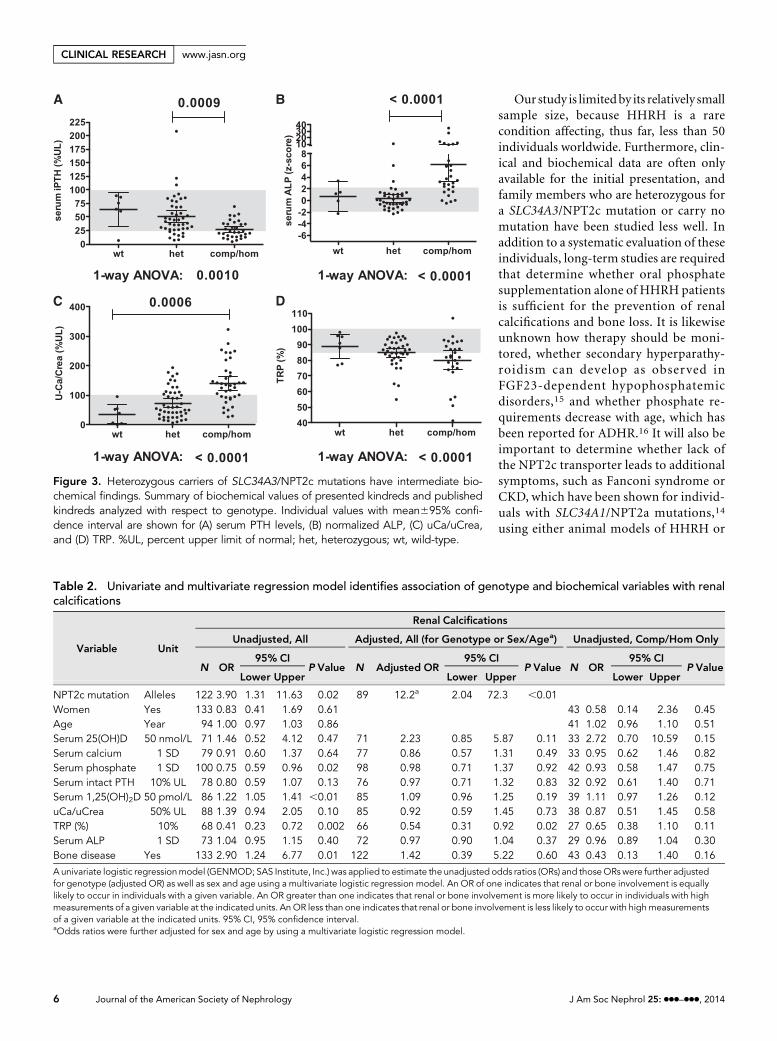
Figure 3. Heterozygous carriers of SLC34A3/NPT2c mutations have intermediate bio-chemical findings. Summary of biochemical values of presented kindreds and publishedkindreds analyzed with respect to genotype. Individual values with mean695% confi-dence interval are shown for (A) serum PTH levels, (B) normalized ALP, (C) uCa/uCrea,and (D) TRP. %UL, percent upper limit of normal; het, heterozygous; wt, wild-type.
Table 2. Univariate and multivariate regression model identifies association of genotype and biochemical variables with renalcalcifications
Variable Unit
Renal Calcifications
Unadjusted, All Adjusted, All (for Genotype or Sex/Agea) Unadjusted, Comp/Hom Only
N OR95% CI
P Value N Adjusted OR95% CI
P Value N OR95% CI
P ValueLower Upper Lower Upper Lower Upper
NPT2c mutation Alleles 122 3.90 1.31 11.63 0.02 89 12.2a 2.04 72.3 ,0.01Women Yes 133 0.83 0.41 1.69 0.61 43 0.58 0.14 2.36 0.45Age Year 94 1.00 0.97 1.03 0.86 41 1.02 0.96 1.10 0.51Serum 25(OH)D 50 nmol/L 71 1.46 0.52 4.12 0.47 71 2.23 0.85 5.87 0.11 33 2.72 0.70 10.59 0.15Serum calcium 1 SD 79 0.91 0.60 1.37 0.64 77 0.86 0.57 1.31 0.49 33 0.95 0.62 1.46 0.82Serum phosphate 1 SD 100 0.75 0.59 0.96 0.02 98 0.98 0.71 1.37 0.92 42 0.93 0.58 1.47 0.75Serum intact PTH 10% UL 78 0.80 0.59 1.07 0.13 76 0.97 0.71 1.32 0.83 32 0.92 0.61 1.40 0.71Serum 1,25(OH)2D 50 pmol/L 86 1.22 1.05 1.41 ,0.01 85 1.09 0.96 1.25 0.19 39 1.11 0.97 1.26 0.12uCa/uCrea 50% UL 88 1.39 0.94 2.05 0.10 85 0.92 0.59 1.45 0.73 38 0.87 0.51 1.45 0.58TRP (%) 10% 68 0.41 0.23 0.72 0.002 66 0.54 0.31 0.92 0.02 27 0.65 0.38 1.10 0.11Serum ALP 1 SD 73 1.04 0.95 1.15 0.40 72 0.97 0.90 1.04 0.37 29 0.96 0.89 1.04 0.30Bone disease Yes 133 2.90 1.24 6.77 0.01 122 1.42 0.39 5.22 0.60 43 0.43 0.13 1.40 0.16
A univariate logistic regressionmodel (GENMOD; SAS Institute, Inc.) was applied to estimate the unadjusted odds ratios (ORs) and thoseORs were further adjustedfor genotype (adjusted OR) as well as sex and age using a multivariate logistic regression model. An OR of one indicates that renal or bone involvement is equallylikely to occur in individuals with a given variable. An OR greater than one indicates that renal or bone involvement is more likely to occur in individuals with highmeasurements of a given variable at the indicated units. AnOR less than one indicates that renal or bone involvement is less likely to occur with highmeasurementsof a given variable at the indicated units. 95% CI, 95% confidence interval.aOdds ratios were further adjusted for sex and age by using a multivariate logistic regression model.
6 Journal of the American Society of Nephrology J Am Soc Nephrol 25: ccc–ccc, 2014
CLINICAL RESEARCH www.jasn.org

large HHRH kindreds, such as those kindreds described byTieder et al.4,5
In summary, we here present five previously unreportedHHRH kindreds and two individuals with IH in whomSLC34A3/NPT2c nucleotide sequence analysis identifiedknown or novel mutations. Review of the clinical presentationof these kindreds and previously published HHRH kindredssuggests that renal calcifications and/or renal stones may be animportant, often unrecognized initial symptom in carriers ofcomp/hom SLC34A3/NPT2c mutations. Even heterozygouscarriers can be affected by nephrocalcinosis and nephrolithia-sis, which is consistent with their intermediate biochemicalprofile. Serum 1,25(OH)2D, phosphate, and TRP may be pre-dictors of renal calcifications, and future studies will help deter-mine whether these biochemical parameters are independentof genotype and can guide therapy to prevent nephrocalcinosis,nephrolithiasis, and potentially CKD.
CONCISE METHODS
Laboratory AssaysWith the exception of genetic analyses, all laboratory studies were
performed at laboratories used by the different investigators (normal
ranges are provided in parentheses after each value). 25-Hydroxy
vitamin D levels were measured by liquid chromatography with
tandemmass spectroscopy or chemiluminescence immunoassay, and
1,25(OH)2D levels were measured by radioimmunoassay or ELISA.
Serum intact PTH levels were determined by electrochemiluminescence
immunoassay, and FGF23 levelsweremeasuredby anELISA that detects
the intact hormone and C-terminal fragments thereof (Immutopics)
as well as intact FGF23 (Kainos). The renal TRP (in percentage) was
calculated using the following equation: 1003(12(urine phosphorus3
serum creatinine)/(serum phosphorus3urine creatinine); when
serum phosphorus is below the reference range for age, TRP should
be above 90. Tubular maximum phosphate reabsorption per
GFR (TmP/GFR) was estimated using the Walton and Bijvoet
nomogram.43
SLC34A3 Genetic AnalysesMutational and haplotype analysis of SLC34A3 was performed after
informed written consent was obtained using forms approved by the
Institutional Review Board of the Massachusetts General Hospital
(MGH). The entire SLC34A3 gene, including approximately 800 bp
59 of the transcriptional start site, all intervening sequences, and ap-
proximately 200 bp of the 39-untranslated region, was amplified by
PCR from genomic DNA of the index cases followed by nucleotide
sequence analysis at the MGH DNA Sequencing Core Facility or
Genewiz, Inc. as described.1 PCR assays to confirm the findings in
index cases and analyze family members and controls were per-
formed as described1 using Qiagen reagents or the Expand Long
Template PCR System (Roche) at standard PCR cycling conditions
followed by restriction enzymatic digest or nucleotide sequence anal-
ysis. Primer sequences are listed in Supplemental Table 4, and con-
ditions for amplification and detection of the mutations are given in
Supplemental Table 1. Searches of the National Center for Biotech-
nology Information–dbSNP24 and the 1000 Genomes Project23 were
negative for the identified novel mutations, with the exception of Het.
c.413C.T(p.S138F) (HGMD: CM060480), which was identified as a
rare single nucleotide polymorphism rs141734934, Het: 0.001 based
on nucleotide sequence analysis of 4352 chromosomes (1000 Ge-
nomes). GenBank accession numbers for SLC34A3 are as follows:
Table 3. Univariate and multivariate regression model identifies association of genotype and biochemical variables with bonedisease
Variable Unit
Bone Disease
Unadjusted Adjusted, All (for Genotype or Sex and Agea) Unadjusted, Comp/Hom Only
N OR95% CI
P Value N Adjusted OR95% CI
P Value N OR95% CI
P ValueLowerUpper Lower Upper Lower Upper
NPT2c mutation Alleles 12220.8 7.84 55.09 ,0.001 89 33.9a 9.52 120.9 ,0.001Women Yes 133 0.71 0.33 1.55 0.39 43 0.43 0.08 2.41 0.34Age Year 94 0.96 0.92 0.99 0.02 41 1.02 0.97 1.08 0.45Serum 25(OH)D 50 nmol/L 71 0.74 0.31 1.79 0.51 71 1.18 0.40 3.49 0.77 33 1.22 0.22 6.79 0.82Serum calcium 1 SD 79 1.32 0.80 2.18 0.28 77 1.41 0.87 2.28 0.16 33 1.19 0.75 1.88 0.45Serum phosphate 1 SD 100 0.39 0.24 0.63 ,0.001 98 0.50 0.30 0.83 ,0.01 42 0.45 0.19 1.03 0.06Serum intact PTH 10% UL 78 0.73 0.62 0.87 ,0.001 76 0.94 0.78 1.14 0.51 32 1.16 0.71 1.88 0.56Serum 1,25(OH)2D 50 pmol/L 86 1.62 1.20 2.20 0.002 85 1.32 1.04 1.69 0.02 39 1.43 1.01 2.02 0.04uCa/uCrea 50% UL 88 2.62 1.36 5.05 0.004 85 1.45 0.66 3.21 0.35 38 1.38 0.51 3.72 0.52TRP(%) 10% 68 0.23 0.13 0.40 ,0.001 66 0.21 0.08 0.56 0.002 27 0.02 0.00 0.94 0.05Serum ALP 1 SD 73 2.46 1.73 3.50 ,0.001 72 2.00 1.24 3.20 0.004 29 1.45 1.04 2.02 0.03Renal calcifications Yes 133 2.90 1.24 6.77 0.01 122 1.39 0.36 5.33 0.63 43 0.43 0.13 1.40 0.16
A univariate logistic regressionmodel (GENMOD; SAS Institute, Inc.) was applied to estimate the unadjusted odds ratios (ORs) and thoseORs were further adjustedfor genotype (adjusted OR) as well as sex and age using a multivariate logistic regression model. An OR of one indicates that renal or bone involvement is equallylikely to occur in individuals with a given variable. An OR greater than one indicates that renal or bone involvement is more likely to occur in individuals with highmeasurements of a given variable at the indicated units. AnOR less than one indicates that renal or bone involvement is less likely to occur with highmeasurementsof a given variable at the indicated units. 95% CI, 95% confidence interval.aOdds ratios were further adjusted for sex and age by using a multivariate logistic regression model.
J Am Soc Nephrol 25: ccc–ccc, 2014 Frequency of Renal Calcifications in HHRH 7
www.jasn.org CLINICAL RESEARCH

genomic contig, NT024000.15; cDNA, NM080877.1; protein,
NP543153.1.
Statistical and Data AnalysesFor our meta-analysis, we used clinical and laboratory findings (LFs)
obtained at presentation before therapy was initiated. For statistical
evaluation, we determined the number of individuals affected by (1)
nephrolithiasis who had history of renal colic or based on imaging
or stone analysis, (2) nephrocalcinosis, in whom renal mineral de-
posits were found on imaging studies (i.e., ultrasound or computed
tomography), (3) rickets/osteomalacia based on history of bowing, cor-
rective surgery, stress fractures, bone pain, or imaging studies (i.e., skel-
etal radiographic survey and 99mTc methylene diphosphonate bone
scan), and (4) osteopenia/osteoporosis based on World Health Or-
ganization bone densitometry criteria. Asymptomatic individuals
were negative for the above renal and bone phenotypes either by
imaging or based on absence of history of symptoms suggesting the
presence of renal calcifications or bone disease. Age-related reference
values (RVs; i.e., 95% confidence intervals) were obtained from http://
www.mayomedicallaboratories.com/, http://cclnprod.cc.nih.gov/
dlm/testguide.nsf/Index/1D336E0232533D3285256B9C0059E-
CEA, and respective clinical laboratories; age-related RVs for TmP/
GFR are in refs. 44–46, and age-related RVs for uCa/uCrea are from
ref. 47. Assuming normal distribution, LFs can be transformed into
z scores using means of age-appropriate RVs (RVmean) and SDs
(where SD=RV/4) according to the formula: z score=(LF2RVmean)/
SD.48 z Scores were used for all statistical analyses with the fol-
lowing exceptions: absolute values are presented for 25-hydroxy
vitamin D, 1,25(OH)2D, TRP, and TmP/GFR measurements. The
data for PTH and age-specific uCa/uCrea are presented as percentage
of the upper limit of normal RV. These exceptions are important,
because for these assays, upper and lower RVs have not been es-
tablished in sufficiently large number of normal age-specific con-
trols.49 We used Prism for OSX, version 5.0d (GraphPad Software
Inc.), to perform t tests for unpaired comparisons, one-way ANOVAs
for multiple comparisons, and linear regression analyses. For uni-
variate and multivariate logistic regression models, we used SAS
(GENMOD; SAS Institute, Inc.) to estimate the odds ratios. To in-
crease sample size, we treated all renal phenotypes (nephrolithiasis and
nephrocalcinosis) or bone phenotypes (rickets, osteomalacia, and
osteopenia/osteoporosis) collectively. Data are expressed asmeans695%
confidence intervals.
ACKNOWLEDGMENTS
This workwas supported byNational Institutes ofHealthGrants R01-
DK46718-20 and P01-DK11794, project IV (to H.J.), National In-
stitutes of Health/National Institute of Diabetes and Digestive and
KidneyDiseases Grant 5K08-DK078361 (to C.B.), Young Investigator
Awards by the National Kidney Foundation, and the American As-
sociation for Clinical Investigation (C.B.).
DISCLOSURESNone.
REFERENCES
1. Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-ZahraH, Frappier D, Burkett K, Carpenter TO, Anderson D, Garabedian M,Sermet I, Fujiwara TM,Morgan K, TenenhouseHS, Jüppner H: SLC34A3mutations in patients with hereditary hypophosphatemic rickets withhypercalciuria predict a key role for the sodium-phosphate cotransporterNaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet 78:179–192, 2006
2. Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-RakoverY, Wagenstaller J, Tiosano D, Gershoni-Baruch R, Albers N, Lichtner P,Schnabel D, Hochberg Z, Strom TM: Hereditary hypophosphatemicrickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet 78: 193–201, 2006
3. Ichikawa S, Sorenson AH, Imel EA, Friedman NE, Gertner JM, EconsMJ: Intronic deletions in the SLC34A3 gene cause hereditary hypo-phosphatemic rickets with hypercalciuria. J Clin Endocrinol Metab 91:4022–4027, 2006
4. Tieder M, Modai D, Samuel R, Arie R, Halabe A, Bab I, Gabizon D,Liberman UA: Hereditary hypophosphatemic rickets with hyper-calciuria. N Engl J Med 312: 611–617, 1985
5. Tieder M, Modai D, Shaked U, Samuel R, Arie R, Halabe A, Maor J,Weissgarten J, Averbukh Z, Cohen N, Edelstein S, Liberman UA: “Idi-opathic” hypercalciuria and hereditary hypophosphatemic rickets. Twophenotypical expressions of a common genetic defect. N Engl J Med
316: 125–129, 19876. Page K, Bergwitz C, Jaureguiberry G, Harinarayan CV, Insogna K: A
patient with hypophosphatemia, a femoral fracture, and recurrent kid-ney stones: Report of a novel mutation in SLC34A3. Endocr Pract 14:869–874, 2008
7. Kremke B, Bergwitz C, Ahrens W, Schütt S, Schumacher M, Wagner V,Holterhus PM, Jüppner H, Hiort O: Hypophosphatemic rickets withhypercalciuria due to mutation in SLC34A3/NaPi-IIc can be masked byvitaminDdeficiency and can be associatedwith renal calcifications.ExpClin Endocrinol Diabetes 117: 49–56, 2009
8. Phulwani P, Bergwitz C, Jaureguiberry G, Rasoulpour M, Estrada E: He-reditary hypophosphatemic rickets with hypercalciuria and nephrolithiasis-identification of a novel SLC34A3/NaPi-IIc mutation.AmJMedGenet A
155A: 626–633, 20119. JaureguiberryG,CarpenterTO,FormanS, JüppnerH,BergwitzC:Anovel
missensemutation in SLC34A3 that causes hereditary hypophosphatemicrickets with hypercalciuria in humans identifies threonine 137 as an im-portant determinant of sodium-phosphate cotransport in NaPi-IIc. Am J
Physiol Renal Physiol 295: F371–F379, 200810. Mejia-Gaviria N, Gil-Peña H, Coto E, Pérez-Menéndez TM, Santos F:
Genetic and clinical peculiarities in a new family with hereditary hy-pophosphatemic rickets with hypercalciuria: A case report.Orphanet J
Rare Dis 5: 1, 201011. Tencza AL, Ichikawa S, Dang A, Kenagy D, McCarthy E, Econs MJ,
Levine MA: Hypophosphatemic rickets with hypercalciuria due to mu-tation in SLC34A3/type IIc sodium-phosphate cotransporter: Pre-sentation as hypercalciuria and nephrolithiasis. J Clin Endocrinol
Metab 94: 4433–4438, 200912. Romero V, Akpinar H, Assimos DG: Kidney stones: A global picture of
prevalence, incidence, and associated risk factors. Rev Urol 12: e86–e96, 2010
13. Schissel BL, Johnson BK: Renal stones: Evolving epidemiology andmanagement. Pediatr Emerg Care 27: 676–681, 2011
14. MagenD, Berger L, CoadyMJ, Ilivitzki A, Militianu D, Tieder M, Selig S,Lapointe JY, Zelikovic I, Skorecki K: A loss-of-functionmutation inNaPi-IIaand renal Fanconi’s syndrome. N Engl J Med 362: 1102–1109, 2010
15. The HYP Consortium: A gene (PEX) with homologies to endopepti-dases is mutated in patients with X-linked hypophosphatemic rickets.Nat Genet 11: 130–136, 1995
8 Journal of the American Society of Nephrology J Am Soc Nephrol 25: ccc–ccc, 2014
CLINICAL RESEARCH www.jasn.org

16. ADHRConsortium: Autosomal dominant hypophosphataemic rickets isassociated with mutations in FGF23. Nat Genet 26: 345–348, 2000
17. Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI,Zhang S, Rios H, DreznerMK, Quarles LD, Bonewald LF, White KE: Lossof DMP1 causes rickets and osteomalacia and identifies a role for os-teocytes in mineral metabolism. Nat Genet 38: 1310–1315, 2006
18. Lorenz-Depiereux B, Bastepe M, Benet-Pagès A, Amyere M,Wagenstaller J, Müller-Barth U, Badenhoop K, Kaiser SM, RittmasterRS, Shlossberg AH, Olivares JL, Loris C, Ramos FJ, Glorieux F, VikkulaM, Jüppner H, Strom TM: DMP1 mutations in autosomal recessivehypophosphatemia implicate a bonematrix protein in the regulation ofphosphate homeostasis. Nat Genet 38: 1248–1250, 2006
19. Lorenz-Depiereux B, Schnabel D, Tiosano D, Häusler G, Strom TM:Loss-of-function ENPP1 mutations cause both generalized arterialcalcification of infancy and autosomal-recessive hypophosphatemicrickets. Am J Hum Genet 86: 267–272, 2010
20. Jaureguiberry G, De la Dure-Molla M, Parry D, Quentric M, HimmerkusN, Koike T, Poulter J, Klootwijk E, Robinette SL, Howie AJ, Patel V,Figueres ML, Stanescu HC, Issler N, Nicholson JK, Bockenhauer D,Laing C, Walsh SB, McCredie DA, Povey S, Asselin A, Picard A,Coulomb A, Medlar AJ, Bailleul-Forestier I, Verloes A, Le Caignec C,Roussey G, Guiol J, Isidor B, Logan C, Shore R, Johnson C, InglehearnC, Al-Bahlani S, Schmittbuhl M, Clauss F, Huckert M, Laugel V,Ginglinger E, Pajarola S, Spartà G, Bartholdi D, Rauch A, Addor MC,Yamaguti PM, Safatle HP, Acevedo AC, Martelli-Júnior H, dos SantosNetos PE, Coletta RD, Gruessel S, Sandmann C, Ruehmann D,Langman CB, Scheinman SJ, Ozdemir-Ozenen D, Hart TC, Hart PS,Neugebauer U, Schlatter E, Houillier P, Gahl WA, Vikkula M, Bloch-Zupan A, Bleich M, Kitagawa H, Unwin RJ, Mighell A, Berdal A, Kleta R:Nephrocalcinosis (enamel renal syndrome) caused by autosomal re-cessive FAM20A mutations. Nephron, Physiol 122: 1–6, 2012
21. White AJ, Northcutt MJ, Rohrback SE, Carpenter RO, Niehaus-SauterMM, Gao Y, Wheatly MG, Gillen CM: Characterization of sarcoplasmiccalcium binding protein (SCP) variants from freshwater crayfish Pro-cambarus clarkii. Comp Biochem Physiol B Biochem Mol Biol 160: 8–14, 2011
22. Yu Y, Sanderson SR, Reyes M, Sharma A, Dunbar N, Srivastava T,Jüppner H, Bergwitz C: Novel NaPi-IIc mutations causing HHRH andidiopathic hypercalciuria in several unrelated families: Long-term follow-up in one kindred. Bone 50: 1100–1106, 2012
23. Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA,Hurles ME, McVean GA; 1000 Genomes Project Consortium: A map ofhuman genome variation from population-scale sequencing. Nature
467: 1061–1073, 201024. Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V,
Church DM, DiCuccio M, Edgar R, Federhen S, Geer LY, Helmberg W,Kapustin Y, Kenton DL, Khovayko O, Lipman DJ, Madden TL, MaglottDR, Ostell J, Pruitt KD, Schuler GD, Schriml LM, Sequeira E, Sherry ST,Sirotkin K, Souvorov A, Starchenko G, Suzek TO, Tatusov R, TatusovaTA,Wagner L, Yaschenko E: Database resources of the National Centerfor Biotechnology Information. Nucleic Acids Res 34[Database issue]:D173–D180, 2006
25. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, BorkP, Kondrashov AS, Sunyaev SR: A method and server for predictingdamaging missense mutations. Nat Methods 7: 248–249, 2010
26. Braithwaite V, Pettifor JM, Prentice A: Novel SLC34A3 mutation caus-ing hereditary hypophosphataemic rickets with hypercalciuria in aGambian family. Bone 53: 216–220, 2013
27. Areses-TrapoteR, López-García JA,Ubetagoyena-ArrietaM, EizaguirreA, Sáez-Villaverde R: Hereditary hypophosphatemic rickets with hy-percalciuria: Case report. Nefrologia 32: 529–534, 2012
28. Hasani-Ranjbar S, Amoli MM, Ebrahim-Habibi A, Dehghan E, Soltani A,Amiri P, Larijani B: SLC34A3 intronic deletion in a new kindred withhereditary hypophosphatemic rickets with hypercalciuria. J Clin Res
Pediatr Endocrinol 4: 89–93, 2012
29. Ichikawa S, Tuchman S, Padgett LR, Gray AK, Baluarte HJ, Econs MJ:Intronic deletions in the SLC34A3 gene: A cautionary tale for mutationanalysis of hereditary hypophosphatemic rickets with hypercalciuria.Bone 59: 53–56, 2014
30. Yamamoto T, Michigami T, Aranami F, Segawa H, Yoh K, Nakajima S,Miyamoto K, Ozono K: Hereditary hypophosphatemic rickets with hy-percalciuria: A study for the phosphate transporter gene type IIc andosteoblastic function. J Bone Miner Metab 25: 407–413, 2007
31. Stechman MJ, Loh NY, Thakker RV: Genetic causes of hypercalciuricnephrolithiasis. Pediatr Nephrol 24: 2321–2332, 2009
32. Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL: Aclinician’s guide to X-linked hypophosphatemia. J Bone Miner Res 26:1381–1388, 2011
33. Langman CB: The molecular basis of kidney stones. Curr Opin Pediatr
16: 188–193, 200434. Gudbjartsson DF, Holm H, Indridason OS, Thorleifsson G, Edvardsson
V, SulemP, de Vegt F, d’Ancona FC, denHeijerM,Wetzels JF, FranzsonL, Rafnar T, Kristjansson K, Bjornsdottir US, EyjolfssonGI, Kiemeney LA,Kong A, Palsson R, Thorsteinsdottir U, Stefansson K: Association ofvariants at UMODwith chronic kidneydisease and kidney stones-role ofage and comorbid diseases. PLoS Genet 6: e1001039, 2010
35. Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U,Misselwitz J, Klaus G, Kuwertz-Bröking E, Fehrenbach H, Wingen AM,Güran T, Hoenderop JG, Bindels RJ, Prosser DE, Jones G, Konrad M:Mutations in CYP24A1 and idiopathic infantile hypercalcemia.NEngl J
Med 365: 410–421, 201136. Tore S, Casula S, Casu G, Concas MP, Pistidda P, Persico I, Sassu A,
Maestrale GB, Mele C, CarusoMR, Bonerba B, Usai P, Deiana I, ThorntonT, Pirastu M, Forabosco P: Application of a new method for GWAS in arelated case/control samplewith knownpedigree structure: Identificationof new loci for nephrolithiasis. PLoS Genet 7: e1001281, 2011
37. Kestenbaum B, Glazer NL, Köttgen A, Felix JF, Hwang SJ, Liu Y,Lohman K, Kritchevsky SB, Hausman DB, Petersen AK, Gieger C, RiedJS, Meitinger T, Strom TM, Wichmann HE, Campbell H, Hayward C,Rudan I, de Boer IH, Psaty BM, Rice KM, Chen YD, Li M, Arking DE,Boerwinkle E, Coresh J, Yang Q, Levy D, van Rooij FJ, Dehghan A,Rivadeneira F, Uitterlinden AG, Hofman A, van Duijn CM, Shlipak MG,KaoWH,Witteman JC, Siscovick DS, Fox CS: Common genetic variantsassociate with serum phosphorus concentration. J Am Soc Nephrol 21:1223–1232, 2010
38. Prié D, Ravery V, Boccon-Gibod L, Friedlander G: Frequency of renalphosphate leak among patients with calcium nephrolithiasis.Kidney Int60: 272–276, 2001
39. HesseM, Fröhlich LF, Zeitz U, Lanske B, Erben RG:Ablation of vitaminDsignaling rescues bone, mineral, and glucose homeostasis in Fgf-23deficient mice. Matrix Biol 26: 75–84, 2007
40. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T,OhyamaY,KurabayashiM,KanameT,KumeE, IwasakiH, IidaA,Shiraki-IidaT, Nishikawa S, Nagai R, Nabeshima YI: Mutation of the mouse klothogene leads to a syndrome resembling ageing.Nature 390: 45–51, 1997
41. Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL: Aclinician’s guide to X-linked hypophosphatemia. J Bone Miner Res 26:1381–1388, 2011
42. Gonlusen G, Akgun H, Ertan A, Olivero J, Truong LD: Renal failure andnephrocalcinosis associated with oral sodium phosphate bowelcleansing: clinical patterns and renal biopsy findings. Arch Pathol Lab
Med 130: 101–106, 200643. Walton RJ, BijvoetOL: A simple slide-rulemethod for the assessment of
renal tubular reaborption of phosphate inman.Clin ChimActa 81: 273–276, 1977
44. Alon U, Hellerstein S: Assessment and interpretation of the tubularthreshold for phosphate in infants and children. Pediatr Nephrol 8:250–251, 1994
45. Brodehl J, Gellissen K, Weber HP: Postnatal development of tubularphosphate reabsorption. Clin Nephrol 17: 163–171, 1982
J Am Soc Nephrol 25: ccc–ccc, 2014 Frequency of Renal Calcifications in HHRH 9
www.jasn.org CLINICAL RESEARCH

46. Kruse K, Kracht U, Göpfert G: Renal threshold phosphate concentration(TmPO4/GFR). Arch Dis Child 57: 217–223, 1982
47. Mitchell DM, Jüppner H: Regulation of calcium homeostasis and bonemetabolism in the fetus and neonate. Curr Opin Endocrinol DiabetesObes 17: 25–30, 2010
48. Boyd JC, Lacher DA: A multi-stage Gaussian transformation algorithmfor clinical laboratory data. Clin Chem 28: 1735–1741, 1982
49. LeeWT, Jiang J: The resurgence of the importance of vitamin D in bonehealth. Asia Pac J Clin Nutr 17[Suppl 1]: 138–142, 2008
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2013101085/-/DCSupplemental.
10 Journal of the American Society of Nephrology J Am Soc Nephrol 25: ccc–ccc, 2014
CLINICAL RESEARCH www.jasn.org




















