
московский государственный университет имени
152
МОСКОВСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ ИМЕНИ М.В.ЛОМОНОСОВА БИОЛОГИЧЕСКИЙ ФАКУЛЬТЕТ, КАФЕДРА БИОИНЖЕНЕРИИ ФЕДЕРАЛЬНЫЙ ИССЛЕДОВАТЕЛЬСКИЙ ЦЕНТР «ФУНДАМЕНТАЛЬНЫЕ ОСНОВЫ БИОТЕХНОЛОГИИ» РОССИЙСКОЙ АКАДЕМИИ НАУК ИНСТИТУТ БИОХИМИИ ИМ. А.Н.БАХА ЛАБОРАТОРИЯ БИОХИМИИ АЗОТФИКСАЦИИ И МЕТАБОЛИЗМА АЗОТА; На правах рукописи ЗЕРНОВ АНТОН ЛАВРЕНТИЕВИЧ МИКРОЧАСТИЦЫ ИЗ БИОСИНТЕТИЧЕСКИХ ПОЛИОКСИАЛКАНОАТОВ ДЛЯ ПРОЛОНГИРОВАННОГО ВЫСВОБОЖДЕНИЯ БЕЛКОВ 03.01.06 – биотехнология (в том числе бионанотехнологии) Диссертационная работа на соискание ученой степени кандидата биологических наук Научный руководитель: к.б.н. Бонарцев А.П. Научный консультант: к.б.н. Бонарцева Г.А Москва 2017
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of московский государственный университет имени

МОСКОВСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ ИМЕНИ
М.В.ЛОМОНОСОВА
БИОЛОГИЧЕСКИЙ ФАКУЛЬТЕТ, КАФЕДРА БИОИНЖЕНЕРИИ
ФЕДЕРАЛЬНЫЙ ИССЛЕДОВАТЕЛЬСКИЙ ЦЕНТР «ФУНДАМЕНТАЛЬНЫЕ
ОСНОВЫ БИОТЕХНОЛОГИИ» РОССИЙСКОЙ АКАДЕМИИ НАУК
ИНСТИТУТ БИОХИМИИ ИМ. А.Н.БАХА
ЛАБОРАТОРИЯ БИОХИМИИ АЗОТФИКСАЦИИ И МЕТАБОЛИЗМА АЗОТА;
На правах рукописи
ЗЕРНОВ АНТОН ЛАВРЕНТИЕВИЧ
МИКРОЧАСТИЦЫ ИЗ БИОСИНТЕТИЧЕСКИХ ПОЛИОКСИАЛКАНОАТОВ
ДЛЯ ПРОЛОНГИРОВАННОГО ВЫСВОБОЖДЕНИЯ БЕЛКОВ
03.01.06 – биотехнология (в том числе бионанотехнологии)
Диссертационная работа на соискание ученой степени
кандидата биологических наук
Научный руководитель:
к.б.н. Бонарцев А.П.
Научный консультант:
к.б.н. Бонарцева Г.А
Москва 2017

2
Оглавление
ВВЕДЕНИЕ.................................................................................................................... 5
ЧАСТЬ 1. ЛИТЕРАТУРНЫЙ ОБЗОР ......................................................................... 9
1.1. Поли(3-гидроксиалканоаты) ................................................................................. 9
1.1.1. Поли(3-гидроксибутират) ................................................................................ 13
1.2. Системы пролонгированного высвобождения белков ..................................... 32
1.2.1. Системы пролонгированного высвобождения в тканевой инженерии ....... 32
1.2.2. Методы получения биополимерных микрочастиц ........................................ 35
1.2.3. Кинетика высвобождения веществ из полимерных микрочастиц ............... 43
1.2.4. Проблема стабильности высвобождаемого белка ......................................... 44
1.2.5. Применение полимерных микрочастиц, загруженных белком в тканевой
инженерии .................................................................................................................... 47
ЧАСТЬ 2. МАТЕРИАЛЫ И МЕТОДЫ ИССЛЕДОВАНИЯ .................................. 55
2.1. ПГБ и сополимер ПГБ-ПЭГ ................................................................................ 55
2.1.1. Синтез гомополимера поли(3-гидроксибутирата) различной молекулярной
массы. ........................................................................................................................... 55
2.1.2. Синтез сополимера поли(3-гидроксибутират)-со-поли(этилен гликоль) ... 56
2.1.3. Выделение поли-3-гидроксибутирата и его сополимеров из биомассы ..... 57
2.1.4. Определение молекулярной массы полимера ................................................ 57
2.1.5. Ядерный магнитный резонанс ......................................................................... 58
2.1.6. ИК-спектроскопия с Фурье преобразованием ............................................... 58
2.1.7. Определение гидрофильности полимерного материала ............................... 59
2.2. Системы пролонгированного высвобождения белков на основе
полиоксиалканоатов. .................................................................................................. 59
2.2.1. Белки, инкапсулируемые в полимерную матрицу микрочастиц ................ 59
2.2.2. Получение микрокапсул по методике W/O/W из ПГБ с
инкапсулированными БСА ........................................................................................ 60
2.2.3. Получение композита декстрана с лизоцимом .............................................. 61
2.2.4. Получение композита поли(3-гидроксибутирата) с лизоцимом. ................. 61
2.2.5. Получение композита нано-гидроксиаппатита с лизоцимом ...................... 62

3
2.2.6. Получение полимерных микрочастиц с инкапсулированным в них
композитом нано-гидроксиаппатита с лизоцимом .................................................. 62
2.2.7. Исследование пролонгированного высвобождения лизоцима и БСА из
полимерных микроструктур in vitro .......................................................................... 64
2.2.8. Спектрофотометрическое определение содержания лизоцима и БСА в
полученных образцах ................................................................................................. 64
2.2.9. Исследование полученных микроструктур с помощью методов
конфокальной микроскопии ...................................................................................... 65
2.2.10. Исследование морфологии полученных микроструктур с помощью
методов сканирующей электронной микроскопии ................................................. 65
2.2.11. Исследование деградации микрочастиц ....................................................... 65
2.3. Исследование стабильности белка высвободившегося из полимерных
микрочастиц ................................................................................................................ 66
2.3.1. Определение целостности первичной структуры лизоцима при помощи
электрофореза в ПААГ в присутствии SDS ............................................................. 66
2.3.2. Исследование вторичной структуры высвободившегося белка методом
кругового дихроизма .................................................................................................. 68
2.3.3. Исследование третичной структуры высвободившегося белка методом
определения его ферментативной активности ......................................................... 68
2.3.4. Выделение мезенхимальных стволовых клеток из костного мозга крыс
линии Wistar ................................................................................................................ 69
2.3.5. Оценка цитотоксичности микрочастиц, загруженных лизоцимом in vitro. 70
ЧАСТЬ 3. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ ................................................... 71
3.1. Материал поли(3-гидроксибутират .................................................................... 71
3.1.1. Получение поли(3-гидроксибутирата) различной молекулярной массы ... 71
3.2. Микрокапсулы для пролонгированной доставки модельного белка БСА .... 81
3.2.1. Получение и характеризация полимерных микрокапсул с
инкапсулированным модельным белком БСА......................................................... 81
3.2.2. Кинетика высвобождения модельного белка из микрокапсул in vitro и
механизм длительного высвобождения белка из структур. ................................... 84

4
3.3. Сплошные микрочастицы для пролонгированной доставки модельного
белка лизоцима ............................................................................................................ 88
3.3.1. Создание композита белка с носителем ......................................................... 89
3.3.2. Создание полимерных микрочастиц, загруженных композитом
гидроксиапатит/лизоцим по методике двухэтапного эмульгирования «твердая
фаза/масляная фаза/водная фаза» .............................................................................. 92
3.3.3. Высвобождение лизоцима из полимерных микрочастиц, основанных на
гомополимере ПГБ 250 кДа ....................................................................................... 96
3.3.4. Биосинтез модифицированных полимеров для эффективного
инкапсулирования лизоцима в полимерные микроструктуры .............................. 99
3.3.5. Получение и характеризация полимерных микрочастиц с
инкапсулированным модельным белком лизоцимом на основе
модифицированных полимеров ............................................................................... 106
3.3.6. Кинетика высвобождения лизоцима из полимерных микрочастиц,
основанных на модифицированных полимерах. Стабильность
высвобождающегося белка ...................................................................................... 109
3.3.7. Исследование полимерных микрочастиц, загруженных лизоцимом на
цитотоксичность in vitro ........................................................................................... 117
ЗАКЛЮЧЕНИЕ ......................................................................................................... 119
ВЫВОДЫ ................................................................................................................... 121
БЛАГОДАРНОСТИ .................................................................................................. 122
СПИСОК СОКРАЩЕНИЙ ...................................................................................... 123
СПИСОК ЛИТЕРАТУРЫ ........................................................................................ 124

5
ВВЕДЕНИЕ
В настоящее время поли(3-гидроксибутират) (ПГБ) и его сополимеры,
получаемые биотехнологичеким путем, привлекают большое внимание как
биосовместимые и биодеградируемые полимеры. Микробиологический синтез
полимера с помощью штамма сверхпродуцента Azotobaacter chroococcum 7Б
позволяет, с одной стороны, получать полимеры разной молекулярной массы, а
с другой вводить в полимерную цепь различные молекулы, получая при этом
сополимеры, варьируя при этом физико-химические свойства материала в
широких пределах. Поэтому такие биосинтетические материалы могут быть
использованы для разработки широкого спектра изделий биомедицинского
назначения, таких как микрочастицы для пролонгированной доставки лекарств,
скаффолды для тканевой инженерии и других медицинских изделий:
кардиоваскулярных стентов, пародонтологических мембран и др. Наибольший
интерес в современной биотехнологии представляют системы доставки,
созданные на основе полимерных микрочастиц, которые могут снижать
недостатки традиционных лекарственных форм медицинских препаратов, а
именно: высокую токсичность, нестабильность и неэффективный расход
действующего начала, сложность введения и другие. Среди таких систем
следует отметить микрочастицы, загруженные низкомолекулярными
веществами (цитостатиками, противовоспалительными средствами,
антибиотиками), макромолекулами, такими как белки, а так же
неорганическими наночастицами. Широкое внедрение в последнее десятилетие
в медицинскую практику препаратов терапевтических белков (гормонов,
цитокинов, факторов роста, моноклональных антител и др.) делает разработку
систем контролируемого высвобождения белков особенно актуальным. Одним
из перспективных подходов для решения этой проблемы является
использование в качестве материала для создания таких систем гидрофобных
полимеров, растворимых в органических растворителях, связывающих белковые
макромолекулы лишь физически, исключая при это химические контакты. Это с
одной стороны позволяет добиваться высвобождения белков из таких

6
гетерогенных систем с заданной кинетикой, а с другой нивелирует негативное
воздействие на нативную структуру белка со стороны полимерного носителя.
Однако создание таких систем с белками является сложной и нетривиальной
задачей. Основная проблема состоит в том, что белок необратимо адсорбируется
на полимерный матрикс в процессе создания таких структур, а также при
диффузии макромолекул сквозь него, денатурирует и теряет свою активность.
Это связано с высокой гидрофобностью полимерных материалов. Для решения
этой проблемы возможно повышение гидрофильности различными методами,
например сополимеризацией с более гидрофильным полимером, либо
снижением молекулярной массы полимера, что приводит к лучшей
биосовместимости полимерного носителя. Создание таких систем рационально
проводить с использованием хорошо охарактеризованных относительно
стабильных и доступных в большом количестве модельных белков, которые,
тем не менее, являются типичными представителями целых классов
терапевтических белков.
Таким образом, целью работы явилось создание системы
пролонгированного высвобождения белков на основе микрочастиц из
полиоксиалканоатов.
В соответствии с целью были сформулированы следующие задачи:
1. Биосинтез полимеров – поли(3-гидроксибутирата) различной
молекулярной массы и его сополимера с поли(этилен гликолем) для создания
систем пролонгированного высвобождения белков.
2. Разработка методики инкапсулирования белков, сывороточного
альбумина и лизоцима, в микрочастицы на основе полученных
полиоксиалканоатов и ее модификация для улучшения эффективности
инкапсулирования белка в получаемые микрочастицы.
3. Исследование морфологии, физико-химических свойств, а так же
механизмов включения и высвобождения белков из полученных структур.
4. Исследование стабильности высвобождающегося белка.
5. Исследование биосовместимости полученных микрочастиц in vitro.

7
Научная новизна. В данной работе был разработан ряд оригинальных
методик инкапсулирования белков в полимерные микрочастицы на основе
поли(3-гидрокибутирата) и его сополимеров. При этом модельный белок
лизоцим включался в полимерную матрицу в составе композита с
гидроксиапатитом.
Впервые для инкапсулирования белков использован биосинтетический
сополимер поли(3-гидроксибутирата) (ПГБ) с поли(этилен гликолем) (ПЭГ), а
также показано, что микрочастицы из сополимера обладают лучшими
параметрами инкапсулирования модельного белка лизоцима. Также впервые
показано, что сополимеризация ПГБ с ПЭГ обеспечивает большую стабильность
высвобождаемого белка по сравнению с гомополимером на протяжении двух
недель высвобождения in vitro.
Для частиц на основе ПГБ-ПЭГ, загруженных модельным белком
лизоцимом показана высокая степень биосовместимости in vitro на
мезенхимальных стволовых клетках крысы.
Практическая значимость работы. Проведено исследование новой
полимерной системы доставки белков на основе микрочастиц из
полиоксиалканоатов, пригодных для медицинского применения. В качестве
модельного белка был выбран лизоцим, обладающий сходным физико-
химическими свойствами с белковыми факторами роста и цитокинами,
используемыми в тканевой инженерии, в частности в инженерии костной ткани.
Для систем пролонгированного высвобождения белков в тканевой инженерии
основной проблемой является низкая стабильность инкапсулированного
вещества – белок может денатурировать, став неактивным, а также вызывать
иммунную реакцию. При помощи биополимерных микрочастиц в данной работе
удалось достичь пролонгированного высвобождения модельного белка в
нативной форме на протяжении 14 суток. Полученные микрочастицы
продемонстрировали хорошую биосовместимость и могут быть использованы в
качестве систем доставки ростовых факторов и цитокинов в тканевой
инженерии.

8
Апробация работы. Основные положения диссертационной работы были
доложены на международной конференции по биоинформатике и
биомедицинской инженерии (Испания, Гранада, 2014), XIII Российско-
Китайского Симпозиума «Новые материалы и технологии» (Россия, Казань,
2015 г). 38м конгрессе Европейского биохимического общества (Россия, Санкт-
Петербург, 2013г.), XVIII, XIX и XX международном молодежном форуме
«Ломоносов» (Россия, Москва, 2011, 2012, 2013гг.), первой и второй
международной научно-практической конференции «Высокие технологии,
фундаментальные и прикладные исследования в физиологии и медицине»
(Россия, Санкт-Петербург, 2010, 2011 гг.),
Публикации. По теме диссертации опубликовано 3 статьи в журналах их
перечня ВАК ведущих рецензируемых научных журналов и изданий, 4 статьи в
зарубежных рецензируемых журналах, 3 статьи в сборниках статей по
материалам конференций (2 отечественные и одна зарубежная), отражающие
основной объем диссертационной работы. Результаты работы также были
представлены на международных и всероссийских конференциях.

9
ЧАСТЬ 1. ЛИТЕРАТУРНЫЙ ОБЗОР
1.1. Поли(3-гидроксиалканоаты)
Поли(3-гидроксиалканоаты) (ПГА) – это биосовместимые и
биодеградируемые полимеры 3-гидроксикарбоновых кислот, встречающиеся в
природе в виде запасного вещества бактерий. В бактериальной клетке они
находятся в виде гранул, расположенных в цитоплазме. Впервые они были
открыты в 1926 году микробиологом Maurice Lemoigne в бактериях рода
Bacillus megaterium [Lemoigne M., 1926], после чего они были обнаружены в
более чем 100 штаммах бактерий. Их общая форма представлена на рисунке 1. В
зависимоси от бокового радикала, различают полигидроксибутират,
полигидроксивалерат, полигидроксигексаноат, полигидроксиалканоат и так
далее (таблица 1). Все они довольно сильно различаются по своим физико-
химическим, механическим и другим свойствам, таким как кристалличность,
температура плавления, температура стеклования, гидрофобность,
пластичность, модуль упругости и другим [Bloembergen S., 1986], [Barham P.J.,
1984], [Scandola M., 1995]. Благодаря тому, что в зависимости от состава,
свойства этих материалов меняются в широких пределах, сферы медико-
биологического применения ПГА могут быть также самыми разными, такими
как хирургия [Sodian R., 2000], регенеративная инженерия мягких и скелетных
тканей организма [Caracciolo P. C., 2009] и многие другие. При этом, являясь
биосовместимыми и биодеградируемыми полимерами, ПОА не вызывают
отторжения внутренней средой организма, а постепенно замещаются на
собственную ткань организма.

10
Рисунок 1. Общая химическая формула поли(3-гидроксиалканоатов).
Таблица 1. Химическое строение и номенклатура основных представителей
полиоксиалкаоатов
Радикал Полное название Сокращенное название
СН3 – Поли(3-гидроксибутират) ПГБ
СН3 – СН2 – Поли(3-гидроксивалерат) ПГВ
СН3 – (СН2)2 – Поли(3-гидроксигексаноат) ПГГк
СН3 – (СН2)2 – Поли(3-гидроксиоктаноат) ПГО
СН3 – (СН2)2 – Поли(3-гидроксидеканоат) ПГД
Поли(3-гидрокси-5-
фенилбутират)
ПГФБ
Среди ПГА наиболее распространенным является поли(3-
гидроксибутират) (ПГБ) - полимер 3-гидроксималсялой кислоты, который и был
открыт первым. Именно его открыл в 1926 году Maurice Lemoigne [Lemoigne M.,
1926]. Впервые использовать в медицинских целях его предложили в 1962 году
в виде рассасываемых шовных нитей и пленок для заживления повреждений
кровеносных сосудов [Baptist J.N., 1962]. Однако в 1974 году Wallen и
Rohwedder смогли идентифицировать другие ПГА помимо ПГБ. Это были
поли(3-гидроксивалерат) и поли(3-гидроксигексаноат) [Wallen L. L., 1974]. Это
открытие значительно подстегнуло интерес к этому классу полимеров. Ведь
гомополимер, ввиду его хрупкости, имеет лишь ограниченное число
применений, в то время, как создание сополимеров с другими 3-
гидроксикарбоновыми кислотами позволило придать совершенно новые
свойства материалу, такие как пластичность, гибкость и так далее. В 1980х

11
годах началось масштабное производство сополимеров [Holmes P. A., 1985]. На
этом этапе удалось создать и охарактеризовать практически все возможные
сочетания мономеров, которые могли быть включены в состав ПГА. Также в это
время расширился и спектр штаммов-продуцентов. Оказалось, что к синтезу
таких полезных для человека запасных веществ способны не только Грамм-
отрицательные бактерии, но и Грамм-положительные, а также такие
таксономические группы, как цианобактерии и археи [Anderson A.J., 1990],
[SteinbuÈchel A., 1991].
Следующим шагом, позволившим еще более глубоко проникнуть в
изучение ПГА, явилось исследование генов, участвующих в их синтезе. С
помощью молекулярно-биологических методов стало возможным
контролировать процесс биосинтеза ПГА, а также синтезировать их с помощью
генетически-модифицированной Escherichia coli [Slater S.C., 1988], [Schubert P.,
1988], [Peoples O.P., 1989] – наиболее удобного на тот момент организма для
проведения ферментаций в промышленных масштабах.
Благодаря этим открытиям и разработкам, начиная с 1990х годов стало
появляться большое количество работ с самыми различными применениями
ПОА: системы доставки, матриксы, подложки, импланты. Однако до сих пор
наиболее распространенным и важным представителем ПГА является поли(3-
гидроксибутират). При этом, хотя материалом для большинства изделий являлся
ПГБ, а также его сополимер поли(3-гидроксибутиат-со-3-гидроксивалерат),
другие материалы также нашли свое применение: поли(3-гидроксиоктаноат-со-
3-гидроксигексаноат) использовался для васкулярных матриксов в сердечно-
сосудистой тканевой инженерии [Shum-Tim D., 1999.–], который был
впоследствии заменен на поли(4-гидроксибутират) – более быстро
деградируемый полимер 4-гидроксимасляной кислоты, примененный для
создания сердечных клапанов [Hoerstrup S.P., 2000], [Sodian R., 2000], и
сосудистых трансплантатов [Hoerstrup S.P., 2001], [Opitz F., 2004]. Отметим, что
последний является на данный момент одним из наиболее перспективных
биоразлагаемых полимеров [Martin D.P., 2003] и прошел исследования

12
Агентством по контролю над пищевыми и медицинскими продуктами США
(FDA) как материал, предназначенный для изготовления эндопротезов и
шовных нитей (Заключения FDA K112733 от 15.02.2012, K082178 от 30.10.2008,
P030050/S2 от 28.07.2009). Полимеры, состоящие из блоков с большим числом
атомов углерода, например поли(3-гидроксибутират-со-3-гидроксигексаноат),
могут быть использованы для создания матриксов и каркасов, обеспечивающих
прохождение остеонеогенеза, а также в инженерии хрящевой ткани [Deng Y.,
2002], [Wang Y.W., 2004]. Данные полимеры изображены на рисунке 2
Рисунок 2. Изображения структурных формул поли(3-гидроксибутирата),
поли(4-гидроксибутирата) и их сополимеров
В этой главе мы в первую очередь остановимся на ПГБ, как наиболее
типичном и широко применяемом ПГА, а также на его сополимерах.
П3ГБ П3ГБ-3ГВ
П4ГБ
П3ГБ-4ГБ
П3ГО-П3ГГ

13
1.1.1. Поли(3-гидроксибутират)
Поли(3-гидроксибутират) – это основной и первый описанный полимер из
группы ПГА. Он является полимером 3-оксималсяной кислоты и встречается в
качестве запасного вещества в большом количестве микроорганизмов [Anderson
A. J., 1990], [Dawes E. A., 1973.]. Будучи биополимером, он включает в себя
только R-форму оксимасляной кислоты (рис.3) и является частично
кристаллическим полиэфиром: выделенный из бактерий ПГБ имеет
кристалличность от 55% до 80% [Holmes P. A., 1998]. Это является полезным
свойством, так как для таких материалов легче контролировать их физико-
химические свойства, в то время как у аморфных соединений подобных свойств
не наблюдается. В конденсированном виде ПГБ имеет супрамолекулярную
ламеллярную структуру [Bagrov D. V., 2012], о чем будет подробнее рассказано
далее.
Рисунок 3. Структурная формула поли(3-гидрокибутирата)
Биосинтез
Поли(3-гидроксибутират) синтезируется сотнями различных
микроорганизмов в виде запасного вещества при определенных условиях
внешней среды. К внутриклеточному накоплению ПГБ способны такие роды
бактерий, как Azotobacter sp., Pseudomonas sp., Bacillus sp. и Methylobacterium sp.
[Forsyth W. G. C., 1958]. Они аккумулируют этот полимер в цитоплазме в виде
гранул, как пул углерода и химической энергии под воздействием азотного
голодания [Dawes E. A., 1973], [Wallen L. L., 1974].
Рассмотрим метаболические пути биосинтеза ПГБ. Эти пути являются
общими практически для всех бактерий-продуцентов. Фермент β-кетотиолаза

14
катализирует образование углерод-углеродной связи двух ацетил-КоА остатков
путем коденсации Кляйзена. Молекулы ацетил-КоА поступают при этом из
гликолиза через образование пирувата. Далее НАДФН-зависимая ацетоацетил-
КоА-редуктаза превращает ацетоацетил-КоА в 3-гидроксибутирил-КоА [Findlay
R. H., 1983]. На следующем этапе молекулы 3-гидроксибутирил-КоА
связываются с ПГБ-полимеразой (рисунок 4). В неактивном состоянии она
представляет собой мономерные субъединицы, которые растворены в
цитоплазме. При связывании субстрата эти субъединицы димеризуются и с
помощью этого гомодимера начинается синтез полимера. Связывание остатков
3-гидроксимасляной кислоты осуществляется конститутивным остатком
цистеина. По мере синтеза полимера и роста цепи, такие комплексы
организуются в гранулы, внутри которых находятся гидрофобная цепь
растущего ПГБ, а снаружи расположены молекулы ПГБ-полимеразы со
вспомогательными белками, которые продолжают синтез на поверхности гранул
(рисунок 5). При этом на одну молекулу мономера – 3-гидроксимасляной
кислоты – приходятся 2 молекулы ацетил-КоА [Shrivastav A., 2013]. ПГБ-
полимераза может существовать в двух формах: свободной, растворенной в
цитоплазме и гидрофобной, связанной с полимерными гранулами. Во время
роста при дефиците углерода фермент существует в растворенном состоянии,
однако при наступлении стрессовой ситуации (например, азотного голодания)
он переходит в ассоциированную с гранулами форму и приступает к синтезу
запасного вещества – ПГБ. Биохимически синтез ПГБ контролируется
соотношением НАДФН к НАДФ+ в цитоплазме. Когда источник азота иссякает,
это соотношение возрастает, что ингибирует ферменты цикла трикарбоновых
кислот. Когда поток ацетил-КоА уменьшается, понижается уровень кофермента
А и снимается ингибирование β-кетотиолазы и начинается синтез полимера
[Lageveen R. G., 1988], [Holmes P. A, 1985]. Однако параллельно с
анаболическими процессами протекают и катаболические реакции, то есть
происходит также непрерывное разложение ПГБ до мономеров ферментов ПГБ-
деполимеразой. Таким образом, регуляция процесса полимерного биосинтеза

15
осуществляется путем смещения равновесия между прямой и обратной
реакциями полимеризации и деполимеризации.
Рисунок 4. Схема биосинтеза ПГБ, проходящего в бактериальных клетках
Рисунок 5. Схема формирования полимерных гранул в цитоплазме
бактериальных клеток при синтезе ПГБ

16
Фермент ПГБ-синтаза стереоспецифичен – только R-изомеры 3-
гидроксимасляной кислоты могут участвовать в реакции. Также, у ПГБ-синтазы
различных организмов могут включать в полимерную цепь не только остатки 3-
гидроксибутирата, но и, например 3-гидроксивалерата и более
длинноцепочечных 3-гидроксикарбоновых кислот [Shiraki M., 2006], [Rehm
B.H., 2002], [Tsuge T., 2000]. Именно за счет этого возможно получение
сополимеров ПГА, обладающих уникальными свойствами.
Физико-химические свойства
Кристаллическая структура ПГБ была установлена с помощью
рентгеноструктурного анализа ориентированных в пространстве фибрилл
[Okamura K., 1967], [Yokouchi M., 1973]. С его помощью были обнаружены
повторяющиеся вдоль полимерной цепи структуры с шагом 0,596 нм,
соответствующие длине двух кислотных остатков. Они оказываются упакованы
в орторомбическую ячейку с размерами 0:576 нм × 1:320 нм × 0.596 нм. Исходя
из конформационного анализа, основанного на расчетах внутримолекулярной
энергии, было установлено, что молекулы ПГБ имеют лево-закрученную 21-
спиральную конформацию. [Okamura K., 1967], [Yokouchi M., 1973], [Cornibert
J., 1972], Рядом исследователей при изучении одиночных кристаллов ПГБ была
показана структура полимера, в которой цепь укладывается примерно по 10
звеньев в специальные структуры – ламели. Однако в то время как
монокристаллы полимера, складываясь, образуют моноламеллярную систему,
реальные полимерные объекты, такие как пленки, изделия и др. образуют
мультиламеллярные кристаллы, складывающиеся в сферолиты [Barham P.J.,
1984], в которых эти кристаллы укладываются в радиальные стеки. Все это
говорит нам о том, что данный тип биополимеров имеет сложную
пространственную укладку, что задает большинство его физико-механических
свойств.
Молекулярная масса синтезируемого полимера колеблется от 10 до 3000
кДа с индексом полидисперсности около. Температура стеклования ПГБ около 4

17
°С, в то время, как температура плавления колеблется в районе значения 180 °С,
что было определено калориметрическим методом. Плотность аморфного и
кристаллического ПГБ составляет, соответственно, 1,26 г/см3 и 1,18 г/см
3.
Модуль Юнга составляет примерно 3,5 ГПа, а предел прочности на разрыв – 43
МПа. При этом растяжение на разрыв составляет всего 5%, что говорит нам о
том, что ПГБ – это довольно жесткий и хрупкий материал. Такие механические
свойства не всегда подходят для решения задач, связанных с использованием
каркасных полимерных структур для создания имплантов. Поэтому было
проведено множество работ, направленных на выяснение природы хрупкости
ПГБ, а также путей изменения его физико-механических свойств. Одним из
наиболее эффективных подходов по изменению механических свойств является
создание сополимеров, например с 3-оксивалерьяновой [Holmes P. A., 1988], 4-
оксимасляной [Doi Y., 1990] или 3-оксигексановой кислотой [Doi Y., 1995].
Также сильное влияние на механические свойства оказывает молекулярная
масса полимера: например, предел прочности на разрыв начинает резко
снижаться при снижении молекулярной массы ниже порогового значения в
районе 100 кДа [Renstadt R., 1998].
Также одним из наиболее важных свойств полимера является его
гидрофобность. Она влияет на взаимодействие полимера с внутренней средой
организма при имплантации, его биосовместимость, а также является очень
важным параметром при разработке систем доставки лекарств, так как
определяет характер взаимодействий действующего вещества с носителем [Li J.,
2006]. Это свойство материала так же зависит от молекулярной массы, степени
кристалличности и множества других факторов.
Таким образом, влияние на структуру поли(3-гидрокибутирата) путем
изменения его молекулярной массы, а так же создания новых сополимеров
позволяет создать материалы с заданными физико-химическими свойствами,
необходимыми для решения определенных задач в создании биомедицинских
изделий.

18
Биосовместимость
Применимость ПГБ для создания изделий биомедицинского назначения, а
также систем доставки лекарств обуславливается, прежде всего, его
биосовместимостью. Этот термин означает, что такие структуры не должны
вызывать сильной иммунной реакции со стороны мягких тканей, а также крови
организма-реципиента как в начале имплантации, так и в процессе его
биодеградации. Следует отметить, что ПГБ встречается не только в виде
запасного вещества в микроорганизмах, а повсеместно встречаются в природе, в
том числе у растений, а также у животных [Reusch R. N., 1989]. Именно поэтому
с точки зрения биосовместимости ПГБ представляет собой отличный материал
для создания имплантатов, каркасных матриксов и так далее. Метаболизм,
выведение из организма ПГБ довольно хорошо изучены. Что касается его
мономера – R-3-гидроксимасляной кислоты – то она водит в катаболизм
жирных кислот, протекающий в клетках печени животных, что говорит нам
также о его нетоксичости [Lee S.Y., 1996]. Это кетоновые тела, которые
продуцируются в митохондриях, которые затем распределяются по организму,
участвуя в дальнейших метаболических превращениях организма. 3-
гидроксибутират – это конститутивный компонент крови, его концентрация
колеблется от 0,3 до 1,3 мМ [Zinn M., 2001]. После обнаружения столь широкого
распространения ПГБ в таких разных организмах, начиная от бактерий и
заканчивая человеком, разработка изделий на основе этого материала стала еще
более привлекательной. В большинстве случаев, низкомолекулярный ПГБ
встречается в виде комплексов с полифосфатом кальция в клеточных мембранах
[Reusch R.N., 1992], [Reusch R. N., 1989].
Итак, различные медицинские приложения ПГБ (системы
пролонгированной доставки цитокинов и ростовых факторов, матриксы для
замещения костной ткани, кардиоваскулярные стенты и многие другие)
получают широкое распространение во многих областях. При этом, каждый раз,
они контактируют с внутренней средой организма – определенной тканью.

19
Рассмотрим для начала биосовместимость ПГБ на низшем уровне организации –
опытах на клетках in vitro [Misra S. K., 2006], [Wu Q., 2009].
Фибробласты мыши NIH 3T3 являются одним из наиболее удобных и
распространенных объектов для определения цитотоксичности материала. При
их культивировании на полимерных мембранах, они показали хорошие
адгезивные качества и высокую выживаемость [Shishatskaya E. I., 2004]. В
другом исследовании, уже на линии фибробластов L929 ПГБ также показал
отличные результаты (было показано, что пролиферативная активность
растущих на этом полимере клеток не снижается), обойдя по этому параметру
широко применяемый поли(L-лактид) (ПМК), являющийся довольно
распространенным материалом и на данный момент одним из самых
популярных биосовместимых полимеров [Wang Y. W., 2005]. Также внесение
различных добавок, как например гидроксиапатита, увеличивает это свойство
[Wang Y. W., 2005].
В данное время ведется множество работ по регенеративной инженерии,
связанной с культивированием мезенхимальных стволовых клеток (МСК). ПГБ
проявляет по отношению к ним отличную биосовместимость: клетки
адгезируют на изделиях из этого материала и не проявляют признаков угнетения
роста. Примером могут послужить исследования, в которых МСК хорошо
растут на пленках из ПГБ [Hu Y. J., 2009]. В этом исследовании Ya-Jun Hu с
соавторами показывает также преимущество ПГБ над все тем же ПЛА, также
улучшение показателей культивации при сополимеризации поли(3-
гидроксибутирата) с 3-гидроксигексаноатом и 3-гидроксивалератом (ПГБВ).
Также следует отметить исследования роста этой культуры клеток, проведенные
в том числе в нашей лаборатории, на электроволокнистых скаффолдах [Zharkova
I. I., 2013], а также на пористых матриксах [Andreeva N. V., 2015], [You M.,
2011]. В последнем случае исследователями из Китая был показан рост клеток в
объеме и постепенное заселение полостей матрикса мезенхимальными
стволовыми клетками, что является очень полезным свойством при
конструировании костных и хрящевых имплантов из ПГБ и его сополимеров.

20
Korsatko с соавторами в своей работе показал отсутствие значительной разницы
в росте различных клеток на изделиях из ПГБ. При этом были использованы
частицы из низкомолекулярного полимера, моделирующие отделяющиеся от
имплантов куски полимера, появляющиеся при его биодеградации in vivo
[Korsatko W., 1983]. В низких концентрациях, в которых они и встречаются в
организме после имплантации, их воздействие одинаково хорошо переносится и
фибробластами, и гепатоцитами, и макрофаками, и Купферовыми клетками.
Последние два типа клеток, при этом, захватывали эти частицы путем
эндоцитоза и последующей их деградацией [Ciardelli G., 1995].
Прежде чем перейти к исследованиям биосовместимости на лабораторных
животных in vivo, остановимся на не менее важном аспекте. В исследовании
полимерных изделий и материалов также очень важна гемосовместимость,
основанная на определении реакции при контакте полимера с кровью
млекопитающих. Было показано, что ПГБ не вызывает ни активации
тромбоцитов, ни активации системы комплемента [Clarotti G., 1992]. Как
считает ряд исследователей, это связано с тем, что белок не денатурирует,
связываясь в поверхностью ПГБ. В случае отсутствия реакции тромбоцитов,
данные были подтверждены изучением адсорбции фибриногена на полимерные
пленки [Iordanskii A.L., 1999]. Однако если в полимере присутствуют остатки
клеточной стенки бактерии-продуцента, может возникнуть резкая иммунная
реакция [Sevastianov V. I., 2003], [Ji G. Z., 2009].
В опытах in vivo используется имплантация полимерных изделий в
различные участки тела подопытного животного. Например, исследователи Juni
и Nakano изучали на крысах введение довольно крупных микрочастиц ПГБ в
мышечную ткань [Juni K., 1986]. Временная острая воспалительная реакция
заканчивалась к седьмым суткам после имплантации. Так же на полимерных
пленках была показана высокая степень биосовместимости ПГБ, находящегося в
композите с частицами гидроксиапатита, что является очень важным фактом,
так как гидроксиапатит – главный минеральный компонент костной ткани –
широко применяется в регенеративной инженерии костной ткани [Ni J., 2002].

21
Биодеградируемость
Одним из важнейших свойств ПГБ, полученного микробиологическим
путем, является его способность к биодеградации в различных условиях. Это
означает, что поли(3-гидроксибутират) может быть полностью разложен на воду
и углекислый газ клетками микроорганизмов, присутствующих практически
повсеместно в окружающей среде [Byrom D., 1987.], например в почве [Mergaert
J., 1993], морской и пресной воде [Doi Y. et al., 1992], [Mergaert J. et al. 1995].
Эти процессы осуществляются с участием как специфичных ферментов – ПГБ-
деполимеразы, так и неспецифичных – липазы и других эстераз. При этом,
процесс зависит от большого числа параметров окружающей среды
(температуры, влажности, рН), и свойств самого полимера (состава,
кристалличности, площади поверхности). Путь разложения этого полимера
заключается в первичном поверхностном ферментативном гидролизе до
мономеров с последующим их включением в свой метаболизм. При проведении
таких исследований исследователи определяют изменения молекулярной массы
полимера и снижение веса полимерного изделия, изменение его прочности и так
далее [Kanesawa Y., 1989], [Bonartsev A.P., 2012]. Фермент, осуществляющий
биодеградацию ПГБ в окружающей среде – ПГБ-деполимераза – является
ферментом из класса гидролаз с молекулярной массой от 37 до 60 кДа
[Jendrossek D., 1993], [Saito T., 1989], [Shinomiya M., 1997]. Он состоит из двух
доменов, соединенных полипептидным линкером, что является
распространенной структурой макромолекулы для деполимераз, разлагающих
природные полимеры [Gilkes N. R., 1991], [Kellett L. E., 1990]. Было показано,
что С-концевой домен – это домен связывания с поверхностью ПГБ, в то время,
как N-концевой – катализирующий. Механизм работы этого фермента
представлен на рисунке 6.

22
Рисунок 6. Механизм разложения монокристалла ПГБ с помощью фермента
деполимеразы
В опытах, проведенных исследователями из Турции, было показано, что
скорость биодеградации ПГБ после имплантации in vivo значительно превышает
скорость его гидролиза in vitro [Piskin E., 1995]. Это означает, что внутри тела
млекопитающих также происходит ферментативный процесс разрушения
полимерной цепи. Однако в мягких тканях млекопитающих нет специфичной
деполимеразы, которая могла бы осуществлять этот процесс. Группа
исследователей из Испании показала, что липазы, встречающиеся во всех
организмах и участвующие в катаболизме триглицеридов, приводит к резкому
снижению молекулярной массы ПГБ, что говорит о проведении гидролиза
полимерной сложноэфирной связи, то есть биодеградации полимера [Alejandra
R. C., 2012]. При этом, липазы действуют неспецифически, и поэтому их
активность может сильно варьировать в зависимости от организма, из которого
они выделены.
Переходя к исследованиям in vivo следует отметить, что в зависимости от
места имплантации полимерных изделий, скорость их деградации сильно
разнится. Также на скорость деградации оказывает сильное влияние способ
изготовления самого импланта. Наиболее частой является форма введения
полимера в виде пленок, вылитых из расплава или полученных осаждением из
раствора [Ishikawa K., 1996]. В нашей лаборатории было проведено
исследование подкожной имплантации пленок из ПГБ разной молекулярной

23
массы [Босхомджиев А.П., 2009], которое показало полное рассасывание для
полимера средней молекулярной массы и почти полное для
высокомолекулярного ПГБ (рис. 7). При этом было отмечено сильное снижение
молекулярной массы полимера уже после 1 месяца после имплантации (рис. 8)
Рисунок 7. Кинетика деградации полимерных пленок при их подкожном
введении крысам
Рисунок 8. Снижение молекулярной массы ПГБ после 1 месяца подкожной
имплантации крысам полимерных пленок.

24
В работе Gogolewski с соавторами показана деградация имплантов,
полученных путем осаждения гомополимера поли(3-гидроксибутирата) из
раствора, в сравнении с его сополимерами при из подкожном введении
[Gogolewski S., 1993]. При этом такая форма, в отличие от литых пленок,
хорошо моделирует конденсированное состояние полимера в матриксах,
скаффолдах и так далее. Хроматографически с помощью гель-фильтрации было
показано снижение молекулярной массы полимера (рис. 9) в течение шести
месяцев после имплантации. Если сравнивать данный полимер с
полилактидами, то ПГБ деградирует значительно более медленно.
Рисунок 9. Снижение молекулярной массы ПГБ после его подкожной
имплантации.
Биомедицинское применение
Будучи биосовместимым и биодеградируемым полимером, поли(3-
гидроксибутират) является перспективными материалом для разработки

25
широкого спектра самых разных биомедицинских изделий (рис. 10). В данной
главе мы остановимся на двух из них, которые являются одними из самых
важных на данный момент: изготовление матриксов и скаффолдов для тканевой
инженерии, а также системы пролонгированной доставки биологически
активных веществ [Chen G. Q., 2005].
Рисунок 10. Разнообразие изделий, изготавливаемых из ПГБ.
Для обеспечения применения в тканевой инженерии у ПГБ есть все
необходимые качества: он биосовместимый, способствует клеточному росту,
нетоксичен и обладает индукцией к образованию ткани из отдельных клеток
[Williams S. F., 1999]. На рисунке 11 продемонстрирована концепция
применения таких матриксов. На нем аутологичные клетки, выделенные из
образцов биопсии самого пациента засеваются на полимерный матрикс с
последующим его обратным вживлением в организм. Однако изготовление
таких матриксов является основной проблемой всех подобных исследований
[Atala A., 1997.]. Значительная часть такой работы связана именно с подбором
свойств для обеспечения пролиферации клеток и образования полноценной
ткани организма. Также необходимо, чтобы к иммобилизованным клеткам был
доступ питательных веществ и кислорода. В идеале, пористый скаффолд должен
обеспечивать клетки необходимыми веществами на расстояниях меньше, чем
300 µм [Choi R.S., 1988]. В противном случае наступает угнетение роста клеток
и снижение их пролиферативной активности.

26
Рисунок 11. Схема введения в организм матрикса из ПГБ, засеянного
аутологичными клетками.
Хорошо известно, что изделия из ПГБ могут быть изготовлены с
использованием широкого спектра традиционных методов, которые включают
литье из растворов, электроспиннинг, вспенивание, и плавильные методы
обработки, такие как экструзия, литье под давлением, а в некоторых случаях
выдувное формирование [Holmes P.A., 1988]. Изготовленные изделия при этом
могут представлять собой пленки, волокна, тканые и нетканые материалы,
трубки, композитные материалы и так далее. При необходимости, ПГБ также
можно объединять или смешивать с другими полимерами и материалами для
улучшения его физико-механических свойств. При этом ПГБ имеет богатый
потенциал для изготовления полимерных матриксов для тканевой инженерии,
запрограммированных для решения необходимых задач в организме путем
осуществления контроля за биологическими и физико-химическими свойствами
изделия. Использование этих методов для изготовления пористых матриксов из
ПГБ позволило не только оптимизировать поддержание жизнеспособности
засеваемых клеток и обеспечение их роста и пролиферации при различных
условиях, но решать конкретные задачи тканевой инженерии. Разберем на
примере полимерных матриксов, изготовленных с помощью метода
выщелачивания. При таком подходе применяется специальный

27
порообразователь, который диспергируется в полимерном растворе, а затем,
посте его застывания, вымывается из готового изделия [Thomson R.C., 1997].
При этом форму изделия можно заранее задать любую. На рисунке 12
представлена трубка, сделанная из такого пористого материала в сравнении с
трубкой, полученной из гладкой пленки. При этом такая трубка может быть
заселена клетками с последующим замещением полимерного материала тканью
организма.
Рисунок 12. Полимерные трубки, полученные из гладкой (слева) и пористой
(справа) полимерных пленок.
В типичной процедуре получения пористой трубки из ПГА с
использованием метода выщелачивания, расплав полимера смешивают с
просеянными частицами соли в весовом соотношении 1:2 до получения
гомогенной смеси. Размеры частиц соли в данном случае были 80 - 180 µм;
Однако, размер частиц, распределение и весовой процент можно варьировать в
зависимости от желаемого размера пор и плотности. Солевая смесь была
спрессована в тонкую пленку и отформована вокруг цилиндрической
тефлоновой заготовки. Затем изделие было подвергнуто полному
выщелачиванию соли для получения пористой полимерной трубки.

28
В нашей лаборатории также проводились исследования пористых
матриксов на основе ПГБ, приготовленных различными способами. Это и
электроспиннинговые маты для культивирования мезенхимальных стволовых
клеток [Andreeva N. V., 2015], и пористые матриксы для инженерии костной
ткани, полученные также методом выщелачивания [Bonartsev A. P., 2016] При
этом была показана высокая клеточная адгезия к полимерным изделиям,
отсутствие цитотоксического действия. В качестве порообразователей
использовались карбонат аммония и сахароза. Данные изделия приведены на
рисунке 13.
Рисунок 13. Пористые матриксы из ПГБ, получаемые в нашей лаборатории
методами электроспиннинга (А) и выщелачивания с использованием карбоната
аммония (В) и сахарозы (С).
В начале 1990-х годов ПГА стали рассматривать в качестве кандидатов на
роль носителей лекарственных веществ в связи с их свойствами
биоразлагаемости и биосовместимости. ПГБ является биологически
совместимым и гидрофобным полимером, который можно использовать для
инкапсулирования различных молекул, используя при этом как гомополимер,
так и различные сополимеры ПГБ. Такие микроструктуры широко используются
для доставки ряда лекарственных средств, таких как анестетики, антибиотики,
противовоспалительные средства, противораковые средства, гормоны,
стероиды, и вакцины [Nobes G. A. R., 1998], [Orts W. J., 2008]. Об использовании
полимерных микросфер в качестве носителей для стероидов упоминается в
работе Gangrade и Price [Gangrade N., 1991]. ПГБ и ПГБВ были использованы
для получения микрочастиц, содержащих прогестерон в качестве модельного
лекарственного средства. Включение прогестерона в микросферы было очень
эффективным: более 80% от теоретического содержания было загружено в

29
полимерную матрицу. Было показано медленное пролонгированное вещества из
структур, с содержанием действующего вещества 9%.
Потенциальное использование ПГБ и его сополимера ПГБВ для доставки
лекарств было оценено в ряде исследований. Они могут быть потенциальным
средством лечения резистентных инфекций, поскольку показали способность
поддержания постоянных концентраций антибиотиков в местах инфекционного
заражения [Gould P. L., 1987], [Gursel I., 2002]. Также эти полимеры
использовались в качестве стержней для местной доставки антибиотиков при
терапии хронического остеомиелита [Yagmurlu M. F., 1999], [Gürsel İ. et al.
2000]. При сравнении высвобождения противоракового агента ломустина in
vitro и in vivo из микрочастиц ПГБ по сравнению с полилактидами было
установлено, что препарат, основанный на микрочастицах ПГБ, обладает
лучшими кинетическими показателями высвобождения [Bissery M. C., 1985].
При этом включение эфиров жирных кислот в микросферы из ПГБ повышало
скорость высвобождения лекарственного вещества [Kubota M. H., 1988]. Sendil с
соавторами исследовали ПГБВ с различным содержанием 3-гидроксивалерата,
загруженный тетрациклиновым антибиотиком, который, как известно,
эффективен против пародонтоза. Были установлены способы влияния на
кинетику высвобождения антибиотика в зависимости от состава полимерной
матрицы [Sendil D., 1999]. При этом тетрациклин был загружен в микросферы из
ПГБВ как в кислой форме так и в нейтральной. Следует отметить, что
высвобождение завершилось без каких-либо наблюдаемых признаков
деградации полимерной матрицы.
Исследование, проведенное с использованием микрочастиц ПГБ,
показало, что высвобождение противоопухолевого препарата рубомицина
ингибирует пролиферативную активность карциномы Эрлиха у мышей
[Shishatskaya E., I. 2008]. Если рассматривать наночастицы из ПГБ, содержащие
преднизолон, приготовленные с использованием гомогенизации под высоким
давлением [Koosha F., 1988], то в этом исследовании была достигнута
двухфазная модель высвобождения. В первые часы высвобождалась основная

30
терапевтическая доза вещества, после чего скорость высвобождения снижалась
и пролонгированное высвобождение гормона обеспечивало доставку
предизолона в поддерживающих дозах. В работе Kawaguchi с соавторами
показана разработка микросфер из ПГБ, содержащего противоопухолевый
препарат 2’,3’-диацил-5-фтор-2’-дезоксиуридин [Kawaguchi T., 1992]. Частицы
показали низкую токичность и хорошую биосовместимость у мышей и крыс.
С работах Shah с соавторами описывается исследование эффективности и
биологической доступности цисплатина – химиотерапевтического препарата,
используемого против различных опухолевых заболеваний – в виде
загруженных цисплатином наночастиц амфифильного блок-сополимера поли(3-
гидроксибутират)-поли(4-гидроксибутират)-поли(этилен гликоль) [Shah M.,
2012]. Профиль высвобождения цисплатина из гидрофобного ядра показал
длительную кинетику выхода вещества. Также с помощью электронной
микроскопии было показано вхождение этих наночастиц в раковые клетки, а
также показал эффект подавления роста опухолевых клеток путем стимуляции
их апоптоза.
1.1.2. Поли(3-гидроксибутират)-ко-поли(этилен гликоль)
Изменение физико-химических свойств полимера – это очень важное
направление в биотехнологии. Ведь с помощью такого подхода можно
направленно задавать свойства материала, снижать его недостатки, привносить
новые функции. При этом, создавая композитные материалы, можно лишь слить
воедино и преимущества, и недостатки смешиваемых полимерных материалов,
в то время как создание новых сополимеров позволяет достичь качественно
новых материалов. Рассмотрим сополимеризацию поли(3-гидроксибутирата) с
поли(этилен гликолем). Последний представляет собой синтетический
амфифильный полимер, растворимый как в органических растворителях, так и в
воде. Его инкорпорирование в полимерную цепь должно, по идее, снизить
гидрофобность ПГБ, а также улучшить его механические свойства, такие как
напряжение на разрыв, удлинение при растяжении и так далее. С одной

31
стороны, такой сополимер можно получить химически, например, под
действием ультрафиолета [Zhijiang C., 2007]. При этом авторами было показано
снижение гидрофобности материала методом измерения контактного угла с
водой, а также методом дифференциальной сканирующей калориметрии (ДСК).
В другом случае химического синтеза гидролизованный ПГБ сшивался с ПЭГ
специальным агентом – гексаметилен диизоцианатом [Ravenelle F., 2002].
Включение ПЭГ в полимерную цепь так же было показано с помощью методов
ядерного магнитного резонанса (ЯМР) и инфракрасной спектроскопии с Фурье-
преобразованием. Однако гораздо более интересные результаты были получены
исследователями из Индии. Путем микробиологического культивирования
бактерий Azotobacter chroococcum MAL-201 они получили данный сополимер
путем лишь внесения поли(этилен гликоля) в культуральную среду спустя 6
часов после начала культивации. При этом, не требовалось более никаких
химических операций для получения этого материала, он получался
экстракцией уже в чистом виде и был готов для дальнейших его исследований
[Saha S. P., 2006]. Также и в нашей лаборатории был получен такой же полимер
с использованием другого штамма A. chroococcum 7Б [Bonartsev A. P., 2016].
При этом были решены сразу две задачи. Во-первых была проведена
сополимеризация с более гидрофильным полимером, а во-вторых, снижена
молекулярная масса для оптимизации кинетики деградации полимера в
организме.
Следует отметить, что для изменения физико-химических и механических
свойств ПГБ полимера в литературе чаще всего применяется создание
композитной смеси ПГБ с ПЭГ. При этом с одной стороны, достигается
необходимый результат [Chan R. T. H., 2011.], например увеличение
гидрофильности [Chan R. T. H., 2013], однако он может элиминироваться по
мере вымывания ПЭГ в окружающую среду. Также при этом невозможно
достичь тех уникальных свойств материала, состоящего из амфифильных
молекул, определяющихся их укладкой при конденсации ПГБ-ПЭГ из раствора
в процессе изготовления изделия [Chen C., 2006].

32
1.2. Системы пролонгированного высвобождения белков
Существует большое количество систем пролонгированного
высвобождения биологически активных веществ. Это могут быть новые формы
лекарственных веществ, системы доставки белков и нуклеиновых кислот и
многие другие. В данной работе мы остановимся на системах
пролонгированного высвобождения белков, одним из основных применений
которых является тканевая инженерия.
1.2.1. Системы пролонгированного высвобождения в тканевой инженерии
В тканевой и регенеративной инженерии остро стоит проблема доставки
ростовых факторов и других веществ, позволяющих стимулировать
направленный рост определенных тканей организма. Это необходимо,
например, для более быстрой регенерации костных дефектов, проведения при
этом остеонеогенеза путем направленной остеодифференцировки
мезенхимальных стволовых клеток, стимуляции ангиогенеза в матриксах,
замещающихся новой тканью [Perets A., 2003] и так далее.
Для начала рассмотрим общие стратегии и подходы для доставки
ростовых и дифференцировочных факторов для тканевой инженерии. Их можно
объединить в две группы:
(1) химическая иммобилизация фактора роста на матрице и (2) физическое
его инкапсулирование в систему доставки.
Первый подход обычно включает химическое связывание или
сорбционное взаимодействие между фактором роста, и клеткой или тканью.
Другой – это инкапсулирование с последующим запрограммированным
высвобождением фактора роста из подложки в окружающие ткани. Стоит
заметить, что эффективность доставки фактора может быть значительно
повышена путем его внесения на трехмерный каркас [Ehrbar M., et al. 2004],
[Ehrbar, M., 2007], [Zisch, A. H., 2001].

33
Методики иммобилизации факторов роста на натуральных или
синтетических биоматериалах и химических веществах весьма разнообразны и
активно разрабатываются множеством авторов. Такие иммобилизованные
факторы становятся доступны для клеток, вступающих в контакт с матриксом,
обеспечивая высоко локализованный сигнал для управления судьбой отдельных
клеток в конкретном месте (например, дифференцировки стволовых клеток;
[Dawson, E., 2007], [Dawson, E., 2008]). Факторы роста могут быть сразу
активными в связанном состоянии или же быть активированы путем
отщепления от матрицы. Сайт-специфическое прикрепление факторов роста и
других биологических молекул позволяет контролировать их
разнонаправленные функции. Существуют две основные стратегии для
презентации факторов роста на внеклеточном матриксе:
(I) адсорбция вследствие белок-белковых водородных связей или
гидрофобных взаимодействий с молекулами наполнителей, часто действующих
как молекулярные шапероны (нековалентный подход);
(II) прямая ковалентная иммобилизация фактора роста или имитирующих
его молекул к матричному каркасу (ковалентный подход). Независимо от того,
какой метод используется, это делает матриксы способными к клеточной
адгезии, что помогает создать более тесный контакт с отдельными клетками.
Ковалентное связывание факторов роста с биополимерным или
искусственным носителем является наиболее широко применяемым подходом,
поэтому мы остановимся на нем более подробно. Эта методика, позволяет
обеспечить более длительное высвобождение биомолекул, чем физическая
иммобилизация. Факторы могут быть конъюгированы с полимерами с помощью
функциональных групп, которые включены в них путем сополимеризации или
химической сшивки [Ito, Y., 2008]. Например, эпидермальный фактор роста
(EGF) можно ковалентно присоединить к амино-силановому стеклу с помощью
звездообразного разветвленного полиэтилен гликоля (star-PEG), что позволяет
лиганду сохранять бòльшую мобильность и более эффективно обеспечивать
пролиферацию первичных гепатоцитов крыс, чем физически адсорбированной

34
EGF [Kuhl P. R., 1996]. Аналогичным образом, трансформирубщий фактор роста
бета (TGF-b1), ковалентно пришитый к гидрогелю из ПЭГ, способствует
увеличению роста внеклетчного матрикса [Mann B. K., 2001]. Тем не менее,
существуют некоторые ограничения этого подхода, а именно специфичность
сайта связывания белка, достичь которой бывает довольно сложно, а значит,
трудно обеспечить селективность связывания конкретных белков. Кроме того,
белки по-прежнему могут потерять свою биологическую активность при
иммобилизации вследствие повреждения биологически активных
функциональных групп и потери нативной структуры, особенно важной для
узнавания лиганда рецептором. Кроме того, с помощью такого подхода
невозможно обеспечение градиентов концентраций биологически активных
веществ (БАВ), настолько важного и перспективного принципа, применяемого в
тканевой инженерии. [Wang Y., 2011].
Другой подход основан на физическом инкапсулировании факторов роста
с предварительно запрограммированным высвобождением и доставкой в
окружающие ткани. Это хорошо изученный подход для локальной доставки
факторов роста в тканевой инженерии, который является привлекательным за
счет своей простоты и доступности, по сравнению с химической конъюгацией.
Можно легко изготовить необходимые материалы для системы доставки,
которые будут пригодны для инъекционного введения, трансплантации, а так же
соответствовать требованиям механической прочности, пористости и скорости
деградации. Для инкапсуляции факторов роста используются различные методы
изготовления медицинских изделий: литье из раствора, выщелачивание,
лиофилизирование, разделение фаз, литье из расплава, фазовое эмульгирование,
полимеризация in situ и газообразование [Lanza R. P., 2007]. Ключевым
вопросом является минимизация жестких воздействий на ростовые факторы в
процессе обработки для того, чтобы защитить активность биомолекул.
Например, методы, основанные на газообразовании, были разработаны, чтобы
избежать воздействия на белки органических растворителей, широко
применяемых во многих других методов (что, зачастую, приводит к

35
денатурации белка [Harris L. D., 1998], [Mooney D. J., 1996], [Sheridan M. H.,
2000]). Углекислый газ, при высоком давлении являющийся жидким,
растворяется в соответствующем полимере, и при уменьшении давления
происходит вспенивание, приводящее к формированию пор, и может привести к
расширению полимерной матрицы. Сочетание методов газового вспенивания с
методом выщелачивания может привести к образованию открытой системы пор
и быть использовано для доставки факторов роста в нативной форме для
формирования новой ткани [Chen R. R., 2003]. Однако применение таких систем
зачастую связано с использованием сложных методик, которые проблематично
будет применить в дальнейшем на практике. Кроме того, срок высвобождения
вещества при применении таких методов обычно не велик и не превышает
нескольких суток, в то время как на стадиях отмывки, выщелачивания и так
далее теряется значительная часть инкапсулированного белка [Chen R. R., 2003].
Наиболее распространенными системами доставки инкапсулированных
белковых макромолекул являются полимерные микрочастицы, полученные
путем эмульгирования. Это простые системы, в процесс изготовления которых
входит не более двух этапов [Andreas K., 2011]. При этом метод легко
масштабируется на большие производственные мощности [Nisisako T., 2008].
По своей эффективности они не уступают, а зачастую превосходят системы,
полученные другим способом [Freytag T., et al. 2000], при этом не связываясь
химически и не модифицируя включенное в них вещество. Время
высвобождения из таких частиц может достигать от нескольких недель для
белков [Andreas K., 2011] до нескольких месяцев для низкомолекулярных
соединений [Bonartsev A. P., 2006]. В данной работе исследуются именно такие
системы, поэтому в дальнейшем речь пойдет именно о них.
1.2.2. Методы получения биополимерных микрочастиц
Для инкапсулирования биологически активных веществ и создания
биополимерных микрочастиц используют самые разнообразные методы. Так,

36
для создания микрочастиц с инкапсулированными лекарственными веществами
используют в основном методы одноэтапного и двухэтапного эмульгирования с
различными модификациями, и методы высушивания при распылении с
модификациями. Способы получения и стабилизации эмульсии обычно
включают одну или несколько следующих стадий: быстрое перемешивание с
помощью верхнеприводной мешалки [Wang S.H., 2005], гомогенизатора [Tsung
M.J., 2001], эмульгирование с использованием ультразвука [Jaganathan K.S.,
2005].
Метод одноэтапного эмульгирования «масляная фаза/водная фаза» (O/W)
применяется для получения полимерных частиц из гидрофобных или
амфифильных полимеров, таких как полимолочная кислота (ПМК),
полигликолевая кислота (ПГК), ПГА и другие. Он заключается в
эмульгировании комбинированного раствора полимера и лекарственного
вещества (ЛВ) в органическом растворителе (масляная фаза) в водной фазе с
сурфактантом, последующем постепенном выпаривании органического
растворителя при интенсивном перемешивании или ультразвуковом
воздействии, осаждении и высушивании биополимерных микрочастиц. Метод
позволяет получать как биополимерные микрочастицы с диаметром от 1 до 100
мкм [Jalil R., 1990], так и биополимерные частицы субмикронных размеров и
наночастицы с диаметром от 50 до 1000 нм [Feng S., 2001], а также
макроскопические частицы, диаметр которых может достигать 1 мм. При этом,
лекарственной вещество должно быть растворимо в том же органическом
растворителе, что и сам полимер и следует избегать его вымывания в конечный
эмульгатор, представляющий собой водную фазу. При одной из модификаций
этого метода применяют диспергирование ЛВ в органическом растворителе в
случае, если ЛВ плохо растворимо как в воде, так и в органических
растворителях [Gupte A., 2004]. Другой модификацией этого метода является
метод гомогенизации под высоким давлением. Метод заключается в
гомогенизации смеси комбинированного раствора полимера и ЛВ в
органическом растворителе и водной фазы с эмульгатором под давлением с

37
использованием гомогенизатора высокого давления, последующем постепенном
выпаривании органического растворителя, осаждении и высушивании
биополимерных микрочастиц. При этом можно достичь значительного
снижения минимального размера получаемых микрочастиц, в том числе
получать наночастицы [Dong Y., 2007]. При другой модификации этого метода
«масляная фаза/масляная фаза» (O/O) вместо водной фазы (водного раствора)
используют масляную фазу (различные масла: хлопковое, силиконовое,
кастроловое, парафин и др.). Эта модификация используется также в случае,
если ЛВ – хорошо растворимо в воде и плохо в органических растворителях
[Ike O., 1992] и позволяет снизить его потери при изготовлении. Если же
водорастворимое вещество, например, белок, инкапсулируется в гидрофильный
полимер по методике одноэтапного эмульгирования, в качестве раствора
эмульгатора используют раствор сурфактанта в органическом растворителе,
например гексане. Такой метод называется «водная фаза/масляная фаза» (W/O).
При этом для дальнейшего использования таких частиц в водной среде, то есть,
чтобы они не растворялись сразу при контакте с водой, используют перешивку
полимера-носителя специальными кросс-линкерами. Например, в работе Englert
[Englert C., 2007] исследуются различные способы перешивки
модифицированного коллагена, в том числе глутаровым альдегидом,
карбодиимидом и другими агентами (рис 14). При использовании такого
биополимера как альгиновая кислота применяется ионная перешивка с
помощью ионов двухвалентных металлов, например кальция [Moebus K., 2009].
Данный вид перешивки основан на взаимодействии двух карбоксильных групп
на соседних полимерных цепочках с кальцием, с последующим образованием
ионного мостика (рис 15).

38
Рисунок 14. Методы перешивания коллагена для снижения его растворимости в
водных растворах.
Рисунок 15. Образование перешивающих ионных мостиков при
взаимодействии альгината с солью кальция.
Недостатком данного метода для инкапсулирования белков является
применение перешивающих агентов, которые могут взаимодействовать не
только с молекулами носителя, но и с инкапсулированным веществом,
модифицируя его функциональные группы и приводя к его инактивации. Таким
образом, при создании таких систем на основе водорастворимых полимеров
необходимо это учитывать и тщательно прорабатывать все возможные
взаимодействия макромолекул белка с кросс-линкерами.

39
Однако вернемся к методам инкапсулирования белков в гидрофобные
полимеры. Здесь нет необходимости в использовании дополнительных
скрепляющих полимерную цепь агентов – микрочастицы формируются лишь за
счет нековалентных контактов между молекуляами полимера, предтавляющими
в конечном счете физически гель. Такая комбинация встречается наиболее часто
за счет удобства работы с биосовместимыми полиэфирами, такими как ПМК,
ПГА и их сополимерами. В таком случае для получения микро-/нанокапсул и
микро-/наночастиц используется метод двухэтапного эмульгирования. В этом
случае лекарственное вещество (ЛВ), например белок, не растворяется в том же
растворителе, что и биополимер, а используется его водный раствор, например в
буфере. Существует несколько модификаций этого метода, различающихся по
последовательности эмульгирования в различных фазах и по составу этих фаз.
Метод двухэтапного эмульгирования «водная фаза/масляная фаза/водная фаза»
(W/O/W) заключается в 1) эмульгировании водного раствора ЛВ в растворе
полимера в органическом растворителе; 2) эмульгировании полученной
эмульсии в водной фазе. Конечный эмульгатор удаляют с помощью
дистиллированной воды и выделяют микрочастицы [Maysinger D., 1994],
[Anderas K., 2011]. При этом в конечном счете инкапсулируемое вещество
находится внутри полых микрокапсул в виде раствора, а сами капсулы
представляют собой полые структуры, ограниченные полимерной стенкой [Choi
D. H., 2010], достаточно сильно различающиеся по своему строению (рис 16).
Рисунок 16. Разнообразие строения микрокапсул в зависимости от способа их
изготовления.

40
Метод двухэтапного эмульгирования «масляная фаза/масляная
фаза/водная фаза» (O/O/W) заключается в 1) приготовлении отдельных
растворов двух различных полимеров; 2) диспергировании ЛВ в одном из
растворов; 3) эмульгировании первого раствора полимера и ЛВ во втором
растворе полимера; 4) эмульгировании полученной эмульсии в водной фазе.
Этот метод позволяет получать микрочастицы с инкапсулированными
водорастворимыми ЛВ, которые имеют дополнительную оболочку из чистого
полимера [Lee T.H., 2002], [Anderas K., 2011]. Модификацией этого метода
можно считать метод покрытия полимерных микрочастиц дополнительной
оболочкой из другого полимера. Метод заключается в том, что микрочастицы
диспергируют в растворе такого биополимера, который не растворяется в том
же растворителе, что и полимер, из которого были созданы микрочастицы, а
сами микрочастицы при этом не растворяются в этом растворе. Полученную
дисперсию эмульгируют в масле или жидком парафине при перемешивании.
Микрочастицы затем выделяют центрифугированием и промывают гексаном
[Krishnamachari Y., 2007]. Метод двухэтапного эмульгирования «водная
фаза/масляная фаза/масляная фаза» (W/O/O) также предназначен для
инкапсулирования водорастворимых веществ и заключается в 1)
эмульгировании водного раствора ЛВ в растворе полимера в органическом
растворителе; 2) эмульгировании полученной эмульсии в различных маслах
(хлопковом, силиконовом, кастроловом, парафине и др.) [Blanco-Prı´eto M.J.,
2004]. При этом не происходит вымывания инкапсулированного вещества в
масло, так как оно не растворимо в нем. Это позволяет существенно повысить
эффективность инкапсулирования вещества в полимерные микрочастицы. При
этом возможно также добавление стабилизирующих агентов в одну из фаз
[Sandor M. 2002].
Особое внимание следует уделить методикам двухэтапного
эмульгирования, в которых лекарственное вещество присутствует в виде
твердой фазы. Метод «твердая фаза/масляная фаза/водная фаза» (S/O/W)
применяется для создания микрочастиц и микрокапсул и инкорпорированным

41
ЛВ, представляющим собой либо нерастворимое ни в воде, ни в органическом
растворителе, любо нерастворимые только в органическом растворителе БАВ,
например, белки [Leach W.T., 2005]. Он заключается в 1) приготовлении
дисперсии наночастиц композита БАВ с наполнителем (используется при
изготовлении самих наночастиц); 2) приготовлении раствора полимера в
органическом растворителе; 3) смешивании полученных растворов; 4)
эмульгировании полученной эмульсии в водной фазе [Castellanos I.J., 2003],
[Anderas K., 2011], [Wang X, 2012]. За счет отсутствия контакта водного
раствора белка с органическим растворителем, таким как хлороформ или
дихлорметан, белок в большей мере сохраняет свою стабильность и не
подвергается денатурации [Griebenow, K., 1996]. Также в этом случае возможно
использование твердых носителей, которые сами по себе имеют биологическую
активность, таких как гидроксиапатит и другие [Liao S. 2005].
Метод двухэтапного эмульгирования «твердая фаза/масляная
фаза/масляная фаза» (S/O/O) подобен методу S/O/W, однако в роли конечного
эмульгатора используются различные масла, например хлопковое [Tobio M.,
1999], что также приводит к снижению потерь инкапсулируемого вещества при
изготовлении микрочастиц.
Размер частиц, получаемых методами эмульгирования, определяет ряд
параметров, главными из которых являются скорость перемешивания,
концентрация эмульгатора и раствора полимера, из которого изготавливают
микрочастицы. Обычно средний диаметр микрочастиц уменьшается с ростом
скорости перемешивания эмульсии, а также с увеличением концентрации
эмульгатора и уменьшением концентрации раствора полимера [Kumar R., 2000].
Основными проблемами, которые возникают при инкапсулировании
белков в микросистемы из гидрофобных полимеров по методике двухэтапного
эмульгирования возникает спектр проблем, связанных со сложностью строения
белков, таких как их денатурация в органическом растворителе, необратимая
адсорбция на полимерную поверхность с нарушением нативной структуры, и т.
д. Большинство из них легко решаются применением различных наполнителей,

42
экранирующих белковые молекулы в составе систем высвобождения, или
обладающих в композите с белками иными физическими свойствами. Также,
применяя твердофазные методики S/O/W и S/O/O можно избежать контакта
водного раствора белка с органическим растворителеа, а белок с твердом
состоянии не способен к денатурации, о чем далее будет рассказано более
подробно [Griebenow K., 1996].
Итак, одним из путей решения проблем, возникающей при
микрокапсулировании белковых ЛВ с помощью методов двухэтапного
эмульгирования, является изготовление твердофазной формы белка. Так как
белки в сухом виде представляют собой вещество, которое плохо образует
гомогенные микро- и наночастицы, необходимые для применения этих методик,
актуальным представляется использование различных носителей и
наполнителей, облегчающих этот процесс [Cleland J. L., 1996]. В литературе
встречается много примеров, когда изготавливаются наночастицы композита
белка с носителем. При этом последний может представлять собой как
инертное, так и биологически активное вещество. В качестве наполнителя
может использоваться поливиниловый спирт (ПВС). Суть методики
заключается в 1) диспергировании водных растворов ПВС и белка на
верхнеприводной мешалке при высоких оборотах в силиконовом масле; 2)
нескольких циклах замораживания-оттаивания полученной эмульсии; 3)
отмывании наноструктур с белковыми включениями ацетоном и последующее
удаление растворителя лиофилизацией [Wang N., 1999]. Таким же наполнителем
может быть ПЭГ. Для создания композита ПЭГ-белок используется лиофильное
высушивание их растворов. Далее полученный порошок используют в качестве
твердой фазы в методике двухэтапного эмульгирования [Castellanos I.J., 2003].
Также многие авторы применяют сложные системы изготовления белковых
наночастиц, основанные на использовании размораживания, оттаивания,
лиофилизации и систем экструзии через форсунки и фильтраций [Constantino
H.R., 2000]. В качестве носителей также зачастую используются наночастицы
неорганических веществ, таких как оксид кремния [Azioune A., 2004],

43
гидроксиапатита [Kandori K., 2010] и других материалов. Связывание белка при
этом может происходить как за счет адсорбции на поверхность, так и за счет
химического связывания носителем протеиновых макромолекул.
1.2.3. Кинетика высвобождения веществ из полимерных микрочастиц
Изучение кинетики высвобождения инкапсулированных веществ из
полимерных микрочастиц является основной характеристикой таких систем.
Действительно, характер выхода вещества из структур определяет основные
терапевтические свойства новой лекарственной формы и характеризует области
ее применения. Длительность, равномерность подачи, эффективность
высвобождения – все это говорит нам о том, насколько длительную терапию
можно осуществлять с помощью разрабатываемой системой.
Вещество внутри микрочастиц находится либо в виде агломератов, либо
диффузно распространено по всему объему. Высвобождение обычно
описывается первым и вторым законами Фика [Narasimhan B., 2000]. На
высвобождение оказывает влияние огромное количество факторов, таких как
температура, рН, загрузка вещества, его растворимость, площадь поверхности и
форма микрочастиц и многие другие [Donbrow M., 1992]. Большинство из них
сводятся к процессу вымывания вещества из полимерного матрикса в среду, в
которую и диффундирует инкапсулированное вещество. При этом следует
учитывать такие особенности микрочастиц, как способность к деградации
полимерного матрикса, набухание, разрушение частиц [Donbrow M., 1992]. При
этом частицы не являются правильными шариками одинакового диаметра –
существует распределение по размерам и форме, различия в количестве
включенного вещества и так далее. Суммарное высвобождение, при этом, будет
складываться из быстрого высвобождения из маленьких частиц, сменяющегося
продолжительным выходом из больших частиц [Borgquist P., 2004].
Теоретически, нужно просто интегрировать высвобождение из частиц всех
размеров, однако зависимость обычно является не линейной и процессы

44
высвобождения из частиц разного диаметра могут идти совершенно по-разному
[Yang S., 2005], [Donbrow M., 1992]. Например, в некоторых случаях при
увеличении диаметра скорость может вырасти, несмотря на увеличившееся
расстояние, которое должны продиффундировать молекулы
инкапсулированного вещества [Siepmann J. 2004]. Варьирование других
параметров также вносит, зачастую, непредсказуемые изменения в кинетику
высвобождения веществ из микроструктур. Поэтому в каждом конкретном
случае кинетика высвобождения вещества будет сильно различаться.
1.2.4. Проблема стабильности высвобождаемого белка
Одной из самых главных проблем является потеря стабильности белка при
его адсорбции на полимерный матрикс. При эмульгировании часть белка из
раствора адсорбируется на полимерный матрикс, представляющий собой
гидрофобную поверхность. В случае глобулярных белков, изменение
микроокружения молекулы белка с гидрофильного (связанные диполи воды) на
гидрофобное (полимерный матрикс), влечет за собой высвобождение участков
полипептидной цепи из гидрофобного ядра молекулы, что является более
выгодным с энергетической точки зрения [Севастьянов В.И., 1999]. При этом
может возникнуть 2 типа адсорбции: обратимая и необратимая. В первом случае
энергетический барьер фазового перехода «белок в растворе» /
«адсорбированный белок» небольшой, и молекула легко возвращается в свое
нативное состояние в водном окружении, взаимодействуя с поверхностью
частицы большей частью своими гидрофильными участками. Во втором случае
барьер высок и десорбция затруднена, в то время как из-за обширного
изменения конформации белка произошла его денатурация, являющаяся более
выгодной в данных условиях по энергетическим аспектам фазовых переходов
белок в растворе» / «адсорбированный белок» и «нативный белок /
«денатурированный белок» [Севастьянов В.И., 1999], [Финкельштейн А.В.,
2005, стр. 226-230]. Обоснованность применения общих принципов адсорбции и

45
денатурации белков применительно к проблеме микрокапсулирования белковых
молекул в биосовместимый полимер ПГБ показана на практике [Полищук А.Я.,
1982], [Iordanskii A.L., 1999].
В первую очередь белок может потерять свою нативную структуру в
процессе изготовления микрочастиц. При использовании эмульсионных
методик, подразумевающих эмульгирование водного раствора белка в
органическом растворителе на границе раздела фаз наблюдается денатурация
белка. [Cleland J. L., 1996], [Kim H. K., 1999], [Chen L., 1997]. При этом
используемая для диспергирования обработка ультразвуком, также негативно
сказывается на стабильности инкапсулируемого белка [Suslick K. S., 1986]. Для
того, чтобы снизить потери белка, необходимо минимизировать как площадь
поверхности границы раздела фаз, так и время контакта и обработки
ультразвуком [Zambaux M. F., 1999]. Также исправить ситуацию может
использование более мягких органических растворителей. Например,
применение этилацетата вместо более гидрофобного метиленхлорида позволяет
значительно снизить опосредованную эмульсификацией денатурацию белка
[Alonso M. J., 1994], [Sah H., 1999]. Однако, при использовании технологий
изготовления полимерных микрочастиц, основанных на суспендировании белка
в твердом состоянии в органическом растворе полимера, денатурации на
границе раздела фаз не происходит. Klibanov с соаворами в своей работе
показал, что конформация сухих белков, находящихся в органическом
растворителе находится в «кинетической ловушке» [Griebenow K., 1996].
Именно поэтому разворачивание белковой молекулы при этом происходит
настолько медленно, что молекулы не успевают денатурировать в процессе
приготовления микроструктур. Однако, в таком случае можно столкнуться с
другой проблемой, когда белок частично растворим в гидрофобном
растворителе. В таком случае его молекулы начинает расплетаться и он теряет
свою стабильность [Griebenow K., 1997], [Knubovets T., 1999].
Также важным моментом с точки зрения сохранения нативной структуры
белка является процесс его высвобождения из полимерных микрочастиц.

46
Обычно это связано с ковалентным или нековалентным взаимодействием белка
с носителем, его гидролизом вследствие закисления, а также необратимой
адсорбции белка на полимер. Например, при использовании таких
биополимеров, как ПМК и ПМГК, в процессе их биодеградации возникают
продукты их гидролиза молочная и гликолевая кислоты, которые могут вызвать
локальное закисление внутри микрочастиц, вызвав при этом денатурацию белка
[Wang W., 1999]. Также эти полимеры имеют концевые карбоксильные связи,
которые могут взаимодействовать с амидными связями аминокислотных
остатков в макромолекуле белка как электростатически, так и с образованием
ковалентной связи [Kim H. K., 1999], [Crotts G., 1997], [Park T. G., 1998].
Гидрофобные контакты также представляют опасность для сохранения белковой
стабильности. Они приводят к агрегированию белковых макромолекул, вызывая
их необратимую адсорбцию на полимерный матрикс [Zambaux M. F. 1999], [Lu
W., 1995], [Butler S. M., 1999]. Из-за этого белок высвобождается не в полном
объеме и зачастую вышедшие макромолекулы уже не имеют своей
ферментативной активности.
Решение этой проблемы является актуальным на данный момент и
существует несколько способов ее решения. Среди них следует отметить
несколько. Во-первых, экранирование структурных доменов белковых молекул
с помощью высокомолекулярных соединений, образующих композитные смеси
с белком. То есть при приготовлении первичной фазы, содержащей белок, нее
добавляется модификатор, образующий в дальнейшем композит белком. Среди
таких веществ следует отметить в первую очередь высокомолекулярный
полиэтиленгликоль (ПЭГ), декстран – полисахарид, не имеющий токсического
действия на организм млекопитающих [Cadee J.A., 2001], а также хитозан –
полисахарид, несущий на себе положительно заряженные аминогруппы и
являющийся денатурированным хитином [Lee E.S., 2007]. Показано, что эти
вещества подходит для предотвращения необратимой адсорбции белка на
полимерный матрикс, однако не решают проблему «burst-effect» [Ito F., 2008].
Также возможно комбинирование этих двух наполнителей, то есть

47
использование и ПЭГ и декстрана одновременно [Geng Y., 2008]. Также это
вещество помогает решить другую проблему микрокапсулирования: взрывной
выброс вещества в первые часы пролонгированного высвобождения – «burst-
effect» [Pai S.S., 2009]. Однако применение ПЭГ ограничивается его
растворимость в таких органических растворителях, как наиболее части
применимые в методах двухэтапного эмульгирования хлороформе и
дихлорметане, что вызывает его вымывание на стадии приготовления
первичной суспензии белка в растворе конечного гидрофобного полимера с
последующей агрегацией белка. В случае же хитозана следует учитывать тот
факт, что этот полисахарид растворим лишь в кислой среде, что также способно
дестабилизировать загружаемые на него макромолекулы.
Во-вторых, для снижения необратимой адсорбции возможно снижение
гидрофобности самого полимера путем введения в его состав композитных
добавок [Weert M., 2002]. Чаще всего для этих целей применяется амфифильный
полимер поли(этилен гликоль). Такой вид модификации снижает необратимую
адсорбцию белка на полимерный матрикс частиц, так как снижает
гидрофобность самого полимера. Другим подходом является модификация
самого полимера, например снижение молекулярной массы или
сополимеризация с более гидрофильными полимерами, например с тем же
самым ПЭГ [Saha S. P., 2006]. С помощью таких подходов можно достаточно
эффективно влиять на сохранение стабильности высвобождающегося белка в
течение длительного времени.
1.2.5. Применение полимерных микрочастиц, загруженных белком в
тканевой инженерии
В инженерии различных тканей человека постоянно возникает
необходимость доставки ростовых факторов, факторов дифференцировки и
других белковых молекул, стимулирующих рост определенной ткани в
необходимом месте. Для этого идеально подходят полимерные системы
пролонгированного высвобождения белков, которые затем можно

48
иммобилизовать на полимерном каркасном матриксе для создания градиентов
таких макромолекул в пространстве [Wang Y., 2011], [Perets A., 2003].
Существует множество работ, описывающих микрокапсулы из поли-(L-
молочной кислоты)-ко-поли(гликолевой ксилоты) (ПМГК), содержащих
факторы роста, полученные по методике двухэтапного эмульгирования, и они
широко применяются в регенеративной инженерии [Wenk E., 2009], [Meinel L.,
2001]. Рядом авторов показано, например, что пролонгированная доставка
фактор роста эндотелия сосудов (VEGF), заключенного в микрочастицы из
ПМГК, может активизировать ангиогенез [Rocha F. G., 2008]. Степень
инкапсулирования и механизм вхождения фактора роста зависят от
гидрофобных или гидрофильных взаимодействий между молекулами и
полимерами. Создание композитов из нескольких материалов позволяет решать
более сложные задачи и создавать усложненные структуры с
инкапсулированными белками. Например, микросферы, основанные на смеси
ПМГК и ПГБВ, полученные с помощью методики испарения растворителя при
эмульгировании представляют собой структуру ядро-оболочка. Молекулы
ПГБВ, которые являются более гидрофобными и имеют меньшую способность к
биорезорбции, были распределены внутри оболочки, что предотвращает потерю
факторов роста во время изготовления и процессов очистки, обеспечивая более
качественную защиту инкапсулируемого материала, чем микрочастицы,
состоящие только из ПМГК или ПГБВ [Zhu, X. H., 2009]. Частицы на основе
липосом представляют собой еще один подход к инкапсуляции факторов роста
клеток [Jain R. A., 2000]; [Anderson J. M., 2012]. Преимуществом этих структур
является то, что можно избежать использования жестких органических
растворителей в процессе их изготовления, потенциально повышающий
биосовместимость. Одним из последних примеров является работа,
демонстрирующая улучшение сердечной функции за счет липосомальной
доставки VEGF на модели инфаркта миокарда [Scott R. C., 2009]. При этом,
недостатком по сравнению с полимерными частицами, является короткий
период высвобождения. Синтетические полимерные носители также могут быть

49
использованы для изучения комбинированного воздействия ростовых факторов
на формирование ткани. Такие синергетические эффекты между различными
факторами роста были продемонстрированы в нескольких работах [Chen, R. R.,
2007]; [Chen R. R., 2007]; [Pepper M. S., 1992]. Например, был разработан
трехмерный матрикс из ПМГК, который постепенно выделяет фактор роста
эндотелия сосудов (VEGF), а затем тромбоцитарный фактор роста (TGF). Такая
комбинация привела как к значительному увеличению локальной плотности
сосудов, так и к созреванию новообразованных сосудов [Richardson T. P., 2001].
Эти два фактора роста были включенные вместе в один и тот же матрикс путем
смешивания полимерных микросфер, содержащих преинкапсулированный ТФР
с лиофилизированной VEGF до иммобилизации их на матриксе. Введение этих
факторов по отдельности практически не вызвало реваскуляризации.
Микросферы из желатина и его комбинаций с глицидил
метакрилированным декстраном были успешно использованы для доставки
костных факторов роста, в том числе костного морфогенетичсекого фактора-2
(BMP-2) [Chen F. M., 2009]. Среди множества преимуществ, однако, мы опять
сталкиваемся с проблемой нестабильности полученных частиц. Дело в том, что
время высвобождения белков из таких частиц сопоставимо с липосомальными
формами и является слишком коротким. Так же при работе с такими
материалами сложно контролировать физико-химические свойства полученных
изделий и систем [Fournier E., 2003]. Для решения этой проблемы исследователи
обратились к частичному окислению полимерных цепей с помощью периодата
натрия, достигая при этом гидролитической деградации [Bouhadir K. H., 2001] и
изменения молекулярно-массового распределения полимера [Boontheekul T.,
2005].
Отдельно хочется сказать про такую область регенеративной медицины,
как инженерия костной ткани. Исследования в этой области направлены на
разработку систем вживления аутологичных мезенхимальных стволовых клеток
в организм пациента с последующей их дифференцировкой в костную ткань.
Основным белком, регулирующим эти процессы, является костный

50
морфогенетичсекий фактор (BMP). Одной из его функций является индукция
дифференцировки мезенхимальных стволовых клеток в хондрогенные и
остеогенные клетки, а также их пролиферация [Canalis E., 1985]. Это приводит к
генерации костной ткани на месте перелома кости in vivo. При этом на
клеточном уровне запускается пролиферация клеток в надкостнице, костном
мозге и мышцах, окружающих перелом и недифференцированные
мезенхимальные клетки начинают мигрировать в место перелома. Было
показано, что BMP (BMP-2 и/или BMP-4) выделяется на начальной стадии
процесса заживления переломов и, по-видимому, участвуют в запуске серии
каскадов реакций процесса восстановления костей.
В 1965 году Urist описал BMP, как биологически активное вещество,
которое может создать новую гетеротопическую кость из декальцинированной
костной ткани in vivo [Urist M. R., 1965], [Urist M. R., 1973]. Затем, в 1988 году
Wozney с соавторами получили кДНК BMP-2 и BMP-4. Они обнаружили, что
белок образует димер с молекулярной массой около 32000 [Wozney J. M., 1988].
Итак, так как BMP является белком, ответственным за остеогенное воздействие
в первую очередь, рассмотрим его иммобилизацию на носителях в первую
очередь. В ряде работ показано, как иммобилизованный BMP может запускать
образование новой гетеротопиской кости с кроветворной системой костного
мозга и трабекулами in vivo.
В первую очередь следует отметить исследования, в которых на роль
носителя был выбран гидроксиапатит (ГА). Это основная минеральная
составляющая костной ткани. Было проведено большое количество
исследований таких гибридных систем с BMP на животных. При этом когда
материалы, изготовленные из гидроксиапатита погружали в раствор BMP, была
показана хороша аффинность белка в минералу.
Исследования на крысах показали, что композит ГА/BMP
продемонстрировал образование кости на месте дефекта и обширный остеогенез
по сравнению с традиционной терапией. [Sachse A, 2005], [Alam M. I., 2001].
Интересно, что некоторые исследования говорят о том, что BMP-2 остается

51
стабильным при температурах до 70°C, 27, что делает возможным включение
BMP в матрицу из гидроксиапатита непосредственно во время его синтеза из
раствора.
Rohanizadeh в своей работе [Rohanizadeh R., 2011] сравнил 3 метода
загрузки BMP-2 в ГА: (1) включение BMP во время формирования ГА в
растворе при кальцинировании, (2) погружение порошка ГА в раствор BMP, и
(3) включение BMP в дигидрат фосфата кальций с последующим
преобразованием в ГА. Сравнение этих методов проводилось по степени
загруженности BMP в носитель, профилю его высвобождения из
гидроксиапатита in vitro, и влияние белка на физико-химические и
морфологические свойства кристаллов гидроксиапатита в носителе.
Было показано, что ГА способен к поглощению белка в широком
диапазоне и, высвобождаясь, изменяет его физико-химические свойства. При
условиях, наиболее близких к физиологическим, включение белка снижало
кристалличность матрикса из гидрокиапатита, а так же размер кристаллитов.
Само же высвобождение белка из структур показало хороший
пролонгированный эффект на протяжении 14 суток. Наилучшие показатели в
данной работе продемонстрировал метод, основанный на преципитации ГА.
Наибольший задел по доставке факторов роста сделан с использованием
поли(молочной кислоты), а так же ее сополимера поли-(лактид-со-гликолида)
Saitoh и др. исследовали пригодность поли(молочной кислоты) в качестве
носителя для BMP [Saitoh H. 1994]. Частицы поли(молочной кислоты) были
смешаны с деминерализованной костью и имплантированы подкожно на
межреберные мышцы крыс. Гистологическое исследование показало,
образование хряща за две недели, образования новой костной ткани за четыре
недели, а также формирование костного мозга в течение 24 недель. Частицы
поли(молочной кислоты) постепенно рассасывались и полностью исчезли через
24 недели, путем из замещения на соединительную ткань.
Майер с коллегами сообщили о восстановлении верхнечелюстной
трещины у собак при использовании микрочастиц из поли-(лактид-ко-

52
гликолида), загруженных BMP-2 [Mayer M. 1996]. При этом были использованы
полимерные частицы в диапазоне размеров от 125 до 250 мкм. Гистологическое
исследование на протяжении двух месяцев не показало никакого остаточного
влияния поли-(лактид-ко-гликолида) (ПЛГК) и никакого клеточного ответа,
указывающего на присутствие частиц. Turk с соавторами исследовали
заживление критических дефектов черепа с использованием загруженных
остеоиндуктивными белками шариками трех различных композиций
сополимеров молочной и гликолевой кислот [Turk A. E., 1993]. Сополимеры с
различными молярными соотношениями молочной и гликолевой кислоты
показали вариативность резорбции от шести недель до шести месяцев. Кроме
того, гранулы так же покрывали поли-2-гидроксиметил-метакрилатом.
Гистологическая оценка показала наличие образования новой костной ткани и
наличие макрофагов и гигантских клеток инородных тел в реакции на шарики.
Отдельный интерес представляет работа Basmanav, в которой была
создана система с остеогенной активностью на МСК [Basmanav F. B., 2008].
BMP был включен в микросферы полиэлектролитных комплексов альгиновой
кислоты и поли(4-винилпиридина) с целью достижения инкапсуляции в очень
мягких условиях. Дело в том, что альгиновая кислота может быть легко
перешита в водном растворе раствором CaCl2. При этом мы избегаем контакта
белка с органическим растворителем и любыми химическими жесткими
воздействующими факторами, которые могут нанести вред и снизить
биологическую активность факторов роста. Однако следует отметить, что BMP
белок, несущий положительный заряд, а кинетику высвобождения из микросфер
изучали с помощью модельного белка БСА, несущего отрицательный заряд. В
связи с этим, не понятно, как целевой белок будет выходить из системы и не
будет ли он реагировать с полианионным носителем. Тем не менее,
пролиферативные и дифференцировочные эффекты BMP-2 и BMP-7 на МСК
были зафиксированы.
Дальнейшая судьба таких частиц – иммобилизация на матриксе, который
в свою очередь уже будет имплантирован. Это показано в работах того же

53
Basmanav, а так же Selcan Gungor-Ozkerim [Basmanav F. B., 2008], [Selcan
Gungor-Ozkerim P., 2014] При этом, частицы иммобилизовались между слоями
фибрилл электроспиннингового матрикса из ПМГК, либо, как в других работах
[Li L., 2015], использующих, права, уже наночастицы, непосредственно в сами
фибриллы. В последнем случае, кстати, Li с соавторами показал значительное
ускорение остеогенеза после введения еще одного вещества – дексаметазона, – в
паре с BMP-2. Этот низкомолекулярный, жирорастворимый агент так же
хорошо известен своим действием на МСК.
Напоследок, хотелось бы рассказазать о подходе, предложенном Park. Он
с коллегами показал путь остеонеогенеза на подкожной модели животного без
матрикса, а лишь используя сами частицы [Park J. S., 2010]. Для этого он создал
ПМГК микрочастицы, загруженные BMP-2, а затем ковалентно пришил на их
поверхность пептид с последовательностью аминокислот Arg-Gly-Asp, который
является молекулой адгезии для человеческих мезенхимальных стволовых
клеток. Далее он смешал частицы с МСК и ввел подкожно крысе. При этом
наблюдался обширный остеонеогенез, причем материалом, скрепляющим
частицы были сами клетки.
Следует отметить, что полимерные микрочастицы пролонгированной
доставки белков являются в большинстве своем уникальными системами, при
разработке которых необходим длительный подбор условий, зависящих как от
физико-химических и механических свойств используемого полимера, так и
технологических аспектов процесса создания полимерных микрочастиц.
Поэтому изначально такие системы разрабатываются на модельных белках.
Выбор модельного белка при этом обусловлен дальнейшей областью
применения таких систем. Так как наиболее перспективной областью
применения систем пролонгированного высвобождения белков на основе
полимерных микрочастиц является доставка ростовых и дифференцировочных
факторов для тканевой инженерии, а так же учитывая, что большинство таких
факторов являются небольшими полипептидами, имеющими положительный
заряд [Scheufler C., 1999], [Pellegrini L., 2000], [Love M., 2012], наш выбор пал на

54
лизоцим, как наиболее доступный из модельных белков, имеющий легко
определяемую ферментативную активность, pI 11,35 и ММ 14,3 кДа [Kundrot C.,
1987]

55
ЧАСТЬ 2. МАТЕРИАЛЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
2.1. ПГБ и сополимер ПГБ-ПЭГ
2.1.1. Синтез гомополимера поли(3-гидроксибутирата) различной
молекулярной массы.
В данной работе использовался поли(3-гидроксибутират) (ПГБ) различной
молекулярной массы: 826, 255 и 25 кДа. В первых двух случаях, полимер
получали микробиологическим путем с использованием штамма-продуцента
ПГБ Azotobacter chroococcum 7Б. В случае низкомолекулярного гомополимера
синтезированный ранее ПГБ в виде хлороформенного раствора подвергался
кислому гидролизу с использованием соляной кислоты в качестве
гидролитического агента [Yu G., Marchessault 2000].
Штамм-продуцент A. chroococcum 7B, азотфиксирующий микроорганизм,
способный накапливать ПГБ в количестве 80% от своего сухого веса, был
использован для синтеза высокомолекулярного гомополимера 3-
гидроксимасляной кислоты – поли(3-гидроксибутирата). Данный штамм,
полученный в нашей лаборатории, был выделен из ризосферы пшеницы
(дерново-подзолистая почва). Штамм культивировали на среде Эшби,
содержащей 0.2 г/л K2HPO4 × 3H2O, 0.2 г/л МgSO4 × 7H2O, 0.2 г/л NaCl, 0.006 г/л
Na2MoO4 × 2 H2O, 5.0 г/л CaCO3, 20 г/л сахарозы и 20 г/л агара. Для
сверхсинтеза ПГБ использовалась среда Берка, содержащая: 0,4 г/л МgSO4 ×
7H2O; 0,01 г/л FeSO4 × 7H2O; 0,006 г/л Na2MoO4 × 2H2O; 0,5 г/л цитрат натрия;
0,1 г/л CaCl2; 1,05 г/л K2HPO4 × 3H2O; 0,2 г/л KH2PO4 × 3H2O и сахарозу в
количестве 20 г/л в роли первичного источника углерода. Все эксперименты
проводились в лабораторных условиях при перемешивании в
микробиологических колбах. Молекулярная масса ПГБ, выделяемого из
бактериальных клеток, достигает более 1700 кДа. Она была снижена путем
введения в ростовую среду специальных агентов, ингибирующих синтез
полимера клетками A. сhroococcum: поли(пропилен гликоля) (ППГ, ММ 0,4 кДа)
и ацетата натрия. В первом случае, для получения полимера молекулярной

56
массой около 800 кДа в среду был добавлен ППГ в концентрации 150 мМ спустя
12 часов после начала ферментационного процесса. Для снижения
молекулярной массы ПГБ до значения около 250 кДа в среду добавлялся ацетат
натрия в концентрации 5 г/л (60 мМ). Такие концентрации агентов, снижающего
молекулярный вес, были выбраны исходя из работ, сделанных ранее в нашей
лаборатории [Myshkina V. L., 2008] [Bonartsev A. P., 2016]
Для дальнейшего снижения молекулярной массы был использован
химический способ, основанный на кислом гидролизе выделенного полимера в
растворе хлороформа [Marchessault Yu. G., 2000]. Раствор ПГБ титровался
хлороформенным раствором соляной кислоты при постоянном перемешивании.
В пробах, отбиравшихся с заданным временным интервалом,
вискозиметрически определялась молекулярная масса полимера с помощью
уравнения Марка-Куна-Хаувинка [Ol’khov A. A., 2016], [Akita S., 1976].
Гидролиз был остановлен нейтрализацией раствора по достижении значения
молекулярной массы 25 кДа.
2.1.2. Синтез сополимера поли(3-гидроксибутират)-со-поли(этилен гликоль)
Для биосинтеза сополимера также использовали штамм-продуцент A.
chroococcum 7Б, выращенный на среде Берка с сахарозой в качестве первичного
источника углерода. Затем, спустя 12 часов с начала культивации, в
культуральную среду добавляли дополнительный источник углерода –
поли(этилен гликоль) (ПЭГ, ММ 0,3 кДа) в концентрации 150 мМ. Было
показано, что дальнейшее увеличение концентрации ПЭГ ингибирует рост,
снижает синтез ПГА и его молекулярную массу. Таким образом, была
использована оптимальная концентрация ПЭГ [Saha S. P., 2006]. Эксперимент
проводился в течение 72 часов. Оптическую плотность контролировали путем
нефелометрии.

57
2.1.3. Выделение поли-3-гидроксибутирата и его сополимеров из биомассы
Для выделения полимера из биомассы использовался метод экстракции с
последующей преципитацией. При этом также происходила очистка полимера
от примесей, содержащихся в биомассе A. chroococcum. Процесс включал в себя
следующие стадии:
1) Экстракция полимера хлороформом на качалке в течение 12 часов при
37°C;
2) Отделение полимерного раствора от клеточных остатков
фильтрованием;
3) Преципитация полимера из раствора хлороформа изопропиловым
спиртом;
4) Очистка полимера многократным повторением стадий
растворения/преципитации;
5) высушивание при 60° С.
2.1.4. Определение молекулярной массы полимера
Молекулярная масса ПГБ, а также его сополимера ПГБ-ПЭГ была
определена методом гельпроникающей хроматографии с использованием насоса
Waters 1525, подключенного к четырем столбцам Waters styragel (Styragel HT
6E, 4,6 × 300 мм). Система детекции полимера состояла из дифференциального
детектора показателя преломления Waters 2414 и УФ-детектора. Хлороформ
элюировался при скорости потока 1,0 мл/мин. Стандартный объем образцов
раствора полимера (2 мг/мл) составлял 50 мкл. Для определения молекулярного
веса использовалась универсальная калибровочная кривая, построенная при
помощи стандартов ограниченной полидисперсности полистиролов (Sigma-
Aldrich, Германия). При этом, делалась поправка на изменение объема
расходованного элюента. Также молекулярная масса определялась
вискозиметрически: вязкость хлороформенного раствора полимера измеряли
при 30°C на вискозиметре RT RheoTec (RheoTec, Германия); молекулярная

58
масса была рассчитана с использованием уравнения Марка-Куна-Хаувинка
[Myshkina V. L., 2008], [Akita S., 1976]. При этом достигалась высокая степень
корреляции данных молекулярной массы полимера, полученных
вискозиметрически и с помощью гельпроникающей хроматографии.
2.1.5. Ядерный магнитный резонанс
Изучение строения полимеров проводилось с помощью метода 1H-ЯМР
спектроскопии. Растворы ПГБ, а так же ПГБ-ПЭГ в дейтерированном
хлороформе исследовались на спектрометре MSL-300 (Bruker, Германия) при
рабочей частоте 400 МГц. Химические сдвиги в ppm (parts per million) считались
от остаточного сигнала протонов в дейтерированном хлороформе (7,27 ppm).
Исследование проводилось при 40°С со временем измерения 2,5 сек в растворе с
концентрацией 1% при ширине спектра 4 кГц. Содержание мономерных звеньев
этилен гликоля в сополимере ПГБ-ПЭГ рассчитывали на основании отношения
суммы интегральных интенсивностей сигналов группы EG–CH2- (3,61; 3,70;
3,66; 3,73; 4,24 ppm) к сумме интегральных интенсивностей сигналов метильной
группы 3-оксибутирата (1,27 ppm) [Bonartsev A. P., 2016].
2.1.6. ИК-спектроскопия с Фурье преобразованием
Возрастание количества концевых гидрофильных групп при химическом
снижении молекулярной массы регистрировалось с помощью ИК-
спектроскопии с Фурье преобразованиями. Спектры полимерных образцов
снимались на Фурье спектрометре IFS-66 v/s (Bruker, Германия) в области 4000-
600см-1
(кристалл Ge). Разрешающая способность составляла 2 см-1. Для этого
исследования использовались полимерные образцы молекулярной массой 250
кДа, а также 25 кДа [Li S. D., 2003].

59
2.1.7. Определение гидрофильности полимерного материала
Гидрофильность поверхности полимера определялась путем измерения
динамического угла натекания на цифровом углометре Drop Shape Analysis
System - DSA100 (KRÜSS, GmbH, Германия). Для этого были изготовлены
полимерные пленки, нанесенные на предметное стекло. Двести микролитров
раствора полимера в хлороформе с концентрацией 30 мг/мл наносились на
стекло. Затем, после полного испарения органического растворителя, образец
помещался в поле зрения видеокамеры прибора перед источником света. При
измерении система дозирования подавала тестируемую жидкость
(деаэрированную дистиллированную воду), на поверхность подготовленных
образцов каплями, увеличивающимися в объеме от 1,5 до 5 мкл с постоянной
скоростью (200 µл/мин). Динамическое равновесие в системе наступало через
некоторое время, когда угол контакта становился постоянным. Таким образом
были получены углы смачивания для исследуемых матералов. Аппроксимация
формы капли осуществляется методом Лапласа-Юнга.
2.2. Системы пролонгированного высвобождения белков на основе
полиоксиалканоатов.
2.2.1. Белки, инкапсулируемые в полимерную матрицу микрочастиц
Для изучения систем пролонгированного высвобождения в данной работе
был использован лизоцим (Sigma-Aldrich, Германия) – фермент мурамидаза,
полученный из яичного белка. Его молекулярная масса равняется 14,3 кДа, рI =
11,35. Таким образом, он является небольшим положительно заряженным
белком. Помимо лизоцима в работе был использован бычий сывороточный
альбумин (БСА) (Sigma Aldrich, Германия) – белок плазмы крови, молекулярная
масса равняется 66,4 кДа, рI 4,7. Для исследований на конфокальном
микроскопе был использован флуоресцентно меченный БСА (ФИТЦ-БСА) и
лизоцим (ФИТЦ-лизоцим) (Sigma Aldrich, Германия).

60
2.2.2. Получение микрокапсул по методике W/O/W из ПГБ с
инкапсулированными БСА
Поли-3-гидроксибутират (ПГБ) (ММ 826 кДА);
Хлороформ (трихлорметан, ОСЧ) (Экос-1, РФ);
Бычий сывороточный альбумин (Sigma Aldrich, Германия);
ФИТЦ-меченный бычий сывороточный альбумин (ФИТЦ-БСА)
(Sigma Aldrich, Германия);
Поливиниловый спирт (ПВС) (Sigma Aldrich, Германия);
Калий-фосфатный буфер рН 7,4 (Химмед, РФ);
Микрокапсулы получали методом «водная фаза/масляная фаза/водная
фаза» [Geng Y., 2008].
Для приготовления эмульсии водного раствора белка в хлороформенном
растворе полимера, 50 μл 20% (масса/объем) раствор белка в калий-фосфатном
буфере добавляли по каплям к 4 мл хлороформенного раствора ПГБ (ММ = 826
кДа) с концентрацией 10 мг/мл. После длительного встряхивания на вортексе
полученную эмульсию добавляли по каплям к 50 мл водного раствора ПВС с
концентрацией 1,5% (масса/объем). Перемешивание проводили с помощью
верхнеприводной мешалки R2R 2021 (Heidolph, Германия) при 2000 об/мин.
После полного испарения органического растворителя, микрокапсулы отделяли
центрифугированием (10 мин при 4400 об/мин) с использованием центрифуги
5707R (Eppendorf, Германия), а затем 3 раза промывали дистиллированной
водой до полного удаления эмульгатора. Затем микрокапсулы
лиофилизировали, используя Freeze dryer ALPHA 1-2 LDplus (Германия),
предварительно заморозив их в жидком азоте.
Аналогичным образом были приготовлены микрокапсулы с
инкапсулированным ФИТЦ-меченным БСА для исследований на конфокальном
микроскопе.

61
2.2.3. Получение композита декстрана с лизоцимом
Лизоцим (Sigma Aldrich, Германия);
Декстран (нГА) (Sigma Aldrich, Германия).
Калий-фосфатный буфер рН 7,4 (Химмед, РФ);
Микрочастицы были приготовлены по методике пьзоэлектрического
мембранного распылительного высушивания [Van S. N., 2013]. На первом этапе
декстран и лизоцим были растворены в 25 мМ калий-фосфатном буфере в
соотношении 90% биополимера к 10% белка. Конечная концентрация этой
композиции была 50 мг/мл. Полученный раствор был профильтрован и загружен
в распылительную сушилку Buchi Nano Spray Dryer B-90 (Buchi, Швейцария).
Скорость осушающего воздуха была установлена на значении 120 л/мин,
внутреннее давление – 51 мбар, температура осушающего воздуха не
превышала 40 °С.
2.2.4. Получение композита поли(3-гидроксибутирата) с лизоцимом.
Лизоцим (Sigma Aldrich, Германия);
Хлороформ (трихлорметан, ОСЧ) (Экос-1, РФ);
Калий-фосфатный буфер рН 7,4 (Химмед, РФ);
Поли(3-гидроксибутират) (ПГБ) 25 кДа;
Твин 80 (Sigma Aldrich, Германия).
Полимерные микрочастицы диаметром до 3 μм для последующей
адсорбции белка на них были приготовлены по методике пьзоэлектрического
мембранного распылительного высушивания в бескислородной атмосфере
[Heng D., 2011]. ПГБ молекулярной массой 25 кДа был растворен в хлороформе
в концентрации 50 мг/мл. В качестве сурфактанта был добавлен твин-80 в
соотношении 1:1000 (вес.). Полученный раствор был профильтрован и загружен
в распылительную сушилку Buchi Nano Spray Dryer B-90 (Buchi, Швейцария).
Для создания бескислородной среды были использованы газы N2 и CO2
категории ОСЧ. Скорость осушающего воздуха была установлена на значении

62
120 л/мин, внутреннее давление – 53 мбар. Далее микрочастицы были
суспедированы в фосфатном буфере, содержащем лизоцим. Соотношение
белок/ПГБ равнялось 1/9. Полученная суспензия была заморожена в жидком
азоте с последующим лиофилизированием.
2.2.5. Получение композита нано-гидроксиаппатита с лизоцимом
Лизоцим (Sigma Aldrich, Германия);
Нано-гидроксиапатит (ГА) (Sigma Aldrich, Германия).
Калий-фосфатный буфер, рН 7,4 (Химмед,РФ);
Композит нано-гидроксиаппатита (ГА) с лизоцимом получали с помощью
лиофильного высушивания дисперсии частиц гидроксиапатита в растворе
соответствующего белка, предварительно замороженной в жидком азоте,
используя Freeze dryer ALPHA 1-2 LDplus (Германия). Данная методика
представляет собой комбинацию техник изготовления композиции
лекарственного вещества с носителем [Castellanos I. J., 2003], [Wang Y., 2007],
Соотношение количества белка и количества ГА было выбрано как 1:9 по массе.
Для этого навеску 100 мг лизоцима растворяли в 1 мл деионизированной воды.
Затем в нее добавляли 900 мг ГА и интенсивно перемешивали на вортексе. Не
допуская оседания частиц нано-гидроксиаппатита, полученную дисперсию
немедленно помещали в жидкий азот. После заморозки, ее помещали в камеру и
лофилизировали в течение ночи.
Аналогичным образом были приготовлены композиты с ФИТЦ-меченным
лизоцимом для исследований на конфокальном микроскопе.
2.2.6. Получение полимерных микрочастиц с инкапсулированным в них
композитом нано-гидроксиаппатита с лизоцимом
Поли-3-гидроксибутират (ПГБ) (ММ ≈ 25 кДа и 255);

63
Сополимер поли(3-гидроксибутира)т-со-поли(этилен гликоль) (ПГБ-
ПЭГ) (ММ ≈ 250 кДа);
Хлороформ (трихлорметан, ОСЧ) (Экос-1, РФ);
Поливиниловый спирт (ПВС) (Sigma Aldrich, , Германия);
Микроструктуры получали при помощи использования метода «твердая
фаза/масляная фаза/водная фаза» [Castellanos I. J., 2003], заключающегося в
покрытии частиц белка с носителем оболочкой из полимера. В качестве
наполнителя был выбран ГА, сам по себе обладающий биологической
активностью. Весовое соотношение полимер:наногидроксиапатит:лизоцим было
выбрано как 90:9:1 (вес.). Была приготовлена дисперсия композита ГА с белком
объемом 1 мл и концентрацией 15 мг/мл (масса/объем) в органическом
растворителе. Затем был приготовлен раствор ПГБ молекулярной массы 25 и
255 кДа, либо сополимера ПГБ-ПЭГ с концентрацией 25 мг/мл (масса/объем) в
органическом растворителе объемом 5,4 мл. Полученную дисперсию и раствор
слили вместе и тщательно перемешали на вортексе. Конечный коллоидный
раствор по каплям добавляли к 100 мл раствора ПВС с концентрацией 1,5 %
(масса/объем). Перемешивание проводили на верхнеприводной мешалке R2R
2021 (Heidolph, Германия) при 700 об/мин. После полного испарения
органического растворителя микрочастицы осаждали центрифугированием (10
мин при 4400 об/мин) с использованием центрифуги 5707R (Eppendorf,
Германия), а затем 3 раза промывали дистиллированной водой для полного
удаления эмульгатора. Затем микрочастицы лиофилизировали, используя Freeze
dryer ALPHA 1-2 LDplus (Германия), предварительно заморозив их в жидком
азоте.
Аналогичным образом были приготовлены микрочастицы с ФИТЦ-
меченым лизоцимом для исследований на конфокальном микроскопе.

64
2.2.7. Исследование пролонгированного высвобождения лизоцима и БСА из
полимерных микроструктур in vitro
Пролонгированное высвобождение белка из полученных микроструктур
проводили согласно стандартной методике [Bonartsev A. P., 2007], [Bonartsev A.
P., 2006] при 37ºС в термостате ТС 1/20 (Россия) в 25 мМ калий-фосфатном
буфере (рН = 7,4) с добавлением антибактериального агента азида натрия с
концентрацией 0,004% (вес/объем): по 10 мг микрокапсул в 1 мл буфера
перемешивали при 350 об/мин на орбитальном шейкере OS-10 (BIOSAN,
Латвия). При исследовании кинетики высвобождения белка, через заданные
интервалы времени полимерные микрочастицы отделяли от буфера
центрифугированием при 10 000 об/мин на центрифуге MiniSpin (Eppendorf,
Германия). В полученном супернатанте спектрофотометрически определяли
количество содержащегося в нем белка, а оставшиеся частицы заливали 1 мл
свежего буфера и продолжали инкубировать в термостате для дальнейшего
высвобождения.
2.2.8. Спектрофотометрическое определение содержания лизоцима и БСА в
полученных образцах
Содержание белка в полученных образцах определяли
спектрофотометрически с окрашиванием по Бредфорду, а так же методом
определения концентрации белка в растворе при длине волны 280 нм [Bradford
M. M. 1976], [Whitaker J. R., 1980]. Измерения проводились на
спектрофотометре UV-1601PC (SHIMADZU, Япония) при λ = 595 нм и λ = 280
нм для каждого метода соответственно. Для определения каждого белка
строились отдельные калибровочные кривые зависимости концентрации от
величины оптического поглощения для лизоцима и для БСА.

65
2.2.9. Исследование полученных микроструктур с помощью методов
конфокальной микроскопии
Чтобы определить характер включения белка в структуры, были
изготовлены микрокапсулы и микрочастицы с включенным в них ФИТЦ-БСА.
Максимум поглощения флуорофора – 495 нм, максимум испускания
флуоресценции – 520 нм. Приготовленную дисперсию микроструктур в воде
исследовали с помощью конфокального микроскопа Zeiss LSM 510 META
(Германия) с объективом C-Apochromat63x/1.2 W corr. Возбуждение
проводилось лазером с длиной волны 488 нм, радиус конфокальной диафрагмы
был равен 1 диску Эри (112 мкм). Использовался фильтр LP 505 нм.
2.2.10. Исследование морфологии полученных микроструктур с помощью
методов сканирующей электронной микроскопии
Для получения данных по морфологии частиц был использован
сканирующий электронный микроскоп – Quanta 200 3D (FEI Company, США)
без напыления, а также JEOL JSM-638 OLA (FEI Company, США) с напылением
палладия на образцы.
2.2.11. Исследование деградации микрочастиц
Деградация полученных микрочастиц изучалась на протяжении всего
опыта по высвобождению белка in vitro. Для этого микрочастицы,
инкубировавшиеся в фосфатно-солевом буфере, отбирали на разных сроках
инкубации, осаждали на центрифуге MiniSpin (Eppendorf, Германия) и
лиофилизировались с помощью Freeze dryer ALPHA 1-2 LDplus (Германия).
Полученные образцы были изучены с помощью электронного микроскопа
Quanta 200 3D (FEI Company, США) без напыления.

66
2.3. Исследование стабильности белка высвободившегося из полимерных
микрочастиц
2.3.1. Определение целостности первичной структуры лизоцима при
помощи электрофореза в ПААГ в присутствии SDS
Для определения целостности первичной структуры лизоцима,
высвобождаемого из полимерных микрочастиц, была проведена стандартная
процедура электрофореза по Лэммли в ПААГ в присутствии SDS [Laemmli U.
K., 1970] и с использованием камеры BG-verMINI (Baygene, Китай).
Для приготовления образцов высвободившегося белка микрочастицы в
количестве 100 мг инкубировали при 37ºС в термостате ТС 1/20 (Россия) в 25
мМ калий-фосфатном буфере (рН = 7,4) с добавлением антибактериального
агента азида натрия с концентрацией 0,004% (вес/объем) при постоянном
перемешивании на орбитальном шейкере OS-10 (BIOSAN, Латвия). Через
заданные интервалы времени полимерные микрочастицы отделяли от буфера
центрифугированием при 10 000 об/мин на центрифуге MiniSpin (Eppendorf,
Германия). Осажденные частицы заливали свежим буфером, а в полученном
супернатанте повышали концентрацию с помощью мембранных центрифужных
концентраторов (Millipore, Германия). Так как пределом чувствительности
метода данного метода является ~1 мкг белка в пробе [Сафронова М.И., 2009],
необходимо было проводить концентрирование образцов до достижения
значения 1 мкг в наносимой пробе.
Для проведения электрофореза были приготовлены растворы для
концентрирующего и разделяющего гелей и буферные растворы:
1. Разделяющий гель
Раствор для приготовления разделяющего геля готовился на основе 4х-
кратного Трис-НСl буфера (1,5М; рН=8,8; 0,8% SDS) и содержал 14%
акриламида (АА) и 0,8% метиленбисакриламида (МБА). Раствор для
приготовления геля доводили до нужного объема деионизированной водой.
2. Концентрирующий гель

67
Раствор для приготовления концентрирующего геля готовился на основе
4х-кратного Трис-НСl буфера (0,5 М; рН=6,8; 0,8% SDS) и содержал 7%
акриламида (АА) и 0,4% метиленбисакриламида (МБА). Раствор для
приготовления геля доводили до нужного объема деионизированной водой.
3. Электродный буфер
В качестве электродного буфера использовали Трис-глициновый буфер с
добавлением додецил-сульфата натрия (25 мМ Трис-НСl; 192 мМ глицина; 0,1%
SDS; рН 8,6).
4. Буфер для образцов
Для нанесения образцов был приготовлен специальный 2-х кратный буфер
(Трис-НСl 0,5 М; 4% SDS; 0,04% бромфеноловый синий; 10% глицерин; 4% β-
меркаптоэтанол; рН=6,8)
5. Раствор для фиксации и промывки гелей
Раствор, содержащий 10% уксусной кислоты и 30% изопропанола в воде
6. Раствор для окрашивания гелей
0,1% раствошпр красителя Кумасси G-250, содержащий 10% уксусной
кислоты и 30% изопропанола в воде
Непосредственно перед заливкой в гели добавляли TEMED (0,1%) и
персульфат аммония (PSA) на кончике шпателя для полимеризации гелей. В
ячейку камеры заливали около 6 мл разделяющего геля, полимеризация
проводилась при 37 °C в термостате. Затем поверх разделяющего, заливали
около 1,5 мл концентрирующего геля и, вставив гребенку, ставили в термостат
для прохождения реакции полимеризации.
На дорожки наносились следующие образцы: лизоцим, высвободившийся
из микрочастиц различного состава, контрольный раствор лизоцима, белки-
маркеры. Исследуемые образы смешивали в равных объемах с буфером для
образцов и подвергались кипячению 3-5 мин. После этого они наносились на
дорожки, камера подключалась к току (10 мА на один гель) и проводился
электрофорез. После вхождения образцов на 3-5 мм в разделяющий гель ток
переключали на 25 мА. Когда краситель достигал нижней границы геля,

68
отключали ток от камеры, вынимали гель, промывали его водой и помещали в
раствор для фиксации на 15 мин. За это время белки фиксировались на геле,
избыток додецил-сульфата натрия вымывался из него. Затем гель помещали в
окрашивающий раствор и инкубировали в течение ночи. Затем гель отмывали
раствором для промывки гелей при нагревании.
2.3.2. Исследование вторичной структуры высвободившегося белка
методом кругового дихроизма
Для определения целостности вторичной структуры лизоцима,
высвобождаемого из полимерных микрочастиц, белок был исследован с
помощью метода кругового дихроизма на дихроиметре Chirascan (Applied
Photophysics, Великоблитания).
Для приготовления образцов высвободившегося белка микрочастицы в
количестве 100 мг инкубировали при 37ºС в термостате ТС 1/20 (Россия) в 25
мМ натрий-фосфатном буфере (рН = 7,4) без добавления азида натрия в
стерильных условиях при постоянном перемешивании на орбитальном шейкере
OS-10 (BIOSAN, Латвия). Через 14 суток инкубации полимерные микрочастицы
отделяли от буфера центрифугированием при 10 000 об/мин на центрифуге
MiniSpin (Eppendorf, Германия). Осажденные частицы заливали свежим
стерильным буфером, а в полученном супернатанте повышали концентрацию с
помощью мембранных центрифужных концентраторов (Millipore, Германия). В
качестве контрольного образца сравнения исследовался свежеприготовленный
раствор лизоцима с концентрацией 0,33 мкг/мл, а так же эталонный спектр
лизоцима, полученный из литературных данных [Matsuo K., 2004]
2.3.3. Исследование третичной структуры высвободившегося белка
методом определения его ферментативной активности
Определение ферментативной активности белка осуществляли на
спектрофотометре UV-1601PC (SHIMADZU, Япония). В качестве субстрата для

69
лизоцима использовали лиофилизированную культуру Micrococcus luteus
[Mckenzie H. A., 1986], [Van-Thanh T., 2012]. Для этого была приготовлена
суспензия данной культуры с концентрацией 0,2 мг/мл в Трис-HCl буфере. Два
миллилитра раствора помещали в кювету, к ним добавляли 0,3 мл раствора
высвободившегося лизоцима, проводили измерение при длине волны 450 нм и
строили зависимость поглощения от времени реакции. Для построения
калибровочной прямой зависимости изменения поглощения в единицу времени
от концентрации лизоцима также использовались растворы лизоцима с
концентрациями от 1 до 100 мкг/мл. Активность оценивалась как отношение к
контрольному образцу нативного лизоцима.
2.3.4. Выделение мезенхимальных стволовых клеток из костного мозга
крыс линии Wistar
Для тестирования биосовместимости полимерных микрочастиц была
использована линия мезенхимальных стволовых клеток (МСК), полученных из
костного мозга крысы. [Bonartsev A. P., 2016], [Maniatopoulos C., 1988]. Для
этого были использованы трехдневные крысы линии Вистар. Забой проводился
декапитацией. После этого из крысы были извлечены бедренные кости, которые
были очищены от мышечной ткани, простерилизованы и промыты. Отрезанные
от них эпифизы были помещены в керамическую ступку с ростовой средой –
модифицированной средой Игла (DMEM, Invitrogen, США) с высоким
содержанием глюкозы (4,5 г/л), содержащей 10% эмбриональную телячью
сыворотку, антибиотик (100 МЕ/мл пенициллина и 100 мкг/мл раствора
стрептомицина) и фермент для расщепления внеклеточного матрикса
коллагеназу. После гомогенизации, полученный материал был помещен в
чашках Петри на шейкер для инкубации в течение часа. Затем, после замены
среды, клеточный материал был помещен в термостат при 37°С в увлажненной
атмосфере 5% CO2. После суток инкубации, остатки костей были удалены. На
вторые сутки инкубации на чашках Петри был получен монослой клеток.

70
2.3.5. Оценка цитотоксичности микрочастиц, загруженных лизоцимом in
vitro
Для оценки цитотоксичности микрочастиц, загруженных композитом
ГА/лизоцим, микрочастицы инкубировали с МСК крысы [Santos H. A., 2010].
Клетки инкубировали в термостате при 37°С в увлажненной атмосфере 5% CO2
в ростовой среде, содержащей модифицированную среду Игла (DMEM,
Invitrogen, США) с высоким содержанием глюкозы (4,5 г/л), 10%
эмбриональную телячью сыворотку, антибиотик (100 МЕ/мл пенициллина и 100
мкг/мл раствора стрептомицина). После трех пассажей, клетки были сняты
трипсином, отцентрифугированы, помыты фосфатно-солевым буфером (БФС), и
рассажены в 96-ти луночный планшет в количестве 104 клеток на лунку. Объем
ростовой среды на лунку равнялся 200 µл. После инкубации МСК в течение 24
часов и образования монослоя, при замене среды к клеткам были добавлены
стерильные микрочастицы, загруженные композитом ГА/лизоцим в различных
концентрациях (от 1 до 20 мг/мл), а также пустые частицы из соответствующих
полимеров в качестве контроля. После 36 часов инкубации клетки были
отделены от частиц, и их выживаемость была оценена методом ХТТ с
использованием набора реагентов ХТТ-proliferation kit (Biological Industries,
Израиль). Этот метод основан на способности активных митохондрий живых
клеток превращать соли тетразолия в соли формазана. Для этого в конце
времени культивации, после отмывки клеток от микрочастиц, в каждую лунку
добавляли 50 µл раствора ХТТ-реагента и 50 µл бессывороточной среды DMEM.
Планшеты инкубировали при 37°С еще в течение 4 часов. После этого
поглощение образцов измеряли спектрофотометрически при 450 нм, вычитая
фоновое поглощение при 620 нм. Количество жизнеспособных клеток на
пленках затем определяли по калибровочной кривой на основе их XTT-
реакционной способности. Результаты были представлены в виде процента
жизнеспособных клеток по отношению к положительному контролю, в который
микрочастицы не добавлялись.

71
ЧАСТЬ 3. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
3.1. Материал поли(3-гидроксибутират
3.1.1. Получение поли(3-гидроксибутирата) различной молекулярной массы
На данном первичном этапе работы был получен поли(3-гидроксибутират)
(ПГБ) с заданной молекулярной массой – 255 кДа и 826 кДа, – необходимой для
разработки систем пролонгированного высвобождения. Это полимер 3-
гидроксималяной кислоты, биосовместимый и биодеградируемый полиэфир,
свойства которого зависят в первую очередь от его молекулярной массы. Среди
таких свойств следует отметить его физико-химические, такие как
гидрофобность, кристалличность, прочность на разрыв, модуль Юнга,
температура стеклования, температура плавления, способность к
пленкообразованию [Myshkina V. L., 2008], [Boskhomdzhiev A. P., 2010], а также
биохимические – скорость биорезорбции, биосовместимость, взаимодействие с
биомакромолекулами внутренней среды организма и т.д. [Босхомджиев А. П.,
2009]. При этом полимер, получаемый из биомассы при использовании
сахарозы в качестве единственного источника углерода, обладает высокой
молекулярной массой – около 1700 кДа. Однако его свойства не подходят для
создания систем пролонгированного высвобождения белков. Дело в том, что
полимер такой молекулярной массы обладает высокой степенью
гидрофобности, а скорость деградации такого материала как in vitro так и in vivo
мала: будучи имплантирован подкожно крысам, за два месяца масса
полимерной пленки снижается лишь на 20% [Босхомджиев А. П., 2009]. Также,
следует отметить, что работать с таким полимером крайне трудно ввиду
высокой вязкости его растворов даже при низких концентрациях.
Для биосинтеза гомополимера ПГБ заданной молекулярной массы нами
была разработана методика, основанная на добавлении различных агентов,
ингибирующих рост биомассы Azotobacter chroococcum 7B. Данный штамм-
продуцент, выделенный в нашей лаборатории из ризосферы пшеницы (дерново-
подзолистая почва) является наиболее продуктивным среди A.chroococcum, и

72
способен накапливать до 80% от сухого веса клеток с хорошими параметрами
роста биомассы [Myshkina V. L., 2010], [Bonartsev A. P., 2011]. Итак, для
разработки различных систем пролонгированной доставки белков были
синтезтрованы образцы следующих молекулярных масс: 826 кДа (условно
обозначенный как ПГБ 800), 255 кДа (условно обозначенный как ПГБ 250), и 25
кДа. К сожалению, микробиологически мы смогли получить лишь первые два, в
то время как низкомолекулярный полимер получался последующей химической
модификацией одного из них [Yu G., Marchessault 2000].
Для понимания процесса снижения молекулярной массы полимера
различными агентами, обратимся к механизму биосинтеза поли-3-
гидроксибутирата. Поли(гидроксибутират)-полимераза (ПГБ-полимераза) – это
фермент, в активном состоянии представляющий собой гомодимер [Bernd H. A.,
2001], [Wodzinska J., 1996], в активном центре которого находится остаток
цистеина. Механизм действия фермента ПГБ-полимеразы представлен на
рисунке 17. Он заключается в связывании молекул 3-гидроксибутирил-КоА с
тиольной группой цистеина в активном центре каждой субъединицы, что
приводит к димеризации фермента. Далее, на стадии элонгации цепи,
консервативный остаток аспаргиновой кислоты одной из субъединиц
активирует гидроксильную группу у третьего атома углерода и она атакует
тиоэфирную группу второй молекулы связанного 3-гидроксибутирильного
остатка. Таким образом, растущая полимерная цепь как бы перебрасывается с
одной из субъединиц ПГБ-полимеразы на другую, каждый раз удлиняясь на
один блок [Witkowski A., 1997]

73
Рисунок 17 Механизм действия фермента ПГБ-полимеразы
При внесении в ростовую среду альтернативных источников углерода,
помимо сахарозы, происходит ингибирование процесса биосинтеза полимера. За
счет этого молекулярная масса полимера значительно снижается. При этом, в
некоторых случаях возможно встраивание агентов – молекул предшественников
– в полимерную цепь, как это показано в ряде работ по получению поли(3-
гидроксибутират-ко-3-гидроксивалерата) (ПГБВ) [Iadevaia S., 2007], [Slater S.,
1998], [Мышкина В.Л., 2010], [Bonartsev A. P., 2007].
В данной работе для получения полимера молекулярной массой около 800
кДа был использован поли(пропилен гликоль) (ППГ), как агент, снижающий
молекулярный вес полимера [Simon-Colin C., 2009]. Он вносился в
культуральную среду в количестве 150 мМ через 12 часов после начала
культивирования A.chroococcum. Однако в отличие от своих аналогов, таких как
поли(этилен гликоль) (ПЭГ) [Ashby R., 2002], [Ashby R. D., 1997], он не

74
встраивался в полимерную цепь [Bonartsev A. P., 2016]. По-видимому, при
внесении ППГ в бактериальную ростовую среду, как в аналогичном случае с
ПЭГом, происходит его связывание с активным центром фермента и
ингибирование его активности, но не происходит сополимеризации, так как
гидроксильная группа ППГ не подходит для переброски растущей полимерной
цепи ПГБ на молекулу поли(пропилен гликоля) [Bernd H. A., 2001], [Wodzinska
J., 1996]. Молекулярная масса полученного полимера равнялась 826 кДа.
Для получения поли(3-гидроксибутирата) средней молекулярной массы в
культуральную среду добавлялся ацетат натрия (АН). Нами было
продемонстрировано, что его добавление в различных концентрациях (от 10 до
100 мМ) при использовании сахарозы в качестве основного источника углерода
и при оптимальных параметрах биосинтеза приводило не только к уменьшению
выхода биомассы и содержания полимера в клетках, но и к уменьшению
молекулярной массы ПГБ, синтезированного A.chroococcum 7Б. Причем,
уменьшение молекулярной массы полимера было обратно пропорционально
концентрации ацетата натрия, добавляемого в культуральную среду (в
интервале от 20 до 60 мМ) [Myshkina V. L. 2008]; [Bonartseva, G. A., 2001], как
показано в таблице 2. Следует отметить, что добавление ацетата натрия
приводило к значительному изменению молекулярной массы при относительно
небольшом подавлении процесса самого биосинтеза. Именно поэтому это
вещество можно эффективно использовать для регуляции молекулярной массы
синтезируемого полимера. Эти более полные данные подтверждают ранее
полученные в нашей лаборатории результаты [Николаева Д. А. 2004.].
Механизм действия ацетата натрия на биосинтез ПГБ можно объяснить
следующим образом: при повышении внутриклеточной концентрации
ацетильных групп возрастает активность ацетоацетил-КоА редуктазы (КФ 1.1-
1.36), вследствие чего, повышается концентрация гидроксибутирил-КоА,
который является субстратом ПГБ-полимеразы. Дело в том, что, строго говоря,
ферментативный синтез ПГБ идет не во всем объеме цитоплазмы, а на
поверхности полимерных гранул. Объясняется данный феномен

75
нерастворимостью гидрофобного продукта такого биосинтеза – ПГБ. Среди
существующих моделей, описывающих данный механизм, наиболее близкой к
действительности является так называемая «мицеллярная модель» (рисунок 18).
Согласно ей, растворенные в цитоплазме мономерные субъединицы ПГБ-
полимеразы начинают димеризоваться при появлении субстрата для них. Такие
точки называются центрами полимеризации. При этом молекулы
водорастворимого мономера – 3-гидроксимасляной кислоты – полимеризуются
в нерастворимый в воде гидрофобный продукт – поли(3-гидроксибутират).
Образующийся при этом комплекс фермента с растущей цепью ПГБ имеет
амфифильные свойства и самопроизвольно собирается в мицеллы, в которых
гидрофобным ядром является полимер, а оболочкой – гидрофильные молекулы
ПГБ-полимеразы. При этом синтез полимерной цепи не прекращается, а белки
обкладки гранул формируют плотную оболочку гранулы [Gerngross T. U., 1995],
[Tian J., 2005]. Следует отметить, что неосинтезированный полимер имеет
ламеллярную укладку цели [Bagrov D. V. 2012], а в молекуле ПГБ-полимеразы
есть домен, обеспечивающий прочное связывание ПГБ-полимеразного
комплекса на поверхности зрелых полимерных гранул за счет нахождения в нем
короткой аминокислотной последовательности, предназначенной именно для
этого и обладающей высокой специфичностью.
Рисунок 18 Схема формирования внутриклеточных гранул ПГБ согласно
мицеллярной модели
Таким образом, при высоких концентрациях ионов ацетата образуется
большое число молекул гидроксибутирил-КоА, а значит и количество центров
полимеризации, и количество начальных фрагментов полимерных цепей так же

76
растет, что приводит к синтезу ПГБ с более низкой молекулярной массой.
Следует отметить также, что другие продуценты ПГБ, такие как Ralstonia
eutropha, используют соли уксусной кислоты как основной источник углерода,
тогда как, как можно видеть из этих данных, высокие концентрации ацетата
натрия (выше 20 мМ) ингибируют рост штамма-продуцента A.chroococcum 7Б и
биосинтез им полимера [Myshkina V. L., 2008];.
Итак, при добавлении ацетата натрия в культуральную среду,
молекулярная масса ПГБ в различной мере снижалась в зависимости от
количества добавленного АН (таблица 2). Из нее мы видим, что наиболее
значительного снижения молекулярной массы можно достичь лишь после
добавления АН в количестве, превышающем 60 мМ, однако при концентрациях,
больших, чем 60 мМ наступает сильное ингибировние роста бактерий и
значение урожая биомассы с одного литра культуральной среды падает более
чем в 2 раза. Таким образом, для получения ПГБ средней молекулярной массы,
оптимальным количеством ацетата натрия является 60 мМ. При этом
молекулярная масса полимера составила 255 кДа (условно обозначим его как
ПГБ 250), что отвечает поставленной задаче.
Таблица 2 Параметры синтеза ПГБ штаммом-продуцентом A. chroococcum 7Б в
культуральной среде с сахарозой в качестве основного источника углерода и
ацетатом натрия в качестве дополнительного источника углерода
Концентрация
ацетата
натрия, мМ
Урожай
биомассы,
г/л среды
Содержание ПОА в
биомассе, % от
сухого веса клеток
Молекулярная
масса ПОА,
×106 Да
0 5,8±0,6 83,4±3,1 1,71
20 5,2±0,5 78,8±3,4 1,13
35 4,3±0,5 71,7±3,2 0,44
60 3,9±0,6 66,9±3,0 0,25
100 2,1±0,8 61,3±3,5 0,19
Также в данной работе предполагалось использовать низкомолекулярный
полимер. Для его получения стоковый раствор ПГБ 250 кДа был подвергнут
кислому гидролизу [Marchessault Yu G., 2000]. В качестве гидролитического

77
агента была выбрана соляная кислота. Кинетика изменения молекулярной массы
в процессе гидролиза приведена на рисунке 19. Высокая скорость прохождения
гидролиза полимерной цепи связана с тем, что сложноэфирные связи при
нахождении ПГБ в растворе открыты, в то время как в твердом
конденсированном состоянии полимерная цепь ПГБ упакована в виде
ламеллярных структур [Bagrov D. V., 2012]. По прошествии 75 часов гидролиз
был остановлен добавлением эквимолярного количества гидроксида натрия для
нейтрализации соляной кислоты. Таким образом, был получен ПГБ 25 кДа,
обладающий необходимыми свойствами и характеристиками, необходимыми
для создания систем пролонгированного высвобождения белков.
Рисунок 19 Ход гидролитического разложения высокомолекулярного ПГБ. На
вставке более крупно изображен начальный промежуток времени.
Снижение молекулярной массы, а также увеличение количества
гидрофильных концевых групп показано с помощью ИК-спектроскопии с
Фурье-преобразованиями. На рисунке 20 представлены ИК спектры полимеров
ПГБ 250 и ПГБ 25. Качественно спектры – идентичны. Концевые
0
200
400
600
800
1000
1200
1400
1600
1800
0 20 40 60 80
Время гидролиза (час)
Мо
лекул
яр
ная
масса П
ГБ
(кД
а)
0
10
20
30
40
50
60
70
80
90
100
10 20 30 40 50 60 70 80
Время гидролиза (час)
Мо
лекул
яр
ная
масса П
ГБ
(кД
а)

78
гидроксильные группы обычно проявляются острой полосой в области 3480см-1
.
В данном случае – это широкая размытая полоса с максимумом при 3240 см-1.
Такое происходит, если концевые гидроксильные группы ассоциированы
водородными связями либо между собой, либо со спиртом или водой. Тем не
менее расчеты относительных интенсивностей этих полос для спектров
отнормированных по полосе метильных групп 1380 см-1 показали, что
интенсивность полосы 3240см-1
в образце ПГБ 250 кДа составляет 0,87 ед., а в
образце ПГБ 25 кДа – 1, 21 ед., что однозначно указывает на увеличение
содержания концевых –ОН групп и снижении молекулярной массы.
600 800 1000 1200 1400 1600 2800 3000 3200 3400 3600 3800
0,0
0,2
0,4
0,6
250кДа
25кДа
Int.
cm-1
Рисунок 20. Сравнение ИК- спектров образцов ПГБ разной молекулярной
массы.
На рисунке 21 видно, что полосы от одинарных С–О связей имеющие
лишь перегибы в спектре образца высокомолекулярного полимера спектре
образца низкомолекулярного и дает новые полосы 1043 и 1261 см-1
от С–ОН и
С(О)–ОН групп соответственно. Полоса от связей С=О (рис 22) в спектре
низкомолекулярного образца сдвинута в область длинных волн по сравнению со
спектром образца 250 кДа. Это также является указанием присутствия в образце

79
ПГБ 25 кДа большего количества концевых карбоксильных групп, так как
сложно-эфирные группы основной цепи имеют полосу от С=О при 1730-1740см-
1, а карбоксилатные группы при 1700-1710см
-1.
1000 1200 1400
0,0
0,2
0,4
#4 250кДа
#7 25кДа
Int.
cm-1
1043
1261
Рисунок 21. Сравнение спектров ПГБ разной мол.массы в области поглощения
связей С-О.
1600 1650 1700 1750 1800
0,0
0,2
0,4
0,6
Int.
cm-1
1723
Рисунок 22. Сравнение спектров ПГБ разной молекулярной массы в области
поглощения связей С=О.

80
Системы пролонгированного высвобождения белков на основе
микрочастиц из ПГБ и его сополимеров
Разработка систем пролонгированного высвобождения белков является
очень важной задачей современной биотехнологии. Среди широкого спектра
приложений таких систем следует отметить в первую очередь тканевую
инженерию, как наиболее перспективное направление регенеративной
медицины. Для решения проблемы дифференцировки стволовых клеток в
клетки органов и тканей применяются различные подходы доставки ростовых
факторов, программирующих дальнейшее развитие клетки, в том числе и
полимерные микрочастицы. Однако, перед тем, как создавать такие системы,
необходимо построить модель, на которой будут определены параметры,
влияющие на характер высвобождения, количества высвобождающегося
вещества. Так как доставляемые молекулы являются белками, макромолекулами
с четко заданной укладкой полипептидной цепи, определяющей их
функциональность, вопрос стабильности инкапсулируемого вещества так же
очень важен. Ниже описан постепенный подбор морфологии, материалов и
других параметров создания полимерных микрочастиц, необходимых для
максимально эффективной доставки белков с минимальной потерей их
функции. На первом этапе исследовались полые микрокапсулы, далее их
сменили сплошные композитные микрочастицы.
На первом этапе работы применялся широко распространенный
модельный белок БСА, на котором определялись в первую очередь,
кинетические параметры систем пролонгированной доставки белков. В
дальнейшем, для оценки стабильности высвобождаемого белка, исследовались
микрочастицы с инкапсулированным модельным белком лизоцимом –
положительно заряженным белком малого размера, подобным большому
количеству цитокинов и ростовых факторов, применяемых в тканевой
инженерии.

81
3.2. Микрокапсулы для пролонгированной доставки модельного белка БСА
3.2.1. Получение и характеризация полимерных микрокапсул с
инкапсулированным модельным белком БСА
В первой части работы были получены микрокапсулы из ПГБ 800 кДа,
загруженные модельным белком БСА. Примеры полученных микрокапсул
можно видеть на микрофотографии, приведенной на рисунке 23. Хорошо
заметна их шероховатая поверхность с множеством тяжей. Следует обратить
внимание, что микрокапсулы представляют собой полые частицы, внутрь
которых инкорпорирован белок, что хорошо видно при рассмотрении частицы,
рассеченной пучком ионов при исследовании на микроскопе Quanta 200 3D –
рисунок 24. На нем видна полость внутри частицы, ограниченная монолитной
стенкой, в которой и находится белок. Распределение по размерам частиц (рис.
25) показывает характерный размер частиц от 10 до 25 мкм.
Рисунок 23 Микрофотография микрокапсул из ПГБ с инкапсулированным БСА

82
Рисунок 24. Микрофотография сечения образца микрокапсул из ПГБ с
инкапсулированным БСА.
0
20
40
60
80
100
120
140
0 5 10 15 20 25 30 35 40 45 50размер микрокапсулы (мкм)
ча
сто
та
Рисунок 25. Распределение микрокапсул из ПГБ с инкапсулированным БСА по
размерам.
Для описания характера включений белка в полимерную матрицу был
выбран метод конфокальной микроскопии. Для этого были изготовлены
частицы с ФИТЦ-меченым БСА. Результаты исследования представлены на
рисунках 26 и 27. На этих изображениях хорошо видно диффузное
расположение белка внутри микрокапсул, в то время как на поверхности
микрочастиц находятся скопления адсорбированного белка, показанные
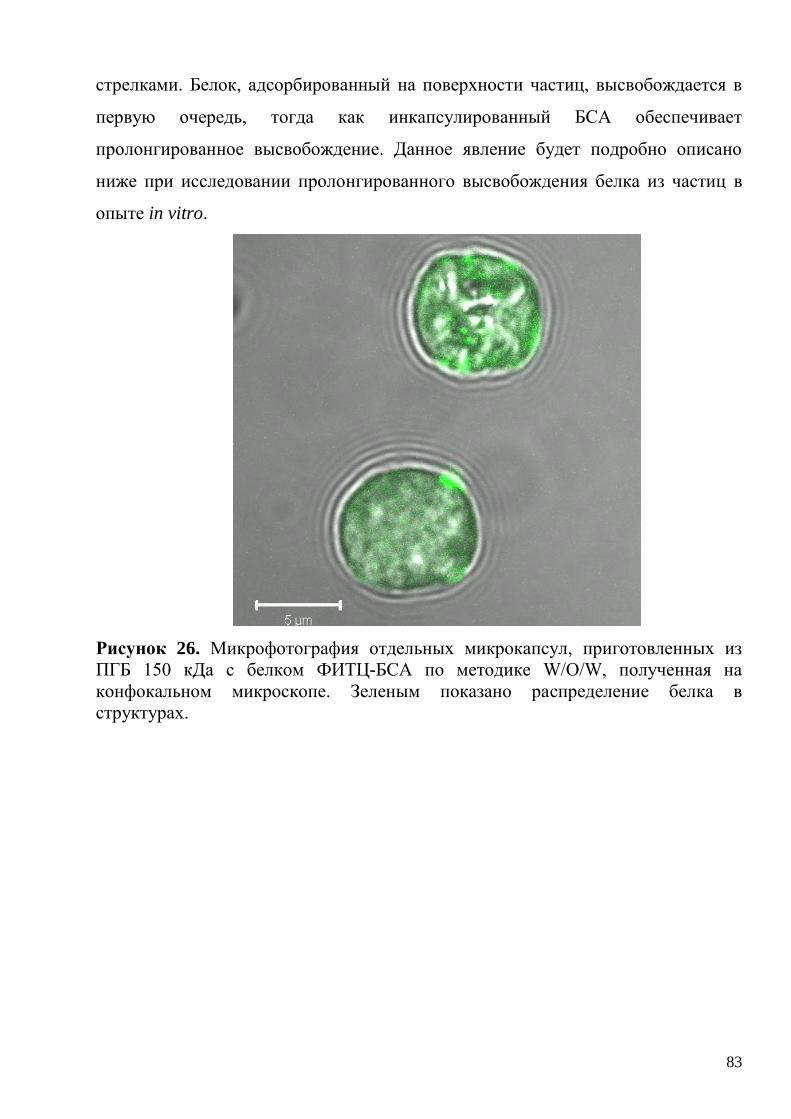
83
стрелками. Белок, адсорбированный на поверхности частиц, высвобождается в
первую очередь, тогда как инкапсулированный БСА обеспечивает
пролонгированное высвобождение. Данное явление будет подробно описано
ниже при исследовании пролонгированного высвобождения белка из частиц в
опыте in vitro.
Рисунок 26. Микрофотография отдельных микрокапсул, приготовленных из
ПГБ 150 кДа с белком ФИТЦ-БСА по методике W/O/W, полученная на
конфокальном микроскопе. Зеленым показано распределение белка в
структурах.

84
Рисунок 27. Реконструкция 3D модели по Z-стопке отдельных микрокапсул,
приготовленных из ПГБ 1000 кДа с белком ФИТЦ-БСА по методике W/O/W,
полученная с помощью конфокального микроскопа. Стрелками показаны
участки скопления белка.
3.2.2. Кинетика высвобождения модельного белка из микрокапсул in vitro и
механизм длительного высвобождения белка из структур.
Полученные частицы ПГБ с загруженным белком были использованы в
опыте по высвобождению БСА in vitro. Результаты представлены на рисунке
28. Он проводился в течение более чем 190 часов (8 суток) в модельных
физиологических условиях до того момента, когда выходящий белок перестал
фиксироваться. При этом из-за невозможности расчета всего
инкапсулированного белка (по причине того, что при необратимой адсорбции
белка ПГБ и БСА образуют агломерат, не растворимый ни в воде, ни в
хлороформе) за 100% был взят весь высвободившийся белок. Иными словами,
нас интересовал только «полезный» белок, обратимо адсорбированный на
полимер, а затем высвободившийся из полимерных структур.
Денатурированным, необратимо адсорбированным БСА мы пренебрегали.

85
0
10
20
30
40
50
60
70
80
90
100
0 50 100 150 200
Время (часы)
Ко
ли
че
ств
о в
ыс
во
бо
ди
вш
его
ся
бе
ка
(%
)
Рисунок 28. Кинетический профиль высвобождения БСА из микрокапсул ПГБ.
По данным опыта были рассчитаны параметры инкапсулирования БСА.
Данные приведены в таблице 3
Таблица 3. Параметры инкапсулирования структур из ПГБ
Степень
загруженности %
Выход микрокапсул
%
Величина burst-
effect %
1,92 ± 0,51 46,62 ± 5,96 49,94 ± 3,50
Как мы видим, степень загруженности является довольно низкой – около
1,92 %. Это обуславливается особенностями методики, в которой белок,
изначально находящийся в виде раствора, подвергается сильным воздействиям.
Именно поэтому данная методика может быть в дальнейшем применена лишь
для белковых препаратов, получение которых относительно дешево и просто,
таким как лизоцим, инсулин и другие. По другим параметрам частицы являются
довольно приемлемыми. Значение бёрст-эффекта (burst-effect),
характеризующего начальное взрывное высвобождение белка из полученных
частиц, находящееся в районе 50% означает, что частицы смогут обеспечить как
целевую первичную доставку препарата в требуемой дозе, так и последующее

86
длительное поддержание постоянной терапевтической концентрации в
окружающей среде. Значение выхода полимерных микроструктур так же
является приемлемым.
Но вернемся к рассмотрению рисунка 28. На нем отчетливо видно, что
кинетический профиль высвобождения можно разделить на два этапа. На
первом (от 0 до 5 часов) этапе высвобождения виден так называемый бёрст-
эффект, характеризующийся сильным начальным выбросом белка.
Предположительно, в это время происходит десорбция вещества с поверхности
частиц, охарактеризованная ранее (рис.27). Затем (5 – 190 часы) наблюдалось
пролонгированное высвобождение белка. Этот временной промежуток
представляет собой наибольший интерес, так как процессы, происходящие на
нем, и обеспечивают пролонгированное высвобождение БСА из полимерных
микрокапсул. Для исследования механизма высвобождения были проведены
морфологические исследования частиц на протяжении всего времени их
инкубации. Для этого после завершения опыта по высвобождению in vitro
образцы частиц были заморожены, а затем лиофилизированы. Их исследование
с помощью сканирующей электронной (рисунок 29), а также конфокальной
(рисунок 30) микроскопии показало, что механизм высвобождения белка из
микроструктур, полученных с помощью данной методики, представляет собой
разрыв стенок полимерного матрикса с дальнейшим выходом белка из него. При
этом белок легко выходит в окружающую среду. Более интересны данные,
полученные с помощью конфокального микроскопа. На рисунке 30 видно
отверстие, через которое начал высвобождаться ФИТЦ-меченый БСА. Однако,
флуоресценция в месте разрыва полимерной оболочки гораздо выше, чем
внутри частицы. Это связано, по-видимому, с возникновением дефектов
материала, имеющего, как известно, полукристаллическую структуру. В связи с
изменением укладки цепей ПГБ адсорбция выходящего белка на материал
локально повышается и возникает картина, представленная на
микрофотографии, а так же ортогональных проекциях данной микрочастицы

87
(рисунок 30). Подобный процесс пока подробно не описан и требует
дополнительного изучения.
Рисунок 29 Образцы микрокапсул из ПГБ с инкапсулированным БСА после
проведения опыта in vitro в течение 200 часов.
Рисунок 30. Микрофотография и ортогональные проекции отдельной
микрокапсулы.
Таким образом, на первом этапе работы была разработана оригинальная
методика инкапсулирования модельного белка в частицы из биоразлагаемого
полимера. Полученные микрокапсулы из ПГБ, представляют собой весьма
удачный пример создания пролонгированной формы препарата белковой

88
природы. Однако низкий процент вхождения белка в капсулы вносит
ограничение на создание на их основе новой пролонгированной формы
терапевтического белка, которое возможно лишь при условии выделения не
включившихся в полимер молекул и повторного их использования. Такой
процесс требует больших затрат, а также связан с возможной денатурацией
рециклизируемого белкового препарата. В связи с этим, в дальнейшем,
методика создания микрочастиц, загруженных белком, была кардинально
изменена, что позволило достичь существенных успехов.
3.3. Сплошные микрочастицы для пролонгированной доставки модельного
белка лизоцима
Во второй части работы была разработана методика инкапсулирования
белка в сплошные полимерные микрочастицы. Целью данного этапа являлось
устранение недостатков системы пролонгированного высвобождения белков,
основанной на микрокапсулах, а именно низкой степени загруженности, а также
высокого уровня начального взрывного высвобождения – бёрст эффекта.
Модельный белок был заменен на лизоцим. Это было обусловлено тем, что БСА
не имеет ферментативной активности, с помощью который можно судить о
степени разрушения белка в процессе его высвобождения из полимерных
структур, а также их изготовления. Лизоцим же является ферментом –
муреимидазой, активность которого определяется достаточно просто по
изменению поглощения суспензии лиофилизированных бактерий Micrococcus
lysodecticus [McKenzie H. A., 1986].

89
3.3.1. Создание композита белка с носителем
Как было описано ранее, потери белка в процессе изготовления
микроструктур во многом связаны с его денатурацией. Именно поэтому
наиболее важным аспектом инкапсулирования белков является сохранение его
стабильности на каждом этапе. Известно, что метод двухэтапного
эмульгирования «водная фаза/масляная фаза/водная фаза» приводит к
денатурации белка на границе раздела фаз вода – органический растворитель
[van de Weert M., 2000], [Kwon Y.M., 2001]. Чтобы обойти эту стадию
приготовления микрочастиц, белок вносился в органический растворитель в
твердом состоянии по методике двухэтапного эмульгирования «твердая
фаза/масляная фаза/водная фаза». Он заключался в поэтапном эмульгировании
сначала твердых частиц белка, нанесенного на носитель в хлороформенном
растворе ПГБ, а затем полученной суспензии в водном растворе конечного
эмульгатора – поли(винилового спирта) (ПВС). Для осуществления этой
методики сначала были изготовлены композитные структуры, состоящие из
лизоцима, адсорбированного на наночастицы носителя. Изначально для этого
были использованы декстран, а также частицы из полигидроксибутирата малого
размера. В случае декстрановых частиц использовалось пьезоэлектрическое
спрей-высушивание на специальной установке Nano Spray Dryer b-90 (Buchi,
Швейцария). На нее загружался водный раствор декстрана и лизоцима.
Полученный в итоге композит представлен на рисунке 31. Полученные частицы
имели правильную сферическую форму и диаметр около 1-2 µм. Во втором
случае по той же технологии были изготовлены частицы ПГБ, на которые
впоследствии белок адсорбировался путем лиофилизирования суспензии
полимерных микрочастиц в растворе лизоцима (рис. 31). Однако эти попытки
оказались неудачными: декстрановые частицы растворялись в водном растворе
конечного эмульгатора, а частицы из ПГБ в органическом растворителе.

90
Рисунок 31. Композиции модельного белка лизоцима с различными
носителями. А – декстрановые микрочастицы, загруженные лиоцимом; Б –
микрочастицы низкомолекулярного ПГБ с адсорбированным на них лизоцимом.
Таким образом, необходим был носитель, нерастворимый ни в воде, ни в
хлороформе, при этом обладающий биосовместимостью и способностью
связывать белок. Наш выбор пал на наночастицы гидроксиапатита – основного
неорганического компонента костной ткани. Таким образом, сначала были
изготовлены композитные структуры, состоящие из лизоцима,
адсорбированного на наночастицы носителя – гидроксиапатита [Zhang N., 2015].
Гидроксиапатит был выбран для решения этой задачи, потому что, являясь
главным неорганическим компонентом костной ткани, он проявляет высокую
биосовместимость и имеет широкое применение в костной инженерии и
регенеративной медицине [Zhou H., 2011]. На электронных микрофотографиях
(рис.32) мы видим нанокристаллы лизоцима, образующие агломераты, диаметр
которых приблизительно равен 3 μm. После загрузки белка на носитель мы уже
не наблюдали подобной картины из-за слоя белка, покрывавшего наночастицы.
Композитные структуры также имели размер около 3 μm.

91
Рисунок 32. Электронные микрофотографии наноГА (А) и композита
наноГА/лизоцим (Б)
Количество лизоцима, загруженного на наночастицы гидроксиапатита,
ограничено емкостью данного носителя. При превышении максимального
значения, белок перестает связываться и происходит его смывание конечным
эмульгатором. В таблице 4 представлены данные по эффективности
инкапсулирования лизоцима загруженного на наночастицы гидроксиапатита в
различном массовом соотношении. Как мы видим на рисунке 33, при
увеличении количества используемого лизоцима более чем 10% по отношению
к наночастицам гидроксиапатита, содержание белка внутри микрочастиц
возрастает незначительно, так как при этом резко падает эффективность
инкапсулирования. Начиная с 15% степень загруженности почти не растет, в то
время, так как эффективность инкапсулирования заметно снижается и доходит
до 15,6% при равном использовании белка и ГА для создания загружаемого
композита. Поэтому для создания микрочастиц из ПГБ с загруженным
композитом ГА/лизоцим, их массовое соотношение было выбрано как 90/10.
Таблица 4. Значения эффективности инкапсулирования лизоцима в полимерные
частицы с использованием различных соотношений белок/носитель
Соотношение
ГА/лизоцим 99/1 95/5 90/10 85/15 80/20 50/50
Эффективность
инкапсулирования 41.29% 41.43% 39.10% 31.72% 26.53% 15,60%

92
0
10
20
30
40
50
60
99/1 95/5 90/10 85/15 80/20 50/50
Соотношение ГА/лизоцим
Эф
фе
ктивно
сть
инка
псул
ир
ова
ни
я (
%)
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1
Сте
пе
нь з
агр
уж
енно
сти
(%
)
Эффективноссть инкапсулирования(%)Степень загруженности (%)
Рисунок 33. Эффективность инкапсулирования лизоцима в полимерные
частицы и их степень загруженности при различных соотношениях ГА/лизоцим
3.3.2. Создание полимерных микрочастиц, загруженных композитом
гидроксиапатит/лизоцим по методике двухэтапного эмульгирования
«твердая фаза/масляная фаза/водная фаза»
На следующем этапе приготовления микрочастиц мы поместили композит
нГА и лизоцима в полимерный матрикс. Для этого использовалась методика
двухэтапного эмульгирования «твердая фаза/масляная фаза/водная фаза». В
качестве сурфактанта был выбран поливиниловый спирт. Этот метод
практически не оказывает разрушающего воздействия на белок, так как
последний контактирует с хлороформом лишь в сухом виде, а, следовательно,
его денатурация, опосредованная растворителем, невозможна. Дело в том, что
белок в безводном органическом растворителе проявляет меньшую
конформационную подвижность, чем в водных растворах, чем и объясняется эта
устойчивость к денатураций при контакте с хлороформенным раствором
полимера-носителя. [Griebenow K., 1996]. Схема всего процесса представлена
на рисунке 34.

93
Рисунок 34. Схема приготовления полимерных микрочастиц, загруженных
композитом наногидроксиапатита с лизоцимом.
Для разработки самого метода инкапсулирования, а так же для
установления механизмов высвобождения белка из полученных структур мы
выбрали гомополимер ПГБ 250 кДа. Это гомополимер средней молекулярной
массы, достаточно быстро биодеградирующий и обладающий необходимыми
физико-химическими свойствами, такими как среднее значение гидрофобности
и достаточно низкое значение приведенной вязкости, необходимое для
осуществления процесса изготовления микрочастиц. Электронные
микрофотографии полученных микрочастиц приведены на рисунке 35. Частицы
с характерным размером 24,3 ± 8,4 μм (рис.36) имели правильную округлую
форму. С помощью большого увеличения удалось определить, что они имеют
пористую поверхность с диаметром пор около 1 μм. Это свойство поверхности
является положительной характеристикой для дальнейшего применения таких
частиц в костной инженерии, так как известно, что адгезия клеток к
шероховатым поверхностям значительно выше, чем к гладким, и клетки смогут
легко прикрепляться к поверхности микроструктур, загруженных белком
[Deligianni D. D., 2000]. Это позволяет в дальнейшем использовать такие
микрочастицы в тканевой инженерии, обеспечивая адгезию и пролиферацию
клеток на поверхности самих микрочастиц [Park J. S., 2010]. Рассчитанные
степень загруженности и эффективность инкапсулирования составляли
соответственно 39,10% и 0,39%.

94
Рисунок 35. Электронные микрофотографии загруженных лизоцимом
микрочастиц, основанных на ПГБ 250 кДа (А) и их поверхности (Б)
0
0,05
0,1
0,15
0,2
0,25
0 5 10 15 20 25 30 35 40 45
0
0,2
0,4
0,6
0,8
1
1,2
Рисунок 36. Распределение микрочастиц загруженных лизоцимом микрочастиц,
основанных на ПГБ 250 кДа по размерам
Вхождение лизоцима в полимерный матрикс микрочастиц было
подтверждено с помощью сканирующей лазерной конфокальной микроскопии.
На рисунке 37 представлен анализ флуоресценции вдоль линии сечения образца.
По характерным пикам, видно, что белок включается в микрочастицы в виде
агломератов, что является положительной характеристикой, так как белок,
находящийся в агрегатах более устойчив к влиянию гидрофобного окружения
полимерного матрикса и менее склонен к денатурации. Дело в том, что
денатурация может наступать при необратимой адсорбции лизоцима на ПГБ

95
[Van Tassel P.R., 1998] в процессе приготовления частиц, однако белок внутри
агломерата не контактирует с полимером и является защищенным.
Рисунок 37. Распределение флуоресценции вдоль линии сечения в образце
микрочастиц, полученных из ПГБ 250 кДа, загруженных композитом
нГА/лизоцим по методике S/O/W. Серой линией показан сигнал канала
проходящего света, зеленой – сигнал флуоресценции, линией на
микрофотографии – сечение в образце. По оси абсцисс отложена координата
сечения в образце (мкм), по оси ординат – интенсивность флуоресценции (усл.
ед.).
Следует также отметить, что соотношение ПГБ к композиту
гидроксиапатита с лизоцимом, выбранное как 90/10 является максимальным,
при котором происходит полное связывание микрочастиц композита. На
микрофотографиях частиц, содержащих по загрузке 10% комплексов белка с
ГА присутствуют лишь сами микрочастицы, в то время как на изображениях
структур, содержащих по загрузке 20% и более встречаются также и
несвязавшиеся агломераты композита лизоцим/ГА (рисунок 38). Таким образом,
соотношение полимера к композиту носителя с белком 90/10 является
оптимальным, так как при нем связываются все частицы композита, и при этом
загрузка белка в микроструктуры максимальна. Финальная композиция
микрочастиц составляет по загрузке ПГБ/наногидроксиапатит/лизоцим – 90/9/1
по массе.

96
Рисунок 38. Изображения микрочастиц, полученных при загрузке различных
количеств композита ГА/лизоцим: А – 10% по загрузке, Б – 20% по загрузке.
3.3.3. Высвобождение лизоцима из полимерных микрочастиц, основанных
на гомополимере ПГБ 250 кДа
Высвобождение белка из микроструктур продолжалось в течение более
чем 25 суток. Кинетический профиль высвобождения лизоцима из полимерных
микрочастиц, основанных на ПГБ 250 кДа, показано на рисунке 39. На нем
видно, что выход белка характеризовался высоким уровнем начального
взрывного высвобождения, в то время, как на этапе пролонгированного
высвобождения высвобождалась лишь малая его часть.
А Б

97
0
5
10
15
20
25
30
35
0 2 4 6 8 10 12 14 16 18 20 22 24 26
время (сутки)
су
мм
ар
но
е в
ыс
во
бо
жд
ен
ие
ли
зо
ци
ма
%
Рисунок 39. Кинетический профиль высвобождения лизоцима из микрочастиц,
основанных на ПГБ 250 кДа
Одной из основных теоретических моделей высвобождения белка из
биодеградируемых микрочастиц и разложения полимерной матрицы является
предложенная Langer с соавторами [Batycky R. P., 1997]. По мере того, как
частица деградирует, загруженное вещество высвобождается путем десорбции и
диффузии. Предполагается, что на первичной стадии начального взрывного
высвобождения происходит десорбция макромолекул с поверхности
микрочастиц, а также из мезопор, связанных с поверхностью, в то время как
диффузия затрудняется индукционным временем, необходимым для того, чтобы
сделать возможным сообщение микропор областей в поверхностном слое
микрочастицы с ее внутренними областями через микропористый матрикс. В
ходе гидратации и деградации биоразлагаемой полимерной основы системы
средний размер пор, ведущих в окружающее пространство, растет. Необходимое
для этого время называется временем индукции. После этого наступает фаза
пролонгированного высвобождения вещества из полимерных микрочастиц. То
есть высвобождение белка из полимерных микрочастиц проходит по двум
параллельным механизмам: посредством диффузии, а также при деградации

98
полимерной матрицы [Johnson O. F. L., 1996]. Для подтверждения этой теории
применительно к нашим микрочастицам, были поставлены два эксперимента. В
первом случае, для моделирования диффузионных процессов, протекающих при
высвобождении лизоцима, пустые полимерные микрочастицы инкубировались
в концентрированном растворе флуоресцентно меченого белка на протяжении
нескольких часов. С помощью конфокальной микроскопии было показано
диффузионное вхождение белка внутрь полимерного матрикса (рис 40), а
значит, наблюдается и обратный процесс. Таким образом, с помощью этого
инвертированного эксперимента было показано, что белок может
высвобождаться из полимерной матрицы посредством диффузии. Во втором
случае, для подтверждения деградации микрочастиц, загруженных композитом
гидроксиапатита с лизоцимом, они инкубировались в буферном растворе в
течение длительного времени. При этом на каждом этапе отбирались пробы,
которые после лиофильного высушивания исследовались с помощью
электронной микроскопии. После одних суток инкубации частицы не поменяли
своей морфологии, в то время как начальные морфологические признаки
деградации были отмечены уже после 20 суток инкубации, а после 40 дней
наблюдалась практически полная деструкция полимерной матрицы
микрочастиц (рис 41). Это означает, что сначала высвобождение белка
происходит лишь посредством диффузии, но со временем к ней добавляется еще
и деградация самих микрочастиц. Следует также отметить, что для
исследования морфологии деградированных микрочастиц применялась
сканирующая электронно-ионная микроскопия, что позволило наблюдать
реальную морфологию структур без использования напыления.
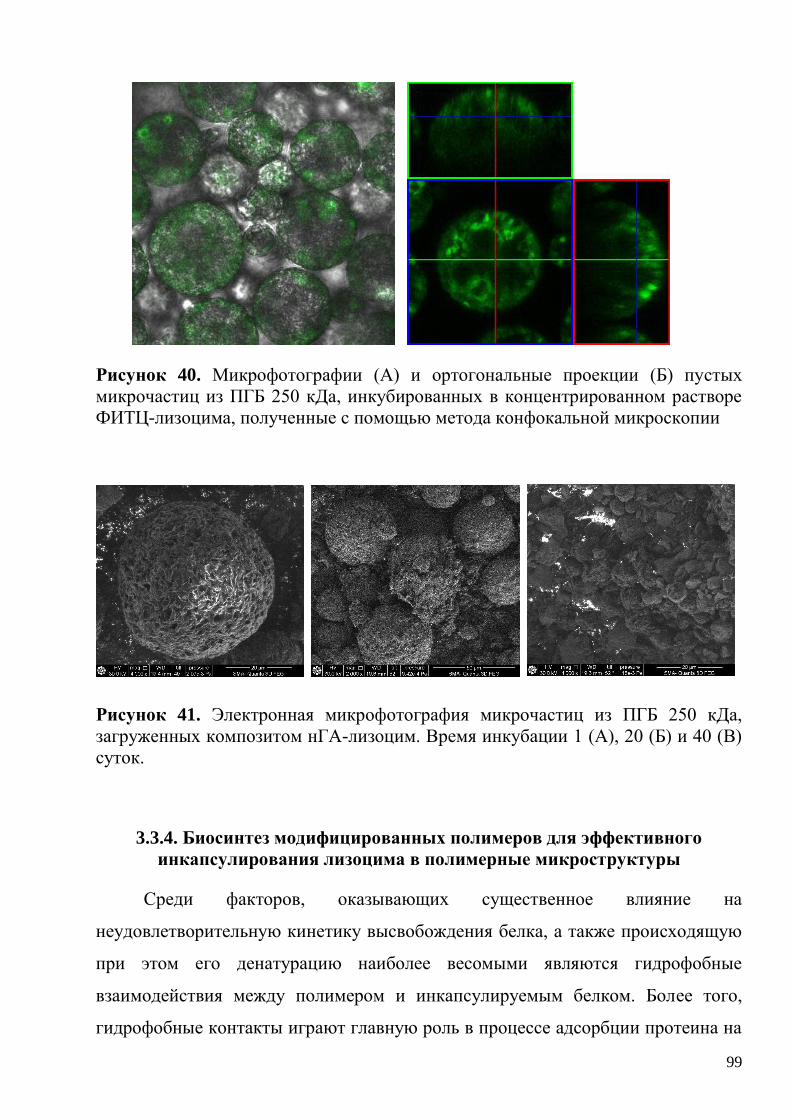
99
Рисунок 40. Микрофотографии (А) и ортогональные проекции (Б) пустых
микрочастиц из ПГБ 250 кДа, инкубированных в концентрированном растворе
ФИТЦ-лизоцима, полученные с помощью метода конфокальной микроскопии
Рисунок 41. Электронная микрофотография микрочастиц из ПГБ 250 кДа,
загруженных композитом нГА-лизоцим. Время инкубации 1 (А), 20 (Б) и 40 (В)
суток.
3.3.4. Биосинтез модифицированных полимеров для эффективного
инкапсулирования лизоцима в полимерные микроструктуры
Среди факторов, оказывающих существенное влияние на
неудовлетворительную кинетику высвобождения белка, а также происходящую
при этом его денатурацию наиболее весомыми являются гидрофобные
взаимодействия между полимером и инкапсулируемым белком. Более того,
гидрофобные контакты играют главную роль в процессе адсорбции протеина на

100
полимерный матрикс [Gombotz W.R., 1995]. Ранее мы использовали поли(3-
гидроксибутират) как базовый носитель для доставки лизоцима. Это
гидрофобный гомополимер 3-гидроксимасляной кислоты с молекулярной
массой 250 кДа, которая была получена путем добавления в процессе
биосинтеза специального снижающего молекулярный вес агента (ацетата
натрия) в культуральную среду штамма Azotobacter chroococcum 7Б. К
сожалению, ПГБ 250 кДа все равно являлся слишком сильно гидрофобным
полимером. Это является неудовлетворительным свойством для стабильности и
высвобождения инкапсулированного гидрофильного белка. Поэтому мы решили
повысить гидрофильность полимера двумя путями (рис. 42). В первом случае
мы химически снизили молекулярный вес полимера с использованием кислого
гидролиза эфирной связи полимера [Yu G., 2000]. При этом число
гидрофильных концевых групп возросло (см. рис. 20-22), что привело к
снижению гидрофобности. Однако главным недостатком такого пути является
возрастающее при этом количество карбоксильных групп. Дело в том, что
взаимодействие белка с иммобилизованными заряженными группами может
привести к деградации и денатурации третичной структуры белка, а также к
локальному закислению среды как это отмечается в ряде работ [Lai M. C., 1999].
Одним из таких взаимодействий является формирование амидной связи между
карбоксилом на полимерной цепи и первичной аминогруппой белка [Domb A.J.,
1994]. Поэтому, во втором случае мы использовали биосинтетическую
сополимеризацию ПГБ с поли-(этилен гликолем) (ПГБ-ПЭГ).

101
Рисунок 42. Схема решения проблемы высокой гидрофобности носителя ля
частиц, загруженных лизоцимом: снижение молекулярной массы полимера (А)
и сополимеризация с поли(этилен гликолем). (В) – значения контактного угла с
водой для пленок ПГБ 25 кДа, ПГБ 250 кДа и ПГБ-ПЭГ.
Таблица 5. Значения контактного угла с водой для пленок, изготовленных из
модифицированных полимерных материалов.
Полимер ПГБ 250 кДа ПГБ 25 кДа ПГБ-ПЭГ
Значение контактного угла 93.99° ± 0.56 83,30° ± 1.79 85,79° ± 2.21
Полученные низкомолекулярный гомополимер, а также сополимер
показали значительно более низкую гидрофобность, которую мы определяли с
помощью измерения контактного угла с водой (табл. 5) – его значение упало на
~10° и составило 83,3° для ПГБ 25 кДа и 85,8° для ПГБ-ПЭГ. Полученные
данные свидетельствуют о том, что и снижение молекулярного веса, и
сополимеризация с поли(этилен гликолем) (ПЭГ) привели к значительному
повышению гидрофильности носителя. Таким образом, мы создали два новых

102
материала, из которых в дальнейшем будут изготовлены микрочастицы,
загруженные лизоцимом, а также будет проведено исследование стабильности
белка, высвобождающегося из этих структур.
Остановимся более подробно на биосинтезе сополимера. Он был получен
биотехнологически, путем добавления в ростовую среду Azotobacter
chroococcum ПЭГ. Кривая роста биомассы показывает снижение пролиферации
клеток при добавлении в культуральную среду ПЭГ. На рисунке 43
представлены кривые роста биомассы штамма продуцента, а также
микрофотографии клеток при различных условиях культивации. Так, мы видим,
что при добавлении ПЭГ рост бактерий замедляется, а их морфология сильно
видоизменяется. Бактериальные клетки становятся сильно вытянутыми и
длинными. Для подтверждения прохождения сополимеризации был получен 1Н
спектр, показывающий включение в полимерную цепь ПЭГа. Наблюдалось пять
слабых ЯМР 1H сигналов при 3,66 ppm (самый высокий сигнал) и при 3,61, 3,70,
3,73 и 4,24 ppm, которые соответствуют протонам повторяющихся мономеров
ЭГ (рис.44). Сигналы на 4,24 и 3,73 ppm соотносятся с протонами,
обозначенными на рисунке а и b, соответственно, эфирного сегмента цепи ПЭГ;
пики при 3,61 и 3,70 ppm – протоны e и d концевых свободных гидроксильных
групп ЭГ. Самый высокий пик является суммой сигналов от протонов средней
части ПЭГ. При этом ПГБ, синтезированный при отсутствии ПЭГ, не
показывает 1H ЯМР-сигналов в спектральной области 3,6-3,8 ppm [Мышкина
В.Л., 2010]. Такой вид спектра согласуется с предположением, что ПЭГ садится
на один конец полимерной цепи ПГБ, где находится карбоксильная группа с
образованием ковалентной сложноэфирной связи. Чтобы понять механизм
синтеза, вернемся к рисунку 17, на котором изображена схема работы фермента
ПГБ-полимеразы – гомодимерного фермента, осуществляющего синтез нашего
полимера. На стадии элонгации цепи происходит перебрасывание растущей
полимерной цепочки на гидроксигруппу в β-положении остатка оксимасляной
кислоты. При добавлении ПЭГ в культуральную жидкость этот полимер, по-
видимому, связывается с реакционным центром фермента и происходит

103
неспецифическое образование сложноэфирной связи между ПГБ и ПЭГ. При
этом происходит обрыв цепи и дальнейший рост становится невозможен
[Bonartsev A. P., 2016], так как полимерная цепь больше не связана с остатком
цистеина активного центра ПГБ-полимеразы (рис.45). Следует отметить, что
ранее рядом авторов было показано формирование сополимеров ПОБ-ПЭГ
низкой молекулярной массы, что может быть объяснено взаимодействием ПЭГ
непосредственно с молекулами ПГА на других микроорганизмах-продуцентах
ПГА, как это было в случае А. eutropha [Shi F., 1996a] и P. oleovorans [Ashby
R.D., 2002].
0
1
2
3
4
5
6
0 10 20 30 40 50 60
Время (ч)
Би
ом
асса (
г сухо
й б
ио
массы
/ л
)
сахароза
+ ПЭГ
сахароза
Рисунок 43. А – кривая роста Azotobacter chroococcum в среде без добавления и
с добавлением поли(этиленгликоля), В – микрофотография Azotobacter
chroococcum в среде без добавления поли(этиленгликоля) (72 ч), С –
микрофотография Azotobacter chroococcum в среде с добавлением
поли(этиленгликоля) (72 ч)
Таблица 6. Биосинтез сополимера ПГБ-ПЭГ штаммом-продуцентом A.
chroococcum 7Б в культуральной среде с сахарозой в качестве основного
источника углерода и ПЭГ в качестве предшественника для биосинтеза
сополимера.

104
Субстрат Сахароза, 50 мМ Сахароза, 50 мМ +
ПЭГ 300, 150 мМ
Время добавления ПЭГ в
культуральную среду, часы - 12
Урожай биомассы, г/л среды 4,7 3,6
Содержание полимера в биомассе,
% от сухого веса клеток 81,2 52,4
Молекулярная масса полимера, кДа 1630 218
Содержание ЭГ в сополимере, 0 0,15
Молярное отношение ПЭГ/ПГБ 0 0,63
Рисунок
44. ЯМР спектр сополимера ПГБ-ПЭГ

105
Ри
сунок 45. Механизм обрыва цепи поли(этилен гликолем) при прохождении
сополимеризации ПГБ с ПЭГ

106
3.3.5. Получение и характеризация полимерных микрочастиц с
инкапсулированным модельным белком лизоцимом на основе
модифицированных полимеров
Как уже было отмечено, первоначально полученные микрочастицы из
ПГБ 250 кДа проявили неудовлетворительную кинетику высвобождения
вещества из них. Она характеризовалась низким количеством лизоцима,
высвободившегося пролонгировано, в то время как в период начального
взрывного высвобождения вышло более 50% белка (табл. 7). По-видимому, эта
проблема объяснялась следующим образом: полимер и белок имеют очень
разную природу. ПГБ это гидрофобный полукристалличный гомополимер, не
имеющий в цепи заряженных групп, в то время как белок – гетерополимер,
растворимый в воде, включающий в свой состав заряженные группы и
имеющий низкую устойчивость к физико-химическим воздействиям [van de
Weert M., 2000]. Ввиду этой разницы поли(3-гидроксибутират) не может
удержать белок, а может лишь необратимо адсорбировать его на свою
поверхность. Как мы видим на микрофотографии частиц, загруженных
флуоресцентно меченым белком, и инкубированных in vitro в течение 90 суток
до полного прекращения высвобождения лизоцима, большое количество белка
все равно остается адсорбированным на полимерный матрикс частиц (рис 46).
Более того, по имеющимся данным, гидрофобность полимера плохо влияет на
стабильность инкапсулированного белка. [Lu W., 1995], и он остается
необратимо адсорбированным, неактивным и денатурированным агломератом,
связанным с полимером.

107
Рисунок 46. Микрочастицы ПГБ 250 кДа после полного завершения
высвобождения белка, инкубированные более 90 суток в калий-фосфатном
буфере. Конфокальная микрофотография
Итак, для создания новых частиц, загруженных лизоцимом, мы
использовали ПГБ 25 кДа и ПГБ-ПЭГ в качестве носителей с
усовершенствованными физико-химическими свойствами. Микрофотографии
полученных частиц, а так же их поверхности показаны на рисунке 47.
Рассчитанные на основании данных сканирующей электронной микроскопии
размеры частиц составляли 22.4 ± 6.4 μм и 25.1 ± 7.2 μм соответственно для ПГБ
25 кДа и ПГБ-ПЭГ. Поверхность частиц так же не отличалась от
первоначальных. Степень загруженности (СЗ), эффективность
инкапсулирования (ЭИ), а также величина бёрст эффекта (БЭ) приведены в
таблице 7. Из этих данных видно, что низкомолекулярный гомополимер
обладает низким сродством к полимеру – степень загруженности частиц ПГБ 25
кДа ниже остальных почти в два раза. Для сополимера этот показатель оказался
примерно равен первичным частицам, основанным на высокомолекулярном
гомополимере. То есть при снижении молекулярной массы на порядок,
наблюдалась картина, обратная ожидаемой: вместо увеличения сродства
полимера к белку ввиду повышения его гидрофильности происходило снижение

108
способности ПГБ связывать лизоцим. Это может объясняться повышением
кристалличности по отношению к высокомолекулярному полимеру [Perego G.,
1996], [Barham P. J., 1984], что приводит в дальнейшем к снижению
эффективности инкапсулирования, показанному рядом авторов [Schugens C.,
1994]. При проведении сополимеризации с поли(этилен гликолем) этого не
происходит и эффективность инкапсулирования остается высокой.
Рисунок 47. Морфология микрочастиц, основанных на ПБ 25 кДа, ПГБ 250 кДа,
ПГБ-ПЭГ

109
Таблица 6. Параметры инкапсулирования лизоцима в частицы, основанные на
различных полимерах
Полимер ЭИ % СЗ % БЭ %
ПГБ 250 кДа 39.10 0.39 64,65
ПГБ 25 кДа 20.53 0.21 43,55
ПГБ-ПЭГ 38.13 0.38 21,82
3.3.6. Кинетика высвобождения лизоцима из полимерных микрочастиц,
основанных на модифицированных полимерах. Стабильность
высвобождающегося белка
Для исследования влияния полимерного носителя на кинетику
высвобождения лизоцима из микроструктур, мы провели опыт in vitro для
частиц на основе ПГБ 250 кДа, ПГБ 25 кДа и ПГБ-ПЭГ. На рисунке 48 видно,
что кинетические профили высвобождения лизоцима из частиц на основе
модифицированных полимерах, значительно более пологие, а фаза
пролонгированного высвобождения выражена значительно в большей степени,
чем на микрочастицах из высокомолекулярного гомополимера. На гистограмме,
изображенной на рисунке 49, представлены соотношения белка
высвободившегося в первые часы (бёрст-эффект) и белка, высвободившегося
пролонгированно в течение длительного времени. Из него хорошо видно, что
наилучшими характеристиками обладает сополимер с поли(этилен гликолем):
более 80% белка высвобождается из таких частиц пролонгировано, в то время
как лишь малая его часть выходит в первые часы. Низкомолекулярный полимер
так же продемонстрировал хорошую кинетику высвобождения, однако
начальный взрывной выброс вещества по-прежнему вносил слишком
существенный вклад: более 40% белка высвободилось в первые часы. Таким
образом, суммируя вышеперечисленные показатели, а так же имея в виду, что
эффективность инкапсулирования лизоцима в микрочастицы из ПГБ-ПЭГ
достаточно высока, можно говорить о том, что по кинетике высвобождения

110
белка сополимер является наилучшим носителем для лизоцима. Как мы
предполагаем, данные результаты связаны с амфифильностью молекул
сополимера. При затвердевании полимерного раствора в водной эмульсии
молекулы, предположительно поворачиваются гидрофильной частью – блоком
поли(этилен гликоля) – наружу, в то время как гидрофобная часть молекулы –
блок поли(3-гидроксибутират) – преимущественно должна быть ориентирована
вовнутрь полимерного матрикса (рисунок 50). В сторону гидрофильного белка
также должна быть ориентирована более гидрофильная часть – блок
поли(этилен гликоля). По-видимому, это и определяет более «плотные»
взаимодействия между белком и полимерным носителем, что в свою очередь
предотвращает первоначальное взрывное высвобождение белка.
0
5
10
15
20
25
30
35
40
45
0 2 4 6 8 10 12 14 16 18 20 22 24 26
Время (сутки)
су
мм
ар
но
е в
ысв
об
ож
де
ни
е л
изо
ци
ма
%
ПГБ 250 кДа
ПГБ 25 кДа
ПГБ-ПЭГ
Рисунок 48. Кинетический профиль высвобождения лизоцима из микрочастиц,
основанных на ПГБ 250 кДа, ПГБ 25 кДа и ПГБ-ПЭГ
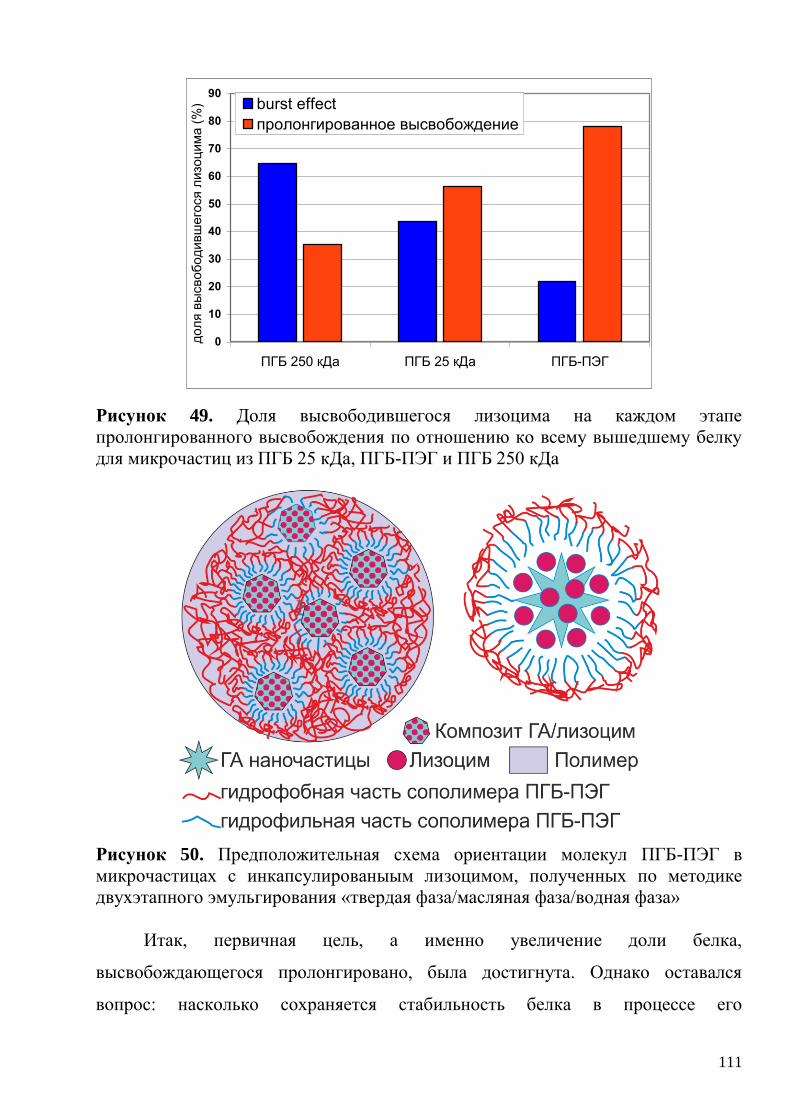
111
0
10
20
30
40
50
60
70
80
90
ПГБ 250 кДа ПГБ 25 кДа ПГБ-ПЭГ
дол
я в
ысвоб
од
ивш
его
ся л
изо
цим
а (
%) burst effect
пролонгированное высвобождение
Рисунок 49. Доля высвободившегося лизоцима на каждом этапе
пролонгированного высвобождения по отношению ко всему вышедшему белку
для микрочастиц из ПГБ 25 кДа, ПГБ-ПЭГ и ПГБ 250 кДа
Рисунок 50. Предположительная схема ориентации молекул ПГБ-ПЭГ в
микрочастицах с инкапсулированыым лизоцимом, полученных по методике
двухэтапного эмульгирования «твердая фаза/масляная фаза/водная фаза»
Итак, первичная цель, а именно увеличение доли белка,
высвобождающегося пролонгировано, была достигнута. Однако оставался
вопрос: насколько сохраняется стабильность белка в процессе его

112
высвобождения из полимерных микрочастиц? Ведь, как мы уже говорили,
гидрофобные контакты белка в процессе его диффузии через полимерный
матрикс приводят к дестабилизации структуры белковой молекулы [van de
Weert M., 2000], а значит, снижение гидрофобности носителя должно
благотворно сказаться на сохранении нативной структуры белка.
Для решения этого вопроса, стабильность высвободившегося лизоцима
была исследована на трех уровнях: первичной, вторичной и третичной
структуры белка. Для проверки целостности первичной структуры белка был
проведен анализ образцов с помощью электрофореза по Лэммли. Для этого
использовались пробы, отобранные после 3 часов, 7 и 14 суток инкубации в
буфере при постоянном перемешивании при 37°С. Полученная
электрофореграмма приведена на рисунке 51. На ней видны полосы лизоцима и
не видно полос белка меньшей молекулярной массы, а значит, полипептидная
цепь лизоцима не разрушалась.
Исследование на уровне вторичной структуры белка проводилось при
помощи кругового дихроизма. Как известно, на вид этого спектра влияет
соотношение элементов вторичной структуры, а именно α-спирали, β-укладки и
неупорядоченных участков цепи. Каждому типу соответствует определенная
область спектра. На рисунке 52 приведены характерные спектры для каждого
типа вторичной структуры белка [Финкельштейн А.В., 2005, стр. 94]. Спектры
кругового дихроизма проб белка, высвободившегося из микрочастиц после 14
суток инкубации, приведены на рисунке 53. Как мы видим, спектры белка из
частиц ПГБ 250 кДа и ПГБ-ПЭГ практически совпадают с эталонным, взятым из
литературных источников [Matsuo K., 2004]. Однако спектр лизоцима,
высвободившегося из микроструктур, основанных на низкомолекулярном
гомополимере ПГБ 25 кДа, сильно отличается в диапазоне 180-200 нм. В этом
диапазоне основной вклад в вид спектра имеют как раз упорядоченные участки
вторичной структуры белка: α-спираль и β-укладка (см рис.52), – а значит,
лизоцим при взаимодействии с ПГБ 25 в процессе высвобождения изменяет
свою вторичную структуры, и, следовательно, денатурирует. Как мы знаем,

113
подобный эффект могут вызвать межмолекулярные взаимодействия участков
полипептидной цепи с карбоксильными группами ПГБ, которых в
низкомолекулярном полимере на порядок больше, чем в высокомолекулярном
гомополимере [Domb A. J., 1994]. При этом, возвращаясь к сополимеру, следует
отметить, что в нем все карбоксильные группы экранированы путем
ПЭГилирования (рисунок 44), а значит подобные взаимодействия между ним и
полипептидной цепочкой исключены.
Рисунок 51. Электрофореграммы высвободившегося белка на различных этапах
высвобождения. Дорожки: 1-ПГБ25, 2-ПГБ250, 3-ПГБ-ПЭГ, 4-лизоцим, 5-белки-
маркеры

114
Рисунок 52. Примеры спектров кругового дихроизма различной вторичной
структуры белка: α – альфа спираль, β – бета укладка, r – неупорядоченная
полипептидня цепь.

115
-15
-10
-5
0
5
10
15
20
25
30
180 200 220 240
Длина волны (нм)
[θ]M
RW
*10
^(-
4),
de
gr*
cm
^2
*dm
ol^
(-1
)
ПГБ 25 кДа ПГБ 250 кДа
ПГБ-ПЭГ контроль
Рисунок 53. Спектры кругового дихроизма лизоцима высвободившегося после
14 суток икубации микрочастиц, полученных из полимеров ПГБ 2150 кДаб ПГБ
25 кДа и ПГБ-ПЭГ
Но самым важным параметром стабильности белка является его
ферментативная активность, которая напрямую связана с его третичной
структурой [Финкельштейн А.В., 2005, стр. 138-147]. Лизоцим является
ферментом – муреимидазой, гидролизующей муреин в составе клеточной стенки
бактерий. Как известно, белки проявляют ферментативную активность в
большинстве случаев лишь в нативном состоянии, поэтому на заключительном,
третьем этапе исследования стабильности высвободившегося белка, была
установлена способность лизоцима к разложению клеточной стенки бактерий
Micrococcus luteus. На рисунке 54 видно, что активность белка из микрочастиц
ПГБ 25 кДа падает после 2 недель инкубации более чем в два раза. Полученные
данные хорошо коррелируют с информацией со спектров кругового дихроизма,
показавшей денатурацию вторичной структуры белка, высвободившегося
именно из этого низкомолекулярного полимера. По-видимому, с увеличенным

116
количеством концевых карбоксильных групп, ПГБ 25 кДа, как уже говорилось,
вступает в межмолекулярные взаимодействия с белковыми макромолекулами, а
также вызывает локальное закисление среды внутри микрочастиц, что приводит
к повышенной нестабильности белковых макромолекул [Lai M. C., 1999],
[Domb A. J., 1994]. В свою очередь, высокомолекулярные гомо- и сополимер не
оказывают столь губительного воздействия высвобождающийся белок: около
80% молекул белка сохраняют свою способность к разложению муреина. Это
говорит о том, что фермент, загруженный в микрочастицы на основе ПГБ-ПЭГ,
сохраняет свою нативную форму в течение двух недель, что является
важнейшей характеристикой для системы пролонгированной доставки белков в
организм.
0
20
40
60
80
100
120
1 сутки 7 суток 14 суток
% а
кти
вно
го б
ел
ка о
т все
го
вы
сво
бо
ди
вш
его
ся
ПГБ 25 кДа
ПГБ 150 кДа
ПГБ-ПЭГ
Рисунок 54. Доля активного белка высвободившегося на разных этапах
высвобождения по отношению ко всему высвободившемуся белку для
микрочастиц, полученных из полимеров ПГБ 250 кДа, ПГБ 25 кДа и ПГБ-ПЭГ
Подводя промежуточный итог, необходимо отметить, что основываясь на
эффективности инкапсулирования и кинетике высвобождения лизоцима из
микросфер, наилучшими показателями обладает сополимер поли(3-
гидроксибутирата) с поли(этилен гликолем), продемонстрировавший так же

117
наилучшие показатели по сохранению стабильности, нативной структуры и
ферментативной активности высвобождающегося белка.
3.3.7. Исследование полимерных микрочастиц, загруженных лизоцимом на
цитотоксичность in vitro
Основным показателем материалов в тканевой инженерии является их
биосовместимость. Так как исследуемые в данной работе микрочастицы
планируется применять в инженерии костной ткани, нас интересовала их
цитотоксическая реакция по отношению к культуре мезенхимальных стволовых
клеток (МСК), полученных из костной мозга мыши. МСК широко используют в
инженерии костной ткани, т.к. с их помощью можно регенерировать
критические дефекты кости, в частности, с помощью аутологических МСК,
выделяемых из костного мозга самой крысы, костный дефект которой и
предполагается в дальнейшем заживлять [Liu Y., 2008], [Im J. Y., 2013], [Zhou Y.
S., 2006]. Клетки выделялись из эпифизов бедренной кости 3-дневной крысы
линии Wistar, после чего культивировались вместе с микрочастицами
различного состава, загруженными лизоцимом. В качестве контрольных
образцов использовались пустые микрочастицы из соответствующего полимера,
а также наночастицы гидроксиапатита и лизоцим. При этом, концентрация
лизоцима и гидроксиапатита была выбрана в соответствии с содержанием этих
веществ в полимерных микросферах. На рисунке 55 показано, что после 36
часов инкубации частицы не угнетают пролиферацию клеток в диапазоне
концентраций до 20 мг/мл, чего нельзя сказать о наночастицах гидроксиапатита.
Как мы знаем, наночастицы гидроксиапатита вызывают остеогенную
дифференцировку мезенхимальных стволовых клеток [Mygind T., 2007]. Более
того, матриксы из ПГБ, содержащие наночастицы гидроксиапатита, могут
вызывать спонтанную дифференцировку МСК в остеогенном направлении
[Shishatskaya E. I., 2006]. Метод определения выживаемости клеток ХТТ
основан на способности дегидрогеназного комплекса дыхательной цепи
митохондрий живых клеток восстанавливать тетразолиевую соль (2,3-бис-(2-

118
метокси-4-нитро-5-сульфонил)-2H-тетразолиум-5-карбоксанилид) [Mosmann T.,
1983]. Известно, что при остеогенной дифференцировке МСК их
митохондриальная активность снижается вследствие снижения экспрессии
митохондриальных ферментов, несмотря на то, что общее число митохондрий
растет [Chen C. T., 2008]. Можно предположить, что именно поэтому при их
обработке наночастицами гидроксиапатита, тест показывает снижение
выживаемости клеток, хотя на самом деле происходит их
остеодифференцировка. Таким образом, исследование полученных микрочастиц
не выявило цитотоксичности по отношению к мезенхимальным стволовым
клеткам, что говорит об их высокой биосовместимости.
0
20
40
60
80
100
120
140
ПГБ
25
кДа
ПГБ
25
кДа
ПГБ
-ПЭГ
ПГБ
25
кДа/лизо
цим
ПГБ
250
кДа/
лизоцим
ПГБ
-ПЭГ/
лизоцим
нано-Г
А
лизоцим
(0,2
мг/мл)
"+" ко
нтроль
5 мг/мл
10 мг/мл
20 мг/мл
Рисунок 55. Исследование на цитотоксичность к МСК микрочастиц из ПГБ 25
кДа, 250 кДа и ПГБ-ПЭГ, загруженных лизоцимом, а также пустых полимерных
микрочастиц, наночастиц гидроксиапатита и чистого лизоцима. В качестве
положительного контроля использовалась среда с клетками.

119
ЗАКЛЮЧЕНИЕ
В данной работе представлено исследование высвобождения модельного
белка лизоцима из полимерных микрочастиц на основе полиоксибутирата. Была
разработана оригинальная методика инкапсулирования модельного белка
лизоцима в микрочастицы из гомополимера с использованием наночастиц
гидроксиапатита как носителя белка по методике двухэтапного эмульгирования
S/O/W, а так же исследовано пролонгированное высвобождение белка из
полученных структур. В качестве полимерной матрицы был использован как
гомополимер ПГБ различной молекулярной массы, так и его блок-сополимер
ПГБ-ПЭГ, который был впервые использован для создания систем
пролонгированного высвобождения белков. Гомополимеры необходимой
молекулярной массы, а также сополимер были получены микробиологическим
синтезом с использованием штамма Azotobacter chroococcum 7Б.
Частицы диаметром около 25 мкм имели правильную округлую форму и
пористую поверхность с диаметром пор до 1 мкм, что показано методом
сканирующей электронной микроскопии. Включение и распределение белка в
виде агломератов внутри полимерного матрикса показано с помощью
конфокальной микроскопии. Высвобождение белка из микрочастиц
осуществлялось in vitro в фосфатном буфере (pH 7,4) при 37° С. Также в ходе
эксперимента было показано, что высвобождение белка из микрочастиц
происходит посредством двух процессов: диффузии молекул сквозь
полимерный матрикс и деградации самих микрочастиц.
Опыты по проведению пролонгированного высвобождения белка показали,
что кинетический профиль микрочастиц, основанных на сополимере, сильно
отличался от кинетического профиля гомополимерных микроструктур, а именно
большая часть белка высвобождалась равномерно в течение более чем двух
недель при низком начальном выбросе вещества. Кроме того, по эффективности
инкапсулирования белка данный сополимер также имел преимущество.
Модификация частиц путем снижения молекулярной массы полимера принесла

120
не столь ощутимые плоды, однако микроструктуры так же продемонстрировали
улучшение кинетических параметров высвобождения белка из них.
Также было показано, что модельный белок, высвобождающийся из
микрочастиц, основанных на сополимере ПГБ-ПЭГ, сохранял свою нативную
структуру, а также активность на протяжении более чем двух недель. Опыты на
биосовместимость in vitro показали, что данные микрочастицы не имеют
ингибирующего действия на мезенхимальные стволовые клетки.
Полученные в данной работе результаты говорят о том, что созданная
система пролонгированного высвобождения белков имеет огромный потенциал
для использования уже не с модельными белками, а с терапевтическими при
создании конструкций для тканевой инженерии, например для регенеративной
инженерии костной ткани.

121
ВЫВОДЫ
1. Методом контролируемого биосинтеза и химическим путем на основе
гидрофобного высокомолекулярного поли(3-гидроксибутирата) получены
полимеры, обладающие большей гидрофильностью: низкомолекулярный
поли(3-гидроксибутират) и его сополимер с поли(этилен гликолем).
2. Разработана оригинальная методика инкапсулирования белков в
сплошные полимерные микрочастицы с использованием гидроксиапатита, как
носителя. Полученные частицы имеют сферическую форму, размер ~25 µм,
пористую поверхность с размером пор до 1 мкм. Эффективность
инкапсулирования составляет около 40%, белок расположен в виде агломератов.
Впервые получены микрочастицы из биосинтетического сополимера поли(3-
гидроксибутирата) с поли(этилен гликолем), загруженные белком.
3. Высвобождение лизоцима из полимерного матрикса полученных
микроструктур осуществляется по двум механизмам: деградации микрочастиц и
диффузии белка из них.
4. Наилучшими параметрами высвобождения лизоцима обладали
микрочастицы на основе биосинтетического сополимера поли(3-
гидроксибутирата) с поли(этилен гликолем). Лизоцим сохранял свою
стабильность на протяжении двух недель высвобождения белка из таких частиц
in vitro.
5. У микрочастиц отсутствует цитотоксичность по отношению к
мезенхимальным стволовым клеткам крысы на протяжении 36 часов инкубации
in vitro.

122
БЛАГОДАРНОСТИ
Автор выражает глубокую благодарность своим научному руководителю
Бонарцеву Антону Павловичу, а также научному консультанту Бонарцевой
Гарине Александровне всестороннюю поддержку во всех начинаниях; центру
общего пользования ФИЦ «Биотехнологии» РАН в лице Шубина В.В. за
помощь в спектроскопических методах исследования, а также исследованиях
методом кругового дихроизма; кафедре электронной микроскопии в лице
Давидовича Н.Д. за помощь в исследованиях методом сканирующей
электронной микроскопии, а также центру коллективного пользования
Института кристаллографии им. А.В.Шубникова РАН; Феофанову А.В. за
помощь в работе с использованием метода конфокальной микроскопии.
Работа была выполнена при финансовой поддержке Российского Фонда
Фундаментальных исследований (грант офи-м № 15-29-04856) в части
разработки методик получения микрочастиц с использованием метода
мембранного пьезоэлектрического распылительного высушивания и
эмульгирования и при финансовой поддержке Российского Научного Фонда
(грант № 15-15-10014) в части исследования цитотоксичности микрочастиц с
использованием мезенхимальных стволовых клеток и получения микрочастиц
из альгината натрия.

123
СПИСОК СОКРАЩЕНИЙ
КoA кофермент А;
ЛВ лекарственное вещество;
ММ молекулярная масса;
НАД никотинамидадениндинуклеотид;
НАДФ никотинамидадениндинуклеотидфосфат;
ПГК поли(гликолиевая кислота);
ПМК поли(L-молочная кислота);
ПМГК сополимер полимолочной и полигликолевой кислот;
ПГА полигидроксиалканоаты;
ПГБ, П3ГБ поли(3-гидроксибутират);
ПГБВ, ПГВ поли(3-гидроксибутират-со-3-оксивалерат);
ПСКВ (СКВ) полимерная система контролируемого высвобождения;
ПЭГ полиэтиленгликоль;
ПГБ-ПЭГ сополимер поли(3-гидроксибутират)-поли(этилен гликоль);
ФИТЦ флуоресцеин-изотиоцианат;
ГА гидроксиапатит;
BMP костный морфогенетичсекий фактор
FGF фактор роста фибробластов
VEGF фактор роста эпителия сосудов
TGF тромбоцитарный фактор роста
БСА бычий сывороточный альбумин
МСК мезенхимальные стволовые клетки
ЭИ эффективность инкапсулирования
СЗ степень загруженности
БЭ бёрст эффект

124
СПИСОК ЛИТЕРАТУРЫ
1. Бонарцева Г.А., Мышкина В.Л., Загреба Е.Д., Николаева Д.А.
Способ получения поли-β-оксибутирата заданной молекулярной массы. Патент
РФ № 2201453/18.10.2001;
2. Босхомджиев А. П. и др. Сравнительное изучение кинетики
биодеградации биополимерных систем на основе поли-3-оксибутирата
//Биомедицинская химия. – 2009. – Т. 55. – №. 6. – С. 702-712;
3. М.И.Сафронова, Н.Н. Зайцева, А.М.Рубцов, В.Г. Гривенникова, А.С.
Рыжавская, Н.Б. Гусев. Основы практической биохимии белка, Мосвка, 2009;
4. Мышкина В.Л. и др. Биосинтез сополимера поли(3-
гидроксиьутирата)-(3-гидроксивалерата) штаммом Azotobacter chroococcum 7Б
//Прикладная биохимия и микробиология. – 2010. – Т. 46. – №. 3. – С. 315-323;
5. Николаева Д. А. Биосинтез поли-3-гидроксибутирата разной
молекулярной массы культурой Azotobacter chroococcum и его биодеградация :
дис. – М. : Ин-т биохимии им. АН Баха РАН, 2004;
6. Полищук А.Я., Иорданский А.Л., Заиков Г.Е.. Диффузионно-
кинетическая модель адсорбции плазменных белков на гидрофобной
полимерной поверхности. Доклады АН СССР, 1982, Т. 264, С. 1431-1435;
7. Севастьянов В.И. Биосовместимость, Адсорбция белков и
гемосовместимость изделий, ГУП «Информационный центр ВНИИгеосистем» ,
Москва, 1999, стр. 88-94;
8. Севастьянов В.И. Биосовместимость, Адсорбция белков и
гемосовместимость изделий, ГУП «Информационный центр ВНИИгеосистем» ,
Москва, 1999, стр. 94-111;
9. Финкельштейн А.В., Птицын О.Б. Физика белка, КДУ, Москва,
2005, стр. 226-230;
10. Финкельштейн А.В., Птицын О.Б. Физика белка, КДУ, Москва,
2005, стр. 94;

125
11. Финкельштейн А.В., Птицын О.Б. Физика белка, КДУ, Москва,
2005, стр. 138-147;
12. Akita S. et al. Solution properties of poly (d-β-hydroxybutyrate). 1.
Biosynthesis and characterization //Macromolecules. – 1976. – Т. 9. – №. 5. – С. 774-
780;
13. Alam M. I. et al. Evaluation of ceramics composed of different
hydroxyapatite to tricalcium phosphate ratios as carriers for rhBMP-2 //Biomaterials.
– 2001. – Т. 22. – №. 12. – С. 1643-1651;
14. Alejandra R. C., Margarita C. M., Soledad M. C. M. Enzymatic
degradation of poly (3-hydroxybutyrate) by a commercial lipase //Polymer
degradation and stability. – 2012. – Т. 97. – №. 11. – С. 2473-2476;
15. Alonso M. J. et al. Biodegradable microspheres as controlled-release
tetanus toxoid delivery systems //Vaccine. – 1994. – Т. 12. – №. 4. – С. 299-306;
16. Anderson A. J., Dawes E. A. Occurrence, metabolism, metabolic role,
and industrial uses of bacterial polyhydroxyalkanoates //Microbiological reviews. –
1990. – Т. 54. – №. 4. – С. 450-472;
17. Anderson J. M., Shive M. S. Biodegradation and biocompatibility of PLA
and PLGA microspheres //Advanced drug delivery reviews. – 2012. – Т. 64. – С. 72-
82;
18. Andreas K. et al. Biodegradable insulin-loaded PLGA microspheres
fabricated by three different emulsification techniques: investigation for cartilage
tissue engineering //Acta biomaterialia. – 2011. – Т. 7. – №. 4. – С. 1485-1495;
19. Andreas K. et al. Biodegradable insulin-loaded PLGA microspheres
fabricated by three different emulsification techniques: investigation for cartilage
tissue engineering //Acta biomaterialia. – 2011. – Т. 7. – №. 4. – С. 1485-1495;
20. Andreeva N. V. et al. Culturing of mouse mesenchymal stem cells on
poly-3-hydroxybutyrate scaffolds //Bulletin of experimental biology and medicine. –
2015. – Т. 159. – №. 4. – С. 567;
21. Ashby R. D., Shi F., Gross R. A. Use of poly (ethylene glycol) to control
the end group structure and molecular weight of poly (3-hydroxybutyrate) formed by

126
Alcaligenes latus DSM 1122 //Tetrahedron. – 1997. – Т. 53. – №. 45. – С. 15209-
15223;
22. Ashby R., Solaiman D., Foglia T. Poly (ethylene glycol)-mediated molar
mass control of short-chain-and medium-chain-length poly (hydroxyalkanoates) from
Pseudomonas oleovorans //Applied microbiology and biotechnology. – 2002. – Т. 60.
– №. 1-2. – С. 154-159;
23. Atala A., Mooney D. J. (ed.). Synthetic biodegradable polymer scaffolds.
– Springer Science & Business Media, 1997;
24. Azioune A. et al. Synthesis and Characterization of Active Ester-
Functionalized Polypyrrole− Silica Nanoparticles: Application to the Covalent
Attachment of Proteins //Langmuir. – 2004. – Т. 20. – №. 8. – С. 3350-3356;
25. Bagrov D. V. et al. Amorphous and semicrystalline phases in ultrathin
films of poly (3-hydroxybutirate) //TechConnect World NTSI-Nanotech 2012
Proceedings. – 2012. – С. 602-605;
26. Baptist J.N., US Patent, 3044942.– 1962;
27. barbitone// J. Microencap., 1990, v. 7, p. 53-66;
28. Barham P. J. et al. Crystallization and morphology of a bacterial
thermoplastic: poly-3-hydroxybutyrate //Journal of Materials Science. – 1984. – Т. 19.
– №. 9. – С. 2781-2794;
29. Basmanav F. B., Kose G. T., Hasirci V. Sequential growth factor delivery
from complexed microspheres for bone tissue engineering //Biomaterials. – 2008. – Т.
29. – №. 31. – С. 4195-4204;
30. Batycky R. P. et al. A theoretical model of erosion and macromolecular
drug release from biodegrading microspheres //Journal of pharmaceutical sciences. –
1997. – Т. 86. – №. 12. – С. 1464-1477;
31. Bernd H. A. et al. Matrix-assisted in vitro refolding of Pseudomonas
aeruginosa class II polyhydroxyalkanoate synthase from inclusion bodies produced in
recombinant Escherichia coli //Biochemical Journal. – 2001. – Т. 358. – №. 1. – С.
263-268;

127
32. Bissery M. C., Valeriote F. A., Thies C. Therapeutic efficacy of CCNU-
loaded microspheres prepared from poly (D, L) lactide (PLA) or poly-B-
hydroxybutyrate (PHB) against Lewis lung (LL) carcinoma //Proceedings of the
American Association for Cancer Research. – PUBLIC LEDGER BLDG, SUITE 816,
150 S. INDEPENDENCE MALL W., PHILADELPHIA, PA 19106 : AMER ASSOC
CANCER RESEARCH, 1985. – Т. 26. – №. MAR. – С. 355-355;
33. Blanco-Prı eto M. J. et al. Importance of single or blended polymer types
for controlled in vitro release and plasma levels of a somatostatin analogue entrapped
in PLA/PLGA microspheres //Journal of Controlled Release. – 2004. – Т. 96. – №. 3.
– С. 437-448;
34. Bloembergen S. et al. Studies of composition and crystallinity of bacterial
poly (β-hydroxybutyrate-co-β-hydroxyvalerate) //Macromolecules. – 1986. – Т. 19. –
№. 11. – С. 2865-2871;
35. Bonartsev A. et al. Degradation of poly (3-hydroxybutyrate) and its
derivatives: characterization and kinetic behavior. – 2012;
36. Bonartsev A. P. et al. 3D-Scaffolds from Poly (3-hydroxybutyrate) Poly
(ethylene glycol) Copolymer for Tissue Engineering //Journal of Biomaterials and
Tissue Engineering. – 2016. – Т. 6. – №. 1. – С. 42-52;
37. Bonartsev A. P. et al. Biosynthesis of poly (3-hydroxybutyrate)
copolymers by Azotobacter chroococcum 7B: A precursor feeding strategy
//Preparative Biochemistry and Biotechnology. – 2016. – С. 1-12.,
38. Bonartsev A. P. et al. Biosynthesis, biodegradation, and application of
poly (3-hydroxybutyrate) and its copolymers-natural polyesters produced by
diazotrophic bacteria //Communicating Current Research and Educational Topics and
Trends in Applied Microbiology. – 2007. – Т. 1. – С. 295-307;
39. Bonartsev A. P. et al. Controlled release profiles of dipyridamole from
biodegradable microspheres on the base of poly (3-hydroxybutyrate) //Express
Polymer Letters. – 2007. – Т. 1. – №. 12. – С. 797-803;

128
40. Bonartsev A. P. et al. New poly (3-hydroxybutyrate)-based systems for
controlled release of dipyridamole and indomethacin //Applied biochemistry and
microbiology. – 2006. – Т. 42. – №. 6. – С. 625-630;
41. Bonartsev A. P. et al. Poly (3-hydroxybutyrate) and poly (3-
hydroxybutyrate)-based biopolymer systems //Biochemistry (Moscow) Supplement
Series B: Biomedical Chemistry. – 2011. – Т. 5. – №. 1. – С. 10-21;
42. Boontheekul T., Kong H. J., Mooney D. J. Controlling alginate gel
degradation utilizing partial oxidation and bimodal molecular weight distribution
//Biomaterials. – 2005. – Т. 26. – №. 15. – С. 2455-2465;
43. Borgquist P. et al. Simulation of the release from a multiparticulate
system validated by single pellet and dose release experiments //Journal of controlled
release. – 2004. – Т. 97. – №. 3. – С. 453-465;
44. Boskhomdzhiev A. P. et al. Biodegradation kinetics of poly (3-
hydroxybutyrate)-based biopolymer systems //Biochemistry (Moscow) Supplement
Series B: Biomedical Chemistry. – 2010. – Т. 4. – №. 2. – С. 177-183;
45. Bouhadir K. H. et al. Degradation of partially oxidized alginate and its
potential application for tissue engineering //Biotechnology progress. – 2001. – Т. 17.
– №. 5. – С. 945-950;
46. Bradford M. M. A rapid and sensitive method for the quantitation of
microgram quantities of protein utilizing the principle of protein-dye binding
//Analytical biochemistry. – 1976. – Т. 72. – №. 1-2. – С. 248-254;
47. Butler S. M., Tracy M. A., Tilton R. D. Adsorption of serum albumin to
thin films of poly (lactide-co-glycolide) //Journal of Controlled release. – 1999. – Т.
58. – №. 3. – С. 335-347.
48. Byrom D. Polymer synthesis by microorganisms: technology and
economics //Trends in Biotechnology. – 1987. – Т. 5. – №. 9. – С. 246-250;
49. Cadee J. A. et al. A comparative biocompatibility study of microspheres
based on crosslinked dextran or poly (lactic‐co‐glycolic) acid after subcutaneous
injection in rats //Journal of biomedical materials research. – 2001. – Т. 56. – №. 4. –
С. 600-609;

129
50. Canalis E. Effect of growth factors on bone cell replication and
differentiation //Clinical orthopaedics and related research. – 1985. – Т. 193. – С. 246-
263;
51. Caracciolo P. C. et al. Electrospinning of novel biodegradable poly (ester
urethane) s and poly (ester urethane urea) s for soft tissue-engineering applications
//Journal of Materials Science: Materials in Medicine. – 2009. – Т. 20. – №. 10. – С.
2129-2137;
52. Castellanos I. J., Griebenow K. Improved α-chymotrypsin stability upon
encapsulation in PLGA microspheres by solvent replacement //Pharmaceutical
research. – 2003. – Т. 20. – №. 11. – С. 1873-1880;
53. Chan R. T. H. et al. Application of polyethylene glycol to promote
cellular biocompatibility of polyhydroxybutyrate films //International Journal of
Polymer Science. – 2011. – Т. 2011;
54. Chan R. T. H. et al. Poly (ethylene glycol)‐modulated cellular
biocompatibility of polyhydroxyalkanoate films //Polymer International. – 2013. – Т.
62. – №. 6. – С. 884-892;
55. Chen C. et al. Biodegradable nanoparticles of amphiphilic triblock
copolymers based on poly (3-hydroxybutyrate) and poly (ethylene glycol) as drug
carriers //Biomaterials. – 2006. – Т. 27. – №. 27. – С. 4804-4814;
56. Chen C. T. et al. Coordinated changes of mitochondrial biogenesis and
antioxidant enzymes during osteogenic differentiation of human mesenchymal stem
cells //Stem Cells. – 2008. – Т. 26. – №. 4. – С. 960-968;
57. Chen F. M. et al. In vitro cellular responses to scaffolds containing two
microencapulated growth factors //Biomaterials. – 2009. – Т. 30. – №. 28. – С. 5215-
5224;
58. Chen G. Q., Wu Q. The application of polyhydroxyalkanoates as tissue
engineering materials //Biomaterials. – 2005. – Т. 26. – №. 33. – С. 6565-6578;

130
59. Chen L., Apte R. N., Cohen S. Characterization of PLGA microspheres
for the controlled delivery of IL-1α for tumor immunotherapy //Journal of controlled
release. – 1997. – Т. 43. – №. 2. – С. 261-272;
60. Chen R. R. et al. Integrated approach to designing growth factor delivery
systems //The FASEB Journal. – 2007. – Т. 21. – №. 14. – С. 3896-3903;
61. Chen R. R. et al. Spatio–temporal VEGF and PDGF delivery patterns
blood vessel formation and maturation //Pharmaceutical research. – 2007. – Т. 24. –
№. 2. – С. 258-264;
62. Chen R. R., Mooney D. J. Polymeric growth factor delivery strategies for
tissue engineering //Pharmaceutical research. – 2003. – Т. 20. – №. 8. – С. 1103-1112;
63. Choi D. H. et al. Fabrication of core–shell microcapsules using PLGA
and alginate for dual growth factor delivery system //Journal of Controlled Release. –
2010. – Т. 147. – №. 2. – С. 193-201;
64. Choi RS, Vacanti JP. In: Patrick C. W., Mikos A. G., McIntire L. V.
(ed.). Frontiers in tissue engineering. – Elsevier, 1998;
65. Ciardelli G. et al. Phagocytosis and biodegradation of short-chain poly
{(R)-3-hydroxybutyric acid} particles in macrophage cell line //Journal of Materials
Science: Materials in Medicine. – 1995. – Т. 6. – №. 12. – С. 725-730;
66. Clarotti G. et al. Modification of the biocompatible and haemocompatible
properties of polymer substrates by plasma-deposited fluorocarbon coatings
//Biomaterials. – 1992. – Т. 13. – №. 12. – С. 832-840;
67. Cleland J. L., Jones A. J. S. Stable formulations of recombinant human
growth hormone and interferon-γ for microencapsulation in biodegradable
mircospheres //Pharmaceutical research. – 1996. – Т. 13. – №. 10. – С. 1464-1475;
68. Cobntbekt J., Mabchessault R. H. Physical properties of poly-β-
hydroxybutyrate: IV. Conformational analysis and crystalline structure //Journal of
molecular biology. – 1972. – Т. 71. – №. 3. – С. 735-756;
69. Costantino H. R. et al. Protein spray-freeze drying. Effect of atomization
conditions on particle size and stability //Pharmaceutical research. – 2000. – Т. 17. –
№. 11. – С. 1374-1382;

131
70. Crotts G., Park T. G. Stability and release of bovine serum albumin
encapsulated within poly (D, L-lactide-co-glycolide) microparticles //Journal of
Controlled Release. – 1997. – Т. 44. – №. 2. – С. 123-134;
71. Dawes E. A., Senior P. J. The role and regulation of energy reserve
polymers in micro-organisms //Advances in microbial physiology. – 1973. – Т. 10. –
С. 135-266;
72. Dawson E. et al. Biomaterials for stem cell differentiation //Advanced
drug delivery reviews. – 2008. – Т. 60. – №. 2. – С. 215-228;
73. Deligianni D. D. et al. Effect of surface roughness of hydroxyapatite on
human bone marrow cell adhesion, proliferation, differentiation and detachment
strength //Biomaterials. – 2000. – Т. 22. – №. 1. – С. 87-96;
74. Deng Y. et al. Study on the three-dimensional proliferation of rabbit
articular cartilage-derived chondrocytes on polyhydroxyalkanoate scaffolds
//Biomaterials. – 2002. – Т. 23. – №. 20. – С. 4049-4056;
75. Doi Y. et al. Biodegradation of microbial polyesters in the marine
environment //Polymer degradation and stability. – 1992. – Т. 36. – №. 2. – С. 173-
177;
76. Doi Y., Kitamura S., Abe H. Microbial synthesis and characterization of
poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) // Macromolecules.– 1995.– Т. 28.–
С. 4822–4828;
77. Doi Y., Segawa A., Kunioka M. Biosynthesis and characterization of
poly (3-hydroxybutyrate-co-4-hydroxybutyrate) in Alcaligenes eutrophus
//International Journal of Biological Macromolecules. – 1990. – Т. 12. – №. 2. – С.
106-111;
78. Domb A. J., Turovsky L., Nudelman R. Chemical interactions between
drugs containing reactive amines with hydrolyzable insoluble biopolymers in aqueous
solutions //Pharmaceutical research. – 1994. – Т. 11. – №. 6. – С. 865-868;
79. Donbrow M. The relation of release profiles from ensembles to those of
individual microcapsules and the influence of types of batch heterogeneity on release

132
kinetics. In: Donbrow M. Microcapsules and nanoparticles in medicine and pharmacy.
– CRC press, 1991;
80. Dong Y., Feng S. S. Poly (D, L-lactide-co-glycolide)(PLGA)
nanoparticles prepared by high pressure homogenization for paclitaxel chemotherapy
//International journal of pharmaceutics. – 2007. – Т. 342. – №. 1. – С. 208-214;
81. Ehrbar M. et al. Cell-demanded liberation of VEGF121 from fibrin
implants induces local and controlled blood vessel growth //Circulation research. –
2004. – Т. 94. – №. 8. – С. 1124-1132;
82. Ehrbar M. et al. Enzymatic formation of modular cell-instructive fibrin
analogs for tissue engineering //Biomaterials. – 2007. – Т. 28. – №. 26. – С. 3856-
3866;
83. Englert C. et al. Bonding of articular cartilage using a combination of
biochemical degradation and surface cross-linking //Arthritis research & therapy. –
2007. – Т. 9. – №. 3. – С. R47;
84. Feng S., Huang G. Effects of emulsifiers on the controlled release of
paclitaxel (Taxol®) from nanospheres of biodegradable polymers //Journal of
Controlled Release. – 2001. – Т. 71. – №. 1. – С. 53-69;
85. Findlay R. H., White D. C. Polymeric beta-hydroxyalkanoates from
environmental samples and Bacillus megaterium //Applied and Environmental
Microbiology. – 1983. – Т. 45. – №. 1. – С. 71-78;
86. Forsyth W. G. C., Hayward A. C., Roberts J. B. Occurrence of poly-β-
hydroxybutyric acid in aerobic gram-negative bacteria //Nature. – 1958. – Т. 182. –
№. 4638. – С. 800-801;
87. Fournier E. et al. Biocompatibility of implantable synthetic polymeric
drug carriers: focus on brain biocompatibility //Biomaterials. – 2003. – Т. 24. – №. 19.
– С. 3311-3331;
88. Freytag T. et al. Improvement of the encapsulation efficiency of
oligonucleotide-containing biodegradable microspheres //Journal of Controlled
Release. – 2000. – Т. 69. – №. 1. – С. 197-207;

133
89. Gangrade N., Price J. C. Poly (hydroxybutyrate-hydroxyvalerate)
microspheres containing progesterone: preparation, morphology and release properties
//Journal of microencapsulation. – 1991. – Т. 8. – №. 2. – С. 185-202;
90. Geng Y. et al. Formulating erythropoietin-loaded sustained-release PLGA
microspheres without protein aggregation //Journal of Controlled Release. – 2008. –
Т. 130. – №. 3. – С. 259-265;
91. Gerngross T. U., Martin D. P. Enzyme-catalyzed synthesis of poly {(R)-
(-)-3-hydroxybutyrate}: formation of macroscopic granules in vitro //Proceedings of
the National Academy of Sciences. – 1995. – Т. 92. – №. 14. – С. 6279-6283;
92. Gilkes N. R. et al. Domains in microbial beta-1, 4-glycanases: sequence
conservation, function, and enzyme families //Microbiological reviews. – 1991. – Т.
55. – №. 2. – С. 303-315;
93. Gogolewski S. et al. Tissue response and in vivo degradation of selected
polyhydroxyacids: Polylactides (PLA), poly (3‐hydroxybutyrate)(PHB), and poly
(3‐hydroxybutyrate-co-3-hydroxyvalerate)(PHB/VA) //Journal of Biomedical
Materials Research Part A. – 1993. – Т. 27. – №. 9. – С. 1135-1148;
94. Gombotz, W. R., Pettit D. K. Biodegradable polymers for protein and
peptide drug delivery// Bioconjugate chemistry. – 1995. – Т 6. – № 4. – С. 332-351;
95. Gould P. L., Holland S. J., Tighe B. J. Polymers for biodegradable
medical devices. IV. Hydroxybutyrate-valerate copolymers as non-disintegrating
matrices for controlled-release oral dosage forms //International journal of
pharmaceutics. – 1987. – Т. 38. – №. 1-3. – С. 231-237;
96. Griebenow K., Klibanov A. M. Can conformational changes be
responsible for solvent and excipient effects on the catalytic behavior of subtilisin
Carlsberg in organic solvents? //Biotechnology and Bioengineering. – 1997. – Т. 53. –
№. 4. – С. 351-362;
97. Gupte A., Ciftci K. Formulation and characterization of Paclitaxel, 5-FU
and Paclitaxel+ 5-FU microspheres //International journal of pharmaceutics. – 2004. –
Т. 276. – №. 1. – С. 93-106;

134
98. Gursel I. et al. In vitro antibiotic release from poly (3-hydroxybutyrate-
co-3-hydroxyvalerate) rods //Journal of microencapsulation. – 2002. – Т. 19. – №. 2. –
С. 153-164;
99. Gürsel İ. et al. In vivo application of biodegradable controlled antibiotic
release systems for the treatment of implant-related osteomyelitis //Biomaterials. –
2000. – Т. 22. – №. 1. – С. 73-80;
100. Harris L. D., Kim B. S., Mooney D. J. Open pore biodegradable matrices
formed with gas foaming. – 1998;
101. Heng D. et al. The nano spray dryer B-90 //Expert opinion on drug
delivery. – 2011. – Т. 8. – №. 7. – С. 965-972;
102. Hoerstrup S. P. et al. Functional living trileaflet heart valves grown in
vitro //Circulation. – 2000. – Т. 102. – №. suppl 3. – С. Iii-44-Iii-49;
103. Hoerstrup S. P. et al. Tissue engineering of small caliber vascular grafts
//European journal of cardio-thoracic surgery. – 2001. – Т. 20. – №. 1. – С. 164-169;
104. Holmes P. A. Biologically produced (R)-3-hydroxy-alkanoate polymers
and copolymers //Developments in crystalline polymers. – Springer Netherlands,
1988. – С. 1-65;
105. Holmes PA, Wright LF, Colins SH. Β-hydroxybutyrate polymers.
European patent application. EP 52, 459;
106. Holmes PA. In: Bassett D. C. (ed.). Developments in crystalline
polymers. – London : Applied Science Publishers, 1982. – Т. 1. – С. 151;
107. Hu Y. J. et al. Biocompatibility of poly (3-hydroxybutyrate-co-3-
hydroxyvalerate-co-3-hydroxyhexanoate) with bone marrow mesenchymal stem cells
//Acta biomaterialia. – 2009. – Т. 5. – №. 4. – С. 1115-1125;
108. Iadevaia S., Mantzaris N. V. Synthesis of PHBV block copolymers
driven by an oscillatory genetic network //Journal of biotechnology. – 2007. – Т. 128.
– №. 3. – С. 615-637;
109. Ike O. et al. Controlled cisplatin delivery system using poly (d, l-lactic
acid //Biomaterials. – 1992. – Т. 13. – №. 4. – С. 230-234.;

135
110. Im J. Y. et al. Bone regeneration of mouse critical-sized calvarial defects
with human mesenchymal stem cells in scaffold //Laboratory animal research. – 2013.
– Т. 29. – №. 4. – С. 196-203;
111. Iordanskii A. L. et al. Desorption of human serum albumin and human
fibrinogen from the poly (3-hydroxybutyrate) surface //Journal of applied polymer
science. – 1999. – Т. 74. – №. 3. – С. 595-600;
112. Ishikawa K. Flexible member for use as a medical bag: пат. 5480394
США. – 1996;
113. Ito F., Fujimori H., Makino K. Factors affecting the loading efficiency of
water-soluble drugs in PLGA microspheres //Colloids and Surfaces B: Biointerfaces.
– 2008. – Т. 61. – №. 1. – С. 25-29;
114. Ito Y. Covalently immobilized biosignal molecule materials for tissue
engineering //Soft matter. – 2008. – Т. 4. – №. 1. – С. 46-56;
115. Jaganathan K. S. et al. Development of a single dose tetanus toxoid
formulation based on polymeric microspheres: a comparative study of poly (d, l-
lactic-co-glycolic acid) versus chitosan microspheres //International journal of
pharmaceutics. – 2005. – Т. 294. – №. 1. – С. 23-32;
116. Jain R. A. The manufacturing techniques of various drug loaded
biodegradable poly (lactide-co-glycolide)(PLGA) devices //Biomaterials. – 2000. – Т.
21. – №. 23. – С. 2475-2490;
117. Jalil R., Nixon J. R. Microencapsulation using poly (L-lactic acid) IV:
Release properties of microcapsules containing phenobarbitone //Journal of
microencapsulation. – 1990. – Т. 7. – №. 1. – С. 53-66;
118. Jamie Tsung M., Burgess D. J. Preparation and characterization of gelatin
surface modified PLGA microspheres //The AAPS Journal. – 2001. – Т. 3. – №. 2. –
С. 14-24;
119. Jendrossek D., Müller B., Schlegel H. G. Cloning and characterization of
the poly (hydroxyalkanoic acid)-depolymerase gene locus, phaZ1, of Pseudomonas
lemoignei and its gene product //European journal of biochemistry/FEBS. – 1993. – Т.
218. – №. 2. – С. 701-710;

136
120. Ji G. Z., Wei X., Chen G. Q. Growth of human umbilical cord Wharton's
jelly-derived mesenchymal stem cells on the terpolyester poly (3-hydroxybutyrate-co-
3-hydroxyvalerate-co-3-hydroxyhexanoate) //Journal of Biomaterials Science,
Polymer Edition. – 2009. – Т. 20. – №. 3. – С. 325-339;
121. Johnson O. F. L. et al. A month–long effect from a single injection of
microencapsulated human growth hormone //Nature medicine. – 1996. – Т. 2. – №. 7.
– С. 795-799;
122. Juni K., Nakano M. Poly (hydroxy acids) in drug delivery //Critical
reviews in therapeutic drug carrier systems. – 1986. – Т. 3. – №. 3. – С. 209-232;
123. Kandori K. et al. Preparation of calcium hydroxyapatite nanoparticles
using microreactor and their characteristics of protein adsorption //The Journal of
Physical Chemistry B. – 2010. – Т. 115. – №. 4. – С. 653-659;
124. Kanesawa Y. et al. Hydrolytic degradation of microbial poly
(hydroxyalkanoates) //Macromolecular Rapid Communications. – 1989. – Т. 10. – №.
5. – С. 227-230;
125. Kawaguchi T. et al. Control of drug release with a combination of
prodrug and polymer matrix: Antitumor activity and release profiles of 2′, 3′-diacyl-5-
fluoro-2′-deoxyuridine from poly (3-hydroxybutyrate) microspheres //Journal of
pharmaceutical sciences. – 1992. – Т. 81. – №. 6. – С. 508-512;
126. Kellett L. E. et al. Xylanase B and an arabinofuranosidase from
Pseudomonas fluorescens subsp. cellulosa contain identical cellulose-binding domains
and are encoded by adjacent genes //Biochemical Journal. – 1990. – Т. 272. – №. 2. –
С. 369-376;
127. Kim H. K., Park T. G. Microencapsulation of human growth hormone
within biodegradable polyester microspheres: protein aggregation stability and
incomplete release mechanism //Biotechnology and bioengineering. – 1999. – Т. 65. –
№. 6. – С. 659-667;
128. Knubovets T., Osterhout J. J., Klibanov A. M. Structure of lysozyme
dissolved in neat organic solvents as assessed by NMR and CD spectroscopies
//Biotechnology and bioengineering. – 1999. – Т. 63. – №. 2. – С. 242-248;

137
129. Koosha F., Muller R. H., Davis S. S. A continuous flow system for in
vitro evaluation of drug-loaded biodegradable colloidal barriers //J Pharm Pharmacol.
– 1988. – Т. 40. – С. 131P;
130. Korsatko W. et al. poly-d-(-)-3-hydroxybutyric acid-a biodegradable
carrier for long-term medication dosage. 2. The biodegradation in animal organism
and invitro-invivo correlation of the liberation of pharmaceuticals from parenteral
matrix retard tablets //Pharmazeutische Industrie. – 1983. – Т. 45. – №. 10. – С. 1004-
1007;
131. Krishnamachari Y., Madan P., Lin S. Development of pH-and time-
dependent oral microparticles to optimize budesonide delivery to ileum and colon
//International journal of pharmaceutics. – 2007. – Т. 338. – №. 1. – С. 238-247;
132. Kubota M., Nakano M., Juni K. Mechanism of enhancement of the
release rate of aclarubicin from poly-beta-hydroxybutyric acid microspheres by fatty
acid esters //Chemical & pharmaceutical bulletin. – 1988. – Т. 36. – №. 1. – С. 333-
337;
133. Kuhl P. R., Griffith-Cima L. G. Tethered epidermal growth factor as a
paradigm for growth factor–induced stimulation from the solid phase //Nature
medicine. – 1996. – Т. 2. – №. 9. – С. 1022-1027;
134. Kumar M. Nano and microparticles as controlled drug delivery devices
//J. Pharm. Pharm. Sci. – 2000. – Т. 3. – №. 2. – С. 234-258;
135. Kundrot C. E., Richards F. M. Crystal structure of hen egg-white
lysozyme at a hydrostatic pressure of 1000 atmospheres //Journal of molecular
biology. – 1987. – Т. 193. – №. 1. – С. 157-170;
136. Kwon Y. M. et al. In situ study of insulin aggregation induced by water-
organic solvent interface //Pharmaceutical research. – 2001. – Т. 18. – №. 12. – С.
1754-1759;
137. Laemmli U. K. Glycine-SDS-PAGE for separation of proteins //Nature. –
1970. – Т. 227. – №. 5;
138. Lageveen R. G. et al. Formation of polyesters by Pseudomonas
oleovorans: effect of substrates on formation and composition of poly-(R)-3-

138
hydroxyalkanoates and poly-(R)-3-hydroxyalkenoates //Applied and Environmental
Microbiology. – 1988. – Т. 54. – №. 12. – С. 2924-2932;
139. Lai M. C., Topp E. M. Solid‐state chemical stability of proteins and
peptides //Journal of pharmaceutical sciences. – 1999. – Т. 88. – №. 5. – С. 489-500;
140. Lanza R., Langer R., Vacanti J. P. (ed.). Principles of tissue engineering.
– Academic press, 2011;
141. Leach W. T. et al. Encapsulation of protein nanoparticles into uniform-
sized microspheres formed in a spinning oil film //AAPS PharmSciTech. – 2005. – Т.
6. – №. 4. – С. E605-E617;
142. Lee E. S. et al. Glycol chitosan as a stabilizer for protein encapsulated
into poly (lactide-co-glycolide) microparticle //International journal of pharmaceutics.
– 2007. – Т. 338. – №. 1. – С. 310-316;
143. Lee S. Y. Bacterial polyhydroxyalkanoates //Biotechnology and
bioengineering. – 1996. – Т. 49. – №. 1. – С. 1-14;
144. Lee T. H., Wang J., Wang C. H. Double-walled microspheres for the
sustained release of a highly water soluble drug: characterization and irradiation
studies //Journal of controlled release. – 2002. – Т. 83. – №. 3. – С. 437-452;
145. Lemoigne M. Produit de déshydratation et de polymérisation de l’acide β-
oxybutyrique //Bull Soc Chim Biol. – 1926. – Т. 8. – С. 770-782;
146. Li J. et al. Self-assembled supramolecular hydrogels formed by
biodegradable PEO–PHB–PEO triblock copolymers and α-cyclodextrin for controlled
drug delivery //Biomaterials. – 2006. – Т. 27. – №. 22. – С. 4132-4140;
147. Li L. et al. Controlled dual delivery of BMP-2 and dexamethasone by
nanoparticle-embedded electrospun nanofibers for the efficient repair of critical-sized
rat calvarial defect //Biomaterials. – 2015. – Т. 37. – С. 218-229;
148. Li S. D. et al. Thermal degradation of poly (3‐hydroxybutyrate) and
poly (3‐hydroxybutyrate‐co‐3‐hydroxyvalerate) as studied by TG, TG–FTIR,
and Py–GC/MS //Journal of applied polymer science. – 2003. – Т. 89. – №. 6. – С.
1530-1536;

139
149. Liao S. et al. A three-layered nano-carbonated
hydroxyapatite/collagen/PLGA composite membrane for guided tissue regeneration
//Biomaterials. – 2005. – Т. 26. – №. 36. – С. 7564-7571;
150. Liu Y. et al. Injectable tissue-engineered bone composed of human
adipose-derived stromal cells and platelet-rich plasma //Biomaterials. – 2008. – Т. 29.
– №. 23. – С. 3338-3345;
151. Love M. et al. Solution structure of CCL21 and identification of a
putative CCR7 binding site //Biochemistry. – 2012. – Т. 51. – №. 3. – С. 733-735;
152. Lu W., Park T. G. Protein release from poly (lactic-co-glycolic acid)
microspheres: protein stability problems //PDA Journal of Pharmaceutical Science and
Technology. – 1995. – Т. 49. – №. 1. – С. 13-19;
153. Maniatopoulos C., Sodek J., Melcher A. H. Bone formation in vitro by
stromal cells obtained from bone marrow of young adult rats //Cell and tissue
research. – 1988. – Т. 254. – №. 2. – С. 317-330;
154. Mann B. K., Schmedlen R. H., West J. L. Tethered-TGF-β increases
extracellular matrix production of vascular smooth muscle cells //Biomaterials. –
2001. – Т. 22. – №. 5. – С. 439-444;
155. Martin D. P., Williams S. F. Medical applications of poly-4-
hydroxybutyrate: a strong flexible absorbable biomaterial //Biochemical Engineering
Journal. – 2003. – Т. 16. – №. 2. – С. 97-105;
156. Matsuo K., Yonehara R., Gekko K. Secondary-structure analysis of
proteins by vacuum-ultraviolet circular dichroism spectroscopy //Journal of
biochemistry. – 2004. – Т. 135. – №. 3. – С. 405-411;
157. Mayer M. et al. Maxillary alveolar cleft repair in dogs using recombinant
human bone morphogenetic protein-2 and a polymer carrier //Plastic and
reconstructive surgery. – 1996. – Т. 98. – №. 2. – С. 247-259;
158. Maysinger D., Filipović-Grčić J., Alebić-Kolbah T. Preparation,
characterization and release of microencapsulated bromodeoxyuridine //Life sciences.
– 1994. – Т. 54. – №. 1. – С. 27-34;

140
159. McKenzie H. A., White F. H. Determination of lysozyme activity at low
levels with emphasis on the milk enzyme //Analytical biochemistry. – 1986. – Т. 157.
– №. 2. – С. 367-374;
160. McKenzie H. A., White F. H. Determination of lysozyme activity at low
levels with emphasis on the milk enzyme //Analytical biochemistry. – 1986. – Т. 157.
– №. 2. – С. 367-374;
161. Meinel L. et al. Stabilizing insulin-like growth factor-I in poly (D, L-
lactide-co-glycolide) microspheres //Journal of controlled release. – 2001. – Т. 70. –
№. 1. – С. 193-202;
162. Mergaert J. et al. In situ biodegradation of poly (3-hydroxybutyrate) and
poly (3-hydroxybutyrate-co-3-hydroxyvalerate) in natural waters //Canadian Journal
of Microbiology. – 1995. – Т. 41. – №. 13. – С. 154-159;
163. Mergaert J. et al. Microbial degradation of poly (3-hydroxybutyrate) and
poly (3-hydroxybutyrate-co-3-hydroxyvalerate) in soils //Applied and environmental
microbiology. – 1993. – Т. 59. – №. 10. – С. 3233-3238;
164. Misra S. K. et al. Polyhydroxyalkanoate (PHA)/inorganic phase
composites for tissue engineering applications //Biomacromolecules. – 2006. – Т. 7. –
№. 8. – С. 2249-2258;
165. Moebus K., Siepmann J., Bodmeier R. Alginate–poloxamer
microparticles for controlled drug delivery to mucosal tissue //European Journal of
Pharmaceutics and Biopharmaceutics. – 2009. – Т. 72. – №. 1. – С. 42-53;
166. Mooney D. J. et al. Novel approach to fabricate porous sponges of poly
(D, L-lactic-co-glycolic acid) without the use of organic solvents //Biomaterials. –
1996. – Т. 17. – №. 14. – С. 1417-1422;
167. Mosmann T. Rapid colorimetric assay for cellular growth and survival:
application to proliferation and cytotoxicity assays //Journal of immunological
methods. – 1983. – Т. 65. – №. 1-2. – С. 55-63;
168. Mygind T. et al. Mesenchymal stem cell ingrowth and differentiation on
coralline hydroxyapatite scaffolds //Biomaterials. – 2007. – Т. 28. – №. 6. – С. 1036-
1047;

141
169. Myshkina V. L. et al. Biosynthesis of poly (3-hydroxybutyrate-co-3-
hydroxyvalerate) copolymer by Azotobacter chroococcum strain 7B //Applied
biochemistry and microbiology. – 2010. – Т. 46. – №. 3. – С. 289-296;
170. Narasimhan B. Accurate models in controlled drug delivery systems. In:
Wise DL, Brannon-Peppas L, Klibanow AM, Mikos AG, Peppas NA, Trantolo DJ,
Wnek GE, Yaszemski MJ, eds. Handbook of Pharmaceutical Controlled Release
Technology. New York: Marcel Dekker, 2000:155–181;
171. Ni J., Wang M. In vitro evaluation of hydroxyapatite reinforced
polyhydroxybutyrate composite //Materials Science and Engineering: C. – 2002. – Т.
20. – №. 1. – С. 101-109;
172. Nisisako T., Torii T. Microfluidic large-scale integration on a chip for
mass production of monodisperse droplets and particles //Lab on a Chip. – 2008. – Т.
8. – №. 2. – С. 287-293;
173. Nobes G. A. R., Maysinger D., Marchessault R. H.
Polyhydroxyalkanoates: materials for delivery systems //Drug Delivery. – 1998. – Т.
5. – №. 3. – С. 167-177;
174. Okamura K, Marchessault RH. X-ray structure of poly-b-
hydroxybutyrate. In: Ramachandran GM, editor. Conformation of biopolymers. –Т. 2.
New York: Academic Press, 1967. p. 709-720;
175. Ol’khov A. A. et al. Electrospinning of biodegradable poly-3-
hydroxybutyrate. Effect of the characteristics of the polymer solution //Russian
Journal of Physical Chemistry B. – 2016. – Т. 10. – №. 5. – С. 830-838;
176. Opitz F. et al. Tissue engineering of ovine aortic blood vessel substitutes
using applied shear stress and enzymatically derived vascular smooth muscle cells
//Annals of biomedical engineering. – 2004. – Т. 32. – №. 2. – С. 212-222;
177. Orts W. J. et al. Poly (hydroxyalkanoates): biorefinery polymers with a
whole range of applications. The work of Robert H. Marchessault //Canadian Journal
of Chemistry. – 2008. – Т. 86. – №. 6. – С. 628-640;

142
178. Pai S. S., Tilton R. D., Przybycien T. M. Poly (ethylene glycol)-modified
proteins: implications for poly (lactide-co-glycolide)-based microsphere delivery
//The AAPS journal. – 2009. – Т. 11. – №. 1. – С. 88-98;
179. Park J. S. et al. Osteogenic differentiation of human mesenchymal stem
cells using RGD-modified BMP-2 coated microspheres //Biomaterials. – 2010. – Т.
31. – №. 24. – С. 6239-6248;
180. Park T. G., Lee H. Y., Nam Y. S. A new preparation method for protein
loaded poly (D, L-lactic-co-glycolic acid) microspheres and protein release
mechanism study //Journal of Controlled Release. – 1998. – Т. 55. – №. 2. – С. 181-
191;
181. Pellegrini L. et al. Crystal structure of fibroblast growth factor receptor
ectodomain bound to ligand and heparin //Nature. – 2000. – Т. 407. – №. 6807. – С.
1029-1034;
182. Peoples O. P., Sinskey A. J. Poly-beta-hydroxybutyrate biosynthesis in
Alcaligenes eutrophus H16. Characterization of the genes encoding beta-ketothiolase
and acetoacetyl-CoA reductase //Journal of Biological Chemistry. – 1989. – Т. 264. –
№. 26. – С. 15293-15297;
183. Pepper M. S. et al. Potent synergism between vascular endothelial growth
factor and basic fibroblast growth factor in the induction of angiogenesis in vitro
//Biochemical and biophysical research communications. – 1992. – Т. 189. – №. 2. –
С. 824-831;
184. Perego G., Cella G. D., Bastioli C. Effect of molecular weight and
crystallinity on poly (lactic acid) mechanical properties //Journal of Applied Polymer
Science. – 1996. – Т. 59. – №. 1. – С. 37-43;
185. Perets A. et al. Enhancing the vascularization of three‐dimensional
porous alginate scaffolds by incorporating controlled release basic fibroblast growth
factor microspheres //Journal of Biomedical Materials Research Part A. – 2003. – Т.
65. – №. 4. – С. 489-497;
186. Piskin E. Biodegradable polymers as biomaterials //Journal of
Biomaterials Science, Polymer Edition. – 1995. – Т. 6. – №. 9. – С. 775-795;

143
187. Ravenelle F., Marchessault R. H. One-step synthesis of amphiphilic
diblock copolymers from bacterial poly (R-3-hydroxybutyric acid)
//Biomacromolecules. – 2002. – Т. 3. – №. 5. – С. 1057-1064;
188. Rehm B. H. A. et al. Molecular characterization of the poly (3-
hydroxybutyrate)(PHB) synthase from Ralstonia eutropha: in vitro evolution, site-
specific mutagenesis and development of a PHB synthase protein model //Biochimica
et Biophysica Acta (BBA)-Protein Structure and Molecular Enzymology. – 2002. – Т.
1594. – №. 1. – С. 178-190;
189. Renstad R., Karlsson S., Albertsson A. C. The influence of processing
conditions on the properties and the degradation of poly (3-hydroxybutyrate-co-3-
hydroxyvalerate) //Macromolecular Symposia. – Hüthig & Wepf Verlag, 1998. – Т.
127. – №. 1. – С. 241-249;
190. Reusch R. N. Poly-β-hydroxybutyrate/calcium polyphosphate complexes
in eukaryotic membranes //Experimental Biology and Medicine. – 1989. – Т. 191. –
№. 4. – С. 377-381;
191. Reusch R. N., Sparrow A. W., Gardiner J. Transport of poly-β-
hydroxybutyrate in human plasma //Biochimica et Biophysica Acta (BBA)-Lipids and
Lipid Metabolism. – 1992. – Т. 1123. – №. 1. – С. 33-40;
192. Richardson T. P. et al. Polymeric system for dual growth factor delivery
//Nature biotechnology. – 2001. – Т. 19. – №. 11. – С. 1029-1034;
193. Rocha F. G. et al. The effect of sustained delivery of vascular endothelial
growth factor on angiogenesis in tissue-engineered intestine //Biomaterials. – 2008. –
Т. 29. – №. 19. – С. 2884-2890;
194. Rohanizadeh R., Chung K. Hydroxyapatite as a carrier for bone
morphogenetic protein //Journal of Oral Implantology. – 2011. – Т. 37. – №. 6. – С.
659-672;
195. Sachse A. et al. Osteointegration of hydroxyapatite-titanium implants
coated with nonglycosylated recombinant human bone morphogenetic protein-2
(BMP-2) in aged sheep //Bone. – 2005. – Т. 37. – №. 5. – С. 699-710;

144
196. Sah H. Protein instability toward organic solvent/water emulsification:
implications for protein microencapsulation into microspheres //PDA Journal of
Pharmaceutical Science and Technology. – 1999. – Т. 53. – №. 1. – С. 3-10;
197. Saha S. P., Patra A., Paul A. K. Incorporation of polyethylene glycol in
polyhydroxyalkanoic acids accumulated by Azotobacter chroococcum MAL-201
//Journal of Industrial Microbiology and Biotechnology. – 2006. – Т. 33. – №. 5. – С.
377-383;
198. Saito T. et al. Cloning, nucleotide sequence, and expression in
Escherichia coli of the gene for poly (3-hydroxybutyrate) depolymerase from
Alcaligenes faecalis //Journal of bacteriology. – 1989. – Т. 171. – №. 1. – С. 184-189;
199. Saitoh H. et al. Effect of polylactic acid on osteoinduction of
demineralized bone: preliminary study of the usefulness of polylactic acid as a carrier
of bone morphogenetic protein //Journal of oral rehabilitation. – 1994. – Т. 21. – №. 4.
– С. 431-438;
200. Sandor M. et al. Effect of lecithin and MgCO 3 as additives on the
enzymatic activity of carbonic anhydrase encapsulated in poly (lactide-co-
glycolide)(PLGA) microspheres //Biochimica et Biophysica Acta (BBA)-General
Subjects. – 2002. – Т. 1570. – №. 1. – С. 63-74;
201. Santos H. A. et al. In vitro cytotoxicity of porous silicon microparticles:
effect of the particle concentration, surface chemistry and size //Acta biomaterialia. –
2010. – Т. 6. – №. 7. – С. 2721-2731;
202. Scandola M. Polymer blends based on bacterial poly (3-hydroxybutyrate)
//Canadian journal of microbiology. – 1995. – Т. 41. – №. 13. – С. 310-315.);
203. Scheufler C., Sebald W., Hülsmeyer M. Crystal structure of human bone
morphogenetic protein-2 at 2.7 Å resolution //Journal of molecular biology. – 1999. –
Т. 287. – №. 1. – С. 103-115;
204. Schubert P., Steinbüchel A., Schlegel H. G. Cloning of the Alcaligenes
eutrophus genes for synthesis of poly-beta-hydroxybutyric acid (PHB) and synthesis
of PHB in Escherichia coli //Journal of Bacteriology. – 1988. – Т. 170. – №. 12. – С.
5837-5847;

145
205. Schugens C. et al. Effect of the emulsion stability on the morphology and
porosity of semicrystalline poly l-lactide microparticles prepared by w/o/w double
emulsion-evaporation //Journal of Controlled Release. – 1994. – Т. 32. – №. 2. – С.
161-176;
206. Scott R. C. et al. Targeting VEGF-encapsulated immunoliposomes to MI
heart improves vascularity and cardiac function //The FASEB Journal. – 2009. – Т.
23. – №. 10. – С. 3361-3367;
207. Selcan Gungor-Ozkerim P. et al. Incorporation of growth factor loaded
microspheres into polymeric electrospun nanofibers for tissue engineering
applications //Journal of Biomedical Materials Research Part A. – 2014. – Т. 102. –
№. 6. – С. 1897-1908;
208. Sendil D. et al. Antibiotic release from biodegradable PHBV
microparticles //Journal of Controlled Release. – 1999. – Т. 59. – №. 2. – С. 207-217;
209. Sevastianov V. I. et al. Production of purified polyhydroxyalkanoates
(PHAs) for applications in contact with blood //Journal of Biomaterials Science,
Polymer Edition. – 2003. – Т. 14. – №. 10. – С. 1029-1042;
210. Shah M. et al. Amorphous amphiphilic P (3HV-co-4HB)-b-mPEG block
copolymer synthesized from bacterial copolyester via melt transesterification:
nanoparticle preparation, cisplatin-loading for cancer therapy and in vitro evaluation
//European Journal of Pharmaceutics and Biopharmaceutics. – 2012. – Т. 80. – №. 3.
– С. 518-527;
211. Sheridan M. H. et al. Bioabsorbable polymer scaffolds for tissue
engineering capable of sustained growth factor delivery //Journal of Controlled
Release. – 2000. – Т. 64. – №. 1. – С. 91-102;
212. Shi F., Ashby R., Gross R. A. Use of poly (ethylene glycol) s to regulate
poly (3-hydroxybutyrate) molecular weight during Alcaligenes eutrophus cultivations
//Macromolecules. – 1996. – Т. 29. – №. 24. – С. 7753-7758;
213. Shinomiya M. et al. Cloning of the gene for poly (3-hydroxybutyric acid)
depolymerase of Comamonas testosteroni and functional analysis of its substrate-
binding domain //FEMS microbiology letters. – 1997. – Т. 154. – №. 1. – С. 89-94;

146
214. Shiraki M., Endo T., Saito T. Fermentative production of (R)-(−)-3-
hydroxybutyrate using 3-hydroxybutyrate dehydrogenase null mutant of Ralstonia
eutropha and recombinant Escherichia coli //Journal of bioscience and bioengineering.
– 2006. – Т. 102. – №. 6. – С. 529-534;
215. Shishatskaya E. I. et al. Evaluation of antitumor activity of rubomycin
deposited in absorbable polymeric microparticles //Bulletin of experimental biology
and medicine. – 2008. – Т. 145. – №. 3. – С. 358-361;
216. Shishatskaya E. I., Khlusov I. A., Volova T. G. A hybrid PHB–
hydroxyapatite composite for biomedical application: production, in vitro and in vivo
investigation //Journal of Biomaterials Science, Polymer Edition. – 2006. – Т. 17. –
№. 5. – С. 481-498;
217. Shishatskaya E. I., Volova T. G. A comparative investigation of
biodegradable polyhydroxyalkanoate films as matrices for in vitro cell cultures
//Journal of Materials Science: Materials in Medicine. – 2004. – Т. 15. – №. 8. – С.
915-923;
218. Shrivastav A., Kim H. Y., Kim Y. R. Advances in the applications of
polyhydroxyalkanoate nanoparticles for novel drug delivery system //BioMed research
international. – 2013. – Т. 2013;
219. Shum-Tim D. et al. Tissue engineering of autologous aorta using a new
biodegradable polymer //The Annals of thoracic surgery. – 1999. – Т. 68. – №. 6. – С.
2298-2304;
220. Siepmann J. et al. Effect of the size of biodegradable microparticles on
drug release: experiment and theory //Journal of Controlled Release. – 2004. – Т. 96. –
№. 1. – С. 123-134;
221. Simon-Colin C. et al. Biosynthesis of medium chain length poly (3-
hydroxyalkanoates)(mcl PHAs) from cosmetic co-products by Pseudomonas
raguenesii sp. nov., isolated from Tetiaroa, French Polynesia //Bioresource
technology. – 2009. – Т. 100. – №. 23. – С. 6033-6039;
222. Slater S. C., Voige W. H., Dennis D. E. Cloning and expression in
Escherichia coli of the Alcaligenes eutrophus H16 poly-beta-hydroxybutyrate

147
biosynthetic pathway //Journal of bacteriology. – 1988. – Т. 170. – №. 10. – С. 4431-
4436;
223. Slater S. et al. Multiple β-ketothiolases mediate poly (β-
hydroxyalkanoate) copolymer synthesis in Ralstonia eutropha //Journal of
bacteriology. – 1998. – Т. 180. – №. 8. – С. 1979-1987;
224. Sodian R. et al. Evaluation of biodegradable, three-dimensional matrices
for tissue engineering of heart valves //Asaio Journal. – 2000. – Т. 46. – №. 1. – С.
107-110;
225. Sodian R. et al. Evaluation of biodegradable, three-dimensional matrices
for tissue engineering of heart valves //Asaio Journal. – 2000. – Т. 46. – №. 1. – С.
107-110;
226. Steinbüchel A. Polyhydroxyalkanoic acids //Biomaterials. – Palgrave
Macmillan UK, 1991. – С. 123-213;
227. Suslick K. S., Hammerton D. A., Cline R. E. Sonochemical hot spot
//Journal of the American Chemical Society. – 1986. – Т. 108. – №. 18. – С. 5641-
5642;
228. Thomson RC, Yaszemski MJ, Mikos AG. In: Lanza R., Langer R.,
Vacanti J. P. (ed.). Principles of tissue engineering. – Academic press, 2011. – С.:261;
229. Tian J., Sinskey A. J., Stubbe J. A. Kinetic studies of
polyhydroxybutyrate granule formation in Wautersia eutropha H16 by transmission
electron microscopy //Journal of bacteriology. – 2005. – Т. 187. – №. 11. – С. 3814-
3824;
230. Tobío M. et al. A novel system based on a poloxamer/PLGA blend as a
tetanus toxoid delivery vehicle //Pharmaceutical research. – 1999. – Т. 16. – №. 5. –
С. 682-688;
231. Tran V. T. et al. Protein-loaded PLGA–PEG–PLGA microspheres: a tool
for cell therapy //European Journal of Pharmaceutical Sciences. – 2012. – Т. 45. – №.
1. – С. 128-137;
232. Tsuge T. et al. Molecular cloning of two (R)-specific enoyl-CoA
hydratase genes from Pseudomonas aeruginosa and their use for

148
polyhydroxyalkanoate synthesis //FEMS microbiology letters. – 2000. – Т. 184. – №.
2. – С. 193-198;
233. Turk A. E. et al. Enhanced healing of large cranial defects by an
osteoinductive protein in rabbits //Plastic and reconstructive surgery. – 1993. – Т. 92.
– №. 4. – С. 593-600;
234. Urist M. R. Bone: formation by autoinduction //Science. – 1965. – Т.
150. – №. 3698. – С. 893-899;
235. Urist M. R., Iwata H. Preservation and biodegradation of the
morphogenetic property of bone matrix //Journal of theoretical biology. – 1973. – Т.
38. – №. 1. – С. 155-167;
236. van de Weert M. et al. The effect of a water/organic solvent interface on
the structural stability of lysozyme //Journal of controlled release. – 2000. – Т. 68. –
№. 3. – С. 351-359;
237. van de Weert M., Hennink W. E., Jiskoot W. Protein instability in poly
(lactic-co-glycolic acid) microparticles //Pharmaceutical research. – 2000. – Т. 17. –
№. 10. – С. 1159-1167;
238. van de Weert M., Hennink W. E., Jiskoot W. Protein instability in poly
(lactic-co-glycolic acid) microparticles //Pharmaceutical research. – 2000. – Т. 17. –
№. 10. – С. 1159-1167;
239. Van S. N., Minh H. D., Anh D. N. Study on chitosan nanoparticles on
biophysical characteristics and growth of Robusta coffee in green house //Biocatalysis
and Agricultural Biotechnology. – 2013. – Т. 2. – №. 4. – С. 289-294;
240. Van Tassel P. R. et al. A particle-level model of irreversible protein
adsorption with a postadsorption transition //Journal of colloid and interface science. –
1998. – Т. 207. – №. 2. – С. 317-323;
241. Wallen L. L., Rohwedder W. K. Poly-beta-hydroxyalkanoate from
activated sludge //Environmental science & technology. – 1974. – Т. 8. – №. 6. – С.
576-579;
242. Wang N., Wu X. S., Li J. K. A heterogeneously structured composite
based on poly (lactic-co-glycolic acid) microspheres and poly (vinyl alcohol) hydrogel

149
nanoparticles for long-term protein drug delivery //Pharmaceutical research. – 1999. –
Т. 16. – №. 9. – С. 1430-1435;
243. Wang S. H. et al. Controlled release of levonorgestrel from biodegradable
poly (D, L-lactide-co-glycolide) microspheres: in vitro and in vivo studies
//International journal of pharmaceutics. – 2005. – Т. 301. – №. 1. – С. 217-225;
244. Wang W. Instability, stabilization, and formulation of liquid protein
pharmaceuticals //International journal of pharmaceutics. – 1999. – Т. 185. – №. 2. –
С. 129-188;
245. Wang X. et al. Poly (lactide-co-glycolide) encapsulated hydroxyapatite
microspheres for sustained release of doxycycline //Materials Science and
Engineering: B. – 2012. – Т. 177. – №. 4. – С. 367-372;
246. Wang Y. et al. Fabrication, characterization and long-term in vitro release
of hydrophilic drug using PHBV/HA composite microspheres //Materials Letters. –
2007. – Т. 61. – №. 4. – С. 1071-1076;
247. Wang Y. W. et al. Effect of composition of poly (3-hydroxybutyrate-co-
3-hydroxyhexanoate) on growth of fibroblast and osteoblast //Biomaterials. – 2005. –
Т. 26. – №. 7. – С. 755-761;
248. Wang Y. W. et al. Evaluation of three-dimensional scaffolds made of
blends of hydroxyapatite and poly (3-hydroxybutyrate-co-3-hydroxyhexanoate) for
bone reconstruction //Biomaterials. – 2005. – Т. 26. – №. 8. – С. 899-904;
249. Wang Y. W., Wu Q., Chen G. Q. Attachment, proliferation and
differentiation of osteoblasts on random biopolyester poly (3-hydroxybutyrate-co-3-
hydroxyhexanoate) scaffolds //Biomaterials. – 2004. – Т. 25. – №. 4. – С. 669-675;
250. Wang Y., Irvine D. J. Engineering chemoattractant gradients using
chemokine-releasing polysaccharide microspheres //Biomaterials. – 2011. – Т. 32. –
№. 21. – С. 4903-4913;
251. Wenk E. et al. Microporous silk fibroin scaffolds embedding PLGA
microparticles for controlled growth factor delivery in tissue engineering
//Biomaterials. – 2009. – Т. 30. – №. 13. – С. 2571-2581;

150
252. Whitaker J. R., Granum P. E. An absolute method for protein
determination based on difference in absorbance at 235 and 280 nm //Analytical
biochemistry. – 1980. – Т. 109. – №. 1. – С. 156-159;
253. Williams S. F. et al. PHA applications: addressing the price performance
issue: I. Tissue engineering //International journal of biological macromolecules. –
1999. – Т. 25. – №. 1. – С. 111-121;
254. Witkowski A., Joshi A. K., Smith S. Characterization of the interthiol
acyltransferase reaction catalyzed by the β-ketoacyl synthase domain of the animal
fatty acid synthase //Biochemistry. – 1997. – Т. 36. – №. 51. – С. 16338-16344;
255. Wodzinska J. et al. Polyhydroxybutyrate synthase: evidence for covalent
catalysis //Journal of the American Chemical Society. – 1996. – Т. 118. – №. 26. – С.
6319-6320;
256. Wozney J. M. et al. Novel regulators of bone formation: molecular clones
and activities //Science. – 1988. – Т. 242. – №. 4885. – С. 1528-1535;
257. Wu Q., Wang Y., Chen G. Q. Medical application of microbial
biopolyesters polyhydroxyalkanoates //Artificial Cells, Blood Substitutes, and
Biotechnology. – 2009. – Т. 37. – №. 1. – С. 1-12;
258. Yagmurlu M. F. et al. Sulbactam-cefoperazone polyhydroxybutyrate-co-
hydroxyvalerate (PHBV) local antibiotic delivery system: In vivo effectiveness and
biocompatibility in the treatment of implant-related experimental osteomyelitis
//Journal of Biomedical Materials Research Part A. – 1999. – Т. 46. – №. 4. – С. 494-
503;
259. Yang S., Washington C. Drug release from microparticulate systems
//Microencapsulation: Methods and Industrial Applications, Second Edition. –
Informa Healthcare, 2005. – С. 183-211;
260. Yokouchi M. et al. Structural studies of polyesters: 5. Molecular and
crystal structures of optically active and racemic poly (β-hydroxybutyrate) //Polymer.
– 1973. – Т. 14. – №. 6. – С. 267-272;
261. You M. et al. Chondrogenic differentiation of human bone marrow
mesenchymal stem cells on polyhydroxyalkanoate (PHA) scaffolds coated with PHA

151
granule binding protein PhaP fused with RGD peptide //Biomaterials. – 2011. – Т. 32.
– №. 9. – С. 2305-2313;
262. Yu G., Marchessault R. H. Characterization of low molecular weight poly
(β-hydroxybutyrate) s from alkaline and acid hydrolysis //Polymer. – 2000. – Т. 41. –
№. 3. – С. 1087-1098;
263. Zambaux M. F. et al. Preparation and characterization of protein C-
loaded PLA nanoparticles //Journal of controlled release. – 1999. – Т. 60. – №. 2. – С.
179-188;
264. Zhang N. et al. Environmental pH-controlled loading and release of
protein on mesoporous hydroxyapatite nanoparticles for bone tissue engineering
//Materials Science and Engineering: C. – 2015. – Т. 46. – С. 158-165;
265. Zharkova I. I. et al. Biocompatibility of electrospun poly (3-
hydroxybutyrate) and its composites scaffolds for tissue engineering
//Biomeditsinskaia khimiia. – 2013. – Т. 60. – №. 5. – С. 553-560;
266. Zhijiang C., Zhihong W. Preparation of biodegradable poly (3-
hydroxybutyrate)(PHB) and poly (ethylene glycol)(PEG) graft copolymer //Journal of
materials science. – 2007. – Т. 42. – №. 14. – С. 5886-5890;
267. Zhou H., Lee J. Nanoscale hydroxyapatite particles for bone tissue
engineering //Acta biomaterialia. – 2011. – Т. 7. – №. 7. – С. 2769-2781;
268. Zhou Y. S., Liu Y. S., Tan J. G. Is 1, 25-dihydroxyvitamin D3 an ideal
substitute for dexamethasone for inducing osteogenic differentiation of human adipose
tissue-derived stromal cells in vitro? //Chinese medical journal. – 2006. – Т. 119. – №.
15. – С. 1278-1286;
269. Zhu X. H., Wang C. H., Tong Y. W. In vitro characterization of
hepatocyte growth factor release from PHBV/PLGA microsphere scaffold //Journal of
Biomedical Materials Research Part A. – 2009. – Т. 89. – №. 2. – С. 411-423;
270. Zinn M., Witholt B., Egli T. Occurrence, synthesis and medical
application of bacterial polyhydroxyalkanoate //Advanced drug delivery reviews. –
2001. – Т. 53. – №. 1. – С. 5-21;

152
271. Zisch A. H. et al. Covalently conjugated VEGF–fibrin matrices for
endothelialization //Journal of Controlled Release. – 2001. – Т. 72. – №. 1. – С. 101-
113;




















