
Monte Carlo simulation of homopolymer chains. I. Second virial coefficient
13
Monte Carlo simulation of homopolymer chains. I. Second virial coefficient Ian M. Withers Department of Chemistry, University of North Carolina, Chapel Hill, North Carolina 27599-3290 Andrey V. Dobrynin Institute of Materials Science and Department of Physics, University of Connecticut, Storrs, Connecticut 06269-3136 Max L. Berkowitz and Michael Rubinstein Department of Chemistry, University of North Carolina, Chapel Hill, North Carolina 27599-3290 ~Received 7 August 2002; accepted 13 December 2002! The second virial coefficient, A 2 , is evaluated between pairs of short chain molecules by direct simulations using a parallel tempering Monte Carlo method where the centers of mass of the two molecules are coupled by a harmonic spring. Three off-lattice polymer models are considered, one with rigid bonds and two with flexible bonds, represented by the finitely extensible nonlinear elastic potential with different stiffness. All the models considered account for excluded volume interactions via the Lennard-Jones potential. In order to obtain the second virial coefficient we calculate the effective intermolecular interaction between the two polymer chains. As expected this intermolecular interaction is found to be strongly dependent upon chain length and temperature. For all three models the u temperature ( u n ), defined as the temperature at which the second virial coefficient vanishes for chains of finite length, varies as u n 2u ‘ } n 21/2 , where n is the number of bonds in the polymer chains and u ‘ is the u point for an infinitely long chain. Introducing flexibility into the model has two effects upon u n ; the u temperature is reduced with increasing flexibility, and the n dependence of u n is suppressed. For a particular choice of spring constant an n -independent u temperature is found. We also compare our results with those obtained from experimental studies of polystyrene in decalin and cyclohexane, and for poly~methyl methacrylate! in a water and tert-butyl alcohol mixture, and show that all the data can be collapsed onto a single universal curve without any adjustable parameters. We are thus able to relate both A 2 and the excluded volume parameter v , to the chain interaction parameter z , in a way relating not only the data for different molecular weights and temperatures, but also for different polymers in different solvents. © 2003 American Institute of Physics. @DOI: 10.1063/1.1543940# I. INTRODUCTION The behavior of homopolymer chains at very low con- centrations has attracted considerable attention. 1–4 In particu- lar, Monte Carlo ~MC! simulations have been used to gener- ate self-avoiding random walks for various model homopolymer systems at infinite dilution. 5–14 In all of these models the monomer–monomer interactions are taken to be of van der Waals type; consisting of a hard-core repulsion and a short-ranged attraction. At high temperatures, or good solvent conditions, repulsive interactions dominate and the polymer swells relative to a random coil. Alternatively at low temperatures, or poor solvent conditions, attractive interac- tions dominate and chains collapse into dense globules. Ob- servation of isolated macromolecules in the collapsed state in experimental studies is very difficult since in poor solvent conditions the polymer is no longer soluble and precipitates from solution. Phase separation can, in some cases, be avoided by considering systems which are sufficiently dilute. 15 At intermediate temperatures, between the good and poor solvent regimes, the polymer is in a u-solvent condition, i.e., at temperatures close to the u point. The true u point ( u ‘ ) is defined as the temperature at which the second virial coefficient between two infinitely long chains is zero. 1 This definition is, of course, not practi- cal since neither experiments nor computer simulations can be performed upon infinitely long chains, and the most com- mon empirical definition of the u point is the temperature at which the second virial coefficient between chains of finite length is zero 4 ~we use the notation u n to represent the u temperature of a chain of finite length!. A temperature is thus obtained which may depend upon the number of bonds in the chain, n , and the true u temperature is obtained in the limit u ‘ 5 lim n →‘ u n . ~1! Previously, several computer simulation studies have evalu- ated the u point following this definition of the u temperature. 6 – 8,10,12,14 These models display behavior differ- ent from that observed in experiment; namely a strong n dependence of u n , whereas experimental studies find either a very weak n dependence 16 or no n dependence. 17 Renormal- ization group calculations 4 show that n dependence of u n is to be expected, with a stronger n dependence being predicted with increasing three-body interaction parameter. The mod- els typically used 6 – 8,10,12 in computer simulation studies have a much smaller aspect ratio of their Kuhn segments than real polymers do, resulting in the three-body repulsions being an JOURNAL OF CHEMICAL PHYSICS VOLUME 118, NUMBER 10 8 MARCH 2003 4721 0021-9606/2003/118(10)/4721/12/$20.00 © 2003 American Institute of Physics
Transcript of Monte Carlo simulation of homopolymer chains. I. Second virial coefficient

JOURNAL OF CHEMICAL PHYSICS VOLUME 118, NUMBER 10 8 MARCH 2003
Monte Carlo simulation of homopolymer chains. I. Second virial coefficientIan M. WithersDepartment of Chemistry, University of North Carolina, Chapel Hill, North Carolina 27599-3290
Andrey V. DobryninInstitute of Materials Science and Department of Physics, University of Connecticut, Storrs,Connecticut 06269-3136
Max L. Berkowitz and Michael RubinsteinDepartment of Chemistry, University of North Carolina, Chapel Hill, North Carolina 27599-3290
~Received 7 August 2002; accepted 13 December 2002!
The second virial coefficient,A2 , is evaluated between pairs of short chain molecules by directsimulations using a parallel tempering Monte Carlo method where the centers of mass of the twomolecules are coupled by a harmonic spring. Three off-lattice polymer models are considered, onewith rigid bonds and two with flexible bonds, represented by the finitely extensible nonlinear elasticpotential with different stiffness. All the models considered account for excluded volumeinteractions via the Lennard-Jones potential. In order to obtain the second virial coefficient wecalculate the effective intermolecular interaction between the two polymer chains. As expected thisintermolecular interaction is found to be strongly dependent upon chain length and temperature. Forall three models theu temperature (un), defined as the temperature at which the second virialcoefficient vanishes for chains of finite length, varies asun2u`}n21/2, wheren is the number ofbonds in the polymer chains andu` is theu point for an infinitely long chain. Introducing flexibilityinto the model has two effects uponun ; theu temperature is reduced with increasing flexibility, andthe n dependence ofun is suppressed. For a particular choice of spring constant ann-independentu temperature is found. We also compare our results with those obtained from experimental studiesof polystyrene in decalin and cyclohexane, and for poly~methyl methacrylate! in a water andtert-butyl alcohol mixture, and show that all the data can be collapsed onto a single universal curvewithout any adjustable parameters. We are thus able to relate bothA2 and the excluded volumeparameterv, to the chain interaction parameterz, in a way relating not only the data for differentmolecular weights and temperatures, but also for different polymers in different solvents. ©2003American Institute of Physics.@DOI: 10.1063/1.1543940#
n
ere
oiooothwraO
tente,tlan
atly
-canm-tite
thet
alu-
r-
r a
dod-erealan
I. INTRODUCTION
The behavior of homopolymer chains at very low cocentrations has attracted considerable attention.1–4 In particu-lar, Monte Carlo~MC! simulations have been used to genate self-avoiding random walks for various modhomopolymer systems at infinite dilution.5–14 In all of thesemodels the monomer–monomer interactions are taken tof van der Waals type; consisting of a hard-core repulsand a short-ranged attraction. At high temperatures, or gsolvent conditions, repulsive interactions dominate andpolymer swells relative to a random coil. Alternatively at lotemperatures, or poor solvent conditions, attractive intetions dominate and chains collapse into dense globules.servation of isolated macromolecules in the collapsed staexperimental studies is very difficult since in poor solveconditions the polymer is no longer soluble and precipitafrom solution. Phase separation can, in some casesavoided by considering systems which are sufficiendilute.15 At intermediate temperatures, between the goodpoor solvent regimes, the polymer is in au-solvent condition,i.e., at temperatures close to theu point.
The trueu point (u`) is defined as the temperaturewhich the second virial coefficient between two infinite
4720021-9606/2003/118(10)/4721/12/$20.00
-
-l
bende
c-b-intsbeyd
long chains is zero.1 This definition is, of course, not practical since neither experiments nor computer simulationsbe performed upon infinitely long chains, and the most comon empirical definition of theu point is the temperature awhich the second virial coefficient between chains of finlength is zero4 ~we use the notationun to represent theutemperature of a chain of finite length!. A temperature is thusobtained which may depend upon the number of bonds inchain,n, and the trueu temperature is obtained in the limi
u`5 limn→`
un . ~1!
Previously, several computer simulation studies have evated the u point following this definition of the utemperature.6–8,10,12,14These models display behavior diffeent from that observed in experiment; namely a strongndependence ofun , whereas experimental studies find eithevery weakn dependence16 or non dependence.17 Renormal-ization group calculations4 show thatn dependence ofun isto be expected, with a strongern dependence being predictewith increasing three-body interaction parameter. The mels typically used6–8,10,12in computer simulation studies hava much smaller aspect ratio of their Kuhn segments thanpolymers do, resulting in the three-body repulsions being
1 © 2003 American Institute of Physics

in
e
tehalhaa
in-thr
res
telle
th
bl-hire
Intinpotisrruinsethon
onf n
cic
eed
nt
rager
tor
woto
ionnter--
wo
singmhise an
ol-eryo
er.ingili-but
hishisnt-lu-o
ectofy of
m
toe-
ionsth
heingo
thea-
4722 J. Chem. Phys., Vol. 118, No. 10, 8 March 2003 Withers et al.
order of magnitude stronger in the model systems.18 The ex-perimentally observedn independence ofun could, there-fore, be due to relatively weak three-body interactions bepresent in real polymeric materials.
It is, therefore, a worthy task to attempt to find a modpolymer chain which exhibits ann independent or weaklyn-dependentu temperature, such that the results of compusimulations will be closer to experimental ones. We note tdetermination of theu temperature from conformationaproperties of lattice models of polymer chains indicates tthe n dependence ofun may be suppressed by consideringmodel with flexible bonds.19 Introducing a variable bondlength into the model is computationally simpler thancreasing the aspect ratio of the Kuhn segments; allowingbond length to vary requires only one bonded energy tewhile increasing the chain stiffness requires two or thbonded terms~the bond length, the valence angle and posbly the torsion angle!. In this paper we, therefore, evaluathe second virial coefficient,A2 , for three types of modepolymer chains, one with rigid bonds and two with flexibbonds of different stiffnesses, containing (n11)52, 4, 8,and 16 monomers, using a new method inspired by theoretical work of Grosberg and Kuznetsov.20 It will be shownthat an appropriate choice of parametrization of the flexibond may lead to ann-independentu temperature. The results obtained from a study of the chain dimensions of tmodel polymer, along with comparison with theoretical pdictions, are presented in Paper II.18
The remainder of this paper is organized as follows:the next section we describe our new method for evaluathe second virial coefficient. In Sec. III we consider the aplication of the method to the simple case of a pairLennard-Jones particles where results obtained by simulaare compared with theexact numerical evaluation. Resultobtained for short homopolymer chains, along with compason with previous simulation studies of similar models, apresented in Sec. IV. We also compare our numerical resof the second virial coefficient with experimental resultsSec. IV, and show that the simulation data may be collaponto a universal curve with the experimental data withoutuse of any adjustable parameters. Finally our conclusiand discussion are given in Sec. V.
II. DESCRIPTION OF THE METHOD
The second virial coefficient,A2 , of a polymer solutionis an important property, since it describes the interactibetween pairs of molecules, and has been the subject omerous theoretical and experimental studies.3,4 An expres-sion for the second virial coefficient in terms of the interation potentials can be obtained from standard statistmechanics as21
A2522pNA
M2 E0
`
R2FexpS 2U~R!
kBT D21GdR, ~2!
whereM is the molar mass of the molecules andU(R) isconsidered as an effective intermolecular interaction betwthe chain molecules whose centers of mass are separatea distanceR, i.e., a potential of mean force which is solve
g
l
rt
t
emei-
e-
e
s-
g-fon
i-elts
des
su-
-al
nby
and temperature dependent and corresponds to an aveover all possible chain conformations~i.e., the average oveall internal conformations of each chainand over all orien-tations of each chain relative to the intermolecular vecbetween the centers of mass of the two molecules,R!. Thetask of evaluating the second virial coefficient between tchain molecules by computer simulation thus reducesevaluatingU(R).
The most popular methods of evaluatingU(R) is basedupon the idea that the effective intermolecular interactmay be expressed as the average of the intermolecular iaction energy,U12(R,a1 ,a2), between chain 1 in conformation a1 and chain 2 in conformationa2 ,3
U~R!52kBT lnK expS 2U12~R,a1 ,a2!
kBT D La1 ,a2
, ~3!
where^ &a1 ,a2denotes an average over conformations of t
chains without interchain interactions. The average in Eq.~3!is typically evaluated6,7 by generating many pairs of chainof the desired length independently and calculatU12(R,a1 ,a2) between the two chains at many randoseparations and orientations relative to each other. Tmethod has become popular since it has been shown to befficient method for evaluatingU(R) in good solvent condi-tions. In poor solvent conditions, where the two chains clapse into a single dense globule, this method is not vefficient due to the very low probability of generating twchains independently which will entwine with each othFurthermore this method is inefficient for systems containsolvent, since the solvent particles would have to be equbrated around the two chains after they are generatedbefore calculating the intermolecular interaction energy. Tstrategy would be very time consuming and render tmethod highly inefficient for examining the effects of solveproperties~for example, density! upon the second virial coefficient. It is therefore desirable to have a method for evaating U(R) during the course of a direct simulation of twpolymer chains.
To overcome the sampling problem we performed dirsimulations of the two interacting polymers which centermasses are coupled via a spring such that the probabilitfinding the two chains at a distanceR apart becomes
Ps~R!5C exp~2~U~R!1Us~R!!/kBT!, ~4!
where C is the normalization constant andUs(R)5kR2/2kBT is the contribution to the potential energy frothe spring with spring constantk. The introduction of theadditional interaction potential between chains allows ussample the configurational spaces of the two chains in Mtropolis sense—generating the most probable configuratof two interacting chains.22 Since the energy associated withe spring increases rapidly with increasingR, it is possibleto choose a value of the spring constant,k, such that theprobability of finding the two chains beyond the range of tintermolecular interaction decreases rapidly with increasR, thus placing a restriction upon how far apart the twchains may move from each other. This restriction allowsprobability Ps(R) to be accumulated by computer simul

nciseo
re
etiode
te
irrd
p
a
an
agte
hi
th-ac
wl
brela
alt
r-tive
ing.
-eel
ling
el
4723J. Chem. Phys., Vol. 118, No. 10, 8 March 2003 Monte Carlo simulation of homopolymer chains
tions whose run times are not prohibitively excessive, siwe are now only interested in accumulating data up to dtances where the probability of finding the two chains bcomes negligibly small. It should be noted that the choicethe spring constant,k, is very important since too stiff aspring will perturb the chain conformations, altering thesulting form of the intermolecular interactionU(R). Alter-natively, too weak a spring will allow the polymers to movfurther apart than is necessary, leading to longer simularuns to obtain the probability histograms with a desiredgree of accuracy.
III. LENNARD-JONES PARTICLES
To demonstrate the validity of this method to calculathe effective intermolecular interaction,U(R), and the sec-ond virial coefficient,A2 , we apply the new method to a paof particles interacting via the truncated-shifted LennaJones potential,
ULJ~r !
5H 4eF S s
r D 12
2S s
r D 6G24eF S s
r cD 12
2S s
r cD 6G , r ,r c ,
0, r>r c ,
~5!
where r is the distance between the two particles,s is the~effective! diameter of the particle,e is the depth of the at-tractive minima, andr c is the cutoff radius (52.5s). Sincethis is a particularly simple problem@U(R) should equalULJ(r )] it is our aim in this section to demonstrate the aplicability of this method for evaluation ofU(R) by com-puter simulation, while comparing our results with analyticfunctions and theexactnumerical integration of Eq.~2!.
To accumulate the probabilityPs(R) we use theMetropolis Monte Carlo method.22 Trial conformations weregenerated by randomly choosing one of the two particlesthen displacing it in thex, y, and z directions by randomdistances. The maximum displacement was kept fixed0.1s throughout the simulations. The spring constant,k,linking the two particles was chosen such that the averdistance between particles without the Lennard-Jones inaction was 5s. The probabilityPs(R) was collected by ac-cumulating the distance between the two particles into atogram whose bins were of widthdR50.02s. In order toimprove the sampling process we have also employedparallel tempering method.23 Our simulations were performed on nine processors with the temperature of esimulation being linearly distributed over the rangekBT/e51.0 to 5.0. Temperature exchanges between systemsattempted every 23103 attempted particle moves. In tota23107 trial moves were attempted at each temperature.
In Fig. 1 we illustrate the procedure by which the proability, Ps(R), obtained from a simulation at temperatukBT/e53.0 was converted into the effective intermolecuinteraction. In Fig. 1~a! the points showPs(R) obtained fromthe simulation and the solid line represents the analyticexpected probability distribution@Eq. ~4!#. The agreemenbetween the simulation data and the analytical function
e--f
-
n-
-
-
l
d
at
er-
s-
e
h
ere
-
r
ly
is
excellent, indicating that a sufficient number of trial confomations have been generated. In order to obtain the relaprobability of finding two particles at a distanceR apart duesolely to the effective intermolecular interaction,
P~R!5expS 2U~R!
kBT D , ~6!
it is necessary to remove the contribution due to the spr
FIG. 1. Manipulation of the simulated probability distributionPs(R) toobtain the intermolecular interactionU(R) for a pair of Lennard-Jones particles at a temperature ofkBT/e53.0. Results from the simulation arshown as points (1) and analytic functions are shown by solid lines. Pan~a! shows the probability distribution functionPs(R) obtained from thesimulation, the dashed line shows the contribution from the spring coupthe centers of masses together, panel~b! shows the probability distributionfunction P(R) after removal of the contribution from the spring, and pan~c! shows the resulting intermolecular potential,U(R). Error bars aresmaller than the point size.

gto
oe
e
is
th
ia
inoioeais
ncia
et.
wcof
ly-metial
on-nd
n
ly;tial,ted
as-no-rd-r
on
a-idr atthethe-
tedby
tion,rd-
d ang
ctsbeareherre-ter-nwas
gy-be-an
tant
m
t t
4724 J. Chem. Phys., Vol. 118, No. 10, 8 March 2003 Withers et al.
To accomplish this it is noted that beyond the ranof the intermolecular interaction the only contributionPs(R) comes from the spring term, i.e.,Ps(R)5C exp(2Us(R)/kBT) for R.r c , whereC is a constant ofproportionality arising due to the difference between the nmalization of the total probability and the probability duonly to the spring. The numerical value ofC may be ob-tained by a least-squares-fitting procedure24 to Ps(R) overthe rangeR.r c @as shown by the dashed line in Fig. 1~a!#.Division of Ps(R) by C exp(2Us(R)/kBT) gives the relativeprobability of finding the two particles at distanceR apartdue to the effective intermolecular interaction,P(R) @Fig.1~b!#, which is normalized correctly to allow the effectivintermolecular interaction to be obtained, i.e.,P(R)51 forlarge R. Finally, the effective intermolecular interactiongiven by the relationU(R)52kBT ln(P(R)). Figure 1~c!clearly shows that this process has lead us back toLennard-Jones potential given by Eq.~5! @the points showthe simulation data and the solid line shows Eq.~5!#.
In Table I we present the values of the second vircoefficient obtained by numerical integration of Eq.~2!,24
where U(R) is given analytically@Eq. ~5!# and has beendetermined from simulation. The step size between poused for theexactnumerical integration was taken to be tworders of magnitude smaller than that used in the simulatThe error estimates quoted for the simulation results wobtained by performing the numerical integration usingintermolecular interaction obtained from the probability dtributions functionPS(R)6s(R) @wheres(R) is the statisti-cal error obtained by the block averaging technique#.22 Theresults quoted in Table I clearly demonstrate the equivalewithin the statistical error, of the values of the second vircoefficient obtained from the simulation and by theexactnumerical integration, illustrating that this method providan accurate route to calculate the second virial coefficien
IV. SHORT POLYMER CHAINS
A. Model
Having demonstrated the applicability of the nemethod for determining the effective intermolecular interation and the second virial coefficient for the simple casetwo Lennard-Jones particles, we now present the results
TABLE I. Values of the second virial coefficient (A2) in units of s3 for apair of Lennard-Jones particles evaluated at various temperatures by nucal integration of Eq.~2! whereU(R) is evaluated from Eq.~5! and fromsimulation. Numbers in parentheses in the simulation column represenstatistical error to the last two decimal places.
kBT/e A2M2/NA ~analytical! A2M2/NA ~simulation!
1.0 24.167 24.183(75)1.5 21.778 21.779(62)2.0 20.771 20.748(61)2.5 20.224 20.204(41)3.0 0.115 0.113~41!3.5 0.343 0.323~61!4.0 0.506 0.510~26!4.5 0.627 0.628~17!5.0 0.720 0.730~30!
e
r-
e
l
ts
n.ren-
e,l
s
-for
two short polymer chains. We consider three models of pomer chains. All the models account for the excluded voluinteraction by the truncated-shifted Lennard-Jones poten@Eq. ~5!#. The difference between these models is how cnectivity of the chain is assured. In the first case all bolengths are kept fixed ats. This type of model has beeconsidered previously by Harismiadis and Szleifer,7 althoughit should be noted that their model differs from ours slightwe employ the truncated-shifted Lennard-Jones potenwhile Harismiadis and Szleifer employed only the truncaLennard-Jones potential, i.e., an energy discontinuity wpresent in Ref. 7 atr 5r c . The other two models have flexible bonds. The interaction energy between adjacent momers is given by the sum of the truncated-shifted LennaJones potential@Eq. ~5!# and the finitely extensible nonlineaelastic~FENE! potential,
UFENE~r !521
2kFENER0
2 lnF12r 2
R02G , ~7!
wherekFENE is an adjustable spring constant andR0 is themaximum extension of the bond, at which the interactienergy becomes infinite. In the present study we usedR0
52s andkFENE510.0e/s2 and 30.0e/s2.Different methods are used to generate trial conform
tions for each of these models. For the model with rigbonds a trial move is generated by selecting a monomerandom and then rotating it by a random amount aboutaxis formed by the line that joins the centers of mass oftwo adjacent sites~or the next two innermost sites if a monomer at the end of the chain was selected!. The extent of therotation was chosen uniformly in the interval 0 to 2p. For themodel with flexible bonds trial conformations are generaby selecting a monomer at random and then displacing itrandom amounts in thex, y, and z directions, generateduniformly between 20.1s and 0.1s. As with the twoLennard-Jones particles considered in the preceding secthe trial move for both models is accepted or rejected accoing to the Metropolis acceptance criterion.22
To choose the value of the spring constant,k, connectingthe centers of mass of the two polymers we conductepreliminary series of simulations, for all three models usithe longest chain length, with chains containingn515bonds, at the highest dimensionless temperature,kBT/e55.0, considered in this work, since the unfavorable effeassociated with the use of too strong a spring are likely tomore noticeable in good solvent conditions as the chainslarger ~swollen! and are expected to repel each other. Tsimulations were started with a small spring constant, cosponding to an average extension of the spring without inmolecular interactions of 10s. The spring constant was theincreased, such that the average extension of the springreduced by integer values ofs, until deviations were ob-served in the average end-to-end distance and radius ofration. For all three models considered, such deviationscame apparent for a spring constant corresponding toaverage extension of the spring of 3s. The remainder of thesimulations were therefore performed using a spring consgiving an average spring extension of 4s. It is also noted that
eri-
he

omth
veato
ttio
pt
0dritois
hs--lydto
eo
neo-
c-beo
rlycA
sseaitivr
A
sreinfo
-
b-eglyior
r
litythad-or
s aentmalleen
rof
sionthe
essem-soci-lso
on
s.ondthein-epa-lyly,
ionan
l the
4725J. Chem. Phys., Vol. 118, No. 10, 8 March 2003 Monte Carlo simulation of homopolymer chains
the root-mean-square radius of gyration for then515 chainsat this temperature is approximately 2.25s.
The simulations were started by generating two randself-avoiding chain conformations independently, whereinitial bond length was taken ass for all the models. The twochains were placed such that their centers of mass weresapart. The system was then checked for intermolecular olap ~an overlap is defined as two monomers being separby a distance smaller thans! and if monomers were found toverlap then two new conformations were generated andprocess was repeated until a nonoverlapping conformawas generated. The system was then equilibrated for 105 MCsweeps, where one MC sweep consists of one attemmove per monomer, i.e., 23(n11) moves. The probabilitydistribution,Ps(R), was accumulated over an additional 17
MC sweeps. Parallel tempering was once again employeimprove the efficiency of the sampling process. The majoof the simulations were performed using nine processwith the temperature of each simulation being linearly dtributed over the rangekBT/e51.0 to 5.0. An additionalthree processors~12 in total! were used for the chains witkFENE510.0e/s2, with these extra simulations being asigned temperatures very close to theu temperature to confirm the observation of preliminary simulations using onnine processors. Temperature exchanges were attempteery 103 MC sweeps. Removal of the spring contributionPs(R) was once again achieved by fittingPs(R) toC exp(2Us(R)/kBT) for distances beyond the range of thintermolecular interaction. Since the range of the intermlecular potential is not knowna priori, we perform fits in therangeR.r l , where r l is the lowest integer multiple ofswhich yields the same value of the coefficientC, withinerror estimates, when compared with the values obtaifrom fits performed with larger lower bounds. Via this prcedure, we findr l53s, 4s, 5s, and 7s for n51, 3, 7, and15, respectively.
B. Intermolecular potential
In Fig. 2 we show the effective intermolecular interation between polymer chains as a function of distancetween their centers of mass for chains with rigid bondslengthn515 at five different temperatures. It can be cleaseen that the strength, and form, of the effective intermolelar interaction shows a strong variation with temperature.the highest temperature considered,kBT/e55.0, U(R) ispositive for all R, i.e., the intermolecular interaction ipurely repulsive, indicative of good solvent conditionwhere the chains swell and repel each other. As has breported previously,6,7 the interaction energy at zero separtion between the centers of mass of the two chains is posand finite. This behavior occurs since, unlike the monomecase, it is possible for the centers of mass of polymersoverlap without resulting in an overlap of any monomers.the lowest temperature considered,kBT/e51.0, U(R) isnegative for all R, i.e., the intermolecular interaction ipurely attractive, indicative of poor solvent conditions whethe monomers attract each other and the chains collapsea globular state. In this case, it is interesting to note thatseparations smaller than;1.5s the intermolecular interac
e
5r-
ed
hen
ed
toyrs-
ev-
-
d
-f
u-t
,en-e
ictot
tor
tion is approximately constant, with no upturn being oserved with decreasingR. This behavior tends to indicatthat the two polymer chains attract each other so stronthat they coalesce in a single globule. This type of behavhas been inferred previously,6 however, the statistical erroobserved in this previous study~see Fig. 5 of Ref. 6! isconsiderably worse than ours, due to the very low probabiof growing two chains independently that will entwine wieach other in a single dense globule, illustrating a clearvantage of employing our method to study behavior in posolvent conditions. That said, the current method yieldlarger statistical error for small separations in good solvconditions, since the interchain repulsion makes these sseparations difficult to sample. For temperatures betwthese two limiting cases,U(R) displays behavior similar tothat observed previously;7 an increase in the intermoleculainteraction between molecules with overlapping centersmass with increasing temperature, arising since the repulbetween the chains increases as the chains swell, andobservation of an attractive minimum, which becomes ldeep and occurs at larger separations with increasing tperature, once again due to the increased repulsion asated with increasing temperature. Similar behavior is aobserved for the models with flexible bonds.
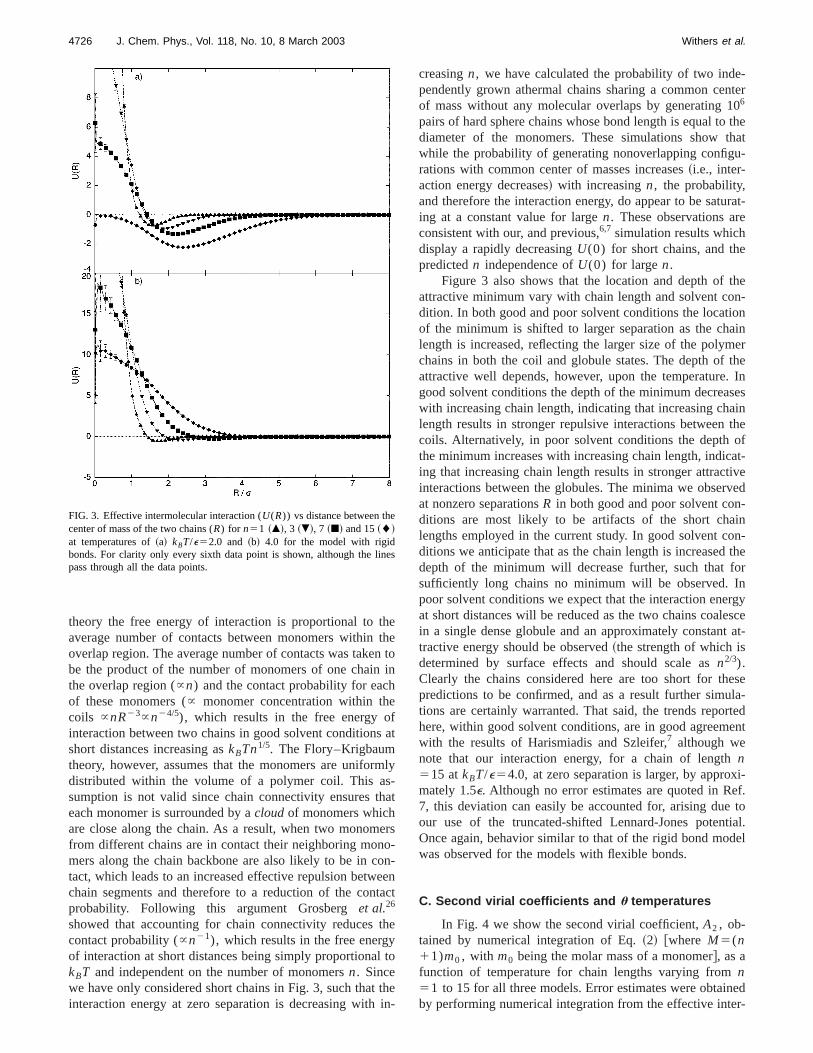
Figure 3 shows the effective intermolecular interactibetween two chains at temperatures ofkBT/e52.0 and 4.0for the model with rigid bonds and different chain lengthAs will be confirmed shortly, these temperatures correspto poor and good solvent conditions, respectively, for allchain lengths considered. For both solvent conditions,creasing the chain length reduces the repulsion at zero sration from infinity for the monomeric case to approximate11e and 0 for good and poor solvent conditions, respectivefor the longest chain considered here,n515. This behaviorhas been observed previously in computer simulatstudies,6,7 but is, however, unexpected following the mefield theory of Flory and Krigbaum.25 In this mean field
FIG. 2. Effective intermolecular interaction (U(R)) vs distance between thecenter of mass of the two chains (R) for n515 with rigid bonds at tempera-tures ofkBT/e51.0 ~d!, 2.0 ~m!, 3.0 ~.!, 4.0 ~j!, and 5.0~l!. For clarityonly every sixth data point is shown, although the lines pass through aldata points.
hethni
hef
s a
rms-ha
eooneta
thyl t
thi
-nter0the
thatu-
urat-e
then-onainer
the. Insesaintheofat-tiveved-inon-the
forInrgy
escet at-
esela-
rtedent
xi-ef.
totial.el
edr-
ne
4726 J. Chem. Phys., Vol. 118, No. 10, 8 March 2003 Withers et al.
theory the free energy of interaction is proportional to taverage number of contacts between monomers withinoverlap region. The average number of contacts was takebe the product of the number of monomers of one chainthe overlap region (}n) and the contact probability for eacof these monomers (} monomer concentration within thcoils }nR23}n24/5), which results in the free energy ointeraction between two chains in good solvent conditionshort distances increasing askBTn1/5. The Flory–Krigbaumtheory, however, assumes that the monomers are unifodistributed within the volume of a polymer coil. This asumption is not valid since chain connectivity ensures teach monomer is surrounded by acloudof monomers whichare close along the chain. As a result, when two monomfrom different chains are in contact their neighboring monmers along the chain backbone are also likely to be in ctact, which leads to an increased effective repulsion betwchain segments and therefore to a reduction of the conprobability. Following this argument Grosberget al.26
showed that accounting for chain connectivity reducescontact probability (}n21), which results in the free energof interaction at short distances being simply proportionakBT and independent on the number of monomersn. Sincewe have only considered short chains in Fig. 3, such thatinteraction energy at zero separation is decreasing with
FIG. 3. Effective intermolecular interaction (U(R)) vs distance between thecenter of mass of the two chains (R) for n51 ~m!, 3 ~.!, 7 ~j! and 15~l!at temperatures of~a! kBT/e52.0 and ~b! 4.0 for the model with rigidbonds. For clarity only every sixth data point is shown, although the lipass through all the data points.
eton
t
ly
t
rs--
enct
e
o
en-
creasingn, we have calculated the probability of two independently grown athermal chains sharing a common ceof mass without any molecular overlaps by generating 16
pairs of hard sphere chains whose bond length is equal todiameter of the monomers. These simulations showwhile the probability of generating nonoverlapping configrations with common center of masses increases~i.e., inter-action energy decreases! with increasingn, the probability,and therefore the interaction energy, do appear to be sating at a constant value for largen. These observations arconsistent with our, and previous,6,7 simulation results whichdisplay a rapidly decreasingU(0) for short chains, and thepredictedn independence ofU(0) for largen.
Figure 3 also shows that the location and depth ofattractive minimum vary with chain length and solvent codition. In both good and poor solvent conditions the locatiof the minimum is shifted to larger separation as the chlength is increased, reflecting the larger size of the polymchains in both the coil and globule states. The depth ofattractive well depends, however, upon the temperaturegood solvent conditions the depth of the minimum decreawith increasing chain length, indicating that increasing chlength results in stronger repulsive interactions betweencoils. Alternatively, in poor solvent conditions the depththe minimum increases with increasing chain length, indicing that increasing chain length results in stronger attracinteractions between the globules. The minima we obserat nonzero separationsR in both good and poor solvent conditions are most likely to be artifacts of the short chalengths employed in the current study. In good solvent cditions we anticipate that as the chain length is increaseddepth of the minimum will decrease further, such thatsufficiently long chains no minimum will be observed.poor solvent conditions we expect that the interaction eneat short distances will be reduced as the two chains coalin a single dense globule and an approximately constantractive energy should be observed~the strength of which isdetermined by surface effects and should scale asn2/3).Clearly the chains considered here are too short for thpredictions to be confirmed, and as a result further simutions are certainly warranted. That said, the trends repohere, within good solvent conditions, are in good agreemwith the results of Harismiadis and Szleifer,7 although wenote that our interaction energy, for a chain of lengthn515 atkBT/e54.0, at zero separation is larger, by appromately 1.5e. Although no error estimates are quoted in R7, this deviation can easily be accounted for, arising dueour use of the truncated-shifted Lennard-Jones potenOnce again, behavior similar to that of the rigid bond modwas observed for the models with flexible bonds.
C. Second virial coefficients and u temperatures
In Fig. 4 we show the second virial coefficient,A2 , ob-tained by numerical integration of Eq.~2! @where M5(n11)m0 , with m0 being the molar mass of a monomer#, as afunction of temperature for chain lengths varying fromn51 to 15 for all three models. Error estimates were obtainby performing numerical integration from the effective inte
s

4727J. Chem. Phys., Vol. 118, No. 10, 8 March 2003 Monte Carlo simulation of homopolymer chains
FIG. 4. Second virial coefficient (A2) as a function oftemperature for~a! the model with rigid bonds,~b! themodel with flexible bonds andkFENE530e/s2, and~c!the model with flexible bonds andkFENE510e/s2 forvarious chain lengths,n51 ~d!, 3 ~m!, 7 ~j!, and 15~l!.
is-
alghrthd
or-rial
he
molecular interactions determined from the probability dtributionsPs(R)6s(R). The qualitative behavior of the second virial coefficient with temperature is the same formolecular weights and is similar for all three models. At hitemperatures the second virial coefficient is positive, cleashowing that repulsive interactions dominate betweentwo chains.A2 decreases with decreasing temperature, in
-
l
lyei-
cating that attractive interactions start to play a more imptant role. All the curves show a point where the second vicoefficient vanishes. This is theu temperature,un . For themodels with rigid bonds and flexible bonds withkFENE
530e/s2, un is dependent upon the chain length, but for tmodel with flexible bonds andkFENE510e/s2, un remainsconstant ~within numerical error! independent of chain

la
ew
d
dle
dlynpo
icedmoteoth
h
reenri
lltrao
ed
ac-of
ent
andain
s
is
ol-
to
4728 J. Chem. Phys., Vol. 118, No. 10, 8 March 2003 Withers et al.
length. Below theu temperature,A2 displays a negativevalue, indicative of poor solvent conditions where molecuattraction is dominant.
In order to examine the chain length dependence of thupoint we note that previous simulation studies have shothat un varies asn21/2,8,27,28 although more sophisticatescaling relations have also been used.10 In Fig. 5 we plotun
versusn21/2 for all three models. For the model with rigibonds, linear dependence is clearly observed, and asquares fit of the form
kBun /e5Bn21/21kBu` /e ~8!
yields values of B520.3160.03 and kBu` /e53.5260.01. Our value of theu temperature for an infinitely longchain is considerably less than that obtained by Harismiaand Szleifer7 (;3.9), however, the discrepancy seems liketo be due to our use of the truncated-shifted Lennard-Jopotential, since, as we have already shown, the shiftedtential enhances the repulsive contribution to the intermlecular interaction, in turn lowering the temperature at whthe second virial coefficient vanishes. That said, we concthat this estimate ofu` has been obtained using data froshort chains, and that the value quoted above may be mfied by considering longer chains and/or a more sophisticascaling analysis. Figure 5 also shows the introductionflexible bonds into the model results in a reduction of boun and u` , such thatkBu` /e53.18360.008 and 2.85660.006 forkFENEs
2/e530 and 10, respectively, along wita suppression of then dependence ofun , B520.1460.01and 0.0160.01 forkFENEs
2/e530 and 10, respectively.To compare our simulation results with experimental
sults we attempt to collapse our data for the three differmodel polymers onto a single universal curve with expemental data. To achieve this we note that in theu regime theinteraction energy between two overlapping chains is smathan the thermal energy, such that the chains interpeneeach other and the monomers interact directly. The secvirial coefficient is therefore proportional to the excludvolumev of a Kuhn segment,31
FIG. 5. u temperature (kBun /e) as a function of chain length (n21/2) for themodel with rigid bonds~d! and the model with flexible bonds andkFENE
530e/s2 ~m! andkFENE510e/s2 ~j!. The lines show least squares fitsthe simulation data.
r
n
ast
is
eso--
he
di-df
-t
-
erte
nd
A25NAv
2M02 , ~9!
whereM0 is the molar mass of a Kuhn segment andNA isthe Avogadro’s number. Let us introduce the chain intertion parameterzth that is the square root of the numberthermal blobs per chain
zth5S N
NthD 1/2
, ~10!
where
Nth5S 2C1
b3
v D 2
~11!
is the number of Kuhn segments in a thermal blob andC1 isa numerical constant. This interaction parameterzth cantherefore be written in terms of the length of a Kuhn segmb and its excluded volumev
zth51
2C1
vb3 N1/2. ~12!
Combining this expression with Eq.~9! one can obtain therelation between second virial coefficientA2 and the interac-tion parameterzth ,
A25C1
NAb3
M03/2M1/2zth for zth,1. ~13!
In good solvents the chains repel each other stronglydo not interpenetrate. Since the volume excluded by a chis of the order of its pervaded volume,R3,
A2}NAR3
M2 , ~14!
and the relative swelling of the chains in good solvents i
R
bN1/2}zth0.176 ~15!
the second virial coefficient in the good solvent regimegiven by
A2}NAb3
M03/2M1/2zth
0.528 for zth.1. ~16!
Combining Eqs.~13! and~16! and rearranging to obtaindimensionless quantities one can write
A2M1/2M0
3/2
NAb3 5C1H zth zth,1 ~u solvent!,
zth0.528 zth.1 ~good solvent!.
~17!
Taking into account the linear dependence of excluded vume parameterv on the effective temperature
v52C1C2b3S T2un
T D ~18!
we can relate the interaction parameterzth to the reducedtemperature
zth5C2N1/2T2un
T, ~19!
exie
ne
e
t
-. Ith
be
ldudynto
rri-
ere
tion
ri-er-
reeble
l,ndads
am-
ionnde-ep-ibleondvey-onic
ilityof
inghetheo-
ofre-
is-s ofion
re
s
4729J. Chem. Phys., Vol. 118, No. 10, 8 March 2003 Monte Carlo simulation of homopolymer chains
whereC2 is another numerical constant. Therefore onepects the universal relation between second virial coefficA2 and reduced temperature.
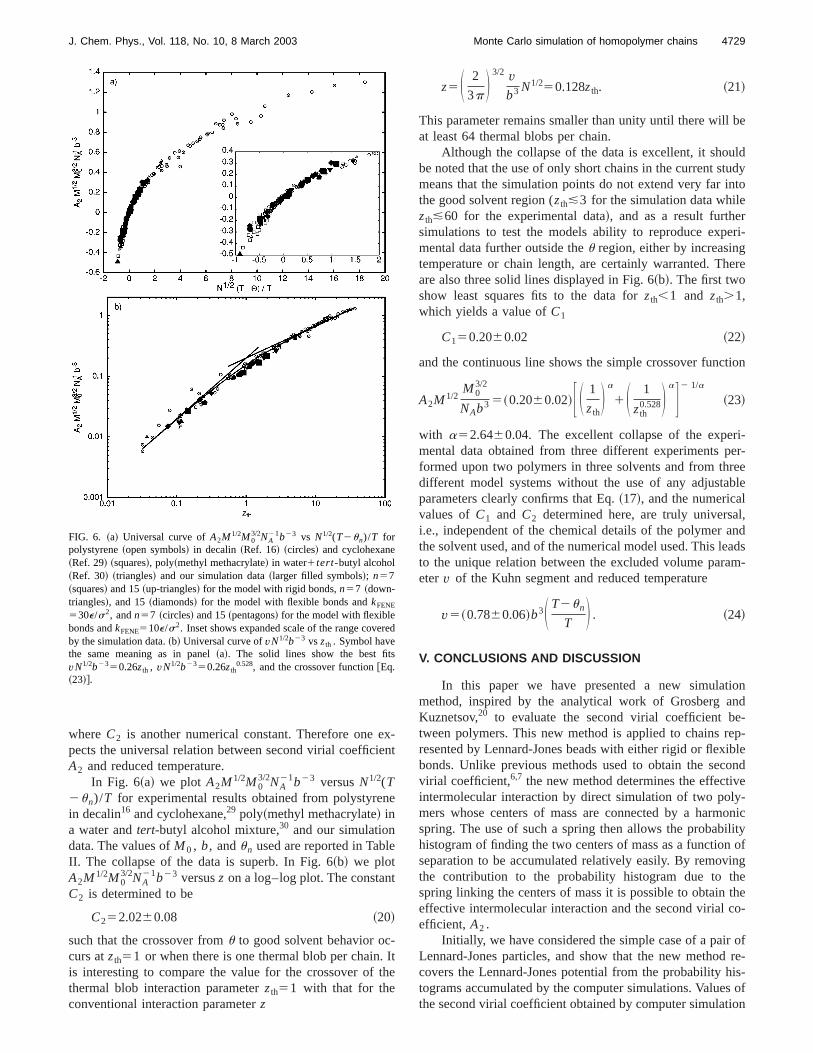
In Fig. 6~a! we plot A2M1/2M03/2NA
21b23 versusN1/2(T2un)/T for experimental results obtained from polystyrein decalin16 and cyclohexane,29 poly~methyl methacrylate! ina water andtert-butyl alcohol mixture,30 and our simulationdata. The values ofM0 , b, andun used are reported in TablII. The collapse of the data is superb. In Fig. 6~b! we plotA2M1/2M0
3/2NA21b23 versusz on a log–log plot. The constan
C2 is determined to be
C252.0260.08 ~20!
such that the crossover fromu to good solvent behavior occurs atzth51 or when there is one thermal blob per chainis interesting to compare the value for the crossover ofthermal blob interaction parameterzth51 with that for theconventional interaction parameterz
FIG. 6. ~a! Universal curve ofA2M 1/2M03/2NA
21b23 vs N1/2(T2un)/T forpolystyrene~open symbols! in decalin~Ref. 16! ~circles! and cyclohexane~Ref. 29! ~squares!, poly~methyl methacrylate! in water1tert-butyl alcohol~Ref. 30! ~triangles! and our simulation data~larger filled symbols!; n57~squares! and 15~up-triangles! for the model with rigid bonds,n57 ~down-triangles!, and 15~diamonds! for the model with flexible bonds andkFENE
530e/s2, andn57 ~circles! and 15~pentagons! for the model with flexiblebonds andkFENE510e/s2. Inset shows expanded scale of the range coveby the simulation data.~b! Universal curve ofvN1/2b23 vs zth . Symbol havethe same meaning as in panel~a!. The solid lines show the best fitvN1/2b2350.26zth , vN1/2b2350.26zth
0.528, and the crossover function@Eq.~23!#.
-nt
te
z5S 2
3p D 3/2 vb3 N1/250.128zth. ~21!
This parameter remains smaller than unity until there willat least 64 thermal blobs per chain.
Although the collapse of the data is excellent, it shoube noted that the use of only short chains in the current stmeans that the simulation points do not extend very far ithe good solvent region (zth&3 for the simulation data whilezth&60 for the experimental data!, and as a result furthesimulations to test the models ability to reproduce expemental data further outside theu region, either by increasingtemperature or chain length, are certainly warranted. Thare also three solid lines displayed in Fig. 6~b!. The first twoshow least squares fits to the data forzth,1 and zth.1,which yields a value ofC1
C150.2060.02 ~22!
and the continuous line shows the simple crossover func
A2M1/2M0
3/2
NAb3 5~0.2060.02!F S 1
zthD a
1S 1
zth0.528D aG2 1/a
~23!
with a52.6460.04. The excellent collapse of the expemental data obtained from three different experiments pformed upon two polymers in three solvents and from thdifferent model systems without the use of any adjustaparameters clearly confirms that Eq.~17!, and the numericalvalues ofC1 and C2 determined here, are truly universai.e., independent of the chemical details of the polymer athe solvent used, and of the numerical model used. This leto the unique relation between the excluded volume pareterv of the Kuhn segment and reduced temperature
v5~0.7860.06!b3S T2un
T D . ~24!
V. CONCLUSIONS AND DISCUSSION
In this paper we have presented a new simulatmethod, inspired by the analytical work of Grosberg aKuznetsov,20 to evaluate the second virial coefficient btween polymers. This new method is applied to chains rresented by Lennard-Jones beads with either rigid or flexbonds. Unlike previous methods used to obtain the secvirial coefficient,6,7 the new method determines the effectiintermolecular interaction by direct simulation of two polmers whose centers of mass are connected by a harmspring. The use of such a spring then allows the probabhistogram of finding the two centers of mass as a functionseparation to be accumulated relatively easily. By removthe contribution to the probability histogram due to tspring linking the centers of mass it is possible to obtaineffective intermolecular interaction and the second virial cefficient,A2 .
Initially, we have considered the simple case of a pairLennard-Jones particles, and show that the new methodcovers the Lennard-Jones potential from the probability htograms accumulated by the computer simulations. Valuethe second virial coefficient obtained by computer simulat
d
ata
4730 J. Chem. Phys., Vol. 118, No. 10, 8 March 2003 Withers et al.
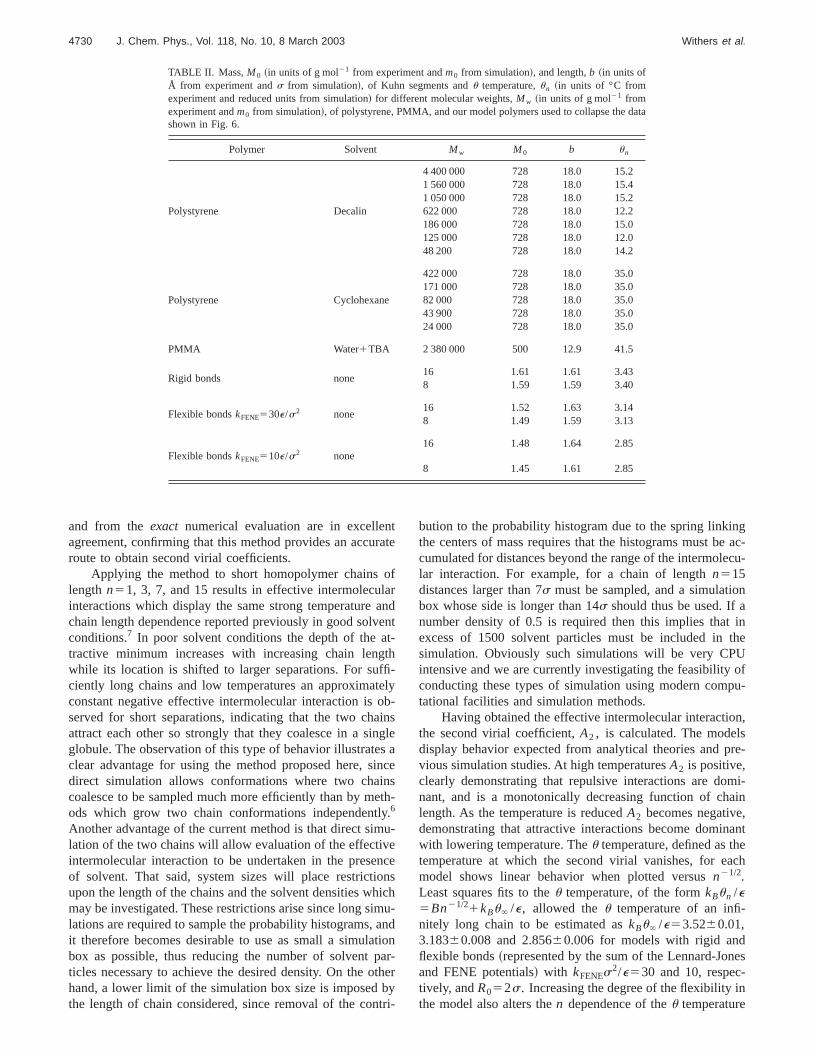
TABLE II. Mass,M 0 ~in units of g mol21 from experiment andm0 from simulation!, and length,b ~in units ofÅ from experiment ands from simulation!, of Kuhn segments andu temperature,un ~in units of °C fromexperiment and reduced units from simulation! for different molecular weights,Mw ~in units of g mol21 fromexperiment andm0 from simulation!, of polystyrene, PMMA, and our model polymers used to collapse the dshown in Fig. 6.
Polymer Solvent Mw M0 b un
4 400 000 728 18.0 15.21 560 000 728 18.0 15.41 050 000 728 18.0 15.2
Polystyrene Decalin 622 000 728 18.0 12.2186 000 728 18.0 15.0125 000 728 18.0 12.048 200 728 18.0 14.2
422 000 728 18.0 35.0171 000 728 18.0 35.0
Polystyrene Cyclohexane 82 000 728 18.0 35.043 900 728 18.0 35.024 000 728 18.0 35.0
PMMA Water1TBA 2 380 000 500 12.9 41.5
Rigid bonds none16 1.61 1.61 3.438 1.59 1.59 3.40
Flexible bondskFENE530e/s2 none16 1.52 1.63 3.148 1.49 1.59 3.13
Flexible bondskFENE510e/s2 none16 1.48 1.64 2.85
8 1.45 1.61 2.85
tra
oaravet-
gtffiteobin
ngsin
insetly.
uencn
hicimanti
path
bytr
gac-
cu-
n
intheUofpu-
n,sre-
mi-ain,anteach
es-n
and from theexact numerical evaluation are in excellenagreement, confirming that this method provides an accuroute to obtain second virial coefficients.
Applying the method to short homopolymer chainslengthn51, 3, 7, and 15 results in effective intermoleculinteractions which display the same strong temperaturechain length dependence reported previously in good solconditions.7 In poor solvent conditions the depth of the atractive minimum increases with increasing chain lenwhile its location is shifted to larger separations. For suciently long chains and low temperatures an approximaconstant negative effective intermolecular interaction isserved for short separations, indicating that the two chaattract each other so strongly that they coalesce in a siglobule. The observation of this type of behavior illustrateclear advantage for using the method proposed here, sdirect simulation allows conformations where two chacoalesce to be sampled much more efficiently than by mods which grow two chain conformations independent6
Another advantage of the current method is that direct simlation of the two chains will allow evaluation of the effectivintermolecular interaction to be undertaken in the preseof solvent. That said, system sizes will place restrictioupon the length of the chains and the solvent densities wmay be investigated. These restrictions arise since long slations are required to sample the probability histograms,it therefore becomes desirable to use as small a simulabox as possible, thus reducing the number of solventticles necessary to achieve the desired density. On the ohand, a lower limit of the simulation box size is imposedthe length of chain considered, since removal of the con
te
f
ndnt
h-ly-sleace
h-
-
eshu-d
onr-er
i-
bution to the probability histogram due to the spring linkinthe centers of mass requires that the histograms must becumulated for distances beyond the range of the intermolelar interaction. For example, for a chain of lengthn515distances larger than 7s must be sampled, and a simulatiobox whose side is longer than 14s should thus be used. If anumber density of 0.5 is required then this implies thatexcess of 1500 solvent particles must be included insimulation. Obviously such simulations will be very CPintensive and we are currently investigating the feasibilityconducting these types of simulation using modern comtational facilities and simulation methods.
Having obtained the effective intermolecular interactiothe second virial coefficient,A2 , is calculated. The modeldisplay behavior expected from analytical theories and pvious simulation studies. At high temperaturesA2 is positive,clearly demonstrating that repulsive interactions are donant, and is a monotonically decreasing function of chlength. As the temperature is reducedA2 becomes negativedemonstrating that attractive interactions become dominwith lowering temperature. Theu temperature, defined as thtemperature at which the second virial vanishes, for emodel shows linear behavior when plotted versusn21/2.Least squares fits to theu temperature, of the formkBun /e5Bn21/21kBu` /e, allowed theu temperature of an infi-nitely long chain to be estimated askBu` /e53.5260.01,3.18360.008 and 2.85660.006 for models with rigid andflexible bonds~represented by the sum of the Lennard-Jonand FENE potentials! with kFENEs
2/e530 and 10, respectively, andR052s. Increasing the degree of the flexibility ithe model also alters then dependence of theu temperature

s
eo-tinu
ecge
ohal
t
ex
isa
lu-
-ethn
thoweteth
nt,
ethe
rlyere-f ithe
TCee-o.
rthmensr
hys.
-
4731J. Chem. Phys., Vol. 118, No. 10, 8 March 2003 Monte Carlo simulation of homopolymer chains
with the absolute value ofB decreasing, such thatB520.3160.03 for the model with rigid bonds,20.1460.01 and 0.0160.01 for the models with flexible bondandkFENEs
2/e530 and 10, respectively.The reason for the observedn independence of theu
temperature for the model with flexible bonds andkFENE
510e/s2 is not clear since the second virial coefficient btween two chains is obtained from a renormalization of twthree-, etc., body interaction.4 Such calculations predict thathe n dependence ofun should be suppressed by decreasthe strength of three-body repulsions. However, the introdtion of flexible bonds into our model results in the aspratio of the Kuhn segments remaining essentially unchan(b51.61s for the model with rigid bonds compared withb51.64s for the model with flexible bonds andkFENE
510e/s2), and it is therefore expected that the strengththree-body repulsions will be similar in these models. Tsaid, determination of theu temperature from conformationaproperties of the bond-fluctuation model19 also indicates thathe introduction of flexible bonds suppresses then depen-dence ofun .
The suppression of then dependence ofun for the modelpolymer chain with flexible bonds andkFENE510e/s2 makesthis model an ideal candidate for further study since inperimental systems theu temperature either displays nondependence or very weakn dependence. Furthermore, thmodel is ideal for a detailed examination of the conformtional properties since the location ofu temperature is nowknown for all chain lengths. Thus comparison of the vakBun /e5kBu` /e52.85660.006 obtained here with methods typically used to determine theu temperature from conformational properties, along with comparison with theorical predictions, may be made with relative ease. Sucstudy has been undertaken and the results will be presein Paper II.18
Finally, we have compared our numerical results ofsecond virial coefficient with experimental results and shthat the simulation data may be collapsed onto the expmental data without the use of any adjustable parameThis process leads to the following universal functions ofsecond virial coefficient,A2 ,
A2M1/2M0
3/2
NAb3
5H ~0.3960.03!S N1/2T2un
T D ~u solvent!
~0.2860.01!S N1/2T2un
T D 0.528
~good solvent!
~25!
5~0.2060.02!H zth ~zth,1,u solvent!
zth0.528 ~zth.1, good solvent!
~26!
5~0.2060.02!F S 1
zthD 2.6
1S 1
zth0.528D 2.6G2 1/2.6
, ~27!
-,
gc-td
ft
-
-
e
-a
ted
e
ri-rs.e
whereM0 andb are the mass and length of a Kuhn segmerespectively. Furthermore, Eqs.~25! and ~9! leads to an ex-cluded volume of a Kuhn segment,
v5~0.7860.06!b3S T2un
T D . ~28!
In Paper II18 we will show that this relation, used within thmean-field Flory theory, successfully predicts the size ofmodel polymer chains at temperatures close to theu tem-perature.
The observation of superb collapse of the data cleashows that these are truly universal functions and may thfore be applied to any linear homopolymer independent ochemical details and type of solvent without alteration of tprefactors in Eqs.~25!–~27!.
ACKNOWLEDGMENTS
The material is based upon work supported by the SProgram of the National Science Foundation under Agrment No. CHE-9876674, and by the NSF under Grant NDMR-0102267. We also wish to acknowledge the NoCarolina Super Computer Center for an award of CPU tion the IBM SP. This work has benefitted from discussiowith Dr. M. Adam and Dr. R. Colby, and we thank them fotheir contributions.
1P. J. Flory,Principles of Polymer Chemistry~Cornell University Press,Ithaca, NY, 1967!.
2P. J. Flory,Statistical Mechanics of Chain Molecules~Interscience, NewYork, 1969!.
3H. Yamakawa,Modern Theory of Polymer Solution~Harper and Row,New York, 1971!.
4J. des Cloizeaux and G. Jannink,Polymers in Solution: Their Modelingand Structure~Claredon, Oxford, 1990!.
5Refs. 1–28 of Paper II~Ref. 18!.6J. Dautenhahn and C. K. Hall, Macromolecules27, 5399~1994!.7V. I. Harismiadis and I. Szleifer, Mol. Phys.81, 851 ~1994!.8P. Grassberger and R. Hegger, J. Chem. Phys.102, 6881~1995!.9A. M. Rubio and J. J. Freire, Macromolecules29, 6946~1996!.
10A. M. Rubio and J. J. Freire, J. Chem. Phys.106, 5638~1997!.11L. Leu and J. M. Prausnitz, Macromolecules30, 6650~1997!.12Y. C. Chiew and V. Sabesan, Fluid Phase Equilib.155, 75 ~1999!.13C. Vega, J. M. Labaig, L. G. MacDowell, and E. Sanz, J. Chem. Phys.113,
10398~2000!.14P. G. Bolhuis, A. A. Loius, J. P. Hansen, and E. J. Meijer, J. Chem. P
114, 4296~2001!.15G. Swislow, S.-T. Sun, I. Nishio, and T. Tanaka, Phys. Rev. Lett.44, 796
~1980!.16G. C. Berry, J. Chem. Phys.44, 4550~1966!.17Y. Nakamura, T. Norisuya, and A. Teramoto, Macromolecules24, 4904
~1991!.18I. M. Withers, A. V. Dobrynin, and M. Rubinstein, J. Chem. Phys.~to be
published!.19M. Wittkop, S. Kreitmeier, and D. Go¨ritz, J. Chem. Phys.104, 3373
~1996!.20A. Y. Grosberg and D. V. Kuznetsov, J. Phys. II2, 1327~1992!.21D. A. McQuarrie,Statistical Thermodynamics~Harper & Row, New York,
1976!.22D. Frenkel and B. Smit,Understanding Molecular Simulation—From Al
gorithms to Applications~Academic, London, 1996!.23E. Marinari and G. Parisi, Europhys. Lett.19, 451 ~1992!.

.,
4732 J. Chem. Phys., Vol. 118, No. 10, 8 March 2003 Withers et al.
24W. T. Vetterling W. H. Press, S. A. Teukolsky, and B. P. Flannery,Numeri-cal Recipies, 2nd ed.~Cambridge University Press, Cambridge, 1992!.
25P. J. Flory and W. R. Krigbaum, J. Chem. Phys.18, 1086~1950!.26A. Y. Grosberg, P. G. Khalatur, and A. R. Khokhlov, Makromol. Chem
Rapid Commun.3, 709 ~1982!.27A. Baumgartner, J. Chem. Phys.72, 871 ~1980!.
28I. Szleifer, E. M. O’Toole, and A. Z. Panagiotopoulos, J. Chem. Phys.97,6802 ~1992!.
29R. Perzynski. Ph.D. thesis, Universite Paris VI, 1984.30M. Nakata, Phys. Rev. E51, 5770~1995!.31L. H. Sperling, Introduction to Physical Polymer Science~Wiley, New
York, 1992!.





















