
Development of a Sample Environment for Neutron Diffraction ...
Upload
independentCategory
view
3download
0
www.elsevier.com/locate/molstruc
Journal of Molecular Structure 842 (2007) 93–99
Molecular association in deuterated liquidN-methylacetamide–water complex: Neutron diffraction
and density functional theory study
Sahbi Trabelsi a, Salah Nasr a,*, Mohamed Bahri b, Marie-Claire Bellissent-Funel c
a Laboratoire Physico-Chimie des Materiaux, Departement de Physique, Faculte des Sciences de Monastir, Monastir 5019, Tunisieb Laboratoire de Spectroscopie Atomique, Moleculaire et Applications, Departement de Physique, Faculte des Sciences,
Universite Tunis-El Manar, 1060 Tunis, Tunisiec Laboratoire Leon Brillouin (CEA-CNRS) CEA-Saclay, 91191 Gif-sur-Yvette Cedex, France
Received 16 October 2006; received in revised form 13 December 2006; accepted 16 December 2006Available online 11 January 2007
Abstract
A structural investigation of fully deuterated liquid N-methylacetmide–water complexes (NMA-W) is performed at 308 K and atmo-spheric pressure by using neutron diffraction in combination with a density functional theory (DFT). The data are analyzed to yield themolecular form factor F1(Q) and the distinct pair correlation function gL(r). DFT calculations are performed to examine some possibleclusters that may describe the local arrangement in the liquid state. Neutron measurements can be interpreted in term of trans-NMA-3D2O complex. In this model there are two water molecules hydrogen bonded to the C@O oxygen and one to the N–H group. The inter-molecular structure factor extracted from this cluster fairly agrees with the experimental one beyond Q = 3 A�1.� 2007 Elsevier B.V. All rights reserved.
Keywords: NMA-W complex; Local order; H-bonding; Neutron diffraction; DFT
1. Introduction
The trans secondary amide function, also called the pep-tide group is a basic unit of biological macromolecules suchas peptides and proteins [1]. For understanding the struc-tural properties and biological functions of proteins as wellas the effects of the solvent on the stabilization of proteinstructures in solution, it is essential to know how the pep-tide group interacts in an aqueous environment throughhydrogen bonding. N-Methylacetamide (NMA) is one ofthe simplest model compounds having the peptide bond,providing a good basis for the study of large hydrogen-bonded systems such as proteins [2–4]. This molecule hastwo sites for hydrogen bonding; the hydrogen atom ofthe amino group acts as a proton donor and the oxygenatom of the carbonyl group acts as a proton acceptor. In
0022-2860/$ - see front matter � 2007 Elsevier B.V. All rights reserved.
doi:10.1016/j.molstruc.2006.12.028
* Corresponding author.E-mail address: [email protected] (S. Nasr).
order to understand the hydrogen bonding strengths andtheir effect on the structure of NMA, Guo and Karplus[5] have carried out a computational chemical study onthe NMA–water complexes and have found that a trans-NMA interacts preferentially with three water molecules.They have also found that water molecules bind to the pep-tide group and the hydrogen bond increases the methylrotational barriers. Other analyses [6–9], including simula-tion studies, have focused on the ability of water moleculesto compete with peptide hydrogen bonds. Previous investi-gations including NMR data result [10], UV resonanceRaman experimental [11], and ab initio calculations [12]have indicated that NMA in aqueous solution is mainlytrans conformation. Subsequently, Han and Subai [13]have performed DFT calculations to investigate the prop-erties of several trans- and cis-NMA–water complexes.They have concluded that, the cooperative effects of hydro-gen bonding is found in the trans-NMA–water complexesand have suggested that cis-NMA in aqueous solution

94 S. Trabelsi et al. / Journal of Molecular Structure 842 (2007) 93–99
(water) is unstable. However, none of earlier studies wasinterested in experimentally characterizing the intermolecu-lar arrangement in NMA-W complex. This is the reasonwhy the problem of local order in this system is reconsid-ered in the present study. We particularly aim to investigatethe intermolecular arrangement in NMA-W complex byusing neutron diffraction data in combination with aDFT calculation. The later technique is qualified to be amore adapted method for geometry prediction and physi-cochemical properties investigation in amides [4]. In oursystem the number of D2O molecules is chosen for beingthree times more important than the number of NMA onesto test particularly the formation of the more probablecluster predicted by Guo and Karplus [5]. First, scatteringdata are analyzed to yield the total structure and the inter-molecular structure factors. Second, the theoretical intra-molecular distances for the NMA and D2O monomersare considered to construct the molecular form factor.Third, to examine the intermolecular interactions due tothe contribution of the hydrogen-bond quantities that aregenerally difficult to determine experimentally, the latterare deduced here theoretically via a DFT calculation ofthe minimum energy structure for some NMA–water clus-ters. Finally, the computed intermolecular pair correlationfunction for each model is then compared with that extract-ed from experimental data.
2. Experiment details
The fully deuterated N-methylacetamide–water complex(NMA-W) was supplied by C/D/N isotopes (chemicalpurity 99% +). In the experiments, NMA-W was usedwithout further purification. The sample holders were filledand sealed in a glove box in order to avoid contaminationby atmospheric water.
The neutron scattering experiments were performed atthe reactor Orphee of the Laboratoire Leon Brillouin, atSaclay (France) using the 7C2 spectrometer. All detailsabout this spectrometer have been reported by Ambroise,Bellissent-Funel, and Bellissent [14,15]. This spectrometeris equipped with a BF3 position sensitive detector with640 cells; the angular step between two adjacent cells isequal to 0.2�, which leads to a maximum diffraction angleof 128.7�. We selected an incident wavelength ofk = 0.711 A by means of a Cu (111) monochromator.The sample is contained inside a vanadium cell, having adiameter of 8 and 0.1 mm wall thickness. All experimentswere carried out at 308 K. The usual measurements ofthe vanadium rodðIexp t
v Þ, the cadmium rodðIexp tcd Þ and the
empty cryostat ðIexp tvacuumÞ were also performed.
3. Theoretical considerations
3.1. Neutron scattering theory
Neutron scattering by molecular liquids has been dis-cussed in many papers [16–20]. The data treatment and
the fitting procedures can also be found in Refs. [21,22].Only the necessary details will be reported here.
The coherent contribution of differential scatteringcross-section of an assembly of N molecules each with m
atoms is given, in the static approximation, by [16]
drdX
� �coh
¼ 1
N
XN
j;k¼1
Xm
a;b¼1
bjabkb exp iQðrja � rkbÞ( )* +
; ð1Þ
where bja is the coherent scattering length of the nucleus afteraveraging over all isotopes and nuclear spins; Q = k0 � k isthe scattering wave vector, where k0 and k are, respectively,the wave vectors of incident and scattered radiations.(jQj ¼ Q ¼ 4p sin H
k in the case of elastic scattering), 2H is thescattering angle, k is the wavelength of the radiation and rja
is the position vector of the atom a in the molecule j. TheÆ æ brackets denote an average over the grand canonicalensemble.
The interference function or total structure factor SM(Q)of liquid NMA-W is given by:
SMðQÞ ¼drdX
� �coh
� �� Xm
a¼1
ba
!2
: ð2Þ
At large Q, SM(Q) goes to the asymptotic valueSMð1Þ ¼ ð
Pab2
aÞ=ðP
abaÞ2. For Q fi 0, limQ!0SMðQÞ ¼qKBTvT, where q is the molecular number density; KB, theBoltzmann’s constant; T, the absolute temperature; and,vT, the isothermal compressibility.
For molecular liquids, the total structure factor SM(Q)can be separated into two terms arising from intramolecu-lar and intermolecular terms, such as [21]:
SMðQÞ ¼ F 1ðQÞ þ DMðQÞ; ð3Þwhere F1(Q) is the molecular form factor which is given by[21]
F 1ðQÞ ¼Xm
a;b¼1
babb
sinðQrabÞQrab
exp �hDr2abi
Q2
2
� �, Xm
a¼1
ba
!2
:
ð4ÞThe distinct structure factor DM(Q) which contains the
information on the intermolecular structure of the liquidusually decays to zero very rapidly at large Q-values. How-ever, for hydrogen-bonded structures, this function contin-ues to oscillate in the high Q-range; therefore, it can be splitinto two parts [20,21]
DMðQÞ ¼ DHBM ðQÞ þ DNHB
M ðQÞ ð5Þwhere DHB
M ðQÞ represents the H-bonds contribution of agiven NMA molecule linked to D2O ones and DNHB
M ðQÞcontains the intermolecular correlations other than theH-bonded interactions.
The intermolecular pair correlation function ginter(r),may be expressed by
ginterðrÞ ¼ 1þ 1
2p2qr
Z 1
0
QDMðQÞ sinðQrÞdQ; ð6Þ
where q is the molecular number density of the liquid.
0
100000
200000
0 40 80 120
12
16
20
Iexp
t (cou
nts)
(A1)
(A2) (A3) (A4)
(A5)
2θ (deg)
( )Qd
d
Ωσ
P(θ)
a
b
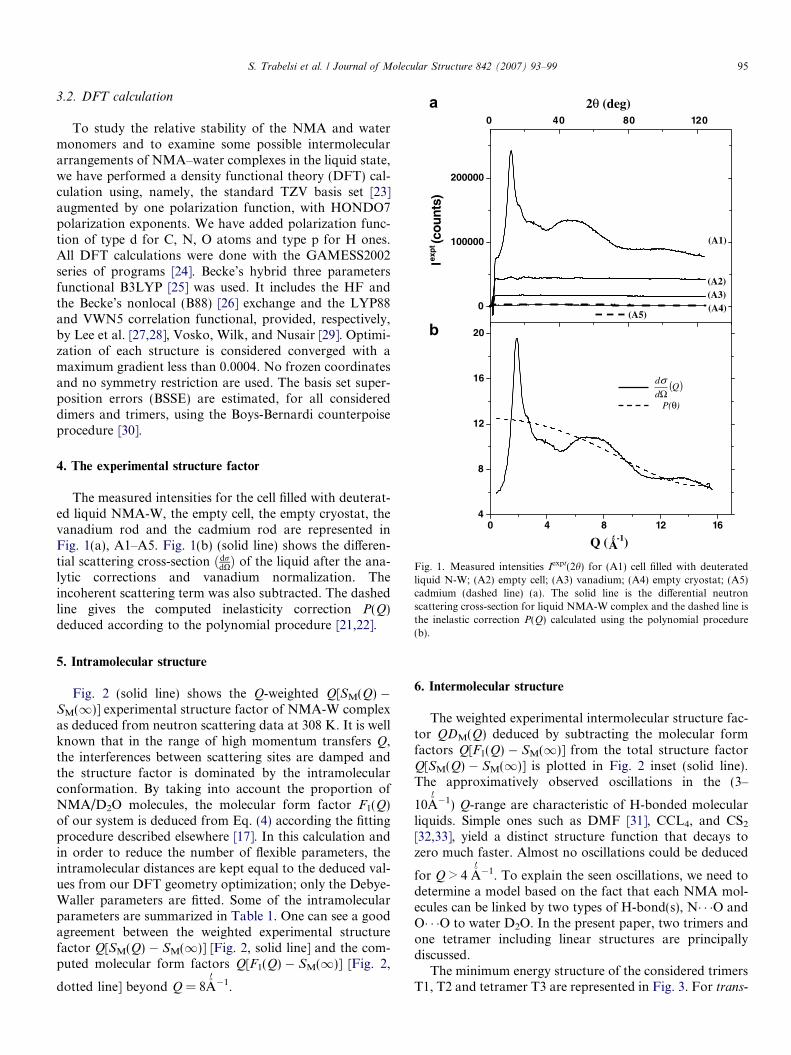
S. Trabelsi et al. / Journal of Molecular Structure 842 (2007) 93–99 95
3.2. DFT calculation
To study the relative stability of the NMA and watermonomers and to examine some possible intermoleculararrangements of NMA–water complexes in the liquid state,we have performed a density functional theory (DFT) cal-culation using, namely, the standard TZV basis set [23]augmented by one polarization function, with HONDO7polarization exponents. We have added polarization func-tion of type d for C, N, O atoms and type p for H ones.All DFT calculations were done with the GAMESS2002series of programs [24]. Becke’s hybrid three parametersfunctional B3LYP [25] was used. It includes the HF andthe Becke’s nonlocal (B88) [26] exchange and the LYP88and VWN5 correlation functional, provided, respectively,by Lee et al. [27,28], Vosko, Wilk, and Nusair [29]. Optimi-zation of each structure is considered converged with amaximum gradient less than 0.0004. No frozen coordinatesand no symmetry restriction are used. The basis set super-position errors (BSSE) are estimated, for all considereddimers and trimers, using the Boys-Bernardi counterpoiseprocedure [30].
0 4 8 12 164
8
Q ( -1)
Fig. 1. Measured intensities Iexpt(2h) for (A1) cell filled with deuteratedliquid N-W; (A2) empty cell; (A3) vanadium; (A4) empty cryostat; (A5)cadmium (dashed line) (a). The solid line is the differential neutronscattering cross-section for liquid NMA-W complex and the dashed line isthe inelastic correction P(Q) calculated using the polynomial procedure(b).
4. The experimental structure factor
The measured intensities for the cell filled with deuterat-ed liquid NMA-W, the empty cell, the empty cryostat, thevanadium rod and the cadmium rod are represented inFig. 1(a), A1–A5. Fig. 1(b) (solid line) shows the differen-tial scattering cross-section ðdr
dXÞ of the liquid after the ana-lytic corrections and vanadium normalization. Theincoherent scattering term was also subtracted. The dashedline gives the computed inelasticity correction P(Q)deduced according to the polynomial procedure [21,22].
5. Intramolecular structure
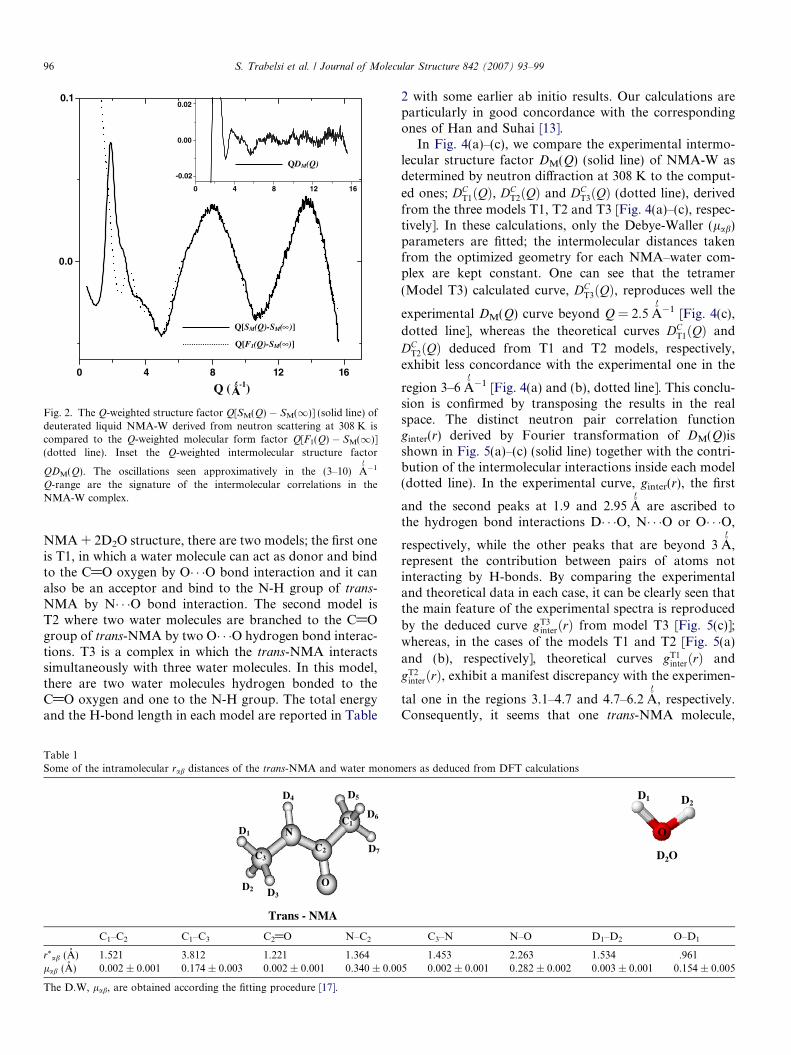
Fig. 2 (solid line) shows the Q-weighted Q[SM(Q) �SM(1)] experimental structure factor of NMA-W complexas deduced from neutron scattering data at 308 K. It is wellknown that in the range of high momentum transfers Q,the interferences between scattering sites are damped andthe structure factor is dominated by the intramolecularconformation. By taking into account the proportion ofNMA/D2O molecules, the molecular form factor F1(Q)of our system is deduced from Eq. (4) according the fittingprocedure described elsewhere [17]. In this calculation andin order to reduce the number of flexible parameters, theintramolecular distances are kept equal to the deduced val-ues from our DFT geometry optimization; only the Debye-Waller parameters are fitted. Some of the intramolecularparameters are summarized in Table 1. One can see a goodagreement between the weighted experimental structurefactor Q[SM(Q) � SM(1)] [Fig. 2, solid line] and the com-puted molecular form factors Q[F1(Q) � SM(1)] [Fig. 2,
dotted line] beyond Q = 8�A0�1.
6. Intermolecular structure
The weighted experimental intermolecular structure fac-tor QDM(Q) deduced by subtracting the molecular formfactors Q[F1(Q) � SM(1)] from the total structure factorQ[SM(Q) � SM(1)] is plotted in Fig. 2 inset (solid line).The approximatively observed oscillations in the (3–
10�A0�1) Q-range are characteristic of H-bonded molecular
liquids. Simple ones such as DMF [31], CCL4, and CS2
[32,33], yield a distinct structure function that decays tozero much faster. Almost no oscillations could be deduced
for Q > 4 �A0�1. To explain the seen oscillations, we need to
determine a model based on the fact that each NMA mol-ecules can be linked by two types of H-bond(s), N� � �O andO� � �O to water D2O. In the present paper, two trimers andone tetramer including linear structures are principallydiscussed.
The minimum energy structure of the considered trimersT1, T2 and tetramer T3 are represented in Fig. 3. For trans-
0 4 8 12 16
0.0
0.1
0 4 8 12 16
-0.02
0.00
0.02
Q ( -1)
Q[SM(Q)-SM( )]
Q[F1(Q)-SM( )]
QDM(Q)
∞
∞
Fig. 2. The Q-weighted structure factor Q[SM(Q) � SM(1)] (solid line) ofdeuterated liquid NMA-W derived from neutron scattering at 308 K iscompared to the Q-weighted molecular form factor Q[F1(Q) � SM(1)](dotted line). Inset the Q-weighted intermolecular structure factor
QDM(Q). The oscillations seen approximatively in the (3–10) �A0�1
Q-range are the signature of the intermolecular correlations in theNMA-W complex.
96 S. Trabelsi et al. / Journal of Molecular Structure 842 (2007) 93–99
NMA + 2D2O structure, there are two models; the first oneis T1, in which a water molecule can act as donor and bindto the C@O oxygen by O� � �O bond interaction and it canalso be an acceptor and bind to the N-H group of trans-NMA by N� � �O bond interaction. The second model isT2 where two water molecules are branched to the C@Ogroup of trans-NMA by two O� � �O hydrogen bond interac-tions. T3 is a complex in which the trans-NMA interactssimultaneously with three water molecules. In this model,there are two water molecules hydrogen bonded to theC@O oxygen and one to the N-H group. The total energyand the H-bond length in each model are reported in Table
Table 1Some of the intramolecular rab distances of the trans-NMA and water monom
C2
C1
C3
O
ND1
D2 D3
D4 D5
D6
D7
Trans - NMA
C1–C2 C1–C3 C2@O N–C2
r*ab (A) 1.521 3.812 1.221 1.364
lab (A) 0.002 ± 0.001 0.174 ± 0.003 0.002 ± 0.001 0.340 ± 0.00
The D.W, lab, are obtained according the fitting procedure [17].
2 with some earlier ab initio results. Our calculations areparticularly in good concordance with the correspondingones of Han and Suhai [13].
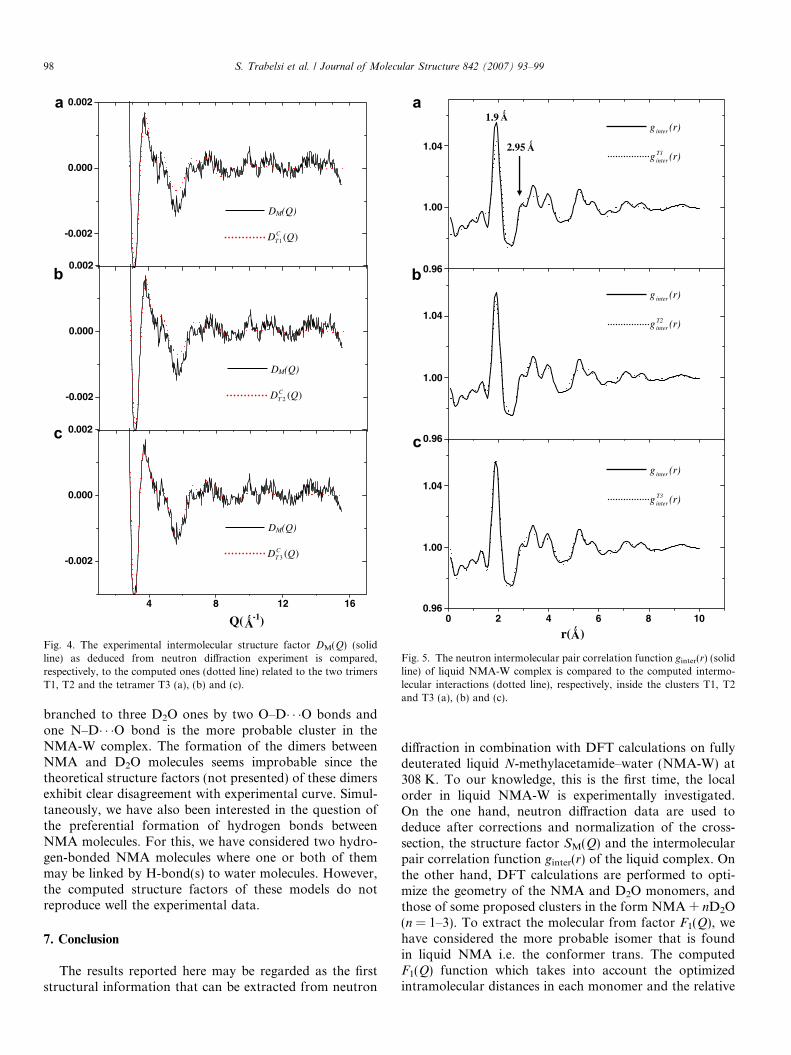
In Fig. 4(a)–(c), we compare the experimental intermo-lecular structure factor DM(Q) (solid line) of NMA-W asdetermined by neutron diffraction at 308 K to the comput-
ed ones; DCT1ðQÞ, DC
T2ðQÞ and DCT3ðQÞ (dotted line), derived
from the three models T1, T2 and T3 [Fig. 4(a)–(c), respec-tively]. In these calculations, only the Debye-Waller (lab)parameters are fitted; the intermolecular distances takenfrom the optimized geometry for each NMA–water com-plex are kept constant. One can see that the tetramer
(Model T3) calculated curve, DCT3ðQÞ, reproduces well the
experimental DM(Q) curve beyond Q = 2.5 �A0�1 [Fig. 4(c),
dotted line], whereas the theoretical curves DCT1ðQÞ and
DCT2ðQÞ deduced from T1 and T2 models, respectively,
exhibit less concordance with the experimental one in the
region 3–6 �A0�1 [Fig. 4(a) and (b), dotted line]. This conclu-
sion is confirmed by transposing the results in the realspace. The distinct neutron pair correlation functionginter(r) derived by Fourier transformation of DM(Q)isshown in Fig. 5(a)–(c) (solid line) together with the contri-bution of the intermolecular interactions inside each model(dotted line). In the experimental curve, ginter(r), the first
and the second peaks at 1.9 and 2.95 �A0
are ascribed tothe hydrogen bond interactions D� � �O, N� � �O or O� � �O,
respectively, while the other peaks that are beyond 3 �A0,
represent the contribution between pairs of atoms notinteracting by H-bonds. By comparing the experimentaland theoretical data in each case, it can be clearly seen thatthe main feature of the experimental spectra is reproducedby the deduced curve gT3
interðrÞ from model T3 [Fig. 5(c)];whereas, in the cases of the models T1 and T2 [Fig. 5(a)and (b), respectively], theoretical curves gT1
interðrÞ and
gT2interðrÞ, exhibit a manifest discrepancy with the experimen-
tal one in the regions 3.1–4.7 and 4.7–6.2 �A0, respectively.
Consequently, it seems that one trans-NMA molecule,
ers as deduced from DFT calculations
D1 D2
D2O
O
C3–N N–O D1–D2 O–D1
1.453 2.263 1.534 .9615 0.002 ± 0.001 0.282 ± 0.002 0.003 ± 0.001 0.154 ± 0.005
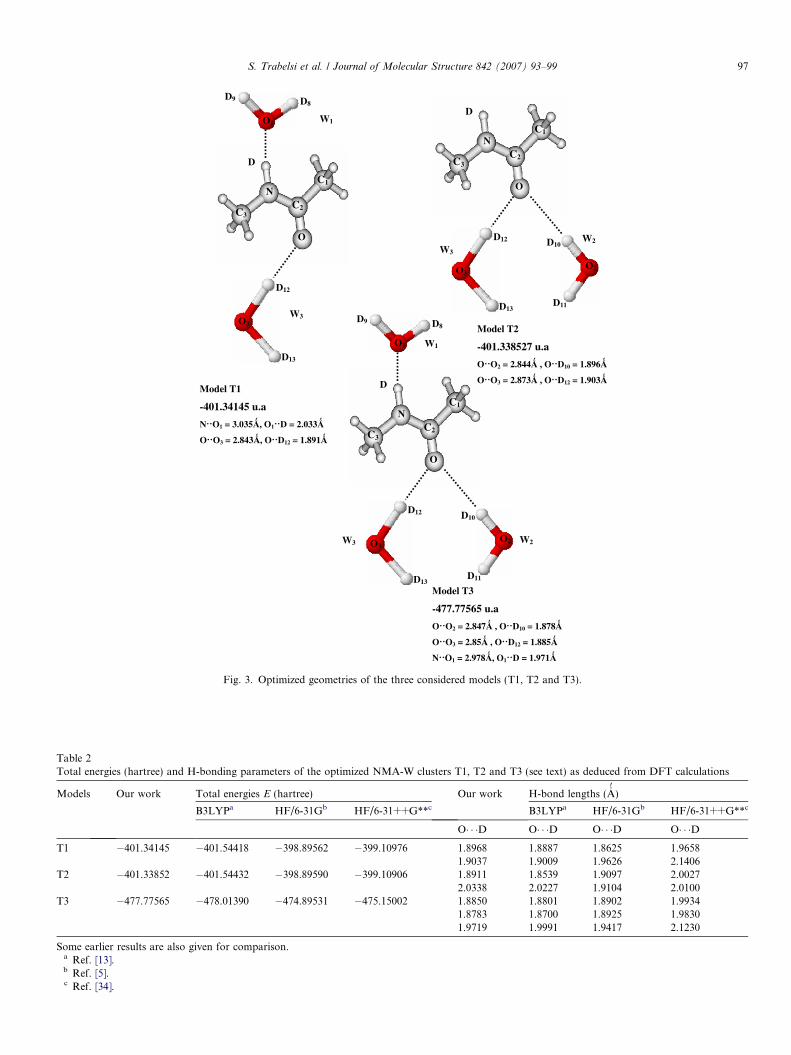
Fig. 3. Optimized geometries of the three considered models (T1, T2 and T3).
Table 2Total energies (hartree) and H-bonding parameters of the optimized NMA-W clusters T1, T2 and T3 (see text) as deduced from DFT calculations
Models Our work Total energies E (hartree) Our work H-bond lengths (�A0)
B3LYPa HF/6-31Gb HF/6-31++G**c B3LYPa HF/6-31Gb HF/6-31++G**c
O� � �D O� � �D O� � �D O� � �DT1 �401.34145 �401.54418 �398.89562 �399.10976 1.8968 1.8887 1.8625 1.9658
1.9037 1.9009 1.9626 2.1406T2 �401.33852 �401.54432 �398.89590 �399.10906 1.8911 1.8539 1.9097 2.0027
2.0338 2.0227 1.9104 2.0100T3 �477.77565 �478.01390 �474.89531 �475.15002 1.8850 1.8801 1.8902 1.9934
1.8783 1.8700 1.8925 1.98301.9719 1.9991 1.9417 2.1230
Some earlier results are also given for comparison.a Ref. [13].b Ref. [5].c Ref. [34].
S. Trabelsi et al. / Journal of Molecular Structure 842 (2007) 93–99 97
4 8 12 16
-0.002
0.000
0.002
-0.002
0.000
0.002
-0.002
0.000
0.002
Q( -1)
DM(Q)
)(1 QDCT
DM(Q)
)(2 QDCT
DM(Q)
)(3 QDCT
a
b
c
Fig. 4. The experimental intermolecular structure factor DM(Q) (solidline) as deduced from neutron diffraction experiment is compared,respectively, to the computed ones (dotted line) related to the two trimersT1, T2 and the tetramer T3 (a), (b) and (c).
0.96
1.00
1.04
0.96
1.00
1.04
0 2 4 6 8 100.96
1.00
1.04
r( )
(r)ginter
(r)g T3inter
(r)ginter
(r)g T1inter
(r)ginter
(r)g T2inter
1.9
2.95
a
b
c
Fig. 5. The neutron intermolecular pair correlation function ginter(r) (solidline) of liquid NMA-W complex is compared to the computed intermo-lecular interactions (dotted line), respectively, inside the clusters T1, T2and T3 (a), (b) and (c).
98 S. Trabelsi et al. / Journal of Molecular Structure 842 (2007) 93–99
branched to three D2O ones by two O–D� � �O bonds andone N–D� � �O bond is the more probable cluster in theNMA-W complex. The formation of the dimers betweenNMA and D2O molecules seems improbable since thetheoretical structure factors (not presented) of these dimersexhibit clear disagreement with experimental curve. Simul-taneously, we have also been interested in the question ofthe preferential formation of hydrogen bonds betweenNMA molecules. For this, we have considered two hydro-gen-bonded NMA molecules where one or both of themmay be linked by H-bond(s) to water molecules. However,the computed structure factors of these models do notreproduce well the experimental data.
7. Conclusion
The results reported here may be regarded as the firststructural information that can be extracted from neutron
diffraction in combination with DFT calculations on fullydeuterated liquid N-methylacetamide–water (NMA-W) at308 K. To our knowledge, this is the first time, the localorder in liquid NMA-W is experimentally investigated.On the one hand, neutron diffraction data are used todeduce after corrections and normalization of the cross-section, the structure factor SM(Q) and the intermolecularpair correlation function ginter(r) of the liquid complex. Onthe other hand, DFT calculations are performed to opti-mize the geometry of the NMA and D2O monomers, andthose of some proposed clusters in the form NMA + nD2O(n = 1–3). To extract the molecular from factor F1(Q), wehave considered the more probable isomer that is foundin liquid NMA i.e. the conformer trans. The computedF1(Q) function which takes into account the optimizedintramolecular distances in each monomer and the relative

S. Trabelsi et al. / Journal of Molecular Structure 842 (2007) 93–99 99
proportion of each molecule reproduces correctly the struc-ture factor SM(Q) at high Q-range.
The residual difference observed between the experimen-tal structure factors SM(Q) and F1(Q) functions areassigned to the intermolecular correlations. To analyzethese interactions, we have considered in this study differ-ent clusters. The model T3 formed by one N-methylaceta-mide molecule with 3D2O ones is the more probablecluster. The theoretical intermolecular structure factorand the intermolecular pair correlation function deducedfrom this model are in good agreement with the experimen-tal data. Consequently, our measurements lead to the con-clusion that, in liquid NMA-W, hydrogen bonds give riseto the formation of tetramer structures made up of onetrans-NMA with 3D2O molecules.
Acknowledgment
We thank Moncef Rassas for his help to improve the lin-guistic aspect of our paper.
References
[1] R. Ludwig, J. Mol. Liq. 118 (2000) 65.[2] H. Hiramatsu, H. Hamaguchi, Chem. Phys. Lett. 361 (2002) 457.[3] S. Trabelsi, S. Nasr, J. Chem. Phys. 121 (2004) 6380.[4] S. Trabelsi, M. Bahri, S. Nasr, J. Chem. Phys. 122 (2004) 024502.[5] H. Guo, M. Karplus, J. Phys. Chem. 96 (1992) 7273.[6] H. Guo, M. Karplus, J. Phys. Chem. 98 (1994) 7104.[7] A.J. Doig, D.H. Willams, J. Am. Chem. Soc. 114 (1992) 338.[8] J. Tirado-Rives, W.L. Jorgensen, Biochemistry 30 (1991) 3864.[9] K.V. Soman, A. Carimi, D.A. Case, Biopolymers 31 (1991) 1352.
[10] A. Radzicka, L. Pedersen, R. Wolfenden, Biochemistry 27 (1988) 4538.[11] S. Song, S.A. Asher, S.J. Krimm, Am. Chem. Soc. 113 (1991) 1155.[12] N.G. Mirkin, S. Krimm, J. Am. Chem. Soc. 112 (1990) 9016;
N.G. Mirkin, S. Krimm, J. Am. Chem. Soc. 113 (1991) 9742.[13] W. Han, S. Suhai, J. Phys. Chem. 100 (1996) 3949.[14] J.P. Ambroise, R. Bellissent, in: P. Convert, J.B. Forsyth (Eds.),
Position Sensitive Detection of Thermal Neutrons, Academic, NewYork, 1983.
[15] J.P. Ambroise, M.-C. Bellissent-Funel, R. Bellissent, Rev. Phys. Appl.19 (1984) 731.
[16] H. Bertagnolli, P. Chieux, M.D. Zeidler, Mol. Phys. 32 (1976) 759.[17] M.-C. Bellissent-Funel, S. Nasr, L. Bosio, J. Chem. Phys. 106 (1997)
7913.[18] T. Pfleiderer, I. Waldner, H. Bertagnolli, K. Todheide, H.E. Fischer,
J. Chem. Phys. 113 (2000) 3690.[19] M.-C. Bellissent-Funel, J. Teixeira, L. Bosio, J. Chem. Phys. 87 (1987)
2231.[20] M.-C. Bellissent-Funel, L. Bosio, J. Chem. Phys. 102 (1995) 3727.[21] F. Hammami, S. Nasr, M.-C. Bellissent-Funel, J. Chem. Phys. 122
(2005) 064505.[22] S. Nasr, M.-C. Bellissent-Funel, R. Cortes, J. Chem. Phys. 110 (1999)
10945.[23] T.H. Dunning, J. Chem. Phys. 55 (1971) 716.[24] M.W. Schmid, K.K. Baldrige, J.A. Boatz, J. Comput. Chem. 14
(1993) 1347.[25] R.H. Hertwig, W. Koch, Chem. Phys. Lett. 268 (1998) 345.[26] A.D. Becke, Phys. Rev. A 38 (1988) 3098.[27] C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 785.[28] B. Miehlich, A. Savin, H. Stoll, H. Preuss, Chem. Phys. Lett. 157
(1989) 200.[29] S.H. Vosko, L. Wilk, M. Nusair, Can. J. Phys. 58 (1980) 1200.[30] S.F. Boys, F. Bernardi, Mol. Phys. 19 (1970) 553.[31] T. Radnai, S. Itoch, H. Ohtaki, Bull. Chem. Soc. Jpn. 61 (1988)
3845.[32] A.H. Narten, J. Chem. Phys. 65 (1976) 573.[33] S.I. Sandler, A.H. Narten, Mol. Phys. 32 (1994) 1543.[34] H. Torii, M. Tasumi, T. Tasumi, J. Raman Spectrosc. 29 (1998) 537.
Copyright © 2022 FDOKUMEN












![Arrangement of ceramide [EOS] in a stratum corneum lipid model matrix: new aspects revealed by neutron diffraction studies](https://static.fdokumen.com/doc/165x107/631f0e12198185cde200ea75/arrangement-of-ceramide-eos-in-a-stratum-corneum-lipid-model-matrix-new-aspects.jpg)







