
22q13.3 deletion syndrome: Clinical and molecular analysis using array CGH
Upload
khangminh22Category
view
0download
0
From the Department of Women’s and Children’s Health
Karolinska Institutet, Stockholm, Sweden
MOLECULAR AND CLINICAL STUDIES OF INTESTINAL MALROTATION
Karin Salehi Karlslätt
Stockholm 2022

Cover by Karin Salehi Karlslätt Pictures in the thesis are generated from open access sources with general permissions or illustrated by Karin Salehi Karlslätt, if nothing else is stated. All previously published papers were reproduced with permission from the publisher. Published by Karolinska Institutet. Printed by Universitetsservice US-AB, 2022 © Karin Salehi Karlslätt, 2022 ISBN 978-91-8016-526-6

Molecular and clinical studies of intestinal malrotation THESIS FOR DOCTORAL DEGREE (PhD)
By
Karin Salehi Karlslätt
Principal Supervisor:
Agneta Nordenskjöld, professor
Karolinska Institutet
Department of Women’s and Children’s Health
Division of Pediatric Oncology and Pediatric
Surgery
Co-supervisors:
Britt Husberg, med dr
Karolinska Institutet
Division of clinical sciences
Department of Surgery at Ersta Hospital
Ersta Hospital
Anna Lindstrand, professor
Karolinska Institutet
Department of Molecular Medicine and Surgery
Division of Rare Diseases
Tomas Wester, professor
Karolinska Institutet
Department of Women’s and Children’s Health
Division of Pediatric Oncology and Pediatric
Surgery
Opponent:
Martin Salö, docent
Lund University
Department of Clinical Sciences
Division of Pediatric Surgery
Examination Board:
Greger Lindberg, professor
Karolinska Institutet
Department of Medicine
Division of Medical Gastroenterology and
Hepatology
Catharina Lavebratt, docent
Karolinska Institutet
Department of Molecular Medicine and Surgery
Division of Neurogenetics
Anders Elfvin, docent
Sahlgrenska Academy, University of Gothenburg
Division of Clinical Sciences
Department of Pediatrics
Division of Neonatology


To David, Arvid and Jennifer
Human beings are members of a whole,
in creation of one essence and soul.
If one member is inflicted with pain,
other members uneasy will remain.
If you have no sympathy for human pain,
the name of human you cannot retain.
Saadi Shirazi (1210-1292) from Gulistan, 1258
Translation from Persian by M. Aryanpoor


POPULAR SCIENCE SUMMARY OF THE THESIS
Intestinal malrotation (IM) is a congenital malformation of the intestines characterized by
deficient attachment of the intestines to the posterior abdominal wall and increases the risk
for midgut volvulus. It may be as common as in 1/500 individuals. Volvulus requires
immediate surgery to derotate the intestines. The symptoms can also be of a more chronic
character due to a mechanical obstruction of the duodenum that may cause abdominal
discomfort, sometimes during several years. This has mainly been described in children, but
during recent years also among adults. The treatment is Ladd’s procedure, when the
intestines are placed in the abdomen in an organized way so that an intestinal strangulation
cannot happen and also embryonal adhesive bands, called Ladd’s bands, that can obstruct
the intestines are removed.
The knowledge that also adults can have symptoms from intestinal malrotation is relatively
new and not well-studied so we wanted to study all age groups regarding symptoms,
diagnostics and outcomes and compare them to better understand the variants of the
disease. In addition, the molecular knowledge on how IM develops is limited. There are
familial cases as well as patients with malrotation and genetic syndromes or different
chromosomal aberrations. These data together speak for genetic causes in at least some
cases of malrotation.
We performed two clinical studies (study I and II) on 138 children and 39 adults with
intestinal malrotation and we compared initial symptoms, other associated malformations,
treatment and the outcome after treatment. Other malformations were common and
symptoms were age-dependent to some extent. We found that in children up to five years of
age the main symptom was vomiting, and that is was bile-stained up to the same age. From
five years of age abdominal pain is the main symptom at diagnosis. Children born
extremely preterm and patients with severely affected circulation of the intestines had a
dismal outcome in the pediatric study. In addition, it is more common with a need for
emergency surgery in children but recurrence of volvulus was more prevalent in the adult
group. Longer periods of diffuse or intermittent acute abdominal discomfort before
diagnosis was often described among adult patients and it was not uncommon with residual
abdominal discomfort also after surgery.
We also performed two molecular projects (study III and IV) to investigate molecular
causes for malrotation. In study III we did ¨array comparative genomic hybridization¨
(array CGH), in order to find submicroscopic chromosome changes. We found six such
variants in five of the 47 DNA samples that we examined, whereof four were disease

causing. Among these patients, all had one or several other malformations or a
developmental disability, which lead us to recommend that a genetic investigation should
be done in intestinal malrotation with an additional malformation or developmental delay.
In study IV we performed DNA sequencing of all genes in ten patients with only intestinal
malrotation, to look for possibly pathological variants among a few hundred candidate
genes. One patient had two gene variants in the same gene, the TTC7A-gene. Further
studies showed that the mother also had the same variants as well as a milder form of
malrotation. Interestingly, TTC7A-deficiency is a rare disorder that is autosomal
recessively inherited, meaning that you can inherit the disorder if you get one pathogenic
variant from each of your parents. Generally, these patients have several stops in the
intestines (atresias) in combination with an immunodeficiency, and also some patients have
been born with intestinal malrotation. We hypothesize that intestinal malrotation is a mild
manifestation of TTC7A-deficiency and these findings call for further studies in a larger
material.
In summary this thesis describes symptoms and treatment outcome in patients with
intestinal malrotation of all ages and also molecular studies are performed using DNA from
patients in order to elucidate the background for the malformation.
NORMAL INTESTINES
INTESTINAL MALROTATION MIDGUT VOLVULUS

POPULÄRVETENSKAPLIG SAMMANFATTNING
Malrotation av tarmen är en missbildning av tarmens vidhäftning mot bakre bukväggen
som medför en ökad risk att utveckla tarmvred. Det finns olika uppgifter om hur vanligt
malrotation är, men det förekommer hos ungefär 1/500 och ungefär 1/6000 spädbarn
uppvisar symptom. Av tarmvred, får man generellt akut magsmärta, men även kräkningar,
vilket är speciellt påtagligt hos spädbarn och man behöver opereras akut. Symptombilden
kan även variera i svårighetsgrad med kroniska besvär som tidig mättnadskänsla eller
återkommande buksmärta. Behandlingen är Ladds operation, då tarmarna placeras på ett
organiserat sätt i buken så att inte tarmvred ska kunna utvecklas, samt att embryonala
bindvävsstråk som kan obstruera tarmen tas bort.
Vi ville kartlägga hur symptomen skiljer sig genom olika skeden i livet, som generellt mest
varit kända under neonatalperioden och vi ville även se över diagnostik, behandling och
postoperativa komplikationer i de olika åldersgrupperna. Kunskapen om malrotation är
begränsad och det finns familjära fall som tyder på att det kan vara ärftligt, vilket vi också
ville utforska.
I avhandlingen presenteras två kliniska studier (I och II), där 138 barn respektive 39 vuxna,
som fått diagnosen malrotation, studerades med avseende på symptom, andra
missbildningar, behandling samt kliniska resultat efter operationen. Andra samtidigt
förekommande missbildningar är vanliga och uppvisade symptom är till viss del
ålderstypiska. Vi fann att hos barn upp till fem års ålder var kräkningar det främsta
symptomet. Från fem år till vuxen ålder dominerade symptomet buksmärta.
Extremprematurer och patienter med kraftig cirkulationspåverkan på tarmen hade störst risk
för komplikationer i barnstudien, där var det även dubbelt så många pojkar som drabbades.
Det var även vanligare med ett akut behov av operation i barnstudien och de uppvisade mer
sällan recidiv efter operation jämfört med den vuxna gruppen. Hos äldre barn och vuxna var
det vanligt att man haft långa perioder av oklara magbesvär innan man fick sin diagnos och
för vuxna var det inte ovanligt med kvarvarande lite mer diffusa magbesvär även efter
operation.
Två molekylära studier (III och IV) har också gjorts, för att leta efter en möjlig orsak till
malrotation. I studie III gjorde vi ¨array comparative genomic hybridization¨ (array CGH),
som är en metod att leta efter submikroskopiska kromosomförändringar, där vi fann sex
förändringar hos fem av 47 patienter, varav 4 av dem var direkt sjukdomsorsakande. De
som hade sjukdomsorsakande fynd hade alla någon ytterligare missbildning eller
utvecklingsförsening, vilket ledde till slutsatsen att patienter med malrotation i kombination

med någon annan missbildning eller utvecklingsavvikelse borde genomgå en genetisk
utredning. Vi gjorde även helgenomsekvensering på tio patienter som bara hade malrotation
utan annan missbildning, för att leta efter avvikelser i kandidatgener hos dessa patienter. Av
dessa fann vi en patient som hade två genavvikelser i samma gen, TTC7A-genen. Utredning
visade att även mamman hade samma genavvikelser och en liknande mildare avvikelse av
tarmrotationen. TTC7A-brist är ovanligt och recessivt nedärvt, dvs man får normalt sett
sjukdomen om ens båda arvsanlag är skadade dvs har varsin mutation. Denna sjukdom
innebär att man föds med stopp (atresier) på flera ställen i tunntarmen och immunbrist, men
även fall med malrotation är rapporterade. I detta fall var det alltså bara en av de två
genkopiorna som bar på avvikelserna och i association med rotationsavvikelser i tarmen.
Detta behöver studeras ytterligare i större omfång.
Sammanfattningsvis beskriver vi symptom och behandlingsutfall för patienter med
malrotation i alla åldrar och vi beskriver våra molekylära studier där vi med DNA från
patienterna letat efter submikroskopiska kromosomförändringar och mer specifika
gensekvensförändringar.

ABSTRACT
Intestinal malrotation (IM) can present as a potentially life-threatening condition with
midgut volvulus and need of immediate surgery, but also as a chronic condition with long-
lasting symptoms. It is a well-known neonatal condition but it is not as well “explored” up
to adulthood. Familial cases and chromosomal aberrations have been described, as well as
genetic syndromes associated with intestinal malrotation, but the molecular cause is not
known.
The aim of study I and II was to describe symptoms, management and outcomes of IM
throughout all age groups by scrutinizing medical records and through interviews. In study
III and IV the aim was to study the molecular background of IM.
In study I, 39 adult patients, 15-67 years old, were included. 31 (79%) underwent a Ladd’s
procedure and among them 12 (31%) as an emergency procedure. A triple contrast CT scan
confirmed the diagnosis and many patients, who often had symptoms for several years were
relived of symptoms after Ladd’s procedure, even though six needed re-do surgery because
of recurrence.
In study II, 138 children were included based on strict inclusion criteria, 1-15 years old.
125 (91%) had Ladd’s procedure and only one had recurrence needing re-do surgery.
The major findings were that the most common diagnostic symptom up to 5 years of age
was vomiting, while abdominal pain was a more relevant symptom in older children and
adults. Complications were mainly detected among extremely premature children, with an
overall mortality of 50%, and also among children who had severely affected circulation.
Four children had a chronic intestinal failure, two because of midgut volvulus and 14
patients (11%) had adhesive bowel obstruction.
In study III, by using array CGH we detected six different copy number variants (CNVs)
in five patients of the 47 DNA samples that were analyzed. Five were found in patients with
other malformations or developmental delay and one in a patient with isolated IM had a
variant of unknown significance in the GALNT14 gene.
In study IV, we performed whole genome sequencing (WGS) in ten children with severe
isolated IM, and found two maternally inherited allelic mutations in the TTC7A gene in a
boy with isolated IM. Further studies showed that the mother also had a rotational
abnormality. The rare autosomal recessively inherited TTC7A deficiency disorder causes
multiple intestinal atresia and immunodeficiency, but also IM in some case reports.

In conclusion, the results of this thesis show that IM should be regarded as a malformation
affecting all age groups, and midgut volvulus can occur in all ages. The symptoms vary
with age and calls for an active strategy for diagnosis and treatment. Extreme prematurity
as well as midgut volvulus with severely affected circulation, increase the risk for short-
term complications after surgery. We recommend genetic screening for CNVs in cases with
IM and other associated malformations or a neurodevelopmental disorder and the TTC7A
gene needs to be further studied in larger groups of cases with IM.

LIST OF SCIENTIFIC PAPERS
I. Britt Husberg, Karin Salehi, Trevor Peters, Ulf Gunnarsson, Margareta
Michanek, Agneta Nordenskjöld, Karin Strigård. Congenital intestinal
malrotation in adolescent and adult patients: a 12-year clinical and
radiological survey. Springerplus. 2016; 5: 245
II. Karin Salehi Karlslätt, Britt Husberg, Ulla Ullberg, Agneta Nordenskjöld,
Tomas Wester. Intestinal malrotation in children: Clinical presentation and
outcomes. Manuscript.
III. Karin Salehi Karlslätt*, Maria Pettersson*, Nina Jäntti, Przemyslaw
Szafranski, Tomas Wester, Britt Husberg, Ulla Ullberg, Pawel Stankiewicz,
Ann Nordgren, Johanna Lundin, Anna Lindstrand, Agneta Nordenskjöld.
Rare copy number variants contribute pathogenic alleles in patients with
intestinal malrotation. Molecular Genetics and Genomic Medicine. 2019;
7(3): e549
IV. Karin Salehi Karlslätt, Maria Pettersson, Kristina Lagerstedt-Robinson,
Ulla Ullberg, Anna Lindstrand, Agneta Nordenskjöld. Gene panel-based
screening of patients with isolated intestinal malrotation: An extended
phenotype for mutations in the TTC7A gene. Manuscript.
* Equal contribution


CONTENTS
1 INTRODUCTION ......................................................................................................... 5 1.1 Historical Background ...................................................................................... 5 1.2 Embryological development of the midgut ....................................................... 7
Definitions of intestinal rotation abnormalities ................................... 10 Secondary malrotation ......................................................................... 12
1.3 Clinical perspectives on IM ............................................................................ 12 Associated anomalies .......................................................................... 13 Radiological diagnostics ..................................................................... 13 Management and surgical treatment ................................................... 16 Outcomes ............................................................................................. 18 Adult patients and IM .......................................................................... 18
1.4 Molecular perspectives on IM ......................................................................... 19 Human genetics ................................................................................... 19 Experimental data ................................................................................ 21 Familial cases ...................................................................................... 22 Chromosomal aberrations associated with IM .................................... 22 Monogenic IM ..................................................................................... 24 Ciliopathies .......................................................................................... 24
2 RESEARCH AIMS ..................................................................................................... 27 3 CLINICAL STUDIES I & II ....................................................................................... 29
3.1 Methods ........................................................................................................... 29 Study design, Setting and Participants ................................................ 29 Variables .............................................................................................. 30 Statistical analysis ............................................................................... 31
3.2 Main results and discussion ............................................................................ 32 Study I inclusion .................................................................................. 32 Study II inclusion and variants of IM .................................................. 32 Associated malformations ................................................................... 33 Gender and age at diagnosis ................................................................ 34 Clinical presentation ............................................................................ 35 Diagnostic procedures ......................................................................... 37 Management ........................................................................................ 39 Outcome .............................................................................................. 40 Adult patients and IM .......................................................................... 42
4 MOLECULAR STUDIES III & IV ............................................................................ 43 4.1 Methods ........................................................................................................... 43
Study design, Setting and Participants ................................................ 43 Molecular analyses .............................................................................. 44
4.2 Main results and discussion Study III ............................................................. 48 Inclusion .............................................................................................. 48 Reported CNVs ................................................................................... 48 Clinical relevance ................................................................................ 50
4.3 Main results and discussion Study IV: ............................................................ 51 Inclusion .............................................................................................. 51 Reported variants ................................................................................. 51 The TTC7A-gene and IM ..................................................................... 52 Clinical relevance ................................................................................ 53
5 STRENGTHS AND LIMITATIONS ......................................................................... 55 6 ETHICAL CONSIDERATIONS ................................................................................ 56

7 CONCLUSIONS ......................................................................................................... 57 8 POINTS OF PERSPECTIVE ...................................................................................... 59 9 ACKNOWLEDGEMENTS ........................................................................................ 60 10 REFERENCES ............................................................................................................ 65

LIST OF ABBREVIATIONS
A
ALL Array CGH
C CAD
CADD CI
CID
Adenine
Acute lymphoblastic leukemia Array comparative genomic hybridization
Cytosine Congenital alveolar dysplasia
Combined Annotation Dependent Depletion Confidence Interval
Combined immunodeficiency CDH
CLDS CLMP
CNV CNS
CT DNA
Congenital diaphragmatic hernia
Cornelia de Lange syndrome CXADR Like Membrane Protein
Copy number variants Central nervous system
Computed tomography Deoxyribonucleic acid
EOIBD FLNA
FOXF1 FTR
G GA
GALNT14 HPO
IM IMA
Early onset inflammatory bowel disease Filamin A
Forkhead Box F1 Failure to rescue
Guanine Gestational Age
Polypeptide N-Acetylgalactosaminyltransferase 14 Human Phenotype Ontology
Intestinal malrotation Inferior mesenteric artery
IRA IUGR
kb MIA
MRI mRNA
MZ OMIM
OR
Intestinal rotation abnormalities Intrauterine growth retardation
kilobase multiple intestinal atresias
Magnetic Resonance Imaging Messenger ribonucleic acid
Monozygotic Online Mendelian Inheritance in Man
Odds Ratio

PI4IIIα-complex
PN SBS
SMA SMV
SNV SV
T TPR
TTC7A/B UGI study
VOUS VUR
VSD WGS
3D
Phosphatidylinositol 4-kinase IIIα-complex
Parenteral nutrition Short bowel syndrome
Superior mesenteric artery Superior mesenteric vein
Single nucleotide variant structural variants
Thymine Tetratricopeptide Repeat
Tetratricopeptide Repeat Domain 7A/7B Upper gastrointestinal contrast study
Variant of uncertain significance Vesicoureteral reflux
Ventricular septal defect Whole genome sequencing
Three-dimensional

5
1 INTRODUCTION Intestinal malrotation means that the intestines are positioned abnormally in the abdomen
constituting a predisposition for midgut volvulus, which causes obstruction and ischemia of
the intestines. This is a potentially life-threatening condition and needs immediate surgery.
1.1 HISTORICAL BACKGROUND
The historical background of intestinal malrotation displays the beginning of a thrilling
story on how a malformation is discovered, diagnosed, treated and later studied concerning
molecular background. This thesis pursues to be a small chapter of this adventure, and a
summary on how far this expedition has come, which still has a long way to go until it is
finished.
To define a malformation, the normal anatomy must first be understood. Václav Treitz
(1819-1872), worked as a professor in anatomy in Prague and published "Ueber einen
neuen Muskel am Duodenum des Menschens" (On a New Muscle in the Duodenum of Man)
in 1853 where he described the ligament of Treitz. This suspensory muscle of duodenum is
the fixating unit of the duodeno-jejunal junction in the left upper quadrant, and its absence
has become a hallmark for intestinal malrotation (IM) (Lampl 2009).
In 1898, Mall was the first to describe the normal rotation and fixation of the intestine
during embryogenesis (Mall 1898), a theory which has remained unchanged until recently.
IM was described before 1900 from autopsy- and surgical findings, and in 1923, Dott
pointed out the relationship between incomplete embryological intestinal rotation and its
need for surgical treatment because of its predisposition to midgut volvulus (Dott 1923).
The Halifax Disaster (1917) during the First World War was an accidental collision of two
ships in the Halifax harbor in Canada. The explosion killed 2000 people and over 9000
people were harmed, including children. William E. Ladd (1880-1967), as well as other
surgeons, were sent there to help. He was strongly affected by the disaster, especially by the
wounded children, so after returning to his hospital in Boston, USA, he devoted his career
to treat children (Lampl 2009). He has since contributed to pediatric surgery in many ways
but his name is immortal due to the Ladd´s procedure that he described in 1932 on how to
treat intestinal malrotation and volvulus, which is still the gold standard treatment for
intestinal malrotation (Ladd 1932, Ladd 1936).
The cause of the deviating embryonal development is not known, but it is thought to be
partly environmental and partly genetic in cause. The understanding of genetic causes of

6
human disease started first through the possibility to study chromosomes by karyotyping
from 1959 and then later the possibility to look for single nucleotide variants (SNVs) in the
genome.
In the 1990s, Sanger sequencing was clinically used. In 2001, the entire human genome was
sequenced from the important insight of a need to have a common, unbiased reference for
scientists throughout the world in order to accelerate biomedical research (Lander 2001).
Mass sequencing and softwares to compare and interpret changes in the genome has turned
the knowledge of about 300 rare monogenic diseases in 1990 to 7000 in 2022 and complex
diseases from zero to almost 70 000.
In order to study the genetic backgrounds different methods are used that can discover
different genetic mechanisms. Karyotyping when chromosomes are sorted and studied in a
light microscope has a resolution of down to 50-100 genes (≈5-10 Mb) (Figure 1). Array
CGH that is a method we used in this thesis can detect submicroscopic changes, sometimes
down to part of genes. In order to examine smaller gene variants different methods for
DNA sequencing are used, like here whole genome sequencing (WGS) although we only
examined a subset of all genes, regarded as candidates to cause a certain disease. Thus
different molecular methods can be used for different research questions and have different
advantages and disadvantages. Methods can also be used together. For example, if DNA
from a patient with a rare disease shows a chromosomal aberration, this can indicate that
disease genes are located in that specific chromosome region and those genes can further be
studied with other genetic methods like sequencing.
Figure 1: Normal male 46, XY human karyotype, 44 autosomes and 2 sex chromosomes
(here X and Y). The entire genome is packed into 23 pairs of chromosomes with one copy
from each parent. (Wessex Reg. Genetics Centre. Attribution 4.0 International (CC BY
4.0))

7
1.2 EMBRYOLOGICAL DEVELOPMENT OF THE MIDGUT The different stages of embryologic development are important when studying the etiology
of intestinal malrotation. Current knowledge is based on experimental observations and
hitherto not completely revealed.
Rotation of the intestines occurs between the fourth and twelfth weeks of gestation. The
mechanism leading to the “intestinal rotation” and the exact order of events has been
discussed since the 19th century based on observations of the normal development and of
malformations such as IM and abdominal wall defects (Mall 1889, Frazer 1915). These
concepts are recently reviewed in human and rat fetuses and are now considered to occur
somewhat differently (Soffers 2015, Kluth 2003, Metzger 2011, Ginzel 2021).
The embryology of gut rotation has been described to start during the fourth gestational
week when the primitive gut develops into three sub-divisions: Foregut, midgut and
hindgut (Figure 2). The midgut will develop into the future duodenum, jejunum, ileum,
ascending colon and right two thirds of the transverse colon. At four to five weeks of
gestation the midgut is a linear tube lying straight in the midline. The midgut elongates
Figure 2. Schematic drawing of an embryo at week 5. The gut-tube is marked in yellow and
the midgut is highlighted in light yellow. The arteries branching from the aorta from
cranial to caudal are 1) the celiac trunk, 2) the superior mesenteric artery (SMA) and 3) the
inferior mesenteric artery (IMA).
3
2
1

8
rapidly compared to the embryo’s growth. As the midgut grows, it turns into a loop with a
sagittal orientation, the primary loop, with the superior mesenteric artery as its axis, which
is connected to the yolk sac by the vitelline duct (Figure 2, figure 3D). The loop herniates
into the base of the umbilical cord during the sixth week of gestation. It is hypothesized that
the herniation takes place due to the small size of the abdominal cavity of the embryo. As
the midgut loop herniates into the umbilical cord, in the sixth week, it appears to turn 90°
counterclockwise (viewed from the front) with the superior mesenteric artery as its axis
(Figure 3E1). It is probably the adjacent surrounding structures that move that cause the
“rotation”, rather than an actual primary rotation of the SMA and the dorsal mesentery.
Figure 3. A) The primary loop herniating into the umbilical ring. B) The duodenum
forming its secondary loop under SMA, progressing towards the final position of the
duodenojejunal junction. C) The embryo is not in a straight line, rather in a chiral
formation. D-E) demonstrating the formation of the primary, secondary and tertiary loops,
as well as the return of the intestines into the abdominal cavity. F) Final position of the
colon with the cecum in the right lower quadrant. (Soffers 2015) 3: SMA, 7: Right vitelline
vein, 8: Vitelline duct, 9: Duodenum, Dp: Dorsal pancreas, Vp: Ventral pancreas, Gb:
Gallbladder, Li: Liver, St: Stomach.
Experiments using 3D visualization techniques on serial sections from human embryos
have shown that the embryo actually has a chiral formation (Figure 3C), with the caudal
C
A
B

9
part being located on the right side of the cranial part (Ramasubramanian 2013). When the
midline structures (upper abdominal and thoracic) in the embryo descend, the cranial part
of the intestinal loop will descend rightwards and be located to the right side of the SMA
and the caudal part of the loop goes to the left where after the loop then attains a transverse
position (Soffers 2015) referred to as “rotating” 90° (Figure 3E1).
As the intestines are herniated, the secondary loops form. The first secondary loop is
located in the abdominal cavity, comprising the duodenum and first part of jejunum. It
expands rightward and caudally on the right side of the SMA. During the eighth week of
gestation, prior to the return of the intestines, the first secondary loop, (the duodenal part)
grows substantially and continues to expand below the SMA towards the left approaching
its final position of the duodenojejunal junction (Metzger 2011, Soffers 2015) (Figure 3B).
The second to fourth secondary loop forms in the herniated part (Figure 3 E2). Of the
colon, only the cecum and most proximal part is herniated. The intestines grow further and
tertiary loops develop within the areas of the secondary loops, but not in the first secondary
loop (the duodenal part) (Figure 3 E3). The primary and secondary loops seem
predetermined, and the second secondary loop will always for unknown reasons, position
itself to the left in the herniation when developing the tertiary loops (Soffers 2015, Ginzel
2021).
The return of the intestines occurs in a cranial-to-caudal manner into the abdominal cavity
as it expands, at 8.5 – 9.5 weeks of gestation, previously explained as rotating 180°
counterclockwise, en bloc. First the second secondary loop to the left, then the third in the
midline, and the fourth in a ventral position in the abdomen. Previously, it was understood
that the cecum is the last portion to return into the abdominal cavity, however the cecum
returns before the distal part of the ileum, indicating that the colon is co-migrating (Ginzel
2021, Soffers 2015). In week 10, the cecum has taken its definitive position in the right
fossa (Soffers 2015). The cause of the return has many theories, among them that there is a
traction of the vessels, and that the umbilical ring opens up to allow for an intestinal return
(Ginzel 2021).
Altogether, a 270° counterclockwise rotation is said to occur, even though this is not true
for all parts of the midgut. The ascending colon and most of the descending colon fuse with
the posterior abdominal wall and becomes retroperitoneal during the fourth and fifth month
of gestation (Langer 2017). Thereby the duodenojejunal junction, marked by the ligament
of Treitz, and the cecum in the right lower quadrant has maximum distance in the abdomen
in a diagonal manner, leading to a wide dorsal mesentery (dotted line), which carries the
superior mesenteric artery and vein to the intestines (Figure 4).

10
Figure 4. Normal intestines. The dotted line demonstrates a wide dorsal mesenteric base.
Kluth et al showed that it is rather the elongation of the intestines that seem to push them to
rotate than an actual rotational process. Also, the duodenojejunal junction is pushed under
the mesentery through lengthening and ends up in its correct position. The development of
the duodenum is crucial in IM, and interestingly the duodenum is not being a part of the
herniating and rotating process. The driving force is not an actual rotation, but rather
elongation in selective parts of the intestines at different time periods. The elongation of the
intestines with anisotropic growth is shown to be self-consistent by smooth muscle
contractions; peristalsis (Khalipina 2019) and the stereotypical looping pattern seem to be
driven by growth as well (Savin 2011).
1.2.1 Definitions of intestinal rotation abnormalities A disturbance in this embryological process is thought to cause intestinal malrotation and
other intestinal rotation abnormalities (IRA) (Langer 2017). There are some concepts that
are used in pediatric surgery in order to describe these intestinal rotational abnormalities.
When there is “no rotation” around the superior mesenteric artery, this is called
“nonrotation”. The cranial part of the midgut, including duodenojejunal junction and most
Duodenojejunal junction
Cecum

11
part of the small intestine, will lay to the right in the abdomen, while the caudal part of the
midgut, including the colon and terminal ileum, lays to the left in the abdomen (Nehra
2011). “Incomplete rotation” or intestinal malrotation (IM) occurs when there is only a
“partial rotation”. As a result, the cecum will stay located in the right upper quadrant, often
with adhesive peritoneal bands overriding the duodenum, fixed to the right sided abdominal
wall and sometimes also to the gallbladder and duodenum. These embryonic fibrous bands
constitute a failed attempt of cecal fixation, also referred to as Ladd’s bands. In both
incomplete rotation and non-rotation, duodenum goes straight caudally without overriding
to the left of the vertebral spine towards the position of the ligament of Treitz. IM
predisposes to midgut volvulus due to a narrow mesenteric base and non-fixated intestines.
There is also a risk of duodenal obstruction due to Ladd’s bands causing more chronic
symptoms.
Figure 5. Intestinal malrotation (A) and Midgut volvulus (B)
In non-rotation, the risk of midgut volvulus might be smaller due to a wider mesenteric
base (Langer 2017) though there is still the element of non-fixated intestines. Other
rotational abnormalities, not necessarily included in the topic but worth mentioning, are
“reversed rotation”, “para-duodenal hernias” and atypical malrotation. Reversed rotation
occurs when there is a 90° clockwise rotation from the original sagittal plane, leading to
colon located behind the superior mesenteric vessels and the duodenum in front. Para-
A B

12
duodenal hernias occur when the right or left mesocolon fails to fuse to the posterior body
wall. There will be an opening into which the small intestine can herniate, causing potential
obstruction. Atypical malrotation is defined as the duodenojejunal junction being positioned
to the left of the midline, but below the level of the pylorus (Graziano 2015). All grades
may exist, from atypical malrotation to nonrotation.
1.2.2 Secondary malrotation IM can also arise as an intrinsic component of congenital diaphragmatic hernia,
omphalocele or gastroschisis. This is also called secondary malrotation, since it is
secondary to another major abdominal malformation interrupting the normal developmental
process of the midgut. This is generally in the form of non-rotation.
1.3 CLINICAL PERSPECTIVES ON IM
The true incidence of intestinal malrotation is not known, from radiological studies, it is
reported to be up to 0.2% (Kantor 1934), while autopsy studies estimate an incidence of
some form of intestinal rotation abnormalities (IRA) up to 1% (Kapfer 2004). The
incidence of symptomatic IM (in neonates) is reported to be 1/2500-6000 infants (Adams
2014, Kapfer 2004) Traditionally it has been reported that up to 90% of patients present
within the first year of life, usually within the first month (Ford 1992, Stewart 1976).
However a recent study suggests that 60% present below one year of age (Aboagye 2014).
The risk for midgut volvulus in patients with intestinal malrotation is present regardless of
age. Midgut volvulus is a clockwise rotation of the base of the dorsal mesentery,
obstructing the blood- and lymphatic flow to and from the intestines through the superior
mesenteric vessels. If not treated, it will lead to intestinal gangrene and death. The
symptoms are usually acute bilious vomiting, abdominal discomfort and distension. The
abdominal symptoms, followed by metabolic acidosis and hypovolemic shock, will get
worse within hours if not treated. Since the blood supply to and from the damaged
intestines is strangulated, sometimes lab-tests, such as leukocytes, lactate and electrolytes
can be normal, even if the damage is severe.
Other less acute symptoms of intestinal malrotation can arise from intermittent duodenal
obstruction due to Ladd’s bands, intermittent volvulus or chronic volvulus. In addition,
there are dysmotility disorders that have been shown to sometimes coexist with intestinal
malrotation (Coombs 1991, Erez 2001, Devane 1992). Duodenal obstruction causes

13
symptoms like gastroesophageal reflux, failure to thrive, feeling of fullness after small
meals and mild abdominal discomfort. Chronic volvulus can show similar symptoms, due
to malfunctioning nutrient absorption resulting from insufficient blood supply and chronic
irritation.
1.3.1 Associated anomalies Associated anomalies are relatively common; intestinal atresia, 5-26%; imperforate anus, 0-
9%; cardiac anomalies, 7-13%; duodenal web, 1-2%; Meckel’s diverticulum, 1-4%;
inguinal hernia, 0-7%; trisomy 21, 3-10% (Little 2010). Hirschsprung’s disease and CNS-
anomalies are also associated with intestinal malrotation (Stratulat-Chiriac 2016). Intestinal
rotation abnormalities are known to coexist with heterotaxy syndrome. Volvulus in utero
may also cause multiple intestinal atresias (Budd 1988).
1.3.2 Radiological diagnostics The clinical symptoms of IM are very variable, and IM may present as an incidental finding
during a radiographic evaluation of another diagnosis or with peritonitis and shock from a
midgut volvulus.
A plain abdominal radiograph is usually the first step to investigate abdominal symptoms.
This can be inconclusive, but especially in new-borns there can be a “double bubble”,
which corresponds to a distended ventricle and a dilated proximal part of the duodenum.
This can appear in duodenal atresia, presence of Ladd’s bands or midgut volvulus. In
volvulus, there might be an edema and thickening of the bowel wall. Free gas in the
abdomen is a sign of intestinal perforation and requires emergency surgery.
The next step in children to establish the course of the duodenum is an upper
gastrointestinal contrast study, which was introduced in the 1960s and is the gold standard
to diagnose IM (Binu 2021). Barium enema or iodine-based water-soluble contrast is given
orally or through a nasogastric tube. Before that, when suspecting midgut volvulus, barium
enemas were used, looking for the position of the cecum (Lampl 2009). Normally, the
duodenojejunal junction is in the anatomical position of the ligament of Treitz to the left of
the spine at the level of the duodenal bulb (McVay 2007). In IM, the duodenojejunal
junction will be located on the right side of the spine (Figure 5A). In patients with duodenal
obstruction, the suspicion of midgut volvulus should be high. Other pathological findings
are “corkscrew” appearance of the duodenum (Figure 5B) and “bird’s beak” appearance of
the duodenum, both being signs of midgut volvulus. There can also be the “coil spring”

14
sign, which is a sign of intramural hematoma or edema and blood infiltration (Freeman
2008). In patients with signs of duodenal obstruction and deteriorating general condition,
midgut volvulus cannot be excluded which calls for an acute surgical intervention. Saline-
aided doppler ultrasound can also be used to show a dilated duodenum and an inversion of
the superior mesenteric artery and vein in midgut volvulus (Chen 2022).
Figure 5. UGI studies, demonstrating A) Intestinal malrotation B) Corkscrew sign
In older children and adults, a computed tomography (CT) with double contrast,
intravenous and oral, is recommended. The course of the duodenum can be followed, as
well as the colon if “triple” contrast investigation is done. In addition, the abnormal
positioning of the superior mesenteric artery and vein can be seen (Strouse 2004) (Figure
6). In midgut volvulus the “whirlpool sign”, a clockwise rotation of the superior mesenteric
vein, bowel, and the mesentery around the superior mesenteric artery, can be visualized
(Strouse 2004, Brinkley 2016) (Figure 7A). If strangulation of the SMA is complete, the
“whirlpool sign” might not be seen, so one also has to look beyond the signs. In children
the “whirlpool sign” can be demonstrated by Doppler ultrasound with a 97% positive
predictive value (Brinkley 2016, Orzech 2006), also prenatal diagnosis of IM with midgut
volvulus was performed from 25 weeks GA with Doppler ultrasonography by the
“whirlpool sign” (Yang 2022).
A B

15
Figure 6. A) A CT-scan, coronal section, demonstrating the normal position of SMA (red)
to the left of SMV (blue). SMV and the splenic vein join to form the portal vein. B) IM
demonstrating the SMA on the right side of the SMV.
Figure 7. A) Whirlpool sign in an adult patient, coronal secion B) Normal finding of SMA
in the same section as aorta C) SMA drifting towards the right, not found in the same
section as the aorta. Sometimes seen in intestinal malrotation and more common in midgut
volvulus.
A B
A
B C

16
1.3.3 Management and surgical treatment In the typical case with acute symptoms and radiological signs of intestinal malrotation an
operative treatment is indicated, often as an emergency procedure. The volvulus is reduced
by de-rotation and Ladd’s procedure prevents volvulus from recurring. The Ladd’s bands
are divided, the small intestine mesentery is widened to avoid relapse, the small intestines
are placed to the right and the large intestines to the left in the abdominal cavity (Swenson
1945). Usually an appendectomy is performed, since the atypical position may delay a later
diagnosis of appendicitis.
When the radiographic examination is inconclusive and the symptoms are mild, generally
follow-up or repeated contrast study is recommended. In asymptomatic patients with
incidental findings, there is a lack of evidence on whether to operate. Prophylactic surgery
on asymptomatic patients could be considered in younger patients with low risk of
postoperative complications, while older asymptomatic patients could benefit from follow-
up and information (Graziano 2015).
The surgical procedure can also be performed laparoscopically, which was first reported in
1995 in a 7-days old neonate with midgut volvulus (van der Zee 1995). Laparoscopic
approach may be preferred in elective cases when radiological examinations are
inconclusive and the diagnosis needs to be confirmed. A four-port technique is usually
applied. In acute volvulus, the accessibility can be limited due to bowel edema and chylous
ascites. Comparative retrospective studies have shown that the laparoscopic Ladd’s
procedure takes a longer time, but shortens the time to full enteral feeding and hospital stay.
Open surgery was related to more post-operative complications, however, midgut volvulus
after Ladd’s surgical procedure was encountered in 3.5% of patients in the laparoscopic
group and 1.4% in the open group (Catania 2016).

17
Figure 9. Ladd’s procedure with photographs taken during surgical procedures on adults (Brungardt 2021 with permission, photos by Britt Husberg and from study I)
Midgut volvulus
Ladd’s bands Widening of the mesentery and placement of the small intestines to the right, and the large intestines to the lef.

18
1.3.4 Outcomes After Ladd’s procedure, adhesive small bowel obstruction is the most common
complication, with a reported incidence of 5-10% (Fischer 2011). Recurrence of midgut
volvulus may occur and is encountered in 0.5-7% (Dilley 2000, El-Gohary 2010, Feitz
1997). The mortality of midgut volvulus depends on the damage on the small bowel and if
there is systemic shock. The overall mortality rate after surgical correction of intestinal
malrotation is 3-9%. The risk increases if there is intestinal necrosis, other abnormalities, or
prematurity (Fischer 2011). In one study, the mortality rate in patients undergoing surgery
for small bowel volvulus were 10-35%. In cases of gangrenous bowel, the mortality
increased to 20-60% (Iwuagwu 1999).
IM with volvulus is one of the most common etiologies of intestinal failure in children.
This condition is a malabsorptive state often following an extensive resection of the small
intestine (Vanderhoof 1997). The ability to absorb nutrients and balance fluid excretion is
disrupted and adaption and recovery depend on the amount and function of the remaining
small intestine. Generally, the ileum has a greater adaptive ability than the jejunum and the
importance of preserving the ileocecal valve has been put forward. Supportive therapy is a
combination of enteral feeding, parenteral nutrition, medical therapies and occasionally
intestinal transplantation.
In adults, the most common cause of intestinal failure is mesenteric ischemia due to
thromboembolic events whereas in children, it is necrotizing enterocolitis (Fischer 2011).
Intestinal failure among neonates is caused by volvulus in 10% (Wales 2010).
Approximately 2% of children and adults with intestinal volvulus develop intestinal failure,
in adults most commonly caused by intra-abdominal adhesions, and in children due to IM
(Koffeman 2003).
The overall incidence of intestinal failure is 24.5 per 100 000 live births (Wales 2004) and
the risk for intestinal failure is higher in low birth-weight infants, as well as in live births
before 37 weeks of gestation (Wales 2010). The mortality rate in pediatric intestinal failure
was previously reported to be between 20-40% (Wales 2010) but more recent advances
have increased the pediatric intestinal failure survival rate to >90% (Duggan 2017). The
mortality depends primarily on the need of parenteral nutrition, which can lead to central
venous catheter infections and intestinal failure-associated liver disease.
1.3.5 Adult patients and IM Intestinal malrotation has until recently been considered a strict pediatric diagnosis and was
rarely diagnosed in adult patients. Nowadays, it has become more accepted that it is an

19
important diagnosis also in the adult population. Symptoms like abdominal pain, early
satiety and dyspepsia can be caused by malrotation. Surgery may also prevent development
of an acute volvulus, even though the risk generally seems to decrease with age (Nehra
2011). The incidence of midgut volvulus in adults is reported to be between 0.00001% to
0.19% (Leow 2016).
1.4 MOLECULAR PERSPECTIVES ON IM
1.4.1 Human genetics
Figure 10. DNA transcription
and translation into a protein (a
folded polypeptide consisting of
amino acids). Only about 1% of
the human genome consists of
coding genes and they are
approximately 20 000 in number.
The genome is composed of
deoxyribonucleic acid (DNA),
which consists of different
nucleotides, adenine (A), thymine
(T) cytosine (C) and guanine (G).
They are referred to as bases and
pair as A-T and C-G, called base pairs. (Picture used with license from shutterstock.com)
The DNA is to a very high degree identical between individuals especially concerning the
parts of the DNA that codes for proteins. Overall there are still non-disease-causing base
variants that can be used for “finger printing”. When you study a disorder and find a genetic
variant you need to evaluate how damaging the variant is, for example how deleterious
(harming to the protein product) it is, if it is conserved over different species and how
common it is in the general population as well as its original function compared to the
phenotype of the carrier of the gene. Some specific sequences are rather constant, and also
between species (conserved), and they are generally more important for normal
development and function and a sequence variant in these regions are generally more
damaging.

20
We have two sets of each gene, one of each of the two autosomal chromosome pairs. If a
genetic disease is caused by a mutation in one of the genes, it is an autosomal dominant
disease that can be inherited in 50% of the offspring. If both genes need to be mutated for
the patient to be affected by a disease, it is an autosomal recessive inheritance, meaning that
each parent is a carrier of one mutation and then 25% of the offspring will get the disorder.
Additionally, 50% will be carriers.
Figure 11. Autosomal dominant, autosomal recessive and X-linked recessive inheritance
patterns. (Picture used with license from shutterstock.com)
1.4.1.1 Genetic variants
Genes that code for proteins consist of intervals of DNA called exons and introns, where
only the exons are translated to proteins. In this thesis we focused on the protein coding
genome. Smaller changes in the DNA can have different consequences on proteins. There
are synonymous variants that do not change an amino acid and the final protein will not be
altered. Missense variants will code for a different amino acid, possibly altering the
function of the protein and nonsense variants changes the code into a stop codon, resulting
in premature termination of translation. Frameshift variants are deletions, duplications,
insertions, or a combination, that alter the number of bases usually causing lots of changes
in the protein or more commonly a premature stop codon. All these variants will generally
be found with sequencing methods such as whole genome sequencing (WGS).
Larger deletions or duplications or insertions, referred to as copy number variants
(CNVs) are unbalanced structural variants (SVs), meaning the total gene dosage will
change. For example it may either increase (if a duplication) or decrease (if a deletion) the
expression of a gene which can affect regulatory mechanisms. If a SV is balanced it means

21
that there is an inversion or translocation, not changing the gene dosage. Only unbalanced
SVs can be detected when performing array CGH.
1.4.2 Experimental data A disturbance in left-right axis development in embryogenesis might be a possible cause of
intestinal malrotation, even though gut tube elongation is also considered to be involved. It
is known that Nodal is the start of laterality, probably through Pitx2 that activates Wnt5a.
Examples of genes that are important for left-right asymmetry in humans are for example
LEFTYA, CFC1, ZIC3, NKX2.5, AVCR2B (Maclean 2004). Sequence variants in those
genes have been found in patients with heterotaxy, sometimes in combination with IM
(Bamford 2000). Furthermore, the motile cilia are important in establishing normal LR
asymmetry of the body. Hundreds of genes are involved in cilia formation and function and
many of those have been linked to human disease, collectively called ciliopathies (Table 2).
The molecular landscape during embryologic development of the gut has been investigated
extensively in chicken embryos and in mice. In those model systems, rotation of the midgut
is initiated by differences in the cellular architecture of the right and left side in the dorsal
mesentery (Davis 2008). Different cascades of transcription factors cause the epithelial
(columnar shape) and mesenchymal cells on the left side of the dorsal mesentery to become
more condensed whilst the right-side epithelium is cuboidal and the mesenchyme remains
in an uncondensed state. A few key components are Nodal, regulating factors downstream
and is expressed only on the left side, also Pitx2, Fzd4, Fzt8, Gpc3, Daam2 and Isl1, are
only expressed on the left side and Tbx18 is expressed on the right side. It is suggested that
Wnt5 activates expression of the left-sided genes including Daam2, that regulates cell-cell
contacts to create a condensed state of the mesenchyme. Right-sided expression inhibits the
Wnt5a-pathway by for instance Sfrp1 and Sfrp2. Therefore, Daam2 is not activated and the
cells on the right side are not condensed. The result will be a leftward tilt of the dorsal
mesentery, suggesting the beginning of rotation of the midgut. (Davis 2008) (Figure 12).
Wnt5a is not expressed in the dorsal mesentery, but in the gut tube, and is important for
elongation of the tube (Sadler 2004, Welsh 2013). Also, Foxf1 is important in the formation
of the dorsal mesentery and is also a target for sonic hedgehog signaling, that is essential in
embryonic development (Mahlapuu & Enerbäck 2001, Mahlapuu & Ormestad 2001).

22
Figure 12. Transverse section of an embryo, left from 30 days of gestation and in the right
picture the dorsal mesentery shows a leftward tilt. The dorsal mesentery in red and the gut-
tube in yellow. (Martin 2010)
1.4.3 Familial cases Even though most IM cases are isolated and sporadic there are several reports on familial
cases (Erez 2001, Smith 1972, Beaudoin 2005, Budd 1988), mainly siblings, and thus
malrotation is regarded as a complex disorder depending on genetic as well as
environmental factors. Familial cases with IM together with other malformations have been
reported. One known familial disorder is congenital short bowel associated with
malrotation, intestinal dysmotility and short bowel (Erez 2001) and hereditary MIA and
malrotation is also previously reported (Gibson 1987).
1.4.4 Chromosomal aberrations associated with IM In the literature autosomal dominant, autosomal recessive, X-linked and chromosomal
forms of intestinal malrotation have been reported, mostly as syndromes with unknown
genetic etiology (Martin 2010) (Table 1).

23
Involved chromosome region
Associated malformations apart from IM Reference
Del 13q32 and dup 4q25
IUGR, cleft lip-palate, absent thumbs, bicuspid pulmonary valve, pulmonary hypoplasia. Father and paternal grandmother and great-grandmother with balanced translocations
Najafzadeh 1983
Ring chromosome 15 IUGR, microcephaly, a large VSD, dysmorphic ears, fingers and feet
Otto 1984
Partial distal 6p trisomy Abnormal lung lobulation, renal hypoplasia, crossed renal ectopia. Fetus 19 week GA, Maternal balanced translocation.
Fryns 1986
Del 13q21.2q32.3 Hirschsprung disease, developmental delay Lamont 1989
Mosaic tetrasomy in 8p Developmental disabilities, skeletal anomalies, rectal atresia, horse-shoe kidney, VUR. I(8p) in 68% of lymphocytes and 44% in skin fibroblasts.
Fisher 1993
Del 13q14.3q22.1 Hirschsprung disease, developmental delay Shanske 2001
1p36 del syndrome Growth delay, seizures, craniofacial dysmorphism and annular pancreas
Minama 2005
Mosaic trisomy 22, de novo
Total colon aganglionosis, congenital hearing loss, IUGR
Hall 2009
Mosaic trisomy 18 VACTERL including renal agenesis, cardiac laterality defect
Harewood 2010
t(2:6)(p23:q14), de novo Renal agenesis, cardiac and respiratory laterality defect
Harewood 2010
Partial trisomy 2p and monosomy 16p.
Dysmorphic features and developmental delay. Developed a neuroblastoma
Morgenstern 2013
3p14 interstitial del, 6.88 Mb, de novo
MZ twins, intellectual disability, autism, facial dysmorphism
Okumura 2014
Distal 15q25 dup and 12p13 del
Dysmorphic, craniosynostosis, horse-shoe kidney, pyloric stenosis
McLaughlin 2015
22q11.1q11.22 dup 6.6 Mb de novo
Dysmorphic face, CHD and later developed AML
Vaisvilas 2018
13q21.31q33.1 interstitial del (38.8 Mb)
Craniofacial dysmorphism, Mb Hirschsprung and hearing loss
Chatmetakul 2019
12p del syndrome Not specified Oliveira 2020
Mosaic trisomy 12 Dysmorphic face, bicornuate uterus, broad thumbs, partial anomalous pulmonary venous return. TOP week 22.
Bonasoni 2020
Table 1. Examples of chromosomal aberrations reported in the literature. Del deletion, dup
duplication, GA gestational age, IUGR intrauterine growth retardation, VSD ventricular
septal defect, VUR vesicoureteral reflux, MZ monozygotic twins, CHD congenital heart
defects, ALL acute lymphoblastic leukemia, TOP termination of pregnancy.

24
1.4.5 Monogenic IM One syndrome associated with IM and with known genetic etiology is congenital alveolar
dysplasia (CAD) due to mutations in the FOXF1 gene located in chromosome 16q24 that
usually leads to death within the first months after birth. This syndrome also presents with
intestinal malrotation, congenital short bowel, duodenal stenosis, annular pancreas,
pulmonary isomerism, cranio-facial features, cardiovascular and genitourinary
malformations (Stankiewicz 2009). Another syndrome is pediatric idiopathic intestinal
pseudo-obstruction, associated with intestinal malrotation, pyloric stenosis, hydronephrosis,
undescended testis, patent ductus arteriosus and thrombocytopenia. It is caused by an X-
linked recessive inheritance pattern due to mutations in the FLNA gene at Xq28 (Martin
2010, Gargiulo 2007). So far there are, to our knowledge, no such reports on patients with
isolated IM.
1.4.6 Ciliopathies Cilia are found on almost every human cell. There are motile and primary (non-motile)
sensory cilia (Wheway 2018, Fliegauf 2007). Examples of motile cilia are those found in
the airways to mobilize mucous and particles that need to be excreted. Also, the Node is an
important transient embryological structure that initiates the process of laterality through
motile cilia (Sharma 2008, Dasgupta 2016). Primary cilia (non-motile, sensory cilia) are
found on almost every other human cell that does not carry motile cilia and mediates
signals through sensation or signaling pathways (Reiter 2017). Primary cilia are important
during embryonic development and are involved in the Wnt-signaling pathway and in
Hedgehog signaling, both being associated with laterality defects in animal models
(Hildebrandt 2011).
A defect in the cilia, or in proteins important for the function of the cilia, can result in a
ciliopathy (Badano 2006, Fliegauf 2007, Reiter 2017), many of which include situs defects
(Table 2). Intestinal rotational abnormalities and heterotaxy are known to coexist, the latter
being associated to ciliopathies. Interestingly, there seem to be a male predominance in IM,
and the male to female ratio of heterotaxy is 2:1 (Ware 2011). There are cases with
ciliopathies presenting with IM, for example in a case with Jeune asphyxiating thoracic
dystrophy, hypothesizing that intestinal malrotation may be a ciliopathy (Hall 2009).

25
Table 2. Primary ciliopathies where LR axis defects have been described (Pollara 2022).
Disease Main clinical phenotypes Main gene(s) Inheritance
Nephronophthisis tubulointerstitial fibrosis, liver fibrosis, situs inversus, cardiac defects
NPHP1-4, IQCB1, CEP290, GLIS2, RPGRIP1L, NEK8,
SDCCAG8, TMEM67, TTC21B, WDR19, ZNF423, CEP164,
ANKS6, CEP83, INVS, DCDC2, MAPKBP1
Autosomal recessive
Bardet-Biedl syndrome
Obesity, polydactyly, retinitis pigmentosa, anosmia, congenital heart defects, situs inversus
BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, TTC8, BBS9, BBS10, TRIM32, BBS12, MKS1, CEP290, C2ORF86, IFT74, CFAP418, LZTFL1, IFT172, IFT27, SDCCAG8, PTHB1, BBIP1, TRIM32, WDPCP
Autosomal recessive
Meckel syndrome Cystic kidneys, encephalocele, polydactyly, liver fibrosis, congenital heart defects, situs defects
MKS1, TMEM216, TMEM67, CEP290, RPGRIP1L, CC2D2A,
NPHP3, B9D2, TMEM231, TCTN2, TMEM107, KIF14
Autosomal recessive
Joubert syndrome Cerebellar-brainstem malformation (molar tooth sign), developmental delay, ataxia, ocular-motor apraxia, retinal dystrophy, polydactyly, nephronophthisis
INPP5E, TMEM216, AHI1, NPHP1, CEP290, TMEM67, RPGRIP1L, ARL13B, CC2D2A, CPLANE1, OFD1, ZNF423, B9D2, TMEM107, CEP41, TMEM237, CSPP1, PDE6D, PIBF1, TCTN2, KIAA0586, KIAA0556, KIAA0753, CEP104, CEP120, TMEM231, TCTN1, ARMC9, ARL3, B9D1, MKS1, FAM149B1, SUFU
Autosomal recessive, X linked recessive (OFD1)
Alström syndrome Dilated cardiomyopathy, obesity, sensorineural hearing loss, retinitis pigmentosa, endocrine, renal and hepatic defects
ALMS1 Autosomal recessive
Jeune asphyxiating thoracic dystrophy/Short rib polydactylies
Skeletal dysplasia, polydactyly, cystic kidneys, retinal degeneration, cardiac, liver and brain anomalies
NEK1, DYNC2H1, DYNC2LI1, KIAA0586, WDR35, WDR60, IFT52, IFT43, IFT81, IFT80, WDR34, IFT140, TCTEX1D2, TTC21B, IFT172, WDR19, CEP120, CSPP1, DYNC2I1-2
Autosomal recessive


27
2 RESEARCH AIMS
The overall aim of this thesis was to better understand the molecular background and the
clinical spectrum of IM especially according to age and to increase the knowledge about the
outcome after treatment.
Specific aims:
- To understand the symptoms, diagnostics, treatments and outcomes of IM
throughout life. (Study I and Study II)
- To examine the prevalence of structural genomic variants in IM. (Study III)
- To examine underlying genetic causes of isolated IM. (Study IV)


29
3 CLINICAL STUDIES I & II
3.1 METHODS
3.1.1 Study design, Setting and Participants
To understand and describe the symptoms of IM throughout life we have described IM in
both adult and pediatric patients.
3.1.1.1 Study I – Adult patients
This was a retrospective case series at the Department of Surgery at Karolinska University
Hospital, where patients diagnosed and treated for IM between 2002 and 2013 from 15
years of age were included. Patients were continuously (prospectively) recruited into the
study during the time-period when diagnosed with IM. Inclusion criteria was IM, including
secondary IM.
3.1.1.2 Study II – Pediatric patients
This was a retrospective case series at the Department of Pediatric Surgery at Karolinska
University Hospital with children 0-15 years old who were diagnosed and treated for IM
from January 1983 to December 2016 were included. The hospital’s administrative
database was used, applying the ICD-codes for IM (ICD-10 Q433, ICD-9 751.4/751E) and
volvulus (ICD-10 K562, ICD-9 560.2/560C) and additional searches in the operation
planning system were made. We paired the temporary social security numbers and removed
double registrations. The medical records were reviewed and unclear cases were discussed
further with an experienced pediatric radiologist and two experienced pediatric surgeons.
When going through this large material we realized that many different phenotypes were
included in this group of patients. Every medical record needed extended scrutiny together
with evaluation of the radiologic findings to understand the specific malformation of each
patient for a valid inclusion.
Inclusion criteria was IM. Exclusion criteria were incorrect diagnose code, insufficient
information, incomplete variants of IM and cases with volvulus and no IM. Also patients
older than 15 years at onset of symptoms and patients with secondary IM were excluded.

30
3.1.2 Variables Data were retrieved from the medical records concerning age, sex, associated
malformations, symptoms, diagnostic and surgical procedures, previous disorders and
outcomes.
The patients were further divided into age groups.
Study II: <1 month, 1 month-1 year, 1-5 years, 6-15 years
Study I: 15-20 years, 21-50 years, 51-67 years
3.1.2.1 Study I, outcomes and follow-up
The patients were assessed 6 weeks, 6 months and 12 months postoperatively with a
possibility to resume contact if they had further complaints. Semi-structured telephone
interviews were performed 2012-2013 by an experienced research nurse regarding the
patients’ current symptoms as well as onset of symptoms, such as pain, nausea, vomiting,
constipation, and if they regarded their general physical condition as improved to a high
degree, improved with some reservation, or without any notable improvement.
3.1.2.2 Study II, outcomes and follow-up
Clavien-Dindo classification (Clavien 2009) was used to evaluate short-term complications
(<30 days) where grade IIIb or higher were taken into consideration (Table 3).
The patients were further divided into two groups based on severity in order to assess if
there was a greater risk for complications. The severely affected group still had
compromised bowel after de-rotation of midgut volvulus, defined by needing intestinal
resection or rescue strategies such as “second look” or “open abdomen” procedures.
Long term outcomes were collected from medical records: surgical procedures, intestinal
failure and mortality. In addition, a postoperative follow-up telephone interview was
conducted by an experienced research nurse with patients or parents according to a semi-
structured questionnaire. The questions were focused on the onset of symptoms, as well as
later gastrointestinal symptoms such as abdominal pain, nausea, vomiting, bloating,
constipation or diarrhea. If no symptoms were reported, the patients were considered “free
from symptoms”. Questions were also asked about perceived health after surgery and
current disorders.

31
Grade Definition
Grade I Any deviation from the normal postoperative course without the need for pharmacological treatment or surgical, endoscopic and radiological interventions.
Grade II Requiring pharmacological treatment with drugs other than such allowed for grade I complication.
Grade IIIa Requiring surgical, endoscopic or radiological intervention, not under general anesthesia.
Grade IIIb Requiring surgical, endoscopic or radiological intervention, under general anesthesia.
Grade Iva Life-threatening complication (including CNS complications) requiring IC/ICU management with single organ dysfunction.
Grade Ivb Life-threatening complication (including CNS complications) requiring IC/ICU management with multiorgan dysfunction.
Grade V Death of a patient
Table 3. Clavien-Dindo classification of surgical complications. (Clavien 2009)
3.1.2.3 Radiological diagnostics
Two experienced radiologists analyzed the radiological examinations, one in all adult
patients (Study I) and one in the difficult pediatric cases (Study II) respectively. In the adult
cases most patients had a CT scan with triple-contrast; per oral, intravenous, and intrarectal
contrast. This allowed us to visualize and evaluate the position of the duodenum, but also
the orientation of the superior mesenteric artery and vein as well as the position of the
cecum.
3.1.3 Statistical analysis Categorical variables are presented as frequencies and percentages in all studies. In Study
II, Chi-square test or Fisher’s exact test was used to analyze the association between age-
groups and symptoms, diagnosis and treatments. Univariable, as well as stepwise logistic
regression was used to analyze whether Clavien-Dindo complications could be caused by
variables that we assessed as risk factors. Results are presented as odds ratios (OR) and
95% confidence intervals (CI). P-values <0.05 were considered statistically significant.

32
3.2 MAIN RESULTS AND DISCUSSION
3.2.1 Study I inclusion
Between 2002 to 2013, 39 patients aged between 15 and 67 years were diagnosed with IM
and were all included in the study.
3.2.2 Study II inclusion and variants of IM
There were 319 patients taken into consideration for the study through ICD-codes after
doubles were removed and temporary social security numbers were matched, 138 patients
were included in the study. One hundred eighty-one patients were excluded.
Cause of exclusion N =
Secondary IM 18
Incomplete variants of IM (IRA) 47
Volvulus without IM 49
Insufficient information 25
No IM, IRA or volvulus 34
Age >15 years at onset 8
Total: 181
Table 4. Patients excluded in Study II.
Many excluded patients had volvulus without IM because the ICD-code for volvulus was
used in the search and the most common type was partial small intestinal volvulus (n=37).
A large group of the patients with the ICD-code Q43.3, that were excluded had intestinal
rotation abnormalities (IRA), but not IM. Thirty-nine had mobile cecum, six had an
abnormal variant of duodenums position, but did not fulfill the criteria for IM and two had
both. An interesting aspect, especially from a molecular perspective, is that patients with
IRA harbored almost identical rates of associated malformations, except for the CNS-
malformations (p=0.018) (Table 5). Within the IRA group one patient had a midgut
volvulus, another patient had a partial small intestinal volvulus and a third patient had a
cecal volvulus. All three had a normally positioned duodenum. Nine patients had
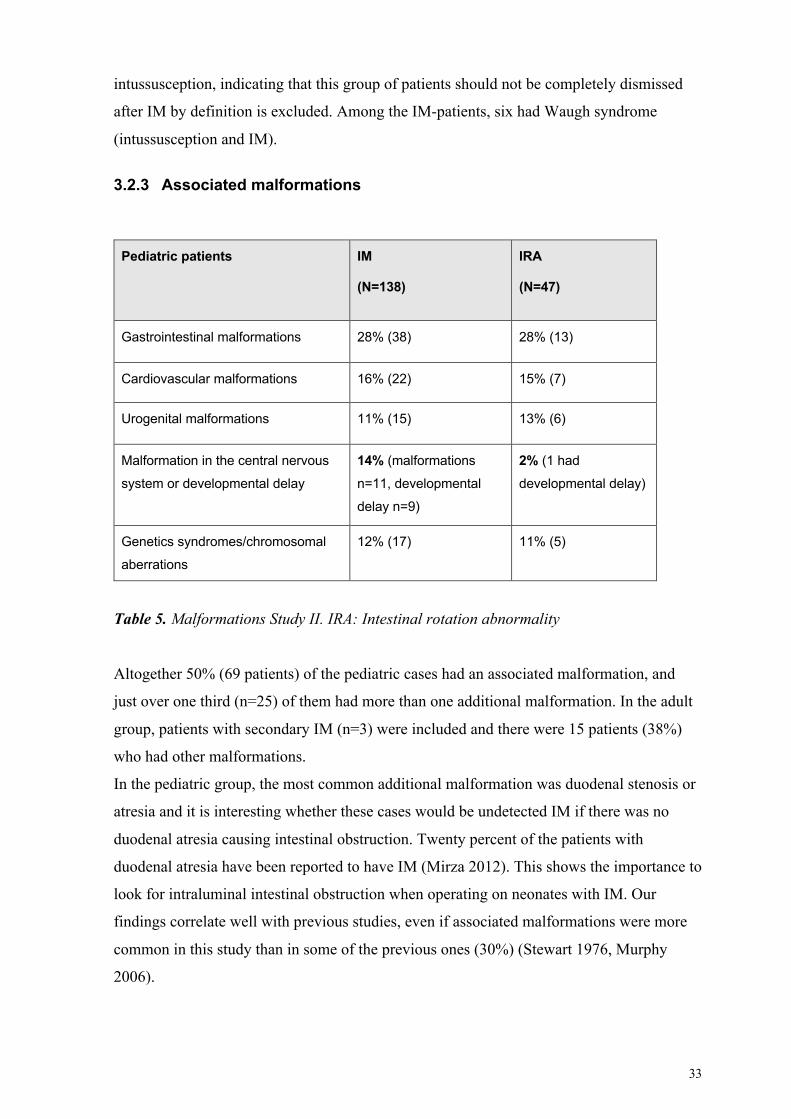
33
intussusception, indicating that this group of patients should not be completely dismissed
after IM by definition is excluded. Among the IM-patients, six had Waugh syndrome
(intussusception and IM).
3.2.3 Associated malformations
Pediatric patients IM
(N=138)
IRA
(N=47)
Gastrointestinal malformations 28% (38) 28% (13)
Cardiovascular malformations 16% (22) 15% (7)
Urogenital malformations 11% (15) 13% (6)
Malformation in the central nervous
system or developmental delay
14% (malformations
n=11, developmental
delay n=9)
2% (1 had
developmental delay)
Genetics syndromes/chromosomal
aberrations
12% (17) 11% (5)
Table 5. Malformations Study II. IRA: Intestinal rotation abnormality
Altogether 50% (69 patients) of the pediatric cases had an associated malformation, and
just over one third (n=25) of them had more than one additional malformation. In the adult
group, patients with secondary IM (n=3) were included and there were 15 patients (38%)
who had other malformations.
In the pediatric group, the most common additional malformation was duodenal stenosis or
atresia and it is interesting whether these cases would be undetected IM if there was no
duodenal atresia causing intestinal obstruction. Twenty percent of the patients with
duodenal atresia have been reported to have IM (Mirza 2012). This shows the importance to
look for intraluminal intestinal obstruction when operating on neonates with IM. Our
findings correlate well with previous studies, even if associated malformations were more
common in this study than in some of the previous ones (30%) (Stewart 1976, Murphy
2006).

34
Among the adult patients, six had gastrointestinal motility disturbances and among the
pediatric patients, two cases of gastrointestinal dysmotility were reported. Both groups
included one case of Hirschsprung.
Many patients in the pediatric group had a known syndrome or chromosomal aberration,
which also was present in the adult group. IM with multiple malformations was also noted
in previously reported cohorts, indicating that syndromes among these patients is probably
not that uncommon (Ford 1992, Martin 2010).
In the pediatric group hepatobiliary malformations were present, like biliary tract
malformations in five and pancreas annulare in three children. Maybe variants in the
hepatobiliary system could partly explain why eight of the adult patients had a history of
disease in the hepatobiliary and pancreatic system, including four cases of pancreatitis.
3.2.4 Gender and age at diagnosis
In the pediatric group (up to 15 years) the male:female ratio was 2.1:1, with no significant
difference between age groups. However, in the adult group the sex ratio was 0.8:1. At the
same time previous reports have shown a male to female ratio 1.29:1 – 2:1 (Spigland 1990,
Feitz 1997, El-Gohary 2010, Nagdeve 2012, Kulayalat 2015, Butterworth 2018, Black
2020) indicating a male predominance.
In the pediatric group 91/138 (66%) presented before one year of age, which is in
accordance with another recent study suggesting that 60% present under one year of age
(Aboagye 2014) and differs from previous reports on 90%. Interestingly, recently acquired
figures from the National Board of Health and Welfare show that 645 patients received the
diagnosis Q4.33 in Sweden between 2008 and 2020 and of them, only 37% were diagnosed
under one year of age and 31% being over 15 years when diagnosed (data received through
personal communication). In patients up to 15 years, 53% presented under one year of age,
which does not deviate as much compared to our findings in the pediatric study. The sex
ratio was equal, as well as in the IRA-group. It must be taken into consideration that the
diagnose code Q4.33 includes IM, and milder variants. The numbers may therefore differ
due to other diagnosis than strict IM.
Among the adult patients, presenting symptoms did not correlate with age. Even though
some patients had experienced symptoms for a long period of time, the symptoms seemed
to be aggravated at some point and needed assessment in all age groups.

35
3.2.4.1 Preterm birth and IM
In the pediatric group 33/138 (24%) were born preterm, the median gestational age among
preterm infants was 32 weeks (range 23-36 weeks, one missing data). One third of the
infants diagnosed with IM within the first month, as well as in the first year of life, were
born preterm. The reason for the high number of preterm patients could be the general
increased incidence of malformations in preterm infants (Rasmussen 2001).
Eight were born extremely preterm with a median gestational age 24.5 weeks; range 23-27
weeks and five of them presented within the first year of life. Half of the extremely preterm
infants died related to surgery, and two of them had necrotic bowel. There are many
comorbidities in extremely preterm patients and there is a higher risk to attain infections. It
can be a challenge to differentiate midgut volvulus from other conditions such as
necrotizing enterocolitis and sepsis. At the same time, in a recent study symptomatic IM
was diagnosed in 1.4% of 514 extremely premature infants, while only in 0.2% born after
28 weeks of gestation. They were all admitted to a neonatal intensive care unit, thus not
representing all newborns, but indicating an increased representation among the extremely
preterm patients (Mishra 2021).
3.2.5 Clinical presentation
A previous study divided 170 IM-patients into three groups: under one year of age, one to
18 years of age, and adults (Nehra 2011). We further divided the pediatric and adult groups
in order to more specifically describe the symptoms at presentation according to age. The
presented data shows that symptoms of IM will vary with age (Figure 13). The most
significant finding was that vomiting is the predominant symptom until five years of age,
whereas abdominal pain is the most common presenting symptom from 6 years of age and
through adulthood.
Bile-stained vomiting was seen up to five years of age and was the cardinal sign in the
neonatal group, but also common in the age group from one month to one year together
with abdominal pain and distended abdomen. This confirmed Nehra’s findings showing
that 93% of patients presented with vomiting if they were less than 1 year of age. He also
described that abdominal pain was the most common symptom in the age group one to18
years. In the present study, it was abdominal pain and vomiting that predominated from one
to five years of age, but also faltering growth was reported among one third. From 6 years
and through adulthood abdominal pain was the predominant symptom, even if vomiting, or
in the adult group as signs of intestinal obstruction, was common until 20 years of age.

36
Another recent publication on children with IM over one year of age (Dekonenko 2019)
reported abdominal pain, bilious vomiting and feeding intolerance as the most common
symptoms which confirmed our findings even though they reported bilious vomiting as
common up to 18 years of age, which differed from the present study. Blood-stained stool
was present in about 10% until 5 years of age and was then an alarming sign, all having
strangulating midgut volvulus except for one patient with chronic constipation.
Figure 13. Symptoms in children and adults related to age (%). *Referred to as obstruction
in adults.
Figure 14. Duration of symptoms. Pediatric patients up to 15 years diagnosed 1983-2016,
and adult patients from 15 years diagnosed 2002-2013.
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
<1 month 1 month-1year
1-5 years 6-15 years 15-20 years 21-50 years >50 years
Hours/days Weeks Months Years
0
10
20
30
40
50
60
70
80
90
100
<1 month 1 month-1 year 1-5 years 6-15 years 15-20 years 21-50 years >50 years
Vomiting* Bilious vomiting Abdominal pain Faltering growth

37
Infants had an early diagnosis, older children and adults, however, often had long-lasting
symptoms, in many cases for several years, which correlates with previous reports (Nehra
2011) (Figure 14). Almost half of the adult patients had symptoms during childhood, many
of them ongoing since. Other adult patients experienced a relatively sudden onset of
abdominal pain, interestingly sometimes associated with stressful events or life changes,
developing into chronic symptoms and intermittent abdominal pain.
The reason why the diagnosis is more rapidly established among neonates could be that IM
is a more recognized diagnosis in in that age-group who generally present emergency
symptoms. In the older age-groups, often with less alarming and more chronic symptoms,
probably other diagnoses were primarily taken into consideration. In addition,
gastroenterologists and surgeons treating adult patients with abdominal symptoms,
probably do not have a congenital malformation as a primary differential diagnosis.
In both studies, incidental findings were rare, but they were more common in the pediatric
group with 16%, mostly because of prenatal diagnosis of intestinal obstruction by duodenal
atresia in the neonatal period, as compared to other studies with incidental findings up to
36% (Dekonenko 2019, Nehra 2011).
3.2.6 Diagnostic procedures
The gold standard UGI-study were most often used in the pediatric cases, and often led to
diagnosis, given that the examination was done properly. In adults a multi-detector
computed tomography with triple contrast was used and was preferred as other diseases
generally are suspected as a cause for intestinal obstruction.
3.2.6.1 Upper gastrointestinal series
In the pediatric study, UGI-series were done in 83 patients and in 56 of them IM was
confirmed by the exam, nine were inconclusive, two were not available and one was
assessed as having normal anatomy. This demonstrates the importance of not excluding IM
based on a normal UGI-study. Fourteen patients had a non-acute duodenal obstruction, 15
patients had signs of midgut volvulus, such as a bird’s beak (n=5) or corkscrew appearance
(n=5) of the intestines, and in all but one of them malrotation was also described. One
patient had a follow-up CT-scan with oral and IV contrast after an inconclusive UGI study.
Another option to obtain a diagnose after an inconclusive UGI study, was a diagnostic
laparoscopic procedure.

38
3.2.6.2 Computed Tomography
The pediatric patients who had a CT-scan were given intravenous contrast, sometimes oral
contrast, and revealed cases of midgut volvulus but not always IM. Among the 19 patients
who had a CT-scan, 14 were 6 years or older. According to the medical records, the
abnormal position of the duodenum was confirmed only in three, inversion of SMA and
SMV in five, abnormal positioning of colon in eight, and signs of midgut volvulus in five
(with whirlpool sign in three of them and strangulation of the SMA in the other two).
In the 33 adult patients who had a CT-scan (one had an MRI) the duodenum was located on
the right side of the abdomen, with the duodenojejunal junction not traversing the vertical
spine, except for in one case, where the duodenal course initially crossed the midline to the
left but as a loop turned back again to the right side. The cecum had an abnormal position in
22 cases and SMA and SMV were mispositioned in 26 cases, whereas all three signs were
confirmed in 19 cases (Figure 15). This confirms that the positioning of the duodenum is
the most reliable diagnostic feature to look for in a CT-scan as in coherence with previous
studies (Zissin 1999).
Figure 15. Radiological findings of the 33 adult cases who had a CT-scan with triple-
contrast (one had MRI). AC- Ascending colon. SMA- Superior mesenteric artery. SMV-
Superior mesenteric vein.
Interestingly, “whirlpool sign” was noted in seven of the adult patients, not just in
emergency cases, but also in chronic ones, and none of them needed intestinal resection.
The three children with “whirlpool sign” did not have an affected circulation, two were
noted as incidental findings in a trauma and in a case of appendicitis and one had chronic
symptoms from midgut volvulus. The two children who showed midgut volvulus by
strangulation of the SMA, had severely affected circulation at exploration as expected.
SMA and SMV n= 26
Duodenum n= 33
Cecum n= 22
n= 19
100%
AC mobile at surgery

39
3.2.7 Management
The treatment of IM was either surgery, using Ladd’s procedure or clinical follow-up
depending on comorbidities and symptoms. Diagnostic laparoscopy was sometimes
performed in the pediatric group (n=10), and when diagnose was established, converted to
open surgery in six cases. Laparoscopic Ladd’s procedure was performed in the remaining
four cases. The same open surgical procedure was performed in pediatric and adult cases.
Ninety-one % (125/138) of the pediatric group underwent Ladd’s procedure and 79%
(31/39) in the adult group. In the pediatric group 75 patients only had Ladd’s procedure,
additional procedures were done in the remaining 50 because of duodenal atresia or
stenosis, small bowel atresia, resection of Meckel’s diverticulum, other malformations,
planned gastrostomy and reduction of intussusception. In the adult group Ladd’s surgery
was the only procedure.
One third of the adult cases underwent emergency surgery, compared to half of the
pediatric cases. Of the pediatric cases there were 24 who had a midgut volvulus with
impaired bowel circulation. Midgut volvulus with severely compromised bowel was found
in eight patients, they needed intestinal resection, second look or open abdomen rescue to
reevaluate bowel vitality and to prevent compartment syndrome. Among the adult patients
there were eight who had midgut volvulus with compromised circulation, with one needing
resection of the entire necrotic small bowel and three who needed a minor resection. Two
of these patients were over 50 years old.
3.2.7.1 Appendectomy
An incidental appendectomy was performed in all the adult patients and in 58% of the
pediatric patients. One patient had acute appendicitis eight years after Ladd’s procedure. It
has been tradition to perform an appendectomy as part of Ladd’s procedure because it
decreases the risk of misdiagnosing appendicitis. The different recent approach to save the
appendix is based on reasonings such as diagnosing appendicitis radiologically has become
more accessible and an appendectomy is not free from complications. Furthermore, some
children with pseudo-obstruction (that is related to IM) may need a future appendicostomy
for antegrade enemas (Healy 2016).

40
3.2.8 Outcome
3.2.8.1 Recurrent midgut volvulus
Recurrence was more common in the adult group with six patients, compared to one patient
in the pediatric group. There are studies supporting that it is more common among adult
patients (Ferreira 2020) and one could speculate that a cause could be that the intestines
have grown and settled into a certain way, and sometimes into a chronic rotated state that it
returns to. If that is the case, this is also a reason to diagnose and treat IM early.
3.2.8.2 Intestinal failure
In the pediatric group four patients developed chronic intestinal failure, defined as having
the need for parenteral nutrition for at least 90 days (Duggan 2017). Two because of short
bowel syndrome from a midgut volvulus, one because of necrotizing enterocolitis and one
because of intestinal dysmotility. Two patients were weaned off parenteral nutrition after
nine and 24 months, one patient needed intestinal transplantation and the one patient with
necrotizing enterocolitis died at 11 months of age. Among the adult patients, one patient
died due to short bowel after midgut volvulus. Another 17 of 124 patients (one missing
data) needed parenteral nutrition for more than two weeks postoperatively in the pediatric
group and two of 31 in the adult group.
3.2.8.3 Short-term complications (Study I)
The Clavien-Dindo classification scale was used to look for short-term complications
within 30 days. Postoperative data on one of the 125 patients who had surgery were missing
and only 124 cases could be assessed. Complications from type IIIb-V were considered
because of the difficulty to ensure that the correct data would be collected on grade I-IIIa
from the retrospective cohort.
The risk to develop short term complications grade IIIb-V were 20% (25/124), which was
similar compared to a previous study where 16% developed complications graded III-V
(Salö 2019). Predictive factors to develop complications were midgut volvulus with
severely affected circulation, OR 8 (95% CI 1.77-36.22). Of the eight patients who had a
severely affected circulation, defined by intestinal resection of necrotic intestines, “second
look” and “open abdomen” procedure, 63% (n=5) had a complication with 25% (n=2)
mortality, while in the other group with Ladd’s procedure with or without midgut volvulus,
17% (n=20) had a complication, with 2% (n=2) mortality.
Another predictive factor was extreme prematurity (gestational age < week 28), OR 30.95
(95% CI 3.52-271.93), which is in accordance with a recent large study showing that short

41
term postoperative complications and failure to rescue (FTR) increases with decreasing
gestational age (GA) (Mehl 2021), even though the OR to develop complications among
patients born week 28 – 37 was not significant in the present study.
3.2.8.4 Long-term complications
In the pediatric study 11% developed small bowel obstruction which correlates with
previous studies (Fredriksson 2016, Salö 2019). In the adult group it was only 8%, maybe
partly because no other surgical procedures were done simultaneously.
The pediatric and adult patients who had Ladd’s surgical procedure were followed-up by a
telephone interview. In the adult group 26 patients were available for an interview 1-12
years after surgery, and in the pediatric group 61 patients 1-36 years after surgery (median
8 years).
Adult group
The symptom of acute intermittent pain that was present before surgery in almost all of the
interviewed patients was mainly gone postoperatively, leaving only one patient who did not
experience a relieve. However, abdominal pain of a more chronic character remained in 12
patients (46%). Among 13 patients with constipation, it remained in eight postoperatively.
When asked about their general life quality, it was improved in all but one patient
postoperatively.
Pediatric group
Nineteen patients reported constipation prior to surgery and 12 patients postoperatively
where 10 claimed need of pharmacological treatment. Constipation in one fifth of the
patients who had surgery for IM, is in coherence with previous reports on constipation
associated with IM (Salö 2019) and may be expected as motility disturbances are associated
with IM. Seventeen patients (28%) still had abdominal pain after surgery and of them it was
four who experienced the same pain as before the surgery.
The telephone interview created an opportunity for the parents to express their experiences
and in 45 of the cases they described it as a traumatic experience with words as “horrific”,
“a hopeless situation” and “shocking”.
3.2.8.5 Mortality
In the adult group one patient died because of intestinal failure after midgut volvulus with
total bowel necrosis, four other patients died due to other comorbidities. In the pediatric
group, four died within five days postoperatively, they were all born extremely preterm,
were aged between two to six months and had a short history of abdominal symptoms. Two
of them had midgut volvulus with necrotic intestines at exploration. Five patients,

42
previously operated for IM died due to other comorbidities. The mortality rate was
relatively high, but within previous reported data, 2-9%, and the cause of death as in our
case, usually were not related to IM (El-Gohary 2010, Murphy 2006). Also, there was an
increased mortality <30 days postoperatively among the patients with severely affected
bowel (P=0.021).
3.2.9 Adult patients and IM
With increased knowledge about IM, the diagnosis needs to be considered as a possible
cause of acute and chronic symptoms also in adults, which has also been pointed out in
previous studies (Coe 2015, Nehra 2011). In this group, concurrent developmental
disorders and lower age, were red flags and should raise suspicion. Due to age-related
comorbidities and a seemingly decreased risk for midgut volvulus in the older age group, a
more conservative approach regarding surgery could be considered, even though other
studies recommend surgery on incidental adult cases (Moldrem 2008). It is impossible to
predict who will develop a strangulating midgut volvulus in the future, especially
considering the oldest patient being 62 years old in this material.

43
4 MOLECULAR STUDIES III & IV
4.1 METHODS
4.1.1 Study design, Setting and Participants
To study the molecular background of IM we both screened for submicroscopic structural
chromosome variants and performed whole genome sequencing (WGS). In the clinical
studies, patients were asked to donate blood for molecular studies on DNA and were in the
semi-structured interviews asked about if there was any knowledge about affected relatives.
Among the children, also the parents were asked to donate blood.
4.1.1.1 Study III
A custom designed array CGH, targeting 1989 genes involved in ciliopathies and other
genes important for early development was used, to screen for small intragenic and large
deletions and duplications in a single experiment. The study included data from two
cohorts.
Cohort 1: All adult and pediatric patients with IM, who had donated blood for molecular
studies n=42.
Cohort 2: Five patients with an IM diagnosis that had undergone clinical genetic
investigation at the Department of Clinical Genetics (Karolinska University Hospital).
Using the terms “intestinal malrotation” or “midgut volvulus” they were identified in the
internal database of >5000 clinical array CGH analyses.
4.1.1.2 Molecular Study IV
We also wanted to investigate if there was a genetic predisposition in patients with isolated
IM without any other comorbidities. We used WGS to look for sequence variants in
candidate genes, but also used the data to look for larger structural variants as in Study III.
This study included pediatric patients with IM, who had donated blood for further
molecular studies. Inclusion criteria was isolated IM with early onset midgut volvulus
where surgery had also verified the diagnosis and whole blood for DNA extraction was
collected from both parents.

44
4.1.2 Molecular analyses
4.1.2.1 Array comparative genomic hybridization (array CGH)
array CGH is a cytogenetic technique to detect submicroscopic chromosomal copy number
variants (CNVs) with a high resolution and covering the whole genome. In clinical
diagnostics, the resolution is usually about 25-50 kilo-bases, but with custom made targeted
array designs the resolution can be a few hundred base pairs in specific regions (Lindstrand
2016).
Figure 16. array CGH. DNA from patient and control is labelled with different fluorescent
dyes. When the labelled and denatured single stranded DNA hybridizes to the array it will
bind to the matching oligonucleotide probes in a dose dependent way. The patient DNA is
labelled in green (cy3) and the control DNA in red (cy5) and when they bind in an equal
amount the signal will be yellow. When there is a deletion, there will be more cy5-labeled
control-DNA causing emission of red fluorescence and when there is a duplication there
will be green fluorescence as there is more cy3-labeled DNA. The ratio of the two
fluorescent signals is quantified by a scanner and visualized as a log2 plot in an array
analysis software. Two examples are shown, a duplication and a deletion.

45
Databases with CNVs that are often present in healthy controls, are used to filter out
common CNVs, regarded as normal variants, found in the patient.
In Study III, cohort 1, array CGH analysis was performed in accordance with a previously
published method (Pettersson 2017). A custom designed microarray, consisting of 400 000
oligonucleotide probes per patient was used. The probes target 1989 genes, including all
genes in the cilia proteome and genes involved in malformation syndromes. Within the
targeted genes, intragenic variants can be detected, with the resolution 1 probe per 100 base
pairs (bp) in the coding regions and 1 probe per 500 pb in the noncoding regions. The
remaining probes, cover the genome with an approximate resolution of 25-50 kilobases
(kb).
Genomic DNA from the patients and reference DNA were labeled and hybridization were
performed according to protocols provided by Agilent Technologies. The slides were
scanned, using a DNA Microarray Scanner. The scanned file was processed and then
analyzed using Cytosure Interpret Software.
In cohort 2, array CGH analyses were done due to clinical reasons at the Department of
Clinical Genetics at Karolinska University Hospital, where a 180K microarray was used
with whole genome coverage.
Classification Description
Benign Not in individuals with certain phenotypes, reported in multiple peer-reviewed publications and repeatedly found in the normal population.
VOUS Contains genes of interest, but little is known about them.
Likely pathogenic
Described previously in a patient with a similar phenotype, or a gene has an associated function to the patient’s phenotype.
Pathogenic Well documented as clinically significant in multiple peer-reviewed publications.
Table 6. Classification of CNVs at a glance. VOUS, Variant of uncertain significance

46
4.1.2.2 Whole genome sequencing (WGS)
Whole genome sequencing (WGS) methods sequence the whole genome and the main
difference from array CGH is that SNVs can be identified, as well as SVs not targeted by
the array CGH, such as smaller duplications and deletions and insertions. In addition, some
chromosomal abnormalities are not seen in array CGH, such as balanced translocations.
WGS provides an opportunity to look for specific candidate genes, and since the data is
stored additional candidate genes can be analyzed later on. Detection of structural variants
is based on read depth. Lower read depth would indicate a deletion and vice versa.
Sequence variants are detected using filtering programs that first compare the reads to the
human reference genome, and then filter against the common variants seen in the general
population.
Figure 17: Whole genome sequencing. Briefly a DNA-library is prepared with DNA
fragmented to around 300bp and use short signature tags of approximately 20-30
nucleotides on both the 5’ and 3’ end of the DNA-fragment for paired-end sequencing. The
DNA-fragments are introduced to a flow cell, a glass slide full of oligonucleotides, that the
DNA can bind to and will be used for further sequencing. For the fluorescent signal to be
strong, cluster amplification is the next step, generating multiple clonal DNA-strands in a
cluster on the flow cell by a polymerase. In the beginning of the amplification-step the
paired-end reads will bind on both ends as an upside-down U and strands from both sides
will be copied and further amplified. The sequencing takes place as fluorescently tagged
nucleotides attach to the DNA-strand and emit a characteristic fluorescent signal that is
read by a laser scanner. This happens in multiple clusters of DNA at the same time and is
scanned in a parallel manner. For each cycle a fluorescently tagged nucleotide adapts to
the chain and the number of cycles will determine the length of the read. All scanned reads
will be further processed for data analysis.
Librarypreparation
Cluster amplification
Sequencing
Data analysis

47
In Study IV, DNA from whole blood from each patient was sequenced using the national
genomics infrastructure (NGI) with an average read depth of 30x with Illumina TruSeq
PCR-free paired end protocol on a Illumina NovaSeq 6000. In brief, Scout v63.0 software
was used for calling variants among the 429 candidate genes through a built-in ranking
pipeline, prioritizing the variants from most to least interesting (Stranneheim 2021). Then
vcf2cytosure (Lindstrand 2019) was used to visualise SVs in a similar way as in the
previous array CGH in the four patients who did not previously have an array CGH done.
4.1.2.3 Candidate genes
HPO-panels for IM (HP:0002566) and heterotaxy (HP:0030853) was used (143 genes), as
well as gene panel for cilipathies (204 genes). Through literature review in PubMed and
searches in OMIM, 103 additional genes involved in intestinal development and IM were
identified, as well as 22 genes that are paralogues to or interact with the TTC7A and TTC7B
genes.
4.1.2.4 Sanger sequencing
Sequence variants were confirmed, and carrier testing of parental DNA was performed in
some cases.

48
4.2 MAIN RESULTS AND DISCUSSION STUDY III
4.2.1 Inclusion
Cohort 1
Forty-two patients were included, treated between 2012 and 2015, 20 male and 22 female.
Median age at inclusion were 20.5 years and forty patients had abdominal surgery. 52%
(n=22) had isolated IM. Associated malformations were: Situs inversus n=1, cardiac
malformations n=4 (including one heterotaxy syndrome), stenosis or atresia of the intestine
n=6, urogenital malformations n=2, CNS-malformations n=5, cleft lip palate n=1. At least
five patients were born preterm, four patients had intellectual disability or developmental
delay and three had cerebral palsy. Secondary malrotation in three cases was caused by
omphalocele, gastroschisis and congenital diaphragmatic hernia (CDH). All patients in the
study had symptomatic IM.
Cohort 2
Five patients were included, none had isolated IM. Two patients had cardiac malformations,
one patient had omphalocele and Meckel’s diverticulum, and one had duodenal atresia.
Two patients had developmental delay or intellectual disability.
4.2.2 Reported CNVs
We found six rare CNVs in five patients.
Cohort Array result
Coordinates Affected gene/ loci Classification IM previously reported
1 Deletion 2p23.1 GALNT14 VOUS No
1 Duplication 7q11.23 7q11.23 microduplication syndrome
Likely pathogenic
Yes
Duplication 16p13.11 16p13.11 microduplication syndrome
VOUS
1 Deletion 18q22.1q23 18q deletion syndrome
Pathogenic No
1 Deletion Xq13.1q13.2 HDAC8 (Cornelia de Lange typ 5)
Pathogenic No
2 Duplication 16q24 FOXF1 Pathogenic Yes
Table 7. Genotype of patients with rare CNVs and IM. VOUS, Variant of uncertain
Significance.

49
Patient one had isolated IM and had an intragenic heterozygous deletion in the GALNT 14-
gene, inherited from the healthy father and not previously implicated in human disease, and
thus was considered as of uncertain significance. The GALNT14 gene was of interest
because it is important during embryonal development and overexpression increases
mRNA expression of N-cadherin (Huanna 2015). It is a cell adhesion protein important in
asymmetric cell-formations in the dorsal mesentery (Davis 2008, Plageman 2011, Welsh
2013). Knockdown of the gene inhibits cellular migration and affects cell morphology
(Wang 2013). This patient was the only one of 22 patients with isolated IM that harboured a
rare CMV.
Patient two, had two heterozygous rare de novo separate microduplications, both described
as having varying penetrance (Kirov 2014). The 7q11.23 microduplication syndrome
(MIM#609757) is known and causes language and developmental delay, also intellectual
disability with an estimated 44% penetrance (Kirov 2014). In addition, congenital heart
defects, neonatal hypotonia and cryptorchidism are common. One case of congenital
diaphragmatic hernia and one case of umbilical hernia was reported (van der Aa 2009) and
one previous report with IM (Morris 2015). This patient had IM, expressional language
disorder and cryptorchidism which fits the above features. The other duplication, located in
the 16p13.11 region is associated with congenital anomalies and neurodevelopmental
disorders, however only exhibiting a 8.4% penetrance (Kirov 2014, Sahoo 2011). The
patient’s phenotype was likely caused by a combinatory effect of both detected
rearrangements.
Patient three had a heterozygous terminal deletion 18q22.1-q23, previously associated with
many congenital anomalies (Strathdee 1995). The patient had congenital nystagmus,
delayed myelination, global developmental delay, left-sided clubfoot, and umbilical hernia
which was in concordance with the 18q deletion syndrome (MIM#601808). The patient
also had IM and an accessory spleen which was not previously reported in patients with 18q
terminal deletions (Cody 1999).
Patient four had a de novo heterozygous deletion, Xq13.1-q13.2, involving HDAC8
(MIM#300269), associated with X-linked dominant Cornelia de Lange syndrome (CDLS)
type 5 (MIM#300882). The patient had IM, delayed psychomotor development, speech
delay, strabismus, dysmorphic facial features, nevus flammeus, brachydactyly and

50
hirsutism, all in concordance with CDLS. CDLS is associated with gastrointestinal
malformations, but only 2.3% has IM (Kapoor 2014) and to our knowledge, this is the first
published case of HDAC8-related CDLS with IM.
Patient five had a large de novo duplication on 16q24.1, including the FOXF1 gene. The
patient had IM, mild intellectual disability, short stature, mild pulmonary artery stenosis,
mild dysmorphic facial features. Duplications involving the FOXF1 gene, from 15kb to
1,7Mb in size have previously been described in four families with intellectual disability,
speech delay, and gastrointestinal abnormalities including IM, thus IM in a syndromic
context (Dharmadhikari 2014).
4.2.3 Clinical relevance
We could present, to our knowledge, the first cases of 18q terminal deletion syndrome and
CDLS type 5 with IM which could be rare phenotypes. Since IM can be asymptomatic, it
can have been missed in previous cases. In addition, these findings could be incidental,
since the cause of IM is probably often multifactorial.
We identified likely pathogenic or pathogenic CNVs in 8,5% (4/47) patients with IM. All
with those findings had associated malformations and neurological symptoms. Therefore,
we suggest that genetic investigation with CNV screening is indicated in cases with IM and
additional extra-gastrointestinal malformations or developmental delay.
We did not find any SVs in ciliopathy genes thus we found no support for the theory that
IM is a ciliopathy.
To fully understand the impact on rare CNVs on IM, further genetic studies on larger
cohorts are needed. For this, array CGH is an effective and well-established tool to identify
disease causing genes in both isolated and syndromic gastrointestinal malformations
(Dworschak 2015, Genesio 2018, Tsai 2015). Further advances have led to more accessible
whole genome sequencing (WGS). With tools, to find structural variants (SVs), it is an
appealing approach to also be able to screen for SNVs in gene panels and to be able to
reanalyze data when discovering future candidate genes.

51
4.3 MAIN RESULTS AND DISCUSSION STUDY IV:
4.3.1 Inclusion
This was a pilot study of WGS in ten patients with isolated IM and no other malformations
or comorbidities, except for one who had intrauterine growth retardation and was born in
week 35+0. Of the ten, nine were male. Early symptoms were a prerequisite to attain a
severe phenotype and five had symptoms since birth. Seven were diagnosed within the first
month, two were one year old and one patient was four years old when diagnosed and
treated. All had midgut volvulus at the time of diagnosis and surgery, except for one who
had recurrent midgut volvulus later on and needed redo-surgery.
4.3.2 Reported variants
After filtering and ranking the WGS data on all ten cases, we identified two rare allelic
variants in the Tetratricopeptide Repeat Domain 7A, TTC7A gene: c.433G>A p.Ala145Thr
and c.1057G>A p.Glu353Lys in one patient (Table 8).
Variant c.433G>A p.(Ala145Thr) c.1057G>A p.(Glu353Lys)
Exon 3 8
Allele frequency (GnomAD) 0.0004 0.000008
Conservation Conserved Conserved
CADD score 22.8 25.9
TPR-domain Yes No
Table 8: Description of the discovered variants in TTC7A.
The male patient had surgery when he was 16 days old, and was followed up to 14 years of
age. No other malformations or comorbidities were reported except for gluten intolerance
as well as some abdominal complaints with fecal urgency.
Carrier testing showed that both variants were inherited from the mother, who has an
autoimmune disease and a mild intestinal rotation variant with the cecum pathologically
located in the right upper quadrant discovered when an experienced radiologist analyzed

52
previous radiologic investigations due to other causes. She also suffers from fecal urgency,
as does her sister. The maternal grandmother also has an autoimmune disease (Figure 19).
Figure 19. Pedigree of the family with two variants in cis in the TTC7A-gene (from study IV).
Familial cases with IM can easily be missed so several affected could be more common
than reported. Abdominal complaints are not uncommon but with a family history of IM,
further investigation could be indicated.
4.3.3 The TTC7A-gene and IM
The TTC7A gene is expressed in the intestines and involved in intestinal development. It
has 20 exons, is located on chromosome 2p21 and contains nine tetratricopeptide repeat-
domains (TPR-domains) that are important in protein-protein interactions.
TTC7A-deficiency is an autosomal recessive condition, causing multiple intestinal atresias
(MIA) and immune defects; combined immunodeficiency (CID) (Lemoine 2014, Woutsas
2015, Yang 2015). Another expression of the disease is early onset inflammatory bowel
disease (EOIBD). So far 55 cases are reported, with homozygous or compound
heterozygous mutations in the TTC7A gene (Jardine 2019).
IM has been reported in at least five cases with MIA, in two of the cases, who also had
CID, genetic testing was done and showing smaller homozygous deletions in the TTC7A

53
gene; c.53344_53347del, and c.313_216delTATC respectively (Bilodeau 2004, Mandiá
2018, Guanà 2014).
4.3.4 Clinical relevance
In this first confirmed familial case in our cohort, the mother who has an IRA, also has an
autoimmune disease which could cohere with a mild phenotype of TTC7A-deficiency.
Our findings could be incidental, but there are examples of autosomal recessive disorders
where carriers express a milder phenotype. (Wigley 1994, Nordenström 2019). Even
though carriers of TTC7A variants generally do not have symptoms, it is interesting to
speculate that IM could be a mild manifestation of a milder form of TTC7A-deficiency,
when in this family both carriers have a rotational anomaly and the mother also manifests
an autoimmune disease.
Possible mechanisms connecting TTC7A to IM are the Phosphatidylinositol 4-kinase IIIα-
complex (PI4IIIα-complex) where TTC7A is one of four proteins. PI4IIIα is another protein
in the complex, and translates from the PI4KA gene. A homozygous missense mutation in
the PI4KA gene was reported in a consanguineous family, with 13 patients with MIA and
inflammatory changes in the intestines. Two of them also had IM and the variant had a
negative impact on PI4KIIIα and TTC7A-interaction as identified by biochemical studies
(Salter 2021). A TTC7A protein defect also leads to worsened interaction, and causes the
same disease, including some cases with IM. Also, the TTC7A protein regulates the
RhoA/ROCK-signaling pathway (Ras homolog family member A/Rho-associated protein
kinase signaling pathway). Studies on Xenopus have shown that the gut-tube formation and
elongation is dependent on Rho/ROCK/MyosinII activity where mild cases had IM and
more severe cases had elongation deformities (Reed 2009). This would support the theory
that embryological development and “rotation” of the midgut is driven by elongation. The
full role of the PIKIIIα-complex and of TTC7A in the RhoA/ROCK pathway is however
still under investigation. The involvement in elongation of the gut tube is interesting in the
light of the embryological development.
Genes in fetal development not only associated to laterality and dorsal mesentery, but also
genes associated with intestinal elongation, function and development have been high-
lighted. For example, CLMP and FLNA genes are associated with short bowel and
malrotation (Negri 2020, Wang 2021), also pseudo-obstruction and IM is associated with the
FLNA-gene (Clayton-Smith 2009). These genes were included in our gene panels and without
any findings. Pathogenic CNVs in patients with IM are found in association with other

54
malformations and not in isolated cases. We do not believe that IM is a monogenic disease
possibly due to many compensating systems in fetal development. Clinically, there are
isolated cases (in the pediatric study 50%) and the rest have additional malformations
showing a failed embryonal mechanism, affecting several organs. The isolated cases could
derive from either variants that have well established compensatory mechanisms in other
organs but fail in the intestinal development. Alternatively, they act locally on the intestines,
or due to local insults such as ischemia or environmental factors acting at a critical stage of
intestinal development.

55
5 STRENGTHS AND LIMITATIONS The clinical studies (I and II) consist of well-defined large cohorts considering that IM is an
uncommon disease, so they are a valuable addition to previous studies in the field. The
purpose of the statistical analysis is to simplify interpretation of the data, and its limitations
should to be taken into consideration such as an increased risk to miss or to overestimate an
association. Previous studies are also valuable in order to validate our findings. One could
consider a selection bias considering so many excluded patients in the clinical study in
children. However, we used a wide search in an effort not to miss anyone and defined the
diagnosis through strict inclusion criteria from surgical records and radiological findings. It
was also important to have a defined phenotype for further molecular studies.
Like in other retrospective cohorts, limitations such as recall- and misclassification bias, as
well as missing data, should be considered and an attempt to counterbalance this somehow
was by having a strict attitude when assessing the data.
In study III the median age at inclusion is high compared to the typical IM-patient and at
the time of the study the response rates among older patients were higher, explaining the
skewed median age. This can be a strength since malformations and comorbidities, not
discovered at birth, have been revealed by time. For the molecular studies we have well
characterized phenotypes, and it is a relatively large group with a diagnosis that has not
previously been studied this way. We have the possibility to follow up the families and
gather blood samples for further studies on relatives. During the studies we came to realize
that identifying familial cases is extremely challenging since the spectrum of intestinal
rotation varies and some are asymptomatic. Some families that we thought we had
identified had to be excluded for different reasons. Further comorbidities can develop over
time, that we have not yet detected so the estimation of associated comorbidities may be
understated. Array CGH is a well-established method to look for unbalanced SVs, even
though balanced SVs, such as balanced translocations and inversions, are missed. In study
IV there are challenges that arise when analyzing WGS, we screened for candidate genes
and for larger duplications or deletions in the genome. Bad reads could have caused us to
exclude potentially interesting findings, also balanced SVs might be missed as well as
findings in the genes we did not yet screen for. Study IV was a small pilot study and will
need further confirmation in larger materials.

56
6 ETHICAL CONSIDERATIONS
The studies were approved by the Regional Ethical Review Board, Dnr; 2008/670,
2012/957-31/3, 2013/944-31, and they were designed in accordance with the Declaration of
Helsinki/Good Clinical Practice. Written informed consent was collected from all
participants or parents/legal guardians where blood was collected or interviews were
performed.
Parents to patients with malformations want to know the cause, possible treatments and
how the future for the patient will be affected. The aim of these studies was to learn how to
better diagnose and treat IM, as well as to understand the genetic background. Such
increased knowledge can sometimes ease the worry that parents to patients with a
malformation have, like a feeling of guilt when they do not know the cause of the disease.
Earlier experiences are that patients, and their parents, generally are positive to these
studies and see it as an opportunity to increase knowledge about their disease.
This is a rather rare disease and patients who have been treated might feel that they can
recognize themselves in the studies. This is avoided by grouping the results and be as
general as possible when describing a phenotype.
Blood was donated from patients and relatives, they received written and oral information
and it was voluntary with clear information on that they could change their decision about a
consent at any time. If possible, blood was collected in association with other blood tests.
The advancement in mass sequencing has opened up new possibilities in many ways,
among rare disease and cancer treatment strategies. One possible dilemma may be
incidental findings of a hereditary diagnosis. The analyzed data is always coded or double-
coded and is only used in relation to the specific research question/ diagnosis.

57
7 CONCLUSIONS
The studies have contributed to an expanded knowledge or confirmed previous finding on
the clinical picture and outcome from birth to adulthood, as well as increased knowledge on
the molecular etiology of IM.
In study I, we concluded that IM should be regarded as a malformation affecting all age-
groups, with acute obstruction by midgut volvulus occurring in all ages. Surgery generally
relieves symptoms, even long-lasting pain and a contrast enhanced computed tomography
reveals the diagnosis. The most common presenting symptom is abdominal pain.
In study II, we concluded that vomiting is the most common presenting symptom until 5
years of age, and abdominal pain from six years of age. About 60% of pediatric cases
present before one year of age, and older children often have delayed diagnosis, as in the
adult cases. Risk factors to develop complications after surgery are extreme prematurity and
midgut volvulus with severely affected circulation of the intestines.
In study III, based on our findings, we suggest that genetic investigation with CNV
screening should be performed in cases with IM and additional malformations or
intellectual disability. We could also present, to our knowledge, the first cases of 18q
terminal deletion syndrome and CDLS type 5 with IM.
In study IV, we suggest that IM can be a mild manifestation of TTC7A-deficiency. The
TTC7A gene needs to be studied further in DNA from a larger cohort of patients born with
IM.


59
8 POINTS OF PERSPECTIVE
It was recently put to light that also adult patients can develop acute and chronic symptoms
from IM and not much was known about this group of patients. They are rare and the
elective treatment is centralized to surgeons who have also had training in pediatric surgery.
The emergency cases are not centralized and knowledge on how many patients who are
diagnosed in these settings is unknown. If the diagnosis is not detected, the outcome may be
fatal, emphasizing the importance to ensure that general surgeons are aware about the
condition. Minimally invasive surgical methods are becoming more established in the
clinical practice and further evaluation on their efficiency is needed.
Among pediatric patients we found that extremely preterm patients have a dismal outcome
and the only way to avoid complications is to have a low threshold for suspicion and trying
to avoid a severely affected circulation, which would increase the risk further. Knowledge,
clinical skills and experience seem to be the most important factors in diagnosing and
treating IM, so it is important to keep discussing and providing information about the
malformation. When investigating abdominal complaints, it is especially important to have
the diagnosis in mind when encountering a patient with other malformations,
developmental disorders or who might for other reasons lack the ability to express clinical
symptoms. A workup can relieve years of future discomfort or even be lifesaving. Further if
the diagnosis is confirmed genetic investigation is suggested.
Recently a better knowledge on the embryological development has contributed to a better
understanding of the pathomechanism and new techniques for molecular studies such as
WGS provide favorable conditions to further investigate the molecular cause of IM. Larger
studies are needed to confirm our recent findings, in addition, using WGS in trios we could
more easily look for autosomal recessive disorders as well as de novo mutations.
I look forward to use my obtained knowledge from this project in my clinical work, to
recognize the pediatric patients who would benefit from genetic counseling, and to
contribute to the important collaboration between the clinical work setting and the research
lab. Here, the exciting story about intestinal malrotation has come to an end, but it is only a
chapter of this endless adventure.

60
9 ACKNOWLEDGEMENTS
First of all I would like to thank the patients and their families for participating in the
studies.
I would like to express my sincere gratitude to all researchers, clinicians, co-authors and
friends who helped and supported me during these years. We made it through the
pandemic. Many people have made this possible and especially I would like to thank
Agneta Nordenskjöld, my main supervisor for seeing potential in me a long time ago and
helping me grow in the field of research but also on a personal level with kindness,
enthusiasm and experience as leading lights. Words cannot express my gratitude. You are a
fantastic person as well as researcher and clinician. You always found a solution where
there was none and believed in me when I did not. Thank you for taking me in as your
PhD-student.
Anna Lindstrand my co-supervisor and a visionary with a contagious enthusiasm,
problem solving and point of view angles none else noticed. Thank you for being patient
with me when I was struggling with how to handle research, clinical work and life and
found ways to make it possible.
Britt Husberg my co-supervisor for your insightful creative conversations, always bringing
me new knowledge, and good spirit. You are always supportive and make time for me.
Tomas Wester my co-supervisor for always being so wise and always having a great
refence up your sleeve. You can see what I don’t see and help me see it too, and you have
always been so supportive in research as well as in the clinic.
I also would like to thank all my co-authors and my unofficial co-supervisors
Ulla Ullberg, for always being there when I need to discuss radiology or life and bringing
the laughter. As soon I am done with my thesis I will read a book about breathtaking huge
fungus thanks to you. Maria Pettersson for always being so kind and supportive and
always takes time for me when I need to discuss genetics, and explains it in the best way.
You patiently led me through your areas of expertise and taught me a lot.

61
Margareta Michanek for our road trips when meeting families and collecting blood and
for all the personal talks.
Bo-Michael Bellander, my mentor who I knew was there as a support if needed.
The wonderful malformations genetics group and tissue engineering group that has
changed a lot over the years, for example when we moved from CMM to BioClinicum. Jia
Cao, who have been such a good friend and so patient with me during lab work, taught me
to make my own mistakes, and then helped me to get on the right path again. Christina
Nyström, who taught me how to extract DNA from blood and had such nice conversations
at our lunches. Wonderful Clara Chamorro always putting a smile on my face, and
fantastic adventurous Magdalena Fossum always with new exciting stories. Also Anna
Skarin Nordenvall, Samara Arkani, Lottie Phillips and Nikolai Juul for contributing
with good spirits.
Everyone in Rare diseases and Clincal Genetics Research group for your open
welcoming arms while I ran back and forth between clinical work and the lab. Nina Jäntti
with your great humor, for all the lunch talks and who always has a solution. I would also
like to thank you for the times you held my hand in the lab. Johanna Lundin and Kristina
Lagerstedt who saved me when I struggled with genetics. Fulya Taylan for your great
knowledge and for sharing it. Ann Nordgren with great enthusiasm, knowledge and
positive spirit, as well as Gierdre Grigelioniene and Isabel Tapia Paez. All fellow PhD-
students, especially Cecilia Arthur, Dominyka Batkovskyte, Jakob Schuy and Sofia
Frisk, you are positive and kind and share the struggle. Jesper Eisfeldt, you bring
happiness and laughter and make the most difficult things sound easy. Raquel Vas, you
have the best zebra fish stories and are always being kind and supportive.
All my friends and colleagues at the department of Pediatrics ALB, including the
subspecialities. A special thank you to my boss Jakob Frie, for making it easy to ask for
help when I was struggling, and schemaläggare Agnar Andrésson who made it possible
for me to get here. Anna Undeman Asarnoj for looking after ST and Charlotte Löttiger
Jansten for making everything so smooth and always saving me. Pediatric resident
colleagues, you have great spirits and are inspiring as well as hard working, empathetic and
always fun to work with. My clinical supervisor Elena Palleri who is so supportive, always
making time and want me to move forward. Marco Bartocci who hired me as a junior

62
doctor and all Doctors and Nurses at Neo Danderyd who let me into the world of
neonatology, made everyone feel like family and who always had research and learning in
mind. Anna Ekesbo Freisinger and Wouk Stannervik for allowing me into the warm
arms of pediatrics.
All friends and former colleagues at the department of pediatric surgery ALB. You all
contributed to my great memories at the pediatric surgery department. Especially I would
like to thank Björn Frenckner and Nader Ghaffarpour for introducing me to pediatric
surgery and for all the talks. Carmen Mesas Burgos for letting me be your tail when I was
a student. Henrik Ehrén for sharing your knowledge and giving me confidence during
surgical procedures, Pär-Johan Svensson for your wisdom. Katarina Bitkover for being a
great friend and Jakob Stenman for your expertise and great ring tone “I just can’t get
enough” that always made my day.
All friends and former colleagues at the department of pediatric surgery at
Akademiska Sjukhuset in Uppsala. Gisela Reinfeldt Engberg, Anders Stenbäck,
Elisabet Gustafson, Johan Danielson and Rolf Christofferson and the whole fantastic
gang, I loved to experience pediatric surgery with you.
KI:s Forskarskolan i molekylärmedicin, providing me with such invaluable insights and
also great new friends in the field.
Magnus Nordenskjöld for your support, insightful comments, and spot on jokes. And for
having the patience to hear Agnetas and I´s conversations on face time in the background
all day long during the pandemic.
Rune Hedlund for believing in that I could become an orthopedic back surgeon many
years ago and introducing me to research.
My friends, who stay by my side no matter what and especially Camilla, Therese, Beda
and Caroline for always making me laugh in the darkest of times. Parto, you are the
bravest person I know, and Mojgan soon we will go and buy plants together again.
Shervin, Ellen, Despina, Eleftheria and Bushra you were my dream team during the
university years and Ewonne, you had my back since I was 2 years old. Christine, I am so
glad we found each other again.

63
My parents, Annette and Behzad for always helping out with the children, bringing large
amounts of homemade meatballs, giving supportive talks and joining me for long evening
walks. Also my sister Irene who shares my weird humor and who always believes in me
when I don’t.
Finally I want to thank the best of the best, David for sticking with me even though I have
been married to work, teaching me the best phrase ever “det löser sig”, and my children for
reminding me of all the other wonderful things in life and bringing me some perspective.
In memory of Tina Granholm, always enthusiastic and knowledgeable about research.
You awoke my interest in neonatology and you were always supportive.
Researchers, clinicians, co-authors, family and friends who have contributed to the projects
in this thesis, without you I would not be here, and for all your hard work I am forever
grateful.


65
10 REFERENCES Aboagye J, Goldstein SD, Salazar JH, Papandria D, Okoye MT, Al-Omar K, Stewart D, Lukish J, Abdullah F. Age at presentation of common pediatric surgical conditions: Reexamining dogma. J Pediatr Surg. 2014 Jun;49(6):995-9. Adams SD, Stanton MP. Malrotation and intestinal atresias. Early Hum Dev. 2014 Dec;90(12):921-5.
Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125-48. Bamford RN, Roessler E, Burdine RD, Saplakoğlu U, dela Cruz J, Splitt M, Goodship JA, Towbin J, Bowers P, Ferrero GB, Marino B, Schier AF, Shen MM, Muenke M, Casey B. Loss-of-function mutations in the EGF-CFC gene CFC1 are associated with human left-right laterality defects. Nat Genet. 2000 Nov;26(3):365-9. Beaudoin S, Mathiot-Gavarin A, Gouizi G, Bargy F. Familial malrotation: report of three affected siblings. Pediatr Surg Int. 2005 Oct;21(10):856-7. Bilodeau A, Prasil P, Cloutier R, Laframboise R, Meguerditchian AN, Roy G, Leclerc S, Péloquin J. Hereditary multiple intestinal atresia: thirty years later. J Pediatr Surg. 2004 May;39(5):726-30. Binu V, Nicholson C, Cundy T, Gent R, Piotto L, Taranath A, Goh DW. Ultrasound imaging as the first line of investigation to diagnose intestinal malrotation in children: Safety and efficacy. J Pediatr Surg. 2021 Dec;56(12):2224-2228. Black AJ, Lu DY, Yefet LS, Baird R. Sex differences in surgically correctable congenital anomalies: A systematic review. J Pediatr Surg. 2020 May;55(5):811-820. Bonasoni P, Tonni G, Comitini G, Barbieri V, Rinaldini M, Marinelli M. Mosaic Trisomy 12: Prenatal Diagnosis at Amniocentesis and Molecular Genetic Analysis on Fetal Tissues. Fetal Pediatr Pathol. 2022 Apr;41(2):299-305. Brinkley MF, Tracy ET, Maxfield CM. Congenital duodenal obstruction: causes and imaging approach. Pediatr Radiol. 2016 jul;46(8):1084-95. Brungardt JG, Liebscher SC, Schropp KP. Malrotation Correction in the Adult Population. World J Surg. 2021 Jan;45(1):141-147. Budd JS, Powley PH. Small bowel volvulus in two siblings. Br Med J (Clin Res Ed). 1988 Jun 4;296(6636):1572. Butterworth WA, Butterworth JW. An adult presentation of midgut volvulus secondary to intestinal malrotation: A case report and literature review. Int J Surg Case Rep. 2018;50:46-49. Catania VD, Lauriti G, Pierro A, Zani A. Open versus laparoscopic approach for intestinal malrotation in infants and children: a systematic review and meta-analysis. Pediatr Surg Int. 2016 Dec;32(12):1157-1164. Chatmethakul T, Phaltas R, Minzes G, Martinez J, Bhat R. A Rare Co-occurrence of Intestinal Malrotation and Hirschsprung's Disease in a Neonate with 13q21.31q33.1 Interstitial Deletion Including the EDNRB Gene. J Pediatr Genet. 2019 Sep;8(3):142-146. Chen S, Chen D, Zhong W, Jiang H, Li Y, Yang B, Chen H, Shan Q, Zhong Z, Zhou W, Wang H, Ma X, Liu J, Sun K, Xie X, Zhou L. Saline-Aided Ultrasound Versus Upper Gastrointestinal Series in Neonates and Infants With Suspected Upper Gastrointestinal Obstruction: A Prospective Multicenter Comparative Study. AJR Am J Roentgenol. 2022 Mar;218(3):526-533. Clavien PA, Barkun J, de Oliveira ML, Vauthey JN, Dindo D, Schulick RD, de Santibañes E, Pekolj J, Slankamenac K, Bassi C, Graf R, Vonlanthen R, Padbury R, Cameron JL, Makuuchi M. The Clavien-Dindo classification of surgical complications: five-year experience. Ann Surg. 2009 Aug;250(2):187-96.

66
Clayton-Smith J, Walters S, Hobson E, Burkitt-Wright E, Smith R, Toutain A, Amiel J, Lyonnet S, Mansour S, Fitzpatrick D, Ciccone R, Ricca I, Zuffardi O, Donnai D. Xq28 duplication presenting with intestinal and bladder dysfunction and a distinctive facial appearance. Eur J Hum Genet. 2009 Apr;17(4):434-43. Cody JD, Ghidoni PD, DuPont BR, Hale DE, Hilsenbeck SG, Stratton RF, Hoffman DS, Muller S, Schaub RL, Leach RJ, Kaye CI. Congenital anomalies and anthropometry of 42 individuals with deletions of chromosome 18q. Am J Med Genet. 1999 Aug 27;85(5):455-62. Coe TM, Chang DC, Sicklick JK. Small bowel volvulus in the adult populace of the United States: results from a population-based study. Am J Surg. 2015 Aug;210(2):201-210.e2. Coombs RC, Buick RG, Gornall PG, Corkery JJ, Booth IW. Intestinal malrotation: the role of small intestinal dysmotility in the cause of persistent symptoms. J Pediatr Surg. 1991 May;26(5):553-6. Dasgupta A, Amack JD. Cilia in vertebrate left-right patterning. Philos Trans R Soc Lond B Biol Sci. 2016 Dec 19;371(1710):20150410. Davis NM, Kurpios NA, Sun X, Gros J, Martin JF, Tabin CJ. The chirality of gut rotation derives from left-right asymmetric changes in the architecture of the dorsal mesentery. Dev Cell. 2008 Jul;15(1):134-45. Dekonenko C, Sujka JA, Weaver K, Sharp SW, Gonzalez K, St Peter SD. The identification and treatment of intestinal malrotation in older children. Pediatr Surg Int. 2019 Jun;35(6):665-671. Devane SP, Coombes R, Smith VV, Bisset WM, Booth IW, Lake BD, Milla PJ. Persistent gastrointestinal symptoms after correction of malrotation. Arch Dis Child. 1992 Feb;67(2):218-21. Dharmadhikari AV, Gambin T, Szafranski P, Cao W, Probst FJ, Jin W, Fang P, Gogolewski K, Gambin A, George-Abraham JK, Golla S, Boidein F, Duban-Bedu B, Delobel B, Andrieux J, Becker K, Holinski-Feder E, Cheung SW, Stankiewicz P. Molecular and clinical analyses of 16q24.1 duplications involving FOXF1 identify an evolutionarily unstable large minisatellite. BMC Med Genet. 2014 Dec 4;15:128. Dilley AV, Pereira J, Shi EC, Adams S, Kern IB, Currie B, Henry GM. The radiologist says malrotation: does the surgeon operate? Pediatr Surg Int. 2000;16(1-2):45-9. Dott NM. Anomalies of intestinal rotation: their embryology and surgical aspects: with report of five cases. Br J Surg. 1923; 24:251-286. Duggan CP, Jaksic T. Pediatric Intestinal Failure. N Engl J Med. 2017 Aug 17;377(7):666-675. Dworschak GC, Draaken M, Hilger AC, Schramm C, Bartels E, Schmiedeke E, Grasshoff-Derr S, Märzheuser S, Holland-Cunz S, Lacher M, Jenetzky E, Zwink N, Schmidt D, Nöthen MM, Ludwig M, Reutter H. Genome-wide mapping of copy number variations in patients with both anorectal malformations and central nervous system abnormalities. Birth Defects Res A Clin Mol Teratol. 2015 Apr;103(4):235-42. El-Gohary Y, Alagtal M, Gillick J. Long-term complications following operative intervention for intestinal malrotation: a 10-year review. Pediatr Surg Int. 2010 Feb;26(2):203-6. Erez I, Reish O, Kovalivker M, Lazar L, Raz A, Katz S. Congenital short-bowel and malrotation: clinical presentation and outcome of six affected offspring in three related families. Eur J Pediatr Surg. 2001 Oct;11(5):331-4. Feitz R, Vos A. Malrotation: the postoperative period. J Pediatr Surg. 1997 Sep;32(9):1322-4. Ferreira MS, Simões J, Folgado A, Carlos S, Carvalho N, Santos F, Costa PM. Recurrent midgut volvulus in an adult patie–t - The case for pexy? A case report and review of the literature. Int J Surg Case Rep. 2020;66:91-95. Fischer JE, Jones DB, Pomposelli FB, Upchurch G, Fischer JE, Jones D, et al. Fischer’s Mastery of Surgery.
6th ed. Philadelphia: Wolters Kluwer Health; 2011.

67
Fisher AM, Barber JC, Crolla JA, James RS, Lestas AN, Jennings I, Dennis NR. Mosaic tetrasomy 8p: molecular cytogenetic confirmation and measurement of glutathione reductase and tissue plasminogen activator levels. Am J Med Genet. 1993 Aug 1;47(1):100-5. Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007 Nov;8(11):880-93. Ford EG, Senac MO Jr, Srikanth MS, Weitzman JJ. Malrotation of the intestine in children. Ann Surg. 1992 Feb;215(2):172-8. Frazer JE, Robbins RH. On the Factors concerned in causing Rotation of the Intestine in Man. J Anat Physiol. 1915 Oct;50(Pt 1):75-110. Freeman AH, Sala E. Radiology of the Stomach and Duodenum. 1st ed. 2008. Berlin, Heidelberg: Springer Berlin Heidelberg; 2008. Fredriksson F, Christofferson RH, Lilja HE. Adhesive small bowel obstruction after laparotomy during infancy. Br J Surg. 2016 Feb;103(3):284-9. Fryns JP, Kleczkowska A, Moerman F, van den Berghe K, van den Berghe H. Partial distal 6p trisomy in a malformed fetus. Ann Genet. 1986;29(1):53-4. Gargiulo A, Auricchio R, Barone MV, Cotugno G, Reardon W, Milla PJ, Ballabio A, Ciccodicola A, Auricchio A. Filamin A is mutated in X-linked chronic idiopathic intestinal pseudo-obstruction with central nervous system involvement. Am J Hum Genet. 2007 Apr;80(4):751-8. Genesio R, Maruotti GM, Saccone G, Mormile A, Conti A, Cicatiello R, Sarnataro V, Sirico A, Izzo A, Martinelli P, Nitsch L. Prenatally diagnosed distal 16p11.2 microdeletion with a novel association with congenital diaphragmatic hernia: a case report. Clin Case Rep. 2018 Feb 9;6(4):592-595. Gibson MF. Familial multiple jejunal atresia with malrotation. J Pediatr Surg. 1987 Nov;22(11):1013-4. Ginzel M, Martynov I, Haak R, Lacher M, Kluth D. Midgut development in rat embryos using microcomputed tomography. Commun Biol. 2021 Feb 12;4(1):190. Graziano K, Islam S, Dasgupta R, Lopez ME, Austin M, Chen LE, Goldin A, Downard CD, Renaud E, Abdullah F: Asymptomatic malrotation: Diagnosis and surgical management: An American Pediatric Surgical Association outcomes and evidence based practice committee systematic review. J Pediatr Surg 2015, 50(10):1783-1790. Guanà R, Garofano S, Teruzzi E, Vinardi S, Carbonaro G, Cerrina A, Morra I, Montin D, Mussa A, Schleef J. The complex surgical management of the first case of severe combined immunodeficiency and multiple intestinal atresias surviving after the fourth year of life. Pediatr Gastroenterol Hepatol Nutr. 2014 Dec;17(4):257-62. Hall T, Bush A, Fell J, Offiah A, Smith V, Abel R. Ciliopathy spectrum expanded? Jeune syndrome associated with foregut dysmotility and malrotation. Pediatr Pulmonol. 2009 Feb;44(2):198-201. Hall T, Samuel M, Brain J. Mosaic trisomy 22 associated with total colonic aganglionosis and malrotation. J Pediatr Surg. 2009 Jan;44(1):e9-e11. Harewood L, Liu M, Keeling J, Howatson A, Whiteford M, Branney P, Evans M, Fantes J, Fitzpatrick DR. Bilateral renal agenesis/hypoplasia/dysplasia (BRAHD): postmortem analysis of 45 cases with breakpoint mapping of two de novo translocations. PLoS One. 2010 Aug 25;5(8):e12375. Healy JM, Olgun LF, Hittelman AB, Ozgediz D, Caty MG. Pediatric incidental appendectomy: a systematic review. Pediatr Surg Int. 2016 Apr;32(4):321-35. Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011 Apr 21;364(16):1533-43.

68
Huanna T, Tao Z, Xiangfei W, Longfei A, Yuanyuan X, Jianhua W, Cuifang Z, Manjing J, Wenjing C, Shaochuan Q, Feifei X, Naikang L, Jinchao Z, Chen W. GALNT14 mediates tumor invasion and migration in breast cancer cell MCF-7. Mol Carcinog. 2015 Oct;54(10):1159-71. Iwuagwu O, Deans GT. Small bowel volvulus: a review. J R Coll Surg Edinb. 1999 Jun;44(3):150-5. Jardine S, Dhingani N, Muise AM. TTC7A: Steward of Intestinal Health. Cell Mol Gastroenterol Hepatol. 2019;7(3):555-570. Kantor JL. Anomalies of the colon: their roentgen diagnosis and clinical significance. Radiology. 1934;23:651-661. Kapfer SA, Rappold JF. Intestinal malrotation-not just the pediatric surgeon's problem. J Am Coll Surg. 2004 Oct;199(4):628-35. Kapoor S. Intestinal volvulus and its increased incidence in patients with Cornelia de Lange syndrome. J Crohns Colitis. 2014 Apr;8(4):331. Khalipina D, Kaga Y, Dacher N, Chevalier NR. Smooth muscle contractility causes the gut to grow anisotropically. J R Soc Interface. 2019 Oct 31;16(159):20190484. Kirov G, Rees E, Walters JT, Escott-Price V, Georgieva L, Richards AL, Chambert KD, Davies G, Legge SE, Moran JL, McCarroll SA, O'Donovan MC, Owen MJ. The penetrance of copy number variations for schizophrenia and developmental delay. Biol Psychiatry. 2014 Mar 1;75(5):378-85. Kluth D, Jaeschke-Melli S, Fiegel H. The embryology of gut rotation. Semin Pediatr Surg. 2003 Nov;12(4):275-9. Koffeman GI, van Gemert WG, George EK, Veenendaal RA. Classification, epidemiology and aetiology. Best Pract Res Clin Gastroenterol. 2003 Dec;17(6):879-93. Kulaylat AN, Hollenbeak CS, Engbrecht BW, Dillon PW, Safford SD. The impact of children's hospital designation on outcomes in children with malrotation. J Pediatr Surg. 2015 Mar;50(3):417-22. Ladd WE. Congenital obstruction of the duodenum in children. New Engl J Med. 1932;206(6):277-83. Ladd WE. Surgical diseases of the alimentary tract in infants. New Engl J Med. 1936;215(16):705-8. Lamont MA, Fitchett M, Dennis NR. Interstitial deletion of distal 13q associated with Hirschsprung's disease. J Med Genet. 1989 Feb;26(2):100-4. Lampl B, Levin TL, Berdon WE, Cowles RA. Malrotation and midgut volvulus: a historical review and current controversies in diagnosis and management. Pediatr Radiol. 2009 Apr;39(4):359-66. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann Y, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A,

69
Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blöcker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowki J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ, Szustakowki J; International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001 Feb 15;409(6822):860-921. Langer JC. Intestinal Rotation Abnormalities and Midgut Volvulus. Surg Clin North Am. 2017 Feb;97(1):147-159. Lemoine R, Pachlopnik-Schmid J, Farin HF, Bigorgne A, Debré M, Sepulveda F, Héritier S, Lemale J, Talbotec C, Rieux-Laucat F, Ruemmele F, Morali A, Cathebras P, Nitschke P, Bole-Feysot C, Blanche S, Brousse N, Picard C, Clevers H, Fischer A, de Saint Basile G. Immune deficiency-related enteropathy-lymphocytopenia-alopecia syndrome results from tetratricopeptide repeat domain 7A deficiency. J Allergy Clin Immunol. 2014 Dec;134(6):1354-1364.e6.
Leow JJ, Huey T, Low JK. Primary adult midgut volvulus mimicking acute appendicitis: A case report and review of the literature. Int J Surg Case Rep. 2016;24:182-4.
Lindstrand A, Eisfeldt J, Pettersson M, Carvalho CMB, Kvarnung M, Grigelioniene G, Anderlid BM, Bjerin O, Gustavsson P, Hammarsjö A, Georgii-Hemming P, Iwarsson E, Johansson-Soller M, Lagerstedt-Robinson K, Lieden A, Magnusson M, Martin M, Malmgren H, Nordenskjöld M, Norling A, Sahlin E, Stranneheim H, Tham E, Wincent J, Ygberg S, Wedell A, Wirta V, Nordgren A, Lundin J, Nilsson D. From cytogenetics to cytogenomics: whole-genome sequencing as a first-line test comprehensively captures the diverse spectrum of disease-causing genetic variation underlying intellectual disability. Genome Med. 2019 Nov 7;11(1):68. Lindstrand A, Frangakis S, Carvalho CM, Richardson EB, McFadden KA, Willer JR, Pehlivan D, Liu P, Pediaditakis IL, Sabo A, Lewis RA, Banin E, Lupski JR, Davis EE, Katsanis N. Copy-Number Variation Contributes to the Mutational Load of Bardet-Biedl Syndrome. Am J Hum Genet. 2016 Aug 4;99(2):318-36. Little DC Smith SD chapter 32 Malrotation. In: Holcomb GW, Murphy JP, Ostlie DJ (ed) Ashcraft's Pediatric Surgery, 5th edn. W.B. Saunders; 2010, Pages 416-424. Maclean K, Dunwoodie SL. Breaking symmetry: a clinical overview of left-right patterning. Clin Genet. 2004 Jun;65(6):441-57. Mahlapuu M, Ormestad M, Enerbäck S, Carlsson P. The forkhead transcription factor Foxf1 is required for differentiation of extra-embryonic and lateral plate mesoderm. Development. 2001 Jan;128(2):155-66. Mahlapuu M, Enerbäck S, Carlsson P. Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development. 2001 Jun;128(12):2397-406. Mall FP: Development of the human intestine and its position in the adult. Bulletin of the Johns Hopkins Hospital. 1898;9(90-91):197-208. Mandiá N, Pérez-Muñuzuri A, López-Suárez O, López-Sanguos C, Bautista-Casanovas A, Couce ML. Congenital intestinal atresias with multiple episodes of sepsis: A case report and review of literature. Medicine (Baltimore). 2018 Jun;97(23):e10939. Martin V, Shaw-Smith C. Review of genetic factors in intestinal malrotation. Pediatr Surg Int. 2010 Aug;26(8):769-81.

70
McLaughlin BM, Hufnagel RB, Saal HM. Small bowel malrotation in distal 15q duplication: evidence for a rare association. Clin Dysmorphol. 2015 Apr;24(2):65-7. McVay MR, Kokoska ER, Jackson RJ, Smith SD. Jack Barney Award. The changing spectrum of intestinal malrotation: diagnosis and management. Am J Surg. 2007 Dec;194(6):712-7; discussion 718-9. Mehl SC, Portuondo JI, Pettit RW, Fallon SC, Wesson DE, Shah SR, Vogel AM, Lopez ME, Massarweh NN. Association of prematurity with complications and failure to rescue in neonatal surgery. J Pediatr Surg. 2021 Nov 6:S0022-3468(21)00766-1. Metzger R, Metzger U, Fiegel HC, Kluth D. Embryology of the midgut. Semin Pediatr Surg. 2011 Aug;20(3):145-51. Minami K, Boshi H, Minami T, Tamura A, Yanagawa T, Uemura S, Takifuji K, Kurosawa K, Tsukino R, Izumi G, Yoshikawa N. 1p36 deletion syndrome with intestinal malrotation and annular pancreas. Eur J Pediatr. 2005 Mar;164(3):193-4. Mirza B, Sheikh A. Multiple associated anomalies in patients of duodenal atresia: a case series. J Neonatal Surg. 2012 Apr 1;1(2):23. Mishra PR, Stringer MD. Intestinal malrotation in extremely premature infants: a potential trap. Pediatr Surg Int. 2021 Nov;37(11):1607-1612. Moldrem AW, Papaconstantinou H, Broker H, Megison S, Jeyarajah DR. Late presentation of intestinal malrotation: an argument for elective repair. World J Surg. 2008 Jul;32(7):1426-31. Morgenstern DA, Soh SY, Stavropoulos DJ, Bowdin S, Baruchel S, Malkin D, Meyn MS, Irwin MS. Metachronous neuroblastoma in an infant with germline translocation resulting in partial trisomy 2p: a role for ALK? J Pediatr Hematol Oncol. 2014 Apr;36(3):e193-6 Morris CA, Mervis CB, Paciorkowski AP, Abdul-Rahman O, Dugan SL, Rope AF, Bader P, Hendon LG, Velleman SL, Klein-Tasman BP, Osborne LR. 7q11.23 Duplication syndrome: Physical characteristics and natural history. Am J Med Genet A. 2015 Dec;167A(12):2916-35. Murphy FL, Sparnon AL. Long-term complications following intestinal malrotation and the Ladd's procedure: a 15 year review. Pediatr Surg Int. 2006 Apr;22(4):326-9. Negri E, Coletta R, Morabito A. Congenital short bowel syndrome: systematic review of a rare condition. J Pediatr Surg. 2020 Sep;55(9):1809-1814. Nagdeve NG, Qureshi AM, Bhingare PD, Shinde SK. Malrotation beyond infancy. J Pediatr Surg. 2012 Nov;47(11):2026-32. Najafzadeh TM, Littman VA, Dumars KW. Familial t(4;13) with abnormal offspring in three generations. Am J Med Genet. 1983 Sep;16(1):15-22 Nehra D, Goldstein AM. Intestinal malrotation: varied clinical presentation from infancy through adulthood. Surgery. 2011 Mar;149(3):386-93. Nordenström A, Svensson J, Lajic S, Frisén L, Nordenskjöld A, Norrby C, Almqvist C, Falhammar H. Carriers of a Classic CYP21A2 Mutation Have Reduced Mortality: A Population-Based National Cohort Study. J Clin Endocrinol Metab. 2019 Dec 1;104(12):6148-6154. Okumura A, Yamamoto T, Miyajima M, Shimojima K, Kondo S, Abe S, Ikeno M, Shimizu T. 3p interstitial deletion including PRICKLE2 in identical twins with autistic features. Pediatr Neurol. 2014 Nov;51(5):730-3. Oliveira JT, Marques P, Preza Fernandes JM, Teixeira T, Santos MD, Povo A, Castro Alves E. Cecum perforation in intestinal malrotation setting in a patient with chromosome 12p deletion syndrome: A case report. Int J Surg Case Rep. 2020;66:342-345. Orzech N, Navarro OM, Langer JC. Is ultrasonography a good screening test for intestinal malrotation? J Pediatr Surg. 2006 May;41(5):1005-9.

71
Otto J, Back E, Fürste HO, Abel M, Böhm N, Pringsheim W. Dysplastic features, growth retardation, malrotation of the gut, and fatal ventricular septal defect in a 4-month-old girl with ring chromosome 15. Eur J Pediatr. 1984 Aug;142(3):229-31. Pettersson M, Viljakainen H, Loid P, Mustila T, Pekkinen M, Armenio M, Andersson-Assarsson JC, Mäkitie O, Lindstrand A. Copy Number Variants Are Enriched in Individuals With Early-Onset Obesity and Highlight Novel Pathogenic Pathways. J Clin Endocrinol Metab. 2017 Aug 1;102(8):3029-3039. Plageman TF Jr, Zacharias AL, Gage PJ, Lang RA. Shroom3 and a Pitx2-N-cadherin pathway function cooperatively to generate asymmetric cell shape changes during gut morphogenesis. Dev Biol. 2011 Sep 1;357(1):227-34. Pollara L, Sottile V, Valente EM. Patient-derived cellular models of primary ciliopathies. J Med Genet. 2022 Feb 19:jmedgenet-2021-108315. Ramasubramanian A, Chu-Lagraff QB, Buma T, Chico KT, Carnes ME, Burnett KR, Bradner SA, Gordon SS. On the role of intrinsic and extrinsic forces in early cardiac S-looping. Dev Dyn. 2013 Jul;242(7):801-16. Rasmussen SA, Moore CA, Paulozzi LJ, Rhodenhiser EP. Risk for birth defects among premature infants: a population-based study. J Pediatr. 2001 May;138(5):668-73. Reed RA, Womble MA, Dush MK, Tull RR, Bloom SK, Morckel AR, Devlin EW, Nascone-Yoder NM. Morphogenesis of the primitive gut tube is generated by Rho/ROCK/myosin II-mediated endoderm rearrangements. Dev Dyn. 2009 Dec;238(12):3111-25. Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. 2017 Sep;18(9):533-547. Sadler TW, Langman J, Ecker PM. Langman’s medical embryology. 9. ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2004. Sahoo T, Theisen A, Rosenfeld JA, Lamb AN, Ravnan JB, Schultz RA, Torchia BS, Neill N, Casci I, Bejjani BA, Shaffer LG. Copy number variants of schizophrenia susceptibility loci are associated with a spectrum of speech and developmental delays and behavior problems. Genet Med. 2011 Oct;13(10):868-80. Salter CG, Cai Y, Lo B, Helman G, Taylor H, McCartney A, Leslie JS, Accogli A, Zara F, Traverso M, Fasham J, Lees JA, Ferla MP, Chioza BA, Wenger O, Scott E, Cross HE, Crawford J, Warshawsky I, Keisling M, Agamanolis D, Ward Melver C, Cox H, Elawad M, Marton T, Wakeling MN, Holzinger D, Tippelt S, Munteanu M, Valcheva D, Deal C, Van Meerbeke S, Walsh Vockley C, Butte MJ, Acar U, van der Knaap MS, Korenke GC, Kotzaeridou U, Balla T, Simons C, Uhlig HH, Crosby AH, De Camilli P, Wolf NI, Baple EL. Biallelic PI4KA variants cause neurological, intestinal and immunological disease. Brain. 2021 Dec 31;144(12):3597-3610. Salö M. Is There a Need for Bowel Management after Surgery for Isolated Intestinal Malrotation in Children? Pediatr Gastroenterol Hepatol Nutr. 2019 Sep;22(5):447-452. Savin T, Kurpios NA, Shyer AE, Florescu P, Liang H, Mahadevan L, Tabin CJ. On the growth and form of the gut. Nature. 2011 Aug 3;476(7358):57-62. Shanske A, Ferreira JC, Leonard JC, Fuller P, Marion RW. Hirschsprung disease in an infant with a contiguous gene syndrome of chromosome 13. Am J Med Genet. 2001 Aug 15;102(3):231-6. Sharma N, Berbari NF, Yoder BK. Ciliary dysfunction in developmental abnormalities and diseases. Curr Top Dev Biol. 2008;85:371-427. Smith SL. Familial midgut volvulus. Surgery. 1972 Sep;72(3):420-6. Soffers JH, Hikspoors JP, Mekonen HK, Koehler SE, Lamers WH. The growth pattern of the human intestine and its mesentery. BMC Dev Biol. 2015 Aug 22;15:31.

72
Spigland N, Brandt ML, Yazbeck S. Malrotation presenting beyond the neonatal period. J Pediatr Surg. 1990 Nov;25(11):1139-42. Stankiewicz P, Sen P, Bhatt SS, Storer M, Xia Z, Bejjani BA, Ou Z, Wiszniewska J, Driscoll DJ, Maisenbacher MK, Bolivar J, Bauer M, Zackai EH, McDonald-McGinn D, Nowaczyk MM, Murray M, Hustead V, Mascotti K, Schultz R, Hallam L, McRae D, Nicholson AG, Newbury R, Durham-O'Donnell J, Knight G, Kini U, Shaikh TH, Martin V, Tyreman M, Simonic I, Willatt L, Paterson J, Mehta S, Rajan D, Fitzgerald T, Gribble S, Prigmore E, Patel A, Shaffer LG, Carter NP, Cheung SW, Langston C, Shaw-Smith C. Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet. 2009 Jun;84(6):780-91. Stranneheim H, Lagerstedt-Robinson K, Magnusson M, Kvarnung M, Nilsson D, Lesko N, Engvall M, Anderlid BM, Arnell H, Johansson CB, Barbaro M, Björck E, Bruhn H, Eisfeldt J, Freyer C, Grigelioniene G, Gustavsson P, Hammarsjö A, Hellström-Pigg M, Iwarsson E, Jemt A, Laaksonen M, Enoksson SL, Malmgren H, Naess K, Nordenskjöld M, Oscarson M, Pettersson M, Rasi C, Rosenbaum A, Sahlin E, Sardh E, Stödberg T, Tesi B, Tham E, Thonberg H, Töhönen V, von Döbeln U, Vassiliou D, Vonlanthen S, Wikström AC, Wincent J, Winqvist O, Wredenberg A, Ygberg S, Zetterström RH, Marits P, Soller MJ, Nordgren A, Wirta V, Lindstrand A, Wedell A. Integration of whole genome sequencing into a healthcare setting: high diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med. 2021 Mar 17;13(1):40. Stankiewicz P, Sen P, Bhatt SS, Storer M, Xia Z, Bejjani BA, Ou Z, Wiszniewska J, Driscoll DJ, Maisenbacher MK, Bolivar J, Bauer M, Zackai EH, McDonald-McGinn D, Nowaczyk MM, Murray M, Hustead V, Mascotti K, Schultz R, Hallam L, McRae D, Nicholson AG, Newbury R, Durham-O'Donnell J, Knight G, Kini U, Shaikh TH, Martin V, Tyreman M, Simonic I, Willatt L, Paterson J, Mehta S, Rajan D, Fitzgerald T, Gribble S, Prigmore E, Patel A, Shaffer LG, Carter NP, Cheung SW, Langston C, Shaw-Smith C. Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet. 2009 Jun;84(6):780-91. Stewart DR, Colodny AL, Daggett WC. Malrotation of the bowel in infants and children: a 15 year review. Surgery. 1976 Jun;79(6):716-20. Strathdee G, Zackai EH, Shapiro R, Kamholz J, Overhauser J. Analysis of clinical variation seen in patients with 18q terminal deletions. Am J Med Genet. 1995 Dec 4;59(4):476-83. Stratulat-Chiriac I, Mc Laughlin D, Antao B. Rare Association of Extended Total Colonic Aganglionosis and Intestinal Malrotation. J Neonatal Surg. 2016 Oct 10;5(4):61. Strouse PJ. Disorders of intestinal rotation and fixation ("malrotation"). Pediatr Radiol. 2004 Nov;34(11):837-51. Swenson O, Ladd WE. Surgical emergencies of the alimentary tract of the newborn. N Engl J Med. 1945 Nov 29;233:660-3. Tsai EA, Grochowski CM, Falsey AM, Rajagopalan R, Wendel D, Devoto M, Krantz ID, Loomes KM, Spinner NB. Heterozygous deletion of FOXA2 segregates with disease in a family with heterotaxy, panhypopituitarism, and biliary atresia. Hum Mutat. 2015 Jun;36(6):631-7. Van der Aa N, Rooms L, Vandeweyer G, van den Ende J, Reyniers E, Fichera M, Romano C, Delle Chiaie B, Mortier G, Menten B, Destrée A, Maystadt I, Männik K, Kurg A, Reimand T, McMullan D, Oley C, Brueton L, Bongers EM, van Bon BW, Pfund R, Jacquemont S, Ferrarini A, Martinet D, Schrander-Stumpel C, Stegmann AP, Frints SG, de Vries BB, Ceulemans B, Kooy RF. Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur J Med Genet. 2009 Mar-Jun;52(2-3):94-100. Vanderhoof JA, Langnas AN. Short-bowel syndrome in children and adults. Gastroenterology. 1997 Nov;113(5):1767-78. van der Zee DC, Bax NM. Laparoscopic repair of acute volvulus in a neonate with malrotation. Surg Endosc. 1995 Oct;9(10):1123-4.

73
Wales PW, de Silva N, Kim J, Lecce L, To T, Moore A. Neonatal short bowel syndrome: population-based estimates of incidence and mortality rates. J Pediatr Surg. 2004 May;39(5):690-5. Wales PW, Christison-Lagay ER. Short bowel syndrome: epidemiology and etiology. Semin Pediatr Surg. 2010 Feb;19(1):3-9. Wang R, Yu C, Zhao D, Wu M, Yang Z. The mucin-type glycosylating enzyme polypeptide N-acetylgalactosaminyltransferase 14 promotes the migration of ovarian cancer by modifying mucin 13. Oncol Rep. 2013 Aug;30(2):667-76. Wang Y, Chen S, Yan W, Lu L, Tao Y, Xiao Y, Cai W. Congenital Short-Bowel Syndrome: Clinical and Genetic Presentation in China. JPEN J Parenter Enteral Nutr. 2021 Jul;45(5):1009-1015.
Ware SM, Aygun MG, Hildebrandt F. Spectrum of clinical diseases caused by disorders of primary cilia. Proc Am Thorac Soc. 2011 Sep;8(5):444-50. Vaisvilas M, Dirse V, Aleksiuniene B, Tamuliene I, Cimbalistiene L, Utkus A, Rascon J. Acute Pre-B Lymphoblastic Leukemia and Congenital Anomalies in a Child with a de Novo22q11.1q11.22 Duplication. Balkan J Med Genet. 2018 Oct 29;21(1):87-91. Welsh IC, Thomsen M, Gludish DW, Alfonso-Parra C, Bai Y, Martin JF, Kurpios NA. Integration of left-right Pitx2 transcription and Wnt signaling drives asymmetric gut morphogenesis via Daam2. Dev Cell. 2013 Sep 30;26(6):629-44. Wheway G, Nazlamova L, Hancock JT. Signaling through the Primary Cilium. Front Cell Dev Biol. 2018 Feb 8;6:8. Wigley WC, Prihoda JS, Mowszowicz I, Mendonca BB, New MI, Wilson JD, Russell DW. Natural mutagenesis study of the human steroid 5 alpha-reductase 2 isozyme. Biochemistry. 1994 Feb 8;33(5):1265-70. Woutsas S, Aytekin C, Salzer E, Conde CD, Apaydin S, Pichler H, Memaran-Dadgar N, Hosnut FO, Förster-Waldl E, Matthes S, Huber WD, Lion T, Holter W, Bilic I, Boztug K. Hypomorphic mutation in TTC7A causes combined immunodeficiency with mild structural intestinal defects. Blood. 2015 Mar 5;125(10):1674-6. Yang L, Chen H, Lv G, Li F, Liao J, Ke L. Evaluation of ultrasonography in fetal intestinal malrotation with midgut volvulus. Ginekol Pol. 2022 Feb 14. Yang W, Lee PP, Thong MK, Ramanujam TM, Shanmugam A, Koh MT, Chan KW, Ying D, Wang Y, Shen JJ, Yang J, Lau YL. Compound heterozygous mutations in TTC7A cause familial multiple intestinal atresias and severe combined immunodeficiency. Clin Genet. 2015 Dec;88(6):542-9. Zissin R, Rathaus V, Oscadchy A, Kots E, Gayer G, Shapiro-Feinberg M. Intestinal malrotation as an incidental finding on CT in adults. Abdom Imaging. 1999 Nov-Dec;24(6):550-5.
Copyright © 2022 FDOKUMEN














![Molecular Imaging of Murine Intestinal Inflammation With 2-Deoxy-2-[ 18F]Fluoro- d-Glucose and Positron Emission Tomography](https://static.fdokumen.com/doc/165x107/6344fff26cfb3d4064097a1a/molecular-imaging-of-murine-intestinal-inflammation-with-2-deoxy-2-18ffluoro-.jpg)





