
Clinical, biochemical, and molecular findings in three patients with 3-hydroxyisobutyric aciduria
7
334 q 2001 Blackwell Science Ltd Clinical, biochemical and molecular findings in a series of families with hereditary hyperferritinaemia–cataract syndrome Domenico Girelli, 1 Claudia Bozzini, 1 Gabriella Zecchina, 1 Elisa Tinazzi, 1 Sandra Bosio, 2 Alberto Piperno, 3 Ugo Ramenghi, 4 Jutta Peters, 5 Sonia Levi, 6 Clara Camaschella 2 and Roberto Corrocher 1 1 Department of Clinical and Experimental Medicine, University of Verona, Verona, 2 Department of Clinical and Biological Sciences, University of Turin, Turin, 3 Institute of Biomedical Sciences, Monza, 4 Department of Paediatric Sciences, University of Turin, Turin, Italy, 5 Department of Gastroenterology and Infectious Disease, University of Berlin, Berlin, Germany, and 6 DIBIT S. Raffaele Hospital, Milan, Italy Received 5 April 2001; accepted for publication 27 June 2001 Summary. Hereditary hyperferritinaemia–cataract syn- drome (HHCS) is an autosomal dominant disease caused by mutations in the iron responsive element (IRE) of the l- ferritin gene. Despite the elucidation of the genetic basis, the overall clinical spectrum of HHCS has been less well studied as, to date, only individual case reports have been described. Therefore, we studied a total of 62 patients in 14 unrelated families, with nine different mutations. No relevant symptoms other than visual impairment were found to be associated with the syndrome. A marked phenotypic variability was observed, particularly with regard to ocular involvement (i.e. age range at which cataract was diagnosed in 16 subjects with the C39T: 6–40 years). Similarly, serum ferritin levels varied substantially also within subjects sharing the same mutation (i.e. range for the A40G: 700– 2412 mg/l). We followed an HHCS newborn in whom well- defined lens opacities were not detectable either at birth or at 1 year. The lens ferritin content was analysed in two subjects who underwent cataract surgery at different ages, with different cataract morphology. Values were similar and about 1500-fold higher than in controls. These observations suggest that: (i) in HHCS the cataract is not necessarily congenital; (ii) in addition to the IRE genotype, other genetic or environmental factors may modulate the phenotype, especially the severity of the cataract. Keywords: hyperferritinaemia–cataract syndrome, ferritin, cataract, iron responsive element, lens. Ferritins, which are ubiquitous iron storage proteins, are multimer shells made up of 24 heavy (H, Mr 21 000) or light (L, Mr 19 000) subunits, tightly regulated by iron, and assembled in variable proportions (Theil, 1983). In 1995, our group (Girelli et al, 1995a) and a French group (Bonneau et al, 1995) simultaneously described a new autosomal dominant disease, namely, hereditary hyperferri- tinaemia–cataract syndrome (HHCS). The hallmark of HHCS is the presence of persistently elevated levels of serum ferritin independent of the amount of iron stores. Under physiological conditions, ferritin synthesis is finely regulated at the translational level by iron availability. This mechanism is part of a co-ordinated control of the expression of other proteins involved in iron homeostasis, such as erythroid d-aminolevulinate synthase (d-ALA), and transferrin receptor (TfR) (Klausner et al, 1993). It is achieved by the high–affinity interaction between cytoplas- mic mRNA-binding proteins, known as iron regulatory proteins (IRPs), and a well-characterized family of cis-acting non-coding mRNA stem-loop structures, known as iron responsive elements (IREs) (Theil, 1994). The IREs are located in the untranslated regions (UTRs) of the mRNAs. A single IRE is located on the 5 0 UTR of ferritins and d-ALA mRNAs, where binding of IRPs represses translation. In contrast, multiple IREs are present in the 3 0 UTR of the mRNA for TfR, where binding of IRPs stabilizes the mRNA. Recently, a single IRE has been found also in the 3 0 UTR of the gene for divalent metal transporter 1 (DMT1), a protein involved in iron absorption (Fleming et al, 1999). Intracel- lular iron levels modulate the affinity of the IRPs for the IREs. When iron is low, the IRP/IRE high-affinity binding prevents iron utilization and storage by blocking the translation of ferritin and d-ALA, whereas iron uptake is promoted by increasing the synthesis of TfR and DMT1. Conversely, in conditions of iron repletion, the IRP1 acquires a [4Fe-4S] cluster that prevents the binding to IREs, and the British Journal of Haematology , 2001, 115, 334–340 Correspondence: Domenico Girelli, MD, PhD, Department of Clinical and Experimental Medicine, Policlinico G.B.Rossi, 37134 Verona, Italy. E-mail: [email protected]
Transcript of Clinical, biochemical, and molecular findings in three patients with 3-hydroxyisobutyric aciduria

334 q 2001 Blackwell Science Ltd
Clinical, biochemical and molecular findings in a series of
families with hereditary hyperferritinaemia±cataract syndrome
Domenico Girelli,1 Claudia Bozzini,1 Gabriella Zecchina,1 Elisa Tinazzi,1 Sandra Bosio,2
Alberto Piperno,3 Ugo Ramenghi,4 Jutta Peters,5 Sonia Levi,6 Clara Camaschella2 and
Roberto Corrocher1 1Department of Clinical and Experimental Medicine, University of Verona, Verona,2Department of Clinical and Biological Sciences, University of Turin, Turin, 3Institute of Biomedical Sciences, Monza,4Department of Paediatric Sciences, University of Turin, Turin, Italy, 5Department of Gastroenterology and Infectious
Disease, University of Berlin, Berlin, Germany, and 6DIBIT S. Raffaele Hospital, Milan, Italy
Received 5 April 2001; accepted for publication 27 June 2001
Summary. Hereditary hyperferritinaemia±cataract syn-drome (HHCS) is an autosomal dominant disease causedby mutations in the iron responsive element (IRE) of the l-ferritin gene. Despite the elucidation of the genetic basis, theoverall clinical spectrum of HHCS has been less well studiedas, to date, only individual case reports have been described.Therefore, we studied a total of 62 patients in 14 unrelatedfamilies, with nine different mutations. No relevantsymptoms other than visual impairment were found to beassociated with the syndrome. A marked phenotypicvariability was observed, particularly with regard to ocularinvolvement (i.e. age range at which cataract was diagnosedin 16 subjects with the C39T: 6±40 years). Similarly, serumferritin levels varied substantially also within subjects
sharing the same mutation (i.e. range for the A40G: 700±2412 mg/l). We followed an HHCS newborn in whom well-defined lens opacities were not detectable either at birth orat 1 year. The lens ferritin content was analysed in twosubjects who underwent cataract surgery at different ages,with different cataract morphology. Values were similar andabout 1500-fold higher than in controls. These observationssuggest that: (i) in HHCS the cataract is not necessarilycongenital; (ii) in addition to the IRE genotype, other geneticor environmental factors may modulate the phenotype,especially the severity of the cataract.
Keywords: hyperferritinaemia±cataract syndrome, ferritin,cataract, iron responsive element, lens.
Ferritins, which are ubiquitous iron storage proteins, aremultimer shells made up of 24 heavy (H, Mr 21 000) orlight (L, Mr 19 000) subunits, tightly regulated by iron, andassembled in variable proportions (Theil, 1983). In 1995,our group (Girelli et al, 1995a) and a French group(Bonneau et al, 1995) simultaneously described a newautosomal dominant disease, namely, hereditary hyperferri-tinaemia±cataract syndrome (HHCS). The hallmark ofHHCS is the presence of persistently elevated levels ofserum ferritin independent of the amount of iron stores.Under physiological conditions, ferritin synthesis is finelyregulated at the translational level by iron availability. Thismechanism is part of a co-ordinated control of theexpression of other proteins involved in iron homeostasis,such as erythroid d-aminolevulinate synthase (d-ALA), andtransferrin receptor (TfR) (Klausner et al, 1993). It is
achieved by the high±affinity interaction between cytoplas-mic mRNA-binding proteins, known as iron regulatoryproteins (IRPs), and a well-characterized family of cis-actingnon-coding mRNA stem-loop structures, known as ironresponsive elements (IREs) (Theil, 1994). The IREs arelocated in the untranslated regions (UTRs) of the mRNAs. Asingle IRE is located on the 5 0 UTR of ferritins and d-ALAmRNAs, where binding of IRPs represses translation. Incontrast, multiple IREs are present in the 3 0 UTR of themRNA for TfR, where binding of IRPs stabilizes the mRNA.Recently, a single IRE has been found also in the 3 0 UTR ofthe gene for divalent metal transporter 1 (DMT1), a proteininvolved in iron absorption (Fleming et al, 1999). Intracel-lular iron levels modulate the affinity of the IRPs for theIREs. When iron is low, the IRP/IRE high-affinity bindingprevents iron utilization and storage by blocking thetranslation of ferritin and d-ALA, whereas iron uptake ispromoted by increasing the synthesis of TfR and DMT1.Conversely, in conditions of iron repletion, the IRP1 acquiresa [4Fe-4S] cluster that prevents the binding to IREs, and the
British Journal of Haematology, 2001, 115, 334±340
Correspondence: Domenico Girelli, MD, PhD, Department of Clinicaland Experimental Medicine, Policlinico G.B.Rossi, 37134 Verona,
Italy. E-mail: [email protected]

IRP2 is degraded so that the synthesis of ferritin and d-ALAincreases, whereas that of TfR and DMT1 decreases.Molecular studies have demonstrated that HHCS is causedby mutations in the IRE of the l-ferritin gene onchromosome 19q13.3-qter, which disrupt the negativecontrol of l-ferritin synthesis at the translational levelthrough IRE±IRP interaction (Girelli et al, 1995b; Beau-mont et al, 1995). The constitutive up-regulation of ferritinl-chains results in both hyperferritinaemia and the accu-mulation of ferritin in the cells, mainly in the form of H0-L24
homopolymers (Levi et al, 1998). These homopolymers areunable to incorporate detectable amounts of iron becausethey lack the ferroxidase activity which is specific to the H-chain (Santambrogio et al, 1996; Corsi et al, 1998). Sincethe first reports in 1995, other families with HHCS have alsobeen described in the UK (Mumford et al, 1998), Germany(Merkt, 1997), Spain (Balas et al, 1999; Perez de Nanclareset al, 2001) and North America (Barton et al, 1998; Kato &Casella, 1999). Thus, it is suggested that HHCS may bedistributed widely throughout the world and not sporadic,whereas its prevalence remains to be established. In spite ofthe elucidation of the genetic basis, the overall spectrum ofthe clinical manifestations of HHCS appears to have beenstudied less thoroughly as, to date, the literature publishedconsists essentially of individual case reports. In particular, anumber of items remain to be established: (i) whether thecataract is congenital or not; (ii) apart from ocularinvolvement, the possible association of HHCS with othersymptoms or signs with lower penetrance; (iii) the extent bywhich the variability reported in the severity of visualsymptoms and/or ferritin levels could be explained in termsof genotype/phenotype correlation. Recently, an elegant invitro study (Allerson et al, 1999) based on the thermo-denaturation profile and dissociation constant of the IRE±IRP complex performed for several mutated IREs hasprovided evidence for a possible correlation betweenheterogeneous IRE mutations and serum ferritin levels. Onthe other hand, the in vivo relevance of these conclusionshas not been determined completely.
Here we present data regarding a total of 62 affectedsubjects belonging to 14 unrelated HHCS families. To thebest of our knowledge, this is the largest series of HHCSpatients reported to date.
PATIENTS AND METHODS
The probands were diagnosed in our centres (five in Verona,seven in Turin, two in Monza) over a 5-year period fromMay 1995 to December 2000, to which they were referredmainly because of `suspected haemochromatosis', caused bypersistently high ferritin levels. Serum ferritin and transfer-rin saturation were determined using standard methods. Athorough medical history was collected for each proband,with particular reference to factors known to affect ironstatus such as blood donations, conditions associated withblood loss, pregnancy, abnormalities of the menstrual cycle,etc. A review of the subject's clinical records was also madeat each of the referral centres. The ophthalmological
evaluations were performed by an ophthalmologist using aslit-lamp.
Detection of IRE mutations. Informed consent was obtainedfrom all subjects for genetic studies. DNA was extracted fromperipheral lymphocytes using phenol±chloroform protocol.The entire IRE sequence of the l-ferritin gene was amplifiedby polymerase chain reaction (PCR) using the flankingprimers (forward, 5 0-TCCTTGCCACCGCAGATTG-3 0; reverse,5 0-TTGGCAAGAAGGAGCTAACC-3 0) and then sequenced asdescribed previously (Girelli et al, 1995b).
Determination of the ferritin content in the lens. Recently,two of our patients underwent cataract surgery using thephacoemulsification technique with posterior chamberintraocular lens implantation, which is used widely inmodern cataract surgery (Ohrloff & Zubcov, 1997). Duringthe procedure, after opening the anterior capsule, the lens isfragmented by ultrasound and then aspirated in a closedirrigation±aspiration system. The recovered lens materialconsisted of a suspension in a buffered-saline solution.Soluble homogenates of the solid phases, which wereamorphous and unsuitable for histological examination,were obtained by incubation in 0´2% NP40 followed bycentrifugation. These samples were analysed for H and Lferritin content using enzyme-linked immunosorbent assay(ELISA) as previously described (Levi et al, 1998). Eachmeasurement was carried out in triplicate.
RESULTS
Tables I and II summarize the clinical, biochemical andmolecular findings in each of the probands and in thecorresponding relatives. Figure 1 represents the normalstem-loop IRE sequence located in the 5' UTR of the l-ferritin gene and the nine mutations found in our familyseries.
Overall, the review of the clinical records of 62 affectedsubjects failed to detect any clinically relevant symptomsassociated with the HHCS other than the visual impairment.The majority of the probands came to clinical observation inour haematology units because of findings of unexplainedhyperferritinaemia during routine blood examinations.Three of them (Table I) were referred to us because ofmild iron-deficient anaemia with `paradoxical' hyperferritin-aemia. They became anaemic during pregnancy or soonafter a few phlebotomies were performed because of amistaken diagnosis of `haemochromatosis' (proband 1). Thereview of the clinical records of the relatives also showed ahistory of iron-deficient anaemia in four of them (Table II),though in the presence of a condition known to beassociated with blood loss (blood donations, erosive gastritis,tendency to hypermenorrhoea). A full evaluation of ironstores was possible only in the proband from family 1, inwhom both bone marrow aspiration and liver biopsyrevealed no stainable iron. In the other subjects it was notcarried out as they refused the invasive procedures, and/orthe diagnosis of iron-deficient anaemia was retrospective,with normal haematological parameters at the time of ourobservation.
Recently, we had an opportunity to obtain new important
q 2001 Blackwell Science Ltd, British Journal of Haematology 115: 334±340
Clinical Findings in HHCS 335
information during the follow-up of the members of a familywith what is apparently one of the most severe genotypes,i.e. the 29 bp deletion (Girelli et al, 1997). The extendedpedigree with the newly observed subjects is reported inFig 2A. We were able to follow the ophthalmological patternof an affected newborn from this family. The newborn(patient IV-1 in Fig 2A) had serum ferritin levels persistentlyhigher than 1000 mg/l on three different occasions (1920±2860±1448 mg/l, at birth, 1 month, and 1 year of age). Onthe other hand, the ophthalmological examination did notreveal definite lens opacities, either at birth or at the 1 yearexamination. Moreover, two family members, aged 11 and31 years, respectively, had cataract surgery recently usingthe phacoemulsification technique. The ophthalmologicalpictures of these subjects are shown in Fig 2(B and C). Thelens material was recovered and analysed for ferritincontent. The values were similar in the 11 year-old boywith `pulverulent' morphology (sparse, dust-like deposits) ofthe cataract (1763 ^ 254 ng/mg of protein) and in his31 year-old aunt (1588 ^ 217 ng/mg) with a `sunflower'
morphology. These values were also similar to the ones wehad found previously in a 25 year-old female carrying theG41C mutation (Levi et al, 1998), which were about 1500-fold higher than those in lenses from age-matched controls(mean value in controls 161 ng/mg). Similarly, the contentof the H-chain in the two new samples was reducedcompared with controls, with an L:H ratio ranging from397 to 440 (range in lenses from controls 2´7±6),consistent with the accumulation of non-functional H0-L24
homopolymers in the HHCS lens (Levi et al, 1998).In most of the families (13 of 14) the mutation was a
single base substitution. Three different point mutationsinvolved the CAGUGX hexanucleotide loop: C39T, A40Gand G41C. The A40G and the C39T were recurrent, each ofthem being detected in four and three unrelated familiesrespectively. In one family, the C39T was demonstratedpreviously to be a de novo mutation (Arosio et al, 1999). Onemutation involved the highly conserved, unpaired cytosineat position 133 (C33T). Three different base substitutionsinvolved the guanine at position 132, immediately below
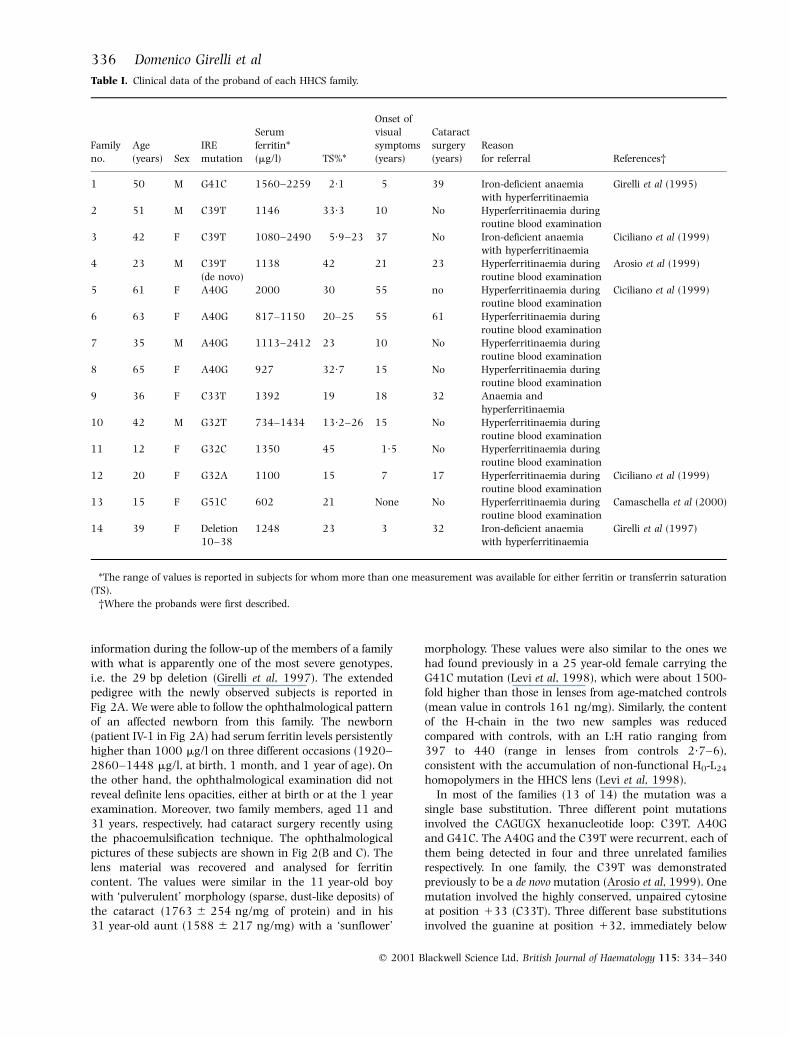
Table I. Clinical data of the proband of each HHCS family.
Familyno.
Age(years) Sex
IREmutation
Serum
ferritin*(mg/l) TS%*
Onset of
visual
symptoms(years)
Cataract
surgery(years)
Reasonfor referral References²
1 50 M G41C 1560±2259 2´1 5 39 Iron-deficient anaemia
with hyperferritinaemia
Girelli et al (1995)
2 51 M C39T 1146 33´3 10 No Hyperferritinaemia during
routine blood examination
3 42 F C39T 1080±2490 5´9±23 37 No Iron-deficient anaemiawith hyperferritinaemia
Ciciliano et al (1999)
4 23 M C39T
(de novo)
1138 42 21 23 Hyperferritinaemia during
routine blood examination
Arosio et al (1999)
5 61 F A40G 2000 30 55 no Hyperferritinaemia duringroutine blood examination
Ciciliano et al (1999)
6 63 F A40G 817±1150 20±25 55 61 Hyperferritinaemia during
routine blood examination
7 35 M A40G 1113±2412 23 10 No Hyperferritinaemia duringroutine blood examination
8 65 F A40G 927 32´7 15 No Hyperferritinaemia during
routine blood examination9 36 F C33T 1392 19 18 32 Anaemia and
hyperferritinaemia
10 42 M G32T 734±1434 13´2±26 15 No Hyperferritinaemia during
routine blood examination11 12 F G32C 1350 45 1´5 No Hyperferritinaemia during
routine blood examination
12 20 F G32A 1100 15 7 17 Hyperferritinaemia during
routine blood examination
Ciciliano et al (1999)
13 15 F G51C 602 21 None No Hyperferritinaemia during
routine blood examination
Camaschella et al (2000)
14 39 F Deletion10±38
1248 23 3 32 Iron-deficient anaemiawith hyperferritinaemia
Girelli et al (1997)
*The range of values is reported in subjects for whom more than one measurement was available for either ferritin or transferrin saturation(TS).
²Where the probands were first described.
336 Domenico Girelli et al
q 2001 Blackwell Science Ltd, British Journal of Haematology 115: 334±340

the bulged cytosine (G32A, G32T, and G32C). One familywas found to have a substitution of the guanine at position151 on the 3 0 side (G51C). Finally, one family had a largedeletion spanning 29 nucleotides (D 10±38), removingmost of the IRE sequence.
A substantial phenotypic variability was noted among
subjects sharing the same mutation, whether they belongedto the same family or not. This was particularly evident forthe severity of the ocular involvement, in terms of either theage at which cataract was diagnosed or the age at whichsubjects were submitted to cataract surgery. For example, in14 subjects from the four families carrying the A40Gmutation, the age at which the cataract was diagnosedranged from 9 to 55 years. In 16 subjects carrying theC39T mutation the corresponding age range was 6±40 years.
Serum ferritin levels were $ 700 mg/l in each subjectcarrying a mutation in the IRE CAGUGX hexaloop, whereasthey varied substantially within subjects sharing the samemutation. For example, ferritin ranged from 700 to2412 mg/l in 14 subjects carrying the A40G mutation,and from 823 to 2490 mg/l in 16 subjects carrying theC39T mutation. A remarkable variation of ferritin levelswas also noted in several patients for whom serummeasurements on different occasions were available(Tables I and II).
DISCUSSION
HHCS is an interesting and almost unique model of humantranslational pathology, in which an altered gene expressionis caused by mutations in a translational regulatoryelement. The large number of different mutations detectedby us and Milon & Beaumont (1998), confirm the molecularheterogeneity of this genetic disease. Because some of these
Table II. Clinical data of the affected relatives from each HHCS family.
Family number
(IRE mutation)
Number
of affected
relatives
Age
range
(years)
Sex
(M/F)
Serum ferritin
range (mg/l)²
TS%
range
Onset of visual
symptoms)
(years
Cataract
surgery
(years)
Iron-deficient
anaemia
1 (G41C) 2 24±25 1/1 1186±2138
(1036±2138)
4±6 1 21 Yes (both)
2 (C39T) 6 16±73 2/4 823±2230(1388±2230)
35±36 6 45±62 Yes (1 female,history of)
3 (C39T) 7 30±47 3/4 918±1615 30±31 39±40 40* No
4 (C39Tde novo)
None
5 (A40G) 7 20±38 4/3 1000±1240 15±32 19±30 45±70 No
6 (A40G) None
7 (A40G) 2 22* 0/2 700±1580 Not available 12 31 No8 (A40G) 1 31 1/0 1000 27´2 9 no No
9 (C33T) 1 9 0/1 653 25 9 60 No
10 (G32T) 6 4±70 4/2 800±1362
(1150±1362)
27* 3±40 40±45 No
11 (G32C) 6 7±69 3/3 1270±2036 20±33 1´5±36 no Yes (1 female,
history of)
12 (G32A) 3 55* 1/2 928±1275 26* 18±20 19±50 No13 (G51C) 1 50 1/0 854 21 None no No
14 (deletion
10±38)
6 3±64 3/3 763±2860
(1448±2860)
13´8±17´6 before the
third year
11±60 No
*Data available for only one subject.
²When available, the ferritin range for repeated measurement in an individual patient is reported between brackets.
Fig 1. The normal stem-loop IRE sequence located in the 5 0-UTR of
l-ferritin gene and the nine mutations found in our series. The
numbering of the nucleotide positions is with respect to thetranscriptional start site.
q 2001 Blackwell Science Ltd, British Journal of Haematology 115: 334±340
Clinical Findings in HHCS 337

mutations are de novo (1 of 14 in our series), HHCS shouldbe included in the differential diagnosis of hyperferritinae-mia even without a positive family history. In this relativelylarge series of HHCS patients, the main finding is thesubstantial phenotypic variability we noted, especially interms of severity of the ocular involvement. This variabilitycannot be explained on the basis of the different mutationsbecause it was also evident among subjects sharing thesame mutation, whether they belonged to the same familyor not. These findings suggest that, besides the l-ferritin IREgenotype, additional factors are likely to modulate the lensinvolvement and the rate of progression to severe cataract inHHCS patients. Environmental factors known to influencethe extremely common age-related cataract, such assmoking, diet, exposure to sunlight and oestrogen status(Livingston et al, 1995; Dolin, 1998) may play a role. Also,interactions with other genes may occur because, recently,an important genetic effect has been established even for theage-related cataract (Hammond et al, 2000), possiblyinvolving `mild' mutations in the same genes which havebeen recognized as giving rise to congenital cataracts whenmutated in a severe fashion (Francis et al, 1999). Initially,the cataract was thought to be `congenital', as the probandsfrom the first families reported severe visual symptoms from
infancy (Bonneau et al, 1995; Girelli et al, 1995a). However,the lack of well-defined lens opacities in an affected newbornfrom the family with the 29 bp deletion removing most ofthe 5' IRE sequence, indicates that cataract in HHCS is notnecessarily congenital, even in subjects carrying what isapparently the most severe genotype. This is in accordancewith data obtained in transgenic mice overexpressing l-ferritin and having a 10-fold increase in the lens l-ferritincontent that did not develop cataract, even after 18 months(C. Beaumont, personal communication). In the samefamily, the ophthalmological pattern showed a progressionfrom `pulverulent' cataract in an 11 year-old boy to well-defined `sunflower' opacities in his 31 year-old aunt. Thelatter morphology was quite similar to the morphology weobserved using slit-lamp examination in non-operated,symptomatic adults from other families, and also to thatreported by Arnold et al (1997). Taken together, thesemorphological observations suggest that an age-relatedevolution of the lens opacities occurs in HHCS. A possiblemechanism accounting for this phenomenon could be anage-related progressive accumulation of l-ferritin in thelens. However, when we analysed the lens specimens of theabove-mentioned patients, we failed to detect any substan-tial difference in the l-ferritin content, despite the different
Fig 2. (A) Pedigree of Family 14 carrying one of the apparently most severe genotype, a 29 bp deletion removing most of the IRE. The arrow
denotes the proband. The red circle denotes a newborn (serum ferritin . 1400 mg/l on three different occasions) in whom the
ophthalmological examination did not detect well-definite lens opacities either at birth or at 1 year of age.(B and C) Slit-lamp examination (afew days before cataract surgery) of subject IV-3 (11 years old) with `pulverulent' cataract and III-2 (31 years old), with `sunflower' cataract
respectively. The l-ferritin content in the lenses was 1763 ^ 254 and 1588 ^ 217 ng/mg protein respectively. See also the Results for details.
338 Domenico Girelli et al
q 2001 Blackwell Science Ltd, British Journal of Haematology 115: 334±340

age and cataract morphology. Whereas the ultimatemechanism leading to cataract formation in HHCS remainspuzzling, this observation once again suggests a role foradditional factors, either environmental or genetic, inmodulating the progression of lens involvement.
In this series of HHCS patients, we failed to detect anyother symptoms related to the syndrome. This is consistentwith the concept that overexpression of l-ferritin has nofunctional effects in most cell types with the exception of thelens. As mentioned above, some subjects had a documentedhistory of mild iron-deficient anaemia, which appeared as aconsequence of relatively slight stimuli, such as blooddonations or pregnancy. It is known that the H-ferritinchain is essential for adequate iron uptake into the ferritincavity, because of its ferroxidase activity not shared by the l-chain (Levi et al, 1988, 1994). Previous studies on HHCSlymphoblastoid cell lines demonstrated an accumulation ofnon-functional H0-L24 homopolymers, and a shift of theremaining pool of isoferritins toward l-chain-rich moleculeswith a suboptimal content of H-chains (Levi et al, 1998).Thus, it is possible to speculate that the formation of irondeposits in HHCS individuals may be at least slightly lessefficient than in normal subjects, depending on the degree ofl-chain overproduction. Unfortunately, most of our clinicaldata were collected retrospectively, without the possibility ofany systematic evaluation of iron stores. Whether ourobservations reflect a true tendency of HHCS patients tobecome iron-deficient in certain conditions because of thealtered H:L ratio of tissue isoferritins remains to beelucidated.
As regards serum ferritin levels, we found importantdifferences among subjects sharing the same mutationswhether or not they belonged to the same family. Also,fluctuations with time for a given individual were observed.This could be caused in part by transient influences of non-genetic factors such as infection or inflammation (Theil,1983) and/or to the heterogeneous sensitivity of differentlaboratory methods of quantifying ferritin values clearlyoutside the normal range. In any event, similar to thatobserved for cataracts, no clear-cut genotype-phenotypecorrelation was apparent.
One relative limitation of our study is that it included onlysubjects with mutations in the hexanucleotide loop or in theupper stem of the IRE sequence. These regions are directlyinvolved in the sequence-specific contact with the IRPs(Basilion et al, 1994). On the other hand, the integrity of thelower stem also seems important to ensure an adequateinteraction with the IRPs, as mutations affecting its lengthand/or G-C content are predicted to affect the stability of theIRE (Allerson et al, 1999). Only one HHCS family carrying adouble mutation in the lower stem (involving positions 118and 122) has been described to date (Cazzola et al, 1997).In vitro experiments have shown that this mutation retaineda nearly normal affinity for the IRPs but resulted insubstantial thermodynamic changes, suggesting an alteredmRNA stability (Allerson et al, 1999). Interestingly, carriersof this peculiar genotype had only mild elevations of serumferritin (ranging from 350 to 650 mg/l) and no visualsymptoms. It is reasonable to assume that most of the
subjects with such a mild phenotype escape clinicalobservation. Thus, the inclusion of an appropriate numberof subjects with mutations in the lower stem of the IREwould probably lead to a more comprehensive definition ofthe whole HHCS biochemical spectrum and a clearergenotype-phenotype correlation according to the in vitrostudies.
In summary, the observations from this series of patientssuggest that: (i) in HHCS the cataract is not necessarilycongenital; (ii) in addition to the IRE genotype, other geneticor environmental factors may modulate the phenotype,especially in terms of lens involvement and rate ofprogression to severe cataract; (iii) no relevant symptomsother than visual impairment seem to be associated with thedisease.
ACKNOWLEDGMENTS
This work was supported by Telethon Italy, grant no. E.749,Cariverona Foundation and MURST 60%.
REFERENCES
Allerson, C.R., Cazzola, M. & Rouault, T.A. (1999) Clinical severity
and thermodynamic effects of iron-responsive element mutationsin hereditary hyperferritinemia±cataract syndrome. Journal of
Biological Chemistry, 274, 26439±26447.
Arnold, J.D., Mumford, A.D., Lindsay, J.O., Hedge, U., Hagan, M. &
Hawkins, J.R. (1997) Hyperferritinemia in the absence of iron
overload. Gut, 41, 408±410.
Arosio, C., Fossati, L., Vigano, M., Trombini, P., Cazzaniga, G. &Piperno, A. (1999) Hereditary hyperferritinemia cataract syn-
drome: a de novo mutation in the iron responsive element of the
l-ferritin gene. Haematologica, 84, 560±561.
Balas, A., Aviles, M.J., Garcia-Sanchez, F. & Vicario, J.L. (1999)Description of a new mutation in the l-ferritin iron-responsive
element associated with hereditary hyperferritinemia±cataract
syndrome in a Spanish family. Blood, 93, 4020±4021.
Barton, J.C., Beutler, E. & Gelbart, T. (1998) Coinheritance of alleles
associated with hemochromatosis and hereditary hyperferritine-mia±cataract syndrome. Blood, 92, 4480.
Basilion, J.P., Rouault, T.A., Massinople, C.M., Klausner, R.D. &
Burgess, W.H. (1994) The iron-responsive element-binding
protein: localization of the RNA-binding site to the aconitase
active-site cleft. Proceedings of the National Academy of Sciences ofthe United States of America, 91, 574±578.
Beaumont, C., Leneuve, P., Devaux, I., Scoazec, J.Y., Berthier, M.,
Loiseau, M.N., Grandchamp, B. & Bonneau, D. (1995) Mutation
in the iron responsive element of the L ferritin mRNA in a family
with dominant hyperferritinaemia and cataract. Nature Genetics,11, 444±446.
Bonneau, D., Winter-Fuseau, I., Loiseau, M.N., Amati, P., Berthier,
M., Oriot, D. & Beaumont, C. (1995) Bilateral cataract and high
serum ferritin: a new dominant genetic disorder? Journal ofMedical Genetics, 32, 778±779.
Camaschella, C., Zecchina, G., Lockitch Roetto, A., Campanella, A.,
Arosio, P. & Levi, S. (2000) A new mutation (G51C) in the iron-
responsive element (IRE) of l-ferritin associated with hyperferri-
tinaemia±cataract syndrome decreases the binding affinity of themutated IRE for iron regulatory proteins. British Journal of
Haematology, 108, 480±482.
Cazzola, M., Bergamaschi, G., Tonon, L., Arbustini, E., Grasso, M.,
q 2001 Blackwell Science Ltd, British Journal of Haematology 115: 334±340
Clinical Findings in HHCS 339

Vercesi, E., Barosi, G., Bianchi, P.E., Cairo, G. & Arosio, P. (1997)
Hereditary hyperferritinemia±cataract syndrome: relationship
between phenotypes and specific mutations in the iron-responsiveelement of ferritin light-chain mRNA. Blood, 90, 814±821.
Ciciliano, M., Zecchina, G., Roetto, A., Bosio, S., Infelise, V., Stefani,
S., Mazza, U. & Camaschella, C. (1999) Recurrent mutations inthe iron regulatory element of l-ferritin in hereditary iperferri-
tinemia±cataract syndrome. Haematologica, 84, 489±492.
Corsi, B., Perrone, F., Bourgeois, M., Beaumont, C., Panzeri, M.C.,
Cozzi, A., Sangregorio, R., Santambrogio, P., Albertini, A., Arosio,P. & Levi, S. (1998) Transient overexpression of human H and L
ferritin chains in COS cells. Biochemical Journal, 330, 315±320.
Dolin, P.J. (1998) Epidemiology of cataract. The Epidemiology of Eye
Disease. (ed. by G.J. Johnson, D.C Minassian & R.A Weale), pp.103±108. Chapman & Hall Medical London.
Francis, P.J., Berry, V., Moore, A.T. & Bhattacharya, S. (1999) Lens
biology: development and human cataractogenesis. Trends inGenetics, 15, 143±196.
Fleming, R.E., Migas, M.C., Zhou, X.Y., Jiang, J., Britton, R.S., Brunt,
E.H., Tomatsu, S., Waheed, A., Bacon, B.R. & Sly W.S. (1999)
Mechanism of increased iron absorption in murine model ofhereditary hemachromatosis: increased duodenal expression
of the iron transporter DMT1. Proceedings of the National Academy
of Sciences of the United States of America, 96, 3143±3148.
Girelli, D., Olivieri, O., De Franceschi, L., Corrocher, R., Berga-
maschi, G. & Cazzola, M. (1995a) A linkage between hereditary
hyperferritinaemia not related to iron overload and autosomal
dominant congenital cataract. British Journal of Haematology, 90,931±934.
Girelli, D., Corrocher, R., Bisceglia, L., Olivieri, O., De Franceschi, L.,
Zelante, L. & Gasparini, P. (1995b) Molecular basis for the
recently described hereditary hyperferritinemia±cataract syn-drome: a mutation in the iron-responsive element of ferritin l-
subunit gene (the `Verona mutation'). Blood, 86, 4050±4053.
Girelli, D., Corrocher, R., Bisceglia, L., Olivieri, O., Zelante, L.,Panozzo, G. & Gasparini, P. (1997) Hereditary hyperferritinemia
cataract syndrome caused by a 29-base paired deletion in the
iron responsive element of ferritin l-subunit gene. Blood, 90,
2084±2088.
Hammond, C.J., Snieder, H., Spector, T.D. & Gilbert, C.E. (2000)
Genetic and environmental factors in age-related nuclear
cataracts in monozygotic and dizygotic twins. New England
Journal of Medicine, 342, 1786±1790.
Kato, G.J. & Casella, F. (1999) l-ferritin-Baltimore-1: a novel
mutation in the iron responsive element (C32G) as a cause of the
hyperferritinemia±cataract syndrome [abstract]. Blood, 94, 407a.
Klausner, R.D., Rouault, T.A. & Hardford, J.B. (1993) Regulating the
fate of mRNA: the control of cellular iron metabolism. Cell, 72,
19±28.
Levi, S., Luzzago, A., Cesareni, G., Cozzi, A., Franceschinelli, F.,Albertini, A. & Arosio, P. (1988) Mechanism of ferritin iron
uptake: activity of the H-chain and deletion mapping of the ferro-
oxidase site. A study of iron uptake and ferro-oxidase activity ofhuman liver, recombinant H-chain ferritins, and of two H-chain
deletion mutants. Journal of Biological Chemistry, 263, 18086±
18092.
Levi, S., Santambrogio, P., Cozzi, A., Rovida, E., Corsi, B., Tamborini,E., Spada, S., Albertini, A. & Arosio, P. (1994) The role of the l-
chain in ferritin iron incorporation. Studies of homo and
heteropolymers. Journal of Molecular Biology, 238, 649±654.
Levi, S., Girelli, D., Perrone, F., Pasti, M., Beaumont, C., Corrocher,R., Albertini, A. & Arosio, P. (1998) Analysis of ferritin in
lymphoblastoid cells lines and in the lens of subjects with
hereditary hyperferritinemia±cataract syndrome. Blood, 91,4190±4197.
Livingston, P.M., Carson, C.A. & Taylor, H.R. (1995) The
epidemiology of cataract: a review of the literature. Ophthalmic
Epidemiology, 2, 151±164.Merkt, J. (1997) Hereditary hyperferritinemia±cataract syndrome.
Deutsche Medizinische Wochenschrift, 122, 504±506.
Milon, B. & Beaumont, C. (1997) Molecular genetics of hereditary
hyperyperferritinemia syndrome Annales de Biologie et de Clinique(Paris) 56, 361±40.
Mumford, A.D., Vulliamy, T., Lindsay, J. & Watson, A. (1998)
Hereditary hyperferritinemia±cataract syndrome: two novel
mutations in the l-ferritin iron-responsive element. Blood, 91,367±368.
Ohrloff, C. & Zubcov, A.A. (1997) Comparison of phacoemulsifica-
tion and planned extracapsular extraction. Ophthalmologica, 211,8±12.
Perez de Nanclares, G., Castano, L., Marul, P., Rica, I., Vela, A.,
Sanjurjo, P., Aldamiz-Echevarria, K., Martinez, R. & Sarrionan-
dia, M.J. (2001) Molecular analysis of hereditary hyperferritine-mia±cataract syndrome in a large Basque family. Journal of
Pediatric Endocrinology and Metabolism, 14, 295±300.
Santambrogio, P., Levi, S., Cozzi, A., Corsi, B. & Arosio, P. (1996)
Evidence that the specificity of iron incorporation into homo-polymers of human ferritin L- and H-chains is conferred by the
nucleation and ferroxidase centers. Biochemical Journal, 314,
139±144.Theil, E.C. (1983) Ferritin: structure, function, and regulation. Iron
Binding Proteins Without Cofactors or Sulfur Clusters (ed. by E.C
Theil,. G.L.Eichhorn & L.GMarzil),pp.1. Elsevier, New York.
Theil,E.C. (1994) Iron regulatory elements (IREs): a family of mRNAnon-coding sequences. Biochemical Journal, 304, 1±11.
340 Domenico Girelli et al
q 2001 Blackwell Science Ltd, British Journal of Haematology 115: 334±340




















