
Modulation of cytokine-induced cyclooxygenase 2 expression by PPARG ligands through NFkappaB signal...
20
Modulation of Cytokine-Induced Cyclooxygenase 2 Expression by PPARG Ligands Through NFκB Signal Disruption in Human WISH and Amnion Cells 1 William E. Ackerman IV 3 , Xiaolan L. Zhang 5 , Brad H. Rovin 5 , and Douglas A. Kniss 3,4,2 3Department of Obstetrics and Gynecology, Laboratory of Perinatal Research and Division of Maternal-Fetal Medicine, 4Center for Biomedical Engineering, and 5Dorothy M. Davis Heart and Lung Research Institute and Department of Internal Medicine, The Ohio State University, Columbus, Ohio 43210 Abstract Cyclooxygenase (COX) activity increases in the human amnion in the settings of term and idiopathic preterm labor, contributing to the generation of uterotonic prostaglandins (PGs) known to participate in mammalian parturition. Augmented COX activity is highly correlated with increased COX2 (also known as prostaglandin-endoperoxide synthase 2, PTGS2) gene expression. We and others have demonstrated an essential role for nuclear factor κB (NFκB) in cytokine-driven COX2 expression. Peroxisome proliferator-activated receptor gamma (PPARG), a member of the nuclear hormone receptor superfamily, has been shown to antagonize NFκB activation and inflammatory gene expression, including COX2. We hypothesized that PPARG activation might suppress COX2 expression during pregnancy. Using primary amnion and WISH cells, we evaluated the effects of pharmacological (thiazolidinediones) and putative endogenous (15-deoxy-Δ 12 , 14 -prostaglandin J 2 , 15d-PGJ 2 ) PPARG ligands on cytokine-induced NFκB activation, COX2 expression, and PGE 2 production. We observed that COX2 expression and PGE 2 production induced by tumor necrosis factor alpha (TNF) were significantly abrogated by 15d-PGJ 2 . The thiazolidinediones rosiglitazone (ROSI) and troglitazone (TRO) had relatively little effect on cytokine-induced COX2 expression except at high concentrations, at which these agents tended to increase COX2 abundance relative to cells treated with TNF alone. Interestingly, treatment with ROSI, but not TRO, led to augmentation of TNF-stimulated PGE 2 production. Mechanistically, we observed that 15d-PGJ 2 markedly diminished cytokine-induced activity of the NFκB transcription factor, whereas thiazolidinediones had no discernable effect on this system. Our data suggest that pharmacological and endogenous PPARG ligands use both receptor-dependent and -independent mechanisms to influence COX2 expression. Keywords cytokines; gene regulation; parturition; placenta; pregnancy 1 Supported in part by NIH grant RO1 HD35881 (D.A.K.) and The Ohio State University Perinatal Research and Development Fund. Portions of this work were presented at the 51st annual meeting of the Society for Gynecologic Investigation, Houston, Texas, March 24–27, 2004. 2 Correspondence: Douglas A. Kniss, Laboratory of Perinatal Research, Department of Obstetrics and Gynecology, The Ohio State University, 5th Floor Means Hall, 1654 Upham Drive, Columbus, OH 43210. FAX: 614 293 5728; e-mail:[email protected]. NIH Public Access Author Manuscript Biol Reprod. Author manuscript; available in PMC 2006 September 1. Published in final edited form as: Biol Reprod. 2005 September ; 73(3): 527–535. NIH-PA Author Manuscript NIH-PA Author Manuscript NIH-PA Author Manuscript
Transcript of Modulation of cytokine-induced cyclooxygenase 2 expression by PPARG ligands through NFkappaB signal...

Modulation of Cytokine-Induced Cyclooxygenase 2 Expression byPPARG Ligands Through NFκB Signal Disruption in Human WISHand Amnion Cells1
William E. Ackerman IV3, Xiaolan L. Zhang5, Brad H. Rovin5, and Douglas A. Kniss3,4,23Department of Obstetrics and Gynecology, Laboratory of Perinatal Research and Division ofMaternal-Fetal Medicine,
4Center for Biomedical Engineering, and
5Dorothy M. Davis Heart and Lung Research Institute and Department of Internal Medicine, TheOhio State University, Columbus, Ohio 43210
AbstractCyclooxygenase (COX) activity increases in the human amnion in the settings of term and idiopathicpreterm labor, contributing to the generation of uterotonic prostaglandins (PGs) known to participatein mammalian parturition. Augmented COX activity is highly correlated with increased COX2 (alsoknown as prostaglandin-endoperoxide synthase 2, PTGS2) gene expression. We and others havedemonstrated an essential role for nuclear factor κB (NFκB) in cytokine-driven COX2 expression.Peroxisome proliferator-activated receptor gamma (PPARG), a member of the nuclear hormonereceptor superfamily, has been shown to antagonize NFκB activation and inflammatory geneexpression, including COX2. We hypothesized that PPARG activation might suppress COX2expression during pregnancy. Using primary amnion and WISH cells, we evaluated the effects ofpharmacological (thiazolidinediones) and putative endogenous (15-deoxy-Δ12,14-prostaglandin J2,15d-PGJ2) PPARG ligands on cytokine-induced NFκB activation, COX2 expression, and PGE2production. We observed that COX2 expression and PGE2 production induced by tumor necrosisfactor alpha (TNF) were significantly abrogated by 15d-PGJ2. The thiazolidinediones rosiglitazone(ROSI) and troglitazone (TRO) had relatively little effect on cytokine-induced COX2 expressionexcept at high concentrations, at which these agents tended to increase COX2 abundance relative tocells treated with TNF alone. Interestingly, treatment with ROSI, but not TRO, led to augmentationof TNF-stimulated PGE2 production. Mechanistically, we observed that 15d-PGJ2 markedlydiminished cytokine-induced activity of the NFκB transcription factor, whereas thiazolidinedioneshad no discernable effect on this system. Our data suggest that pharmacological and endogenousPPARG ligands use both receptor-dependent and -independent mechanisms to influence COX2expression.
Keywordscytokines; gene regulation; parturition; placenta; pregnancy
1Supported in part by NIH grant RO1 HD35881 (D.A.K.) and The Ohio State University Perinatal Research and Development Fund.Portions of this work were presented at the 51st annual meeting of the Society for Gynecologic Investigation, Houston, Texas, March24–27, 2004.2Correspondence: Douglas A. Kniss, Laboratory of Perinatal Research, Department of Obstetrics and Gynecology, The Ohio StateUniversity, 5th Floor Means Hall, 1654 Upham Drive, Columbus, OH 43210. FAX: 614 293 5728; e-mail:[email protected].
NIH Public AccessAuthor ManuscriptBiol Reprod. Author manuscript; available in PMC 2006 September 1.
Published in final edited form as:Biol Reprod. 2005 September ; 73(3): 527–535.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

INTRODUCTIONProstaglandins (PGs) that elicit myometrial contractions and cervical maturation are pivotal tothe onset and maintenance labor in humans and all other mammalian species that have beeninvestigated [1]. The exact triggers for the onset of parturition at term are incompletelyunderstood, but there is a growing consensus that cytokines, such as interleukin 1β (IL1B) andtumor necrosis factor alpha (TNF), are instrumental in unleashing vigorous biosynthesis ofPGE2 and PGF2α within intrauterine tissues, leading to active labor and birth [2]. In addition,it appears that many of the fundamental biological forces governing labor at term are alsoevoked during episodes of preterm labor elicited by inflammatory insults to the mother [3].
While the majority of parturition research has focused on the feed-forward mechanisms throughwhich uterine stimulants are generated, a relevant question remains unanswered: Whatbiological inputs keep the uterus from contracting and the cervix from ripening during the vastmajority of pregnancy? It is reasonable to hypothesize that tonically active signals prohibitmyometrial activation before term and that these biomolecules are of fundamental importancefor a thorough understanding of the mechanisms governing parturition. In this regard, it hasrecently been reported that PGs of the J2 series may exert anti-inflammatory activity in a varietyof circumstances [4]. Peroxisome proliferator-activated receptor gamma (PPARG) is a nuclearreceptor whose ligands include PGs of the J2 series (particularly 15-deoxy-Δ12,14-prostaglandin J2, 15d-PGJ2) in addition to polyunsaturated fatty acids, products oflipoxygenase metabolism, and oral hypoglycemic agents (thiazolidinediones) [5,6]. Severalgroups have reported that PPARG antagonizes the expression of inflammatory signals in abroad array of experimental contexts [7,8], including human gestational tissues [9]. Moreover,some PPARG ligands inhibit the expression of inflammatory gene products, such as induciblenitric oxide synthase (also known as nitric oxide synthase 2A, NOS2A), cyclooxygenase 2(COX2, also known as prostaglandin-endoperoxide synthase 2, PTGS2), and microsomalprostaglandin E synthase-1 (also known as prostaglandin E synthase, PTGES) by mechanismsthat may be PPARG-dependent and/or -independent [7,8,10,11].
A common theme among the proinflammatory enzymes cited above is their regulation at thetranscriptional level by nuclear factor κB (NFκB) [12,13]. The genes encoding theseinflammatory proteins harbor one or more copies of a κB-binding motif through whichtranscription may be enhanced [12]. For example, cytokines use NFκB within intrauterine cellsto stimulate expression of the COX2 gene [14–17], whose product catalyzes the committingand rate-limiting step in uterotonic PG formation [18]. Recent evidence suggests that COX2-mediated synthesis of PGD2 metabolites (including the PPARG ligand, 15d-PGJ2) may providea mechanism for feedback control of PG biosynthesis [4,19]. Furthermore, we recently reportedthat a reciprocal relationship exists between the expression of COX2 and PPARG proteins infetal membranes obtained from women before the onset of labor compared with tissuescollected following delivery [20]. Thus, in the present study, we examined the mechanism bywhich known PPARG ligands govern COX2 expression in WISH cells and primary culturesof human amnion.
MATERIALS AND METHODSMaterials
Recombinant human TNF was purchased from R&D Systems (Minneapolis, MN). Antibodiesagainst inhibitory factor κBα (IκBα, also known as nuclear factor of kappa light polypeptidegene enhancer in B-cells inhibitor alpha, NFKBIA), IκB kinase α (IKKα, also known asconserved helix-loop-helix ubiquitous kinase, CHUK), IκB kinase β (IKKβ, also known asinhibitor of kappa light polypeptide gene enhancer in B-cells kinase beta, IKBKB), COX2, andNFκB subunits p65 (also known as reticuloendotheliosis viral oncogene homolog A, RELA),
Ackerman et al. Page 2
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

p50 (also known as nuclear factor of kappa light polypeptide gene enhancer in B-cells 1,NFKB1), p52 (also known as nuclear factor of kappa light polypeptide gene enhancer in B-cells 2, NFKB2), cRel (also known as reticuloendotheliosis viral oncogene homolog, REL),and RelB (also known as reticuloendotheliosis viral oncogene homolog B, RELB) wereobtained from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse anti-rabbitglyceraldehyde-3-phosphate dehydrogenase (GAPD) antibody, which is cross-reactive withthe human isoform, was purchased from Chemicon International (Temecula, CA). Antibodiesrecognizing phosphorylated IκBα (Ser32) and IKKα (Ser180)/IKKβ (Ser181) were from CellSignaling Technology (Beverly, MA), as were rabbit anti-human PPARG antibodies. A secondPPARG antibody was purchased from Affinity Bioreagents (Golden, CO). The 1.8-kilobase(kb) cDNA fragment used for COX2 mRNA Northern blotting was a kind gift from Dr. TimothyHla (University of Connecticut, Farmington, CT). Arachidonic acid, all PGs, rosiglitazone,and PGE2 ELISA kits were obtained from Cayman Chemical (Ann Arbor, MI). Troglitazonewas from BIOMOL (Plymouth Meeting, PA). DIG Nucleic Acid Detection and DIG-HighPrime kits were purchased from Roche Diagnostics (Indianapolis, IN). Assays-on-Demandgene expression target assay mix (Hs00153133 m1), 18S rRNA assay mix, and TaqManUniversal Master Mix were obtained from Applied Biosystems (Foster City, CA). SuperSignalchemiluminescent detection reagents were obtained from Pierce Biotechnology (Rockford,MA). Prolong antifade mounting reagent and Alexa Fluor-594-conjugated goat anti-rabbitantibodies were purchased from Molecular Probes (Eugene, OR). The NFκB consensusoligonucleotide was obtained from Promega (Madison, WI). All other reagents, unlessotherwise specified, were obtained from Sigma (St. Louis, MO).
Cell CulturesHuman WISH cells were obtained from the American Type Culture Collection (CCL-25) andmaintained in Ham F-12/Dulbecco modified Eagle medium (F-12/DMEM, Invitrogen,Carlsbad, CA) supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, and 10% (v/v)newborn calf serum. Cells were grown at 37°C in a humidified atmosphere of 95% air/5%CO2 and used for experiments between the 3rd and 25th passages.
Primary cultures of human amnion cells were obtained at the time of uncomplicated scheduledcesarean delivery at term in the absence of labor or membrane rupture. There were no clinicalsignals of infection in any of these subjects. Informed consent was obtained in all casesfollowing a protocol approved by the Institutional Review Board of The Ohio State University.Cultures were established according to the method of Okita et al. [21], with modifications.Briefly, amnion tissue was dissected from the choriodecidua within minutes of delivery, placedin sterile Hanks buffered salt solution (HBSS), and transported to our laboratory for asepticprocessing. Amnion tissue was washed in ice-cold HBSS until free of blood, minced into ~1-cm2 fragments, and digested for 30 min in HBSS containing 0.4% (w/v) trypsin at 37°C withconstant rocking. Undigested material was recovered and fresh trypsin solution was added.Following an additional 30-min incubation, cells were strained through 60 mesh and the flow-through was collected. This process was repeated using the remaining undigested tissue. Theflow-through from the second and third digestions was pooled and cells were collected bycentrifugation at 600 × g for 10 min. Cells were then washed, plated into tissue culture dishes,and grown in F-12/DMEM supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, 50μg/ml gentamicin sulfate, and 10% (v/v) fetal bovine serum. Within 7–10 days, cultures hadattained confluence and were used for experiments.
Northern Blot AnalysisTo assess COX2 mRNA expression in WISH cells following treatments, total RNA wasextracted using TRIZOL according to manufacturer’s specifications (Invitrogen). Total RNA (20μg/lane) was fractionated on a 1% (w/v) agarose gel containing 2% (v/v) formaldehyde,
Ackerman et al. Page 3
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

transferred to a nylon membrane, and immobilized using ultraviolet irradiation. Membraneswere then probed with digoxigenin (DIG)-containing cDNA probes labeled using the DIG-High Prime kit. A 1.8-kb cDNA fragment encoding human COX2 and a 0.6-kb cDNA fragmentencoding human GAPD were used for detection. Blots were hybridized overnight at 42°C in7% (w/v) SDS, 50% (v/v) formamide, 325 mM sodium chloride, 32.5 mM sodium citrate, 50mM sodium phosphate (pH 7.0), 0.1% (w/v) N-lauroylsarcosine, 50 μg/ml sheared salmonsperm DNA, and 2% (w/v) DIG Blocking Reagent (Roche). Bound probes were identifiedusing the DIG Nucleic Acid Detection kit (Roche). Chemiluminescent signals were detectedusing the VersaDoc Imaging System and analyzed using Quantity One software (Bio-RadLaboratories, Hercules, CA).
Real-Time Reverse Transcriptase-Polymerase Chain ReactionsFollowing treatments, total RNA was extracted from amnion cell cultures using TRIzol(Invitrogen). Because total RNA yields from amnion cells were typically ~10% that of theWISH cells, quantities were insufficient to allow for detection by Northern blotting using therelatively insensitive DIG-labeled cDNA probes. Therefore, relative abundance of COX2mRNA was performed by real-time reverse transcriptase-polymerase chain reactions (RT-PCR) detection using an ABI PRISM 7700 sequence detector (Applied Biosytems, Foster City,CA). Two μg/reaction of total RNA were reverse transcribed to cDNA using random hexamerprimers and SuperScript II reverse transcriptase (Invitrogen), according to instructionsprovided by the manufacturer. Human COX2 expression was detected using the Assays-on-Demand gene expression target assay mix (Hs00153133 m1; Applied Biosystems), whichcontained a mixture of specific primers and a hydrolysis probe containing the fluorescent dye6-FAM. As the reporter probe sequence was designed to detect a region spanning multipleintron-exon junctions, target cDNA was detected selectively, irrespective of the presence ofcontaminant genomic DNA. A primer/VIC-labeled probe set for detecting 18S rRNA (AppliedBiosystems) was used as an internal control in each reaction. Amplification mixtures contained2.5 μl 20× target assay mix (Applied Biosystems), 1 μl first-strand cDNA synthesis mixture(corresponding to 50 ng reverse transcribed RNA), 0.5 μl of 18S rRNA control mix (AppliedBiosystems), 25 μl of 2× TaqMan Universal Master Mix (Applied Biosystems), and nuclease-free distilled deionized water to a total volume of 50 μl. Amplification was performed over 40cycles of denaturation at 95°C for 15 sec and annealing/extension at 60°C for 1 min.
For each treatment condition, duplicate amplifications were performed. Relative abundance ofCOX2 mRNA was normalized to the 18S rRNA internal standard and calculated as 2−ΔCT,where ΔCT denotes difference between the sample CT (cycle threshold) and that of thereference message present in cells under nonstimulated conditions. Amplification curves wereanalyzed using software provided by the manufacturer (Applied Biosystems).
Electrophoretic Mobility Shift AssayCells were treated with the test substances indicated and nuclear protein extracts were prepared[22]. Nuclear protein extracts (10 μg/reaction) were then mixed with an oligonucleotide (5′-AGTTGAGGGGACTTTCC CAGGC; Promega) representing the consensus sequence of theκB-binding motif (underlined) labeled with [32P]-ATP (approximately 104 cpm/tube) aspreviously described [22]. Control incubations contained either no nuclear extracts, a 20-foldexcess of an unlabeled, irrelevant oligonucleotide (5′-GGCGAAACTTCTGGAATATTCCCGAACTTTCAG, a sequence from the human heatshock 70-kDa protein 1 [HSPA1A] gene promoter [23]), or a 20-fold excess of unlabeled κBoligonucleotide. After a 20-min incubation at room temperature, the extracts were fractionatedon nondenaturing 5% (w/v) polyacrylamide gels in 0.25 M Tris-boric acid-EDTA buffer. Gelswere then dried and exposed to x-ray film at −80°C.
Ackerman et al. Page 4
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

To conduct supershift assays, samples were prepared for electrophoretic mobility shift assay(EMSA) as described above and to each tube 2 μg of anti-p65, anti-p50, anti-p52, anti-RelB,or anti-cRel was added for 30 min at room temperature. Control incubations contained the sameconcentration of isotype-specific IgG. Samples were fractionated on nondenaturing gels andautoradiographed as described above.
Immunoblot AnalysisCellular proteins were extracted as previously described [24]. Proteins (30 μg/lane) wereresolved by SDS-PAGE and transferred to nitrocellulose. Immunoblotting was performedusing antibodies directed against native and phosphorylated IκBα, native and phosphorylatedIKKα/β, PPARG, COX2, and GAPD in Tris-buffered saline (pH 8.0) containing 0.05% (v/v)Tween-20 and 5% (w/v) BSA. Following exposure to horse-radish peroxidase-conjugatedsecondary antibodies, chemiluminescent signals were revealed using SuperSignalchemiluminescent detection reagents (Pierce). Immunoreactive proteins were visualized usingthe VersaDoc Imaging System and analyzed using Quantity One software (Bio-RadLaboratories).
ImmunofluorescenceCells were seeded onto flame-sterilized glass coverslips placed in a 24-well tissue culture plate.Following treatments, cells were fixed for 1 h in 4% (w/v) paraformaldehyde/PBS, madepermeable by the addition of 0.2% (v/v) Triton X-100/PBS for 15 min, and blocked in 5% (v/v) horse serum/PBS overnight at 4°C before the addition of antibodies. Monoclonal antibodiesdirected against p65 (sc-8008; Santa Cruz) were then applied. In control experiments, anequivalent concentration of isotype-specific normal mouse IgG was substituted for the primaryantibody. After stringent washing in PBS, the coverslips were exposed to fluorochrome-conjugated secondary antibodies (Molecular Probes). Nuclei were counterstained with 5 μg/ml 4′,6-diamidino-2-phenylindole (DAPI, Sigma). Cells were mounted using the ProLongAntifade kit (Molecular Probes) and visualized using an epifluorescence microscope (NikonInstruments, Melville, NY).
Prostaglandin E2 ELISACells were plated in 48-well tissue culture plates in serum-free culture medium supplementedwith 5 μM arachidonic acid. Following 6-h incubations with test substances, the media werecollected and frozen at −80°C until assays were performed. Media were analyzed for PGE2content using commercially available ELISA kits (Cayman Chemical). Cells were solubilizedwith 1 N NaOH, total protein was measured, and the data were expressed as nanograms ofPGE 2 produced/mg protein. In all ELISA experiments, 4–6 replicates were performed perassay and the experiments were repeated a minimum of twice. The intra- and interassaycoefficients of variation of this assay were <10% and the limit of detection was 15 pg/ml(corresponding to approximately 12.5 pg/mg when adjusted for the average protein content/well).
Statistical AnalysisELISA and densitometric data were assessed by one-way analysis of variance (ANOVA)followed by the Tukey-Kramer multiple comparisons post hoc test; P < 0.05 was consideredstatistically significant.
Ackerman et al. Page 5
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

RESULTSPPARG Ligands Modulate PGE2 Biosynthesis
Among several potential endogenous ligands for PPARG, 15-deoxy-Δ12,14-PGJ2 (15d-PGJ2),a nonenzymatically-derived terminal metabolite of PGD2, has been extensively studied [19,23,25–28]. We examined the effects of this cyclopentenone in relation to pharmacologicalPPARG agonists of the thiazolidinedione class (rosiglitazone [ROSI] and troglitazone [TRO]).WISH cells were preincubated for 30 min in the absence or presence of 15d-PGJ2, ROSI, orTRO and then challenged with TNF (25 ng/ml) for 6 h. Control cultures were incubated with0.1% (v/v) ethanol (vehicle for the thiazolidinediones) or 0.8% (v/v) methyl acetate (MeAc,vehicle for 15d-PGJ2). To exclude the possibility of cross-reactivity between the antibodiesand the reagents, in some cases, we spiked aliquots of test medium with the highestexperimental concentrations of 15d-PGJ2, ROSI, or TRO and subjected these to ELISA. Noneof these compounds were found to interfere with the assay system. Expression of the PPARGreceptor protein in WISH and primary amnion cells was confirmed by immunoblotting (Fig.1A).
In the presence of 30 μM 15d-PGJ2, TNF-stimulated PGE2 production was diminishedcompared with cells preincubated with vehicle alone (Fig. 1B and data not shown). At 3 μMof 15d-PGJ2, this response was less marked, and no effect was evident when 0.3 μM of 15d-PGJ2 was used (Fig. 1B). By contrast, preincubation with ROSI or TRO did not abrogate PGgeneration (Fig. 1B). In fact, in preliminary experiments, it was found that preincubation with10 μM ROSI led to a significant enhancement of cytokine-induced PGE2 release, whereas 10μM TRO had no discernable effect (data not shown). When tested at an expanded range ofconcentrations (0.1–10 μM), it was discovered that cytokine-mediated PGE2 synthesis wasconsistently enhanced in the presence of ROSI at concentrations of 1 μM or greater (Fig. 1B).There was no enhancement of basal PGE2 production when these cells were incubated withROSI or TRO in the absence of TNF (data not shown). These data indicated that, while both15d-PGJ2 and thiazolidinediones are activators of PPARG, they exert differential effects onPGE2 formation in this model system.
15-deoxy-Δ12,14-PGJ2 Prohibits TNF-Induced COX2 AccumulationThe mechanisms through which 15d-PGJ2 exerts its anti-inflammatory effects remaincontroversial, as both PPARG-dependent and -independent actions have been reported [10,28,29]. Like the other PGD2 derivatives (PGJ2 and Δ12-PGJ2), 15d-PGJ2 has a cyclopentenonering structure that is characterized by one or two α,β-unsaturated carbonyl moieties withelectrophilic carbons that are very susceptible to nucleophilic addition reactions (reviewed in[30], also see Fig. 2A). Many of the PPARG-independent effects of 15d-PGJ2 appear to besimilar to those seen with other cyclopentenone PGs, and may arise through the ability of thesecompounds to form thiol conjugates with cysteinyl residues in a variety of cellular proteins[30,31].
To determine whether diminished PGE2 biosynthesis was due to decreased COX2 proteinfollowing cytokine stimulation, we preincubated WISH cells either with 15d-PGJ2 (30 μM),ROSI (0.1–10 μM), TRO (0.1–10 μM), or vehicles and then challenged the cells with TNF for4 h. Additionally, to compare the effects of 15d-PGJ2 with other cyclopentenone PGs, cellswere also treated with equivalent concentrations (30 μM) of the following: PGA1, PGD2,PGJ2, Δ12-PGJ2, 15d-PGA1, 15d-PGA2, and 15d-PGD2. Cell extracts were subjected toimmunoblotting with anti-COX2 antibodies. In the absence of cytokine treatment,immunoreactive COX2 was barely detectable. By contrast, treatment with TNF elicited adramatic increase in COX2 protein (Fig. 2, B and C, lane 2), which was consistent with anearlier report from our group [24]. When cells were preincubated with 15d-PGJ2, TNF did not
Ackerman et al. Page 6
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

stimulate an increase in COX2 production (Fig. 2B, compare lanes 2 and 7). Interestingly,among cells pretreated with related cyclopentenones (PGA1, PGJ2, Δ12-PGJ2) or their 15-deoxy derivatives (15d-PGA1, 15d-PGA2, 15d-PGD2), only Δ12-PGJ2 inhibited COX2 proteinexpression, and this effect was modest (Fig. 2D). Neither ROSI nor TRO inhibited TNF-induced COX2 protein expression (Fig. 2C, in which it should be noted that the apparentdiminution of immunoreactive COX2 in lane 4 was not a consistent finding). Rather, for bothROSI and TRO at 10 μM, we observed a consistent trend toward increased COX2 protein (Fig.2E).
15-Deoxy-Δ12,14-PGJ2 and Thiazolidinediones Differentially Regulate COX2 mRNAExpression
After demonstrating that 15d-PGJ2 inhibits TNF-stimulated COX2 protein expression in WISHcells, we determined whether 15d-PGJ2 or thiazolidinediones attenuate COX2 mRNAupregulation following cytokine exposure. In Northern blots, COX2 mRNA wascharacteristically upregulated following a 60-min challenge with TNF (Fig. 3A, lane 2). WhenWISH cells were preincubated with 15d-PGJ2 for 30 min before cytokine stimulation, we notednearly complete abrogation of COX2 mRNA accumulation (Fig. 3A, compare lanes 1, 2, and7). A similar, but less substantial, effect was noted for Δ12-PGJ2, a second PGD2 metabolitethat has been shown to exhibit both PPARG-dependent and -independent activities [31,32].With respect to the other PGs, PGA1, 15d-PGA1, and 15d-PGA2 all exhibited similar effectsin attenuating TNF-elicited COX2 mRNA expression (Fig. 3, A and B). By contrast, neitherPGD2, PGJ2, nor 15d-PGD2 produced a discernable effect on COX2 mRNA levels in responseto cytokine challenge, despite the presence of electrophilic carbons on PGJ2 and 15d-PGD2(Fig. 3, A and B). RNA from WISH cells that had been pretreated with ROSI or TRO followedby TNF showed no diminution in COX2 mRNA (Fig. 3A). Instead, at thiazolidinedioneconcentrations of 10 μM, there was a modest but reproducible enhancement of cytokine-mediated COX2 mRNA accumulation (Fig. 3A, lanes 11 and 13).
To examine whether the observations in WISH cells were also seen in amnion cells culturedfrom full-term fetal membranes, we preincubated amnion cell monolayers for 30 min withvehicle, 15d-PGJ2, ROSI, or TRO and then challenged cells for 1 h with TNF. Total RNA wasextracted and assayed by real-time RT-PCR using a human COX2 primer pair/probe set. Figure3C demonstrates that TNF caused a nearly fivefold increase in COX2 mRNA expressionfollowing cytokine treatment. In contrast, amnion cells preincubated with 15d-PGJ2 manifestedan approximately 40% decrease in COX2 mRNA when challenged with cytokine (Fig. 3C).When amnion cells were preincubated with ROSI or TRO and then TNF, there was anenhancement of cytokine-induced COX2 mRNA accumulation (Fig. 3C). Thus, both WISHand primary amnion cells exhibited decreased expression of COX2 mRNA when preincubatedwith 15d-PGJ2, while there was no such diminution when cells were pretreated withthiazolidinediones. In fact, thiazolidinedione pretreatment elicited a modest and consistentelevation in COX2 mRNA expression following cytokine stimulation in both cell types.
Cyclopentenones Inhibit TNF-Stimulated Activation of NFκBProinflammatory cytokines use the NFκB pathway as a principal means to elicit COX2expression within intrauterine cells [15–17]. Under basal conditions, NFκB dimers (which canbe composed of the subunits p65, p50, p52, cRel, and RelB) are bound to an inhibitor (IκB)[12]. In the presence of IκB, the net intracellular distribution of NFκB is cytoplasmic; however,when cells are stimulated, IκB is phosphorylated by IκB kinases (IKKs), which results in thedestruction of this inhibitor by the 26 S proteasome complex [12]. Once this occurs, theNFκB complex translocates to the nucleus, binds to response elements within the promotersof target genes, and activates transcription.
Ackerman et al. Page 7
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

To examine the biochemical mechanisms that underlie cyclopentenone inhibition of cytokinesignaling via NFκB, WISH cells were preincubated either with PGD2, cyclopentenones,thiazolidinediones, or vehicles and then stimulated with TNF (25 ng/ml) for 15 min (a timepoint at which the signaling events responsible for NFκB activation were at their peak, asestablished previously [17]). Immunoblots were probed with antibodies recognizing the nativeand phosphorylated forms of IKKα, IKKβ, and IκBα.
TNF elicited phosphorylation of IKKα and IKKβ, which was followed by phosphorylation andrapid degradation of IκBα (data not shown). This resulted in the loss of IκBα immunoreactivityobserved by 15 min poststimulation (Fig. 4A, compare lanes 1 and 2). As previouslydemonstrated [17], this loss in IκBα immunoreactivity could be blocked with MG-132, aninhibitor of protein degradation by the 26 S proteasome complex (data not shown). NeitherPGD2, the cyclopentenones, nor the thiazolidinediones were found to affect signaling eventsleading to TNF-mediated phosphorylation of IKKα or IKKβ (Fig. 4, A and B). However, inthe presence of 15d-PGJ2, cytokine-induced phosphorylation of IκBα was completely blocked,and there was no evidence of lost IκBα immunoreactivity (Fig. 4A, lane 7). Several othercyclopentenones, including PGA1, PGJ2, Δ12-PGJ2, 15d-PGA1, and 15d-PGA2, causedretention of phosphorylated IκBα following TNF stimulation, consistent with disruption ordelay in the events leading to NFκB activation (Fig. 4A). The findings of inhibited IκBαphosphorylation and/or degradation were congruous with prior reports, in which the activitiesof both IKK and the proteasome complex were found to be directly inhibited by thecyclopentenone PGs [28,33–35]. By contrast, neither PGD2, 15d-PGD2, nor thethiazolidinediones blocked the phosphorylation/degradation of IκBα.
We next conducted immunolocalization studies to evaluate whether 15d-PGJ2 attenuatedcytosolic to nuclear translocation of the NFκB subunit p65. WISH cells were preincubated for30 min either with vehicle, ROSI, or 15d-PGJ2. To test the potential specificity of 15d-PGJ2,we also preincubated some cultures with a second cyclopentenone, PGA1. The cells were thenchallenged with TNF and fixed for immunostaining with anti-p65 at various intervals from 5to 60 min. As previously observed with IL1B [17], TNF stimulation resulted in translocationof p65 from cytosol to nucleus within 15 min (Fig. 4C, arrows in panel 2). ROSI had no effecton the movement of p65 to the nuclear compartment (Fig. 4C, arrows in panel 3). 15d-PGJ2,however, completely abrogated the ability of TNF to elicit p65 translocation to the nucleus,even through the 60-min time period tested (the effect of this agent at 15 min poststimulationis shown in Fig. 4C, panel 4). PGA1, though somewhat less efficacious, was able to delay p65nuclear localization (data not shown). By 15 min following TNF challenge in the presence ofPGA1, nuclear p65 immunoreactivity was observed in a minority of cells (Fig. 4C, panel 5).Similar to the data obtained in WISH cells, we also noted in primary amnion cells that 15d-PGJ2, but not ROSI, inhibited TNF-stimulated p65 nuclear translocation, while the effects ofPGA1 were incomplete (Fig. 4C, panels 6–10).
We next examined whether 15d-PGJ2 could prevent binding of NFκB to radiolabeledoligonucleotides representing the consensus κB motif, similar to those found in the 5′-promoterregion of the human COX2 gene [36]. In initial EMSA experiments, it was observed that nuclearextracts prepared from WISH cells stimulated for 30 min with TNF led to the restricted mobilityof two specific bands (I and II in Fig. 5, A and B). A third specific band migrating above bandII was noted in some cases (Fig. 5B). Super-shifting with polyclonal antibodies directed againstNFκB subunits p65 and p50 demonstrated that the DNA binding activity of band I likelyconsisted of p50/p50 homodimers, while that of band II likely consisted of p65/p50heterodimers. We ruled out the likely participation of other NFκB members using antibodiesto p52, cRel, and RelB (data not shown). Following a 30-min preincubation with 30 μM of15d-PGJ2 or 10 μM of ROSI, we observed that TNF failed to elicit protein binding to the κBmotif in the presence of 15d-PGJ2 only (Fig. 5C). Collectively, these data demonstrated that
Ackerman et al. Page 8
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

15d-PGJ2, like some of the other cyclopentenones (PGA1, PGJ2, Δ12-PGJ2, 15d-PGA1, and15d-PGA2), but in contrast with the pharmacological PPARG ligands, was able to attenuatecytokine-stimulated NFκB activation.
DISCUSSIONIn this investigation, we evaluated the ability of PPARG ligands to downregulate the COX2gene and subsequent formation of PGE 2 in WISH and primary amnion cells. Our datademonstrated an interesting dichotomy between 15d-PGJ2, a cyclopentenone PG, and thethiazolidinediones, which are pharmacological PPARG activators. Whereas 15d-PGJ2treatment of amnion cells in primary culture or WISH cells almost completely abrogated TNF-driven COX2 expression, the thiazolidinediones partially upregulated COX2 expression. Thesedata render a straightforward mechanism through ligation of nuclear PPARG unlikely andinstead invoke a more complex level of COX2 regulation by these agonists.
In this study, we employed WISH cells in many experiments, as these cells are easilymanipulated genetically and manifest many phenotypic features of amnion epithelial cells[37–39]. Although WISH cells have been used as reliable and reproducible facsimile foramnion epithelial cells for many years [14,17,24,27,40], it must be acknowledged that thesecells contain HeLa chromosome markers, as we have recently reported [41]. We verified theapplicability of our data by repeating key experiments using primary amnion cell cultures,which in these cases showed similar results. Thus, to a degree, WISH cells remain a veryrealistic surrogate for molecular mechanistic studies in amnion cells.
In the case of 15d-PGJ2, suppression of cytokine-elicited COX2 expression appears to havearisen to a large extent through interference with NFκB activation. In amnion cell models, theNFκB signaling cascade appears to be the predominant mechanism through which cytokinesstimulate COX2 gene expression [14,16,17]. Moreover, our group has reported that NFκB isa prominent transcription factor governing COX2 gene expression in ED27 cells [22]. Likeother cyclopentenone PGs, 15d-PGJ2 has been shown to impede NFκB signaling at multiplesteps: it may interfere with the phosphorylation of IκB by suppressing IKK activity [33], itmay interfere with IκB degradation by inhibiting activity of the 26 S proteasome complex[34], and it may interfere directly with the ability of NFκB subunits to bind to their cognateDNA response elements [28]. These effects are thought to occur independently of PPARG,through the formation of covalent adducts between 15d-PGJ2 and critical cysteinyl residueswithin these proteins [28,35]. Consistently, we observed that other cyclopentenone PGs bearingα,β-unsaturated ketone groups also interfered with cytokine-mediated NFκB activation, albeitfar less potently, whereas thiazolidinediones did not alter this signaling pathway. Althoughprior reports have lead to the speculation that PPARG might interfere with NFκB activitythrough direct protein-protein interactions [8] or through competition for limiting amounts ofcoactivator proteins [42], our data suggest that, in WISH and in amnion cells cultured in vitro,PPARG may not be involved directly in the antagonism of NFκB activation. In support of this,Lindstrom and Bennett have recently reported that antagonism of cytokine-elicited NFκBactivation by 15d-PGJ2 occurs independently of PPARG in amnion and myometrial cells[43].
In addition to inhibiting NFκB, cyclopentenone PGs have also been shown to suppress theactivity of other transcription factors involved in cytokine-stimulated COX2 expression, suchas activator protein-1 (AP-1) [8,44]. By impeding such signaling cascades, it is likely 15d-PGJ2 and other cyclopentenones suppress TNF-stimulated COX2 expression. In general, theability of the cyclopentenones to attenuate COX2 induction was correlated with the number ofelectrophilic β-carbons present within the molecules. Both 15d-PGJ2 and Δ12-PGJ2, with cross-conjugated α,β- unsaturated ketones, exhibited greater potency relative to the cyclopentenones
Ackerman et al. Page 9
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

having only a simple α,β-unsaturated carbonyl moiety, which is consistent with priorobservations [35]. However, given that 15d-PGJ2 suppressed cytokine-induced COX2expression to a greater degree than did Δ12-PGJ2, our results suggest that the number of reactiveβ-carbons alone is insufficient to explain the effects of these agents. It is therefore noteworthythat 15d-PGD2, which bears an exocyclic α,β-unsaturated ketone, was unable to suppress eithercytokine-mediated NFκB activation or COX2 expression. Although we do not yet have asuitable explanation for this observation, we speculate that endocyclic β-carbons might exhibitgreater reactivity than exocyclic β-carbons for the formation of cyclopentenone proteinconjugates.
Many previous studies have demonstrated proapoptotic actions of cyclopentenones such as15d-PGJ2 in several cell types [45,46]. Keelan et al. [27] reported that 15d-PGJ2 elicitedprogrammed cell death in WISH cells. However, in the present study, we treated WISH cellsand primary amnion cells for relatively brief periods (<12 h) and observed little evidence ofapoptotic cell death (data not shown). Thus, it is unlikely that cytotoxicity is an explanationfor the observed decreases in COX2 gene expression and subsequent PGE2 biosynthesisreported here. We cannot exclude the possibility that longer incubations may lead to apoptosis,however.
In contrast with 15d-PGJ2 and other cyclopentenones, we observed a tendency forthiazolidinediones, when given at high concentrations, to augment TNF-stimulated COX2expression. This effect was particularly evident when low concentrations of TNF (5 ng/ml)were used (data not shown). While this finding was unexpected, there is precedence thatPPARG might positively regulate COX2 gene expression in certain cell types [47]. The humanCOX2 gene contains a PPAR response element located approximately 3900 bp upstream ofthe translational start site, through which ligand-bound PPARG may promote transcriptionalactivation [47]. However, in numerous experiments, we failed to detect any stimulation in basalCOX2 expression by thiazolidinediones (including troglitazone, rosiglitazone, andpioglitazone, data not shown) in the absence of cytokine stimulation. Furthermore, despite thefact that TRO and ROSI enhanced cytokine-elicited COX2 expression to similar degrees, onlyROSI was found to augment TNF-stimulated PGE2 production. In ongoing experiments, weare examining whether this effect might have occurred through differential regulation of anenzyme downstream of COX2 in the PGE2 metabolic pathway (such as PTGES), or througha more global change in cellular arachidonic acid utilization. It is noteworthy that these effectswere seen only when high concentrations of these agents were employed. While it is not unusualfor thiazolidinediones to be used at concentrations of 10 μM (or higher) in cell cultureexperiments [9,10,48], it is nevertheless true that such amounts are often in excess of what isrequired for saturation of the PPARG receptor (which, in the case of ROSI, is below 1 μM[49]). Therefore, we speculate that some of the pharmacological effects observed may haveoccurred independently of PPARG. We are currently conducting experiments to discern thePPARG dependency of these effects in a comprehensive manner, but the present data providea vital backdrop for these investigations.
The true physiological significance of these data awaits further experimental clarification.While it has been demonstrated that cells of the fetal membranes, including the amnion, expressthe PPARG receptor [20], the existence of endogenous PPARG ligands in these tissues remainsunclear. It has been established that the placenta is the major source of PGD2, the precursorfor PGs of the J series (PGJ2, Δ12-PGJ2, and 15d-PGJ2) within the uterus [50]. Furthermore,amniotic fluid contains PGD2 [51], and there is some evidence to suggest that 15d-PGJ2 alsomight be present [52], although this latter finding is controversial. Therefore, it is notunreasonable to suggest that substances such as the PGs of the J2 might serve a physiologicalrole during gestation. However, there is a growing body of literature suggesting that 15d-PGJ2 is not the only potential endogenous PPARG ligand; polyunsaturated fatty acids (PUFAs)
Ackerman et al. Page 10
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

and products of lipoxygenase metabolism have also been shown to activate PPARG [6,53],and a novel, as yet uncharacterized, endogenous PPARG ligand has recently been discovered[54]. These and potentially other unidentified ligands may comprise a mileau of anti-inflammatory biomolecules that are present endogenously throughout normal human gestation,as suggested by the recent findings of Waite and colleagues [55]. Members of our laboratoryare currently developing screening systems to allow for the detection and identification of suchPPARG ligands within amniotic fluid, maternal plasma, and gestational tissues.
In summary, we have demonstrated in WISH and primary amnion cells that reported ligandsfor PPARG (cyclopentenone PGs and thiazolidinediones) exert differential effects on COX2gene expression. Inasmuch as these two classes of PPARG agonists do not act in precisely thesame manner, we have proposed a working model to delineate the potential mechanismsthrough which these agents govern COX2 expression and PG biosynthesis (Fig. 6).
Acknowledgements
We thank Dr. Timothy Hla for his generous donation of the human COX2 cDNA probe.
References1. Olson DM, Mijovic JE, Sadowsky DW. Control of human parturition. Semin Perinatol 1995;19:52–
63. [PubMed: 7754411]2. Kniss DA, Iams JD. Molecular effectors of human preterm parturition. In: Seifer DB, Samuels P, Kniss
DA (eds.), Physiologic Basis of Gynecology and Obstetrics. Philadelphia, PA: Lippincott Williamsand Wilkins; 2001:442–455.
3. Gibb W. The role of prostaglandins in human parturition. Ann Med 1998;30:235–241. [PubMed:9677008]
4. Colville-Nash PR, Gilroy DW. COX-2 and the cyclopentenone prostaglandins—a new chapter in thebook of inflammation? Prostaglandins Other Lipid Mediat 2000;62:33–43. [PubMed: 10936414]
5. Murphy GJ, Holder JC. PPAR-gamma agonists: therapeutic role in diabetes, inflammation and cancer.Trends Pharmacol Sci 2000;21:469–474. [PubMed: 11121836]
6. Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D,Glass CK. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature 1999;400:378–382. [PubMed: 10432118]
7. Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatorycytokines. Nature 1998;391:82–86. [PubMed: 9422509]
8. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998;391:79–82. [PubMed: 9422508]
9. Lappas M, Permezel M, Georgiou HM, Rice GE. Regulation of proinflammatory cytokines in humangestational tissues by peroxisome proliferator-activated receptor-gamma: effect of 15-deoxy-delta(12,14)-PGJ(2) and troglitazone. J Clin Endocrinol Metab 2002;87:4667–4672. [PubMed: 12364456]
10. Cheng S, Afif H, Martel-Pelletier H, Pelletier J-P, Li X, Farrajota K, Lavigne M, Fahmi H. Activationof peroxisome proliferator-activated protein γ inhibits interleukin-1β-induced membrane-associatedprostaglandin E2 synthase-1 expression in human synovial fibroblasts by interfering with Egr-1. JBiol Chem 2004;279:22057–22065. [PubMed: 15023995]
11. Petrova TV, Akama KT, Van Eldik LJ. Cyclopentane prostaglandins suppress activation of microglia:downregulation of inducible nitric oxide syntase by 15-deoxy-delta12,14-prostaglandin J2. Proc NatlAcad Sci U S A 1999;96:4668–4673. [PubMed: 10200320]
12. Baeuerle PA. IkappaB-NF-kappaB structures: at the interface of inflammation control. Cell1998;95:729–731. [PubMed: 9865689]
13. Poligone B, Baldwin AS. Positive and negative regulation of NF-kappaB by COX-2: roles of differentprostaglandins. J Biol Chem 2001;276:38658–38664. [PubMed: 11509575]
Ackerman et al. Page 11
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

14. Allport VC, Slater DM, Newton R, Bennett PR. NF-kappaB and AP-1 are required for cyclooxygenase2 gene expression in amnion epithelial cell line (WISH). Mol Hum Reprod 2000;6:561–565.[PubMed: 10825375]
15. Belt AR, Baldassare JJ, Molnar M, Romero R, Hertelendy F. The nuclear transcription factor NF-kappaB mediates interleukin-1beta-induced expression of cyclooxygenase-2 in human myometrialcells. Am J Obstet Gynecol 1999;181:359–366. [PubMed: 10454683]
16. Yan X, Wu XC, Sun M, Tsang BK, Gibb W. Nuclear factor kappa B activation and regulation ofcyclooxygenase type-2 expression in human amnion mesenchymal cells by interleukin-1beta. BiolReprod 2002;66:1667–1671. [PubMed: 12021045]
17. Ackerman WE IV, Rovin BH, Kniss DA. Epidermal growth factor and interleukin-1beta use divergentsignaling pathways to synergistically upregulate cyclooxygenase-2 gene expression in humanamnion-derived WISH cells. Biol Reprod 2004;71:2079–2086. [PubMed: 15329330]
18. Kniss DA. Cyclooxygenases in reproductive medicine and biology. J Soc Gynecol Invest 1999;6:285–292.
19. Cuzzocrea S, Wayman NS, Mazzon E, Dugo L, Di Paola R, Serraino I, Britti D, Chatterjee PK, CaputiAP, Thiemermann C. The cyclopentenone prostaglandin 15-deoxy-Δ12,14-prostaglandin J2attenuates the development of acute and chronic inflammation. Mol Pharmacol 2002;61:997–1007.[PubMed: 11961117]
20. Dunn-Albanese LR, Ackerman WE 4th, Xie Y, Iams JD, Kniss DA. Reciprocal expression ofperoxisome proliferator-activated receptor-gamma and cyclooxygenase-2 in human term parturition.Am J Obstet Gynecol 2004;190:809–816. [PubMed: 15042019]
21. Okita JR, Sagawa N, Casey ML, Snyder JM. A comparison of human amnion tissue and amnion cellsin primary culture by morphological and biochemical criteria. In Vitro 1983;19:117–126. [PubMed:6826195]
22. Kniss DA, Rovin B, Fertel RH, Zimmerman PD. Blockade NF-kappaB activation prohibits TNF-alpha-induced cyclooxygenase-2 gene expression in ED27 trophoblast-like cells. Placenta2001;22:80–89. [PubMed: 11162356]
23. Zhang X, Lu L, Dixon C, Wilmer W, Song H, Chen X, Rovin BH. Stress protein activation by thecyclopentenone prostaglandin 15-deoxy-delta12,14-prostaglandin J2 in human mesangial cells.Kidney Int 2004;65:798–810. [PubMed: 14871400]
24. Perkins DJ, Kniss DA. Tumor necrosis factor-alpha promotes sustained cyclooxygenase-2 expression:attenuation by dexamethasone and NSAIDs. Prostaglandins 1997;54:727–743. [PubMed: 9440135]
25. Hinz B, Brune K, Pahl A. 15-Deoxy-Δ12,14-prostaglandin J2 inhibits the expression ofproinflammatory genes in human blood monocytes via a PPAR-γ-independent mechanism. BiochemBiophys Res Commun 2003;302:415–420. [PubMed: 12604364]
26. Janabi N. Selective inhibition of cyclooxygenase-2 expression by 15-deoxy-Δ12,14-prostaglandinJ2 in activated human astrocytes, but not in human brain macrophages. J Immunol 2002;168:4747–4755. [PubMed: 11971025]
27. Keelan JA, Helliwell RJA, Nijmeijer BE, Berry EBE, Sato TA, Marvin KW, Mitchell MD, GilmourRS. 15-Deoxy-Δ12,14-prostaglandin J2-induced apoptosis in amnion-like WISH cells.Prostaglandins Other Lipid Mediat 2001;66:265–282. [PubMed: 11785780]
28. Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, Sengchanthalangsy LL, Ghosh G,Glass CK. 15-Deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signalingpathway. Proc Natl Acad Sci U S A 2000;97:4844–4849. [PubMed: 10781090]
29. Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-gamma dependent andindependent effects on macrophage gene expression in lipid metabolism and inflammation. Nat Med2001;7:48–52. [PubMed: 11135615]
30. Fukushima M. Biological activities and mechanisms of action of PGJ2 and related compounds: anupdate. Prostaglandins Leukot Essent Fatty Acids 1992;47:1–12. [PubMed: 1438462]
31. Atsmon J, Sweetman BJ, Baertschi SW, Harris TM, Roberts LJ. Formation of thiol conjugates of 9-deoxy-delta 9,delta 12(E)-prostaglandin D2 and delta 12(E)-prostaglandin D2. Biochemistry1990;29:3760–3765. [PubMed: 2340271]
Ackerman et al. Page 12
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

32. Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell 1995;83:803–812. [PubMed: 8521497]
33. Castrillo A, Diaz-Guerra MJ, Hortelano S, Martin-Sanz P, Bosca L. Inhibition of IkappaB kinase andIkappaB phosphorylation by 15-deoxy-Delta(12,14)-prostaglandin J(2) in activated murinemacrophages. Mol Cell Biol 2000;20:1692–1698. [PubMed: 10669746]
34. Mullally JE, Moos PJ, Edes K, Fitzpatrick FA. Cyclopentenone prostaglandins of the J series inhibitthe ubiquitin isopeptidase activity of the proteasome pathway. J Biol Chem 2001;276:30366–30373.[PubMed: 11390388]
35. Verbitski SM, Mullally JE, Fitzpatrick FA, Ireland CM. Punaglandins, chlorinated prostaglandins,function as potent Michael receptors to inhibit ubiquitin isopeptidase activity. J Med Chem2004;47:2062–2070. [PubMed: 15056003]
36. Tazawa R, Xu XM, Wu KK, Wang LH. Characterization of the genomic structure, chromosomallocation and promoter of human prostaglandin H synthase-2 gene. Biochem Biophys Res Commun1994;203:190–199. [PubMed: 8074655]
37. Hayflick L. The establishment of a line (WISH) of human amnion cells in continuous cultivation.Exp Cell Res 1961;23:14–20. [PubMed: 13712490]
38. Kniss DA, Zimmerman PD, Fertel RH, Iams JD. Proinflammatory cytokines interact synergisticallywith epidermal growth factor to stimulate PGE2 production in amnion-derived cells. Prostaglandins1992;44:237–244. [PubMed: 1410528]
39. Bry K. Epidermal growth factor and transforming growth factor-alpha enhance the interleukin-1- andtumor necrosis factor-stimulated prostaglandin E2 production and the interleukin-1 specific bindingon amnion cells. Prostaglandins Leukot Essent Fatty Acids 1993;49:923–928. [PubMed: 8140119]
40. Hansen WR, Drew A, Helsby N, Keelan JA, Sato TA, Mitchell MD. Regulation of cytosolicphospholipase A2 expression by cytokines in human amnion cells. Placenta 1999;20:303–308.[PubMed: 10329351]
41. Kniss DA, Xie Y, Li Y, Kumar S, Linton EA, Cohen P, Fan-Havard P, Redman CW, Sargent IL. ED(27) trophoblast-like cells isolated from first-trimester chorionic villi are genetically identical to HeLacells yet exhibit a distinct phenotype. Placenta 2002;23:32–43. [PubMed: 11869090]
42. Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors in inflammationcontrol. J Endocrinol 2001;169:453–459. [PubMed: 11375115]
43. Lindstrom TM, Bennett PR. 15-deoxy-delta(12,14)-prostaglandin J2 inhibits IL-1beta-induced NF-kappaB in human amnion and myometrial cells: mechanisms and implications. J Clin EndocrinolMetab 2005;90:3534–3543. [PubMed: 15755849]
44. Perez-Sala D, Cernuda-Morollon E, Canada FJ. Molecular basis for the direct inhibition of AP-1DNA binding by 15-deoxy-delta(12,14)-prostaglandin J2. J Biol Chem 2003;278:51251–51260.[PubMed: 14532268]
45. Bishop-Bailey D, Hla T. Endothelial cell apoptosis induced by the peroxisome proliferator-activatedreceptor (PPAR) ligand 15-deoxy-delta 12,14-prostaglandin J2. J Biol Chem 1999;274:17042–17048.[PubMed: 10358055]
46. Kawahito Y, Kondo M, Tsubouchi Y, Hashiramoto A, Bishop-Bailey D, Inoue K, Kohno M, YamadaR, Hla T, Sano H. 15-deoxy-Δ12,14-PGJ2 induces synoviocyte apoptosis and suppresses adjuvant-induced arthritis in rats. J Clin Invest 2000;106:189–197. [PubMed: 10903334]
47. Meade EA, McIntyre TM, Zimmerman GA, Prescott SM. Peroxisome proliferators enhancecyclooxygenase-2 expression in epithelial cells. J Biol Chem 1999;274:8328–8334. [PubMed:10075740]
48. Subbaramaiah K, Lin DT, Hart JC, Dannenberg AJ. Peroxisome proliferator-activated receptorgamma ligands suppress the transcriptional activation of cyclooxygenase-2. Evidence forinvolvement of activator protein-1 and CREB-binding protein/p300 . J Biol Chem 2001;276:12440–12448. [PubMed: 11278336]
49. Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabeticthiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma(PPAR gamma). J Biol Chem 1995;270:12953–12956. [PubMed: 7768881]
Ackerman et al. Page 13
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

50. Mitchell MD, Kraemer DL, Strickland DM. The human placenta: a major source of prostaglandinD2. Prostaglandins Leukot Med 1982;8:383–387. [PubMed: 6955806]
51. Berryman GK, Strickland DM, Hankins GD, Mitchell MD. Amniotic fluid prostaglandin D2 inspontaneous and augmented labor. Life Sci 1987;41:1611–1614. [PubMed: 3476816]
52. Marvin KW, Eykholt RL, Keelan JA, Sato TA, Mitchell MD. The 15-deoxy-delta(12,14)-prostaglandin J(2)receptor, peroxisome proliferator activated receptor-gamma (PPARgamma) isexpressed in human gestational tissues and is functionally active in JEG3 choriocarcinoma cells.Placenta 2000;21:436–440. [PubMed: 10833383]
53. Glass CK. Potential roles of the peroxisome proliferator-activated receptor-γ in macrophage biologyand atherosclerosis. J Endocrinol 2001;169:461–464. [PubMed: 11375116]
54. Tzameli I, Fang H, Oilero M, Shi H, Hamm JK, Kievitt P, Hollenberg AN, Flier JS. Regulatedproduction of a peroxisome proliferator-activated receptor-g ligand during an early period ofadipocyte differentiation in 3T3-L1 adipocytes. J Biol Chem 2004;279:36093–36102. [PubMed:15190061]
55. Waite LL, Louie RE, Taylor RN. Circulating activators of PPARs are reduced in preeclampticpregnancy. J Clin Endocrinol Metab 2004;90:620–626. [PubMed: 15562025]
Ackerman et al. Page 14
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
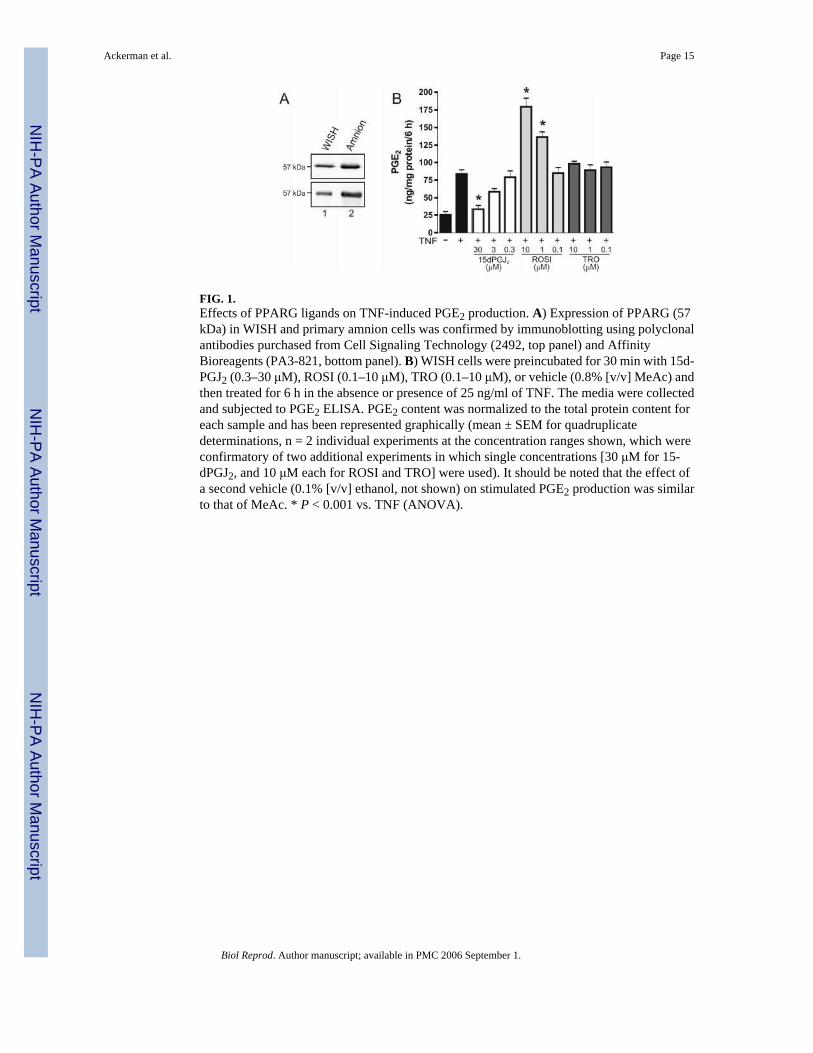
FIG. 1.Effects of PPARG ligands on TNF-induced PGE2 production. A) Expression of PPARG (57kDa) in WISH and primary amnion cells was confirmed by immunoblotting using polyclonalantibodies purchased from Cell Signaling Technology (2492, top panel) and AffinityBioreagents (PA3-821, bottom panel). B) WISH cells were preincubated for 30 min with 15d-PGJ2 (0.3–30 μM), ROSI (0.1–10 μM), TRO (0.1–10 μM), or vehicle (0.8% [v/v] MeAc) andthen treated for 6 h in the absence or presence of 25 ng/ml of TNF. The media were collectedand subjected to PGE2 ELISA. PGE2 content was normalized to the total protein content foreach sample and has been represented graphically (mean ± SEM for quadruplicatedeterminations, n = 2 individual experiments at the concentration ranges shown, which wereconfirmatory of two additional experiments in which single concentrations [30 μM for 15-dPGJ2, and 10 μM each for ROSI and TRO] were used). It should be noted that the effect ofa second vehicle (0.1% [v/v] ethanol, not shown) on stimulated PGE2 production was similarto that of MeAc. * P < 0.001 vs. TNF (ANOVA).
Ackerman et al. Page 15
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

FIG. 2.Effects of cyclopentenone PGs and thiazolidinediones on TNF-elicited COX2 proteinexpression. A) Chemical structures of PGD2 and the cyclopentenone PGs used in this studyare shown in relation to that of the parent compound, cyclopentenone. Electrophilic carbonsare denoted by gray asterisks. B, C) Representative immunoblots prepared from WISH celllysates 4 h following challenge with TNF (25 ng/ml) alone or in combination with PGD2 (30μM), cyclopentenone PGs (PGA1, PGJ2, Δ12-PGJ2, 15d-PGJ2, 15d-PGA1, 15d-PGA2, and15d-PGD2, all at 30 μM) or thiazolidinediones (ROSI and TRO, 0.1–10 μM). Control cellsreceived either 0.8% (v/v) MeAc (B) or 0.1% (v/v) ethanol (C). D, E) Immunoreactive COX2(72 kDa) was quantified by densitometric scanning. Data were normalized to GAPD (34 kDa)protein levels for each treatment group and have been represented graphically as ratios ofarbitrary densitometric units (mean ± SEM, n = 2 [D] or 3 [E] individual experiments). * P <0.01 vs. TNF (AN-OVA). ** P < 0.05 vs. TNF (ANOVA).
Ackerman et al. Page 16
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

FIG. 3.Effects of cyclopentenone PGs and thiazolidinediones on cytokine-induced COX2 mRNAexpression. A) Representative Northern blot demonstrating COX2 mRNA levels in WISH cells1 h following treatment with TNF (25 ng/ml) in the absence or presence of PGD2 (30 μM),cyclopentenone PGs (PGA1, PGJ2, Δ12-PGJ2, 15d-PGJ2, 15d-PGA1, 15d-PGA2, and 15d-PGD2, all at 30 μM) or thiazolidinediones (ROSI and TRO, 1–10 μM). Control cells receivedeither 0.8% (v/v) MeAc or 0.1% (v/v) ethanol, with similar effects. B) Chemiluminescentdetection of the major COX2 mRNA band (4.5 kb) was quantified by densitometric scanning.Data were normalized to GAPD mRNA levels in each treatment group and have beenrepresented graphically as a ratio of arbitrary densitometric units (mean ± SEM, n = 3 individualexperiments). C) Real-time RT-PCR was used to assess COX2 mRNA expression in primaryamnion cells following treatment with vehicle alone, vehicle with TNF (25 ng/ml), or TNF inthe presence of 15d-PGJ2 (30 μM), ROSI (10 μM), or TRO (10 μM). The threshold cycle(CT) at which COX2 mRNA was detected was normalized to that of the 18S rRNA internalcontrol. The ratios of COX2 mRNA/18S rRNA have been expressed graphically as the foldincrease in relation to control cells, for which the level of COX2 mRNA expression was set to1 (mean ± SEM for duplicate determinations, n = 2 individual experiments). * P < 0.01 vs.TNF (ANOVA). ** P < 0.05 vs. TNF (AN-OVA).
Ackerman et al. Page 17
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

FIG. 4.Effects of cyclopentenone PGs and thiazolidinediones on cytokine-stimulated NFκBactivation. A, B) WISH cells were challenged for 15 min with TNF (25 ng/ml) in the absenceor presence of PGD2 (30 μM), cyclopentenone PGs (PGA1, PGJ2, Δ12-PGJ2, 15d-PGJ2, 15d-PGA1, 15d-PGA2, and 15d-PGD2, all at 30 μM) or thiazolidinediones (ROSI and TRO, 0.1–10 μM). Control cells received either 0.8% (v/v) MeAc (A) or 0.1% (v/v) ethanol (B).Activation of NFκB was assessed using antibodies detecting native and phosphorylated formsof IKKα (85 kDa), IKKβ (87 kDa), and IκBα (39 kDa). These blots are representative of theresults of two independent experiments. C) Intracellular localization of NFκB subunit p65 wasdetected by immunofluorescence in WISH (panels 1–5) or primary amnion (panels 6–12) cellstreated with or without 25 ng/ml of TNF for 15 min in the absence (vehicle only) or presenceof ROSI (10 μM, panels 3 and 8), 15d-PGJ2 (30 μM, panels 4 and 9), or PGA1 (30 μM, panels5 and 10). Arrows indicate cell nuclei. A representative control experiment is shown (panel11) in which an equivalent amount of normal mouse IgG1 was substituted for the primaryantibody; DAPI staining within this same field demonstrates the location of cell nuclei (panel12). Bars = 20 μm.
Ackerman et al. Page 18
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
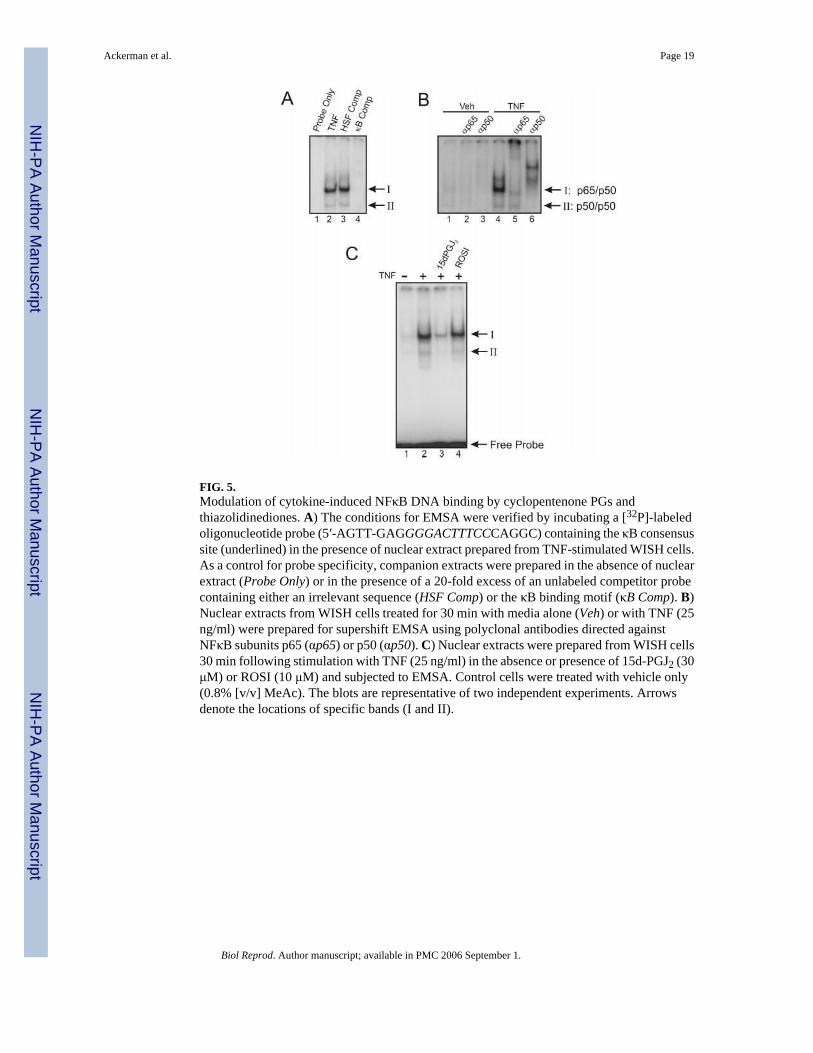
FIG. 5.Modulation of cytokine-induced NFκB DNA binding by cyclopentenone PGs andthiazolidinediones. A) The conditions for EMSA were verified by incubating a [32P]-labeledoligonucleotide probe (5′-AGTT-GAGGGGACTTTCCCAGGC) containing the κB consensussite (underlined) in the presence of nuclear extract prepared from TNF-stimulated WISH cells.As a control for probe specificity, companion extracts were prepared in the absence of nuclearextract (Probe Only) or in the presence of a 20-fold excess of an unlabeled competitor probecontaining either an irrelevant sequence (HSF Comp) or the κB binding motif (κB Comp). B)Nuclear extracts from WISH cells treated for 30 min with media alone (Veh) or with TNF (25ng/ml) were prepared for supershift EMSA using polyclonal antibodies directed againstNFκB subunits p65 (αp65) or p50 (αp50). C) Nuclear extracts were prepared from WISH cells30 min following stimulation with TNF (25 ng/ml) in the absence or presence of 15d-PGJ2 (30μM) or ROSI (10 μM) and subjected to EMSA. Control cells were treated with vehicle only(0.8% [v/v] MeAc). The blots are representative of two independent experiments. Arrowsdenote the locations of specific bands (I and II).
Ackerman et al. Page 19
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

FIG. 6.Working model for the effects of 15d-PGJ2 and thiazolidinediones on TNF-stimulated NFκBactivation and COX2 expression. Depicted in this schematic diagram is the NFκB signalingpathway, through which TNF and other cytokines stimulate COX2 gene expression in WISHand amnion cells [17]. Upon binding to its cellular receptor, TNF invokes a signaling cascadewhereby the IκB kinase (IKK) complex becomes activated. IKK acts to phosphorylate IκB,leading to the destruction of this protein by the 26 S proteasome complex. Once this occurs,NFκB can migrate to the nucleus, bind to response elements within the 5′-promoter region ofthe COX2 gene, and activate transcription. Independently of PPARG, 15d-PGJ2 may act atmultiple steps in this pathway to impede NFκB activation, either by inhibiting of IKK activity(1), or by attenuating destruction of IκB by the 26 S proteasome complex (2). The effects of15d-PGJ2 through activation of the PPARG receptor are unclear (3); however, our current dataindicate that pharmacological activation of PPARG by thiazolidinediones (TZDs) might leadto enhanced COX2 transcription.
Ackerman et al. Page 20
Biol Reprod. Author manuscript; available in PMC 2006 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript




















