
MODIFICATION OF NUCLEOTIDE EXCISION REPAIR ... - QSpace
153
MODIFICATION OF NUCLEOTIDE EXCISION REPAIR ACTIVITY BY 4-(METHYLNITROSAMINO)-1-(3-PYRIDYL)-1- BUTANONE (NNK) AND SULFORAPHANE by Christopher Matthew Harris A thesis submitted to the Department of Biomedical and Molecular Sciences In conformity with the requirements for the degree of Doctor of Philosophy Queen’s University Kingston, Ontario, Canada (January, 2017) Copyright ©Christopher Matthew Harris, 2017
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of MODIFICATION OF NUCLEOTIDE EXCISION REPAIR ... - QSpace

MODIFICATION OF NUCLEOTIDE EXCISION REPAIR
ACTIVITY BY 4-(METHYLNITROSAMINO)-1-(3-PYRIDYL)-1-
BUTANONE (NNK) AND SULFORAPHANE
by
Christopher Matthew Harris
A thesis submitted to the Department of Biomedical and Molecular Sciences
In conformity with the requirements for
the degree of Doctor of Philosophy
Queen’s University
Kingston, Ontario, Canada
(January, 2017)
Copyright ©Christopher Matthew Harris , 2017

ii
Abstract
The studies described in this thesis investigated the relationship between the DNA repair
pathway nucleotide excision repair (NER, which is critical for protecting against carcinogenesis),
and two exogenous chemicals, the tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-
butanone (NNK) and the nutraceutical sulforaphane.
NER activity in female A/J mouse liver nuclear extracts decreased by 46% 12 h post
NNK treatment and increased by 48% 24 h post NNK. These changes in NER activity were not
attributed to alterations in levels of the NER proteins XPC, XPA, XPB and p53. However, the
binding of hepatic XPA and XPB to damaged DNA at both timepoints was increased, while the
binding of XPC to damaged DNA was unchanged at 12 h and decreased at 24 h.
A 63% decrease in NER activity in female A/J mouse lung nuclear protein extracts 24 h
post NNK treatment was not attributed to changes in levels of XPC, XPA, XPB or p53.
However, the binding of lung XPA and XPB to damaged DNA was decreased and the binding of
XPC to damaged DNA was increased at 24 h. Hence, NNK-induced NER changes are time and
organ dependent and are associated with changes in the binding activities, but not the levels, of
specific recognition and early excision NER proteins.
A dose (100 mg/kg) and timepoint (6 h) of maximal effect of sulforaphane on female
CD-1 mouse hepatic and pulmonary mRNA levels of two genes regulated by Nrf2, a major
signalling pathway of sulforaphane mediated-effects, were found. A 50% increase in NER
activity was observed in liver nuclear extracts 12 h post sulforaphane treatment, while lung NER
at 12 h and liver NER at 6 h were not affected by sulforaphane. The increase in hepatic NER
activity at 12 h was not associated with changes in levels of XPC, XPA, XPB or p53 or in the
binding of hepatic XPC, XPA and XPB to damaged DNA. These results suggest that the impact
of sulforaphane on repair-associated processes is time and organ dependent and is not associated

iii
with changes in the levels or activities of these specific recognition and early excision NER
proteins.

iv
Co-Authorship
This research was conducted by the candidate Christopher M. Harris, under the
supervision of Dr. Thomas E. Massey. Natalie Chow provided the qRT-PCR work contained in
chapter 4.

v
Acknowledgements
I would like to begin by thanking Dr. Thomas Massey, my supervisor, for his guidance
and support throughout the work that is presented here. I would like to thank you for your
patience and encouragement through this whole process; without you I would be lost. Also thank
you to Dr. Kanji Nakatsu for helping me learn how to write scientifically.
Thank you to Natalie Chow, who provided much of the hard work comprising Chapter 4.
Your work is so appreciated, and rewards you every day.
I would also like to thank the inimitable Sandra Graham for technical assistance on this
project, and Drs. Jeanne Mulder and Pamela Brown for helpful suggestions. Jeanne and Sandy,
thanks to you especially for your positivity and your kindness in dealing with my flaws and me.
Thanks to my friends and family in Toronto, London, New York and Kingston- Jessi,
David, Roz, Jon, Kelcey, Gab and all of the Queen’s people- you know who you are. I love you
all; you are my bottomless well of support and motivation. Mia and James- I love every moment
I get to spend with you. I love seeing you grow and learn, I can’t wait to see you fix this world.
Mia- I am sure there is a typo in here, I will be blaming that on you.
Thank you to my parents and in-laws: you are always understanding about the things I
say and do that you don’t understand. I am so fortunate to have two families that only want for
me what I want for myself. My gift to you is this is the only section of this book I expect you to
read.
Finally, thanks to my peerless partner, Erica. My life would be so quiet without you, in
so many ways. You are the reason our life together is colourful. You are my good spirit, my
better self; I love jumping over bad times, holding your hand.
This work was supported by the Canadian Institutes of Health Research and the Natural
Sciences and Engineering Research Council of Canada.

vi
Table of Contents
Abstract ............................................................................................................................................ ii
Co-Authorship ................................................................................................................................. iv
Acknowledgements .......................................................................................................................... vList of Figures .................................................................................................................................. x
List of Abbreviations .................................................................................................................... xiv
Chapter 1 General Introduction ....................................................................................................... 1 Statement of the research problem ......................................................................................... 11.1
Chemical carcinogenesis ........................................................................................................ 31.2
DNA repair and nucleotide excision repair ........................................................................... 51.3
1.3.1 DNA repair ...................................................................................................................... 51.3.2 Nucleotide excision repair .............................................................................................. 6
1.3.2.1 Triple-check recognition .......................................................................................... 8
1.3.2.2 Incision/excision ...................................................................................................... 91.3.2.3 Gap filling .............................................................................................................. 10
1.3.3 Changes in NER activity ............................................................................................... 101.3.3.1 Rate limiting NER proteins .................................................................................... 11
1.3.3.2 p53 and NER .......................................................................................................... 121.3.3.3 Conditions that increase NER activity. .................................................................. 131.3.3.4 Conditions that decrease NER activity .................................................................. 14
NNK ..................................................................................................................................... 151.4
1.4.1 Occurrence and source of NNK .................................................................................... 151.4.2 Biotransformation of NNK ........................................................................................... 181.4.3 Detoxification of NNK ................................................................................................. 20
1.4.4 Carcinogenicity of NNK in experimental animals ........................................................ 20
1.4.5 NNK and human cancers .............................................................................................. 21
1.4.6 DNA repair and NNK ................................................................................................... 24
Sulforaphane ........................................................................................................................ 261.5
1.5.1 Glucosinolates ............................................................................................................... 26
1.5.2 Sulforaphane properties and kinetics ............................................................................ 291.5.3 Sulforaphane as a cancer chemopreventive, cytotoxic and cytoprotective chemical ... 30
1.5.4 Nrf2 ............................................................................................................................... 321.5.5 The actions of Nrf2 ....................................................................................................... 35

vii
1.5.6 Sulforaphane mechanisms not associated with Nrf2 .................................................... 36
1.5.7 Sulforaphane and DNA repair ...................................................................................... 37 Research hypotheses and objectives .................................................................................... 391.6
Chapter 2 Time course of effects of NNK on nucleotide excision repair activity on A/J mouse
liver and possible mechanisms of change ...................................................................................... 42 Introduction .......................................................................................................................... 422.1
Materials and methods ......................................................................................................... 462.2
2.2.1 Animal treatments ......................................................................................................... 462.2.2 Preparation of cell-free whole tissue nuclear protein extract ........................................ 46
2.2.3 Preparation of damaged plasmid DNA ......................................................................... 47
2.2.4 In vitro DNA repair synthesis assay ............................................................................. 482.2.5 Immunoblot analysis of NER protein levels in mouse liver nuclear protein extracts .. 49
2.2.6 Analysis of binding of NER incision proteins to damaged DNA ................................. 492.2.7 Data analysis ................................................................................................................. 51 Results .................................................................................................................................. 512.3
2.3.1 Time course of effects of NNK treatment on mouse liver NER activity ...................... 512.3.2 The effect of in vivo treatment of mice with NNK on levels of key NER proteins in
mouse liver nuclear protein extracts ...................................................................................... 532.3.3 Effect of in vivo treatment of mice with NNK on binding of liver NER proteins to
damaged DNA ....................................................................................................................... 56 Discussion ............................................................................................................................ 592.4
Chapter 3 Investigation of the mechanisms by which NNK decreases nucleotide excision repair
activity in mouse lung .................................................................................................................... 63 Introduction .......................................................................................................................... 633.1
Materials and methods ......................................................................................................... 663.2
3.2.1 Animal treatments ......................................................................................................... 66
3.2.2 Preparation of cell-free whole tissue nuclear protein extract ........................................ 67
3.2.3 Preparation of damaged plasmid DNA ......................................................................... 673.2.4 In vitro DNA repair synthesis assay ............................................................................. 67
3.2.5 Immunoblot analysis of NER protein levels in mouse lung nuclear protein extracts ... 673.2.6 Analysis of binding of NER recognition proteins to damaged DNA ........................... 67
3.2.7 Data analysis ................................................................................................................. 67
Results .................................................................................................................................. 683.3

viii
3.3.1 Effect of treatment of mice with NNK on repair synthesis of mouse lung nuclear
protein extracts ....................................................................................................................... 683.3.2 Effect of in vivo treatment of mice with NNK on levels of key NER proteins in mouse
lung nuclear protein extracts .................................................................................................. 70
3.3.3 Effect of in vivo treatment of mice with NNK on key NER protein DNA binding in
mouse lung nuclear protein extracts ...................................................................................... 72
Discussion ............................................................................................................................ 743.4
Chapter 4 Investigation into the effects of in vivo sulforaphane treatment on nucleotide excision
repair activity and nucleotide excision repair recognition proteins ............................................... 77
Introduction .......................................................................................................................... 774.1
Materials and methods ......................................................................................................... 814.2
4.2.1 Animal treatment for dose response and time course of sulforaphane effects ............. 81
4.2.2 Analysis of mRNA from Nrf2-regulated genes ............................................................ 814.2.3 Preparation of cell-free whole tissue nuclear protein extracts ...................................... 824.2.4 Preparation of damaged plasmid DNA ......................................................................... 82
4.2.5 In vitro DNA repair synthesis assay ............................................................................. 824.2.6 Immunoblot analysis of NER incision protein levels in mouse liver nuclear protein
extracts ................................................................................................................................... 824.2.7 Analysis of binding of NER recognition proteins to damaged DNA ........................... 83
4.2.8 Data analysis ................................................................................................................. 83 Results .................................................................................................................................. 834.3
4.3.1 Response of two Nrf2 regulated genes to in vivo treatment with sulforaphane ............ 83
4.3.2 Timecourse of responses of two Nrf2 regulated genes to 100 mg/kg of sulforaphane . 854.3.3 Effect of in vivo treatment of mice with sulforaphane on repair synthesis activity in
mouse liver and lung nuclear protein extracts ....................................................................... 87
4.3.4 Effects of in vivo treatment of mice with sulforaphane on levels of NER proteins in
mouse liver nuclear protein extracts ...................................................................................... 89
4.3.5 Effect of in vivo treatment of mice with sulforaphane on key NER protein DNA
binding in mouse liver nuclear protein extracts ..................................................................... 91
Discussion ............................................................................................................................ 934.4
Chapter 5 General Discussion ........................................................................................................ 97
Overall conclusions .............................................................................................................. 975.1
Future directions .................................................................................................................. 995.2

ix
5.2.1 Post-translational modifications of NER proteins as a basis for observed changes in
GG-NER activity ................................................................................................................... 995.2.1.1 XPC binding to damaged DNA ........................................................................... 101
5.2.1.2 XPA binding to damaged DNA ........................................................................... 101
5.2.1.3 TFIIH binding and activity .................................................................................. 1025.2.2 Effects of sulforaphane and NNK on other NER proteins .......................................... 103
5.2.2.1 RPA ...................................................................................................................... 103
5.2.2.2 PCNA ................................................................................................................... 1045.2.2.3 Post-translational modifications of p53 ............................................................... 105
5.2.3 Protein adducts ............................................................................................................ 106
5.2.4 The effects of sulforaphane are mediated through HDAC inhibition and Nrf2 activation
.............................................................................................................................................. 107
5.2.5 The effects of cigarette smoking on NER activity in humans .................................... 107References ....................................................................................... Error! Bookmark not defined.Appendix A The effect of DMSO on nucleotide excision repair activity in A/J mouse lung and
liver .............................................................................................................................................. 131

x
List of Figures
Figure 1.1 Simplified mechanism of global genome nucleotide excision repair. Adapted from Li
et al. (2015b) and Marteijn et al. (2015). ......................................................................................... 7
Figure 1.2 Structures of the three most common tobacco-specific nitrosamines formed by the
nitrosation of nicotine. Abbreviations in text. .............................................................................. 17
Figure 1.3 Pathways of NNK metabolism to reactive metabolites that * methylate,☆
pyridyloxobutylate or ★ pyridylhydroxybutylate (© Brown, 2008). ........................................... 19
Figure 1.4 The hydrolytic conversion of glucoraphanin to sulforaphane by myrosinase. ............. 28
Figure 1.5 Model of Nrf2 activation mechanism. In the absence of stressors, Nrf2 is bound to
dimeric Keap1 and serves as a substrate for ubiquitin ligase to target Nrf2 for ubiquitination and
proteasomal degradation. Activation occurs via oxidant or electrophilic reaction with cysteine
residues on Keap1, leading to stabiliziation of Nrf2 through an unclear mechanism (reviewed by
Baird & Dinkova-Kostova, 2011) and eventual translocation into the nucleus where Nrf2
dimerizes with small Mafs and activates ARE-dependent transcription of several cytoprotective
enzymes. NAD(P)H:quinone oxidoreductase 1 (NQO1), phosphoglycerate dehydrogenase
(PGD), glutamate-cysteine ligase catalytic subunit (GCLC), heme oxygenase 1 (HO-1), ferritin
light chain (FTL). ........................................................................................................................... 34
Figure 2.1. Effects of in vivo NNK or saline treatment on in vitro DNA repair activity of mouse
liver extracts towards NNKOAc-induced pyridyloxobutyl DNA damage. Extracts were prepared
from mice treated with either 10 µmol NNK or saline and killed 2, 4, 12, 24 or 72 h post-dosing.
Data are presented as mean + SD. * significantly different from control (p<0.05; n=4 pools of 4
mouse livers each.) ......................................................................................................................... 52
Figure 2.2 a) Relative density of each key NER protein in mouse liver nuclear protein extracts
isolated from saline and NNK-treated mice 12 h post-dosing, relative to a constant internal
standard extract. Data are presented as mean ± SD. b) Representative immunoblots of the

xi
relative levels of each key NER protein in mouse liver extracts. S= nuclear protein extracts from
saline treated mice, N=nuclear protein extracts from NNK treated mice; n=4 pools of 4 mouse
livers each. ..................................................................................................................................... 54
Figure 2.3 a) Relative density of each key NER protein in mouse liver nuclear protein extracts
isolated from saline and NNK-treated mice 24 h post-dosing, relative to a constant internal
standard extract. Data are presented as ean + SD. b) Representative immunoblots of the relative
levels of each key NER protein in mouse liver extracts. S= nuclear protein extracts from saline
treated mice, N=nuclear protein extracts from NNK treated mice; n=4 pools of 4 mouse livers
each. ............................................................................................................................................... 55
Figure 2.4 Effects of in vivo NNK treatment on in vitro NER protein binding to NNKOAc-
induced pyridyloxobutyl DNA damage 12h post treatment. Data are presented as mean + SD. *
Significantly different from control (p<0.05; n=4 pools of 4 mouse livers each.) ........................ 57
Figure 2.5 Effects of in vivo NNK treatment on in vitro NER protein binding to NNKOAc-
induced pyridyloxobutyl DNA damage 24h post treatment. Data are presented as mean + SD. *
Significantly different from control (p<0.05; n=4 pools of 4 mouse livers each.) ........................ 58
Figure 3.1 Effects of in vivo NNK or saline treatment on in vitro DNA repair activity of A/J
mouse lung extracts towards NNKOAc-induced pyridyloxobutyl DNA damage. Extracts were
prepared from mice treated with either sterile saline or 10 µmol NNK and sacrificed 24h post-
dosing. Data are presented as mean + SD. (*) Significantly different from control (n=4 pools of 4
mice per pool, p<0.05). .................................................................................................................. 69
Figure 3.2 a) Relative density of each key NER protein in A/J mouse lung nuclear protein extracts
isolated from saline or NNK-treated mice 24h post-dosing, relative to a constant internal standard
extract. Data are presented as the mean + SD presented. b) Representative immunoblots of the
relative levels of each key NER protein in mouse lung extracts. S= nuclear protein extracts from
saline treated mice, N=nuclear protein extracts from NNK treated mice; ; n=4 pools of lungs of 4
mice per pool. ................................................................................................................................. 71
Figure 3.3 Effects of in vivo NNK treatment on in vitro NER protein binding to NNKOAc-
derived pyridyloxobutyl DNA damage. The data are presented as mean + SD. (*) Significantly

xii
different from control (XPA n=5 pools of lungs of 4 mice per pool; XPC and XPB n=6 pools of
lungs of 4 mice per pool, p<0.05). ................................................................................................. 73
Figure 4.1 mRNA levels for dose-response relationship of two Nrf2 regulated genes relative to
control, GCLC and HO-1; 3 h after treatment with sulforaphane (SF; n=4). The data are
presented as mean + SD. (*) Significantly different from control (p<0.05). ................................. 84
Figure 4.2 Timecourse of effects for two Nrf2 regulated genes HO-1 and GCLC in lung and liver
after treatment with 100mg/kg of sulforaphane (SF) or corn oil (n=4). The data are presented as
mean + SD. (*) Significantly different from control (p<0.05). ..................................................... 86
Figure 4.3 Effects of in vivo sulforaphane treatment on in vitro DNA repair activity of mouse
liver and lung nuclear protein extracts towards NNKOAc-induced pyridyloxobutyl DNA damage.
Extracts were prepared from mice treated with either 100mg/kg sulforaphane or corn oil and
killed 6 h or 12 h post-dosing (n=4 pools of lungs of 4 mice per pool or n=4 livers). Data are
presented as mean + SD. (*) Significantly different from control (p<0.05). ................................ 88
Figure 4.4 a) Relative density of each NER protein in mouse liver extracts isolated from corn oil
or sulforaphane-treated mice 12 h post-dosing, relative to a constant internal standard extract
(CTL). The data are presented as mean + SD. b) Representative immunoblots of the relative
levels of each key NER protein in mouse liver extracts. S= nuclear protein extracts from
sulforaphane treated mice, C=nuclear protein extracts from corn oil treated mice; all experiments
n=4. ................................................................................................................................................ 90
Figure 4.5 Effect of in vivo sulforaphane treatment on in vitro NER protein binding to
pyridyloxobutyl-adducted DNA. All data are presented as mean + SD. (all n=4, p>0.05) .......... 92
Figure A.1 Effects of in vivo DMSO treatment on in vitro DNA repair activity of mouse lung or
liver extracts towards NNKOAc-induced pyridyloxobutyl DNA damage. Extracts were prepared
from mice treated with either 0.1mL sterile saline or 0.04mL DMSO and killed 2 h post-dosing.
Data are presented as mean + SD. (*) Significantly different from control (n=4 pools of liver or
lungs of 4 mice per pool, p<0.05). ............................................................................................... 134

xiii

xiv
List of Abbreviations
AGT O6-alkylguanine-DNA alkyltransferases
ARE antioxidant response element
ATP adenosine triphosphate
ATR ataxia-telangiectasia mutated and Rad3-related
BAX bcl-2-like protein 4
BER base excision repair
°C degree(s) Celsius
CAK cyclin-dependent kinase-activating kinase
cDNA complementary DNA
CYP cytochrome p450
dATP deoxyadenosine triphosphate
dCTP deoxycytidine triphosphate
DDR direct damage reversal
dGTP deoxyguanosine triphosphate
DNA deoxyribonucleic acid
dsDNA double-stranded DNA
DTT dithiothreitol
dTTP deoxythymidine triphosphate
EDTA ethylenediaminetetraacetic acid
EGTA ethylene glycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid
EpRE electrophile response elements
et al. et alia (and others)
FTL ferritin light chain
g gram(s)

xv
GAPDH glyceraldehyde 3-phosphate dehydrogenase
GCLC glutamate-cysteine ligase catalytic subunit
GG-NER global genome nucleotide excision repair
GSH glutathione
GST glutathione-S-transferase
h hour(s)
HDAC histone deacetylase
HEPES-KOH 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid-potassium hydroxide
HO-1 heme oxygenase 1
HPB 4-hydroxy-1-(3-pyridyl)-1-butanone
HPLC high performance liquid chromatography
HRP horseradish peroxidase
i.e. id est (that is)
in vitro outside the living body
in vivo inside the living body
i.p intraperitoneal injection
ITC isothiocyanate
KEAP1 kelch-like ECH-associated protein 1
L litres
M molar
mRNA messenger RNA
mM millimolar
mg milligram
mL millilitre(s)
µCi microCurie(s)

xvi
µg microgram
µmol micromoles
µL microlitres
NAB 1-nitrosoanabasine
NAT N’-nitrosoanatabine
NER nucleotide excision repair
ng nanograms
NNAL 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol
NNK 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
NNKOAc 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone
NNN N’-nitrosonornicotine
NQO1 NAD(P)H:quinone oxidoreductase 1
Nrf2 NF-E2-related factor 2
p53 tumour suppressor protein p53
PAH polycyclic aromatic hydrocarbons
PBS phosphate-buffered saline
PCR polymerase chain reaction
PCNA proliferating cell nuclear antigen
PGD phosphoglycerate dehydrogenase
PhIP 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine
PheT r-1,t-2,3,c-4-tetrahydroxy-1,2,3,4-tetrahydrophenanthrene
POB pyridyloxobutyl
PVDF polyvinylidene fluoride
RNA ribonucleic acid
ROS reactive oxygen species

xvii
RPA replication protein A
SD standard deviation
SDS sodium dodecyl sulfate
SDS-PAGE sodium dodecyl sulfate polyacrylamide gel electrophoresis
SF sulforaphane
SF-GSH SF-glutathione
SF-NAC SF-N-acetylcysteine
SF-Cys SF-cysteine
SIRT1 sirtuin 1
ssDNA single stranded DNA
SUMO small ubiquitin-like modifiers
TC-NER transcription coupled nucleotide excision repair
TFIIH transcription factor II human
TGF-β1 Transforming growth factor β-1
TSNA tobacco specific nitrosamines
U enzymatic unit(s)
UV ultra-violet
UV-DDB UV-damaged DNA-binding protein
V volt(s)
XP xeroderma pigmentosum
XPA xeroderma pigmentosum class A protein
XPB xeroderma pigmentosum class B protein
XPC xeroderma pigmentosum class C protein- human homologue of RAD23B
XPD xeroderma pigmentosum class D protein

xviii
XPF-ERCC1 xeroderma pigmentosum class F protein- excision repair cross-complementing
repair class 1 protein
XPG xeroderma pigmentosum class G protein

1
Chapter 1
General Introduction
Statement of the research problem 1.1
Cancer is a major cause of morbidity and mortality worldwide. In Canada, cancer was
responsible for 30.2% of all deaths in 2012, with an average annual per patient cost of care
estimated at over $39,000 (Boyle & Levin, 2009). These direct financial costs represent only a
small portion of the impact of cancer on Canadian society. This class of diseases decreases
quality of life for patients and their families, places emotional and financial burdens on the
families of those afflicted with the diseases (e.g. loss of income, medical expenditures), and
represents a major demand on the medical system. Indirect and direct costs accrue in spite of the
application of contemporary surgical, radiological and chemotherapeutic practices. Clearly, it is
essential that biomedical research characterizes the mechanisms involved in cancer genesis and
progression in order to develop novel strategies to decrease morbidity and mortality associated
with this class of diseases.
Generally, cancer is characterized by unrestricted cell growth through the loss of
regulatory mechanisms that manage homeostasis and normal growth of cells. This transformation
to an abnormal, cancerous cell is dependent on the accumulation of a number of genetic and
epigenetic changes. Genomic integrity is constantly under threat from both exogenous and
endogenous chemicals and processes. Environmental agents and reactive chemicals generated by
cellular metabolic processes can attack DNA, causing various types of damage, including bulky
adducts and single-nucleotide alterations. Persistence of DNA damage can lead to mutations that
can be replicated and these alterations can change gene function or regulation of gene expression

2
and possibly lead to progression to a carcinogenic phenotype. DNA repair processes exist in
order to ensure genomic stability through excision or direct repair of damaged sequences.
Conversion of environmental carcinogens into their DNA-damaging forms (i.e. metabolic
activation or bioactivation) and the ensuing DNA damage have been the subjects of considerable
investigation, but the effects of environmental carcinogens on the DNA repair pathways have
only more recently been studied in detail. 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone
(NNK) is one of the most potent carcinogens tested in experimental animals and is present in
large quantities in burned and unburned tobacco. Once in the body, NNK is bioactivated into
several highly reactive DNA-binding species that form DNA adducts. Genomic stability after
genetic insult such as DNA adduction is achieved through several forms of DNA repair: direct
reversal, excision repair, post-replication repair and recombination repair. Nucleotide excision
repair (NER) and base-excision repair (BER) pathways respond to NNK-induced DNA damage.
Of particular relevance to NNK-induced DNA damage is NER, which recognizes and removes
bulky, helix-distorting genetic damage, and is modulated after treatment of experimental animals
with NNK. NER comprises three overlapping stages: recognition, incision/excision and gap
filling. Recently, regulation of these three stages has been shown to be complex and multiple
compounds appear to affect the regulation of their activities. Particular focus has been on ‘early
phase’ NER, comprising the recognition and early incision stages. However, the mechanisms of
NNK-induced NER activity changes are still not understood.
The development of novel chemopreventive strategies and effective cancer treatments
that affect NER necessitates a thorough understanding of the mechanisms governing NER
activity. In the absence of a comprehensive model for NER activity, adverse outcomes can result
from treatment. For example, preconditioning a tumour cell line with a nonlethal dose of UV-
radiation enhances NER following damage induced by cisplatin treatment (Choi et al., 2015).

3
However, UV-radiation causes DNA damage on its own; therefore, the optimal approach to
enhancing NER would be to increase NER without causing toxicity including additional DNA
damage. One promising compound in this regard is sulforaphane, a phytochemical found in high
concentrations in broccoli sprouts and other cruciferous vegetables. Sulforaphane has multiple
beneficial actions including the activation and enhancement of detoxification pathways
(Tortorella et al., 2015). Sulforaphane may have an effect on NER (Abassi Joozdani et al., 2015;
Piberger et al., 2014), although no studies have investigated whether these effects occur in vivo.
This thesis investigates the effects of in vivo treatment of mice with NNK or sulforaphane
on pulmonary and hepatic NER activities.
Chemical carcinogenesis 1.2
Carcinogenesis is the genesis of cancer, whereby healthy cells transform into abnormal
cells characterized by unrestricted growth and the potential to spread to other tissues. There are
four classes of carcinogens: chemical, physical (radiation), biological, and genetic (Pitot &
Dragan, 1991). The most diverse and well-studied class of carcinogens is the chemical
carcinogens, compounds that, after exposure to living tissue, result in neoplasms, or new and
abnormal growth of tissue (for review, see Loeb & Harris, 2008). The 18th century surgeon
Percival Pott published the first link between chemical exposure and carcinogenesis when he
reported a high incidence of scrotal cancer in chimney sweeps (Oliveira et al., 2007). It took over
200 years of incremental research before the connection between the chemicals in soot and
carcinogenesis were understood, from confirmation of the role of coal tar in the experimental
induction of cancer in rabbits to identification of benzo[a]pyrene-induced adducts in DNA and
that damage-induced DNA mutations were important in understanding the mechanisms of
carcinogenesis (Loeb & Harris, 2008). It is often the case that the link between carcinogenesis
and chemical exposure is described well before the mechanism of carcinogenesis is understood.

4
In fact, many mechanisms still have not been described or have only recently been discovered
(Pitot & Dragan, 1991; Loeb & Harris, 2008).
In the most widely accepted model of carcinogenesis, cancers develop in three distinct
stages: initiation, promotion and progression (Pitot & Dragan, 1991). Initiation via a genetic
carcinogen occurs when DNA damage alters the expression of genes involved in key regulatory
pathways. In order for the damage to result in a neoplasm it must be converted to a permanent
mutation through replication and persistence of the mutated gene (Pitot & Dragan, 1991). One
study estimated 3 to 10 instances of genetic damage were necessary to allow a cell to progress to
the promotion stage (Barrett, 1993). Promotion increases the proliferation rate at the
preneoplastic foci by altering gene expression, and results in the formation of a neoplastic
growth; there is no direct genetic mutation at this stage (Pitot & Dragan, 1991; Oliveira et al.,
2007). Promotion is the only stage of carcinogenesis that is reversible. There is no direct
structural genetic mutation in this stage; rather there is an alteration in gene expression.
Progression is the last stage of carcinogenesis and is irreversible. Neoplastic cells develop a
malignant phenotype that is largely driven by a stimulation of cell growth pathways and
inhibition of cell death pathways. Progression is characterized by permanent changes to the
genome and epigenome that dysregulate cellular metastatic capability, growth rate and
invasiveness of the cell genome (Oliveira et al., 2007). Pathologists identify differences in these
parameters between healthy and cancerous cells as part of the diagnostic process.
Mechanistically, carcinogens induce either structural modification of DNA or alteration
of cell metabolism, growth or death pathways in order to initiate neoplastic development (Pitot &
Dragan, 1991). Changes to DNA structure occur most often and cause mutations by perturbing
normal copying and replication of DNA and as discussed above, these mutations can be important
for both initiation and progression (Perera et al., 1982). DNA damage can be repaired through

5
one of six DNA repair pathways, and these pathways are imperative for reduction of carcinogenic
risk.
DNA repair and nucleotide excision repair 1.3
1.3.1 DNA repair
DNA damage ranges from mismatched deoxynucleotides to single or double strand
breaks and to the addition of bulky adducts. Organisms have evolved six processes to repair
damaged DNA (Branzei & Foiani, 2008): homologous recombination, non-homologous end-
joining, mismatch repair, base excision repair (BER), direct damage reversal (DDR) and
nucleotide excision repair (NER). Damage of endogenous origin caused by metabolic and other
biochemical reactions is repaired by BER, DDR, mismatch repair and strand-break repair
pathways (De Bont, 2004; Branzei & Foiani, 2008; Bates, 2010), while single and double strand
breaks are repaired through homologous recombination or non-homologous end-joining. The
DDR and BER pathways involve the removal and repair of small single-nucleotide adducts such
as oxidized DNA bases formed by endogenous reactive oxygen species (ROS), while mistakes in
DNA replication resulting in mismatched base-pairings are repaired by mismatch repair. Some
forms of DNA damage caused by exposure to exogenous agents are repaired by BER, DDR,
mismatch repair and strand-break repair pathways. However, helix-distorting DNA lesions
caused by bulky DNA adducts derived from a number of exogenous agents and pyrimidine
dimers caused by ultra-violet (UV) irradiation, are repaired largely by NER (Branzei & Foiani,
2008). NER is a multi-step process that involves multiple proteins working in concert to excise
large bulky adducts and patch the excised region with new nucleotides. NER plays an important
role in preventing cancer and attenuating the effects of chemical carcinogens; this pathway is
described in detail below.

6
1.3.2 Nucleotide excision repair
NER progresses through three steps: recognition, wherein the lesion is recognized and
repair begins; incision/excision, wherein several proteins work in concert to cut and remove the
single DNA strand with the lesion; and gap filling, wherein DNA replication machinery fills in
the gap left by the excised strand. NER is divided into two sub-pathways, distinguished by how
the damage is recognized: global genome NER (GG-NER) and transcription coupled NER (TC-
NER). In GG-NER, damage is recognized by the complex xeroderma pigmentosum (XP) class C
protein-human homologue of RAD23B (XPC; Sugasawa et al., 1998). It is only this recognition
step that differs between the two sub-pathways; in fact, apparently the only difference between
GG-NER and TC-NER is the lack of the recognition protein XPC in TC-NER. However,
mutations repaired by GG-NER are a better predictive marker for carcinogen-induced
tumourigenesis than mutations repaired through TC-NER, suggesting GG-NER plays a more
important role in protection against carcinogenesis (Balajee & Bohr, 2000). Figure 1.1 displays
the three overlapping steps of GG-NER, which are described below.

7
Figure 1.1 Simplified mechanism of global genome nucleotide excision repair. Adapted
from Li et al. (2015b) and Marteijn et al. (2015).

8
1.3.2.1 Triple-check recognition
The rate-limiting step of GG-NER is lesion recognition, which proceeds through three
checkpoints, as discussed by Marteijn et al. (2015). The recognition protein XPC binds to DNA
with helix-distorting lesions or thermodynamically destabilized sites such as base-base
mismatches. XPC can often bind indiscriminately leading to false positives, making additional
recognition checkpoints important (Li et al., 2015b; Sugasawa et al., 1998). Additionally, XPC
poorly recognizes UV-induced cyclobutane pyrimidine dimers, and an additional protein, UV-
damaged DNA-binding protein (UV-DDB; also known as XPE) binds to these adducts first and
facilitates XPC loading (Sugasawa et al., 2001; Sugasawa et al., 2005). The final two recognition
checks are transcription factor II H (TFIIH) and XP class A protein (XPA). TFIIH is a multi-
subunit complex comprised of a three-subunit cyclin-dependent kinase (CDK)-activating kinase
(CAK) module and Core7, a seven subunit complex made up of two ATP-dependent DNA
helicases (3’ to 5’ XP class B protein (XPB) and 5’ to 3’ XP class D protein (XPD)) p62, p52,
p44, p34 and p8 (Park et al., 1995; Li et al., 1998; Li et al., 2015b; Habraken et al., 1996;
Bradsher et al., 2000). DNA bubble-bound XPC recruits TFIIH, which then scans for lesions
through bi-directional helicase strand threading of XPB and XPD in which TFIIH moves along
the DNA strand with XPB scanning the 3’-5’ strand and XPD scanning the 5’-3’ strand (Li et al.,
2015b). XPA binds altered nucleotides in a single-strand DNA (ssDNA) context, confirming
damage, but also is integral to stimulating the release of the CAK subcomplex from TFIIH which
inhibits the helicase function (Camenisch et al., 2006; Coin et al., 2008; Li et al., 2015b). Thus,
the triple check system proceeds via: (1) sensing of base-pairing disruptions by XPC; (2) TFIIH-
mediated, XPA-accelerated strand threading for translocation-inhibiting injuries; and (3)
recognition of chemically altered nucleotide by XPA (Li et al., 2015b; Marteijn et al., 2015;
Sugasawa et al., 1998). TC-NER is initiated when a lesion blocks RNA polymerase II during

9
elongation (Fousteri & Mullenders, 2008; Hanawalt & Spivak, 2008), TFIIH and XPA are
recruited to the site of damage as above, and initiate the incision/excision step (Fousteri &
Mullenders, 2008; Damsma et al., 2007; Sarker et al., 2005). In GG-NER progression into the
incision/excision phase is marked by the binding of replication protein A (RPA) and the
uncoupling of XPC.
1.3.2.2 Incision/excision
At the beginning of the incision/excision phase, the helicases XPB and XPD have halted
at the lesion and their action has opened up the area around the lesion forming a
‘bubble’(Bradsher et al., 2000; Evans et al., 1997). Herein, XPA and RPA act as scaffolds,
keeping the complementary strands from re-annealing, protecting ssDNA from degradation and
act to recruit the other necessary proteins (Missura et al., 2001; de Laat et al., 1998; Park et al.,
1995; Shivji et al., 1995; Rademakers et al., 2003; Yokoi, 2000). The release of XPC allows for
the loading of XP class G protein (XPG) followed by XP class F protein- excision repair cross-
complementing repair class 1 protein (XPF-ERCC1) to the lesion-carrying strand (Araújo et al.,
2001; O’Donovan et al., 1994; Riedl et al., 2003). These two proteins are nucleases that incise
the damaged strand; XPG cuts at the 3’ end of the lesion strand, and XPF-ERCC1 attaches to and
cuts at the 5’ end of the strand; the double cut allows for displacement and excision of the
damaged strand (Araújo et al., 2001; O’Donovan et al., 1994). RPA continues to protect the
undamaged ssDNA from degradation, and the damaged strand is released by the helicases
(Matsunaga et al., 1995; Moggs et al., 1996; Mu et al., 1996; Constantinou et al., 1999;
O’Donovan et al., 1994).

10
1.3.2.3 Gap filling
The final step of NER, gap filling, begins before XPF-ERCC1 has excised the damaged
strand; DNA polymerase δ or ε attaches to the undamaged strand and begins replication with the
help of proliferating cell nuclear antigen (PCNA), and the newly synthesized patch of DNA is
ligated to preexisting DNA through DNA ligase I (Shivji et al., 1995; Moser et al., 2007; Ogi &
Lehmann, 2006).
1.3.3 Changes in NER activity
Most of the NER repair proteins are named for the rare genetic disorder they were
discovered in, xeroderma pigmentosum (XP). XP is typified by an inability to repair DNA
damage caused by UV light, resulting in severe sunburn when exposed to only small amounts of
sunlight, and often followed by skin malignancies at a very young age. There are several types of
XP; the most common types are attributable to a mutation in one of seven necessary NER genes:
XPC, XPA, XPB, XPD, ERCC4 (XPF), RAD2/ERCC5 (XPG) or a gene required to replicate
DNA with unrepaired damage POLH (XPV; DNA polymerase eta). The disease can have various
degrees of severity, dependent on the mutation type and the protein mutated (Lehmann et al.,
2011). XPC and XPE are only required for GG-NER and not TC-NER, so individuals who are
deficient in one of these two genes do not have as strong a sunburn response as individuals with
another polymorphism, likely due to the activity of TC-NER (Lehmann et al., 2011). The effects
shown by XP patients are exemplary of the importance of each of these NER proteins, and
inhibition of each can have effects on NER activity.
There are examples of XP-like phenotypes in cells as well. Testicular germ cell tumours
are especially susceptible to cisplatin treatment, due to a significant reduction in the levels of
XPA protein (Köberle et al., 1999). In counterpoint, several cancer cell types have developed
resistance to DNA-damaging chemotherapeutics associated with upregulation of certain NER

11
proteins including upregulation of DDB2 and XPC in malignant melanoma cells (Barckhausen et
al., 2014) and XPF- ERCC1 upregulation in ovarian cancer cells (Reed, 1998). Research has
shown there are many ways NER activity can be affected, but in each case the alterations are
attributable to effects on a single protein involved in the NER pathway.
1.3.3.1 Rate limiting NER proteins
XPC was previously identified as the rate-limiting protein of NER activity, due to its
importance in lesion recognition; however the binding affinity of XPC for lesions does not always
fully explain the difference in excision activity for cellular extracts (Lee et al., 2014; Li et al.,
2015b). Also, an overabundance of XPC protein is actually detrimental to cell viability, likely
due to false-positive binding (Aboussekhra et al., 1995). Recent understanding of the lesion
recognition process discussed above (“triple check recognition”) may partially explain these
phenomenon, with XPC functioning as a “blunt tool”, binding to many false-positives sites, with
TFIIH and XPA acting as the final confirmation for NER to proceed (Li et al., 2015b; Marteijn et
al., 2015). These rate-limiting proteins are important, as it has been shown that exposure to
genotoxic carcinogens can cause significant changes in NER activity through changes to specific
proteins.
Changes to NER activity appear to occur through the change of NER protein levels in the
nucleus, through post-translational modifications that change NER protein binding to damaged
DNA and through mutations to genes encoding for the NER proteins (Coin et al., 2004; Fan &
Luo, 2010; Kang et al., 2011; Lehmann et al., 2011; Li et al., 2015b; Li et al., 2011b; van Cuijk
et al., 2015). Defective XP protein functioning causes a cancer-prone phenotype, with increased
DNA mutation loads and tumour incidences (Lehmann et al., 2011). The causes of these changes
are not necessarily mutually exclusive, as can be observed in the circadian rhythm of XPA, which
appears to be caused both by changes to NER protein levels and post-translational modifications

12
(Kang et al., 2010b; Kang et al., 2011). As discussed in subsection 1.3.2, the protein XPA
provides support and additional confirmation in lesion recognition and structural support. At
homeostasis, XPA protein level exhibits circadian rhythmicity, achieved by transcriptional
control and degradation via post-translational modification by HERC2 E3 ubiquitin ligase (Kang
et al., 2010b; Kang et al., 2011). In addition to control of protein level in the nucleus,
protein/protein interactions of XPA involve both phosphorylation by the checkpoint kinase
ataxia-telangiectasia mutated and Rad3-related (ATR) and deacetylation by SIRT1 which control
binding activity of XPA to damaged DNA (Fan & Luo, 2010; Wu et al., 2006). Together, these
lead to diurnal variation in overall NER activity (Kang et al., 2010b; Kang et al., 2011).
1.3.3.2 p53 and NER
In response to genotoxic stresses, tumour suppressor protein p53 (p53) has multiple
effects that serve to enhance cancer prevention; these include promotion of apoptosis, cell cycle
arrest, and DNA repair activity increases (Seo & Jung, 2004). Although p53 is not essential for
NER when replicating the pathway in vitro using purified protein, there is still some unclear link
between NER and p53 (Aboussekhra et al., 1995; Adimoolam & Ford, 2003). p53 appears to
affect both TC-NER and GG-NER in several cell types (Dregoesc et al., 2007); however, Li-
Fraumani human fibroblasts with decreased p53 function are deficient in GG-NER with no effect
observed in TC-NER (Ford & Hanawalt, 1995). One theory is that p53 stimulates chromatin
decondensation prior to lesion recognition, but the most likely role p53 plays within NER is
regulation of lesion recognition (Rubbi & Milner, 2003; Adimoolam & Ford, 2003).
After UV-induced damage, p53 is translocated from the cytosol to the nucleus, where it
increases the expression of genes integral to NER (DDB2, XPC and XPA; Chang et al., 2008;
Hwang et al., 1999; Barckhausen et al., 2014; Liu et al., 2012). p53 is also involved in the
translocation of XPA into the nucleus through the ATR kinase activity in vitro, and this action

13
may explain why both TC-NER and GG-NER are increased by p53 induction (Li et al., 2011a).
p53 can also bind directly to damaged DNA (Chang et al., 2008) and p53 enhances repair of UV-
induced damage, probably by direct recruitment of TFIIH factors XPB and XPD. Deletion of the
C-terminus of p53 leads to decreased recruitment of TFIIH for repair of UV-induced adducts
(Léveillard et al., 1996; Chang et al., 2008; Hoffmann et al., 1996). Additionally, p53 is
apparently necessary for repair of adducts of the tobacco carcinogen metabolites benzo[a]pyrene
diol-epoxide and benzo(g)chrysene diol-epoxide, since these adducts persisted in p53 null human
fibroblast cells (Lloyd & Hanawalt, 2000; Wani et al., 2002). Also, deficient p53 significantly
decreases NER of UV-induced DNA damage and benzo[a]pyrene-induced DNA adducts (Zhu et
al., 2000; Ikehata et al., 2010; Wani et al., 2002). However, there appear to be some
compensatory mechanisms that are yet to be understood, as DNA damage responses appear to be
mechanistically different between p53 wildtype and deficient cells (Li et al., 2011b). Overall,
p53 is integral to the response of cells to injury by exogenous chemicals, and in vitro it appears to
have a role in NER but there is no universal understanding of this role.
1.3.3.3 Conditions that increase NER activity.
DNA-damage induced changes in NER activity were first observed in cell extracts which
gain a period of increased NER activity after treatment with UV light (Protić et al., 1988). This
increase is mediated by Sirt1 deacetylation and ATR-p53 import of XPA into the nucleus (Fan &
Luo, 2010; Ming et al., 2010; Donninger et al., 2015; Choi et al., 2015). Additionally, XPC and
DDB2 may induce phosphorylation of XPA through recruitment of ATR kinase to the damaged
site (Ray et al., 2013). This phosphorylation of XPA also appears to be UV-induced, and
inhibiting XPA phosphorylation decreases cell viability after UV-damage (Wu et al., 2006). A
low level dose of UV light has even been used for chemoprevention against cisplatin exposure in
lung cancer cells (Choi et al., 2015). Although not fully understood, it appears p53 is important

14
for the increase in NER activity after exposure to several carcinogens. Mulder et al. (2014)
showed that mouse liver p53 deficiency resulted in attenuation of the NER increased activity
observed after treatment with the carcinogen aflatoxin B1 (Bedard et al., 2005). Additionally, it
is possible that p53 is responsible for the chemopreventive effects selenium compounds have
against colon and mammary cancers in rodent models (Fischer et al., 2006; Seo et al., 2002). In
human and mouse fibroblasts, selenium in the form of selenomethionine significantly induces
NER, which likely involves interaction with p53 induction (Seo et al., 2002; Fischer et al., 2006).
The nicotine-derived carcinogen NNK also significantly increases NER activity specifically in
the livers of A/J mice (Brown & Massey, 2009), and its effects are one of the topics of this thesis.
1.3.3.4 Conditions that decrease NER activity
Decreases in NER activity are observed after treatment with certain chemicals. In human
skin fibroblasts, arsenic significantly decreases the expression of XPC and XPE, leading to a
significant decrease in repair of 6-4 photoproducts, which are bulky DNA adducts induced by
UVC exposure (Nollen et al., 2009). In human alveolar epithelial cells, hypochlorous acid, a
byproduct of the inflammatory response, also decreases the transcription of XPC, leading to a
significant decrease in NER (Güngör et al., 2007). Cyclosporin A causes a significant decrease in
XPA and XPG expression, leading to a significant decrease in NER in human fibroblasts
(Kuschal et al., 2011). However, some of the causes of NER activity decreases are still not all
understood, including the inhibition of NER by the plant toxin arecoline in HEp-2 squamous
carcinoma cells, and particulate matter in human lung adenocarcinoma cells (Mehta et al., 2008;
Huang et al., 2016). Several metals including nickel (in human fibroblasts), cadmium and
chromium (in chinese hamster ovary cells) cause significant decreases in NER (Hu et al., 2004b;
Fatur et al., 2003; Hu et al., 2004c). The mechanisms of the effects of these metals on NER are
not yet understood, but it appears that they inhibit NER in the lesion recognition-early incision

15
step. The nicotine-derived carcinogen NNK also significantly decreases NER activity
specifically in the lungs of A/J mice (Brown & Massey, 2009), and its effects are one of the
topics of this thesis.
NNK 1.4
1.4.1 Occurrence and source of NNK
According to the World Health Organization, tobacco kills 50% of its users,
approximately 6 million people a year worldwide; 5 million users and 1 million due to second
hand smoke inhalation (Organization, 2012). Tobacco use is declining in most industrialized
countries, but overall consumption is increasing worldwide due in part to population growth and
economic development (Jatoi et al., 2009). Tobacco products contain a diverse array of
chemicals, including nicotine and several carcinogens. These carcinogens are derived from
various chemicals including polycyclic aromatic hydrocarbons (PAH), tobacco-specific
nitrosamines (TSNAs), aromatic amines, heterocyclic aromatic amines, aldehydes, aza-arenes and
other organic and inorganic compounds (Hecht, 1999). Nicotine is the addictive component of
tobacco and tobacco smoke; it is not a complete carcinogen, but it is known to activate various
signaling pathways related to tumour promotion (Hukkanen et al., 2005; Warren & Singh, 2013).
Nicotine also undergoes chemical conversions into carcinogenic TSNAs such as NNK, 4-
(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL), iso-NNAL, 1-nitrosoanabasine (NAB), 4-
(methylnitrosamino)-4-(3-pyridyl)butyric acid, N’-nitrosonornicotine (NNN) and N’-
nitrosoanatabine (NAT). TSNAs are formed through the nitrosation of nicotine and related
tobacco alkaloids. Nitrosation reactions occur by replacement of N-H with N-N=O in the case of
secondary amines or via oxidative cleavage of carbon-nitrogen bonds of tertiary amines (Figure
1.2). Of the TSNAs, NNN, NNK and NAT are found in the highest quantities in both burned and

16
unburned tobacco (Appleton et al., 2013; Borgerding et al., 2012; Counts et al., 2004; Stanfill et
al., 2011). The complexity of tobacco smoke prevents definitive assignment of cause and effect,
but NNK, NNAL and NNN appear to play a significant role in cancer induction (Hecht et al.,
1993a; Hecht, 1998; Hecht, 1999). TSNAs are also present in significant amounts in unburned
tobacco, and NNK and NNN are likely to play a major role in oral cancer induction (Hecht et al.,
1993a; Hecht, 1998; Hecht, 1999). NNK is also present in many e-cigarette preparations, but at
much lower concentrations than in cigarettes (Farsalinos et al., 2015; Goniewicz et al., 2014).
NNK is the most potent TSNA in terms of tumourigenicity in experimental animals (Hecht et al.,
1993a; Hecht, 1998). TSNAs induce tumours in the esophagus, lung, liver, pancreas and bladder,
but the major target organ of effect is substantially affected by chemical structure and animal test
species (Hecht et al., 1993a; Hecht, 1998; Hecht, 1999). The major organ of NNK carcinogenesis
is the lung, and a single dose of NNK can cause lung and respiratory tract cancers in experimental
animals (Hecht & Hoffmann, 1989).

17
Figure 1.2 Structures of the three most common tobacco-specific nitrosamines formed by
the nitrosation of nicotine. Abbreviations in text.

18
1.4.2 Biotransformation of NNK
NNK is a procarcinogen, and it has multiple metabolic pathways as illustrated in Figure
1.3. Carbonyl reduction and α-carbon hydroxylation are the two most important reactions leading
to the ultimate carcinogenic metabolites (Hecht, 1998). Carbonyl reduction of NNK results in the
formation of NNAL, which can undergo bioactivation similar to that of NNK. NNAL can be
oxidized to NNK, and this NNK reconversion may be important in NNK carcinogenicity (Hecht,
1998). Both NNK and NNAL can be α-carbon hydroxylated, which form DNA-reactive species
that are mutagenic through methylation and pyridyloxobutylation of DNA.
NNK α-methyl carbon hydroxylation generates 4-(hydroxymethylnitrosamino)-1-(3-
pyridyl)-1-butanone (1), which decomposes to 4-oxo-4-(3-pyridyl)-1-butanediazohydroxide (2),
which can pyridyloxobutylate DNA. Reaction of 2 with water produces 4-hydroxy-1-(3-pyridyl)-
1-butanone (Keto Alcohol; III). NNAL α-methyl carbon hydroxylation generates a reactive
metabolite (3) which then produces 4-hydroxy-4-(3-pyridyl)-1-butanediazohydroxide (4) which is
further metabolized to 4-hydroxy-1-(3-pyridyl)-1-butanol (Diol, IV) ;4 can
pyridylhydroxybutylate DNA. NNK α-methylene carbon hydroxylation leads to formation of 4-
hydroxy-4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (5), which decomposes to methane
diazohydroxide (6), which can methylate DNA, and keto aldehyde which is further oxidized to
for 4-oxo-4-(3-pyridyl) butyric acid (keto acid; IV). NNAL α-methylene carbon hydroxylation
goes through similar metabolism forming methane diazohydroxide (8) and hydroxy aldehyde
which is further oxidized to 4-hydroxy-4-(3-pyridyl) butyric acid (hydroxy acid; V).
In the laboratory, DNA can be pyridyloxobutylated (POB) by a chemically activated
form of NNK, 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone (NNKOAc), and
NNKOAc has been used previously to show the importance of POB damage (Brown et al., 2008;
Peterson et al., 2001).

19
Figure 1.3 Pathways of NNK metabolism to reactive metabolites that * methylate,☆
pyridyloxobutylate or ★ pyridylhydroxybutylate (© Brown, 2008).
N
NCH3
O NO
N
NCH3
NO
OH
NN CH2OHN O
OH
N
N CH3
N OOH
OH
N
NCH3
NO
O
O
NN CH2OHN O
O
NN CH3
NO
O
OH
N
NCH3
NO
OH
O
N
NO
NOH
CHOH
CO2
N
OHO
N
HO
OCH3N NOH
CH3OH
N
OHO
O
NO OH CH3N NOH
CH3OH
N
OH
O
OH
N
N NOHOH
CHOH
CO2
N
OHOH
OH2 OH2 OH2OH2
NNK NNAL
NNK-N-Oxide NNAL-N-Oxide
Reduction
+
Keto Alcohol
+
Keto Aldehyde
Keto Acid
Lactol
+
Hydroxy Acid
+
Diol
Detoxification Detoxification
α -HydroxylationMetabolic Activation
Pathways
+ + + +
I II
III IV V VI
α α
(1)
(2) (4)
(3)(5)
(6)
(7)
(8)
* *

20
1.4.3 Detoxification of NNK
The detoxification of NNK and NNAL occurs mainly through N-oxidation, resulting in
the formation of excretable N-oxides (Figure 1.3; Hecht, 1998). In addition, NNAL is
glucuronidated to form both β-O-[4-(methylnitrosamino)-1-(3-pyridyl)-1-but-1-yl]-D-
glucosiduronic acid and β-N-[4-(methylnitrosamino)-1-(3-pyridyl)-1-but-1-yl]-D-glucosiduronic
acid, depending on whether the glucuronidation occurs at the carbinol group or the pyridine
nitrogen (Morse et al., 1990; Hecht et al., 1993b; Hecht et al., 2008). These appear to be
important detoxification products of NNK as they are readily excreted. 4-Oxo-4-(3-pyridyl)-1-
butanediazohydroxide undergoes O-glucuronidation in rats and may be another important
detoxification product of NNK.
DNA damage caused by NNK is removed via several DNA repair pathways, discussed in
subsection 1.4.6.
1.4.4 Carcinogenicity of NNK in experimental animals
In experimental animals, NNK is the most potent carcinogenic TSNA, and in rodents it
causes almost exclusively lung adenocarcinomas regardless of route of exposure (Hecht, 1998;
Hoffmann et al., 1996). NNAL, an NNK metabolite and a carcinogen itself, has approximately
30-70% the potency of NNK, and also leads largely to lung tumours when administered directly
to animals (Hecht et al., 1990; Hoffmann et al., 1996). F344 rats develop lung tumours after
exposure to NNK in drinking water, or via subcutaneous injection, gavage, oral swab or
intravenous administration (Hecht et al., 1980; Hecht, 1998; Hecht, 1999). Lung tumours are
always predominant over local (e.g. esophageal) tumours, with the nasal cavity being the second
most common site. Lung tumours are always adenomas or adenocarcinomas. Liver tumours are
observed only in high subcutaneous doses, and these are hepatocellular carcinomas and
hemangiosarcomas (Hecht, 1998; Hecht, 1999). In hamster, NNK causes lung adenomas and

21
adenocarcinomas predominantly, with some squamous and adenosquamous carcinomas (Hecht,
1998). Tracheal tumours are multiple papillomas or NNK administration obliterates the tracheal
lumen, while nasal cavity cancers and liver tumours are not observed. Both sensitive and
resistant mouse strains develop lung tumours after NNK treatment; liver and forestomach
tumours can also develop but are infrequent (Hecht, 1998; Hecht, 1999). Overall, NNK-induced
tumour incidence and multiplicity are usually lower in resistant mouse strains, and the time to
neoplasms depends on the resistance of the strain (Hecht, 1998).
A/J mice have been used extensively in studies of lung tumour induction by NNK. In the
most common assay, a single i.p. dose of 10 µmol of NNK per mouse results in 7-12 lung
adenoma or adenocarcinoma tumours per mouse after 16 weeks (Hecht & Hoffmann, 1989).
This model is championed for its ease of use, and the obvious separation of initiation, promotion
and progression. In a second assay, oral NNK administration in drinking water over 7 weeks
(total dose 44 µmol) also leads to lung tumours, but forestomach tumours also develop.
1.4.5 NNK and human cancers
The TSNA NNK is present in high concentration in cigarette and smokeless tobacco
(Appleton et al., 2013; Ashley et al., 2003; IARC Working Group on the Evaluation of
Carcinogenic Risks to Humans, 2007). In animal studies, NNK is not only the most potent
individual TSNA, but also one of the most potent individual carcinogens in tobacco. The over 60
carcinogens found in cigarette smoke make the connection difficult, but research in experimental
animals has implicated NNK as a carcinogen likely to be important in human lung cancer (Hecht,
2014; Hecht et al., 2016).
NNK levels in mainstream smoke have a direct association with total urinary NNAL
levels; as discussed in section 1.4.1, NNAL is a carcinogenic NNK metabolite (Ashley et al.,
2010). Smokers and nonsmokers exposed to secondhand tobacco smoke have NNAL in their

22
urine, and total NNAL has been widely used as a biomarker of NNK exposure (Ashley et al.,
2010; Hecht et al., 2016). In fact, NNAL is the only tobacco carcinogen biomarker consistently
elevated in nonsmokers exposed to secondhand smoke (Hecht, 2014). Several studies have
shown a statistically significant association between total NNAL level and lung cancer
(Hoffmann et al., 1996; Yuan et al., 2011; Yuan et al., 2014; Appleton et al., 2014). They
illustrate the relationship between NNK and lung cancer, and the importance of the study of
NNK-induced carcinogenesis. The first case-controlled study looked at three biomarkers of
cigarette smoking: cotinine, a metabolite of nicotine, NNAL, a biomarker of NNK exposure, and
r-1,t-2,3,c-4-tetrahydroxy-1,2,3,4-tetrahydrophenanthrene (PheT), a biomarker of PAH exposure,
using 100 randomly selected lung cancer cases and 100 controls from the Prostate, Lung,
Colorectal and Ovarian Cancer Screening Trial (Church et al., 2009). They found a significant
association between lung cancer risk and total urinary NNAL with no association between lung
cancer risk and any other tobacco constituent biomarker tested, including PAH, and volatile
organic hydrocarbons acrolein, benzene, 1,3-butadiene, crotonaldehyde and ethylene oxide (Yuan
et al., 2014). Additionally, adenocarcinoma risk was significantly associated with total urinary
NNAL, but not non-adenocarcinoma lung tumours. In larger cohort studies, total urinary NNAL
has consistently been associated with lung cancer, specifically adenocarcinoma risk. Using the
18,244 person Shanghai cohort, Stephanov et al. (2014) and Yuan et al. (2011). showed total
urinary NNAL is strongly associated with lung, but not esophageal cancers after correction for
NNN exposure again, there were no associations between lung cancer risk and all other tobacco
urinary biomarkers tested. These results are consistent with carcinogenicity studies in
experimental animals treated with NNK as discussed above, making NNAL the most important
tobacco-associated cancer risk biomarkers available (Hecht et al., 2016).

23
TSNA levels are highly variable between tobacco brands and tobacco-derived smoke
worldwide (Appleton et al., 2013; Ashley et al., 2003; 2007). In a study across 35 years, NNK in
mainstream smoke ranged from 8.7 to 868 ng/cigarette with the highest concentrations coming
from cigarettes manufactured in the 1980’s (Appleton et al., 2013). NNK levels are highest in
U.S.-branded cigarettes, when compared to all other local brands, while Canadian brands have
some of the lowest levels (Ashley et al., 2003; Hecht, 2014). The level of TSNAs in e-cigarette
replacement liquids and smoke are considerably lower than that of tobacco cigarettes (Kim &
Shin, 2013; Farsalinos et al., 2015; Goniewicz et al., 2014). One study found that NNK was
present only in trace amounts in vapour, undetectable to 28.3 ng per e-cigarette (Goniewicz et al.,
2014). Replacement of cigarette smoking with e-cigarette use may be an effective harm
reduction strategy by smokers, but the presence of TSNAs in e-cigarette vapour suggests that the
risk is not completely abrogated. TSNAs NNK, NNN, NNAL and NAB are all found in
smokeless tobacco, and are considered the most harmful toxicants present in those products
(IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, 2007; Hoffmann &
Djordjevic, 1997). There is some debate on the amounts, but since 1980, TSNA levels in
cigarettes have decreased substantially through different curing, processing and storing
procedures; however they are still found in high concentrations (Hecht, 2014; Gunduz et al.,
2016). A global survey of smokeless tobacco products found NNK in concentrations between 4.5
and 516,000 ng/g product (Stanfill et al., 2011), and NNK has historically been found in
concentrations of 7,870,000 ng/g of product (2007). Additionally, urinary NNAL levels are
higher in smokeless tobacco users than in persons using smoked tobacco.
Worldwide, incidence of adenocarcinomas has been gradually increasing in number over
a 20 year period, and it is possible that NNK plays a significant role in this increase (Devesa et
al., 2005; Houston et al., 2014). Between 2004 and 2009 in a U.S. census, adenocarcinoma rose

24
1.3% in men and 2.8% in women while overall incidence rates of lung cancer have decreased
(Houston et al., 2014). This is despite declining cigarette use in most Western countries and
shifts to filtered/low tar cigarettes worldwide. Two explanations have been proposed for these
increases, one concerning cigarette design and a second concerning NNK concentrations in
tobacco (Burns et al., 2011; Hoffmann et al., 1996). The introduction of filters and other design
features resulted in deeper inhalation by smokers and delivery of smoke constituents to the distal
lung, leading to an increase in adenocarcinomas which are peripheral lung tumours (Burns et al.,
2011; Hoffmann et al., 1997). As discussed above, NNK induces adenocarcinomas in
experimental animals and the second explanation revolves around a relative increase in NNK
exposure. There is some debate about whether the trends of NNK levels in mainstream smoke,
smokeless and cigarette tobacco have decreased worldwide (Hecht, 2014). However, the levels
of benzo[a]pyrene and other PAHs have significantly decreased since the 1950s (Counts et al.,
2004; Hoffmann et al., 1997). PAHs tend to induce squamous cell carcinomas of the lung
(Humans, 2010) which have decreased in frequency, and it is possible that the increase in
adenocarcinomas is due to an increase in the ratio of NNK to benzo[a]pyrene, as discussed by
Hecht et al. (2014).
1.4.6 DNA repair and NNK
The genotoxic effects of NNK are mediated through mutations created due to the
formation of pyridyloxobutylated and methyl DNA adducts. Methyl adducts are repaired by
alkyltransferases, enzymes that catalyze single-step reactions whereby the methyl group is
transferred to an amino acid residue on the transferase itself. Pyridyloxobutylation occurs at the
N7- and O6- positions of guanine, and the O2- position of cytosine and thymine. O6-
Pyridyloxobutylated guanine and O2-pyridyloxobutylated thymine are the most stable of these
four adducts, and make up most of the damage caused after NNK exposure (Peterson, 2010).

25
Under hydrolytic conditions, unstable POB adducts decompose, returning the DNA to its normal
state (Hecht et al., 1993a; Peterson et al., 1990). O6-Methylguanine, the most common methyl
adduct, is repaired by O6-alkyl-guanine-DNA-alkyltransferase (Peterson & Hecht, 1991). O6-
Pyridyloxobutylated guanine is primarily repaired by O6-alkyl-guanine-DNA-alkyltransferase
(AGT); however, knockdown or depletion of AGT does not appear to significantly increase cell
death in vitro or tumour incidence in vivo, meaning NER may play an important additional role
(Peterson, 2010; Urban et al., 2012). The other POB adducts are repaired primarily by NER
(Wang et al., 1997; Brown et al., 2008; Hecht et al., 1993a; Peterson & Hecht, 1991). Treatment
of A/J mice with the pyridyloxobutylating agent, NNKOAc, showed the lack of a clear
relationship between a single POB adduct and the incidence of lung tumour formation (Urban et
al., 2012). Three adducts persisted in mouse lung DNA in significant levels (O6-
pyridyloxobutylated guanine, 7-pyridyloxobutylated guanine and O2-pyridyloxobutylated
thymine), indicating multiple adducts may contribute to the tumourigenic properties of NNK
(Urban et al., 2012).
As discussed above, no single DNA damage repair pathway can deal with all of the
diverse types of DNA adducts formed after most carcinogenic exposure. Repair of the damage
caused by NNK proceeds by NER, BER and DDR, which have been discussed previously in
section 1.3.1. Without these DNA repair mechanisms, cell death or the formation of neoplasms is
inevitable. Therefore, any change in DNA repair activity will have an effect on the potential for
carcinogenesis. So, it is of note that NNK treatment of A/J mice results in a significant change
AGT and NER activities in the liver and lung (Brown & Massey, 2009; Peterson et al., 2001).
The results showed significant decreases in AGT and NER activities of lung extracts, significant
increase of NER activity in liver extracts and no effect on AGT activity in liver extracts (Brown
& Massey, 2009; Peterson et al., 2001). The increase of NER activity in the liver could be

26
explained as a homeostatic response to alleviate the damage caused by the carcinogens on DNA,
and an increase in DNA repair capacity has been observed in several situations discussed above.
However, the mechanism behind the changes to NER activity after treatment with NNK is still
inconclusive. The decrease of NER activity in the lungs is especially important, as adducts
caused by the carcinogens may persist in the lungs, thereby increasing the probability of
neoplastic growth. Again, a decrease in NER activity after treatment with a chemical has been
observed in the situations described above, but the mechanism causing NNK-mediated change in
activity has yet to be revealed.
Sulforaphane 1.5
1.5.1 Glucosinolates
Epidemiological studies in several countries have shown an apparent protective effect of
consumption of cruciferous vegetables leading to reduction of cancer risk (Higdon et al., 2007;
Royston & Tollefsbol, 2015). They are among the dominant food crops worldwide and include
such diet staples as cauliflower, Brussels sprouts, cabbage, bok choy and broccoli (Verkerk et al.,
2009). Cruciferous vegetables are high in a number of vitamins, nutrients, and phytochemicals
and are especially high in a group of sulfur-containing phytochemicals, the glucosinolates
(Verkerk et al., 2009).
Glucosinolates are responsible for the odour and spicy or bitter taste of cruciferous
vegetables. Over 100 glucosinolates have been identified in the cruciferae, many of which form
biologically active compounds after hydrolysis (Fahey et al., 2001). The glucosinolate molecule
comprises two parts: a variable side chain and a sulfur-linked β-D-glucopyranose moiety (Figure
1.4; Fahey et al., 2001). This hydrolysis is most frequently catalyzed by myrosinase, an enzyme
found in the cruciferae which is released and brought in contact with the glucosinolates when the

27
plants are cut or chewed; although there appears to be some myrosinase activity by human gut
bacteria as well (Bones & Rossiter, 2006). Myrosinase catalyzes the hydrolysis of the glucosidic
bond, yielding glucose and an unstable thiohydroxamate-o-sulfonate which undergoes
spontaneous rearrangement to form a number of possible products dependent on the original
glucosinolate side chain structure, presence of metal ions and reaction conditions such as pH
(Bones & Rossiter, 2006). At a pH of 6 to7, the major products are isothiocyanates (ITCs; Bones
& Rossiter, 2006). Over 120 ITCs have been described, but relatively few are found in the
cruciferae. Of the ITCs, one with a high concentration in broccoli sprout extract, sulforaphane,
has become a focus of interest for its anti-carcinogenic effects.

28
Figure 1.4 The hydrolytic conversion of glucoraphanin to sulforaphane by myrosinase.

29
1.5.2 Sulforaphane properties and kinetics
Significant amounts of the glucosinolate glucoraphanin, the precursor to sulforaphane,
are found in Brassicaceae, and it is found in extremely high amounts in broccoli sprouts (16.6
µmol/g in young sprouts; 1.08 µmol/g in mature broccoli; Fahey et al., 1997; Fahey et al., 2001).
Glucoraphanin is relatively stable, and hydrolysis to sulforaphane is mainly mediated by
myrosinase after mastication/blending or conversion by human microbiota in the colon (Fahey et
al., 2001). Method of preparation of cruciferous vegetables can also have a major impact on
sulforaphane formation: boiling, steaming and microwaving can inactivate myrosinase (Pérez et
al., 2014; Li et al., 2013a). Raw florets can be eaten and 60-80% of the glucosinolate content is
converted to sulforaphane-nitrile, not the ITC, due to combined effects of a non-catalytic protein
cofactor (epithiospecifer protein) and myrosinase (Juge et al., 2007; Matusheski et al., 2006).
However, mild cooking can denature the cofactor while preserving myrosinase, resulting in
almost 100% conversion of glucoraphanin into sulforaphane (Juge et al., 2007). Consequently,
several different preparations of broccoli sprouts have been developed to increase availability of
sulforaphane (Li et al., 2013a). In humans, sulforaphane is rapidly absorbed, metabolized and
excreted, with 80% of parent compound and metabolites found in the urine 12-24 h after
administration (Atwell et al., 2015; Shapiro et al., 1998; Ye et al., 2002; Hu et al., 2004a; Clarke
et al., 2011; Li et al., 2013a). In humans and mice, peak plasma concentrations are found 0.5-1 h
after consumption of sulforaphane or broccoli sprout extract (Petri et al., 2003; Ye et al., 2002; Li
et al., 2013a). However, the conversion to sulforaphane by the gut microflora can take more
time, leading to a secondary peak 2 h after consumption of a broccoli sprout extract (Li et al.,
2013a).
Sulforaphane is the most hydrophobic of all dietary ITCs and rapidly diffuses into the
intestinal epithelium. Sulforaphane then undergoes metabolism conjugation with glutathione

30
(SF-GSH), catalyzed by glutathione-S-transferase (GST) enzymes, followed by subsequent
formation of SF-N-acetylcysteine (SF-NAC) and SF-cysteine (SF-Cys). An autoradiographic
whole-body study showed sulforaphane-derived radioactivity in the GI tract, liver, kidney and
blood (Tortorella et al., 2015). In CD-1 and ICR mice, the greatest concentrations of
sulforaphane, SF-GSH, SF-Cys and SF-NAC were measured between 0.5-2 h in the liver, kidney,
lung, heart and muscle (Clarke et al., 2011; Li et al., 2013a). In F344 rats, the plasma
concentration of sulforaphane reaches its peak at 4 h after oral dosing (Hu et al., 2004a).
There is general consensus that sulforaphane can cause biologically opposite effects
depending on its concentration (Sestili & Fimognari, 2015). At low doses, it exerts
chemopreventative, indirect antioxidant and cytoprotective effects, whereas at high doses it exerts
cytotoxic and antitumour effects (Sestili & Fimognari, 2015; Zanichelli et al., 2012a; Zanichelli
et al., 2012b). After treatment with sulforaphane, gene expression profiles in mice, rats and
cancer cell lines show effects at a wide range of timepoints, with significant changes from 3-24 h
(Bhamre et al., 2009; Hu et al., 2006). In a gene activity profile studying 39,000 genes in CD-1
livers from mice treated with 90 mg/kg sulforaphane, Hu et al. (2006) found significantly
elevated gene expression at both 3 h and 12 h, with the greater number of individual gene
inductions occurring at 12 h.
1.5.3 Sulforaphane as a cancer chemopreventive, cytotoxic and cytoprotective chemical
As reviewed by Tortorella et al. (2015), sulforaphane has gained particular interest as a
cancer chemopreventative chemical, particularly with respect to inhibition of phase I enzymes
and production of DNA adducts, induction of phase II enzymes, and anti-inflammatory and anti-
oxidant activities.
Sulforaphane can inhibit phase I enzymes, significantly reducing the burden of many
procarcinogens through inhibiting the enzymes that bioactivate them. Thus, inhibition of phase I

31
enzymes is an important mechanism of blocking chemical carcinogenesis. Sulforaphane can
inhibit phase I metabolism through direct interactions with cytochrome P450 enzymes (CYP) or
by regulating transcript levels (Juge et al., 2007). Specifically, sulforaphane inhibits CYP1A1
and 2B1/2 in a concentration dependent manner in rat hepatocytes and CYP3A4 in human
hepatocytes (Mahéo et al., 1997). However, most research has focused on the effect of
sulforaphane on phase II enzymes and production of carcinogen detoxification products.
Recent evidence illustrates the protective effects of sulforaphane against a wide range of
cytotoxic and carcinogenic chemicals in several cell lines and animal models. Prior treatment
with sulforaphane and several other ITCs caused decreases in lung adenocarcinoma incidences
and malignant lung tumour multiplicity in A/J mice treated with a mixture of benzo[a]pyrene and
NNK (Conaway et al., 2005). Co-treatment of sulforaphane with 14C-labelled 2-amino-1-methyl-
6-phenylimidazo[4,5-b]pyridine (PhIP) caused a significant decrease in PhIP-generated DNA
adducts, associated with increases in phase II detoxification enzymes (Bacon et al., 2003).
Sulforaphane treatment increases total GST activity, which acts to detoxify the hepatocarcinogen
aflatoxin B1-8,9-epoxide (Gao et al., 2010). Sulforaphane inhibits acetaminophen-induced
hepatotoxicity, apparently at least partially through induction of heme oxygenase 1 (HO-1)
induced antioxidant effects (Noh et al., 2015). A similar mechanism likely reduces doxorubicin-
induced oxidative stress (Li et al., 2015a; Singh et al., 2015). In addition, sulforaphane can
protect fibroblasts from ionizing radiation (Mathew et al., 2014). UV-radiation induced skin
carcinogenesis in SKH-1 hairless mice was substantially inhibited by topical treatment with a
broccoli sprout extract containing sulforaphane (Dinkova-Kostova & Abramov, 2015). Few
clinical sulforaphane studies exist; however, pretreatment of human skin with sulforaphane
causes a significant decrease in UV radiation-induced apoptosis and a significant increase in the
antioxidant enzyme catalase (Kleszczyński et al., 2013; Knatko et al., 2015).

32
Research on sulforaphane has focussed largely on its effects after concurrent treatment
with a chemical or other damaging agents, but sulforaphane can have multiple other effects in
models of disease. For example, diabetes-induced testicular cell death and aortic damage can be
abrogated through the antioxidant effects of sulforaphane (Wu et al., 2015; Wang et al., 2014b;
Wang et al., 2014a). Additionally, sulforaphane pretreatment causes a decrease in blood brain
barrier disruption and neurological dysfunction following ischemic stroke in Sprague-Dawley rats
(Alfieri et al., 2013). Sulforaphane can also have cytotoxic and antiproliferative effects in certain
cancer cells; for example, colon cancer cell counts were significantly reduced after sulforaphane
treatment (Rajendran et al., 2013). Sulforaphane attenuates the proteolysis and mRNA
degradation of programmed cell death 4, a tumour suppressor protein in HepG2 cells (Cho et al.,
2014). Sulforaphane acts as an antiproliferative agent in mammary cancer cells, apparently by
perturbation of mitotic microtubules thereby halting cell cycle exit from metaphase (Jackson &
Singletary, 2004). Many of these actions, anti-cancer and other are mediated through
sulforaphane activation of the transcription factor NF-E2-related factor 2 (Nrf2).
1.5.4 Nrf2
Nrf2 is a transcription factor involved in regulation of several cytoprotective genes in
response to oxidative and electrophilic stresses (Itoh et al., 1997; Kwak & Kensler, 2010;
Tortorella et al., 2015). The hundreds of target genes of Nrf2 are activated through antioxidant
response elements (ARE) or electrophile response elements (EpRE) in their promoter regions
(Itoh et al., 2004). Unstressed, Nrf2 is regulated through a ubiquitin-proteasome pathway via
kelch-like ECH-associated protein 1 (Keap1) association and CR6-interacting factor 1 promoted
ubiquitin degradation (Figure 1.5;Kang et al., 2010a). Inactivated Nrf2 is held in the cytoplasm
through binding of Keap1 dimer (Itoh et al., 2004; Kang et al., 2010a). Nrf2 is released by
Keap1 in the presence of ROS or electrophiles through one of two pathways (Baird & Dinkova-

33
Kostova, 2011). In the first pathway, reactive cysteines in Keap1 dimers are modified after
reaction with electrophiles, thereby releasing the bound Nrf2 (Wakabayashi et al., 2004). In the
second pathway, secondary sensor proteins activate protein kinase signaling pathways,
phosphorylating Nrf2 causing release of Keap1 (Huang et al., 2002). After release of Keap1 and
translocation into the nucleus, Nrf2 dimerizes with multiple small Maf proteins and binds to ARE
or EpRE promoter regions, inducing target gene expression (Motohashi et al., 2004; Itoh et al.,
2004). Further control of Nrf2 effects have also been discovered: alcohol can decrease Nrf2
induction through an increase in transforming growth factor β-1 (TGF-β1) expression, which
appears to impair Nrf2-ARE signalling (Sueblinvong et al., 2014). There is also further control
of Nrf2-mediated transcription of target genes: cyclic adenosine monophosphate-response
element-binding protein can enhance transcription, whereas Nrf2 target genes can be negatively
regulated through the actions of p53 and CCAAT enhancer-binding protein alpha (Faraonio et al.,
2006; Ikeda et al., 2006; Shen et al., 2004).

34
Figure 1.5 Model of Nrf2 activation mechanism. In the absence of stressors, Nrf2 is bound
to dimeric Keap1 and serves as a substrate for ubiquitin ligase to target Nrf2 for
ubiquitination and proteasomal degradation. Activation occurs via oxidant or electrophilic
reaction with cysteine residues on Keap1, leading to stabiliziation of Nrf2 through an
unclear mechanism (reviewed by Baird & Dinkova-Kostova, 2011) and eventual
translocation into the nucleus where Nrf2 dimerizes with small Mafs and activates ARE-
dependent transcription of several cytoprotective enzymes. NAD(P)H:quinone
oxidoreductase 1 (NQO1), phosphoglycerate dehydrogenase (PGD), glutamate-cysteine
ligase catalytic subunit (GCLC), heme oxygenase 1 (HO-1), ferritin light chain (FTL).

35
1.5.5 The actions of Nrf2
Nrf2 increases the synthesis of GST and other electrophile conjugating enzymes, direct
acting antioxidants and reducing equivalents as well as the 20S proteasome catalytic core causing
the ubiquitin-independent degradation of oxidized proteins (Itoh et al., 2010; Jaramillo & Zhang,
2013; Kwak & Kensler, 2010). Creation of the Nrf2 knockout mouse allowed for an
understanding of the wide range of effects displayed in Figure 1.5 (Ramos-Gomez et al., 2001;
Ramos-Gomez et al., 2003). Microarray data has shown Nrf2 is involved in the induction of
antioxidant proteins, NADPH-generating enzymes, drug metabolizing enzymes, drug
transporters, metal binding proteins and stress response proteins as reviewed by Hayes et al.
(2010). Nrf2 is integral for protection against chemical carcinogenesis as many genotoxic
carcinogens are either directly DNA reactive or are bioactivated to electrophilic DNA-reactive
species. The importance of Nrf2 signaling in reduction of electrophilic species was first shown
with acetaminophen toxicity which, although not genotoxic, produces toxicity via formation of a
protein-binding electrophile (Chan et al., 2001; Enomoto et al., 2001). An increase in Nrf2
signalling increases the synthesis of GSH through increased expression of glutamate-cysteine
ligase catalytic subunit (GCLC) and phosphoglycerate dehydrogenase (PGD). Other detoxifying
molecules such as NAD(P)H:quinone oxidoreductase 1 (NQO1) increase the conjugation and
excretion of reactive metabolites. Ferritin light chain (FTL) is also induced and prevents
uncontrolled surges in the intracellular free concentration of the highly reactive, poorly soluble
ferric iron (Kensler et al., 2007). In addition, metallothionein, a potent antioxidant, is induced
with Nrf2 and results in the reduction of DNA damage, and can decrease diabetic neuropathy
(Chubatsu & Meneghini, 1993; Wu et al., 2015). Knockout or reduction of Nrf2 results in the
infiltration of inflammatory cells, increase in pro-inflammatory cytokines and an increase in
oxidative damage. Active Nrf2 decreases the development of aberrant crypt foci, thought to be

36
an early marker of colorectal cancer, in a model of colitis-associated colorectal cancer
(Paszkowska-Szczur et al., 2015). Oxidative stress is implicated in multiple disease states linked
to exposure of environmental toxicants. The role of Nrf2 in protecting against oxidative stress
appears to occur at multiple levels. During periods of non-stress, the effect of Nrf2 on basal
levels of reactive oxygen species (ROS) is minimal, but this effect is integral to mitochondrial
membrane potential, fatty acid oxidation, availability of substrates for respiration and ATP
synthesis (Dinkova-Kostova & Abramov, 2015). During periods of high oxidative stress, Nrf2
signalling becomes vital for the decrease of ROS within mitochondria and in the rest of cell
(Dinkova-Kostova & Abramov, 2015; Gong & Cederbaum, 2006). However, the overall action
of Nrf2 remains to be understood, as several studies have indicated results in which Nrf2 action
was associated with pro-carcinogenic activity; one study using urethane to initiate lung tumours
in mice showed resistance to tumour growth in Nrf2 deficient mice (Satoh et al., 2013).
Additionally, heterozygous Nrf2 mice treated with dimethylhydrazine had a reduced tumour
burden when compared to wildtype mice (Rajendran et al., 2015).
1.5.6 Sulforaphane mechanisms not associated with Nrf2
In addition to the downstream effects of Nrf2 induction discussed above, sulforaphane is
associated with several actions independent of Nrf2 induction including inhibition of phase I
biotransformation and acts as a histone deacetylase (HDAC) inhibitor. HDACs remove acetyl
groups on nuclear histones, leading to condensed chromatin and repressed transcription. The
action of sulforaphane as an HDAC inhibitor leads to an increase in acetylated histones and hence
overall transcriptional activation. Importantly, HDAC inhibition occurs in cells lacking Nrf2
expression, indicating an action independent of Nrf2 (Dashwood & Ho, 2007; Ho et al., 2009).
Whereas the Nrf2 effects and phase I biotransformation effects are largely due to direct

37
interaction with sulforaphane, the HDAC inhibitor effects appear to largely be mediated by SF-
NAC and SF-Cys (Myzak et al., 2006).
Sulforaphane treatment can inhibit both class I and class II HDACs, and can affect a wide
range of genes that are neutral, beneficial or detrimental for chemoprevention (Baier et al., 2014).
An increase in HDAC activity is observed in a number of cancer types, and can result in the
repression of cell cycle and apoptotic mechanisms, among others. The HDAC inhibitory activity
of sulforaphane was first reported by Myzak et al. (2004). Sulforaphane also significantly
increased acetylated H3 and H4 histones (Myzak et al., 2006). Acetylated H3 and H4 appear to
have opposing effects, and the extent to which HDAC inhibition affects transcriptional control is
still not fully understood (Gansen et al., 2015). However, research in tumour cell lines showed
that H4 acetylation is tied to the transcriptional regulation of important chemopreventive proteins
p21 and bcl-2-like protein 4 (BAX) (Myzak et al., 2006). BAX is a pro-apoptotic protein, while
the effects of p21 are variable; it has roles in cell cycle progression, motility, apoptosis and DNA
repair (Murphy et al., 2002). BAX and p21 are repressed in several tumour cells, and their
derepression after treatment with sulforaphane has added to the growing literature on its
chemopreventive and chemotherapeutic effects (Dashwood & Ho, 2007; Ho et al., 2009).
1.5.7 Sulforaphane and DNA repair
Sulforaphane can have direct and indirect effects on DNA repair. Several studies have
shown that pretreatment with sulforaphane can lead to a decrease in the effect of genotoxic
carcinogens; however the mechanisms of the beneficial effects of sulforaphane are not fully
understood. Several examples of the anti-genotoxic effect of sulforaphane have shown effects
largely due to phase II detoxification. For example, the decrease in aflatoxin B1-induced DNA
adducts with sulforaphane pretreatment appears to be largely mediated through the Nrf2-
induction of GST (Techapiesancharoenkij et al., 2015). In HepG2 human hepatoma cells, co-

38
treatment of sulforaphane with PhIP leads to a significantly reduced amount of PhIP-DNA
adducts (Bacon et al., 2003). However, treatment with sulforaphane after treatment with PhIP
does not lead to any significant reduction in PhIP-DNA adducts, leading the authors to conclude
that sulforaphane does not have a significant effect on DNA repair activities (Bacon et al., 2003).
On the other hand, benzo[a]pyrene- and 1,6-dinitropyrene-DNA adducts are significantly reduced
in human mammary epithelial cells after treatment with sulforaphane, but the reduction could not
be explained by increased detoxification (Singletary & MacDonald, 2000). Furthermore, Nrf2
inhibition caused a significant decrease in UV-B induced homologous recombination repair in
A549 cells (Jayakumar et al., 2015). Also, sulforaphane-enhanced H3 histone acetylation may
facilitate DNA repair, as relaxation of chromatin allows for binding of repair proteins (Duan &
Smerdon, 2014).
Jayakumar et al. (2015) showed that Nrf2 activation is directly involved in homologous
recombination repair, the first example of a direct action in DNA repair. p21, a downstream
target of p53 that is believed to exert a number of the actions of p53 including regulation of DNA
repair pathways, has been implicated with Nrf2 induction and stability (Chen et al., 2009). On
the other hand, p53 is directly and indirectly involved in many DNA repair pathways, and it is
known to be inversely related to the action of Nrf2 (Faraonio et al., 2006). A hypothesis for these
actions was presented by Jaramillo and Zheng, whereby strong induction of p53 inhibits Nrf2 by
an undefined mechanism, decreasing antioxidant defense and cell survival pathways; promoting
apoptosis (2013). Mild stress only weakly induces p53, but induces downstream effector proteins
including p21, stalling the cell cycle at G1/S-phase checkpoint, inducing DNA repair pathways
and the Nrf2 mediated cytoprotective response (Jaramillo & Zhang, 2013).
As of yet, there has not been a report of in vivo data on the effect of sulforaphane
treatment on NER activity. However, an in vitro experiment showed that sulforaphane

39
pretreatment caused a significant decrease in repair of (+)-anti-benzo[a]pyrene-7,8-diol-9,10-
epoxide- induced adducts in HCT 116 cells (Piberger et al., 2014). This decrease appeared to be
caused by a direct interaction between sulforaphane and repair protein XPA through a release of
the XPA zinc-finger domain. This resulted in an inactivated protein complex and a decrease in
XPA- damaged DNA binding (Piberger et al., 2014).
Research hypotheses and objectives 1.6
As discussed, genetic instability caused by persistence of bulky DNA adducts can lead to
apoptosis and carcinogenesis. Treatment with exogenous chemicals not only cause bulky DNA
adducts, but can also have significant effects on NER activity. However, the mechanisms of
these NER changes are not fully understood. In addition, the treatment with an exogenous
chemical that can increase NER activity without causing DNA damage would be important for
the modification of carcinogenic risk after exposure to a DNA damaging agent. However, no
agent that causes an increase in NER activity without concomitant DNA damage has been
discovered. In this thesis, the effects of the DNA damaging carcinogen NNK and the
chemoprotective phytochemical sulforaphane on the mechanisms of changes to NER activity are
studied.
Rationale: Studies using NNK showed that administration to a mouse resulted in a change to the
NER activity in the liver and lung (Brown & Massey, 2009). These effects were organ-
dependent; there was a decrease in lung NER activity, but an increase in liver NER activity
(Brown & Massey, 2009). It was hypothesized that the increase in NER activity protected the
liver from carcinogenesis, while the decrease in NER activity in the lung makes that organ more
susceptible to carcinogenesis; however, the mechanism(s) involved in these changes in activity
are still not understood. Also, the time course of effects on NER activity in the liver after
treatment with NNK has not been described.

40
Hypothesis 1: The effects of in vivo NNK treatment on in vitro NER activity of liver nuclear
extracts occur at times other than 24 h after treatment and are associated with alterations in levels
of specific proteins in the NER pathway.
Objective 1. To determine the effects of NNK treatment on NER activity in female A/J mouse
liver at 2, 4, 12, 24 and 72 h post treatment.
Objective 2. To determine the effects of NNK treatment on nuclear levels of the recognition and
early-incision NER proteins XPC, XPA, XPB and p53 as well as on binding activity to damaged
DNA in female A/J mouse liver.
Hypothesis 2: Decreased NER activity in lung from NNK-treated female A/J mice is associated
with effects on specific proteins in the NER pathway.
Objective 1. To determine the effects of NNK treatment on nuclear protein levels of the
recognition and early-incision NER proteins XPC, XPA, XPB and p53 as well as on binding
activity to damaged DNA in female A/J mouse lung.
Rationale: Sulforaphane treatment leads to activation of multiple genes involved in
chemopreventative pathways. Pre-treatment with sulforaphane before several types of DNA-
damaging carcinogens also significantly decreases the amount of DNA adducts in a sample and
sulforaphane causes significant changes to proteins associated with NER. However, recent work
indicates that in vitro exposure of human colon cancer (HCT 116) cells to sulforaphane causes a
decrease in NER activity through inhibition of XPA binding activity (Piberger et al., 2014). NER
is integral to the chemoprotective response to genotoxic chemicals, and sulforaphane is a current
candidate for use as a chemoprotective agent; thus it is important to understand the effect of
sulforaphane on NER.
Hypothesis 3: Sulforaphane treatment causes changes to NER activity in female CD-1 mouse
liver and lung that are through changes to specific NER proteins.

41
Objective 1. To determine a dose of sulforaphane that affects expression of Nrf-2 regulated genes
in female CD-1 mouse liver and lung.
Objective 2. To determine a time point that affects Nrf-2 regulated genes HO-1 and GCLC in
female CD-1 mouse liver and lung.
Objective 3. To determine whether NER is affected at this time point and dose of sulforaphane.
Objective 4. To determine the effects of sulforaphane treatment on nuclear levels of the
recognition and early-incision NER proteins XPC, XPA, XPB and p53 as well as on binding
activity to damaged DNA.

42
Chapter 2
Time course of effects of NNK on nucleotide excision repair activity on
A/J mouse liver and possible mechanisms of change
After in vivo treatment with the tobacco specific nitrosamine 4-(methylnitrosamino)-1-(3-
pyridyl)-1-butanone (NNK), mouse liver has higher levels of DNA adducts than does mouse
lung, but mouse lung is the major target of NNK-induced carcinogenesis. One potential
explanation for the lack of liver carcinogenesis is an adaptive increase in nucleotide excision
repair (NER) activity in mouse liver. However, the timecourse and mechanisms of this increase
are not well understood. A 46% decrease in NER activity was observed in liver nuclear extracts
12 h post NNK treatment and a 48% increase was observed at 24 h post NNK. These changes in
NER activity were not attributed to changes in levels of the key recognition and early incision
NER proteins XPC, XPA, XPB and p53. However, the binding of hepatic XPA and XPB to
damaged DNA at both timepoints was increased. The binding of XPC to damaged DNA was
unchanged at 12 h and decreased at 24 h. The impact of NNK on mouse liver NER -associated
processes is time dependent and associated with changes in the binding activities of specific
recognition and early excision NER proteins to damaged DNA.
Introduction 2.1
Nicotine-derived nitrosamino ketone (NNK) or 4-(methylnitrosamino)-1-(3-pyridyl)-1-
butanone is one of the tobacco-specific nitrosamines which play an important role in
carcinogenesis. NNK is one of the most potent cancer-causing tobacco-specific nitrosamines in
all animal species tested, and is believed to be a major contributor to the induction of lung

43
adenocarcinoma in humans and animals (Hecht et al., 1993a; Hecht, 1998). NNK is present in
unburned tobacco and tobacco smoke but must be metabolically activated to be carcinogenic. α-
Methylene carbon hydroxylation leads to formation of 4-hydroxy-4-(methylnitrosamino)-1-(3-
pyridyl)-1-butanone, which decomposes to methanediazohydroxide, which methylates DNA
bases (Figure 1.3). α-Methyl carbon hydroxylation generates 4-(hydroxymethylnitrosamino)-1-
(3-pyridyl)-1-butanone, which decomposes to 4-oxo-4-(3-pyridyl)-1-butanediazohydroxide,
which can pyridyloxobutylate DNA (Figure 1.3). Pyridyloxobutylation (POB) occurs at the N7-
and O6- positions of guanine, and the O2- position of cytosine and thymine.
Bioactivation and formation of DNA adducts are critical determinants of mutagenicity
and the maintenance of genetic integrity depends on the ability to repair damaged DNA. DNA
methylation is repaired by direct reversal by O6-alkylguanine-DNA alkyltransferases (AGT)
while POB adducts are repaired primarily by nucleotide excision repair (NER; Brown et al.,
2008; Kotandeniya et al., 2013; Li et al., 2009; Peterson et al., 2001; Peterson, 2010; Urban et al.,
2012). Previous reports have shown that treating mice with a tumourigenic dose of NNK causes
a 290% increase in liver NER activity in an in vitro assay (Brown & Massey, 2009). It is possible
the significant increase in NER activity causes a chemopreventive effect in mouse liver, which is
resistant to carcinogenesis after NNK exposure (Hecht et al., 1993a). However, no study has yet
examined the timecourse of effects of NNK exposure on liver NER activity.
NER can be divided into two subpathways: global genome repair (GG-NER) and
transcription coupled repair (TC-NER). TC-NER removes lesions from transcribed strands of
active genes, whereas GG-NER removes lesions from non-transcribed strands of transcribed
genes and non-transcribed regions of the genome. NER proceeds through three sequential steps:
recognition, incision/excision and DNA resynthesis (Figure 1.1). The damage recognition step is
the distinctive difference between GG-NER and TC-NER, whereas the two subpathways employ

44
a common set of proteins for the remaining steps. We have focused our work on GG-NER
because mutations in nontranscribed genes or genes on the nontranscribed strand of active genes
are better predictive markers for carcinogen-induced tumourigenesis than mutations in actively
transcribed genes (Balajee & Bohr, 2000). Recent focus in this field has been on the regulation of
“early stage repair” which encompasses the recognition and early incision stages of NER.
Lesion recognition is a ‘triple-check’ process, where a bulky adduct is first recognized by
the xeroderma pigmentosum (XP) group C (XPC) complex, then verified by the multi-subunit
transcription factor II H (TFIIH) and XP group A (XPA) (Marteijn et al., 2015). XPC does not
bind directly to the bulky adduct, but to the undamaged ssDNA opposite the lesion (Trego &
Turchi, 2006). XPC recruits TFIIH, which then scans for lesions bidirectionally via the two
helicases XPB and XPD (Li et al., 2015b). XPA further verifies damage, sensing chemically
altered nucleotides and displaces the cyclin-dependent kinase-activating kinase (CAK)
subcomplex from TFIIH (Camenisch et al., 2006; Coin et al., 2008). The release of XPC and
binding of replication protein A (RPA) are the events which lead to conclusion of early stage
repair; XPA and RPA then form stable structures for downstream NER factors. Tumour
suppressor protein 53 (p53) also appears to be involved in early stage NER; functional p53 is
required for NER but its exact role remains elusive (Lakin & Jackson, 1999; Zhu et al., 2000).
One theory is that p53 stimulates chromatin decondensation prior to lesion recognition, but the
most likely role p53 plays within NER is regulation of lesion recognition (Rubbi & Milner, 2003;
Adimoolam & Ford, 2003). p53 is known to interact directly with TFIIH in repair of UV light-
induced cyclobutane-pyrimidine dimers and pyrimidine-pyrimidone photoproducts (Chang et al.,
2008) and can transcriptionally regulate XPA nuclear import (Li et al., 2011a) and levels of XPC
(Adimoolam & Ford, 2003).

45
Previous research showed that the presence or absence of each of the more than 30 NER
proteins could increase or decrease NER activity (Nocentini et al., 1997; Araújo et al., 2001;
Hoogervorst et al., 2004; Koberle et al., 2006; Coin et al., 2008; Brown & Massey, 2009; Brown
et al., 2010; Kang et al., 2010b; Fan & Luo, 2010; Melis et al., 2011; Li et al., 2011a; Fadda,
2013; Rossner et al., 2013; Li et al., 2013b; Ziani et al., 2014; Mulder et al., 2014; Choi et al.,
2015; Donninger et al., 2015; Li et al., 2015b). Only recently have specific proteins been
identified that induce changes in NER activity without changes in protein levels, generally
through post-translational modifications involving ubiquitin or ubiquitin-like compounds (Huang
& D’Andrea, 2006; Kang et al., 2010b; Gali et al., 2012; van Cuijk et al., 2015). Early stage
NER, that is the recognition and early incision stages, has recently been identified as a key stage
for induction of NER after carcinogen exposure (Marteijn et al., 2015; Choi et al., 2015;
Donninger et al., 2015; Li et al., 2015b).
It was previously shown there is a significant increase in NER activity in mouse liver
tissue after a tumourigenic dose of NNK (Brown & Massey, 2009). This response is not unique,
in that several other studies have shown that an increase in NER activity is caused by treatment of
cell lines with a DNA damaging agent, either UV light (Choi et al., 2015; Tomicic et al., 2011),
or mitomycin C (Protić et al., 1988), and in vivo with aflatoxin B1 (Bedard et al., 2005). The
upregulation of NER is consistent with an adaptive response that protects cells from mutagens,
and a recent study showed that pre-treatment with a nonlethal dose of UV-light caused a
significant increase in NER of subsequent cisplatin damage in lung cancer cells (Choi et al.,
2015). However, these studies have been carried out in cell culture and/or are all shown at one or
two timepoints following carcinogen treatment, so persistence of the effect in vivo has not been
established.

46
In this study, there is a significant change in NER activity at two time points in mouse
liver after treatment with NNK. These effects are associated with changes in the binding
activities of NER proteins XPC, XPA, XPB or p53 in liver nuclear protein extracts to damaged
DNA and are not attributable to a change in the nuclear levels of these proteins.
Materials and methods 2.2
2.2.1 Animal treatments
Female A/J mice, aged 8-11 weeks (Taconic Labs, Hudson, NY) were housed with a 12 h
light/dark cycle and provided food and water ad libitum. A/J mice were chosen for this study
since NNK carcinogenicity studies have been carried out predominantly in this strain (Hecht,
1998). Mice were treated with saline (0.1 mL, i.p.) or a single dose of 10 µmol NNK (Toronto
Research Chemicals, Toronto, ON) in saline (0.1 mL, i.p.), a dose of NNK that induces a
significant increase in multiplicities of lung adenocarcinomas in A/J mice (Hecht et al., 1989).
Due to concerns over mouse NER activity diurnal variation (Kang et al., 2010b), treatments were
carried out at the same time every day, starting at 0900 h. Two, 4, 12, 24, or 72 h following NNK
administration, mice were killed by cervical dislocation. Livers were perfused with 10mM
Tris/1mM EDTA (pH 7.9), excised, finely chopped, frozen in liquid nitrogen, and stored at -80°C
until extract preparation. All animals were cared for in accordance with the guidelines of the
Canadian Council on Animal Care and the experimental procedures involving animals were
approved by the Queen’s University Animal Care Committee.
2.2.2 Preparation of cell-free whole tissue nuclear protein extract
Nuclear protein extracts from 1.0 to 1.3 g of tissue were prepared from pooled livers of
four mice as described (Wood et al., 1988; Wood et al., 1995) with modifications (Bedard et al.,
2005). Briefly, chopped tissue pieces frozen in liquid nitrogen were pulverized into a fine

47
powder with mortar and pestle. Hypotonic lysis of pulverized tissue was done for 30 minutes on
ice under gentle rotation in 4 mL hypotonic lysis buffer (10 mM Tris-HCl (pH 7.9), 1 mM EDTA,
5 mM dithiothreitol). Following the addition of protease inhibitors (0.5 mM phenyl methyl
sulfonyl fluoride, 4.7 mM leupeptin, 1.6 mM aprotinin and 1.5 mM pepstatin), lysate was
homogenized using a Dounce homogenizer (20 strokes). With gentle stirring on ice, 5 mL of
sucrose-glycerol solution (50 mM Tris-HCl (pH 7.9), 10 mM MgCl2, 2 mM dithiothreitol, 25 %
sucrose, 50 % (v/v) glycerol) and 1 mL saturated ammonium sulfate (neutralized with NaOH)
were added dropwise, and homogenate was gently stirred for an additional 30 minutes.
Homogenate was centrifuged for 3 h at 100,000 g at 4°C and the supernatant was collected,
leaving 1 mL above pellet. With gentle stirring on ice, supernatant was adjusted to 0.33 g/mL
with ammonium sulfate, 10 µL of 1 M NaOH per gram of ammonium sulfate added and
supernatant was stirred for an additional 30 min. Precipitate was collected by centrifugation at
15,000 g at 4°C for 20 min. Pellet was dissolved in 200 µL of dialysis buffer (20 mM HEPES-
KOH (pH 7.9), 100 mM KCl, 12.5 mM MgCl2, 0.1 mM EDTA, 17 % (v/v) glycerol, 2 mM
dithiothreitol) and dialysis was performed against three changes of 200mL (per Slide-A-Lyzer
cassette, ThermoFisher, Waltham, MA, USA) of buffer for 18 h and changed after 1 and 16 h of
dialysis. The dialysate was clarified by centrifugation at 10,000 g for 3 min, and stored in
aliquots at -80°C. Protein contents of extracts were determined by the Lowry method as modified
by Peterson et al. (1977). Extracts from livers contained between 10 to 15 mg/mL protein.
Extracts were frozen in liquid nitrogen and stored at -80°C.
2.2.3 Preparation of damaged plasmid DNA
Pyridyloxobutylated plasmid DNA was prepared as described (Brown et al., 2008). In
the laboratory, DNA can be pyridyloxobutylated by a chemically activated form of NNK, 4-
(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone (NNKOAc). Briefly, NNKOAc (20mM,

48
Toronto Research Chemicals, North York, ON, Canada) was reacted with 1.04 mg/mL
pBluescript SK+ plasmid DNA (Stratagene, La Jolla, CA, USA) in a 250 µL reaction mixture
containing sodium citrate buffer (final concentration, 15 mM sodium citrate, pH 7.0, 1 mM
EDTA) and 4 U/mL porcine liver esterase (Sigma-Aldrich Co., St. Louis, MO, USA) for 1 h at
room temperature. Plasmid DNA damaged by this method contained approximately 10-12
adducts/plasmid as determined by HPLC analysis of 4-hydroxy-1-(3-pyridyl)-1-butanone release
with equal proportions of the four major POB adducts, 7-[4-(3-pyridyl)-4-oxobut-1-yl]guanine,
O2-[4-(3-pyridyl)-4-oxobut-1-yl]cytosine, O2-[4-(3-pyridyl)-4-oxobut-1-yl]thymidine and O6-[4-
(3-pyridyl)-4-oxobut-1-yl]-2´-deoxyguanosine as determined by mass spectrometry (Brown et al.,
2008).
2.2.4 In vitro DNA repair synthesis assay
The repair synthesis assay was performed as described (Brown & Massey, 2009). With
minor changes: each 50 µL reaction mixture contained 400 ng of pyridyloxobutylated plasmid
DNA or undamaged plasmid DNA, 40 mM HEPES-KOH (pH 7.8), 0.5 mM DTT, 4.0 µmol/L
dATP, 20µM of each of dGTP, dCTP, and dTTP, 23 mM phosphocreatine, 18 µg bovine serum
albumin (nuclease-free, Sigma-Aldrich Co., St. Louis, MO, USA), 2.5 µg creatine
phosphokinase, 2.0 mM ATP, 5.0 mM MgCl2, 0.4 mM EDTA, 100mM KCl, 100µg protein
extract and 2.0 µCi [α32P]dATP (Perkin Elmer, Waltham, MA, USA). The repair synthesis assay
was performed in duplicate for each nuclear protein extract and results are presented as the
mean± SD (n=4). To determine damage-specific repair activity, radioisotope incorporation into
undamaged plasmid DNA was subtracted from repair synthesis activity calculated with POB
damaged DNA.

49
2.2.5 Immunoblot analysis of NER protein levels in mouse liver nuclear protein extracts
Rabbit polyclonal antibodies anti-human XPA (1/2,000; ab10905), anti-human XPB
(1/4,000; ab53198), anti-human XPC (1/2,000; ab78064) and mouse monoclonal antibody anti-
human p53 (1:1,000; ab28) were purchased from Abcam Inc. (Cambridge, MA, USA). HRP-
conjugated polyclonal goat anti-rabbit IgG (1/5,000; sc-2004) and HRP-conjugated polyclonal
donkey anti-mouse IgG (1/5,000; sc-2314) were purchased from Santa Cruz Biotechnology Inc.
(Santa Cruz, CA, USA).
Nuclear protein extracts (5µg) were subjected to sodium dodecyl sulfate polyacrylamide
gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes
(Millipore, Billerica, MA, USA). Membranes were incubated in 10% skim milk overnight at 4°C
and proteins immunoblotted with above antibodies for 1 h at room temperature. After incubating
membranes with secondary antibodies conjugated to horseradish peroxidase for 1 h at room
temperature, specific proteins were visualized as immunoreactive bands by the enhanced
chemiluminescence detection system (Perkin Elmer, Waltham, MA, USA). To normalize for
protein loading, α-actinin was used as a housekeeping protein (1/5,000; sc-15335; Santa Cruz
Biotechnology, Santa Cruz, CA, USA) as a loading control. Films were scanned and processed
with Licor® Image Studio™ Lite (Mandel Scientific Company, Guelph, ON, Canada). Total
values were converted to relative densities using a standard that was prepared by pooling tissues
from 16 animals. Immunoblots were performed in duplicate for each nuclear protein extract and
results are presented as the relative densities ± SD (n=4).
2.2.6 Analysis of binding of NER incision proteins to damaged DNA
An assay to assess the binding of NER proteins to damaged DNA was performed as
described (Frit et al., 1998; Li et al., 1998; Salles et al., 1995a; Salles et al., 1995b; Trego &
Turchi, 2006) with slight modifications. Briefly, 50 ng of POB-adducted or undamaged plasmid

50
DNA in 10 mM phosphate buffer (pH 7.0) were adsorbed on Microlite II Microtitre 96 well
plates (Thermo Scientific, Waltham, MA, USA) that were pre-treated with 100 µL poly-L-lysine
hydrochloride (1 mg/mL) in phosphate-buffered saline (10mM phosphate buffer (pH 7.2)/ 150
mM NaCl) at 30°C for 30 min with gentle shaking. Picogreen dsDNA quantitation kit
(Invitrogen Molecular Probes, Carlsbad, CA, USA) was used to verify the amount of plasmid
DNA adsorbed to wells with a microplate reader. The protein binding reaction was carried out in
50 µL per well and contained mouse lung (100 µg) or liver (120 µg) nuclear protein extract in 100
mM KCl, 40 mM HEPES-KOH (pH 7.6), 7.0 mM MgCl2, 2.0 mM ATP, 0.5 mM DTT, 10 mM
phosphocreatine, 2.5 µg creatine phosphokinase, 2.0 mM EGTA, and 18 µg bovine serum
albumin (nuclease-free, Sigma-Aldrich Co., St. Louis, MO, USA). After 3 h at 30 ºC, wells were
washed three times with 50 µL of PBS plus 0.01 % Tween-20, and proteins bound to immobilized
DNA were eluted with 25 µL of sample buffer (62.5 mM Tris-HCl (pH 6.8), 4 M urea, 10 % (v/v)
glycerol, 2 % (w/v) SDS, 5 % (v/v) β-mercaptoethanol, 0.003 % (w/v) bromophenol blue) for 30
min at 30 ºC with shaking, followed by 15 min at 85 ºC. The proteins were then resolved by
SDS-PAGE and analyzed as described above (Subsection 2.2.5) for immunoblot analyses with
minor modifications. Primary antibodies were incubated overnight and at higher concentrations
(XPC 1/1,000; XPA 1/2,000; XPB 1/4000). Each DNA bound protein sample was comprised of
reactions pooled from 3 wells. Protein binding reaction samples were conducted in duplicate for
each nuclear protein extract. Films and stained blots were scanned and processed with Adobe
Photoshop 6.0 Software, and densitometry of the scanned image was performed using Li-Cor
Biosciences Image Studio Lite 5.2.5 software. Relative density for undamaged-DNA- and
damaged-DNA-bound NER proteins was calculated and protein levels were normalized by total
amount of protein bound based on Amido Black staining. Binding factor was calculated by

51
subtracting the average relative protein level of undamaged DNA bound protein from the average
relative protein level of damaged-DNA-bound protein.
2.2.7 Data analysis
Statistically significant differences in repair activity and NER protein levels between
treatment groups were determined by unpaired Student’s t-tests (Graphpad Prism 6 software).
Due to inter-experiment variability, Mann-Whitney U test was used comparing DNA-associated
NER protein binding (Subsection 2.2.6). P<0.05 was considered significant in all cases.
GraphPad Prism 6 was used to graph all results.
Results 2.3
2.3.1 Time course of effects of NNK treatment on mouse liver NER activity
To observe the effect of NNK treatment over time, A/J mice were treated with a
tumourigenic dose of NNK and sacrificed at 2, 4, 12, 24, or 72 h. NNK treatment caused a 46%
decrease in activity at 12 h and a 48 % increase in NER activity at 24 h (Figure 2.1). The 2 h
timepoint had higher control and treated NER activities than other timepoints, this is possibly
attributable to diurnal variation in in vivo NER activity (Kang et al., 2010b; Kang et al., 2011).
This variation was not the focus of this study and was not pursued further.

52
Figure 2.1. Effects of in vivo NNK or saline treatment on in vitro DNA repair activity of
mouse liver extracts towards NNKOAc-induced pyridyloxobutyl DNA damage. Extracts
were prepared from mice treated with either 10 µmol NNK or saline and killed 2, 4, 12, 24
or 72 h post-dosing. Data are presented as mean + SD. * significantly different from
control (p<0.05; n=4 pools of 4 mouse livers each.)
2h 4h 12h 24h 72h0
5000
10000
15000
Mouse Liveramol
[α32
P] d
ATP
inco
rpor
ated
/µg
DN
ASalineNNK
*
*

53
2.3.2 The effect of in vivo treatment of mice with NNK on levels of key NER proteins in
mouse liver nuclear protein extracts
The effect of NNK on levels of key NER proteins in mouse liver extracts was examined.
At 12 h (Figure 2.2) and 24 h (Figure 2.3) post-treatment liver extracts showed no significant
changes in key NER protein levels.

54
Figure 2.2 a) Relative density of each key NER protein in mouse liver nuclear protein
extracts isolated from saline and NNK-treated mice 12 h post-dosing, relative to a constant
internal standard extract. Data are presented as mean ± SD. b) Representative
immunoblots of the relative levels of each key NER protein in mouse liver extracts. S=
nuclear protein extracts from saline treated mice, N=nuclear protein extracts from NNK
treated mice; n=4 pools of 4 mouse livers each.

55
Figure 2.3 a) Relative density of each key NER protein in mouse liver nuclear protein
extracts isolated from saline and NNK-treated mice 24 h post-dosing, relative to a constant
internal standard extract. Data are presented as ean + SD. b) Representative immunoblots
of the relative levels of each key NER protein in mouse liver extracts. S= nuclear protein
extracts from saline treated mice, N=nuclear protein extracts from NNK treated mice; n=4
pools of 4 mouse livers each.

56
2.3.3 Effect of in vivo treatment of mice with NNK on binding of liver NER proteins to
damaged DNA
To determine if NNK affects binding of NER proteins to damaged DNA, an in vitro assay
was used that measures the binding of repair proteins to damaged DNA. There was a significant
increase in the binding of XPB (12 h: 2,137% increase; 24 h: 98% increase) and XPA (12 h:
461% increase; 24 h: 962% increase) after NNK treatment at both 12 h (Figure 2.4) and 24 h
(Figure 2.5), whereas there was no significant change in XPC binding at 12 h (Figure 2.4) and a
significant decrease in XPC binding at 24 h (45% decrease; Figure 2.5).

57
Figure 2.4 Effects of in vivo NNK treatment on in vitro NER protein binding to NNKOAc-
induced pyridyloxobutyl DNA damage 12h post treatment. Data are presented as mean +
SD. * Significantly different from control (p<0.05; n=4 pools of 4 mouse livers each.)
XPC XPB XPA0.0
0.5
1.0
1.5
2.0
2.5
12h Liver Extracts
Bin
ding
Fac
tor
SalineNNK
*
*

58
Figure 2.5 Effects of in vivo NNK treatment on in vitro NER protein binding to NNKOAc-
induced pyridyloxobutyl DNA damage 24h post treatment. Data are presented as mean +
SD. * Significantly different from control (p<0.05; n=4 pools of 4 mouse livers each.)
XPC XPB XPA0.0
0.5
1.0
1.5
2.0
2.5
24h Liver Extracts
Bin
ding
Fac
tor
SalineNNK
*
*
*

59
Discussion 2.4
This study examined the effect of NNK treatment on liver NER activity at several
timepoints post exposure. Previously, NNK had been shown to increase NER activity in mouse
liver after 4 h and 24 h (Brown & Massey, 2009). Here, we expanded to a time course of effects
of NNK treatment on NER activity, encompassing 2, 4, 12, 24 and 72 h post-treatment. We
showed that NNK affects the repair of POB-DNA adducts and begin to understand how it effects
NER regulation.
We confirm a significant increase in liver NER activity at 24 h post-treatment the
timepoint of maximal hepatic POB adduct levels in A/J mouse lung (Peterson & Hecht, 1991).
Although the timecourse of hepatic NNK-derived DNA adducts for A/J mice has not been
reported, POB adduct levels are consistently greater in the liver than in the lung (Hecht et al.,
1993a; Peterson & Hecht, 1991). We did not confirm the significant effect at 4 h, and we see a
smaller effect than that observed previously; Brown and Massey showed a 200% increase in NER
activity 4 h post-treatment and 290% induction 24 h post-treatment (2009). Here, we found no
significant effect on NER activity (p = 0.4) 4 h post NNK treatment, but a 48% increase in
activity 24 h post-NNK (Figure 2.1). The reason for these inconsistencies with the previous study
is not readily apparent. We made two changes to the protocol used previously. In the previous
study, livers from 4 different mice were used for NER assays, while in the present study, we used
a pool of livers from four NNK- treated A/J mice for each of four samples, in order to be
consistent with the lung study, where a pool of 4 lungs is required for a sufficient nuclear protein
extract concentration for NER activity assays (Chapter 3). Additionally, the manufacturer no
longer produces the radiolabelled nucleotide used in the previous study, [α32P]dTTP, and this was
replaced in this study with [α32P]dATP. It is unlikely these changes affected the observations at 4

60
h. Several years have transpired between the studies, and it is possible that genetic drift has
resulted in a somewhat blunted NNK response in the A/J mice.
Notably, we found that NNK treatment inhibited NER at 12 h post-treatment. This is the
first time we have found a carcinogen-mediated decrease in NER activity after carcinogen
treatment in mouse liver. Since there were no significant changes in NER activity at other
timepoints, subsequent mechanistic experiments focused on the 12 and 24 h timepoints.
Dysregulation of many of the more than 30 proteins involved in NER can change repair
activity (Araújo et al., 2001; Chang et al., 2008; Li et al., 1998; Melis et al., 2011; Ray et al.,
2013). Of particular importance are the early stage repair recognition and early incision phase
proteins XPC, XPA, TFIIH and p53. Several in vitro studies have shown that alterations in these
proteins appear to cause a change in NER activity after exposure to other DNA-damaging agents.
p53 may contribute to the binding of XPA to damaged DNA (Li et al., 2011a), and the
increase in NER activity caused by chronic low doses of aflatoxin B1 was diminished in mice that
were heterozygous for defective p53 (Mulder et al., 2014). Although p53 status is important in
NNK-mediated carcinogenesis (Ellinger-Ziegelbauer et al., 2004), the lack of effect of NNK on
levels of the proteins XPC and XPA, whose expression is regulated by p53, or on XPB (a subunit
of TFIIH) suggests that the changes in NER activity we have observed with NNK treatment are
not attributable to p53-mediated changes in expression of these components of NER.
A recent study has shown that exposure of cultured human lung cancer cells to UV light
increases NER activity without a significant increase in repair protein levels, but with an increase
in XPA binding activity (Choi et al., 2015). The binding of NER proteins XPC, XPA and TFIIH
(XPB) has previously been used to predict the activity of NER (Frit et al., 1998; Li et al., 1998;
Salles et al., 1995a; Koberle et al., 2006). In partial agreement, we observed that the binding of
XPA and XPB to damaged DNA was altered by NNK treatment at 12 h (Figure 2.4) and XPA,

61
XPB and XPC at 24 h (Figure 2.5) post-treatment with NNK. It was expected that the binding of
XPA and XPB to damaged DNA would closely reflect NER activity; this was the case at 24h, but
at 12 h there was a decrease in NER activity with NNK treatment, which was opposite to effects
on XPA and XPB binding.
The decrease in XPC binding to damaged DNA at 24h post NNK may have been a
consequence of the increase of TFIIH (XPB) and XPA, as after these proteins and RPA bind
within the repair complex, XPC is displaced to allow it to sense other DNA damage (Li et al.,
2015b; Melis et al., 2011). In fact, failure of XPC release has been associated with a decrease in
NER activity (Li et al., 2015b; Long et al., 2015). It is also possible that inhibition of NER
activity seen at 12 h with NNK is due to effects on late stage repair. Late-stage NER protein such
as RPA, XPG, XPF or PCNA might be inhibited, either through covalent binding of an NNK
metabolite or a decrease in protein synthesis. Previously, it has been theorized that NNK causes
covalent modifications to key NER proteins (Brown & Massey, 2009) and recent research has
shown that RPA is particularly susceptible to damage (Guven et al., 2015). It is possible that
NNK inhibits NER at 12 h through damage or covalent binding to RPA. In future studies it may
be valuable to assess the binding of RPA, as this protein stays bound as the linkage between early
and late stage NER (Gourdin et al., 2014). Furthermore, perturbation of either XPF or XPG
inhibits NER (Araújo et al., 2001; McNeil & Melton, 2012; Su et al., 2012; Tomicic et al., 2011).
However, the cycle of synthesis and degradation of these proteins is not well understood, so the
potential ability of XPF and XPG activities to recover quickly after insult, is not known.
Alternatively, the importance of post-translational modifications of key NER proteins in
response to bulky DNA damage is now recognized (Beli & Jackson, 2015; Dou et al., 2010; Gali
et al., 2012; Hoege et al., 2002; Huang & D’Andrea, 2006; Kang et al., 2010b; Poulsen et al.,
2013; van Cuijk et al., 2015). In UV-induced NER, regulation of XPC, XPA and TFIIH

62
damaged-DNA binding is controlled by these post-translational modifications. The lack of any
significant NNK-mediated changes in XPC, XPB or XPA protein levels at 12 h (Figure 2.2) or 24
h (Figure 2.3) suggests that the observed changes in NER activity could be caused by post-
translational modifications, and it may be of value to examine such modifications to NER
proteins in the future.
Overall, we show the effects of NNK treatment on NER activity across several
timepoints. At the two timepoints with significant and opposite changes of NER activity, 12 h
and 24 h, we also evaluated changes in pre-incision NER protein levels and binding to damaged
DNA. We found that NNK treatment caused an increase in binding of XPA and XPB at both
timepoints, no change in binding of XPC at 12 h and a significant decrease of XPC binding at 24
h. It is likely that NNK increases the activity of early-stage NER at 24 h and, while early stage
NER is upregulated at 12 h, the transition step between early and late stage could be inhibited, or
another protein downstream of XPC release could be inhibited, causing the decrease in NER
activity. Further research to determine the specific mechanism responsible for this phenomenon
is required and will help provide an understanding of the processes by which NER protects
against genotoxic insults.

63
Chapter 3
Investigation of the mechanisms by which NNK decreases nucleotide
excision repair activity in mouse lung
After in vivo exposure to the tobacco specific nitrosamine 4-(methylnitrosamino)-1-(3-
pyridyl)-1-butanone (NNK), mouse lung is the major target of NNK-induced carcinogenesis.
One explanation for this susceptibility to carcinogenesis is a decrease in nucleotide excision
repair (NER) activity in mouse lung. However, the mechanisms of this decrease are not well
understood. A 63% decrease in NER activity was observed in lung nuclear protein extracts 24 h
post NNK. This change in NER activity were not attributed to changes in levels of key
recognition and early incision NER proteins XPC, XPA, XPB or p53. However, the binding of
lung XPA and XPB to damaged DNA was decreased and the binding of XPC to damaged DNA
was increased at 24 h. These results suggest that the impact of NNK on repair-associated
processes are associated with changes in the activities of specific recognition and early excision
NER proteins.
Introduction 3.1
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) is one of the key tobacco-
specific nitrosamines that play an important role in smoking-induced lung carcinogenesis. NNK
is one of the most potent cancer-causing tobacco-specific nitrosamine in all animal species tested,
and is has been correlated with the induction of lung adenocarcinoma in both humans and animals
(Hecht et al., 1993a; Hecht, 1998). NNK is found in unburned tobacco and tobacco smoke but
must be metabolically activated as described in subsection 1.4.2. Metabolic activation results in
the formation of methyl and pyridyloxobuylated DNA.

64
Genetic integrity depends on the ability to repair damaged DNA, bioactivation and
formation of DNA adducts are critical determinants of mutagenicity. DNA methylation is
repaired by O6-alkylguanine-DNA alkyltransferases (AGT), while POB adducts are repaired
largely by nucleotide excision repair (NER) (Peterson et al., 2001). Previous reports have shown
that treating mice with a tumourigenic dose of NNK causes a significant decrease in NER activity
in mouse lung nuclear extracts 24 h after treatment (Brown & Massey, 2009). A/J mice treated
with NNK form a large number of lung adenomas (Hecht et al., 1993a), and it is possible that the
decrease in NER activity is a significant contributor to lung tumourigenicity after NNK exposure.
However, the mechanism of the decrease in NER is not yet understood.
As discussed in previous chapters, NER can be divided into two subpathways: global
genome repair (GG-NER) and transcription coupled repair (TC-NER). TC-NER removes lesions
from transcribed strands of active genes, whereas GG-NER removes lesions from non-transcribed
strands of transcribed genes and non-transcribed regions of the genome. NER can be divided into
three steps: recognition, incision/excision and DNA resynthesis. The damage recognition protein
xeroderma pigmentosum (XP) group C (XPC) complex is the distinctive difference between GG-
NER and TC-NER, and they employ a common set of proteins for the remaining steps. Balajee
and Bohr displayed mutations in genes on the nontranscribed strand or nontranscribed genes may
be better predictive markers than genes that are actively transcribed, thus we focussed our work
on GG-NER (2000).
Removal of many of the more than 30 GG-NER proteins will lead to disruption of the
NER pathways, however, changes in NER activity appear to be mediated largely by the proteins
that carry out lesion recognition and early incision (Li et al., 2015b; Marteijn et al., 2015).
Lesion recognition is a ‘triple-check’ process, where a bulky adduct is first recognized by XPC,
then verified by XP group A (XPA) and the multi-subunit transcription factor II Human (TFIIH)

65
(Marteijn et al., 2015). XPC does not bind directly to the bulky adduct, but rather to the
undamaged single stranded DNA opposite the lesion. XPA further verifies damage, binding to
chemically altered nucleotides, and its presence displaces the cyclin-dependent kinase-activating
kinase -subcomplex (CAK) from TFIIH (Coin et al., 2008). The presence of XPC attracts TFIIH,
which then scans for lesions bidirectionally via the two helicases XP group B (XPB) and XP
complementation group D (XPD), and this action is significantly increased after CAK
disassociation (Li et al., 2015b). The binding of an additional scaffolding protein, replication
protein A (RPA), signals the end of the recognition stage; XPA and RPA form stable structures
for downstream NER factors and protect the DNA strand from degradation. Tumour suppressor
protein 53 (p53) also appears to be involved in early stage NER, and functional p53 is required
for NER (Lakin & Jackson, 1999; Zhu et al., 2000) but its exact role in NER remains elusive
(Momot et al., 2014; Loewer et al., 2013; Li et al., 2011b; Ikehata et al., 2010). Studies have
shown that p53 is involved both directly and indirectly in NER; it is known to interact directly in
repair of UV-DNA adducts (carried out by NER) likely through interaction with the XPB subunit
of TFIIH(Chang et al., 2008), and it can induce XPC protein synthesis (Adimoolam & Ford,
2003) and can transcriptionally regulate nuclear import of XPA (Li et al., 2011a).
Classical understanding of NER regulation dealt with increasing or decreasing nuclear
protein levels of key NER proteins in order to increase or decrease NER activity (Aboussekhra et
al., 1995). Many of the NER proteins are named for xeroderma pigmentosum, a class of genetic
diseases wherein the protein is inactivated, thereby inhibiting NER activity. Additionally, diurnal
variation in NER appears to be caused by diurnal variation in degradation of XPA protein (Kang
et al., 2010b; Kang et al., 2011). More recently, research on NER activity after UV-induced
DNA damage and NER factor post-translational modifications have added another layer on to
NER regulation (Huang & D’Andrea, 2006; Dou et al., 2010; Kang et al., 2011; van Cuijk et al.,

66
2015; Li et al., 2015b). The binding of XPC (Ming et al., 2010), XPA (Fan & Luo, 2010; Kang
et al., 2011) and TFIIH (Coin et al., 2008) to the NER complex is all mediated by post-
translational modifications. Induction of these pathways is implicated in the induction of NER
activity without the increase of nuclear levels of the key NER proteins (Choi et al., 2015; Fan &
Luo, 2010; Li et al., 2015b). Disruption of any of these pathways has also been implicated in the
inhibition of NER activity (Choi et al., 2015; Gaddameedhi et al., 2015; Li et al., 2011a; Marteijn
et al., 2015; Ray et al., 2013).
There is a decrease in NER activity in mouse lung tissue after a tumourigenic dose of
NNK (Brown & Massey, 2009). This effect is not unique to NNK several other studies have
shown a decrease in NER activity after chemical treatment with the anticancer drug STI571
(Sliwinski et al., 2008), toposiomerase I/II inhibitor F11782 in vitro (Kruczynski et al., 2004) and
the mycotoxin aflatoxin B1 in vivo(Bedard et al., 2005). The combination of an NNK-mediated
decrease in NER activity and NNK-mediated DNA damage is hypothesized to make the mouse
lung especially susceptible to carcinogenesis.
In this study, we show that the decrease in mouse lung NER activity that occurs at 24 h
after treatment with NNK is not due to a change in the amounts of key early phase NER proteins
in the nuclear protein extracts, but we demonstrate changes in their binding activities to damaged
DNA.
Materials and methods 3.2
3.2.1 Animal treatments
Animals were treated as described in section 2.2.1.

67
3.2.2 Preparation of cell-free whole tissue nuclear protein extract
Nuclear protein extracts were prepared from pooled lungs of four mice as described in
section 2.2.2. Extracts from lungs contained between 5 to 8 mg/mL protein. Extracts were frozen
in liquid nitrogen and stored at -80°C.
3.2.3 Preparation of damaged plasmid DNA
Pyridyloxobutylated plasmid DNA was prepared as described in section 2.2.3.
3.2.4 In vitro DNA repair synthesis assay
The repair synthesis assay was performed as described in section 2.2.4.
3.2.5 Immunoblot analysis of NER protein levels in mouse lung nuclear protein extracts
The immunoblot analysis of NER protein levels in mouse lung nuclear protein extracts
was performed as described in section 2.2.5.
3.2.6 Analysis of binding of NER recognition proteins to damaged DNA
An assay to assess the binding of NER proteins to damaged DNA was performed as
described in section 2.2.6 using mouse lung nuclear protein extracts.
3.2.7 Data analysis
Statistically significant differences in repair activity and NER protein levels between
treatment groups were determined by unpaired Student’s t-test (Graphpad Prism 6 software). Due
to inter-experiment variability Mann-Whitney U test was used comparing DNA-associated NER
protein binding (section 3.2.6). P<0.05 was considered significant in all cases. GraphPad Prism 6
was used to graph all results.

68
Results 3.3
3.3.1 Effect of treatment of mice with NNK on repair synthesis of mouse lung nuclear
protein extracts
At 24 h following in vivo NNK treatment, repair synthesis activity of mouse lung extracts
toward POB damage was decreased by 63% (Figure 3.1). This is consistent with a previous result
from Brown and Massey (2009).

69
Figure 3.1 Effects of in vivo NNK or saline treatment on in vitro DNA repair activity of A/J
mouse lung extracts towards NNKOAc-induced pyridyloxobutyl DNA damage. Extracts
were prepared from mice treated with either sterile saline or 10 µmol NNK and sacrificed
24h post-dosing. Data are presented as mean + SD. (*) Significantly different from control
(n=4 pools of 4 mice per pool, p<0.05).
Saline NNK0
1000
2000
3000
24h Lung Extractsamol
[α32
P] d
ATP
inco
rpor
ated
/µg
DN
A
*

70
3.3.2 Effect of in vivo treatment of mice with NNK on levels of key NER proteins in mouse
lung nuclear protein extracts
The effect of NNK on levels of key NER proteins in mouse lung nuclear protein extracts
(the same extracts as were used for NER assays) was examined (Figure 3.2). Twenty-four hours
post NNK, no significant changes in specific NER protein levels were observed, relative to
control at the same time point.

71
Figure 3.2 a) Relative density of each key NER protein in A/J mouse lung nuclear protein
extracts isolated from saline or NNK-treated mice 24h post-dosing, relative to a constant
internal standard extract. Data are presented as the mean + SD presented. b)
Representative immunoblots of the relative levels of each key NER protein in mouse lung
extracts. S= nuclear protein extracts from saline treated mice, N=nuclear protein extracts
from NNK treated mice; ; n=4 pools of lungs of 4 mice per pool.

72
3.3.3 Effect of in vivo treatment of mice with NNK on key NER protein DNA binding in
mouse lung nuclear protein extracts
To determine if NNK was affecting the interactions between key NER proteins and
damaged DNA, an in vitro assay was used that measures the binding of repair proteins to
damaged DNA. There was a significant decrease in the binding of XPB (53% decrease) and XPA
(83% decrease) after NNK treatment at 24 h (Figure 3.3), whereas there was a significant increase
in XPC binding at 24 h (115% increase; Figure 3.3).

73
Figure 3.3 Effects of in vivo NNK treatment on in vitro NER protein binding to NNKOAc-
derived pyridyloxobutyl DNA damage. The data are presented as mean + SD. (*)
Significantly different from control (XPA n=5 pools of lungs of 4 mice per pool; XPC and
XPB n=6 pools of lungs of 4 mice per pool, p<0.05).
XPC XPA XPB0.0
0.5
1.0
1.5
2.0
24h Lung Extracts
Bin
ding
Fac
tor
SalineNNK
* * *

74
Discussion 3.4
In this study, we examined the effect of NNK treatment on lung NER activity at 24 h post
exposure, the timepoint of maximal POB adduct levels in A/J mouse lung (Peterson & Hecht,
1991). We confirm the previously described decrease in NER activity at 24 h (Brown & Massey,
2009); this inhibition of NER may be a major contributor to the high incidence of A/J mouse lung
tumours observed after NNK treatment. Previously, reports have shown that mutation or removal
of many of the more than 30 proteins involved in NER can decrease repair activity (Araújo et al.,
2001; Chang et al., 2008; Li et al., 1998; Melis et al., 2011; Ray et al., 2013). Of particular
importance are the early stage repair recognition and early incision phase proteins XPC, XPA,
TFIIH and p53 and we examined these proteins in NNK-treated mouse lung nuclear protein
extracts first via immunoblotting for protein levels, as well as by the ability to bind to damaged
DNA.
NNK had no significant effect on levels of p53, XPC, XPA or XPB proteins (Figure 3.2).
As discussed above, the contributions of p53 to NER has been under investigation for a number
of years (Vélez-Cruz & Johnson, 2012; Zhu et al., 2000); functional p53 is required for NER but
its exact role in NER has not yet been fully elucidated (Momot et al., 2014; Loewer et al., 2013;
Li et al., 2011b; Ikehata et al., 2010). Studies have shown that p53 acts both directly and
indirectly in NER, and NER activity induction caused by exposure to low-doses of aflatoxin B1
was abrogated with heterozygous mutant p53 (Mulder et al., 2014). Although p53 status is
important in NNK-mediated lung tumourigenesis (Ellinger-Ziegelbauer et al., 2004), it appears
that this is largely due to processes other than NER. UV light exposure increased NER activity in
A549 and H460 human lung cancer cells without a significant increase in levels of NER proteins
XPC, XPA, XPB, XPD, XPE, XPF and XPG (Choi et al., 2015). Rather, the elevated NER was
associated with increased binding of XPA to damaged DNA (Choi et al., 2015), linked to post-

75
translational modifications of key NER proteins in response to UV-light exposure (Marteijn et al.,
2015; Dou et al., 2010; Gali et al., 2012; Hoege et al., 2002; Huang & D’Andrea, 2006; Kang et
al., 2010b; Poulsen et al., 2013; van Cuijk et al., 2015). Here, we show a significant decrease in
the binding activity of XPA and XPB in lung nuclear extracts 24 h post-treatment with NNK,
which coincides with the decrease in NER activity in the same extracts (Figure 3.1 and Figure
3.3).
XPC, XPB and XPA all bind together to the damaged DNA strand, and XPC only
increases the likelihood of XPB and XPA binding, so it is unlikely that the significant increase in
XPC binding is the cause of the significant decrease in XPA and XPB binding (Figure 3.3). The
binding of XPA and TFIIH, however, are intricately tied together, as XPA catalyzes the release of
the CAK subcomplex of TFIIH, which increases TFIIH binding activity to damaged DNA (Coin
et al., 2008; Li et al., 2015b). XPA binding activity is affected not only by protein levels in the
nuclear extract, but also through deacetylation via sirtuin 1 (SIRT1). Recently, it has been shown
that, after UV exposure, induction of SIRT1 via the Ras association domain-containing protein 1
tumour suppressor appears to decrease the level of acetylated XPA in several tumour cell lines
(Donninger et al., 2015). Deacetylated XPA is required for efficient repair of bulky adducts
(Donninger et al., 2015; Kang et al., 2011), and knockdown of SIRT1 significantly decreases the
NER activity in vitro (Fan & Luo, 2010). SIRT1 levels are lowered in smokers (Rajendrasozhan
et al., 2008), and in A/J mouse lung extracts treated with NNK plus nicotine (Iskandar et al.,
2013). The lack of any significant change in XPC, XPB or XPA protein levels following NNK
treatment (Figure 3.2) would be consistent with effects on NER being caused by the post-
translational modifications discussed above.
The observed NNK-induced increase in XPC binding could be due to a damage response
in the lung; after UV light damage, XPC is ubiquitylated, which increases its affinity for damaged

76
DNA in vitro (Sugasawa et al., 2005). It is probable that NNK causes a similar effect, leading to
an increase in the binding activity of XPC. Additionally, XPC release is mediated by
SUMOylation of the ubiquitylated XPC in vitro (van Cuijk et al., 2015). NER is inhibited if XPC
is not released before XPG and XPF/ERCC1 can bind to the repair complex, so it is possible that
disruption of this SUMOylation of XPC contributes to the decrease in NER activity observed here
(Riedl et al., 2003; Long et al., 2015; van Cuijk et al., 2015). However, the role of these
modifications in NNK-induced XPC binding has yet to be confirmed.
It is possible that the activity of an NER protein such as XPG or XPF, whose activity we
did not assess, is significantly inhibited by NNK treatment, either through covalent binding of an
NNK metabolite or a decrease in protein synthesis. Indeed, decreased levels or modified
structures of either XPF or XPG can inhibit repair (Araújo et al., 2001; McNeil & Melton, 2012;
Su et al., 2012; Tomicic et al., 2011). However, both XPG and XPF are reliant on the release of
XPC and the binding of TFIIH and would be expected to be significantly inhibited by the changes
in binding activities we have shown here (Araújo et al., 2001; van Cuijk et al., 2015; Ziani et al.,
2014).
We found that NNK treatment caused a significant decrease in binding activity of XPA
and XPB and a significant increase in binding of XPC at 24 h in mouse lung. Previously, similar
alterations in the binding activities of XPC and XPA have been connected to decreases in NER
activity (Choi et al., 2015; Donninger et al., 2015; Li et al., 2015b; Liu et al., 2005; Saijo et al.,
2004; van Cuijk et al., 2015). It is possible the change in NER activity is due exclusively to the
changes in protein binding observed here, but further research is required to elucidate the
mechanisms that lead to the changes in binding activity.

77
Chapter 4
Investigation into the effects of in vivo sulforaphane treatment on
nucleotide excision repair activity and nucleotide excision repair
recognition proteins
The phytochemical sulforaphane has gained interest recently for its apparent connection
with the reduction of cancer risk and other cytoprotective properties. Nucleotide excision repair
(NER) is an important repair pathway for mitigating carcinogenesis from exogenous DNA
damaging agents; however, it is unclear whether sulforaphane affects NER in vivo. We first
found a dose and time point of apparent maximal effect of sulforaphane in treated mouse lung and
liver using two genes regulated by Nrf2, one of the major signalling pathways of sulforaphane
mediated-effects. A 99% increase in NER activity was observed in liver nuclear extracts 12 h
post sulforaphane treatment. This change in NER activity was not attributed to changes in levels
of key recognition and early incision NER proteins XPC, XPA, XPB or p53. Additionally, the
binding of hepatic XPC, XPA and XPB to damaged DNA was not affected by sulforaphane
treatment. These results suggest that the impact of sulforaphane on repair-associated processes is
time and organ-dependent and is not associated with changes in the levels or damaged-DNA
binding activities of these specific recognition and early excision NER proteins.
Introduction 4.1
Epidemiological studies in several countries have shown a correlation between high
consumption of cruciferous vegetables and reduction of cancer risk (Higdon et al., 2007;
Tortorella et al., 2015). Cruciferous vegetables are major food crops worldwide and include such
diet staples as cauliflower, Brussels sprouts, cabbage, bok choy and broccoli (Verkerk et al.,

78
2009). They are high in a number of vitamins, nutrients, phytochemicals and are especially high
in a group of sulfur-containing phytochemicals, the glucosinolates (Bones & Rossiter, 2006;
Fahey et al., 2001; Verkerk et al., 2009).
Glucosinolates are responsible for the odour and spicy or bitter taste of these vegetables,
and have gained interest due to their various functions after they are activated. Glucosinolates are
hydrolyzed into biologically active compounds by the enzyme myrosinase, which is found in a
separate cellular compartment from the glucosinolate; the glucosinolates and myrosinase come
into contact after the plant is crushed or chewed (Bones & Rossiter, 2006). One of the best
characterized glucosinolates is glucoraphanin which is hydrolyzed to sulforaphane (Figure 1.4).
Sulforaphane is the most hydrophobic of all dietary ITCs and thus is highly lipophilic and is
rapidly diffused into the intestinal epithelium. After diffusion, sulforaphane undergoes
metabolism via the mercapturic acid pathway through conjugation with glutathione (SF-GSH).
Subsequent steps result in the generation of sulforaphane-cysteine (SF-Cys) followed by
sulforaphane-N-acetylcysteine (SF-NAC). Sulforaphane and its metabolites have shown anti-
inflammatory, antibiotic, antioxidant and anticarcinogenic actions (Alumkal et al., 2015; Atwell
et al., 2015; Clarke et al., 2008; Hunakova et al., 2014; Knatko et al., 2015; Li et al., 2014; Noh
et al., 2015).
The many actions of sulforaphane are mediated largely by activation of the Kelch-like
ECH-associated protein 1 (Keap1)/nuclear factor erythroid 2-related factor 2 (Nrf2) signalling
pathway (Baier et al., 2014; Hu et al., 2006). However, sulforaphane metabolites SF-Cys and
SF-NAC can also have direct actions as HDAC inhibitors. Sulforaphane treatment is associated
with increased apoptosis and decreases in metastasis, proliferation and angiogenesis in tumour
cells and decreases in DNA lesions after treatment with carcinogens in experimental animals and

79
tumour cell lines (Alumkal et al., 2015; Aoki et al., 2001; Clarke et al., 2008; Gao et al., 2010;
Kim et al., 2015; Noh et al., 2015). Nrf2 null mice have increased tumour incidence (Aoki et al.,
2001; Ramos-Gomez et al., 2001; Ramos-Gomez et al., 2003; Saw et al., 2014) and many of the
actions of sulforaphane are abrogated in Nrf2 null mice (Fahey et al., 2001; Kwak & Kensler,
2010; Ramos-Gomez et al., 2001). The HDAC inhibitory effects are not well characterized, but
have been linked to the acetylation of histones H3 and H4 and tied to the induction of important
tumour suppressor and pro-apoptosis proteins p21 and Bax (Gansen et al., 2015; Myzak et al.,
2006)
Sulforaphane appears to decrease the number of DNA adducts after pretreatment with
several genotoxic carcinogens: diesel exhaust (Aoki et al., 2001), 2-amino-1-methyl-6-
phenylimidazo(4,5-b)pyridine (Bacon et al., 2003), benzo[a]pyrene (Ramos-Gomez et al., 2003)
and aflatoxin B1 (Gao et al., 2010). However, its use is contraindicated with certain DNA-
damaging chemotherapeutic drugs as it can protect tumours from the chemotherapeutics
(Alumkal et al., 2015; Hunakova et al., 2014; Kim et al., 2015; Li et al., 2015a; Li et al., 2014).
Multiple studies have shown that sulforaphane-mediated induction of phase II biotransformation
pathways contributes to the decrease of reactive metabolites causing DNA adducts, but few
studies thus far have looked at sulforaphane effect on DNA repair (Jayakumar et al., 2015;
Piberger et al., 2014). In HepG2 human hepatoma cells, co-treatment of sulforaphane with PhIP
leads to a significantly reduced amount of PhIP-DNA adducts (Bacon et al., 2003). However,
treatment with sulforaphane after treatment with PhIP does not lead to any significant reduction
in PhIP-DNA adducts, leading the authors to conclude sulforaphane does not have a significant
effect on DNA repair activities (Bacon et al., 2003). Benzo[a]pyrene- and 1,6-dinitropyrene-
DNA adducts are significantly reduced in human mammary epithelial cells after treatment with
sulforaphane, but the reduction could not be explained by increased detoxification (Singletary &

80
MacDonald, 2000). Additionally, Nrf2 inhibition caused a significant decrease in UV-B induced
homologous recombination repair in A549 cells (Jayakumar et al., 2015). Also, sulforaphane-
induced H3 histone acetylation may facilitate DNA repair, as relaxation of the chromatin allows
for binding of repair proteins (Duan & Smerdon, 2014).
DNA damage caused by exposure to exogenous chemicals is repaired largely by
nucleotide excision repair (NER; Rechkunova et al., 2011). This multi-step process involves over
30 proteins which excise and replace adducted nucleotides. Persistence of adducts may lead to
mutations and apoptosis or carcinogenesis. NER can be induced by many exogenous chemicals
through multiple pathways via increased expression of key proteins (Kang et al., 2011; Lefkofsky
et al., 2015) or by post-translational modifications such as acetylation, ubiquitylation and
SUMOylation (Beli & Jackson, 2015; Choi et al., 2015; Coin et al., 2004; Donninger et al., 2015;
Kang et al., 2011; Li et al., 2015b; Li et al., 2011a; Li et al., 2013b; Poulsen et al., 2013; Silver et
al., 2011; van Cuijk et al., 2015). Decreased NER activity can be caused by decreases in key
NER proteins through adducts or mutations in regulatory genes (Lefkofsky et al., 2015). There
has yet to be a report of sulforaphane’s effects on NER activity per se in vivo. However, in vitro,
sulforaphane directly displaced the zinc-finger domain of a key NER protein, XPA, inhibiting its
ability to bind to damaged DNA (Piberger et al., 2014). Sulforaphane interacts with many
proteins that are involved in NER regulation including p53 (Kim et al., 2015; Li et al., 2011a;
Piberger et al., 2014; Saw et al., 2014) and p21 (Atwell et al., 2015; Colton et al., 2006; Dutto et
al., 2016; Mocquet et al., 2008).
To our knowledge, this is the first study to examine the in vivo effect of sulforaphane
treatment on NER activity, and it includes an assessment of levels and activities of several key
proteins in the NER pathway. First we found the dose and time point of maximal sulforaphane

81
effect on Nrf2 regulated gene induction. We then measured the change in NER activity using an
in vitro NER activity assay. Finally, we measured the change in key NER protein levels and the
ability of key NER proteins to bind to damaged DNA.
Materials and methods 4.2
4.2.1 Animal treatment for dose response and time course of sulforaphane effects
Female CD-1 mice, aged 6-9 weeks (Charles River Canada Inc., St. Constant, PQ,
Canada), were housed with a 12-hour light/dark cycle. CD-1 mice were chosen for this study due
to their use in a previous NER activity study (Bedard et al., 2005), and their use in sulforaphane
kinetics and in vivo studies on Nrf2 regulated genes (Li et al., 2013a; Philbrook & Winn, 2014).
Animals were provided AIN-76A purified diet (BIOSERV, Flemington, New Jersey, USA) and
water ad libitum. Dose of 0, 10, 20, 50, or 100 mg/kg sulforaphane was given in a volume of 200
µL corn oil by gavage. After 3 hours, mice were killed by cervical dislocation, and livers and
lungs were removed and flash frozen in liquid nitrogen. After the dose was determined, for the
time course, mice were treated with 100 mg/kg sulforaphane in 200 µL corn oil or 200 µL corn
oil by gavage. In the timecourse, mice were killed after 1, 3, 6, 12 or 24 h; livers and lungs were
removed and flash frozen in liquid nitrogen. All animals were cared for in accordance with the
guidelines of the Canadian Council on Animal Care and the experimental procedures involving
animals were approved by the Queen’s University Animal Care Committee.
4.2.2 Analysis of mRNA from Nrf2-regulated genes
Total RNAs from mouse lungs or livers of control and sulforaphane-treated mice were
isolated from 20-30 mg tissue with the RNeasy mini kit (Qiagen, Mississauga, ON, Canada).
Total RNA quality was confirmed by formaldehyde-agarose gel electrophoresis. cDNAs were
synthesized from 5 µg of total RNA following the protocol for TaqMan High Capacity cDNA

82
Reverse Transcription (Applied Biosystems, Grand Island, NY, USA). PCR reactions were
carried out using 200 ng cDNA product, each tube containing 7 µL RNase-free water, 10 µL
TaqMan Universal PCR Master Mix and 1µL TaqMan Gene Expression assay kit for each of the
genes of interest (Cat# 4331182; HO-1: Mm00516005_m1; GCLC: Mm01253561_m1; GAPDH:
Mm99999915_g1). Levels of reverse transcription product were measured using fluorescein
amidite fluorescence collected during real-time PCR on a BIORAD CFX96 thermal cycler
(Hercules, CA, USA). Relative mRNA levels were quantified by normalization with reference
gene GAPDH and expressed using the delta-delta Ct method.
4.2.3 Preparation of cell-free whole tissue nuclear protein extracts
Nuclear protein extracts from 1.0 to 1.3 g of tissue were prepared from livers or pooled
lungs of four mice as described in section 2.2.2. Extracts from lungs or livers contained between
5 to 20 mg/mL protein. Extracts were frozen in liquid nitrogen and stored at -80°C.
4.2.4 Preparation of damaged plasmid DNA
Pyridyloxobutylated plasmid DNA was prepared as described in section 2.2.3.
4.2.5 In vitro DNA repair synthesis assay
The repair synthesis assay was performed as described in section 2.2.4.
4.2.6 Immunoblot analysis of NER incision protein levels in mouse liver nuclear protein
extracts
The immunoblot analysis of NER protein levels in mouse liver nuclear protein extracts
was performed as described in section 2.2.5.

83
4.2.7 Analysis of binding of NER recognition proteins to damaged DNA
An assay to assess the binding of NER repair proteins to damaged DNA was performed
as described in section 2.2.6 using mouse liver nuclear protein extracts.
4.2.8 Data analysis
For dosage study, Kruskal-Wallis test was used to analyze qRT-PCR activity. In the time
point study, multiple Mann-Whitney U tests were used to analyze qRT-PCR activity data at the
individual time points. Statistically significant differences in repair activity and NER protein
levels between treatment groups were determined by unpaired Student’s t-test. Due to inter-
experiment variability, Mann-Whitney U test was used comparing DNA-associated NER protein
binding (Subsection 4.2.7). P<0.05 was considered significant in all cases. GraphPad Prism 6
was used to graph all results.
Results 4.3
4.3.1 Response of two Nrf2 regulated genes to in vivo treatment with sulforaphane
To assess activation of Nrf2 signaling, expression of two Nrf2 regulated genes, heme
oxygenase-1 (HO-1) and glutamate-cysteine ligase catalytic subunit (GCLC), was measured.
mRNA levels for each gene were normalized to values obtained for glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) mRNA. At three hours post treatment, GCLC mRNA levels were
elevated with 100 mg/kg sulforaphane in lung tissue, and were increased with 50 and 100mg/kg
sulforaphane in liver tissue (Figure 4.1). HO-1 mRNA levels were elevated with 50 and 100
mg/kg sulforaphane in liver only (Figure 4.1). It was decided that 100 mg/kg sulforaphane would
be used for the timecourse, as it most consistently increased mRNA levels of these Nrf2 reporter
genes.

84
Figure 4.1 mRNA levels for dose-response relationship of two Nrf2 regulated genes relative
to control, GCLC and HO-1; 3 h after treatment with sulforaphane (SF; n=4). The data are
presented as mean + SD. (*) Significantly different from control (p<0.05).
GCLC lung
0 10 20 50 100
0
2
4
6
8
Dose mg SF/kg mouse
GC
LC m
RN
A (re
lativ
e to
con
trol)
*
GCLC liver
0 10 20 50 100
0
2
4
6
8
Dose mg SF/kg mouse
GC
LC m
RN
A (re
lativ
e to
con
trol)
* *
HO-1 lung
0 10 20 50 100
0
2
4
6
8
Dose mg SF/kg mouse
HO
-1 m
RN
A (re
lativ
e to
con
trol)
HO-1 liver
0 10 20 50 100
0
2
4
6
8
Dose mg SF/kg mouse
HO
-1 m
RN
A (re
lativ
e to
con
trol)
**

85
4.3.2 Timecourse of responses of two Nrf2 regulated genes to 100 mg/kg of sulforaphane
Sulforaphane (100mg/kg) in 200 µL corn oil was administered to female CD-1 mice that
were killed at 1, 3, 6, 12, or 24 h later. In mouse lung, there was a significant increase in HO-1
mRNA at 3 h (Figure 4.2; p<0.05) and an apparent trend at 6 h (Figure 4.2; p>0.05), as well as a
significant increase of GCLC at 3 and 6 h (Figure 4.2; p<0.05). In mouse liver, there was
significant increase of HO-1 at 6 h (Figure 4.2; p<0.05) and a trend at 3 h (Figure 4.2; p>0.05);
and a significant increase of GCLC at 1, 3 and 6 h (Figure 4.2; p<0.05).

86
Figure 4.2 Timecourse of effects for two Nrf2 regulated genes HO-1 and GCLC in lung and
liver after treatment with 100mg/kg of sulforaphane (SF) or corn oil (n=4). The data are
presented as mean + SD. (*) Significantly different from control (p<0.05).
HO-1 lung
corn
oil SF
corn
oil SF
corn
oil SF
corn
oil SF
corn
oil SF0
2
4
6
8
10
Treatment
Rel
ativ
e Ex
pres
sion
1hr
3hr
6hr
12hr
24hr*
HO-1 liver
corn
oil SF
corn
oil SF
corn
oil SF
corn
oil SF
corn
oil SF0
2
4
6
8
10
Treatment
Rel
ativ
e Ex
pres
sion
1hr
3hr
6hr
12hr
24hr
*
GCLC lung
corn
oil SF
corn
oil SF
corn
oil SF
corn
oil SF
corn
oil SF0
2
4
6
8
10
Treatment
Rel
ativ
e Ex
pres
sion
1hr
3hr
6hr
12hr
24hr
*
*GCLC liver
corn
oil SF
corn
oil SF
corn
oil SF
corn
oil SF
corn
oil SF0
2
4
6
8
10
Treatment
Rel
ativ
e Ex
pres
sion
1hr
3hr
6hr
12hr
24hr***

87
4.3.3 Effect of in vivo treatment of mice with sulforaphane on repair synthesis activity in
mouse liver and lung nuclear protein extracts
The timepoints 6 h and 12 h post-treatment were chosen for NER activity studies because
protein synthesis can sometimes lag behind increases in mRNA transcripts (Greenbaum et al.,
2003). At 6 h following in vivo sulforaphane treatment, repair synthesis activity of mouse liver
extracts toward POB damage showed no significant change relative to control (Figure 4.3).
However, repair activity increased by 99% at 12 h post in vivo dosing in mouse liver (Figure 4.3).
Due to the concentration of nuclear protein extract required for the repair assay, and the amount
of lung tissue needed for nuclear protein extracts, we were unable to treat sufficient animals to
test 6 h lung NER activity. There was no significant change in NER activity in 12 h mouse lung
nuclear extract (Figure 4.3).
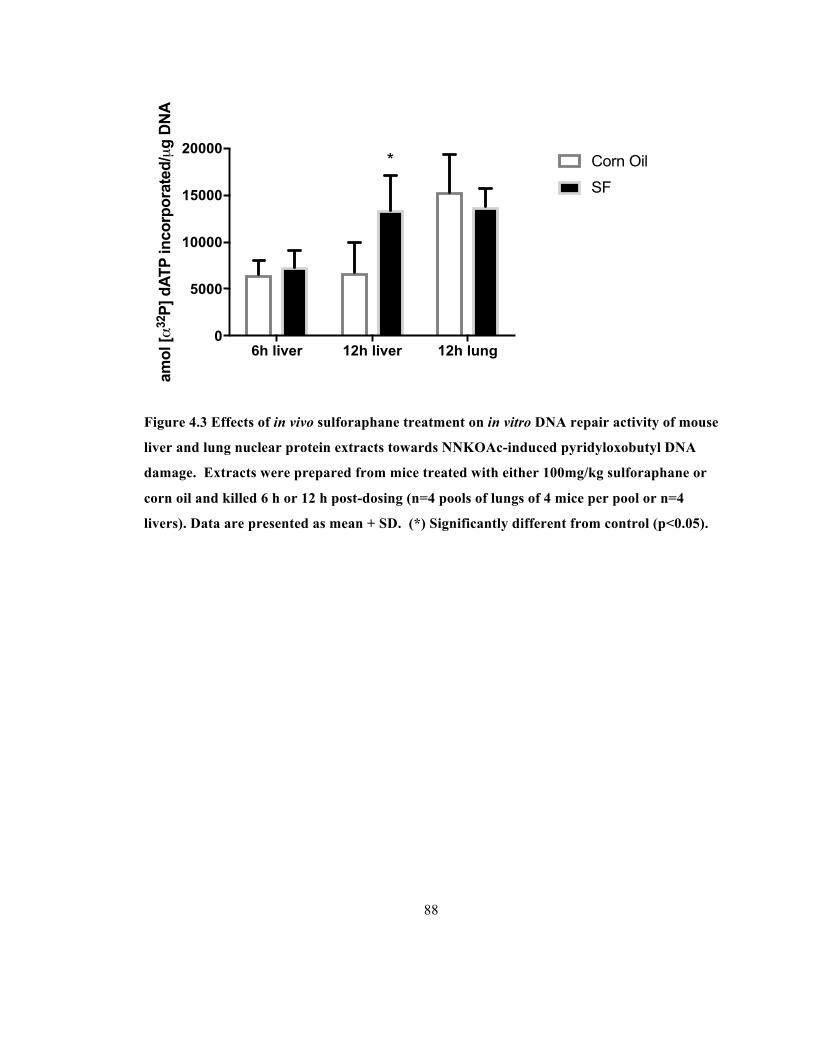
88
Figure 4.3 Effects of in vivo sulforaphane treatment on in vitro DNA repair activity of mouse
liver and lung nuclear protein extracts towards NNKOAc-induced pyridyloxobutyl DNA
damage. Extracts were prepared from mice treated with either 100mg/kg sulforaphane or
corn oil and killed 6 h or 12 h post-dosing (n=4 pools of lungs of 4 mice per pool or n=4
livers). Data are presented as mean + SD. (*) Significantly different from control (p<0.05).
6h liver 12h liver 12h lung0
5000
10000
15000
20000am
ol [α
32P
] dAT
P in
corp
orat
ed/µ
g D
NA
Corn Oil
SF
*

89
4.3.4 Effects of in vivo treatment of mice with sulforaphane on levels of NER proteins in
mouse liver nuclear protein extracts
Since sulforaphane had a significant effect on NER activity in mouse liver extracts, levels
of recognition NER proteins in mouse liver extracts following sulforaphane treatment were
examined (Figure 4.4). At 12 h post-treatment, liver extracts showed no significant changes in
early phase NER recognition and incision proteins XPC, XPA, XPB and p53.

90
Figure 4.4 a) Relative density of each NER protein in mouse liver extracts isolated from
corn oil or sulforaphane-treated mice 12 h post-dosing, relative to a constant internal
standard extract (CTL). The data are presented as mean + SD. b) Representative
immunoblots of the relative levels of each key NER protein in mouse liver extracts. S=
nuclear protein extracts from sulforaphane treated mice, C=nuclear protein extracts from
corn oil treated mice; all experiments n=4.

91
4.3.5 Effect of in vivo treatment of mice with sulforaphane on key NER protein DNA
binding in mouse liver nuclear protein extracts
To determine if sulforaphane affects the interactions between key NER proteins and
damaged DNA, an assay was used that measures the binding of NER repair proteins to damaged
DNA during the NER reaction. Binding of the three recognition and incision NER proteins was
not significantly different between liver extracts from sulforaphane treated and control animals at
12 h after treatment (Figure 4.5).

92
Figure 4.5 Effect of in vivo sulforaphane treatment on in vitro NER protein binding to
pyridyloxobutyl-adducted DNA. All data are presented as mean + SD. (all n=4, p>0.05)
XPCXPA
XPB0
1
2
3
4
12h Liver Extracts
Rel
ativ
e B
indi
ng
Corn OilSulforaphane

93
Discussion 4.4
In this study, it was confirmed that sulforaphane increases expression of two genes
known to be induced by Nrf2 activation: GCLC and HO-1. Furthermore, at 100 mg/kg,
sulforaphane increased NER activity by 99% in liver tissue in an in vitro NER activity assay 12 h
after treatment. Recently, sulforaphane has been under investigation for its multiple effects
against cancer (Lenzi et al., 2014). Sulforaphane is known to inhibit cancer initiation after
treatment with a carcinogen, as well as promotion and progression of tumour cells (Clarke et al.,
2008; Kwak & Kensler, 2010). Sulforaphane has been used prophylactically to decrease DNA
adducts after treatment with several DNA damaging agents (Aoki et al., 2001; Bacon et al., 2003;
Gao et al., 2010; Ramos-Gomez et al., 2003). In many cases, it was theorized that sulforaphane
induces detoxification pathways and clears metabolites before they can have a genotoxic effect,
as sulforaphane induces phase II biotransformers and phase III transporters (Atwell et al., 2015;
Clarke et al., 2008; Knatko et al., 2015; Rajendran et al., 2013). Here, we describe another
mechanism that can decrease genotoxicity—upregulation of NER, thereby enhancing removal of
adduct-damaged DNA regions. This is the first report of an effect of in vivo treatment of
sulforaphane on NER activity.
The induction of established Nrf2 regulated genes with 100 mg/kg sulforaphane at 3 to 6
h post-treatment is consistent with previous work (Clarke et al., 2011; Hu et al., 2006; Keum,
2011; Li et al., 2013a). Sulforaphane treatment of C57BL/6J mice showed a similar effect- a
significant increase of HO-1 at 3 h and no significant increase at 12 h in treated mouse liver (Hu
et al., 2006). Additionally, Philbrook and Winn showed a significant increase in GCLC mRNA
transcripts in mouse liver of pregnant and non-pregnant CD-1 mice only after 6 h of sulforaphane
treatment (Philbrook & Winn, 2014). Initially, only the liver tissue could be used for the NER
activity experiments due to the amount of tissue required for lung nuclear protein extracts (n of 1

94
requires 4 whole mouse lungs). As a result, we chose the 6 h timepoint for the NER activity
assay, as both liver HO-1 and GCLC are significantly increased (Figure 4.2). Metabolism and
clearance of sulforaphane are rapid in experimental animals and humans (Clarke et al., 2011;
Petri et al., 2003; Shapiro et al., 2006). Female CD-1 mice receiving a large bolus dose of
sulforaphane-enriched broccoli extract (approximately 2.5 mg/g) excreted the sulforaphane by 24
h (Li et al., 2013a). Small concentrations of sulforaphane appear to have important effects on
Nrf2 regulation and HDAC inhibition as sulforaphane can have different and opposite effects at
high and low concentrations (Alfieri et al., 2013; Zanichelli et al., 2012a). As a result, we also
chose to look at the effect of sulforaphane treatment on NER activity at the 12 h timepoint, which
is after the effects of Nrf2 on HO-1 and GCLC have subsided.
There are more than 30 proteins involved in NER, and multiple studies have shown these
proteins can influence NER activity through post-translational ubiquitylations, SUMOylations
and increased or decreased abundance (Kang et al., 2010b; Kang et al., 2011; Huang &
D’Andrea, 2006; Poulsen et al., 2013; Silver et al., 2011; van Cuijk et al., 2015). Recently, three
key proteins have been identified as the triple check, rate-limiting step in NER (discussed in
section 1.3.2.1): xeroderma pigmentosum (XP) class C (XPC), XP class A (XPA) and
transcription factor II H (TFIIH) (Marteijn et al., 2015; Li et al., 2015b). As discussed in
Marteijn et al., NER progresses sequentially after initial sensing of the damage by XPC (2015).
XPC recruits XPB-containing TFIIH and XPA, which both verify the DNA damage and protect
the DNA strand. Other research has shown a significant change in the ability of XPC, XPA and
XPB to bind to damaged DNA in an in vitro, damaged-DNA NER protein binding assay after
exposure to the carcinogen NNK (Chapter 2 and Chapter 3). Here, we show a significant
sulforaphane-induced increase in liver NER activity at 12 h without a significant change in levels
or damaged DNA binding activities of specific NER proteins (Figure 4.3-Figure 4.5). The

95
increased NER activity may be due to a mechanism occurring downstream of XPA and TFIIH
binding of the damaged DNA strand (Figure 1.1). There are other, less understood NER proteins
downstream of TFIIH that may be involved in the increased NER activity, including proliferating
cell nuclear antigen (PCNA; Cazzalini et al., 2014) and XPF-ERCC1 (Su et al., 2012; Tomicic et
al., 2011). PCNA is recruited to the DNA strand after DNA unwinding by TFIIH, and binds to
RPA, which acts to stabilize the replication complex (Overmeer et al., 2011). XPF-ERCC1 is a
nuclease that makes the incision to the 5’ of the DNA lesion, and binds to XPA (Fadda, 2013;
Matsunaga et al., 1995). Thus, these are important proteins to examine in the future.
Sulforaphane has the potential to activate antioxidant response elements and multiple
pathways that are involved in NER regulation (Rajendran et al., 2013). p53 is important for cell
cycle arrest allowing for prolonged repair before replication of damaged DNA and presence of
p53 appears to affect NER activity in some in vitro repair assays (Chang et al., 2008). However,
p53 was not a cellular target of sulforaphane in HCT116 and XP12RO cells (Piberger et al.,
2014) and we show there is no significant effect on p53 protein amount in sulforaphane-treated
liver nuclear protein extracts (Figure 4.4). Therefore, it is unlikely the significant increase in
NER activity in sulforaphane treated mouse liver at 12 h (Figure 4.4) is due to p53.
Another of the proteins induced by sulforaphane is p21, which has multiple roles as a
cell-cycle regulator and a regulator of transcription, cell motility, apoptosis and DNA repair. This
protein is likely induced via the HDAC inhibitory actions of sulforaphane metabolites SF-NAC
and SF-Cys and these effects are sustained (Myzak et al., 2004; Myzak et al., 2006). In fact,
Myzak et al. (2006) showed the HDAC inhibitory effects of sulforaphane persisted to 48h in the
colonic mucosa of Apcmin mice after a single bolus dose of either sulforaphane or SF-NAC.
Specifically, p21 appears to have an effect on NER; however there are multiple reports that p21
has positive, neutral or negative effects on total NER activity (Dutto et al., 2016).

96
One previous study reported a significant decrease in TC-NER activity after in vitro
treatment of HCT116 and XP12RO human colon cancer and fibroblast cells with sulforaphane
(Piberger et al., 2014). That study concluded sulforaphane caused an uncoupling of the zinc-
finger domain of XPA which abrogates damaged-DNA binding of XPA (Piberger et al., 2014).
Here, rather than an impairment of in vitro NER activity, we observed a 99% increase in NER
activity in liver tissue 12 h post in vivo treatment with sulforaphane (Figure 4.3). We also see no
significant change in the binding of XPA to damaged DNA at 12 h post-treatment (Figure 4.5).
Piberger et al. concluded that only early-stage repair (TC-NER) was affected by sulforaphane in
these cell lines, whereas our in vitro repair assay is optimized to measure effects on late-stage
repair (global-genome NER (GG-NER)). We focus on GG-NER because nontranscribed genes or
genes on the nontranscribed strand of active genes (both repaired by GG-NER) are better
predictive markers for carcinogen-induced tumourigenesis than mutations in actively transcribed
genes (Balajee & Bohr, 2000).
It is known that carcinogens can increase NER activity (Bedard et al., 2005; Brown &
Massey, 2009; Choi et al., 2015; Protić et al., 1988). As a chemopreventive strategy, it could be
valuable to identify a non-genotoxic chemical that can cause similar effects. Although it is
unclear whether sulforaphane is completely non-genotoxic (Baasanjav-Gerber et al., 2011), our
findings show it may be a candidate for this purpose.

97
Chapter 5
General Discussion
Overall conclusions 5.1
Endogenously formed metabolic products as well as environmental chemicals and
radiation constantly threaten DNA integrity. DNA damage loads may total 104-105 DNA lesions
per mammalian cell per day, and it is this damage that can initiate carcinogenesis (Swenberg et
al., 2011). It is therefore imperative that the damage is removed and the DNA repaired in a
timely and error-free manner through one of several DNA repair pathways (Bates, 2010). It has
been established experimentally that there are several endogenous pathways and exogenous
agents that can affect the activity of DNA damage repair pathways (Bates, 2010). Additionally,
defects in DNA repair pathways predispose individuals to cancer; for example, individuals with
xeroderma pigmentosum have a high incidence of skin cancer. GG-NER is one DNA damage
repair pathway whose activity is changed with exposure to certain exogenous agents (Bedard et
al., 2005; Brown & Massey, 2009; Cho, 2002; Choi et al., 2015; Fatur et al., 2003; Li et al.,
2015b; Marteijn et al., 2015; Mulder et al., 2014; Nollen et al., 2009). Several successive steps
are important for error-free repair through GG-NER, and only recently has our understanding of
the successive steps of GG-NER and their regulation expanded. Additionally, this research into
GG-NER regulation has exclusively been done in vitro. In this thesis, we focussed on one part of
the GG-NER pathway, the recognition and early incision phase, and the effects of in vivo
treatment with NNK and sulforaphane on specific protein levels and activities.
Earlier studies showed that treatment of mice with tumourigenic doses of NNK or
aflatoxin B1increased NER activity in mouse liver and decreased it in lung (Bedard et al., 2005;
Brown & Massey, 2009). Additionally, p53 status altered aflatoxin B1 mediated change of NER

98
(Mulder et al., 2014). However, the specific role of NER recognition proteins, p53 and the
change of NER activity across a 72 h period had not been investigated after treatment with NNK.
There was a significant decrease in NER activity at 12 h post NNK treatment and a
significant increase in NER activity at 24 h in mouse liver (Chapter 2) and a significant decrease
in NER activity at 24 h in mouse lung (Chapter 3). NNK treatment caused no significant change
in levels of NER proteins XPC, XPA, XPB and p53 in mouse liver or lung nuclear protein
extracts at 12 and 24 h (Chapter 2, liver; Chapter 3, lung). At 12 h in liver, there was no change
in XPC binding to damaged DNA, but decreases in XPA and XPB binding (Chapter 2).
Conversely, at 24 h in mouse liver, there was a decrease in XPC binding and increases in XPA
and XPB binding (Chapter 2). In comparing the damaged-DNA binding activities of recognition
proteins at 24 h in mouse lung, there was a significant increase in XPC binding, and significant
decreases in XPA and XPB binding (Chapter 3). These results indicated NNK can have time-
and tissue-dependent effects on NER and the ability of specific NER proteins to bind to damaged
DNA, and it is likely this effect is due in part to changes in recognition-protein damaged-DNA
binding activity rather than altered levels of the proteins.
Following a single dose of DMSO, there was a significant decrease in NER activity at 2 h
in mouse liver relative to an i.p. saline treatment (Appendix A). DMSO is often used as a vehicle
for in vivo and in vitro administration of chemicals, but its effects are often not described. The
mechanism by which DMSO affects NER activity was not pursued further as DMSO was not
used in this thesis, but it shows the importance of understanding the vehicle used in future studies.
Sulforaphane has been under investigation for multiple effects leading to a decrease in
DNA adducts, including those repaired by NER (Gerhauser, 2013; Singletary & MacDonald,
2000; Topè & Rogers, 2009). However, the effect of in vivo treatment with sulforaphane on NER
activity had not been studied previously. In order to gauge the effect of sulforaphane treatment

99
on Nrf2, mRNA levels of two Nrf2 regulated genes were assayed in a dose and time course study.
We found only the 100 mg/kg sulforaphane dose and the 6 h time point consistently caused an
increase in GCLC and HO-1 mRNA levels in mouse liver and lung (Chapter 4). There was no
effect of sulforaphane on NER activity at 6 h in mouse liver or at 12 h in mouse lung nuclear
protein extracts, but there was a significant increase in NER activity in mouse liver at 12 h
(Chapter 4). Sulforaphane treatment caused no significant change in levels of NER proteins
XPC, XPA, XPB and p53 in 12 h mouse liver nuclear protein extract and there was no significant
change in the damaged-DNA binding activities of recognition proteins XPC, XPA and XPB
(Chapter 4). This indicates that, despite increasing NER activity at 12 h, sulforaphane doesn’t
appear to do so by increasing the ability of these recognition proteins to bind to damaged DNA,
and the cause of the increase of NER activity remains to be elucidated. Suggestions for possible
mechanisms are discussed below.
Future directions 5.2
5.2.1 Post-translational modifications of NER proteins as a basis for observed changes in
GG-NER activity
Only recently have specific proteins been identified that induce changes in DNA repair
activity without changes in protein levels, and this generally occurs through post-translational
modifications involving ubiquitin or ubiquitin-like compounds (Gali et al., 2012; Huang &
D’Andrea, 2006; Kang et al., 2010b; van Cuijk et al., 2015). The covalent attachment of the 76
amino acid residue protein ubiquitin and the approximately 100 residue small ubiquitin-related
modifier (SUMO) is the most common. Although ubiquitylation and SUMOylation were initially
discovered as mechanisms targeting the degradation of proteins by the proteasome, their actions
more recently have been recognized to include regulation of protein activity, localization and
interactions (Bergink & Jentsch, 2009; Komander & Rape, 2012; Jackson & Durocher, 2013).

100
Other post-translational modifications such as phosphorylation and acetylation can also affect
DNA repair (Cazzalini et al., 2014; Donninger et al., 2015; Duan & Smerdon, 2014; Fan & Luo,
2010; Gansen et al., 2015). In fact, all DNA repair pathways appear to be regulated in some way
by phosphorylation, acetylation, ubiquitylation and/or SUMOylation, and recent work has shown
that several steps in the regulation of the GG-NER pathway are dependent on acetylation,
ubiquitylation and SUMOylation (Rüthemann et al., 2016).
Early stage NER has recently been identified as a key stage for NER activity changes
after carcinogen exposure (Choi et al., 2015; Donninger et al., 2015; Li et al., 2015c; Marteijn et
al., 2015), and recent in vitro work has indicated the importance of post-translational
modifications of key NER proteins in response to bulky DNA damage (Beli & Jackson, 2015;
Dou et al., 2010; Gali et al., 2012; Hoege et al., 2002; Huang & D’Andrea, 2006; Kang et al.,
2010b; Poulsen et al., 2013; van Cuijk et al., 2015). The cascade of effects after carcinogen
exposure is still being revealed, but recent research into the effects of exposure of cultured cells
to UV light has allowed for a better understanding of the sequence of events and regulation of
these events (Marteijn et al., 2015). However, until now this research has not been expanded
with other agents of DNA damage, or after in vivo exposure. It is possible that the differences in
NER protein damaged-DNA binding observed after treatment with NNK (Chapters 2 and 3)
could be caused by changes to post-translational modifications of XPC, XPA and TFIIH (XPB).
Additionally, it is possible the modification of another component of the NER pathway causes the
increase in NER activity observed after treatment with sulforaphane (Chapter 4). Expansion of
our understanding of the NER regulation events after in vivo exposure sulforaphane or NNK will
be an important priority in the future.

101
5.2.1.1 XPC binding to damaged DNA
Recognition protein XPC can be ubiquitylated at multiple sites and SUMOylated (van
Cuijk et al., 2015). After UV exposure, XPC is both ubiquitylated at lysine 48 and SUMOylated,
which increase XPC damaged-DNA binding affinity and protect XPC from degradation,
respectively (Sugasawa et al., 2005; Wang et al., 2005). XPC release from damaged DNA is then
mediated by RNF111, a ubiquitin ligase that ubiquitylates XPC at lysine 63 and is a critical
component of the TGF-β signaling pathway (Miyazono & Koinuma, 2011; Poulsen et al., 2013;
van Cuijk et al., 2015). Release of the XPC protein allows the incorporation of the endonucleases
XPG and ERCC1/XPF into the repair complex, and removal of RNF111 stalls in vitro NER of
UV-induced DNA lesions (Poulsen et al., 2013; van Cuijk et al., 2015). No study has examined
the effect of NNK on RNF111, but abrogation of the TGF-β signalling pathway significantly
increases NNK-induced lung tumour frequency (Kanehira et al., 2005). Additionally, it is
unclear whether XPC binding to POB DNA damage is affected by these post-translational
modifications as has been observed for UV-induced DNA damage. XPC binding to damaged
DNA increased in lung nuclear protein extracts 24 h post NNK, but decreased in liver extracts 24
h post NNK (Chapter 2 and Chapter 3). It is possible the ubiquitylation/ SUMOylation of XPC is
important in the organ and time dependent changes in XPC binding activity after NNK treatment,
and this should be a focus of future research.
5.2.1.2 XPA binding to damaged DNA
The recognition protein XPA can be ubiquitylated and acetylated (Donninger et al., 2015;
Kang et al., 2011). In mice, XPA degradation is controlled by ubiquitylation in a diurnal cycle
and as a result NER activity varies throughout the day (Kang et al., 2010b; Kang et al., 2011).
XPA binding to damaged DNA, however, is regulated by deacetylation (Fan & Luo, 2010; Kang
et al., 2011). After UV exposure, induction of sirtuin 1 (SIRT1) via the RASSF1A tumour

102
suppressor appears to decrease the level of acetylated XPA (Donninger et al., 2015). SIRT1 is an
important mammalian deacetylase with roles in the regulation of gene expression, cell survival,
proliferation, differentiation, metabolism, immune response and carcinogenesis (Wang & Chen,
2013). Deacetylated XPA is required for efficient repair of bulky adducts (Donninger et al.,
2015; Kang et al., 2011), and knockdown of the SIRT1 gene significantly decreases NER activity
in vitro (Fan & Luo, 2010). There was an increase in XPA binding in liver extracts 12 h and 24 h
post NNK treatment, and a decrease in XPA binding in lung extracts 24 h post NNK treatment
(Chapter 2 and Chapter 3). It is possible that the acetylation of XPA is important in the observed
changes in binding activity after NNK treatment, and this should be a focus of future research.
SIRT1 can be detected using an antibody against SIRT1.
5.2.1.3 TFIIH binding and activity
TFIIH binding activity is regulated by the binding of XPC and XPA to damaged DNA
(Coin et al., 2008; Choi et al., 2015; Donninger et al., 2015; Fan & Luo, 2010; Li et al., 2015b).
Whereas the presence of XPC recruits TFIIH to the damaged site, the presence of XPA stimulates
the release of the CAK subunit from the TFIIH subunit (Coin et al., 2008) which significantly
increases the binding efficiency of TFIIH to the area near the lesion (Choi et al., 2015; Donninger
et al., 2015; Fan & Luo, 2010). It appears that the presence of XPA and the release of the CAK
subunit are more important than XPC presence for TFIIH binding activity, so any change to XPA
binding can cause a change in TFIIH binding (Li et al., 2015b). Additionally, the lack of any
significant change in XPB or XPA protein levels (Chapters 2 and 3) suggests that the changes in
XPB binding activity could be the result of the change in XPA binding activity. Also, XPB and
XPD are helicases, and it is unclear whether protein binding is concomitant with helicase activity.
TFIIH helicase activity is closely tied with CAK release, which is another important connection
between TFIIH and XPA (Coin et al., 2008; Li et al., 2015b). However, there is no information

103
on whether NNK or sulforaphane affect the helicase activity of XPB or XPD. It is possible to
quantify the activity of XPB and XPD as helicases in an assay (Shin et al., 2005), but this assay
has never been optimized for use with whole tissue protein extracts. Therefore, it is also
important for future studies to examine the effects of NNK and sulforaphane on TFIIH binding
and XPB and XPD helicase activity.
5.2.2 Effects of sulforaphane and NNK on other NER proteins
5.2.2.1 RPA
In NER, RPA forms the linkage between pre-incision factors such as XPC, XPA and
TFIIH and post-incision factors such as proliferating cell nuclear antigen (PCNA), DNA
polymerases and ligases, protecting the single stranded DNA after damage excision (Overmeer et
al., 2011; Gourdin et al., 2014; Riedl et al., 2003). Like other NER proteins, RPA is subject to
multiple post-translational modifications, including phosphorylation, poly-ADP ribosylation,
ubiquitylation and SUMOylation (Dou et al., 2010; Maréchal & Zou, 2015; Hass et al., 2012;
Binz et al., 2004) and is involved in both DNA repair and replication. After cellular DNA
damage such as UV-induced damage, RPA is hyper-phosphorylated, which down-regulates its
role in DNA replication and appears to promote its activity in DNA repair (Binz et al., 2004).
However, the binding activity of RPA after DNA damage has never been quantified. In the
future, the damaged-DNA NER protein binding assay used in this thesis could be used to probe
for RPA binding activity. The other post-translational modifications appear to regulate the
activation of the ATR checkpoint, which controls cell cycle arrest after DNA damage, and
promote largely homologous recombination DNA repair; however, their roles in NER specifically
are not yet understood (Gourdin et al., 2014; Hass et al., 2012; Maréchal & Zou, 2015). RPA is
integral for NER, and any damage or dysregulation of RPA can have a detrimental effect on NER

104
activity (Guven et al., 2015). Therefore, it is important to understand the role of post-
translational modifications on RPA binding to NER proteins and ssDNA in NER.
5.2.2.2 PCNA
During NER, PCNA is recruited to the DNA strand after DNA unwinding by TFIIH, and
binds to RPA, which acts to stabilize the replication complex (Overmeer et al., 2011). PCNA
binds the dually incised DNA and recruits polymerases for DNA elongation. PCNA can be
ubiquitylated, SUMOylated and acetylated (Cazzalini et al., 2014; Gali et al., 2012; Kumar &
Saha, 2015; Mocquet et al., 2008; Riedl et al., 2003). Ubiquitylation and SUMOylation were the
first post-translational modifications to be unambiguously identified for PCNA (Hoege et al.,
2002), and these modifications are important for DNA replication and maintaining genomic
integrity (Gali et al., 2012; Hoege et al., 2002; Kannouche et al., 2004; Kumar & Saha, 2015;
Kubota et al., 2013). On the other hand, acetylation appears to be important in NER regulation
(Cazzalini et al., 2014). PCNA acetylation occurs in proliferating cells and after UV damage, and
may be partially controlled by p21 (Cazzalini et al., 2014). p21 has multiple roles as a cell-cycle
regulator, a regulator of transcription, and in cell motility, apoptosis and DNA repair (Cazzalini et
al., 2010; Colton et al., 2006; Dutto et al., 2016; Lee et al., 2009). Specifically, p21 appears to
have an effect in NER; however, different studies have reported positive, neutral or negative
effects on total NER activity (Cazzalini et al., 2010; Dutto et al., 2016). Many of these effects
may be due to varied experimental conditions and cell types, and a current hypothesis is that in
vivo p21 has positive effects on NER activity through its interactions with PCNA (Dutto et al.,
2016).
The results in Chapter 4 show that there is an increase in DNA repair activity in mouse
liver 12 h after sulforaphane exposure, but this was not accompanied by changes in levels of
XPC, XPA, XPB or p53 or in the binding of XPC, XPA or XPB to damaged DNA. The

105
explanation for upregulated NER activity in the absence of changes in these NER proteins
remains to be elucidated, but PCNA acetylation could conceivably play a role. The effect of
sulforaphane on PCNA acetylation has not been reported, but the protein p21 is affected by
sulforaphane treatment (Chen et al., 2009; Cho et al., 2014; Ho et al., 2009). Future experiments
should examine the role of p21 in the sulforaphane-mediated NER activity increase reported in
Chapter 4.
5.2.2.3 Post-translational modifications of p53
As discussed in previous chapters, p53 has multiple effects on NER including indirect
effects on the transcription of NER proteins DDB2 and XPC and direct effects through binding to
damaged DNA and to NER proteins (Adimoolam & Ford, 2003; Bhana et al., 2008; Chang et al.,
2008; Dregoesc et al., 2007; Lakin & Jackson, 1999). Additionally, p53 is frequently mutated in
human tumours (Rodin & Rodin, 2000). In this thesis, treatment with NNK or sulforaphane
caused no significant change in levels of p53 protein in nuclear protein extracts (Chapters 2-4).
However, p53 is known to be controlled by many post-translational modifications including
phosphorylation, dephosphorylation and acetylation, which mediate its cell cycle arrest, apoptosis
and DNA damage repair effects (Lakin & Jackson, 1999; Ashcroft et al., 1999). TFIIH is likely
involved in the effects mediated by p53; both XPB and XPD appear to bind to p53 (Chang et al.,
2008; Hou et al., 2003; Léveillard et al., 1996). p53 presence and XPB/XPD binding appear to
enhance NER activity (Gillet & Scharer, 2006; Léveillard et al., 1996; Wang, 2003), but the
mechanism is not clear. Also, there is no report of whether post-translational modifications of
p53 are important for the effects of p53 on NER activity. In addition, the decreases observed in
NER activity (Chapters 2 and 3) could be due to covalent modifications of p53 either in the C-
terminal amino acid of p53, or in the DNA binding region, thereby inhibiting the increased NER
efficiency provided by p53. Protein modifications can be observed after protein fractionation by

106
2-D SDS-PAGE and mass-spectrometry after immunoblotting. Western blotting with antibodies
for p53 phosphorylation and acetylation can be used to test whether these post-translational
modifications have any role in the significant changes to NER activity after sulforaphane and
NNK treatment.
The research in this thesis does not entirely rule out a role for p53 as a cause of the
effects observed after treatment with NNK or sulforaphane. Unfortunately, the binding of p53 to
damaged DNA could not be assessed reliably using the nuclear protein extracts isolated in this
thesis. Optimization of p53 in the damaged DNA-NER protein binding assay could be an
important next step to fully explain the importance of p53 in NNK- and sulforaphane-mediated
NER activity changes.
5.2.3 Protein adducts
It has previously been theorized that NNK causes covalent modifications to key NER
proteins (Brown, 2008). HPB-releasing hemoglobin adducts have been detected in smokers and
in rats treated with NNK, but it is not known if NNK also forms adducts with NER proteins
(Hecht et al., 1993a; Peterson et al., 1990). It is possible that the effects shown in mouse lung
after treatment with NNK are due to a covalent adduction of NER proteins. There are known
differences in bioactivation between A/J mouse lung and liver, and the organs may differ in their
abilities to respond to adducted proteins or it is possible that NER proteins may be adducted only
in the lung (Zheng & Takano, 2011; Zhou et al., 2012). Covalent protein adducts in any of the
binding regions of NER proteins XPC, XPA or XPB would affect their abilities to bind to
damaged DNA. Other NER proteins such as RPA, XPF or XPG could be affected in a similar
way by covalent modifications. RPA is particularly susceptible to damage (Guven et al., 2015),
and work has shown that modified structures of both XPF and XPG can inhibit repair (Araújo et
al., 2001; McNeil & Melton, 2012; Su et al., 2012; Tomicic et al., 2011). Potentially adducted

107
NER proteins can be separated by 2D SDS-PAGE, identified by immunoblotting and subjected to
mass spectrometric analysis.
5.2.4 The effects of sulforaphane are mediated through HDAC inhibition and Nrf2
activation
It is unclear whether the Nrf2 downstream effects or the histone deacetylase inhibitor
effects of sulforaphane mediate the increase in hepatic NER activity reported in Chapter 4. The
time point of maximal effect for our chosen Nrf2 genes was before 12 h; however, effects of
sulforaphane can occur at various timepoints in vivo (Hu et al., 2004a; Hu et al., 2006). In the
future, repeating the experiment in the Nrf2 knockout mouse will allow for better understanding
of the effects of sulforaphane on NER. If the effect of sulforaphane on NER is mediated
exclusively by the activation of Nrf2, then it is expected the effect on NER will not be observed
in the Nrf2 knockout mouse. Whereas, if the effect of sulforaphane on NER activity is due to
HDAC inhibition, then the effects observed in this thesis will persist in the Nrf2 knockout mouse.
The sulforaphane metabolites, SF-Cys and SF-NAC, are the major HDAC inhibitors (Myzak et
al., 2004; Myzak et al., 2006), and these can be investigated if it appears the NER effect is due to
the HDAC inhibitory effect of sulforaphane.
5.2.5 The effects of cigarette smoking on NER activity in humans
NNK is a known carcinogen, and it appears to be important in smoking-induced human
lung cancers with increased urinary NNK metabolites linked to increased risk for lung
adenocarcinomas (Hecht et al., 2016; Stepanov et al., 2014; Yuan et al., 2011; Yuan et al., 2014).
However, no study has been reported regarding the effects of smoking on NER activity. It has
been established that NNK inhibits NER in mouse lung (Brown, 2008), and it is possible smoking
can cause a similar effect in human lung. Nuclear protein can be extracted from human lung

108
tissue with a similar protocol to the one used in this thesis, and these extracts can be used to
assess DNA repair activities on POB adducts using the same NER activity assay used in this
thesis. If there is an effect of smoking on NER, approaches used and described in this thesis
could be used to determine mechanisms.

109
References
Abassi Joozdani, F., Yari, F., Abassi Joozdani, P., & Nafisi, S. (2015). Interaction of sulforaphane with DNA and RNA. PLoS One, 10(6), e0127541.
Aboussekhra, A., Biggerstaff, M., Shivji, M. K., Vilpo, J. A., Moncollin, V., Podust, V. N. et al. (1995). Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell, 80(6), 859-868.
Adimoolam, S., & Ford, J. M. (2003). p53 and regulation of DNA damage recognition during nucleotide excision repair. DNA Repair, 2(9), 947-954.
Alfieri, A., Srivastava, S., Siow, R. C., Cash, D., Modo, M., Duchen, M. R. et al. (2013). Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood-brain barrier disruption and neurological deficits in stroke. Free Radic Biol Med, 65, 1012-1022.
Alumkal, J. J., Slottke, R., Schwartzman, J., Cherala, G., Munar, M., Graff, J. N. et al. (2015). A phase II study of sulforaphane-rich broccoli sprout extracts in men with recurrent prostate cancer. Invest New Drugs, 33(2), 480-489.
Aoki, Y., Sato, H., Nishimura, N., Takahashi, S., Itoh, K., & Yamamoto, M. (2001). Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol Appl Pharmacol, 173(3), 154-160.
Appleton, S., Olegario, R. M., & Lipowicz, P. J. (2013). TSNA levels in machine-generated mainstream cigarette smoke: 35 years of data. Regul Toxicol Pharmacol, 66(2), 197-207.
Appleton, S., Olegario, R. M., & Lipowicz, P. J. (2014). TSNA exposure from cigarette smoking: 18 years of urinary NNAL excretion data. Regul Toxicol Pharmacol, 68(2), 269-274.
Araújo, S. J., Nigg, E. A., & Wood, R. D. (2001). Strong functional interactions of TFIIH with XPC and XPG in human DNA nucleotide excision repair, without a preassembled repairosome. Mol Cell Biol, 21(7), 2281-2291.
Ashcroft, M., Kubbutat, M. H., & Vousden, K. H. (1999). Regulation of p53 function and stability by phosphorylation. Mol Cell Biol, 19(3), 1751-1758.
Ashley, D., Beeson, M., Johnson, D., McCraw, J., Richter, P., Pirkle, J. et al. (2003). Tobacco-specific nitrosamines in tobacco from U.S. brand and non-U.S. brand cigarettes. Nicotine & Tobacco Research, 5(3), 323-331.
Ashley, D. L., O’Connor, R. J., Bernert, J. T., Watson, C. H., Polzin, G. M., Jain, R. B. et al. (2010). Effect of differing levels of tobacco-specific nitrosamines in cigarette smoke on the levels of biomarkers in smokers. Cancer Epidemiol Biomarkers Prev, 19(6), 1389-1398.
Atwell, L. L., Hsu, A., Wong, C. P., Stevens, J. F., Bella, D., Yu, T. W. et al. (2015). Absorption and chemopreventive targets of sulforaphane in humans following consumption of broccoli sprouts or a myrosinase-treated broccoli sprout extract. Mol Nutr Food Res, 59(3), 424-433.
Baasanjav-Gerber, C., Hollnagel, H. M., Brauchmann, J., Iori, R., & Glatt, H. (2011). Detection of genotoxicants in Brassicales using endogenous DNA as a surrogate

110
target and adducts determined by 32P-postlabelling as an experimental end point. Mutagenesis, 26(3), 407-413.
Bacon, J. R., Williamson, G., Garner, R. C., Lappin, G., Langouët, S., & Bao, Y. (2003). Sulforaphane and quercetin modulate PhIP-DNA adduct formation in human HepG2 cells and hepatocytes. Carcinogenesis, 24(12), 1903-1911.
Baier, S. R., Zbasnik, R., Schlegel, V., & Zempleni, J. (2014). Off-target effects of sulforaphane include the derepression of long terminal repeats through histone acetylation events. J Nutr Biochem, 25(6), 665-668.
Baird, L., & Dinkova-Kostova, A. T. (2011). The cytoprotective role of the Keap1–Nrf2 pathway. Arch Toxicol, 85(4), 241-272.
Balajee, A. S., & Bohr, V. A. (2000). Genomic heterogeneity of nucleotide excision repair. Gene, 250(1-2), 15-30.
Barckhausen, C., Roos, W. P., Naumann, S. C., & Kaina, B. (2014). Malignant melanoma cells acquire resistance to DNA interstrand cross-linking chemotherapeutics by p53-triggered upregulation of DDB2/XPC-mediated DNA repair. Oncogene, 33(15), 1964-1974.
Barrett, J. C. (1993). Mechanisms of Multistep Carcinogenesis and Carcinogen Risk Assessment. Environ Health Perspect, 100, 9-20.
Bates, S. E. (2010). DNA repair: a reinvigorated therapeutic target. Clin Cancer Res, 16(18), 4510.
Bedard, L. L., Alessi, M., Davey, S., & Massey, T. E. (2005). Susceptibility to aflatoxin B1-induced carcinogenesis correlates with tissue-specific differences in DNA repair activity in mouse and in rat. Cancer Res, 65(4), 1265-1270.
Beli, P., & Jackson, S. P. (2015). Ubiquitin regulates dissociation of DNA repair factors from chromatin. Oncotarget, 14727-14728.
Bergink, S., & Jentsch, S. (2009). Principles of ubiquitin and SUMO modifications in DNA repair. Nature, 458(7237), 461-467.
Bhamre, S., Sahoo, D., Tibshirani, R., Dill, D. L., & Brooks, J. D. (2009). Temporal changes in gene expression induced by sulforaphane in human prostate cancer cells. Prostate, 69(2), 181-190.
Bhana, S., Hewer, A., Phillips, D. H., & Lloyd, D. R. (2008). p53-dependent global nucleotide excision repair of cisplatin-induced intrastrand cross links in human cells. Mutagenesis, 23(2), 131-136.
Binz, S. K., Sheehan, A. M., & Wold, M. S. (2004). Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair (Amst), 3(8-9), 1015-1024.
Bones, A. M., & Rossiter, J. T. (2006). The enzymic and chemically induced decomposition of glucosinolates. Phytochemistry, 67(11), 1053-1067.
Borgerding, M. F., Bodnar, J. A., Curtin, G. M., & Swauger, J. E. (2012). The chemical composition of smokeless tobacco: a survey of products sold in the United States in 2006 and 2007. Regul Toxicol Pharmacol, 64(3), 367-387.
Boyle, P., & Levin, B. (Eds.). (2009). World Cancer Report 2008. Lyon, France. Bradsher, J., Coin, F., & Egly, J.-M. (2000). Distinct Roles for the Helicases of TFIIH in

111
Transcript Initiation and Promoter Escape. Journal of Biological Chemistry, 275(4), 2532-2538.
Branzei, D., & Foiani, M. (2008). Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol, 9(4), 297-308.
Brown, K. L., Roginskaya, M., Zou, Y., Altamirano, A., Basu, A. K., & Stone, M. P. (2010). Binding of the human nucleotide excision repair proteins XPA and XPC/HR23B to the 5R-thymine glycol lesion and structure of the cis-(5R,6S) thymine glycol epimer in the 5’-GTgG-3’ sequence: destabilization of two base pairs at the lesion site. Nucleic Acids Res, 38(2), 428-440.
Brown, P. (2008). Biotransformation and DNA repair in 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced pulmonary carcinogenesis. Queen’s University, Kingston, Ontario.
Brown, P. J., Bedard, L. L., & Massey, T. E. (2008). Repair of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced DNA pyridyloxobutylation by nucleotide excision repair. Cancer Lett, 260(1-2), 48-55.
Brown, P. J., & Massey, T. E. (2009). In vivo treatment with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induces organ-specific alterations in in vitro repair of DNA pyridyloxobutylation. Mutat Res, 663(1-2), 15-21.
Burns, D. M., Anderson, C. M., & Gray, N. (2011). Do changes in cigarette design influence the rise in adenocarcinoma of the lung. Cancer Causes Control, 22(1), 13-22.
Camenisch, U., Dip, R., Schumacher, S. B., Schuler, B., & Naegeli, H. (2006). Recognition of helical kinks by xeroderma pigmentosum group A protein triggers DNA excision repair. Nat Struct Mol Biol, 13(3), 278-284.
Camici, G. G., Steffel, J., Akhmedov, A., Schafer, N., Baldinger, J., Schulz, U. et al. (2006). Dimethyl sulfoxide inhibits tissue factor expression, thrombus formation, and vascular smooth muscle cell activation: a potential treatment strategy for drug-eluting stents. Circulation, 114(14), 1512-1521.
Cazzalini, O., Scovassi, A. I., Savio, M., Stivala, L. A., & Prosperi, E. (2010). Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat Res, 704(1-3), 12-20.
Cazzalini, O., Sommatis, S., Tillhon, M., Dutto, I., Bachi, A., Rapp, A. et al. (2014). CBP and p300 acetylate PCNA to link its degradation with nucleotide excision repair synthesis. Nucleic Acids Res, 42(13), 8433-8448.
Chan, K., Han, X. D., & Kan, Y. W. (2001). An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci U S A, 98(8), 4611-4616.
Chang, Y. C., Jan, K. Y., Cheng, C. A., Liao, C. B., & Liu, Y. C. (2008). Direct involvement of the tumor suppressor p53 in nucleotide excision repair. DNA Repair (Amst), 7(5), 751-761.
Chen, W., Sun, Z., Wang, X.-J., Jiang, T., Huang, Z., Fang, D. et al. (2009). Direct Interaction between Nrf2 and p21Cip1/WAF1 Upregulates the Nrf2-Mediated Antioxidant Response. Molecular Cell, 34(6), 663-673.

112
Cho, J. H., Kim, Y. W., Choi, B. Y., & Keum, Y. S. (2014). Sulforaphane inhibition of TPA-mediated PDCD4 downregulation contributes to suppression of c-Jun and induction of p21-dependent Nrf2 expression. Eur J Pharmacol, 741, 247-253.
Cho, S. (2002). Evidence That Nucleotide Excision Repair Is Attenuated in Bax-Deficient Mammalian Cells Following Ultraviolet Irradiation. Experimental Cell Research, 278(2), 158-165.
Choi, J. Y., Park, J. M., Yi, J. M., Leem, S. H., & Kang, T. H. (2015). Enhanced nucleotide excision repair capacity in lung cancer cells by preconditioning with DNA-damaging agents. Oncotarget, 6(26), 22575-22586.
Chubatsu, L. S., & Meneghini, R. (1993). Metallothionein protects DNA from oxidative damage. Biochem J, 291(Pt 1), 193-198.
Church, T. R., Anderson, K. E., Caporaso, N. E., Geisser, M. S., Le, C. T., Zhang, Y. et al. (2009). A prospectively measured serum biomarker for a tobacco-specific carcinogen and lung cancer in smokers. Cancer Epidemiol Biomarkers Prev, 18(1), 260-266.
Clarke, J. D., Dashwood, R. H., & Ho, E. (2008). Multi-targeted prevention of cancer by sulforaphane. Cancer Lett, 269(2), 291-304.
Clarke, J. D., Hsu, A., Williams, D. E., Dashwood, R. H., Stevens, J. F., Yamamoto, M. et al. (2011). Metabolism and tissue distribution of sulforaphane in Nrf2 knockout and wild-type mice. Pharm Res, 28(12), 3171-3179.
Coin, F., Auriol, J., Tapias, A., Clivio, P., Vermeulen, W., & Egly, J. M. (2004). Phosphorylation of XPB helicase regulates TFIIH nucleotide excision repair activity. EMBO J, 23(24), 4835-4846.
Coin, F., Oksenych, V., Mocquet, V., Groh, S., Blattner, C., & Egly, J. M. (2008). Nucleotide excision repair driven by the dissociation of CAK from TFIIH. Mol Cell, 31(1), 9-20.
Colton, S. L., Xu, X. S., Wang, Y. A., & Wang, G. (2006). The involvement of ataxia-telangiectasia mutated protein activation in nucleotide excision repair-facilitated cell survival with cisplatin treatment. J Biol Chem, 281(37), 27117-27125.
Conaway, C. C., Wang, C. X., Pittman, B., Yang, Y. M., Schwartz, J. E., Tian, D. et al. (2005). Phenethyl isothiocyanate and sulforaphane and their N-acetylcysteine conjugates inhibit malignant progression of lung adenomas induced by tobacco carcinogens in A/J mice. Cancer Res, 65(18), 8548-8557.
Constantinou, A., Gunz, D., Evans, E., Lalle, P., Bates, P. A., Wood, R. D. et al. (1999). Conserved Residues of Human XPG Protein Important for Nuclease Activity and Function in Nucleotide Excision Repair. Journal of Biological Chemistry, 274(9), 5637-5648.
Counts, M. E., Hsu, F. S., Laffoon, S. W., Dwyer, R. W., & Cox, R. H. (2004). Mainstream smoke constituent yields and predicting relationships from a worldwide market sample of cigarette brands: ISO smoking conditions. Regul Toxicol Pharmacol, 39(2), 111-134.
Damsma, G. E., Alt, A., Brueckner, F., Carell, T., & Cramer, P. (2007). Mechanism of transcriptional stalling at cisplatin-damaged DNA. Nat Struct Mol Biol, 14(12),

113
1127-1133. Dashwood, R. H., & Ho, E. (2007). Dietary histone deacetylase inhibitors: from cells to
mice to man. Semin Cancer Biol, 17(5), 363-369. De Bont, R. (2004). Endogenous DNA damage in humans: a review of quantitative data.
Mutagenesis, 19(3), 169-185. de Laat, W. L., Appeldoorn, E., Sugasawa, K., Weterings, E., Jaspers, N. G., &
Hoeijmakers, J. H. (1998). DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev, 12(16), 2598-2609.
Devesa, S. S., Bray, F., Vizcaino, A. P., & Parkin, D. M. (2005). International lung cancer trends by histologic type: male:female differences diminishing and adenocarcinoma rates rising. Int J Cancer, 117(2), 294-299.
Dinkova-Kostova, A. T., & Abramov, A. Y. (2015). The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med, 88, 179-188.
Donninger, H., Clark, J., Rinaldo, F., Nelson, N., Barnoud, T., Schmidt, M. L. et al. (2015). The RASSF1A tumor suppressor regulates XPA-mediated DNA repair. Mol Cell Biol, 35(1), 277-287.
Dou, H., Huang, C., Singh, M., Carpenter, P. B., & Yeh, E. T. (2010). Regulation of DNA repair through deSUMOylation and SUMOylation of replication protein A complex. Mol Cell, 39(3), 333-345.
Dregoesc, D., Rybak, A. P., & Rainbow, A. J. (2007). Increased expression of p53 enhances transcription-coupled repair and global genomic repair of a UVC-damaged reporter gene in human cells. DNA Repair (Amst), 6(5), 588-601.
Duan, M. R., & Smerdon, M. J. (2014). Histone H3 lysine 14 (H3K14) acetylation facilitates DNA repair in a positioned nucleosome by stabilizing the binding of the chromatin Remodeler RSC (Remodels Structure of Chromatin). J Biol Chem, 289(12), 8353-8363.
Dutto, I., Tillhon, M., & Prosperi, E. (2016). Assessing Cell Cycle Independent Function of the CDK Inhibitor p21(CDKN¹A) in DNA Repair. Methods Mol Biol, 1336, 123-139.
Ellinger-Ziegelbauer, H., Stuart, B., Wahle, B., Bomann, W., & Ahr, H. J. (2004). Characteristic expression profiles induced by genotoxic carcinogens in rat liver. Toxicol Sci, 77(1), 19-34.
Enomoto, A., Itoh, K., Nagayoshi, E., Haruta, J., Kimura, T., O’Connor, T. et al. (2001). High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci, 59(1), 169-177.
Evans, E., Fellows, J., Coffer, A., & Wood, R. D. (1997). Open complex formation around a lesion during nucleotide excision repair provides a structure for cleavage by human XPG protein. EMBO J, 16(3), 625-638.
Fadda, E. (2013). Conformational determinants for the recruitment of ERCC1 by XPA in the nucleotide excision repair (NER) Pathway: structure and dynamics of the XPA binding motif. Biophys J, 104(11), 2503-2511.
Fahey, J. W., Zalcmann, A. T., & Talalay, P. (2001). The chemical diversity and

114
distribution of glucosinolates and isothiocyanates among plants. Phytochemistry, 56(1), 5-51.
Fahey, J. W., Zhang, Y., & Talalay, P. (1997). Broccoli sprouts: an exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc Natl Acad Sci U S A, 94(19), 10367-10372.
Fan, W., & Luo, J. (2010). SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol Cell, 39(2), 247-258.
Faraonio, R., Vergara, P., Di Marzo, D., Pierantoni, M. G., Napolitano, M., Russo, T. et al. (2006). p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J Biol Chem, 281(52), 39776-39784.
Farsalinos, K. E., Gillman, G., Poulas, K., & Voudris, V. (2015). Tobacco-Specific Nitrosamines in Electronic Cigarettes: Comparison between Liquid and Aerosol Levels. Int J Environ Res Public Health, 12(8), 9046-9053.
Fatur, T., Lah, T. T., & Filipič, M. (2003). Cadmium inhibits repair of UV-, methyl methanesulfonate- and N-methyl-N-nitrosourea-induced DNA damage in Chinese hamster ovary cells. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 529(1-2), 109-116.
Fischer, J. L., Lancia, J. K., Mathur, A., & Smith, M. L. (2006). Selenium protection from DNA damage involves a Ref1/p53/Brca1 protein complex. Anticancer Res, 26(2A), 899-904.
Ford, J. M., & Hanawalt, P. C. (1995). Li-Fraumeni syndrome fibroblasts homozygous for p53 mutations are deficient in global DNA repair but exhibit normal transcription-coupled repair and enhanced UV resistance. Proc Natl Acad Sci U S A, 92(19), 8876-8880.
Fousteri, M., & Mullenders, L. H. (2008). Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res, 18(1), 73-84.
Frit, P., Calsou, P., Chen, D. J., & Salles, B. (1998). Ku70/Ku80 protein complex inhibits the binding of nucleotide excision repair proteins on linear DNA in vitro. J Mol Biol, 284(4), 963-973.
Gaddameedhi, S., Selby, C. P., Kemp, M. G., Ye, R., & Sancar, A. (2015). The circadian clock controls sunburn apoptosis and erythema in mouse skin. J Invest Dermatol, 135(4), 1119-1127.
Gali, H., Juhasz, S., Morocz, M., Hajdu, I., Fatyol, K., Szukacsov, V. et al. (2012). Role of SUMO modification of human PCNA at stalled replication fork. Nucleic Acids Res, 40(13), 6049-6059.
Galvao, J., Davis, B., Tilley, M., Normando, E., Duchen, M. R., & Cordeiro, M. F. (2014). Unexpected low-dose toxicity of the universal solvent DMSO. FASEB J, 28(3), 1317-1330.
Gansen, A., Tóth, K., Schwarz, N., & Langowski, J. (2015). Opposing roles of H3- and H4-acetylation in the regulation of nucleosome structure––a FRET study. Nucleic Acids Res, 43(3), 1433-1443.
Gao, S. S., Chen, X. Y., Zhu, R. Z., Choi, B. M., & Kim, B. R. (2010). Sulforaphane

115
induces glutathione S-transferase isozymes which detoxify aflatoxin B(1)-8,9-epoxide in AML 12 cells. Biofactors, 36(4), 289-296.
Gerhauser, C. (2013). Cancer chemoprevention and nutriepigenetics: state of the art and future challenges. Top Curr Chem, 329, 73-132.
Gillet, L. C., & Scharer, O. D. (2006). Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev, 106(2), 253-276.
Gong, P., & Cederbaum, A. I. (2006). Nrf2 is increased by CYP2E1 in rodent liver and HepG2 cells and protects against oxidative stress caused by CYP2E1. Hepatology, 43(1), 144-153.
Goniewicz, M. L., Knysak, J., Gawron, M., Kosmider, L., Sobczak, A., Kurek, J. et al. (2014). Levels of selected carcinogens and toxicants in vapour from electronic cigarettes. Tob Control, 23(2), 133-139.
Gourdin, A. M., van Cuijk, L., Tresini, M., Luijsterburg, M. S., Nigg, A. L., Giglia-Mari, G. et al. (2014). Differential binding kinetics of replication protein A during replication and the pre- and post-incision steps of nucleotide excision repair. DNA Repair (Amst), 24, 46-56.
Greenbaum, D., Colangelo, C., Williams, K., & Gerstein, M. (2003). Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol, 4(9), 117.
Gunduz, I., Kondylis, A., Jaccard, G., Renaud, J. M., Hofer, R., Ruffieux, L. et al. (2016). Tobacco-specific N-nitrosamines NNN and NNK levels in cigarette brands between 2000 and 2014. Regul Toxicol Pharmacol, 76, 113-120.
Güngör, N., Godschalk, R. W., Pachen, D. M., Van Schooten, F. J., & Knaapen, A. M. (2007). Activated neutrophils inhibit nucleotide excision repair in human pulmonary epithelial cells: role of myeloperoxidase. FASEB J, 21(10), 2359-2367.
Guven, M., Brem, R., Macpherson, P., Peacock, M., & Karran, P. (2015). Oxidative Damage to RPA Limits the Nucleotide Excision Repair Capacity of Human Cells. J Invest Dermatol, 135(11), 2834-2841.
Habraken, Y., Sung, P., Prakash, S., & Prakash, L. (1996). Transcription factor TFIIH and DNA endonuclease Rad2 constitute yeast nucleotide excision repair factor 3: implications for nucleotide excision repair and Cockayne syndrome. Proc Natl Acad Sci U S A, 93(20), 10718-10722.
Hakura, A., Mochida, H., & Yamatsu, K. (1993). Dimethyl sulfoxide (DMSO) is mutagenic for bacterial mutagenicity tester strains. Mutat Res, 303(3), 127-133.
Hanawalt, P. C., & Spivak, G. (2008). Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol, 9(12), 958-970.
Hass, C. S., Chen, R., & Wold, M. S. (2012). Detection of posttranslational modifications of replication protein A. Methods Mol Biol, 922, 193-204.
Hayes, J. D., McMahon, M., Chowdhry, S., & Dinkova-Kostova, A. T. (2010). Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid Redox Signal, 13(11), 1713-1748.
Hecht, S. S. (1998). Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol, 11(6), 559-603.

116
Hecht, S. S. (1999). Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst, 91(14), 1194-1210.
Hecht, S. S. (2014). It is time to regulate carcinogenic tobacco-specific nitrosamines in cigarette tobacco. Cancer Prev Res (Phila), 7, 639-647.
Hecht, S. S., Carmella, S. G., Foiles, P. G., Murphy, S. E., & Peterson, L. A. (1993a). Tobacco-specific nitrosamine adducts: studies in laboratory animals and humans. Environ Health Perspect, 99, 57-63.
Hecht, S. S., Carmella, S. G., Stepanov, I., Jensen, J., Anderson, A., & Hatsukami, D. K. (2008). Metabolism of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone to its biomarker total NNAL in smokeless tobacco users. Cancer Epidemiol Biomarkers Prev, 17(3), 732-735.
Hecht, S. S., & Hoffmann, D. (1989). The relevance of tobacco-specific nitrosamines to human cancer. Cancer Surv, 8(2), 273-294.
Hecht, S. S., Stepanov, I., & Carmella, S. G. (2016). Exposure and Metabolic Activation Biomarkers of Carcinogenic Tobacco-Specific Nitrosamines. Acc Chem Res, 49(1), 106-114.
Hecht, S. S., Trushin, N., Reid-Quinn, C. A., Burak, E. S., Jones, A. B., Southers, J. L. et al. (1993b). Metabolism of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in the patas monkey: pharmacokinetics and characterization of glucuronide metabolites. Carcinogenesis, 14(2), 229-236.
Hecht, S. S., Chi-hong, B. C., Ohmori, T., & Hoffmann, D. (1980). Comparative carcinogenicity in F344 rats of the tobacco-specific nitrosamines, N -nitrosonornicotine and 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone. Cancer research, 40(2), 298-302.
Hecht, S. S., Jordan, K. G., Choi, C.-I., & Trushin, N. (1990). Effects of deuterium substitution on the tumorigenicity of 4-(metlhylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in A/J mice. Carcinogenesis, 11(6), 1017-1020.
Hecht, S. S., Morse, M. A., Amin, S., Stoner, G. D., Jordan, K. G., Choi, C.-I. et al. (1989). Rapid single-dose model for lung tumor induction in A/J mice by 4-(methylnitrosainino)-1-(3-pyridyl)-1-butanone and the effect of diet. Carcinogenesis, 10, 1901-1904.
Higdon, J. V., Delage, B., Williams, D. E., & Dashwood, R. H. (2007). Cruciferous vegetables and human cancer risk: epidemiologic evidence and mechanistic basis. Pharmacol Res, 55(3), 224-236.
Ho, E., Clarke, J. D., & Dashwood, R. H. (2009). Dietary sulforaphane, a histone deacetylase inhibitor for cancer prevention. J Nutr, 139(12), 2393-2396.
Hoege, C., Pfander, B., Moldovan, G. L., Pyrowolakis, G., & Jentsch, S. (2002). RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature, 419(6903), 135-141.
Hoffmann, D., & Djordjevic, M. V. (1997). Chemical composition and carcinogenicity of smokeless tobacco. Adv Dent Res, 11(3), 322-329.
Hoffmann, D., Djordjevic, M. V., & Hoffmann, I. (1997). The changing cigarette. Prev

117
Med, 26(4), 427-434. Hoffmann, D., Rivenson, A., & Hecht, S. S. (1996). The Biological Significance of Tobacco-
SpecificN- Nitrosamines: Smoking and Adenocarcinoma of the Lung . Critical reviews in toxicology, 26(2), 199-211. Hoogervorst, E. M., van Oostrom, C. T., Beems, R. B., van Benthem, J., Gielis, S.,
Vermeulen, J. P. et al. (2004). p53 heterozygosity results in an increased 2-acetylaminofluorene-induced urinary bladder but not liver tumor response in DNA repair-deficient Xpa mice. Cancer Res, 64(15), 5118-5126.
Hou, S. M., Ryk, C., Kannio, A., Angelini, S., Fält, S., Nyberg, F. et al. (2003). Influence of common XPD and XRCC1 variant alleles on p53 mutations in lung tumors. Environ Mol Mutagen, 41(1), 37-42.
Houston, K. A., Henley, S. J., Li, J., White, M. C., & Richards, T. B. (2014). Patterns in lung cancer incidence rates and trends by histologic type in the United States, 2004-2009. Lung Cancer, 86(1), 22-28.
Hu, R., Hebbar, V., Kim, B. R., Chen, C., Winnik, B., Buckley, B. et al. (2004a). In vivo pharmacokinetics and regulation of gene expression profiles by isothiocyanate sulforaphane in the rat. J Pharmacol Exp Ther, 310(1), 263-271.
Hu, R., Xu, C., Shen, G., Jain, M. R., Khor, T. O., Gopalkrishnan, A. et al. (2006). Gene expression profiles induced by cancer chemopreventive isothiocyanate sulforaphane in the liver of C57BL/6J mice and C57BL/6J/Nrf2 (-/-) mice. Cancer Lett, 243(2), 170-192.
Hu, W., Feng, Z., & Tang, M. S. (2004b). Nickel (II) enhances benzo[a]pyrene diol epoxide-induced mutagenesis through inhibition of nucleotide excision repair in human cells: a possible mechanism for nickel (II)-induced carcinogenesis. Carcinogenesis, 25(3), 455-462.
Hu, W., Feng, Z., & Tang, M. S. (2004c). Chromium(VI) enhances (+/-)-anti-7beta,8alpha-dihydroxy-9alpha,10alpha-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene-induced cytotoxicity and mutagenicity in mammalian cells through its inhibitory effect on nucleotide excision repair. Biochemistry, 43(44), 14282-14289.
Huang, H. C., Nguyen, T., & Pickett, C. B. (2002). Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem, 277(45), 42769-42774.
Huang, J. L., Lu, H. H., Lu, Y. N., Hung, P. S., Lin, Y. J., Lin, C. C. et al. (2016). Enhancement of the genotoxicity of benzo[a]pyrene by arecoline through suppression of DNA repair in HEp-2 cells. Toxicol In Vitro, 33, 80-87.
Huang, T. T., & D’Andrea, A. D. (2006). Regulation of DNA repair by ubiquitylation. Nat Rev Mol Cell Biol, 7(5), 323-334.
Hukkanen, J., Jacob, P., & Benowitz, N. L. (2005). Metabolism and disposition kinetics of nicotine. Pharmacol Rev, 57(1), 79-115.
Hunakova, L., Gronesova, P., Horvathova, E., Chalupa, I., Cholujova, D., Duraj, J. et al. (2014). Modulation of cisplatin sensitivity in human ovarian carcinoma A2780 and SKOV3 cell lines by sulforaphane. Toxicol Lett, 230(3), 479-486.
Hwang, B. J., Ford, J. M., Hanawalt, P. C., & Chu, G. (1999). Expression of the p48

118
xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proceedings of the National Academy of Sciences, 96(2), 424-428.
IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. (2007). Smokeless tobacco and some tobacco-specific N-nitrosamines. 89. Lyon, France: International Agency for Research on Cancer, WHO
IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. (2010). Some non-heterocyclic polycyclic aromatic hydrocarbons and some related exposures. 92. Lyon, France: International Agency for Research on Cancer, WHO
Ikeda, H., Omoteyama, K., Yoshida, K., Nishi, S., & Sakai, M. (2006). CCAAT enhancer-binding protein alpha suppresses the rat placental glutathione S-transferase gene in normal liver. J Biol Chem, 281(10), 6734-6741.
Ikehata, H., Okuyama, R., Ogawa, E., Nakamura, S., Usami, A., Mori, T. et al. (2010). Influences of p53 deficiency on the apoptotic response, DNA damage removal and mutagenesis in UVB-exposed mouse skin. Mutagenesis, 25(4), 397-405.
Iskandar, A. R., Liu, C., Smith, D. E., Hu, K. Q., Choi, S. W., Ausman, L. M. et al. (2013). β-cryptoxanthin restores nicotine-reduced lung SIRT1 to normal levels and inhibits nicotine-promoted lung tumorigenesis and emphysema in A/J mice. Cancer Prev Res (Phila), 6(4), 309-320.
Itoh, K., Chiba, T., Takahashi, S., Ishii, T., Igarashi, K., Katoh, Y. et al. (1997). An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun, 236(2), 313-322.
Itoh, K., Mimura, J., & Yamamoto, M. (2010). Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid Redox Signal, 13(11), 1665-1678.
Itoh, K., Tong, K. I., & Yamamoto, M. (2004). Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med, 36(10), 1208-1213.
Jackson, S. J., & Singletary, K. W. (2004). Sulforaphane: a naturally occurring mammary carcinoma mitotic inhibitor, which disrupts tubulin polymerization. Carcinogenesis, 25(2), 219-227.
Jackson, S. P., & Durocher, D. (2013). Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell, 49(5), 795-807.
Jaramillo, M. C., & Zhang, D. D. (2013). The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev, 27(20), 2179-2191.
Jatoi, I., Cummings, K. M., & Cazap, E. (2009). Global tobacco problem getting worse, not better. J Oncol Pract, 5(1), 21-23.
Jayakumar, S., Pal, D., & Sandur, S. K. (2015). Nrf2 facilitates repair of radiation induced DNA damage through homologous recombination repair pathway in a ROS independent manner in cancer cells. Mutat Res, 779, 33-45.
Jia, Z., Zhu, H., Li, Y., & Misra, H. P. (2010). Potent inhibition of peroxynitrite-induced DNA strand breakage and hydroxyl radical formation by dimethyl sulfoxide at very low concentrations. Exp Biol Med (Maywood), 235(5), 614-622.
Juge, N., Mithen, R. F., & Traka, M. (2007). Molecular basis for chemoprevention by

119
sulforaphane: a comprehensive review. Cell Mol Life Sci, 64(9), 1105-1127. Kanehira, T., Tani, T., Takagi, T., Nakano, Y., Howard, E. F., & Tamura, M. (2005).
Angiotensin II type 2 receptor gene deficiency attenuates susceptibility to tobacco-specific nitrosamine-induced lung tumorigenesis: involvement of transforming growth factor-beta-dependent cell growth attenuation. Cancer Res, 65(17), 7660-7665.
Kang, H. J., Hong, Y. B., Kim, H. J., & Bae, I. (2010a). CR6-interacting factor 1 (CRIF1) regulates NF-E2-related factor 2 (NRF2) protein stability by proteasome-mediated degradation. J Biol Chem, 285(28), 21258-21268.
Kang, T. H., Lindsey-Boltz, L. A., Reardon, J. T., & Sancar, A. (2010b). Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc Natl Acad Sci U S A, 107(11), 4890-4895.
Kang, T. H., Reardon, J. T., & Sancar, A. (2011). Regulation of nucleotide excision repair activity by transcriptional and post-transcriptional control of the XPA protein. Nucleic Acids Res, 39(8), 3176-3187.
Kannouche, P. L., Wing, J., & Lehmann, A. R. (2004). Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell, 14(4), 491-500.
Kensler, T. W., Wakabayashi, N., & Biswal, S. (2007). Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol., 47(1), 89-116.
Keum, Y. S. (2011). Regulation of the Keap1/Nrf2 system by chemopreventive sulforaphane: implications of posttranslational modifications. Ann N Y Acad Sci, 1229, 184-189.
Kim, H. J., & Shin, H. S. (2013). Determination of tobacco-specific nitrosamines in replacement liquids of electronic cigarettes by liquid chromatography-tandem mass spectrometry. J Chromatogr A, 1291, 48-55.
Kim, T., Kim, Y. J., Han, I. H., Lee, D., Ham, J., Kang, K. S. et al. (2015). The synthesis of sulforaphane analogues and their protection effect against cisplatin induced cytotoxicity in kidney cells. Bioorg Med Chem Lett, 25(1), 62-66.
Kleszczyński, K., Ernst, I. M., Wagner, A. E., Kruse, N., Zillikens, D., Rimbach, G. et al. (2013). Sulforaphane and phenylethyl isothiocyanate protect human skin against UVR-induced oxidative stress and apoptosis: role of Nrf2-dependent gene expression and antioxidant enzymes. Pharmacol Res, 78, 28-40.
Knatko, E. V., Ibbotson, S. H., Zhang, Y., Higgins, M., Fahey, J. W., Talalay, P. et al. (2015). Nrf2 Activation Protects against Solar-Simulated Ultraviolet Radiation in Mice and Humans. Cancer Prev Res (Phila), 8(6), 475-486.
Köberle, B., Masters, J. R., Hartley, J. A., & Wood, R. D. (1999). Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours. Curr Biol, 9(5), 273-276.
Koberle, B., Roginskaya, V., & Wood, R. D. (2006). XPA protein as a limiting factor for nucleotide excision repair and UV sensitivity in human cells. DNA Repair (Amst), 5(5), 641-648.

120
Komander, D., & Rape, M. (2012). The ubiquitin code. Annu Rev Biochem, 81, 203-229. Kotandeniya, D., Murphy, D., Yan, S., Park, S., Seneviratne, U., Koopmeiners, J. S. et al.
(2013). Kinetics of O(6)-pyridyloxobutyl-2’-deoxyguanosine repair by human O(6)-alkylguanine DNA alkyltransferase. Biochemistry, 52(23), 4075-4088.
Kruczynski, A., Barret, J. M., Van Hille, B., Chansard, N., Astruc, J., Menon, Y. et al. (2004). Decreased nucleotide excision repair activity and alterations of topoisomerase IIalpha are associated with the in vivo resistance of a P388 leukemia subline to F11782, a novel catalytic inhibitor of topoisomerases I and II. Clin Cancer Res, 10(9), 3156-3168.
Kubota, T., Nishimura, K., Kanemaki, M. T., & Donaldson, A. D. (2013). The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol Cell, 50(2), 273-280.
Kumar, D., & Saha, S. (2015). HAT3-mediated acetylation of PCNA precedes PCNA monoubiquitination following exposure to UV radiation in Leishmania donovani. Nucleic Acids Res, 43(11), 5423-5441.
Kuschal, C., Thoms, K. M., Boeckmann, L., Laspe, P., Apel, A., Schön, M. P. et al. (2011). Cyclosporin A inhibits nucleotide excision repair via downregulation of the xeroderma pigmentosum group A and G proteins, which is mediated by calcineurin inhibition. Exp Dermatol, 20(10), 795-799.
Kwak, M. K., & Kensler, T. W. (2010). Targeting NRF2 signaling for cancer chemoprevention. Toxicol Appl Pharmacol, 244(1), 66-76.
Lakin, N. D., & Jackson, S. P. (1999). Regulation of p53 in response to DNA damage. Oncogene, 18(53), 7644-7655.
Lee, J. Y., Kim, H. S., Kim, J. Y., & Sohn, J. (2009). Nuclear translocation of p21(WAF1/CIP1) protein prior to its cytosolic degradation by UV enhances DNA repair and survival. Biochem Biophys Res Commun, 390(4), 1361-1366.
Lee, Y. C., Cai, Y., Mu, H., Broyde, S., Amin, S., Chen, X. et al. (2014). The relationships between XPC binding to conformationally diverse DNA adducts and their excision by the human NER system: is there a correlation. DNA Repair (Amst), 19, 55-63.
Lefkofsky, H. B., Veloso, A., & Ljungman, M. (2015). Transcriptional and post-transcriptional regulation of nucleotide excision repair genes in human cells. Mutat Res, 776, 9-15.
Lehmann, A. R., McGibbon, D., & Stefanini, M. (2011). Xeroderma pigmentosum. Orphanet J Rare Dis, 6, 70.
Lenzi, M., Fimognari, C., & Hrelia, P. (2014). Sulforaphane as a promising molecule for fighting cancer. Cancer Treat Res, 159, 207-223.
Léveillard, T., Andera, L., Bissonnette, N., Schaeffer, L., Bracco, L., Egly, J. M. et al. (1996). Functional interactions between p53 and the TFIIH complex are affected by tumour-associated mutations. EMBO J, 15(7), 1615-1624.
Li, B., Kim, D. S., Yadav, R. K., Kim, H. R., & Chae, H. J. (2015a). Sulforaphane prevents doxorubicin-induced oxidative stress and cell death in rat H9c2 cells. Int J Mol Med, 36(1), 53-64.

121
Li, C. L., Golebiowski, F. M., Onishi, Y., Samara, N. L., Sugasawa, K., & Yang, W. (2015b). Tripartite DNA Lesion Recognition and Verification by XPC, TFIIH, and XPA in Nucleotide Excision Repair. Mol Cell, 59(6), 1025-1034.
Li, J., Shen, F., Guan, C., Wang, W., Sun, X., Fu, X. et al. (2014). Activation of Nrf2 protects against triptolide-induced hepatotoxicity. PLoS One, 9(7), e100685.
Li, L., Perdigao, J., Pegg, A. E., Lao, Y., Hecht, S. S., Lindgren, B. R. et al. (2009). The influence of repair pathways on the cytotoxicity and mutagenicity induced by the pyridyloxobutylation pathway of tobacco-specific nitrosamines. Chem Res Toxicol, 22(8), 1464-1472.
Li, R. Y., Calsou, P., Jones, C. J., & Salles, B. (1998). Interactions of the transcription/DNA repair factor TFIIH and XP repair proteins with DNA lesions in a cell-free repair assay. J Mol Biol, 281(2), 211-218.
Li, Y., Zhang, T., Li, X., Zou, P., Schwartz, S. J., & Sun, D. (2013a). Kinetics of sulforaphane in mice after consumption of sulforaphane-enriched broccoli sprout preparation. Mol Nutr Food Res, 57(12), 2128-2136.
Li, Y. M., Wang, H. B., Zheng, J. G., Bai, X. D., Zhao, Z. K., Li, J. Y. et al. (2015c). Dimethyl sulfoxide inhibits zymosan-induced intestinal inflammation and barrier dysfunction. World J Gastroenterol, 21(38), 10853-10865.
Li, Z., Musich, P. R., Cartwright, B. M., Wang, H., & Zou, Y. (2013b). UV-induced nuclear import of XPA is mediated by importin-α4 in an ATR-dependent manner. PLoS One, 8(7), e68297.
Li, Z., Musich, P. R., Serrano, M. A., Dong, Z., & Zou, Y. (2011a). XPA-mediated regulation of global nucleotide excision repair by ATR Is p53-dependent and occurs primarily in S-phase. PLoS One, 6(12), e28326.
Li, Z., Musich, P. R., & Zou, Y. (2011b). Differential DNA damage responses in p53 proficient and deficient cells: cisplatin-induced nuclear import of XPA is independent of ATR checkpoint in p53-deficient lung cancer cells. Int J Biochem Mol Biol, 2, 138-145.
Liu, J., Lin, M., Zhang, C., Wang, D., Feng, Z., & Hu, W. (2012). TAp63γ enhances nucleotide excision repair through transcriptional regulation of DNA repair genes. DNA Repair (Amst), 11(2), 167-176.
Liu, Y., Liu, Y., Yang, Z., Utzat, C., Wang, G., Basu, A. K. et al. (2005). Cooperative interaction of human XPA stabilizes and enhances specific binding of XPA to DNA damage. Biochemistry, 44(19), 7361-7368.
Lloyd, D. R., & Hanawalt, P. C. (2000). p53-dependent global genomic repair of benzo[a]pyrene-7,8-diol-9,10-epoxide adducts in human cells. Cancer Res, 60(3), 517-521.
Loeb, L. A., & Harris, C. C. (2008). Advances in chemical carcinogenesis: a historical review and prospective. Cancer Res, 68(17), 6863-6872.
Loewer, A., Karanam, K., Mock, C., & Lahav, G. (2013). The p53 response in single cells is linearly correlated to the number of DNA breaks without a distinct threshold. BMC Biol, 11, 114.
Long, X. D., Huang, H. D., Huang, X. Y., Yao, J. G., & Xia, Q. (2015). XPC codon 939

122
polymorphism is associated with susceptibility to DNA damage induced by aflatoxin B1 exposure. Int J Clin Exp Med, 8(1), 1197-1204.
Mahéo, K., Morel, F., Langouët, S., Kramer, H., Le Ferrec, E., Ketterer, B. et al. (1997). Inhibition of cytochromes P-450 and induction of glutathione S-transferases by sulforaphane in primary human and rat hepatocytes. Cancer Res, 57(17), 3649-3652.
Maréchal, A., & Zou, L. (2015). RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res, 25(1), 9-23.
Marteijn, J. A., Hoeijmakers, J. H., & Vermeulen, W. (2015). Check, Check …Triple Check: Multi-Step DNA Lesion Identification by Nucleotide Excision Repair. Mol Cell, 59(6), 885-886.
Mathew, S. T., Bergström, P., & Hammarsten, O. (2014). Repeated Nrf2 stimulation using sulforaphane protects fibroblasts from ionizing radiation. Toxicol Appl Pharmacol, 276(3), 188-194.
Matsunaga, T., Mu, D., Park, C. H., Reardon, J. T., & Sancar, A. (1995). Human DNA repair excision nuclease. Analysis of the roles of the subunits involved in dual incisions by using anti-XPG and anti-ERCC1 antibodies. J Biol Chem, 270(35), 20862-20869.
Matusheski, N. V., Swarup, R., Juvik, J. A., Mithen, R., Bennett, M., & Jeffery, E. H. (2006). Epithiospecifier protein from broccoli (Brassica oleracea L. ssp. italica) inhibits formation of the anticancer agent sulforaphane. J Agric Food Chem, 54(6), 2069-2076.
McNeil, E. M., & Melton, D. W. (2012). DNA repair endonuclease ERCC1-XPF as a novel therapeutic target to overcome chemoresistance in cancer therapy. Nucleic Acids Res, 40(20), 9990-10004.
Mehta, M., Chen, L. C., Gordon, T., Rom, W., & Tang, M. S. (2008). Particulate matter inhibits DNA repair and enhances mutagenesis. Mutat Res, 657(2), 116-121.
Melis, J. P., Luijten, M., Mullenders, L. H., & van Steeg, H. (2011). The role of XPC: implications in cancer and oxidative DNA damage. Mutat Res, 728(3), 107-117.
Ming, M., Shea, C. R., Guo, X., Li, X., Soltani, K., Han, W. et al. (2010). Regulation of global genome nucleotide excision repair by SIRT1 through xeroderma pigmentosum C. Proc Natl Acad Sci U S A, 107(52), 22623-22628.
Missura, M., Buterin, T., Hindges, R., Hübscher, U., Kaspárková, J., Brabec, V. et al. (2001). Double-check probing of DNA bending and unwinding by XPA-RPA: an architectural function in DNA repair. EMBO J, 20(13), 3554-3564.
Miyazono, K., & Koinuma, D. (2011). Arkadia--beyond the TGF-β pathway. J Biochem, 149(1), 1-3.
Mocquet, V., Lainé, J. P., Riedl, T., Yajin, Z., Lee, M. Y., & Egly, J. M. (2008). Sequential recruitment of the repair factors during NER: the role of XPG in initiating the resynthesis step. EMBO J, 27(1), 155-167.
Moggs, J. G., Yarema, K. J., Essigmann, J. M., & Wood, R. D. (1996). Analysis of Incision Sites Produced by Human Cell Extracts and Purified Proteins during Nucleotide Excision Repair of a 1,3-Intrastrand d(GpTpG)-Cisplatin Adduct.

123
Journal of Biological Chemistry, 271(12), 7177-7186. Momot, D., Nostrand, T. A., John, K., Ward, Y., Steinberg, S. M., Liewehr, D. J. et al.
(2014). Role of nucleotide excision repair and p53 in zidovudine (AZT)-induced centrosomal deregulation. Environ Mol Mutagen, 55(9), 719-726.
Morse, M. A., Eklind, K. I., Toussaint, M., Amin, S. G., & Chung, F. L. (1990). Characterization of a glucuronide metabolite of 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and its dose-dependent excretion in the urine of mice and rats. Carcinogenesis, 11(10), 1819-1823.
Moser, J., Kool, H., Giakzidis, I., Caldecott, K., Mullenders, L. H., & Fousteri, M. I. (2007). Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol Cell, 27(2), 311-323.
Motohashi, H., Katsuoka, F., Engel, J. D., & Yamamoto, M. (2004). Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc Natl Acad Sci U S A, 101(17), 6379-6384.
Mu, D., Hsu, D. S., & Sancar, A. (1996). Reaction Mechanism of Human DNA Repair Excision Nuclease. Journal of Biological Chemistry, 271(14), 8285-8294.
Mulder, J. E., Bondy, G. S., Mehta, R., & Massey, T. E. (2014). Up-regulation of nucleotide excision repair in mouse lung and liver following chronic exposure to aflatoxin B(1) and its dependence on p53 genotype. Toxicol Appl Pharmacol, 275(2), 96-103.
Murphy, M., Mabruk, M. J., Lenane, P., Liew, A., McCann, P., Buckley, A. et al. (2002). Comparison of the expression of p53, p21, Bax and the induction of apoptosis between patients with basal cell carcinoma and normal controls in response to ultraviolet irradiation. J Clin Pathol, 55(11), 829-833.
Myzak, M. C., Dashwood, W. M., Orner, G. A., Ho, E., & Dashwood, R. H. (2006). Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. FASEB J, 20(3), 506-508.
Myzak, M. C., Karplus, P. A., Chung, F. L., & Dashwood, R. H. (2004). A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res, 64(16), 5767-5774.
Nocentini, S., Coin, F., Saijo, M., Tanaka, K., & Egly, J.-M. (1997). DNA damage recognition by XPA protein promotes efficient recruitment of transcription factor II H. Journal of Biological Chemistry, 272(37), 22991-22994.
Noh, J. R., Kim, Y. H., Hwang, J. H., Choi, D. H., Kim, K. S., Oh, W. K. et al. (2015). Sulforaphane protects against acetaminophen-induced hepatotoxicity. Food Chem Toxicol, 80, 193-200.
Nollen, M., Ebert, F., Moser, J., Mullenders, L. H., Hartwig, A., & Schwerdtle, T. (2009). Impact of arsenic on nucleotide excision repair: XPC function, protein level, and gene expression. Mol Nutr Food Res, 53(5), 572-582.
Nouspikel, T. (2009). DNA repair in mammalian cells : Nucleotide excision repair: variations on versatility. Cell Mol Life Sci, 66(6), 994-1009.
O’Donovan, A., Davies, A. A., Moggs, J. G., West, S. C., & Wood, R. D. (1994). XPG

124
endonuclease makes the 3’ incision in human DNA nucleotide excision repair. Nature, 371(6496), 432-435.
Ogi, T., & Lehmann, A. R. (2006). The Y-family DNA polymerase kappa (pol kappa) functions in mammalian nucleotide-excision repair. Nat Cell Biol, 8(6), 640-642.
Oliveira, P. A., Colaço, A., Chaves, R., Guedes-Pinto, H., De-La-Cruz P, L. F., & Lopes, C. (2007). Chemical carcinogenesis. An Acad Bras Cienc, 79(4), 593-616.
Overmeer, R. M., Moser, J., Volker, M., Kool, H., Tomkinson, A. E., van Zeeland, A. A. et al. (2011). Replication protein A safeguards genome integrity by controlling NER incision events. J Cell Biol, 192(3), 401-415.
Pal, R., Mamidi, M. K., Das, A. K., & Bhonde, R. (2012). Diverse effects of dimethyl sulfoxide (DMSO) on the differentiation potential of human embryonic stem cells. Arch Toxicol, 86(4), 651-661.
Park, C. H., Mu, D., Reardon, J. T., & Sancar, A. (1995). The general transcription-repair factor TFIIH is recruited to the excision repair complex by the XPA protein independent of the TFIIE transcription factor. J Biol Chem, 270(9), 4896-4902.
Parkin, J., Shea, C., & Sant, G. R. (1997). Intravesical dimethyl sulfoxide (DMSO) for interstitial cystitis--a practical approach. Urology, 49(5A Suppl), 105-107.
Paszkowska-Szczur, K., Scott, R. J., Górski, B., Cybulski, C., Kurzawski, G., Dymerska, D. et al. (2015). Polymorphisms in nucleotide excision repair genes and susceptibility to colorectal cancer in the Polish population. Mol Biol Rep, 42(3), 755-764.
Perera, F. P., Poirier, M. C., Yuspa, S. H., Nakayama, J., Jaretzki, A., Curnen, M. M. et al. (1982). A pilot project in molecular cancer epidemiology: determination of benzo[a]pyrene--DNA adducts in animal and human tissues by immunoassays. Carcinogenesis, 3(12), 1405-1410.
Pérez, C., Barrientos, H., Román, J., & Mahn, A. (2014). Optimization of a blanching step to maximize sulforaphane synthesis in broccoli florets. Food Chem, 145, 264-271.
Peterson, G. L. (1977). A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem, 83(2), 346-356.
Peterson, L. A. (2010). Formation, repair, and genotoxic properties of bulky DNA adducts formed from tobacco-specific nitrosamines. J Nucleic Acids, 2010, 284935.
Peterson, L. A., Carmella, S. G., & Hecht, S. S. (1990). Investigations of metabolic precursors to hemoglobin and DNA adducts of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis, 11(8), 1329-1333.
Peterson, L. A., & Hecht, S. S. (1991). O6-methylguanine is a critical determinant of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone tumorigenesis in A/J mouse lung. Cancer Res, 51(20), 5557-5564.
Peterson, L. A., Thomson, N. M., Crankshaw, D. L., Donaldson, E. E., & Kenney, P. J. (2001). Interactions between methylating and pyridyloxobutylating agents in A/J mouse lungs: implications for 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis. Cancer Res, 61(15), 5757-5763.
Petri, N., Tannergren, C., Holst, B., Mellon, F. A., Bao, Y., Plumb, G. W. et al. (2003).

125
Absorption/metabolism of sulforaphane and quercetin, and regulation of phase II enzymes, in human jejunum in vivo. Drug Metab Dispos, 31(6), 805-813.
Philbrook, N. A., & Winn, L. M. (2014). Sub-chronic sulforaphane exposure in CD-1 pregnant mice enhances maternal NADPH quinone oxidoreductase 1 (NQO1) activity and mRNA expression of NQO1, glutathione S-transferase, and glutamate-cysteine ligase: potential implications for fetal protection against toxicant exposure. Reprod Toxicol, 43, 30-37.
Piberger, A. L., Köberle, B., & Hartwig, A. (2014). The broccoli-born isothiocyanate sulforaphane impairs nucleotide excision repair: XPA as one potential target. Arch Toxicol, 88(3), 647-658.
Pitot, H. C., & Dragan, Y. P. (1991). Facts and theories concerning the mechanisms of carcinogenesis. FASEB J, 5(9), 2280-2286.
Poulsen, S. L., Hansen, R. K., Wagner, S. A., van Cuijk, L., van Belle, G. J., Streicher, W. et al. (2013). RNF111/Arkadia is a SUMO-targeted ubiquitin ligase that facilitates the DNA damage response. J Cell Biol, 201(6), 797-807.
Protić, M., Roilides, E., Levine, A. S., & Dixon, K. (1988). Enhancement of DNA repair capacity of mammalian cells by carcinogen treatment. Somat Cell Mol Genet, 14(4), 351-357.
Rademakers, S., Volker, M., Hoogstraten, D., Nigg, A. L., Mone, M. J., van Zeeland, A. A. et al. (2003). Xeroderma Pigmentosum Group A Protein Loads as a Separate Factor onto DNA Lesions. Molecular and Cellular Biology, 23(16), 5755-5767.
Rajendran, P., Dashwood, W. M., Li, L., Kang, Y., Kim, E., Johnson, G. et al. (2015). Nrf2 status affects tumor growth, HDAC3 gene promoter associations, and the response to sulforaphane in the colon. Clin Epigenetics, 7(1), 102.
Rajendran, P., Kidane, A. I., Yu, T. W., Dashwood, W. M., Bisson, W. H., Löhr, C. V. et al. (2013). HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics, 8(6), 612-623.
Rajendrasozhan, S., Yang, S. R., Kinnula, V. L., & Rahman, I. (2008). SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med, 177(8), 861-870.
Ramos-Gomez, M., Dolan, P. M., Itoh, K., Yamamoto, M., & Kensler, T. W. (2003). Interactive effects of nrf2 genotype and oltipraz on benzo[a]pyrene-DNA adducts and tumor yield in mice. Carcinogenesis, 24(3), 461-467.
Ramos-Gomez, M., Kwak, M. K., Dolan, P. M., Itoh, K., Yamamoto, M., Talalay, P. et al. (2001). Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A, 98(6), 3410-3415.
Ray, A., Milum, K., Battu, A., Wani, G., & Wani, A. A. (2013). NER initiation factors, DDB2 and XPC, regulate UV radiation response by recruiting ATR and ATM kinases to DNA damage sites. DNA Repair (Amst), 12(4), 273-283.
Rechkunova, N. I., Krasikova, Y. S., & Lavrik, O. I. (2011). Nucleotide excision repair:

126
DNA damage recognition and preincision complex assembly. Biochemistry (Moscow), 76(1), 24-35.
Reed, E. (1998). Nucleotide excision repair and anti-cancer chemotherapy. Cytotechnology, 27(1-3), 187-201.
Riedl, T., Hanaoka, F., & Egly, J. M. (2003). The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J, 22(19), 5293-5303.
Rodin, S. N., & Rodin, A. S. (2000). Human lung cancer and p53: the interplay between mutagenesis and selection. Proc Natl Acad Sci U S A, 97(22), 12244-12249.
Rossner, P., Mrhalkova, A., Uhlirova, K., Spatova, M., Rossnerova, A., Libalova, H. et al. (2013). Nucleotide excision repair is not induced in human embryonic lung fibroblasts treated with environmental pollutants. PLoS One, 8(7), e69197.
Royston, K. J., & Tollefsbol, T. O. (2015). The Epigenetic Impact of Cruciferous Vegetables on Cancer Prevention. Curr Pharmacol Rep, 1(1), 46-51.
Rubbi, C. P., & Milner, J. (2003). Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. The EMBO journal, 22, 6068-6077.
Rüthemann, P., Balbo Pogliano, C., & Naegeli, H. (2016). Global-genome Nucleotide Excision Repair Controlled by Ubiquitin/Sumo Modifiers. Front Genet, 7, 68.
Saijo, M., Matsuda, T., Kuraoka, I., & Tanaka, K. (2004). Inhibition of nucleotide excision repair by anti-XPA monoclonal antibodies which interfere with binding to RPA, ERCC1, and TFIIH. Biochem Biophys Res Commun, 321(4), 815-822.
Salles, B., Frit, P., Provot, C., Jaeg, J.-P., & Calsou, P. (1995a). In vitro eukaryotic DNA excision repair assays: an overview. Biochimie, 77(10), 796-802.
Salles, B., Provot, C., Calsou, P., Hennebelle, I., Gosset, I., & Fourni?, G. J. (1995b). A chemiluminescent microplate assay to detect DNA damage induced by genotoxic treatments. Analytical biochemistry, 232(1), 37-42.
Santos, N. C., Figueira-Coelho, J., Martins-Silva, J., & Saldanha, C. (2003). Multidisciplinary utilization of dimethyl sulfoxide: pharmacological, cellular, and molecular aspects. Biochem Pharmacol, 65(7), 1035-1041.
Sarker, A. H., Tsutakawa, S. E., Kostek, S., Ng, C., Shin, D. S., Peris, M. et al. (2005). Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: insights for transcription-coupled repair and Cockayne Syndrome. Mol Cell, 20(2), 187-198.
Satoh, H., Moriguchi, T., Takai, J., Ebina, M., & Yamamoto, M. (2013). Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res, 73(13), 4158-4168.
Saw, C. L., Yang, A. Y., Huang, M. T., Liu, Y., Lee, J. H., Khor, T. O. et al. (2014). Nrf2 null enhances UVB-induced skin inflammation and extracellular matrix damages. Cell Biosci, 4, 39.
Seo, Y. R., & Jung, H. J. (2004). The potential roles of p53 tumor suppressor in nucleotide excision repair (NER) and base excision repair (BER). Exp Mol Med, 36(6), 505-509.
Seo, Y. R., Sweeney, C., & Smith, M. L. (2002). Selenomethionine induction of DNA

127
repair response in human fibroblasts. Oncogene, 21(23), 3663-3669. Sestili, P., & Fimognari, C. (2015). Cytotoxic and Antitumor Activity of Sulforaphane:
The Role of Reactive Oxygen Species. Biomed Res Int, 2015, 402386. Shapiro, T. A., Fahey, J. W., Dinkova-Kostova, A. T., Holtzclaw, W. D., Stephenson, K.
K., Wade, K. L. et al. (2006). Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isothiocyanates: a clinical phase I study. Nutr Cancer, 55(1), 53-62.
Shapiro, T. A., Fahey, J. W., Wade, K. L., Stephenson, K. K., & Talalay, P. (1998). Human metabolism and excretion of cancer chemoprotective glucosinolates and isothiocyanates of cruciferous vegetables. Cancer Epidemiol Biomarkers Prev, 7(12), 1091-1100.
Shen, G., Hebbar, V., Nair, S., Xu, C., Li, W., Lin, W. et al. (2004). Regulation of Nrf2 transactivation domain activity. The differential effects of mitogen-activated protein kinase cascades and synergistic stimulatory effect of Raf and CREB-binding protein. J Biol Chem, 279(22), 23052-23060.
Shin, J. H., Reeve, J. N., & Kelman, Z. (2005). Use of a restriction enzyme-digested PCR product as substrate for helicase assays. Nucleic Acids Res, 33(1), e8.
Shivji, M. K., Podust, V. N., Hübscher, U., & Wood, R. D. (1995). Nucleotide excision repair DNA synthesis by DNA polymerase epsilon in the presence of PCNA, RFC, and RPA. Biochemistry, 34(15), 5011-5017.
Silver, H. R., Nissley, J. A., Reed, S. H., Hou, Y. M., & Johnson, E. S. (2011). A role for SUMO in nucleotide excision repair. DNA Repair (Amst), 10(12), 1243-1251.
Singh, P., Sharma, R., McElhanon, K., Allen, C. D., Megyesi, J. K., Beneš, H. et al. (2015). Sulforaphane protects the heart from doxorubicin-induced toxicity. Free Radic Biol Med, 86, 90-101.
Singletary, K., & MacDonald, C. (2000). Inhibition of benzo[a]pyrene- and 1,6-dinitropyrene-DNA adduct formation in human mammary epithelial cells bydibenzoylmethane and sulforaphane. Cancer Lett, 155(1), 47-54.
Sliwinski, T., Czechowska, A., Szemraj, J., Morawiec, Z., Skorski, T., & Blasiak, J. (2008). STI571 reduces NER activity in BCR/ABL-expressing cells. Mutat Res, 654(2), 162-167.
Stanfill, S. B., Connolly, G. N., Zhang, L., Jia, L. T., Henningfield, J. E., Richter, P. et al. (2011). Global surveillance of oral tobacco products: total nicotine, unionised nicotine and tobacco-specific N-nitrosamines. Tob Control, 20(3), e2.
Stepanov, I., Sebero, E., Wang, R., Gao, Y. T., Hecht, S. S., & Yuan, J. M. (2014). Tobacco-specific N-nitrosamine exposures and cancer risk in the Shanghai Cohort Study: remarkable coherence with rat tumor sites. Int J Cancer, 134(10), 2278-2283.
Su, Y., Orelli, B., Madireddy, A., Niedernhofer, L. J., & Schärer, O. D. (2012). Multiple DNA binding domains mediate the function of the ERCC1-XPF protein in nucleotide excision repair. J Biol Chem, 287(26), 21846-21855.
Sueblinvong, V., Tseng, V., Smith, T., Saghafi, R., Mills, S. T., Neujahr, D. C. et al. (2014). TGFβ1 mediates alcohol-induced Nrf2 suppression in lung fibroblasts.

128
Alcohol Clin Exp Res, 38(11), 2731-2742. Sugasawa, K., Ng, J. M., Masutani, C., Iwai, S., van der Spek, P. J., Eker, A. P. et al.
(1998). Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell, 2, 223-232.
Sugasawa, K., Okamoto, T., Shimizu, Y., Masutani, C., Iwai, S., & Hanaoka, F. (2001). A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev, 15(5), 507-521.
Sugasawa, K., Okuda, Y., Saijo, M., Nishi, R., Matsuda, N., Chu, G. et al. (2005). UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell, 121(3), 387-400.
Swenberg, J. A., Lu, K., Moeller, B. C., Gao, L., Upton, P. B., Nakamura, J. et al. (2011). Endogenous versus exogenous DNA adducts: their role in carcinogenesis, epidemiology, and risk assessment. Toxicol Sci, 120 Suppl 1, S130-45.
Techapiesancharoenkij, N., Fiala, J. L., Navasumrit, P., Croy, R. G., Wogan, G. N., Groopman, J. D. et al. (2015). Sulforaphane, a cancer chemopreventive agent, induces pathways associated with membrane biosynthesis in response to tissue damage by aflatoxin B1. Toxicol Appl Pharmacol, 282(1), 52-60.
Tomicic, M. T., Reischmann, P., Rasenberger, B., Meise, R., Kaina, B., & Christmann, M. (2011). Delayed c-Fos activation in human cells triggers XPF induction and an adaptive response to UVC-induced DNA damage and cytotoxicity. Cell Mol Life Sci, 68(10), 1785-1798.
Topè, A. M., & Rogers, P. F. (2009). Evaluation of protective effects of sulforaphane on DNA damage caused by exposure to low levels of pesticide mixture using comet assay. J Environ Sci Health B, 44(7), 657-662.
Tortorella, S. M., Royce, S. G., Licciardi, P. V., & Karagiannis, T. C. (2015). Dietary Sulforaphane in Cancer Chemoprevention: The Role of Epigenetic Regulation and HDAC Inhibition. Antioxid Redox Signal, 22(16), 1382-1424.
Trego, K. S., & Turchi, J. J. (2006). Pre-steady-state binding of damaged DNA by XPC-hHR23B reveals a kinetic mechanism for damage discrimination. Biochemistry, 45(6), 1961-1969.
Urban, A. M., Upadhyaya, P., Cao, Q., & Peterson, L. A. (2012). Formation and repair of pyridyloxobutyl DNA adducts and their relationship to tumor yield in A/J mice. Chem Res Toxicol, 25(10), 2167-2178.
van Cuijk, L., van Belle, G. J., Turkyilmaz, Y., Poulsen, S. L., Janssens, R. C., Theil, A. F. et al. (2015). SUMO and ubiquitin-dependent XPC exchange drives nucleotide excision repair. Nat Commun, 6, 7499.
Vélez-Cruz, R., & Johnson, D. G. (2012). E2F1 and p53 transcription factors as accessory factors for nucleotide excision repair. Int J Mol Sci, 13(10), 13554-13568.
Verkerk, R., Schreiner, M., Krumbein, A., Ciska, E., Holst, B., Rowland, I. et al. (2009). Glucosinolates in Brassica vegetables: the influence of the food supply chain on intake, bioavailability and human health. Mol Nutr Food Res, 53 Suppl 2, S219.
Wakabayashi, N., Dinkova-Kostova, A. T., Holtzclaw, W. D., Kang, M. I., Kobayashi, A., Yamamoto, M. et al. (2004). Protection against electrophile and oxidant stress

129
by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci U S A, 101(7), 2040-2045.
Wang, C. C., Lin, S. Y., Lai, Y. H., Liu, Y. J., Hsu, Y. L., & Chen, J. J. (2012). Dimethyl sulfoxide promotes the multiple functions of the tumor suppressor HLJ1 through activator protein-1 activation in NSCLC cells. PLoS One, 7(4), e33772.
Wang, L., Spratt, T. E., Liu, X. K., Hecht, S. S., Pegg, A. E., & Peterson, L. A. (1997). Pyridyloxobutyl adduct O6-[4-oxo-4-(3-pyridyl)butyl]guanine is present in 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone-treated DNA and is a substrate for O6-alkylguanine-DNA alkyltransferase. Chem Res Toxicol, 10(5), 562-567.
Wang, Q. (2003). Tumor suppressor p53 dependent recruitment of nucleotide excision repair factors XPC and TFIIH to DNA damage. DNA Repair, 2(5), 483-499.
Wang, Q. E., Zhu, Q., Wani, G., El-Mahdy, M. A., Li, J., & Wani, A. A. (2005). DNA repair factor XPC is modified by SUMO-1 and ubiquitin following UV irradiation. Nucleic Acids Res, 33(13), 4023-4034.
Wang, Y., Zhang, Z., Guo, W., Sun, W., Miao, X., Wu, H. et al. (2014a). Sulforaphane reduction of testicular apoptotic cell death in diabetic mice is associated with the upregulation of Nrf2 expression and function. Am J Physiol Endocrinol Metab, 307(1), E14-23.
Wang, Y., Zhang, Z., Sun, W., Tan, Y., Liu, Y., Zheng, Y. et al. (2014b). Sulforaphane attenuation of type 2 diabetes-induced aortic damage was associated with the upregulation of Nrf2 expression and function. Oxid Med Cell Longev, 2014, 123963.
Wang, Z., & Chen, W. (2013). Emerging Roles of SIRT1 in Cancer Drug Resistance. Genes Cancer, 4(3-4), 82-90.
Wani, M. A., El-Mahdy, M. A., Hamada, F. M., Wani, G., Zhu, Q., Wang, Q. E. et al. (2002). Efficient repair of bulky anti-BPDE DNA adducts from non-transcribed DNA strand requires functional p53 but not p21(waf1/cip1) and pRb. Mutat Res, 505(1-2), 13-25.
Warren, G. W., & Singh, A. K. (2013). Nicotine and lung cancer. J Carcinog, 12, 1. Wood, R. D., Biggerstaff, M., & Shivji, M. K. K. (1995). Detection and measurement of
nucleotide excision repair synthesis by mammalian cell extracts in vitro. Methods, 7(2), 163-175.
Wood, R. D., Robins, P., & Lindahl, T. (1988). Complementation of the xeroderma pigmentosum DNA repair defect in cell-free extracts. Cell, 53(1), 97-106.
World Health Organization. (2012). WHO Global Report: Mortality Attributable to Tobacco. Geneva, Switzerland: World Health Organization
Wu, H., Kong, L., Cheng, Y., Zhang, Z., Wang, Y., Luo, M. et al. (2015). Metallothionein plays a prominent role in the prevention of diabetic nephropathy by sulforaphane via up-regulation of Nrf2. Free Radic Biol Med, 89, 431-442.
Wu, X., Shell, S. M., Yang, Z., & Zou, Y. (2006). Phosphorylation of nucleotide excision repair factor xeroderma pigmentosum group A by ataxia telangiectasia mutated and Rad3-related-dependent checkpoint pathway promotes cell survival in response to

130
UV irradiation. Cancer Res, 66(6), 2997-3005. Yasuda, G., Nishi, R., Watanabe, E., Mori, T., Iwai, S., Orioli, D. et al. (2007). In vivo
destabilization and functional defects of the xeroderma pigmentosum C protein caused by a pathogenic missense mutation. Mol Cell Biol, 27(19), 6606-6614.
Ye, L., Dinkova-Kostova, A. T., Wade, K. L., Zhang, Y., Shapiro, T. A., & Talalay, P. (2002). Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin Chim Acta, 316(1-2), 43-53.
Yokoi, M. (2000). The Xeroderma Pigmentosum Group C Protein Complex XPC-HR23B Plays an Important Role in the Recruitment of Transcription Factor IIH to Damaged DNA. Journal of Biological Chemistry, 275(13), 9870-9875.
Yuan, J. M., Butler, L. M., Stepanov, I., & Hecht, S. S. (2014). Urinary tobacco smoke-constituent biomarkers for assessing risk of lung cancer. Cancer Res, 74(2), 401-411.
Yuan, J. M., Gao, Y. T., Murphy, S. E., Carmella, S. G., Wang, R., Zhong, Y. et al. (2011). Urinary levels of cigarette smoke constituent metabolites are prospectively associated with lung cancer development in smokers. Cancer Res, 71(21), 6749-6757.
Zanichelli, F., Capasso, S., Cipollaro, M., Pagnotta, E., Cartenì, M., Casale, F. et al. (2012a). Dose-dependent effects of R-sulforaphane isothiocyanate on the biology of human mesenchymal stem cells, at dietary amounts, it promotes cell proliferation and reduces senescence and apoptosis, while at anti-cancer drug doses, it has a cytotoxic effect. Age (Dordr), 34(2), 281-293.
Zanichelli, F., Capasso, S., Di Bernardo, G., Cipollaro, M., Pagnotta, E., Cartenì, M. et al. (2012b). Low concentrations of isothiocyanates protect mesenchymal stem cells from oxidative injuries, while high concentrations exacerbate DNA damage. Apoptosis, 17(9), 964-974.
Zheng, H. C., & Takano, Y. (2011). NNK-Induced Lung Tumors: A Review of Animal Model. J Oncol, 2011, 635379.
Zhou, X., D’Agostino, J., Xie, F., & Ding, X. (2012). Role of CYP2A5 in the bioactivation of the lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in mice. J Pharmacol Exp Ther, 341(1), 233-241.
Zhu, Q., Wani, M. A., El-Mahdy, M., & Wani, A. A. (2000). Decreased DNA repair efficiency by loss or disruption of p53 function preferentially affects removal of cyclobutane pyrimidine dimers from non-transcribed strand and slow repair sites in transcribed strand. J Biol Chem, 275(15), 11492-11497.
Ziani, S., Nagy, Z., Alekseev, S., Soutoglou, E., Egly, J. M., & Coin, F. (2014). Sequential and ordered assembly of a large DNA repair complex on undamaged chromatin. J Cell Biol, 206(5), 589-598.

131
Appendix A
The effect of DMSO on nucleotide excision repair activity in A/J mouse
lung and liver
Introduction
Dimethyl sulfoxide (DMSO) is an organic solvent with a rich pharmacological history
(Santos et al., 2003). Initially known in the 19th century as a byproduct of the wood industry, it is
frequently used for clinical and laboratory purposes (Santos et al., 2003). In the laboratory it is a
utility solvent, as it can dissolve both polar and nonpolar compounds, whereas DMSO received
approval from the US Food and Drug Administration for treatment of interstitial cystits in 1978
(Parkin et al., 1997). It has been used most successfully in treatment of leukemia, and proven to
have multiple anticancer properties in cells and in vivo (Camici et al., 2006; Jia et al., 2010; Pal et
al., 2012). In invasive lung adenocarcinoma cells, DMSO stimulated the tumour suppressor gene
HLJ1, thereby inhibiting cell invasion, migration and proliferation (Wang et al., 2012). DMSO
appears to cause retinal cell death at low doses in vivo, inducing apoptosis by inhibiting
mitochondrial respiration (Galvao et al., 2014). It is also known to be mutagenic in two Ames
test strains (Hakura et al., 1993).
Damage caused by endogenous reactive metabolites and exogenous factors can cause
multiple kinds of damage to a cell. Several methods of clearing and repairing DNA damage exist,
depending on the structure or type of damage (Branzei & Foiani, 2008). Repair of bulky adducts,
most commonly associated with damage caused by exposure to exogenous chemicals, is
performed by nucleotide excision repair (Branzei & Foiani, 2008). No less than 30 proteins are
involved in the multi-step process of lesion excision and repair in nucleotide excision repair

132
(Gillet & Scharer, 2006; Wood et al., 1988), and their importance can be understood by the lack
of viability of cells lacking active proteins (Nouspikel, 2009; Yasuda et al., 2007).
In the laboratory, many compounds important in DNA repair studies are dissolved
exclusively in DMSO. Studies on AFB1 are of specific interest in DNA repair studies, as
previously described a tissue-specific alteration of nucleotide excision repair (NER) activity after
intraperitoneal dosage of AFB1 (Bedard et al., 2005). Briefly, after treating mice with AFB1,
Bedard et al. found treated liver tissue extracts had increased nucleotide excision repair activity,
whereas treated lung tissue extracts had decreased nucleotide excision repair activity (2005).
Here, we were interested in comparing two commonly used solvents, sterile saline and DMSO.
In this experiment, we observed a decrease of DMSO treated liver NER activity, and no change in
lung NER activity.
Materials and methods
Animal treatments
Female A/J mice (JAX Laboratories), aged 7-10 weeks, were housed in a 12 h light/dark
cycle and provided with food and water ad libitum. Mice were treated with saline (0.1mL, i.p.) or
DMSO (0.04 mL, i.p.). Two hours following this administration, mice were killed by cervical
dislocation. Lungs and livers were perfused with 10mM Tris/1mM EDTA (pH 7.9), excised,
finely chopped, frozen in liquid nitrogen, and stored at -80ºC until extract preparation. All
animals were cared for in accordance with the guidelines of the Canadian Council on Animal
Care. Experimental procedures involving animals and animal care were approved by the Queen’s
University Animal Care Committee.
Preparation of cell-free whole tissue nuclear protein extract

133
Nuclear protein extracts were prepared from pooled lungs or pooled livers of four mice as described in section 2.2.2.
Preparation of damaged plasmid DNA
Pyridyloxobutylated plasmid DNA was prepared as described in section 2.2.3.
In vitro DNA repair synthesis assay
The repair synthesis assay was performed as described in section 2.2.4.
Results
Effect of in vivo treatment of mice with DMSO on DNA repair synthesis of mouse liver and
lung nuclear protein extracts
Following in vivo DMSO treatment after 2 h, repair synthesis activity of mouse lung
extracts toward POB DNA damage showed no significant change relative to control (Figure A.1).
However, DNA repair activity decreased by 39% at 2 h post in vivo dosing in mouse liver (Figure
A.1).

134
Figure A.1 Effects of in vivo DMSO treatment on in vitro DNA repair activity of mouse lung
or liver extracts towards NNKOAc-induced pyridyloxobutyl DNA damage. Extracts were
prepared from mice treated with either 0.1mL sterile saline or 0.04mL DMSO and killed 2
h post-dosing. Data are presented as mean + SD. (*) Significantly different from control
(n=4 pools of liver or lungs of 4 mice per pool, p<0.05).
Lung Liver0
5000
10000
15000am
ol [α
32P
] dA
TP in
corp
orat
ed/µ
g D
NA
SalineDMSO
*

135
Conclusions
DMSO is an important laboratory solvent, and has found use in several pharmacological
actions. In this study, we show DMSO has a significant negative effect on NER activity in
treated liver after 2 hours, but not in treated lungs at the same timepoint. We have previously
shown the effects of exogenous chemicals on NER activity can at least partially be explained by
effects on specific NER proteins (Chapter 2 and Chapter 3). Considering how prevalent the use
of DMSO is in the laboratory, investigations into the molecular causes of this significant decrease
in NER activity are warranted.




















